

Abstract
Background
Intravenous immunoglobulin (IVIG) IQYMUNE® is a highly purified 10% IVIG that was assessed using the new stringent definition of response described in the revised guideline on the clinical investigation of IVIG. The efficacy and the safety of IQYMUNE® were investigated in adult patients with chronic primary immune thrombocytopenia (ITP).
Methods
In this phase III multinational, multicentre, prospective, uncontrolled, open-label, single-arm study, adult patients with a baseline platelet count < 30 × 109/L were treated with IVIG 10% at a dose of 2 g/kg body weight administered over 2 consecutive days. The primary endpoint was Response over the study period and was defined according to the recent and most stringent European Medicines Agency guidelines (platelet count ≥ 30 × 109/L and a ≥ 2-fold increase from baseline, no new bleeding, and no concomitant treatment with drugs that affect platelet count and/or induce bleeding cessation).
Results
Thirty-eight patients were enrolled; 73 infusions were administered (38 on Day 1 and 35 on Day 2). Response was reached by 24 patients corresponding to 63.2% of patients in the full analysis set (95% CI: 46.0; 78.2) and 68.6% of patients in the per-protocol set (95% CI: 50.7; 83.1). The median time to Response was 1 day. The median duration of Response was 13.5 days. Reasons for non-response were failure to reach the required platelet count (n = 12), a new bleeding event (n = 1), and forbidden medication use (n = 1). Among the 23 patients with a baseline platelet count ≤ 20 × 109/L, 19 patients (82.6%) achieved a platelet count ≥ 50 × 109/L at least once before Day 5 (previous European Medicines Agency definition of response). Treatment was well tolerated even in patients with a high flow rate (≥ 6 mL/kg/h in 40% of patients). Headache (34.2%), pyrexia (15.8%), and creatinine renal clearance decrease, including one case of decrease in glomerular filtration rate (10.5%) were the most frequently reported drug-related adverse events.
Conclusions
Administration of IQYMUNE® for 2 consecutive days at a dose of 2 g/kg was safe and efficacious. These results support the treatment of adult patients with chronic ITP with IQYMUNE®.
Keywords: Intravenous immunoglobulin, High dose, Immune thrombocytopenia, Response, European Medicines Agency guidelines, Platelet count, Bleeding assessment, Infusion rate
Introduction
Immune thrombocytopenia (ITP) is an autoimmune disorder characterized by a platelet count lower than 100 × 109/L and an increased risk of bleeding [1]. Patients with ITP mostly experience mild mucocutaneous hemorrhages such as purpura or ecchymosis [2]. Occasionally, when the platelet count falls below 20 × 109/L, life-threatening hemorrhages, such as gastrointestinal or intracranial bleeding, may occur [3].
Immune thrombocytopenia develops when autoantibodies target platelet glycoproteins, such as GPIIb/IIIa, and megakaryocytes. This leads to platelet destruction and suboptimal platelet production [4, 5]. Initial treatment for ITP, which is intended to increase the platelet count rapidly, includes glucocorticoids, intravenous immunoglobulins (IVIGs), and intravenous anti-D (Rho) globulin in Rh (+) patients [6]. The mechanism of action of IVIG in ITP includes mainly the blockade of Fcγ receptor on macrophages, which spares opsonized platelets from Fcγ receptor-mediated phagocytosis and induces the acceleration of anti-platelet antibody elimination linked to the saturation of neonatal Fc receptor [7].
Since the initial 1981 report by Imbach et al [8], several studies have confirmed the efficacy of IVIG in patients with ITP [9-13]. IVIGs remain of critical importance in the treatment of ITP including in the current protocols, in which the introduction of rituximab and thrombopoietin-receptor agonists (TPO-RA) have contributed to a more effective treatment of this disorder. IVIGs have proven to result in a more rapid control of bleeding manifestations in ITP presenting with wet purpura; and its use, associated with corticosteroids, is a common clinical practice in the management of “acute” situations [14]. Similarly, IVIG administration remains the most effective and rapid rescue treatment in patients who are no longer responsive to treatment with other agents, including TPO-RA. Finally, during pregnancy, IVIG has proven to be safe, not associated with any fetal damage, and the only available treatment in case of intolerance to corticosteroids [6, 15]. Bleeding control after IVIG infusion has been observed even in cases not showing a proportional platelet increase, thus possibly indicating that platelets are consumed at the patients’ bleeding sites [16, 17].
IVIG IQYMUNE® is a highly purified, sugar-free, salt-free, and ready-to-use liquid IVIG that was developed using the Quality by Design approach. This standard of quality, which has been recommended by the United States Food and Drug Administration and the European Medicines Agency (EMA) since 2011 [18], was introduced in the IQYMUNE® manufacturing process in 2007. Three purification steps by precipitation or chromatography and two dedicated viral removal steps were specifically developed to drive the IQYMUNE® purification process and to meet the five key objectives: 1) Removal of activated coagulation factors to reduce the risk of thromboembolic adverse events; 2) Removal of IgA to avoid immune responses (anaphylactic reactions) in patients with IgA deficiency; 3) Removal of anti-A and anti-B hemagglutinins to prevent hemolysis events in patients with A, B, or, AB blood groups; 4) Reduction of aggregates to avoid adverse events through complement activation; 5) Reduction of potential blood-borne pathogens.
In this phase III study, the efficacy and safety of IQYMUNE® were investigated in adult patients with chronic primary ITP. To our knowledge, this study is one of the first studies to be designed using the revised 2010 EMA guideline on the clinical investigation of human immunoglobulins for intravenous administration [19].
Materials and Methods
This prospective, single-arm, multicentre, open-label, phase III study was conducted in Europe from February 2012 to May 2013 in order to investigate the efficacy and safety of IQYMUNE® (LFB Biomedicaments, Les Ulis, France) over 30 days in adult patients with chronic primary ITP and a platelet count < 30 × 109/L. The protocol was designed based on the revised EMA guidelines on clinical investigation of IVIG dated July 2010 [19] and was approved by the Independent Ethics Committees of each center. The study was conducted in accordance with the principles of the Declaration of Helsinki, ICH Good Clinical Practice, and relevant national laws. Written informed consent was obtained from each patient.
Patients were treated with IQYMUNE® at a total dose of 2 g/kg body weight administered over 2 consecutive days (1 g/kg on Day 1 and 1 g/kg on Day 2). The infusion starting rate was 1 mL/kg/h for 30 min. If well tolerated, the rate was increased gradually to a maximum of 4 mL/kg/h during the first infusion and to a maximum of 8 mL/kg/h during the second infusion.
Study patients
Adult patients were eligible for enrolment in the study if they were between 18 and 65 years of age and had been diagnosed with primary ITP for at least 12 months prior to study entry. At the time of inclusion, platelet count had to be < 30 × 109/L. Immune thrombocytopenia had to have been originally diagnosed according to three standard criteria: 1) Isolated thrombocytopenia with a platelet count < 100 × 109/L and no abnormality of cells of other hematological lineages; 2) A normal bone marrow aspirate or a history of response to ITP treatment (corticosteroids, IVIG, anti-D); and 3) Absence of other causes of thrombocytopenia. Patients on long-term stable or decreasing corticosteroid therapy over the previous 4 weeks or with refractory ITP were allowed.
The main exclusion criteria were severe hemorrhagic syndrome at inclusion; splenectomy within the previous 2 months, history of allergy or serious adverse reaction to immunoglobulin therapy, presence of anti-immunoglobulin A antibodies, a glomerular filtration rate (GFR) < 80 mL/min/1.73 m2 according to the Modification of Diet in Renal Disease (MDRD) formula, levels of alanine aminotransferase or aspartate aminotransferase > 3 times the upper limit of normal, levels of total bilirubin > 2 times the upper limit of normal, protein-losing enteropathy or nephrotic syndrome, history of thrombosis within the past 12 months, history of hemolysis or hemolytic anemia with IVIG therapy, pregnancy, and breastfeeding.
Concomitant treatments that could interfere with the efficacy or safety assessment were not allowed. Such drugs included loop diuretics within the previous week and anticoagulants within 15 days prior to study drug infusion; cyclosporin A, IVIGs, anti-D antibody, immunosuppressors such as azathioprine and cyclophosphamide, thrombopoietin receptor agonists, and immunomodulators during the previous month; rituximab during the previous 4 months; and danazol during the previous 6 months. Premedication prior to the first infusion was forbidden. Paracetamol, antihistamines (chlorpheniramine), hydroxyzine, and antiemetics were allowed before the second infusion in patients who experienced an adverse reaction during or soon after the first infusion.
Efficacy endpoints
The primary efficacy endpoint was Response defined as a platelet count ≥ 30 × 109/L and a ≥ 2-fold increase from baseline at two visits that were at least 7 days apart; the absence of new bleeding between the end of the last infusion and the second visit in which platelet count criteria were met (at least 7 days after the first visit); and no use of forbidden medication (medication that could increase platelet count and/or induce bleeding cessation) between enrolment and the second visit in which platelet count criteria were met (at least 7 days after the first visit).
The secondary efficacy endpoints were Complete Response, using the same definition as Response but with a threshold platelet count ≥ 100 × 109/L; the time to Response/Complete Response (number of days between the start of the first infusion and the first day Response/Complete Response was observed); the duration of Response/Complete Response (number of days between the first day of Response/Complete Response and the last day when Response/Complete Response was observed); the maximum platelet count; the time to maximum platelet count; and response according to the previous EMA guideline definition (baseline platelet count ≤ 20 × 109/L and a platelet count ≥ 50 × 109/L on at least one evaluation on or before Day 5) [20]. Other secondary efficacy parameters included regression of baseline bleeding.
Blood samples for platelet determination were drawn on Day 1 pre-infusion, Day 2 post-infusion, on Days 3, 4, 5, 6, 7, 9, 11, Day 14 ± 1, Day 21 ± 1, and Day 30 ± 1. Bleeding was assessed at the hospital by the physician at baseline (Day 1) and on Days 2, 14, and 30 using the World Health Organization and the Khellaf bleeding scores [21]. In addition, patients kept a daily home diary record of all bleeding symptoms. The use of forbidden concomitant medication, defined as any medications that could induce a platelet count increase and/or interfere with the assessment of bleeding, was documented at each hospital visit.
Safety evaluation
Patients were monitored for safety at each hospital visit and between visits using home diaries. Treatment-emergent adverse events (TEAE) were defined as any adverse event (AE) occurring between the start of the first infusion and the end of the study. Temporally associated adverse events (TAAEs) were defined as AEs occurring between the start of infusion and 72 h after the end of infusion.
Vital signs were assessed before the start of infusions, during infusions, and within 30 min after the end of infusions. Blood samples were collected for routine safety hematology and biochemistry parameters on Day 1 pre-infusion, on Day 2 post-infusion, on Day 14, and at the last study visit.
Statistical analysis
In accordance with the EMA guidelines [19], a cohort of 30 adults with chronic primary ITP patients and a baseline platelet count of < 30 × 109/L is required to evaluate the efficacy and safety of an investigational IVIG. In order to obtain at least 30 evaluable patients, assuming a 25% drop-out rate, 40 patients needed to be included.
Primary and secondary efficacy variables were evaluated in the full analysis set, which was defined as all patients who received the study drug at least once and who had a platelet count < 30 × 109/L at baseline and at least one subsequent platelet count during the study period. For robustness, the primary efficacy variable was also evaluated in the per-protocol population, which was defined as all patients in the full analysis set who had no major deviations from the protocol. Major protocol deviations included major inclusion/exclusion criteria violations and deviations preventing the evaluation or leading to an incorrect analysis of the primary efficacy endpoint. Safety variables were evaluated in the population of all enrolled patients who received the study drug at least once.
Demographic variables and patient characteristics were analysed using descriptive statistics. The primary efficacy endpoint was analysed using descriptive statistics along with exact 95% Clopper-Pearson confidence intervals (CI). The secondary efficacy endpoints were analysed using descriptive summaries. Duration of Response/Complete Response was analysed by the Kaplan-Meier method and a time-to-event curve. The maximum platelet count and the time to maximum platelet count were estimated using the Hodges-Lehmann estimator and Kaplan-Meier method, respectively. The proportion of patients who responded to treatment according to the previous EMA guideline was calculated using descriptive statistics along with exact 95% Clopper-Pearson CIs. Missing observations were imputed using the last observation carried forward. All analyses were performed using SAS software version 9.1.3 (SAS Institute Inc., Cary, NC, USA).
Results
Nineteen centres in eight European countries enrolled 38 adult patients with chronic ITP. All 38 patients met the criteria of the full analysis set.
Patients age ranged from 18 to 60 years (mean of 37.2 ± 11.8 years) (Table 1). Fourteen patients were male (36.8%). Six patients had refractory ITP defined as the failure to achieve a response or loss of response after splenectomy. Patients had, on average, a history of 0.74 ± 2.03 acute episodes per month. The median time between the last acute episode and study enrolment was 3 months. A total of 35 patients (92.1%) had been previously treated for ITP, including 11 patients (28.9%) who had received IVIG.
Table 1. Demographics and Baseline Characteristics of Patients (N = 38 Patients).
| Parameter | Results |
|---|---|
| Demographics | |
| Sex (n (%)) | |
| Male | 14 (36.8) |
| Female | 24 (63.2%) |
| Age (years) | |
| Mean ± SD | 37.2 ± 11.8 |
| Median (range) | 35.0 (18, 60) |
| Weight (kg) | |
| Mean ± SD | 67.5 ± 17.0 |
| Median (range) | 68.5 (43.0, 115.0) |
| Body mass index (kg/m2) | |
| Mean ± SD | 23.8 ± 4.2 |
| Median (range) | 23.8 (17.0, 34.9) |
| Medical condition/history reported by ≥ 3 patients (n (%)) | |
| Hypertension | 6 (15.8) |
| Anemia | 6 (15.8) |
| Tonsillectomy | 5 (13.2) |
| Gastritis | 5 (13.2) |
| Appendectomy | 4 (10.5) |
| History of ITP | |
| Time from diagnosis (years) | |
| Mean ± SD | 7.55 ± 8.63 |
| Median (range) | 3.65 (1.0, 40.7) |
| Platelet count at diagnosis (109/L) | |
| Mean ± SD | 24.4 ± 24.7 |
| Median (range) | 17.0 (0.7, 101.0) |
| Frequency of acute* episodes per month (n) | |
| Mean ± SD | 0.74 ± 2.03 |
| Median (range) | 0.30 (0.0, 12.0) |
| Time from previous acute* episode (months) | |
| Mean ± SD | 16.67 ± 36.36 |
| Median (range) | 3.00 (0.0, 157.0) |
| Platelet count at baseline (109/L) | |
| Mean ± SD | 16.7 ± 8.00 |
| Median (range) | 18.1 (1, 29) |
| Patients with a platelet count ≤ 20 × 109/L at baseline (n (%)) | 23 (60.5) |
| Refractory ITP and splenectomy (n (%)) | 6 (15.8) |
| Bleeding (n (%)) | |
| Bleeding history | 30 (78.9) |
| Life-threatening bleeding history | 3 (7.9) |
| Bleeding history reported in ≥ 3 patients | |
| Cutaneous | 27 (71.1) |
| Mucosal | 17 (56.7) |
| Genitourinary tract | 7 (23.3) |
| Bleeding at baseline | 29 (76.3) |
| Prior ITP medication in ≥ 3 patients (n (%)) | 35 (92.1) |
| Corticosteroids, systemic | 32 (84.2) |
| Immunoglobulins | 11 (28.9) |
| Azathioprine | 3 (7.9) |
| Eltrombopag | 2 (5.3) |
| Rituximab | 2 (5.3) |
| Other blood products | 2 (5.3) |
*Defined as a drop in platelet count to ≤ 50 × 109. ITP: immune thrombocytopenia.
Thirty patients (78.9%) had a history of bleeding. Three patients had experienced at least one life-threatening bleeding episode since diagnosis. Bleeding was present at baseline in 29 patients (40 events) and was categorized as mild in 25 patients (34 events), moderate in five patients (five events), and severe in one patient (extensive purpura).
A total of 73 infusions were administered during the study, 38 on the first day and 35 on the second day. Three patients received only one infusion of 1 g/kg due to moderate pyrexia/headache/back pain (one patient), mild GFR decrease (one patient), and protocol deviation (one patient). One patient was inadvertently underdosed and received 0.3 g/kg/day on both infusion days; and one patient received 2.4 g/kg over 2 days. All other patients received the planned dose. The mean total dose was 1.89 ± 0.37 g/kg (range 0.6 to 2.4).
On Day 1, all the infusions were started at a flow rate of 0.5 mL/kg/h. The flow rate was subsequently increased to a mean maximum of 3.65 ± 0.80 mL/kg/h (range from 1 to 4 mL/kg/h), resulting in a mean infusion time of 4.87 ± 1.97 h. On Day 2, 14 infusions were administered at a maximum flow rate of ≤ 4 mL/kg/h, 11 infusions at a flow rate > 4 to < 6 mL/kg/h, and 14 infusions at a flow rate ≥ 6 - 8 mL/kg/h (14 patients (40%) of which eight patients (23%) reached a flow rate of 8 mL/kg/h). The Day 2 mean maximum infusion rate was 5.33 ± 1.83 mL/kg/h and mean infusion time was 4.10 ± 1.57 h. Modifications of flow rate due to adverse event included a decrease in rate due to mild pyrexia that occurred at 6.0 mL/kg/h (one patient), a temporary interruption of infusion due to a mild anaphylactoid reaction (one patient) and an interruption of infusion due to recurrent moderate headache (one patient).
Efficacy
Three patients were excluded from the per-protocol analysis. Reasons for exclusion were hydrocortisone injections on Day 1 and 2 (one patient); premature withdrawal from the study on Day 2 due to drug-related fever, headache and back pain (one patient); and secondary ITP diagnosis obtained after completion of the study based on a seropositive human immunodeficiency virus (HIV) test performed at study entry.
The primary efficacy endpoint, Response (Table 2), was reached by 24 patients corresponding to 63.2% of patients in the full analysis set (95% CI: 46.0; 78.2) and 68.6% of patients in the per-protocol set (95% CI: 50.7; 83.1). The median time to Response was 1 day (range 1 to 6 days). The estimated median duration of Response was 13.5 days (95% CI: 10; 20).
Table 2. Efficacy Endpoints (Full Analysis Set).
| Efficacy variable | N = 38 |
|---|---|
| Response, n (%); (Clopper-Pearson 95% CI) | 24 (63.2); (46.0; 78.2) |
| Time to Response (days), median (range) | 1 (1 - 6) |
| Duration of Response (days), estimate (Kaplan Meier 95% CI) | 13.5 (10.0; 20.0) |
| Loss of Response, n (%); (Clopper-Pearson 95% CI) | 17 (70.8); (48.9; 87.4) |
| No Response, n (%); (Clopper-Pearson 95% CI) | 14 (36.8); (21.8; 54.0) |
| Complete Response, n (%); (Clopper-Pearson 95% CI) | 11 (28.9); (15.4; 45.9) |
| Time to Complete Response (days), median (range) | 2 (1 - 8) |
| Duration of Complete Response (days), estimate (Kaplan Meier 95% CI) | 12 (10.0; 18.0) |
| Loss of Complete Response, n (%); (Clopper-Pearson 95% CI) | 9 (81.8); (48.2; 97.7) |
| No Complete Response, n (%); (Clopper-Pearson 95% CI) | 27 (71.1); (54.1; 84.6) |
| Maximum platelet count (× 109)*, mean (Hodges-Lehmann 95% CI) | 165 (113; 189) |
| Time to maximum platelet count (days)†, median (Kaplan Meier 95% CI) | 4.0 (3.0; 6.0) |
Response was defined as a platelet count ≥ 30 × 109/L and a 2-fold increase from baseline, no new bleeding events, and no intake of forbidden medications. Complete Response was defined as a platelet count ≥ 100 × 109/L, no new bleeding events, and no intake of forbidden medications. *Determined in the full analysis set N = 38. †N = 37; exclusion of one patient who took a forbidden treatment on Day 1. CI: confidence interval.
A Complete Response (Table 2) was observed in 11 patients (28.9% of patients in the full analysis set; 95% CI: 15.4; 45.9). The median time to Complete Response was 2 days (range 1 to 8 days). The estimated median duration of Complete Response was 12 days (95% CI: 10; 18 days). Among the 6 refractory patients, four patients achieved Complete Response and one patient only achieved Response. Among the 14 non-responders, 12 patients failed to reach the required platelet count, one patient had a new bleeding event (mild epistaxis on Day 3), and one patient received a forbidden medication (hydrocortisone on Day 1 and 2).
Platelet assessments
Mean platelet count increased from 16.7 × 109/L at baseline to 45.4 × 109/L on Day 2 post-infusion, reached a mean peak value of 139.4 × 109/L on Day 7, then decreased to 81.8 × 109/L on Day 14, and continued to decrease gradually until Day 21 (Fig. 1). The maximum platelet count per patient (Table 2) was 165 × 109/L (95% CI: 113; 189) and was reached in a median time of 4 days (range 1 to 29 days). Among the 23 patients with a baseline platelet count ≤ 20 × 109/L, 19 of them (82.6%; 95% CI: 61.2; 95.0) achieved a platelet count ≥ 50 × 109/L at least once by Day 5.
Figure 1.
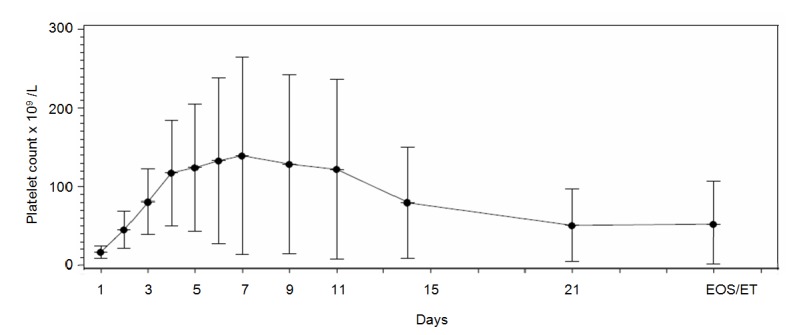
Platelet count over time. Blood samples for platelet determination were drawn on Day 1 pre-infusion (hospital), Day 2 post-infusion (hospital), Days 3, 4, 5, 6, 7, 9, 11 (outpatient/visiting nurse), Day 14 ± 1 (hospital), Day 21 ± 1 (outpatient/visiting nurse) and Day 30 ± 1 (hospital). Means and standard deviations are presented. EOS/ET: end of study/early termination.
Bleeding assessments
Twenty-one of the 29 patients (72.4%) with bleeding at baseline improved their Khellaf score by Day 14. In 16 patients (55.2%), bleeding completely resolved; in six patients (20.7%) bleeding was stable; and in two patients, bleeding worsened. In one of these two patients, worsening led to red blood cell transfusions on Day 5 due to an aggravation of chronic genital bleeding. The number of patients with no bleeding varied from 9/37 (23.7%) at baseline to 11/36 (30.6%) on Day 2, 21/31 patients (67.7%) on Day 14, and 18/29 patients (62.1%) at the end-of-study visit.
A total of 25 new hemorrhages in 15 patients (39.5%) occurred between the second infusion and the end-of-study visit, of which 20 occurred on or after Day 14. New bleeding events were mild (23 events; 13 patients) and moderate (two events; two patients). New hemorrhages that occurred in more than one patient were purpura/petechiae (eight events; six patients), epistaxis (five events; three patients), skin hemorrhage (three events; three patients), and contusion (two events; two patients).
Safety
A total of 89 TEAEs were reported in 31 patients (81.6%) (Table 3). Sixty-nine TEAEs were of mild intensity (77.5% of TEAEs; 18 patients), 18 were moderate (20.2% of TEAEs; 11 patients), and two were severe (2.2% of TEAEs; two patients). The two severe TEAEs were not drug-related and consisted of an unintentional underdose of study medication (one patient) and a positive HIV test (one patient).
Table 3. Safety Evaluation: Adverse Events and Serious Adverse Events (38 Patients, 73 Infusions).
| Patients (N (% patients)) | Adverse event (N) | |
|---|---|---|
| Treatment emergent adverse events, total | 31 (81.6) | 89 |
| Treatment emergent drug-related adverse events, total | 25 (65.8) | 66 |
| Treatment emergent drug-related adverse events, in ≥ two patients | ||
| Headache | 13 (34.2) | 21 |
| Pyrexia | 6 (15.8) | 6 |
| Creatinine renal clearance decrease or GFR decrease | 4 (10.5) | 6 |
| Systolic blood pressure increase | 3 (7.9) | 4 |
| Vomiting | 3 (7.9) | 3 |
| Body temperature increase | 2 (5.3) | 2 |
| Influenza-like illness | 2 (5.3) | 2 |
| Nausea | 2 (5.3) | 2 |
| Arthralgia | 2 (5.3) | 2 |
| Temporally associated adverse events, total | 27 (71.1) | 64 |
| Discontinuation of study drug due to adverse event | 2 (5.3) | 4 |
| Interruption of infusion due to adverse event | 2 (5.3) | 2 |
| Flow rate decrease due to mild pyrexia | 1 (2.6) | 1 |
| Serious adverse events, total | 7 (18.4) | 8 |
| Study drug overdose | 1 (2.6) | 1 |
| Study drug underdose | 1 (2.6) | 1 |
| Decrease in glomerular filtration rate | 1 (2.6) | 1 |
| Aseptic meningitis | 1 (2.6) | 1 |
| Influenza-like illness | 1 (2.6) | 1 |
| Pyrexia | 1 (2.6) | 1 |
| Headache, recurrent | 1 (2.6) | 1 |
| Positive human immunodeficiency virus test | 1 (2.6) | 1 |
GFR: glomerular filtration rate.
A total of 67 drug-related TEAEs were observed in 25 patients. The most common drug-related TEAEs were headache (34.2% of patients), pyrexia (15.8%), creatinine renal clearance decrease or GFR decrease (10.5%), systolic blood pressure increase (7.9%), vomiting (7.9%), influenza-like illness (5.3%), arthralgia (5.3%), and nausea (5.3%).
In addition, in one patient, a transient anaphylactoid reaction consisting of swallowing disorder, acuphenia, and dyspnea was observed during the first infusion and resolved without sequelae after a temporary interruption of the infusion and intravenous hydrocortisone. Lastly, in one patient, a case of aseptic meningitis associating headache, nausea, and vomiting was reported. Recovery was complete after treatment of symptoms with medication.
Eight TEAEs, which occurred in seven patients, were serious adverse events (SAEs). Six of these SAEs were drug-related: aseptic meningitis (one patient), influenza-like illness (one patient), accidental overdose (one patient), underdose (one patient), in addition one patient experienced two concomitant SAEs which were vascular encephalopathy with hydrocephalus syndrome. All patients recovered without sequelae. The two other SAEs (previously mentioned) were positive HIV test (one patient) and creatinine renal clearance decreased (one patient) which were assessed as not related to study drug by physicians.
Sixty-four of the TEAEs, which occurred in 27 patients (71.1%), were TAAEs. Overall, 36 of the 73 infusions (49.3%) were associated with at least one TAAE. No noteworthy differences were observed between Day 2 infusions (18 of 35 infusions; 51.4%) and Day 1 infusions (18 of 38 infusions; 47.4%). The percentage of infusions associated with at least one TAAE was similar regardless of flow rate: 50% (10 of 20) of infusions with a maximum flow rate > 4 mL/kg/h and 49.1% (26 of 53) of infusions with a maximum flow rate ≤ 4 mL/kg/h. No relationship between the type, frequency, or intensity of TAAEs and the infusion flow rate was observed, with the exception of a trend towards more frequent headaches at flow rates > 6 mL/kg/h.
The analysis of laboratory parameters did not show any new safety signals. Positive direct antiglobulin tests without biological evidence of hemolysis were observed in two patients.
Discussion
This prospective, single-arm, open-label, multicentre, phase III study is one of the first clinical trials to evaluate the efficacy of an IVIG in patients with chronic ITP using the stringent definition of Response from the revised EMA guideline dated July 2010. It takes into account the new standardization and definition criteria proposed by an International Working Group [1, 18]. IQYMUNE® showed a Response rate of 63.2% in the full analysis set (95% CI: 46.0; 78.2), a median time to Response of 1 day, and a median duration of platelet response of 13.5 days.
Using the definition of response of the previous EMA guideline [19], a rate of 82.6% (95% CI: 61.2; 95.0) was obtained. This result is similar to that reported for other IVIG 10% preparations: 76.1% (95% CI: 63.5 - 86.0) for Flebogamma Dif [12], 80.0% (95% CI: 72.7 - 87.3) for Octagam [13], 80.7% (95% CI: 69.2 - 89.3) for Privigen [17], 91.7% (95% CI: 73.0 - 99.0) for Intratect [9], and 73.9% (95% CI 53.5 to 87.7) for Kiovig [18]. As expected, the revised endpoint resulted in a lower response rate since it requires a platelet count ≥ 30 × 109/L with at least a 2-fold increase in the baseline level, the sustainability of this increase in platelet count for at least 7 days, and the absence of even mild new bleeding events.
The median time to Response of 1 day observed in the current study was shorter than the median times of 2 - 4 days reported in the previously mentioned studies [9, 11-13, 22]. These data show that IQYMUNE® can rapidly increase the platelet count in patients presenting with, or at high risk of, severe bleeding. The median duration of Response of 13.5 days was within the range of 10 to 25.5 days reported in the literature [9, 11, 12, 22]. This duration of the response is enough to overcome a bleeding episode and to manage potentially hemorrhagic surgery.
Improvement of the bleeding status also provides evidence of the therapeutic effect of IVIGs [3]. Bleeding events reported in this study were representative of the disease and mainly related to subcutaneous tissue disorders (purpura, petechiae, and skin hemorrhage) or epistaxis. New bleeding events occurred with progressively increasing frequency and severity over the study period; they were mild and infrequent until Day 14 (five mild new events) and increased thereafter (18 mild and two moderate new events) platelet response decreased in some patients.
IQYMUNE® was well tolerated. Safety evaluations, including AEs, laboratory values, and vital signs did not uncover any unexpected safety issues. A total of 89 AEs were reported in 81.6% of patients. Although in our study perfusion rates were high (≥ 6 - 8 mL/kg/h) in 40% of patients; comparable frequencies have been reported in the literature after administration of high-dose IVIG in ITP patients [9, 11, 23]. The most common treatment-related adverse reactions, which were similar to that found with other IVIG 10%, were mild or moderate headache and fever [11, 24, 25]. As pre-medication to prevent AEs was not allowed, it is likely that our design resulted in increased rates of headache, nausea, and vomiting compared to the rate that would be expected in clinical practice. The other frequent reactions were creatinine renal clearance decrease and systolic blood pressure increase, both of which are commonly noted after treatment with high dose IVIG. No cases of thrombosis, hemolysis, or renal failure were observed during the course of the study.
Overall, our results show that some manageable and reversible side effects due to IVIG may still occur. These side effects, which arise despite the progress made in the manufacturing procedures and strict quality control implementation, tend to be milder and less frequent in comparison with the previous generations of product. In fact, no treatments are devoid of risk in ITP, including TPO-RA, the most widely used agents. Up to 30% (or more) of patients are unresponsive or cannot tolerate these treatments and recent reports have highlighted a significant treatment-related morbidity [26].
In summary, IQYMUNE® administered at a dose of 1 g/kg/day on 2 consecutive days was effective in increasing platelet count and preventing bleeding events. It was well tolerated in adult patients with chronic ITP.
Acknowledgments
We thank Martin Kappler, statistician (Statalpha, Baziege, France). We thank Galien Health Publishing and Nezha Chambron (LFB SA) for medical writing support. We also thank the investigators for their contribution of this study: Zoltan Boda, Gianluca Gaidano, Andrzej Pluta, Nuria Bermejo, Gyorgy Blasko, Mohamed Hamidou, Andrzej Lange, Elena Michailova Volodicheva, Agnes Nagy, Hanna Oliynyk, Yaroslava Vyhovska.
Financial Disclosures
Francesco Rodeghiero: Member of Advisory Boards for LFB, Amgen, Argenx, Novartis; Dariusz Woszczyk: No conflict of interest. Borhane Slama: No conflict of interest. Anait Melikyan: No conflict of interest. Jean-Francois Viallard: Participation in advisory boards for: LFB, GSK, AMGEN, NOVARTIS; speakers at symposia for: NOVARTIS, GSK, AMGEN, LFB, SHIRE, CSL-BEHRING; Rabye Ouaja: LFB Biomedicaments; Ousmane Alfa Cisse: LFB Biomedicaments; Abdulgabar Salama: No conflict of interest.
References
- 1.Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, Bussel JB. et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–2393. doi: 10.1182/blood-2008-07-162503. [DOI] [PubMed] [Google Scholar]
- 2.Bolton-Maggs P. Severe bleeding in idiopathic thrombocytopenic purpura. J Pediatr Hematol Oncol. 2003;25(Suppl 1):S47–51. doi: 10.1097/00043426-200312001-00011. [DOI] [PubMed] [Google Scholar]
- 3.Neunert CE. Individualized treatment for immune thrombocytopenia: predicting bleeding risk. Semin Hematol. 2013;50(Suppl 1):S55–57. doi: 10.1053/j.seminhematol.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 4.McMillan R. The pathogenesis of chronic immune (idiopathic) thrombocytopenic purpura. Semin Hematol. 2000;37(1 Suppl 1):5–9. doi: 10.1016/S0037-1963(00)90111-2. [DOI] [PubMed] [Google Scholar]
- 5.Stasi R. Pathophysiology and therapeutic options in primary immune thrombocytopenia. Blood Transfus. 2011;9(3):262–273. doi: 10.2450/2010.0080-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA, American Society of H. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190–4207. doi: 10.1182/blood-2010-08-302984. [DOI] [PubMed] [Google Scholar]
- 7.Jin F, Balthasar JP. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum Immunol. 2005;66(4):403–410. doi: 10.1016/j.humimm.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 8.Imbach P, Barandun S, d'Apuzzo V, Baumgartner C, Hirt A, Morell A, Rossi E. et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981;1(8232):1228–1231. doi: 10.1016/S0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- 9.Colovic M, Dimitrijevic M, Sonnenburg C, Suvajdzic N, Donfrid M, Bogdanovic A. Clinical efficacy and safety of a novel intravenous immunoglobulin preparation in adult chronic ITP. Hematol J. 2003;4(5):358–362. doi: 10.1038/sj.thj.6200303. [DOI] [PubMed] [Google Scholar]
- 10.Godeau B, Lesage S, Divine M, Wirquin V, Farcet JP, Bierling P. Treatment of adult chronic autoimmune thrombocytopenic purpura with repeated high-dose intravenous immunoglobulin. Blood. 1993;82(5):1415–1421. [PubMed] [Google Scholar]
- 11.Julia A, Kovaleva L, Loria S, Alberca I, Hernandez F, Sandoval V, Sierra J. et al. Clinical efficacy and safety of Flebogammadif, a new high-purity human intravenous immunoglobulin, in adult patients with chronic idiopathic thrombocytopenic purpura. Transfus Med. 2009;19(5):260–268. doi: 10.1111/j.1365-3148.2009.00945.x. [DOI] [PubMed] [Google Scholar]
- 12.Kovaleva L, Apte S, Damodar S, Ramanan V, Loriya S, Navarro-Puerto J, Khojasteh A. et al. Safety and efficacy of a 10% intravenous immunoglobulin preparation in patients with immune thrombocytopenic purpura: results of two international, multicenter studies. Immunotherapy. 2016;8(12):1371–1381. doi: 10.2217/imt-2016-0088. [DOI] [PubMed] [Google Scholar]
- 13.Robak T, Mainau C, Pyringer B, Chojnowski K, Warzocha K, Dmoszynska A, Straub J. et al. Efficacy and safety of a new intravenous immunoglobulin 10% formulation (octagam(R) 10%) in patients with immune thrombocytopenia. Hematology. 2010;15(5):351–359. doi: 10.1179/102453310X12719010991867. [DOI] [PubMed] [Google Scholar]
- 14.Godeau B, Chevret S, Varet B, Lefrere F, Zini JM, Bassompierre F, Cheze S. et al. Intravenous immunoglobulin or high-dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: a randomised, multicentre trial. Lancet. 2002;359(9300):23–29. doi: 10.1016/S0140-6736(02)07275-6. [DOI] [PubMed] [Google Scholar]
- 15.Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, Chong BH. et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115(2):168–186. doi: 10.1182/blood-2009-06-225565. [DOI] [PubMed] [Google Scholar]
- 16.Mayer B, Depre F, Ringel F, Salama A. New aspects on the efficacy of high-dose intravenous immunoglobulins in patients with autoimmune thrombocytopenia. Vox Sang. 2017;112(1):64–69. doi: 10.1111/vox.12467. [DOI] [PubMed] [Google Scholar]
- 17.Salama A. Emerging drugs for immune thrombocytopenia (ITP) Expert Opin Emerg Drugs. 2017;22(1):27–38. doi: 10.1080/14728214.2017.1294158. [DOI] [PubMed] [Google Scholar]
- 18. United States Food and Drug Administration, European Medicines Agency. Conclusions of FDA-EMA parallel assessment of quality-by-design elements of marketing applications. 15 Guidance for Industry Q8(R2) Pharmaceutical Development - November 2009 ICH Revision 2. [cited 2017; Available from: http://www.fda.gov/Drugs/DrugSafety/ucm365524.htm.
- 19. European Medicines Agency. Guideline on the clinical investigation of human normal immunoglobulin for intravenous administration (IVIg). EMA/CHMP/BPWP/94033/2007 rev. 2.; 2010.
- 20. European Agency for the Evaluation of Medicinal Products. Note for Guidance on the clinical investigation of human normal immunoglobulin for intravenous administration (IVIg). CMPM/BPWG/388/95 rev. 1. 2000.
- 21.Khellaf M, Michel M, Schaeffer A, Bierling P, Godeau B. Assessment of a therapeutic strategy for adults with severe autoimmune thrombocytopenic purpura based on a bleeding score rather than platelet count. Haematologica. 2005;90(6):829–832. [PubMed] [Google Scholar]
- 22.Robak T, Salama A, Kovaleva L, Vyhovska Y, Davies SV, Mazzucconi MG, Zenker O. et al. Efficacy and safety of Privigen, a novel liquid intravenous immunoglobulin formulation, in adolescent and adult patients with chronic immune thrombocytopenic purpura. Hematology. 2009;14(4):227–236. doi: 10.1179/102453309X439773. [DOI] [PubMed] [Google Scholar]
- 23.Borte M, Davies S, Touraine J-L, Farber C, Lipsic T, Adams C, Spath P. et al. Clinical properties of a novel liquid intravenous immunoglobulin: studies in patients with immune thrombocytopenic purpura and primary immunodeficiencies. Transfus Med Hemother. 2004;31:126–134. doi: 10.1159/000079071. [DOI] [Google Scholar]
- 24.Varga G, Volkova Z, Leibl H, Gasztonyi Z, Hlusi A, Mayer J, Chojnowski K. et al. Efficacy and safety of the new intravenous immunoglobulin IGIV 10% in adults with chronic idiopathic thrombocytopenic purpura. Transfus Med Hemother. 2006;33:509–514. doi: 10.1159/000095764. [DOI] [Google Scholar]
- 25.Julia A. The experience of Flebogammadif(R) in primary immune thrombocytopenia. Clin Exp Immunol. 2011;164(Suppl 2):12–15. doi: 10.1111/j.1365-2249.2011.04389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Depre F, Aboud N, Ringel F, Salama A. Thrombopoietin receptor agonists are often ineffective in immune thrombocytopenia and/or cause adverse reactions: results from one hand. Transfus Med Hemother. 2016;43(5):375–379. doi: 10.1159/000446195. [DOI] [PMC free article] [PubMed] [Google Scholar]