

SUMMARY
Biogenesis of the human telomerase RNA (hTR) involves a complex series of posttranscriptional modifications, including hypermethylation of the 5′ mono-methylguanosine cap to a tri-methylguanosine cap (TMG). How the TMG cap affects hTR maturation is unknown. Here, we show that depletion of trimethylguanosine synthase 1 (TGS1), the enzyme responsible for cap hypermethylation, increases levels of hTR and telomerase. Diminished trimethylation increases hTR association with the cap-binding complex (CBC) and with Sm chaperone proteins. Loss of TGS1 causes an increase in accumulation of mature hTR in both the nucleus and the cytoplasm compared with controls. In TGS1 mutant cells, increased hTR assembles with telomerase reverse transcriptase (TERT) protein to yield elevated active telomerase complexes and increased telomerase activity, resulting in telomere elongation in cultured human cells. Our results show that TGS1-mediated hypermethylation of the hTR cap inhibits hTR accumulation, restrains levels of assembled telomerase, and limits telomere elongation.
Graphical Abstract
In Brief
hTR, the RNA component of telomerase, acquires a trimethylguanosine cap synthesized by Trimethylguanosine synthase 1 (TGS1). Chen et al. show that TGS1 and cap hypermethylation control hTR abundance and intracellular distribution. Loss of TGS1 results in elevated hTR levels, increased telomerase activity and telomere elongation.
INTRODUCTION
Telomere homeostasis is a major determinant for replicative life-span, cellular senescence, and tumor progression (Blackburn et al., 2015). Human telomeres consist of arrays of short repetitive sequences at chromosome ends and are shielded from the DNA repair machinery by specialized capping complexes (Palm and de Lange, 2008). Telomere repeats are added by telomerase, an enzyme whose catalytic core is comprised of the telomerase reverse transcriptase (TERT) catalytic subunit and the human telomerase RNA (hTR) template RNA. While hTR is broadly expressed, the expression of TERT is restricted to stem cells and progenitor cells (Wright et al., 1996); telomere elongation occurs only in cells expressing active telomerase (Cristofari and Lingner, 2006). Haploinsufficiency of either TERT or hTR causes pathologic telomere shortening and leads to the stem cell disease dyskeratosis congenita and other telomere-related diseases (Armanios and Blackburn, 2012; Armanios et al., 2005; Batista et al., 2011; Marrone et al., 2004), suggesting that not only the TERT level but also the hTR level is a limiting factor for telomerase activity. Defining the mechanisms that regulate hTR biogenesis and its assembly into telomerase is critically important for our understanding of telomere-related pathologies and telomerase regulation in cancer (Rousseau and Autexier, 2015).
Human hTR is a 451 nt RNA synthesized by RNA polymerase II (Pol II) that acquires a monomethylguanosine (MMG) cap during the early stages of transcription. This MMG cap is further methylated to a N2, 2, 7 trimethylguanosine (TMG) cap, by trimethylguanosine synthase 1 (TGS1), an evolutionarily conserved enzyme that modifies several classes of noncoding RNAs, including small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNas), some viral RNAs, and selenoprotein mRNAs (Mouaikel et al., 2002; Pradet-Balade et al., 2011; Wurth et al., 2014; Yedavalli and Jeang, 2010). Unlike classical Pol II transcripts, hTR lacks a canonical polyadenylation signal and is processed to generate a defined 3′ end.
The 3′ end of hTR contains an H/ACA motif consisting of two hairpins and two single-stranded regions, the hinge and the ACA containing tail (Kiss et al., 2006; Mitchell et al., 1999). The H/ACA motif, which is found also in small Cajal body RNAs (scaRNAs) and in some snoRNAs, is bound cotranscriptionally by the dyskerin (DKC1)-NOP10-NHP2-NAF1 complex that defines the 3′ end of hTR and stabilizes hTR transcripts (Fu and Collins, 2007; MacNeil et al., 2019; Shukla et al., 2016). Mutations in DKC1, NOP10, or NHP2 lead to dyskeratosis congenita (DC), by impairing telomerase and causing telomere shortening (Armanios and Blackburn, 2012).
hTR is initially transcribed as an extended precursor that is trimmed by 3′−5′ RNA exonucleases to generate its mature 451 nt form. hTR transcripts as long as 1,500 nt have been detected, although it is unclear whether these ultra-long transcripts are processed to mature hTR or whether they are aberrantly terminated transcripts removed by nuclear RNA surveillance through the RNA exosome (Nguyen et al., 2015; Tseng et al., 2015, 2018). Many hTR precursors have 8–10 nt genomically encoded 3′ extensions and are trimmed to generate mature hTR (Goldfarb and Cech, 2013; Roake et al., 2019). These precursors are primarily oligoadenylated by the noncanonical poly(A)polymerase PAPD5 (Moon et al., 2015; Tseng et al., 2015). Oligoadenylated hTR intermediates can either be degraded by the RNA exosome or have their A tails removed by the poly(A)ribonuclease PARN. Patients with biallelic germline mutations in PARN develop DC and idiopathic pulmonary fibrosis (IPF), downstream of telomere shortening (Moon et al., 2015; Stuart et al., 2015; Tummala et al., 2015). In the absence of PARN, oligoadenylated hTR precursors accumulate; the maturation rate of hTR slows, and stalled hTR precursors are degraded causing an overall loss of hTR and telomerase. However, in the absence of both PARN and PAPD5, the maturation of hTR precursors normalizes, indicating that oligoadenylation of hTR precursors governs the maturation rate of hTR (Roake et al., 2019).
Oligoadenylated hTR precursors that are not processed to mature hTR are subject to degradation by the exosome, an RNase complex composed of ten core proteins including the 3′−5′ exonuclease Dis3 and the endonuclease Rrp44. The core exosome also interacts with the 3′−5′ exonuclease Rrp6, which functions similarly to Dis3 (Chlebowski et al., 2013). Exosome substrates are recruited by the nuclear exosome targeting complex (NEXT) together with the cap binding complex (CBC), which includes the CBP20, CBP80, and ARS2 subunits (Andersen et al., 2013; Mitchell, 2014). Loss of CBC, exosome subunits, or some specific NEXT components, causes an increase in hTR levels, suggesting that this pathway removes a subset of maturing hTR transcripts (Gable et al., 2019; Shukla et al., 2016; Tseng et al., 2015). In addition, the RNA exosome is involved in degradation of improperly assembled hTR complexes, because exosome knockdown can rescue the hTR loss caused by dyskerin deficiency or mutations (Boyraz et al., 2016; Fok et al., 2019; Shukla et al., 2016).
Beyond its role in hTR biogenesis and stability, the H/ACA domain of hTR also contains a 3-nt sequence called the CAB box that binds additional proteins. TCAB1 binds the CAB box and directs localization of hTR to Cajal bodies (CBs), nuclear structures devoted to RNA modification and assembly. TCAB1 binding to the CAB box is required for full catalytic activity of telomerase (Chen et al., 2018; Freund et al., 2014; Tycowski et al., 2009; Zhong et al., 2011) and for telomerase recruitment at telomeres during the S phase (Cristofari et al., 2007; Tomlinson et al., 2006; Venteicher et al., 2009). The CAB box also associates with a subset of the seven Sm proteins (Fu and Collins, 2006), which form a heteroheptameric ring that encircles and stabilizes several coding and noncoding RNA species, and SmB directly interacts with TGS1 (Mouaikel et al., 2003a, 2003b). In fission yeast, the Sm proteins bind telomerase RNA and contribute to its maturation and stability (Tang et al., 2012).
CBs are also the sites in which hypermethylation of hTR by TGS1 is thought to occur (Fu and Collins, 2006; Girard et al., 2008; Jády et al., 2004). The role of the cap hypermethylation step in telomerase biogenesis and/or trafficking is unknown. Human TGS1 exists as two isoforms, a long (TGS1-LF) and a short (TGS1-SF) isoform (Girard et al., 2008). The TGS1 LF C-terminal portion contains a highly conserved methyltransferase domain and is present both in the cytoplasm and the nuclear CBs (Girard et al., 2008). The short isoform consists only of the C terminus of the protein and it is exclusively enriched in CBs. The two isoforms have different repertoires of RNA targets: TGS1 LF is thought to hypermethylate snRNAs, while TGS1 SF is believed to be specific for snoRNAs. TGS1 LF has been also implicated in the trafficking of these noncoding RNAs through its interaction with the nuclear export factor CRM1 (Boulon et al., 2004; Verheggen and Bertrand, 2012). The TMG cap synthesized by TGS1 LF and the Sm core proteins bound to SMN form a bipartite nuclear targeting signal that directs import of snRNAs and SMN (Hamm et al., 1990; Narayanan et al., 2004). In addition, it has been proposed that CRM1 modulates the interaction between TGS1 LF and hTR (Pradet-Balade et al., 2011).
Here, we investigate the role of TGS1 in regulating hTR biogenesis, stability, and assembly into functional telomerase. By introducing frameshift mutations in the TGS1 gene in human cancer cells using CRISPR/Cas9 genome editing, we create cells depleted of the TGS1 protein. We use these cells to understand how hTR 5′ cap trimethylation controls hTR biogenesis, telomerase levels and telomere length.
RESULTS
TGS1 Is Required for Proper CB Organization and hTR Subcellular Localization
To investigate the role of cap hypermethylation in hTR biosynthesis and telomerase assembly in HeLa cells, we used CRISPR/Cas9 genomic editing to introduce insertions and deletions within both the first and the eighth TGS1 exon, which affect both TGS1 isoforms. We isolated three clones (TGS1 CRISPR M1, M2, and M3) displaying strongly reduced expression of TGS1. Western blotting showed that in these three clones the TGS1 levels are less than 10% of the level observed in parental HeLa cells (Figure 1A). Stable lentiviral expression of FLAG-tagged TGS1 restored TGS1 to levels that approximate those of the endogenous TGS1 protein (M1R, M2R, and M3R correspond to M1, M2 and M3 knockout clones rescued with FLAG-TGS1) (Figure 1A).
Figure 1. TGS1 Loss Affects hTR Localization.

(A) Western blot (WB) with an anti-TGS1 antibody on extracts from a TGS1-proficient HeLa cell line, and three independent CRISPR-derived TGS1 mutant clones (M1, M2, and M3) expressing a FLAG-TGS1 rescue construct at a similar level as endogenous TGS1 (endo-TGS1). b-tubulin is a loading control (CTR). See also Table S1.
(B and C) Examples of TGS1 mutant cells (M1) showing reductions in hTR (B) and scaU93 RNA (C) foci compared to TGS1 M1 cells expressing the FLAG-TGS1 rescue construct (M1R), and accumulation in the nucleoli of hTR and scaU93, which were detected by RNA FISH (red); nucleoli appear as DAPI-dim areas. (D–G) Average numbers of hTR (D) and U93 (F) foci observed in M (M1, M2, and M3), MR (M1R, M2R, and M3R), and CTR (HeLa parental line) cells. Frequencies of cells with nucleolar accumulations of hTR (E) and U93 (G) in the same cell samples. Data are the means of 3 mutant clones and 3 CTR cell samples. *p < 0.05; ****p < 0.0001, one-way ANOVA.
Because previous studies had shown that TGS1 depletion disrupts CB formation in HeLa cells (Lemm et al., 2006; Roithová et al., 2018), we first tested our TGS1 CRISPR cells for the presence and integrity of CBs using immunofluorescence with antibodies against coilin and TCAB1, which co-localize in CBs (Venteicher et al., 2009) (Figure S1A). While TGS1-proficient HeLa cells showed an average of 2.5 coilin/TCAB1 foci per cell (n = 473), TGS1-deficient cells exhibited only 1.2 coilin/TCAB1 foci/cell (n = 942). The reduction in the CB number was rescued by stable expression of FLAG-TGS1 (2 coilin/TCAB1 foci/cell; n = 1,115) (Figure S1B). In TGS1 mutant cells, the coilin/TCAB1 signals were more diffuse than in controls and formed aggregates of irregular size and shape. These aggregates colocalized with nucleoli in 20% of the nuclei, whereas only 4% of control nuclei displayed nucleolar Coilin/TCAB1 signals (Figures S1A and S1B). These results show that TGS1 is required for CB integrity, likely reflecting an underlying defect in snRNA biogenesis caused by TGS1 loss (Lemm et al., 2006).
We next performed RNA fluorescence in situ hybridization (FISH) to determine the effects of TGS1 depletion on the subcellular localization of hTR, which is normally strongly enriched in the CBs of HeLa cells. Consistent with previous results (Zhu et al., 2004), we found that parental HeLa cells exhibit an average of 2.7 hTR foci/nucleus (n = 142), whereas TGS1 mutant cells (M1 plus M2; n = 263) contain 1.0 hTR focus per nucleus (Figures 1B and 1D). However, TGS1-deficient cells displayed prominent hTR accumulation in nucleoli (Figures 1B, 1E, and S1C). Specifically, while 41% of TGS1 mutant cells showed enrichments of hTR in the nucleoli, only 4% and 13% of control and rescued cells showed similar enrichments, respectively (Figure 1E). TGS1-deficiency also affected localization of scaRNAs. The U93 scaRNA was enriched in CBs of both control cells (2.4 foci/cell, n = 118) and TGS1-rescued cells (2.8 foci/cell n = 344) (Figures 1C and 1F), but showed diminished CB localization (0.7 foci/cell, n = 363) and nucleolar mislocalization in TGS1 M1 and M2 cells (Figures 1F and 1G). Disruption of TGS1 in TGS1 M1 cells led to loss of CB localization of the U2 snRNA (Figure S2A). As expected, normal localization of small nucleolar RNA (snoRNA) in the nucleoli was not affected by TGS1 loss (Figure S2B).
TGS1 Regulates the Abundance of hTR Molecules
To determine whether TGS1 loss affects the abundance of hTR molecules, we analyzed hTR levels by northern blotting using total RNA isolated from either TGS1-proficient cells (parental HeLa cells or TGS1 M1R cells) or TGS1 mutants cells (TGS1 M1, M2). These analyses revealed that the levels of hTR in TGS1 mutant cells increase by 2-fold compared to control or rescued cells (Figure 2A). Similar results were obtained by analyzing hTR levels using quantitative RT-PCR (qRT-PCR). hTR levels were increased on average by 1.8-fold in TGS1 mutant clones (M1, M2, and M3) expressing FLAG-GFP compared with their rescued counterparts stably expressing FLAG-TGS1 (Figure 2B).
Figure 2. Mutations in TGS1 Increase the hTR Levels.

(A) Northern blots (NBs) of total RNA from TGS1-proficient (CTR), TGS1 mutant (M1, M2), and TGS1 M1-rescued (M1R) probed against hTR or U1 snRNA. Ethidium bromide (EtBr)-stained rDNA is a loading CTR. Bottom panel: quantification of the hTR levels normalized to U1snRNA. The hTR doublet represents different folding states of mature hTR (Mitchell et al., 1999). Error bars represent standard deviation derived from three independent NBs.
(B) hTR levels determined by qRT-PCR on total RNA prepared from TGS1 mutant (M1, M2, M3) cells expressing either FLAG-GFP or FLAG-TGS1. Bars represent means from 3 independent experiments, are relative to parental HeLa cells (set to 1) and are normalized to GAPDH (**p < 0.01; ***p < 0.001; one-way ANOVA).
(C) WB showing the TGS1 abundance in UMUC3 and BJ-HELT cells treated with non-targeting (CTR) or TGS1 siRNA for 6 days. Tubulin is a loading CTR.
(D) Quantification of hTR and TGS1 transcripts by qRT-PCR performed on total RNA prepared from UMUC3 or BJ-HELT cells at the indicated days following siRNA treatment; data are relative to cells treated with CTR siRNA (set to 1); hTR levels are normalized to the GAPDH transcript. (**p < 0.01; ***p < 0.001, ns, not significant; one-way ANOVA, left panel; Student’s t test, right panel.)
We also determined whether loss of TGS1 upregulates hTR in other human cell lines and whether the degree of hTR upregulation depends on the basal hTR level in the cell line before TGS1 inactivation. We performed RNAi-mediated TGS1 knockdown in the UMUC3 bladder cancer line and in BJ-HELT cells (BJ fibroblasts stably expressing TERT and SV40 large T antigen) (Hahn et al., 1999; Xu and Blackburn, 2007). TGS1 RNAi efficiently reduced the levels of both TGS1 mRNA and TGS1 protein, resulting in increased hTR levels in each cellular context (Figures 2C and 2D). Together, these results indicate that loss of TGS1 increases hTR levels in human cell lines.
TGS1 Catalyzes Formation of the TMG Cap in hTR Molecules Associated with Telomerase
We next asked whether TGS1 catalyzes formation of the hTR TMG cap in the hTR molecules that associate with telomerase and whether loss of this cap affects hTR incorporation into functional telomerase complexes. To address these questions, we used a two-step immunopurification procedure (Figure 3A). We first precipitated endogenous telomerase complexes from nuclear extracts of either parental cells or TGS1 mutant clones (M1 and M2) using an anti-TERT antibody (Venteicher et al., 2009). We then purified hTR from hTR-TERT complexes using an antibody that specifically recognizes the trimethylated guanosine cap (TMG) (Bringmann et al., 1983). Northern blotting analysis of TERT-IP eluates confirmed that hTR is more abundant in TGS1 mutant cells than in TGS1-proficient control cells (Figure 3B), indicating that loss of TGS1 increases the abundance of both hTR and hTR-TERT complexes in HeLa cell nuclei. qRT-PCR of RNA precipitated by the anti-TMG antibody further showed that hTR molecules are more abundant in control cell precipitates than in precipitates from TGS1 mutant cells (Figure 3C), suggesting that in TGS1-deficient cells most hTR molecules do not contain a TMG cap. These results clearly show that TGS1 catalyzes formation of the hTR TMG cap and that hTR molecules devoid of this cap can efficiently associate with TERT.
Figure 3. Mutations in TGS1 Impair TMG Capping of hTR but Do Not Affect Its Ability to Interact with TERT and Form an Active Telomerase Both In Vitro and In Vivo.

(A) Schematic representation of the experiments. hTR was precipitated from nuclear extracts with an anti-TERT antibody, and purified TERT-hTR complexes were subjected to IP using an anti-TMG cap antibody.
(B) hTR associated with TERT eluates from TGS1 M1 and two TGS1-proficient CTR cells was detected by NB. IgG IP is a CTR for nonspecific binding. EtBr-stained rRNA is a loading CTR. Note that hTR is more abundant in TGS1 mutant cells than in CTR cells.
(C) RNA IP with an anti-TMG antibody from TERT-hTR complexes. qRT-PCR on eluates indicates that TERT-associated hTR is not hypermethylated. The monomethylated beta-actin RNA is a negative CTR. Error bars represent standard deviations derived from two independent RNA IP experiements.
(D) Top: telomeric repeat amplification protocol (TRAP) performed in 33 dilutions on extracts from cells of the indicated genotypes. IC, internal CTR. TGS1 M1 and M2 cells exhibit higher telomerase activities than both TGS1-proficient CTR cells and TGS1 M1 rescued cells (M1R). Bottom: quantification of TRAP activity. Error bars, SEM. a.u., arbitrary units.
(E) Mutations in TGS1 (C1, C2) were generated by CRISPR/Cas9 in the UMUC3 bladder tumor cell line. WB with anti-TGS1 or anti-FLAG antibodies shows reduced levels of endogenous TGS1 in mutant cells and the expression of the TGS1-FLAG rescue construct. See also Table S1.
(F) qRT-PCR showing that TGS1 CRISPR clones stably expressing FLAG-GFP exhibit increased hTR abundance compared to the same clones stably expressing the rescue construct FLAG-TGS1. Data are from three biological replicates, are normalized to GAPDH and relative to the parental cell line (*p < 0.05; **p < 0.01; one-way ANOVA).
(G) TGS1 loss induces telomere lengthening in UMUC3 cells. Telomere restriction fragment (TRF) analysis was performed on genomic DNA extracted from TGS1 mutant (C1, C2) and CTR cell lines kept in culture for the indicated time. PD, population doublings. The growth kinetics were similar for all lines (average doubling time: 1.8 days).
Loss of TGS1 Increases Telomerase Activity and Telomere Length
To understand whether telomerase complexes containing hTR molecules with a hypomethylated cap are catalytically active, we performed a telomeric repeat amplification protocol (TRAP) assay with nuclear extracts from parental control cells, TGS1 mutant cells (M1 and M2), and TGS1 rescued cells (M1R). TRAP showed that telomerase activity is 2-fold higher in TGS1 mutant cells than in either parental control cells or TGS1 M1R cells (Figure 3D). These results are consistent with previous work showing that an increase of the hTR level augments telomerase activity in HeLa cells (Cristofari and Lingner, 2006; Pickett et al., 2009; Xi and Cech, 2014; Zhong et al., 2012). Thus, the presence of a TMG cap is dispensable for the assembly of functional telomerase, and loss of TGS1 increases both the formation of hTR molecules and active TERT/hTR complexes.
To determine whether loss of TGS1 leads to telomere elongation in cultured cells, we exploited the UMUC3 cell line that has a high TERT level but a low hTR content that limits telomerase biosynthesis and telomere elongation (Xu and Blackburn, 2007). We used CRISPR/Cas9 mutagenesis to generate two UMUC3 cell clones (C1 and C2) with mutations in TGS1 and reduced TGS1 expression (Figure 3E). The C1 and C2 lines exhibit a 2.5- and 1.7-fold increase in the hTR level compared to the same lines expressing FLAG-TGS1, respectively (Figure 3F). Telomere restriction fragment (TRF) analysis performed on genomic DNA of these lines grown in culture for several population doublings showed that telomere length increases with time in TGS1 mutant cells (C1 and C2 clones) but not in control cells (Figure 3G). Thus, the increased hTR level and enhanced telomerase activity of TGS1 mutant cells results in telomere elongation.
Loss of TGS1 Increases hTR Accumulation but Does Not Alter hTR Oligoadenylation
Processing of hTR proceeds from genomically templated transcripts that extend up to 10 nt beyond the mature 3′ terminus at position 451 (hTR-451). These extended precursor RNAs are processed through steps that include oligoadenylation and deadenylation by PAPD5 and PARN, respectively (Nguyen et al., 2015; Roake et al., 2019; Tseng et al., 2015). In addition, qRT-PCR detected less abundant longer transcripts from the locus that are degraded by the exosome complex (Nguyen et al., 2015; Tseng et al., 2015). We asked whether TGS1 loss affects the formation of these long hTR transcripts. We performed qRT-PCR with reverse primers positioned at 159 or 445 bp downstream of position 451 (Figure 4A). We found that hTR long molecules are increased 2- to 3-fold in all three TGS1 mutant HeLa clones compared to the corresponding rescued cell lines (Figure 4B). A similar increase in long hTR transcripts was observed in TGS1 small interfering RNA (siRNA)-treated UMUC3 cells compared to scrambled dsRNA-treated controls (Figure 4B). These results may reflect either a defect in transcriptional termination or diminished surveillance of these long transcripts by the RNA exosome.
Figure 4. Mutations in TGS1 Induce Formation of Extended hTR Molecules but Do Not Affect 3′ Processing.

(A) Diagram indicating the localization of the primers used to detect the extended hTR species (EXT1 and EXT2) generated by transcription readthrough beyond the 3′ end of mature transcript (nt 451).
(B) qRT-PCR showing increased expression of EXT1 and EXT2 in TGS1-depleted cells compared to TGS1-expressing cells: TGS1 M versus TGS1 MR, average of three different mutant clones compared to their rescued counterparts; siRNA for TGS1 (siTGS1) or non-targeting CTR (siCTR) in UMUC3 cells. Data are normalized to GAPDH. TGS1 M1 and M2 data are relative to parental cells (set to 1); UMUC3 siTGS1 data are relative to siCTR cells (set to 1). TGS1 M and TGS1 MR bars are means of three clones. UMUC3 siTGS1 are means of two treatments (*p < 0.05; **p < 0.01; one-way ANOVA).
(C and D) 3′RACE-seq was performed to determine the abundance of hTR precursors terminating 1–10 nt beyond the 3′ end of the hTR mature transcript (>451)
(C) and oligoadenylated hTR molecules (OligoA) (D). No significant differences in these two types of precursors were observed in TGS1 M cells compared to CTR or TGS1 M1R cells. PAPD5-KO cells display an 80% reduction in OligoA species and a 75% reduction in >451 nt hTR molecules (C), as previously observed (Roake et al., 2019). In PARN-KO cells there is a 3-fold increase in both OligoA reads and >451 nt hTR reads (D). >35,000 reads per sample were analyzed.
To understand whether the increase in hTR seen in TGS1 mutant cells occurs because of altered 3′ maturation, we quantified hTR precursors and oligoadenylation patterns using 3′RACE RNA sequencing (RNA-seq) in both TGS1 mutant lines and corresponding rescued lines. The previously generated PAPD5-KO and PARN-KO mutant cell lines (Roake et al., 2019) were used as controls for reduced and increased oligoadenylation, respectively. In parental control cells, 20% of hTR molecules were extended precursors, and 17% of hTR precursors showed an oligo-A tail. The proportion of extended precursors and their degree of oligoadenylation were unchanged in TGS1 mutant cells (Figures 4C and 4D). Thus, the increase in hTR levels seen in TGS1 mutants is not caused by altered 3′ processing. These observations are consistent with the view that trimming of hTR 3′ ends and exosomal-mediated decay of long hTR transcripts are governed by distinct pathways (Roake et al., 2019).
TGS1 Deficiency Causes Cytoplasmic Accumulation of hTR
TGS1 has been shown to control the intracellular distribution of snRNAs and snoRNAs (Roithová et al., 2018; Verheggen and Bertrand, 2012). To test whether TGS1 plays a similar role in regulating hTR trafficking, we asked whether loss of TGS1 affects relative hTR accumulation in the nucleus versus the cytoplasm. To characterize hTR partitioning, we analyzed both nuclear and cytoplasmic fractions isolated from three TGS1 mutant clones (TGS1 M1–3), from TGS1-proficient cells (CTR, TGS1 M1R, CTR-R) and from PAPD5-KO and PARN-KO cell lines. Northern blotting analysis showed an increased abundance of both cytoplasmic and nuclear hTR in all three TGS1 mutant cell lines compared to PAPD5-KO, PARN-KO, or TGS1-proficient control cells (Figures 5A and 5B). We quantified the relative hTR amounts in the cytoplasm and the nucleus by normalizing the hTR levels with those of U1 and U6 snRNAs. In TGS1 mutant cells, the relative cytoplasmic enrichment in hTR molecules (cytoplasm/nucleus + cytoplasm ratio, abbreviated as C/N+C) was significantly higher (1.6-fold) than in the other cell lines (lanes 4, 6, 8 versus lines 2, 14, 16 of Figures 5A–5C and S3A–S3D). qRT-PCR confirmed an overall increase of the hTR level in TGS1 mutant cells compared to PAPD5-KO, PARN-KO, or control cells (Figure 5D).
Figure 5. TGS1 Mutants Have a High Cytoplasmic Level of hTR that Is Not Assembled with Telomerase.

(A) NB on nuclear (N) or cytoplasmic (C) RNA, extracted with hypo-osmotic lysis buffer (Chen et al., 2014) from parental HeLa cells (CTR); TGS1 M1, M2, and M3 mutant cells; CTR-R and TGS1 M1R cells expressing the FLAG-TGS1 rescue construct; PAPD5-KO and PARN-KO cells. Membranes were probed for hTR, U1, and U6 snRNA, and U3 snoRNA. Note that U6 runs as a doublet in TGS1 mutants. EtBr staining is a loading CTR.
(B) Quantification of the blots in (A) by densitometric analysis. The bars represent the nuclear and cytoplasmic abundance of hTR normalized to U1snRNA. The numbers on the orange bars are the percentages of cytoplasmic hTR over total RNA [C/(N+C]) (see also C and Figures S3A–S3D). TGS1 M1, M2, and M3 cells show higher hTR cytoplasmic fractions compared to CTR and TGS1 M1R cells; the hTR cytoplasmic fractions in PAPD5-KO cells are comparable to those observed in TGS1-proficient cells, or in PARN-KO cells.
(C) Bar plot showing the mean percentage of cytoplasmic hTR (indicated as % of cytoplasmic hTR over total hTR) in the indicated cell lines, relative to CTR (set to 1) (see also A and Figure S3B).The p value shown is relative to CTR, one-way ANOVA.
(D) qRT-PCR quantification of the hTR levels in total RNA from the indicated cell lines, values are normalized to GAPDH and relative to the parental HeLa cells (CTR). p values are relative to CTR, one-way ANOVA.
(E) qRT-PCR quantification of the hTR levels in cytoplasmic extracts prepared from TGS1 M and TGS1 MR cells according to (Méndez and Stillman, 2000). The hTR levels are normalized to GAPDH or U6, and are relative to CTR cells (set to 1). Data are from three different TGS1 mutant clones (M1–M3). p values, Student’s t test.
(F) Cytoplasmic extracts (S100) from TGS1-proficient (CTR) and TGS1 mutant cells (M1, M2) were precipitated with an anti-TERT antibody; purified RNA was assayed by NB with an hTR probe. IgGs were used as an IP CTR. Nuclear extracts (NE) from CTR cells were used as positive CTR (as in Figure 3B). Note the elevated hTR level in the cytoplasm of TGS1 mutant cells, and TERT-hTR complexes are absent in the cytoplasm of both TGS1-proficient and TGS1 mutant cells. EtBr staining is a loading CTR.
Cytoplasmic fractions were also obtained with a different fractionation method (Méndez and Stillman, 2000), and the hTR levels relative to GAPDH or U6 snRNA were determined by qRT-PCR (Figures 5E and S3E). This independent approach confirmed a two-fold increase in the cytoplasmic/nuclear hTR ration in TGS1 mutant cells compared to corresponding TGS1-rescued cells (Figure 5E). Importantly, we never observed relative increases in the cytoplasmic hTR fraction in PAPD5 mutant cells (Figures 5A, 5C, and S3A–S3D), which also exhibit an increase in total hTR accumulation (Figure 5D) (Roake et al., 2019). Thus, the increase in cytoplasmic hTR observed in TGS1-depleted cells is a specific consequence of TGS1 deficiency, and is not secondary to increased level of nuclear hTR.
We also asked whether the nuclear export factor CRM1 has a role in cytoplasmic accumulation of hTR in TGS1 mutants. Treating cells with the CRM1 inhibitor leptomycin B (LMB) caused an increase in the total hTR level in TGS1-proficient but not in TGS1 mutant cells (Figure S3F), suggesting that this increase, which parallels the increase induced by TGS1 deficiency, is TGS1-dependent. However, treatment with LMB did not substantially alter the distribution of hTR between the nucleus and the cytoplasm in either control or mutant cells. In TGS1 mutant cells neither the nuclear hTR level nor the cytoplasmic hTR level was significantly changed (Figure S3F). These results suggest that hTR export to the cytoplasm in TGS1 mutant cells is not CRM1-dependent. The increased hTR level found in LMB-treated cells could be related to the role of CRM1 in the intranuclear traffic of hTR (Pradet-Balade et al., 2011), but molecular mechanism remains uncertain.
Finally, RNA immunoprecipitation (RNA-IP) with an anti-TERT antibody failed to detect an enrichment in hTR molecules in precipitates from cytoplasmic extracts of TGS1-deficient cells (Figure 5F), indicating that cytoplasmic hTR is not incorporated into assembled telomerase complexes. This is consistent with the notion that telomerase assembly is a process restricted to the nucleus.
TGS1 Loss Leads to Increased Association of hTR and the Cap Binding Complex
In both yeast and human cells, the TMG caps of snRNAs are not recognized by the conserved CBC, which instead binds monomethylated caps with high affinity (Schwer et al., 2011). In wild-type budding yeast, mature snRNAs are TMG-capped and not associated with CBC, but are monomethylated and ectopically bound by CBC in Tgs1 mutants (Schwer et al., 2011). Because CBC binding to snRNAs regulates their nuclear/cytoplasm trafficking (Ohno et al., 2000), we tested the possibility that the increased cytoplasmic retention of hTR in TGS1 mutant cells is a consequence of altered CBC binding affinity toward hypomethylated hTR cap.
We first asked whether the methylation status of the hTR cap affects its affinity for the CBC complex. We isolated RNAs associated with the endogenous larger subunit of the CBC complex, CBP80, using an anti-CBP80 antibody and immunoprecipitation of both nuclear and cytoplasmic extracts from TGS1 mutant cells (M1) and TGS1-rescued cells (M1R) (Figure 6A). RNA precipitates were analyzed by northern blotting for hTR, and the U1 snRNA was used for comparison. U1 snRNA copurified with CBC more efficiently from nuclear extracts of TGS1 mutant cells compared to TGS1-rescued cells (Figure 6A, lanes 1 and 3). No significant difference in U1 abundance was observed in CBC-immunopurified RNA from cytoplasmic extracts of mutant and rescued cells (Figure 6A, lanes 2 and 4). The amount of hTR recovered by CBP80 IP in cytoplasmic extracts of both TGS1 mutant and TGS1-rescued cells was below the detection level by northern blotting (Figure 6A, lanes 2 and 4). However, northern blotting detected hTR in nuclear extracts from TGS1 mutants (Figure 6A, lane 3), showing that TGS1 depletion increases the amount of nuclear hTR associated with CBP80. Consistent with northern blotting results, hTR was significantly enriched in CBC complexes from the nuclei of TGS1 mutant cells compared with those of their rescued counterparts, as shown by qRT-PCR performed on RNA recovered by CBP IP on nuclear extracts (Figure 6B). For comparison, the 5.8S ribosomal RNA, that lacks a 5′ cap structure, did not copurify with CBP80 under either TGS1 mutant or TGS1-proficient conditions (Figure 6B).
Figure 6. Mutations in TGS1 Increase the Association of hTR with the CBC and the Sm Complexes.

(A) NBs on RNA from nuclear (N) or cytoplasmic (C) extracts precipitated with an anti-CBP80 antibody from TGS1 M1 and TGS1 M1R cells. The membranes were probed for hTR and U1snRNA; EtBr staining is a loading CTR. Note the increase in both U1 and hTR CBC-associated RNAs in nuclear extracts from TGS1 M cells.
(B) qRT-PCR on RNA purified by CBP IP. The uncapped 5.8S RNA was used as negative CTR. Error bars, SEM.
(C) qRT-PCR on RNA purified by SmB IP. The uncapped 5.8S RNA is a negative CTR. TGS1 deficiency significantly increases recovery of hTR but not of U1 snRNA in SmB complexes. p value, one-way ANOVA.
(D) NB on the RNA fractions purified by RNA IP with an anti-FLAG antibody from nuclear or cytoplasmic extracts from TGS1-proficient (CTR) or TGS1 M1 cells expressing: FLAG-TERT, FLAG-TERT DTRBD (mutated in the hTR binding domain), and FLAG-CBP80. NBs were probed for hTR; EtBr staining is a loading control. WB with anti-FLAG and anti-CBP20 antibodies on purified proteins verifies the recovery efficiency of the FLAG-tagged proteins and of the entire CBC complex. Note that CBC complex weakly interacts with telomerase. IgG light chain (IgG L.C.) is an IP CTR.
(E) qRT-PCR on RNA co-precipitating with the indicated FLAG-tagged proteins. Error bars, SEM.
To consolidate this result, we studied association between hTR and FLAG-CBP80 by generating TGS1-proficient and TGS1-deficient cells stably expressing FLAG-CBP80 using lentiviral transduction. As positive controls we used cell lines expressing FLAG-tagged wild-type TERT (FLAG-TERT) or FLAG-tagged TCAB1; a FLAG-tagged TERT, carrying a deletion in the T motif of the telomerase RNA binding domain (FLAG-TERT DTRBD), was used as negative control. FLAG IP was carried out on nuclear extracts, and RNA and protein precipitates were analyzed by northern and western blotting (Figure 6D). As expected, FLAG-TERT and FLAG-TCAB1, but not FLAG-TERT DTRBD, efficiently precipitated hTR from extracts of both TGS1 mutant and TGS1-proficient cells (Figure 6D, lanes 1–4). Whereas hTR was undetectable by northern blotting in FLAG-CBP80 complexes from control cells, hTR was identified in FLAG-CBP80 complexes from TGS1-deficient cells (compare lanes 5 and 6 of Figure 6D). Using qRT-PCR on the RNAs co-immunoprecipitated with FLAG-CBP80, we found that hTR association with FLAG-CBP80 was increased by more than 2-fold in TGS1 mutant cells compared to controls (Figure 6E). U1 snRNA showed a greater than 5-fold increased association with FLAG-CBP80 in TGS1 mutant cells compared to controls (Figure S4A). Western blotting for CBP80 and CBP20 revealed that both subunits of the CBC complex are recovered in immunoprecipitates with TERT and that this interaction is weakened by mutations in the TERT RNA binding domain (Figure S4B, left panel). Furthermore, the TERT/CBC interaction was disrupted by treatment with RNase (Figure S4B, right panel), suggesting that a fraction of CBC bound-hTR is associated with assembled telomerase. These results indicate hypomethylated hTR molecules bind the CBC complex, which in TGS1 mutant cells is enriched in hTR, compared to TGS1-proficient cells.
TGS1-Deficiency Promotes Increased Association between hTR and the SmB Protein
In both yeast and human cells, telomerase RNA associates with the Sm protein complex during its processing into a mature form (Franke et al., 2008; Fu and Collins, 2006; Tang et al., 2012). To test whether TGS1 affects the binding of the Sm complex to hTR in human cells, we used an antibody directed against the SmB component of the Sm complex to perform RNA IP on cytoplasmic and nuclear extracts from TGS1 mutant or TGS1-rescued cells, and quantified the precipitated RNAs by qRT-PCR (Figures 6C and S4C). In these experiments, we used U1 snRNA for comparison and 5.8S ribosomal RNA as a negative control. While U1 association with SmB was unaffected by TGS1 deficiency, there was a 2-fold enrichment of hTR in SmB immunoprecipitates from nuclear extracts, of TGS1 mutant cells. However, this enrichment was not found in cytoplasmic extracts (Figures 6C and S4C). Thus, the interaction of hTR with the SmB protein in the nucleus is regulated by TGS1-mediated cap hypermethylation.
DISCUSSION
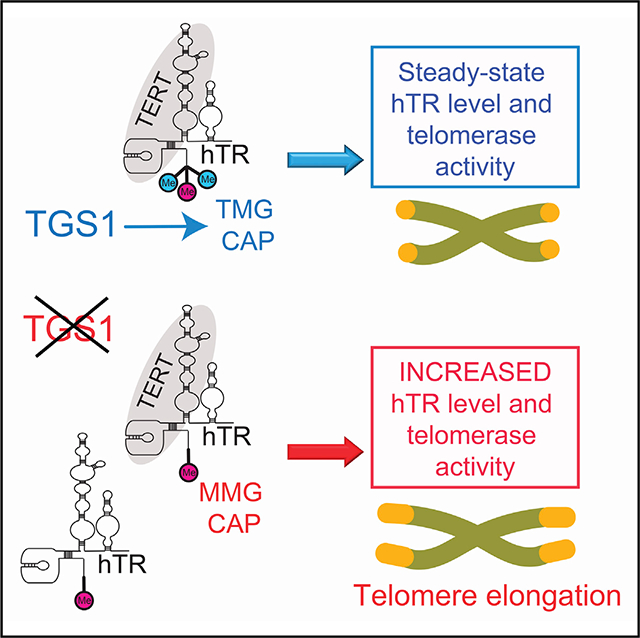
Cap hypermethylation has been implicated in nuclear import for snRNAs, but the role of cap hypermethylation for hTR had remained unknown. To understand the role of 5′ cap trimethylation in telomerase function, we generated cancer cell clones depleted of TGS1 through genome editing. Our data indicate that after hTR is transcribed and capped at the 5′ end, TGS1 hypermethylates the cap in a process that likely occurs in CBs (Figure 7). Surprisingly, hypermethylation of the hTR cap serves to suppress levels of hTR and to contain hTR in the nucleus and in CBs. In cells lacking TGS1, levels of hTR and levels of assembled active telomerase enzyme both increase, resulting in telomere elongation in vivo. Loss of trimethylation leads to increased association between hTR and CBP, which may facilitate inappropriate export to the cytoplasm. With CBs disrupted, hTR accumulates in nucleoli and escapes from the nucleus to the cytoplasm, where it gains increased association with the Sm complex. Thus, formation of a TMG cap on hTR is critical for limiting telomerase accumulation and facilitating nuclear retention (Figure 7).
Figure 7. Model Illustrating the Possible Role of TGS1 in hTR Biogenesis.

In TGS1-proficient cells (TGS1+), TGS1 restricts hTR interaction with the CBC and Sm complexes. hTR is enriched in CBs and excluded from nucleoli. In TGS1 mutant cells (TGS1−), hTR is monomethylated and acquires higher affinity for the CBC and Sm complexes, it is no longer enriched in CBs, relocates to nucleoli, and a fraction is exported into the cytoplasm. Total hTR levels increase, possibly because mislocalization sequesters hTR from the RNA degradation machinery, including the RNA exosome. A fraction of hTR may be reimported back into the nucleus, as it occurs for snRNAs and may be incorporated into telomerase. In the absence of TGS1, hTR is incorporated into functional telomerase complexes and telomerase activity is increased, proportional to hTR increase.
TGS1 Controls Levels of hTR and Telomerase
Several classes of small noncoding RNAs are hypermethylated by TGS1. This post-transcriptional modification of the 5′-CAP may represent a means of regulating subsequent downstream steps, altering the interaction with other RNA binding proteins and determining the composition of the RNP. Each class of noncoding RNA may be altered by CAP hypermethylation in a distinct manner. In the case of hTR, CAP trimethylation suppresses hTR accumulation and alters association with specific RNA binding proteins. One means of controlling hTR levels involves the rate of 3′ processing and maturation of precursor transcripts (Nguyen et al., 2015; Roake et al., 2019; Shukla et al., 2016; Tseng et al., 2015, 2018). Extended hTR precursors containing a few genomically templated nucleotides beyond the annotated 451-nt end are converted into the mature hTR form either directly by exonucleolytic processing or following an alternate pathway in which they are first oligoadenylated by PAPD5 followed by subsequent trimming by PARN (Roake et al., 2019). We found that TGS1 deficiency does not affect the frequency of the extended hTR precursors carrying short genomically templated tails, suggesting that the hTR increase observed in TGS1 mutant cells is not due to variations in the level of these precursors.
Previous studies have shown that PARN deficiency results in accumulation of oligoadenylated hTR precursors and reduction in mature hTR (Boyraz et al., 2016; Moon et al., 2015; Roake et al., 2019; Shukla et al., 2016; Tummala et al., 2015). Oligoadenylated precursors also accumulate in cells depleted of the TOE1 exonuclease, which acts redundantly to PARN to promote hTR maturation (Deng et al., 2019; Son et al., 2018). A subset of oligo-adenylated transcripts are degraded by the exosome (Tseng et al., 2015), and depletion of the exosome components leads to an increase in mature hTR (Nguyen et al., 2015; Tseng et al., 2015). Our finding that in TGS1-deficient cells the fraction of oligoadenylated species is comparable to that found in TGS1-proficient cells suggests that the increase in mature hTR is not mediated by variations in the maturation kinetics of hTR short precursors.
We found that TGS1 depletion leads to an increased abundance of hTR transcripts with very long tails, which are usually degraded by the exosome (Tseng et al., 2015). These long molecules may accumulate due to defective transcription termination, defective clearance by the exosome, or a combination of both. Depletion of exosome components leads to increases in both long hTR transcripts and mature hTR transcripts. However, longer transcripts are largely degraded, suggesting that they are not the primary hTR precursors (Tseng et al., 2015). Moreover hTR molecules with short genomic tails (12 nt) have been shown to comprise the bulk of nascent hTR molecules, and no enrichment of hTR molecules carrying extended tail sequences longer than 12 nt has been observed by nascent RNA sequencing (Roake et al., 2019). Furthermore, we showed that in TGS1 mutant cells the bulk of hTR molecules assembled with TERT are mature forms. Collectively, these results suggest that neither abnormally processed RNA precursors, nor ultra-long hTR transcripts substantially contribute to the increase in mature hTR in TGS1 mutant cells.
Trimethylation Suppresses Cytoplasmic hTR Localization
One relevant difference between control and TGS1-depleted cells is in the proteins that associate with hTR and in the cytoplasmic enrichment in hTR. TGS1-deficient cells exhibit an increased concentration of hTR molecules in the cytoplasm compared to control cells. We found that cytoplasmic hTR is not increased in PAPD5-depleted cells that also exhibit an increase in the nuclear hTR abundance (Shukla et al., 2016). This suggests that the cytoplasmic enrichment of hTR in TGS1 mutant cells is not a mere outcome of the increased abundance of nuclear hTR but is instead a specific consequence of TGS1 deficiency. It has been hypothesized that DKC1 might play a role in counteracting ectopic accumulation of hTR in the cytoplasm (Shukla et al., 2016). In TGS1 mutant cells, cytoplasmic hTR molecules are not bound by TERT. However, it is likely that these mature hTR molecules are associated with DKC1, which binds hTR cotranscriptionally (Darzacq et al., 2006). DKC1 might favor hTR reimport into the nucleus and assembly with TERT (Shukla et al., 2016).
Consistent with the high CBC affinity for monomethylated 5′ caps, we found that in the nuclei of TGS1-deficient cells there is an increase in CBC-bound hTR molecules. We also observed an increase in hTR molecules associated with the SmB protein. In S. pombe, immature telomerase RNA (TER) associates with the Sm protein complex, that favors its processing into mature form; the Sm complex is then replaced by the Lsm complex, following TER hypermethylation by Tgs1. The Sm/Lsm switch is defective in S. pombe Tgs1 mutants, in which the association between Sm proteins and hTR is more stable than in wild-type (Tang et al., 2012). Although in S. pombe, loss of Tgs1 ultimately leads to a reduction in the telomerase level, it is conceivable that in human cells the fraction of hTR molecules bound to Sm is more stable and possibly more resistant to exosome-mediated degradation (see model in Figure 7).
Trimethylation Controls Trafficking
We found that loss of TGS1 disrupts CB organization and favors hTR accumulation in the nucleoli. Subnuclear localization of hTR has been implicated in telomerase biogenesis and recruitment at telomeres (Bizarro et al., 2019; Jády et al., 2006; Schmidt et al., 2016; Tomlinson et al., 2006; Venteicher et al., 2009; Xi et al., 2015). However, CB integrity is not required for telomerase assembly (Chen et al., 2015, 2018) and hTR accumulation in the nucleoli does occur also when the CBs are not disrupted and the overall hTR level is normal (Freund et al., 2014; Venteicher et al., 2009). Studies conducted in different mutant backgrounds did not reveal any obvious relationships between the status of the CB, hTR subnuclear localization, and its steady-state level (Table S2). However, our findings that in TGS1 mutant cells both hTR and the scaRNA U93 aberrantly accumulate in the nucleolus, and previous work showing that TGS1 negatively regulates nucleolar localization of snoRNAs (Verheggen and Bertrand, 2012), suggest the possibility that TGS1 may have a master regulatory role in directing the intranuclear trafficking of small RNAs, including hTR, scaRNAs, and snoRNAs.
TGS1 has different RNA targets that differ in cellular localization and trafficking. For example, human snoRNAs are TMG capped in the nucleus and directly associate with TGS1, which is thought to prevent their transit to the nucleolus until it is displaced by the CRM1 nuclear export factor (Pradet-Balade et al., 2011; Verheggen and Bertrand, 2012). The hypothesis that TGS1 prevents transit to nucleoli is consistent with the observation that the U93 scaRNA is ectopically redirected to nucleoli in the absence of TGS1. snRNAs bind CBC in the nucleus and are exported to the cytoplasm through an interaction between CBC and the CRM1 and PHAX export factors (Ohno et al., 2000). Once in the cytoplasm, snRNAs bind the Sm complex that physically associates with TGS1. Following TMG cap formation and dissociation of the CBC complex, the snRNAs are reimported into the nucleus. We propose that hTR binding by CBC favors its export to the cytoplasm. It is possible that a fraction of this cytoplasmic hTR is reimported into the nucleus, contributing to the increased hTR abundance seen in TGS1 mutant cells. The change in the hTR subcellular localization induced by TGS1 deficiency might also protect a fraction of the hTR transcripts from degradation (a model illustrating these hypotheses is shown in Figure 7).
TGS1 deficiency leads to an increase in mature hTR, increased telomerase activity and telomere elongation. In TGS1-deficient cells, hTR is hypomethylated compared to controls, and yet associates with TERT resulting in a functional telomerase, providing direct evidence that formation of a TMG cap on human hTR is dispensable for telomerase activity. Previous studies showed that mutations in Tgs1 lead to an increase in telomere length in S. cerevisiae, although this increase has not been linked to augmented hTR abundance (Franke et al., 2008). In contrast, loss of Tgs1 induces reduction of telomerase RNA in S. pombe, possibly due to its different biogenesis mechanism of telomeric RNA (Tang et al., 2012). Thus, while the structure and the hypermethylase function of TGS1 are evolutionarily conserved (Hausmann et al., 2008), its role in telomerase regulation appears to be species-specific.
In conclusion, we would like to note that TGS1 deficiency is one of the few conditions that increase hTR abundance and telomerase activity, resulting in telomere extension. Recently, it has been shown that PAPD5 inhibition leads to an increase of hTR and telomerase activity (Roake et al., 2019), rescuing telomere length in dyskeratosis congenita embryonic stem cells (Fok et al., 2019). TGS1 downregulation might be exploited to devise additional therapeutic approaches for telomerase insufficiency in telomere syndromes caused by critical reductions in hTR and in disorders associated with human aging.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further requests for information should be directed to the Lead Contact, Grazia Daniela Raffa, at graziadaniela.raffa@uniroma1.it. All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HeLa, HEK293 and BJ-HELT cells were cultured in DMEM supplemented with 10% fetal bovine serum and Penicillin-Streptomycin at 37°C, in 5% CO2. UMUC 3 cells were cultured in EMEM EBSS supplemented with 2mM Glutamine, 0.1mM Non Essential Amino Acids, 10% fetal bovine serum, 1.5g/L sodium bicarbonate, 1mM sodium pyruvate.
METHOD DETAILS
Cell Culture, Transfections, Generation of TGS1-CRISPR HeLa Cell Lines, and Transductions
Lipofectamine 2000 (Life Technologies) was used for all cDNA transfection experiments. TGS1-CRISPR cells were generated by transfection of HeLa cells with pSpCas9–2A-GFP (PX458) plasmid (Ran et al., 2013) encoding 3x FLAG Cas9-T2A-GFP and guide RNAs to the TGS1 locus. Sequences of guide RNAs are: TGS1–1: AGAGAAACATTTCCGCCACG; TGS1–2: TGTCAGAGCGTATCTT CAGC. GFP-positive cells were single-cell sorted into 96 well plates by FACS, and clones carrying mutations affecting TGS1 expression were screened by immunoblotting using anti TGS1 polyclonal antibody (see below).
To generate TGS1 mutants cells stably expressing FLAG-TGS1 (TGS1 rescued cells), we transfected 293T cells with the pCDH-CMV-PURO-3xFLAG-TGS1 construct (FLAG-TGS1) and packaging constructs; 48 hours later, viral supernatant was collected and concentrated using Retro-X (Clontech). HeLa TGS1 mutant clones were transduced in the presence of 5 μg/ml polybrene and selected in 2 μg/mL puromycin. erated TGS1 mutant cells expressing FLAG-GFP, were generated in a similar way using a pCDH-CMV-PURO-3xFLAG-GFP construct.
For TGS1 knockdown, UMUC 3 cells (Xu and Blackburn, 2007) or (BJ-HELT) fibroblasts (expressing hTERT and SV40 early region) (Hahn et al., 1999) were treated for up to 10 days (every 72 hr) with 50 nM SMARTpool: siGENOME TGS1 siRNA or ON-TARGET plus Non-targeting siRNA using Dharmafect I (Dharmacon, Horizon). The PARN KO and PAPD5 KO cell lines are described in Roake et al. (2019)
Immunofluorescence Staining (IF) and RNA Fluorescence In Situ Hybridization (FISH)
IF experiments were carried out on cells grown on coverslips. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS. Coverslips were incubated with primary antibodies in 3%BSA for 1 hr at room temperature: anti-Coilin (Abcam, AB11822, 25ng/mL); anti-TCAB1 (Venteicher et al., 2009, 25 ng/mL). Coverslips were washed 3x with PBS and incubated with secondary Alexa Fluor conjugated antibodies (Jackson Immunoresearch). Coverslips were washed 3x in PBS, counterstained in a 300 nM DAPI solution and mounted in ProLong Gold Anti-fade Mountant.
RNA FISH experiments to detect hTR, U93scaRNA, U2 snRNA or U13 snoRNA were performed with Custom Stellarisâ FISH Probes (SMF-1000 Series; Quasar 570 conjugated, 75 nM working concentration) from Biosearch Technologies, following the manufacturer’s protocol. Probe sequences are listed in the Key Resources Table. Coverslips were mounted in 2x SSC for analysis. Images were captured on a Leica wide-field fluorescence microscope and processed using Leica LAS AF and Photoshop. Error bars, S.E.M, and significance were calculated by one-way ANOVA with GraphPad Prism.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Coilin mouse monoclonal antibody | Abcam | Cat# ab11822, RRID: AB_2081428 |
| TCAB1 rabbit polyclonal antibody | Venteicher et al., 2009 | N/A |
| TERT (T421) rabbit polyclonal antibody | Venteicher et al., 2009 | N/A |
| TMG cap specific (R1131) polyclonal antibody | Synaptic Systems | Cat# 201 002 |
| SmB mouse monoclonal antibody | A gift from Livio Pellizzoni | Carissimi et al., 2006 |
| CBP20/NCBP2 rabbit antibody | Bethyl Laboratories | Cat# A302–553A; RRID: AB_2034872 |
| CBP80/NCBP1 rabbit polyclonal antibody | Bethyl Laboratories | Cat# A301–793A; RRID: AB_1211224 |
| Actinin (clone AT6/172) mouse monoclonal antibody | Millipore | Cat# MAB1682; RRID: AB_94325 |
| Tubulin mouse monoclonal antibody | Santa Cruz | Cat# SC-5274; RRID: AB_2288090 |
| Lamin B (M21) antibody | Santa Cruz | Cat# sc-6217; RRID: AB_648158 |
| pimt/TGS1 rabbit polyclonal antibody | Bethyl Laboratories | Cat#A300–814A (lot 1) |
| FLAG (clone M2) mouse monoclonal antibody | Sigma | Cat# F1804; RRID: AB_262044 |
| DKC1 | A gift from Philip J. Mason | Mochizuki et al., 2004 |
| Rabbit Anti-Fibrillarin antibody - (ab5821) | abcam | Abcam Cat#ab5821; RRID:AB_2105785 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| InSolution Leptomycin B, Streptomyces sp. - CAS 87081-35-4 | Millipore | Cat# 431050 |
| Critical Commercial Assays | ||
| Kapa Hyperprep DNA Library Prep Kit | Kapa | Cat# KK8502 |
| Kapa Single-Indexed adapters | Kapa | Cat# KK8700 |
| Deposited Data | ||
| RNAend-Seq data | This study | https://www.ncbi.nlm.nih.gov/sra/PRJNA594795 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | Cat# CCL-2.2 |
| HEK293 | ATCC | Cat# CRL-11268 |
| UMUC3 | Xu and Blackburn, 2007 | N/A |
| BJ-HELT | Hahn et al., 1999 | N/A |
| Oligonucleotides | ||
| Sequence | Description | Oligo Name |
| AGAGAAACATTTCCGCCACG | CRISPR guide RNA | sgTGS1–1 |
| TGTCAGAGCGTATCTTCAGC | CRISPR guide RNA | sgTGS1–2 |
| ATGCAGTCGAGTTTCCCACAT | NB probe | U1 Probe |
| GGCCATGCTAAATGTTGTTCTTCTCTGTATCGTTCCAATT | NB probe | U6 probe |
| CGTGTAGAGCACCGAAAACC | NB probe | U3 probe |
| CGCTGTTTTTCTCGCTGACT | qRTPCR primer | hTR F |
| GCTCTAGAATGAACGGTGGAA | qRTPCR primer | hTR R |
| AGCCACATCGCTCAGACAC | qRTPCR primer | GAPDH F |
| GCCCAATACGACCAAATCC | qRTPCR primer | GAPDH R |
| TCAAGATCATTGCTCCTCCTGAG | qRTPCR primer | actin B F |
| ACATCTGCTGGAAGGTGGACA | qRTPCR primer | actin B R |
| GGCTATTACATCAGAGACAGTG | qRTPCR primer | TGS1 F |
| GAATCAAGTTCACTTTCATCCA | qRTPCR primer | TGS1 R |
| CCATGATCACGAAGGTGGTTT | qRTPCR primer | U1 F |
| ATGCAGTCGAGTTTCCCACAT | qRTPCR primer | U1 R |
| GGTGGATCACTCGGCTCGT | qRTPCR primer | 5.8S F |
| GCAAGTGCGTTCGAAGTGTC | qRTPCR primer | 5.8S R |
| AGTTCGCTTTCCTGTTGGTG | qRTPCR primer | hTR ext 1F |
| ATTCATTTTGGCCGACTTTG | qRTPCR primer | hTR ext 1R |
| GGGTGTGGGAGAACAGTCAT | qRTPCR primer; Tseng et al., 2015 | hTR ext 2F |
| ACCTCTGGCATAAACCGATG | qRTPCR primer; Tseng et al., 2015 | hTR ext 2R |
| CTACGTAACGATTGATGGTGCCTACAG | 3′ RACE primer | Univ RT |
| CACCGCGAAGAGTTGGGCTCTG | 3′ RACE primer | Univ RT |
| CCCTAACCCTAACCCTAA | 32P End labeled oligonucleotide probe for TRF | TRF primer |
| AATCCGTCGAGCAGAGTT | 32P End labeled TS primer for TRAP | TS* primer |
| ATTGTCCTCGGATAGAGGAC | RNA FISH | U2 snRNA FISH-1 |
| ATACCAGGTCGATGCGTGGA | RNA FISH | U2 snRNA FISH-2 |
| CACCGTTCCTGGAGGTACTG | RNA FISH | U2 snRNA FISH-3 |
| GCACATCTCACACAAGCGTA | RNA FISH | U13 snoRNA FISH-1 |
| GTCGTAACAAGGTTCAAGGG | RNA FISH | U13 snoRNA FISH-2 |
| GTCAGACGGGTAATGTGCCC | RNA FISH | U13 snoRNA FISH-3 |
| GCCCTTCTCAGTTAGGGTTA | RNA FISH | hTR FISH-1 |
| AAGTCAGCGAGAAAAACAGC | RNA FISH | hTR FISH-2 |
| TCTAGAATGAACGGTGGAAG | RNA FISH | hTR FISH-3 |
| CCAGCAGCTGACATTTTTTG | RNA FISH | hTR FISH-4 |
| GCTGACAGAGCCCAACTCTT | RNA FISH | hTR FISH-5 |
| GTCCCACAGCTCAGGGAATC | RNA FISH | hTR FISH-6 |
| CATGTGTGAGCCGAGTCCTG | RNA FISH | hTR FISH-7 |
| cggctccaagactacagatt | RNA FISH | U93 scaRNA FISH-1 |
| aacagctggctctcgagcag | RNA FISH | U93 scaRNA FISH-2 |
| atcagaggaaaattgcacat | RNA FISH | U93 scaRNA FISH-3 |
| gtggcaacagtgaccagaaa | RNA FISH | U93 scaRNA FISH-4 |
| aatgacatagcccagtcatt | RNA FISH | U93 scaRNA FISH-5 |
| ctcttactgttggcggatag | RNA FISH | U93 scaRNA FISH-6 |
| caatatctcgactgcaaagc | RNA FISH | U93 scaRNA FISH-7 |
| cttgtggcagtacttagtgt | RNA FISH | U93 scaRNA FISH-8 |
| Recombinant DNA | ||
| pSpCas9–2A-GFP (PX458) | N/A | RRID:Addgene_48138 |
| pCDH-CMV-PURO-3xFLAG-TGS1 | This study | N/A |
| pCDH-CMV-PURO-3xFLAG-GFP | This study | N/A |
| pCDH-CMV-PURO-3xFLAG-TERT | Chen et al., 2018 | N/A |
| pCDH-CMV-PURO-3xFLAG-TERT-TRBD | Chen et al., 2018 | N/A |
| pCDH-CMV-PURO-3xFLAG-CBP80 | This study | N/A |
| Software and Algorithms | ||
| NGS tools fastq-join and FastQC | Galaxy | https://usegalaxy.org/ |
| Custom python scripts for 3′-RACE | This study | https://cmroake.people.stanford.edu/links-python-scripts |
| GraphPad Prism Software | Graphpad | GraphPad Prism, RRID:SCR_002798 |
| Adobe Photoshop | Adobe | Adobe Photoshop, RRID:SCR_014199 |
| Other | ||
| SMART pool: ON-TARGETplus TGS1 siRNA | Dharmacon-Horizon Discovery | Cat#:L-017151-00-0005 |
| ON-TARGETplus Non-targeting Control Pool | Dharmacon-Horizon Discovery | Cat#:D-001810-10-05 |
Northern Blotting
RNA was extracted with TRIzol (Life Technologies,15596–026), separated by electrophoresis on a 5% urea TBE gel, transferred onto Hybond-N membrane (GE Healthcare, RPN303N), and hybridized with ULTRAhyb (Ambion, AM8670). hTR probe was generated by random oligo-labeling of DNA encoding full-length hTR (1–451) with Prime-It II Random Primer Labeling Kit (Agilent, 300385). U1, U6 and U3 probes were generated by end labeling antisense oligos (listed in Key Resources Table) using T4 polynucleotide kinase.
RNA immunoprecipitation and Immunopurification of Endogenous Telomerase RNPs and TMG Capped RNA
Nuclear and cytoplasmic extracts were prepared according to Chen et al. (2014). Endogenous telomerase RNPs were purified by incubating nuclear or cytoplasmic extract with 30 μg Rabbit polyclonal anti-TERT antibody (T421) (Venteicher et al., 2009). Immunocomplexes were precipitated by addition of protein A-agarose Fast Flow (P3476 Sigma) and washed extensively with Lys450 (20 mM HEPES-NaOH pH7.9, 450 mM NaCl, 0.5% Triton X-100, 10 mM KCl, 4 mM MgCl2, 0.2mM EDTA, 10% Glycerol, freshly add 1 mM DTT, 200 μM PMSF, and Protease Inhibitor Cocktail [P8340 Sigma]). One half of the sample was subjected to Northern blot analysis, while the other half was incubated with Trizol reagent for RNA extraction. Purified RNA was then subjected to IP with the R1131 anti-TMG cap specific antibody (Bringmann et al., 1983) (Synaptic Systems). One tenth of the sample was saved as input RNA. For RNA immunoprecipitation of naked RNA, 50 μl protein G beads were washed with PBS and blocked with 20 μg tRNA and 20 μg BSA, then washed with NT2 buffer (50mM Tris/HCl pH 7.5; 150mM NaCl; 1mM MgCl2; 0.05% NP40; 1μM DTT; 100U/mL RNasin; 400μM VRC, Vanadyl ribonucleoside complexes) and coupled O/N to either 20 μL of anti-TMG cap antibody or 1 μg IgG in 250 μL NT2 buffer. Beads were then briefly washed with NT2 buffer and incubated with Trizol-purified RNA in 250 μL NT2 buffer for 2 hr at 4°C, while rotating. Beads were then washed 6 times for 2′ while rotating with NT2 buffer and precipitated RNA was Trizol-extracted. Both TMG-immunopurified and input RNAs were then treated with DNase and subjected to reverse transcription and qRT PCR. For immunopurification of SmB or CBC-interacting RNAs, extracts prepared as above were precipitated with the following antibodies: monoclonal mouse anti-SmB (a gift from Livio Pellizzoni, Columbia University) (Carissimi et al., 2006); Rabbit anti-CBP80/NCBP1 (Cat# A301–793A) and anti-CBP20/NCBP2 (Cat# A302–553A) both obtained from Bethyl Laboratories, INC.
For Immunopurification of RNAs interacting with FLAG-tagged proteins, lysates were prepared as described above from HeLa cells expressing either of the following FLAG-tagged proteins (from pCDH-CMV-3xFLAG lentiviral vectors): FLAG TERT, FLAG TERT-TRBD (hTERT mutant containing deletion of aa 542–593, the conserved Motif T); FLAG CBP80. 40 μl of anti-FLAG M2 affinity gel (Sigma, A2220) was washed 3x with PBS then added to HeLa lysates (2–4 mg) diluted in 500 ml NP40 buffer (see above), and incubated for 1 hr at 4°C with rotation. Resin was pelleted (5,000 xg, 30 s) and washed 4x with 1 mL NP-40 lysis buffer, for 10 min at 4°C while rotating. The final pellet was divided into two fractions for protein and RNA analyses. For protein analysis, pellet was eluted in 30 μl 2x SDS-PAGE sample buffer and subjected to western blot. For RNA analyses, pellet was extracted with TRIzol, and RNA was recovered in 20 μl RNase-free water. A recovery control (0.2 ng of in vitro transcribed TERC fragment, bases 1–170) was added to each RNA sample before beginning TRIzol extraction. 10 μl were subjected to Northern Blot analysis, the remaining RNA was reverse-transcribed and used for qRTPCR.
Preparation of Cell Extracts and Telomere Repeats Amplification Protocol (TRAP)
Cells were lysed in NP40 buffer (25 mM HEPES-KOH ([pH 7.5]), 150 mM KCl, 1.5 mMMgCl2, 0.5% NP40, 10% Glycerol supplemented with protease inhibitors (ROCHE, 04693159001) for 20 min on ice. Lysate was clarified by centrifugation at 16,000 g for 10 min and quantified with the Bio-Rad Protein Assay Kit (Bio-Rad, 5000002). For TRAP assay, Telomerase activity was assayed with a two-step TRAP procedure, according to Kim and Wu (1997). Cell extracts prepared as above were incubated with telomeric primers for extension reaction in a thermocycler for 30 min at 30°C, and 5 min at 72°C. 1 μL of the reaction was subjected to PCR amplification (24 cycles of 30 s at 94°C, followed by 30 s at 59°C) in the presence of 32P end-labeled telomeric primers (purified using a micro-spin G-25 column; GE healthcare, 27-5325-01). The PCR reactions were resolved by 9% polyacrylamide gel electrophoresis at room temperature. The gel was exposed to a phosphor-imager and digital images of autoradiography were acquired with a Typhoon scanner (GE Healthcare). The scanned images were quantitated using the TotalLab Quant software. Multiple biological replicates (from independently derived cell clones) and technical replicates were included in each assay.
Telomere Restriction Fragment Analysis
To measure telomere lengths by telomere restriction fragment analysis, cells were harvested and digested with Proteinase K at 6 μg/mL overnight. DNA was extracted by the phenol-chloroform method and digested overnight with Hinfl and RsaI before electrophoresis and Southern blotting with end labeled (CCCTAA)4 oligonucleotide probe.
Cell Fractionation and Western Blotting
Nuclear and cytoplasmic extracts were obtained according to Chen et al. (2014). Alternatively, cells were lysed according to Méndez and Stillman (2000) in 10 mM HEPES, [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1 mM phenylmethylsulfonyl fluoride. Triton X-100 (0.1%) was added, and the cells were incubated for 5 min on ice. The cytoplasmic fraction was collected by centrifugation at 1300g for 4 min. After washing with the buffer above, the pellet (containing nuclei) was suspended in 3 mM, EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and protease inhibitors, and incubated for 30 min. RNA was Trizol extracted from both fractions. For western blotting, 10–50 μg were separated by SDS-PAGE, transferred onto PVDF membrane (GE Healthcare, RPN303F), and blotted according to standard procedures. 5% milk in PBST (0.1% Tween) was used for all blocking and antibody incubation steps; PBST (0.1% Tween) was used for all washes. Commercially available antibodies used for WB are listed in the Key Resources Table. The anti-DKC1 antibody is a Gift from Philip J. Mason (Mochizuki et al., 2004)
Quantitative Real Time PCR (qRTPCR)
Total RNA was extracted with Trizol reagent (Ambion), treated with Ambion DNase I (RNase-free) extracted with phenol/chloroform. The integrity of RNA samples was evaluated by gel electrophoresis. 1 μg of intact RNA (with a 28S:18S rRNA ratio = 2:1) was reverse transcribed with the RevertAid H Minus Reverse Transcriptase kit (Thermo Scientific, EP0451). Real-time PCR reactions were performed with the Brilliant II SYBR® Green QPCR Master Mix (Agilent, 600828)
The relative quantification in gene expression was carried out using the 2-ΔΔCt method (Livak and Schmittgen, 2001). Using this method, we obtained the fold changes in gene expression normalized to the GAPDH or the indicated reference genes (the amplification efficiencies were not significantly different for target and reference among all samples). A total of 3 experiments were performed for three biological replicates and the significance was assessed by either the Student’s t tests or the one-way ANOVA: For qRTPCR of immunoprecipitated RNA, samples were processed as above; Fold change was calculated by normalizing each RIP sample to the relative input.
3′ RACE-Sequencing
600 ng of RNA was ligated to 5 uM of 5′ adenylated, 3′ blocked adaptor (Universal miRNA cloning linker, NEB S1315S) with 250 units of T4 RNA ligase truncated KQ (NEB M0373S), 25% PEG 8000, and 1 μL RnaseOUT (ThermoFisher 10777019) in a 20 μL reaction at 25 degrees for 16 hours. Ligated RNA was cleaned up with RNA clean and concentrator columns (Clontech 740955.50) and DNase treatment, cDNA was synthesized with universal primer and SuperScript III (ThermoFisher 18080093). Amplification was carried out with Phusion (New England Biosystems M0530) and primer sets universal/TERCR1 (listed in the Key Resources Table). PCR products were directly run on an 8% PAGE gel and visualized with SYBR Gold (ThermoFisher S-11494), or subjected to AMPure XP beads (Beckman Coulter A63881) for PCR cleanup and library preparation. Libraries were prepped using Kapa Hyperprep Kit (Kapa KK8504), quantified with Qubit and bioanalyzer, and run on Illumina miSeq at the Stanford Functional Genomics Facility. Reads were paired using fastq-join tool at Galaxy (http://usegalaxy.org). Reads were binned into the various forms of hTR using custom python scripts (https://cmroake.people.stanford.edu/links-python-scripts) and the number of reads in each bin was normalized to total hTR reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with GraphPad Prism Software. Error bars presented represent S.E.M; p < 0.05 was defined as significant. Statistical details can be found in Figure legends and in the Results section.
DATA AND CODE AVAILABILITY
The RNAend-Seq data have been deposited in Sequence Read Archive (BioProject accession number: PRJNA594795).
Supplementary Material
Highlights.
Trimethylguanosine synthase 1 (TGS1) catalyzes formation of a TMG cap on hTR
Loss of TGS1 results in hTR mislocalization to nucleoli and the cytoplasm
TGS1 depletion increases hTR and telomerase levels, leading to telomere elongation
ACKNOWLEDGMENTS
We thank Livio Pellizzoni for the gift of the anti-SmB antibody. This work was supported by NIH (AG056575 and CA197563 to S.E.A.), Telethon (GPP13147 to G.D.R.), and AIRC (IG 20528 to M.G.). L.C. was supported by a Stanford Cancer Institute 2018 Fellowship Award. C.M.R. was supported by MSTP Training Grant GM007365 and by a Gerald J. Lieberman Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.004.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. (2013). The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat. Struct. Mol. Biol 20, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, and Blackburn EH (2012). The telomere syndromes. Nat. Rev. Genet 13, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, and Greider CW (2005). Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl. Acad. Sci. USA 102, 15960–15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, et al. (2011). Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474, 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizarro J, Bhardwaj A, Smith S, and Meier UT (2019). Nopp140-mediated concentration of telomerase in Cajal bodies regulates telomere length. Mol. Cell Biol 30, 3136–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH, Epel ES, and Lin J (2015). Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 350, 1193–1198. [DOI] [PubMed] [Google Scholar]
- Boulon S, Verheggen C, Jady BE, Girard C, Pescia C, Paul C, Ospina JK, Kiss T, Matera AG, Bordonné R, and Bertrand E (2004). PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol. Cell 16, 777–787. [DOI] [PubMed] [Google Scholar]
- Boyraz B, Moon DH, Segal M, Muosieyiri MZ, Aykanat A, Tai AK, Cahan P, and Agarwal S (2016). Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J. Clin. Invest 126, 3377–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann P, Reuter R, Rinke J, Appel B, Bald R, and Lührmann R (1983). 5′-terminal caps of snRNAs are accessible for reaction with 2,2,7-trimethylguanosine-specific antibody in intact snRNPs. J. Biol. Chem 258, 2745–2747. [PubMed] [Google Scholar]
- Carissimi C, Saieva L, Gabanella F, and Pellizzoni L (2006). Gemin8 is required for the architecture and function of the survival motor neuron complex. J. Biol. Chem 281, 37009–37016. [DOI] [PubMed] [Google Scholar]
- Chen L, Ooi SK, Conaway JW, and Conaway RC (2014). Biochemical assays for analyzing activities of ATP-dependent chromatin remodeling enzymes. J. Vis. Exp (92), e51721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng Z, Jiang S, Hu Q, Liu H, Songyang Z, Ma W, Chen S, and Zhao Y (2015). Human cells lacking coilin and Cajal bodies are proficient in telomerase assembly, trafficking and telomere maintenance. Nucleic Acids Res. 43, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Roake CM, Freund A, Batista PJ, Tian S, Yin YA, Gajera CR, Lin S, Lee B, Pech MF, et al. (2018). An Activity Switch in Human Telomerase Based on RNA Conformation and Shaped by TCAB1. Cell 174, 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski A, Lubas M, Jensen TH, and Dziembowski A (2013). RNA decay machines: the exosome. Biochim. Biophys. Acta 1829, 552–560. [DOI] [PubMed] [Google Scholar]
- Cristofari G, and Lingner J (2006). Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 25, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofari G, Adolf E, Reichenbach P, Sikora K, Terns RM, Terns MP, and Lingner J (2007). Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol. Cell 27, 882–889. [DOI] [PubMed] [Google Scholar]
- Darzacq X, Kittur N, Roy S, Shav-Tal Y, Singer RH, and Meier UT (2006). Stepwise RNP assembly at the site of H/ACA RNA transcription in human cells. J. Cell Biol 173, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng T, Huang Y, Weng K, Lin S, Li Y, Shi G, Chen Y, Huang J, Liu D, Ma W, and Songyang Z (2019). TOE1 acts as a 3′ exonuclease for telomerase RNA and regulates telomere maintenance. Nucleic Acids Res. 47, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok WC, Shukla S, Vessoni AT, Brenner KA, Parker R, Sturgeon CM, and Batista LFZ (2019). Posttranscriptional modulation of TERC by PAPD5 inhibition rescues hematopoietic development in dyskeratosis congenita. Blood 133, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke J, Gehlen J, and Ehrenhofer-Murray AE (2008). Hypermethylation of yeast telomerase RNA by the snRNA and snoRNA methyltransferase Tgs1. J. Cell Sci 121, 3553–3560. [DOI] [PubMed] [Google Scholar]
- Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, and Artandi SE (2014). Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 159, 1389–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, and Collins K (2006). Human telomerase and Cajal body ribonucleo-proteins share a unique specificity of Sm protein association. Genes Dev. 20, 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, and Collins K (2007). Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol. Cell 28, 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gable DL, Gaysinskaya V, Atik CC, Talbot CC Jr., Kang B, Stanley SE, Pugh EW, Amat-Codina N, Schenk KM, Arcasoy MO, et al. (2019). ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 33, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard C, Verheggen C, Neel H, Cammas A, Vagner S, Soret J, Bertrand E, and Bordonné R (2008). Characterization of a short isoform of human Tgs1 hypermethylase associating with small nucleolar ribonucleoprotein core proteins and produced by limited proteolytic processing. J. Biol. Chem 283, 2060–2069. [DOI] [PubMed] [Google Scholar]
- Goldfarb KC, and Cech TR (2013). 3′ terminal diversity of MRP RNA and other human noncoding RNAs revealed by deep sequencing. BMC Mol. Biol 14, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, and Weinberg RA (1999). Inhibition of telomerase limits the growth of human cancer cells. Nat. Med 5, 1164–1170. [DOI] [PubMed] [Google Scholar]
- Hamm J, Darzynkiewicz E, Tahara SM, and Mattaj IW (1990). The trimethylguanosine cap structure of U1 snRNA is a component of a bipartite nuclear targeting signal. Cell 62, 569–577. [DOI] [PubMed] [Google Scholar]
- Hausmann S, Zheng S, Costanzo M, Brost RL, Garcin D, Boone C, Shuman S, and Schwer B (2008). Genetic and biochemical analysis of yeast and human cap trimethylguanosine synthase: functional overlap of 2,2,7-trimethylguanosine caps, small nuclear ribonucleoprotein components, pre-mRNA splicing factors, and RNA decay pathways. J. Biol. Chem 283, 31706–31718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jády BE, Bertrand E, and Kiss T (2004). Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J. Cell Biol 164, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jády BE, Richard P, Bertrand E, and Kiss T (2006). Cell cycle-dependent recruitment of telomerase RNA and Cajal bodies to human telomeres. Mol. Biol. Cell 17, 944–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, and Wu F (1997). Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res. 25, 2595–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, Fayet E, Jády BE, Richard P, and Weber M (2006). Biogenesis and intranuclear trafficking of human box C/D and H/ACA RNPs. Cold Spring Harb. Symp. Quant. Biol 71, 407–417. [DOI] [PubMed] [Google Scholar]
- Lemm I, Girard C, Kuhn AN, Watkins NJ, Schneider M, Bordonné R, and Lührmann R (2006). Ongoing U snRNP biogenesis is required for the integrity of Cajal bodies. Mol. Biol. Cell 17, 3221–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- MacNeil DE, Lambert-Lanteigne P, and Autexier C (2019). N-terminal residues of human dyskerin are required for interactions with telomerase RNA that prevent RNA degradation. Nucleic Acids Res. 47, 5368–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone A, Stevens D, Vulliamy T, Dokal I, and Mason PJ (2004). Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood 104, 3936–3942. [DOI] [PubMed] [Google Scholar]
- Méndez J, and Stillman B (2000). Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (2014). Exosome substrate targeting: the long and short of it. Biochem. Soc. Trans 42, 1129–1134. [DOI] [PubMed] [Google Scholar]
- Mitchell JR, Cheng J, and Collins K (1999). A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol 19, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki Y, He J, Kulkarni S, Bessler M, and Mason PJ (2004). Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc. Natl. Acad. Sci. USA 101, 10756–10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, Tai AK, and Agarwal S (2015). Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat. Genet 47, 1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouaikel J, Verheggen C, Bertrand E, Tazi J, and Bordonné R (2002). Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol. Cell 9, 891–901. [DOI] [PubMed] [Google Scholar]
- Mouaikel J, Bujnicki JM, Tazi J, and Bordonné R (2003a). Sequencestructure-function relationships of Tgs1, the yeast snRNA/snoRNA cap hypermethylase. Nucleic Acids Res. 31, 4899–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouaikel J, Narayanan U, Verheggen C, Matera AG, Bertrand E, Tazi J, and Bordonné R (2003b). Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep. 4, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Achsel T, Lührmann R, and Matera AG (2004). Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol. Cell 16, 223–234. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Grenier St-Sauveur V, Bergeron D, Dupuis-Sandoval F, Scott MS, and Bachand F (2015). A Polyadenylation-Dependent 3′ End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. Cell Rep. 13, 2244–2257. [DOI] [PubMed] [Google Scholar]
- Ohno M, Segref A, Bachi A, Wilm M, and Mattaj IW (2000). PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell 101, 187–198. [DOI] [PubMed] [Google Scholar]
- Palm W, and de Lange T (2008). How shelterin protects mammalian telomeres. Annu. Rev. Genet 42, 301–334. [DOI] [PubMed] [Google Scholar]
- Pickett HA, Cesare AJ, Johnston RL, Neumann AA, and Reddel RR (2009). Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 28, 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradet-Balade B, Girard C, Boulon S, Paul C, Azzag K, Bordonné R, Bertrand E, and Verheggen C (2011). CRM1 controls the composition of nucleoplasmic pre-snoRNA complexes to licence them for nucleolar transport. EMBO J. 30, 2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roake CM, Chen L, Chakravarthy AL, Ferrell JE Jr., Raffa GD, and Artandi SE (2019). Disruption of Telomerase RNA Maturation Kinetics Precipitates Disease. Mol. Cell 74, 688–700.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roithová A, Klime sová K, Pánek J, Will CL, Lührmann R, Stanek D, and Girard C (2018). The Sm-core mediates the retention of partially-assembled spliceosomal snRNPs in Cajal bodies until their full maturation. Nucleic Acids Res. 46, 3774–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau P, and Autexier C (2015). Telomere biology: Rationale for diagnostics and therapeutics in cancer. RNA Biol. 12, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Zaug AJ, and Cech TR (2016). Live Cell Imaging Reveals the Dynamics of Telomerase Recruitment to Telomeres. Cell 166, 1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Erdjument-Bromage H, and Shuman S (2011). Composition of yeast snRNPs and snoRNPs in the absence of trimethylguanosine caps reveals nuclear cap binding protein as a gained U1 component implicated in the cold-sensitivity of tgs1D cells. Nucleic Acids Res. 39, 6715–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Schmidt JC, Goldfarb KC, Cech TR, and Parker R (2016). Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat. Struct. Mol. Biol 23, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son A, Park JE, and Kim VN (2018). PARN and TOE1 Constitute a 3′ End Maturation Module for Nuclear Non-coding RNAs. Cell Rep. 23, 888–898. [DOI] [PubMed] [Google Scholar]
- Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. (2015). Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet 47, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Kannan R, Blanchette M, and Baumann P (2012). Telomerase RNA biogenesis involves sequential binding by Sm and Lsm complexes. Nature 484, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson RL, Ziegler TD, Supakorndej T, Terns RM, and Terns MP (2006). Cell cycle-regulated trafficking of human telomerase to telomeres. Mol. Biol. Cell 17, 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, and Baumann P (2015). Human Telomerase RNA Processing and Quality Control. Cell Rep. 13, 2232–2243. [DOI] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Schroeder MR, and Baumann P (2018). The H/ACA complex disrupts triplex in hTR precursor to permit processing by RRP6 and PARN. Nat. Commun 9, 5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, Powell J, Cooper N, Foster A, Mohammed S, et al. (2015). Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Invest 125, 2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycowski KT, Shu MD, Kukoyi A, and Steitz JA (2009). A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol. Cell 34, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, and Artandi SE (2009). A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheggen C, and Bertrand E (2012). CRM1 plays a nuclear role in transporting snoRNPs to nucleoli in higher eukaryotes. Nucleus 3, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, Piatyszek MA, Rainey WE, Byrd W, and Shay JW (1996). Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet 18, 173–179. [DOI] [PubMed] [Google Scholar]
- Wurth L, Gribling-Burrer AS, Verheggen C, Leichter M, Takeuchi A, Baudrey S, Martin F, Krol A, Bertrand E, and Allmang C (2014). Hypermethylated-capped selenoprotein mRNAs in mammals. Nucleic Acids Res. 42, 8663–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi L, and Cech TR (2014). Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res. 42, 8565–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi L, Schmidt JC, Zaug AJ, Ascarrunz DR, and Cech TR (2015). A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 16, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, and Blackburn EH (2007). Human cancer cells harbor T-stumps, a distinct class of extremely short telomeres. Mol. Cell 28, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedavalli VS, and Jeang KT (2010). Trimethylguanosine capping selectively promotes expression of Rev-dependent HIV-1 RNAs. Proc. Natl. Acad. Sci. USA 107, 14787–14792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, and Artandi SE (2011). Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 25, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, and Artandi SE (2012). TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell 150, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tomlinson RL, Lukowiak AA, Terns RM, and Terns MP (2004). Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol. Biol. Cell 15, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAend-Seq data have been deposited in Sequence Read Archive (BioProject accession number: PRJNA594795).