

Abstract
BRCAness is considered a predictive biomarker to platinum and poly(ADP‐ribose) polymerase (PARP) inhibitors. However, recent trials showed that its predictive value was limited in triple‐negative breast cancer (TNBC) treated with platinum. Moreover, tumors with mutations of DNA damage response (DDR) genes, such as homologous recombination (HR) genes, could be sensitive to platinum and PARP inhibitors. Thus, we aim to explore the relationship between mutation status of DDR genes and BRCAness in TNBC. We sequenced 56 DDR genes in 120 TNBC and identified BRCAness by array comparative genomic hybridization. The sequencing results showed that 13, 14, and 14 patients had BRCA, non‐BRCA HR, and non‐HR DDR gene mutations, respectively. Array comparative genomic hybridization revealed that BRCA‐mutated and HR gene‐mutated TNBC shared similar BRCAness features, both having higher numbers and longer length of large‐scale structural aberration (LSA, >10 Mb) and similar altered chromosomal regions of LSA. These suggested non‐BRCA HR gene‐mutated TNBC shared similar characteristics with BRCA‐mutated TNBC, indicating non‐BRCA HR gene‐mutated TNBC sensitive to platinum and PARP inhibitors. Among tumors with mutation of non‐HR DDR genes, 3 PTEN and 1 MSH6 mutation also contained significant LSAs (BRCAness); however, they had different regions of genomic alteration to BRCA and HR gene‐mutated tumors, might explain prior findings that PTEN‐ and MSH6‐mutated cancer cells not sensitive to PARP inhibitors. Therefore, we hypothesize that the heterogeneous genomic background of BRCAness indicates different responsiveness to platinum and PARP inhibitors. Direct sequencing DDR genes in TNBC should be applied to predict their sensitivity toward platinum and PARP inhibitors.
Keywords: BRCAness, DNA damage response, PARP inhibitor, platinum, triple‐negative breast cancer
High‐grade genomic instability (BRCAness) can be present in triple‐negative breast cancer with BRCA, non‐BRCA HR gene, PTEN and MSH6 mutation. We hypothesize that the heterogeneous genomic background of BRCAness indicates different responsiveness to platinum and PARP inhibitors.

Abbreviations
- aCGH
array comparative genomic hybridization
- ACMG
American College of Medical Genomics and Genetics
- CI
confidence interval
- CNV
copy number variation
- DDR
DNA damage response
- DSB
double‐strand break
- ER
estrogen receptor
- HER2
human epidermal growth factor receptor 2
- HR
homologous recombination
- HRD
homologous recombination deficiency
- LOH
loss of heterozygosity
- LSA
large‐scale genomic structural aberration
- NGS
next‐generation sequencing
- PARP
poly(ADP‐ribose) polymerase
- PR
progesterone receptor
- RFS
relapse‐free survival
- TNBC
triple‐negative breast cancer
1. INTRODUCTION
Triple‐negative breast cancer, which lacks expression of ER, PR, and HER2, is an aggressive subtype associated with poor prognosis.1 An important genetic feature of TNBC is the deleterious mutation of BRCA1 and BRCA2. Mutations of BRCA genes cause HRD, which leads to an increased genomic instability.2 Cancer cells with BRCA1/2 mutations have displayed sensitivity to platinum‐based chemotherapy and PARP inhibitors.3 Trials of PARP inhibitors indicated that the survival rate was significantly improved in patients with BRCA‐mutated ovarian and breast cancer.4, 5, 6 However, only approximately 10% of TNBC patients carry BRCA1/2 mutations. Therefore, many studies have attempted to explore whether a subset of non‐BRCA‐mutated tumors responded to platinum and PARP inhibitors.
Prior studies have reported that a subset of non‐BRCA‐mutated cancers show high‐grade genomic instability, resembling tumors originated from germline BRCA‐mutated carriers. These tumors show HRD as well, and these traits are collectively referred to as BRCAness.7, 8 Array comparative genomic hybridization was used to classify BRCAness and non‐BRCAness by assessing chromosome aberrations and measuring the severity of LOH, telomeric allelic imbalance, and large‐scale state transition as an HRD score.2, 9 The BRCAness feature was considered as a marker to predict responsiveness of platinum‐ and PARP inhibitors.2, 10 However, the TNT phase III study showed that BRCAness did not correlate with the therapeutic response of carboplatin in TNBC.11 Another recent clinical trial showed that BRCAness could only modestly predict the treatment outcome of PARP inhibitor‐based therapy.12 Therefore, it is uncertain whether BRCAness is an adequate marker for selecting breast cancer patients for treatment with platinum and PARP inhibitors.
Recently, preclinical experiments have indicated that platinum and PARP inhibitors can kill cancer cells with mutations in several DDR genes, such as ATM, RAD51C, and other genes involved in the HR repair pathway for DSB.13, 14, 15 In a phase II trial of prostate cancer, patients carrying deleterious mutation of HR genes had a higher response rate and a better survival rate when treated with PARP inhibitors.16 Furthermore, mutations of non‐HR DDR genes, such as MLH1, PTEN, and TP53, causes an increase in genomic instability that resembled BRCAness features in pancreatic, ovarian, prostate, and other cancer types.17, 18, 19, 20 Yet, survival was not improved in PTEN‐mutated ovarian cancer treated with platinum‐based chemotherapy.21 These results suggested that mutations in HR genes could be used as biomarkers to predict sensitivity to PARP inhibitors. Whereas other DDR mutations might show BRCAness, their tumors do not necessarily respond to platinum and PARP inhibitors. In TNBC, the mutation incidence of HR genes and DDR genes is not known. Also, the relationship between BRCAness features and DDR gene mutations is uncertain. To explore these uncertainties, we sequenced DDR genes (including HR, non‐homologous end‐joining, base‐excision repair, nucleotide‐excision repair, mismatch repair, and polymerases involved in DNA repair pathways) in TNBC, then we assessed their BRCAness by undertaking aCGH. Finally, we investigated the relationship between BRCAness and the mutation status of DDR genes.
2. MATERIALS AND METHODS
2.1. Patients
This study enrolled patients who were diagnosed as stage I‐III TNBC and had received surgical intervention in our hospital between 2003 and 2010. The clinical and pathologic characteristics were retrospectively recorded. Estrogen receptor, PR, and HER2 were determined by immunohistochemical staining. Estrogen receptor or PR was considered negative when less than 5% of tumor cells showed positive staining. For HER2 staining, a score of 0 or 1+ was considered negative; specimens with a score of 2+ were confirmed by FISH analysis. In addition, the tumor histological grade was defined using the Nottingham combined histological grading system. Finally, the study was approved by the Institutional Review Board of National Taiwan University Hospital (Taipei, Taiwan).
2.2. DNA extraction and library preparation
After a patient had signed an agreement, their tumor sample was stored in either a liquid nitrogen tank or a −80°C refrigerator. As reported previously, DNA was extracted from resected tumors and used for library construction.22 Briefly, the purity and concentration of tumor DNA was checked by agarose gel electrophoresis, OD ratio, and a Qubit 2.0 Fluorometer (Life Technologies), and this was followed by a Covaris fragmentation. Size distribution of the fragmented DNA was confirmed using a Bioanalyzer 2100 (Agilent Technologies). Then DNA libraries were generated with the Truseq DNA Library Prep kits (Illumina), according to the manufacturer's manual. Finally, target gene libraries, containing a customized gene panel of 56 genes selected from DDR genes (Table S1), including HR, nonhomologous end‐joining, base‐excision repair, nucleotide‐excision repair, mismatch repair, and polymerases involved in DNA repair pathways, were generated using a SeqCap EZ Target Enrichment System (Roche NimbleGen).22, 23 Finally, the libraries were sequenced on an Illumina NextSeq 500 that generated paired‐end reads of approximately 300 nt in lengths.
2.3. Sequence data analysis
Previously, we have described the post‐NGS bioinformatics.24, 25 The raw sequencing data were aligned to the reference human genome (Feb. 2009, GRCh37/hg19) by using BWA software (version 0.5.9) and SAMtools (version 0.1.18). Picard (version 1.54) was used to undertaken the necessary data conversion, sorting, and indexing. Genome Analysis Toolkit (version 3.5) was used for variant calling through the parameters of UnifiedGenotyper, HaplotypeCaller, and VariantFiltration. Pindel (version 0.2.4) software was utilized to find structural variants, such as deletions, insertions, and duplications. Finally, ANNOVAR was used to annotate the genetic variants. Variants interpretation were based on the guideline of the ACMG.26 Briefly, the frameshift insertion or deletion (indel), nonsense, uncorrected splice‐site variants, large‐scale deletions, and missense mutations with impaired protein function by functional assays were considered as pathogenic or like pathogenic variants. We used “deleterious mutation” to represent the “pathogenic” and “likely pathogenic” variants by the guidelines of the ACMG.26 Only the deleterious mutations were used in the further analysis; variants of uncertain significance were not.
Large‐scale deletions were detected by CNV analysis.27 For the 56 genes, 15 samples without CNV were selected to construct the CNV reference.22 Briefly, the mean depth of each sample was normalized to the amount of DNA library loaded on the sequencing flow cells, target enrichment rate, and the total output of each sequencing run on Nextseq 500. If the reading depth lied outside the 95% confidence interval of the same region in the reference (Appendices S1 and S2, Figure S1), an exon region was considered CNV.
2.4. Checking mutation origin
Germline DNA was extracted from PBMCs of patients. For the deleterious mutations (small insertion, deletion, or nonsynonymous variants) harbored within tumors, we used Sanger sequencing to check whether the mutations existed in the germline DNA in the same patients. If the blood cells carried the same mutations, the mutation was categorized to be originated from the germline.
2.5. Assessment of genomic instability
Genomic patterns of breast carcinomas were determined by aCGH; the array contained approximately 60 000 probes covering the whole genome with an average spacing of 40 kb. Briefly, fragmented DNA was labeled using the Agilent SureTaq Genomic DNA Labeling Kit (Agilent Technologies) with Cy5‐dUTP (for the experimental sample) and Cy3‐dUTP (for the reference) (PerkinElmer), according to the manufacturer's instructions. After cleaning and denaturing, the labeled samples were hybridized to a SurePrint G3 Custom CGH Microarray, 8 × 60K (G4126A; Agilent Technologies). After drying, the hybridized arrays were scanned on an Agilent DNA microarray scanner at 535 nm for Cy3 and 625 nm for Cy5 at a resolution of 2 μm. To quantify signal intensities and normalize the data for each feature, the scanned images were analyzed on the Agilent Genomic Workbench (Agilent Technologies) using the Feature extraction 10.5.1.1 software (Agilent Technologies).
The allelic frequency of gain or loss for each chromosomal region was calculated by the ratio thresholds of 0.25 and −0.25, respectively. We defined the regions of gains and losses that were >10 Mb as the hallmark of LSA.2 The presence of LSA was considered as evidence of genomic instability. The genomic aberrations of regions <10 Mb were not included for further analysis and are not shown in Figure 1 or Figure S2. The number, length, and specific chromosomal regions of LSA were analyzed.
Figure 1.
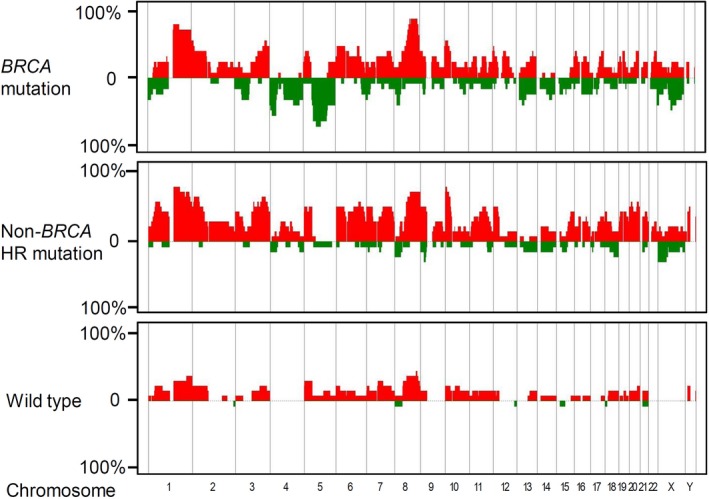
Three panels that show the frequencies of gain (red) or loss (green) of large‐scale structural aberration in BRCA‐mutated, non‐BRCA homologous recombination (HR) gene‐mutated, and control breast cancers
2.6. Statistical analysis
The χ2 test and Fishers exact test were used to calculate the significance of variances between each group. Survival was estimated by Kaplan‐Meier analysis. All P values are 2‐sided, and P values <.05 are considered statistically significant.
3. RESULTS
3.1. Patients and mutation analysis
A total of 120 TNBC patients were analyzed. Based on the mutation status, the 120 TNBC patients were classified into 4 groups: control, BRCA mutations, non‐BRCA HR gene mutations, and other DDR gene mutations. The demographic information is shown in Table 1. Their clinical characteristics were similar in age, stage and grade distribution, and the use of adjuvant therapy. Patients with BRCA mutation had a higher prevalence of family history of cancer, but patients with non‐BRCA HR gene mutations did not, compared with that of controls. Deleterious mutation of TP53 was identified in 92 patients. The proportion of patients carrying TP53 mutation was significantly higher among patients with mutations in either BRCA or other HR genes than those without HR mutation (P = .011).
Table 1.
Clinical and pathologic characteristics of patients with triple‐negative breast cancer stratified by gene mutation
| Characteristic | Without HR mutation (n = 79) | BRCA mutation (n = 13) | Non‐BRCA HR mutation (n = 14) | Other mutation (n = 14) | P value |
|---|---|---|---|---|---|
| Age (mean ± SD) | 53.2 ± 11.9 | 51.7 ± 8.9 | 52.0 ± 9.9 | 56.9 ± 10.3 | .589 |
| T | |||||
| T1 | 38 | 6 | 3 | 5 | .433 |
| T2 | 36 | 7 | 10 | 7 | |
| T3‐4 | 5 | 0 | 1 | 2 | |
| N | |||||
| N0 | 41 | 10 | 6 | 12 | .324 |
| N1 | 20 | 2 | 4 | 1 | |
| N2 | 6 | 0 | 2 | 1 | |
| N3 | 12 | 1 | 2 | 0 | |
| Grade | |||||
| 1 | 3 | 2 | 0 | 2 | .746 |
| 2 | 21 | 3 | 5 | 3 | |
| 3 | 49 | 7 | 8 | 8 | |
| Undetermined | 6 | 1 | 1 | 1 | |
| TP53 mutation | |||||
| Wild type | 29 | 0 | 2 | 7 | .011 |
| Mutation | 50 | 13 | 12 | 7 | |
| Family history | |||||
| All cancer | 33 | 9 | 3 | 5 | .085 |
| Breast/ovarian | 15 | 7 | 0 | 2 | .004 |
| Adjuvant chemotherapy | |||||
| No | 11 | 0 | 1 | 3 | .335 |
| Yes | 68 | 13 | 13 | 11 | |
| Anthracycline | 22 | 8 | 7 | 5 | |
| Taxane | 2 | 0 | 1 | 0 | |
| Combination | 43 | 5 | 5 | 3 | |
| Others | 1 | 0 | 0 | 3 | |
Abbreviations: Adjuvant chemotherapy: anthracycline alone; taxane, taxane‐containing chemotherapy (taxane + anthracycline allowed).
HR, homologous recombination.
There were 25 deleterious mutations in HR genes, including 8 in BRCA1, 3 in BRCA2, 2 in ATM, 2 in BARD1, 1 in CHEK2, 1 in FANCA, 2 in FANCB, 1 in FANCI, 1 in PALB2, 2 in RAD50, and 2 in RAD51D (Table 2). Most BRCA and HR gene‐mutated tumors originated from the germline. Among patients carrying insertion/deletion in non‐BRCA HR genes, 2 patients carried mutations in other cancer‐predisposing genes as well. Copy number variation analysis showed that 3 patients had large‐scale somatic deletion in BRCA1, 4 deletions in BRCA2, and 11 deletions in non‐BRCA HR genes. Overall, 13 patients had BRCA mutations and 14 patients had non‐BRCA HR gene mutations. Beyond the BRCA and HR gene mutation, 14 patients had deleterious mutations in ERCC4, ERCC8, MSH3, MSH6, POLH, POLK, PTEN, STK11, and XPC.
Table 2.
Pathogenic mutations in this cohort of patients with triple‐negative breast cancer
| Patient | Genetic variants | Large deletion | ||
|---|---|---|---|---|
| Type | Gene | Location | Gene | |
| BRCA | ||||
| #1 | Frameshift | BRCA1 | NM_007300:exon10:c.3770_3771del:p.E1257fs | |
| #2 | Stopgain | BRCA1 | NM_007300:exon10:c.T3257G:p.L1086X | |
| #3 | Frameshift | BRCA1 | NM_007300:exon10:c.3228_3229del:p.R1076fs | FANCB |
| #4 | Frameshift | BRCA1 | NM_007300:exon2:c.66dupA:p.E23fs | FANCB, FANCG |
| #5 | Splicing | BRCA1 | NM_007300:exon22:c.5395+1G>A | FANCG |
| #6 | Stopgain | BRCA1 | NM_007300:exon10:c.C928T:p.Q310X | |
| #7 | Frameshift | BRCA2 | NM_000059:exon22:c.8894delA:p.D2965fs | BRCA2, FANCI |
| #8 | Frameshift | BRCA2 | NM_000059:exon10:c.1806dupA:p.G602fs | |
| Stopgain | BRCA2 | NM_000059:exon11:c.C3109T:p.Q1037X | ||
| #9 | Frameshift | BRCA1 | NM_007300:exon12:c.4356delA:p.K1452fs | |
| Frameshift | BRCA1 | NM_007300:exon10:c.865_866del:p.S289fs | ||
| #10 | Splicing | FANCA | NM_000135:exon21:c.1777‐1G>C | BRCA2 |
| #11 | BRCA1, FANCB, FANCC | |||
| #12 | BRCA1, BRCA2, FANCM, RAD51C | |||
| #13 | BRCA1, BRCA2, FACI, ATM, CHEK2 | |||
| Non‐BRCA HR | ||||
| #1a | Frameshift | BARD1 | NM_001282543:exon3:c.943_944del:p.K315fs | |
| #2 | Splicing | FANCB | NM_152633:exon7:c.1327‐2‐>T | |
| #3b | Frameshift | FANCB | NM_152633:exon2:c.483delT:p.V161fs | |
| #4 | Frameshift | RAD51D | NM_001142571:exon4:c.331_332insTA:p.K111fs | |
| #5 | Frameshift | RAD51D | NM_001142571:exon4:c.331_332insTA:p.K111fs | |
| #6 | Frameshift | PALB2 | NM_024675:exon6:c.2555_2567del:p.P852fs | FANCG, FANCM |
| #7 | Frameshift | ATM | NM_000051:exon35:c.5313delA:p.R1771fs | |
| #8 | Frameshift | BARD1 | NM_000465:exon4:c.628dupA:p.T210fs | |
| Frameshift | CHEK2 | NM_001005735:exon14:c.1580delC:p.P527fs | ||
| #9 | Frameshift | FANCI | NM_001113378:exon22:c.2252_2291del:p.E751fs | |
| #10 | Frameshift | RAD50 | NM_005732:exon13:c.2157delA:p.L719fs | |
| Frameshift | ATM | NM_000051:exon56:c.8249_8268del:p.L2750fs | ||
| #11 | Frameshift | RAD50 | RAD50:NM_005732:exon6:c.783_784del:p.N261fs | |
| #12 | FANCB | |||
| #13 | FANCA | |||
| #14 | FANCM | |||
| Other DDR genes | ||||
| #1 | Frameshift | ERCC4 | NM_005236:exon4:c.607_608del:p.F203fs | |
| #2 | Frameshift | ERCC4 | NM_005236:exon5:c.891_892del:p.Y297fs | |
| #3 | Stopgain | MSH3 | NM_002439:exon4:c.C724T:p.Q242X | |
| #4 | Splicing | MSH3 | NM_002439:exon13:c.1764‐2A>G | |
| #5 | Stopgain | MSH6 | NM_000179:exon4:c.C3013T:p.R1005X | |
| #6 | Frameshift | MSH6 | NM_000179:exon5:c.3254delC:p.T1085fs | |
| #7 | Frameshift | POLH | NM_006502:exon8:c.978delA:p.P326fs | |
| #8 | Frameshift | POLK | NM_016218:exon13:c.1631delA:p.E544fs | |
| #9 | Frameshift | PTEN | NM_000314:exon5:c.489_492del:p.K163fs | |
| #10 | Frameshift | PTEN | NM_000314:exon8:c.955dupA:p.L318fs | |
| #11 | Stopgain | PTEN | NM_000314:exon5:c.G448T:p.E150X | |
| #12 | Frameshift | PTEN | NM_000314:exon4:c.244dupA:p.F81fs | |
| #13 | Frameshift | STK11 | NM_000455:exon5:c.684delG:p.L228fs | |
| #14 | Frameshift | XPC | NM_001145769:exon1:c.75dupC:p.K26fs | |
We checked the origin of the deleterious mutations to determine whether they originated from the germline or if they were only acquired (somatic) in tumor cells (Figure S3). Peripheral blood DNA was available in 8 of the 9 BRCA‐mutated patients, and Sanger sequencing of those DNA showed that 8 of the BRCA1/2 mutations came from germline origin (all heterozygous). We then analyzed whether LOH occurred in tumor cells. The increased variant allele frequency was observed in patients #1 to #7 (Table S2), and LOH was highly suspected.28 Patient #8 had 2 BRCA2 mutations in tumor cells; 1 came from the germline, and the other was somatic. Double mutations might have suggested biallelic inactivation of BRCA2 in tumors as well. Patient #9 also had double mutations in the BRCA gene. Therefore, we expected biallelic inactivation of BRCA1 or BRCA2 in these 9 tumors. For non‐BRCA HR genes, peripheral blood DNA was available in 8 patients. The mutations in patients #2, #4, #5, and #6 originated from the germline, and LOH was also observed in their tumors (Table S2). This result is compatible with the previous finding that mutation of HR genes related to TNBC.29, 30
3.2. Genomic patterns of non‐BRCA HR gene‐mutated tumors (vs BRCA‐mutated)
We studied the genomic patterns by aCGH in the 13 BRCA‐mutated, 14 non‐BRCA HR gene‐mutated, and 14 age‐ and clinical stage‐matched control tumors without such mutations. Compared to the control tumors, large‐scale (more than 10 Mb) gain or loss at specific chromosome locations were characterized in tumors with BRCA mutations and non‐BRCA HR gene mutations; profiles are shown in Figure 1. We analyzed the numbers and lengths of LSA in each group, and we discovered that the LSA numbers and the lengths were significantly higher in the BRCA‐mutated tumors than those in the control tumors (both P < .001, Figure 2A,B). Moreover, we observed that the LSA spectrum was similar between BRCA‐mutated and non‐BRCA HR gene‐mutated tumors, either in LSA numbers or lengths (number, P = .946; length, P = .918; Figure 2A,B). These tumors contained significantly more and longer LSAs compared with the control tumors (number, P = .035; length, P = .022; Figure 2A,B). Subsequently, we compared the LSA patterns in hereditary tumors with patterns in tumors with acquired somatic mutations. The LSA spectrums of the 4 cases with somatic BRCA mutations were like those of the 9 cases with germline BRCA mutations (number, P = .460; length, P = .950; Figure 2C,D). Also, the LSA spectrums showed no statistical differences between tumors carrying germline and tumors carrying somatic alterations of non‐BRCA HR genes (number, P = .678; length, P = .472; Figure 2C,D). Both hereditary mutations and acquired HR gene mutations of tumors had significantly higher LSA numbers and lengths than those that do not have such mutation (all P value <.05) (Figure 2C,D). These results indicated that tumors with mutations in non‐BRCA HR genes show a high grade of genomic instability, like those with BRCA1/2 mutations.
Figure 2.
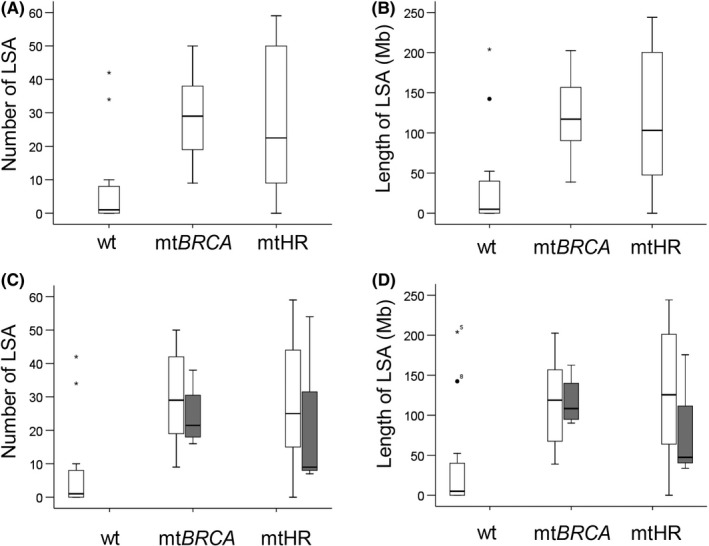
Box and whisker diagrams comparing the numbers and lengths of large‐scale structural aberration (LSA) among BRCA‐mutated, non‐BRCA homologous recombination (HR) gene‐mutated, and control breast cancers. A, B, Tumors with BRCA mutations and non‐BRCA HR gene mutations showed significantly higher numbers and lengths of LSA than those of the control tumors (BRCA vs WT, both P < .001; non‐BRCA HR vs WT, number P = .035; length P = .022). C, D, Tumors with mutation were stratified by germline and acquired somatic mutation. Numbers and lengths of LSA were not different either between germline and somatic BRCA mutations, or between germline and somatic non‐BRCA HR gene mutations
The chromosomal regions of significant gains or losses between the 3 groups are highlighted in Table 3. Compared with control tumors, BRCA‐mutated and non‐BRCA HR gene‐mutated tumors were associated with loss of Xp and gains of 1q, 2p21‐25, 3q21‐29, 8q, 10p, and 22q. Tumors with non‐BRCA HR gene mutations contained more gains of 20p and 4q than the control tumors did, but BRCA‐mutated tumors did not. While only BRCA‐mutated tumors were significantly linked to gain of 12p, tumors with non‐BRCA HR gene mutations did not show significant association with a gain of this region. These results suggest that, although some alterations were specific to TNBC tumors with either BRCA or non‐BRCA HR gene mutations, numerous chromosomal alterations are shared.
Table 3.
Significant chromosomal regions between triple‐negative breast tumors with BRCA‐mutated, non‐BRCA homologous recombination (HR) gene mutations, and WT
| BRCA (%) | Non‐BRCA HR (%) | WT (%) | P value | |||
|---|---|---|---|---|---|---|
| BRCA vs HR | BRCA vs wt | HR vs wt | ||||
| Loss | ||||||
| Xp | 38.46% | 28.57% | 0.00% | .586 | .01 | .031 |
| Gain | ||||||
| 1q21‐25 | 76.92% | 78.57% | 28.57% | .918 | .012 | .008 |
| 2p21‐25 | 53.85% | 64.29% | 21.43% | .581 | .081 | .022 |
| 3q21‐29 | 53.85% | 71.43% | 21.43% | .345 | .081 | .008 |
| 8q | 84.62% | 78.57% | 35.71% | .686 | .025 | .053 |
| 10p | 53.85% | 71.43% | 21.43% | .345 | .081 | .008 |
| 22q | 38.46% | 28.57% | 0.00% | .586 | .01 | .042 |
| 20p | 15.38% | 57.14% | 14.29% | .025 | .936 | .018 |
| 4q | 0.00% | 35.71% | 0.00% | .017 | 1 | .014 |
| 12q | 38.46% | 7.14% | 0.00% | .05 | .01 | .309 |
3.3. Cancer genes at LSA regions
We inspected the LSA regions listed in Table 3 for known cancer genes. A gain of chromosome 1q21 results in the amplification of S100 calcium‐binding protein (S100A) family members PI4KB, SHC1, and NCSTN, which have been previously suggested as driver oncogenes of basal‐type breast cancer.31 In addition, this amplification of 1q21 loci was linked to breast cancer recurrence (Figure 3A).31, 32 Indeed, the S‐100 family was amplified at a higher proportion among the tumors with BRCA mutations (N = 10, 76.9%) and those with non‐BRCA HR gene mutations (N = 11, 78.6%) (P = .030). The gain of chromosomal 8q also caused an amplification of the c‐MYC oncogene. This was discovered significantly in the tumors with BRCA mutations (N = 11, 84.6%) and tumors with non‐BRCA HR gene mutations (N = 11, 78.6%) (Figure 3B), compared to the control tumors (N = 5, 35.7%; P = .039). CRKL, an oncogene located in 22q11.1 that mediates cellular proliferation and metastasis, was amplified in both BRCA‐mutated (N = 5, 38.5%) and non‐BRCA HR gene‐mutated (N = 4, 28.6%) tumors (P = .042, Figure 3C). The gain of 3q and 10p and loss of Xp, which are significantly associated with BRCA and non‐BRCA HR gene mutations, were related to carcinogenesis and reduced survival of patients.33
Figure 3.
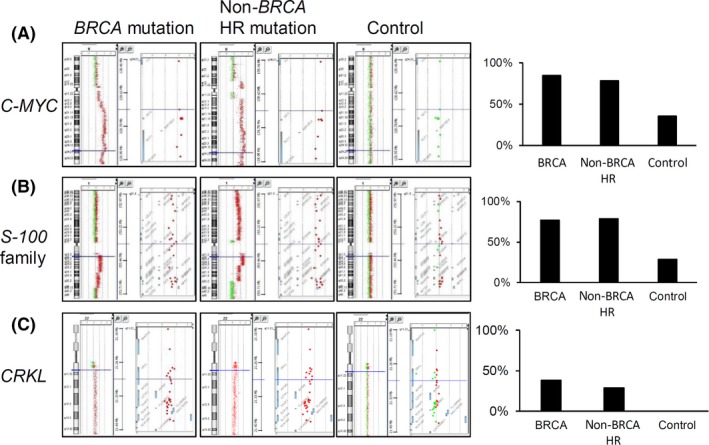
Representative array comparative genomic hybridization (aCGH) profiles of cancer‐related genes in chromosome regions significantly different between BRCA‐mutated, non‐BRCA HR gene‐mutated, and control breast cancers. (A) MYC, (B) S100 family, and (C) CRKL were significantly amplified in BRCA‐mutated and non‐BRCA homologous recombination (HR) gene‐mutated tumors, compared to those in control tumors. Green dot, loss; red dot, gain. Bar graphs represent the percentages of tumors containing amplifications of the target genes in each group
3.4. Genomic patterns and cancer genes of other DDR gene‐mutated tumors
Genomic patterns were examined in 10 tumors with mutations of non‐HR genes and tumor suppressor genes as well (Table 2 and Figure S2). Three of the 4 tumors carrying PTEN mutations showed high numbers of LSA, similar to those with BRCA (number P = .106; length, P = .263) or other HR gene mutations (number, P = .294; length, P = .477). MSH6 mutation was observed in 1 tumor, and it contained large numbers of LSA. Nevertheless, tumors with mutations in XPC, ERCC8, or MSH3 did not.
Among the frequently altered chromosomal regions of BRCA and HR‐mutated tumors, PTEN‐mutated tumors were not associated with loss of Xp or gains of 2p21‐25, 3q21‐29, and 22q (Figure S2). Two of the 4 PTEN‐mutated tumors contained amplified chromosome 1q21 (S100 family) and 8q (c‐MYC); the other 2 did not. Among the 4 PTEN‐mutated tumors, the CRKL gene was not amplified. In the MSH6‐mutated tumor, frequent chromosomal loss was observed, such as 1p, 11p, and 12p. In contrast, they were not altered in BRCA and HR‐mutated tumors. These results might suggest different biologic characteristics of PTEN‐mutated and MSH6‐mutated tumors, compared to BRCA and HR‐mutated tumors. Nevertheless, all those types of tumors had high‐grade genomic instability.
3.5. Clinical relevance
The 5‐year RFS was 78.7% (95% CI, 70.9%‐86.5%) in the overall cohort. The RFS was 85.3% (95% CI, 73.3%‐97.2%), 83.3% (95% CI, 72.1%‐94.4%), and 57.1% (95% CI, 36.9%‐77.2%) in patients with stage I, II, and III TNBC (P = .006), respectively. The 5‐year RFS in the 27 patients with BRCA and non‐BRCA HR gene mutations was 73.5 (95% CI, 56.6%‐90.4%), which was nonsignificantly inferior to those without the mutations (RFS = 80.4%; 95% CI, 71.6%‐89.2%) (Figure S4).
4. DISCUSSION
Through direct sequencing, our study showed an overall 34.2% mutation rate in TNBC (10.8% of BRCA, 11.7% of non‐BRCA HR gene, and 11.7% non‐HR DDR gene). As tumors with mutations of 2 non‐HR DDR genes, PTEN and MSH6, show BRCAness and are not sensitive to PARP inhibitors or platinum, we hypothesize that the heterogeneous genomic background of BRCAness indicates different responsiveness to platinum and PARP inhibitors.
Prior studies considered “BRCAness”, a characteristic with high‐grade genomic instability, as a marker to guide treatment of PARP inhibitors and platinum in ovarian cancer.34 Both TNBC with mutations of BRCA or non‐BRCA HR genes were found to show BRCAness features in our study, which could explain why some non‐BRCA‐mutated BRCAness tumors respond to platinum and PARP inhibitors. However, recent studies reported that BRCAness only possesses a modest value in positively predicting outcomes of breast cancer treated with PARP inhibitors.12 In addition, carboplatin was an effective treatment in BRCA‐mutated metastatic TNBC in the TNT phase III trial, but carboplatin was not an effective treatment for non‐BRCA‐mutated tumors with BRCAness.35 Therefore, the clinical utility of BRCAness, in predicting the response rate to platinum and PARP inhibitors in breast cancer, still needs to be clarified. One possible explanation was that an epigenetic inactivation of the BRCA1 gene, such as promoter methylation, caused a subset of BRCAness tumors.12, 35, 36 The status of methylation is changeable, so these tumors might not actually consistently respond to platinum. In this study, we found that BRCAness features not only resulted from HR/BRCA alteration, but also resulted from non‐HR DDR gene mutations, like MSH6 and PTEN‐mutated tumors (Figure S2). Although PTEN mutation was previously reported as a marker to predict sensitivity to PARP inhibitors,37 more recent evidence shows that PTEN mutation will cause an activation of the PI3K pathway. Hence, PTEN‐mutated tumors would not respond to monotherapy of PARP inhibitors.38, 39 Neither are PTEN‐mutated tumors sensitive to platinum‐based treatment in chemotherapy.21 These clinical features were significantly dissimilar to HR gene‐mutated tumors, suggesting different biologic characteristics between PTEN‐ and HR‐mutated tumors.21 Furthermore, we found different regions of chromosomal gain or loss between PTEN‐ and HR gene‐mutated tumors. MSH6 mutation was only found in 2 cases of our cohort, and their pathology was compatible with previous reports (ER‐negative, high histologic grade, and hypermutated).40 Mutation of Lynch syndrome‐related genes (MSH6, MLH1, MSH2, and PMS2) were rare in breast cancer.40 As only 1 MSH6‐mutated breast cancer was analyzed by aCGH (DNA from the other tumor was not adequate for aCGH), it is not conclusive that all MSH6‐mutated breast tumors have high‐grade genomic instability. Still, MSH6 mutation might not be a marker for PARP inhibitor, because olaparib was not effective to treat colon cancer with Lynch syndrome.41 The case that the genomic background of BRCAness was heterogeneous could provide another rationale for why certain BRCAness TNBC did not respond well to platinum and PARP inhibitors.
In addition to genomic instability assessed by aCGH, various methods have been described to identify BRCAness, including gene expression, specific rearrangement signature, and RAD51 foci analysis by immunostaining.2, 42, 43, 44 Currently, the concordance of mentioned methods is uncertain, and it is unclear which method most appropriately predicts the therapeutic response. A gene expression profile could identify those tumors with BRCAness, but the predictive value of the therapeutic response to PARP inhibitors would only be moderate.45 “Signature 3”, a specific rearrangement signature, is a marker associated with the mutations of BRCA1/2, PALB2, and RAD51C promotor methylation.44 Although tumors that contain mutation of ATM or CHEK2 could respond to PARP inhibitors, “Signature 3” is not associated with the mutations of ATM or CHEK2.42 As RAD51 foci deficiency indicates that the tumor cells have lost the ability to repair DSB, RAD51 could be a good marker to guide treatment with PARP inhibitors. Biallelic alteration of HR genes was significantly associated with RAD51 foci deficiency (P < .001),43 indicating that HR gene‐mutated breast tumors lost the ability to repair DSB. However, assay of RAD51 foci requires a primary cancer cell culture treated with radiation and other medications, which is difficult to undertake in clinical practice. Therefore, to identify patients who can benefit from PARP inhibitors and platinum‐based chemotherapy, direct sequencing of all HR genes would be a more appropriate method. In the era of precision medicine and with the availability of NGS tools, sequencing is a highly rewarding strategy that can be applied to various cancer types.16, 46
A recent meta‐analysis suggested that BRCA mutation is associated with worse overall survival.47 Data from The Cancer Genome Atlas database showed that breast cancer patients with mutations in 12 DDR genes were associated with poor prognosis.48 Our study found that patients with mutations of HR genes had a higher trend of recurrence. Even though the underlying molecular mechanism of the poor prognosis is still not well understood, our study revealed that both BRCA‐mutated and non‐BRCA HR‐mutated tumors were associated with amplification of oncogenes. For example, MYC, S‐100 family genes, and CRKL were all related to poor prognosis. Hence, they might be some of the important reasons for worse disease outcome.31, 32, 49, 50
Our study indicated that the genomic background of BRCAness was heterogeneous. Certain DDR gene mutations led to BRCAness features. Yet, their clinical and biologic characteristics were different from BRCA and HR gene‐mutated tumors. To identify TNBC patients who might benefit from treatment with platinum and PARP inhibitors, the direct sequencing of HR genes could lead to a more accurate prediction of the responsiveness to platinum and PARP inhibitors (Figure S5). Thus, this strategy should be considered in precision medicine.
DISCLOSURE
The authors have no conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported, in part, by research grants from the National Taiwan University Hospital (NTUH. 104‐N2901 and 105‐N3285) and the Ministry of Science and Technology (MOST 103‐2314‐B‐002‐005‐MY2, MOST 104‐2314‐B‐002‐106‐MY3 and MOST 106‐2314‐B‐002‐114‐MY3). We also thank the National Applied Research Laboratories for providing access to their high‐performance computer to analyze the post‐NGS data.
Lin P‐H, Chen M, Tsai L‐W, et al. Using next‐generation sequencing to redefine BRCAness in triple‐negative breast cancer. Cancer Sci. 2020;111:1375–1384. 10.1111/cas.14313
REFERENCES
- 1. Palma G, Frasci G, Chirico A, et al. Triple negative breast cancer: looking for the missing link between biology and treatments. Oncotarget. 2015;6:26560‐26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014;16:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2‐deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14:3916‐3925. [DOI] [PubMed] [Google Scholar]
- 4. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523‐533. [DOI] [PubMed] [Google Scholar]
- 5. Matulonis UA, Penson RT, Domchek SM, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016;27:1013‐1019. [DOI] [PubMed] [Google Scholar]
- 6. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum‐sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375:2154‐2164. [DOI] [PubMed] [Google Scholar]
- 7. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110‐120. [DOI] [PubMed] [Google Scholar]
- 8. Turner N, Tutt A, Ashworth A. Hallmarks of 'BRCAness' in sporadic cancers. Nat Rev Cancer. 2004;4:814‐819. [DOI] [PubMed] [Google Scholar]
- 9. Lips EH, Debipersad RD, Scheerman CE, et al. BRCA1‐mutated estrogen receptor‐positive breast cancer shows BRCAness, suggesting sensitivity to drugs targeting homologous recombination deficiency. Clin Cancer Res. 2017;23:1236‐1241. [DOI] [PubMed] [Google Scholar]
- 10. Muggia F, Safra T. 'BRCAness' and its implications for platinum action in gynecologic cancer. Anticancer Res. 2014;34:551‐556. [PMC free article] [PubMed] [Google Scholar]
- 11. Tutt A, Tovey H, Cheang MCU, et al. Carboplatin in BRCA1/2‐mutated and triple‐negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med. 2018;24:628‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Telli ML, Jensen KC, Vinayak S, et al. Phase II study of gemcitabine, carboplatin, and iniparib as neoadjuvant therapy for triple‐negative and BRCA1/2 mutation‐associated breast cancer with assessment of a tumor‐based measure of genomic instability: PrECOG 0105. J Clin Oncol. 2015;33:1895‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koppensteiner R, Samartzis EP, Noske A, et al. Effect of MRE11 loss on PARP‐inhibitor sensitivity in endometrial cancer in vitro. PLoS ONE. 2014;9:e100041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Min A, Im S‐A, Yoon Y‐K, et al. RAD51C‐deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther. 2013;12:865‐877. [DOI] [PubMed] [Google Scholar]
- 15. Weston VJ, Oldreive CE, Skowronska A, et al. The PARP inhibitor olaparib induces significant killing of ATM‐deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578‐4587. [DOI] [PubMed] [Google Scholar]
- 16. Mateo J, Carreira S, Sandhu S, et al. DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697‐1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang M, Zhuang G, Sun X, et al. TP53 mutation‐mediated genomic instability induces the evolution of chemoresistance and recurrence in epithelial ovarian cancer. Diagn Pathol. 2017;12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hubbard GK, Mutton LN, Khalili M, et al. Combined MYC activation and pten loss are sufficient to create genomic instability and lethal metastatic prostate cancer. Cancer Res. 2016;76:283‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Javid M, Sasanakietkul T, Nicolson NG, et al. DNA mismatch repair deficiency promotes genomic instability in a subset of papillary thyroid cancers. World J Surg. 2018;42:358‐366. [DOI] [PubMed] [Google Scholar]
- 20. Thiffault I, Saunders C, Jenkins J, et al. A patient with polymerase E1 deficiency (POLE1): clinical features and overlap with DNA breakage/instability syndromes. BMC Med Genet. 2015;16:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20:764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin P‐H, Kuo W‐H, Huang A‐C, et al. Multiple gene sequencing for risk assessment in patients with early‐onset or familial breast cancer. Oncotarget. 2016;7:8310‐8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knijnenburg TA, Wang L, Zimmermann MT, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23:239‐254 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin P‐H, Li H‐Y, Fan S‐C, et al. A targeted next‐generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: implications for clinical practice. Cancer Med. 2017;6:349‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chou W‐C, Lin P‐H, Yeh Y‐C, et al. Genes involved in angiogenesis and mTOR pathways are frequently mutated in Asian patients with pancreatic neuroendocrine tumors. Int J Biol Sci. 2016;12:1523‐1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng Y, Chen D, Wang GL, Zhang VW, Wong LJ. Improved molecular diagnosis by the detection of exonic deletions with target gene capture and deep sequencing. Genet Med. 2015;17:99‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khiabanian H, Hirshfield KM, Goldfinger M, et al. Inference of germline mutational status and evaluation of loss of heterozygosity in high‐depth, tumor‐only sequencing data. JCO Precis Oncol. 2018;2018:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi Y, Jin J, Ji W, Guan X. Therapeutic landscape in mutational triple negative breast cancer. Mol Cancer. 2018;17:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple‐negative breast cancer. Can Res. 2013;73:2025‐2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Silva GO, He X, Parker JS, et al. Cross‐species DNA copy number analyses identifies multiple 1q21‐q23 subtype‐specific driver genes for breast cancer. Breast Cancer Res Treat. 2015;152:347‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goh JY, Feng M, Wang W, et al. Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med. 2017;23:1319‐1330. [DOI] [PubMed] [Google Scholar]
- 33. Milioli HH, Tishchenko I, Riveros C, Berretta R, Moscato P. Basal‐like breast cancer: molecular profiles, clinical features and survival outcomes. BMC Med Genomics. 2017;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107:1776‐1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gampenrieder SP, Rinnerthaler G, Greil R. SABCS 2016: systemic therapy for metastatic breast cancer. Memo. 2017;10:86‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Isakoff SJ, Mayer EL, He L, et al. TBCRC009: a multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple‐negative breast cancer. J Clin Oncol. 2015;33:1902‐1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dedes KJ, Wetterskog D, Mendes‐Pereira AM, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2:53ra75. [DOI] [PubMed] [Google Scholar]
- 38. Bian X, Gao J, Luo F, et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP‐PI3K inhibition but not PARP inhibition as monotherapy. Oncogene. 2018;37:341‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gonzalez‐Billalabeitia E, Seitzer N, Song SJ, et al. Vulnerabilities of PTEN‐TP53‐deficient prostate cancers to compound PARP‐PI3K inhibition. Cancer Discov. 2014;4:896‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walsh MD, Buchanan DD, Cummings MC, et al. Lynch syndrome‐associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16:2214‐2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leichman L, Groshen S, O'Neil BH, et al. Phase II study of olaparib (AZD‐2281) after standard systemic therapies for disseminated colorectal cancer. Oncologist. 2016;21:172‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Polak P, Kim J, Braunstein LZ, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49:1476‐1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mutter RW, Riaz N, Ng CKY, et al. Bi‐allelic alterations in DNA repair genes underpin homologous recombination DNA repair defects in breast cancer. J Pathol. 2017;242:165‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nik‐Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature. 2016;534:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wolf DM, Yau C, Sanil A, et al. DNA repair deficiency biomarkers and the 70‐gene ultra‐high risk signature as predictors of veliparib/carboplatin response in the I‐SPY 2 breast cancer trial. NPJ breast cancer. 2017;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takagi M, Yoshida M, Nemoto Y, et al. Loss of DNA damage response in neuroblastoma and utility of a PARP inhibitor. J Natl Cancer Inst. 2017;109:djx062. [DOI] [PubMed] [Google Scholar]
- 47. Zhong Q, Peng HL, Zhao X, Zhang L, Hwang WT. Effects of BRCA1‐ and BRCA2‐related mutations on ovarian and breast cancer survival: a meta‐analysis. Clin Cancer Res. 2015;21:211‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu C, Chang H, Li X‐H, et al. Network meta‐analysis on the effects of DNA damage response‐related gene mutations on overall survival of breast cancer based on TCGA database. J Cell Biochem. 2017;118:4728‐4734. [DOI] [PubMed] [Google Scholar]
- 49. Qian J, Chen H, Ji X, et al. A 3q gene signature associated with triple negative breast cancer organ specific metastasis and response to neoadjuvant chemotherapy. Sci Rep. 2017;7:45828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grushko TA, Dignam JJ, Das S, et al. MYC is amplified in BRCA1‐associated breast cancers. Clin Cancer Res. 2004;10:499‐507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials