

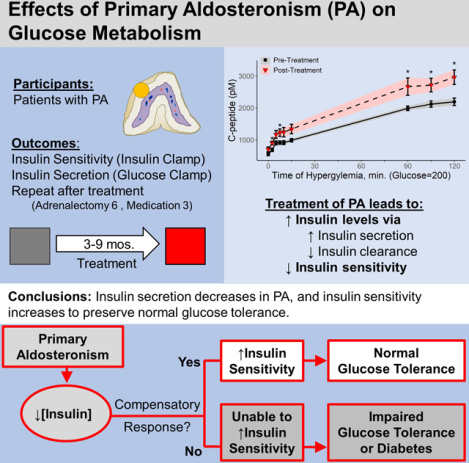
Abstract
Primary aldosteronism is a frequent cause of resistant hypertension and is associated with an increased risk of developing diabetes. Aldosterone impairs insulin secretion in isolated islets and insulin secretion is increased in aldosterone synthase deficient mice. We hypothesized that treatment for primary aldosteronism increases insulin secretion and insulin sensitivity in humans. We conducted a prospective cohort study in patients with primary aldosteronism, with assessment of glucose metabolism before and 3–12 months after treatment. Participants underwent treatment for primary aldosteronism with adrenalectomy or a mineralocorticoid receptor antagonist at the discretion of their treating physician. We assessed insulin secretion and insulin sensitivity by hyperglycemic and hyperinsulinemic-euglycemic clamps, respectively, on two study days after a 5-day standardized diet. After treatment, the C-peptide and insulin response during the hyperglycemic clamp increased compared to pre-treatment (ΔC-peptide at 90–120 min +530.5 ± 384.1 pmol/L, p=0.004; ΔInsulin 90–120 min +183.0 ± 122.6, p=0.004). During hyperinsulinemic-euglycemic clamps, insulin sensitivity (M/I) decreased after treatment (30.7 ± 6.2 vs 18.5 ± 4.7 nmol•kg−1•min−1•pmol−1•L; p=0.02). Insulin clearance decreased after treatment (872.8±207.6 vs 632.3±178.6 mL/min, p=0.03), and disposition index was unchanged. We conclude that the insulin response to glucose increases, and insulin clearance decreases after treatment for primary aldosteronism, and these effects were not due to alterations in creatinine clearance or plasma cortisol. These studies may provide further insight into the mechanism of increased diabetes risk in primary aldosteronism.
Keywords: Aldosterone, mineralocorticoids, Insulin sensitivity, Insulin secretion, Hypertension
Graphical Abstract
INTRODUCTION
Primary aldosteronism is the most common identifiable cause of resistant hypertension and is associated with increased risk of cardiovascular complications compared to essential hypertension (1). Primary aldosteronism is also associated with glucose intolerance and an increased risk of type 2 diabetes (2–4), and diabetes treatment guidelines list primary aldosteronism as a potential secondary cause of diabetes. The mechanisms responsible for this association remains unclear.
JW Conn observed diabetes in nearly half of his initial cohort of patients with hyperaldosteronism, and ascribed this to an insulin secretory defect induced by hypokalemia (5). In his series, however, potassium repletion did not completely restore glucose tolerance and insulin secretion to the same extent as surgical cure of patients with aldosterone-producing adenomas. We recently demonstrated that aldosterone directly impairs insulin secretion in isolated murine pancreatic islets, and that genetic aldosterone deficiency markedly increases insulin secretion in mice (6, 7). Similarly, short-term stimulation of the renin-angiotensin-aldosterone system during sodium restriction impairs insulin secretion in humans (8). In patients with aldosterone-producing adenomas, insulin secretion and disposition index assessed with intravenous glucose tolerance tests increase after adrenalectomy, suggesting that aldosterone impairs insulin secretion directly in humans (9).
Insulin sensitivity and secretion should ideally be determined separately by hyperinsulinemic-euglycemic clamp and by hyperglycemic clamp studies utilizing C-peptide measurement, respectively. We conducted a prospective study of patients presenting with hyperaldosteronism using these methods to determine the effect of adrenalectomy or medical treatment on glucose metabolism.
METHODS
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion Guidelines. The data that support the findings of this study are available from the corresponding author on reasonable request.
Study protocol
All study procedures and protocols were approved by the Vanderbilt University Medical Center IRB and Brigham and Women’s Hospital IRB prior to enrollment, and the study was conducted in accordance with the Declaration of Helsinki. This study was registered at clinicaltrials.gov prior to the enrollment of the first participant ( NCT02362308). Patients age 18 to 70 years with primary aldosteronism were identified prospectively using physician referral and medical record search. After informed consent, participants underwent a screening history and physical exam and were included if they met the criteria for primary aldosteronism. Diagnosis was established by 1) screening plasma aldosterone ≥ 15 ng/dL and aldosterone-to-renin ratio of ≥40 (or ≥30 if on ACE inhibitor) and 2) confirmatory testing by either failure to suppress aldosterone to <7ng/dL after intravenous saline infusion or urinary aldosterone excretion to < 12 μg/day when urine sodium excretion is ≥200 mmol/day. Females of child-bearing potential were excluded unless they were willing to use adequate birth-control and undergo pregnancy testing throughout the study. Other exclusion criteria included prior diagnosis of T2DM or T1DM or use of anti-diabetic medication, potassium >5.5 mmol/L, sodium <135 mmol/L, allergy to study medications, or major cardiovascular, renal, hepatic, pulmonary, neurologic, or psychiatric disorders. Metabolic assessment at baseline was performed after withdrawal of mineralocorticoid receptor antagonists or amiloride for at least six weeks.
To standardize sodium intake in both study periods, participants were provided a calorie-controlled diet containing 160 mmol/d sodium, 100 mmol/d potassium, and 1000 mg/d calcium for 5 days prior to assessment. The calorie content of the diet was calculated for weight maintenance with 55% of calories from carbohydrates, 25% from fats, and 20% from proteins. During the last day of diet, participants collected their urine for measurement of electrolytes and creatinine. A hyperglycemic clamp and hyperinsulinemic-euglycemic clamp were performed on day 5 and day 6 (at Brigham and Women’s Hospital) or day 5 and day 7 (at Vanderbilt) of study diet, respectively, after participants had fasted overnight and reported to the Clinical Research Center. After 30 minutes in the supine position on each study day, blood pressure was measured three times and the average was calculated (Dinamap, Critikon, Carlsbad, CA). Clamp studies were then performed. Anti-hypertensive medications were prescribed according to routine medical care at the discretion of the treating physician but use of a mineralocorticoid receptor antagonist or potassium-sparing diuretic was prohibited for at least 6 weeks prior to the initial study day. After completion of the pretreatment clamp studies, participants underwent either unilateral adrenalectomy or initiation of mineralocorticoid receptor antagonist treatment according to standard medical care and repeated the detailed metabolic assessment after 3 to 12 months.
Hyperglycemic clamps
After an overnight fast, primed continuous glucose was infused for 2 hours via an antecubital vein, as previously described.(8, 10–13) Arterialized blood was obtained through an indwelling venous cannula inserted into a hand vein. The hand was warmed by placing the hand under a heating pad or in a plexiglass box warmed to 60°C. An intravenous priming dose (0.2 g/kg) of 20% dextrose monohydrate was given over 10 minutes at time zero, and plasma glucose was measured every 2.5 to 5 minutes. The glucose infusion rate (20% dextrose) was then adjusted according to protocolized calculations to maintain plasma glucose near 200 mg/dL for 150 minutes (13). At 120 minutes, intravenous 5 g L-arginine hydrochloride was administered over 5 minutes to assess non-glucose stimulated insulin release.
Glucose, insulin, and C-peptide concentrations at −10 and −1 minutes prior to glucose administration were averaged to calculate baseline (t=0) measurements. The acute insulin and C-peptide responses (ΔInsulin0–10, ΔC-peptide0–10) were defined as the maximal change within 0–10 minutes of the initial glucose loading, and the baseline-subtracted area-under-the-curve from 0–10 minutes were calculated for insulin (AUC ΔInsulin0–10) and C-peptide (AUC ΔC-peptide0–10). The second phase insulin response was calculated as the incremental change in average plasma insulin during steady state (time period 90 to 120 minutes) above the baseline value. L-Arginine stimulated insulin secretion was similarly calculated as the incremental change 0–10 minutes after L-Arginine injection above the average insulin from time period 90–120 min (ΔInsulinL-Arginine). The hyperglycemic clamp-derived insulin sensitivity index was calculated by dividing the steady state glucose infusion rate from 90–120 minutes (μmol/kg/min) by the averaged insulin concentration at 90–120 minutes (pmol/mL) (8, 11). We selected individuals without PA from a database of previously conducted hyperglycemic clamps to match our primary aldosteronism participants by age, gender, race, and body mass index using the pairmatch function (R package optmatch, v 0.9–10).
Hyperinsulinemic-Euglycemic clamps
Insulin was administered at a low dose (20 mU/m2/min) followed by a high dose (120 mU/m2/min) for 2 hours at each infusion rate. Plasma glucose was measured every 5 minutes and the exogenous infusion rate was adjusted to maintain plasma glucose at a 95 mg/dL (5.3 mM) (13, 14). The disposition index (DI) was calculated as the product of hyperglycemic clamp-derived acute insulin response (AUC ΔInsulin0–10) and hyperinsulinemic clamp-derived insulin sensitivity index (M/I) (11). Insulin clearance was calculated as the insulin infusion rate divided by average insulin concentration during the last 30 minutes of each insulin infusion period, adjusted for C-peptide to account for endogenous insulin secretion (15).
Body Composition and energy expenditure.
Body composition was measured by Dual Energy X-ray Absorptiometry (DEXA) once during each study period, with a GE Lunar model IDXA Absorptiometer. Resting energy expenditure was measured using indirect calorimetry with a ventilated canopy device (CPX/D system, Medical graphics Corporation, St Paul, MN) (16).
Assays
Blood for plasma glucose was immediately centrifuged and analyzed using the glucose oxidase method (YSI 2300 STAT Plus Glucose Analyzer YSI Life Sciences; Yellow Springs, IL). Blood was collected in EDTA and plasma was separated and stored at −80°C until assayed. Aprotinin was added to plasma for measurement of insulin and C-peptide by radioimmunoassay. Insulin, C-peptide, and ACTH were determined by RIA in the Vanderbilt Diabetes Research Translational Center Hormone Assay laboratory. Plasma corticosteroids were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) at the University of Michigan as previously described (17, 18). Fructosamine was measured using a nitroblue tetrazolium-based colorimetric assay (Abcam kit ab228558).
Statistics
Primary outcomes were prespecified to be insulin secretion, insulin sensitivity, and disposition index determined during the clamps. The primary insulin secretion measures obtained from hyperglycemic clamps were acute insulin response (ΔC-peptide0–10), second phase (ΔC-peptide90–120), and L-arginine insulin response (ΔInsulinL-Arginine). Insulin sensitivity measures obtained from the hyperinsulinemic clamp were the insulin sensitivity index (M/I). Differences between treatment periods were compared using Wilcoxon signed-rank test and differences between groups using the Wilcoxon rank-sum test. A two-sided p-value <0.05 was considered significant. All data analysis was performed using the open source statistical package R (version ≥ 3.5.1) (19).
In hyperglycemic clamps, insulin measurements were unavailable at a total of four time points of two participants and C-peptide results were unavailable at two time points in two participants (samples unavailable). In hyperinsulinemic clamps, insulin measurements were unavailable at a total of three time points of three subjects, and unavailable after 120 min for one subject (sample unavailable). We imputed the missing insulin values, except for the last subject mentioned above. If the signs of the slopes of the two lines formed by the preceding two points and by the subsequent two points are the same, the missing value was imputed by the time-weighted average of its two closest neighbors. If the signs are opposite, the missing value was imputed by the average of the extrapolation of the two lines at this point. Results are presented with imputed values.
RESULTS
Research Participants
Ten patients consented to the study procedures, and 1 participant dropped out due to scheduling difficulty before any metabolic assessment. One participant consented for and completed only the hyperglycemic clamp assessments. Six patients underwent adrenalectomy for treatment of an aldosterone-producing adenoma and three patients with idiopathic hyperaldosteronism were treated with a mineralocorticoid receptor antagonist. Subject baseline characteristics and laboratory evaluation at the time of initial evaluation are shown in Table 1, stratified by treatment. There were no clear differences in screening measurements based on treatment choice. The duration between assessment periods was similar (6.8±2.8 and 5.0±0.8 months in adrenalectomy and medically-treated participants, respectively; p=0.58).
Table 1.
Participant characteristics obtained at time of screening
| Variable | Adrenalectomy (n=6) | MRA (n=3) | All (n=9) |
|---|---|---|---|
| Age (years) | 52.4 ± 7.8 | 44.0 ± 12.2 | 49.6 ± 9.6 |
| Gender, female (%) | 3 (50%) | 1 (33%) | 4/9 (44%) |
| Race, n (%) | |||
| White | 5/6 (83%) | 1/3 (33%) | 6/9 (67%) |
| African American | 1/6 (17%) | 2/3 (67%) | 3/9 (33%) |
| Body mass index (kg/m2) | 26.5 ± 2.8 | 33.9 ± 6.3 | 29.0 ± 5.3 |
| Systolic blood pressure (mmHg) | 139.0 ± 16.4 | 144.3 ± 5.5 | 140.8 ± 13.5 |
| Diastolic blood pressure (mmHg) | 86.0 ± 7.2 | 82.3 ± 10.3 | 84.8 ± 7.9 |
| Heart rate (bpm) | 66.0 ± 12.0 | 73.0 ± 7.2 | 68.3 ± 10.7 |
| Aldosterone (ng/dL) | 39.2 ± 40.7 | 21.0 ± 3.8 | 33.2 ± 33.5 |
| Plasma Renin Activity (ng/mL/hr) | 0.2 ± 0.2 | 0.4 ± 0.1 | 0.3 ± 0.2 |
| Aldosterone:PRA ratio | 197.9 ± 81.8 | 41.2 ± 13.5 | 153.2 ± 101.7 |
Results are mean ± SD.
Effect of treatment on Blood Pressure and Potassium
Averaged values obtained on the study days for systolic blood pressure (131.1 ± 10.8 vs 124.3 ± 23.1 mmHg for pre- vs post-treatment, respectively; p=0.43), serum potassium (3.67 ± 0.24 vs 4.12 ± 0.35 mEq/L; p=0.063), and creatinine (1.09 ± 0.16 vs 1.21 ± 0.38 mg/dL; p=0.35) did not markedly change after treatment. Additional changes after treatment are presented in Table 2, and changes in steroid hormones are presented in Table S1. Potassium supplementation was prescribed less often after treatment (7/9 before vs 1/9 after treatment; p=0.04), and the mean daily potassium chloride supplementation decreased accordingly (80.0 ± 64.0 vs 2.2 ± 6.7 mEq/d after treatment; p=0.02). Blood pressure medications were prescribed to a similar proportion of participants (8/9 before vs 6/9 after treatment; p=0.62), but the number of non-mineralocorticoid receptor antagonist antihypertensive medications was reduced (2.5 ± 1.3 before vs 1.3 ± 1.1 medications after treatment; p=0.049). The use of individual antihypertensive medication classes was similar (Table S2).
Table 2.
Baseline and post-treatment assessment
| Measure | Pre-Treatment | Post-Treatment | Mean within-subject change | p-value |
|---|---|---|---|---|
| Weight (kg) | 87.7 ± 23.0 | 89.3 ± 22.1 | 1.6 ± 3.2 | 0.25 |
| Systolic blood pressure (mmHg) | 131.1 ± 10.8 | 124.3 ± 23.1 | −6.9 ± 22.5 | 0.43 |
| Diastolic blood pressure (mmHg) | 81.7 ± 9.0 | 77. 8 ± 13.8 | −3.9 ± 14.2 | 0.41 |
| Heart Rate (bpm) | 61.3 ± 7.2 | 59.6 ± 6.2 | −1.6 ± 4.6 | 0.30 |
| Serum Potassium (mmol/L) | 3.67 ± 0.24 | 4.12 ± 0.35 | 0.4 ± 0.5 | 0.06 |
| Creatinine (mg/dL) | 1.09 ± 0.16 | 1.21 ± 0.38 | 0.12 ± 0.39 | 0.35 |
| Glucose, fasting (mmol/L) | 5.18 ± 0.60 | 5.22 ± 0.58 | 0.0 ± 0.4 | 0.82 |
| Insulin, fasting (pmol/L) | 61.21 ± 16.13 | 83.18 ± 37.09 | 22.0 ± 46.7 | 0.13 |
| C-peptide, fasting (pmol/L) | 525.34 ± 105.12 | 693.59 ± 224.70 | 168.2 ± 260.5 | 0.13 |
| C-peptide/Insulin ratio | 9.20 ± 2.95 | 9.11 ± 2.64 | −0.1 ± 2.6 | 1.0 |
| Fructosamine (μmol/L) | 461.2±169.1 | 457.1±101.6 | −4.1±94.2 | 0.94 |
| Aldosterone (ng/dL) | 20.4 ± 15.2 | 6.2 ± 3.7 | −14.3 ± 16.6 | 0.02 |
| 11-deoxycorticosterone (pg/mL) | 47.4 ± 34.1 | 16.6 ± 19.3 | −30.9 ± 41.3 | 0.02 |
| Cortisol (μg/dL) | 9.7±2.7 | 9.2±2.6 | −0.6±3.4 | 0.57 |
| Corticotropin (pg/mL) | 40.4 ± 7.4 | 46.6 ± 16.0 | 6.2 ± 12.9 | 0.25 |
| Resting Energy Expenditure (kcal/day) | 1723 ± 434 | 1597 ± 284 | −126 ± 207 | 0.15 |
| Respiratory Quotient | 0.81 ± 0.04 | 0.84 ± 0.07 | 0.03 ± 0.04 | 0.14 |
| DEXA Measurements | ||||
| Fat free mass (kg) | 53.6 ± 13.9 | 53.9 ± 13.9 | 0.3 ± 1.8 | 0.65 |
| Fat mass (kg) | 30.9 ± 12.4 | 32.3 ± 12.0 | 14.7 ± 2.1 | 0.07 |
| fat (%) | 35.3 ± 6.5 | 36.7 ± 6.5 | 1.4 ± 1.8 | 0.06 |
| fat, Gynoid (%) | 39.0 ± 9.0 | 39.2 ± 9.9 | 0.1 ± 2.1 | 1.0 |
| fat, Android (%) | 40.4 ± 9.2 | 43.2 ± 9.7 | 2.8 ± 2.6 | 0.02 |
| fat Android/Gynoid ratio | 1.07 ± 0.30 | 1.15 ± 0.35 | 0.08 ± 0.07 | 0.004 |
Results are mean ± SD. Wilcoxon signed-rank test
Average results reported for variables that were assessed on both study days (blood pressure, heart rate, glucose, insulin C-peptide, and insulin/C-peptide ratio).
Effect of treatment on insulin secretion
During hyperglycemic clamps, glucose was infused to achieve a target glycemia of 200 mg/dl (11.1 mmol/L), and the average achieved glucose at 90–120 minutes was similar (10.8±0.2 vs 10.9±0.3 mmol/L before vs after treatment, p=0.20). The glucose-stimulated insulin response increased markedly after treatment (Figure 1A–B and Table 3), despite similar glucose infusion rate to achieve equivalent hyperglycemia (29.9±10.9 vs 42.0±41.9 μmol/kg/min; p=0.36). To establish that altered insulin secretion rather than insulin metabolism contributed to this difference, we measured the C-peptide response which was similarly increased after treatment as compared with before treatment (Figure 1C–D and Table 3). The acute insulin and C-peptide responses were highly correlated (Spearman’s rho=0.79, p < 0.001). The fasting C-peptide/insulin ratio was similar between treatment periods (Table 2). The C-peptide/insulin ratio at 90–120 minutes decreased from the fasting value was significantly decreased after treatment (average decrease of −2.9±3.0 from pre-treatment value, p=0.020), suggesting that insulin clearance was increased prior to treatment in PA, in addition to insulin secretion.
Figure 1.
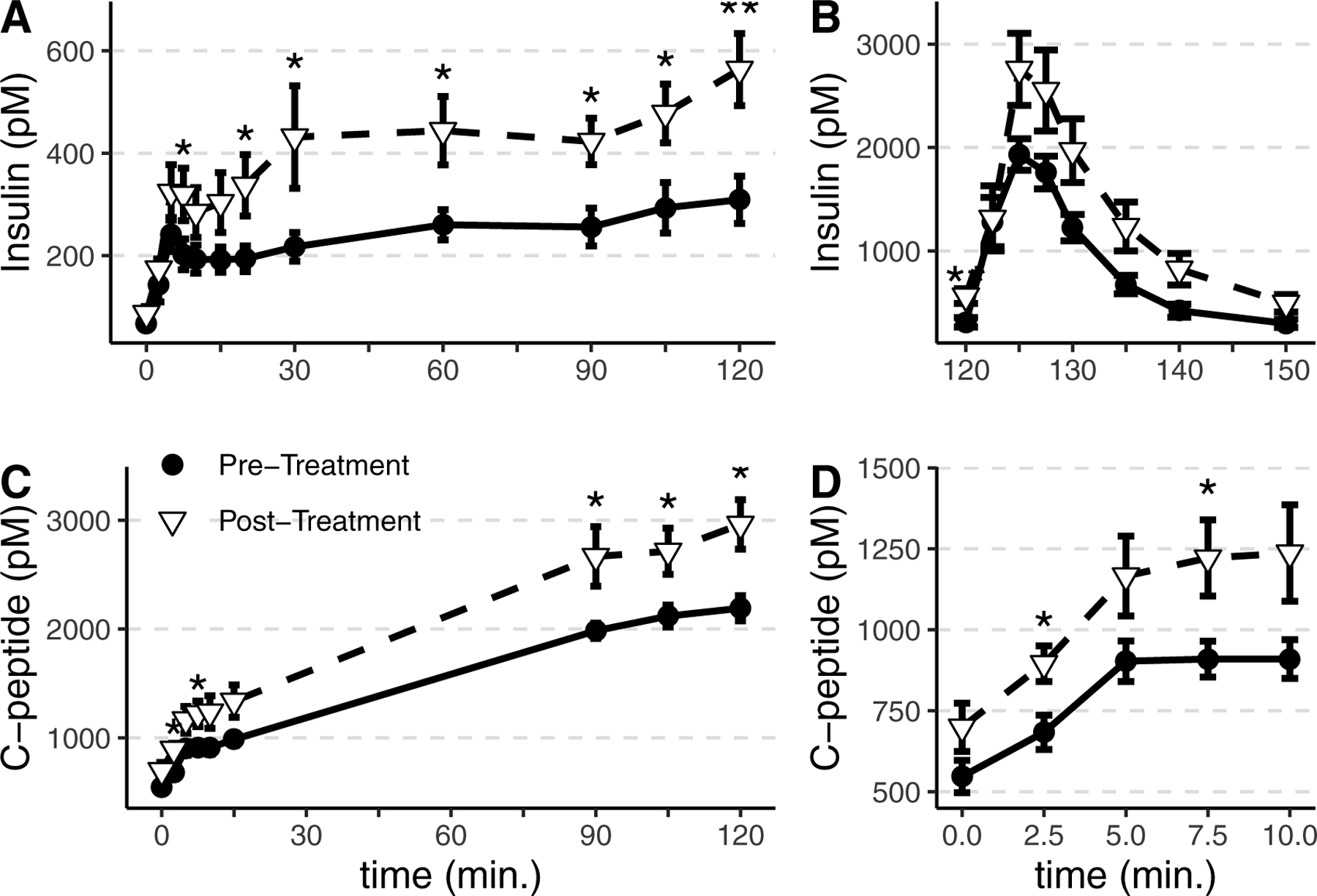
Summary data from hyperglycemic clamps to assess glucose- and L-arginine-stimulated insulin secretion in participants with primary aldosteronism before treatment (●) and post-treatment (▽). Glucose was infused at a similar rate and glucose was maintained at 200mg/dL in both treatment periods. Insulin increased to a greater extent in participants post-treatment in response to hyperglycemia (A) and after L-arginine administration (B). C-peptide increased to a greater extent after treatment in response to hyperglycemia (C, D). The acute insulin response during the initial 0–10 minutes of the clamp is expanded (D). *p<0.05, **p<0.01, ***p<0.001. Group mean and SEM are shown.
Table 3.
Hyperinsulinemic and hyperglycemic clamp summary measures
| Measure | Pre-Treatment | Post-Treatment | Mean within-subject change | p-value |
|---|---|---|---|---|
| Hyperglycemic Clamp | ||||
| ΔInsulin0–10 (pmol/L) | 185.6 ± 104.3 | 256.2 ± 134.8 | 70.7 ± 110.5 | 0.130 |
| AUC ΔInsulin0–10 (pmol • L−1 • min) | 1115.9 ± 734.1 | 1639.5 ± 914.6 | 523.5 ± 699.5 | 0.098 |
| ΔC-peptide0–10 (pmol/L) | 417.2 ± 228.4 | 593.8 ± 336.3 | 176.7 ± 246.6 | 0.074 |
| AUC ΔC-peptide0–10 (pmol • L−1 • min) | 2593.0 ± 1761.7 | 3638.1 ± 2090.0 | 1045.1 ± 2026.6 | 0.130 |
| ΔInsulin90–120 (pmol/L) | 218.6 ± 119.4 | 401.5 ± 126.4 | 183.0 ± 122.6 | 0.004 |
| ΔC-peptide90–120 (pmol/L) | 1551.4 ± 240.2 | 2081.9 ± 561.1 | 530.5 ± 384.1 | 0.004 |
| ΔC-peptide:Insulin ratio90–120 | 0.2 ± 1.9 | −2.7 ± 1.5 | −2.9 ± 3.0 | 0.020 |
| ΔInsulinL-Arginine (pmol/L) | 1730.3 ± 392.2 | 2331.1 ± 963.9 | 600.8 ± 655.9 | 0.039 |
| AUC ΔInsulinL-Arginine (pmol • L−1 • min) | 11483.5 ± 2597.0 | 14840.6 ± 6271.6 | 3357.1 ± 4931.2 | 0.098 |
| Insulin Sensitivity Index (GIR/Ins) | 11.3 ± 3.5 | 8.7 ± 6.4 | −2.6 ± 7.5 | 0.160 |
| Hyperinsulinemic Clamp | ||||
| Glucose90–120 (mM) | 5.3 ± 0.1 | 5.3 ± 0.1 | −0.0 ± 0.1 | 0.890 |
| Glucose210–240 (mM) | 5.3 ± 0.1 | 5.2 ± 0.1 | −0.1 ± 0.2 | 0.380 |
| Insulin90–120 (pmol/L) | 188.9 ± 39.3 | 304.1 ± 115.9 | 115.2 ± 110.2 | 0.023 |
| Insulin210–240 (pmol/L) | 1770.5 ± 272.9 | 2452.6 ± 360.9 | 637.2 ± 460.3 | 0.031 |
| M/ILow (nmol • kg−1 • min−1 • pmol−1 • L) | 81.5 ± 32.7 | 43.0 ± 26.1 | −38.4 ± 28.4 | 0.016 |
| M/IHigh (nmol • kg−1 • min−1 • pmol−1 • L) | 30.7 ± 6.2 | 18.5 ± 4.7 | −11.2 ± 6.6 | 0.016 |
| Insulin Clearance-Low (mL • m−2 BSA • min−1) | 1852.7 ± 445.9 | 1245.8 ± 558.5 | −607.0 ± 471.1 | 0.023 |
| Insulin Clearance-High (mL • m−2 BSA • min−1) | 872.8 ± 207.6 | 632.3 ± 178.6 | −200.3 ± 168.6 | 0.031 |
| Disposition Index | ||||
| Disposition Index (AUC ΔC-peptide0–10 • GIR/I)* | 30,591 ± 21,325 | 39,783 ± 58,374 | 9,192 ± 52,453 | 0.91 |
| Disposition Index (AUC ΔC-peptide0–10 • M/IHigh)† | 153,093 ± 118,577 | 180,011 ± 165,140 | 1,944 ± 10,2206 | 1.0 |
Results are mean ± SD. p-value from Wilcoxon signed-rank test.
Disposition index based on Hyperglycemic clamp only.
Disposition index calculated by insulin secretion during hyperglycemic clamp and insulin sensitivity from hyperinsulinemic clamp result
We compared insulin secretory responses in the current study to a group of control participants (matched for age, gender, race, and BMI) from a dataset of previously-conducted hyperglycemic clamp studies. Controls were well-matched for age, gender, race, BMI, and blood pressure. The post-treatment insulin response was similar to the control comparator results (Table S3 and Figure S1). Similar results were observed in insulin response in surgically- vs medically-treated participants, although differences in C-peptide appear to be more prominent in surgically treated participants (Figure S2).
Effect of treatment on insulin sensitivity
Low and high dose hyperinsulinemic-euglycemic clamps were conducted to assess insulin sensitivity. Plasma glucose concentrations maintained during the clamps were equivalent before and after treatment during both the low-dose insulin (Figure 2A, 5.3±0.1 vs 5.3±0.1 mmol/L; p=0.89) and high-dose insulin (5.3±0.1 vs 5.2±0.1; p=0.38). The achieved insulin concentration was significantly higher post-treatment during both low-dose (Figure 2C, average 188.9 ± 39.3 vs 304.1 ± 115.9 pmol/L for pre- vs post-treatment; p=0.023) and high-dose insulin infusion (average 1770.5 ± 272.9 vs 2452.6 ± 360.9 pmol/L for pre- vs post-treatment; p=0.031). Despite the higher insulin levels post-treatment, the glucose infusion rate (M) required to maintain euglycemia was significantly decreased after treatment during low-dose insulin (Figure 2B, 15.4±7.7vs 12.1±7.2 μmol/kg/min for pre- vs post-treatment; p=0.008), and was non-significantly lower during high-dose insulin (53.5±9.0 vs 45.0±12.7 μmol/kg/min for pre- vs post-treatment; p=0.11). The insulin sensitivity index (glucose infusion rate for achieved insulin concentration, or M/I) was significantly lower after treatment during both low-dose (81.5±32.7 vs 43.0±26.1 nmol/kg/min per pmol/L for pre- vs post-treatment; p=0.016) and high-dose insulin infusion (30.7±6.2 vs 18.5±4.7 nmol/kg/min per pmol/L for pre- vs post-treatment; p=0.016). This decrease in insulin sensitivity is also represented in a right-ward shift in the insulin concentration-response curve post-treatment (Figure 2D). The Disposition index (AUC ΔC-peptide0–10 × Insulin sensitivity [M/I]), a measure that accounts for the hyperbolic relationship between insulin sensitivity and insulin secretion, was similar during both treatment periods, and similar results were obtained by using insulin sensitivity determined during the hyperglycemic clamp (GIR/Insulin at 90–120 minutes) or during the hyperinsulinemic clamp (M/I at end of the clamp, Table 3).
Figure 2.
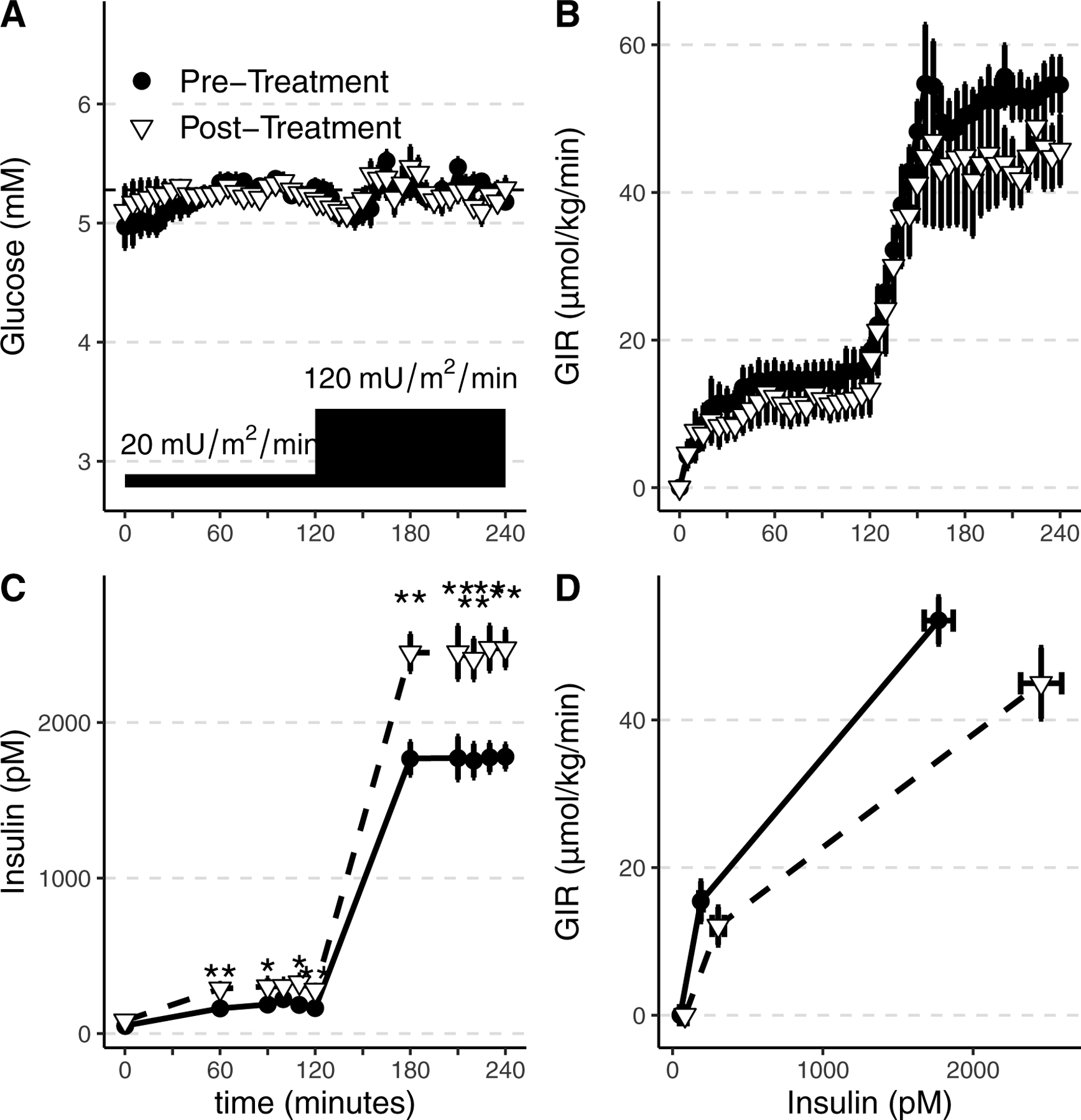
Summary data from the hyperinsulinemic euglycemic clamps to assess insulin sensitivity in participants with primary aldosteronism before treatment (●) and post-treatment (▽). Glucose was infused to maintain euglycemia ~5.28 mmol/L (95 mg/dL) during low and high insulin infusion rates (20 and 120 mU/m2/min) (A). Glucose infusion rates (GIR, B) required to maintain euglycemia were similar on both study days (B). Achieved insulin concentrations during the clamp were increased after treatment compared to baseline assessment, despite identical insulin infusion rates (C). The resulting glucose infusion rate versus achieved plasma insulin curve was shifted rightward, indicating insulin resistance after treatment (D). Group mean and SEM are shown. *p<0.05, **p<0.01, ***p<0.001
Effect on insulin clearance
The increase in achieved insulin concentrations during the post-treatment clamp, despite identical insulin infusion rates, suggests that insulin clearance is altered in primary aldosteronism. Accordingly, we calculated insulin clearance. Insulin clearance was higher in pre-treated than in treated participants (872.8±207.6 vs 632.3±178.6 mL/m2/min; p=0.031). This difference was similar and significant regardless of adjustment for endogenous insulin secretion (15), and the adjusted value is presented.
Effect of treatment on body composition and energy expenditure
We further examined the relationship between body composition and insulin sensitivity and secretion measures. Body weight, lean body percentage, and total fat percentage were unchanged after treatment. The android fat percentage, but not gynoid fat percentage increased significantly after treatment, resulting in an increased android/gynoid ratio (Table 2, p=0.004). The resting energy expenditure and respiratory quotient were unchanged.
Relationships between insulin sensitivity and body composition
Because android fat percentage increased after treatment, we explored the relationship between outcome measures and body composition. In the overall data (combined pre- and post-treatment), insulin sensitivity correlated with android fat percentage (rho=−0.62, p=0.013), body mass index (rho=−0.60, p=0.013), and plasma aldosterone (rho=−0.60, p=0.019). The change in glucose-induced insulin concentration from pre- to post-treatment correlated with change in total fat mass (ΔInsulin90–120 rho = 0.79, p=0.01; ΔInsulinL-Arginine rho=0.79, p=0.012) and the change in the android/gynoid fat ratio (ΔInsulin90–120 rho=0.87, p=0.002; ΔInsulinL-Arginine rho=0.78, p=0.013). Change in C-peptide measures did not correlate with these measures, however.
DISCUSSION
Primary Aldosteronism, a common cause of hypertension affecting up to 20% of patients with resistant hypertension, is associated with an increased risk of diabetes by uncertain mechanisms. Since insulin secretion and sensitivity are key factors governing glycemic control, we assessed these factors using gold-standard hyperglycemic and euglycemic hyperinsulinemic clamp techniques in individuals with primary aldosteronism without diabetes before and three-to-twelve months after treatment with surgery or mineralocorticoid receptor blockade. Treatment of primary aldosteronism led to improved insulin secretion, reduced insulin clearance and reduced insulin sensitivity accompanied by increased adiposity. Thus, the present study suggests that impaired glucose metabolism in primary aldosteronism is due to reductions in insulin secretion (i.e., impaired beta cell function) accompanied by increased insulin clearance.
When examined in isolation during hyperglycemic clamps, our data demonstrate that insulin secretion is decreased in patients with primary aldosteronism versus post-treatment. This decrease in insulin secretion is observed both in the acute insulin response and the steady-state insulin response. It is unclear from our studies if the change in insulin secretion is a direct effect of aldosterone, although this hypothesis is supported by several lines of evidence. Aldosterone impairs insulin secretion in isolated rodent islets and insulinoma cell lines, and conversely aldosterone deficiency in mice increases insulin secretion in vivo without altering insulin sensitivity (6). Furthermore, insulin secretion in matched control subjects was similar to patients after treatment. Other potentially confounding characteristics of primary aldosteronism and control subjects were similar, including blood pressure, body mass index, age, and insulin sensitivity. Insulin secretion should not be viewed in isolation, however, and the reduced insulin secretion in primary aldosteronism pre-treatment was accompanied by an opposing increase in insulin sensitivity.
Altered insulin sensitivity in the present study may be compensatory for the change in insulin secretion, such that individuals remain on the same disposition index curve and maintain glucose homeostasis. Overall, the disposition index was unchanged before versus after treatment of primary aldosteronism, reflecting this dynamic response (20). Alternatively, the alterations in insulin secretion and insulin sensitivity could be influenced by increased angiotensin II concentration after treatment. Chronically elevated angiotensin II in the Ren-2 hypertensive rat worsens skeletal muscle insulin sensitivity via reactive oxygen species (21), and treatment with angiotensin receptor antagonists improve insulin sensitivity and ameliorates the adverse metabolic effects of thiazide treatment (22, 23). Treatment of primary aldosteronism restores renin into the low normal range rather than producing angiotensin II excess, and therefore contribution of angiotensin II to the observed effects remains uncertain. Participants in our study were selected to be relatively healthy and did not have diabetes, but worsening glucose homeostasis could be precipitated in primary aldosteronism patients with underlying insulin resistance if decreases in insulin secretion due to primary aldosteronism did not lead to compensatory improvements in insulin sensitivity. Our studies do not demonstrate a mechanism for improvements in insulin sensitivity in primary aldosteronism, but we cannot rule out a direct beneficial effect of aldosterone on insulin sensitivity.
Hypercortisolemia may coexist with primary aldosteronism, and subclinical cortisol excess could contribute to worsening insulin resistance and type 2 diabetes risk in primary aldosteronism (24, 25). For example, cortisol and total glucocorticoid metabolite excretion in 24-hour urine collections is increased in primary aldosteronism and the glucocorticoid metabolome, but not the mineralocorticoid metabolome, correlates with fasting estimates of insulin resistance (26). Recently, the German Conn’s registry demonstrated that autonomous cortisol secretion (defined by a single abnormal result on one of the following tests: dexamethasone suppression test, midnight salivary cortisol, or urinary cortisol excretion) was present in 76% of primary aldosteronism patients, and was associated with the diagnosis of type 2 diabetes (24). In a Japanese cohort of primary aldosteronism patients, diabetes was more commonly diagnosed than in the general population, and subclinical hypercortisolism was identified as a risk factor (25). Somatic mutations within a limited set of genes (KCNJ5, CACNA1D, ATPA1A, and ATP2B3) have been implicated as causative aldosterone-driver mutations in aldosterone-producing adenomas (27–29). Notably, mutations in KCNJ5 are associated with a florid hyperaldosteronism phenotype (30), co-secretion of cortisol (31) and an increased circulating 18-oxocortisol and 18-hydroxycortisol concentration (32, 33). In the present study, 8AM cortisol and ACTH were unchanged after treatment, although cortisol suppression tests were not routinely obtained. Further, steroid measurements by LC-MS did not reveal a change in other glucocorticoids after treatment of primary aldosteronism. The question of combined glucocorticoid and mineralocorticoid excess requires more rigorous assessment in primary aldosteronism, as both factors could contribute to impaired glucose tolerance in a given individual.
Our finding of decreased insulin secretion in primary aldosteronism is consistent with a prior study which used a combined intravenous glucose tolerance test-hyperinsulinemic clamp to assess insulin secretion and insulin sensitivity in primary aldosteronism (9). However, conducting hyperinsulinemic clamps on separate days may have permitted us to detect altered insulin clearance and insulin sensitivity in addition to impaired insulin secretion. Although potassium and blood pressure did not match perfectly during the two study periods, differences were minimal between the two periods and adjustment for serum potassium and alpha-blocker use did not affect the results. However, potassium is an insulin secretagogue and lower potassium could have contributed to the reduced insulin secretion in pre-treated primary aldosteronism patients despite minimal differences between pre- and post-treatment potassium values and no association between insulin secretion and serum potassium (34). The comparison to matched historical controls suggests that altered insulin secretion is diminished in primary aldosteronism, rather than being accounted for by small differences in blood pressure. The present study was an observational study, because it was not deemed feasible to perform a randomized trial, and so proving causation is difficult. It is possible that insulin secretion changes over time, but we have found that the within-subject correlation is very high in prior studies utilizing hyperglycemic clamps.
Our data demonstrates that insulin clearance is increased during aldosterone excess, which has not been previously described. Insulin clearance is accomplished by insulin degrading enzyme (IDE) in the liver (up to 80%) and outside the liver by muscle, tissue uptake and the kidney (glomerular filtration, renal tubular reabsorption, and degradation by proteolytic enzymes). Angiotensin II and aldosterone may affect insulin clearance via effects on hepatic IDE expression, but these effects have not been previously investigated. We did not observe a change in renal function after treatment, as assessed by plasma creatinine, that would suggest that altered renal clearance contributes to this effect and change in plasma creatinine did not correlate with any changes in insulin clearance. The present study is not able to determine if the changes in insulin clearance are due to altered peripheral metabolism (e.g. liver, adipose, or muscle tissues), and additional studies are warranted.
Several studies have previously suggested that insulin sensitivity is diminished in primary aldosteronism patients, and improved after treatment (35, 36), but these studies did not assess cortisol or other glucocorticoids. In contrast to these prior studies, we observed decreased rather than increased insulin sensitivity after treatment. Consistent with our findings, a Japanese cohort demonstrated reduced glucose tolerance and insulin secretion during glucose tolerance tests, but increased insulin sensitivity during hyperinsulinemic clamps in patients with primary aldosteronism (37), and an additional group similarly demonstrated reduced insulin sensitivity after treatment (38). Insulin clearance was not formally addressed in these studies, but the findings could be consistent with an alteration in insulin clearance. Furthermore, increased total and gynoid fat mass after treatment of primary aldosteronism was an unexpected finding, but this could be explained by increased insulin secretion after treatment. We previously demonstrated that aldosterone deficient mice developed increased adiposity during high fat diet, which accompanied their markedly increased insulin response to hyperglycemia (7). Aldosterone increases pre-adipocyte differentiation tissue in vivo (39) and increased adipocyte-specific MR expression exacerbates diet-induced weight gain and insulin resistance in mice (40). Long-term hyperaldosteronism may therefore facilitate adipose expansion associated with increased insulin secretion after treatment of primary aldosteronism in the current study.
In summary, this paper suggests that insulin levels in primary aldosteronism are reduced due to reduced beta cell function and increased insulin clearance. This decrease in insulin could lead to glucose intolerance unless patients with hyperaldosteronism can adapt by improving insulin sensitivity.
Supplementary Material
Novelty and Significance.
1. What Is New?
Treatment of PA increases insulin concentrations via increased secretion and decreased clearance.
Treatment of PA also decreases insulin sensitivity, which may be the result of a metabolic compensatory response.
2. What Is Relevant?”
Patients with PA are at increased risk of diabetes, and these findings demonstrate changes in insulin metabolism that may contribute.
The increase in insulin sensitivity in PA may compensate for impaired insulin secretion.
3. Summary:
Primary aldosteronism impairs insulin secretion, and compensatory changes in insulin sensitivity may be necessary to prevent deterioration of glucose tolerance. Patients with insulin resistance may not be able to compensate for alterations in insulin secretion, and this could contribute to the development of diabetes.
Perspectives.
Primary aldosteronism is associated with increased risk of cardiovascular events and diabetes, but it is unclear how aldosterone contributes to increased diabetes risk. This study demonstrates that primary aldosteronism causes decreased insulin secretion, which is compounded by an increased rate of insulin clearance. In response, insulin sensitivity also increased in this relatively healthy study population, such that the disposition index was unchanged and glycemic control was maintained. Patients with primary aldosteronism, obesity, and reduced insulin sensitivity will have an increased risk of developing glucose intolerance and diabetes.
Sources of Funding:
NIH grants DK096994, DK081662 (JML), DK109116 (AFT), K24HL103845 (GKA) and 2019087 from the Doris Duke Charitable Foundation (AFT). The project described was supported by: 1) the National Center for Research Resources, Grant UL1 RR024975-01, and is now at the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06; and 2) Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL 1TR002541) and financial contributions from Harvard University and its affiliated academic healthcare centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, Harvard Catalyst, Harvard University and its affiliated academic healthcare centers. This work utilized the core(s) of the Vanderbilt Diabetes Research and Training Center funded by grant DK020593 from the National Institute of Diabetes and Digestive and Kidney Disease.
Footnotes
Disclosure Summary:
JML reports consultant relationship with Selenity Therapeutics and Relypsa.
GKA reports consultant relationship with Pfizer Pharmaceuticals.
References Cited:
- 1.Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, Stowasser M, Young WF Jr. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab 2016;101(5):1889–1916. [DOI] [PubMed] [Google Scholar]
- 2.Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. The lancet. Diabetes & endocrinology 2018;6(1):51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joseph JJ, Echouffo-Tcheugui JB, Kalyani RR, Yeh HC, Bertoni AG, Effoe VS, Casanova R, Sims M, Correa A, Wu WC, Wand GS, Golden SH Aldosterone, Renin, and Diabetes Mellitus in African Americans: The Jackson Heart Study. J. Clin. Endocrinol. Metab 2016;101(4):1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanslik G, Wallaschofski H, Dietz A, Riester A, Reincke M, Allolio B, Lang K, Quack I, Rump LC, Willenberg HS, Beuschlein F, Quinkler M, Hannemann A, participants of the German Conn’s R Increased prevalence of diabetes mellitus and the metabolic syndrome in patients with primary aldosteronism of the German Conn’s Registry. Eur. J. Endocrinol 2015;173(5):665–675. [DOI] [PubMed] [Google Scholar]
- 5.Conn JW Hypertension, the potassium ion and impaired carbohydrate tolerance. N. Engl. J. Med 1965;273(21):1135–1143. [DOI] [PubMed] [Google Scholar]
- 6.Luther JM, Luo P, Kreger MT, Brissova M, Dai C, Whitfield TT, Kim HS, Wasserman DH, Powers AC, Brown NJ Aldosterone decreases glucose-stimulated insulin secretion in vivo in mice and in murine islets. Diabetologia. 2011;54(8):2152–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo P, Dematteo A, Wang Z, Zhu L, Wang A, Kim HS, Pozzi A, Stafford JM, Luther JM Aldosterone deficiency prevents high-fat-feeding-induced hyperglycaemia and adipocyte dysfunction in mice. Diabetologia. 2013;56(4):901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luther JM, Byrne LM, Yu C, Wang TJ, Brown NJ Dietary sodium restriction decreases insulin secretion without affecting insulin sensitivity in humans. J. Clin. Endocrinol. Metab 2014;99(10):E1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer E, Adolf C, Pallauf A, Then C, Bidlingmaier M, Beuschlein F, Seissler J, Reincke M Aldosterone excess impairs first phase insulin secretion in primary aldosteronism. J. Clin. Endocrinol. Metab 2013;98(6):2513–2520. [DOI] [PubMed] [Google Scholar]
- 10.Ramirez CE, Nian H, Yu C, Gamboa JL, Luther JM, Brown NJ, Shibao CA Treatment with Sildenafil Improves Insulin Sensitivity in Prediabetes: A Randomized, Controlled Trial. J. Clin. Endocrinol. Metab 2015;100(12):4533–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah SS, Ramirez CE, Powers AC, Yu C, Shibao CA, Luther JM Hyperglycemic clamp-derived disposition index is negatively associated with metabolic syndrome severity in obese subjects. Metabolism. 2016;65(6):835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnold AC, Garland EM, Celedonio JE, Raj SR, Abumrad NN, Biaggioni I, Robertson D, Luther JM, Shibao CA Hyperinsulinemia and Insulin Resistance in Dopamine beta-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab 2017;102(1):10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeFronzo RA, Tobin JD, Andres R Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol 1979;237(3):E214–223. [DOI] [PubMed] [Google Scholar]
- 14.Ferrannini E, Mari A How to measure insulin sensitivity. J. Hypertens 1998;16(7):895–906. [DOI] [PubMed] [Google Scholar]
- 15.Straznicky NE, Grima MT, Lambert EA, Sari CI, Eikelis N, Nestel PJ, Phillips SE, Hering D, Karapanagiotidis S, Dixon JB, Schlaich MP, Lambert GW Arterial norepinephrine concentration is inversely and independently associated with insulin clearance in obese individuals with metabolic syndrome. J. Clin. Endocrinol. Metab 2015;100(4):1544–1550. [DOI] [PubMed] [Google Scholar]
- 16.Isbell TR, Klesges RC, Meyers AW, Klesges LM Measurement reliability and reactivity using repeated measurements of resting energy expenditure with a face mask, mouthpiece, and ventilated canopy. JPEN J. Parenter. Enteral Nutr 1991;15(2):165–168. [DOI] [PubMed] [Google Scholar]
- 17.Nanba AT, Rege J, Ren J, Auchus RJ, Rainey WE, Turcu AF 11-Oxygenated C19 Steroids Do Not Decline With Age in Women. J. Clin. Endocrinol. Metab 2019;104(7):2615–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turcu AF, Wannachalee T, Tsodikov A, Nanba AT, Ren J, Shields JJ, O’Day PJ, Giacherio D, Rainey WE, Auchus RJ Comprehensive Analysis of Steroid Biomarkers for Guiding Primary Aldosteronism Subtyping. Hypertension. 2020;75(1):183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R Core Team 2018. R: A Language and Environment for Statistical Computing. In. Vienna, Austria: R Foundation for Statistical Computing [Google Scholar]
- 20.Kahn SE, Hull RL, Utzschneider KM Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. [DOI] [PubMed] [Google Scholar]
- 21.Wei Y, Sowers JR, Clark SE, Li W, Ferrario CM, Stump CS Angiotensin II-induced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am. J. Physiol. Endocrinol. Metab 2008;294(2):E345–351. [DOI] [PubMed] [Google Scholar]
- 22.Aksnes TA, Reims HM, Guptha S, Moan A, Os I, Kjeldsen SE Improved insulin sensitivity with the angiotensin II-receptor blocker losartan in patients with hypertension and other cardiovascular risk factors. J. Hum. Hypertens 2006;20(11):860–866. [DOI] [PubMed] [Google Scholar]
- 23.Zappe DH, Sowers JR, Hsueh WA, Haffner SM, Deedwania PC, Fonseca VA, Keeling L, Sica DA Metabolic and antihypertensive effects of combined angiotensin receptor blocker and diuretic therapy in prediabetic hypertensive patients with the cardiometabolic syndrome. J. Clin. Hypertens. (Greenwich) 2008;10(12):894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerards J, Heinrich DA, Adolf C, Meisinger C, Rathmann W, Sturm L, Nirschl N, Bidlingmaier M, Beuschlein F, Thorand B, Peters A, Reincke M, Roden M, Quinkler M Impaired Glucose Metabolism in Primary Aldosteronism Is Associated With Cortisol Cosecretion. J. Clin. Endocrinol. Metab 2019;104(8):3192–3202. [DOI] [PubMed] [Google Scholar]
- 25.Akehi Y, Yanase T, Motonaga R, Umakoshi H, Tsuiki M, Takeda Y, Yoneda T, Kurihara I, Itoh H, Katabami T, Ichijo T, Wada N, Shibayama Y, Yoshimoto T, Ashida K, Ogawa Y, Kawashima J, Sone M, Inagaki N, Takahashi K, Fujita M, Watanabe M, Matsuda Y, Kobayashi H, Shibata H, Kamemura K, Otsuki M, Fujii Y, Yamamoto K, Ogo A, Okamura S, Miyauchi S, Fukuoka T, Izawa S, Hashimoto S, Yamada M, Yoshikawa Y, Kai T, Suzuki T, Kawamura T, Naruse M, Japan Primary Aldosteronism Study G High Prevalence of Diabetes in Patients With Primary Aldosteronism (PA) Associated With Subclinical Hypercortisolism and Prediabetes More Prevalent in Bilateral Than Unilateral PA: A Large, Multicenter Cohort Study in Japan. Diabetes Care. 2019;42(5):938–945. [DOI] [PubMed] [Google Scholar]
- 26.Arlt W, Lang K, Sitch AJ, Dietz AS, Rhayem Y, Bancos I, Feuchtinger A, Chortis V, Gilligan LC, Ludwig P, Riester A, Asbach E, Hughes BA, O’Neil DM, Bidlingmaier M, Tomlinson JW, Hassan-Smith ZK, Rees DA, Adolf C, Hahner S, Quinkler M, Dekkers T, Deinum J, Biehl M, Keevil BG, Shackleton CH, Deeks JJ, Walch AK, Beuschlein F, Reincke M Steroid metabolome analysis reveals prevalent glucocorticoid excess in primary aldosteronism. JCI insight. 2017;2(8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, Thompson LDR, Cohen DL, Luther JM, Gellert L, Vaidya A, Barletta JA, Else T, Giordano TJ, Tomlins SA, Rainey WE Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension. 2019;73(4):885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, Miller BS, Giordano TJ, Tomlins SA, Rainey WE Targeted Molecular Characterization of Aldosterone-Producing Adenomas in White Americans. J. Clin. Endocrinol. Metab 2018;103(10):3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandes-Rosa FL, Boulkroun S, Zennaro MC Somatic and inherited mutations in primary aldosteronism. J. Mol. Endocrinol 2017; [DOI] [PubMed] [Google Scholar]
- 30.Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, Amar L, Mulatero P, Samson-Couterie B, Hahner S, Quinkler M, Fallo F, Letizia C, Allolio B, Ceolotto G, Cicala MV, Lang K, Lefebvre H, Lenzini L, Maniero C, Monticone S, Perrocheau M, Pilon C, Plouin PF, Rayes N, Seccia TM, Veglio F, Williams TA, Zinnamosca L, Mantero F, Benecke A, Jeunemaitre X, Reincke M, Zennaro MC Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension. 2012;59(3):592–598. [DOI] [PubMed] [Google Scholar]
- 31.Yamada M, Nakajima Y, Taguchi R, Okamura T, Ishii S, Tomaru T, Ozawa A, Shibusawa N, Yoshino S, Toki A, Ishida E, Hashimoto K, Satoh T, Mori M KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas. Endocr. J 2012;59(8):735–741. [DOI] [PubMed] [Google Scholar]
- 32.Williams TA, Peitzsch M, Dietz AS, Dekkers T, Bidlingmaier M, Riester A, Treitl M, Rhayem Y, Beuschlein F, Lenders JW, Deinum J, Eisenhofer G, Reincke M Genotype-Specific Steroid Profiles Associated With Aldosterone-Producing Adenomas. Hypertension. 2016;67(1):139–145. [DOI] [PubMed] [Google Scholar]
- 33.Tezuka Y, Yamazaki Y, Kitada M, Morimoto R, Kudo M, Seiji K, Takase K, Kawasaki Y, Mitsuzuka K, Ito A, Nishikawa J, Asai N, Nakamura Y, Gomez-Sanchez CE, Ito S, Dezawa M, Sasano H, Satoh F 18-Oxocortisol Synthesis in Aldosterone-Producing Adrenocortical Adenoma and Significance of KCNJ5 Mutation Status. Hypertension. 2019;73(6):1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rowe JW, Tobin JD, Rosa RM, Andres R Effect of experimental potassium deficiency on glucose and insulin metabolism. Metabolism. 1980;29(6):498–502. [DOI] [PubMed] [Google Scholar]
- 35.Skrha J, Haas T, Sindelka G, Prazny M, Widimsky J, Cibula D, Svacina S Comparison of the insulin action parameters from hyperinsulinemic clamps with homeostasis model assessment and QUICKI indexes in subjects with different endocrine disorders. J. Clin. Endocrinol. Metab 2004;89(1):135–141. [DOI] [PubMed] [Google Scholar]
- 36.Catena C, Lapenna R, Baroselli S, Nadalini E, Colussi G, Novello M, Favret G, Melis A, Cavarape A, Sechi LA Insulin sensitivity in patients with primary aldosteronism: a follow-up study. J. Clin. Endocrinol. Metab 2006;91(9):3457–3463. [DOI] [PubMed] [Google Scholar]
- 37.Shimamoto K, Shiiki M, Ise T, Miyazaki Y, Higashiura K, Fukuoka M, Hirata A, Masuda A, Nakagawa M, Iimura O Does insulin resistance participate in an impaired glucose tolerance in primary aldosteronism? J. Hum. Hypertens 1994;8(10):755–759. [PubMed] [Google Scholar]
- 38.Tsurutani Y, Sugisawa C, Ishida A, Inoue K, Saito J, Omura M, Nagasaka S, Nishikawa T Aldosterone excess may inhibit insulin secretion: A comparative study on glucose metabolism pre- and post-adrenalectomy in patients with primary aldosteronism. Endocr. J 2017;64(3):339–346. [DOI] [PubMed] [Google Scholar]
- 39.Caprio M, Feve B, Claes A, Viengchareun S, Lombes M, Zennaro MC Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007;21(9):2185–2194. [DOI] [PubMed] [Google Scholar]
- 40.Urbanet R, Nguyen Dinh Cat A, Feraco A, Venteclef N, El Mogrhabi S, Sierra-Ramos C, Alvarez de la Rosa D, Adler GK, Quilliot D, Rossignol P, Fallo F, Touyz RM, Jaisser F Adipocyte Mineralocorticoid Receptor Activation Leads to Metabolic Syndrome and Induction of Prostaglandin D2 Synthase. Hypertension. 2015;66(1):149–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.