

Abstract
The mechanistic target of rapamycin (mTOR) pathway has been implicated in a growing number of malformations of cortical development (MCD) associated with intractable epilepsy. Mutations in single genes encoding mTOR pathway regulatory proteins have been linked to MCD such as focal cortical dysplasia (FCD) types IIa and IIb, hemimegalencephaly (HME), and megalencephaly. Recent studies have demonstrated that the GATOR1 protein complex, comprised of DEPDC5, NPRL3, and NPRL2, plays a pivotal role in regulating mTOR signaling in response to cellular amino acid levels and that mutations in DEPDC5, NPRL3, or NPRL2 are linked to FCD, HME, and seizures. Histopathological analysis of FCD and HME tissue specimens resected from individuals harboring DEPDC5, NPRL3, or NPRL2 gene mutations reveals hyperactivation of mTOR pathway signaling. Family pedigrees carrying mutations in either DEPDC5 or NPRL3 share clinical phenotypes of epilepsy and MCD, as well as intellectual and neuropsychiatric disabilities. Interestingly, some individuals with seizures associated with DEPDC5, NPRL3, or NPRL2 variants exhibit normal brain imaging suggesting either occult MCD or a role for these genes in non-lesional neocortical epilepsy. Mouse models resulting from knockdown or knockout of either Depdc5 or Nprl3 exhibit altered cortical lamination, neuronal dysmorphogenesis, and enhanced neuronal excitability as reported in models resulting from direct mTOR activation through expression of its canonical activator RHEB. The role of the GATOR1 proteins in regulating mTOR signaling suggest plausible options for mTOR inhibition in the treatment of epilepsy associated with mutations in DEPDC5, NPRL3, or NPRL2.
Keywords: DEPDC5, dysplasia, epilepsy, mTOR, NPRL3, Rheb, seizure
1 |. INTRODUCTION
Progress in understanding how gene mutations produce malformations of cortical development (MCDs) and epilepsy is occurring at a rapid pace. Mutations in mechanistic target of rapamycin (mTOR) pathway regulatory genes, for example, phosphotidylinositol-3 kinase (PI3K), protein kinase B (AKT3), STE20-related kinase adapter protein alpha (STRADA), Ras homolog enriched in brain (RHEB), and MTOR, have emerged as playing key roles in producing MCDs that are highly associated with intractable epilepsy. Greater understanding of the mTOR signaling cascade has allowed progress beyond simple genotype-phenotype correlations to determine the consequences of mutations on cell and network function and to define new strategies for precision treatment modalities.
Malformations of cortical development linked to mutations in genes encoding regulators of mTOR cascade activity have been termed “mTORopathies.”1 The paradigm disorder to understand mTORopathies is tuberous sclerosis complex (TSC), in which inherited or sporadic loss-of-function mutations in the mTOR regulators TSC1 or TSC2 result in constitutive hyperactivation of mTOR kinase activity in the fetal brain. As a consequence, focal cortical dysplasias (FCDs; tubers) form during fetal brain development that cause significant network disruption, and are highly associated with both infantile spasms and intractable epilepsy.2 Over the past few years, the phenotypic spectrum of MCDs linked to mTOR cascade mutations has expanded to include a range of MCD subtypes spanning FCD types IIa and IIb to widespread hemispheric and whole brain abnormalities, for example, hemimegalencephaly (HME) and megalencephaly (ME), respectively.3–6 The molecular and cellular mechanisms responsible for these phenotypic differences remain to be fully defined.
The central tenet for conceptualizing mTORopathies is that mutations in distinct genes within the mTOR cascade culminate in common phenotypic features including mTOR signaling pathway activation, abnormal neuronal morphology, aberrant cortical laminar structure, neuronal hyperexcitability, and clinically, seizures.7–9 Indeed, the net cellular effects of mTOR pathway mutations seem to funnel through mTOR complex 1 (mTOR and its binding partner raptor; mTORC1) as a common signaling node, since in vitro and in vivo data demonstrate reversibility of both structural and functional (eg, hyperexcitability) effects of these mutations by pharmacologic mTOR inhibition with rapamycin or related compounds known as rapalogs.8,10
Recently, germline and somatic loss-of-function mutations in DEP domain containing 5 (DEPDC5) and Nitrogen Permease Regulator - Like 3 (NPRL3) genes11 that code for protein components of the GTPase-activating protein (GAP) activity toward Rags 1 (GATOR1) protein complex have been identified in focal epilepsies associated with MCDs4,6,12,13 (Figure 1). Subsequent studies have identified mutations in another GATOR1 component, the Nitrogen Permease Regulator Like - 2 protein (NPRL2,) in focal onset epilepsy, variably associated with MCDs.14–16 DEPDC5, NPRL3, and NPRL2 have been identified as putative tumor suppressor genes that are individually deleted in multiple cancers and cancer cell lines. Whereas TSC1 and TSC2 proteins regulate mTORC1 in response to growth factor signaling, the GATOR1 complex regulates mTORC1 signaling in response to fluctuating cellular amino acid (AA) levels through a complex and elegant protein signaling pathway (see below Figure 2). GATOR1 components function as GAPs that inhibit mTORC1 activity by inactivating the Rag GTPases. When AA levels are low, GATOR1 signals to inhibit mTORC1; when AA levels are adequate, GATOR1 releases mTORC1 inhibition so that mTORC1 can signal in response to growth factors and promote cellular growth.
FIGURE 1.
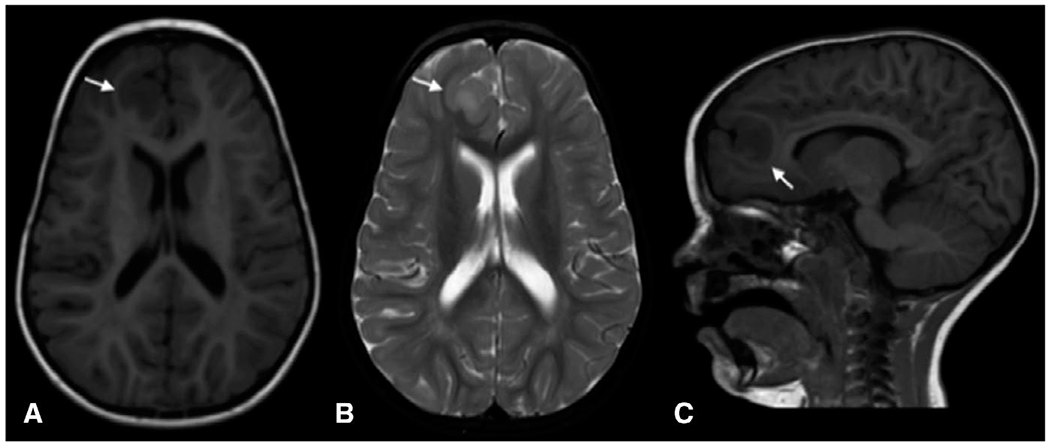
Magnetic resonance imaging (MRI) showing focal cortical dysplasia (arrows) in a 5-year-old child with NPRL3 (c.349delG, p.Glu117LysFS) mutation. A, Axial T1-weighted image; B, axial T2-weighted image; C, Sagittal T1-weighted image
FIGURE 2.
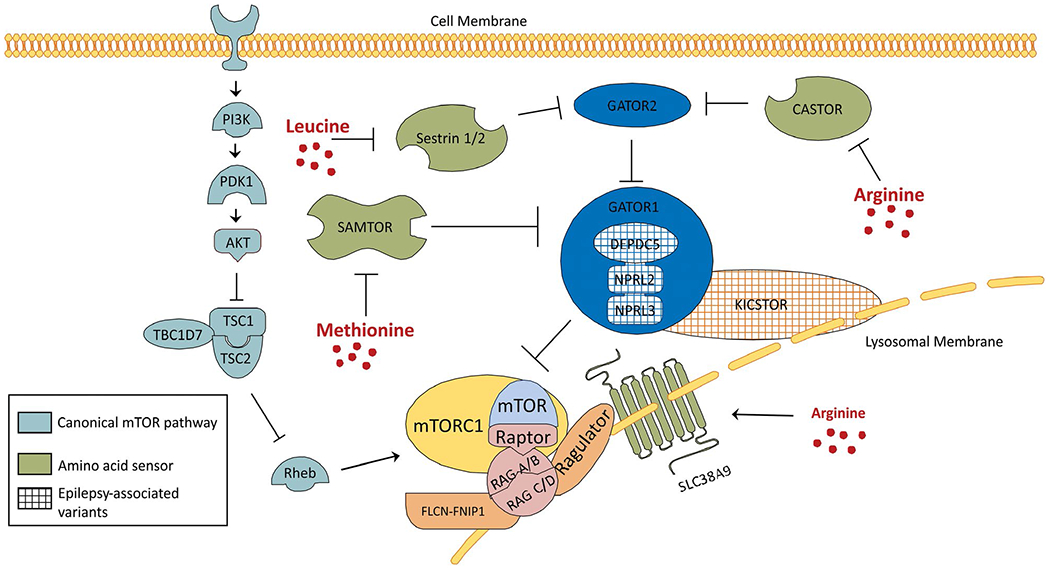
Schematic of the amino acid modulator arm of the mammalian target of rapamycin (mTOR) pathway. The canonical mTOR pathway (gray) and its relationship to the amino acid modulatory arm of the mTOR pathway is depicted. Several amino acid sensors (green) have been defined. The amino acid sensors SAMTOR, Sestrin 1/2, and CASTOR impact mTORC1 signaling by directly or indirectly disinhibiting the GATOR1 complex (blue). SLC38A9 functions as both an amino acid sensor and amino acid transporter coupled to the Ragulator-RAG GTPase complex. Within the amino acid modulatory arm, multiple proteins have been linked to cortical malformations and epilepsy including known variants in the components of GATOR1: DEPDC5, NPRL3, NPRL2 and multiple components of the KICSTOR complex: KPTN, SZT2, and C12 orf66
GTPase-activating proteins activity toward Rags 1 complex mutations are a frequent cause of focal epilepsies and may suggest a unique subset of mTORopathies functionally subclassified as “GATORopathies.” For example, NPRL3 variants have been linked to familial and sporadic FCD type IIa, characterized by cytomegalic dysmorphic neurons and severe dyslamination. DEPDC5 variants have been identified in FCD type IIa and HME as well as a number of different epilepsies including sleep-related focal hypermotor epilepsy, familial focal epilepsy with variable foci, and interestingly, nonlesional neocortical epilepsy.12,14–18 DEPDC5 and NPRL3 gene variants have been linked to seizures, intellectual disability, language delay, anxiety, attention-deficit disorder, and autism spectrum disorder. The exact mechanisms through which GATOR1 complex loss-of-function mutations lead to altered structural assembly of the cerebral cortex and seizures remain to be defined, although a number of preclinical studies using cell culture and mouse models have begun to shed light on these processes.
We recently presented an Investigator’s Workshop (IW) at the 2018 AES meeting in New Orleans, LA, addressing the collective associations between GATOR1 complex gene mutations and human epilepsy syndromes (“GATORopathies”). The genetics of GATOR1 component mutations and individual epilepsy syndromes, the cell biology of the GATOR1 proteins, and mouse models of mTORopathies to investigate the mechanisms of epileptogenesis caused by GATOR1 variants were discussed. This review summarizes the presentations at the IW and proposes new directions for research in epilepsy associated with DEPDC5, NPRL3, and NPRL2.
2 |. GENOTYPE AND PHENOTYPE IN GATOROPATHIES
The initial association between DEPDC5 and epilepsy was reported in several large pedigrees with familial focal epilepsy with variable foci (FFEVF17). In these autosomal dominant pedigrees, there was association of the DEPDC5 variants with distinct epilepsy subtypes, that is, sleep-related focal hypermotor epilepsy and temporal lobe epilepsy, as well as neuropsychiatric abnormalities. Imaging data were not included for these pedigrees.19 A subsequent report identified DEPDC5 variants in association with bottom-of-the-sulcus dysplasia or band heterotopia.18 Because DEPDC5 is a member of the GATOR1 complex, it was not surprising when mutations in the gene encoding the DEPDC5-binding protein NPRL3 were identified in association with focal onset epilepsy and FCDIIa.14–16 Histopathologic analysis of brain tissue resected as part of epilepsy surgery in the DEPDC5 and NPRL3 variant-associated cases revealed hyperactivation of mTOR signaling evidenced by hyperphosphorylation of ribosomal S6 protein, a downstream substrate of mTORC1.12,13 Mutations in NPRL2 have been linked to focal epilepsy with a less clear association with FCD and mTOR pathway activation.14–16
It has been estimated that germline variants in the GATOR1 complex genes (DEPDC5, NPRL3, and NPRL2) are present in approximately 10% of all focal epilepsy cases.20 There have been over 180 unrelated families described to date with GATOR1 complex mutations, making the GATOR1 complex among the most frequently mutated genes in focal epilepsies. A recent comprehensive study highlighted the wide range of phenotypic variability seen in the GATORopathies, analyzing the complete clinical spectrum of known GATOR1 variants.15 In this cohort of 73 unrelated probands, 63 distinct GATOR1 variants were identified, for example, 53 in DEPDC5, 7 in NPRL3, and 3 in NPRL2. Most individuals had focal epilepsy with variable onset foci, that is, frontal, temporal, frontotemporal, occipital, parietal, centrotemporal epilepsies (49%) or sleep-related hypermotor seizures (36%), but some of the probands presented with infantile spasms (10%) or generalized epilepsy (4%). Seizures started in childhood (mean age 4.4 years, range 0-16 years), with onset in the first year of life in 30% of cases. Brain imaging was normal in a majority of probands and MCD was found in only 25% of cases.15 In this cohort, 54% of probands had drug-resistant epilepsy, and 15% underwent resective surgery, with an Engel Class I outcome in 60% of patients. In another report, successful surgical remission was achieved in patients with germline DEPDC5 variants and thus these data demonstrated that detection of germline GATOR1 variants is not a contraindication for epilepsy surgery.15,20,21 A major finding was that sudden unexpected death in epilepsy (SUDEP) occurred in 12% of cases, suggesting that variants in the GATOR1 genes are associated with increased risk of SUDEP.15 In one series, DEPDC5 variants were found in epilepsy with auditory features.22
One challenging mechanistic feature of GATORopathies is that most result from germline mutations identified in large multiplex pedigrees, and yet the radiologic findings demonstrate focal MCDs. This of course begs the interesting question of how a heterozygous germline mutation, affecting all cells in the brain and not causing mTOR hyperactivation, might cause a focal malformation within a restricted brain area associated with hyperactivated mTOR signaling? This problem had previously faced investigators working in TSC and was resolved by finding a somatic “second-hit” mutation in the unaffected gene restricted to cortical tubers.23,24 The somatic mutations plus the germline mutation yields a functionally homozygous condition, fully inactivating the gene and leading to loss-of-function of the encoded protein and mTOR hyperactivation. In FCD associated with germline DEPDC5 mutations, somatic “second-hit” mutations have recently been identified in DEPDC5 suggesting a mechanism like TSC.25 In contrast, an unresolved clinical challenge is that while TSC is a multisystem disorder affecting brain, eye, heart, kidney, skin, and lung, it appears that DEPDC5 or NPRL3 confer a neurologic phenotype, only with no systemic features.
DEPDC5 and NPRL3 variants are associated with a range of phenotypes including FCDIIa, FCDIIb, HME, and nonlesional neocortical epilepsy and, in part, the phenotypic variability in GATORopathies may reflect the genomic diversity within these genes. For example, to date, 140 different variants in the GATOR1 complex genes have been detected in 183 unrelated individuals with epilepsy; 85% DEPDC5, 11% NPRL3, 6% NPRL2 (see reference 17). Among these, 67% of the probands had loss-of-function variants, leading to haploinsufficiency and 27% are missense variants.15 A novel, predicted pathogenic variant in NPRL3 (c.349delG, p.Glu117LysFS) was recently linked to a Mennonite cohort from Ohio that was highly associated with an epilepsy phenotype.26 Ongoing studies are assessing the genotype-phenotype correlations in two large pedigrees in the Mennonite community from Pennsylvania and Ohio of epilepsy resulting from single Founder variants in NPRL3 or DEPDC5. Amazingly, despite the fact that all affected individuals in these pedigrees share the same Founder mutations, there is high clinical variability across epilepsy, intellectual disability, and neuropsychiatric phenotypes, suggesting that intragenic differences within the GATOR1 genes are not likely to be the cause of the wide phenotypic spectrum seen in the GATORopathies (V. Carson, personal communication). Possible explanations include effects of somatic “second-hit” mutations, modifier genes for each phenotype, or contributions from the exposome, that either increase or decrease the likelihood of developing seizures or MCDs within individuals affected by either DEPDC5 or NPRL3 variant.
3 |. CELL BIOLOGY OF THE GATOR1 COMPLEX
The GATOR1 complex is the key regulatory complex within the amino acid modulatory arm of the mTOR cascade composed of DEPDC5, NPRL2, and NPRL3, which regulate mTOR activation in response to ambient cellular amino acid levels11 in concert with several other protein complexes (Figure 2). This portion of the mTOR cascade serves to either permit or inhibit mTORC1 kinase activity when amino acids are replete or reduced within the cell, respectively. GATOR1 alters the nucleotide-loaded state (GTP or GDP) of the Rag proteins on the lysosomal membrane to prevent the mTORC1 complex from localizing to the lysosomal membrane and carrying out its kinase activity in the absence of amino acids. The Rag proteins are heterodimers (Rag A/B and Rag C/D) tethered to the lysosome by the protein complex Regulator that acts as a dock for mTORC1.27 The GATOR1 complex has two effector modes: an inhibitory mode and a “GAP” mode. In the “GAP” mode, the heterodimer of NPRL2-NPRL3 exerts GTPase activity on the Rag A protein.28 In the inhibitory mode, DEPDC5 interacts directly with Rag A and prevents the GTPase activity. While GATOR1 acts on Rags A/B, a separate protein complex, folliculin/folliculin-interacting protein 1 (FLCN-FNIP1), controls the nucleotide binding state of Rags C/D. In this paradigm, when amino acid levels are low, GATOR1 acts on Rags A/B to inactivate it (GDP bound) and FLCN-FNIP1 sits on the lysosomal surface ready to activate Rag C/D. When amino acids are replete, FLCN-FNIP1 will hydrolyze the GTP on Rags C/D thereby activating it and permitting mTORC1 to return to the lysosomal membrane.29,30
To inhibit mTORC1, GATOR1 must be recruited to the lysosomal surface. This is achieved by the four-protein complex KICSTOR composed of Kaptin (KPTN), Seizure Threshold 2 Protein Homolog (SZT2), Chromosome 12 open reading frame 66 (C12orf66), and Integrin Alpha FG-GAP Repeat Containing 2 (ITFG2). Indeed, cells lacking components of the KICSTOR complex are insensitive to amino acid starvation, and GATOR1 and mTORC1 stay localized to the lysosomal surface.31 It remains unknown how KICSTOR recruits and interacts with GATOR1 to chaperone it to the lysosomal surface.
GATOR1 receives signaling cues from amino acid sensors located in both the lysosome and cytoplasm that either inhibit or disinhibit GATOR1 to modulate mTORC1 activity. There are two known mTOR pathway–associated amino acid sensors that detect intracellular arginine, an essential amino acid important for proper cell division. Cytosolic levels of arginine are detected by the protein Cellular Arginine Sensor for mTORC1 (CASTOR1). CASTOR1 is a homodimeric protein complex that binds to GAP Activity Towards Rags 2 (GATOR2; a poorly defined pentameric protein complex) when cytosolic arginine levels are low and allosterically inhibits it.32–34 GATOR2 inhibition results in the release of GATOR1 and mTORC1 signaling is prevented. In arginine replete conditions, binding of arginine to CASTOR1 prevents its association with GATOR2 and thus GATOR1 is inhibited and mTORC1 can localize to the lysosome.32–34
SLC38A9 is an additional arginine sensor that serves to detect intra-lysosomal levels of arginine and aids in the transport of amino acids out of the lysosome.35,36 SLC38A9 modulates mTORC1 kinase activity in a GATOR1-independent fashion, modulating the GTP/GDP bound state of the Rag heterodimers.37 In contrast to GATOR1, which actively inhibits mTORC1 when amino acids are scarce, homeostatic levels of intra-lysosomal arginine actively stimulate mTORC1 via SLC38A9 interaction with the Rag proteins and actively inhibits mTORC1 when arginine is scarce through a series of transmembrane domains. Furthermore, SLC38A9 is an avid transporter of leucine and to a lesser degree transports isoleucine, tyrosine, valine, and phenylalanine out of the lysosome. However, SLC38A9 does not efficiently transport arginine out of the lysosome as evidenced by lack of change in arginine concentration within the lysosome in SLC38A9 null cells. In addition, intra-lysosomal arginine levels are important in facilitating the transport of other amino acids out of the lysosome. In both live cells and in purified lysosomes, arginine deprivation increased lysosomal leucine concentration and stimulation of these systems, and the addition of arginine resulted in a release of leucine from the lysosome. A similar but less potent arginine-dependent release of lysine was also observed.38
Signaling to GATOR1 in response to cytosolic changes in leucine (an essential amino acid important in protein biosynthesis) levels is accomplished through the proteins Sestrins 1 and 2 (Sestrins 1/2).39 Cytosolic leucine binds Sestrins ½, which inhibit its binding to GATOR2 and mTORC1 remains active.40 Sestrins 1/2 also bind amino acids similar to leucine including methionine, isoleucine, and valine, albeit with lesser affinity. Indeed, in instances where leucine is low, Sestrins 1/2 may bind methionine instead.40–42 Methionine, however, has its own amino acids sensor. The S-adenosylmethionine sensor upstream of mTORC1 (SAMTOR; formerly C7orf60) binds directly to, and inhibits, GATOR1 under methionine replete conditions. Indeed, both KICSTOR and GATOR1, but not GATOR2, are required to inhibit mTORC1 signaling during methionine starvation, indicating that SAMTOR acts independently of GATOR2 (unlike Sestrins 1/2 and CASTOR) to inhibit mTORC1.43
Most of the experiments defining the components of the amino acid modulatory arm of the mTOR pathway have been performed in non-neuronal cell lines. Recent studies in neurons, however, have shown how DEPDC5 and NPRL3 may modulate neural activity and epileptogenesis.25,44–46 A recent study demonstrated that knockdown of Depdc5 or Nprl3 caused mTOR-dependent increases in cell size and process outgrowth in Neuro2a cells (N2a cells; a neuroblastoma cell line) and in mouse neural progenitor cell.44 Furthermore, Depdc5 or Nprl3 knockdown altered disrupted colocalization of mTOR to the lysosome after incubation in amino acid-free media (Figure 3). These data demonstrated that GATOR1 regulates mTOR pathway activity base on amino acid availability in neuronal cells. Multiple gene variants within this cascade including NPRL2,14 STZ2,47–49 KPTN,50,51 and C12orf6652 have been associated with epilepsy and have not yet been fully functionally validated in neurons. Progress in our understanding of the neurologic function and pathophysiologic consequences of loss-of-function in these proteins will surely provide novel treatment targets for epilepsy and MCD.
FIGURE 3.
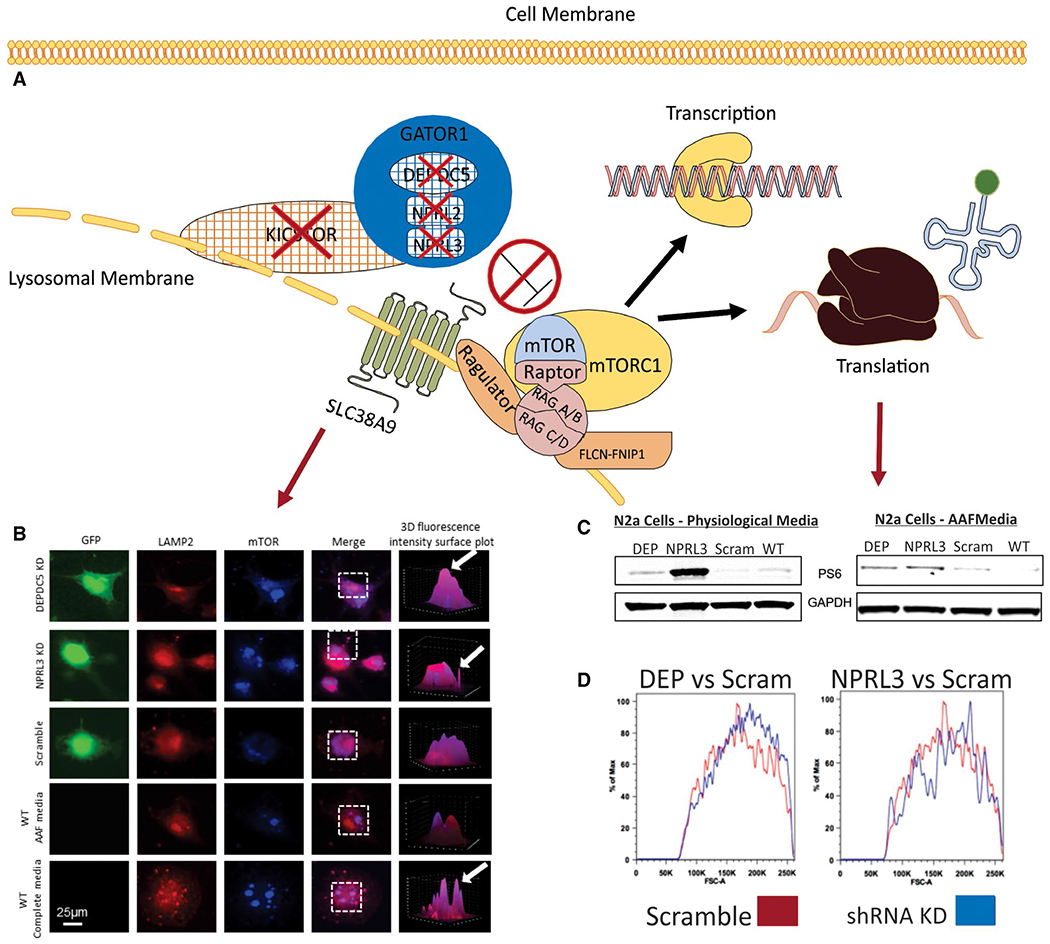
Functional and morphologic changes in neuronal cells after Depde5 or Nprl3 knockdown (modified from reference 44). A, Schematic representation of the consequences (red arrows) of loss-of-function in amino acid modulatory arm proteins (red x’s). Loss-of-function in amino acid regulatory arm protein results in a lack of inhibition of mTORC1 by the GATOR complex (dark blue) resulting in increased localization of mTOR to the lysosomal membrane (B) and thus increased mTOR pathway activation (C) under physiologic and amino acid deprivation conditions. Loss-of-function in these proteins also produces changes in cellular morphology (D). B, Using a fluorescence intensity colocalization assay and three-dimensional (3D) reconstruction of regions of interested (white dashed boxes) where regions of purple (arrows) indicated colocalization between the lysosome (LAMP2, red) and mTOR (light blue), we observed a higher degree of colocalization between the lysosome and mTOR after Depdc5 or Nprl3 KD and incubation in amino acid free media vs scramble and WT cells under the same conditions. WT N2a cells in complete media display similar degrees of colocalization as what was observed in Depdc5 or Nprl3 KD N2a cells in amino acid free media. C, After Depdc5 or Nprl3 knockdown in N2a cells, there is increased mTOR pathway signaling, as measured by ribosomal S6 phosphrylation (PS6), in both physiologic (DMEM, serum free) and amino acid free media compared to scramble control and WT N2a cells. Using FACS forward scatter analysis (D), Depdc5 or Nprl3 KD N2a cells were compared to scramble control transfected cells. Deflections to the right indicate large cells. Depdc5 or Nprl3 KD cells are larger than scramble control transfected cells (n = 100,000 per group). AAF, amino acid free; DEP, Depdc5; GFP, green fluorescent protein; LAMP2, lysosomal-associated membrane protein 2; N2a, Neuro 2a cells; scram, scramble control; WT, wild-type (see text for abbreviations).
4 |. EPILEPTOGENESIS AND MTOR-GATOR SIGNALING
The mechanisms by which loss-of-function mutations in DEPDC5, NPRL2, or NPRL3 cause hyperexcitability and seizures remain to be fully defined. Indeed, ongoing studies are focused on understanding how activation of mTOR kinase whether by a gain-of-function mutation in a pathway activator, that is, PI3KCA, AKT3, RHEB, or MTOR itself versus a loss-of-function mutation in a pathway inhibitor, that is, PTEN, TSC1, TSC2, and STRADA, culminates in MCD and network hyperexcitability. The translational classification of mTORopathies and “GATORopthies” serves to link mechanisms of mTOR activation with potential therapeutic and disease-modifying approaches using not only mTOR inhibitors, but also manipulation of alternative pathways and proteins that might dampen mTOR activation. A number of preclinical rodent model systems have been developed to study the effects of either overexpression of mTOR activators or knockdown (KD) or conditional knockout (KO) of mTOR inhibitors on cortical development, cell morphology, and network excitability (Figure 4). There are interesting commonalities to each model system, although the precise mechanisms culminating in seizures for each may be similar or distinct. Conditional KO of Pten, Tsc1, or Tsc2 in mice leads to dramatic alterations in brain architecture, enhanced neuronal size, and variably, spontaneous seizures.10 Similarly, overexpression of Akt3, Rheb, or Mtor containing activating mutations also causes cytomegaly, cytoarchitectural disorganization in the cortex, and seizures.8,53,54 Targeted KD of Strada causes FCD and cytomegaly in the mouse cortex55,56 but the Strada germline KO exhibits a perinatal lethal phenotype,57 so a seizure phenotype has not been yet reported.
FIGURE 4.
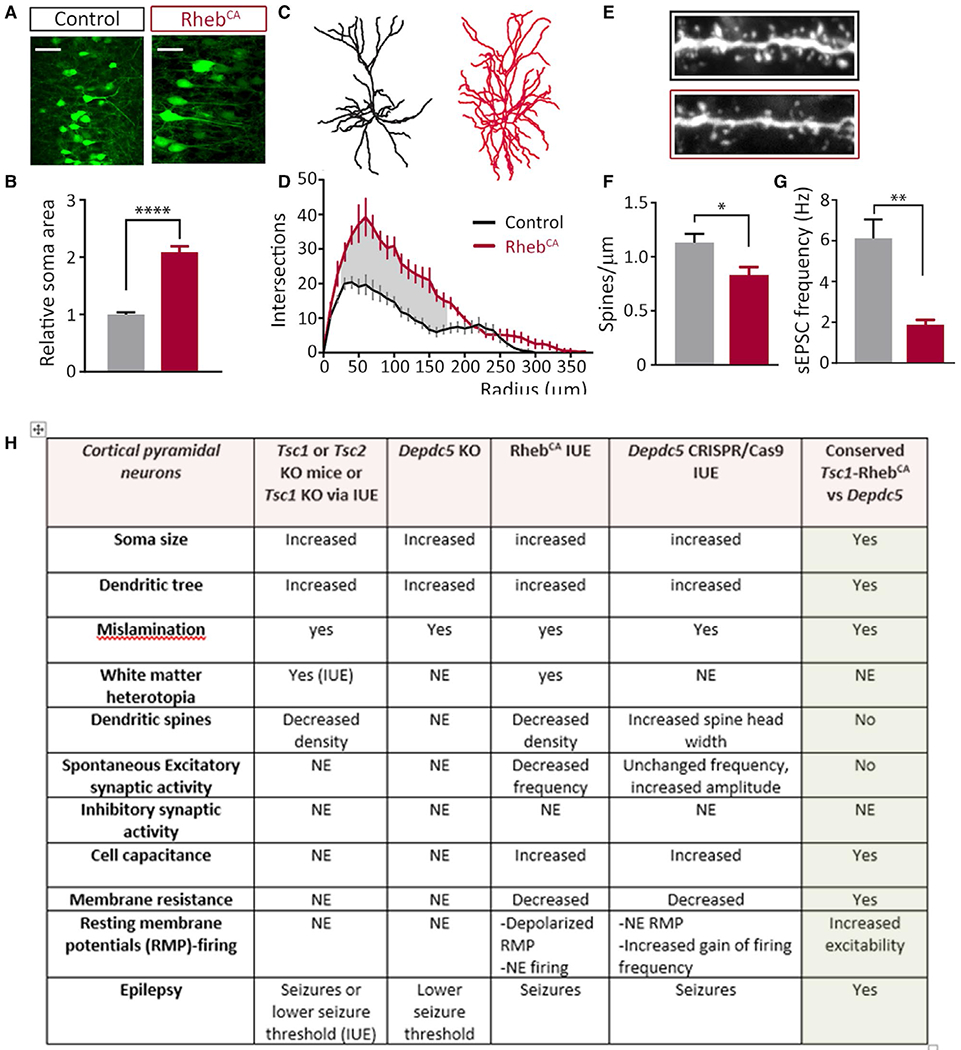
Conserved and nonconserved cellular abnormalities in mTOR-related MCDs (Figure modified from 8, 9). A, Mice were electroporated with GFP (control) or varying concentrations of GFP+ RhebCA plasmids at E15, targeting layer 2/3 neurons in the cortex. Pups were screened for successful electroporation via GFP expression at P0. Mice were monitored with video-EEG recording for 5–7 consecutive days at 2–4 mo of age and afterward euthanized for histologic and immunohistochemical analysis. Scale bar: 50 μm. B, Quantification of soma areas of layer 2/3 GFP+ cells in coronal sections from P28 control and RhebCA-electroporated mice shows neuronal enlargement. Areas were normalized to control soma area (n = 25 cells, three mice per condition, t test). C, D, Sholl analysis and representative traces of dendrites (n = 5-7 cells, three or four mice per condition, RM two-way ANOVA, Bonferroni post hoc). Shaded areas indicate radii where P <.05. E, F, Spine density quantification and images of basal dendrites in control and RhebCA neurons (n = 9-13 branches, three mice per condition, t test) (Magnification: H, 100×). G, Representative traces of spontaneous (s) EPSCs recorded from layer 2/3 neurons for each condition. RhebCA expression decreases sEPSC frequency (n = 8 or 9 neurons from four or five mice per condition). Scale bar: 20 pA per 0.2 s. *P <.05; ***P <.001; ****P <.0001. All data are presented as mean ± SEM. H, Table demonstrating similarities and differences between Tsc1, Tsc2, RhebCA, and Depdc5 animal models systems (see text)
A number of Depdc5 transgenic strains have been described in several species. For example, a zebrafish model generated with antisense morpholino oligonucleotides causing depdc5 loss-of-function KD exhibits hyperkinesia, circular swimming, and increased neuronal activity.58 These phenotypic features were reduced upon treatment with rapamycin. A depdc5 zebrafish knockout model exhibited spontaneous epileptiform activity, increased seizure susceptibility, reduced motor activity, and premature death at 2-3 weeks postfertilization59 in association with hyperactivation of mTOR signaling. A Depdc5 KO rat was generated using TALEN technology.46 Depdc5(−/−) embryos died by embryonic day 14.5 and exhibited constitutive mTORC1 hyperactivation in fibroblasts and brain. Prenatal treatment with rapamycin rescued the phenotype of Depdc5(−/−) embryos. These studies provide strong evidence that biallelic inactivation of DEPDC5 is necessary to cause mTOR hyperactivation, akin to what has been described in human MCD tissue specimens.24,25 Conversely, Depdc5(±) rats developed normally and did not exhibit spontaneous seizures but had altered cortical neuron excitability. Depdc5(±) rats displayed cortical cytomegalic dysmorphic neurons expressing increased phosphorylated S6, indicative of mTORC1 upregulation. A conditional Depdc5flox/flox-Syn1Cre mouse strain exhibits brain enlargement and cytomegalic dysmorphic neurons throughout the cortex60 in association with mTOR hyperactivation. These Depdc5flox/flox-Syn1Cre mice also have lowered seizure thresholds, with decreased latency to seizures after chemo-convulsant exposure and increased mortality following pentylenetetrazol-induced seizures. A Depdc5-null mouse model generated using CRISPR mutagenesis causing a double-stranded break in Depdc5 exon 2 exhibited reduced body size, anemia, diffuse edema, altered cranial structure, and abnormal cardiovascular development in association with mTORC1 hyperactivity and early embryonic lethality.61 Of interest, in the heterozygous mice, no cortical structural abnormalities were identified, and neither spontaneous nor induced seizures were detected.
Two recent studies using CRISPR-editing of Depdc5 in the rodent cerebral cortex demonstrated abnormal electrical bursts and clinical seizures associated with experimentally induced FCD following CRISPR-editing of Depdc5 by in utero electroporation.25,45 In a rat model45 two short guide RNAs (gRNAs) were used to target rat Depdc5 either at exon 8 or exon 12 by in utero electroporation on embryonic day 13. Analysis of these animals on postnatal day 21 revealed enlarged neuronal somata in association with hyperactivated mTORC1. All electroporated rats exhibited interictal electroencephalography (EEG) abnormalities and spontaneous seizures. In a mouse model,25 gRNAs targeting exon 16 of Depdc5 were transfected by in utero electroporation into fetal mouse brains at embryonic day 14.5. These animals showed a clear FCD with migration abnormalities and enlarged cells demonstrating enhanced mTORC1 activity. These animals exhibited spontaneous seizures, interictal EEG abnormalities, and even sudden death (Figures 1–3).
To further study the mechanisms of epileptogenesis and hyperexcitability in mTORopathies vs GATORopathies, a plasmid encoding a constitutively active form of the mTOR activator Rheb (RhebCA,8,9) was introduced to the fetal mouse brain by in utero electroporation. Rheb was selected for comparative genetic manipulation because two recent studies have reported gain-of-function mutations in RHEB leading to MCD associated with epilepsy62,63 and because Rheb integrates both the canonical growth factor signaling vector and the ATP-AMP-AMPK-dependent vector, thus allowing comparison with the amino acid signaling arm of the mTOR pathway. In addition, from a methodologic perspective, RhebCA expression rapidly (6-8 hours) activates the mTOR pathway as compared to other experimental strategies such as conditional transgenic mice, small hairpin RNA (shRNA), or CRISPR/Cas9 vectors because the timing of mTORC1 activation depends on the half-life of the KD/KO protein of interest. Comparing RhebCA and CRISPR/Cas9 Depc5 editing models, both animal groups exhibit Racine grade 4/5 seizures, thus demonstrating how each leads to network hyperexcitability. Both models exhibit similar histopathologic abnormalities including cortical dyslamination with severe neuronal misplacement and white matter heterotopia, and abnormal neuronal morphology characterized by increased soma size and altered dendritic complexity (Figure 4). Patch-clamp recordings in the animals reveal increased cell capacitance and decreased membrane resistance in the RhebCA model. Astrogliosis and microglial activation, and increased axonal length, are observed in the RhebCA-model and in the Depdc5flox/flox-Syn1Cre mouse.8,64,65 Of interest, as in Tsc2 cKO mice,66 the degree of mTOR hyperactivity correlates with the severity of epilepsy and associated neuropathology including soma size and misplacement RhebCA plasmid model.65 These findings suggest that there are commonalities to the effects of mTOR pathway gene variants, but specific gene variants may lead to different levels of mTORC1 activation and thus differentially impact cortical development. As investigators look to foster clinical trials in mTORopathies and GATORopathies, it may be important to evaluate the effects of individual variants on mTOR activity.
In contrast, although mTORopathies and GATORopthies are linked through the mTORC1 kinase signaling node, there are some differences between each of the models. For example, although RhebCA expression or Depc5 loss leads to conserved alterations related to cell misplacement and hypertrophy, there are differences in the effects of membrane properties and synaptic connectivity in the same type of neurons. RhebCA-expressing cortical pyramidal neurons display decreased dendritic spine density and a major (80%) decrease in the frequency of excitatory postsynaptic synaptic currents, that is, functional inputs (Figure 4), although Depc5 mutant neurons displayed a small but significant increase in the frequency of excitatory postsynaptic currents. Although both gene effects culminate in mTORC1 hyperactivity, the differential effects of each may depend on cell-autonomous functions of the different genes being manipulating, such that Rheb and DEPDC5 may affect parallel pathways independent of mTOR. For example, the work on Ptennull neurons compared to Tsc1null neurons recorded in the same culture conditions suggests signaling differences in which loss of Pten led to an increase in both excitatory and inhibitory neurotransmission, while loss of Tsc1 did not affect excitatory neurotransmission and reduced inhibitory transmission.67 Collectively, the impact of mTOR pathway gene deletion or constitutive overactivation on synaptic connectivity and cell excitability will likely share some common features due to signaling through mTOR, but there may also be differences due to differential effects of individual proteins on other signaling cascades. There will likely also be region- and cell-specific effects that yield differential effects in distinct cortical or subcortical regions.
5 |. SUMMARY AND CONCLUSIONS
Mutations in DEPDC5, NPRL3, and NPRL2 are highly associated with medically intractable epilepsy as well as with MCDs including FCD and HME. The effects of these gene mutations are heterogeneous, with multiple phenotypes including seizures, MCD, autism spectrum disorder, and perhaps most important, SUDEP. Because loss-of-function mutations in DEPDC5 or NPRL3 are associated with mTORC1 hyperactivation both in experimental models and in resected human tissue, clinical trials with mTOR inhibitors such as sirolimus or everolimus may provide new avenues for therapy in these individuals.
Key Points.
Mutations in GATOR1 complex genes DEPDC5, NPRL3, NPRL2 are highly associated with focal epilepsy.
Clinical phenotypes associated with DEPDC5, NPRL3, NPRL2 mutations are variable and include both non-lesional cases and cases associated with MCD.
The GATOR1 complex modulates mTOR pathway signaling in response to cellular amino acid levels.
There are a number of unique animal model systems that have been developed to study how DEPDC5 mutations cause network excitability and seizures.
RHEB is a key mTOR regulatory protein that can used to study effects of mTOR hyperactivation on brain structure and neuronal excitability.
ACKNOWLEDGMENTS
P.B.C. is supported by NINDS R01NS094596 and R01NS099452. A.B. is supported by NINDS R01NS093704, DoD W81XWH-16-1-0164, and Swebilius Foundation.
Funding information
U.S. Department of Defense, Grant/Award Number: W81XWH-16-1-0164; National Institute of Neurological Disorders and Stroke, Grant/Award Number: R01NS094596 and R01NS099452
Footnotes
CONFLICTS OF INTEREST
P. Crino and A. Bordey have served as a paid consultant for Evogen, Inc and Asklepios BioPharmaceutical, Inc (AsBio), respectively. A. The remaining authors have no conflicts of interest. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
REFERENCES
- 1.Crino PB. mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17:734–42. [DOI] [PubMed] [Google Scholar]
- 2.Curatolo P Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol. 2015;52:281–9. [DOI] [PubMed] [Google Scholar]
- 3.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen LA, Mirzaa GM, Ishak GE, O’Roak BJ, Hiatt JB, Roden WH, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol. 2015;78:375–86. [DOI] [PubMed] [Google Scholar]
- 6.Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol. 2015;77:675–83. [DOI] [PubMed] [Google Scholar]
- 7.Crino PB. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013;125:317–32. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh LS, Wen JH, Claycomb K, Huang Y, Harrsch FA, Naegele JR, et al. Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat Commun. 2016;7:11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin TV, Hsieh L, Kimura T, Malone TJ, Bordey A. Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc Natl Acad Sci U S A. 2016;113:11330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen LH, Brewster AL, Clark ME, Regnier-Golanov A, Sunnen CN, Patil VV, et al. mTOR inhibition suppresses established epilepsy in a mouse model of cortical dysplasia. Epilepsia. 2015;56:636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sim JC, Scerri T, Fanjul-Fernández M, Riseley JR, Gillies G, Pope K, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol. 2016;79:132–7. [DOI] [PubMed] [Google Scholar]
- 13.Scerri T, Riseley JR, Gillies G, Pope K, Burgess R, Mandelstam SA, et al. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol. 2015;2:575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricos MG, Hodgson BL, Pippucci T, Saidin A, Ong YS, Heron SE, et al. Mutations in the mTOR pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol 2016;79:120–31. [DOI] [PubMed] [Google Scholar]
- 15.Baldassari S, Picard F, Verbeek NE, van Kempen M, Brilstra EH, Lesca G, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019;21:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia. 2016;57:994–1003. [DOI] [PubMed] [Google Scholar]
- 17.Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45:546–51. [DOI] [PubMed] [Google Scholar]
- 18.Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–7. [DOI] [PubMed] [Google Scholar]
- 19.Ishida S, Picard F, Rudolf G, Noé E, Achaz G, Thomas P, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013;45:552–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baldassari S, Licchetta L, Tinuper P, Bisulli F, Pippucci T. GATOR1 complex: the common genetic actor in focal epilepsies. J Med Genet. 2016;53:503–10. [DOI] [PubMed] [Google Scholar]
- 21.Carvill GL, Crompton DE, Regan BM, McMahon JM, Saykally J, Zemel M, et al. Epileptic spasms are a feature of DEPDC5 mTORopathy. Neurol. Genet. 2015;1:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pippucci T, Licchetta L, Baldassari S, Palombo F, Menghi V, D’Aurizio R, et al. Epilepsy with auditory features: a heterogeneous clinico-molecular disease. Neurol Genet. 2015;1:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crino PB, Aronica E, Baltuch G, Nathanson K. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology. 2010;74:1716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 2017;21:3754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest. 2018;128:2452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strauss KA, Gonzaga-Jauregui C, Brigatti KW, Williams KB, King AK, Van Hout C, et al. Genomic diagnostics within a medically underserved population: efficacy and implications. Genet Med. 2018;20:31–41. [DOI] [PubMed] [Google Scholar]
- 27.Sancak Y, Sabatini DM. Rag proteins regulate amino-acid-induced mTORC1 signalling. Biochem Soc Trans. 2009;37:289–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen K, Huang RK, Brignole EJ, Condon KJ, Valenstein ML, Chantranupong L, et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature. 2018;556:64–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng J, Ferguson SM. GATOR1-dependent recruitment of FLCN-FNIP to lysosomes coordinates Rag GTPase heterodimer nucleotide status in response to amino acids. J Cell Biol. 2018;217:2765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature. 2016;536:229–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gai Z, Wang Q, Yang C, Wang L, Deng W, Wu G. Structural mechanism for the arginine sensing and regulation of CASTOR1 in the mTORC1 signaling pathway. Cell Discov. 2016;2:16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165:153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347:188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rebsamen M, Pochini L, Stasyk T, de Aradjo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung J, Genau HM, Behrends C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol. 2015;35:2479–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171(642–654):e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chantranupong L, Wolfson RF, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JS, Ro SH, Kim M, Park HW, Semple IA, Park H, et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep. 2015;5:9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9:1281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017;358:813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iffland PH 2nd, Baybis M, Barnes AE, Leventer RJ, Lockhart PJ, Crino PB. DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol Dis. 2018;114:184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu S, Knowlton RC, Watson BO, Glanowska KM, Murphy GG, Parent JM, et al. Somatic Depdc5 deletion recapitulates electro-clinical features of human focal cortical dysplasia type IIA. Ann Neurol. 2018;84:140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marsan E, Ishida S, Schramm A, Weckhuysen S, Muraca G, Lecas S, et al. Depdc5 knockout rat: a novel model of mTORopathy. Neurobiol Dis. 2016;89:180–9. [DOI] [PubMed] [Google Scholar]
- 47.Naseer MI, Alwasiyah MK, Abdulkareem AA, Bajammal RA, Trujillo C, Abu-Elmagd M, et al. A novel homozygous mutation in SZT2 gene in Saudi family with developmental delay, macrocephaly and epilepsy. Genes Genomics. 2018;40:1149–55. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura Y, Togawa Y, Okuno Y, Muramatsu H, Nakabayashi K, Kuroki Y, et al. Biallelic mutations in SZT2 cause a discernible clinical entity with epilepsy, developmental delay, macrocephaly and a dysmorphic corpus callosum. Brain Dev. 2018;40:134–9. [DOI] [PubMed] [Google Scholar]
- 49.Basel-Vanagaite L, Hershkovitz T, Heyman E, Raspall-Chaure M, Kakar N, Smirin-Yosef P, et al. Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am J Hum Genet. 2013;93:524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pajusalu S, Reimand T, Ounap K. Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am J Med Genet A. 2015;167A:1913–5. [DOI] [PubMed] [Google Scholar]
- 51.Baple EL, Maroofian R, Chioza BA, Izadi M, Cross HE, Al-Turki S, et al. Mutations in KPTN cause macrocephaly, neurodevelopmental delay, and seizures. Am J Hum Genet. 2014;94:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mc Cormack A, Sharpe C, Gregersen N, Smith W, Hayes I, George AM, et al. 12q14 microdeletions: additional case series with confirmation of a macrocephaly region. Case Rep Genet. 2015;2015:192071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baek ST, Copeland B, Yun EJ, Kwon SK, Guemez-Gamboa A, Schaffer AE, et al. An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat Med. 2015;21:1445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 55.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, et al. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med. 2013;5:182ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Veleva-Rotse BO, Smart JL, Baas AF, Edmonds B, Zhao ZM, Brown A, et al. STRAD pseudokinases regulate axogenesis and LKB1 stability. Neural Dev. 2014;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Calbiac H, Dabacan A, Marsan E, Tostivint H, Devienne G, Ishida S, et al. Depdc5 knockdown causes mTOR-dependent motor hyperactivity in zebrafish. Ann Clin Transl Neurol. 2018;5:510–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swaminathan A, Hassan-Abdi R, Renault S, Siekierska A, Riche R, Liao M, et al. Non-canonical mTOR-independent role of DEPDC5 in regulating GABAergic network development. Curr Biol. 2018;28(1924–1937):e1925. [DOI] [PubMed] [Google Scholar]
- 60.Yuskaitis CJ, Jones BM, Wolfson RL, Super CE, Dhamne SC, Rotenberg A, et al. A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis. 2018;111:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hughes J, Dawson R, Tea M, McAninch D, Piltz S, Jackson D, et al. Knockout of the epilepsy gene Depdc5 in mice causes severe embryonic dysmorphology with hyperactivity of mTORC1 signalling. Sci Rep. 2017;7:12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reijnders MRF, Kousi M, van Woerden GM, Klein M, Bralten J, Mancini GMS, et al. Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat Commun. 2017;8:1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salinas V, Vega P, Piccirilli MV, Chicco C, Ciraolo C, Christiansen S, et al. Identification of a somatic mutation in the RHEB gene through high depth and ultra-high depth next generation sequencing in a patient with Hemimegalencephaly and drug resistant Epilepsy. Eur J Med Genet. 2018. in press. [DOI] [PubMed] [Google Scholar]
- 64.Gong X, Zhang L, Huang T, Lin TV, Miyares L, Wen J, et al. Activating the translational repressor 4E-BP or reducing S6K-GSK3beta activity prevents accelerated axon growth induced by hyperactive mTOR in vivo. Hum Mol Genet. 2015;24:5746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen LH, Mahadeo T, Bordey A. mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J Neurosci. 2019;39:2762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan E, Tsai PT, Greene-Colozzi E, Sahin M, Kwiatkowski DJ, Malinowska IA. Graded loss of tuberin in an allelic series of brain models of TSC correlates with survival, and biochemical, histological and behavioral features. Hum Mol Genet. 2012;21:4286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weston MC, Chen H, Swann JW. Loss of mTOR repressors Tsc1 or Pten has divergent effects on excitatory and inhibitory synaptic transmission in single hippocampal neuron cultures. Front Mol Neurosci. 2014;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]