

Abstract
Background
Mycobacterium abscessus is an extensively drug–resistant pathogen that causes pulmonary disease, particularly in cystic fibrosis (CF) patients. Identifying direct patient-to-patient transmission of M. abscessus is critically important in directing an infection control policy for the management of risk in CF patients. A variety of clinical labs have used molecular epidemiology to investigate transmission. However, there is still conflicting evidence as to how M. abscessus is acquired and whether cross-transmission occurs. Recently, labs have applied whole-genome sequencing (WGS) to investigate this further and, in this study, we investigated whether WGS can reliably identify cross-transmission in M. abscessus.
Methods
We retrospectively sequenced the whole genomes of 145 M. abscessus isolates from 62 patients, seen at 4 hospitals in 2 countries over 16 years.
Results
We have shown that a comparison of a fixed number of core single nucleotide variants alone cannot be used to infer cross-transmission in M. abscessus but does provide enough information to replace multiple existing molecular assays. We detected 1 episode of possible direct patient-to-patient transmission in a sibling pair. We found that patients acquired unique M. abscessus strains even after spending considerable time on the same wards with other M. abscessus–positive patients.
Conclusions
This novel analysis has demonstrated that the majority of patients in this study have not acquired M. abscessus through direct patient-to-patient transmission or a common reservoir. Tracking transmission using WGS will only realize its full potential with proper environmental screening, as well as patient sampling.
Keywords: nontuberculous mycobacteria, whole-genome sequencing, transmission, cystic fibrosis, phylogenomics
Whole-genome sequencing should replace the current molecular typing used routinely in clinical microbiology laboratories. The patient-to-patient spread of Mycobacterium abscessus is not common. Environmental screening may provide a better understanding of the acquisition of M. abscessus infections.
Mycobacterium abscessus (recently renamed as Mycobacteroides abscessus) [1], is a group of 3 closely related subspecies: M. abscessus subsp. abscessus, M. abscessus subsp. massiliense, and M. abscessus subsp. bolletii [1, 2]. These rapidly growing, nontuberculous mycobacteria cause chronic pulmonary disease, particularly in patients with cystic fibrosis (CF) and other chronic lung diseases. Mycobacterium abscessus is an important pathogen that has emerged in the CF patient population and that has been associated with poor clinical outcomes, especially following lung transplantation [3–5]. This is due, at least in part, to the extensive antibiotic resistance that makes infections with this organism difficult to treat [2, 6]. CF patients infected with M. abscessus are frequently not listed for transplant; therefore, the acquisition of this pathogen is considered to be a serious complication in this group.
The epidemiology of M. abscessus strains has been studied using Variable Nucleotide Tandem Repeats (VNTR) and Multi Locus Sequence Typing (MLST) [7]. The clustering of globally spread sequence types was confirmed with whole-genome sequencing (WGS) and has provided greater resolution in how the various lineages are related, as well as predicting possible transmission routes [8, 9]. A dominant method of transmission of M. abscessus remains contested [10, 11], with evidence for and against patient-to-patient transmission being the common route [8, 12–14]. Mycobacterium abscessus is ubiquitous in the environment, with its niche hypothesized to be free-living amoeba [15, 16], but due to the difficulties in isolating the organism, little has been done to track environment-to-patient acquisition. The confirmation of direct patient-to-patient transmission is important, as it influences the management of high-risk patients and it could increase the effectiveness of infection control interventions by directing the use of limited resources.
In this retrospective study, we assessed the utility of using WGS to characterize subspecies, antimicrobial resistance (AMR) profiles, and typing of M. abscessus isolates. We also wanted to utilize the data to investigate the scale of patient-to-patient transmission and whether identification of single nucleotide variants (SNVs) by WGS can confirm transmission. To do this, we sequenced the genomes of 145 M. abscessus clinical isolates from a well-characterized cohort of 62 patients from 4 hospitals in 2 countries over 16 years.
METHODS
Patients and Sample Collection
We collected 33 M. abscessus isolates from 30 patients at Hospital de la Santa Creu I Sant Pau (bcn_hsp), Hospital Clínic (bcn_hcl), and Hospital Vall d’Hebron (bcn_hvh) in Barcelona, Spain, and 112 isolates from 32 patients from Great Ormond Street Hospital (GOSH) in London, United Kingdom (Table 1). At GOSH, CF patients were screened for nontuberculous mycobacterial infections when attending clinics, as part of their routine management. In addition to this, CF patients and other patients at all hospitals included in this study were screened for nontuberculous mycobacterial infections when they presented with suggestive clinical symptoms or exacerbations. Demographic and patient location data were obtained from the patient administration system and microbiological data were obtained from the laboratory information management system using Structured Query Language (SQL) and Excel spreadsheets. Additional sources of information included CF and transplant databases. American Thoracic Society consensus guidelines were used to verify evidence of nontubercuolous mycobacterial infections [17]. All investigations were performed in accordance with the Hospitals Research governance policies and procedures.
Table 1.
Study Patient Information
| Patient | Hospital | Subspecies | Sex | Underlying Condition | Source of Isolate | Infection Status at First Contact | Date of First Isolate | Date of First Contact | Date of First Ward Admission |
| bcn_hcl_002 | HCL | abscessus | M | Cystic fibrosis | Lung | Already infected | 30/12/13 | 30/12/13 | 30/12/13 |
| bcn_hcl_004 | HCL | abscessus | F | Bronchiectasis | Lung | Not infected | 09/05/13 | 01/01/12 | 01/01/12 |
| bcn_hcl_005 | HCL | abscessus | F | None | Lung | Already infected | 01/08/14 | 01/08/14 | 01/08/14 |
| bcn_hcl_007 | HCL | abscessus | M | Liver neoplasi | Blood | Not infected | 10/09/14 | 2013 | 2013 |
| bcn_hcl_008 | HCL | massiliense | M | None | Lung | Already infected | 30/06/14 | 30/06/14 | 30/06/14 |
| bcn_hcl_009 | HCL | abscessus | F | Bronchiectasis | Lung | Not infected | 08/05/13 | 2008 | 2008 |
| bcn_hsp_011 | HSP | bolletii | M | None | Lung | Already infected | 01/12/00 | … | … |
| bcn_hsp_012 | HSP | abscessus | F | None | Lung | Already infected | 31/10/08 | 31/10/08 | 31/10/08 |
| bcn_hsp_014 | HSP | abscessus | F | None | Lung | Already infected | 30/01/01 | 30/01/01 | 30/01/01 |
| bcn_hsp_019 | HSP | abscessus | M | None | Lung | Already infected | 23/01/14 | 03/12/04 | 23/01/14 |
| bcn_hsp_021 | HSP | abscessus | F | Chronic bronchial infection | Lung | Not infected | 24/04/15 | 02/02/14 | 02/02/14 |
| bcn_hsp_1 | HSP | abscessus | F | None | Lung | Already infected | 17/09/09 | 17/09/09 | 17/09/09 |
| bcn_hsp_2 | HSP | abscessus | F | None | Lung | Already infected | 24/03/09 | 24/03/09 | 24/03/09 |
| bcn_hsp_3 | HSP | abscessus | F | None | Lung | Already infected | 05/06/07 | 05/06/07 | 05/06/07 |
| bcn_hvh_030 | HVH | abscessus | F | Cystic fibrosis | Lung | … | 09/01/12 | … | … |
| bcn_hvh_031 | HVH | abscessus | F | Cystic fibrosis | Lung | … | 06/05/09 | … | … |
| bcn_hvh_033 | HVH | abscessus | M | Cystic fibrosis | Lung | … | 15/07/13 | … | … |
| bcn_hvh_034 | HVH | massiliense | F | Cystic fibrosis | Lung | … | 23/01/09 | … | … |
| bcn_hvh_035 | HVH | abscessus | F | Cystic fibrosis | Lung | … | 14/02/14 | … | … |
| bcn_hvh_036 | HVH | massiliense | M | Cystic fibrosis | Lung | … | 29/03/11 | … | … |
| bcn_hvh_037 | HVH | abscessus | F | Cystic fibrosis | Lung | … | 30/01/13 | … | … |
| bcn_hvh_038 | HVH | abscessus | F | Cystic fibrosis | Lung | … | 25/07/14 | … | … |
| bcn_hvh_039 | HVH | abscessus | M | Cystic fibrosis | Lung | … | 22/01/07 | … | … |
| bcn_hvh_040 | HVH | massiliense | M | Cystic fibrosis | Lung | … | 04/09/09 | … | … |
| bcn_hvh_041 | HVH | bolletii | F | Lung transplant | Lung | … | 29/08/12 | … | … |
| bcn_hvh_042 | HVH | massiliense | F | Lung transplant | Lung | … | 18/04/12 | … | … |
| bcn_hvh_043 | HVH | abscessus | M | Lung transplant | Lung | … | 23/02/13 | … | … |
| bcn_hvh_045 | HVH | massiliense | M | Lung transplant | Lung | … | 10/04/13 | … | … |
| bcn_hvh_046 | HVH | massiliense | M | Lung transplant | Lung | … | 08/11/13 | … | … |
| bcn_hvh_047 | HVH | abscessus | F | Lung transplant | Lung | … | 28/03/13 | … | … |
| ldn_gos_1 | GOSH | bolletii | F | Cystic fibrosis | Lung | Already infected | 29/06/2004 | 27/06/2004 | 27/06/2004 |
| ldn_gos_11 | GOSH | massiliense | F | Cystic fibrosis | Lung | Not infected | 07/10/2008 | 09/06/1997 | 15/09/1998 |
| ldn_gos_14 | GOSH | massiliense | F | Cystic fibrosis | Lung | Already infected | 25/04/2005 | 24/04/2005 | 24/04/2005 |
| ldn_gos_15 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 26/06/2006 | 29/08/1995 | 26/05/2003 |
| ldn_gos_17 | GOSH | abscessus | M | Cystic fibrosis | Lung | Not infected | 21/04/2009 | 04/07/1995 | 01/01/1997 |
| ldn_gos_18 | GOSH | abscessus | M | Cystic fibrosis | Lung | Not infected | 13/12/2004 | 27/02/2001 | 03/07/2002 |
| ldn_gos_19 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 18/06/2007 | 16/04/1991 | 27/07/1994 |
| ldn_gos_2 | GOSH | abscessus | F | Cystic fibrosis | Lung | Already infected | 18/08/2005 | 15/08/2005 | 15/08/2005 |
| ldn_gos_21 | GOSH | abscessus | M | Cystic fibrosis | Lung | Not infected | 28/10/2008 | 11/11/1996 | 20/10/1997 |
| ldn_gos_22 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 08/05/2008 | 15/02/1994 | 05/01/1998 |
| ldn_gos_23 | GOSH | massiliense | F | Cystic fibrosis | Lung | Not infected | 03/02/2009 | 17/06/2003 | 10/11/2006 |
| ldn_gos_24 | GOSH | abscessus | F | Cystic fibrosis | Lung | Already infected | 30/03/2009 | 30/03/2009 | 30/03/2009 |
| ldn_gos_27 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 16/10/2010 | 07/05/2003 | 07/05/2003 |
| ldn_gos_28 | GOSH | massiliense | F | Cystic fibrosis | Lung | Already infected | 06/06/2011 | 06/06/2011 | 06/06/2011 |
| ldn_gos_3 | GOSH | abscessus | F | Cystic fibrosis | Lung | Already infected | 05/09/2005 | 04/09/2005 | 04/09/2005 |
| ldn_gos_30 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 28/06/2012 | 11/11/1997 | 13/01/1999 |
| ldn_gos_32 | GOSH | abscessus | F | Cystic fibrosis | Lung | Already infected | 08/11/2010 | 08/11/2010 | 08/11/2010 |
| ldn_gos_35 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 21/10/2013 | 05/08/2000 | 05/08/2000 |
| ldn_gos_36 | GOSH | abscessus | F | CNS tumor | Feces | Not infected | 11/01/2014 | 10/10/2013 | N/A |
| ldn_gos_37 | GOSH | abscessus | M | Cystic fibrosis | Lung | Not infected | 20/02/2014 | 01/10/2013 | 28/10/2013 |
| ldn_gos_38 | GOSH | massiliense | M | Cystic fibrosis | Lung | Already infected | 31/07/2014 | 28/04/2014 | 28/04/2014 |
| ldn_gos_39 | GOSH | massiliense | F | Cystic fibrosis | Lung | Already infected | 29/09/2014 | 29/09/2014 | 29/09/2014 |
| ldn_gos_40 | GOSH | abscessus | F | Cystic fibrosis | Lung | Already infected | 29/03/2015 | 16/02/2015 | 16/02/2015 |
| ldn_gos_41 | GOSH | abscessus | M | Cystic fibrosis | Lung | Already infected | 02/06/2015 | 01/06/2015 | 01/06/2015 |
| ldn_gos_42 | GOSH | abscessus | M | SCID | Lung | Not infected | 30/07/2015 | 27/09/2002 | … |
| ldn_gos_43 | GOSH | abscessus | M | Cystic fibrosis | Lung | Not infected | 03/08/2015 | 02/11/1999 | 25/02/2001 |
| ldn_gos_44 | GOSH | massiliense | F | Cystic fibrosis | Lung | Already infected | 27/10/2015 | 26/10/2015 | 26/10/2015 |
| ldn_gos_45 | GOSH | abscessus | M | Cystic fibrosis | Lung | Already infected | 26/02/2016 | 05/01/2015 | 05/01/2015 |
| ldn_gos_46 | GOSH | massiliense | F | Cystic fibrosis | Lung | Not infected | 09/01/2017 | 10/11/2004 | 10/11/2004 |
| ldn_gos_7 | GOSH | massiliense | F | Cystic fibrosis | Lung | Already infected | 17/12/2007 | 17/12/2007 | 17/12/2007 |
| ldn_gos_8 | GOSH | abscessus | M | Cystic fibrosis | Lung | Not infected | 29/06/2007 | 17/06/1997 | 17/06/1998 |
| ldn_gos_9 | GOSH | abscessus | F | Cystic fibrosis | Lung | Not infected | 04/12/2007 | 20/08/2007 | 10/06/2008 |
Abbreviations: CNS, central nervous system; F, female; GOSH, Great Ormond Street Hospital; HCL, Hospital Clínic; HSP, Hospital de la Santa Creu I Sant Pau; HVH, Hospital Vall d’Hebron; M, male; SCID, Severe combined immunodeficiency.
DNA Extraction, Whole-genome Sequencing, and Multi Locus Sequence Typing
Information on DNA extraction, whole-genome sequencing, and MLST are included in the Supplementary Methods.
Read Mapping and Variant Calling
Sequenced reads for all samples were first mapped to M. abscessus subsp. abscessus ATCC 19977 using BBMap v37.90 (Joint Genome Institute). SNVs were called against the reference genome using freebayes v1.2.0 [18], and variants were filtered to only include those at sites with a mapping quality >30, a base quality >30, and at least 5 supporting reads, where the variant was present on at least 2 forward and reverse strand reads and present at the 5’ and 3’ end of at least 2 reads.
Phylogenetic Analysis
Potential regions of recombination were identified from the consensus genome sequences using Gubbins v2.3.1 [19]. Regions within the genome with low coverage (<5x) were masked on a per sample basis and regions with low coverage across 75% of samples were masked across the entire data set. A maximum likelihood tree was inferred from all samples using RAxML v8.2.4 [20], using a General Time Reversible (GTRCAT) model with 99 bootstraps. Subspecies were identified for each sample based on their position upon this tree.
Separate subtrees were also inferred for M. abscessus subsp. massiliense sequences, as well as for M. abscessus subsp. abscessus ST-1 and ST-26 sequences. All samples in each subtree were mapped against a suitable reference. Mycobacterium abscessus subsp. massiliense str. GO 06 was used as the reference sequencing for study massiliense sequences, and the de novo assembly of the earliest ST-26 study sequence (ldn_gos_2_520) was used as a reference for other ST-26 samples. Mycobacterium abscessus subsp. abscessus ATCC 19977 was again used as the reference for ST-1 sequences, as it is the same sequence type. All subtrees were generated using the same method outlined above, apart from the ST-26 subtree, which did not use Gubbins: instead, variants were filtered if 3 SNVs were found within a 100 bp window.
Sequence Clusters
Sequence clusters to infer possible transmission were generated using 3 different methods on each subtree. First, we used an SNV threshold that was based on the upper bounds of all within-patient diversity and was applied to complete linkage hierarchical clustering based on a pairwise SNV matrix. Secondly, we assigned clusters using the R package rPinecone, as it incorporates SNV thresholds and root-to-tip distances and has been useful when applied to clonal populations [21]. Lastly, we also used hierarchical Bayesian Analysis of Population Structure (hierBAPS) [22] to assign clusters; however, due to the fact that all samples are included in the sequence clusters, we found it was not appropriate for this study question. We made the assumption that any strains taken from different patients that were within-sequence clusters constituted possible transmission events.
De Novo Assembly
All samples underwent de novo assembly of bacterial genomes using St. Petersburg genome Assembler (SPAdes) and pilon, wrapped in the Unicycler v0.4.4 package [23]. Assembled contigs were annotated using prokka v1.13 [24] and comparison of the accessory genome was generated using roary v3.12.0 [25]. To generate a list of genes that could be used to differentiate isolates, we filtered the annotated genes to remove coding sequences greater than 8000 bp and less than 250 bp, as well as those only present in a single sample and those present in every sample.
RESULTS
Mycobacterium abscessus Population Distribution
We obtained WGSs for 145 M. abscessus isolates from 62 patients. There were 33 M. abscessus isolates from Barcelona, subdivided into 24 M. abscessus subsp. abscessus, 2 M. abscessus subsp. bolletii, and 7 M. abscessus subsp. massiliense. There were 112 M. abscessus isolates from the United Kingdom, subdivided into 78 M. abscessus subsp. abscessus, 1 M. abscessus subsp. bolletii, and 33 M. abscessus subsp. massiliense. Sample MLST definitions, VNTR definitions, and AMR-associated mutations are shown in Supplementary Table 1.
Possible Transmission Within Mycobacterium abscessus Clusters
To confirm possible transmission between patients, we required their isolate genomes to be clustered together by 2 independent methods and epidemiological evidence that both patients were at the same hospital during the same time period. Using WGS data, we inferred a phylogenetic tree from a reference genome SNV matrix for all patients (Figure 1). We observed 2 low-variant clusters of isolates that corresponded to ST-1 and ST-26 Pasteur MLST profiles (VNTR II and I, respectively), as well as other closely related M. abscessus subsp. massiliense isolates between patients. We used an SNV matrix from mapping against a reference (M. abscessus subsp. abscessus ATCC19977), as well as hierBAPS and rPinecone, to predict sequence clusters. The sequence clusters generated from the single-reference SNV matrix provided no further information than the MLST profiles and, in many cases, provided spurious findings with large groups of isolates clustered with no epidemiological link (Supplementary Figure 1). This included large sequence clusters relating to a single MLST type, which included isolates from different hospitals and countries.
Figure 1.

Maximum likelihood SNV tree, using only the earliest isolated sample from all 62 patients. SNVs were identified from mapping reads to the ATCC19977 Mycobacterium abscessus subsp. abscessus reference genome. Sample names are highlighted in color, based on what hospital they were isolated from: Great Ormond Street Hospital, London, United Kingdom; Hospital Clínic, Barcelona, Spain; Hospital de la Santa Creu i Sant Pau, Barcelona, Spain; and Hospital Vall d’Hebron, Barcelona, Spain. The scale bar represents the number of SNVs and the node bootstrap scores below are shown if below 75. Abbreviation: SNV, single nucleotide variant.
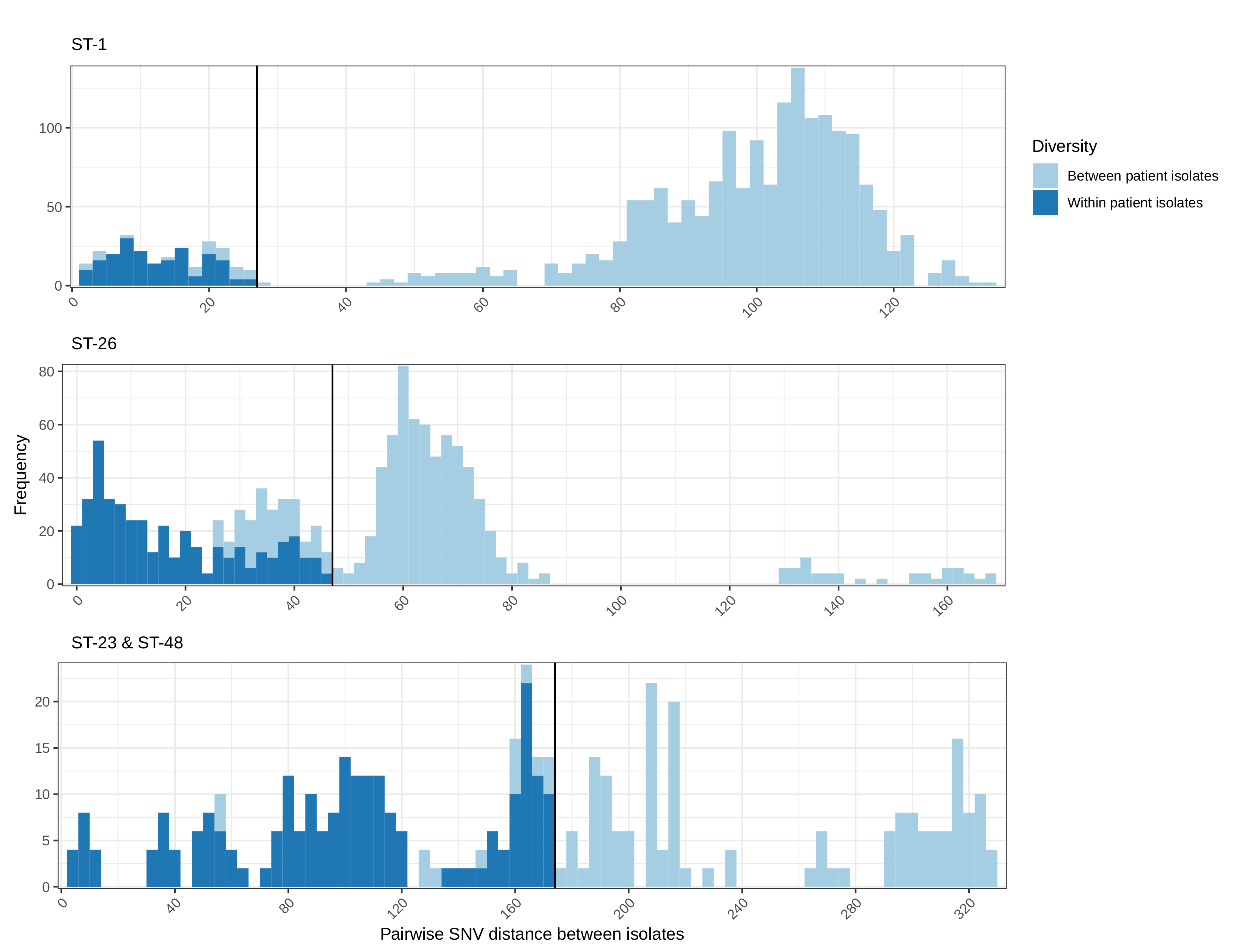
Mapping to a single reference genome led to the inability of a single SNV cut-off, or model, to exclude unrelated isolates from sequence clusters, because the number of pairwise SNV distances varied greatly between both subspecies and specific lineages (Figure 2). For example, the pairwise median SNV distance between just ST-1 isolates was 73 (interquartile range [IQR] 62–81), compared to 29 589 (IQR 27 701–63 703) for all M. abscessus subsp. abscessus isolates. The same differences were seen in M. abscessus subsp. massiliense, as well with a pairwise median SNV distance between ST-23 and ST-48 isolates of 2084 (IQR 960–7274), compared to 70 545 (IQR 59 947–71 891) across all isolates from the subspecies.
Figure 2.

Frequency of pairwise SNV distances between all isolates. SNVs were identified from mapping sequence reads to Mycobacterium abscessus subsp. abscessus ATCC19977. The full plot includes all samples, while the bottom subsidiary plot only includes isolates that have a pairwise difference between 0 and 1000 SNVs. Abbreviations: ABS; Mycobacterium abscessus subsp. abscessus; BOL, Mycobacterium abscessus subsp. bolletii; MAS, Mycobacterium abscessus subsp. massiliense; SNV, single nucleotide variant; ST, sequence type.
Subtree Sequence Clusters
The variation in the scale of diversity within subspecies and sequence types hampered efforts to capture possible transmission events. In order to improve the accuracy of sequence clustering, multiple subtrees were made for closely related isolates using a more suitable reference sequence. We separated M. abscessus subsp. abscessus and M. abscessus subsp. massiliense isolates, as well as further subtrees for ST-1 (VNTR II), ST-26 (VNTR I), and ST-23/ST-48 (VNTR III) isolates. We also integrated the presence of accessory genes when interrogating possible sequence clusters for transmission (Figures 3, 4, & 5). Sequence clusters were assigned for each subtree using both a single SNV threshold (Supplementary Figure 2) and rPinecone. Overall, we found that predicting transmission from the subtrees reduced the number of different patients clustered together from 46 to 19 and reduced the number of possible sequence clusters suggesting patient-to-patient transmission from 11 to 7.
Figure 3.

Maximum likelihood SNV tree for all ST-26 isolates. SNVs were identified from mapping reads to a de novo assembled study isolate genome (ldn_gos_2_520). Samples are highlighted based on inclusion in sequence clusters. The tree is annotated with the presence (black) and absence (white) of accessory genes, as well as the presence of AMR-associated genes and mutations. This includes the presence of a functional erm(41) gene conferring inducible resistance to macrolides; the presence of 2 rrl mutations conferring high-level macrolide resistance; and the presence of a mutation in rrs conferring high-level amikacin resistance. The scale bar represents the number of SNVs and the node bootstrap scores below are shown if below 75. Abbreviations: AMR, antimicrobial resistance; SNV, single nucleotide variant.
Figure 5.

Maximum likelihood SNV tree for all ST-23 and ST-48 isolates. SNVs were identified from mapping reads to Mycobacterium abscessus subsp. massiliense GO 06. Samples are highlighted based on inclusion in sequence clusters. The tree is annotated with the presence (black) and absence (white) of accessory genes, as well as the presence of AMR-associated genes and mutations. This included the presence of a functional erm(41) gene conferring inducible resistance to macrolides; the presence of 2 rrl mutations conferring high-level macrolide resistance; and the presence of a mutation in rrs conferring high-level amikacin resistance. The scale bar represents the number of SNVs and the node bootstrap scores below are shown if below 75. Abbreviations: AMR, antimicrobial resistance; SNV, single nucleotide variant; ST, sequence typing.
A total of 18 sequence clusters (I–XVIII) were identified (listed in Supplementary Table 1): 15 of these were within the subtrees (I–XV), and 7 clusters contained samples from more than 1 patient (IV, V, VI, VIII, XIV, XVI, & XVII). We found no sequence clusters that contained samples from both the United Kingdom and Spain. We found no evidence of transmission between patients within ST-26 (Figure 3). Within ST-1, 4 clusters (IV, V, VI, and VIII) containing samples from more than 1 patient were found. Of these, 3 clusters (IV, V, and VI) contained isolates from 9 patients from multiple hospitals within Barcelona. Only 2 of these patients were in hospital during the same time period (cluster VI: bcn_hcl_009 and bcn_hvh_30), but both were treated in different hospitals. Cluster VIII suggested transmission between 2 patients (ldn_gos_18 and ldn_gos_19) who were siblings and were previously assumed to have been infected either through direct transmission or a common reservoir (Figure 4) [13]. A single cluster (XIV) containing samples from 2 patients (ldn_gos_46 and ldn_gos_7) was found among ST-23 isolates. However, the 2 strains were isolated from samples taken 9 years apart (Figure 5). Patient ldn_gos_7 was already positive for M. abscessus on her first admission to GOSH and the 2 patients were present at the lung function lab within a month of each other on 2 occasions, but were never in the same location on the same day and were never admitted to the same ward.
Figure 4.

Maximum likelihood SNV tree for all ST-1 isolates. SNVs were identified from mapping reads to Mycobacterium abscessus subsp. abscessus ATCC19977. Samples are highlighted based on inclusion in sequence clusters. The tree is annotated with the presence (black) and absence (white) of accessory genes, as well as the presence of AMR-associated genes and mutations. This included the presence of a functional erm(41) gene conferring inducible resistance to macrolides; the presence of 2 rrl mutations conferring high-level macrolide resistance; and the presence of a mutation in rrs conferring high-level amikacin resistance. The scale bar represents the number of SNVs, and the node bootstrap scores below are shown if below 75. Abbreviations: AMR, antimicrobial resistance; SNV, single nucleotide variant.
All samples found within their respective clusters also contained similar accessory gene profiles, with the median shared percentage of accessory genes within a sequence cluster being 89% (IQR 79–94%), compared to 18% (IQR 12–37%) for isolates not in the same sequence cluster.
For the 32 GOSH CF patients included in the study, 16 became infected with M. abscessus after their first visit to the clinic (Table 1); however, transmission could only be confirmed by both WGS and epidemiological data in 1 case (ldn_gos_19), thus suggesting a different route of acquisition for the rest of these patients.
DISCUSSION
This study has shown that WGS of M. abscessus isolates can determine subspecies, identify previously reported AMR-associated mutations, and provide common typing definitions in a single workflow. This single method can replace the multiple existing molecular assays used in clinical microbiology laboratories to provide the same information and could be used to predict novel resistance variants [26]. We used the WGS data to investigate the likelihood of cross-transmission and found 43 (69%) patients had unique isolates that did not cluster with other patients. We identified 7 sequence clusters from the remaining 19 patients, but only 1 pair of patients (ldn_gos_18 and ldn_gos_19) had a plausible epidemiological link to support possible patient-to-patient transmission, as they were siblings. All other patients with genetically similar strains were either isolated in different countries or different hospitals or were isolated from samples that were taken years apart, making direct transmission of these strains extremely unlikely.
Every M. abscessus isolate from a GOSH patient was sequenced, so the data set generated represents a complete picture of M. abscessus infection in this hospital, which is vital for inferring transmission. Most of these patients were only attending clinics at GOSH; therefore, this study has captured all of their M. abscessus isolates and they are unlikely to have been in contact with M. abscessus–positive patients at other hospitals (Table 1). Therefore, if direct patient-to-patient transmission was occurring frequently, we would expect to see evidence of it here. In contrast to this, we found that the majority of patients in this study had unique strains and the majority of sequence clusters were multiple isolates from the same patients. This study confirms previous findings that, despite many M. abscessus–negative patients spending considerable time on the same wards as patients with ongoing M. abscessus infections, they did not subsequently acquire genetically similar isolates [13, 14, 27].
We have, therefore, found that a fixed number of SNVs cannot be reliably used to infer cross-transmission across all M. abscessus isolates, as there seem to be irreconcilable differences in the substitution rate between both subspecies and dominant clones. These difficulties are similar to those seen in Legionella pneumophila outbreaks, where the majority of cases can belong to only a few sequence types [25]. Legionella pneumophila can also display different scales of genetic diversity within different sequences or genotypes, indicating that a single SNV threshold cut-off will not provide sufficient discriminatory power [26]. When using WGS to infer relatedness in M. abscessus, there has previously been an attempt to find an absolute threshold which can rule in or rule out strains in a transmission event. This has previously been placed as below 25–30 SNVs [8, 14, 28, 29]. From our findings, we would advocate using a suitable, genetically similar reference sequence when carrying out core genome SNV calling, especially for the dominant clones, such as ST-1 and ST-26. There is a large amount of variation within the genomes of M. abscessus [30], so the use of a single reference, such as M. abscessus subsp. abscessus ATCC 19977, will mask many differences between strains and generate spurious clusters of genetically similar sequences. Where a suitable reference is not available, we recommend using a high-quality draft de novo assembly of the first isolated sample to compare other isolates against, as in the example of the ST-26 samples in this study (Figure 3).
In addition to conducting the core genome SNV analysis, we also found that the integration of accessory genome information is a useful indicator of relatedness within M. abscessus isolates that can be used to further interrogate assigned sequence clusters. Generally, there was good concordance between the proportion of putative genes shared and the SNV distance between 2 samples. This was helped by using closely related reference sequences to map sequence reads against. We have seen in this study and previously [31] that there is diversity in the accessory genome profiles—as well as in the number of SNPs and AMR-associated mutations—taken from multiple samples from the same patient on the same day. However, we have always found interpatient diversity to be greater than that seen within the same patient. This would suggest that any direct transmission between patients of even minority populations would still be identified by WGS and, taken together, the data suggest that person-to-person transmission of M. abscessus in pediatric patients in our institution is very uncommon. In this study, we have an example of 2 patients with transmission predicted by genomic epidemiology (ldn_gos_7 and ldn_gos_46) that had attended a lung function laboratory on 3 occasions within a month of each other. In this case, the only way transmission could have occurred would be if ldn_gos_7, who was already infected, contaminated the environment and the infection was then transmitted to ldn_gos_46. The predominant view [8] that human-to-human transmission occurs via contamination of fomites by respiratory secretions could explain this, although no other instances of this appeared to have occurred, despite numerous other CF patients attending the unit over many years. What is harder to explain is that, for this to be the case, the interval between exposure and culture positivity was 9 years. It could be that M. abscessus remains present but undetectable by conventional methods for this time period or, intriguingly, could cause a latent infection, like what occurs with Mycobacterium tuberculosis. To the best of our knowledge, this has never been a demonstrated part of the pathogenesis of M. abscessus infection, and may be worthy of further investigation.
In agreement with previous studies, we found an international distribution of M. abscessus–dominant clones [8]. We found WGS to be useful to confirm whether different patients’ strains are unrelated, even within the dominant clones, but it has been far more difficult to reach definite conclusions about cross-transmission. Without environmental samples, we cannot rule out the possibility of intermediate sources of infection; therefore, WGS as a tool for tracking cross-transmission in M. abscessus will only realize its full potential with the proper screening of environmental sources, alongside longitudinal patient sampling.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
Notes
Acknowledgments. The authors thank the Biomedical Scientist team for sample collection at Great Ormond Street Hospital, as well as Dr Julià Gonzalez and Dr Teresa Tórtola for sample collection at Hospital Clinic and Hospital de la Vall d’Hebron, respectively.
Financial support. This work was supported by the National Institute for Health Research (European Molecular Biology Organization Short-Term Fellowship 7307 to M. R.) and the European Association of National Metrology Institutes (grant number 15HLT07 to R. M. D.).
Potential conflicts of interest. The authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Gupta RS, Lo B, Son J. Phylogenomics and comparative genomic studies robustly support division of the genus mycobacterium into an emended genus mycobacterium and four novel genera. Front Microbiol 2018; 9:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griffith DE, Brown-Elliott BA, Benwill JL, Wallace RJ Jr. Mycobacterium abscessus. “Pleased to meet you, hope you guess my name.” Ann Am Thorac Soc 2015; 12:436–9. [DOI] [PubMed] [Google Scholar]
- 3. Chalermskulrat W, Sood N, Neuringer IP, et al. Non-tuberculous mycobacteria in end stage cystic fibrosis: implications for lung transplantation. Thorax 2006; 61:507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang HC, Weigt SS, Derhovanessian A, et al. Non-tuberculous mycobacterium infection after lung transplantation is associated with increased mortality. J Heart Lung Transplant 2011; 30:790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robinson PD, Harris KA, Aurora P, Hartley JC, Tsang V, Spencer H. Paediatric lung transplant outcomes vary with Mycobacterium abscessus complex species. Eur Respir J 2013; 41:1230–2. [DOI] [PubMed] [Google Scholar]
- 6. Rubio M, March F, Garrigó M, Moreno C, Español M, Coll P. Inducible and acquired clarithromycin resistance in the Mycobacterium abscessus complex. PLOS One 2015; 10:e0140166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Macheras E, Konjek J, Roux AL, et al. Multilocus sequence typing scheme for the Mycobacterium abscessus complex. Res Microbiol 2014; 165:82–90. [DOI] [PubMed] [Google Scholar]
- 8. Bryant JM, Grogono DM, Rodriguez-Rincon D, et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 2016; 354:751–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davidson RM, Hasan NA, de Moura VC, Duarte RS, Jackson M, Strong M. Phylogenomics of Brazilian epidemic isolates of Mycobacterium abscessus subsp. bolletii reveals relationships of global outbreak strains. Infect Genet Evol 2013; 20:292–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malcolm KC, Caceres SM, Honda JR, et al. Mycobacterium abscessus displays fitness for fomite transmission. Appl Environ Microbiol 2017; 83:e00562–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pasipanodya JG, Ogbonna D, Ferro BE, et al. Systematic review and meta-analyses of the effect of chemotherapy on pulmonary Mycobacterium abscessus outcomes and disease recurrence. Antimicrob Agents and Chemother 2017; 61:e01206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bryant JM, Grogono DM, Greaves D, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 2013; 381:1551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris KA, Underwood A, Kenna DT, et al. Whole-genome sequencing and epidemiological analysis do not provide evidence for cross-transmission of Mycobacterium abscessus in a cohort of pediatric cystic fibrosis patients. Clin Infect Dis 2015; 60:1007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tortoli E, Kohl TA, Trovato A, et al. Mycobacterium abscessus in patients with cystic fibrosis: low impact of inter-human transmission in Italy. Eur Respir J 2017; 50:1602525. [DOI] [PubMed] [Google Scholar]
- 15. N’Goma JCB, Moigne VL, Soismier N, et al. Mycobacterium abscessus phospholipase C expression is induced during coculture within amoebae and enhances M. abscessus virulence in mice. Infect Immun 2015; 83:780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laencina L, Dubois V, Le Moigne V, et al. Identification of genes required for Mycobacterium abscessus growth in vivo with a prominent role of the ESX-4 locus. Proc Natl Acad Sci USA 2018; 115:E1002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Griffith DE, Aksamit T, Brown-Elliott BA, et al. ; American Thoracic Society Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416. [DOI] [PubMed] [Google Scholar]
- 18. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. Available at: http://arxiv.org/abs/1207.3907. Accessed 11 September 2015.
- 19. Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 2015; 43:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014; 30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wailan AM, Coll F, Heinz E, et al. rPinecone: Define sub-lineages of a clonal expansion via a phylogenetic tree. Microb Genom 2019; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tonkin-Hill G. rhierbaps: R implementation of hierBAPS. Contribute to gtonkinhill/rhierbaps development by creating an account on GitHub. Available at: https://github.com/gtonkinhill/rhierbaps. Accessed 3 September 2018.
- 23. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput Biol 2017; 13:e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014; 30:2068–9.24642063 [Google Scholar]
- 25. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015; 31:3691–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lipworth S, Hough N, Leach L, et al. Whole-genome sequencing for predicting clarithromycin resistance in Mycobacterium abscessus. Antimicrobial Agents and Chemotherapy 2019; 63:e01204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomson R, Tolson C, Sidjabat H, Huygens F, Hargreaves M. Mycobacterium abscessus isolated from municipal water - a potential source of human infection. BMC Infect Dis 2013; 13:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schürch AC, Arredondo-Alonso S, Willems RJL, Goering RV. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene-based approaches. Clin Microbiol Infect 2018; 24:350–4. [DOI] [PubMed] [Google Scholar]
- 29. Yan J, Kevat A, Martinez E, et al. Investigating transmission of Mycobacterium abscessus amongst children in an Australian cystic fibrosis centre. Journal of Cystic Fibrosis 2019. Available at: http://www.sciencedirect.com/science/article/pii/S1569199318309184. Accessed 19 March 2019. [DOI] [PubMed]
- 30. Choo SW, Wee WY, Ngeow YF, et al. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci Rep 2014; 4:4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaw LP, Doyle RM, Kavaliunaite E, et al. Children with cystic fibrosis are infected with multiple subpopulations of Mycobacterium abscessus with different antimicrobial resistance profiles. Clin Infect Dis 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.