

Abstract
The recent pandemic of coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 has raised global health concerns. The viral 3-chymotrypsin-like cysteine protease (3CLpro) enzyme controls coronavirus replication and is essential for its life cycle. 3CLpro is a proven drug discovery target in the case of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). Recent studies revealed that the genome sequence of SARS-CoV-2 is very similar to that of SARS-CoV. Therefore, herein, we analysed the 3CLpro sequence, constructed its 3D homology model, and screened it against a medicinal plant library containing 32,297 potential anti-viral phytochemicals/traditional Chinese medicinal compounds. Our analyses revealed that the top nine hits might serve as potential anti- SARS-CoV-2 lead molecules for further optimisation and drug development process to combat COVID-19.
Keywords: Coronavirus, SARS-CoV-2, COVID-19, Natural products, Protein homology modelling, Molecular docking, Molecular dynamics simulation
Graphical abstract

Highlights
-
•
SARS-CoV-2 3CLpro is conserved, share 99.02% sequence identity with SARS-CoV 3CLpro and together with 12 point-mutations.
-
•
Mutations disrupt important hydrogen bonds and alter the receptor binding site of SARS-CoV-2 3CLpro.
-
•
Medicinal plants phytochemicals were proved potential anti-COVID-19 druggable candidates.
1. Introduction
A novel coronavirus strain linked with fatal respiratory illness was reported in late 2019 [1]. Swift actions were taken by the Centre for Disease Control and Prevention (CDC), Chinese health authorities, and researchers. The World Health Organization (WHO) temporarily named this pathogen 2019 novel coronavirus (2019-nCoV) [2]. On January 10, 2020, the first whole-genome sequence of 2019-nCoV was released, which helped researchers to quickly identify the virus in patients using reverse-transcription polymerase chain reaction (RT-PCR) methods [3]. On January 21, the first article related to 2019-nCoV was published, which revealed that 2019-nCoV belongs to the beta-coronavirus group, sharing ancestry with bat coronavirus HKU9-1, similar to SARS-coronaviruses, and that despite sequence diversity its spike protein interacts strongly with the human ACE2 receptor [1]. On January 30, the WHO announced a Public Health Emergency of International Concern (PHEIC) for the 2019-nCoV outbreak. Later, the human-to-human transmission was confirmed. As of January 31, 51 whole-genome sequences of 2019-nCoV from different laboratories and regions had been submitted to GISAID database [4]. On February 12, the WHO permanently named the 2019-nCoV pathogen as SARS-CoV-2 and the causing disease as coronavirus disease 2019 (COVID-2019). The Chinese government’s swift actions helped them to control COVID-19 in China. However, SARS-CoV-2 affected several countries world-wide. On March 11, the WHO formally recognized the COVID-19 as a pandemic. By March 19, 2020, the global death toll reached 9,913, with 2,42,650 laboratory-confirmed cases. The case fatality rate among infected people varies in different countries. However, global case fatality rate is presently around 3.92% (calculated as deaths/[deaths + laboratory confirmed cases]).
Coronaviruses are single-stranded positive-sense RNA viruses that possess large viral RNA genomes [5]. Recent studies showed that SARS-CoV-2 has a similar genomic organization to that of other beta-coronaviruses, consisting of a 5′-untranslated region (UTR), a replicase complex (orf1ab) encoding non-structural proteins (nsps), a spike protein (S) gene, envelope protein (E) gene, a membrane protein (M) gene, a nucleocapsid protein (N) gene, 3′-UTR, and several unidentified non-structural open reading frames [3]. Although SARS-CoV-2 is classified into the beta-coronaviruses group, it is different from MERS-CoV and SARS-CoV. Recent studies highlighted that SARS-CoV-2 genes share <80% nucleotide identity and 89.10% nucleotide similarity with SARS-CoV genes [6,7]. Usually, beta-coronaviruses produce a ∼800 kDa polypeptide upon transcription of the genome. This polypeptide is proteolytically cleaved to generate various proteins. The proteolytic processing is mediated by papain-like protease (PLpro) and 3-chymotrypsin-like protease (3CLpro). The 3CLpro cleaves the polyprotein at 11 distinct sites to generate various non-structural proteins that are important for viral replication [8]. 3CLpro plays a critical role in the replication of virus particles and unlike structural/accessory protein-encoding genes, it is located at the 3′ end which exhibits excessive variability. Therefore, it is a potential target for anti-coronaviruses inhibitors screening [9]. Structure-based activity analyses and high-throughput studies have identified potential inhibitors for SARS-CoV and MERS-CoV 3CLpro [[10], [11], [12]]. Medicinal plants, especially those employed in traditional Chinese medicine, have attracted significant attention because they include bioactive compounds that could be used to develop formal drugs against several diseases with no or minimal side effects [13]. Therefore, the present study was conducted to gain structural insights into the SARS-CoV-2 3CLpro and to discover potent anti-COVID-19 natural compounds.
2. Materials and methods
2.1. Data collection
Whole-genome sequences of all SARS-CoV-2 isolates available till January 31, 2020, were downloaded from GISAID database (accession numbers and details are given in Table S1) [4]. The genome sequence of BetaCoV/Kanagawa/1/2020 (GISAID: EPI_ISL_402126) was incomplete, and the genome sequence of BetaCoV/bat/Yunnan/RaTG13/2013 (EPI_ISL_402131) was an old sequence (2013); therefore, these sequences were not included in our analyses. Gene sequences of 3CLpro were extracted from the whole-genome sequences and translated into protein sequences using the translate tool of the ExPASy server [14]. The first SARS-CoV-2 sequence (Wuhan-Hu-1; GSAID: EPI_ISL_402125) was used as a reference in our analysis.
2.2. Sequence analyses
In order to identify similar sequences and key/conserved residues, and to infer phylogeny, multiple sequence alignment of SARS-CoV-2 3CLpro followed by phylogenetic tree analyses were performed using T-Coffee [15] and the alignment figure was generated using ESPript3 [16]. Physicochemical parameters of SARS-CoV-2 3CLpro including isoelectric point, instability index, grand average of hydropathicity (GRAVY), and amino acid and atomic composition were investigated using the ProtParam tool of ExPASy [14].
2.3. Structural analyses
To probe the molecular architecture of SARS-CoV-2 3CLpro, comparative homology modelling was performed using Modeller v9.11 [17]. To select closely-related templates for modelling, PSI-BLAST was performed against all known structures in the protein databank (PDB) [18]. Chimera v1.8.1 [19] and PyMOL educational version [20] were used for initial quality estimation, energy minimisation, mutation analyses, and image processing.
2.4. Ligand database preparation and molecular docking
A comprehensive medicinal plant library containing 32,297 potential anti-viral phytochemicals and traditional Chinese medicinal compounds was generated from our previously collected data and studies [13,[21], [22], [23]], and screened against the predicted SARS-CoV-2 3CLpro structure. Molecular operating environment (MOE) [24] was used for molecular docking, ligand-protein interaction and drug likeness analyses. All analyses were performed using the same protocols that are already described in our previous studies [13,25,26]. The qualitative assessment of absorption, deposition, metabolism, excretion and toxicity (ADMET) profile of selected hits were predicted computationally by using ADMETsar server [27].
2.5. Molecular dynamics simulations
Explicit solvent molecular dynamics (MD) simulations were performed to verify docking results and to analyse the binding behaviour and stability of potential compounds using the predicted SARS-CoV-2 3CLpro homology model. GROMACS v5.1.4, GROMOS96 and the PRODRG server were employed to run 50 ns MD simulations [28,29] following the same protocol as described in our previous studies [13,30].
3. Results and discussion
3.1. Sequence and structural analyses
Multiple sequence alignment results revealed that 3CLpro was conserved, with 100% identity among all SARS-CoV-2 genomes. Next, the SARS-CoV-2 3CLpro protein sequence was compared with its closest homologs (Bat-CoV, SARS-CoV, MERS-CoV, Human-CoV and Bovine-CoV). The results revealed that SARS-CoV-2 3CLpro clusters with bat SARS-like coronaviruses and shares 99.02% sequence identity (Fig. 1A). Furthermore, it shares 96.08%, 87.00%, 90.00% and 90.00% sequence identity with SARS-CoV, MERS-CoV, Human-CoV and Bovine-CoV homologs, respectively (Fig. 1B). These findings were consistent with those of initial studies reporting that SARS-CoV-2 is more similar to SARS-CoV than to MERS-CoV, and shares a common ancestor with bat coronaviruses [1,3,31]. Analysis of physicochemical parameters revealed that the SARS-CoV-2 3CLpro polypeptide is 306 amino acids long with a molecular weight of 33,796.64 Da and a GRAVY score of −0.019, categorising the protein as a stable, hydrophilic molecule capable of establishing hydrogen bonds (Table 1).
Fig. 1.

(A) Phylogenetic tree inferred from closest homologs of SARS-CoV-2 3CLpro. The maximum likelihood method was used to construct this tree. (B) Multiple sequence alignment of closest homologs of SARS-CoV-2 3CLpro sharing ≥70% sequence identity. (C) Cartoon representation of the SARS-CoV-2 3CLpro homodimer. Chain-A (protomer-A) is in multicolour and Chain-B (protomer-B) is in dark blue. The N-finger that plays an important role in dimerization maintaining the active conformation is shown in hot pink, domain I is coloured cyan, domain II is shown in green, and domain III is coloured yellow. The N- and C-termini are labelled. Residues of the catalytic dyad (Cys-145 and His-41) are highlighted in yellow and labelled. (D) Cartoon representation of the 3CLpro monomer model (chain/protomer-A) of SARS-CoV-2 superimposed with the SARS-CoV 3CLpro structure. The SARS-CoV 3CLpro template is coloured cyan, the SARS-CoV-2 3CLpro structure is coloured grey, and all identified mutations are highlighted in red. (E) Docking of 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone inside the receptor-binding site of SARS-CoV-2 3CLpro, showing hydrogen bonds with the catalytic dyad (Cys-145 and His-41). The 3CLpro structure is coloured dark blue, the 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone is orange, and hydrogen coloured maroon.
Table 1.
Physicochemical parameters of SARS-CoV-2 3CLpro.
| Parameters | SARS-CoV-2 3CLpro |
|---|---|
| Mol. Weight | 33796.64 Dalton |
| No. of amino acids | 306 |
| Theoretical pI | 5.95 |
| Instability index (II) | 27.65 (stable) |
| No. of negatively charged residues (Asp + Glu) | 26 |
| No. of positively charged residues (Arg + Lys) | 22 |
| Aliphatic index | 82.12 |
| Grand average of hydropathicity (GRAVY) | −0.019 |
| Atomic composition | Carbon-1499; Hydrogen-2318; Nitrogen-402; Oxygen-445; Sulfur-22 |
| Amino acid composition | Ala-17 (5.6%); Arg-11 (3.6%); Asn-21 (6.9%); Asp-17 (5.6%); Cys-12 (3.9%); Gln-14 (4.6%); Glu-9 (2.9%); Gly-26 (8.5%); His-7 (2.3%); Ile-11 (3.6%); Leu-29 (9.5%); Lys-11 (3.6%); Met-10 (3.3%); Phe-17 (5.6%); Pro-13 (4.2%); Ser-16 (5.2%); Thr-24 (7.8%); Trp-3 (1.0%); Tyr-11 (3.6%); Val-27 (8.8%); Pyl-0 (0.0%); Sec-0 (0.0%) |
Next, for comparative modelling, BLAST [32] search identified SARS-CoV 3CLpro (PDB ID: 3M3V) as the best possible match in the PDB, with 100% query coverage, an E-value of 0.00, and 96.08% sequence identity. There were 12-point mutations (Val35Thr, Ser46Ala, Asn65Ser, Val86Leu, Lys88Arg, Ala94Ser, Phe134His, Asn180Lys, Val202Leu, Ser267Ala, Ser284Ala and Leu286Ala) between SARS-CoV and SARS-CoV-2 3CLpro enzymes (Fig. S1). Except for replacement of Leu with Ala at position 286, all other replacements conserve polarity and hydrophobicity. However, these mutations may affect 3CLpro structure and function. Therefore, the 3D structure of SARS-CoV-2 3CLpro was predicted. Firstly, a single chain monomeric model comprising all domains (Domain I = residues 8–100; Domain II = residues 101–183; Domain III = residues 200–303) was built (Fig. S2). N-terminal amino acids 1 to 7 form the N-finger that plays a significant role in dimerization and formation of the active site of 3CLpro. Domains I and II, collectively referred to as the N-terminal domain, include an antiparallel β-sheet structure with 13 β-strands. The binding site for the substrate is situated in a cleft between domains I and II. A loop from residues 184 to 199 joins the N-terminal domain and domain III, which is also called the C-terminal domain and comprises an anti-parallel cluster of five α-helices. The overall molecular architecture of SARS-CoV-2 3CLpro was in consistent with the crystal structure of SARS-CoV (PDB ID: 3M3V); the root mean square deviation (RMSD) between the homology model and the template was 0.629 Å. Structural and Ramachandran plot analyses revealed that 99% of residues are in favourable regions.
After quality assessment, individual chains were combined to form a homodimeric 3D structure, as shown in Fig. 1C. To facilitate other researchers, the predicted 3D model has been submitted to the Protein Model Database (PMDB) [33], and anyone can download/use the SARS-CoV-2 3CLpro final structure using PMDB ID: PM0082635. Furthermore, mutational analyses depicted none of the mutations affected the overall structure of SARS-CoV-2, which fully superimposed on the SARS-CoV 3CLpro structure (Fig. 1D). The results also revealed that SARS-CoV-2 has a Cys-His catalytic dyad (Cys-145 and His-41), consistent with that of SARS 3CLpro (Cys-145 and His-41), TGEV 3CLpro (Cys-144 and His-41) and HCoV 3CLpro (Cys-144 and His-41) [34]. These results revealed that the SARS-CoV-2 3CLpro receptor-binding pocket conformation resembles that of the SARS-CoV 3CLpro binding pocket and raises the possibility that inhibitors intended for SARS-CoV 3CLpro may also inhibit the activity of SARS-CoV-2 3CLpro.
3.2. Molecular docking
To test this hypothesis, we docked (R)-N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)furan-2-carboxamide), a potential noncovalent inhibitor of SARS-CoV 3CLpro named ML188 [35], with the SARS-CoV-2 3CLpro homology model. We also docked ML188 with the SARS-CoV 3CLpro structure (PDB ID: 3M3V) as a reference, and ML188 bound strongly to the receptor binding site of SARS-CoV 3CLpro. The inhibitor targets the Cys-His catalytic dyad (Cys-145 and His-41) along with the other residues, and the docking score (S = −12.27) was relatively high. However, surprisingly, ML188 did not show significant binding to the catalytic dyad (Cys-145 and His-41) of SARS-CoV-2, and the docking score (S = −8.31) was considerably lower (Fig. S3). These results indicated that the 12-point mutations identified at previous step may disrupt important hydrogen bonds and alter the receptor binding site, thereby affecting its ability to bind with the SARS-CoV inhibitors.
Therefore, it is essential to discover novel compounds that may inhibit SARS-CoV-2 3CLpro and serve as potential anti-COVID-19 drug compounds. We developed a library from our previously published studies that contains numerous natural compounds possessing potential anti-viral activities and screened it against the SARS-CoV-2 3CLpro homology model. Recent drug repurposing studies proposed few drugs that target SARS-CoV-2 3CLpro and suggested that they could be used to treat COVID-19. Herein, we selected the best of these (Nelfinavir, Prulifloxacin and Colistin) from three different drug repurposing studies [36,37] and docked them as controls in the present study (Fig. S4). Our analyses identified nine novel non-toxic, druggable natural compounds that are predicted to bind with the receptor binding site and catalytic dyad (Cys-145 and His-41) of SARS-CoV-2 3CLpro (Table 2; Fig. S5). ADMET profiling of the selected hits is given in Table S2. Among these screened phytochemicals, 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone is an isoflavone extracted from Psorothamnus arborescens [38] that exhibited the highest binding affinity (−29.57 kcal/mol) and docking score (S = −16.35), and formed strong hydrogen bonds with the catalytic dyad residues (Cys-145 and His-41) as well as significant interactions with the receptor-binding residues Thr24, Thr25, Thr26, Cys44, Thr45, Ser46, Met49, Asn142, Gly143, His164, Glu166 and Gln189 (Fig. 1E). A literature review revealed that 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone has been successfully used as an anti-leishmanial agent [38], and it is also found in traditional Chinese medicine records [39]. Our screened phytochemicals displayed higher docking scores, stronger binding energies, and closer interactions with the conserved catalytic dyad residues (Cys-145 and His-41) than Nelfinavir, Prulifloxacin and Colistin. These results suggested that natural products identified in our study may prove more useful candidates for COVID-19 drug therapy.
Table 2.
Summary of top ranked phytochemicals screened against SARS-CoV-2 3CLpro receptor binding site with their respective structures, docking score, binding affinity and interacting residues.
| IDs | Phytochemical name | Plant source | Phytochemical structure | Docking score | Binding affinity (kcal/mol) | 3CLpro residues interacting with phytochemical through H-bonding and other interactions |
|---|---|---|---|---|---|---|
| PubChem 11610052 | 5,7,3′,4′-Tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone | Psorothamnus arborescens | 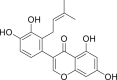 |
−16.35 | −29.57 | His41, Cys145, Thr24, Thr25, Thr26, Cys44, Thr45, Ser46, Met49, Asn142, Gly143, His164, Glu166, Gln189 |
| PubChem 5281673 | Myricitrin | Myrica cerifera | 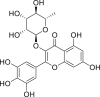 |
−15.64 | −22.13 | Gly143, Cys145, His41, Thr24, Thr25, Thr26, Leu27, Cys44, Ser46, Met49, Leu141, Asn142, Ser144, His163, Glu166, Gln189 |
| PubChem 6479915 | Methyl rosmarinate | Hyptis atrorubens Poit | 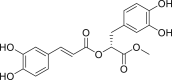 |
−15.44 | −20.62 | Cys145, His41, Thr24, Thr25, Thr26, Cys44, Thr45, Met49, Leu141, Asn142, Gly143, Ser144, His163, His164, Glu166, Gln189 |
| NPACT00105 | 3,5,7,3′,4′,5′-hexahydroxy flavanone-3-O-beta-D-glucopyranoside | Phaseolus vulgaris | 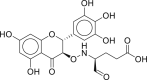 |
−14.42 | −19.10 | Met49, Cys145, His41, Thr24, Thr25, Thr26, Cys44, Ser46, Asn142, His164, Met165, Glu166, Gln189 |
| PubChem 10930068 | (2S)-Eriodictyol 7-O-(6″-O-galloyl)-beta-D-glucopyranoside | Phyllanthus emblica | 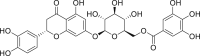 |
−14.41 | −19.47 | Thr24, Thr25, Gly143, Met49, Cys145, His41, Thr26, Cys44, Thr45, Glu166, Leu167, Gln189, Thr190, Ala191, Gln192 |
| PubChem 5273567 | Calceolarioside B | Fraxinus sieboldiana | 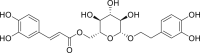 |
−14.36 | −19.87 | His41, Gly143, Cys145, Glu166, Thr24, Thr25, Thr26, Leu27, Ser46, Leu50, Leu141, Asn142, Ser144, His164, Met165, Gln189 |
| PubChem 5318606 | Myricetin 3-O-beta-D-glucopyranoside | Camellia sinensis | 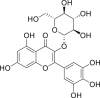 |
−13.70 | −18.42 | Asn142, Glu166, Cys145, His41, Thr24, Thr25, Thr26, Thr45, Ser46, Met49, Leu141, Gly143, Ser144, His163, His164, Met165, Gln189 |
| PubChem 11111496 | Licoleafol | Glycyrrhiza uralensis | 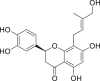 |
−13.63 | −19.64 | Cys145, His41, Thr24, Thr25, Thr26, Cys44, Thr45, Met49, Gly143, His163, Met165, Glu166, Gln189 |
| PubChem 6123095 | Amaranthin | Amaranthus tricolor | 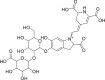 |
−12.67 | −18.14 | Thr26, Glu166, Cys145, His41, Thr24, Thr25, Cys44, Thr45, Asn142, His163, His164, Met165, Glu166, Leu167, Pro168, Gln189 |
| PubChem 64143 | Nelfinavir | Drugs used as control | 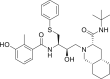 |
−12.20 | −17.31 | Met49, Met165, Glu166, Leu167, Pro168, Gly 170, Gln189, Thr190, Ala191 |
| PubChem 65947 | Prulifloxacin | 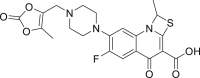 |
−11.32 | −15.40 | Gln192, Leu50, Met165, Glu166, Leu167, Pro168, Arg 188, Gln189, Thr190, Ala191 | |
| PubChem 5311054 | Colistin | 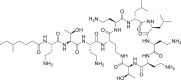 |
−13.73 | −18.57 | Met49, Thr24, Thr25, Thr26, Thr45, Ser46, Glua47, Leu50, Asn142, Gly143, Met165, Glu166, Leu167, Pro168, Gln189, Thr190, Ala191, Gln192 |
∗3CLpro catalytic dyad (Cys-145 and His-41) residues are highlighted with bold font.
3.3. MD simulations
To further investigate the molecular docking results, the top three phytochemical complexes, namely 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone, myricitrin, and methyl rosmarinate, were subjected to 50 ns MD simulation. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (RoG) and hydrogen bond parameters were calculated. RMSD is an indicator of the stability of ligand-protein complexes. None of the complexes showed any obvious fluctuations, and all three were stable, with average RMSD values of 1.6 ± 0.02 Å, 1.5 ± 0.02 Å and 1.7 ± 0.02 Å for 5,7,3′,4′-tetrahydroxy-2’-(3,3-dimethylallyl) isoflavone, myricitrin, and methyl rosmarinate, respectively (Fig. 2A). RMSF is an indicator of residual flexibility. Minimal fluctuations were observed for myricitrin and methyl rosmarinate, and the overall complexes remained stable throughout the simulations. The functionally important catalytic dyad residues (Cys-145 and His-41) displayed stable behaviour, and fluctuations were observed toward the C-terminal end of the SARS-CoV-2 3CLpro molecule (Fig. 2B). RoG is an indicator of protein compactness, stability, and folding, and the results suggested normal behaviour for all three complexes; all remained compact and stable throughout the 50 ns simulations (Fig. 2C). In addition, hydrogen bonds, which are the main stabilising interactions factors in proteins, suggested that the SARS-CoV-2 3CLpro internal hydrogen bonds remain stable throughout the simulation, with no obvious fluctuations (Fig. 2D). These results confirmed our findings and further indicated that these compounds may serve as potential anti-COVID-19 drug sources.
Fig. 2.

(A) Root mean square deviation (RMSD), (B) root mean square fluctuation (RMSF), (C) potential energy and (D) Hydrogen Bond interactions for all three complexes over the 50 ns simulation.
4. Conclusion
In conclusion, our study revealed that 3CLpro is conserved in SARS-CoV-2. It is highly similar to bat SARS-like coronavirus 3CLpro, with some differences from other beta-coronaviruses. We predicted the 3D structure of the SARS-CoV-2 3CLpro enzyme, and the findings may help researchers working on COVID-19 drug discovery. Despite significant overall similarity with the SARS-CoV 3CLpro structure, the SARS-CoV-2 3CLpro substrate binding site had some key differences, which highlighted the need for rapid drug discovery to address the alarming COVID-19 pandemic. Medicinal plant compounds have already been used to successfully treat numerous viral diseases. Herein, we screened a medicinal plant database containing 32,297 potential anti-viral phytochemicals and selected the top nine hits that may inhibit SARS-CoV-2 3CLpro activity and hence virus replication. Further in-vitro and in-vivo analyses are required to transform these potential inhibitors into clinical drugs. We anticipate that the insights gained in the present study may prove valuable for exploring and developing novel natural anti-COVID-19 therapeutic agents in the future.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2020YFC0845600), the Hubei Provincial Natural Science Foundation of China (2019CFA014), the Starting Research Grant for High-level Talents from Guangxi University, Nanning, China and Postdoctoral Research Platform Grant of Guangxi University, Nanning, China. We also acknowledge all the authors and laboratories mentioned in Table S1 for their sampling, analysis, and genome sequencing efforts. In addition, we acknowledge GISAID (https://www.gisaid.org/) for facilitating open data sharing.
Footnotes
Peer review under responsibility of Xi'an Jiaotong University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.03.009.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Xu X., Chen P., Wang J. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji W., Wang W., Zhao X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 2020;92:433–440. doi: 10.1002/jmv.25682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shu Y., McCauley J.J.E. GISAID: Global initiative on sharing all influenza data–from vision to reality. Euro Surveill. 2017;22 doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y., Liu Q., Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Z.-L., Zhou P., Yang X.-L. Discovery of a Novel Coronavirus Associated with the Recent Pneumonia Outbreak in Humans and its Potential Bat Origin. BioRxiv. 2020 doi: 10.1101/2020.01.22.914952. [DOI] [Google Scholar]
- 7.Wu F., Zhao S., Yu B. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand K., Ziebuhr J., Wadhwani P. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Needle D., Lountos G.T., Waugh D.S. Structures of the middle east respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2015;71:1102–1111. doi: 10.1107/S1399004715003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh A.K., Xi K., Ratia K. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 11.Kumar V., Tan K.P., Wang Y.M. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016;24:3035–3042. doi: 10.1016/j.bmc.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pillaiyar T., Manickam M., Namasivayam V. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tahir ul Qamar M., Maryam A., Muneer I. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019;9:1–16. doi: 10.1038/s41598-018-38450-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasteiger E., Gattiker A., Hoogland C. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Notredame C., Higgins D.G., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 16.Gouet P., Courcelle E., Stuart D.I. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 17.Eswar N., Webb B., Marti-Renom M.A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006;15 doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson M., Zaretskaya I., Raytselis Y. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pettersen E.F., Goddard T.D., Huang C.C. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 20.DeLano W. Pymol: An open-source molecular graphics tool. CCP4 Newsletter on protein crystallography. 2002;40:82–92. [Google Scholar]
- 21.Mumtaz A., Ashfaq U.A., Tahir Ul Qamar M. MPD3: a useful medicinal plants database for drug designing. Nat. Prod. Res. 2017;31:1228–1236. doi: 10.1080/14786419.2016.1233409. [DOI] [PubMed] [Google Scholar]
- 22.Ashfaq U.A., Mumtaz A., Tahir Ul Qamar M. MAPS Database: medicinal plant activities, phytochemical and structural database. Bioinformation. 2013;9:993–995. doi: 10.6026/97320630009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mumtaz A., Ashfaq U.A., Tahir ul Qamar M. Addendum, Nat. Prod. Res. 2020 110.1080/14786419.2020.1735129. [Google Scholar]
- 24.Vilar S., Cozza G., Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 25.Tahir Ul Qamar M., Saleem S., Ashfaq U.A. Epitope-based peptide vaccine design and target site depiction against middle east respiratory syndrome coronavirus: an immune-informatics study. J. Transl. Med. 2019;17:362. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tahir Ul Qamar M., Bari A., Adeel M.M. Peptide vaccine against chikungunya virus: immuno-informatics combined with molecular docking approach. J. Transl. Med. 2018;16:298. doi: 10.1186/s12967-018-1672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng F., Li W., Zhou Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 28.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 29.van Aalten D.M., Bywater R., Findlay J.B. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996;10:255–262. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- 30.Muneer I., Tahir Ul Qamar M., Tusleem K. Discovery of selective inhibitors for cyclic AMP response element-binding protein: a combined ligand and structure-based resources pipeline. Anti Canc. Drugs. 2019;30:363–373. doi: 10.1097/CAD.0000000000000727. [DOI] [PubMed] [Google Scholar]
- 31.Zhou P., Yang X.L., Wang X.G. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye J., McGinnis S., Madden T.L. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34 doi: 10.1093/nar/gkl164. W6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castrignanò T., De Meo P.D., Cozzetto D. The PMDB protein model database. Nucleic Acids Res. 2006;34 doi: 10.1093/nar/gkj105. D306-D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H., Yang M., Ding Y. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobs J., Zhou S., Dawson E. National Center for Biotechnology Information (US); Bethesda (MD): 2010. Discovery of Non-covalent Inhibitors of the SARS Main Proteinase 3CLpro, Probe Reports from the NIH Molecular Libraries Program. PMID: 23658941. [PubMed] [Google Scholar]
- 36.Xu Z., Peng C., Shi Y. Nelfinavir Was Predicted to Be a Potential Inhibitor of 2019-nCov Main Protease by an Integrative Approach Combining Homology Modelling, Molecular Docking and Binding Free Energy Calculation. BioRxiv. 2020 doi: 10.1101/2020.01.27.921627. [DOI] [Google Scholar]
- 37.Li Y., Zhang J., Wang N. Therapeutic Drugs Targeting 2019-nCoV Main Protease by High-Throughput Screening. BioRxiv. 2020 doi: 10.1101/2020.01.28.922922. [DOI] [Google Scholar]
- 38.Salem M.M., Werbovetz K.A. Isoflavonoids and other compounds from psorothamnus a rborescens with antiprotozoal activities. J. Nat. Prod. 2006;69:43–49. doi: 10.1021/np0502600. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J., Xie G., Yan X. Vol. 6. Springer; Indexes, New York: 2011. (Encyclopedia of Traditional Chinese Medicines - Molecular Structures, Pharmacological Activities, Natural Sources and Applications). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.