

Abstract
Carbamazepine (CBZ) causes life‐threating T‐cell‐mediated hypersensitivity reactions, including serious cutaneous adverse reactions (SCARs) and drug‐induced liver injury (CBZ‐DILI). In order to evaluate shared or phenotype‐specific genetic predisposing factors for CBZ hypersensitivity reactions, we performed a meta‐analysis of two genomewide association studies (GWAS) on a total of 43 well‐phenotyped Northern and Southern European CBZ‐SCAR cases and 10,701 population controls and a GWAS on 12 CBZ‐DILI cases and 8,438 ethnically matched population controls. HLA‐A*31:01 was identified as the strongest genetic predisposing factor for both CBZ‐SCAR (odds ratio (OR) = 8.0; 95% CI 4.10–15.80; P = 1.2 × 10−9) and CBZ‐DILI (OR = 7.3; 95% CI 2.47–23.67; P = 0.0004) in European populations. The association with HLA‐A*31:01 in patients with SCAR was mainly driven by hypersensitivity syndrome (OR = 12.9; P = 2.1 × 10−9) rather than by Stevens‐Johnson syndrome/toxic epidermal necrolysis cases, which showed an association with HLA‐B*57:01. We also identified a novel risk locus mapping to ALK only for CBZ‐SCAR cases, which needs replication in additional cohorts and functional evaluation.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Carbamazepine (CBZ) is associated with serious, and sometimes fatal, cutaneous and liver adverse reactions. Genomewide profiling has shown that these predisposing factors largely reside in the HLA region (HLA‐B*15:02 and HLA‐A*31:01) consistent with the immune pathogenesis.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What are the genetic predisposing factors in Northern and Southern European populations for CBZ‐induced hypersensitivity reactions affecting both the skin and liver?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Genetic profiling confirmed that HLA‐A*31:01 predisposes to serious cutaneous adverse reaction (SCAR) in both Northern and Southern European populations. HLA‐A*31:01 also seems to predispose to CBZ‐induced liver injury. In addition, an uncommon variant in the ALK gene was associated with an increased risk of CBZ‐SCAR.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Our study adds to the overwhelming evidence of the role of HLA‐A*31:01 in predisposing to a variety of CBZ hypersensitivity phenotypes and highlights the need to implement its pre‐prescription and preemptive use in clinical settings.
Carbamazepine (CBZ) is prescribed for epilepsy, trigeminal neuralgia, and bipolar disorder.1 In 3–10% of patients, CBZ causes a variety of hypersensitivity reactions,2 ranging from mild maculopapular exanthemas to hypersensitivity syndrome, drug reaction with eosinophilia and systemic symptoms (DRESS), acute generalized exanthematous pustulosis (AGEP), Stevens‐Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN); the latter four phenotypes are referred to as serious cutaneous adverse reactions (SCARs). CBZ can also lead to liver injury, which can occur either as part of DRESS or in isolation (the latter is referred to as CBZ‐drug‐induced liver injury (DILI) in this paper).3, 4 These unpredictable clinical phenotypes are T‐cell‐mediated, in which CBZ and/or its metabolites bind specific HLA molecules triggering a T‐cell response.5
CBZ‐induced SJS/TEN is strongly associated with HLA‐B*15:02 in Han Chinese, Thais, and Malays,6 whereas HLA‐A*24:02 has also recently been identified as a risk factor in Han Chinese.7 HLA‐A*31:01 is associated with a variety of CBZ‐SCAR phenotypes and maculopapular exanthema in Japanese,8 Korean,9 and European‐descent populations.10 Several other HLA loci have been suggested as susceptibility loci, including HLA‐A*02:06,8 HLA‐B*15:11,11 and HLA‐B*51:01,12, 13, 14 but the findings have not always been replicated.15 Currently, we have limited knowledge of susceptibility loci for CBZ‐DILI.
The purpose of our study was to perform a meta‐analysis between our previously published CBZ hypersensitivity cohort10 and a newly recruited cohort in order to increase study power and further investigate (i) novel risk loci within and outside the HLA region, and (ii) the role of HLA‐A*31:01 stratifying by European subpopulations and clinical phenotypes, including SCAR and DILI.
Results
Case collection and population structure
Our meta‐analysis included a total of 43 CBZ‐SCAR cases and 10,701 population controls from two genomewide association studies (GWAS) studies: British GWAS (14 cases and 2,263 controls) and broadly European GWAS (29 SCAR cases and 8,438 controls). The British GWAS included Northern European patients from the United Kingdom, which we have reported previously.10 The broadly European GWAS included a newly recruited set of CBZ‐SCAR cases with different clinical phenotypes (Table 1 ) and European ancestries ( Figure S1 ). The inferred population structure of the combined cohorts is shown in Figure 1 . A summary of the clinical characteristics of the SCAR cases is provided in Table 1 . We analyzed 25 patients with DRESS, 16 patients with SJS/TEN, and 2 patients with AGEP. The average age of patients at the time of adverse reaction was 34 years in both study phases. Nearly two‐thirds (63%) of cases were women.
Table 1.
Clinical information of the CBZ‐SCAR and CBZ‐DILI cases included in the studya
| Phenotype | Category | Phase I cases | Phase II cases |
|---|---|---|---|
| SCAR | Total number of cases | 14 | 29 |
| Age at the onset in years (mean) | 34 | 34 | |
| Gender (% female) | 43% | 66% | |
| Allergy (% yes) | 36% | 24% | |
| Clinical subphenotypes | |||
| AGEP | 1 | 1 | |
| Hypersensitivity syndrome (DRESS) | 13 | 12 | |
| SJS | 0 | 5 | |
| SJS/TEN | 0 | 10 | |
| TEN | 0 | 1 | |
| Systemic symptoms | |||
| Eosinophilia | 6 | 6 | |
| Liver involvement | 0 | 10 | |
| Fever | 13 | 22 | |
| Pneumonitis | 2 | 0 | |
| Multi‐organ failure, death | 1 | 0 | |
| Additional evidences for the diagnosis | |||
| Skin biopsy | 1 | 8 | |
| Skin patch testing performed | 1 | 5 | |
| Multiple drug‐induced skin reactions | 3 | 1 | |
| DILI | Total number of cases | 0 | 12 |
| Age at the onset in years (mean) | – | 37.6 | |
| Gender (% female) | – | 67% | |
| Pattern of liver injury | |||
| Cholestatic | – | 3 | |
| Mixed | – | 2 | |
| Hepatocellular | – | 5 | |
| Unknown | – | 2 | |
| Systemic symptoms | |||
| Eosinophilia | – | 0 | |
| Cutaneous rashes | – | 0 | |
| RUCAM scoreb | |||
| 3–5 (possible) | – | 2 | |
| 6–8 (probable) | – | 6 | |
| > 8 (highly probable) | – | 2 | |
AGEP, acute generalized exanthematous pustulosis; CBZ, carbamazepine; DILI, drug‐induced liver injury; DRESS, drug reaction with eosinophilia and systemic symptoms; RUCAM, Roussel Uclaf Causality Assessment Method; SCAR, serious cutaneous adverse reaction; SJS, Stevens‐Johnson syndrome; TEN, toxic epidermal necrolysis.
Each patient can have more than one clinical characteristic; therefore, the numbers do not add up to the number of patients in phases I and II of the study.
RUCAM scores were not possible for two patients recruited previously in the study of McCormack et al.10 because of the lack availability of all clinical information. Causality had been undertaken using temporal relationship to drug intake and exclusion of other causes.
Figure 1.
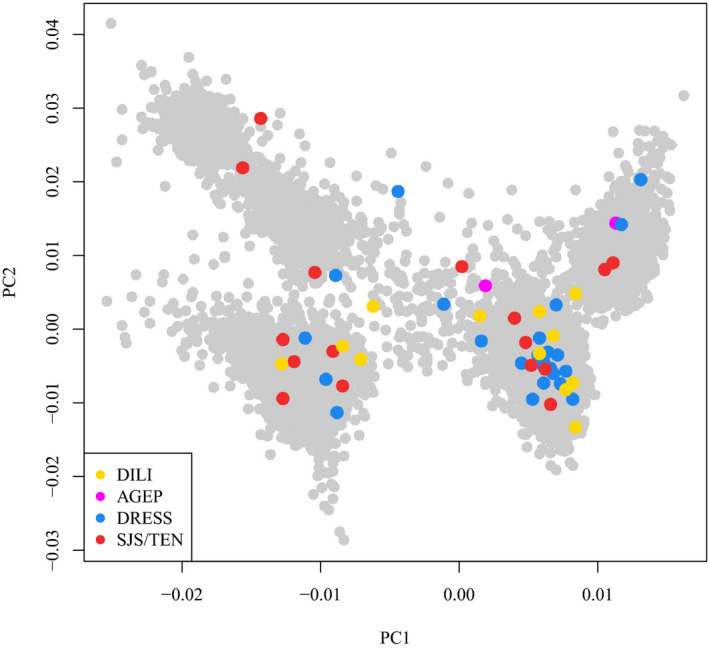
Scatterplot representing the first two principal components (PCs) of the current study cohort. Colored dots are the cases divided by clinical phenotypes and the gray dots are the controls. The controls cluster in four groups representing the Italian, Spanish, United Kingdom, and Swedish major control populations. AGEP, acute generalized exanthematous pustulosis; DILI, drug‐induced liver injury; DRESS, drug reaction with eosinophilia and systemic symptoms; SJS/TEN, Stevens‐Johnson syndrome/toxic epidermal necrolysis.
Separately, we recruited 12 patients with CBZ‐DILI of European descent (Figure 1 ). The type of liver injury in these patients was 50% hepatocellular and 50% cholestatic/mixed. None of them had cutaneous involvement or eosinophilia. Their clinical characteristics are also provided in Table 1 . CBZ‐DILI cases were compared with the 8,438 controls from the broadly European cohort.
Meta‐analysis of CBZ‐SCAR
After quality control of the imputed data, we retained 5,271,349 single nucleotide polymorphisms (SNPs) for association analyses. Our meta‐analysis identified two loci attaining genomewide significance (P < 5 × 10−8):
-
The strongest association signal mapped to the major histocompatibility complex (MHC; lead SNP rs192543598; odds ratio (OR) = 18.1; 95% CI 8.03–40.90; P = 1.7 × 10−12; Pperm < 5 × 10−8; Table 2 , Table S1 , Figure 2 , and Figure S2 ), consistent with our previous result.10
Figure 2.
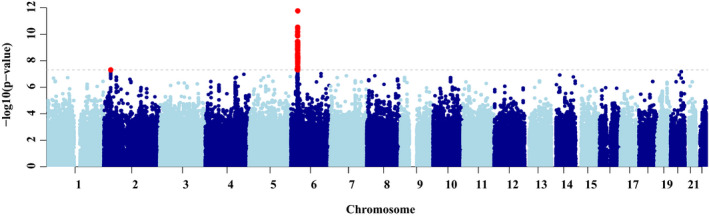 Manhattan plot displaying the results of the meta‐analysis of British and broadly European carbamazepine–serious cutaneous adverse reactions genomewide association study analyses. Single nucleotide polymorphisms in red have a significance level < 5 × 10−8.
Manhattan plot displaying the results of the meta‐analysis of British and broadly European carbamazepine–serious cutaneous adverse reactions genomewide association study analyses. Single nucleotide polymorphisms in red have a significance level < 5 × 10−8. A novel association signal mapping to the ALK gene was observed outside the MHC (lead SNP rs187926838; OR = 12.1; 95% CI 4.94–29.80; P = 4.9 × 10−8; P perm = 1 × 10−7; Table 2 and Figure S3 ). The intronic lead SNP showed a consistent directional effect in both GWAS and was carried by 6% of the cases. The imputation for rs187926838 had high accuracy across the imputation batches (info score > 0.9). The frequency of rs187926838 in our control population was equal to the allele frequency reported in Europeans in gnomad (http://gnomad.broadinstitute.org/) and similar across platforms (Table 1 and Table S1 ), confirming the accuracy of the predicted genotypes. The rs187926838 showed a similar frequency in the SANAD study cohort,2 which has been exposed to a number of anti‐epileptic drugs, including CBZ (allele frequency = 0.005).
Table 2.
Loci attaining genomewide significant evidence of association (P < 5 × 10−8) with CBZ hypersensitivity in a combined meta‐analysis of 43 patients and 10,701 controls of European ancestry
| Locus | Lead SNP | Chr | Position | Alleles | Phase I | Phase II | Combined meta‐analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk | Other | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | Cochran's Q P value | Frequency in controls | Gnomad | ||||
| MHC | rs192543598 | 6 | 29,931,345 | G | A | 127 (28.9–560) | 1.5 × 10−10 | 7.8 (2.95–20.7) | 3.5 × 10−5 | 18.1 (8.03–40.88) | 1.7 × 10−12 | 0.002 | 0.01 | 0.02 |
| ALK | rs187926838 | 2 | 29,818,291 | G | A | 15.3 (2.95–79.7) | 0.001 | 11.0 (3.77–32.1) | 1.1 × 10−5 | 12.1 (4.94–29.80) | 4.9 × 10−8 | 0.74 | 0.007 | 0.008 |
CBZ, carbamazepine; Chr, chromosome; MHC, major histocompatibility complex; OR, odds ratio; SNP, single nucleotide polymorphism.
Dissection of the MHC CBZ‐SCAR association signal
The lead SNP representing the MHC CBZ‐SCAR association signal, rs192543598, was in strong linkage disequilibrium (LD) with HLA‐A*31:01 (r 2 = 0.75). The HLA class I allele also reached genomewide significance in the meta‐analysis (OR = 8.0; 95% CI 4.10–15.80; P = 2.2 × 10−9; Pperm < 5 × 10−8; Table 3 and Table S2 ). The phase II GWAS showed an MHC‐wide HLA‐A*31:01 association, replicating the previously reported signal.
Table 3.
Imputed HLA alleles attaining nominal evidence of association (P < 0.01) with CBZ hypersensitivity in combined meta‐analysis of 43 patients and 10,701 controls of European ancestry
| HLA allele | Phase I | Phase II | Combined meta‐analysis | |||||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | Cochran's Q P value | Frequency in controls | |
| HLA‐A*31:01 | 16.9 (5.40–52.40) | 1.0 × 10−6 | 5.3 (2.29–12.40) | 9.9 × 10−5 | 8.0 (4.10–15.80) | 2.2 × 10−9 | 0.11 | 0.02 |
| HLA‐B*51:01 | 2.8 (0.80–9.60) | 0.10 | 3.7 (1.87–7.28) | 0.0002 | 3.5 (1.91–6.27) | 5.6 × 10−5 | 0.70 | 0.06 |
| HLA‐C*15:02 | 2.1 (0.23–16.5) | 0.47 | 4.1 (1.74–9.92) | 0.001 | 3.7 (1.68–8.35) | 0.002 | 0.56 | 0.02 |
| HLA‐DPB1*09:01 | 4.9 (0.70–35.60) | 0.12 | 4.5 (1.17–17.60) | 0.03 | 4.6 (1.51–14.20) | 0.007 | 0.96 | 0.009 |
CBZ, carbamazepine; OR, odds ratio.
The HLA‐A*31:01 association was less significant in the broadly European cases than in the British cases, probably because of greater heterogeneity in their geographic origin (Figure 1 ). A lower allele frequency was observed in the Spanish cases (6%) compared with other ethnic groups (15% in Northern European and 13% Italian cases) despite an equivalent frequency in the three control groups. However, the effect of HLA‐A*31:01 was conserved in all European populations (ORItalian = 8.7; ORSpanish = 3.66; and ORNorth Europeans = 5.51). There were no differences in allele frequencies of the GWAS variants in control groups despite the different genotyping arrays used.
Reciprocal conditional analyses demonstrated that rs192543598 and HLA‐A*31:01 represent the same underlying association signal ( Table S2 ). After conditioning on the lead SNP, rs192543598, we observed some evidence for residual association (at locus‐wide significance, P < 10−5) with CBZ‐SCAR, mapping to MUC22 (lead SNP rs116071718, OR = 4.0; 95% CI 2.25–7.05; P = 1.5 × 10−6; Table S2 and Figure S4 ). After conditioning on both rs192543598 and rs116071718, the association signal with CBZ‐SCAR in the MHC was fully accounted for ( Figure S4 ).
Marginal associations were also identified with HLA‐B*51:01 and HLA‐C*15:02 (Table 3 ). These HLA alleles along with HLA‐A*31:01 constitute an uncommon haplotype present in European descent individuals (http://www.allelefrequencies.net). Joint carriage of the three alleles had a stronger signal compared with HLA‐A*31:01 alone (OR = 22.1, P = 2.4 × 10−6 vs. OR = 5.7, P = 6.6 × 10−6; Table S3 ).
Assessment of polymorphic amino acid residues in HLA molecules
Predisposition to CBZ‐SCAR may be related to the same essential amino acid (AA) residues in different HLA alleles. Therefore, we performed an aggregated investigation by analyzing the polymorphic AA residues at these proteins. The strongest association was observed for isoleucine73 on the HLA‐A locus (OR = 5.5; 95% CI 2.94–10.40; P = 1.4 × 10−7), but could not better explain the CBZ‐SCAR association than HLA‐A*31:01. The AA is shared by the A*33 and A*31 alleles.16 Conditional analysis to isoleucine73 or to HLA‐A*31:01 revealed that isoleucine80 in the B locus is a new independent factor, and it is shared with HLA‐B*57:01 among other HLA B alleles.16 After conditioning on both the sites, the residual independent association signal was still accounted for by rs116071718 in the MUC22 gene ( Table S4 ). These AA associations have not previously been found to be implicated in genetic predisposition to other adverse drug reactions.
GWAS of CBZ‐SCAR clinical subtypes
We stratified the CBZ‐SCAR cases by clinical phenotypes (AGEP, DRESS, and SJS/TEN; see Methods). The AGEP analysis was not performed because we had only two cases, although it is worth noting that one of the two cases was positive for HLA‐A*31:01.
In the CBZ‐DRESS subgroup, HLA‐A*31:01 and its proxy SNP, rs192543598, reached genomewide significance (P HLA‐A*31:01 = 2.1 × 10−9 and P rs192543598 = 2.4 × 10−13). Consistent with the literature,17 HLA‐A*31:01 showed a stronger effect with CBZ‐DRESS than with SJS/TEN (OR = 12 vs. OR = 2.5, respectively; Table 4 ) with no significant association in the latter group. There was no other phenotype‐specific genomewide significant association for both DRESS and SJS‐TEN ( Figure S5 ).
Table 4.
Association effect size of HLA‐A*31:01 across different clinical phenotypes
| Clinical groups | N | OR (95% CI) | P value | Allele frequency in cases | Allele frequency in controls |
|---|---|---|---|---|---|
| SJS/TEN | 16 | 2.4 (0.55–10.59) | 0.2 | 0.06 | 0.02 |
| DRESS | 25 | 12.9 (5.58–29.78) | 2.1 × 10−9 | 0.14 | 0.02 |
| DILI | 12 | 7.3 (2.47–23.67) | 0.0004 | 0.17 | 0.02 |
| All clinical phenotypes (SCAR and DILI) | 55 | 8.2 (4.56–14.66) | 1.8 × 10−12 | 0.14 | 0.02 |
DILI, drug‐induced liver injury; DRESS, drug reaction with eosinophilia and systemic symptoms; OR, odds ratio; SCAR, serious cutaneous adverse reaction; SJS, Stevens‐Johnson syndrome; TEN, toxic epidermal necrolysis.
The effect of rs187926838 in the ALK gene was conserved among the CBZ‐SCAR clinical subtypes ( Table S5 ). We also found that the SJS/TEN phenotype showed an MHC‐region‐wide significant association with HLA‐B*57:01 (OR = 6.2; 95% CI 2.47–15.37; P = 9.9 × 10−5). HLA‐B*57:01 was in strong LD (r 2 = 0.8) with the most highly associated SNP in the MHC region (rs116347890; OR = 11.0; 95% CI 4.62–26.42; P = 6.6 × 10−8) but was independent of HLA‐A*31:01 (OR = 6.4; 95% CI 2.55–16.11; P = 7.6 × 10−5) after conditioning on HLA‐A*31:01. The polymorphic AA position 97 showed an association when valine was present (OR = 6.0; 95% CI 2.45–15.18; P = 0.0001). Valine97 is specific for HLA‐B*57:01, HLA‐B*57:02, and HLA‐B*57:03 and other very rare B57 alleles.16
GWAS of CBZ‐DILI
GWAS analysis of 12 European CBZ‐DILI cases against 8,438 European controls did not identify a genomewide association because of limited power. However, 33% of cases were HLA‐A*31:01 carriers (OR = 7.3; 95% CI 2.47–23.67; P = 0.0004), although the OR was less than that seen with DRESS (OR = 12.9). Our prediction of the association with HLA‐A*31:01 was fully confirmed by HLA sequencing in 11 cases (DNA was no longer available in one patient). CBZ‐DILI cases did not show an association with rs187926838 (in the ALK gene), distinct from that seen with CBZ‐SCAR cases ( Table S5 ). The most significant AA residues associated with CBZ‐DILI were isoleucine73 (OR = 7.29; 95% CI 2.578–20.6; P = 0.0002) and threonine9 (OR = 5.5; 95% CI 2.31–13.14; P = 0.0001) at the HLA‐A locus. The two residues were in LD (P 73I conditional to 9T = 0.2). Moreover, there was enrichment of HLA‐A*31:01 carriers in the 10 patients with CBZ‐SCAR with liver involvement compared with the remaining cases (25% vs. 12%, respectively).
Taking all the cases of CBZ‐induced hypersensitivity reactions (SCAR and DILI) together, the risk of developing either DILI or SCAR when given CBZ was eightfold higher in cases carrying HLA‐A*31:01 (OR = 8.2; 95% CI 4.56–14.66; P = 1.8 × 10−12; Table 4 ).
Discussion
In the current study, we show that HLA‐A*31:01 is the strongest predictor of CBZ‐SCAR in a European‐ancestry population, extending our previous study in Northern Europeans10 to include Southern Europeans. In addition, we also show that (i) HLA‐A*31:01 seems to predispose to CBZ‐induced liver injury, and (ii) a variant in the ALK gene was associated with an increased risk of CBZ‐SCAR but not CBZ‐DILI.
Our finding of the association between CBZ‐SCAR and HLA‐A*31:01, initially reported in 2011,10 has also been reported by Amstutz et al.18 in a multiethnic North American pediatric cohort and by Genin et al.17 in a European SCAR cohort. In our study, HLA‐A*31:01 was a stronger risk factor for DRESS than SJS/TEN, in line with Genin et al.17 (OR = 57.6 vs. OR = 4.4, respectively) and Amstutz et al.18 (OR = 31.5 vs. OR = 2.8, respectively), but not with our original study (OR = 12 vs. OR = 25, respectively)10 where numbers were much smaller. It is likely that HLA‐A*31:01 is the most important shared risk factor across CBZ hypersensitivity phenotypes, with the strongest predisposition being to DRESS. Indeed, our meta‐analysis shows that HLA‐A*31:01 carriers have eightfold higher risk of developing CBZ‐SCAR than noncarriers. Interestingly, a recent prospective study in Japan was able to show that pre‐prescription genotyping for HLA‐A*31:01 reduces the incidence of CBZ hypersensitivity reactions.19 Taken together, the overwhelming evidence of the role of HLA‐A*31:01 in predisposing to CBZ‐SCAR shows that there is a need to implement its use in clinical settings, as outlined in the recent Clinical Pharmacogenetics Implementation Consortium guideline.20 Independent of the HLA‐A*31:01 association, we identified a residual effect in the MHC region mapping to MUC22 gene (lead SNP rs116071718). MUC22 codes for panbronchiolitis‐related mucin‐like protein 1 and has been associated with SJS/TEN caused by a variety of drugs, including CBZ in European patients.21 Its role in SCAR needs further investigation. We also found that HLA‐B*51:01 was the second most significant HLA allele. HLA‐B*51:01 has already been reported to be marginally associated with CBZ‐SCAR12, 13, 14 and more recently with SCAR due to other drugs.22 HLA‐B*51:01 together with HLA‐A*31:01 and HLA‐C*15:02 constitute a haplotype that has a larger OR than each single allele ( Table S3 ). This is an interesting finding, which suggests that in vivo, the T‐cell response to CBZ‐derived antigens requires cooperativity between different HLA alleles, as we have previously demonstrated in an HLA‐A*31:01‐positive patient.23
We also identified an association between CBZ‐induced SJS/TEN and HLA‐B*57:01. This allele is a well‐known risk factor for other CD8+ T‐cell‐mediated reactions, including abacavir hypersensitivity syndrome24, 25 and flucloxacillin‐induced DILI,26 and more recently, DILI induced by two other drug combinations, pazopanib and a combination of antituberculosis and anti‐HIV drugs.27, 28 Given that the association with CBZ‐induced SJS/TEN was not genomewide significant, it needs replication in other cohorts. Interestingly, our finding of the association of valine97 with CBZ‐SJS/TEN is in line with the association of valine97 with flucloxacillin‐DILI,29 suggesting that the binding site of HLA‐B*57:01 may be promiscuous for a number of drugs, which would be in keeping with the increasing number of reports of immune‐mediated reactions associated with this allele.
We have, we believe, for the first time evaluated whether HLA‐A*31:01 is a risk factor for DILI. Taking all cases of DILI into account, the carriage rate of HLA‐A*31:01 was 33% with an OR of 7, higher than that found in association of HLA class II alleles with lumiracoxib‐induced DILI.30 Interestingly, CBZ‐SCAR and CBZ‐DILI also shared the most significant AA, isoleucine.73 Isoleucine73 is a cryptic epitope specific for A31 and A33 antigens.31 Position 73 is not normally exposed. When the antigen changes its conformation and β2m and peptide dissociate from the heavy chain, isoleucine73 is externalized with the potential to lead to an autoimmune reaction.32 Interestingly, HLA‐A*33:01 and HLA‐A*33:03 have recently been associated with DILI due to several unrelated drugs.33 The association with HLA‐A*31:01, however, was not genomewide significant, which may reflect the small numbers studied. It may also reflect that the mechanism of antigen presentation (be it the parent drug or metabolite) differs between the skin and liver given the major role of the liver in drug metabolism and its ability to form chemically reactive intermediates.34 Further work in larger numbers of patients with CBZ‐DILI, together with mechanistic studies, will be needed to understand the role of drug/metabolites as antigens in the context of different drug metabolizing and antigen presentation capabilities, and, indeed, whether these differences are responsible for the remarkable organ‐specificity of the reactions and their severity seen in different patients.
A tantalizing association that we have identified in our meta‐analysis (that did not pass genomewide significance after permutation) that was observed in the SCAR but not in the DILI cases was with uncommon variants in the ALK gene, which codes for the anaplastic lymphoma kinase gene. Somatic mutations in the ALK gene have been identified in different cancers,35 including lung cancer, which has led to the development of ALK‐inhibitors for therapy. The product of the ALK gene, a receptor tyrosine kinase belonging to the insulin receptor family, seems to be important for the balance between proliferation and apoptosis.36 Furthermore, the associated region falls within a keratinocyte‐specific predicted insulator ( Figure S6 ). Given that ALK is important in cellular proliferation and cell death and shows ubiquitous tissue distribution (http://www.proteinatlas.org/ENSG00000171094-ALK/tissue), it may have an important role in T‐cell proliferation and keratinocyte death, both important in the pathogenesis of SCAR.
In conclusion, we have provided further data regarding genetic factors predisposing to different CBZ adverse reaction phenotypes, which are thought to have an immune pathogenesis, namely SCAR and DILI. We have extended our studies to include Southern Europeans in addition to our previous study in Northern Europeans10 and included an analysis of CBZ‐DILI. We have also identified novel associations with the ALK and MUC22 genes, which require further validation and experimental investigation to determine the mechanisms. It is possible that there are also other genetic factors outside the MHC region, but we may not have had the statistical power to detect them in this study. It is, therefore, important that further work also involves a trans‐ethnic meta‐analysis of the GWAS in diverse populations, which have been undertaken to date, to fully assess differences in the genetic architecture of CBZ adverse reactions between ancestries and, more precisely, localize the underlying causal alleles.
Methods
Study design overview
Our study combines results from two GWAS conducted in subjects with ancestry from Northern Europe, Italy, and Spain. All participants provided written informed consent and each study was approved by the appropriate national or institutional ethical review boards. Because the reactions have a very low prevalence, we used general population samples as study controls.
British CBZ‐SCAR GWAS
The study included 14 patients with CBZ‐hypersensitivity of Northern European descent of the 21 recruited at the University of Liverpool, as described by McCormack et al.10 A total of 2,263 population controls of Northern European descent from the Wellcome Trust Case Control Consortium were utilized. The other Liverpool samples (n = 7) originally described were excluded either because of population stratification issues or because two patients had DILI rather than skin involvement (and so have been included in the liver analysis).
Broadly European CBZ‐SCAR GWAS
The study included a total of 29 CBZ‐SCAR samples obtained from two available sources: 3 cases were from the PGX40001 study37 and 26 were newly recruited between 2009 and 2013 as part of the International Consortium of Drug Hypersensitivity (ITCH) study. The ITCH study was run under the auspices of the International Serious Adverse Event Consortium, involving 12 recruitment centers in Europe, Australia, and South America. Clinical inclusion criteria for all ITCH‐related SCAR cases were as described by our previous phenotype standardization protocol for drug‐induced skin injury.38 All patient phenotypes were independently adjudicated by the co‐authors N.S. and P.F. Further clinical details are provided in Table 1 .
For this group, a total of 8,438 population controls of European descent were selected from the Wellcome Trust Case Control Consortium (not overlapping with samples from the British cohort), the population reference sample (POPRES),39 the PGX40001 study,37 the Spanish Bladder Cohort (dbGaP phs000346.v1),40 the Hypergenes cohort (http://www.hypergenes.eu), the National Spanish DNA Bank cohort (http://www.bancoadn.org/), the Swedish Twin Registry cohort (http://ki.se/en/research/the-swedish-twin-registry), LAM30004 study,37 TSI (Hapmap data), and the iSAEC Italian Penicillin Tolerant Cohort (IPTC).
Broadly European CBZ‐DILI GWAS
As part of the phase II study, we also collected 12 patients of European descent with CBZ‐DILI, in which the liver was involved by itself. The cases were recruited from two large international DILI consortia: iDILIC and DILIN,33 whereas two other cases were as described previously.10 The iDILIC cases were evaluated by application of the Council for International Organizations of Medical Science (CIOMS) scale, also called the Roussel Uclaf Causality Assessment Method (RUCAM), and by expert review by a panel of three hepatologists. Only cases having at least possible causality (score ≥ 3) were included in the study.33 DILIN causality assessment is determined by a panel of three hepatologists who independently assign a causality score ranging from 1 (definite) to 5 (unlikely) as well as a severity score ranging from 1 (mild) to 5 (fatal), as previously described.33 Besides the DILI phenotype, DILIN and iDILIC also collected additional clinical information, including the occurrence of eosinophilia and cutaneous involvement. The cases were compared with the 8,438 phase II population controls of European descent.
Genotyping of cases and controls
Out of the cumulative 43 CBZ‐SCAR cases, three PGX40001 cases were previously genotyped with Illumina 1M Duo chip.41 DNA from the rest of British and broadly European cases was extracted from whole blood and stored in the Wolfson Centre for Personalized Medicine in Liverpool. Genomewide genotyping was profiled by the Illumina Infinium HumanCoreExome Bead Chip for 16 cases and by Illumina HumanOmniExpress Bead Chip for 24 cases at the Broad Institute, Boston. Among the CBZ‐DILI cases, three cases were previously genotyped with Illumina 1M Duo Chip, whereas two cases were newly profiled by HumanCoreExome Bead Chip and seven cases by HumanOmniExpress Bead Chip at the Broad Institute, Boston.33 A total of 10,701 previously genotyped population controls were cumulatively used in British and broadly European cohorts. Information about the genotyping platform used by each of the control cohorts is reported in Table S6 . For each of the sample batches (defined as a set of subjects—either cases or controls—genotyped together by the same array), quality control was conducted at both SNP and subject levels before performing the imputation as previously described.41 Analysis of population structure was performed by the EIGENSTRAT package version 3.0.42 Pre‐phasing and imputation were performed in batches by dividing the cases and controls according to genotyping platform, using SHAPEIT (version v2.r727)43 and IMPUTE2 (version 3)44 with 1000 Genomes Project (release version 3) as reference.43 For downstream analysis, we used best‐guess genotypes retaining imputed genotypes with posterior probability > 0.9. Detailed methods are outlined in the Supplementary Materials.
Association analysis and meta‐analysis
We tested for association of each SNP with CBZ‐SCAR, separately in British and broadly European GWAS, in a logistic regression framework, under an additive genetic model, with adjustment for the principal components from smartPCA to account for population structure using PLINK version 1.07.45 No other additional covariates were included in the model because we did not have clinical information for controls. Association summary statistics from the two phases were combined using effective sample size weighted z‐score fixed‐effects meta‐analysis, implemented in METAL.46 Allelic ORs across the two phases were obtained through inverse‐variance weighting of effect sizes, with heterogeneity assessed with Cochran's Q statistic,47 implemented in METAL. We reported only those SNPs that attained, in addition to genomewide significance, nominal evidence of association (P < 0.05) with the same direction of effect on CBZ‐SCAR in both GWAS phases (internal validation). Furthermore, we tested for association of each SNP with CBZ‐SCAR clinical subtypes: SJS/TEN in phase II GWAS and DRESS across both phases in the same meta‐analysis framework. We also tested for association of each SNP with CBZ‐DILI in phase II GWAS, in a logistic regression framework, under an additive genetic model, with adjustment for the principal components to account for population structure. Genomewide significance was defined using a common threshold of P < 5 × 10−8. To account for the small sample size and the disproportionate case/control ratio, we applied a permutation approach for genomewide significant signals. In particular, we randomly permuted genotypes among individuals within the same phase of the design, tested for association, and then meta‐analyzed. We applied 5 × 108 permutations to demonstrate genomewide significance. All detailed analyses and Manhattan plots were performed with R version 3.0.2.14
HLA imputation, genotyping, and analysis
For each batch, HLA alleles were inferred using HLA genotype imputation with attribute bagging48 using the reference predictor panels specific for the genotyping chip. AA changes were inferred by SNP2HLA using reference data collected by the Type 1 Diabetes Genetics Consortium.49 We tested for association of carriage of each allele/AA/specific HLA haplotypes with CBZ‐SCAR, CBZ‐SCAR subtypes, and CBZ‐DILI using the same methods described above. MHC significance was defined using the Bonferroni correction threshold of P < 0.00025 accounting for 200 observed HLA alleles across loci (0.05/200). Conditional analyses in the MHC region were undertaken and the genotypes at the conditioning SNP(s) were included as covariates under an additive model. Fixed‐effects meta‐analyses across the two phases of GWAS were performed using the methods described above for unconditional analyses. High‐resolution genotyping of HLA loci was performed on all DILI cases by Histogenetics (Ossining, NY), as previously described.33
Funding
This work was supported by the International Serious Adverse Events Consortium (iSAEC). The iSAEC is a nonprofit organization dedicated to identifying and validating DNA‐variants useful in predicting the risk of drug‐related serious adverse events. The Consortium brings together the pharmaceutical industry, regulatory authorities, and academic centers to address clinical and scientific issues associated with the genetics of drug‐related serious adverse events. The iSAEC's current funding members include: Abbott, Amgen, AstraZeneca, Daiichi Sankyo, GlaxoSmithKline, Merck, Novartis, Pfizer, Takeda, and the Wellcome Trust. M.P. is an NIHR Senior Investigator. M.P. and A.A. thank the MRC Centre for Drug Safety Science for support (MR/L006758/1). P.N. was supported by iSAEC. A.P.M. is a Wellcome Trust Senior Fellow in Basic Biomedical Science (under award WT098017). This is a summary of independent research partly (the DILIGEN and iDILIC sample collection) funded by the National Institute for Health Research (NIHR) Nottingham Digestive Diseases Biomedical Research Unit at the Nottingham University Hospitals NHS Trust and University of Nottingham. The DILIN (https://dilin.org/) is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (NIH) as a Cooperative Agreement (U01s). The Spanish DILI Registry is partly funded by the Spanish Medicine Agency, Fondo Europeo de Desarrollo Regional (FEDER; FIS 12/00378, FIS 16/01748). CIBERehd is funded by Instituto de Salud Carlos III. The Swedish case collection SWEDEGENE (www.swedegene.se) has received support from the Swedish Medical Products Agency, the Swedish Society of Medicine (2008‐21619), Swedish Research Council (Medicine 521‐2011‐2440 and 521‐2014‐3370), and Swedish Heart and Lung Foundation (20120557 and 20140291). The EUDRAGENE collaboration received support from the EC 5th Framework program (QLRI‐CT‐2002‐02757). M.M. is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at Guy's and St. Thomas’ NHS Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Conflict of Interest
P.N. is an employee of Sema4 Mount Sinai venture, Stamford, CT, USA. M.R.N. is an employee of GlaxoSmithKline. All other authors declared no competing interests for this work.
Author Contributions
P.N., A.K.D., M.R.N., G.A., A.P.M., A.A., and M.P. wrote the manuscript. P.N., A.P.M., M.R.N., M.J.D., A.K.D., G.A., Y.S., A.A., and M.P. designed the research. P.N., A.S., and A.P.M. analyzed the data. M.P., A.K.D., P.B.W., S.B., L.M., G.A., M.I.L., R.J.A., M.W., P.H., C.S., E.S.B., P.F., K.K., T.L., A.M., M.M., E.P., W.P., A.R., N.S., G.S., L.K.T., and A.F. contributed new reagents/analytical tools.
Supporting information
Supplementary Methods, Figures, and Tables.
Acknowledgments
Special thanks to Arthur Holden for his help and effort in guiding this collaborative work and to the broad genotyping facility for their contribution to the genomewide association studies genotyping. We are grateful to Daniele Cusi (Hypergenes), Patrik K. Magnusson (Swedish Twin Registry), and Javier Martin (Spanish DNA bank) for provision of control data. The data presented in the current publication are also based on the use of study data downloaded from the dbGaP website, under phs000346.v1.p1. We also acknowledge the contribution of all our clinical collaborators and the study participants.
References
- 1. Yip, V.L. , Marson, A.G. , Jorgensen, A.L. , Pirmohamed, M. & Alfirevic, A. HLA genotype and carbamazepine‐induced cutaneous adverse drug reactions: a systematic review. Clin. Pharmacol. Ther. 92, 757–765 (2012). [DOI] [PubMed] [Google Scholar]
- 2. Marson, A.G. et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 369, 1000–1015 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uetrecht, J. & Naisbitt, D.J. Idiosyncratic adverse drug reactions: current concepts. Pharmacol. Rev. 65, 779–808 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bjornsson, E. , Kalaitzakis, E. & Olsson, R. The impact of eosinophilia and hepatic necrosis on prognosis in patients with drug‐induced liver injury. Aliment. Pharmacol. Ther. 25, 1411–1421 (2007). [DOI] [PubMed] [Google Scholar]
- 5. Cheng, C.Y. , Su, S.C. , Chen, C.H. , Chen, W.L. , Deng, S.T. & Chung, W.H. HLA associations and clinical implications in T‐cell mediated drug hypersensitivity reactions: an updated review. J. Immunol. Res. 2014, 565320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung, W.H. et al. Medical genetics: a marker for Stevens‐Johnson syndrome. Nature 428, 486 (2004). [DOI] [PubMed] [Google Scholar]
- 7. Shi, Y.W. et al. HLA‐A*24:02 as a common risk factor for antiepileptic drug‐induced cutaneous adverse reactions. Neurology 88, 2183–2191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ozeki, T. et al. Genome‐wide association study identifies HLA‐A*3101 allele as a genetic risk factor for carbamazepine‐induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 20, 1034–1041 (2011). [DOI] [PubMed] [Google Scholar]
- 9. Kim, S.H. et al. Carbamazepine‐induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Res. 97, 190–197 (2011). [DOI] [PubMed] [Google Scholar]
- 10. McCormack, M. et al. HLA‐A*3101 and carbamazepine‐induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 364, 1134–1143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi, Y.W. et al. Association between HLA and Stevens‐Johnson syndrome induced by carbamazepine in Southern Han Chinese: genetic markers besides B*1502? Basic Clin. Pharmacol. Toxicol. 111, 58–64 (2012). [DOI] [PubMed] [Google Scholar]
- 12. Tassaneeyakul, W. et al. Association between HLA‐B*1502 and carbamazepine‐induced severe cutaneous adverse drug reactions in a Thai population. Epilepsia 51, 926–930 (2010). [DOI] [PubMed] [Google Scholar]
- 13. Niihara, H. , Kakamu, T. , Fujita, Y. , Kaneko, S. & Morita, E. HLA‐A31 strongly associates with carbamazepine‐induced adverse drug reactions but not with carbamazepine‐induced lymphocyte proliferation in a Japanese population. J. Dermatol. 39, 594–601 (2012). [DOI] [PubMed] [Google Scholar]
- 14. Hsiao, Y.H. et al. Genotype‐phenotype association between HLA and carbamazepine‐induced hypersensitivity reactions: strength and clinical correlations. J. Dermatol. Sci. 73, 101–109 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Gonzalez‐Galarza, F.F. et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 43, D784–D788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duquesnoy, R.J. et al. 16th IHIW: a website for antibody‐defined HLA epitope Registry. Int. J. Immunogenet. 40, 54–59 (2013). [DOI] [PubMed] [Google Scholar]
- 17. Genin, E. et al. HLA‐A*31:01 and different types of carbamazepine‐induced severe cutaneous adverse reactions: an international study and meta‐analysis. Pharmacogenomics J. 14, 281–288 (2014). [DOI] [PubMed] [Google Scholar]
- 18. Amstutz, U. et al. HLA‐A 31:01 and HLA‐B 15:02 as genetic markers for carbamazepine hypersensitivity in children. Clin. Pharmacol. Ther. 94, 142–149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mushiroda, T. et al. Association of HLA‐A*31:01 screening with the incidence of carbamazepine‐induced cutaneous adverse reactions in a Japanese population. JAMA Neurol. 75, 842–849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Phillips, E.J. et al. Clinical Pharmacogenetics Implementation Consortium Guideline for HLA genotype and use of carbamazepine and oxcarbazepine: 2017 update. Clin. Pharmacol. Ther. 103, 574–581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Genin, E. et al. Genome‐wide association study of Stevens‐Johnson syndrome and toxic epidermal necrolysis in Europe. Orphanet. J. Rare Dis. 6, 52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang, Y. et al. HLA‐B*51:01 is strongly associated with clindamycin‐related cutaneous adverse drug reactions. Pharmacogenomics J. 17, 501–505 (2017). [DOI] [PubMed] [Google Scholar]
- 23. Lichtenfels, M. et al. HLA restriction of carbamazepine‐specific T‐cell clones from an HLA‐A*31:01‐positive hypersensitive patient. Chem. Res. Toxicol. 27, 175–177 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Mallal, S. et al. Association between presence of HLA‐B*5701, HLA‐DR7, and HLA‐DQ3 and hypersensitivity to HIV‐1 reverse‐transcriptase inhibitor abacavir. Lancet 359, 727–732 (2002). [DOI] [PubMed] [Google Scholar]
- 25. Hetherington, S. et al. Genetic variations in HLA‐B region and hypersensitivity reactions to abacavir. Lancet 359, 1121–1122 (2002). [DOI] [PubMed] [Google Scholar]
- 26. Daly, A.K. et al. HLA‐B*5701 genotype is a major determinant of drug‐induced liver injury due to flucloxacillin. Nat. Genet. 41, 816–819 (2009). [DOI] [PubMed] [Google Scholar]
- 27. Petros, Z. , Kishikawa, J. , Makonnen, E. , Yimer, G. , Habtewold, A. & Aklillu, E. HLA‐B*57 allele is associated with concomitant anti‐tuberculosis and antiretroviral drugs induced liver toxicity in Ethiopians. Front. Pharmacol. 8, 90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu, C.F. et al. HLA‐B*57:01 confers susceptibility to pazopanib‐associated liver injury in patients with cancer. Clin. Cancer Res. 22, 1371–1377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nicoletti, P. et al. Drug‐induced liver injury due to flucloxacillin: relevance of multiple human leukocyte antigen alleles. Clin. Pharmacol. Ther. 106, 245–253 (2019). [DOI] [PubMed] [Google Scholar]
- 30. Singer, J.B. et al. A genome‐wide study identifies HLA alleles associated with lumiracoxib‐related liver injury. Nat. Genet. 42, 711–714 (2010). [DOI] [PubMed] [Google Scholar]
- 31. El‐Awar, N. , Jucaud, V. & Nguyen, A. HLA epitopes: the targets of monoclonal and alloantibodies defined. J. Immunol. Res. 2017, 3406230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lanzavecchia, A. How can cryptic epitopes trigger autoimmunity? J. Exp. Med. 181, 1945–1948 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicoletti, P. et al. Association of liver injury from specific drugs, or groups of drugs, with polymorphisms in HLA and other genes in a genome‐wide association study. Gastroenterology 152, 1078–1089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pirmohamed, M. , Ostrov, D.A. & Park, B.K. New genetic findings lead the way to a better understanding of fundamental mechanisms of drug hypersensitivity. J. Allergy Clin. Immunol. 136, 236–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holla, V.R. et al. ALK: a tyrosine kinase target for cancer therapy. Cold Spring Harb. Mol. Case Stud. 3, a001115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Souttou, B. , Carvalho, N.B. , Raulais, D. & Vigny, M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen‐activated protein kinase pathway. J. Biol. Chem. 276, 9526–9531 (2001). [DOI] [PubMed] [Google Scholar]
- 37. Pirmohamed, M. et al. Investigation into the multidimensional genetic basis of drug‐induced Stevens‐Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics 8, 1661–1691 (2007). [DOI] [PubMed] [Google Scholar]
- 38. Pirmohamed, M. et al. Phenotype standardization for immune‐mediated drug‐induced skin injury. Clin. Pharmacol. Ther. 89, 896–901 (2011). [DOI] [PubMed] [Google Scholar]
- 39. Nelson, M.R. et al. The Population Reference Sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am. J. Hum. Genet. 83, 347–358 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tryka, K.A. et al. NCBI's database of genotypes and phenotypes: dbGaP. Nucleic Acids Res. 42, D975–D979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen, Y. et al. Genome‐wide association study of serious blistering skin rash caused by drugs. Pharmacogenomics J. 12, 96–104 (2012). [DOI] [PubMed] [Google Scholar]
- 42. Price, A.L. , Patterson, N.J. , Plenge, R.M. , Weinblatt, M.E. , Shadick, N.A. & Reich, D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat. Genet. 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 43. Delaneau, O. , Zagury, J.F. & Marchini, J. Improved whole‐chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013). [DOI] [PubMed] [Google Scholar]
- 44. Marchini, J. & Howie, B. Genotype imputation for genome‐wide association studies. Nat. Rev. Genet. 11, 499–511 (2010). [DOI] [PubMed] [Google Scholar]
- 45. Purcell, S. et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willer, C.J. , Li, Y. & Abecasis, G.R. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ioannidis, J.P. , Patsopoulos, N.A. & Evangelou, E. Heterogeneity in meta‐analyses of genome‐wide association investigations. PLoS One 2, e841 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zheng, X. et al. HIBAG–HLA genotype imputation with attribute bagging. Pharmacogenomics J. 14, 192–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8, e64683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Figures, and Tables.