

Abstract
Sepsis induces both intestinal hyperpermeability and epithelial apoptosis. While each has been implicated in mediating sepsis mortality, the relationship between these two processes is unclear. We hypothesized that preventing intestinal apoptosis would prevent gut barrier dysfunction. To test this hypothesis, transgenic mice that overexpress the anti-apoptotic protein Bcl-2 in the gut epithelium (Fabpl-Bcl-2 mice) and wild type (WT) mice were subjected to sham laparotomy or cecal ligation and puncture and orally gavaged with fluorescein isothiocyanate conjugated-dextran (FD-4) five hours before sacrifice. Serum FD-4 concentration was assayed to measure intestinal permeability, and jejunal tight junctions were assayed for mRNA and protein expression. Baseline FD-4 concentration was similar between WT and Fabpl-Bcl-2 mice. Intestinal permeability increased 6, 12, 24 and 48 hours following sepsis in WT mice; however, FD-4 concentration was significantly lower at each timepoint in Fabpl-Bcl-2 mice. In addition, there were no statistically significant changes in permeability between septic and sham transgenic mice. Intestinal mRNA expression of claudin 3, claudin 5 and occludin were lower in septic Fabpl-Bcl-2 mice while claudin 4 mRNA levels were higher in Fabpl-Bcl-2 mice. In contrast, no differences were detected in claudins 2, 7, 15, JAM-A or ZO-1. Protein levels followed the same trend for all tight junction mediators different between WT and Fabpl-Bcl-2 mice except occludin was significantly higher in transgenic mice. Together these results demonstrate that decreasing intestinal epithelial apoptosis prevents hyperpermeability following sepsis via tight junction alterations which may be at least partially responsible for improved survival conferred by Bcl-2 overexpression.
Keywords: Gut, intestine, permeability, apoptosis, sepsis, barrier, claudin, occludin
INTRODUCTION
Sepsis is life-threatening organ dysfunction caused by a dysregulated host response to infection (1). In the United States, sepsis results in between 230,000 and 370,000 deaths annually (2). Despite increasing knowledge of sepsis, if antimicrobial treatment is unsuccessful, management relies upon supportive care, without proven benefit of any adjunctive therapy (3).
The gut is frequently referred to as the “motor” of critical illness (4-6) due to a number of abnormalities that can cause and/or propagate sepsis. Under basal conditions, cellular proliferation is balanced by cell loss by apoptosis and exfoliation of live cells into the gut lumen. However, this balance is altered in critical illness. Sepsis induces intestinal epithelial apoptosis in both preclinical models as well as in human autopsy studies (7). Notably, overexpression of the anti-apoptotic protein Bcl-2 prevents sepsis-induced gut epithelial apoptosis, and this confers a survival advantage in murine models of cecal ligation and puncture (CLP) and Pseudomonas aeruginosa pneumonia (8, 9), although no difference in survival is seen with Bcl-2 overexpression following acute lung injury (10). Although numerous theories have been proposed, the etiology through which prevention of gut apoptosis by Bcl-2 overexpression improves survival in sepsis (as opposed to non-infectious inflammation) is unclear.
Barrier function is maintained by allowing paracellular movement of water, solutes and immune modulating factors while preventing diffusion of potentially harmful components of the microbiome (11). Sepsis induces intestinal hyperpermeability in preclinical models of sepsis and in critically ill patients which can potentially allow components of the pathobiome to escape the gut lumen and cause or worsen systemic illness (12, 13). Sepsis-induced gut barrier dysfunction is mediated via alterations in the apical tight junction (TJ) which alters molecular via two distinct pathways – a high-capacity, size and charge-selective route (pore) and a low-capacity, nonselective route (leak).
The relationship between apoptosis and permeability is complex. While agents that increase apoptosis have been demonstrated to be associated with increased permeability in the small intestine (14), the majority of studies linking these two processes have been performed in cell culture (15-18) and conflicting data exists regarding the relationship between apoptosis and permeability. To determine whether prevention of sepsis-induced gut apoptosis by the anti-apoptotic protein Bcl-2 prevents intestinal hyperpermeability in sepsis in vivo, mice that overexpress Bcl-2 in their gut epithelium (Fabpl-Bcl-2 mice) (8, 9, 19, 20) were assayed for permeability and TJ following CLP.
MATERIALS AND METHODS
Animals
Experiments were performed on six to 12 week old, gender-matched WT FVB/N and Fabpl-Bcl-2 mice, which are on the same genetic background (21). All experiments were performed in the morning to minimize diurnal variation. Mice were maintained on a 12 hour light-dark schedule in a specific pathogen-free environment and received standard laboratory mouse chow and water ad libitum both before and after sepsis. All studies were performed in accordance with the National Institutes of Health Guidelines for the Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Emory University School of Medicine (Protocol 201800033).
Sepsis model
CLP or sham laparotomy was performed according to the method of Baker et al. (22). Under isoflurane anesthesia, a midline abdominal incision was made, and the cecum was exteriorized and ligated below the ileocecal valve to avoid intestinal obstruction. The cecum was then punctured twice with a 25-gauge needle and gently squeezed to extrude a small amount of stool. After replacing the cecum in the abdomen, the abdominal wall was closed in layers. All mice received 1 ml of normal saline via subcutaneous injection to account for insensible fluid losses (23). To mimic clinical management, mice were also given subcutaneous injections of ceftriaxone (50 mg/kg, Sigma-Aldrich, St. Louis, MO) and metronidazole (30 mg/kg, Apotex Corp, Weston, FL) after CLP and every 12 hours thereafter (3). Mice were sacrificed at 6, 12, 24, 48, or 96 hours following CLP. All mice received buprenorphine (0.1mg/kg, McKesson Medical, San Francisco, CA) immediately prior to laparotomy to minimize animal suffering and were re-dosed with buprenorphine post-operatively when deemed appropriate by the staff of the Division of Animal Resources at Emory University.
Intestinal Permeability
Intestinal permeability was measured in vivo by measuring the concentration of FD-4 (22mg/ml, molecular mass 4 KDa, Sigma-Aldrich) in the blood (24, 25). Mice were orally gavaged with 0.5 ml of FD-4 5 hours prior to sacrifice (i.e. 1, 7, 19, 43, or 91 hours following induction of CLP or sham laparotomy). Blood was collected at time of sacrifice and centrifuged at 10,000 rpm at 4°C for 10 minutes. A total of 50 μl of plasma was then diluted with an equal amount of sterile phosphate-buffered saline (pH 7.4), and the concentration of FD-4 was determined using fluorospectrometry (Synergy HT, BioTek, Winooski, VT) using an excitation wavelength of 485 nm and an emission wavelength of 528 nm with serially diluted samples as standards. All samples and standards were run in triplicate.
Quantification of mRNA expression
Segments of jejunum were treated with RNAlater™ Stabilization Solution (Thermo-Fisher Scientific, Waltham, MA) for 24 hours at 4° C and stored at −80° C. Frozen segments of jejunum were thawed and lysed using QIAshredder homogenizer spin columns (QIAGEN, Venlo, Netherlands). Total RNA was extracted using the QIAGEN RNeasy mini kit (QIAGEN) according to the manufacturer’s instructions. Complementary DNA was synthesized from 0.5ug of total RNA using the iScript cDNA Synthesis kit (Bio-rad, Hercules, CA) following the manufacturer’s instruction. Messenger RNA quantification was measured using TaqMan Gene Expression Assays (Thermo-Fisher Scientific, Waltham, MA) using comparative Ct method and calculated in the open-source software package R (26).
Western Blot Analysis
For Western Blots examining TJ, segments of jejunum were snap-frozen in liquid nitrogen and stored at −80° C. These were subsequently weighed and homogenized in 4x volume of ice-cold lysis buffer (50mM Tris HCl; 10mM EDTA; 150mM NaCl; 1% Triton X-100, 1% NaDOC, 1mM EDTA; 0.1% SDS) with protease inhibitor cocktail mix (cOmplete Mini EDTA-free, Roche, Indianapolis, IN) using BulletBlender® Storm homogenizer (BBY24M, Next Advance, Inc, Averill Park, NY) for 2 minutes. Samples were then centrifuged at 10,000 rpm at 4° C for 5 minutes. The supernatant was collected, and total protein concentration was determined using the Pierce 660nm protein assay (Thermo-Fisher Scientific). Protein samples of 30μg and an equal volume of 2x Laemmli buffer (Bio-Rad) were then heated at 95° C for 5 minutes. Samples were loaded on Mini-PROTEAN TGX Stain-Free polyacrylamide gels (Bio-Rad) for 2 hours at 70V. The gel was activated for 5 minutes, and the protein was transferred to polyvinylidene difluoride membrane for 7 minutes using the Trans-blot Turbo system (Bio-Rad). Membranes were then blocked in 5% non-fat milk in Tris-buffered saline with 0.1% Tween 20 (Sigma-Aldrich) at room temperature for 60 minutes and incubated overnight with primary antibody at 4° C. Primary antibodies used were rabbit anti-β-actin (1:2000, 4967S, Cell signaling technology, Danvers, MA), 51-9100, Life Technologies,), rabbit anti-claudin 3 (1:500, 34-1700, Life Technologies, Carlsbad, CA), rabbit anti-claudin 4 (1:2000, PA5-34437, Life Technologies), rabbit anti-claudin 5 (1:2000, 34-1600, Life Technologies), and rabbit anti-occludin (1:250, 710192, Life Technologies). The following day, membranes were washed and incubated for 60 minutes at room temperature with goat anti-rabbit secondary antibody linked to horseradish peroxidase (1:1000, Cell Signaling Technology). Membranes were then striped and probed for actin.
A modification of this technique was performed for Western Blots examining pro- and anti-apoptotic proteins. Samples were loaded on polyacrylamide gels (Bio-Rad) for 45 minutes at 180V. Proteins were detected with a chemiluminescent system (ChemiDoc Touch, Bio-Rad). Resulting bands were analyzed using intensity quantification software (ImageLib 6.0, BioRad). Linear dynamic detection range with stain-free technology was used for lane protein normalization and comparisons (27). Membranes were incubated overnight with primary antibody at 4° C with rabbit anti-Bax (1:1000, Cell Signaling Technology 2772) or rabbit anti-Bcl-xL (1:10000, Cell Signaling Technology, 2762). Protein bands were detected with a chemiluminescent system (BioRad) and visualized with a charged coupled device (ChemiDoc Touch, BioRad). Densitometric analysis was performed by image software (ImageLib 6.0, BioRad). Data are presented as relative protein expression compared with WT mice for all Western blots.
TJ Immunohistochemistry
Slides were deparaffinized, rehydrated and washed with PBS. Samples were incubated with 20% goat serum albumin for 30 minutes and then stained with rabbit anti-claudin-3, anti-claudin-4, anti-claudin-5, or anti-occludin (1:200, Thermo Fisher Scientific) overnight at 4° C. The next day, samples were washed with PBS, then incubated with 1:500 biotinylated anti-rabbit antibody for 1 hour at room temperature. After washing in PBS, the samples were incubated with 1:500 HRP streptavidin for 1 hour at room temperature. After washing in PBS, the samples were counterstained with DAB solution.
Intestinal epithelial apoptosis
Crypt apoptotic cells were quantified in the jejunum by two complementary methods: H&E-staining (morphological analysis) and active caspase-3 staining (functional analysis). Apoptotic cells were identified on H&E-stained sections by characteristic nuclear condensation and fragmentation. For active caspase-3 staining, immunohistochemistry was performed as above and sections were stained for rabbit-anti cleaved caspase 3 (1:100 Cell Signaling Technology 9664). Apoptotic cells were quantified in 100 contiguous well-oriented crypt-villus units per animal by an examiner (TO) blinded to sample identity.
Statistical Analysis
Statistical analyses were performed using Prism (version 7.0, GraphPad Software, San Diego, CA). Data are presented as mean ± SEM. Data were tested for normality by the D’Agostino-Pearson omnibus normality test. Data with a normal distribution were compared using Student’s t-test. Data that did not have a normal distribution were compared using the Mann Whitney-U test. A p value of ≤0.05 was considered to be statistically significant.
RESULTS
Effect of Bcl-2 overexpression in the intestinal epithelium on jejunal apoptosis
Jejunal apoptosis was decreased in Fabpl-Bcl-2 mice compared to WT mice 24 hours after CLP when measured by morphologic criteria (Fig. 1A-C) or by active caspase 3 staining (Fig. 1D-F). This decrease in cell death was not associated with differences in levels of the apoptotic mediators Bax or Bcl-XL in WT and transgenic mice (supplemental figure 1).
FIG. 1. Intestinal apoptosis following CLP.
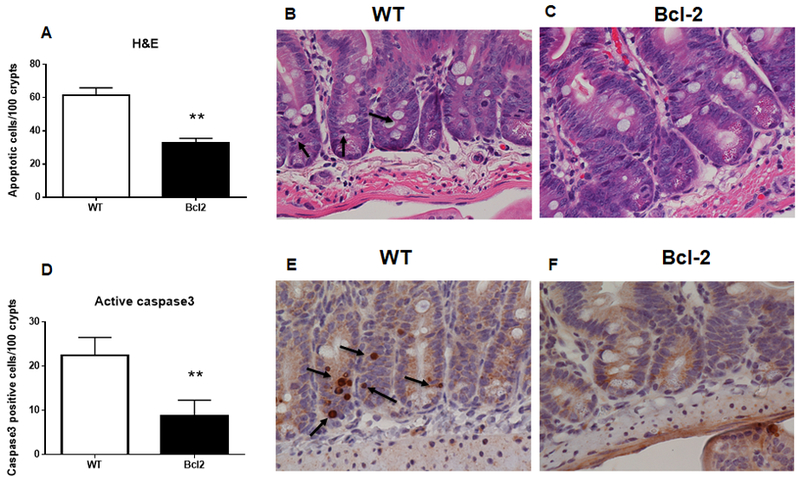
Apoptotic cells were lower in the crypts of Fabpl-Bcl-2 mice than WT mice when measured by H&E (A, p=0.0014) or active caspase 3 (D, p=0.0037), n=10/group. Representative histomicrographs are shown with arrows pointing to apoptotic cells (A, B for H&E, D, E for caspase 3).
Effect of intestinal epithelial apoptosis prevention on sepsis-induced hyperpermeability
Under sham conditions, permeability was similar between WT and Fabpl-Bcl-2 mice (Fig. 2). Sepsis induced intestinal hyperpermeability in WT mice, with increasing FD-4 appearance in the blood detectable at 6 hours persisting out to 48 hours. In contrast, permeability was not significantly different between Fabpl-Bcl-2 mice at any timepoint following CLP compared to sham Fabpl-Bcl-2 mice. In addition, permeability was lower in Fabpl-Bcl-2 mice than WT mice at all timepoints between 6 hours and 48 hours.
FIG. 2. Intestinal permeability by FD-4 following CLP.
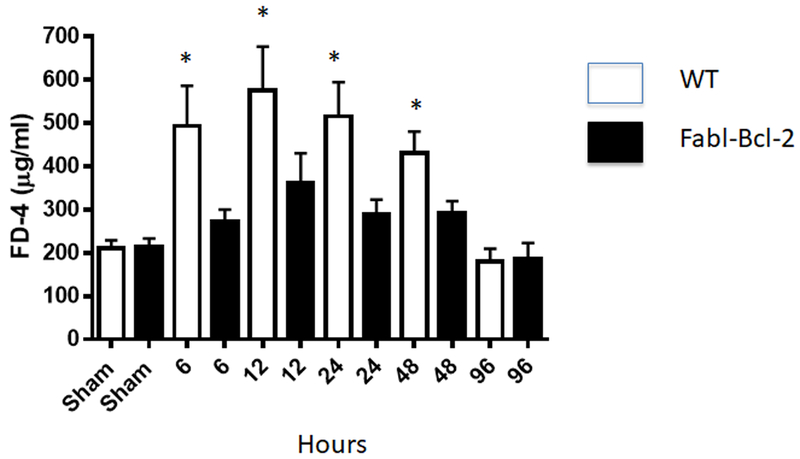
FD-4 levels in the bloodstream were similar following sham laparotomy between WT and Fabpl-Bcl-2 mice (p=0.68). In contrast, permeability was higher in WT mice 6 (p=0.005), 12 (p=0.035), 24 (p=0.031), and 48 (p=0.028) hours after CLP compared to Fabpl-Bcl-2 mice. Permeability was similar between groups 96 hours after CLP (p=0.76), n=7-12 all timepoints except n=4-6 at 96 hours.
Effect of intestinal epithelial apoptosis prevention on TJ mRNA expression
Since permeability was similar in WT mice at multiple timepoints after CLP but apoptosis peaks 24 hours after the onset of sepsis (19), mechanistic studies were performed at 24 hours. Messenger RNA levels were assayed in the jejunum of TJs previously determined to be present in the intestine. Intestinal mRNA expression of claudin 3, claudin 5 and occludin were lower in septic Fabpl-Bcl-2 mice than septic WT mice (Fig. 3). In contrast, claudin 4 mRNA expression was significantly higher in septic Fabpl-Bcl-2 mice. No difference was detected in expression of mRNA expression of claudins 2, 7, 15, JAM-A, and ZO-1 between septic WT and transgenic mice.
FIG. 3. Jejunal mRNA expression following CLP.
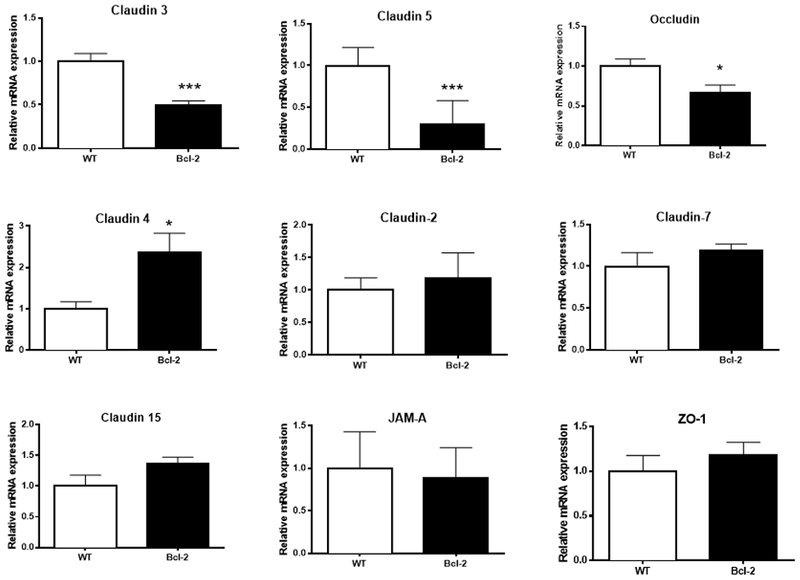
Claudin 3 (p<0.0001), claudin 5 (p=0.004), and occludin (p=0.02) were lower in Fabpl-Bcl-2 mice following CLP whereas Claudin 4 (p=0.01) was higher in transgenic mice. No differences were detected in claudin 2 (p>0,99), claudin 7 (p=0.22), claudin 15 (p=0.08), JAM-A (p=0.84) or ZO-1 (p=0.32), n=10=11 for all TJ assayed.
Effect of intestinal epithelial apoptosis prevention on TJ protein expression and immunohistochemistry
To determine whether transcriptional changes in intestinal TJ protein was associated with changes at the protein level, Western Blots were performed on TJ that were different between WT and transgenic mice at the mRNA level (Fig. 4). Similar trends were noted in claudins 3 and 5 (decreased) and 4 (increased) in Fabpl-Bcl-2 mice 24 hours after CLP. In contrast, occludin showed the opposite expression pattern. While mRNA levels of occludin were lower in transgenic mice following CLP, protein levels were nearly 100 times higher. To confirm the differential expression patterns of intestinal TJ in vivo, immunohistochemistry was performed on WT and transgenic mice 24 hours after CLP (Fig. 5). Patterns identified were generally consistent with that seen on Western Blotting, albeit with differential cellular and geographic specificity along the crypt villus axis depending on TJ assayed.
FIG. 4. Jejunal TJ protein expression following CLP.
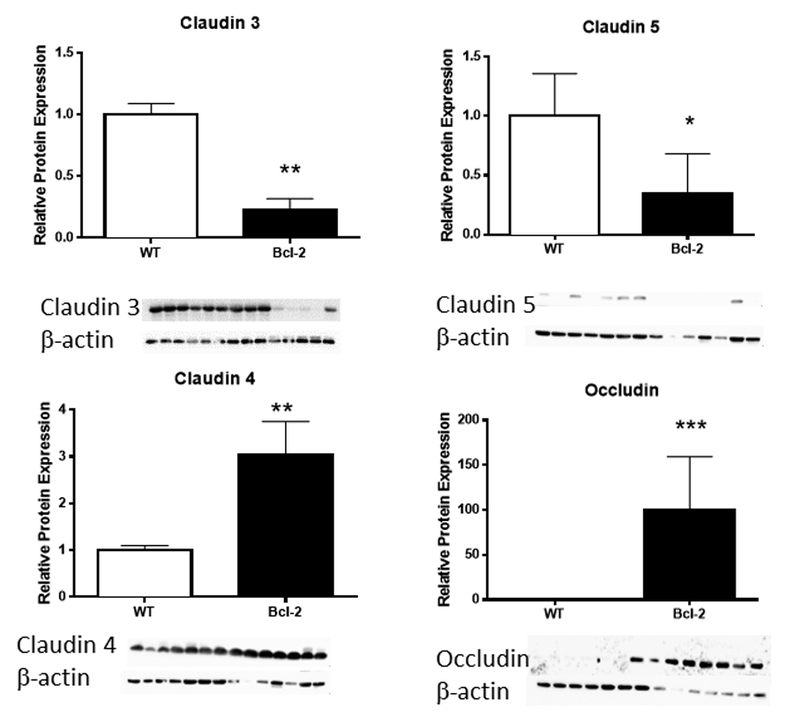
Western blots showed decreased claudin 3 (p<0.0017) and claudin 5 (p=0.05) and increased claudin 4 (p=0.0012) in Fabpl-Bcl-2 mice following CLP. In contrast to mRNA (figure 2), occludin was markedly increased (p=0.0006) in transgenic mice.
FIG. 5. Jejunal immunohistochemistry of TJ following CLP.

Claudin 3 is distributed equally along the crypt-villus axis and staining is more dense in WT mice than Fabp-Bcl-2 mice. Claudin 5 stains more prominently throughout the crypt and base of the villus and is also more prominent in WT mice. Claudin 4 staining is present in spotty areas throughout the crypt and villus of WT mice. This does not change in prominence in transgenic mice; however, staining is more prominent in the lamina propria of Fabp-Bcl-2 mice. Occludin is not detectable in WT mice. Occludin stains heavily in isolated villus cells and intervillus cells in Fabpl-Bcl-2 cells. Representative histomicrographs are shown of n=4 mice/group examined
DISCUSSION
This study demonstrates that while gut barrier function is similar in sham conditions between WT mice and those that overexpress Bcl-2 in their intestinal epithelium, sepsis-induced intestinal hyperpermeability is prevented in transgenic mice. This is associated with alterations in numerous TJ proteins that may mechanistically prevent induction of intestinal hyperpermeability in Fabpl-Bcl-2 mice.
While often thought of as independent processes, the relationship between apoptosis and permeability is complex. Knocking out the TJ protein occludin in normal cells in vitro not only leads to hyperpermeability, it also leads to increased apoptosis via the extrinsic (receptor-mediated) pathway (28). In theory, elimination of dead cells via apoptosis could also cause a gap in an epithelial layer, leading to hyperpermeability. However, as a programmed activity that is evolutionarily conserved from simple multicellular organisms up to humans, tissues have developed methods to eliminate apoptotic cell without altering host homeostasis (29). Sepsis is well known to induce both augmented apoptosis and permeability, in both patients and in animal models. Previous experiments examining a potential link between increased apoptosis and permeability in the intestine have dominantly been performed in cell culture, often with conflicting results. Both Fas and LPS individually induce apoptosis and increase permeability in intestinal epithelial cell monolayers, and permeability is restored when apoptosis is blocked (17, 18). Similarly, apoptosis induced by Giardia labmlia infection in enterocyte monolayers is prevented by pre-treatment with caspase 3 inhibitors (17). In contrast, TNF and IFN-γ-induced gut barrier dysfunction is independent of apoptosis in an epithelial cell line since apoptosis inhibition by the caspase inhibitor z-VAD does not alter paracellular permeability (16). Further, while both TNF and IFN-γ- induce hyperpermeability in a three dimensional cell culture of intestinal epithelial cells, TNF-induced apoptosis contributes to worsened barrier function while IFN-γ’s role does not involve apoptosis (15). The results presented herein expand our understanding of this relationship in a number of ways. First, the results are in vivo. It is essentially impossible to model the complexities of sepsis in a cell culture setting, and the finding that permeability is normalized by Bcl-2 overexpression shows a link between apoptosis and permeability in a setting that is more clinically relevant than in vitro experiments. Next, the findings examine the intrinsic (mitochondrial) pathway of apoptosis. This is mechanistically distinct from the majority of in vitro findings examining the extrinsic (receptor-mediated) pathway. In addition, to the best of our knowledge, our findings represent the most comprehensive analysis of intestinal tight junctions in the setting of gut apoptosis prevention.
The relationship between intestinal TJ proteins and permeability in sepsis is also complex. We have previously demonstrated that CLP induces jejunal claudin 2 and JAM-A while decreasing claudin 5 and occludin (25). Pseudomonas pneumonia impacts the same TJ while also decreasing ZO-1. In contrast, subcellular localization of colonic claudins 1, 3, 4, 5, and 8 all altered and claudin 2 is increased following CLP (30). Intestine-specific overexpression of epidermal growth factor improves gut barrier dysfunction and normalizes sepsis-induced increases in claudin 2 (31). Further, systemic epidermal growth factor decreases intestinal permeability in septic mice with pre-existing chronic alcohol ingestion, associated with increases in claudin 5 and JAM-A (32). In a complementary fashion, this study finds that normalization of intestinal permeability by Bcl-2 overexpression is associated with alterations in claudin 3, claudin 4, claudin 5 and occludin. Paracellular flux through the TJ occurs through two distinct routes – the leak and pore pathways. (11, 33). The leak and pore pathways are modulated by different TJ and TJ-associated proteins -- occludin, MLCK, ZO-1 for leak and the claudin family for pore -- and are activated by different cytokines (34, 35). Notably, claudins can be either pore forming or barrier enhancing depending their structure. Additionally, the different sizes of these pathways are potentially mechanistically important since intact bacteria are too large to translocate through the pore pathway, suggesting intestinal hyperpermeability may play a role in mediating mortality through mechanisms independent of bacterial translocation (although this does not rule out a role for translocation through larger pathways). Putting our results into context suggests that sepsis impacts both the pore and leak pathways, and strategies that improve the gut barrier alter TJ in both pathways. Further, while numerous TJ are present in the intestine, only a subset are altered in sepsis and are thus more likely to play a mechanistic role in gut hyperpermeability, which suggests a targeted approach may be beneficial in efforts to improve the gut barrier in sepsis.
This study has a number of limitations. First, we have used Bcl-2 overexpression as a proxy for apoptosis prevention. Mechanistically, cell death in the intrinsic pathway of apoptosis is modulated by the balance of pro- and anti-apoptotic Bcl-2 family members, and there is extensive mechanistic data for the role of Bcl-2 in modulating apoptosis (36). Despite there being no known role for Bcl-2 in mediating permeability, we cannot absolutely rule out an unknown role of Bcl-2. Further, while our data convincingly demonstrate a role for intestinal Bcl-2 overexpression in modulating gut permeability, this does not mean that all apoptosis mediators impact the gut barrier. In addition, all studies of TJ proteins were carried out only at 24 hours. This experimental design was used because this is the timepoint where maximal apoptosis occurs and also because there was a significant difference in permeability between WT and Fabpl-Bcl-2 mice at this timepoint. Nonetheless, it is possible that we missed important changes in the TJ at earlier or later timepoints by assaying mice only at 24 hours. Further, we cannot conclude which TJs are mechanistically responsible for normalization of permeability in Fabpl-Bcl-2 mice. While numerous TJ proteins were altered in Fabl-Bcl-2 mice compared to WT mice, additional studies either knocking out or augmenting each protein would be required to distinguish which (if any) are causative for the alteration in permeability. It is also unclear why occludin mRNA levels were decreased in Fabpl-Bcl-2 mice whereas protein levels were nearly 100 times higher although we speculate that the differences between mRNA and protein levels were due to post-transcriptional modification. It should be noted that mechanistically increased occludin levels would be expected to be associated with improved permeability, as was seen in transgenic mice. Next, claudins 3, 4 and 5 are all sealing claudin, and, each would be expected to increase if they were mechanistically responsible for improved permeability. However, only claudin 4 was increased while claudins 3 and 5 were decreased in transgenic mice. FD4 has a diameter of 28 Å and measures permeability via the leak pathways (altered by occludin) but not the pore pathway (altered by the claudin family). Identifying the importance (if any) of each of these four TJ proteins in mediating decreased permeability after Bcl-2 overexpression would require future experiments where each TJ was either knocked out or ovreexpressed in transgenic mice. Finally, the cellular and/or subcellular mechanisms through which altering apoptotic machinery leads to changes in permeability require further study.
Despite these limitations, this study demonstrates that apoptosis prevention in the intestine normalizes sepsis-induced hyperpermeability associated with numerous TJ changes. Further mechanistic studies are required in order to determine what role this plays in the survival benefit conferred by intestine-specific Bcl-2 overexpression in sepsis.
Supplementary Material
SUPPLEMENTAL FIG. 1. Jejunal protein expression of Bax and Bcl-xL following CLP. Western blots did not demonstrate a difference between either Bax (p=0.78) or Bcl-xL (p=0.97) 24 hours following CLP.
Acknowledgments
Conflicts of Interest and Source of Funding: This work was supported by funding from the National Institutes of Health (GM072808, GM095442, GM104323, GM113228, AA027396)
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D., Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. : The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, et al. : Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009-2014. JAMA 318:1241–1249, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally ME, et al. : Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit Care Med 45:486–552, 2017/ [DOI] [PubMed] [Google Scholar]
- 4.Mittal R, Coopersmith CM: Redefining the gut as the motor of critical illness. Trends Mol. Med 20: 214–223, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng M, Klingensmith NJ, Coopersmith CM: New insights into the gut as the driver of critical illness and organ failure. Curr. Opin. Crit Care 23:143–148, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fay KT, Ford M., Coopersmith CM : The intestinal microenvironment in sepsis. Biochim. Biophys. Acta 1863:2574–2583, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE: Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 27:1230–1251, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM, Buchman TG, Karl IE, Hotchkiss RS: Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA 287:1716–1721, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Coopersmith CM, Chang KC, Swanson PE, Tinsle KW, Stromberg PE, Buchman TG, Karl IE, Hotchkiss RS: Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med 30:195–201, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Husain KD, Stromberg PE, Javadi P, Buchman TG, Karl IE, Hotchkiss RS, Coopersmith CM: BCL-2 Inhibits Gut Epithelial Apoptosis Induced by Acute Lung Injury in Mice but Has No Effect On Survival. Shock 20:437–443, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Odenwald MA, Turner JR: The intestinal epithelial barrier: a therapeutic target? Nat. Rev. Gastroenterol. Hepatol 14:9–21, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alverdy JC, Krezalek MA: Collapse of the Microbiome, Emergence of the Pathobiome, and the Immunopathology of Sepsis. Crit Care Med 45:337–341, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorentz CA, Liang Z, Meng M, Chen CW, Yoseph BP, Breed ER. Mittal R, Klingensmith NJ, Farris AB, Burd EM: Myosin light chain kinase knockout improves gut barrier function and confers a survival advantage in polymicrobial sepsis. Molecular medicine (Cambridge, Mass.) 23L155–165, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen WY, Wang M, Zhang J, Barve SS, McClain CJ, Joshi-Barve S: Acrolein Disrupts Tight Junction Proteins and Causes Endoplasmic Reticulum Stress-Mediated Epithelial Cell Death Leading to Intestinal Barrier Dysfunction and Permeability. Am J Pathol 187: 2686–2697, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juuti-Uusitalo K, Klunder LJ, Sjollema KA, Mackovicova K, Ohgaki R, Hoekstra D, Dekker J. van Ijzendoorn SC: Differential effects of TNF (TNFSF2) and IFN-gamma on intestinal epithelial cell morphogenesis and barrier function in three-dimensional culture. PloS one 6: e22967, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol 171:6164–6172, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Chin AC, Flynn AN, Fedwick JP, Buret AG: The role of caspase-3 in lipopolysaccharide-mediated disruption of intestinal epithelial tight junctions. Can. J. Physiol Pharmacol 84, 1043–1050, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Abreu MT, Palladino AA, Arnold ET, Kwon RS, McRoberts JA: Modulation of barrier function during Fas-mediated apoptosis in human intestinal epithelial cells. Gastroenterology 119:1524–1536, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Vyas D, Robertson CM, Stromberg PE, Martin JR, Dunne WM, Houchen CW, Barrett TA, Ayala A, Perl M, Buchman TG, et al. : Epithelial apoptosis in mechanistically distinct methods of injury in the murine small intestine. Histol. Histopathol 22:623–630, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stromberg PE, Woolsey CA, Clark AT, Clark JA, Turnbull IR, McConnell KW, Chang KC, Chung CS, Ayala A, Buchman et al. : CD4+ lymphocytes control gut epithelial apoptosis and mediate survival in sepsis. FASEB J 23: 1817–1825, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coopersmith CM, O’Donnell D, Gordon JI: Bcl-2 inhibits ischemia-reperfusion-induced apoptosis in the intestinal epithelium of transgenic mice. Am. J. Physiol 276: G677–G686, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Baker CC, Chaudry IH, Gaines HO, Baue AE: Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery 94: 331–335, 1983. [PubMed] [Google Scholar]
- 23.Osuchowski MF, Ayala A, Bahram S, Bauer M, Boros M, Cavaillon JM, Chaudry IH, Coopersmith CM, Deutschman CS, Drechsler S,et al. : Minimum Quality Threshold in Pre-Clinical Sepsis Studies (MQTiPSS): An International Expert Consensus Initiative for Improvement of Animal Modeling in Sepsis. Shock 50: 377–380, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai PY, Zhang B. He WQ, Zha JM, Odenwald MA, Singh G, Tamura A, Shen L, Sailer A, Yeruva S,et al. : IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host. Microbe 21:671–681, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoseph BP, Klingensmith NJ, Liang Z, Breed E., Burd EM, Mitta R, Dominguez JA, Petrie B, Ford ML, CM Coopersmith Mechanisms of Intestinal Barrier Dysfunction in Sepsis. Shock 46:52–59, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2018. [Google Scholar]
- 27.Gilda JE, Gomes AV: Western blotting using in-gel protein labeling as a normalization control: stain-free technology. Methods Mol. Biol 1295:381–391, 2015 [DOI] [PubMed] [Google Scholar]
- 28.Beeman N, Webb PG, Baumgartner HK Occludin is required for apoptosis when claudin-claudin interactions are disrupted. Cell death & disease 3:e273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nonaka S, Shiratsuchi A, Nagaosa K, Nakanishi Y: Mechanisms and Significance of Phagocytic Elimination of Cells Undergoing Apoptotic Death. Biological & pharmaceutical bulletin 40:1819–1827, 2017. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Zhang Q, Wang C, Liu X, Li N, Li J: Disruption of tight junctions during polymicrobial sepsis in vivo. J. Pathol 218: 210–221, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Clark JA, Gan H, Samocha AJ, Fox AC, Buchman TG, Coopersmith CM: Enterocyte-specific epidermal growth factor prevents barrier dysfunction and improves mortality in murine peritonitis. Am. J. Physiol Gastrointest. Liver Physiol 297:G471–G479, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klingensmith NJ, Yoseph BP, Liang Z, Lyons JD, Burd EM, Margoles LM, Koval M, Ford ML, Coopersmith CM Epidermal Growth Factor Improves Intestinal Integrity and Survival in Murine Sepsis Following Chronic Alcohol Ingestion. Shock 47:184–192, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.France MM, Turner JR: The mucosal barrier at a glance. J. Cell Sci 130, 307–314, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, Wang Y, Wu L. Schneeberger EE, Shen., et al. : Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J. Cell Biol 193:565–582, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR Tight junction pore and leak pathways: a dynamic duo. Annu. Rev. Physiol 73: 283–309, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh R, Letai A, Sarosiek K: Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nature reviews. Molecular cell biology, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL FIG. 1. Jejunal protein expression of Bax and Bcl-xL following CLP. Western blots did not demonstrate a difference between either Bax (p=0.78) or Bcl-xL (p=0.97) 24 hours following CLP.