

Abstract
Background:
Chiari Malformation Type 1 (CM1) affects approximately 1/1000 people symptomatically, although 1/100 meet radiological criteria, making it a common neurological disorder. Diagnosis of underlying conditions has become more sophisticated and new radiologic markers of disease have been described. We sought to determine the prevalence and impact of additional comorbidities and underlying diagnoses in CM1 patients on symptomatology and surgical treatment.
Methods:
Retrospective chart review of 612 pediatric patients with a CM1 diagnosis and imaging data evaluated between 2008–2018 was performed. Because of extensive clinical heterogeneity, patients were separated into four categories based on their primary comorbidities (non-syndromic, central nervous system, skeletal, and multiple congenital anomalies) in order to identify associations with age of onset, radiographic measurements, syringomyelia, and surgical treatment.
Results:
The largest group had non-syndromic CM1 (70%), and had the latest age at diagnosis of any group. In the syndromic group, 6% were diagnosed with a known genetic abnormality, with overgrowth syndromes being the most common. Patients with multiple congenital anomalies had the earliest CM1 onset, most severe tonsillar ectopia and obex position, and were overrepresented among surgical cases. Though there were no statistically significant differences between groups and rates of syrinx, we observed differences in individual diagnoses.
Discussion:
Underlying diagnoses and presence of comorbidities in CM1 patients impacts rates of syringomyelia and surgery. Although most CM1 cases are non-syndromic, clinical evaluation of growth parameters, scoliosis, and joint hypermobility should be routine for all CM1 patients as they are useful to determine syringomyelia risk and may impact treatment.
Keywords: Chiari 1 Malformation, syringomyelia, obex, epidemiology, comorbidities
Introduction
Chiari Malformation Type 1 (CM1) affects 1 in 1000 individuals symptomatically and may be observed radiologically in up to 1%−3.6% of MRIs1–3, making it a common disorder that represents a substantial personal, familial and societal burden. Patients present with a wide array of symptoms from the CM1 itself or from craniovertebral instability, syringomyelia, hydrocephalus, or other causes.4 In fact, approximately 25% of CM1 patients develop syringomyelia, a fluid filled cyst in the spinal cord that can result in motor and sensory deficits and urinary incontinence. Syringomyelia occurs in as many as 75% of surgical CM1 cases as decompressive surgery is often indicated to reduce ongoing spinal cord compression.3 About 20% of CM1 patients develop scoliosis, although this figure increases to 60% when syringomyelia is present.5
Understanding the disorders associated with CM1 is essential to improve surgical and nonsurgical treatment strategies that will translate into improved quality of life for patients. CM1 is typically considered a congenital defect, but it can also rarely be acquired due to trauma.6 Clinical heterogeneity is a major challenge,7 as CM1 often occurs in the context of a myriad of diseases and disorders8 where it may be noted incidentally or a represent a significant pathological focus. The exact frequency of comorbidities or underlying diagnoses in CM1 have not been well established, and CM1 outcomes are not well documented in patients with rare known genetic disease. One study, using MRI examinations to identify abnormalities, reported that approximately 25% of CM1 patients had a syrinx and ~35% had incidental radiographic findings such as Klippel-Feil or platybasia.3 In 2011, Loukas et al. reported more than 50 disorders associated with CM18, suggesting significant genetic heterogeneity. However, over time, both genetic studies and imaging methods have increased in quality become more common, therefore the current data may allow us to provide a better estimate of the number of cases that are non-syndromic.
Here, we report the incidence of non-syndromic CM1 versus CM1 with additional comorbidities or known genetic syndromes, as well as radiographic measures within each subgroup in a large clinical cohort of neurosurgery patients treated at a tertiary academic medical center. We document the extensive heterogeneity of associated conditions that were identified during routine clinical care in the syndromic cases, as we consider it likely that similar underlying biological mechanisms may be involved in the non-syndromic cases. We also sought to determine whether the presence of an associated disorder impacts clinical features, including age of onset, radiographic measures of severity, presence of syringomyelia, and surgical treatment with posterior fossa decompression (PFD). By categorizing CM1 cases based on comorbid symptoms and examining the disease severity in each comorbidity group, we can help elucidate the underlying mechanisms of this disease, which can help guide treatment.
Methods
After IRB approval from Washington University in St. Louis, retrospective chart review of all CM1 patients seen in St. Louis Children’s Hospital from 2008–2018 was performed by screening all pediatric neurosurgery clinics for patients with a CM1 diagnosis (N=612). Diagnosis was made using radiographic criteria of cerebellar tonsil ectopia of at least 5mm9. Obex position was measured on radiographs as the distance of the obex from the foramen magnum as defined by the basion-opisthion line10; 11. MR images were evaluated to determine posterior cranial fossa measurements according to Tubbs et al. using midline sagittal T1-weighted image.10 Additional clinical data collected include sex, ethnicity, presenting signs and symptoms, presence and degree of scoliosis, presence of syringomyelia, and treatment course. Family history was documented and additional medical problems were recorded. Imaging controls (N=50) were identified among patients seen for headaches at pediatric neurology clinics at St. Louis Children’s Hospital who were not found to have tonsillar herniation or other intracranial pathology.
To account for extensive etiological heterogeneity, patients were grouped into four categories based on their comorbid conditions, including central nervous system, skeletal, multiple congenital anomalies, and non-syndromic (Figure 1). The non-syndromic group was defined by not having major comorbidities or underlying diagnoses, although scoliosis and common neuropsychiatric disorders, such as ADHD, anxiety, depression, obsessive-compulsive disorder and oppositional defiant disorder were allowed, as was the presence of syringomyelia, where noted by the treating physician or radiologist. Information on additional comorbidities was considered when grouping the patients. For example, designation of multiple congenital anomalies required more than one comorbidity, such as CNS, skeletal, cardiac, or other major organ system involvement. Patients in the MCA group were the only ones whose additional comorbidities may have been directly to be related to the Chiari (i.e. tethered cord and spina bifida occulta). Patients in the other groups had either no secondary comorbidities, or unrelated comorbidities, such as mental health, dermatological conditions, allergies etc. To determine what proportion of each group presented incidentally or as part of routine work-up for their disease, we reviewed each patient’s indication for MR imaging. The indications for surgery in those having undergone PFD were documented by the treating neurosurgeon after chart review.
Figure 1.
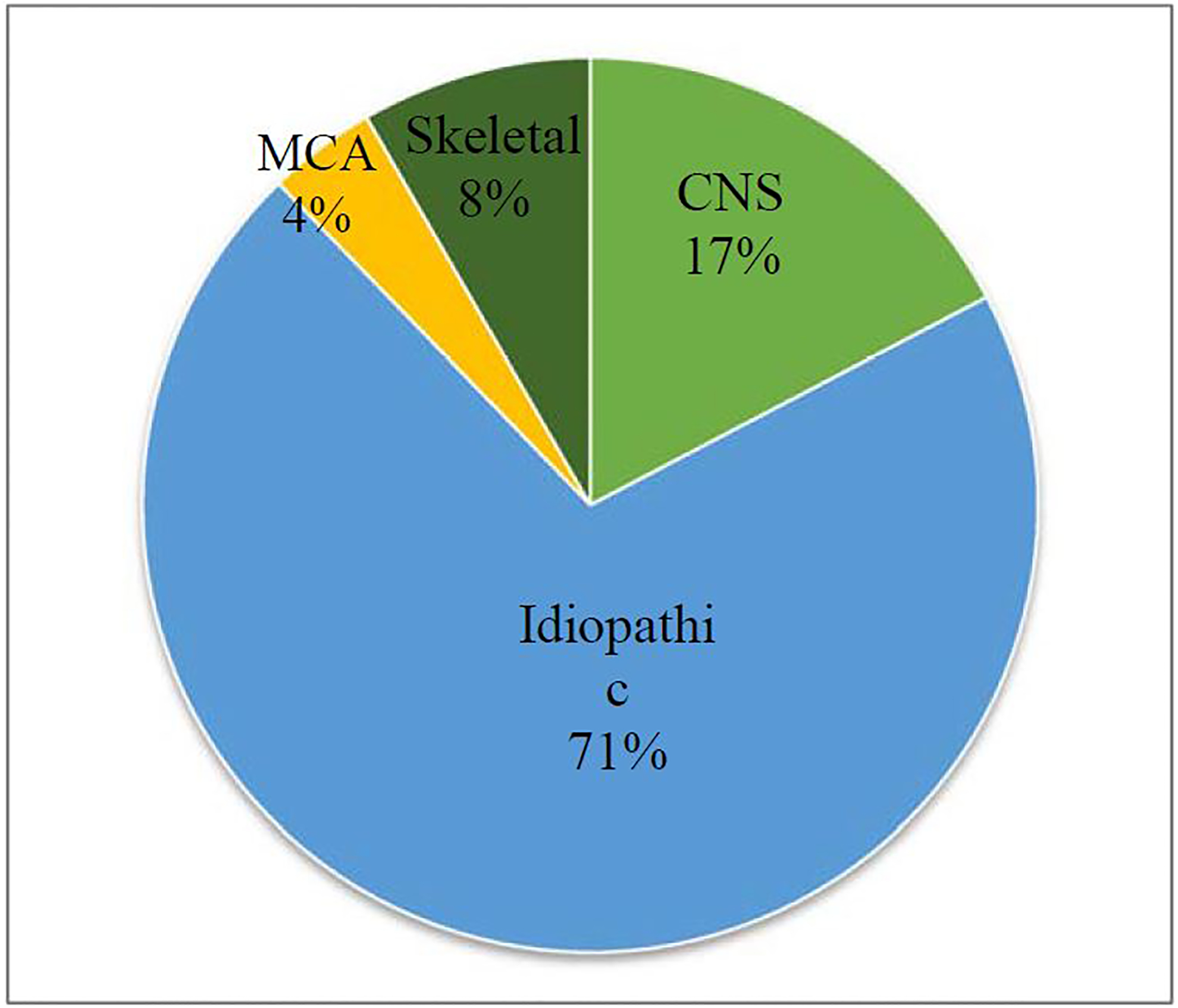
Distribution of comorbidities of patients in the samples
Correlations were made between category and clinical features, including age of onset, presence of syringomyelia, tonsillar ectopia, and obex position. As the data were normally distributed, two-tailed t-tests were performed to determine significance.
Results
Overall, the average age of the 612 patients in this cohort was 15 years with 52% female and 48% male. After reviewing the clinical data, including imaging and genetic test results acquired during routine care, we found that the vast majority of patients (70%) had non-syndromic CM1 (Figure 1). The non-syndromic group was defined by not having major comorbidities or underlying diagnoses, although idiopathic scoliosis and common neuropsychiatric disorders, such as ADHD, anxiety, depression, obsessive-compulsive disorder and oppositional defiant disorder were allowed.
Genetic diagnoses in the sample
When chromosomal abnormalities were included, 6% of our patients were diagnosed with a genetic abnormality as part of their routine clinical evaluations (Table 1). A total of 25 specific genetic conditions or syndromes were observed in at least 1 CM1 patient in our cohort. The largest number of patients had an overgrowth syndrome, which is commonly defined by macrocephaly and either somatic or germline mutations in one of the genes involved in growth12. These patients include those diagnosed with neurofibromatosis type 1 and type 2, tuberous sclerosis complex, Klippel-Trenaunay syndrome, Sturge-Weber syndrome, and several other rare disorders, several of which require sequencing of affected tissues for diagnosis. In addition, Ehlers-Danlos syndrome type 3 (hypermobility type) was the single most common clinical “genetic” diagnosis, though it cannot be tested for genetically as the causative gene or genes have not yet been identified. None of these individual genetic disorders had statistically significantly higher rates of syringomyelia or surgery compared to other comorbidities or underlying diagnoses, possibly due to low sample size. When all patients with underlying genetic conditions or syndromes were combined, 30% had a syrinx and 30% had posterior fossa decompressions.
Table 1.
Prevalence of Genetic Disorders in 612 Patients in the CM1 Cohort
| Genetic Disorders | No. of cases (%) |
|---|---|
| Overgrowth Syndromes | 15 (2.1) |
| Neurofibromatosis Type I (3), Neurofibromatosis Type II (2), Sturge-Weber (2), McCune-Albright, Beckwith-Wiedemann, Cutis Marmorata-Telangiectasia Congenita, Klippel-Trenaunay, PIK3R2, CLOVES, Tatten-Brown-Rahman, Noonan Syndrome, Osler-Weber-Rendu, Tuberous sclerosis | |
| Genomic Copy-Number Variants | 10 (1.6) |
| 2p21p16.2 deletion (2),13p partial trisomy, Pierre Robin sequence, 4q31 deletion, Xp22.33 duplication, 15q33.3 triplicate, 3p25.3 deletion, Trisomy 21, 15q13.2q13.3 deletion | |
| Ehlers Danlos Syndrome Type 3 | 7 (1.1) |
| Loeys-Dietz Syndrome | 2 (0.3) |
| Achondroplasia | 1 (0.2) |
| Alagille Syndrome | 1 (0.2) |
| Congenital Adrenal Hyperplasia | 1 (0.2) |
| Crouzon Syndrome | 1 (0.2) |
| Duane Syndrome | 1 (0.2) |
| Muenke Syndrome | 1 (0.2) |
| Sickle Cell Disease | 1 (0.2) |
Comparison between the groups of CM1 patients
We also sought to determine whether the etiology or the presence of comorbidities resulted in differences in age of onset, radiographic measurements of severity, syringomyelia, or surgical treatment with PFD. To account for extensive heterogeneity and small individual sample size, patients were grouped into four categories based on their comorbid conditions, including central nervous system, skeletal, multiple congenital anomalies, and non-syndromic (Figure 1). Designation of multiple congenital anomalies required more than one comorbidity, such as CNS, skeletal, cardiac, or other major organ system involvement (Table S1). Patients with CNS abnormalities had comorbidities such as hydrocephalus, tethered cord, growth hormone/pituitary abnormalities, cerebral abnormalities such as arachnoid cysts, autism/developmental delay and epilepsy. Skeletal comorbidities include hypermobility/joint laxity, craniosynostosis, platybasia and congenital scoliosis (Table 2). Patients with a known genetic disorder were placed in the category characterized based on their primary comorbid diagnosis.
Table 2.
Primary Comorbidities and Clinical Features of CM1 Patients
| N, % of Sample | Avg. age at CM1 diagnosis (yrs) | Avg. tonsillar ectopia (mm) | Avg. Obex descent (mm) | % with syrinx | % receiving PFD | |
|---|---|---|---|---|---|---|
| Central Nervous System | 113 (18%) | 7.9 | 9.3 | +2.58 | 26 | 30 |
| Epilepsy | 19 | 6.2 | 10.1 | +2.92 | 22 | 17.6 |
| Hydrocephalus | 17 | 2.3 | 10.4 | −0.09 | 29 | 35.3 |
| Autism/Developmental Delay | 14 | 9 | 10.1 | +3.11 | 0 | 20 |
| Growth Hormone/Pituitary Issues | 13 | 7.7 | 10.1 | +3.42 | 18 | 54.5 |
| Arachnoid/Cerebral Cysts | 12 | 10.5 | 8.0 | +3.31 | 18 | 9.1 |
| Ventriculomegaly | 7 | 2 | 7.0 | +5.95 | 40 | 14.2 |
| Tethered Cord/Spinal Anomaly | 7 | 5 | 7.7 | +3.90 | 43 | 57 |
| Pseudotumor Cerebri | 5 | 16 | 9.1 | −0.45 | 20 | 20 |
| Macrocephaly | 5 | 1.2 | 10.2 | +3.10 | 20 | 25 |
| Cerebral Palsy | 4 | 5 | 7.5 | −2.40 | 0 | 75 |
| Cortical Malformations | 4 | 7 | 5.0 | +0.65 | 0 | 0 |
| White Matter Abnormality | 4 | 9.5 | 11.5 | +1.05 | 0 | 50 |
| Stroke/Intraventricular hemorrhage | 2 | 1.5 | 8.5 | +9.65 | 0 | 50 |
| Skeletal | 47 (8%) | 6.8 | 10.1 | +1.06 | 30 | 47 |
| Hypermobility/Joint Laxity | 15 | 8.6 | 9.3 | +3.48 | 40 | 26.6 |
| Craniosynostosis | 8 | 5 | 8.5 | +3.56 | 25 | 50 |
| Platybasia | 7 | 8.4 | 15.3 | −0.76 | 14 | 57.1 |
| Other Bone Malformations | 5 | 3 | 8.8 | −3.60 | 20 | 40 |
| Congenital Scoliosis | 3 | 15.3 | 7.3 | −2.23 | 0 | 67 |
| Congenital Torticollis | 2 | 11 | 22.5 | unknown | 0 | 0 |
| Pyriform Aperture Stenosis | 2 | 5 | 9.0 | −3.65 | 0 | 50 |
| Hemivertebrae | 2 | 3 | 11.0 | −4.45 | 50 | 50 |
| Vitamin D Rickets | 1 | 13 | 13.0 | +1.4 | 0 | 0 |
| Spinal Trauma | 1 | 8 | 17.0 | unknown | 0 | 0 |
| Multiple Congenital Anomalies (MCA) | 26 (4%) | |||||
| Varied | 26 | 5.6 | 12.3 | +5.02 | 27 | 42 |
| Idiopathic | 427 (70%) | |||||
| Varied | 427 | 10.3 | 10.0 | +2.19 | 26 | 37 |
| Overall Sample | 612 (100%) | 9.4 | 10.0 | +2.30 | 25 | 38 |
Age at CM1 diagnosis
Average age at diagnosis was significantly higher for non-syndromic CM1 patients compared to the other three groups. The age at CM1 diagnosis when macrocephaly, hydrocephalus or ventriculomegaly was present was ~2 years, which was the youngest among individual conditions. When groups of conditions were considered, the age at diagnosis was lowest for the MCA group (5.6 years) and highest for the non-syndromic group (10.3 years)(Figure 3). This difference was statistically significant between the non-syndromic group and all three other groups (10−5<p<10−4).
Figure 3. Age at diagnosis is associated with comorbidities and presence of syringomyelia.
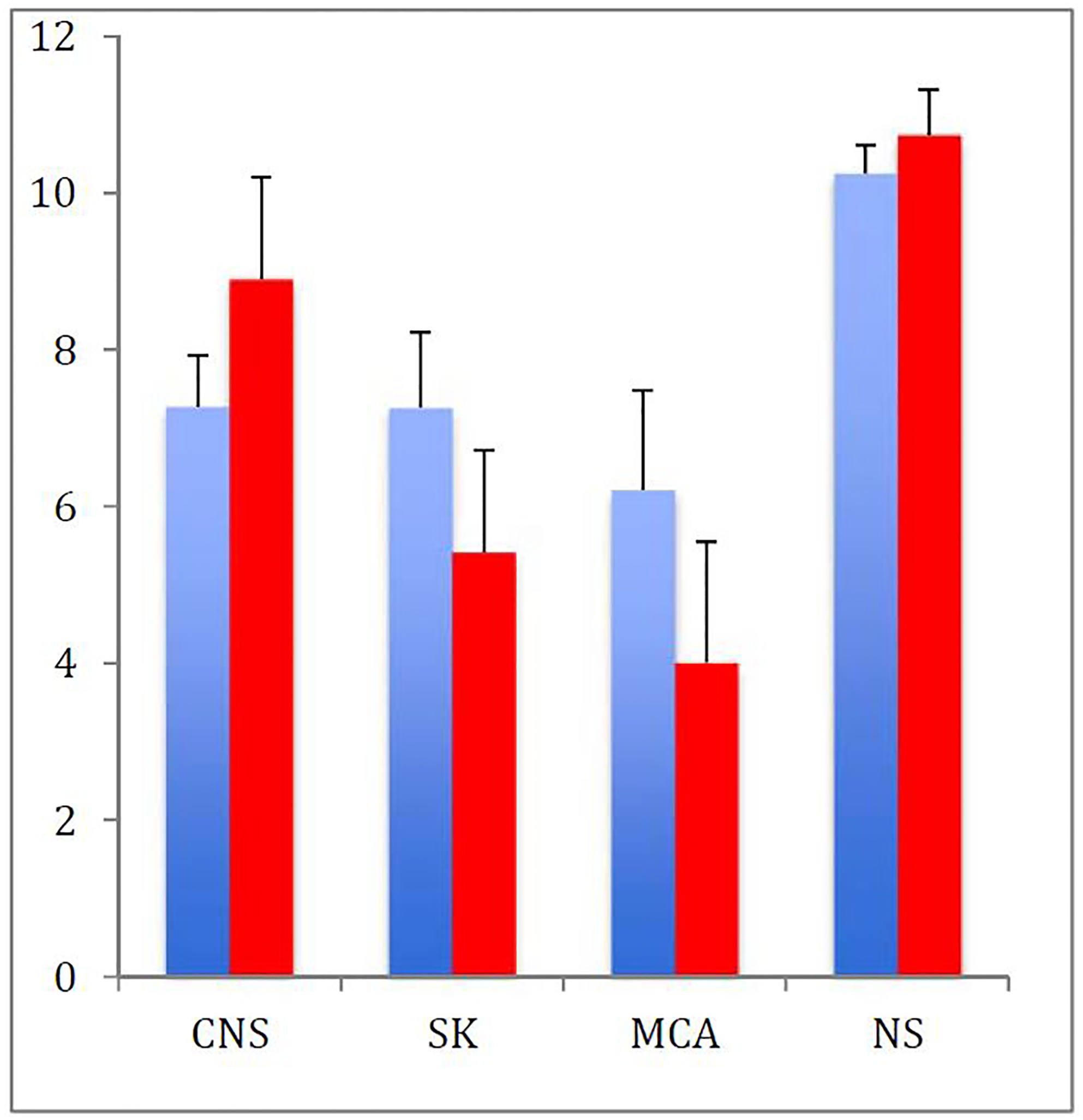
Age at diagnosis of CM1 with (red) and without (blue) a syrinx. Patients with CM1 and multiple congenital anomalies are diagnosed earlier, particularly when a syrinx is present. (CNS=Central nervous system, SK=skeletal, MCA=multiple congenital anomalies, NS=non-syndromic)
Tonsillar ectopia and obex position
To determine if radiographic markers of severity differ among groups, we compared average tonsillar ectopia. Tonsillar descent was greatest in the MCA group (12.3 mm) and lowest in the CNS group (9.3 mm), and the difference between the two was statistically significant (p=0.002). More recently, a recurring complex CM presentation led to the description of the Chiari 1.5 malformation which is associated with obex (brainstem) ectopia below the McRae line (aka basion-opisthion line) and a more severe presentation that often requires PFD surgery10; 11. Obex measurement is shown in Figure 210; 11. To interpret the obex measurement, a smaller number indicates greater caudal descent, with those below the McRae line being the worst and having negative values. The average obex in the entire present cohort was +2.3 mm. In non-Chiari controls (N=50), the average tonsillar position was 9 mm above the foramen magnum and the average obex was +10 mm (manuscript submitted). The skeletal group had a smaller mean obex measurement than all other groups, including the largest proportion with negative measurements of all groups (37%), although the difference was only significant between the skeletal and MCA groups, with the MCA group having a higher average obex position (Table 2). In fact, the MCA group had a higher obex than the sample average.
Figure 2.
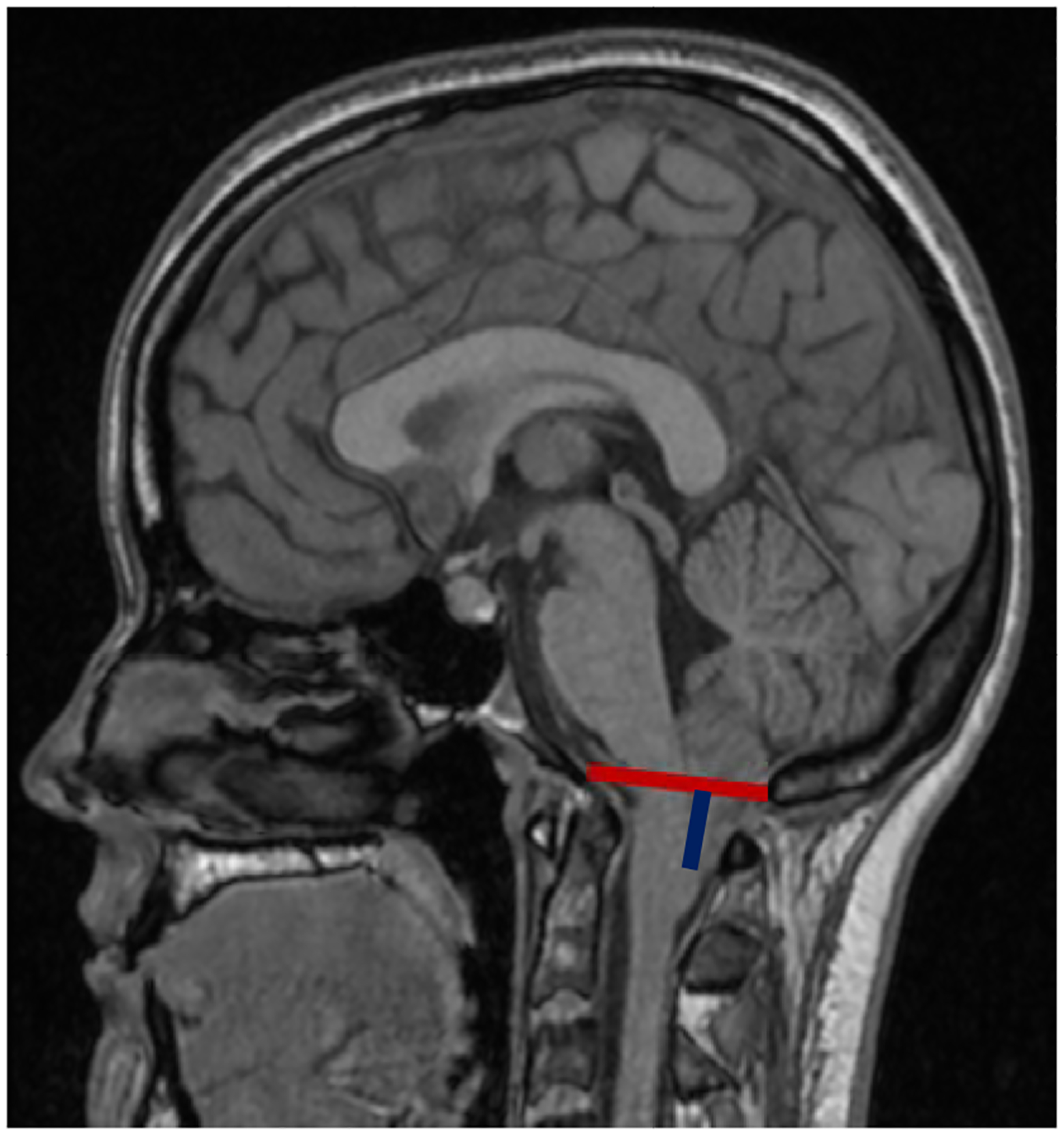
Midline sagittal T1-weighted MRI of a CM1 patient showing McRae’s line a.k.a. basion-opisthion line (red) and obex position (blue).10; 11
Syringomyelia
Because the occurrence of syringomyelia may lead to neurological deficits and is often an indication for surgery13, we sought to determine if there were differences in its prevalence between diagnostic groups. More than 95% of patients with syringomyelia had a spinal MRI available for review.
Syringomyelia was present in 40% of patients with joint hypermobility, as well as in 40% of patients with ventriculomegaly and 29% of patients with hydrocephalus. When data were considered within groups, the skeletal group had a 30% rate of syringomyelia (Table 2), and all others had rates of approximately 25%, although this difference was not statistically significant. Patients within the MCA and skeletal comorbidity groups were diagnosed slightly earlier when a syrinx was present (Figure 3) although this was also not statistically significant.
PFD surgery
We then determined whether the presence of additional comorbidities and underlying diagnoses impacted the proportion of patients receiving posterior decompression surgery for Chiari symptoms (Table 2). Patients with platybasia, tethered cord or other spinal anomalies, growth hormone deficiency, and craniosynostosis all had surgery rates >50%. The skeletal and MCA groups had higher rates of PFD surgery (47% and 42%, respectively) than the overall sample average (38%), although the difference was only statistically significant between the skeletal and CNS subgroups (p=0.05). The two groups with the highest rates of surgery were those with the most severe average tonsillar ectopia (MCA) and obex measurement (skeletal). We then reviewed the indications for PFD surgery and found that in those undergoing PFD surgery, 40% had surgery due to syringomyelia, 40% to headaches, 5% to scoliosis, 2% each to dysphagia and sleep apnea, with neck pain, weakness and vision changes making up the remainder.
Incidental Chiari
Approximately 50% of patients in our cohort had their CM1 discovered incidentally based on original MRI review. Not surprisingly, over half of these were in non-syndromic cases. As CM1 is a neurological disorder, some of these symptoms for which the MRI was ordered may have been related to the Chiari after all and not, in fact, incidental. When removing patients whose MRIs was for symptoms that may be related to CM1, for example scoliosis, syringomyelia, hydrocephalus, tethered cord, craniosynostosis, epilepsy, etc., the number of incidental CM1 cases drops to 27%. Of these incidental cases without any CM1 related symptoms or disorders, 65% were non-syndromic, 20% were CNS, 7% skeletal and 6% MCA. These numbers are nearly identical to the breakdown of patients in the entire sample (Figure 1).
Discussion:
Within our tertiary referral center the vast majority of patients have non-syndromic CM1 (70%) despite the widespread use of sophisticated imaging methods and genetic testing. About 6% of patients had a specific genetic diagnosis, which is higher than earlier estimates of 1%1. When they are present, underlying diagnoses and additional comorbidities are very heterogeneous, suggesting that there are many potential pathways that lead to the cerebellar tonsil ectopia that characterizes CM1.
Connective tissue disease and overgrowth syndromes overrepresented in CM1 cases
Ehlers-Danlos syndrome type 3 (hypermobility type) or self-reported hypermobility was commonly noted in our study, which is consistent with a prior identification of connective tissue disorder in ~10% of CM1 patients14. Unlike the study just referenced, the patients in our retrospective study were not routinely asked about or systematically tested for joint hypermobility, therefore our numbers underestimate its prevalence. Notably, clinical evaluation for joint hypermobility can be performed simply and without special equipment using the Beighton quantitative scoring system15, which should become a standard part of the clinical evaluation of CM1, not only because of the frequency of this comorbidity, but because cardiac and systemic abnormalities often associated with connective tissue disorders can be better managed if the diagnosis is made early. We also identified a large number of patients with overgrowth syndromes, which have recently been shown in some cases to be due to low level somatic mosaicism16; 17, and may not have been diagnosed before the common use of genetic testing. Overgrowth as an etiological cause for CM1 is also supported by the co-occurrence of CM1 in neurofibromatosis type 1, Noonan syndrome, and tuberous sclerosis complex, which are closely related to the somatic disorders due to their growth abnormalities though they are associated with germline genetic variants.
Importance of understanding clinical heterogeneity in CM1
Our outcome data suggests that it is important to understand the underlying etiologies and comorbidities with CM1, as patients may have differing rates of syringomyelia and surgery. For example, compared to the entire CM1 cohort, which had a 25% risk of syringomyelia, we found that patients with joint hypermobility had a slightly higher rate (40%), whereas syringomyelia was not present in any of our patients with unspecified autism/developmental delay. Because the underlying causes of autism/developmental delay are so heterogeneous, however, further study is clearly needed to better predict outcomes. Syringomyelia was slightly higher in patients within the skeletal group of comorbidities. Though we excluded patients with adolescent idiopathic scoliosis from the skeletal group due to its non-syndromic nature, it is well known that patients with CM1 and scoliosis have high rates of syringomyelia18−20, and 8% of presumed pediatric idiopathic scoliosis cases were found upon MRI to have CM121. Among other studies examining comorbid conditions, it was recently determined that among CM1 patients, hydrocephalus and syringomyelia were the most common concurrent diagnoses, followed by scoliosis and tethered cord22.
Patients with multiple congenital anomalies in our sample, not surprisingly, had the earliest diagnosis and the highest rates of PFD surgery, likely reflecting both the severity of disease and earlier imaging that often occurs in children with complex medical conditions. It is likely that earlier symptoms as well as increased number of symptoms in these children drives earlier imaging. Indeed, children with complex chronic conditions that include hydrocephalus have a significantly higher risk of surgical complications19, and this association was especially significant in younger children23. Non-syndromic CM1 may represent a milder form of the disease, which is supported by the latest age at diagnosis of any of the groups, and its underrepresentation among surgical cases. Because our study cohort was recruited primarily from neurosurgery clinic, the rates of surgery in non-syndromic cases may have been even lower if more asymptomatic, non-referred patients were included.
Posterior fossa decompression surgery is costly and invasive, and depending on the cause of the CM1, not always successful.14 Thus, determining which patients are more likely to need surgery is important. We determined that patients with platybasia and patients with growth hormone or pituitary abnormalities, which have both been previously associated with high rates of CM18; 24, are more likely to have surgery. The MCA and skeletal comorbidity groups had the most severe tonsillar ectopia and obex (12.13 mm and 1.29 mm, respectively) and are also overrepresented among surgical cases, suggesting that they have some of the more severe CM1 presentations.
Limitations
A limitation of this study is that, due to its retrospective nature, not all patients received the same testing, whether that was clinical evaluations for joint hypermobility, diagnostic testing for endocrine abnormalities, or genetic testing or spinal cord imaging. Therefore, we may have underestimated the frequency of associated comorbidities. The data in this study also represents a single point in time, therefore some of these patients may have developed syringomyelia or required surgery later in their care, or this may have been performed elsewhere. Additionally, we do not have data on syrinx size for all patients so could not perform association testing on the size of the syrinx itself. We also note that CM1 occurs frequently in patients with craniosynostosis but these cases are underrepresented here because most are typically asymptomatic and, at our center, managed entirely by the craniofacial team. Additionally, there may be a referral bias toward symptomatic patients as these patients were ascertained at a pediatric neurosurgical referral center. Lastly, we note that classification systems of any kind will have their unique issues. We grouped our patients into these four subtypes not only for increased power in analysis, but also as an attempt to address and understand their complex clinical presentations.
Implications for patients
Overall, our data suggests that there are multiple etiological pathways that converge on the Chiari 1 malformation phenotype. Although most patients have non-syndromic CM1 and no underlying genetic abnormality or associated comorbidity, this study suggests that a thorough clinical assessment of CM1 patients should include evaluation of head circumference, height to evaluate for possible endocrine abnormality or connective tissue disorder, clinical scoliosis exam (Adam’s forward bend test), and Beighton test for joint hypermobility. These simple clinical exam findings would aid the diagnosis and recognition of some of the most commonly associated CM1 conditions. Our data suggest that it is important to consider evaluation of underlying etiologies, such as overgrowth syndromes or hypermobility syndromes, among others, as these patients have differing rates of syringomyelia and surgery.
Supplementary Material
Funding:
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases under Award Number R01AR067715, the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number U54 HD087011 to the Intellectual and Developmental Disabilities Research Center at Washington University; The Washington University Institute of Clinical and Translational Sciences grant UL1 TR000448 from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) and The University of Missouri Spinal Cord Injury Research Program 2016-01 to Drs. Gurnett and Limbrick. The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest: Dr. Limbrick receives research support from Medtronic, Inc. and Microbot Medical, Inc. for unrelated research.
References:
- 1.Speer MC, Enterline DS, Mehltretter L, Hammock P, Joseph J, Dickerson M, Ellenbogen RG, Milhorat TH, Hauser MA, and George TM (2003). Review Article: Chiari Type I Malformation with or Without Syringomyelia: Prevalence and Genetics. J Genet Couns 12, 297–311. [DOI] [PubMed] [Google Scholar]
- 2.Markunas CA, Soldano K, Dunlap K, Cope H, Asiimwe E, Stajich J, Enterline D, Grant G, Fuchs H, Gregory SG, et al. (2013). Stratified whole genome linkage analysis of Chiari type I malformation implicates known Klippel-Feil syndrome genes as putative disease candidates. PLoS One 8, e61521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strahle J, Muraszko KM, Kapurch J, Bapuraj JR, Garton HJ, and Maher CO (2011). Chiari malformation Type I and syrinx in children undergoing magnetic resonance imaging. J Neurosurg Pediatr 8, 205–213. [DOI] [PubMed] [Google Scholar]
- 4.Urbizu A, Toma C, Poca MA, Sahuquillo J, Cuenca-Leon E, Cormand B, and Macaya A (2013). Chiari malformation type I: a case-control association study of 58 developmental genes. PLoS One 8, e57241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly MP, Guillaume TJ, and Lenke LG (2015). Spinal Deformity Associated with Chiari Malformation. Neurosurg Clin N Am 26, 579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Alotaibi NM, Samuel N, Ibrahim GM, Fallah A, and Cusimano MD (2016). Acquired Chiari Malformation and Syringomyelia Secondary to Space-Occupying Lesions: A Systematic Review. World Neurosurg. [DOI] [PubMed] [Google Scholar]
- 7.Markunas CA, Lock E, Soldano K, Cope H, Ding CK, Enterline DS, Grant G, Fuchs H, Ashley-Koch AE, and Gregory SG (2014). Identification of Chiari Type I Malformation subtypes using whole genome expression profiles and cranial base morphometrics. BMC Med Genomics 7, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loukas M, Shayota BJ, Oelhafen K, Miller JH, Chern JJ, Tubbs RS, and Oakes WJ (2011). Associated disorders of Chiari Type I malformations: a review. Neurosurg Focus 31, E3. [DOI] [PubMed] [Google Scholar]
- 9.Barkovich AJ, Wippold FJ, Sherman JL, and Citrin CM (1986). Significance of cerebellar tonsillar position on MR. AJNR Am J Neuroradiol 7, 795–799. [PMC free article] [PubMed] [Google Scholar]
- 10.Tubbs RS, Iskandar BJ, Bartolucci AA, and Oakes WJ (2004). A critical analysis of the Chiari 1.5 malformation. Journal of neurosurgery 101, 179–183. [DOI] [PubMed] [Google Scholar]
- 11.Brockmeyer DL, and Spader HS (2015). Complex Chiari Malformations in Children: Diagnosis and Management. Neurosurg Clin N Am 26, 555–560. [DOI] [PubMed] [Google Scholar]
- 12.Keppler-Noreuil KM, Parker VE, Darling TN, and Martinez-Agosto JA (2016). Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet 172, 402–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahn EN, Muraszko KM, and Maher CO (2015). Prevalence of Chiari I Malformation and Syringomyelia. Neurosurg Clin N Am 26, 501–507. [DOI] [PubMed] [Google Scholar]
- 14.Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, and Francomano CA (2007). Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine 7, 601–609. [DOI] [PubMed] [Google Scholar]
- 15.Beighton P, and Horan F (1969). Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br 51, 444–453. [PubMed] [Google Scholar]
- 16.Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, and Darling TN (2017). Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol Clin 35, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pritchard CC, Smith C, Marushchak T, Koehler K, Holmes H, Raskind W, Walsh T, and Bennett RL (2013). A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Genet Med 15, 1004–1007. [DOI] [PubMed] [Google Scholar]
- 18.Strahle J, Smith BW, Martinez M, Bapuraj JR, Muraszko KM, Garton HJ, and Maher CO (2015). The association between Chiari malformation Type I, spinal syrinx, and scoliosis. J Neurosurg Pediatr 15, 607–611. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg JK, Olsen MA, Yarbrough CK, Ladner TR, Shannon CN, Piccirillo JF, Anderson RC, Wellons JC 3rd, Smyth MD, Park TS, et al. (2016). Chiari malformation Type I surgery in pediatric patients. Part 2: complications and the influence of comorbid disease in California, Florida, and New York. J Neurosurg Pediatr 17, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozerdemoglu RA, Denis F, and Transfeldt EE (2003). Scoliosis associated with syringomyelia: clinical and radiologic correlation. Spine (Phila Pa 1976) 28, 1410–1417. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Sha S, Xu L, Liu Z, Qiu Y, and Zhu Z (2016). The prevalence of intraspinal anomalies in infantile and juvenile patients with “presumed idiopathic” scoliosis: a MRI-based analysis of 504 patients. BMC Musculoskelet Disord 17, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horn SR, Shepard N, Vasquez-Montes D, Bortz CA, Segreto FA, De La Garza Ramos R, Goodwin CR, and Passias PG (2018). Chiari malformation clusters describe differing presence of concurrent anomalies based on Chiari type. J Clin Neurosci 58, 165–171. [DOI] [PubMed] [Google Scholar]
- 23.LoPresti MA, Pan IW, Gadgil N, Wagner K, and Lam S (2019). Outcomes and resource utilization in surgery for Chiari I malformation in a national network of children’s hospitals. Childs Nerv Syst 35, 657–664. [DOI] [PubMed] [Google Scholar]
- 24.Klekamp J (2015). Chiari I malformation with and without basilar invagination: a comparative study. Neurosurg Focus 38, E12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.