

Abstract
T-helper 17 (Th17) cells differentiate from naïve CD4+ T cells in response to signals from commensal microbiota and produce cytokines critical for the integrity of mucosal barriers. These cells also disseminate throughout the body, and are key participants in numerous inflammatory processes. A key challenge is to elucidate the mechanisms that govern Th17 cell beneficial versus pathogenic functions, characterized by different cytokine profiles. All Th17 cells require the nuclear hormone receptor RORγt for their differentiation in lymph nodes draining mucosal tissues. Cytokine expression is enabled in select tissues, to which these cells migrate, by external cues, such as the serum amyloid A proteins produced in response to commensal bacteria by epithelial cells In the small intestine. Additional cell intrinsic cues contributing to production of Th17 cytokines during both homeostasis and inflammation include the RORγt-associated DEAD-box RNA helicase DDX5 and lncRNA Rmrp. The helicase activity of DDX5 is required for Rmrp-mediated assembly of the complex and co-localization with RORγt throughout the genome to regulate key Th17 genes. How these are regulated in diverse microenvironments may provide insights for therapeutic intervention in autoimmune disease.
The vertebrate adaptive immune response has evolved to provide protection from a large variety of both pathogenic and non-pathogenic or commensal microbes. An important aspect of its evolution is the diversification of T lymphocytes such that different subsets perform distinct functions that are suited to the microbe and the desired outcome. T-helper-17 cells (Th17) cells exemplify this principle, as they differentiate at mucosal surfaces to perform either homeostatic functions, largely aimed at reinforcing barriers in response to commensal bacteria with defined properties, or protective functions, that limit the growth of subsets of invasive bacteria or fungi. The properties of Th17 cells that render them effective can also cause tissue damage, and hence these cells also participate in a large variety of inflammatory diseases. Th17 cells produce the cytokines IL-17A, IL-17F, and IL-22 (Korn et al. 2009) that regulate central aspects of host immunity, including granulopoiesis, neutrophil recruitment, and the induction of antimicrobial peptides by epithelial cells (Weaver et al. 2007). Under some circumstances, however, after exposure to IL-23, Th17 cells can also contribute to chronic inflammation that results in a number of important human diseases. Such responses often involve IL-23-dependent production of the cytokines IFNγ and GM-CSF (CSF2) and result in auto-inflammatory disease pathologies in animal models of inflammatory bowel disease (e.g. Crohn’s disease), rheumatoid arthritis (RA), psoriasis, and multiple sclerosis (MS) (Furuzawa-Carballeda et al. 2007). Antibody-mediated blockade of IL-23 or IL-17A is highly effective in the treatment of patients with psoriasis and psoriatic arthritis, and blockade of IL-23 has also been reported effective in ankylosing spondylitis and Crohn’s disease, but, paradoxically, IL-17A blockade exacerbated inflammatory bowel disease (Whibley and Gaffen 2015). This paradox may be explained by the dual nature of Th17 cells, which are most abundant in the gastrointestinal (GI) tract, where mutualism between host and microorganisms is critically dependent on a state of balanced immune activation (Honda and Littman 2012).
Th17 cells are dependent on commensal microbiota for their differentiation. In germ-free mice, there are essentially no IL-17-producing CD4+ T cells in the intestine, but colonization with either a diverse commensal flora or with only segmented filamentous bacteria (SFB) results in induction of microbial antigen-specific Th17 cells (Gaboriau-Routhiau et al. 2009; Ivanov et al. 2009; Yang et al. 2014b). Colonization of animals with SFB is linked to exacerbation of Th17-mediated disease in murine autoimmune models of both RA and MS (Wu et al. 2010; Lee et al. 2011). How SFB-induced Th17 cells contribute to systemic autoimmune disease is a major unanswered question.
We recently established a model of acute SFB colonization to investigate the mechanism of Th17 cell induction in a spatiotemporal context (Sano et al. 2015). We found that antigen-specific CD4+ T cells are first induced to express RORγt, a nuclear hormone receptor described as a “master regulator” for the Th17 program, in the draining mesenteric lymph nodes after SFB colonization. While poised to be Th17 cells, these differentiated effector cells migrate to many sites throughout the gut and the rest of the body, but express their signature cytokines, including IL-17A, following migration to the lamina propria in the terminal ileum of the small intestine, the site adjacent to epithelial cells to which SFB attaches. This observation suggested that a trigger(s) restricted to the terminal ileum activates a signaling cascade sensed by the poised Th17 cells to turn on transcription of their effector programs.
Here, we summarize our recent efforts to characterize cell-extrinsic (“triggers”) and cell-intrinsic (“sensors”) mechanisms that modulate Th17 cell effector functions in vivo. We first discuss the role of epithelial serum amyloid A proteins (SAA1 and SAA2) as candidate cell-extrinsic molecules that promote the transcriptional activation of multiple loci which endow Th17 cells with effector functions in the context of SFB colonization in the gut. We then summarize recent studies that revealed critical roles for a RORγt-associated RNA helicase and its long noncoding RNA substrate in the cell-intrinsic sensing of signals that culminate in differentiation of potentially pathogenic Th17 cells.
An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses
SFB colonization of the small intestine results in global transcriptional changes in the host epithelia, including the induction of antimicrobial peptides and stress response genes, such as serum amyloid A (SAA1 and SAA2) (Ivanov et al. 2009). SAAs are typically induced in response to infection and acute injury and can promote inflammation, in part through elicitation of proinflammatory cytokine production and recruitment of granulocytes, monocytes, and T lymphocytes (Uhlar and Whitehead 1999). Interestingly, the expression of epithelial SAAs was abolished in IL-23R-deficient animals, as well as mice treated with anti-IL-22 antibodies. In parallel, RORγt+ Th17 cells patrolling the ileal lamina propria from mice with IL-22 blockade or deficient for SAA1/2 were unaltered in total numbers, but expressed significantly lower levels of IL-17A (Sano et al. 2015). We concluded from these studies that the functional differentiation of intestinal Th17 cells following colonization of mice with SFB occurs in a two-step process: the first step is the priming and polarization of antigen-specific CD4+ T cells in the tissue-draining lymph nodes, resulting in a poised state marked by the expression of RORγt, and the second is the activation of a cytokine gene expression program in a tissue microenvironment in which epithelial cell-derived factors act on poised cells (Figure 1). Absence of the second signal, which in the case of SFB-induced Th17 cells is, at least in part, SAA1/2, does not appear to affect the polarization or dissemination of the RORγt+ Th17 cells induced in the mesenteric lymph nodes (Sano et al. 2015).
Figure 1. Serum amyloid A promotes Th17 effector responses.
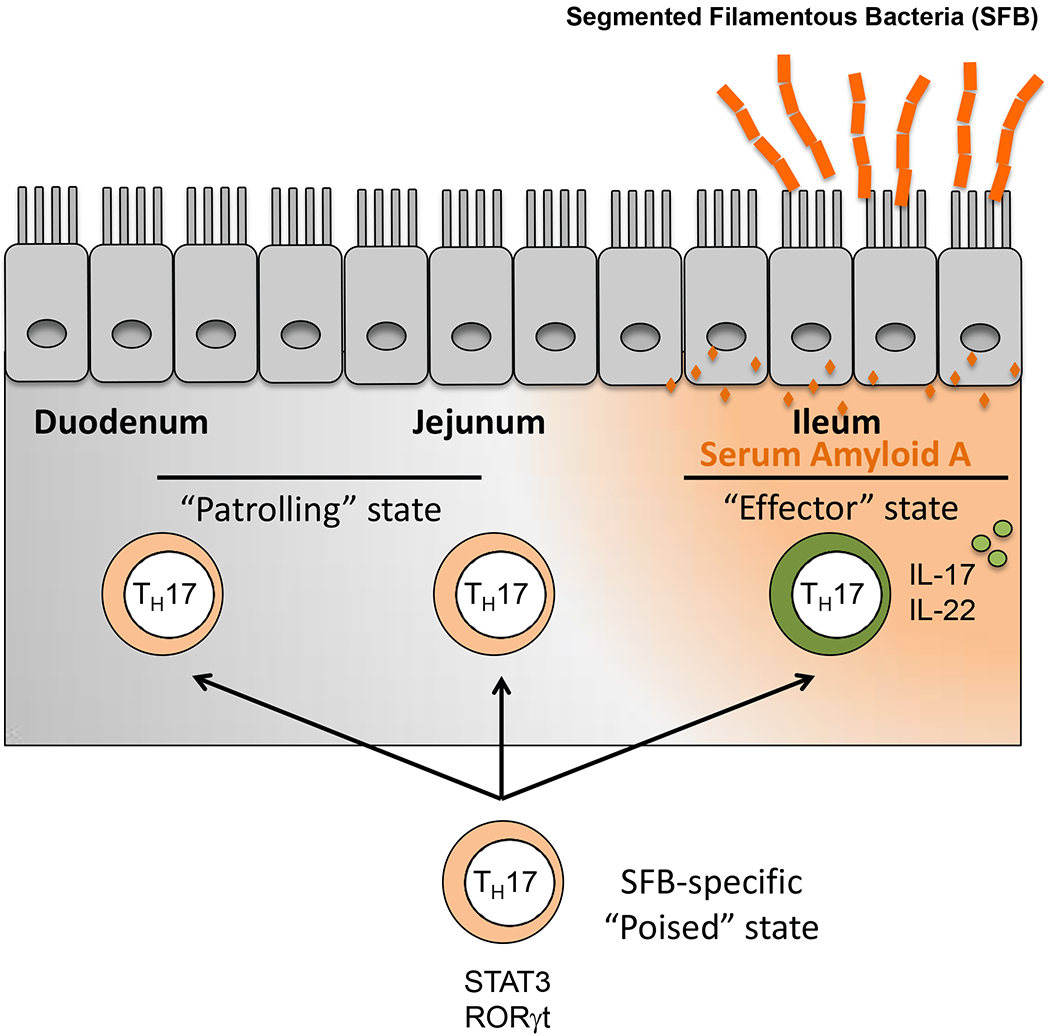
SFB colonizes the ileum in mice. Poised SFB-specific Th17 cells migrate widely, including to the mucosal lamina propria. SAAs, produced by epithelial cells in response to SFB, promote poised Th17 cells to become cytokine-producing effector cells.
SAAs are significantly up-regulated in the joints and serum of rheumatoid arthritis patients (O’Hara et al. 2000) and their levels are correlated with disease progression (Chambers et al. 1983). In mouse models, Th17 cells are also known to contribute to multiple autoimmune diseases (Leppkes et al. 2009; Genovese et al. 2010; Huh et al. 2011), and whether SAA or similar tissue-derived co-stimulatory molecules mediate these processes remains to be elucidated. As microbiota-specific helper T cells, including SFB-specific Th17 cells, circulate widely following their polarization in the gut-associated lymphoid tissues (Hand et al. 2012; Yang et al. 2014b), it is possible that they contribute to autoimmune disease systemically through induction of their effector functions at sites where inflammatory mediators such as SAAs are elevated. An attractive notion is that SAAs may lower the threshold for activation of Th17 cells in inflammatory microenvironments, such that otherwise harmless self-peptide/MHC-reactive T cells would produce the effector cytokines and thus exacerbate tissue damage. It will be of substantial interest to evaluate whether the multi-step process observed with SFB-induced IL-17A-producing Th17 cells in the ileum can be replicated at other sites where there is aberrant SAA production, and whether the resulting cytokine secretion would then contribute to autoimmune pathology. Whether T helper and regulatory lineages that participate in other types of anti-microbial defenses and immune tolerance undergo similar stages of priming/licensing and functional activation is another important question.
Using sorted mouse or human CD4+ T cells cultured under suboptimal Th17 polarizing conditions with limited TGF-β availability, recombinant SAAs substantially enhanced production of IL-17A and IL-17F in RORγt+ Th17 cells (Sano et al. 2015; June-Yong Lee, unpublished). Thus, SAA1/2 appear to act directly on poised T cells, through yet to be identified receptor(s). A specific subset of the Th17 gene expression program was modulated by rmSAA1 treatment, as assessed by RNAseq, and Ingenuity Pathway Analysis predicted RORγt as the top upstream regulatory factor sensitive to SAA stimulation. The DNA-binding domains of RORs recognize ROR response DNA elements (RORE) within promoters and other regulatory regions of their target genes (Jetten et al. 2001). ROR-element driven luciferase assays in Th17 cells confirmed that RORγt transcriptional activity was potentiated during rmSAA1 stimulation in culture. It should be noted that recent work from Kenya Honda’s group (Atarashi et al., 2015), as well as our previous work in collaboration with Honda (Ivanov et al. 2009), failed to show direct stimulation of T cells with recombinant SAA and, instead, found that IL-17 induction required activation of an intermediary myeloid cell. We believe that the discrepancy can be attributed to different preparations of recombinant mouse SAA1, as we can now obtain stimulation of T cells with high confidence.
RORγt and RORγ, the products of the Rorc locus, belong to the nuclear receptor (NR) superfamily. While RORγ is expressed broadly, particularly in liver, muscle, and adipose tissue, RORγt expression is restricted to lymphoid cells. Among lymphocytes, RORγt has multiple other functions in addition to guiding differentiation of Th17 cells and other IL-17-producing T cells, such as γδ17 cells. These include mediating survival of thymocytes and regulating the development of lymphoid tissue inducer (LTi) cells that guide the formation of secondary and tertiary lymphoid tissues, and of type 3 innate lymphoid cells (ILC3) that protect epithelial barriers, largely through production of IL-22 (Sun et al. 2000) (Spits and Di Santo 2011). How RORγ and RORγt function in different cell types to direct distinct transcriptional and functional programs is not understood. As with other members of the family, the ligand-binding domain (LBD) at the C-terminus of RORγ/γt makes these ideal cell intrinsic “sensors” for environmental cues. Ligand binding to RORγ/γt results in recruitment of co-activators, but, as shown with other NRs, there are likely a variety of cofactors and post-translational modifications (PTMs) that modulate transcriptional activity in different cells or tissues and under diverse conditions (reviewed in (Huang and Glass 2010)). We had found that cholesterol biosynthetic intermediates are required for endogenous production of RORγ/γt agonistic ligands (Santori et al. 2015), and it is possible that different ligands can also contribute to tissue-selective activities of RORγ/γt.
There are multiple pharmacologic company programs targeting RORγt for treating diseases such as psoriasis, rheumatoid arthritis, and inflammatory bowel diseases, all of which are thought to be mediated in large part by Th17 cells (Chang et al. 2014). We and others have searched for small molecule antagonists of RORγ/γt transcriptional activity, which allowed for identification of digoxin family glycosides and several other classes of molecules as highly specific RORγ/γt ligand binding domain-interacting antagonists or inverse agonists (Huang et al. 2010; Huh et al. 2011; Huh et al. 2013). While RORγt is indeed an attractive therapeutic target for multiple autoimmune diseases (Huh et al. 2011; Yang et al. 2014a), particularly in light of effective antibody targeting of this pathway, interfering with the shared ligand-binding pocket of RORγ may result in undesired side effects in peripheral organs and tissues. Next, we will summarize our most recent efforts at identifying and examining Th17 cell-intrinsic factors that contribute to RORγt functions in a tissue-specific manner, providing a path towards targeting not only Th17 cells, but also other cell types dependent on RNA helicase-dependent programs.
Regulation of RORγt functions in Th17 cell differentiation by an RNA helicase paired with a long non-coding RNA
To identify cell type-specific partners for RORγt, we applied a combination of proteomics and next-generation RNA sequencing to in vitro polarized mouse Th17 cells. We first identified the DEAD-box RNA helicase DDX5 in a screen for RORγt-associated proteins (Huang et al. 2015). DDX5 functions in multiple cellular processes (Huang and Liu 2002), including transcription and ribosome biogenesis (Lin et al. 2005; Caretti et al. 2006; Jalal et al. 2007; Clark et al. 2008; Fuller-Pace and Moore 2011; Linder and Jankowsky 2011; Arun et al. 2012) in both a helicase activity-dependent and -independent manner. In mice deficient for DDX5 in T cells, expression of multiple RORγt target genes was attenuated and the animals were resistant to disease in models of Th17 cell-mediated autoimmunity, including colitis and multiple sclerosis. Remarkably, conditional inactivation of DDX5 in TCRαβ T cells rendered RORγt+ cells defective for induction of not only IL-17A, but also IFNγ, cytokines associated with auto-inflammatory functions of Th17 cells, in the T cell transfer colitis disease model. Change in IFNγ expression was not observed in classical Th1 cells, which express T-bet but not RORγt. At steady state, the number of RORγt+ T cells in the intestinal lamina propria was similar in mice with T cells sufficient or deficient for DDX5, but there was substantial reduction in the proportion of these cells producing IL-17A and IL-17F in the small intestine. DDX5 hence appears to have a key role in regulating both homeostatic expression of IL-17 in mice colonized with appropriate microbiota and production of additional cytokines by the same cells under inflammatory conditions. The inflammatory functions of Th17 cells are promoted by IL-23, and it is possible that signaling by way of the IL-23R may influence additional functions of RORγt-associated DDX5. Intriguingly, in human studies, a functional distinction has been made between two types of circulating CD4+ T cells that are both dependent on RORγt and express either IFNγ and the chemokine receptors CXCR3 and CCR6 or IL-17 and only CCR6. The former have been designated Th1* cells and are the predominant respondents to mycobacterial antigen, while the latter are Th17 cells (Becattini et al. 2015). Remarkably, patients with homozygous null mutations in RORC (which encodes both isoforms of the nuclear receptor) have defects in both Th17 and Th1* cells and are susceptible to both mucocutaneous candidiasis (controlled by Th17 cells) and dissemination of Bacillus Calmette-Guérin (BCG) after its use as a vaccine (Okada et al. 2015). The Th1* cells may be the human equivalent of the pathogenic IFNγ-producing Th17 cells in mice, and it will hence be of considerable interest to determine the role of DDX5 in their differentiation.
In our structure-function analysis of DDX5, we found that it can enhance IL-17A expression only if its RNA helicase function is intact (Huang et al. 2015). We therefore searched for RNAs associated with DDX5 in primary Th17 cell lysates. By depleting ribosomes and polysome-associated translated RNAs, we enriched for non-coding RNAs in DDX5 and RORγt immunoprecipites, allowing us to identify an associated long noncoding (lnc) RNA, Rmrp, by RNA-seq analysis. Rmrp, RNA component of the mitochondrial RNA-processing endoribonuclease (RNase MRP), is highly conserved between mouse and human and is essential for early murine development (Rosenbluh et al. 2011). Mutations in human RMRP result in the autosomal recessive disease Cartilage Hair Hypoplasia (CHH), characterized by skeletal dysplasia, immune system defects, predisposition to lymphoma, and neuronal dysplasia of the intestine (Makitie et al. 1998; Bonafe et al. 2005). Mutations in humans are never null, but either bi-allelic point mutations or compound heterozygous mutations retaining one transcribed mutant allele, suggesting that, as in mouse, the gene is essential for early development, although several hypomorphic forms of the RNA can be tolerated. We found that, in Th17 cells, Rmrp localizes exclusively to the nucleus, promotes RORγt-DDX5 assembly both in vitro and in vivo, and is recruited to RORγt-occupied genomic loci of critical genes implicated specifically in the Th17 effector program (Huang et al. 2015). The formation of the complex requires ATP-dependent helicase activity and likely involves a conformational change in the lncRNA. Expression of wild-type, but not a CHH mutant, Rmrp promoted Th17 cell differentiation in a DDX5-dependent manner. T cells from mice carrying a single nucleotide change (270 G>T) in Rmrp, corresponding to one found in CHH patients (262 G>T), had a compromised Th17 cell effector program and lost association of DDX5 with RORγt. Rmrp thus acts with DDX5 and RORγt+ to confer target locus-specific activity, enabling the T cell effector programs, including those involved in autoimmune disease (Figure 2. We speculate that defective T cell-dependent immunity in CHH patients may reflect, at least in part, reduced Rmrp-dependent activity at RORγt target genes. It will be of interest to learn whether a subset of CHH patients have RMRP mutations resulting in defective Th1* differentiation with associated candidiasis and susceptibility to BCG vaccination.
Figure 2. RNA helicase DDX5 and its associated lncRNA Rmrp.

DDX5 is required for proper chromatin localization of lncRNA Rmrp in Th17 cells genomewide, and the two together regulate the RORγt-dependent homeostatic and pathogenic Th17 program in vivo. Parallel mechanism of MLE-mediated lncRNA roX remodeling and assembly of the MSL2-DSS complex in X chromosome dosage compensation.
Our results have several implications and raise a number of interesting questions. First, they demonstrate that a lncRNA acts in trans across the genome, regulating RORγt-dependent genes upon interacting with enzymatically active DDX5 helicase. The concept of RNA helicase/lncRNA function in trans-regulation of transcription was first elegantly demonstrated with the lncRNAs roX1 and roX2, substrates of the RNA helicase MLE, that enhance Drosophila male X-chromosome gene expression to achieve dosage compensation (Ilik et al. 2013) (Maenner et al. 2013) (Figure 2). A second example is the role of the DEAD-box helicase DDX21, which acts on the non-coding RNA 7SK to regulate ribosomal gene expression (Calo et al. 2015). While the roX RNAs act broadly on one chromosome, by way of the MSL dosage compensation complex, and 7SK acts at ribosomal gene loci, releasing p-TEFb to allow for transcriptional elongation, Rmrp acts on genes distributed throughout the genome, and thus participates in conferring a distinct differentiation program to T cells (Figure 2). Together, these findings suggest that the role of DDX5 in regulating Th17 cell differentiation may be but one example of how pairing of diverse DEAD-box RNA helicases with distinct lncRNAs allows for cell-specific targeting of DNA or chromatin-binding transcriptional regulators to direct differentiation pathways. Indeed, DDX5 was shown to also associate with MyoD, a key regulatory transcription factor that directs muscle cell differentiation, and co-expression of these factors along with a DDX5-binding putative lncRNA, SRA, resulted in synergistic induction of muscle-specific genes in the myogenic cell line C2C12 (Caretti et al. 2006) (Figure 3). Whether these interactions occur in vivo, and whether SRA is required for DDX5 recruitment to MyoD-occupied chromatin during skeletal muscle development awaits further investigation.
Figure 3. DEAD-box RNA helicases with distinct lncRNAs.

Functional pairing of RNA helicases and their substrates may allow for cell-specific targeting of DNA or chromatin-binding transcriptional regulators to direct differentiation and physiological pathways. TF: transcription factor.
We do not know whether Rmrp and DDX5 act on genes other than RORγt targets to regulate the Th17 cell program, but it appears that they each influence the expression of a set of genes independently of RORγt. This raises the possibility that DDX5 and Rmrp pair with other partners, perhaps targeting other transcriptional complexes (Figure 3). How the association of DDX5 and Rmrp is regulated remains a very interesting puzzle, since both molecules are widely expressed at relatively high levels. For example, DDX5 and Rmrp are abundant in developing thymocytes and in peripheral naïve and differentiated T helper subsets. However, loss of DDX5 in immature thymocytes or replacement of Rmrp with a mutant sequence that impaired Th17 cell differentiation had no apparent effect on thymocyte development, which requires RORγt for cell survival. Development of lymph nodes also appeared normal in mice with mutant Rmrp or with a lymphoid cell-specific inactivation of DDX5, suggesting that lymphoid tissue inducer cells and type 3 innate lymphoid cells, whose development is wholly dependent on RORγt, are intact in such mice. Intriguingly, in contrast to Th17 cell lysates, there was very little Rmrp co-precipitated with DDX5 from thymocyte lysates. If, indeed, DDX5-Rmrp contribute to RORγt-dependent functions only in Th17 cells, there would need to be tissue- or cell type-specific mechanisms that regulate assembly with the transcriptional complexes. There may be Th17 cell-specific chaperones that guide interaction of DDX5 with Rmrp or there may be competition from other lncRNAs or helicases that would govern specific pairing in each cell type. Further characterization of DDX5-associated RNAs in different tissues, including thymus and liver (where RORγ/γt is expressed) and developing muscle (where MyoD is expressed), may provide more insight into this problem.
Implications for targeting Th17 cells in autoimmune disease
The contribution of DDX5-Rmrp to expression of RORγt-dependent genes in T cells suggests that it provides a cell intrinsic function in response to environmental cues. Indeed, SFB-colonized mice had similar numbers of ileal lamina propria RORγt+ Th17 cells regardless of whether the cells expressed DDX5 or wild-type Rmrp. However, deficiencies in these co-factors resulted in reduced proportions of these cells producing IL-17A. This leads us to speculate that epithelium-derived SAAs in the lamina propria act on RORγt+ cells to enable DDX5-Rmrp assembly and/or interaction with RORγt at relevant sites throughout the genome. A critical question is how the homeostatic and pathogenic functions of Th17 cells are regulated by external cues and by cell intrinsic components, such as the RORγt/DDX5/Rmrp complex. Each molecule in the complex is required for both homeostatic function in response to SFB colonization and pathogenicity in autoimmune disease models. Additional signaling pathways may provide either/or digital signals to direct Th17 effector programs, or there may be analog-like differential tuning through RORγt to achieve diverse outcomes. It has been suggested that lipid biosynthetic pathways differ in pathogenic and non-pathogenic Th17 cells and that RORγt binds to different genomic targets under divergent conditions (Wang et al. 2015), but it is not yet known whether the DDX5-Rmrp axis has a role in this process.
A better understanding of how RORγt contributes to Th17 cell programs in different settings is critical for designing effective therapies for the numerous autoinflammatory conditions mediated by these cells. Ideally, it will be possible to target transcriptional pathways that attenuate pathogenic Th17 cells or Th1* cells, without affecting the homeostatic process by which Th17 cells maintain barrier integrity. The discovery of the role of DDX5 and Rmrp in Th17 cell differentiation is a first step on the way to better discriminate RORγt functions in Th17 cells versus other cell types in which this nuclear receptor also performs critical tasks. Elucidation of the mechanisms by which DDX5 alters the conformation of Rmrp to permit subsequent binding to RORγt may reveal features that could be vulnerable to therapeutic intervention. High-throughput screens designed to identify compounds that disrupt the complex may be valuable for developing therapies that selectively inhibit Th17-mediated inflammation, sparing thymocyte and ILC3 development.
Conclusions
The discovery of the roles of DDX5 and Rmrp in T cell differentiation offers multiple opportunities to enhance our understanding of how Th17 cells are regulated and of whether there is a broader role for RNA helicases and their non-coding RNA substrates in directing developmental/differentiation programs. Future investigation of the structural basis of the DDX5-Rmpr-RORγt interactions, of whether DDX5 performs lncRNA-dependent transcriptional functions in other cell types, and of whether related DEAD-box helicases have analogous functions in other cell lineages will likely yield important insights that will guide the development of novel cell type-specific therapies. These studies may also provide a better understanding of the developmental defects observed in CHH patients with different RMRP allelic variants and may reveal new components of the pathway that contribute to either immune deficiency or to autoimmune disease susceptibility.
Acknowledgments
We thank members of our laboratory and our many collaborators for their contributions to the studies described in this overview. The work was supported by NIH grant R01AI080885 (D.R.L.), NIH training grant T32 CA009161 (W.H.), and the Howard Hughes Medical Institute (D.R.L.).
Reference
- Arun G, Akhade VS, Donakonda S, Rao MR. 2012. mrhl RNA, a long noncoding RNA, negatively regulates Wnt signaling through its protein partner Ddx5/p68 in mouse spermatogonial cells. Molecular and cellular biology 32: 3140–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, Fernandez B, Kelderman S, Schumacher TN, Corti D et al. 2015. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 347: 400–406. [DOI] [PubMed] [Google Scholar]
- Bonafe L, Dermitzakis ET, Unger S, Greenberg CR, Campos-Xavier BA, Zankl A, Ucla C, Antonarakis SE, Superti-Furga A, Reymond A. 2005. Evolutionary comparison provides evidence for pathogenicity of RMRP mutations. PLoS genetics 1: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, Wysocka J. 2015. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature 518: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, Fuller-Pace FV, Hoffman EP, Tapscott SJ, Sartorelli V. 2006. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Developmental cell 11: 547–560. [DOI] [PubMed] [Google Scholar]
- Chambers RE, MacFarlane DG, Whicher JT, Dieppe PA. 1983. Serum amyloid-A protein concentration in rheumatoid arthritis and its role in monitoring disease activity. Annals of the rheumatic diseases 42: 665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MR, Rosen H, Griffin PR. 2014. RORs in autoimmune disease. Current topics in microbiology and immunology 378: 171–182. [DOI] [PubMed] [Google Scholar]
- Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ et al. 2008. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res 68: 7938–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller-Pace FV, Moore HC. 2011. RNA helicases p68 and p72: multifunctional proteins with important implications for cancer development. Future oncology 7: 239–251. [DOI] [PubMed] [Google Scholar]
- Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. 2007. Autoimmune inflammation from the Th17 perspective. Autoimmunity reviews 6: 169–175. [DOI] [PubMed] [Google Scholar]
- Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G et al. 2009. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31: 677–689. [DOI] [PubMed] [Google Scholar]
- Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, Sloan-Lancaster J. 2010. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis and rheumatism 62: 929–939. [DOI] [PubMed] [Google Scholar]
- Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO 3rd, Belkaid Y. 2012. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 337: 1553–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Littman DR. 2012. The microbiome in infectious disease and inflammation. Annual review of immunology 30: 759–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Glass CK. 2010. Nuclear receptors and inflammation control: molecular mechanisms and pathophysiological relevance. Arterioscler Thromb Vasc Biol 30: 1542–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Thomas B, Flynn RA, Gavzy SJ, Wu L, Kim SV, Hall JA, Miraldi ER, Ng CP, Rigo FW et al. 2015. DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature 528: 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Huang W, Wang H, Johnson RL, Huang R, Englund EE, Huh J, Littman DR. 2010. Identification of Potent and Selective RORgamma Antagonists in Probe Reports from the NIH Molecular Libraries Program, Bethesda (MD). [Google Scholar]
- Huang Y, Liu ZR. 2002. The ATPase, RNA unwinding, and RNA binding activities of recombinant p68 RNA helicase. The Journal of biological chemistry 277: 12810–12815. [DOI] [PubMed] [Google Scholar]
- Huh JR, Englund EE, Wang H, Huang R, Huang P, Rastinejad F, Inglese J, Austin CP, Johnson RL, Huang W et al. 2013. Identification of Potent and Selective Diphenylpropanamide RORgamma Inhibitors. ACS medicinal chemistry letters 4: 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV et al. 2011. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature 472: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R et al. 2013. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Molecular cell 51: 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV et al. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal C, Uhlmann-Schiffler H, Stahl H. 2007. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic acids research 35: 3590–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten AM, Kurebayashi S, Ueda E. 2001. The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Progress in nucleic acid research and molecular biology 69: 205–247. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and Th17 Cells. Annual review of immunology 27: 485–517. [DOI] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. 2011. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America 108 Suppl 1: 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD et al. 2009. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136: 257–267. [DOI] [PubMed] [Google Scholar]
- Lin C, Yang L, Yang JJ, Huang Y, Liu ZR. 2005. ATPase/helicase activities of p68 RNA helicase are required for pre-mRNA splicing but not for assembly of the spliceosome. Molecular and cellular biology 25: 7484–7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder P, Jankowsky E. 2011. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12: 505–516. [DOI] [PubMed] [Google Scholar]
- Maenner S, Muller M, Frohlich J, Langer D, Becker PB. 2013. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Molecular cell 51: 174–184. [DOI] [PubMed] [Google Scholar]
- Makitie O, Kaitila I, Savilahti E. 1998. Susceptibility to infections and in vitro immune functions in cartilage-hair hypoplasia. European journal of pediatrics 157: 816–820. [DOI] [PubMed] [Google Scholar]
- O’Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. 2000. Acute-phase serum amyloid A production by rheumatoid arthritis synovial tissue. Arthritis research 2: 142–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS et al. 2015. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349: 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Nijhawan D, Chen Z, Wong KK, Masutomi K, Hahn WC. 2011. RMRP is a non-coding RNA essential for early murine development. PloS one 6: e26270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee JY, Ziel JW, Miraldi ER, Domingos AI et al. 2015. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163: 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santori FR, Huang P, van de Pavert SA, Douglass EF Jr., Leaver DJ, Haubrich BA, Keber R, Lorbek G, Konijn T, Rosales BN et al. 2015. Identification of Natural RORgamma Ligands that Regulate the Development of Lymphoid Cells. Cell metabolism 21: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, Di Santo JP. 2011. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature immunology 12: 21–27. [DOI] [PubMed] [Google Scholar]
- Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR. 2000. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288: 2369–2373. [DOI] [PubMed] [Google Scholar]
- Uhlar CM, Whitehead AS. 1999. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265: 501–523. [DOI] [PubMed] [Google Scholar]
- Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M et al. 2015. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 163: 1413–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. 2007. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annual review of immunology 25: 821–852. [DOI] [PubMed] [Google Scholar]
- Whibley N, Gaffen SL. 2015. Gut-Busters: IL-17 Ain’t Afraid of No IL-23. Immunity 43: 620–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. 2010. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32: 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sundrud MS, Skepner J, Yamagata T. 2014a. Targeting Th17 cells in autoimmune diseases. Trends in pharmacological sciences 35: 493–500. [DOI] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X et al. 2014b. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510: 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]