

Summary
A cornerstone of modern synthetic chemistry rests on the ability to manipulate the reactivity of a carbon center by rendering it either electrophilic or nucleophilic. However, accessing a similar reactivity spectrum with boron-based reagents has been significantly more challenging. While classical nucleophilic carbon-based reagents normally do not require steric protection, readily accessible, unprotected boron-based nucleophiles have not yet been realized. Herein, we demonstrate that the bench stable closo-hexaborate cluster anion can engage in a nucleophilic substitution reaction with a wide array of organic and main group electrophiles. The resulting molecules containing B‒C bonds can be further converted to tricoordinate boron species widely used in organic synthesis.
Keywords: Nucleophilic boron, closo-hexaborate cluster, cluster deconstruction, borylation, alkyl bromides, alkyl pseudo halides, stereoinversion, main group electrophiles, boron-heteroatom bonds
Graphical Abstract
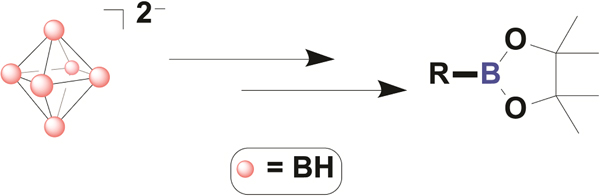
eTOC Blurb
We demonstrate that the bench stable closo-hexaborate cluster dianions can engage in a nucleophilic substitution reaction with a wide array of organic and main group electrophiles without the use of metal catalysts. The resulting molecules containing B‒C bonds can be further converted to tricoordinate boron species widely used in organic synthesis. This strategy showcases a unique substitution/deconstruction reaction sequence with a sterically unprotected nucleophile to afford boronic acid pinacol esters without the use of metal catalysis.
Introduction
Electronic polarization at an atom (e.g. carbon) generally dictates its reactivity profile and determines whether it can undergo electrophilic or nucleophilic substitution chemistry.1 Since the seminal work by Grignard, researchers have been able to generate a variety of useful synthetic reagents featuring an electropositive element interacting with a carbon, rendering the carbon center nucleophilic.2 Given the prevalence of organoboron compounds used in synthesis,3,4 researchers recently have been interested in applying a similar concept of polarity switching commonly used for carbon reagents to boron congeners (Fig. 1a).5 This strategy can potentially diversify the reactivity repertoire beyond the classical electrophilic nature of boron-based reagents.6 However, access to synthetically practical nucleophilic boron compounds remains a significant fundamental challenge. In 2006, Yamashita, Nozaki and co-workers reported the first synthesis and isolation of a well-defined anionic boryllithium 1 (Fig. 1b), which undergoes several reactions with carbon-based electrophiles.7 This discovery was enabled by the use of a sterically encumbering ligand platform that stabilizes the highly reactive nucleophilic boron site, termed as “boryl”. 8 Inspired by the idea of using steric protection, others have targeted the synthesis of nucleophilic boron compounds, generally leveraging electron-donating and sterically bulky ligands to tame the reactivity of the nucleophilic boron species 2 – 5 (Fig. 1b).9–13 Recently, several ligand frameworks were also developed to constrain boron-based centers in a non-traditional electronic environment rendering some of the species (e.g., 6) nucleophilic.14,15 While significant progress has been made in stabilizing nucleophilic boron centers, synthetically demanding protocols and lack of overall benchtop stability have hindered the widespread use of nucleophilic boron reagents in synthetic methodology.
Fig. 1. Development of sterically unprotected cluster-based borylation reagents.

a, General scheme for metal-catalyzed borylation using tricoordinate diborane reagents. b, Representative nucleophilic boron reagents developed previously. c, One of three triply degenerate HOMO representations of B6H62−, synthesis of the B6H71− anion, and a general borylation strategy using sterically unprotected cluster-based nucleophiles developed in this work.
Results and discussion
In this work, we demonstrate a conceptually different approach to stabilize a reactive boron nucleophile, predicated on three-dimensional electron delocalization instead of steric protection enforced by a ligand auxiliary (Fig. 1c). In order to engender the necessary delocalization, we identified a system in which boron atoms are bonded in a cluster-based environment. Specifically, we evaluated polyhedral boranes (BnHn2–, n = 6, 10, 12) (Fig. 1c, top), which are some of the simplest clusters containing catenated boron atoms known to date. In these clusters, electron delocalization across cage boron vertices reduces the otherwise extreme reactivity of atom-centered boryl anions.16 A manifestation of this delocalization can be seen in the example of B12H122−, which has a nucleophilicity as low as that of benzene; neither of these molecules readily react with carbon-based electrophiles.17 On the other hand, smaller boron clusters have been previously suggested to be less inert and could potentially exhibit an increased nucleophilic character compared to B12H122−.18 For example, DFT analysis of B6H62− at the B3LYP-D3/6–31G(d) level of theory suggests that the triply degenerate highest occupied molecular orbitals (HOMO) are delocalized across eight faces of octahedron and that the corresponding energy is 3eV higher than the HOMO of B12H122− (see SI). Ultimately, this begged the question of whether B6H62− can be considered as a competent nucleophile in the context of nucleophilic borylation chemistry to produce substrates that can be efficiently transformed into tricoordinate boron compounds commonly used in synthesis (Fig. 1c, bottom).
Dianionic closo-hexaborate compound 7 (Fig. 2a) can be synthesized on a multi-gram scale in one step from cheap and commercially available NaBH4 and BF3·Et2O. Crude 7 can be directly converted into 8 and 9 from a salt metathesis reaction in water, producing singly-protonated, air-stable closo-hexaborate species soluble in organic solvents (Fig. S1–S10). The protonation state of 8 (NBu4 [B6H6Hfac]) and 9 (MePPh3[B6H6Hfac]) imply that the Brønsted-basic character of B6H62− is readily accessible and could be applied to other electrophiles beyond H+.
Fig. 2. Synthesis and deconstruction studies of perfunctionalized closo-hexaborate anions.

a, Preparation of monoanionic closo-hexaborate cluster reagents (8 and 9) and their reactivity toward model electrophiles. b, Discovery of the selective cage deconstruction process of clusters 10 and 11, leading to the intermediate 13, which can be further degraded with TCNQ to produce tricoordinate alkyl boronate species (14 and 15). c, Oxidation potential variability (see cyclic voltammograms of 10, 11 and 12) of the perfunctionalized clusters leading to the alternative borylation strategy via a monosubstitution using dianionic B6H62− reagent.
To probe whether this cluster could engage in nucleophilic substitution towards organic electrophiles, we conducted reactions between 8 and a series of benzyl bromides in acetonitrile (Fig. 2a; see also Table S1). Using K3PO4 as a base, persubstituted B6-based clusters 10 and 11 can be formed quantitatively as judged by 11B NMR spectroscopy (Table S1; Fig S11). During our investigations into the purification of 10 and 11, we noticed that subjecting these compounds to a slurry of silica gel in dichloroethane at 50 ° C results in the formation of a new cluster-based species as evidenced by a diagnostic change in 11B NMR spectra of the corresponding products (Fig. 2b). From the reaction mixture initially containing 11, we were able to isolate a crystalline solid and subject this material to a series of structural characterization techniques. 1H NMR spectroscopy suggested the formation of new bridging hydrides and implied a loss of one “B– CH2Ar” vertex from starting 11. Both X-ray photoelectron spectroscopy (XPS) and 11B NMR spectroscopy suggest desymmetrization of the cluster precursor and the appearance of two unique boron sites with distinct electronic environments (Fig. S15–S21). Single crystals of the material grown from a concentrated toluene solution were subjected to X-ray diffraction studies and identified the solid as a neutral pentaborane cluster 13 with one benzyl-attached boron vertex removed from parent cluster 11 (Fig. 2b; Table S8–S10). This unprecedented partial cage deconstruction suggests that unlike large closo-borane clusters (e.g., B12H122−),19 peralkylated B6-based clusters can be oxidatively unstable towards B–B bond rupture and that this instability could be leveraged to selectively generate tricoordinate boron reagents through substituted closo-hexaborate cage deconstruction.
To further probe this hypothesis, we investigated the deconstruction of 11 with a range of model oxidants (Table S2; Fig. S22). Upon employing 7,7,8,8-tetracyanoquinodimethane (TCNQ) as single electron oxidant, significant quantities of the corresponding benzyl boronate products (14 and 15) were observed suggesting a successful cage deconstruction under these conditions. While these results are conceptually promising and suggest that one can employ monoanionic 8 or 9 as a nucleophilic boron source to synthesize organic boronates through a substitution/cage deconstruction sequence, several challenges revealed themselves during the course of our investigation that necessitated a modified strategy. Specifically, when cyclic voltammetry (CV) data was compared for 10, 11 and 12 (Fig. S12–S14), significant variability in oxidation potentials, ranging from +0.48 V to 0.03 V (vs. Fc+/Fc), was observed for these compounds (Fig. 2c). This suggests that the nature of the organic substituents dramatically affects the oxidative stability of the perfunctionalized intermediates potentially limiting the synthetic generality of the cage substitution/deconstruction protocol.
We thus hypothesized that partial cage substitution could alleviate these limitations (Fig. 2c). We investigated whether well-defined dianionic B6H62− salts could be used for a base-free nucleophilic substitution, leading to the formation of monoalkylated compounds with similar oxidation potentials (Fig. 2c). The synthesis of the dianion 16 can be accomplished in a straightforward manner on a multi-gram scale by stirring 8 with NBu4OH in the presence of the Na2SO4 (Fig. 3a; Fig. S23–S25). Alternatively, 9 can be deprotonated with a phosphorane reagent (CH2PPh3) producing 17 in a nearly quantitative yield (Fig. 3a; Fig. S26–S30). Both of these dianion-based salts are bench stable solids that can be stored in a desiccator for over 2 months without any noticeable protonation to form the B6H7− monoanion.
Fig. 3. Borylation/cage deconstruction strategy with dianionic closo-hexaborates.

a, Preparation of B6H62− reagents and their subsequent use in borylation of organic electrophiles. b, Substrate scope of alkyl halides. Standard reaction conditions: B6H62− (1.0 equiv), alkyl halide (0.4 mmol, 1.0 equiv), MeCN (2 mL). For unactivated alkyl bromides, 60 °C heating. For benzyl bromides, room temperature. Pinacol (10 equiv), MgSO4 (12 equiv), THF (2 mL), 0 °C to room temperature for deconstruction. Isolated yields are calculated after cage deconstruction. Inset: 11B NMR spectra for the synthesis sequence: closo-hexaborate dianion (top); monosubstituted closo-hexaborate (middle); purified alkyl‒Bpin (bottom). c, Substrate scope of alkyl pseudohalides. Reaction conditions: B6H62− (1.3–2.0 equiv), µwave heating at 140 °C. † Using alkyl iodide (2 equiv). † B6H62− (1.5 equiv) and µwave heating at 110 °C. ‡ Using oil bath heating at 80 °C. ¶ µwave heating at 90 °C. § Using oil bath heating at 60 °C. [N +] = NBu4+, [P+] = MePPh3+
Consistent with our hypothesis, 16 and 17 undergo nucleophilic substitution with a wide variety of alkyl-based electrophiles producing the corresponding mono-substituted [B6H5RHfac]− species (Fig. 3b). Importantly, these reactions take place under mild conditions by simply combining a dianionic reagent together with an electrophile substrate in a variety of non-protic organic solvents (Fig. S31–S32). No other additives are required to forge a B–C bond via this strategy. The unpurified products from these transformations can be directly subjected to oxidative deconstruction of the cluster with TCNQ or nitrosonium tetrafluoroborate (NOBF4) in the presence of pinacol, ultimately producing the corresponding alkyl pinacol boronate esters (Table S3–S4).
Primary alkyl bromides and iodide containing various functional groups such as ester (19), perfluoroalkyl (20), alkyl ether (22, 23) and halide (24) substituents were successfully converted to the corresponding boronic esters using the substitution/cage deconstruction sequence. Phenethyl bromide, which is susceptible to elimination under basic conditions, is tolerated under the developed protocol (21). A substrate bearing a terminal olefin was also converted directly to the boronic ester with no evidence of olefin reduction (e.g., hydroboration) (25). For benzyl bromides containing aryl bromide and iodide substituents, only Csp3‒Br bonds were borylated (15, 28). Interestingly, substrates bearing active sites for nucleophilic aromatic substitution (SNAr) such as pentafluorophenyl and difluorophenyl groups (30, 32) remained intact under the developed conditions (Fig. 3b; Fig S113–S165).
Recently, there have been a growing number of reports using oxygen-containing electrophiles (pseudohalides) to replace alkyl halides as cross-coupling partners.20 However, in metal-catalyzed borylations of alkyl electrophiles, pseudohalides are rarely reported as substrates due to the reduced reactivity of Csp3‒O bonds towards activation by several metal-based catalysts commonly employed under borylation conditions.21 In our case, the substitution of both primary and secondary alkyl‒OTs/OMs substrates by B6H62− (16) proceeded smoothly in the absence of any metal salts or halide additives; subsequent cage deconstruction produced the corresponding pinacol boronate ester compounds (33–38) (Fig. 3c; Fig S166–S180).
To shed light on the nature of the cluster-based nucleophilic substitution, we investigated whether the discovered transformation showed hallmarks of radical reaction pathways. Bromomethylcyclopropane has been previously employed as a standard radical probe in various metal-catalyzed borylations, where in the presence of radical species, ring-opened 3-butenylboronate was isolated as the major product.22,23 In contrast, after subjecting the same electrophile to our optimized reaction conditions, no ring-opened species were observed either in the reaction mixture after monofunctionalization or in the resulting borylated product 26. This observation suggests that a radical mechanism for B‒C bond formation is unlikely (Fig. S74–S75). Additionally, when enantiopure (R)-2-OTs-4-phenylbutane was applied in our protocol, enantio-enriched alkyl boronate 37 was obtained with inversion of stereochemistry (94:6 e.r.), suggesting an apparent SN2 substitution mechanism (Fig. S76–S79). This observation stands in stark contrast to existing metal-catalyzed systems, in which racemic borylated products are observed due to the intermediacy of alkyl radicals, therefore impeding general approaches to stereospecific borylation through chiral induction.23,24 Importantly, this borylation strategy provides a new chiral auxiliary-free pathway to generate enantio-enriched alkyl boronates from chiral alcohol derivatives.25,26
Undesired side reactions, including apparent one-electron reduction chemistry, have been observed with other classes of nucleophilic boron reagents in the presence of alkyl-based electrophiles;27 in addition, some of these species also exhibit reducing behavior toward main group electrophiles.28 Interestingly, during the course of our investigations, we never observed any apparent reduction products stemming from the reactions of closo-hexaborate and alkyl electrophiles, which implies that under the developed conditions this nucleophilic source of boron is significantly less reducing. We therefore were curious whether the nucleophilic behavior of the closo-hexaborate reagent could be extended to reactions with main group electrophiles to forge B-heteroatom bonds (Fig. 4). Traditionally, main group boranes (e.g., phosphinoboranes) are prepared using electrophilic boron-based reagents.29 Interested in reversing the role of boron reagents in these bond formations, we combined 17 and Ph2PCl, resulting in the formation of diphenylphosphine-substituted closo-hexaborate 39 (Fig. 4a), as judged by multinuclear NMR spectroscopy (Fig. S80–S81). Subsequent treatment with allyl bromide followed by workup permitted the isolation of 40 as a zwitterionic closo-hexaborate containing an exopolyhedral P‒B bond (Fig. S82–S88). The methylation of phosphorus, generating zwitterionic 41 (Fig. S89–S92), followed by iodination produced a heteroleptic penta-iodinated cluster 42, which we were able to characterize by multinuclear NMR spectroscopy and single crystal X-ray diffraction, further confirming the proposed B‒P connectivity (Fig. S93–S98; Table S14–S16). These results contrast previous observations of nearly quantitative reduction and formation of the corresponding diphosphine (Ph2P‒PPh2) when 1 was treated with Ph2PCl.28 Consistent with the non-reducing, nucleophilic nature of the closo-hexaborate anion, 17 undergoes a clean transformation with PhSeCl reagent forging a substituted cluster 43 with an exopolyhedral B‒Se bond (Fig. 4b; Fig. S99–S106).
Fig. 4. Reaction of dianionic closo-hexaborate with main group electrophiles.
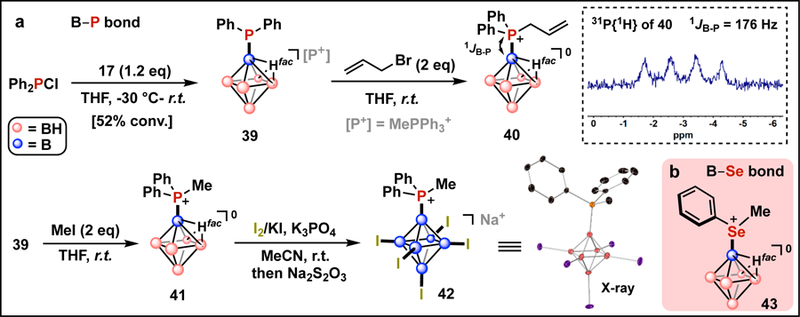
a, Transformations leading to the formation of clusters with B–P bonds (40–42). b, Hexaborate-based cluster featuring a B–Se bond ( 43, see Fig. S80–S106 for synthesis and characterization detail).
Conclusions
Overall, we have discovered that small polyhedral boron clusters featuring a sterically unprotected B6-based cluster core possess a strong, yet non-reducing nucleophilicity that can be leveraged for the borylation of various organic and main group electrophiles. This led us to the development of a simple protocol, whereby carbon-based electrophiles can be transformed into the corresponding tricoordinate boron ester species without the use of metal catalysis. This work highlights how boron-rich clusters can expand the toolkit of main group reagents30–48 ultimately aiding in the development of organic synthesis through new modes of reactivity.
Experimental procedures
Full experimental procedures are provided in the Supplemental Information.
Supplementary Material
Table S7. Bond lengths [Å] and angles [°] for 9
Table S10. Bond lengths [Å] and angles [°] for 13
Table S13. Bond lengths [Å] and angles [°] for 17
Table S16. Bond lengths [Å] and angles [°] for 42
Box 1:
Previously, electrophilic diborane reagents, such as B2Pin2 have been developed to transform alkyl halides to their corresponding boronic acid or boronic esters.
In systems employing these reagents, metals are often added to stabilize the transient boryl species formed during the reaction.
Box 2:
While boron is commonly considered to be electrophilic due to its Lewis acidic character, there have been emerging reports on the preparation of nucleophilic boron species, such as the ones shown here. These compounds largely rely on steric protection of the nucleophilic boron center using bulky ligands. But despite these sterically encumbering ligand frameworks, many of these compounds remain air sensitive and strongly reducing, limiting their applications in synthetic chemistry.
Box 3:
Boron clusters are boron-rich polyhedral compounds and usually display terminal hydrogen atoms at the periphery. The octahedral closo-hexaborate dianion, with 6 boron atoms, exhibits delocalized electron density across all faces of the octahedron. The delocalization of these electrons contributes to nucleophilic character at boron without the need for steric protection as well as pronounced stability to air and moisture.
Box 4:
Boron clusters may seem exotic, but the synthesis of the closo-hexaborate anion is straightforward and can be accomplished using feedstock chemicals on a decagram scale: the combination of BF3•Et 2O with NaBH4 and heat, followed by salt exchange with an ammonium or phosphonium cation, results in the formation of the desired closo-hexaborate.
Box 5:
In this work we use the nucleophilic closo-hexaborate anion to showcase the development of a unique substitution/cage-deconstruction sequence to synthesize boronic esters from their corresponding alkyl halides or pseudohalides without the use of metal catalysts or strong bases. We also extend this chemistry to main group electrophiles and show that the closo-hexaborate anion can be used to forge B-P and B-Se bonds under mild conditions in the absence of any metal catalysts.
Highlights:
Nucleophilic substitution using the closo-hexaborate cluster anions
Boron-based nucleophiles that do not require steric protection
Substitution shown with both carbon-based and main group electrophiles
Catalyst-free production of alkyl boronic esters from alkyl halides
Bigger Picture.
Over the past 50 years, boron-based reagents have been successfully utilized for the production of new chemicals ranging from small molecule drugs to polymer-based materials. In order to expand the practical utility of boron-based synthetic chemistry, emerging efforts have focused on the synthesis of nucleophilic boron species, which rely heavily on bulky ligand protection. These new nucleophilic boron reagents have shown unprecedented reactivity, but generally suffer from intrinsic air instability, limiting their applications in organic synthesis. In this article, we showcase the efficient synthesis of stable nucleophilic closo-hexaborate cluster dianions, which are stabilized via three-dimensional delocalization instead of steric protection. We show that this class of reagents can react with a series of electrophiles without the use of metal catalysts or complex ligand auxiliaries and can be extended by the controlled deconstruction of the substituted clusters, producing tricoordinate alkyl boronic esters.
Acknowledgements
This work has been supported by the NIH (R35GM124746) and the donors of the American Chemical Society Petroleum Research Fund (56562-DNI3). A. M. S. is grateful to the Alfred Sloan foundation for the Sloan Research Fellowship in Chemistry and Research Corporation for Science Advancement (RCSA) for the Cottrell Scholar Award. We are grateful to the National Science Foundation (CHE-1764328) for financial support of this research. Calculations were performed on the Hoffman2 cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575). K. Q. acknowledges the Amgen Foundation for research funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and software availability
The crystallography data have been deposited at the Cambridge Crystallographic Data Center (CCDC) as CCDC: 1909639 (9), CCDC: 1909641 (13), CCDC: 1909643 (17), CCDC: 1909644 (42) and can be obtained free of charge from www.ccdc.cam.ac.uk/getstructures.
Supplemental information
Supplemental Information can be found with this article online at
Declaration of interests
The authors declare no competing financial interests.
REFERENCES AND NOTES
- 1.Smith MB. (2017). “Chapter 11: Carbon-carbon bond-forming reactions: cyanide, alkyne anions, Grignard reagents, and organolithium reagents” in Organic Sy nthesis (Academic Press; ), pp. 547–603. [Google Scholar]
- 2.Grignard V. (1900). Sur quelques nouvelles combinaisons organométaliques du magnésium et leur aplicatione à des synthèses d’alcools et d’hydrocabures. Compt .Rend. 130, 1322–1324. [Google Scholar]
- 3.Suzuki A. (2011). Cross-coupling reactions of organoboranes: an easy way to construct C‒C bonds (Nobel Lecture). Angew. Chem. Int. Ed. 50, 6722–6737. [DOI] [PubMed] [Google Scholar]
- 4.Hall DG. (2005). Boronic acids: Preparation and applications in organic synthesis and medicine. (Wiley-VCH Verlag GmbH; ). [Google Scholar]
- 5.Kubota K., Iwamoto H., and Ito H. (2017). Formal nucleophilic borylation and borylative cyclization of organic halides. Org. Biomol. Chem. 15, 285–300. [DOI] [PubMed] [Google Scholar]
- 6.Neeve EC., Geier SJ., Mkhalid IAI., Westcott SA., and Marder TB. (2016). Diboron(4) compounds: from structural curiosity to synthetic workhorse. Chem. Rev. 116, 9091–9161. [DOI] [PubMed] [Google Scholar]
- 7.Segawa Y., Yamashita M., and Nozaki K. (2006). Boryllithium: isolation, characterization, and reactivity as a boryl anion. Science, 314, 113–115. [DOI] [PubMed] [Google Scholar]
- 8.Braunschweig H. (2007). Lithiumboryl—a synthon for a nucleophilic boryl anion. Angew. Chem. Int. Ed. 46, 1946–1948. [DOI] [PubMed] [Google Scholar]
- 9.Kinjo R., Donnadieu B., Celik MA., Frenking G., and Bertrand G. (2011). Synthesis and characterization of a neutral tricoordinate organoboron isoelectronic with amines. Science, 333, 610–613. [DOI] [PubMed] [Google Scholar]
- 10.Pécharman A-F., Colebatch AL., Hill MS,. McMullin CL., Mahon MF., and Weetman C. (2017). Easy access to nucleophilic boron through diborane to magnesium boryl metathesis. Nature Comm. 8, 15022–15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braunschweig H., Chiu C-W., Radacki K., and Kupfer T. (2010). Synthesis and structure of a carbene-stabilized p-boryl anion. Angew. Chem. Int. Ed. 49, 2041–2044. [DOI] [PubMed] [Google Scholar]
- 12.Kleeberg C., Crawford AG., Batsanov AS., Hodgkinson P., Apperley DC., Cheung MS., Lin Z., and Marder TB. (2012). Spectroscopic and structural characterization of the CyNHC adduct of B2pin2 in solution and in the solid state. J. Org. Chem. 77, 785–789. [DOI] [PubMed] [Google Scholar]
- 13.Braunschweig H., Dewhurst RD., Pentecost L., Radacki K., Vargas A., and Ye Q. (2016). Dative bonding between group 13 elements using a boron-centered Lewis base. Angew. Chem. Int. Ed. 55, 436–440. [DOI] [PubMed] [Google Scholar]
- 14.Kaese T., Trageser T., Budy H., Bolte M., Lerner H-W., and Wagner MA. (2018). Redox-active diborane platform performs C(sp3)–H activation and nucleophilic substitution reacti ons. Chem. Sci. 9, 3881–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sprenger JAP., Landmann J., Drisch M., Ignat’ev N., and Finze M.(2015). Syntheses of tricyanofluoroborates M[BF(CN)3] (M = Na, K): (CH3)3SiCl catalysis, countercation effect, and reaction intermediates. Inorg. Chem. 54, 3403–3412. [DOI] [PubMed] [Google Scholar]
- 16.Balakrishnarajan MM., Hoffman R., Pancharatna PD., and Jemmis ED. (2003). Magic electron counts and bonding in tubular boranes. Inorg. Chem. 42, 4650–4659. [DOI] [PubMed] [Google Scholar]
- 17.Olid D., Núñez R., Viñas C., and Teixidor F.(2013). Methods to produce B–C, B–P, B–N and B–S bonds in boron clusters. Chem. Soc. Rev. 42, 3318–3336. [DOI] [PubMed] [Google Scholar]
- 18.Preetz W., and Peters G. (1999). The hexahydro-closo-hexaborate dianion [B6H6]2− and its derivatives. Eur. J. Inorg. Chem. 1831–1846.
- 19.Axtell JC., Saleh LMA., Qian EA., Wixtrom AI., and Spokoyny AM. (2018). Synthesis and applications of perfunctionalized boron clusters. Inorg. Chem. 57, 2333–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Börjesson M., Moragas T., Gallego D., and Martin R. (2016). Metal-catalyzed carboxylation of organic (pseudo)halides with CO2. ACS Catal., 6, 6739–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang C-T., Zhang Z-Q., Tajuddin H., Wu C-C., Liang J., Liu J-H., Fu Y., Czyzewska M., Steel PG., Marder TB., and Liu L. (2012). Alkylboronic esters from copper-catalyzed borylation of primary and secondary alkyl halides and pseudohalides. Angew. Chem. Int. Ed. 51, 528–532. [DOI] [PubMed] [Google Scholar]
- 22.Bose SK., Brand S., Omoregie HO., Haehnel M., Maier J., Bringmann G., and Marder TB. (2016). Highly efficient synthesis of alkylboronate esters via Cu(II)-catalyzed borylation of unactivated alkyl bromides and chlorides in air. ACS Catal. 6, 8332–8335. [Google Scholar]
- 23.Ito H., and Kubota K. (2012). Copper(I)-catalyzed boryl substitution of unactivated alkyl halides. Org. Lett. 14, 890–893. [DOI] [PubMed] [Google Scholar]
- 24.Yi J., Liu J-H., Liang J., Dai J-J., Yang C-T., Fu Y., and Liu L. (2012). Alkylboronic esters from palladium- and nickel-catalyzed borylation of primary and secondary alkyl bromides. Adv. Synth. Catal. 354, 1685–1691. [Google Scholar]
- 25.Rygus JPG., and Crudden CM. (2017). Enantiospecific and iterative Suzuki−Miyaura cross-cou plings. J. Am. Chem. Soc. 139, 18124–18137. [DOI] [PubMed] [Google Scholar]
- 26.Sandford C., and Aggarwal VK. (2017). Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem. Commun. 53, 5481–5494. [DOI] [PubMed] [Google Scholar]
- 27.Cheung MS., Marder TB., and Lin Z. (2011). Mechanisms of reactions of a lithium boryl with organohalides. Organometallics 30, 3018–3028. [Google Scholar]
- 28.Kaaz M., Bender J., Förster D., Frey W., Nieger M., and Gudat D. (2014). Phosphines with N-heterocyclic boranyl substituents. Dalton Trans. 43, 680–689. [DOI] [PubMed] [Google Scholar]
- 29.Geier SJ., Gilbert TM., and Stephan DW. (2008). Activation of H2 by phosphinoboranes R2PB(C6F5)2. J. Am. Chem. Soc. 130, 12632–12633. [DOI] [PubMed] [Google Scholar]
- 30.Leitao EM., Jurca T., and Manners I. (2013). Catalysis in service of main group chemistry offers a versatile approach to p-block molecules and materials. Nature Chem. 5, 817–829. [DOI] [PubMed] [Google Scholar]
- 31.Geeson MB., and Cummins CC. (2018). Phosphoric acid as a precursor to chemicals traditionally synthesized from white phosphorus. Science 359, 1383–1385. [DOI] [PubMed] [Google Scholar]
- 32.Aldridge S., and Jones C. (2016). Modern main group chemistry. Chem. Soc. Rev. 45, 763–764. [DOI] [PubMed] [Google Scholar]
- 33.Mo Z., Rit A., Campos J., Kolychev EL., and Aldridge S. (2016). Catalytic B−N dehydrogenati on using frustrated Lewis pairs: evidence for a chain-growth coupling mechanism. J. Am. Chem. Soc. 138, 3306–3309. [DOI] [PubMed] [Google Scholar]
- 34.Loh Y-K., Ying L., Fuentes MÀ., Do DCH., and Aldridge S. (2019). An N-heterocyclic boryloxy ligand isoelectronic with N-heterocyclic imines: Access to an acyclic dioxysilylene and its heavier congeners. Angew. Chem. Int. Ed. 58, 4847–4851. [DOI] [PubMed] [Google Scholar]
- 35.Power PP. (2010). Main-group elements as transition metals. Nature 463, 171–177. [DOI] [PubMed] [Google Scholar]
- 36.Power PP., and Chiu C-W. (2017). Preface for the advances in main-group inorganic chemistry forum. Inorg. Chem. 56, 8597–8598. [Google Scholar]
- 37.Lin Y-C., Hatzakis E., McCarthy SM., Reichl KD., Lai T-Y., Yennawar HP., and Radosevich AT. (2017). P−N cooperative borane activation and catal ytic hydroboration by a distorted phosphorous triamide Platform. J. Am. Chem. Soc. 139, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunn NL., Ha M., and Radosevich AT. (2012). Main group redox catalysis: reversible PIII/PV redox cycling at a phosphorus platform. J. Am. Chem. Soc. 134, 11330–11333. [DOI] [PubMed] [Google Scholar]
- 39.Katsuma Y., Wu L., Lin Z., Akiyama S., and Yamashita M. (2019). Reactivity of highly Lewis-acidic diborane(4) toward C≡N and N=N bonds: uncatalyzed addition and N=N bond-cleavage reactions. M. Angew. Chem. Int. Ed. 58, 317–321. [DOI] [PubMed] [Google Scholar]
- 40.Tsukahara N., Asakawa H., Lee K-H., Lin Z., and Yamashita M. (2017). Cleaving dihydrogen with tetra(o-tolyl) diborane(4). J. Am. Chem. Soc. 139, 2593–2596. [DOI] [PubMed] [Google Scholar]
- 41.Fisher SP., Tomich AW., Lovera SO., Kleinsasser JF., Guo J., Asay MJ., Nelson HM., and Lavallo V. (2019). Nonclassical applications of closo-Carborane anions: from main group chemistry and catalysis to energy storage. Chem. Rev. February 1, 2019 DOI: 10.1021/acs.chemrev.8b00551 [DOI] [PubMed]
- 42.Engesser TA., Lichtenthaler MR., Schleep M., and Krossing I. (2016). Reactive p-block cations stabilized by weakly coordinating anions. Chem. Soc. Rev. 45, 789–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawson JR., and Melen RL. (2017). Tris(pentafluorophenyl)borane and beyond: modern advances in borylation chemistry. Inorg. Chem. 56, 8627–8643. [DOI] [PubMed] [Google Scholar]
- 44.Houghton AY., Hurmalainen J., Mansikkamäki A., Piers WE., and Tuononen HM. (2014). Direct observation of a borane–silane complex involved in frustrated Lewis-pair-mediated hydrosilylations. Nat. Chem. 6, 983–988. [DOI] [PubMed] [Google Scholar]
- 45.Geri JB., and Szymczak NK. (2017). Recyclable trifluoromethylation reagents from fluoroform. J. Am. Chem. Soc. 139, 9811–9814. [DOI] [PubMed] [Google Scholar]
- 46.Pan B., and Gabbaï FP. (2014). [Sb(C6F5)4][B(C6F5)4]: An air stable, Lewis acidic stibonium salt that activates strong element-fluorine bonds. J. Am. Chem. Soc. 136, 9564–9567. [DOI] [PubMed] [Google Scholar]
- 47.Lee CF., Diaz DB., Holownia A., Kaldas SJ., Liew SK., Garrett GE., Dudding T., and Yudin AK. (2018). Amine hemilability enables boron to mechanistically resemble either hydride or proton. Nat. Chem. 10, 1062–1070. [DOI] [PubMed] [Google Scholar]
- 48.Qian EA., Wixtrom AI., Axtell JC., Saebi A., Jung D., Rehak P., Han Y., Moully EH., Mosallaei D., Chow S., Messina MS., Wang J., Royappa AT., Rheingold AL., Maynard HD., Král P., and Spokoyny AM. (2017). Atomically precise organomimetic cluster nanomolecules assembled via perfluoroaryl-thiol SNAr chemistry. Nat. Chem. 9, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S7. Bond lengths [Å] and angles [°] for 9
Table S10. Bond lengths [Å] and angles [°] for 13
Table S13. Bond lengths [Å] and angles [°] for 17
Table S16. Bond lengths [Å] and angles [°] for 42