

Summary
Many organisms exhibit visually striking spotted or striped pigmentation patterns. Developmental models predict that such spatial patterns can form when a local autocatalytic feedback loop and a long-range inhibitory feedback loop interact. At its simplest, this self-organizing network only requires one self-activating activator that also activates a repressor, which inhibits the activator and diffuses to neighboring cells. However, the molecular activators and inhibitors fully fitting this versatile model remain elusive in pigmentation systems. Here, we characterize an R2R3-MYB activator and an R3-MYB repressor in monkeyflowers (Mimulus). Through experimental perturbation and mathematical modeling, we demonstrate that the properties of these two proteins correspond to an activator-inhibitor pair in a two-component, reaction-diffusion system, explaining the formation of dispersed anthocyanin spots in monkeyflower petals. Notably, disrupting this pattern impacts pollinator visitation. Thus, subtle changes in simple activator-inhibitor systems are likely essential contributors to the evolution of the remarkable diversity of pigmentation patterns in flowers.
eTOC Blurb
Many species exhibit visually striking spotted or striped pigmentation patterns. By studying mutants that alter the red spots that dapple monkeyflower petals, Ding et al. uncover a simple activator-inhibitor system that conforms to longstanding theoretical predictions for how such patterns self-organize during development.
Graphical Abstract
Introduction
Pigmentation patterns like the stripes of zebras, the patches of giraffes, and the petal spots of many flowers have fascinated biologists and mathematicians for centuries. These patterns often function as visual cues in mate choice and interspecies communications, adapting organisms to their environments and potentially contributing to reproductive isolation among species [1–4]. Unlike previously well characterized pigmentation patterns in insect wings and some flowers that are spatially organized by coinciding expression domains of regulatory genes that serve as position-specific signals [5–11], our understanding of how dispersed pigmentation patterns emerge in concert with or entirely untethered from such positional information is primarily theoretical [12–15]. The reaction-diffusion (RD) model, initially proposed by Turing [16] then independently developed and elaborated by Meinhardt and Gierer [17–19] and further extended by others [12,20–22], postulates that local activation of pattern differentiation factors combined with long-range inhibition of the activity of those factors can produce defined, regularly spaced, tissue-level spot and stripe patterns. In its simplest form, the RD model for self-organizing pattern formation only requires two interacting components: one activator that activates itself as well as a repressor, which diffuses to neighboring cells faster than the activator and inhibits the activator’s function [12,17,18]. However, when the RD interacting network contains more than two nodes, pattern formation does not necessarily require differential diffusivity of the activator and inhibitor as in the two-component RD system [22]. Candidate activators and inhibitors that fit the RD model have been advanced for several developmental patterns ([15,23–27]; but see [13,28,29] for alternative mechanisms not reliant on molecular players). However, the molecular identities and dynamics of the activation and inhibition factors that contribute to self-organized pigmentation patterning have remained elusive, particularly in relevant animal systems where mutant alleles often have deleterious pleiotropic consequences or critical embryonic developmental stages are challenging to study experimentally.
Anthocyanin spots in flower petals provide an excellent empirical system to reveal the molecular basis for the formation and evolution of self-organized pigmentation patterns. These patterns, which are highly diverse in angiosperms even among different varieties of the same species [30,31], are known to serve as critical cues in plant-pollinator interactions [32–34]; and the genetic network controlling anthocyanin pigment production is otherwise well described [35,36]. Although some factors involved in the regulation and natural variation of floral pigment patterning have been identified, these factors act solely through pre-patterning by underlying developmental programs and do not yield periodic or randomly dispersed spatial patterns [7–9]. To reveal developmental mechanisms that contribute to dispersed floral pigment patterns, we have focused on the anthocyanin spots that dot the nectar guides of many species in the monkeyflower genus Mimulus. The presence of these spots facilitates effective pollinator visitation [32], and genomic resources and methods for genetic transformation that foster rigorous genetic and developmental studies have been established for multiple Mimulus species [37–39].
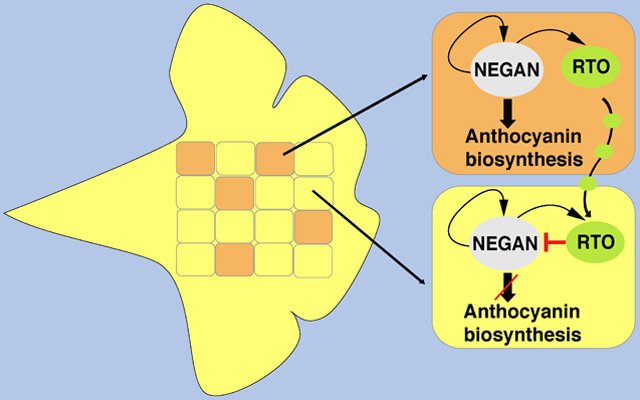
Previously, we identified a self-activating R2R3-MYB transcription factor, NECTAR GUIDE ANTHOCYANIN (NEGAN), that activates anthocyanin spot formation in the monkeyflower species Mimulus lewisii ([40]; Figure 1A). Down-regulation of NEGAN expression abolishes anthocyanin production in the nectar guides (i.e., the two yellow ridges of the ventral petal; Figure 1B). We hypothesized that the dispersed anthocyanin spots could be explained by a simple two-component RD model, which predicts: (i) there should be an inhibitor corresponding to the anthocyanin activator NEGAN, and loss-of-function mutations in the presumptive inhibitor gene should cause expansion of the spots; (ii) the inhibitor is activated by the activator (i.e., NEGAN); (iii) the inhibitor represses the function of the activator; and (iv) the inhibitor can move from cell to cell. Here, through genetic mapping of a chemically induced mutant and natural variants with expanded anthocyanin spots followed by manipulative experiments and computer simulations, we identify a second molecular factor involved in anthocyanin spot patterning in Mimulus petals, reveal how this protein functions as a long-range inhibitor of NEGAN to regulate spot formation, and evaluate the evolutionary and ecological impacts of modulating this self-organizing patterning mechanism.
Figure 1. Dispersed anthocyanin spots in Mimulus lewisii and M. guttatus.

(A–E) Anthocyanin spots on the yellow background of the wild-type M. lewisii (LF10) and various mutants and transgenic lines. D: dorsal; L: lateral; V: ventral. (F–J) Anthocyanin spot patterns of M. guttatus variants segregating in natural populations in Oregon and California. See also Figures S1 and S2.
Results
The R3-MYB gene RED TONGUE is a negative regulator of anthocyanin spot patterning
To search for the putative inhibitor involved in anthocyanin spot formation, we carried out an ethyl methanesulfonate mutagenesis screen using the M. lewisii inbred line LF10 and isolated one candidate mutant, named red tongue (rto). Homozygous rto mutants develop a near uniform patch of anthocyanin color across the entire nectar guide region (Figure 1C). Coincidentally, we discovered naturally occurring mutants with rto-like phenotypes segregating in several geographically distinct populations of the congener M. guttatus (Figures 1F–1J). Complementation crosses among these populations indicate that these natural M. guttatus variants represent different alleles of the same locus (Figure S1A). The rto mutant and rto-like natural alleles behave semi-dominantly; F1s derived by crossing these alleles to corresponding wild-type individuals exhibit intermediate anthocyanin spot number and area phenotypes (Figures 1D, 1G–1I and S2).
Although M. lewisii and M. guttatus diverged about 20 million years ago [41], the similarity and specificity of the altered pigmentation phenotypes led us to suspect that all these mutant phenotypes are caused by lesions in the same gene. To identify the gene responsible for the rto phenotype in each species, we performed bulked segregant analyses by whole genome sequencing on F2 mapping panels segregating for the M. lewisii mutant and the two rto-like M. guttatus alleles found at Sweetwater Creek (SWC) in Oregon (Figure 1G) and Littlerock Dam (LRD) in Southern California (Figure 1H), i.e., rtoSWC and rtoLRD. As expected, the independent mapping experiments all pinpointed the same causal genomic interval (Figures 2A–2C). Inspection of the bulked segregant reads aligning to this ~60 kb region in the wild-type M. lewisii (LF10) genome revealed only one mutation, which causes an aspartic acid-to-glycine replacement at a highly conserved site of a small R3-MYB protein (the ortholog of Migut.B01849) that is 91 amino acids long (Figures 2D and S3A). Furthermore, fine mapping resolved the M. guttatus rtoSWC allele to an interval spanning four genes (Figure S3B), including the same R3-MYB gene Migut.B01849. Sanger sequencing of this gene revealed at least one putative loss-of-function mutation in each of the four M. guttatus rto-like alleles (Figure 2D). Notably, this R3-MYB is closely related to a group of R3-MYBs that are known to repress anthocyanin biosynthesis (Figure S3A), including ROSE INTENSITY1 in M. lewisii [42], MYBx in Petunia [43], and CAPRICE in Arabidopsis [44]. Taken together, these results strongly suggest this R3-MYB gene is the causal gene underlying the RTO locus in both M. lewisii and M. guttatus.
Figure 2. Identification of the RTO gene in Mimulus lewisii and M. guttatus and its relative expression in various mutant and transgenic lines.
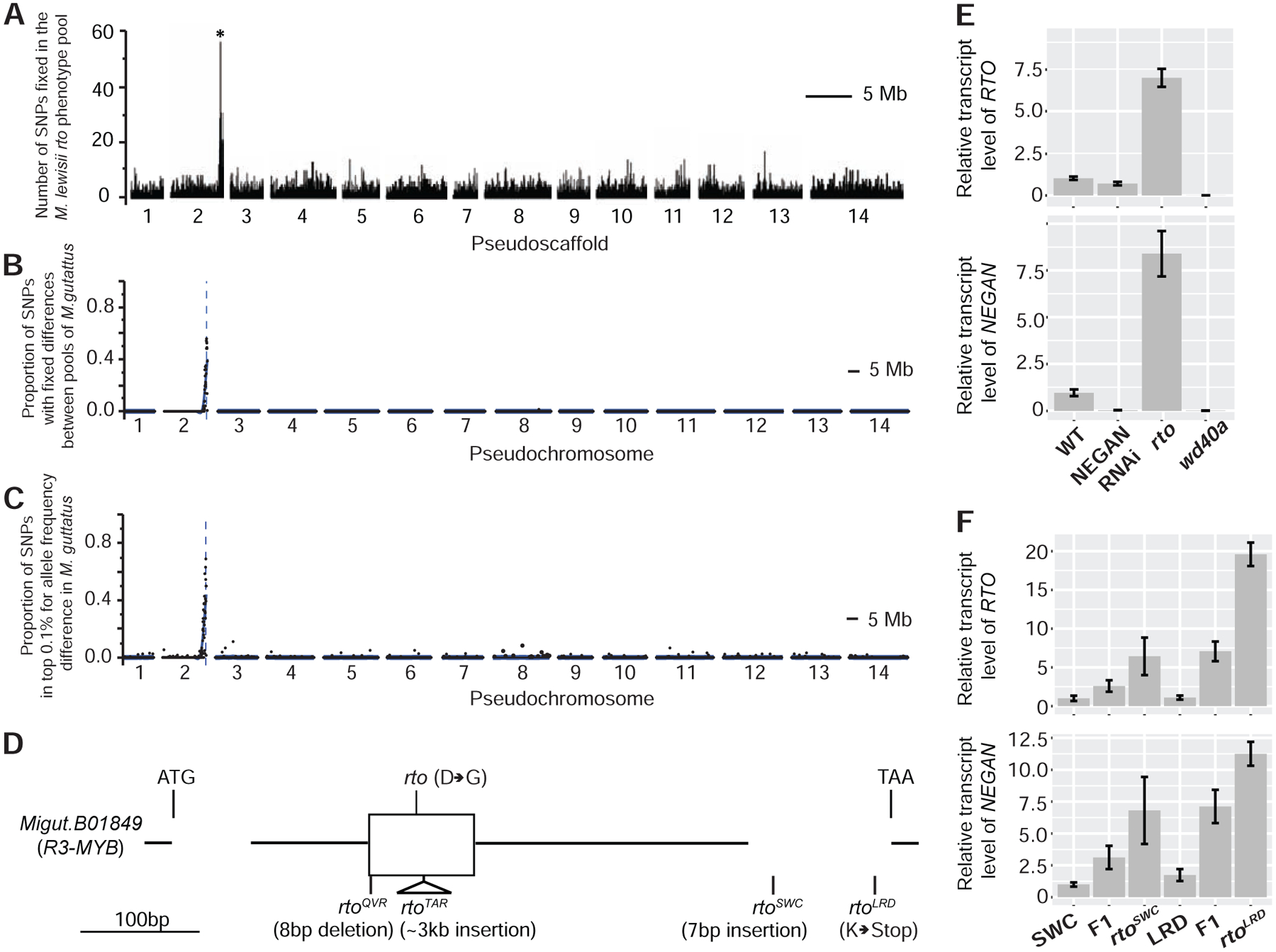
(A–C) Bulked segregant analyses of rto in M. lewisii (A) and rtoSWC (B) and rtoLRD (C) in M. guttatus narrowed RTO down to the same genomic interval. (D) Schematic of the RTO (R3-MYB) gene, showing the molecular lesions of the five mutant alleles. (E and F) Relative transcript level of RTO (upper) and NEGAN (lower) in M. lewisii (E) and M. guttatus (F) as measured by qRT-PCR, standardized to the corresponding wild-type (LF10 for M. lewisii, SWC for M. guttatus). Error bars represent 1 SD from three biological replicates. See also Figures S3 and S4 and Tables S4 and S6.
To verify the function of this R3-MYB gene, we knocked down its expression level in M. lewisii and M. guttatus by RNA interference (RNAi). We obtained multiple independent transgenic lines from both species with severe phenotypes that recapitulate the mutant phenotypes (Figures 3A, 3B and S2A). Some lines in the M. lewisii background have an even stronger phenotype than rto towards the throat of the corolla (e.g., RNAi line 8; Figure 3A), suggesting that rto is likely hypomorphic (partially functional) rather than a null allele. Notably, CRISPR/Cas9-mediated frameshift mutations in MgRTO also recapitulate the rto phenotype (Figures 4A, 4C and S2B), and the targeted mutation does not complement natural variants (Figure 4B), confirming that they are allelic. Together, the association of expanded pigmentation with five independent lesions in RTO and with RNAi knockdown and/or targeted disruptions of RTO in two congeneric species strongly indicates that RTO functions as an inhibitor of anthocyanin spot patterning. As such, we expected that over-expression of RTO driven by the CaMV 35S promoter would attenuate or completely abolish the anthocyanin spots. Indeed, the corollas of strong 35S:MlRTO lines in M. lewisii are completely white and lack anthocyanin spots in the nectar guide region (Figure 3C), confirming that RTO acts as a potent inhibitor of anthocyanin pigmentation.
Figure 3. Functional characterization of RTO in Mimulus lewisii and M. guttatus.
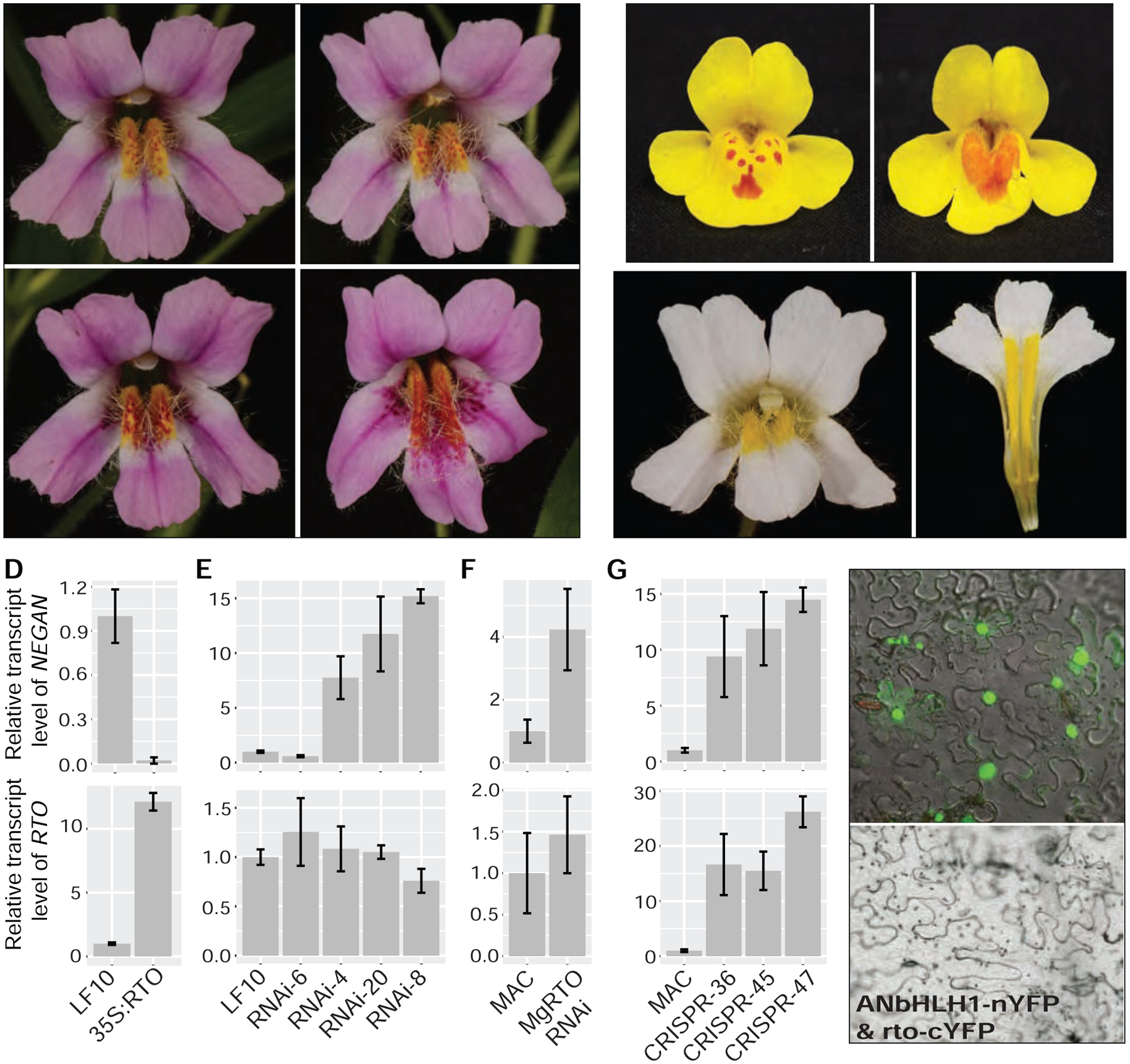
(A) RNAi of RTO in M. lewisii generates a range of anthocyanin spot patterns. (B) RNAi of MgRTO in M. guttatus recapitulates the rto-like phenotype. (C) Over-expression of RTO in M. lewisii abolishes anthocyanin production throughout the corolla. (D–G) Relative expression of NEGAN and RTO in M. lewisii RTO over-expression lines (D), M. lewisii RTO RNAi lines (E), M. guttatus RTO RNAi lines (F), and M. guttatus CRISPR/Cas9 mediated knockout lines (G). All relative transcript levels are measured by qRT-PCR, standardized to the corresponding wild-type (LF10 for M. lewisii, MAC for M. guttatus). Error bars represent 1 SD from three biological replicates. (H) BiFC assay shows that the wild-type RTO protein interacts with ANbHLH1, whereas the D>G amino acid replacement in the mutant rto protein abolishes or attenuates the interaction. See also Figures S2 and S4–S6 and Tables S5 and S6.
Figure 4. CRISPR/Cas9 mediated knockout of MgRTO in Mimulus guttatus.
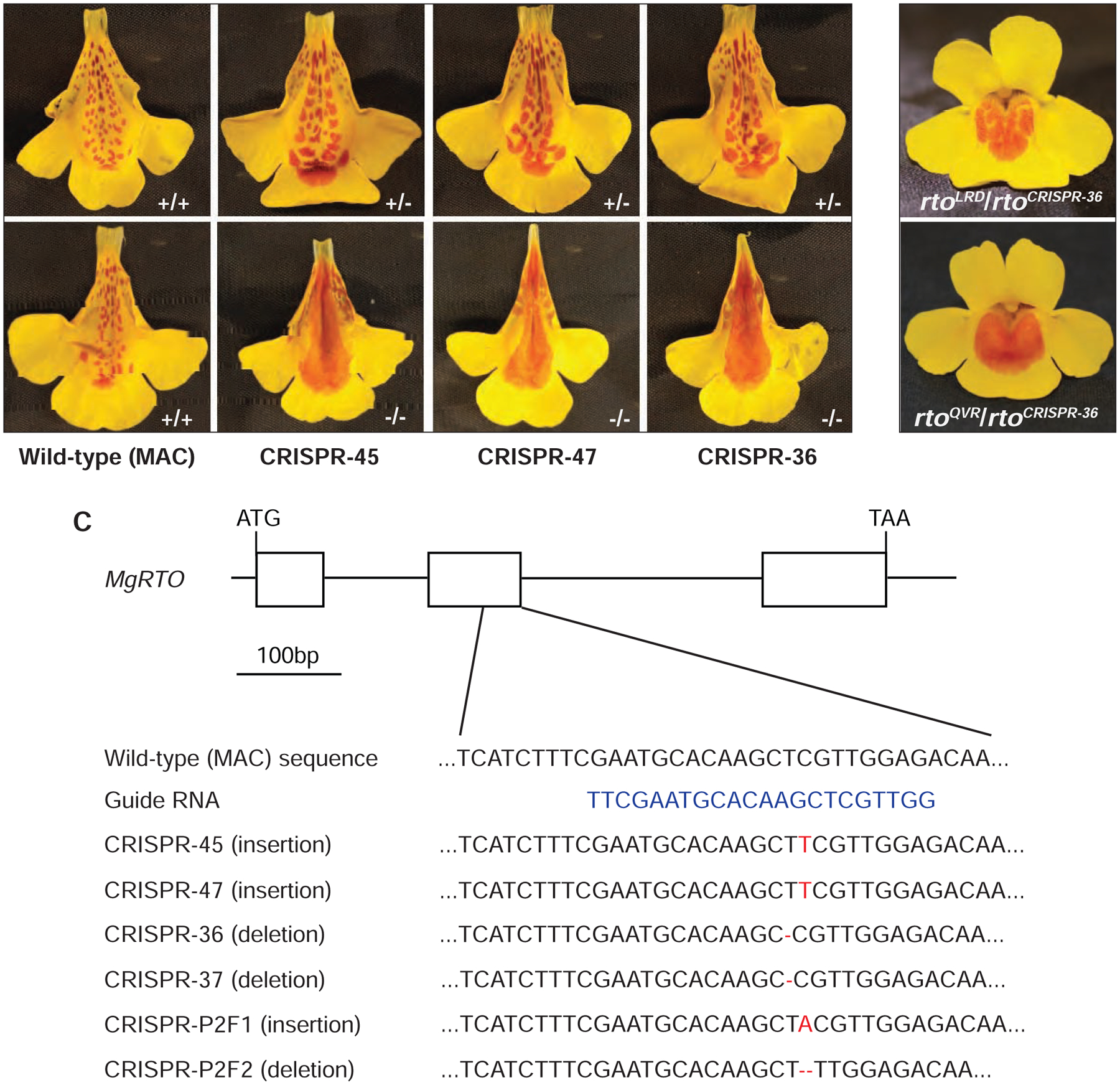
(A) Flower phenotypes of three independent CRISPR knockout lines in their heterozygous (+/−) and homozygous (−/−) conditions, compared to the wild-type (MAC; +/+). (B) Complementation crosses between a CRISPR allele (rtoCRISPR−36) and two natural mutant alleles (rtoLRD and rtoQVR). (C) Examples of six independent CRISPR alleles relative to gRNA position in MgRTO. The primer pair 5’-GTCTTCATATATTCCCATCTCTT-3’ and 5’-CGTGCTCGGTGTAAGTAACG-3’ was used for amplifying and sequencing the target region. See also Figure S2 and Table S5.
NEGAN promotes RTO expression
Having identified the putative inhibitor, we then tested whether NEGAN and RTO function as a local activator/lateral inhibitor pair consistent with the simple two-component RD model [17,18]. In particular, we sought to test whether the self-activating activator NEGAN activates the inhibitor (i.e., RTO), which then diffuses laterally and inhibits the function of the activator along the diffusion path. To test the first prediction that the local activator also activates the inhibitor, we compared RTO gene expression levels between wild-type and NEGAN RNAi plants as well as the M. lewisii wd40a mutant. The wd40a mutant is relevant because in all angiosperms characterized to date, the anthocyanin-activating R2R3-MYB transcription factor (e.g., NEGAN) functions in an obligatory protein complex that also contains a bHLH protein and a WD40 protein [35,36]. In M. lewisii flowers, the bHLH and WD40 components of the complex (MlANbHLH1 and MlWD40a, respectively) are expressed in the entire corolla. The R2R3-MYB component is supplied by two paralogs, NEGAN and PETAL LOBE ANTHOCYANIN (PELAN), that are expressed in nectar guides and petal lobes, respectively [40]. The wd40a mutants completely lack anthocyanins in both their petal lobes and the nectar guides (Figure 1E). Down-regulation of NEGAN by RNAi abolishes the anthocyanin spots in the nectar guides but does not affect the petal lobe color (Figure 1B), whereas the loss-of-function pelan mutant and PELAN RNAi lines lose all the anthocyanin pigments in the petal lobe while the nectar guide spots remain unperturbed ([40]; Figure S4A). If nectar guide expression of RTO is activated by NEGAN acting in a complex with MlANbHLH1 and MlWD40a, we would expect reduced RTO expression in NEGAN RNAi lines and the wd40a mutant compared to wild-type M. lewisii plants.
Indeed, for whole corollas sampled from 10-mm stage flower buds, the developmental stage when both NEGAN and RTO show peak expression level and the anthocyanin spots just start appearing (Figure S4B, S4D, S4F), the RTO transcript level decreased to an undetectable level in the wd40a mutant compared to the wild-type (Figure 2E). NEGAN expression is also abolished in the wd40a mutant (Figure 2E), as previously shown [40], because the ability of NEGAN to self-activate also requires the complete NEGAN-MlANbHLH1-MlWD40a protein complex. In the 10-mm stage whole corollas of M. lewisii NEGAN RNAi lines, however, the RTO transcript level only slightly decreased compared to the wild-type (Figure 2E). We predicted this finding resulted from activation of RTO expression in the petal lobe by PELAN. Indeed, when examined in dissected petal lobes and nectar guides at this stage, petal lobe RTO expression is unaffected in the NEGAN RNAi lines, but is reduced to undetectable level in the pelan mutant; whereas nectar guide RTO expression is unaffected in the pelan mutant, but clearly reduced in the NEGAN RNAi lines (Figure S4C). These results suggest that MlRTO expression is activated by PELAN and NEGAN in the petal lobes and nectar guides, respectively, and requires the same WD40 component in both tissues. That PELAN cannot self-activate [40] may explain the generally uniform anthocyanin pigmentation in the petal lobes, compared to the dispersed spots in the nectar guides. Similarly, when MgNEGAN expression was knocked down in M. guttatus, the anthocyanin spots completely disappeared from nectar guides of strong RNAi lines, and MgRTO expression levels decreased accordingly (Figure S5). Taken together, our results confirm that the activator, NEGAN, activates the expression of the inhibitor, RTO, in the nectar guides of both Mimulus species.
RTO is a negative regulator of NEGAN function
To test whether the inhibitor represses the activator’s function, we assayed relative NEGAN expression levels in the rto mutant and rto-like natural variants as well as various RTO transgenic lines. Since NEGAN expression is self-activated, if RTO inhibits the activity of NEGAN, loss of RTO function should increase NEGAN expression, and over-expression of RTO should do the opposite. Consistent with those predictions, NEGAN transcript abundance increased 6- to 11-fold in rto and the rto-like natural variants (Figures 2E and 2F) and decreased >10-fold in the strong 35S:MlRTO lines (Figure 3D). In the M. lewisii RTO RNAi lines, NEGAN expression increased as much as 15-fold, and NEGAN expression level is positively correlated with the severity of the anthocyanin phenotype (Figures 3A and 3E). Likewise, MgNEGAN expression is also upregulated in the MgRTO RNAi and targeted knockout lines (Figures 3F and 3G). These results strongly indicate that RTO inhibits NEGAN function.
That the transcript level of RTO itself in the RTO RNAi lines is not lower than in the wild-type plants (Figures 3E and 3F) seems surprising at first glance. However, this finding is consistent with the anticipated dynamic interactions between NEGAN and RTO. As shown above, knock-down of RTO transcript levels by RNAi resulted in up-regulation of NEGAN, which in turn would up-regulate RTO, but the increased RTO transcript level would then be counterbalanced by RNAi. Consequently, unlike NEGAN, RTO transcript level showed no clear correlation to the severity of the anthocyanin phenotype in the RTO RNAi lines. Notably, in MgRTO knockout lines, where similar feedback is not expected, RTO levels are conspicuously elevated relative to wild-type plants (Figure 3G).
The mechanism by which R3-MYB repressors inhibit R2R3-MYB activator function is well understood for their homologous proteins in Arabidopsis and Petunia [43–45]. These R3-MYB proteins have neither DNA-binding nor activation domains, but they compete with the R2R3-MYB activator for the limited supply of bHLH proteins, sequestering the bHLH proteins into inactive complexes. Like NEGAN, RTO also contains the bHLH-interacting motif in its R3 domain. The aspartic acid-to-glycine replacement in the mutant rto protein is expected to disrupt the bHLH-interacting motif (Figure S3A). Indeed, in bimolecular fluorescence complementation assays, pairing wild-type MlRTO protein and MlANbHLH1 showed reconstitution of fluorescence, but pairing mutant rto protein and MlANbHLH1 yielded no detectable fluorescence signal (Figures 3H and S6A), indicating that the mutation abolishes or attenuates the interaction between the R3-MYB and the bHLH protein.
RTO exhibits intercellular movement
To test whether RTO exhibits long-range lateral inhibition (i.e., diffusible to neighboring cells), we compared the spatial pattern of RTO transcription with the RTO protein distribution [46]. If RTO moves between cells, RTO proteins should be observable over a broader domain of cells than where RTO is transcribed. To make this comparison, we first examined the spatial pattern of RTO transcription by transforming the wild-type M. lewisii (LF10) with a construct that expresses cyan fluorescent protein (CFP) with an endoplasmic reticulum (ER) retention signal, driven by the RTO promoter. The CFP signal marks the cells with RTO promoter activity, as the CFP-ER product cannot move from the source cell to neighboring cells. Multiple independent RTOpro:CFP-ER lines showed the same pattern; the CFP fluorescence signal co-localizes with the anthocyanin spots (Figure 5A), consistent with the expectation that both RTO and the anthocyanin biosynthetic genes are regulated by the same transcriptional activator (i.e., NEGAN).
Figure 5. Intercellular movement of RTO in Mimulus lewisii.
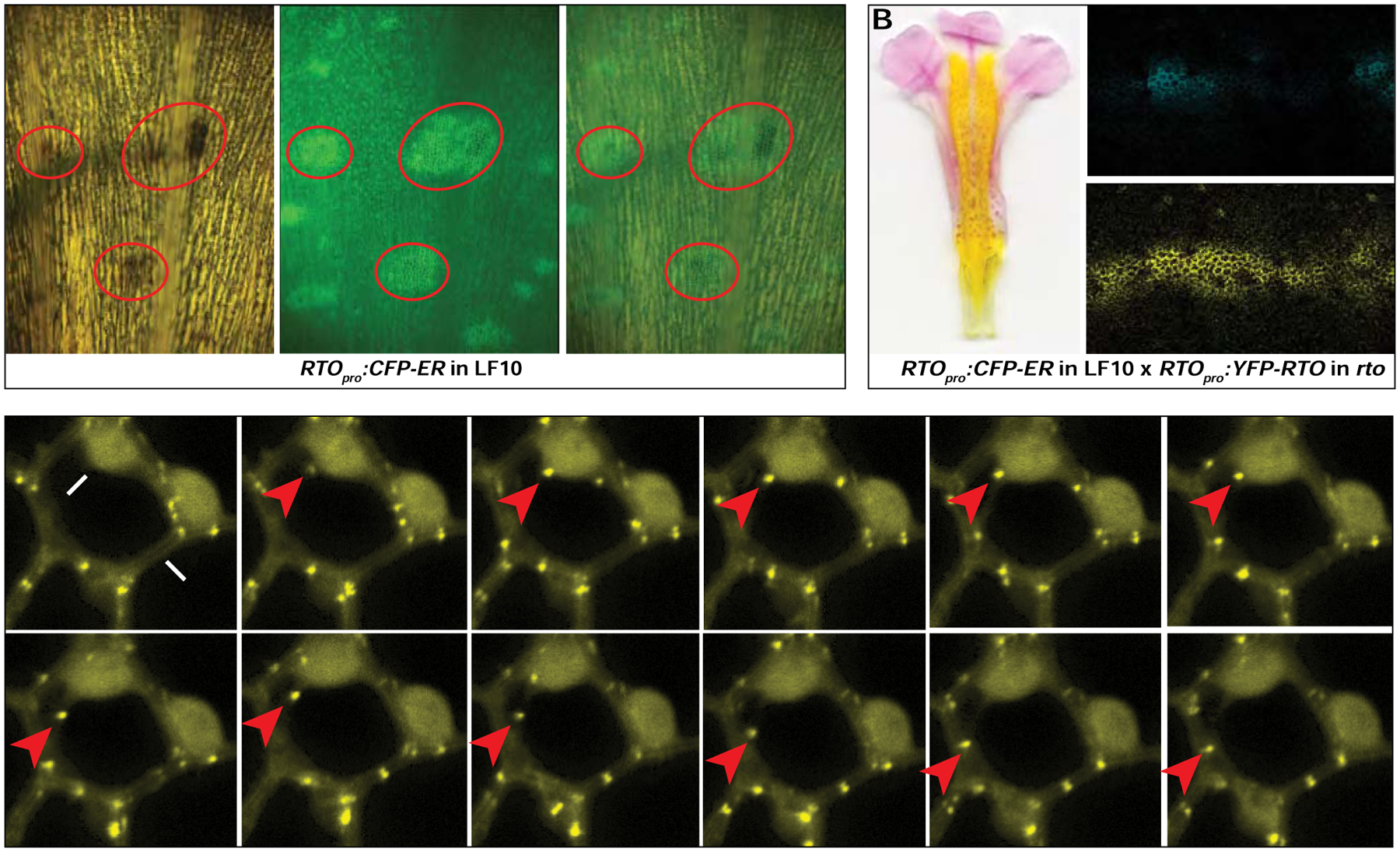
(A) Spatial pattern of RTO promoter activity revealed by the RTOpro:CFP-ER construct. The same nectar guide area was imaged under bright light (left) and the green channel (center). The right image is an overlay between the left and the center. The yellow background under bright light is due to carotenoid pigments, and the anthocyanin spots are red. CFP fluorescence signal (i.e., the green spots in the green channel) foreshadows and co-localizes with anthocyanin production (red ovals). (B) F1 hybrids between RTOpro:CFP-ER line 25 (in the wild-type background) and RTOpro:YFP-RTO line 22 (in the rto background) bear flowers similar in anthocyanin phenotype to the non-transgenic RTO/rto heterozygote and reveal a broader spatial distribution of RTO protein (yellow) than RTO promoter activity (blue). (C) A series of still images from a time-lapse video taken every 30 seconds to track the movement of YFP-RTO from nucleus to cytoplasm in 35S:YFP-RTO petal epidermal cells. See also Figure S6, Table S5, and Video S1.
Next, to reveal the spatial distribution of RTO proteins, we expressed a YFP-RTO fusion protein driven by the same RTO promoter in the rto background. In the majority of the RTOpro:YFP-RTO transformants, the nectar guide anthocyanin pigmentation was reduced and the extent of this “rescue” of the rto phenotype was positively correlated with the transgene expression level (Figures S6B and S6C). Thus, the YFP-RTO fusion protein retains the function of RTO as an anthocyanin repressor in vivo. The complete elimination of nectar guide anthocyanin rather than restoration to the wild-type pattern for a substantial number of RTOpro:YFP-RTO lines in the rto background was unexpected (Figures S6B). One possible explanation is that a recessive epistatic interaction between the hypomorphic rto allele and the transgene shifts the dynamics of the NEGAN-RTO interaction. However, when we crossed representative RTOpro:CFP-ER and RTOpro:YFP-RTO lines to bring the two transgenes together in a RTO/rto heterozygous background, we observed typical RTO/rto anthocyanin patterning rather than the “over-rescue” phenotype (Figure 5B), indicating wild-type NEGAN-RTO dynamics are re-established. In this background, YFP-RTO signal was detected not only in the source cells of the RTO promoter activity, as reflected by the CFP-ER signal, but also in neighboring cells (Figures 5B, S6D and S6E). Taking these transcript and protein localization results together, we conclude that the RTO gene is transcribed in the anthocyanin spot cells but the RTO protein moves to adjacent cells.
To gain further insight into the mechanism of RTO movement, we generated 35S:YFP-RTO transformants in M. lewisii. Like the strong 35S:RTO lines, strong 35S:YFP-RTO transgenic lines produce completely white flowers, confirming again that the YFP-tag does not interfere with RTO function. We found YFP-RTO localized to both the nucleus and cytoplasm. We also found many punctate YFP-RTO signals, mainly in the cytoplasm. These puncta travel from the nucleus to the membrane (Figure 5C and Video S1), reminiscent of the RTO homolog in Arabidopsis, CAPRICE, and other mobile proteins such as SHORT ROOT, which are known to interact with protein transport partners and travel through the endosome system in Arabidopsis roots [47]. These similarities suggest that the same mechanism might be involved in RTO intercellular movement.
The NEGAN/RTO pair fits the simplest two-component reaction-diffusion model
The properties we have described for NEGAN and RTO conform to a two-component activator-inhibitor model, one of the simplest Gierer-Meinhardt topologies underlying pattern formation [17,18]. We therefore simulated this Gierer-Meinhardt activator-inhibitor model (Figure 6A) with the Virtual Cell software [48] to investigate whether tinkering the kinetics of the two molecules in silico can recapitulate the phenotypes that were produced by experimental perturbations. We first determined the parameter values that generated a sparsely spotted pattern resembling wild-type M. lewisii (Figure 6B), guided by previous studies simulating spot pattern formation with similar parameter values that were considered biologically realistic [12,20,49,50]. We then increased the degradation rate of the activator NEGAN (UA), analogous to RNAi-mediated knock-down of NEGAN expression. Consistent with the experimental results (Figures 1B and S5), a 4-fold increase of activator degradation rate is sufficient to abolish the spots in the simulation (Figures 6C, 6I–6L). Similarly, increasing the baseline production rate of the inhibitor RTO (I0), akin to the over-expression of RTO, resulted in the loss of spots (Figures 6D, 6M–6P). Finally, gradually increasing the degradation rate of the inhibitor RTO (UI), mimicking the different degrees of RTO knock-down by RNAi, produced patterns resembling the actual RNAi phenotypes in M. lewisii (Figures 6E–6H). Notably, the seemingly counterintuitive experimental observation that RTO transcript level did not decrease in the RTO RNAi lines (Figures 3E and 3F) is actually predicted by the reaction kinetics of the Gierer-Meinhardt model. Computer simulations showed that increase in inhibitor degradation rate resulted in not only higher average activator level, as expected, but also slightly higher average inhibitor level (Figure S7A). Simulations with another commonly used activator-inhibitor RD model [20,50], as implemented in the software simRDj [12], also generated similar patterns (Figure S7B). Taken together, these simulation results further suggest that the anthocyanin spot patterning in Mimulus is controlled by a simple two-component activator-inhibitor system.
Figure 6. Computer simulation of the anthocyanin spot patterning.

(A) Schematic illustration of a two-component activator-inhibitor model, modified from ref. 18. A: activator; I: inhibitor; DA and DI: diffusion coefficient of the activator and inhibitor, respectively; UA and UI: degradation rates; A0 and I0: background production rates; GA and GI: potency of the activation by the activator. The specific equations are described in the Methods. (B) Simulated pattern resembling the wild-type M. lewisii, with the following parameter values: DA = 0.01; DI = 0.5; UA = 0.03; UI = 0.03; A0 = 0.01; I0 = 0; GA = 0.08; GI = 0.12; (C-P) Simulated patterns that mimic NEGAN RNAi (C and I–L), RTO over-expression (D and M–P), and RTO RNAi lines (E–H). All parameter values are the same as in the wild-type condition (B), except one modification for each perturbation as shown on the lower left corner of each panel. Real flower images of the center of the nectar guides from the corresponding lines are shown on the lower right corner of panel B–H. See also Figure S7.
Alleles disrupting MgRTO occur at non-trivial frequencies in natural populations and impact bumblebee attraction
Having demonstrated that NEGAN and RTO function as an activator-inhibitor pair that pattern anthocyanin spots in the nectar guides, we next explored how variants that alter the NEGAN-RTO interaction dynamics are distributed in natural populations of M. guttatus (Figures 1F–1J) and their potential impacts on plant performance. In two of three population surveys for SWC and LRD, we found that M. guttatus rto-like alleles segregate at low but non-trivial frequencies in the wild (2011 survey: rtoSWC frequency = 0.1, n=10 maternal individuals; 2017 surveys: rtoLRD frequency = 0.02, n=51 and rtoSWC frequency = 0, n=49). Moreover, rto-like plants have been seen in at least two additional populations, each >200km from either SWC or LRD (Figure 1F). In one of these populations, rto-like plants carry an 8-bp deletion that removes a splice acceptor site and causes a frameshift at the start of the second exon of MgRTO (Figure 2D), and we estimate this allele is present at a frequency of ~0.11 in the population based on phenotype (2018 survey: n=200 individuals). In a second population, rto-like plants have a ~3kb insertion that disrupts the second exon and is present at a frequency of ~0.07 (2018 survey: n=15 maternal individuals).
The non-trivial frequencies of rto-like alleles in several M. guttatus populations and the fact that these anthocyanin spots serve as important nectar guides in the closely related M. luteus [32] motivated us to test whether natural variation in the anthocyanin spot patterns could impact pollinator visitation. We carried out a series of bumblebee attraction experiments in a laboratory setting using Bombus impatiens and using the rtoSWC allele since the heterozygous and homozygous genotypes form a broad continuum of spot expansion (Figure 1G). Although this commercially obtainable species is not native to M. guttatus habitats, B. impatiens worker bees are comparable in size to those of native congeners (e.g., B. vosnesenskii and B. californicus), pollinate M. guttatus effectively, and recognize variation in and exert selection on M. guttatus floral characters [51–56]. In a direct comparison between genotypes, naïve bumblebees preferentially visited rtoSWC homozygote flowers over wild-type flowers, and bumblebees also preferred heterozygote to wild-type flowers (Tables 1 and S1–S3). Although we detected significant heterogeneity in attraction among trials for both genotypic contrasts, all trials where one genotype was visited preferentially showed that the rtoSWC containing genotype was favored. This heterogeneity may indicate that individual variation in preference segregates within bumblebee populations. Bees did not discriminate between heterozygote flowers and rtoSWC homozygote flowers, however. Given that corolla UV pigmentation and cell shape do not differ between full-sib RTOSWC homozygous vs. rtoSWC carrier flowers (Figures S1B–S1D), we conclude that altered anthocyanin pigmentation pattern is responsible for preferred visitation to rtoSWC homozygotes and RTOSWC/rtoSWC heterozygotes. Determining whether the rto-like alleles represent nascent but incomplete selective sweeps due to positive impacts on pollination or other functions, are drifting neutrally, or are polymorphisms maintained by frequency-dependent selection or at mutation-selection balance due to costs (e.g., florivore attraction) will require further study in field conditions, particularly as pollinator learning can diminish the influence of innate preferences and thus how the polymorphism impacts individual fitness in natural populations. Nonetheless, these findings demonstrate that in principle, changes in nectar guide patterning that arise by modulating the underlying patterning system can influence fundamental plant-pollinator interactions.
Table 1. Anthocyanin spot patterning affects bumblebee visitation.
Pooled goodness-of-fit test over ten trials per pairwise RTO genotype combination. Significant heterogeneity G-values indicate variation in bee preference among individual trials; nonetheless, in all cases, the effects of genotype in significant individual trials were in the same direction as the pooled test. See also Figure S1 and Tables S1–S3.
| Genotype | Observed Visits | Pooled G-value | d.f. | P | Heterogeneity G-value | d.f. | P |
|---|---|---|---|---|---|---|---|
| RTO/RTO | 246 | 30.99 | 1 | <0.001 | 83.34 | 9 | <0.001 |
| rto/rto | 393 | ||||||
| RTO/RTO | 450 | 17.92 | 1 | <0.001 | 37.01 | 9 | <0.001 |
| RTO/rto | 566 | ||||||
| RTO/rto | 404 | 1.09 | 1 | 0.3 | 14.18 | 9 | 0.12 |
| rto/rto | 420 |
Discussion
In this study, we have shown that a pair of MYB proteins functions as a simple activator-inhibitor system underlying the formation of the dispersed anthocyanin spots that dot the nectar guides of Mimulus flowers. The two-component activator-inhibitor model [12,17,18,20] predicts that the activator self-activates and activates the inhibitor; the inhibitor represses the activator function; and the inhibitor can move from cell to cell. Our experimental evidence is fully consistent with these theoretical predictions. First, the R2R3-MYB activator NEGAN activates its own expression [40]. Second, NEGAN also activates the expression of the R3-MYB inhibitor RTO; when NEGAN or its interacting partner WD40a is knocked down/out by RNAi or chemical mutagenesis, RTO expression is also reduced (Figures 2E and S5). Third, the inhibitor RTO represses the activity of NEGAN. When RTO function is impaired by RNAi or mutations, NEGAN expression level increases (Figures 2E, 2F, and 3E–3G), and when RTO is overexpressed, NEGAN expression is abolished (Figure 3D). Finally, RTO shows intercellular movement, as demonstrated by comparing the spatial pattern of RTO transcription with the RTO protein distribution (Figures 5B, S6D and S6E). To our best knowledge, these findings are the first to reveal a developmental mechanism that generates dispersed pigment spots in plants. In addition, through genetic mapping and knockdown or targeted knockout experiments of MgRTO, our work represents the first instance in which the identity and molecular impact of a specific causal variant underlying natural phenotypic variation have been confirmed by functional perturbation in the evolutionary model system M. guttatus, and the first successful instance of gene editing in the genus Mimulus as a whole.
Computer simulations based on simple activator-inhibitor models [12,17,18,20] provided further support that NEGAN and RTO correspond to an activator-inhibitor pair in a two-component RD system. Gradual increase of the activator degradation rate (UA), inhibitor degradation rate (UI), and background inhibitor production rate (I0) in simulations can recapitulate patterns generated by NEGAN RNAi, RTO RNAi, and RTO over-expression, respectively (Figures 6 and S7B). Furthermore, the simulation results demonstrated that the seemingly surprising observation that RTO transcript level did not decrease in the RTO RNAi lines (Figures 3E and 3F) is actually a predictable outcome of the activator-inhibitor reaction kinetics (Figure S7A). These mathematical models will serve as an important guide for future studies that aim to experimentally determine the relative diffusion rates, molecular half-lives, and potency of activation and inhibition of this protein pair. By extending the tools for studying gene and protein function we have newly applied in Mimulus and adopting additional technologies (e.g., light sheet fluorescent microscopy), we anticipate that these difficult tasks will be achievable.
Although our results strongly suggest that the RD mechanism plays a critical role in the formation of dispersed anthocyanin spots in Mimulus, we cannot rule out the possibility that other mechanisms may also contribute to the pattern formation in this system. An alternative mechanism often invoked to explain spatial patterning during development is the “positional information” (PI) model [14,57–59]. In contrast to the self-organization nature of the RD model, the PI model involves fine orchestration of a hierarchical genetic cascade during development that leads to gradual refinement of discrete boundaries within tissues. The source of the positional information is often associated with anatomical landmarks (e.g., veins of the butterfly wing or flower petals). While historically the PI model and RD model were usually viewed as mutually exclusive [14,58], recent studies suggest that these two mechanisms may act together in pattern formation [25,27,57]. The restriction of spot formation and NEGAN expression to the ventral (lower) petal in Mimulus indicates the existence of upstream positional information that sets the initial conditions for the activator. Future experiments seeking to identify upstream regulators of NEGAN will be necessary to elucidate the putative positional information.
We have shown that an array of anthocyanin spot patterns can be generated by modulating the interaction of a local activator and a lateral inhibitor through experimental manipulations or natural mutations, and altering the spot patterns can impact bumblebee visitation. Thus, subtle changes in this simple interacting network are likely essential contributors to the remarkable diversification of pigmentation patterns in nature, particularly when overlaid on different positional information that imposes unique initial conditions or boundary conditions for the formation of the self-organized patterns. For instance, many Mimulus and other angiosperm species display spotted, blotched, or striped anthocyanin patterns in their nectar guides that may influence interactions with pollinator communities, potentially facilitating reproductive isolation and speciation [7,32,33,60]. A similar R2R3- and R3-MYB pair has been implicated in the periodic patterning of leaf trichomes and root hairs in Arabidopsis [23,61–63], although the R2R3-MYB in that case does not self-activate. The evolution of novel R3-MYB repressors through duplication then degeneration of R2R3-MYB sequences also has been observed [64]. Thus, the origin of two-component RD systems involving an R3-MYB inhibitor that is evolutionarily derived from the R2R3-MYB activator may be a particularly common route to the evolution of pigmentation patterns in flowering plants. We expect that molecular circuits with similar properties do explain various pigmentation patterns in other systems as first envisioned by Turing [16] and Gierer and Meinhardt [17] several decades ago.
STAR ★ METHODS
Lead Contact and Materials Availability
Further information and requests for M. lewisii and M. guttatus resources should be directed to and will be fulfilled by Yao-Wu Yuan (yaowu.yuan@uconn.edu) and Benjamin K. Blackman (bkblackman@berkeley.edu), respectively.
Experimental Model and Subject Details
Plants
Mimulus lewisii plants were grown in the University of Connecticut research greenhouses under natural light supplemented with sodium vapor lamps, ensuring a 16h day length. EMS mutagenesis was performed using the Mimulus lewisii Pursh inbred line LF10 as previously described [65].
Mimulus guttatus individuals with fully or partially expanded nectar guide spots were observed in the selfed offspring of wild plants collected along Sweet Creek Road in Mapleton, OR (SWC; 44°00’51”N, 123°51’36”W) and along Emma Road east of Cheeseboro Road near Littlerock Dam (LRD; 34°30’23”N, 118°01’39”W). SWC plants, F1s, F2s, and 2011 maternal families were grown in Fafard 4P soil in 2.5 square-inch pots in the Duke University greenhouses (18h 21°C days : 6h 18°C nights). After cold stratification (7d at 4°C), plants were misted and bottom watered twice daily on flood benches for 10d and then just bottom watered for the remainder of the experiment. LRD plants, F1s, F2s, and the LRD and SWC 2017 maternal families were grown in Sunshine Mix #1 in 2.5 square-inch pots in the UC Berkeley Oxford Tract Facility greenhouses (16h 24°C : 8h 14°C). Seeds were cold stratified (7d at 4°C), then watered on a mist bench for two weeks as they germinated, and then moved to a standard bench and bottom-watered daily. For both mapping populations, all pots were thinned to one plant per pot. Once plants began flowering, we recorded nectar guide phenotypes and flash froze bud tissue, which was maintained at −80°C until DNA extraction for fine mapping. We also observed rto-like plants in the field or in families grown in the greenhouse from field-collected seed for M. guttatus populations at Quarry Valley Road in the Donald and Sylvia McLaughlin Natural Reserve (QVR; 38°51’37”N, 122°25’15”W) and on CA-3 at Trinity Alps Road (TAR; 40°51’09”N, 122°51’56”W).
Animals
A Natupol hive of Bombus impatiens was purchased from Koppert Biological Systems (Howell, MI). To conduct pollinator choice trials, we constructed a 3’ × 4’ × 2’ flight cage underneath four Reptisun fluorescent bulbs that provided visible and UV light. We connected the hive to the flight cage with a small tube and a series of gates, which allowed us to let an individual bee into the flight cage.
Methods details
Bulked Segregant Analysis in Mimulus lewisii
To identify the causal gene underlying rto, we performed bulked segregant analysis following Yuan et al. [37]. Briefly, the homozygous rto mutant (in the LF10 background) was crossed with and the mapping line SL9. A single F1 individual was then selfed to generate the F2 mapping population (n = 468). DNA samples from 100 F2 segregants displaying the mutant phenotype were pooled with equal representation. A whole genome sequencing library (insert size of ~320 bp) was prepared for the pooled sample, and 100-bp paired-end reads were generated by an Illumina HiSeq 2000 (Illumina, San Diego, CA, USA). The resulting ~196 million short reads were mapped to the SL9 pseudoscaffolds, and the resulting raw SNPs were filtered by depth of coverage, tendency of clustering, and variant frequency. The SL9 pseudoscaffolds were then scanned for regions enriched with homozygous SNPs to determine the candidate gene interval (Figure 2A).
Bulked Segregant Analyses in Mimulus guttatus
For SWC, an individual with a large anthocyanin patch was crossed to an individual with typical nectar guide patterning descending from a maternal family that bred true for that phenotype. A single F1 individual was selfed to generate the F2 mapping population (n = 452 individuals). Similarly, we crossed an LRD individual with expanded anthocyanin spots to a wild-type individual from another maternal line and selfed an F1 descendant to generate an F2 mapping population (n = 631).
For bulked segregant analysis of each mapping panel, we randomly selected 100 wild-type and 100 rto-like plants derived from the corresponding F2 mapping population, and we then collected three equal-sized buds from each plant into one of four 15 mL tubes (25 plants per tube) for each phenotype. We extracted DNA using a modified CTAB protocol [66], including a final cleanup with 2M NaCl, 4% PEG solution. The resulting DNA was quantified using a Quant-iT dsDNA Broad Range Assay Kit (ThermoFisher), and 250 ng from each of the four tubes was pooled for a total of 1 μg per phenotype in 50 μL water. The pooled samples were sonicated for 65s using a Covaris M220 in order to retrieve ~350-bp fragments. AMPure Purification beads were used to remove fragments smaller than 200 bp, and remaining fragments were end-repaired and A-tailed before adaptor ligation using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs). For adaptors, we used the Multiplex Oligos for Illumina Kit (New England Biolabs), and, once indexed, we again size selected for 200–400-bp fragments by bead purification. The libraries were enriched using the Phusion High Fidelity PCR Kit (New England Biolabs), and bead purified once more to remove the smallest fragments. The final whole genome sequencing libraries were run on an Agilent Bioanalyzer to determine fragment size and concentration and then combined in equimolar concentration into one sample. Samples were submitted either to the Institute for Integrative Genome Biology Genomics Core at University of California, Riverside for one lane of 100SR sequencing on an Illumina HiSeq2500 platform or to the QB3 Vincent J. Coates Genome Sequencing Laboratory at University of California, Berkeley for one lane of 100SR sequencing on an Illumina HiSeq4000 platform.
The LRD wild-type and rto-like pools yielded ~184 million and ~172 million reads, respectively. Because the initial sequence data obtained for the two pooled SWC libraries yielded a low number of assignable reads, we constructed new libraries from two pools of individual DNAs from 100 wild-type and from 100 rto-like phenotype plants. Library construction and sequencing were completed as above. Data from both SWC sequencing runs were then combined for analysis (~59 and ~76 million total reads for the wild-type and rto-like pools, respectively).
Fine Mapping of rtoSWC in Mimulus guttatus
We identified recombinants in the candidate genomic interval resulted from the bulked segregant analysis by first genotyping all individuals in the SWC F2 population for four EPIC (exon-primed, intron-capture) markers spanning the region through fragment analysis on the University of Virginia Department of Biology Genomics Core Applied Biosystems 3130 instrument. We then developed additional microsatellite or CAPS (cleaved, amplified polymorphic sequence) markers from polymorphisms distinguishing the parental alleles using the bulked segregant re-sequencing data. These markers were selectively scored on the recombinant individuals using the ABI 3130 or by gel electrophoresis as the region containing the causal locus was iteratively refined. Finally, we confirmed that pigmentation phenotype perfectly co-segregated with the rtoSWC frameshift mutation (Figure 2D) or a 723-bp insertion in the RTO 3’UTR in the SWC and LRD panels, respectively, by gel electrophoresis. Primer sequences for all markers are listed in Table S4.
Plasmid Construction
RNAi plasmids:
We built RTO RNAi constructs by cloning fragments of the M. lewisii or M. guttatus RTO gene into the pFGC5941 vector ([67]; ABRC stock no. CD3–447) in both the sense and antisense directions separated by an intronic spacer. This fragment, and every 12-bp block within it, matched only a single region of the M. lewisii or M. guttatus gene (100% identity), indicating target specificity. Likewise, we built MgNEGAN RNAi constructs following the same procedure.
CRISPR-Cas9 plasmid:
We also undertook a targeted genome editing approach using the Cas9-sgRNA system to recapitulate the RTO loss of function phenotype in M. guttatus. The synthetic guide-RNA (sgRNA) comprised an optimized trans-activating CRISPR RNA (tracrRNA) and a CRISPR RNA (crRNA) that contains a 20-bp guide proximal to an NGG-site recognized by S. pyogenes Cas9. Several sgRNA guides were designed in silico (http://www.e-crisp.org/E-CRISP/) to target an exonic region of MgRTO. The 20-bp guide sequences were verified for its specificity by using a blast algorithm to scan the M. guttatus v2.0 reference genome (phytozome.jgi.doe.gov). The guides were pre-tested for their cleavage efficiency with an in vitro assay system using S. pyogenes Cas9 nuclease (New England Biolabs) to edit a PCR-amplified double-stranded DNA fragment of RTO. A binary plasmid construct expressing plant-codon-optimized Cas9 (pFGC-pcoCas9; Addgene#52256) was modified to express a RTO sgRNA (TTTCGAATGCACAAGCTCGT) that was successful in vitro. We synthesized the cassette of AtU6-polymerase III promoter driving sgRNA and poly-T terminator sequences by PCR. The AtU6-polymerase promoter was amplified from a plasmid pUC119-gRNA (Addgene: 52255). The tracrRNA and crRNA sequence with the poly-T terminator was amplified from plasmid AS-121P-CR3 (provided by A. Schultink, Integrative Genomic Institute, UC Berkeley). The cassette (AtU6p:sgRNA:terminator) for the expression of sgRNA was inserted into plasmid pFGC-pcoCas9 using AscI and PacI restriction sites. Insertion of the sgRNA cassette was confirmed by Sanger sequencing.
RTOpro:
CFP-ER and RTOpro:YFP-RTO plasmids:
To monitor the spatial expression pattern of RTO in the wild-type M. lewisii (LF10), we constructed a plasmid with the RTO promoter (a ~1.7-kb DNA fragment upstream of the translation initiation site) driving CFP with an ER retention signal peptide. To this end, we first cloned the promoter fragment into the pMCS:GW vector ([68]; ABRC stock no. CD3–1933) using the AatII and XhoI restriction enzyme sites and obtained an intermediate vector RTOpro:GW. We then amplified the CFP-ER fragment from the vector ER-cb ([69]; ABRC stock no. CD3–954) and recombined it into the intermediate vector by LR recombination, resulting in the final plasmid RTOpro:CFP-ER. To monitor the RTO protein distribution, we constructed the RTOpro:YFP-RTO plasmid using a similar approach. We first cloned the same promoter fragment into pMCS:YFP-GW (ABRC stock no. CD3–1934) and then recombined the RTO CDS into the intermediate vector through LR recombination, resulting in the final RTOpro:YFP-RTO plasmid.
35S:
RTO and 35S:YFP-RTO plasmids:
To generate the 35S:RTO and 35S:YFP-RTO plasmids, we first amplified the full-length CDS of RTO from the wild-type M. lewisii (LF10) cDNA using the Phusion enzyme (NEB, Ipswich, MA). The amplified fragment was cloned into the pENTR/D-TOPO vector (Invitrogen), and then a linear fragment containing the CDS flanked by the attL1 and attL2 sites was amplified using M13 primers. This linear fragment was subsequently recombined into the Gateway vector pEarleyGate 100 ([70]; ABRC stock no. CD3–724) or pEarleyGate 104 (CD3–686), to generate the final 35S:RTO and 35S:YFP-RTO plasmids, respectively.
BiFC plasmids:
To test the interaction between the wild-type RTO or mutant rto protein with ANbHLH1, bimolecular fluorescence complementation (BiFC) plasmids were constructed by recombining the ANbHLH1 CDS into the destination vector pUBC-nYFP-Dest and the RTO or rto CDS into pUBC-cYFP-Dest [71], following the same procedure as described above for constructing the 35S:RTO plasmid.
Primers used for plasmid constructions are listed in Table S5.
Plant Transformation
Mimulus lewisii transformation followed Yuan et al. [42]. For M. guttatus, we introduced the RTO and NEGAN RNAi constructs and the RTO targeting Cas9-sgRNA construct into the plant through Agrobacterium mediated transformation of callus tissue in tissue culture. To this end, we first transformed the RNAi or Cas9-sgRNA construct into Agrobacterium tumefaciens strain EHA105. Overnight cultures in yeast extract peptone medium supplemented with kanamycin (50 mg/L) and rifampicin (40 mg/L) were resuspended to OD600 = 0.2 with half-strength MS salts supplemented with acetosyringone (200 μM). The resuspended cultures were shaken at 50 rpm in darkness for 4h for virulence induction.
All plant cultures were grown on MS growth medium (Murashige and Skoog salts with Gamborg B5 vitamins, 2% sucrose, 1.3 g/L calcium gluconate, 0.25 g/L 2-N-morpholino ethane sulfonic acid, and 0.25% gelrite), pH 5.6. Plant cultures were incubated under fluorescent lights (200 μmol/m2/s) on 14h light : 10h dark photoperiod, at 21 ± 1°C during transformation and tissue culture. We excised fully expanded leaves with petioles intact from M. guttatus plants, grown in vitro from seeds from inbred lines of the MAC population (Mackville Road, 1–2 mi N of Liberty Road, Sacramento County, CA; 38°14’56”N, 121°05’26”W). Petioles were dipped in the Agrobacterium cultures and co-cultivated on MS growth medium supplemented with 1 mg/L N-(2-Chloro-4-pyridyl)-N′-phenylurea (CPPU) and 100 μM acetosyringone. After 3d of co-cultivation in darkness, the explants were washed with 100 mg/L timentin and 50 mg/L cefotaxime and maintained on the callus induction MS growth medium supplemented with 1 mg/L CPPU, antibiotics, and 6 mg/L phosphinothricin (PPT). We sub-cultured callus tissue that survived selection for 25–28 days, and then transferred callus exhibiting shoot organogenesis to shoot induction MS growth medium supplemented with 0.1 mg/L meta-topolin. We further sub-cultured shoots >1 cm long on half-strength MS growth medium supplemented with 0.1 mg/L 1-napthalene acetic acid and PPT. Rooted plantlets were transplanted to pots filled with soil-less potting medium and hardened on a mist-bench in the green house for four weeks. We confirmed RTO and NEGAN RNAi lines by genotyping and RT-PCR in at least 10 T0 plants per construct and then selfed these individuals to generate T1 full-sib families for phenotyping and expression analyses. For the RTO targeting Cas9-sgRNA experiments, transgenic T0 and T1 plants showing phenotypes resembling RTO/rto heterozygotes were genotyped by Sanger sequencing to verify the mutations in sgRNA target region.
Expression Analysis by RT-qPCR
For M. lewisii, total RNA was isolated from whole corolla or dissected petal lobes and nectar guides of 10 mm flower buds (unless noted otherwise) using the Spectrum Plant Total RNA Kit (Sigma-Aldrich). For each sample we treated 1 μg of total RNA with amplification grade DNaseI (Invitrogen) before cDNA synthesis using the SuperScript III First-Strand Synthesis System (Invitrogen). cDNA samples were diluted 10-fold before quantitative reverse transcriptase PCR (qRT-PCR). All qRT-PCRs were performed using iQ SYBR Green Supermix (Bio-Rad) in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Samples were amplified for 40 cycles of 95°C for 15s and 60°C for 30s. Amplification efficiencies for each primer pair (Table S6) were determined using critical threshold values obtained from a dilution series (1:4, 1:8, 1:16, 1:32) of pooled cDNA. MlUBC was used as a reference gene to normalize expression levels following the delta-delta Ct method.
For M. guttatus, lower lobes of buds measuring approximately 2–3mm (SWC and MAC populations) and 4–5mm (LRD population) were harvested under magnification lenses (Figure S4E). Pools of tissue from least 12–14 buds per each of four biological replicates were frozen in liquid nitrogen and stored in −80°C until processing for RNA extraction. Total RNA was extracted using the Zymo Research Direct-Zol RNA kit and DNase treated with the Zymo Research DNase I Set following the manufacturer protocols. First strand cDNA was synthesized using 500ng of RNA with oligo d(T)15 primer using GoScript™ Reverse Transcriptase System manufacturer kit (Promega Corp. Madison, WI, USA) and diluted 4-fold before qPCR. Three technical replicates per qPCR amplification were conducted in 10μL reactions using PowerUp™ SYBR Green Master Mix (Applied Biosystems). All reactions were run on a 7500 Fast Real-Time PCR System (Applied Biosystems) for 40 cycles of 95°C for 3s and 60°C for 30s with initial UDG activation at 50°C for 2min and polymerase activation at 95°C for 2 min. Amplification efficiencies for each primer pair (Table S6) were determined using critical threshold values obtained from a dilution series (1, 1:2, 1:4, 1:16, 1:64, 1:256) of pooled cDNA. The specificity of each primer pair for its target gene was verified visually by the melt curves obtained with default instrument settings and by Sanger sequencing. The mean Ct values for the three technical replicates per sample-target combination were used to calculate relative expression values. MgUBQ was used as a reference gene to normalize expression levels following the delta-delta Ct method.
BiFC Assay
We transformed the pUBC:ANbHLH1-nYFP, pUBC:RTO-cYFP, and pUBC:rto-cYFP plasmids separately into Agrobacterium strain GV3101. Equal volumes of pUBC:ANbHLH1-nYFP and pUBC:RTO-cYFP cultures or pUBC:ANbHLH1-nYFP and pUBC:rto-cYFP cultures were infiltrated into the abaxial side of Nicotiana benthamiana leaves, following the procedure described previously [72]. The YFP signal was imaged 4–6 days after agroinfiltration. To ensure that the constructs are properly expressed in the infiltrated tobacco leaf tissue, we excised leaf tissue from half of the infiltrated area and stored it in liquid nitrogen until RNA extraction, and we excised the other half for fluorescence imaging. Relative expression levels of ANbHLH1 and RTO were assayed with semi-quantitative RT-PCR. The Nicotiana benthamiana endogenous ACTIN gene was used as the reference gene.
Fluorescence Imaging
For the RTOpro:CFP-ER lines, ventral petals (dissected from 10 mm corolla) were imaged using a Zeiss Axioskop2 microscope with a 5X objective (FLUAR 5X/0.25) under the bright-field for anthocyanin spots (Figure 5A; left) and green channel for CFP signal (Figure 5A; center).
For the F1 hybrids with both RTOpro:CFP-ER and RTOpro:YFP-RTO transgenes, a NIKON A1R confocal laser scanning microscope with a 10X objective was used to image CFP and YFP signals with the excitation wavelength of 457 nm and 514 nm, respectively (Figures 5B, S6D, S6E).
For the BiFC assays, A NIKON A1R confocal laser scanning microscope equipped with a 60X water immersion objective (Plan APO VC 60X/1.20) was used to acquire the images (Figures 3H and S6A) under the transmitted light and the green channel (excitation wavelength 514 nm).
To track the intracellular movement of the YFP-RTO fusion proteins in the 35S:YFP-RTO lines, YFP-RTO fluorescence signals were captured automatically every 30 s within a time span of 15 min using the same confocal laser scanning microscope with the green channel (Figure 5C and Video S1).
UV Photography
We imaged UV-absorbent nectar guides using a Canon T2i Rebel camera converted by LifePixel (Mukilteo, WA) to replace the internal hot mirror filter with a high quality custom manufactured UV band pass filter.
Scanning Electron Microscopy
We dissected fully opened M. guttatus flowers to retain only the ventral petal, fixed tissue with glutaraldehyde, and dehydrated the fixed samples through a graded ethanol series. Following dehydration, the samples were further trimmed to retain the pigmented area of the petal to fit the mounting space, and imaging was completed using the Zeiss Sigma VP HD Field SEM instrument at the University of Virginia School of Medicine Advance Microscope Facility.
Mathematical Modeling
The co-localization of RTO (inhibitor) promoter activity with the anthocyanin spots (Figure 5A) suggests that the activator and the inhibitor have expression patterns that are in-phase, consistent with one of the two simplest Gierer-Meinhardt topologies — the two-component activator-inhibitor model [17,18]. Therefore, we implemented a modified Gierer-Meinhardt model (based on equation 2.1 in [18]) between an autocatalytic activator A (NEGAN) and a rapidly diffusing inhibitor I (RTO) (Figure 6A). A diffuses with a diffusion coefficient DA, degrades with a rate UA. A has a background production rate A0 and is self-activated with a potency GA. The activator production is slowed down by the inhibitor I. A residual value (k = 0.001) was added to the denominator to avoid the situation where the denominator becomes zero.
Similarly, I diffuses with a diffusion coefficient DI, degrades with a rate UI, has a background production rate I0 and is activated by the activator with a potency GI.
Computer Simulations
We implemented the modified Gierer-Meinhardt model in the Virtual Cell modeling and simulation platform [48,73] and performed a number of two-dimensional numerical simulations to explore the pattern formation for different transgenic lines. This required integration of the partial differential equations described above. We used fully implicit, adaptive time step finite volume method. The finite volume method represents partial differential equations as algebraic discretization equations which exactly preserves conservation laws. Values are calculated at discrete places on a meshed geometry. This method employs Sundials stiff solver CVODE for time stepping. 2D discretization is the only source of solution error. According to this error, the time-step is resized during the simulation. All simulations were conducted with a domain size of 100 × 100, a spatial step size of 1.0, an activator initiation concentration of 1.0 randomly distributed across the whole domain, an inhibitor initiation concentration of 0.0, and a zero flux boundary condition.
For simulating patterns resembling the wild-type M. lewisii, we used the following parameter values: DA = 0.01, DI = 0.5, UA = 0.03, UI = 0.03, GA = 0.08, GI = 0.12, A0 = 0.01, and I0 = 0 (Figure 6B). These parameter values were largely adapted from previous studies employing similar two-component activator-inhibitor models [12,20,49,50], and were considered biologically realistic. We then investigated additional cases corresponding to three different types of transgenic manipulations. All parameter values used in these cases were the same as those in the simulation for the wild-type pattern, except one modification for each perturbation, as follows:
NEGAN RNAi, with gradually increased activator degradation rate (UA = 0.06, 0.09, 0.1, 0.11, 0.12, and 0.15; Figure 6C and Figures 6I–L).
RTO over-expression, with gradually increased background production rate for the inhibitor (I0 = 0.01, 0.05, 0.06, 0.07, 0.08, and 0.1; Figure 6D and Figures 6M–P).
RTO RNAi, with gradually increased inhibitor degradation rate (UA = 0.09, 0.15, 0.18, and 0.19; Figures 6E–H).
We used the activator level (A) as the pattern readout [12,20,49,50], and set a threshold value for pattern to appear, with the assumption that the activator should be above a certain threshold level for pigment production. All patterns shown in Figure 6 are based on the threshold value 3.0.
The Virtual Cell model is available in the VCell software (http://vcell.org). After logging in as a guest, search for “Monkeyflower_pigmentation_v2” in VCellDB -> BioModels panel in the left bottom part of the VCell interface. Double click on the model name in the bottom left panel (under Public BioModels) to launch it. Simulation results are available by double clicking on “Pattern_formation” under Applications in the left column and selection of the Simulations tab. To retrieve the simulation results, select the name of a simulation (e.g., WT, RTO_RNAi_0.06), and click the “Native Simulation Results” icon. To view patterns, select “Pattern_threshold_3.0” under “Variables” on the left, and move time slider to see pattern development over time. Similarly, one can see individual variables (e.g., A, I) changes over time. To match simulation results with a pattern visible in plants, we exported VCell images in monochrome Nearly Raw Raster Data (.*nrrd) format into ImageJ, and applied LookUp Table with initial entry yellow (255,255,0) and final entry red (255,0,0).
Besides the VCell simulations, we also performed simulations with another commonly used two-component, activator-inhibitor model ([20]; Figure S7B), as implemented in the java program RDsimJ.jar [12]. The parameter values used to simulate the wild-type (WT) pattern are shown in the screenshot of the graphic interface of RDsimJ.jar (Figure S7). The patterns corresponding to the three types of transgenic manipulations (i.e., NEGAN RNAi, RTO over-expression, and RTO RNAi) are shown in Figure S7B, with the single altered parameter value (compared to the WT values) shown below each simulated pattern.
Allele Frequency Surveys
Mature fruits were collected from 51 maternal plants at the LRD population in May 2017, from 10 maternal plants at the SWC population in June 2011, and from 48 maternal plants at the SWC population in June 2017. Three to seventeen (2011 survey) or sixteen (both 2017 surveys) progeny from each maternal family were then raised in the Duke University greenhouses or the UC Berkeley Oxford Tract Facility as described above. Heterozygosity for the premature stop mutation was confirmed for individuals with nectar guide phenotypes resembling the RTOLRD/rtoLRD heterozygous phenotype (Figure 1) by amplifying, cloning, and Sanger sequencing the PCR products generated with the Migut.B01849 fine mapping primers (Table S4). The QVR population (Quarry Valley Road, Donald and Sylvia McLaughlin Natural Reserve) was phenotypically surveyed in the field. Counts of wild-type, intermediate, and rto-like plants were tallied in 8 patches along a transect through the population (25 randomly selected plants per patch, n = 200 individuals total). We confirmed by Sanger sequencing that rto-like plants (n=4) carry a QVR-specific 8-bp deletion in RTO not observed in the wild-type.
Pollinator Preference Experiments
To determine whether bumblebees preferred flowers of rtoSWC variant carriers to flowers with wild-type nectar guides, we conducted pollinator cage experiments with a commercially obtained hive of Bombus impatiens. Before adding plants to the cage, we counted the total number of open flowers on each plant. We then placed six plants from the SWC F2 mapping population – three of one RTO genotype and three of another RTO genotype – in a regularly-spaced checkerboard pattern based on phenotype such that each plant’s direct neighbors were of a different genotype. We conducted 10 independent trials for each pairwise combination of RTOSWC genotypes.
For each trial, we allowed one bee to enter and forage for 20 minutes. Any bee that did not start foraging within five minutes was removed. To ensure all bees were naïve, any bee which entered the cage, whether or not they successfully foraged, was marked upon removal and returned to the hive. We did not collect any further foraging data from marked bees. If a bee did start foraging, we recorded each visit with the plant that was visited, the time of landing on a flower, the orientation of the visit, and the time of departure. After each successful trial, we replaced the plants. The effect of RTO genotype on pollinator visits was analyzed using G-tests of goodness of fit [74], conducting separate analyses for each pairwise combination of RTOSWC genotypes. Expected visit values were calculated based on the proportion of open flowers of each genotype included in the pollinator cage for each trial.
Quantification and Statistical Analysis
Quantification of Anthocyanin Spots
We quantified the anthocyanin spots (number of spots and relative area in the nectar guides) for both species, including the various mutants and transgenic lines (Figure S2). For M. lewisii, the ventral (lower) petals were scanned using an Epson WorkForce Pro WP-4530 printer. The spots were manually counted for 10 flowers per line from the scanned images with ImageJ. To quantify the relative area of anthocyanin spots in the nectar guides (i.e., the ratio of the areas pigmented by anthocyanin to the total area of the yellow background), we analyzed five scanned flower images for each line. A customized Python script with OpenCV tools was used to perform the analysis. Briefly, we first used the “cv2.findContours” function to isolate nectar guide area from the rest of flower petal images based on color contrast. Then we used “cv2.findContours” again with different argument settings to separate red spots from the yellow background. We also calculated nectar guide area and spots area with the “cv2.contourArea” function.
For M. guttatus, the ventral petals were stretched out to remove the creases, and a transparent tape was used to fix the petal on a sheet of white color paper. Color photographs of the petal were taken along with a ruler to calibrate area estimates. The calibrating ruler was placed close to the petal to minimize errors caused by the distortion of the camera lens. A Canon Rebel T2i digital single-lens reflex (DSLR) camera mounted on a tripod was used for photography. The pictures were imported to ImageJ2, and the scale of the image was set using the metrics on the ruler to obtain area estimates in square centimeters. The free hand tool was used to trace along the petal to measure the area of the whole petal, and individual spots were outlined in a similar approach to measure the area covered by the spots. The ratio of the total area of spots to the whole ventral petal area was calculated. The number of anthocyanin spots was manually counted from each picture. Ten to thirteen plants per genotype were sampled.
Bulked Segregant Analyses for M. guttatus
For each mapping population, sequencing data for each pool were aligned to the M. guttatus v2.0 reference genome (phytozome.jgi.doe.gov) with the aln and samse commands in BWA [75] and converted to sorted BAM files with samtools [76]. A merged BAM file was then converted to mpileup format, and a list of SNPs was called using a custom script that removed indel variants, sites covered by <4 reads or covered by >200 reads, sites that segregated for >2 alleles, and sites where one or both alleles were supported by <2 reads with Illumina base quality scores <4. Pileup files were then generated from the sorted BAM files for each pool, and these were then parsed with a custom script and the SNP list to yield a table listing the allele counts and frequencies for each bulk and the allele frequency difference between the two bulks at each SNP site. SNP sites were filtered for those where each bulk had >10× read coverage and the total read coverage was ≤160×. For the SWC panel, the proportion of SNPs with fixed differences between the two bulk pools was calculated for 50-kb intervals (median = 94 SNPs/interval) in JMP Pro 13 (SAS Institute Inc.). For the LRD panel, the proportion of SNPs in the top 0.1% allele frequency difference (>0.66) was calculated for 50-kb intervals (median = 60 SNPs/interval). Different metrics were calculated and plotted for each panel due to the difficulty of visually distinguishing the LRD panel heterozygote phenotype from the LRD wild-type homozygote phenotype. As a consequence of this ambiguity, heterozygous individuals were included in the wild-type pattern LRD bulked pool at low frequency, leading to a lack of fixed differences overall between the wild-type and rto-like pools.
Data and Code Availability
The datasets and code utilized in this study are available at NCBI SRA: PRJNA481753 and GitHub: https://github.com/BlackmanLabUCB/Bulked-Segregant-Analysis. Many protocols used in this study are available at https://www.protocols.io/groups/mimulus/publications.
Supplementary Material
Video S1: Trafficking of the RTO protein. Related to Figure 5. Images were taken every 30 seconds within a span of 15 mins.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Bacterial and Virus Strains | ||
| Agrobacterium tumefaciens strains EHA105 and GV3101 | Widely distributed | EHA105, GV3101 |
| Biological Samples | ||
| SWC wt × SWC rto-like M. guttatus F2 seed | This Paper | |
| LRD wt × LRD rto-like M. guttatus F2 seed | This Paper | |
| rto × SL9 M. lewisii F2 seed | This Paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-napthalene acetic acid | PhytoTechnology Labs | Cat#N5999 |
| 2-N-morpholino ethane sulfonic acid | Sigma-Aldrich | Cat#M2933 |
| Acetosyringone (4′-Hydroxy-3′,5′-dimethoxyacetophenone) |
Sigma-Aldrich | Cat#D134406 |
| Calcium-D-gluconate | Acros Organics | Cat#299-28-5 |
| Cefotaxime | GoldBio | Cat#C-104–25 |
| Cetyl Trimethyl Ammonium Bromide (CTAB) | Sigma-Aldrich | Cat#H5882 |
| Gelrite (Gellan gum) | Scotts Labs | Cat#4900–1890 |
| Kanamycin | GoldBio | Cat#K-120–10 |
| Meta-topolin | PhytoTechnology Labs | Cat#T-841 |
| Murashige and Skoog salts with Gamborg B5 vitamins | PhytoTechnology Labs | Cat#M404 |
| N-(2-Chloro-4-pyridyl)-N′-phenylurea (CPPU) | PhytoTechnologyLabs | Cat#C279 |
| Phosphinothricin | GoldBio | Cat#P-165–250 |
| Rifampicin | GoldBio | Cat#R-120–1 |
| Timentin™ Ticarcillin / Clavulanate | GoldBio | Cat#T-104–250 |
| S. pyogenes Cas9 nuclease | New England Biolabs | Cat#M0386S |
| Critical Commercial Assays | ||
| AMPure XP beads | Beckman Coulter | Cat#A63880 |
| Quant-iT dsDNA Broad Range Assay Kit | ThermoFisher | Cat#Q33130 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7645S |
| Multiplex Oligos for Illumina Kit | New England Biolabs | Cat#E7335S |
| Phusion High Fidelity PCR Kit | New England Biolabs | Cat#E0553S |
| Spectrum Plant Total RNA Kit | Sigma-Aldrich | Cat#STRN50–1KT |
| Zymo Direct-Zol RNA kit | Zymo Research | Cat#R2055 |
| SuperScript III First-Strand Synthesis System | Invitrogen | Cat#18080051 |
| DNase I | Invitrogen | Cat#18068015 |
| DNase I Set | Zymo Research | Cat#E1010 |
| GoScript Reverse Transcriptase System | Promega | Cat#A5000 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat#A25741 |
| iQ SYBR Green Supermix | Bio-Rad | Cat#1708880 |
| Deposited Data | ||
| Genomic sequence reads generated for bulked segregant analyses | NCBI Sequence Read Archive | SRA: BioProject PRJNA481753 |
| Mimulus guttatus v2.0 reference genome | Phytozome | https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Mguttatus |
| Experimental Models: Organisms/Strains | ||
| Mimulus lewisii | Inbred line LF10 | LF10 |
| Mimulus guttatus | Sweet Creek Road (OR), Littlerock Dam (CA), Trinity Alps Road (CA), Quarry Valley Road (CA), Mackville Road (CA) | SWC, LRD, TAR, QVR, MAC |
| Nicotiana benthamiana | Widely distributed | N/A |
| Bombus impatiens | Koppert Biological Systems | Naturpol |
| Oligonucleotides | ||
| Primers for cloning, sequencing, genotyping, qPCR | This paper (Tables S4–S6) | N/A |
| Recombinant DNA | ||
| pFGC5941 | Arabidopsis Biological Resource Center | CD3–447 |
| pMCS:GW | Arabidopsis Biological Resource Center | CD3–1933 |
| ER-cb | Arabidopsis Biological Resource Center | CD3–954 |
| pMCS:YFP-GW | Arabidopsis Biological Resource Center | CD3–1934 |
| pEarleyGate 100 | Arabidopsis Biological Resource Center | CD3–724 |
| pEarleyGate 104 | Arabidopsis Biological Resource Center | CD3–686 |
| pUC119-gRNA | Addgene | 52255 |
| pFGC-pcoCas9 | Addgene | 52256 |
| pUBC-nYFP-Dest | [71] | N/A |
| pUBC-cYFP-Dest | [71] | N/A |
| pENTR/D-TOPO | Invitrogen | Cat#K240020 |
| AS-121P-CR3 | Alex Schultink, Innovative Genomics Institute | N/A |
| MlRTO RNAi | This paper | N/A |
| MgRTO RNAi | This paper | N/A |
| MlNEGAN RNAi | This paper | N/A |
| MgNEGAN RNAi | This paper | N/A |
| RTO sgRNA+ Cas9 | This paper | N/A |
| RTOpro:CFP-ER | This paper | N/A |
| RTOpro:YFP-RTO | This paper | N/A |
| 35S:MlRTO | This paper | N/A |
| 35S:YFP-MlRTO | This paper | N/A |
| pUBC-cYFP-ANbHLH1 | This paper | N/A |
| pUBC-nYFP-RTO | This paper | N/A |
| pUBC-nYFP-rto | This paper | N/A |
| Software and Algorithms | ||
| bwa | [75] | https://github.com/lh3/bwa |
| samtools | [76] | https://github.com/samtools |
| ImageJ | [77] | https://imagej.nih.gov/ij/ |
| VCell | [48] | http://vcell.org |
| RDsimJ.jar | [12] | Part of the Supporting Online Material |
| JMP Pro 13 | SAS Institute Inc. | https://www.jmp.com/ |
| E-CRISP | [78] | http://www.e-crisp.org/E-CRISP/ |
| Custom Scripts | This Paper | https://github.com/BlackmanLabUCB/Bulked-Segregant-Analysis |
Highlights.
An activator-inhibitor system produces spotted pigment patterning in Mimulus flowers.
Expression of the inhibitor, RTO, is promoted by the activator, NEGAN.
RTO protein is mobile and inhibits NEGAN function in neighboring cells.
Modulating this nectar guide patterning system influences bumblebee visitation.
Acknowledgements
We thank J. Manson for guidance with the pollinator cage experiments; S. Criswell and B. Glover for assistance and advice with SEM; C. O’Connell for assistance with confocal imaging; A. Cowan and F. Morgan for help with ImageJ image processing; E. LoPresti and K. Toll for alerting us to natural rto-like variants; A. Nguyen, J. Weger, and S. McDevitt for assistance with library construction and sequencing; S. Lewis for assistance in M. lewisii mutant screen; T. Rushton, I. Tucker, I. Tan, W. Crannage, and the University of Connecticut, UC Berkeley Oxford Tract Facility, Duke University, and University of Washington research greenhouse staff for plant husbandry and phenotyping assistance; our lab members for feedback on the manuscript; and J. Willis for his generous support. We gratefully acknowledge support from the National Science Foundation (IOS-1558083) and the National Institute of Health (R01GM131055) to Y.W.Y.; by the University of Virginia, the University of California, Berkeley, and the National Science Foundation (IOS-1558035) to B.K.B.; by the National Institute of Health (R24GM134211) to M.L.B; and by the National Institutes of Health (5R01GM088805) to H.D.B. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References
- 1.Hughes KA, Houde AE, Price AC, and Rodd FH (2013). Mating advantage for rare males in wild guppy populations. Nature 503, 108–110. [DOI] [PubMed] [Google Scholar]
- 2.Porter SS (2013). Adaptive divergence in seed color camouflage in contrasting soil environments. New Phytol. 197, 1311–1320. [DOI] [PubMed] [Google Scholar]
- 3.Salis P, Lorin T, Laudet V, and Frédérich B (2019). Magic Traits in Magic Fish: Understanding Color Pattern Evolution Using Reef Fish. Trends Genet. 35, 265–278. [DOI] [PubMed] [Google Scholar]
- 4.Caro T, Izzo A, Reiner RC Jr, Walker H, and Stankowich T (2014). The function of zebra stripes. Nat. Commun 5, 3535. [DOI] [PubMed] [Google Scholar]
- 5.Arnoult L, Su KFY, Manoel D, Minervino C, Magriña J, Gompel N, and Prud’homme B (2013). Emergence and Diversification of Fly Pigmentation Through Evolution of a Gene Regulatory Module. Science 339, 1423–1426. [DOI] [PubMed] [Google Scholar]
- 6.Gompel N, Prud’homme B, Wittkopp P, Kassner V, and Carroll S (2005). Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature 433, 481–487. [DOI] [PubMed] [Google Scholar]
- 7.Shang Y, Venail J, Mackay S, Bailey PC, Schwinn KE, Jameson PE, Martin CR, and Davies KM (2011). The molecular basis for venation patterning of pigmentation and its effect on pollinator attraction in flowers of Antirrhinum. New Phytol. 189, 602–615. [DOI] [PubMed] [Google Scholar]
- 8.Martins TR, Jiang P, and Rausher MD (2017). How petals change their spots: cis-regulatory re-wiring in Clarkia (Onagraceae). New Phytol. 216, 510–518. [DOI] [PubMed] [Google Scholar]
- 9.Jiang P, and Rausher M (2018). Two genetic changes in cis-regulatory elements caused evolution of petal spot position in Clarkia. Nat. Plants 4, 14–22. [DOI] [PubMed] [Google Scholar]
- 10.Lewis JJ, Geltman RC, Pollak PC, Rondem KE, Belleghem SMV, Hubisz MJ, Munn PR, Zhang L, Benson C, Mazo-Vargas A, et al. (2019). Parallel evolution of ancient, pleiotropic enhancers underlies butterfly wing pattern mimicry. Proc. Natl. Acad. Sci. USA doi: 10.1073/pnas.1907068116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner T, Koshikawa S, Williams TM, and Carroll SB (2010). Generation of a novel wing colour pattern by the Wingless morphogen. Nature 464, 1143–1148. [DOI] [PubMed] [Google Scholar]
- 12.Kondo S, and Miura T (2010). Reaction-Diffusion Model as a Framework for Understanding Biological Pattern Formation. Science 329, 1616–1620. [DOI] [PubMed] [Google Scholar]
- 13.Hiscock TW, and Megason SG (2015). Mathematically guided approaches to distinguish models of periodic patterning. Development 142, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green JBA, and Sharpe J (2015). Positional information and reaction-diffusion: two big ideas in developmental biology combine. Development 142, 1203–1211. [DOI] [PubMed] [Google Scholar]
- 15.Marcon L, and Sharpe J (2012). Turing patterns in development: what about the horse part? Current Opinion Genet. Dev 22, 578–584. [DOI] [PubMed] [Google Scholar]
- 16.Turing Alan Mathison (1952). The chemical basis of morphogenesis. Phil. Trans. Roy. Soc. B 237, 37–72. [Google Scholar]
- 17.Gierer A, and Meinhardt H (1972). A theory of biological pattern formation. Kybernetik 12, 30–39. [DOI] [PubMed] [Google Scholar]
- 18.Meinhardt H (2012). Turing’s theory of morphogenesis of 1952 and the subsequent discovery of the crucial role of local self-enhancement and long-range inhibition. Interface Focus 2, 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meinhardt H, and Gierer A (2000). Pattern formation by local self-activation and lateral inhibition. Bioessays 22, 753–760. [DOI] [PubMed] [Google Scholar]
- 20.Kondo S, and Asai R (1995). A reaction–diffusion wave on the skin of the marine angelfish Pomacanthus. Nature 376, 765–768. [DOI] [PubMed] [Google Scholar]
- 21.Maini PK, Woolley TE, Baker RE, Gaffney EA, and Lee SS (2012). Turing’s model for biological pattern formation and the robustness problem. Interface Focus 2, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcon L, Diego X, Sharpe J, and Müller P (2016). High-throughput mathematical analysis identifies Turing networks for patterning with equally diffusing signals. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schellmann S, Schnittger A, Kirik V, Wada T, Okada K, Beermann A, Thumfahrt J, Jürgens G, and Hülskamp M (2002). TRIPTYCHON and CAPRICE mediate lateral inhibition during trichome and root hair patterning in Arabidopsis. EMBO J. 21, 5036–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller P, Rogers KW, Jordan BM, Lee JS, Robson D, Ramanathan S, and Schier AF (2012). Differential Diffusivity of Nodal and Lefty Underlies a Reaction-Diffusion Patterning System. Science 336, 721–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raspopovic J, Marcon L, Russo L, and Sharpe J (2014). Modeling digits. Digit patterning is controlled by a Bmp-Sox9-Wnt Turing network modulated by morphogen gradients. Science 345, 566–570. [DOI] [PubMed] [Google Scholar]
- 26.Muñoz-García J, and Ares S (2016). Formation and maintenance of nitrogen-fixing cell patterns in filamentous cyanobacteria. Proc. Natl. Acad. Sci. USA 113, 6218–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tewary M, Ostblom J, Prochazka L, Zulueta-Coarasa T, Shakiba N, Fernandez-Gonzalez R, and Zandstra PW (2017). A stepwise model of reaction-diffusion and positional information governs self-organized human peri-gastrulation-like patterning. Development 144, 4298–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shyer AE, Rodrigues AR, Schroeder GG, Kassianidou E, Kumar S, and Harland RM (2017). Emergent cellular self-organization and mechanosensation initiate follicle pattern in the avian skin. Science 357, 811–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volkening A, and Sandstede B (2018). Iridophores as a source of robustness in zebrafish stripes and variability in Danio patterns. Nat. Commun 9, 3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamagishi M (2013). How genes paint lily flowers: Regulation of colouration and pigmentation patterning. Sci. Hort 163, 27–36. [Google Scholar]
- 31.Hsu C-C, Chen Y-Y, Tsai W-C, Chen W-H, and Chen H-H (2015). Three R2R3-MYB transcription factors regulate distinct floral pigmentation patterning in Phalaenopsis spp. Plant Physiol. 168, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medel R, Botto-Mahan C, and Kalin-Arroyo M (2003). Pollinator-Mediated Selection on the Nectar Guide Phenotype in the Andean Monkey Flower, Mimulus luteus. Ecology 84, 1721–1732. [Google Scholar]
- 33.Ren Z-X, Li D-Z, Bernhardt P, and Wang H (2011). Flowers of Cypripedium fargesii (Orchidaceae) fool flat-footed flies (Platypezidae) by faking fungus-infected foliage. Proc. Natl. Acad. Sci. USA 108, 7478–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Jager ML, Willis-Jones E, Critchley S, and Glover BJ (2017). The impact of floral spot and ring markings on pollinator foraging dynamics. Evol. Ecol 31, 193–204. [Google Scholar]
- 35.Davies KM, Albert NW, and Schwinn KE (2012). From landing lights to mimicry: the molecular regulation of flower colouration and mechanisms for pigmentation patterning. Functional Plant Biol. 39, 619–638. [DOI] [PubMed] [Google Scholar]
- 36.Glover B (2014). Understanding Flowers and Flowering 2nd ed. (Oxford: Oxford University Press; ). [Google Scholar]
- 37.Yuan Y-W, Sagawa JM, Di Stilio VS, and Bradshaw HD (2013). Bulk segregant analysis of an induced floral mutant identifies a MIXTA-like R2R3 MYB controlling nectar guide formation in Mimulus lewisii. Genetics 194, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hellsten U, Wright KM, Jenkins J, Shu S, Yuan Y, Wessler SR, Schmutz J, Willis JH, and Rokhsar DS (2013). Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl. Acad. Sci. USA 110, 19478–19482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Susič N, Bohanec B, and Murovec J (2014). Agrobacterium tumefaciens-mediated transformation of bush monkey-flower (Mimulus aurantiacus Curtis) with a new reporter gene ZsGreen. Plant Cell Tiss. Organ Cult 116, 243–251. [Google Scholar]
- 40.Yuan Y-W, Sagawa JM, Frost L, Vela JP, and Bradshaw HD (2014). Transcriptional control of floral anthocyanin pigmentation in monkeyflowers (Mimulus). New Phytol. 204, 1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nie Z-L, Sun H, Beardsley PM, Olmstead RG, and Wen J (2006). Evolution of biogeographic disjunction between eastern Asia and eastern North America in Phryma (Phrymaceae). Am. J. Bot 93, 1343–1356. [DOI] [PubMed] [Google Scholar]
- 42.Yuan Y-W, Sagawa JM, Young RC, Christensen BJ, and Bradshaw HD (2013). Genetic dissection of a major anthocyanin QTL contributing to pollinator-mediated reproductive isolation between sister species of Mimulus. Genetics 194, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albert NW, Davies KM, Lewis DH, Zhang H, Montefiori M, Brendolise C, Boase MR, Ngo H, Jameson PE, and Schwinn KE (2014). A Conserved Network of Transcriptional Activators and Repressors Regulates Anthocyanin Pigmentation in Eudicots. Plant Cell 26, 962–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu H-F, Fitzsimmons K, Khandelwal A, and Kranz RG (2009). CPC, a single-repeat R3 MYB, is a negative regulator of anthocyanin biosynthesis in Arabidopsis. Mol. Plant 2, 790–802. [DOI] [PubMed] [Google Scholar]
- 45.Zimmermann IM, Heim MA, Weisshaar B, and Uhrig JF (2004). Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant J. 40, 22–34. [DOI] [PubMed] [Google Scholar]
- 46.Kurata T, Ishida T, Kawabata-Awai C, Noguchi M, Hattori S, Sano R, Nagasaka R, Tominaga R, Koshino-Kimura Y, Kato T, et al. (2005). Cell-to-cell movement of the CAPRICE protein in Arabidopsis root epidermal cell differentiation. Development 132, 5387–5398. [DOI] [PubMed] [Google Scholar]
- 47.Koizumi K, Wu S, MacRae-Crerar A, and Gallagher KL (2011). An Essential Protein that Interacts with Endosomes and Promotes Movement of the SHORT-ROOT Transcription Factor. Curr. Biol 21, 1559–1564. [DOI] [PubMed] [Google Scholar]
- 48.Moraru II, Schaff JC, Slepchenko BM, Blinov ML, Morgan F, Lakshminarayana A, Gao F, Li Y, and Loew LM (2008). Virtual Cell modelling and simulation software environment. IET Syst. Biol 2, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koch AJ, and Meinhardt H (1994). Biological pattern formation: from basic mechanisms to complex structures. Rev. Mod. Phys 66, 1481–1507. [Google Scholar]
- 50.Miura T, and Maini PK (2004). Periodic pattern formation in reaction-diffusion systems: an introduction for numerical simulation. Anat. Sci. Int 79, 112–123. [DOI] [PubMed] [Google Scholar]
- 51.Arathi HS, and Kelly JK (2004). Corolla Morphology Facilitates Both Autogamy and Bumblebee Pollination in Mimulus guttatus. Intl. J. Plant Sci 165, 1039–1045. [Google Scholar]
- 52.Peterson ML, Miller TJ, and Kay KM (2015). An ultraviolet floral polymorphism associated with life history drives pollinator discrimination in Mimulus guttatus. Am. J. Bot 102, 396–406. [DOI] [PubMed] [Google Scholar]
- 53.Carr DE, Roulston TH, and Hart H (2014). Inbreeding in Mimulus guttatus Reduces Visitation by Bumble Bee Pollinators. PLoS ONE 9, e101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ivey CT, and Carr DE (2005). Effects of herbivory and inbreeding on the pollinators and mating system of Mimulus guttatus (Phrymaceae). Am. J. Bot 92, 1641–1649. [DOI] [PubMed] [Google Scholar]
- 55.Bodbyl Roels SA, and Kelly JK (2011). Rapid Evolution Caused by Rapid Pollinator Loss in Mimulus guttatus. Evolution 65, 2541–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meindl GA, Bain DJ, and Ashman T-L (2013). Edaphic factors and plant-insect interactions: direct and indirect effects of serpentine soil on florivores and pollinators. Oecologia 173, 1355–1366. [DOI] [PubMed] [Google Scholar]
- 57.Haupaix N, and Manceau M (2019). The embryonic origin of periodic color patterns. Dev. Biol [DOI] [PubMed] [Google Scholar]
- 58.Wolpert L (1971). Positional Information and Pattern Formation In Current Topics in Developmental Biology, Moscona AA and Monroy A, eds. (Academic Press; ), pp. 183–224. [DOI] [PubMed] [Google Scholar]
- 59.Rogers KW, and Schier AF (2011). Morphogen gradients: from generation to interpretation. Annu. Rev. Cell Dev. Biol 27, 377–407. [DOI] [PubMed] [Google Scholar]
- 60.Jones KN (1996). Pollinator Behavior and Postpollination Reproductive Success in Alternative Floral Phenotypes of Clarkia gracilis (Onagraceae). Intl. J. Plant Sci 157, 733–738. [Google Scholar]
- 61.Schnittger A, Folkers U, Schwab B, Jürgens G, and Hülskamp M (1999). Generation of a Spacing Pattern: The Role of TRIPTYCHON in Trichome Patterning in Arabidopsis. Plant Cell 11, 1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bouyer D, Geier F, Kragler F, Schnittger A, Pesch M, Wester K, Balkunde R, Timmer J, Fleck C, and Hülskamp M (2008). Two-Dimensional Patterning by a Trapping/Depletion Mechanism: The Role of TTG1 and GL3 in Arabidopsis Trichome Formation. PLoS Biol. 6, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wada T, and Tominaga-Wada R (2015). CAPRICE family genes control flowering time through both promoting and repressing CONSTANS and FLOWERING LOCUS T expression. Plant Sci. 241, 260–265. [DOI] [PubMed] [Google Scholar]
- 64.Gates DJ, Olson BJSC, Clemente TE, and Smith SD (2018). A novel R3 MYB transcriptional repressor associated with the loss of floral pigmentation in Iochroma. New Phytol. 217, 1346–1356. [DOI] [PubMed] [Google Scholar]
- 65.Owen CR, and Bradshaw HD (2011). Induced mutations affecting pollinator choice in Mimulus lewisii (Phrymaceae). Arthropod-Plant Inter. 5, 235. [Google Scholar]
- 66.Kelly AJ, and Willis JH (1998). Polymorphic microsatellite loci in Mimulus guttatus and related species. Mol. Ecol 7, 769–774. [Google Scholar]
- 67.Kerschen A, Napoli CA, Jorgensen RA, and Müller AE (2004). Effectiveness of RNA interference in transgenic plants. FEBS Lett. 566, 223–228. [DOI] [PubMed] [Google Scholar]
- 68.Michniewicz M, Frick EM, and Strader LC (2015). Gateway-compatible tissue-specific vectors for plant transformation. BMC Res. Notes 8, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nelson BK, Cai X, and Nebenführ A (2007). A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 51, 1126–1136. [DOI] [PubMed] [Google Scholar]
- 70.Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song K, and Pikaard CS (2006). Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 45, 616–629. [DOI] [PubMed] [Google Scholar]
- 71.Grefen C, Donald N, Hashimoto K, Kudla J, Schumacher K, and Blatt MR (2010). A ubiquitin-10 promoter-based vector set for fluorescent protein tagging facilitates temporal stability and native protein distribution in transient and stable expression studies. Plant J. 64, 355–365. [DOI] [PubMed] [Google Scholar]
- 72.Ding B, and Yuan Y-W (2016). Testing the utility of fluorescent proteins in Mimulus lewisii by an Agrobacterium-mediated transient assay. Plant Cell Rep. 35, 771–777. [DOI] [PubMed] [Google Scholar]
- 73.Slepchenko BM, Schaff JC, Macara I, and Loew LM (2003). Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 13, 570–576. [DOI] [PubMed] [Google Scholar]
- 74.McDonald JH (2014). Handbook of Biological Statistics 3rd ed. (Baltimore: Sparky House Publishing; ). [Google Scholar]
- 75.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup, G.P.D.P. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heigwer F, Kerr G, and Boutros M (2014). E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: Trafficking of the RTO protein. Related to Figure 5. Images were taken every 30 seconds within a span of 15 mins.
Data Availability Statement
The datasets and code utilized in this study are available at NCBI SRA: PRJNA481753 and GitHub: https://github.com/BlackmanLabUCB/Bulked-Segregant-Analysis. Many protocols used in this study are available at https://www.protocols.io/groups/mimulus/publications.