

Abstract
Succinate dehydrogenase (SDH) deficiency is associated with gastrointestinal stromal tumor (GIST) oncogenesis, but the underlying molecular mechanism remains to be further investigated. Here, we show that succinate accumulation induced by SDHB loss of function increased the expression of zinc finger protein 148 (ZNF148, also named ZBP‐89) in GIST cells. Meanwhile, ZNF148 is found to be phosphorylated by ERK at Ser306, and this phosphorylation results in ZNF148 binding to Forkhead box M1 (FOXM1). Through the complex formation at the promoter, ZNF148 facilitates Histone H3 acetylation and FOXM1‐mediated Snail transcription, which eventually promotes cell invasion and tumor growth. The clinical analysis indicates that SDHB deficiency is associated with elevated ZNF148 levels, and ZNF148‐S306 phosphorylation level displays a positive correlation with poor prognosis in GIST patients. These findings illustrate an unidentified molecular mechanism underlying FOXM1‐regulated gene transcription related to GIST cell invasion, which highlights the physiological effects of SDHB deficiency on the invasiveness of GIST.
Keywords: ERK, gastrointestinal stromal tumor, phosphorylation, Succinate dehydrogenase, ZNF148
The article illustrates an unidentified molecular mechanism underlying FOXM1‐regulated gene transcription related to GIST cell invasion, which highlights the physiological effects of SDHB deficiency on the invasiveness of GIST.
1. INTRODUCTION
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors of the gastrointestinal tract. 1 , 2 , 3 GIST are categorized according to the identified genetic mutations, among which the mutually exclusive mutation in the KIT or the PDGFRA gene is frequently observed. 4 , 5 The cases without either KIT or PDGFRA mutation are termed wild‐type (WT) GIST, which are classified into succinate dehydrogenase (SDH)‐deficient and non–SDH‐deficient groups. 6 , 7 , 8 SDH is a mitochondrial enzyme critically involved in the Krebs cycle, which consists of four subunits, SDHA, SDHB, SDHC and SDHD. 9 GIST with genetic alteration in SDH usually present loss of SDHB expression, and immunohistochemical analysis of SDHB is frequently used to determine whether SDH deficiency has occurred. 10 Deficiency of SDH complex function can result in intracellular accumulation of succinate, which is able to competitively inhibit α‐KG‐dependent hypoxia‐inducible factor (HIF) prolyl hydroxylases; this leads to HIF1α stabilization and nuclear accumulation, which is implicated during tumorigenesis. 11 Similarly, succinate accumulation is able to inactivate other α‐KG‐dependent dioxygenases and JmjC domain‐containing histone demethylases (KDM), and these effects are potentially linked with fundamental alteration of cellular physiology in cancer cells.
FOXM1, the Forkhead box protein acting as a transcriptional factor, plays important roles in various cellular events, including cell proliferation, differentiation and migration. 12 , 13 FOXM1 can integrate multiple extracellular stimuli into distinct physiological responses and its deregulation is linked with disease development. 14 Overexpression of FOXM1 is associated with tumor invasion and metastasis in distinct types of human cancers. 13 , 15
ZNF148 is a Krüppel‐type zinc finger family protein that binds to GC‐rich sequences in various gene promoters. 16 , 17 ZNF148 can act as a transcriptional regulator to activate or repress gene expression. 18 Previous studies indicate that ZNF148 potentially acts as a tumor suppressor in HCC, as reflected by its promotional effect on cell apoptosis in a p53‐dependent or p53‐independent manner. 19 , 20 The implications of ZNF148 in tumor development in a genetic context are worthy of further investigation.
In the present study, we show that SDHB deficiency that causes succinate accumulation in GIST cells results in elevated ZNF148 expression. In addition, ZNF148 is phosphorylated by ERK at Ser306, which leads to its interaction with FOXM1 and its recruitment at the promoter. In turn, ZNF148 facilitates FOXM1‐mediated Snail transcription, and thereby promotes GIST cell invasion.
2. MATERIALS AND METHODS
2.1. Cell culture
The GIST882 cell line was established from an untreated human GIST with a homozygous missense mutation in KIT exon 13, and was kindly provided by Dr Fletcher from Harvard Medical School. The GIST‐T1 cell line was purchased from Cosmo Bio. The GIST‐T1 cell line is characterized by a heterozygous deletion of 57 bases in KIT exon 11. GIST882 cells were cultured in RPMI‐1640 (ATCC) supplemented with 15% FBS and 1% l‐glutamine, and GIST‐T1 cells were cultured in DMEM (ATCC) supplemented with 10% FBS.
2.2. DNA constructs and mutagenesis
pGIPZ human SDHB shRNA was generated with the oligonucleotide 5'‐ATGGAGGCAACACTCTAGC‐3'. pGIPZ human ZNF148 shRNA was generated with the oligonucleotide 5'‐AAGCATACTTTGAAACTTTGC‐3' and 5'‐AATTCATTCCTGATCGAAAGC‐3'. pGIPZ human FOXM1 shRNA was generated with the oligonucleotide 5'‐GGACCACUUUCCCUACUUU‐3'. The pGIPZ controls were generated with control oligonucleotide 5'‐GCTTCTAACACCGGAGGTCTT‐3' or 5'‐GCCCGAAAGGGTTCCAGCTTA‐3'. pGIPZ human p300 shRNA was generated with the oligonucleotide 5'‐CTAGAGACACCTTGTAGTA‐3'.
2.3. Materials
Antibodies that recognize H3K36Me2 (ab9049, 1:1000), KDM2A (ab31739), Ac‐H3K9 (ab10812, 1:1000), ZNF148 (ab69933, 1:1000), SDHB 1:1000), FOXM1 (ab180710, 1:1000) and H3 (ab1791, 1:1000) were purchased from Abcam. Antibodies that recognize GST (#T509, 1:1000), Flag (#35535, 1:1000) and His (#T505, 1:1000) were obtained from Signalway Antibody. Antibodies against β‐actin (#4970, 1:2000), p44/42 MAPK (Erk1/2) (#9102, 1:1000) and Phospho‐p44/42 MAPK (Erk1/2) (D13.14.4E, 1:1000) were purchased from Cell Signaling Technology. Rabbit polyclonal ZNF148 pSer‐306 antibody (1:500) was made by Signalway Antibody. A peptide containing ZNF148 pSer‐306 antibody was injected into rabbits. The rabbit serum was collected and purified using an affinity column with non–phosphorylated ZNF148 pSer‐306 peptide to exclude the antibodies for non–phosphorylated ZNF148, followed by an affinity column with phosphorylated ZNF148 pSer‐306 peptide to bind to and purify the ZNF pSer‐306 antibody. U0126, SU6656 and EGF were purchased from Sigma‐Aldrich.
2.4. Immunoprecipitation and immunoblotting analysis
Proteins were extracted from cultured cells using a modified buffer (50 mmol/L Tris‐HCl, 0.5 mmol/L EDTA, 1 mmol/L dithiothreitol, 1% Triton X‐100, 150 mmol/L NaCl, and protease inhibitor cocktail or phosphatase inhibitor cocktail), followed by immunoprecipitation and immunoblotting with the indicated antibodies. The protein concentration was determined using the Bradford assay. Proteins from cellular lysates were separated by SDS‐PAGE, transferred onto PVDF membrane (Millipore Corporation) and probed with the indicated antibodies.
2.5. Recombinant protein purification
Wild‐type and mutant GIST‐ZNF148 were expressed in bacteria and purified. Briefly, constructs were used to transform BL21/DE3 bacteria. The cultures were grown at 37°C, monitored by optical density 600 nm, before isopropyl‐D‐thiogalactopyranoside treatment. Cell pellets were collected and lysed by sonication. Cleared lysates were bound to glutathione‐agarose to obtain GST‐tagged proteins. Elutes were concentrated using Ultrafree‐15 centrifugal filters (Millipore).
2.6. Gene expression analysis
Total RNA were from cells using RNAzol RT (Molecular Research Center) following the manufacturer’s instructions. cDNA was synthesized from 1 μg total RNA using an iScript cDNA Synthesis Kit (Bio‐Rad) and we quantified mRNA levels by real‐time RT‐PCR using SYBR Green (Bio‐Rad). Samples in technical triplicates were calculated, with relative mRNA levels normalized to actin mRNA levels in the same samples. The qPCR primer sequences were: human ZNF148: 5′‐TGATGATGCCATGCAGTTTT‐3′ (forward) and 5′‐TCCCTGCTGTTGTTACT TGCT‐3′ (reverse); Snail: 5′‐CTCTTTCCTCGTCAGGAAGC‐3′ (forward) and 5′‐GG CTGCTGGAAGGTAAACTC‐3′ (reverse).
2.7. In vitro kinase assay
Purified WT and mutant GIST‐ZNF148 were incubated with ERK in the presence of 0.5 μCi of hot ATP (ICN Biochemicals) in 50 μL of kinase buffer that contained 50 mmol/L Tris‐HCl, 4 mmol/L MgCl2 and 10 mmol/L β‐mercaptoethanol for 20 minutes at 30°C.
2.8. ChIP assay
A ChIP assay was performed using an Upstate Biotechnology Kit. Quantitative real‐time PCR was used to measure the amount of bound DNA, and the value of enrichment was calculated. The primers covering putative NF‐κB1 binding consensus of human were used for the real‐time PCR: 5′‐CAGTTTGGTAGATGAAATATGATA GTGC‐3′ (forward) and 5′‐GACAGAACATCATCAAGCAAATCA‐3′ (reverse). The primers covering putative FOXM1 binding consensus of human SNAIL gene promoter region were used for the real‐time PCR: 5′‐AGACAGTAGTTCTGCCCT TCAGGTT‐3′ (forward) and 5′‐ATGGAGCCGTGTTACAGCCT‐3′ (reverse).
2.9. Succinate measurement
Succinate concentrations in cells were determined using the Succinate Assay Kit, purchased from Abcam.
2.10. Cell invasion assay
The indicated cells were seeded in 24‐well invasion chambers (BD Biosciences) with the Matrigel‐coated film insert (8 mm pore). The mixed solution was diluted to a 1 × DMEM solution containing 10% serum. The cells were cultured in the absence or presence of EGF (100 ng/mL) (chemokinesis). After 2 days, cells on the bottom surface of the filter were subjected to staining with DAPI for 1 minute, and then were washed three times with PBS, and the cell number was counted under a fluorescence microscope (Olympus).
2.11. Mouse
All animal experiments were approved by the animal care and use committee of Zhongshan Hospital, Fudan University. Twenty (6‐week‐old) female BALB/c nude mice were divided into two groups (ten mice per group). For the control group, Balb/c nude mice were injected with ZNF148 WT/SDHB shRNA GIST‐T1 cells; for the SDHB‐shRNA group, BALB/c nude mice were injected with ZNF148 mutant/SDHB shRNA GIST‐T1 cells. The prepared cells were injected into the spleen with a needle during an open laparotomy to establish an in vivo mice model. After 8 weeks, mice were killed. Liver tissues were resected, fixed in 4% paraformaldehyde, embedded in paraffin and sectioned at 5 μm. Liver metastasis was confirmed by staining with H&E and CD117.
2.12. Human tissue specimens and immunohistochemical analysis
Human tumor samples were obtained from 67 WT GIST patients treated at the hospital between 2003 and 2013. Written informed consent was obtained from each patient and the investigation was approved by the institutional review board of Zhongshan Hospital, Fudan University, Shanghai, China. Progression free survival time was calculated from the date of surgery to the date of recurrence. Consecutive sections of formalin‐fixed paraffin‐embedded (FFPE) tumors were subjected to immunohistochemistry (IHC) analysis for ZNF148 pSer‐306. Rabbit polyclonal ZNF148 pSer‐306 antibody (Signalway, 1:50) was used. A DAB substrate kit (GTVision Detection System/Mo&Rb Kit) was used according to manufacturer’s instructions. The results were scored by two pathologists blinded to the clinicopathologic data.
2.13. Statistical analysis
Differences between indicated groups were analyzed using the Student t test, the χ2 test, or Spearman’s rank correlation coefficient test. The log‐rank test was used to calculate a P‐value for the significance of divergence of Kaplan‐Meier curves. All probability values were presented as two‐sided. Analyses were conducted with the SPSS version 22.0 and GraphPad Prism 6.02 statistical analysis software. Statistical significance was defined as P < 0.05.
3. RESULTS
3.1. Zinc finger protein 148 is upregulated by loss of SDHB and interacts with Forkhead box M1
To investigate the relevance of FOXM1‐mediated cellular effects to the invasiveness of GIST with SDHB deficiency, Flag‐FOXM1 was stably transfected in GIST cell line GIST‐T1. Immunoprecipitation and the following mass spectrometry analysis indicated that EGF treatment dramatically promoted the binding of ZNF148, a transcriptional factor containing the zinc finger motif, to FOXM1 in SDHB‐silenced but not SDHB‐intact GIST‐T1 cells (Figure 1A and Figure S1A). Intriguingly, SDHB depletion led to the upregulated level of ZNF148, which might explain its positive effect on ZNF148‐FOXM1 interaction (Figure 1A). The interaction between FOXM1 and ZNF148 was verified by co–immunoprecipitation analysis at the endogenous level (Figure 1B). Similarly, SDHB depletion largely enhanced FOXM1‐ZNF148 interaction in GIST cell line GIST882 cells with EGF treatment (Figure S1B). Of note, FOXM1‐ZNF148 complex formation appeared more impressive in SDHB‐silenced GIST882 cells than SDHB‐silenced GIST‐T1 cells in the absence of EGF stimulus (Figure S1C), which would be due to the distinct EGF‐EGFR signaling between two cell lines. With respect to metabolic function, loss of SDHB in GIST‐T1 and GIST882 cells led to a robust accumulation of intracellular succinate, which was not affected by EGF treatment (Figure S1D). Accumulation of succinate can potentially lead to inhibition of α‐KG‐dependent DNA or histone demethylases. Hence, we wondered whether the enhanced ZNF148 expression level resulted from the inhibitory effect of succinate on histone demethylation. As a result, addition of exogenous α‐KG into the cell culture medium significantly repressed ZNF148 expression in SDHB‐silenced GIST‐T1 (Figure 1C) and GIST882 (Figure S1E) cells. In addition, the mRNA and protein level of ZNF148 in SDHB‐silenced GIST‐T1 (Figure 1D,E) and GIST882 (Figure S1F and G) cells was found to be decreased by overexpression of KDM2A, a histone demethylase responsible for H3K36Me2 demethylation, but was not affected by overexpression of KDM4A or KDM7A, which are histone demethylases against H3K36Me3 and H3K27me2, respectively. Interestingly, ChIP analysis further showed that SDHB depletion dramatically increased the accumulation of H3K36Me2 at the ZNF148 promoter region with a putative NF‐κB1 binding site in GIST‐T1 (Figure 1F,G) and GIST882 (Figure S1H,I) cells, which was abrogated by either addition of exogenous α‐KG or overexpression of KDM2A. Meanwhile, KDM2A accumulation at ZNF148 promoter was detected in both cell lines, which was not significantly changed after SDHB depletion (Figure S1J,K). These data indicate that SDHB depletion‐induced succinate accumulation promotes ZNF148 expression through its inhibitory effects on α‐KG‐dependent KDM2A.
Figure 1.
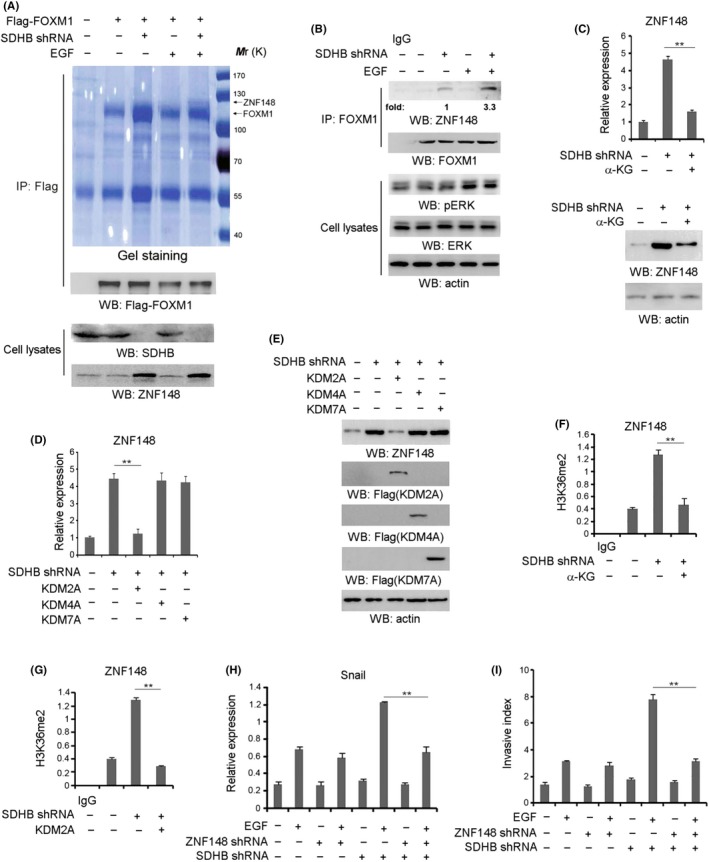
Zinc finger protein 148 (ZNF148) is upregulated by loss of SDHB and interacts with Forkhead box M1 (FOXM1). A, Gastrointestinal stromal tumor (GIST)‐T1 cells transfected with or without plasmid to express SDHB shRNA were treated with or without EGF (100 ng/mL) for 1 h. Coomassie brilliant blue staining of the immunoprecipitates was performed. B, GIST‐T1 cells with or without SDHB shRNA expression were treated with or without EGF (100 ng/mL) for 1 h. C, GIST‐T1 cells with or without SDHB shRNA expression were added with exogenous α‐KG (20 µmol/L). Q‐PCR (upper panel) or immunoblotting analyses (bottom panel) were performed. D, GIST‐T1 cells with or without SDHB shRNA expression were overexpressed with KDM2A, KDM4A or KDM7A. E, GIST‐T1 cells with or without SDHB shRNA expression were overexpressed with KDM2A, KDM4A or KDM7A. Cellular extracts were subjected to immunoblotting analyses. F, GIST‐T1 cells with or without SDHB shRNA expression were added with exogenous α‐KG (20 µmol/L). ChIP analyses with an anti–H3K36me2 antibody were performed. G, GIST‐T1 cells with or without SDHB shRNA expression were overexpressed with KDM2A. ChIP analyses were performed with an anti–H3K36me2 antibody. The primers covering the ZNF148 gene promoter region were used for the RT‐PCR. H, GIST‐T1 cells transfected with or without the plasmid for expressing ZNF148 shRNA and/or SDHB shRNA were treated with or without EGF (100 ng/mL) for 12 h. Q‐PCR analysis was performed. I, GIST‐T1 cells transfected with or without ZNF148 shRNA and/or SDHB shRNA were treated with or without EGF (100 ng/mL). Cell invasion assays were performed. In A‐C, E and H, immunoblotting analyses were performed using the indicated antibodies and data represent 1 out of 3 experiments. In C, D and F‐I, the values are presented as mean ± SEM (n = 3 independent experiments), ** represents P < 0.01 between the indicated groups
To determine the effect of ZNF148 on cell migration, we expressed a ZNF148 shRNA in GIST‐T1 and GIST882 cells (Figure S1L), and found that ZNF148 depletion notably attenuated EGF‐increased mRNA (Figure 1H and Figure S1M) or protein level (Figure S1N and O) of Snail, a FOXM1‐targeted gene, and cell invasion (Figure 1I and Figure S1P) in SDHB‐depleted but not SDHB‐intact cells. In contrast, FOXM1 depletion (Figure S1Q) largely repressed EGF‐induced Snail expression (Figure S1R) and cell invasion (Figure S1S), either in SDHB‐intact or SDHB‐depleted GIST‐T1 cells, which was not ameliorated by ZNF148 depletion (Figure S1T,U). These data demonstrate that increased expression of ZNF148 is required for the enhancement of cell migration by EGF in SDHB‐depleted GIST cells.
3.2. ERK activation is required for zinc finger protein 148‐Forkhead box M1 interaction
We set forth to determine the signaling modulator that is responsible for EGF‐induced ZNF148‐FOXM1 interaction. A time course analysis of co–immunoprecipitation showed that EGF substantially stimulated ZNF148‐FOXM1 complex formation to a peak value around 30 minutes post–treatment in SDHB‐silenced GIST‐T1 cells (Figure 2A). Pretreatment of GIST‐T1 cells with the MEK/ERK inhibitor U0126, Src inhibitor SU6656 and phosphoinositide 3‐kinase inhibitor LY290042 efficiently blocked EGF‐induced phosphorylation of AKT, c‐Src and ERK1/2, respectively (Figure S2), while only inhibition of MEK/ERK significantly inhibited EGF‐induced ZNF148‐FOXM1 interaction (Figure 2B). Of note, EGF could induce the evident increase of ERK in GIST‐T1 cells (Figure 1B), although both cell lines harbor the c‐KIT mutation that might be related to ERK activity and ZNF148‐FOXM1 complex formation detected at basal level (Figure 1B). Subsequently, we expressed the constitutively active MEK1 Q56P mutant in GIST‐T1 cells. As shown in Figure 2C, overexpression of MEK1 Q56P (MEK1 active form) was sufficient for the induction of FOXM1‐ZNF148 interaction. In addition, ZNF148‐FOXM1 interaction induced by either MEK1 Q56P expression (Figure 2C) or EGF stimulation (Figure 2D) was disrupted by expression of the Flag‐ERK2 K52R kinase‐dead mutant, compared with its WT counterpart. These results suggest that ERK activation is required for EGF‐induced interaction between FOXM1 and ZNF148.
Figure 2.
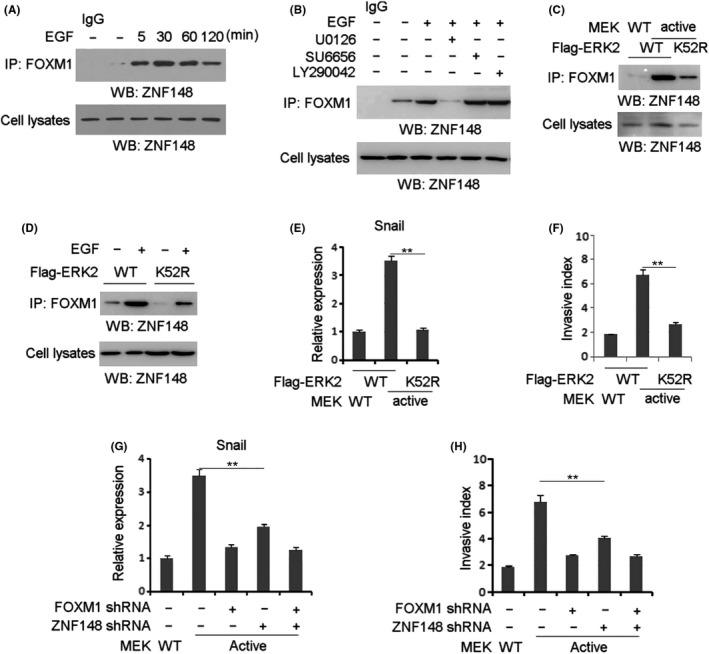
ERK activation is required for zinc finger protein 148 (ZNF148)‐Forkhead box M1 (FOXM1) interaction. A, Gastrointestinal stromal tumor (GIST)‐T1 cells with SDHB depletion were treated with or without EGF (100 ng/mL) for indicated length of time. B, GIST‐T1 cells with SDHB depletion were pretreated with U0126 (20 µmol/L), SU6656 (10 µmol/L) or LY290042 (20 µmol/L) for 1 h, prior to EGF treatment (100 ng/mL) for 1 h. C, GIST‐T1 cells with SDHB depletion were expressed with WT MEK or MEK1 Q56P constitutively active mutant and WT ERK or ERK K52R kinase‐dead mutant. D, GIST‐T1 cells with SDHB depletion were expressed with WT ERK or ERK K52R kinase‐dead mutant. Cells were treated with or without EGF (100 ng/mL) for 1 h. E, GIST‐T1 cells with SDHB depletion were expressed with WT MEK or MEK1 Q56P constitutively active mutant and WT ERK or ERK K52R kinase‐dead mutant. Q‐PCR analysis was performed. F, GIST‐T1 cells with SDHB depletion were expressed with WT MEK or MEK1 Q56P constitutively active mutant and WT ERK or ERK K52R kinase‐dead mutant. Cell invasion assays were performed. G, GIST‐T1 cells with SDHB depletion were expressed with indicated plasmids. Q‐PCR analysis was performed. H, GIST‐T1 cells with SDHB depletion were expressed with indicated plasmids. Cell invasion assays were performed. In A‐D, immunoblotting analyses were performed using the indicated antibodies. In E‐F, the values are presented as mean ± SEM (n = 3 independent experiments), ** represents P < 0.01 between the indicated groups
Subsequently, Snail expression level and cell migration were examined in GIST‐T1 cells with co–expression of MEK1 Q56P and WT ERK2 or ERK K52R. Real‐time PCR (RT‐PCR) analysis showed that MEK1 Q56P expression dramatically increased Snail transcription in GIST‐T1 cells expressed with WT ERK2 but not ERK K52R (Figure 2E). Consistently, the invasive ability of GIST‐T1 cells was enhanced by co–expression of MEK1 Q56P and WT ERK2 (Figure 2F). These results suggest that MEK/ERK signaling is critical to EGF‐enhanced cell invasion in GIST‐T1 cells. Furthermore, either ZNF148 depletion or FOXM1 depletion inhibited MEK1 Q56P‐increased Snail transcription (Figure 2G) and cell invasion (Figure 2H), and ZNF148 depletion did not aggravate the effects from FOXM1 depletion (Figure 2G,H). These results indicate that ZNF148 and FOXM1 promote tumor cell invasion under activation of MEK/ERK signaling.
3.3. ERK phosphorylates zinc finger protein 148 at Ser306
Co–immunoprecipitation analysis further showed that treating precipitates with CIP notably disrupted the EGF‐induced interaction between ZNF148 and FOXM1 (Figure 3A), revealing that ZNF148‐FOXM1 complex formation is phosphorylation‐dependent. To further understand the mechanism underlying ERK‐mediated ZNF148‐FOXM1 interaction, the reciprocal pull‐down assays were performed using purified GST‐ZNF148 or GST‐FOXM1. As a result, GST‐FOXM1 was able to interact with cellular ZNF148 in GIST cells treated with EGF (Figure 3B, the left panel). In contrast, the association of GST‐ZNF148 with cellular FOXM1 was not evident (Figure 3B, the right panel). These data indicate that the EGF‐induced ZNF148‐FOXM1 interaction is more likely reliant on the changed protein status of ZNF148 instead of FOXM1, and raise the possibility that ZNF148 is a substrate of ERK. This assumption was supported by the in vitro protein phosphorylation assay that showed that ZNF148 was phosphorylated by purified activated ERK2 (Figure 3C). Scansite analysis indicated that the ZNF148 amino acid sequence contains multiple consensus phosphorylation motifs presented as S/TP site, and an in vitro protein phosphorylation assay indicated that only the mutation of evolutionarily conserved Ser 306 (Figure 3D, left panel) abolished ZNF148 phosphorylation mediated by ERK2, as demonstrated by autoradiography and immunoblotting analysis with a specific antibody against ZNF148‐S306 phosphorylation (Figure 3D, right panel). Meanwhile, ZNF148‐S306 phosphorylation was remarkably induced by MEK1 Q56P expression, which was blocked by expression of the Flag‐ERK2 K52R kinase‐dead mutant but not its WT counterpart (Figure 3E). These results suggest that ERK2 phosphorylates ZNF148 at Ser306 under EGF stimulation. Subsequently, co–immunoprecipitation analysis showed that EGF notably increased the interaction between FOXM1 and WT ZNF148 but not ZNF148 S306A in SDHB‐depleted GIST‐T1 (Figure 3F) and GIST882 cells (Figure S3A). Furthermore, a GST pull‐down assay showed purified WT ZNF148 but not ZNF148 S306A successfully bound to FOXM1 in the presence of ERK2 (Figure 3G). These results suggest that ZNF148‐S306 phosphorylation by ERK2 is required for ZNF148‐FOXM1 interaction.
Figure 3.
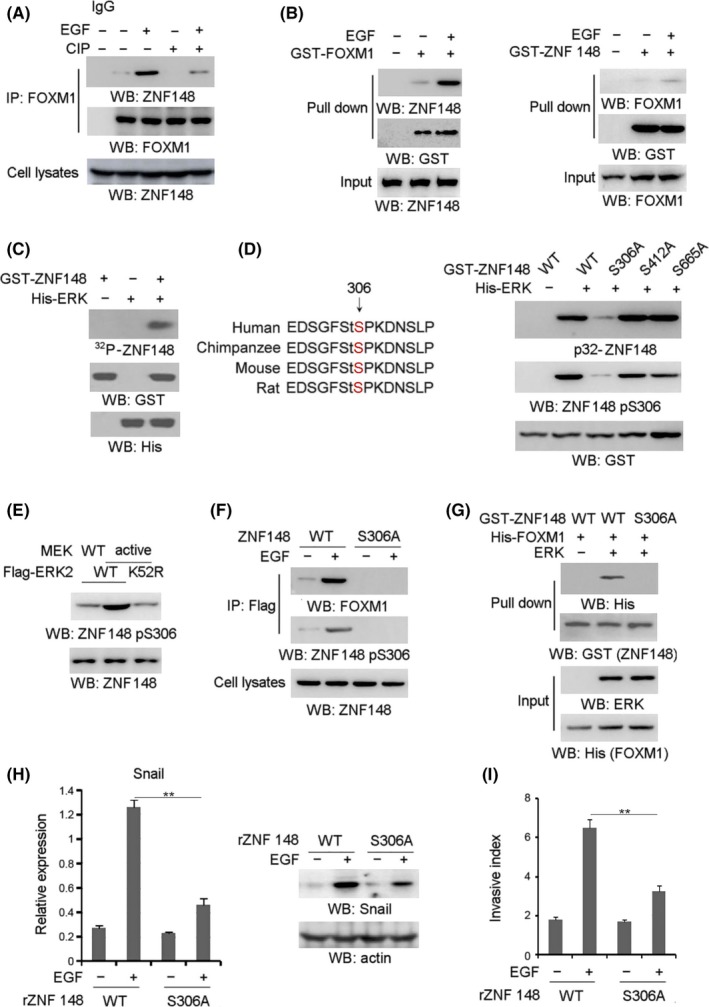
ERK phosphorylates zinc finger protein 148 (ZNF148) at Ser306. A, Gastrointestinal stromal tumor (GIST)‐T1 cells with SDHB depletion were treated with or without EGF (100 ng/mL) for 1 h. The immunoprecipitates were treated with CIP (10 units), followed by immunoblotting analysis. B, Purified GST‐Forkhead box M1 (FOXM1) protein was mixed with cellular extracts from SDHB‐depleted GIST cells treated with EGF (100 ng/mL) for 1 h (left panel). Purified GST‐ZNF148 protein was mixed with cellular extracts from SDHB‐depleted GIST cells treated with EGF (100 ng/mL) for 1 h. C, In vitro phosphorylation analyses were performed by mixing purified active ERK with the indicated purified GST‐ZNF148 proteins in the presence of [γ‐32P] ATP. D, Ser306 of ZNF148 is evolutionarily conserved in the indicated species (left panel). In vitro phosphorylation analyses were performed by mixing the purified active ERK with the indicated purified GST–ZNF148 proteins in the presence of [γ‐32P] ATP (right panel). E, GIST‐T1 cells with SDHB depletion were expressed with WT MEK or MEK1 Q56P constitutively active mutant and WT ERK or ERK K52R kinase‐dead mutant. F, GIST‐T1 cells expressed with Flag‐tagged WT ZNF148 or ZNF148 S306A were treated with or without EGF for 1 h. G, Purified GST‐ZNF148 protein was mixed with or without purified active ERK prior to the incubation with His‐FOXM1 protein. H, GIST‐T1 cells with SDHB depletion, ZNF148 depletion and reconstituted expression reconstituted expression of WT ZNF148 or ZNF148 S306A were treated with or without EGF (100 ng/mL) for 12 h. Q‐PCR (left panel) or immunoblotting analyses (right panel) were performed. I, GIST‐T1 cells with SDHB depletion, ZNF148 depletion and reconstituted expression of WT ZNF148 or ZNF148 S306A were treated with or without EGF (100 ng/mL). Cell invasion assays were performed. In A‐H, immunoblotting analyses were performed using the indicated antibodies. In H and I, the values are presented as mean ± SEM (n = 3 independent experiments), ** represents P < 0.01 between the indicated groups
To test the effect of ZNF148‐S306 phosphorylation on cell invasion, we depleted endogenous ZNF148 in SDHB‐depleted GIST‐T1 (Figure S3B) and GIST882 (Figure S3C) cells, and reconstituted the expression of RNAi‐resistant WT rZNF148 and rZNF148 S306A. In comparison with that in WT rZNF148‐expressing cells, EGF‐induced Snail expression was significantly blocked by rZNF148 S306A expression GIST‐T1 (Figure 3H) or GIST882 (Figure S3D) cells. Consistently, rZNF148 S306A expression apparently blocked EGF‐induced cell invasion, compared with its WT counterpart (Figure 3I and Figure S3E). These results indicated that ZNF148‐S306 phosphorylation is indispensable for EGF‐induced cell invasion in SDHB‐depleted GIST‐T1 cells.
3.4. EGF induced zinc finger protein 148 recruitment to Snail promoter
We further investigated the mechanism of ZNF148‐FOXM1‐regulated Snail transcription. ChIP analysis showed that EGF treatment dramatically induced the enrichment of FOXM1 at the Snail promoter in SDHB‐depleted GIST‐T1 (Figure 4A) and GIST882 (Figure S4A) cells, regardless of ZNF148 depletion, which is consistent with the negative effect of FOXM1 depletion on Snail expression under EGF treatment (Figure S4B). In contrast, we found that EGF treatment significantly promoted the accumulation of ZNF148 at the Snail promoter in SDHB‐depleted GIST‐T1 cells, which was abrogated by either FOXM1 depletion (Figure 4B) or MEK inhibition (Figure 4C). In accordance, WT ZNF148 but not ZNF148 S306A, was found to be substantially enriched at the Snail promoter in GIST (Figure 4D) and GIST882 (Figure S4C) cells treated with EGF. Meanwhile, SDHB‐depleted GIST‐T1 and GIST882 cells showed a remarkable increase of ZNF148‐S306 phosphorylation at the Snail promoter after EGF stimulation, which was blocked by FOXM1 depletion (Figure 4E and Figure S4D). Compared with that in WT rZNF148‐expressing cells, EGF‐induced FOXM1 accumulation at the Snail promoter was not changed by rZNF148 S306A expression in GIST‐T1 (Figure S4E) and GIST882 (Figure S4F) cells. These data reveal that ERK‐mediated ZNF148‐FOXM1 interaction facilitates ZNF148 accumulation at the Snail promoter.
Figure 4.
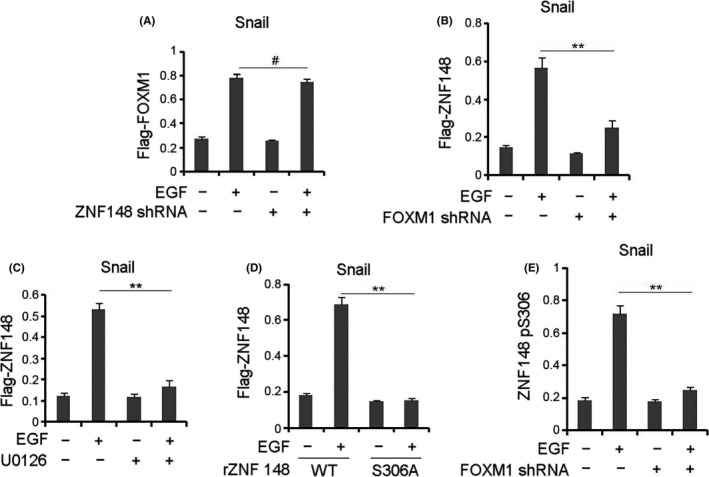
EGF induced zinc finger protein 148 (ZNF148) recruitment to Snail promoter. A, Gastrointestinal stromal tumor (GIST)‐T1 cells with SDHB depletion were transfected with or without the plasmid for expressing ZNF148 shRNA and were treated with or without EGF (100 ng/mL) for 6 h. B, C, GIST‐T1 cells with SDHB depletion were transfected with or without the plasmid for expressing Forkhead box M1 (FOXM1) shRNA (B) or pretreated with U0126 (20 µmol/L) (C). Cells were treated with or without EGF (100 ng/mL) for 6 h. D, GIST‐T1 cells with SDHB depletion, ZNF148 depletion and reconstituted expression of WT ZNF148 or ZNF148 S306A were treated with or without EGF (100 ng/mL) for 6 h. E, GIST‐T1 cells with SDHB depletion were transfected with or without the plasmid for expressing FOXM1 shRNA. Cells were treated with or without EGF (100 ng/mL) for 6 h. In A‐E, ChIP analyses with indicated antibodies were performed. The primers covering the FOXM1 binding region of the Snail gene promoter region were used for the Q‐PCR. The values are presented as mean ± s.e.m. (n = 3 independent experiments), ** represents P < 0.01 between the indicated groups. # represents P > 0.05 between the indicated groups
3.5. Zinc finger protein 148 promotes H3‐K9 acetylation at the Snail promoter
Next, the regulatory effects of ZNF148 on Snail transcription were examined at the epigenetic level. ChIP analysis showed that EGF treatment in SDHB‐silenced GIST‐T1 and GIST882 cells led to an elevated level of H3‐K9 acetylation, a transcriptional marker, at the Snail promoter, which was disrupted by either ZNF148 or FOXM1 depletion (Figure 5A and Figure S5A). EGF‐induced H3‐K9 acetylation at the Snail promoter was evidently compromised by rZNF148 S306A expression in SDHB‐depleted GIST‐T1 cells compared with that in WT counterpart (Figure 5B). In addition, EGF‐induced Snail transcription (Figure 5C) was largely blocked by expression of H3K9R mutant (Figure S5B) in SDHB‐depleted GIST‐T1 cells. These results suggest under EGF stimulation ZNF148 positively regulates H3‐K9 acetylation at the Snail promoter, which is indispensable for the induction of Snail transcription.
Figure 5.
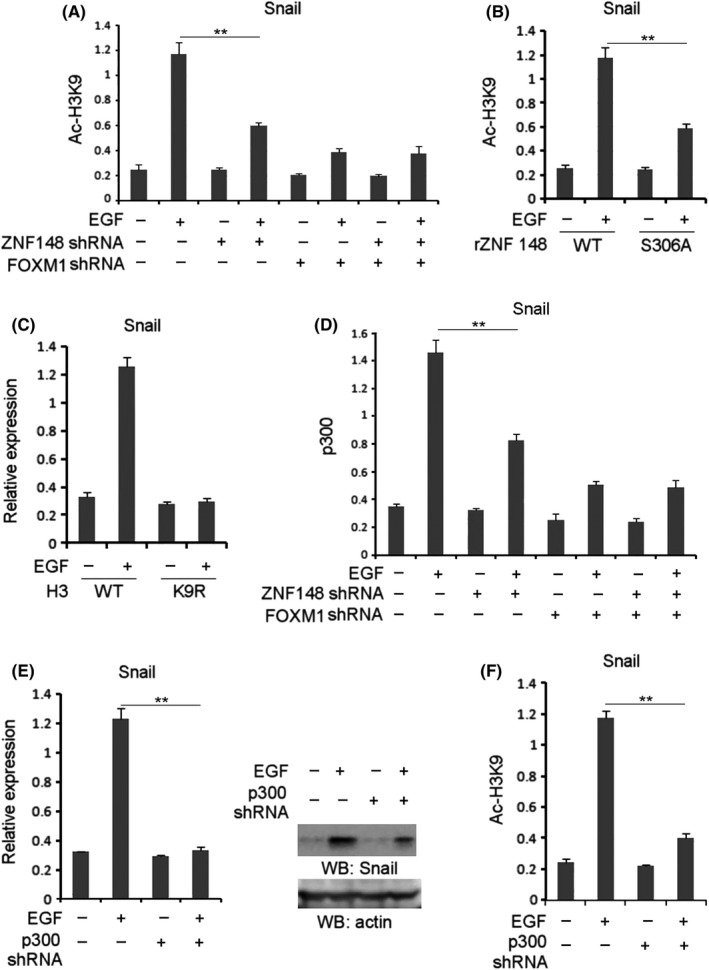
Zinc finger protein 148 (ZNF148) promotes H3‐K9 acetylation at the Snail promoter. A, Gastrointestinal stromal tumor (GIST)‐T1 cells with SDHB depletion were transfected with or without the plasmid for expressing ZNF148 shRNA and/or Forkhead box M1 (FOXM1) shRNA. Cells were treated with or without EGF (100 ng/mL) for 6 h. B, GIST‐T1 cells with SDHB depletion, ZNF148 and reconstituted expression of WT ZNF148 or ZNF148 S306A were treated with or without EGF (100 ng/mL) for 6 h. C, GIST‐T1 cells with SDHB depletion were overexpressed with WT H3 or H3 K9R. Cells were treated with or without EGF (100 ng/mL). Q‐PCR analysis was performed. D, GIST‐T1 cells with SDHB depletion transfected with or without the plasmid for expressing ZNF148 shRNA and/or FOXM1 shRNA were treated with or without EGF (100 ng/mL) for 6 h. E, GIST‐T1 cells with SDHB depletion were transfected with or without the plasmid for expressing p300 shRNA. Cells were treated with or without EGF (100 ng/mL). Q‐PCR (left panel) or immunoblotting analyses (right panel) were performed. F, GIST‐T1 cells with SDHB depletion were transfected with or without the plasmid for expressing p300 shRNA. Cells were treated with or without EGF (100 ng/mL). In A, B, D and F, ChIP analyses with indicated antibodies were performed. The primers covering the FOXM1 binding region of the Snail gene promoter region were used for the Q‐PCR. In A‐F, the values are presented as mean ± SEM (n = 3 independent experiments), ** represents P < 0.01 between the indicated groups
Furthermore, ChIP analysis showed that EGF led to increased enrichment of p300 at the Snail promoter in SDHB‐depleted GIST‐T1 cells, which was inhibited by ZNF148 depletion (Figure 5D). Notably, p300 depletion in SDHB‐silenced GIST‐T1 cells (Figure S5C) abolished EGF‐induced Snail expression (Figure 5E) as well as Snail promoter‐associated H3‐K9 acetylation (Figure 5F), revealing the critical role of p300 in Snail transcription under EGF stimulation. Taken together, these results suggest that FOXM1‐associated ZNF148 mediates EGF‐induced p300 enrichment and the subsequent H3‐K9 acetylation at the Snail promoter.
3.6. Zinc finger protein 148 phosphorylation promotes tumor metastasis and correlates prognosis in SDHB‐deficient gastrointestinal stromal tumor
To determine the implication of ZNF148‐S306 phosphorylation in tumor metastasis, SDHB‐depleted GIST‐T1 cells reconstituted with expression of WT rZNF148 and rZNF148 S306A underwent intrasplenic injection in a liver metastasis mouse model (Figure 6A, left panel). As a result, SDHB‐depleted GIST‐T1 cells with WT rZNF148 expression developed into evident metastatic lesions (Figure 6A, left panel). In contrast, rZNF148 S306A expression apparently inhibited liver metastasis of GIST‐T1 cells (Figure 6A, right panel), suggesting the positive role of ZNF148‐S306 phosphorylation in tumor metastasis.
Figure 6.
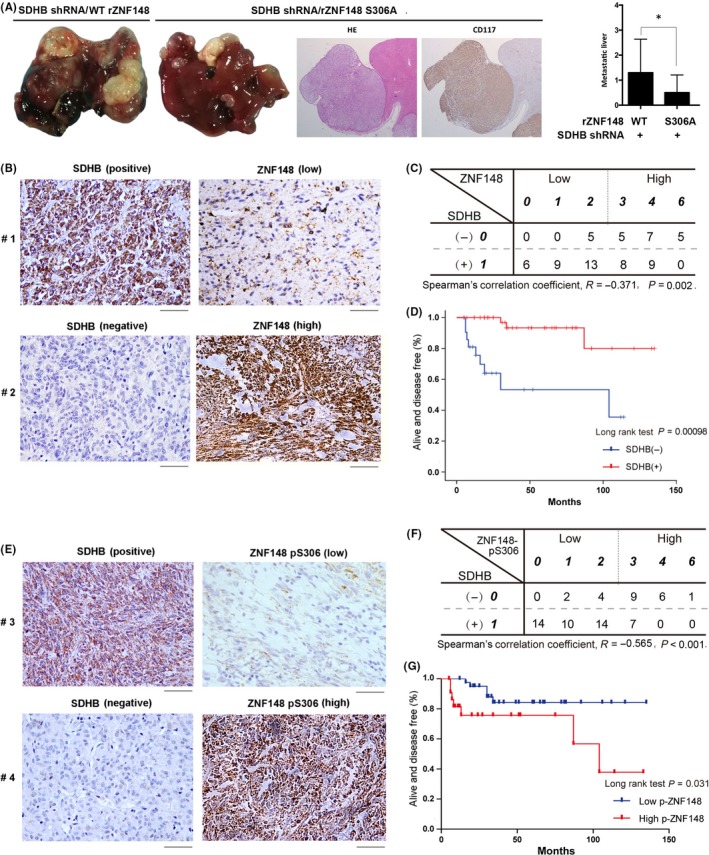
Zinc finger protein 148 (ZNF148) phosphorylation promotes tumor metastasis and correlates prognosis in SDHB‐deficient gastrointestinal stromal tumors (GIST). A, Representative tumor xenografts and liver sections with H&E and CD117 from the rZNF148 S306A group are shown (left panel). The number of visible metastatic lesions in the liver was quantified. Data represent the means ± SEM (n = 10), * represents P < 0.05 (Student’s t test) (right panel). B, Immunohistochemical staining of SDHB and ZNF148 were performed on 67 human GIST specimens. Representative photos are presented. Scale bar: 50 μm. C, Spearman’s rank correlation coefficient test was used to assess the relationship between the expression level of SDHB and ZNF148 (R = −0.371, P = 0.002.). D, The progression free survival (PFS) times for 67 patients with negative SDHB staining (blue curve) versus positive SDHB staining (red curve) were compared. The Kaplan‐Meier method and log‐rank tests indicate the association of SDHB deficiency (P = 0.00096) with inferior PFS. E, Immunohistochemical staining of SDHB and ZNF148pS306 were performed on 67 human GIST specimens. Representative photos are presented. Scale bar: 50 μm. F, Spearman’s rank correlation coefficient test was performed to assess the relationship between the expression level of SDHB and ZNF148 pS306 (R = −0.565, P < 0.001). G, The PFS times for 67 patients with negative low ZNF148 pS306 (blue curve) versus high ZNF148 pS306 (red curve) were compared. The Kaplan‐Meier method and log‐rank tests indicate the association of high ZNF148 pS306 (P = 0.031) with inferior PFS
To examine the clinical relevance of the relationship between SDHB activity and ZNF148 expression, we collected 67 serial sections of WT GIST patient samples and analyzed ZNF148 and SDHB protein levels by IHC staining analysis (Figure 6B). As shown in Figure 6C, ZNF148 is highly expressed in 17 of 22 GIST with negative staining of SDHB, revealing the significant relation of SDHB deficiency to ZNF148 expression levels as reflected in results from cell lines (Figure 1C). Moreover, Kaplan analysis showed that the median progression free survival (PFS) time of GIST patients with tumors with positive SDHB staining (45 cases) was not reached. In contrast, patients with tumors with negative SDHB staining (22 cases) displayed a significantly shorter median PFS of 104 months (Figure 6D).
Subsequently, ZNF148‐S306 phosphorylation was examined in GIST specimens. IHC analysis (Figure 6E) showed that 16 of 22 GIST tumors with loss of SDHB had strong ZNF148‐S306 phosphorylation (Figure 6F), indicating a significant correlation with the high expression level of ZNF148 in SDHB‐deficient samples. Kaplan analysis indicated that GIST patients with low ZNF148 phosphorylation (45 cases) had a median PFS that was not reached, while in tumors with high levels of ZNF148‐S306 phosphorylation (22 cases), the median PFS was reduced to 104 months (Figure 6G). In addition, univariate and multivariate analyses were performed for PFS outcomes using Cox regression modeling (Table 1). The univariate analysis revealed that tumor size, tumor mitotic count and the ZNF148 pS306 level were associated with PFS: (a) tumor size greater than 5 cm was associated with worse outcome (7.063, 95% CI 99.372‐127.057; P = 0.037); (b) high mitosis count was associated with shorter PFS (6.807, 95% CI 100.830‐127.512; P = 0.033); (c) low ZNF148 pS306 level was associated with better outcome (17.534, 95% CI 69.634‐138.366; P = 0.031); and (d) SDH deficiency was strongly associated with shorter PFS (50.950, 95% CI 4.137‐203.863; P < 0.001). Meanwhile, the multivariate analysis indicated that SDH deficiency was an independent prognostic factor for shorter PFS (0.031, 95% CI 0.002‐0.554; P = 0.018). These results reveal the close relation between SDHB deficiency and GIST malignancy and demonstrate the critical role of ZNF148‐S306 phosphorylation in the clinical behavior of human GIST cancer.
Table 1.
Univariate and multivariate analyses for progress free survival in patients with wild‐type gastrointestinal stromal tumor (GIST)
| PFS | ||||
|---|---|---|---|---|
| Univariate analysis | Multivariate analysis | |||
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | |
| Tumor size | ||||
| >5 | 7.063 (99.372‐127.057) | .037 | 3.825 (0.256‐57.101) | .331 |
| ≤5 | ||||
| Tumor mitotic count per 50 HPFs | ||||
| >5 | 6.807 (100.830‐127.512) | .033 | 5.139 (0.595‐44.416) | .137 |
| ≤5 | ||||
| Lymphatic vascular invasion | ||||
| Yes | 6.401 (104.410‐129.504) | .054 | 1.196 (0.129‐11.055) | .875 |
| No | ||||
| SDH deficiency | ||||
| Yes | 50.950 (4.137‐203.863) | <.001 | 0.031 (0.002‐0.554) | .018 |
| No | ||||
| ZNF148 | ||||
| Low | 7.393 (91.475‐120.458) | .235 | 0.546 (0.043‐6.915) | .640 |
| High | ||||
| p‐ZNF148 | ||||
| Low | 17.534 (69.634‐138.366) | .031 | 1.265 (0.139‐11.516) | .835 |
| High | ||||
Bold values indicate that the P < 0.05.
4. DISCUSSION
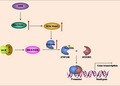
Loss of function of SDHB detected in GIST is found to be associated with tumor malignancy. 9 However, the concrete mechanism underlying the effect of SDHB deficiency on tumor development remains to be clarified. In this study, we found that loss of SDHB leads to succinate accumulation in GIST cells, which leads to enhancement of H3K36me2 at ZNF148 promoter region and, thus, upregulates ZNF148 expression levels. Furthermore, activation of EGFR signaling results in ERK‐mediated ZNF148 phosphorylation at Ser306; this facilitates ZNF148 binding to FOXM1 and its recruitment at the promoter of the FOXM1‐targeted gene, and eventually promotes downstream gene transcription of GIST cells (Figure 7). Together with the results of our clinical analysis, these findings demonstrate the critical role of ZNF148 as a transcriptional co–activator of FOXM1 to drive malignancy progression of GIST with SDHB deficiency.
Figure 7.
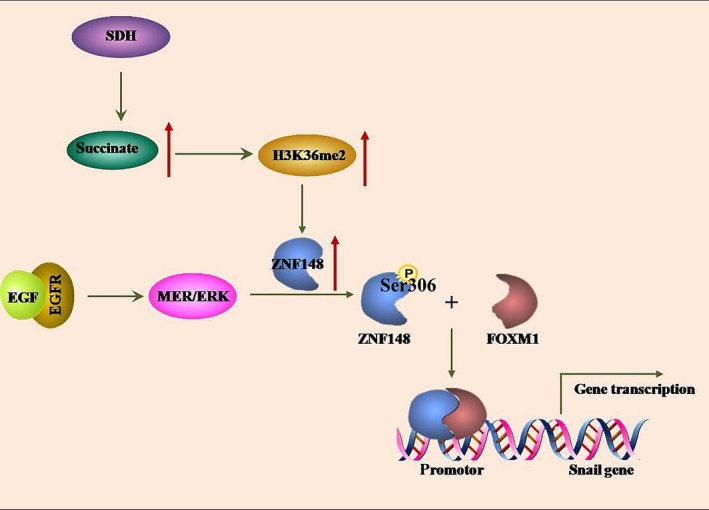
The schematic model showing the cooperative effect between zinc finger protein 148 (ZNF148) and Forkhead box M1 (FOXM1) on Snail transcription in SDHB‐deficient gastrointestinal stromal tumor (GIST) cells. Loss of SDHB function leads to accumulation of intracellular succinate, which results in elevated H3K36me2 levels at ZNF148 promoter region and increased ZNF148 expression. ZNF148 can be phosphorylated by ERK at Ser306; this phosphorylation leads to ZNF148 binding to FOXM1 and, thus, facilitates FOXM1‐targeted Snail transcription that links to tumor cell invasion
Of note, we found that depletion of ZNF148 had no significant effect on the invasion of GIST cells with EGF treatment, while it blocked EGF‐induced tumor cell invasion if with SDHB depletion, which notably increased ZNF148 levels. Thus, it could be inferred that enhanced ZNF148 expression accounts for additive effects of SDHB deficiency on GIST cell invasion under ERK activation induced by EGF stimulus. Meanwhile, the limited impact of ZNF148 depletion shown here indicates that additional effectors are involved in FOXM1‐mediated downstream events. In this study, the investigation of cellular molecular mechanisms is restricted by the poor availability of GIST cell lines. We did not obtain primary GIST cell lines with SDHB loss of function. GIST cells used here displayed a basal level of ERK activation that is most likely attributed to the c‐KIT mutation harbored in cells, which might explain the basal level of ZNF148‐FOXM1 complex detected in GIST cells.
Dysfunction of metabolic enzymes has a fundamental effect on multiple cellular events, including DNA repair and gene transcription. 21 , 22 The physiological effect of SDHB deficiency observed in the present study exemplifies a concrete mechanistic linkage between SDHB metabolic activity and non–metabolic oncogenic signaling and GIST malignancy. Clinically, the molecular mechanism presented here could potentially be applied in the development of therapeutic methods to address certain SDHB‐deficient GIST subtypes with upregulated ZNF148‐FOXM1 signaling.
DISCLOSURE
The authors declare no conflict of interest.
Supporting information
FigS1‐S5
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (31670922, 31470891, 81773006, 81602039 and 81773080).
Gao X, Ma C, Sun X, et al. Upregulation of ZNF148 in SDHB‐deficient gastrointestinal stromal tumor potentiates Forkhead box M1‐mediated transcription and promotes tumor cell invasion. Cancer Sci. 2020;111:1266–1278. 10.1111/cas.14348
Xiaodong Gao and Chunmin Ma contributed equally to this work.
Contributor Information
Yuhui Jiang, Email: yhjiang@shsmu.edu.cn.
Kuntang Shen, Email: shen.kuntang@zs-hospital.sh.cn.
Xian Shen, Email: 13968888872@163.com.
REFERENCES
- 1. Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382:973‐983. [DOI] [PubMed] [Google Scholar]
- 2. Serrano C, George S. Recent advances in the treatment of gastrointestinal stromal tumors. Ther Adv Med Oncol. 2014:115‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laurini JA, Carter JE. Gastrointestinal stromal tumors: a review of the literature. Arch Pathol Lab Med. 2010;134:134‐141. [DOI] [PubMed] [Google Scholar]
- 4. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708‐710. [DOI] [PubMed] [Google Scholar]
- 5. Hirota S, Ohashi A, Nishida T, et al. Gain‐of‐function mutations of platelet‐derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660‐667. [DOI] [PubMed] [Google Scholar]
- 6. Gill AJ, Chou A, Vilain R, et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol. 2010;34:636‐644. [DOI] [PubMed] [Google Scholar]
- 7. Mei L, Smith SC, Faber AC, et al. Gastrointestinal stromal tumors: the GIST of precision medicine. Trends in Cancer. 2018;4:74‐91. [DOI] [PubMed] [Google Scholar]
- 8. Celestino R, Lima J, Faustino A, et al. Molecular alterations and expression of succinate dehydrogenase complex in wild‐type KIT/PDGFRA/BRAF gastrointestinal stromal tumors. Eur J Hum Genet. 2013;21:503‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yantiss RK, Rosenberg AE, Sarran L, Besmer P, Antonescu CR. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol. 2005;18:475‐484. [DOI] [PubMed] [Google Scholar]
- 10. Gaal J, Stratakis CA, Carney JA, et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney‐Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol. 2011;24:147‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. 2018;19:563‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wierstra I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor‐suppressive properties, and target of anticancer therapy. Adv Cancer Res. 2013;119:191‐419. [DOI] [PubMed] [Google Scholar]
- 13. Bai C, Liu X, Qiu C, et al. FoxM1 is regulated by both HIF‐1alpha and HIF‐2alpha and contributes to gastrointestinal stromal tumor progression. Gastric Cancer. 2019;22:91‐103. [DOI] [PubMed] [Google Scholar]
- 14. Saavedra‐García P, Nichols K, Mahmud Z, Fan L‐N, Lam E‐F. Unravelling the role of fatty acid metabolism in cancer through the FOXO3‐FOXM1 axis. Mol Cell Endocrinol. 2018;462:82‐92. [DOI] [PubMed] [Google Scholar]
- 15. Zhang J, Niu Y, Huang C. Role of FoxM1 in the progression and epithelial to mesenchymal transition of gastrointestinal cancer. Recent Pat Anti–Cancer Drug Discov. 2017;12:247‐259. [DOI] [PubMed] [Google Scholar]
- 16. Merchant JL, Iyer GR, Taylor BR, et al. ZBP‐89, a Kruppel‐like zinc finger protein, inhibits epidermal growth factor induction of the gastrin promoter. Mol Cell Biol. 1996;16:6644‐6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang N, Wang S, Yang S‐L, et al. Targeting ZBP‐89 for the treatment of hepatocellular carcinoma. Expert Opin Ther Targets. 2018;22:817‐822. [DOI] [PubMed] [Google Scholar]
- 18. Zhang CZ, Chen GG, Lai PB. Transcription factor ZBP‐89 in cancer growth and apoptosis. Biochem Biophys Acta. 2010;1806:36‐41. [DOI] [PubMed] [Google Scholar]
- 19. Zhang CZY, Cao Y, Yun J‐P, Chen GG, Lai PBS. Increased expression of ZBP‐89 and its prognostic significance in hepatocellular carcinoma. Histopathology. 2012;60:1114‐1124. [DOI] [PubMed] [Google Scholar]
- 20. Bai L, Yoon SO, King PD, Merchant JL. ZBP‐89‐induced apoptosis is p53‐independent and requires JNK. Cell Death Differ. 2004;11:663‐673. [DOI] [PubMed] [Google Scholar]
- 21. Wang T, Yu Q, Li J, et al. O‐GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat Cell Biol. 2017;19:833‐843. [DOI] [PubMed] [Google Scholar]
- 22. Schvartzman JM, Thompson CB, Finley LWS. Metabolic regulation of chromatin modifications and gene expression. J Cell Biol. 2018;217:2247‐2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigS1‐S5