

Abstract
Site-selective functionalization of C–H bonds will ultimately afford chemists transformative tools for editing and constructing complex molecular architectures. Towards this goal, developing strategies to activate C–H bonds that are distal from a functional group is essential. In this context, distinguishing remote C–H bonds on adjacent carbon atoms is an extraordinary challenge due to the lack of electronic or steric bias between the two positions. Herein, we report the design of a catalytic system leveraging a remote directing template and a transient norbornene mediator to selectively activate a previously inaccessible remote C–H bond that is one bond further away. The generality of this approach has been demonstrated with a range of heterocycles, including a complex anti-leukemia agent, and hydrocinnamic acid substrates.
Graphical Abstract
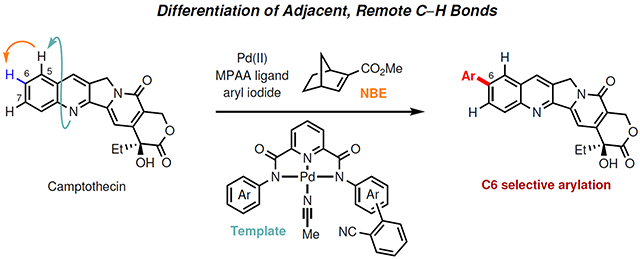
Functionalizing C–H bonds selectively at various locations of molecules will ultimately afford synthetic chemists transformative tools to modify and construct molecular structures1–3. C–H bonds that are remote from functional groups (>6 bonds away) are widespread, and distinguishing these C–H bonds bearing little difference in electronic property is a formidable challenge in C–H activation4–12. For example, benzoazine, especially quinoline, is a ubiquitous motif in natural products, pharmaceuticals, agrochemicals, and functional materials. Multiple C–H bonds on these structures are remote from the chelating nitrogen atom and difficult to distinguish electronically (Fig. 1a). While the selective functionalization of C–H bonds on the azine ring was achieved by taking advantage of strong electronic and steric bias13–19, directed C–H activation on the benzene moiety is limited to C8 position via chelation assisted process20–21. To functionalize the remaining remote C–H bonds, a recoverable and bifunctional template was developed to direct the palladium(II) catalyst selectively to the remote C5 position through the coordination with the nitrogen of quinoline22. This raises a fundamental question of whether such remote directing effect can be exploited to reach other C–H bonds that are one bond further away, thereby significantly expanding our toolkit for functionalizing remote C–H bonds selectively. For example, it would be highly enabling if a directing template can selectively activate the distal C6 or C7 positions of (iso)quinolines which are similarly electron-deficient and unreactive according to the calculated Fukui indices of various heterocycle substrates (see Supplementary Table 11). While engineering the template to match the distance and geometry is potentially feasible, such alternation of the template will inevitably vary with different classes of substrates. Thus, we began to investigate the possibility of combining the remote directing effect with a one-bond relay strategy using a transient mediator (Fig. 1b). We envision that the template directs the initial remote palladation at the C5 position, and subsequently, the norbornene23–27 relays the palladation to the C6 position (Fig. 1c). This strategy could provide a reliable method to distinguish remote C–H bonds which are adjacent to each other and have similar electronic and steric properties (i.e. C6 position and C7 position of quinoline), as well as to override the electronic bias (i.e. the more reactive C5 position and the less reactive C6 position of quinoline).
Figure 1. |. Remote site-selective C–H functionalization.
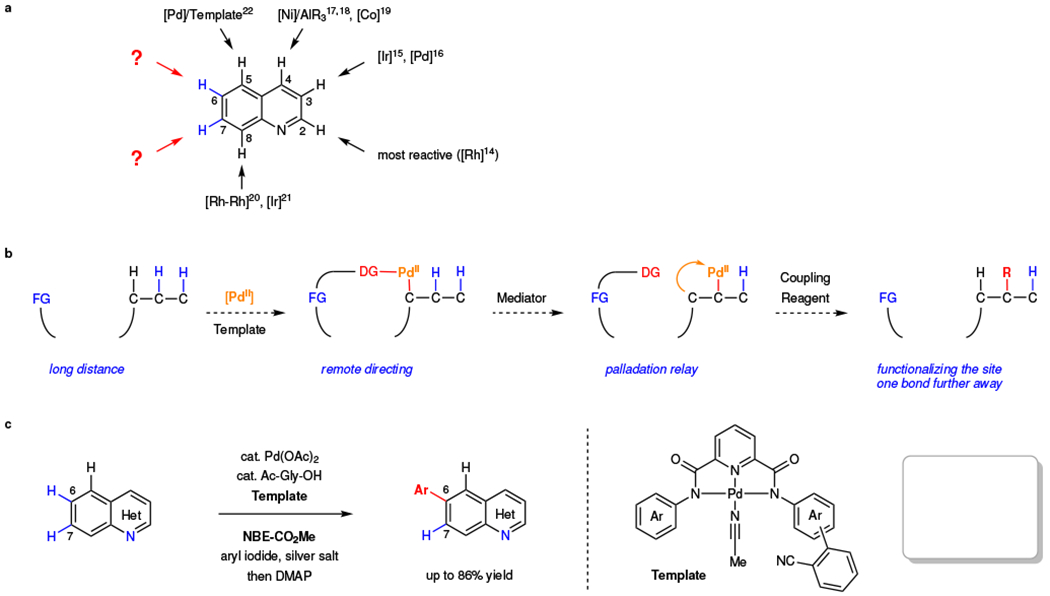
a, Site-selective C–H functionalization of quinolines. C2, C3, C4, C5, and C8 selective functionalizations of quinolones have been realized. Distinguishing adjacent, remote C6 and C7 C–H bonds is still a fundamental challenge due to the lack of electronic or steric bias. Pd, palladium; Ni, nickel; Co, cobalt; Ir, iridium; Rh, rhodium. b, Relay strategy. A bifunctional template directs the initial palladation, and subsequently, a mediator relays the palladium catalyst to the distal position that is one bond further away. FG, functional group; DG, directing group. c, Remote site-selective C–H arylation of benzoazines through the relay strategy that leverages a remote directing template and a norbornene.
To reduce this design into practice, the orchestration of remote directing and the subsequent relaying step faces multiple challenges that must be addressed by carefully engineering molecular structures of the nitrile template, norbornene and ligand. First, the initial C–H palladation directed by the weakly coordinating nitrile template might be prevented by the competitively binding norbornene. On the other hand, the formed macrocyclic C–H palladation intermediate is highly reactive and could undergo the functionalization with aryl iodide prior to the desired norbornene interception and subsequent relay. Second, the norbornene-relayed meta-C–H activation, a highly complex multiple-step sequence, could only proceed with a limited number of previously identified monodentate pyridine and pyridone type of ligands23,25,28. In contrast, the weakly coordinating nitrile template often requires a different set of bidentate ligand for remote directing9,11,22. Finally, the β-carbon elimination step in the norbornene relay has always relied on the ortho-directing group that is adjacent to the initially formed palladium-carbon bond. However, the remote directing group is further away and cannot provide the necessary steric hindrance.
Results and discussion
Owing to the pivotal importance of quinolines and isoquinolines in drug discovery, we selected 3-methyl-isoquinoline as the model substrate to explore the feasibility of remote directing and subsequent relay (Table 1). Exploratory studies were conducted using template T1 that was previously shown to selectively direct C5 C–H palladation of quinolines22. Since pyridone and pyridine type ligands have been shown to be critical for enabling the Pd-catalyst-relay by norbornene analogues23,25,28, we attempted C6 arylation of isoquinoline-T1 substrate using such monodentate ligands (L1, L2, L3, L4). The lack of desired reactivity under these conditions may be attributed to the fact that the weakly coordinating nitrile directing group is prevented from binding to the Pd(II) center in the presence of pyridone and pyridine additives. To ensure the coordination of the nitrile directing group, we decided to evaluate bidentate ligands bearing weakly coordinating group (L5, L6, L7). To our delight, N-acetylglycine (Ac–Gly–OH, L5), one of the mono-N-protected amino acid (MPAA) ligands, turned out to be compatible with the nitrile template, affording the desired C6 arylated isoquinoline-template complex in 21% yield. The recyclable template that bound to the product could be readily removed by in situ treatment with 4-dimethylaminopyridine (see supplementary information, Template recovery). Notably, displacement of the carboxylic acid group of MPAA ligand with a strongly coordinating group, N-heterocycle (bidentate ligand L8, L9), led to loss of reactivity (< 5% yield). These findings demonstrate the importance of matching both the directing template and norbornene with a specific ligand. With this promising lead in hand, we proceeded to evaluate the templates. Firstly, replacing the electron rich left-wing (T1) with electron deficient aryl group (T2) slightly increased the yield to 24%. Switching the directing phenyl nitrile moiety from meta- to para-position on the right-wing (T3) improved the yield markedly (63%). These results are consistent with the notion that site selectivity in remote C–H activation is based on the precise recognition of distance and geometry. Other structural variations of templates reduced the yields (T4, T5, T6). Absence of the nitrile moiety (T7) or the norbornene reagent resulted in loss of the desired reactivity (see Supplementary Table 1 to Table 7 for more optimizations).
Table 1. |.
Exploration of reaction conditions that enables remote site-selective arylation of benzoazins.
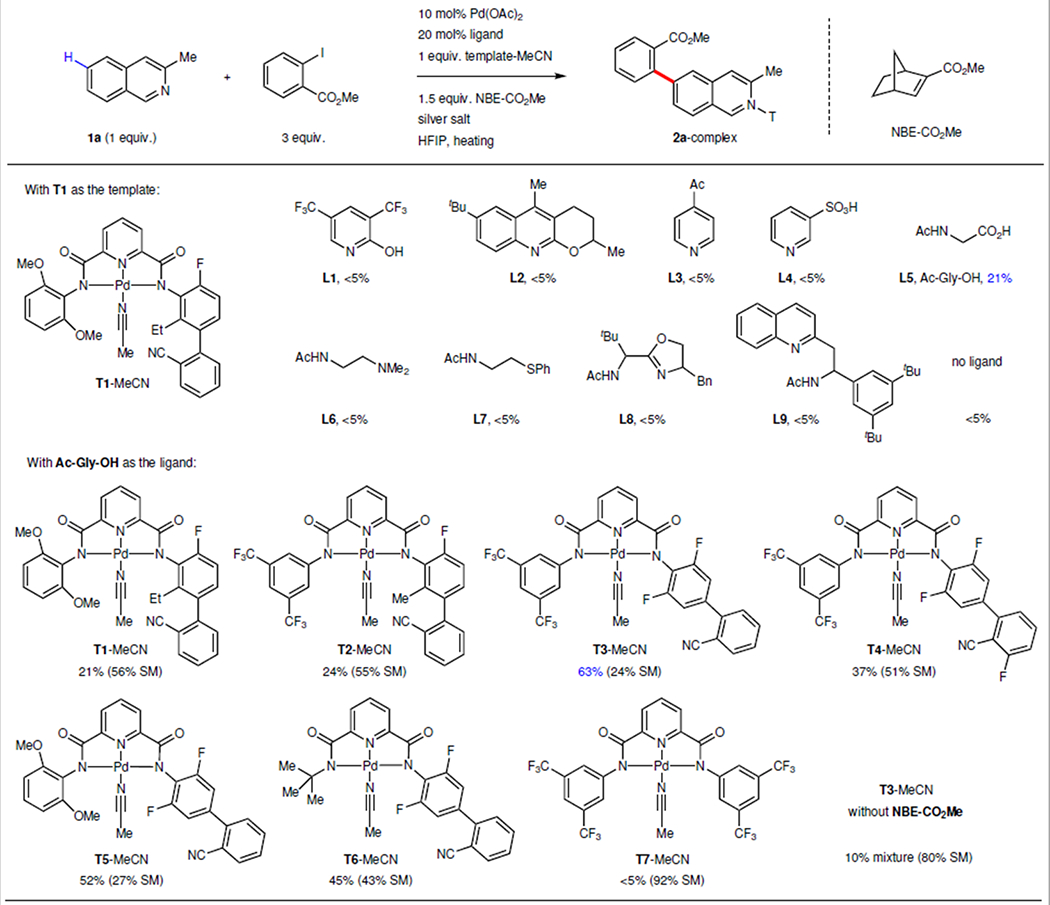 |
10 mol% Pd(OAc)2, 20 mol% ligand, 1 equiv. 3-methyl isoquinoline, 1 equiv. template–MeCN, 1.5 equiv. NBE–CO2Me, 3 equiv. methyl 2-iodobenzoate, 3 equiv. AgOAc, 1 equiv. Ag2CO3, HFIP, 80 °C. The yield was determined by 1H NMR spectroscopy. T, template; HFIP, hexafluoro-2-propanol; Me, methyl group; Et, ethyl group; Ph, phenyl group; Ac, acetyl group; CN, cyano group.
The established remote site-selective C–H arylation protocol was then extended to other pharmaceutically important benzoazines (Table 2). Firstly, we evaluated the isoquinolines and found that a range of derivatives (2a to 2d) were compatible, providing C6 arylation products in moderate yields by using template T3. Notably, bromine and chlorine, which serve as useful synthetic handles for subsequent chemical manipulation, were tolerated (2b, 2d). We were delighted to find that this strategy is also applicable to quinoline family by using template T1 or T2 depending on substitution pattern: T1 for quinolines bearing no functional group on C2-position; T2 for C2-substituted quinolines. The C6 arylation of simple quinoline proceeded smoothly (2e), giving 71% isolated yield. A broad range of quinoline derivatives bearing C2, C3, C4, C7, or C8 substituents (2f-2q) were suitable substrates. Electron-neutral, electron-donating, and electron-withdrawing substituents were all well tolerated, providing yields of up to 86%. Substrates 3-cyano quinoline (2m) and 4-methoxy quinoline (2n) gave lower yields due to the presence of the coordinative nitrile (competing with the template) and the C4 substitution (hindering the C5 palladation) respectively. Disubstituted quinolines were also compatible, affording the desired products in moderate to good yields (2r, 2s, 2t). Notably, polycyclic quinolines also afforded the remote arylation products in good yields (2u, 2v). The broad utility of this method is demonstrated with a wide range of benzoazines including quinoxaline (2w), benzoxazole (2x), benzothiazole (2y, 2z), indazole (2aa), and thienopyridine (2ab). Late-stage modification of an antileukaemic and antitumour alkaloid, camptothecin (2ac), at previously inaccessible site was also successful. In conjunction with our previous methods, we have thus far developed tools to functionalize this highly complex natural product at C5, C6 and C8 with precision demonstrating the unique power of site selective C–H activation22,29. Notably, the structural modifications at these positions through semi-synthesis had led to the discovery of drugs, such as topotecan and irinotecan30.
Table 2. |.
Remote site-selective arylation of benzoazines.
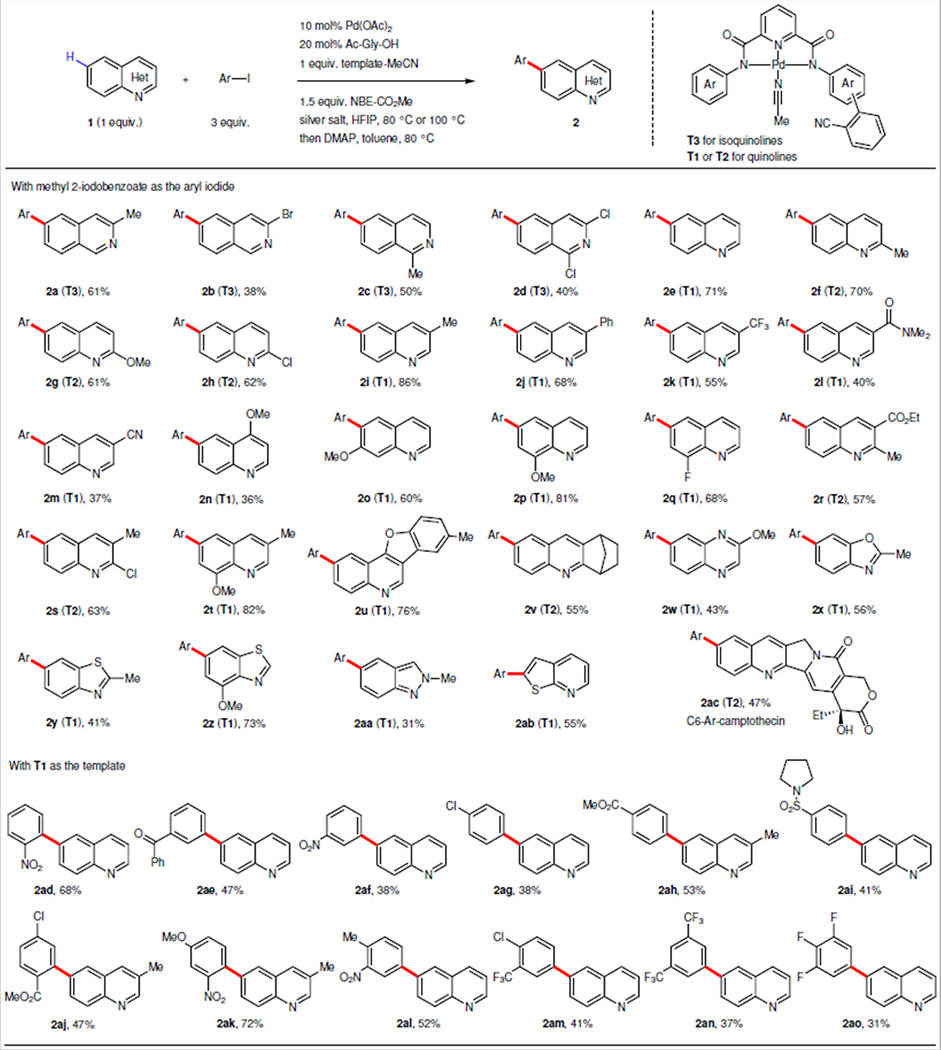 |
Scope of benzoazines: 10 mol% Pd(OAc)2, 20 mol% Ac–Gly–OH, 1 equiv. benzoazine, 1 equiv. template–MeCN, 1.5 equiv. NBE–CO2Me, 3 equiv. aryl iodide, 3 equiv. AgOAc, 1 equiv. Ag2CO3, HFIP, 80 °C, then 3 equiv. DMAP, toluene, 80 °C. For 2n and 2u, 20 mol% Pd(OAc)2 and 40 mol% Ac–Gly–OH were used. Scope of aryl iodides: 20 mol% Pd(OAc)2, 40 mol% Ac–Gly–OH, 1 equiv. benzoazine, 1 equiv. template–MeCN, 1 equiv. NBE–CO2Me, 3 equiv. aryl iodide, 3 equiv. AgOAc, HFIP, 100 °C, then 3 equiv. DMAP, toluene, 100 °C. For 2ad, 2aj and 2ak, 10 mol% Pd(OAc)2, 20 mol% Ac–Gly–OH, 1.5 equiv. NBE–CO2Me, 3 equiv. AgOAc, 1 equiv. Ag2CO3 and 80 °C were used. For each entry number (in bold), data was reported as isolated yield. The structure of 2ah was determined by X-ray crystallography.
We next surveyed the scope of aryl iodides using quinoline as the substrate (Table 2). Ortho-, meta- and para-monosubstituted aryl iodides (2ad to 2ai) were all suitable coupling partners, providing desired products in moderate yields. Furthermore, the di and trisubstituted aryl iodides were compatible under the reaction conditions, giving the desired products in moderate to good yields (2aj to 2ao). Electron rich aryl iodides were not effective coupling partners (less than 15% yields), implying that the oxidative addition of aryl iodides to palladacycle is not facile (see Supplementary Fig. 2 for proposed catalytic cycle). In comparison, the reactivity of these electron-rich aryl iodides can be restored by attaching an electron-withdrawing group (2ak, 72% yield). The regioselectivities observed with various heterocycle substrates and aryl iodide coupling partners are consistently high. In the representative examples (2a, 2e, 2af), either none (2e, 2af) or less than 5% of the over-arylated product (2a’) was detected (see Supplementary Fig. 1). Notably, Fukui indices of most heterocycle substrates show that the electronic density of the remote C6 and C7 sites has little difference, thus further showcasing the unique advantage of our approach using distance and geometry to differentiate remote C–H bonds.
Although the nitrile directed remote C–H palladation9,11,22 and the norbornene relay from ortho- to meta-positions has been previously shown separately23–25, how these two processes are successfully merged in such a complex catalytic cycle is puzzling based on previous mechanistic understanding. Computational studies provided detailed information about the incredibly complex C–H functionalization mechanism which utilizes a bifunctional template coordinated to two Pd metal centers. Fig. 2 highlights the transition state structures for C5 CMD (concerted metalation-deprotonation), norbornene insertion, C6 CMD, and β-carbon elimination steps (see supplementary information, Mechanistic study). The nature of the Pd template allows the nitrile group of the side arm to direct the second Pd catalyst to reach the C5 position of quinoline, but surprisingly the nitrile group has to come on and off of the Pd center in order for the catalytic cycle to proceed. The MPAA ligand promotes the first C5 CMD step but then dissociates in order to provide a vacant coordination site for subsequent norbornene insertion. These studies reveal the extraordinary complexity of merging the nitrile template and the norbornene transient mediator for palladation relay. The ability of both the MPAA ligand and the nitrile group to only associate during certain steps of the catalytic cycle is essential for completing the catalytic cycle.
Figure 2. |. DFT-optimized transition state structures. CMD, concerted metalation-deprotonation; TS, transition state.
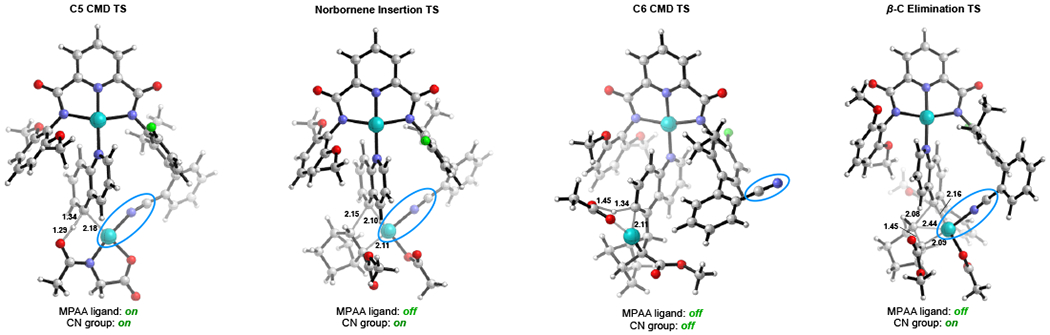
a, CMD transition state for C–H bond at C5 position; b, Transition state for norbornene insertion; c, CMD transition state for C–H bond at C6 position; d, Transition state for β-carbon elimination.
To further test whether this strategy can be broadly used to differentiate remote, adjacent C–H bonds, we embarked on remote site-selective C–H arylation of other arenes bearing covalently attached U-shaped templates (Table 3). We found that tetrahydroisoquinoline can be arylated at the C7 position, which is also one bond further away compared to the previous C8 arylation using remote directing template alone (4a). The reaction conditions tolerated a range of substituents at different positions, including 1-methyl (4b), 3-carboxylate (4c), 5-ethyl (4d), 5-phenyl (4e), and 5-bromo (4f). Finally, para-arylation of phenylpropanoic acid derivatives9 was also realized using this approach (6a, 6b) (Table 4). Various aryl iodides containing mono or disubstitutions were compatible, giving moderate yields (6c to 6f).
Table 3. |.
Remote C7-arylation of tetrahydroisoquinolines.
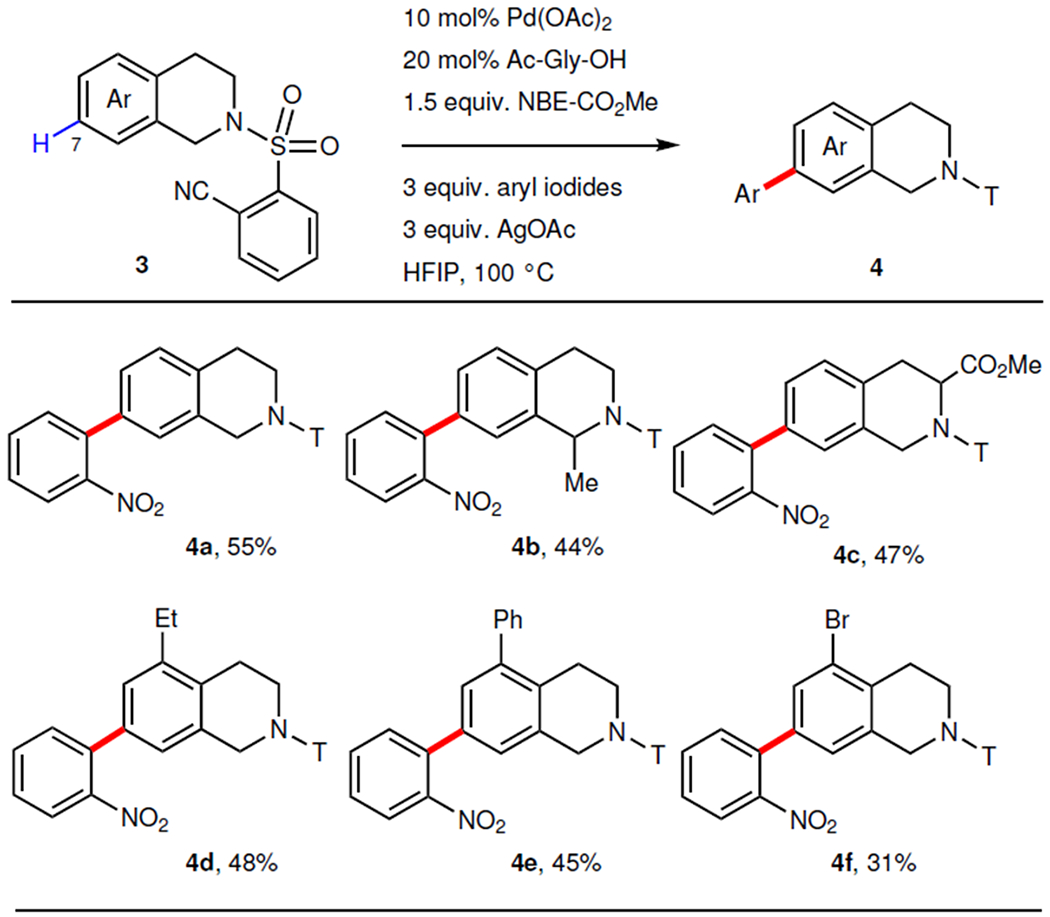 |
Aryl iodide = 1-iodo-2-nitrobenzene. 10 mol% Pd(OAc)2, 20 mol% Ac–Gly–OH, 1 equiv. tetrahydroisoquinolines, 1.5 equiv. NBE–CO2Me, 3 equiv. aryl iodide, 3 equiv. AgOAc, HFIP, 100 °C. For 4c and 4e, 20 mol% Pd(OAc)2 and 40 mol% Ac–Gly–OH were used. For each entry number (in bold), data were reported as isolated yield.
Table 4. |.
Remote para-arylation of phenylpropanoic acid derivatives.
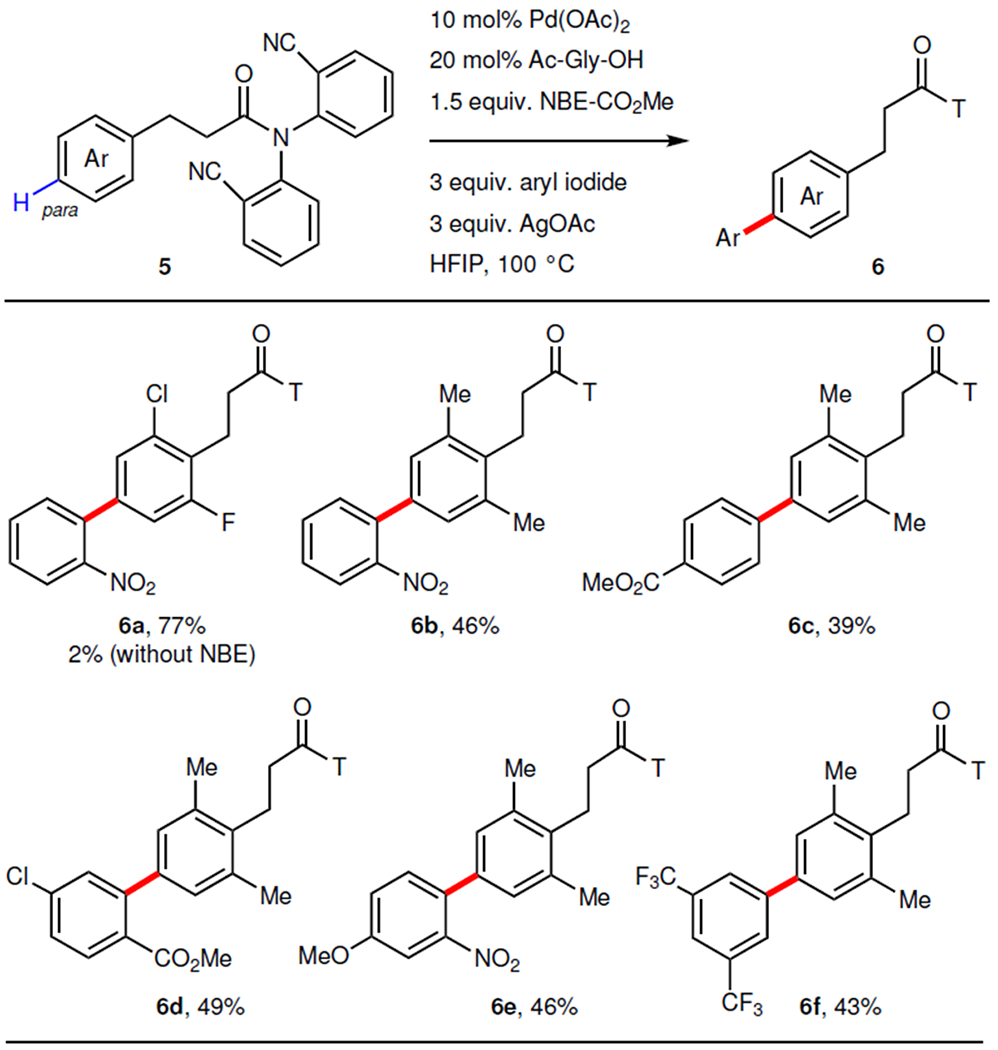 |
10 mol% Pd(OAc)2, 20 mol% Ac–Gly–OH, 1 equiv. arene, 1.5 equiv. NBE–CO2Me, 3 equiv. aryl iodide, 3 equiv. AgOAc, HFIP, 100 °C. For each entry number (in bold), data were reported as isolated yield.
In summary, we have developed a new strategy to distinguish remote C–H bonds within one-bond distance. This new method, along with previously developed template chemistry, allows us to access two different remote C–H bonds with precise control. The established approach is generally applicable to a wide range of heterocycles as well as other classes of synthetically useful substrates. The delicate cooperation of a remote directing template, a transient mediator norbornene and MPAA ligand reveals great potential for developing unprecedented catalytic systems in C–H activation.
Methods
General procedure for the remote site-selective arylation of benzoazines.
A reaction vial (8 mL) was charged with benzoazine (0.10 mmol, 1.0 equiv.), template–MeCN (0.10 mmol, 1.0 equiv.) and 0.2 mL DCM. The mixture was stirred for 5 min at room temperature, and then concentrated in vacuo. Pd(OAc)2 (2.2 mg, 10 μmol, 10 mol%), Ac–Gly–OH (2.3 mg, 20 μmol, 20 mol%), aryl iodide (0.3 mmol, 3 equiv.), AgOAc (50 mg, 0.30 mmol, 3.0 equiv.), Ag2CO3 (27.6 mg, 0.1 mmol, 1.0 equiv.), NBE–CO2Me (22.8 mg, 0.15 mmol, 1.5 equiv.) and HFIP (1.5 ml) were added. The reaction vial was sealed and allowed to stir at 80 °C for 18 h. The reaction mixture was cooled to room temperature. Then a solution of DMAP (36.7 mg, 0.3 mmol, 3 equiv.) in toluene (1.5 mL) was added. The mixture was stirred at 80 °C for 15 min. The reaction mixture was cooled to room temperature, diluted with EtOAc. The mixture was filtered through a short pad of celite and eluted with EtOAc (2 × 2 mL). The filtrate was evaporated under reduced pressure. (If the product release is not complete, a solution of DMAP (18.4 mg, 0.15 mmol, 1.5 equiv.) in toluene (1.5 mL) was added. The solution was stirred at 80 °C for 15 min., and then concentrated.) Purification by preparative TLC afforded the title compound.
Data Availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. Metrical parameters for the structure of 2ah (see supplementary information) are available free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under reference number CCDC 1890836.
Supplementary Material
Acknowledgements
We gratefully acknowledge Scripps Research, the NIH (National Institute of General Medical Sciences grant R01GM102265), the National Science Foundation (CHE-1764328 to K.N.H.) and under the CCI Center for Selective C–H Functionalization (CHE-1700982 to K.N.H) for their financial support. Computation study was also supported by the NSF (OCI-1053575 to K.N.H.). Y. L. and J. W. thank the China Scholarship Council.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Daugulis O, Do H-Q & Shabashov D Palladium- and copper-catalyzed arylation of carbon–hydrogen bonds. Acc. Chem. Res 42, 1074–1086 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyons TW & Sanford MS Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev 110, 1147–1169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abrams DJ, Provencher PA & Sorensen EJ Recent applications of C–H functionalization in complex natural product synthesis. Chem. Soc. Rev 47, 8925–8967 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Li J, Sarkar SD & Ackermann L meta- and para-Selective C–H functionalization by C–H activation. Top. Organomet. Chem 55, 217–257 (2016). [Google Scholar]
- 5.Breslow R Biomimetic control of chemical selectivity. Acc. Chem. Res 13, 170–177 (1980). [Google Scholar]
- 6.Das S, Incarvito CD, Crabtree RH & Brudvig GW Molecular recognition in the selective oxygenation of saturated C–H bonds by a dimanganese catalyst. Science 312, 1941–1943 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Phipps RJ & Gaunt MJ A meta-selective copper-catalyzed C–H bond arylation. Science 323, 1593–1597 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Saidi O, et al. Ruthenium-catalyzed meta sulfonation of 2-phenylpyridines. J. Am. Chem. Soc 133, 19298–19301 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Leow D, Li G, Mei T-S & Yu J-Q Activation of remote meta-C–H bonds assisted by an end-on template. Nature 486, 518–522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuninobu Y Ida H, Nishi M & Kanai M A meta-Selective C–H borylation directed by a secondary interaction between ligand and substrate. Nat. Chem 7, 712–717 (2015) [DOI] [PubMed] [Google Scholar]
- 11.Bag S, et al. Remote para-C–H functionalization of arenes by a D-shaped biphenyl template-based assembly. J. Am. Chem. Soc 137, 11888–11891 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Okumura S, et al. Para-selective alkylation of benzamides and aromatic ketones by cooperative nickel/aluminum catalysis. J. Am. Chem. Soc 138, 14699–14704 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Stephens DE & Larionov OV Recent advances in the C–H-functionalization of the distal positions in pyridines and quinolines. Tetrahedron 71, 8683–8716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman AM, Lewis JC, Bergman RG & Ellman JA Rh(I)-catalyzed direct arylation of pyridines and quinolines. J. Am. Chem. Soc 130, 14926–14927 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takagi J, Sato K, Hartwig JF, Ishiyama T & Miyaura N Iridium-catalyzed C–H coupling reaction of heteroaromatic compounds with bis(pinacolato)diboron: regioselective synthesis of heteroarylboronates. Tetrahedron Lett. 43, 5649–5651 (2002). [Google Scholar]
- 16.Ye M, Gao G-L & Yu J-Q Ligand-promoted C-3 selective C–H olefination of pyridines with Pd catalysts. J. Am. Chem. Soc 133, 6964–6967 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Nakao Y, Yamada Y, Kashihara N & Hiyama T Selective C-4 alkylation of pyridine by nickel/Lewis acid catalysis. J. Am. Chem. Soc 132, 13666–13668 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Tsai C-C, et al. Bimetallic nickel aluminun mediated para-selective alkenylation of pyridine: direct observation of η2,η1-pyridine Ni(0)-Al(III) intermediates prior to C–H bond activation. J. Am. Chem. Soc 132, 11887–11889 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto S, Saga Y, Andou T, Matsunaga S & Kanai M Cobalt-catalyzed C-4 selective alkylation of quinolines. Adv. Synth. Catal 356, 401–405 (2014). [Google Scholar]
- 20.Kwak J, Kim M & Chang S Rh(NHC)-catalyzed direct and selective arylation of quinolines at the 8-Position. J. Am. Chem. Soc 133, 3780–3783 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Konishi S, et al. Site-selective C−H borylation of quinolines at the C8 position catalyzed by a silica-supported phosphane-iridium system. Chem. Asian J 9, 434–438 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Tanaka K & Yu J-Q Remote site-selective C–H activation directed by a catalytic bifunctional template nature. Nature 543, 538–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X-C, et al. Ligand-enabled meta-C–H activation using a transient mediator. Nature 519, 334–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong Z, Wang J & Dong G Simple amine-directed meta-selective C–H arylation via Pd/norbornene catalysis. J. Am. Chem. Soc 137, 5887–5890 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Shen P-X, Wang X-C, Wang P, Zhu R-Y & Yu J-Q Ligand-enabled meta-C–H alkylation and arylation using a modified norbornene. J. Am. Chem. Soc 137, 11574–11577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye J & Lautens M Palladium-catalysed norbornene-mediated C−H functionalization of arenes. Nat. Chem 7, 863–870 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Della Ca’ N., Fontana M, Motti E & Catellani M Pd/Norbornene: a winning combination for selective aromatic functionalization via C–H bond activation. Acc. Chem. Res 49, 1389–1400 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Shi H, Herron AN, Shao Y, Shao Q & Yu J-Q Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 558, 581–586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang P, et al. Ligand-accelerated non-directed C–H functionalization of arenes. Nature 551, 489–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bracher F & Tremmel T From lead to drug utilizing a mannich reaction: the topotecan story. Arch. Pharm. Chem. Life Sci 350, e1600236 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and its Supplementary Information files. Metrical parameters for the structure of 2ah (see supplementary information) are available free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under reference number CCDC 1890836.