

Abstract
Proteasome inhibitors significantly improve cancer outcomes, but their use is eventually followed by proteasome inhibitor resistance and relapse. Current understanding of proteasome inhibitor resistance is limited to cell‐autonomous mechanisms; whether non–autonomous mechanisms can be implicated in the development of proteasome inhibitor resistance is unclear. Here, we show that proteasome inhibitor tolerance can be transmitted non–autonomously through exosome‐mediated intercellular interactions. We revealed that reversible proteasome inhibitor resistance can be transmitted from cells under therapy stress to naïve sensitive cells through exosome‐mediated cell cycle arrest and enhanced stemness in mixed‐lineage leukemia cells. Integrated multi‐omics analysis using the Tied Diffusion through Interacting Events algorithm identified several candidate exosomal proteins that may serve as predictors for proteasome inhibitor resistance and potential therapeutic targets for treating refractory mixed‐lineage leukemia. Furthermore, inhibiting the secretion of exosomes is a promising strategy for reversing proteasome inhibitor resistance in vivo, which provides a novel proof of principle for the treatment of other refractory or relapsed cancers.
Keywords: drug tolerance, exosomes, mixed‐lineage leukemia, non–autonomous, proteasome inhibitor
Using the integrated multi‐omics analysis, our study demonstrates that exosomes derived from MLL cells under therapy stress transmit proteasome inhibitor (PI) tolerance to recipient cells through cell cycle arrest and enhanced stemness. Exosomes can act not only as a mediator of development of PI tolerance but also as a therapeutic target to overcome PI resistance, thereby enhancing clinical benefits of PI therapy in MLL.

1. INTRODUCTION
Mixed‐lineage leukemias (MLL), including acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML), are aggressive hematological malignancies with dismal outcomes.1 Chromosomal translocations involving MLL protein predominantly occur in pediatric patients, including approximately 80% of infant leukemias.2 MLL patients generally have a very poor overall prognosis, and this aggressive disease tends to be refractory to conventional anticancer therapies due to the development of resistance and relapse with established therapies, including chemotherapy and bone marrow transplantation.3, 4 In the last decade, improved molecular understanding of MLL‐fusion proteins has led to the identification of several potential mechanism‐based therapeutic targets, and several candidate therapeutic strategies have emerged, including proteasome inhibition.5, 6 Proteasome inhibitors (PI) are used to inhibit the ubiquitin‐proteasome pathway and act by targeting intracellular protein turnover, thus promoting cancer therapy. We previously reported that the PI bortezomib shows effectiveness in patients with MLL‐rearranged B‐ALL.5 However, the inevitable emergence of resistance imposes limits on bortezomib’s clinical application.
Innate resistance, as well as acquired resistance arising during treatment, prevents cancer therapies from achieving stable and complete responses.7, 8 It is well recognized that cancer cells under therapy stress primarily enter a drug‐tolerant state, which is often achieved through the slowing down of an essential cellular process.9, 10, 11 Increasing evidence implicates the transient drug‐tolerant state as an initial mechanism of the eventual acquisition of drug resistance.9, 10 Cancer cells employ such dynamic survival strategies in which individual cells transiently assume a reversible tolerant state to protect the population from eradication by potentially lethal exposures. Interestingly, patients may remain sensitive to PI after relapse with initial therapy in multiple myeloma, implying that PI resistance is reversible.12, 13, 14 However, how cells under therapy stress transmit this survival strategy to naïve cell populations is largely unknown.
The interplay between intratumor heterogeneity and tumor microenvironment (TME) plays a key role in the emergence of drug resistance.15 Exosomes are small extracellular vesicles (sEV) in the cellular microenvironment that range from 30 to 150 nm in diameter.16 Most types of cells can release exosomes, which harbor various cargoes, including proteins, lipids and nucleic acids.16 Exosomes originating from multi‐vesicular bodies are usually secreted into the extracellular matrix by fusion with cytomembrane.17 According to recent studies, exosomes play essential roles in cellular signaling transduction as well as intercellular communication.18 Prior studies suggest that exosomes contribute to drug resistance and the modification of TME, and promote cancer metastasis.17, 19, 20, 21 Exosomal nucleic acids, including microRNA (miRNA), mRNA and DNA fragments, have been explored for their contribution to cellular immunomodulation, chemotherapy resistance, as well as cancer progression.17, 22 Xu et al (2019) show that exosomal RNA promotes resistance to PI in multiple myeloma.23 However, our mechanistic understanding of the role of exosomal proteins in the physiological and pathological environment, particularly in the acquisition of drug tolerance, remains largely elusive.
Recent advances in bioinformatics allow for the integration of transcriptional, proteomic and other sources of data in the context of prior pathway knowledge to find events that drive tumorigenesis or drug resistance.24 Tied Diffusion through Interacting Events (TieDIE), which uses a network diffusion approach to compute the relationship between proteins, predicts the processes of signal transduction and transcriptomic perturbations.25
Here, we demonstrate that PI resistance in MLL is reversible, referring to this as tolerance. Given the critical roles of exosomes in TME, we reasoned that exosomes transmit PI tolerance in MLL, and exosomal proteins play a causal role in the acquisition of resistance. To investigate this hypothesis, we established RNA sequencing as well as quantitative proteome detection to identify factors regulating the acquisition of PI resistance. Using TieDIE analyses, we identified candidate exosomal proteins through dataset integration, providing potential therapeutic targets for treating the aggressive and otherwise refractory MLL. Together, our study reveals that targeting the secretion of exosomes is a promising strategy for overcoming PI resistance.
2. MATERIALS AND METHODS
2.1. Cell culture
Human pro–B MLL‐rearranged cell lines RS4;11 and SEM were purchased from DSMZ. Cells were cultured in Gibco RPMI 1640 containing 10% exosomes‐free FBS (Gibco, ultracentrifugation at 110 000 g for 16 hours) at 37°C with 5% CO2. Induced drug‐tolerant cells were generated by exposing naïve cells to a sublethal dose of bortezomib for at least 4 weeks, replenishing the inhibitor every 3 days. The remaining cells after the treatment were considered “tolerant” cells and were collected for analysis. Cell culture supernatant was filtered with a 0.22 µmol/L pore filter (Merck) before use. All cultured cells were tested for mycoplasma contamination before use.
2.2. Reagents
Bortezomib (Velcade) and Carfilzomib (PR‐171) were obtained from Selleck Chemicals. GW4869 was obtained from Topscience.
2.3. Exosomes preparation
Approximately 200 mL conditioned medium was harvested from cultured cells (5 × 107 cells for 48 hours). Then, the samples were centrifuged at 300 g for 10 minutes, 2000 g for 10 minutes, 10 000 g for 10 minutes and 110 000 g for 70 minutes at 4°C. Pellets were washed with cold PBS and centrifuged again at 110 000 g for 70 minutes at 4°C. The pelleted exosomes were resuspended in 100 μL PBS with Proteinase Inhibitor Cocktail (Roche) and then stored at −80°C.
2.4. Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was used for calculating the size distribution and concentration of exosomes. NanoSight NS300 equipped with particle‐tracking software was used to analyze the vesicle size and concentration. Parameters were kept constant for all samples.
2.5. Transmission electron microscopy
For transmission electron microscopy (TEM), purified exosomes were directly adsorbed onto a formvar‐carbon‐coated 300 mesh copper grid and stained with 2% phosphotungstic acid. TEM images were obtained using a Philips CM120 transmission electron microscope equipped with a tungsten filament and operated at an acceleration voltage of 120 kV.
2.6. Western blot analysis
Exosomes were lysed with RIPA buffer containing Proteinase Inhibitor Cocktail (Roche) 30 minutes on ice, then centrifuged at 12 000 g for 15 minutes at 4°C. Immunoblot assays have been described previously.26 TSG101, CD63, CD81, CD9, calnexin and GRP78 antibodies were purchased from Abcam. Caspase3 antibody was purchased from Cell Signaling Technology. β‐Actin antibody was obtained from Sigma Aldrich. Antibodies were detected using the enhanced chemiluminescence method (Millipore). Immunoblot signals were acquired with the Amersham Imager 600 (General Electric).
2.7. Liquid chromatography‐mass spectrometry/mass spectrometry
Exosomes isolated from RS4;11‐naïve and bortezomib‐treated RS4;11 cells through ultracentrifugation were dissolved in RIPA buffer, and exosomal proteins were extracted and then digested overnight at 37°C by trypsin (Promega) through using the filter‐aided sample preparation approach. The quantitative label‐free mass spectrometry assays have been described previously.20
2.8. Cell viability and cell proliferation assays
The CellTiter 96 MTS assay (Promega) was used to determine the cytotoxicity of the relevant drugs and cell proliferation, according to the manufacturer’s instructions. Cell viability was measured with MTS assay 24 hours after the addition of bortezomib or carfilzomib with graded concentrations in triplicate.
2.9. Apoptosis and cell cycle assays
Apoptosis and cell cycle were measured using the FITC Annexin V Apoptosis Detection Kit and the APC BrdU Flow Kit from BD Pharmingen as described by the manufacturer, respectively. Cell staining with fluorochromes was acquired using a flow cytometer, and data were analyzed using FlowJo software.
2.10. Total RNA sequencing
Naïve RS4;11 cells treated with 5 μg/mL exosomes derived from DMSO‐treated or bortezomib‐treated RS4;11 cells for 96 hours, respectively, were collected and lysed with the TRIzol Reagent. Total RNA was extracted from TRIzol Reagent according to the manufacturer’s instructions and mRNA‐seq libraries were sequenced using BGISEQ‐500RS.
2.11. Tied Diffusion through Interacting Events pathway analysis
The TieDIE algorithm uses heat diffusion strategies by leveraging different types of inputs to compute “subnetworks” based on the “Multinet” pathway database as a background network 27 with high specificity.24, 25 We performed TieDIE analysis according to the tutorial, to connect differential genes of recipient cells with differential exosomal proteins. The software Cytoscape V3.5.1 is used for integrating biomolecular interaction networks.28
2.12. Mouse studies
NOD‐SCID mice were purchased from Vital River Laboratory; 2 × 107 RS4;11 or 1 × 106 MLL‐rearranged ALL patient‐derived xenograft (MLLr‐PDX) cells were intravenously injected into NOD‐SCID mice. Mice were then administered bortezomib intravenously at 1 mg/kg on a twice‐weekly schedule, or administered GW4869 intraperitoneally at 1.5 mg/kg three times per week, beginning 2 weeks after the xenograft. Mice were treated for 4 consecutive weeks and then monitored for disease and killed when they became moribund. The percentage of leukemic cells in the peripheral blood of mice was monitored using CD133 antibody (CD133‐PE, Miltenyi Biotec) for RS4;11 or CD45 antibody (CD45‐FITC, Biolegend) for MLLr‐PDX.29 Animal care and sacrifice were conducted according to methods approved by the Animal Care and Use Committee of the Center for Animal Experiments of Shanghai Jiao Tong University.
2.13. Statistical analysis
GraphPad Prism 7 statistical software was used for statistical analysis. Student’s t test was used to analyze the differences between the groups. Means were illustrated with error bars representing ± the SD, and statistical relevance was evaluated using the following P‐values: P < 0.05 (*), P < 0.01 (**), or P < 0.001 (***).
3. RESULTS
3.1. Exosomes mediate reversible proteasome inhibitor resistance in mixed‐lineage leukemia cells
We have previously demonstrated that PI shows effectiveness in mouse models and patients with MLL‐rearranged leukemia.5 To determine whether PI resistance in treatment‐refractory MLL cells is reversible, we transplanted RS4;11 cells into NOD‐SCID mice and collected the residual cells from xenograft mice treated with bortezomib or mice in which bortezomib was withdrawn after treatment (Figure 1A). The results revealed that the residual cells surviving bortezomib exposure showed resistance to bortezomib (Figure 1B,C), and the mice in which bortezomib was withdrawn exhibited remarkably enhanced sensitivity to this drug (Figure 1B,C), suggesting that PI resistance is reversible in MLL cells. We further generated MLL‐rearranged patient‐derived xenografts (MLLr‐PDX) and collected the bone marrow cells from xenograft mice treated with bortezomib or from those bortezomib was withdrawn after treatment (Figure S1A). The results revealed that the residual MLLr‐PDX cells surviving bortezomib exposure showed resistance to bortezomib, and subsequent removal of bortezomib diminished resistance in these cells (Figures 1D and S1B), suggesting that PI resistance is reversible in MLLr‐PDX cells.
Figure 1.
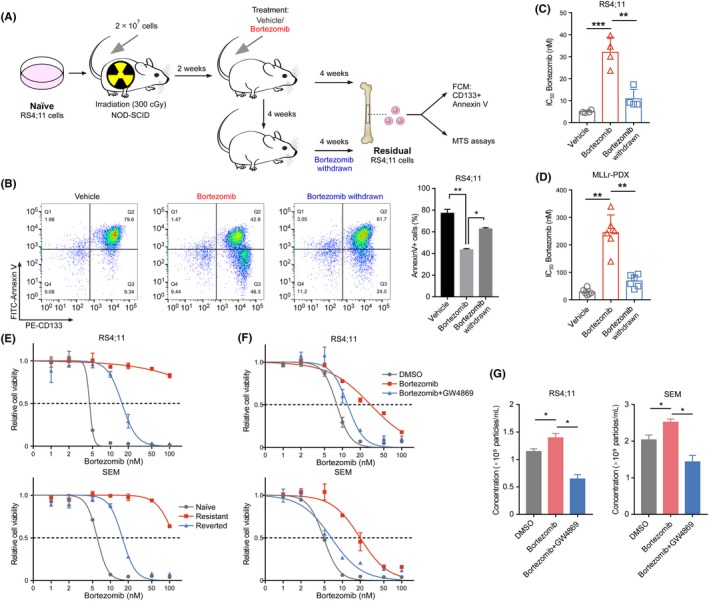
Exosomes mediate reversible proteasome inhibitor (PI) resistance in mixed‐lineage leukemias (MLL) cells. A, Schematic for the different bortezomib treatment strategies of the MLL cells on the xenograft NOD‐SCID mice. B, Bone marrow cells from the xenograft mice were obtained and dead cells were removed. Flow cytometric detection of CD133 expression and apoptosis was measured 16 hr after the addition of bortezomib. Histogram of four independent biological replicates with three technical replicates is shown (right panel). C, Cell viability of bone marrow cells derived from RS4;11 xenograft mice was measured with MTS assay 24 hr after the addition of bortezomib. The IC50 of different cells was quantified. Histogram of four independent biological replicates with three technical replicates is shown. D, Cell viability of bone marrow cells derived from MLLr‐PDX xenograft mice was measured with MTS assay 24 hr after the addition of bortezomib. Histogram of six independent biological replicates with three technical replicates is shown. E, Cell viability of RS4;11 or SEM cells with bortezomib at the indicated concentrations for 24 h. F, RS4;11 or SEM cells were treated with supernatant derived from RS4;11 cells under the indicated treatment (5 nmol/L for bortezomib or 5 μmol/L for GW4869). Cell viability with bortezomib at the indicated concentrations for 24 h was detected. G, The indicated cells were treated with the corresponding cell‐derived supernatant. Nanoparticle tracking analysis of isolated small extracellular vesicles (sEV) was detected. *P < 0.05; **P < 0.01; ***P < 0.001; two‐tailed t test. Data are represented as mean ± SD
To investigate the mechanism underlying the emergence of reversible PI resistance in MLL, we performed sublethal treatment experiments and modeled bortezomib‐resistant cells. MLL cells were exposed to a sublethal dose of bortezomib for 4 weeks, followed by recovery in drug‐free medium for an additional 4 weeks. Notably, sublethal bortezomib‐treated cells were more tolerant to the subsequent treatment (Figure 1E). Besides, RS4;11‐resistant cells exhibited cross–resistance to carfilzomib, another PI (Figure S1C). Subsequent removal of bortezomib allowed these cells to revert to a relatively PI‐sensitive state (Figures 1E and S1C). We also evaluated another pro–B MLL cell line, SEM, and a similar state of drug tolerance was observed (Figures 1E and S1D,E). These results confirmed the reversibility of PI resistance in MLL. Furthermore, our results indicate that these PI‐resistant cells are not inherently resistant to PI but, rather, exist in a transient and reversible drug‐tolerant state.
Increasing evidence implies that the transient drug‐tolerant state acquired by cancer cells under therapy stress is an initial mechanism of the eventual acquisition of drug resistance.9, 10 To evaluate whether the TME under therapy stress mediates the acquisition of PI resistance in naïve cells, we cultured bortezomib‐sensitive cells with cell culture supernatant of DMSO‐treated or bortezomib‐treated RS4;11 cells after filtration (0.22 μm), respectively. The results showed that bortezomib‐treated cell‐derived supernatant significantly increased the resistance of naïve cells to bortezomib (Figure 1F), suggesting the effect of extracellular factors in drug resistance. The neutral sphingomyelinase inhibitor GW4869 partially inhibits the secretion of exosomes through the ceramide pathway (Figure S1F).30 Compared with cells exposed to bortezomib‐treated cell‐derived supernatant, we saw an increased cell sensitivity when using supernatant from the combination of bortezomib and GW4869‐treated RS4;11 cells, which contained fewer exosomes (Figure 1F,G), indicating that the effects on cell viability seen with the addition of PI‐treated cell supernatant are at least partly due to the presence of exosomes, and that these exosomes can be eliminated by treating the cells with GW4869. These results suggest that exosomes produced by cells under treatment stress mediate drug resistance in recipient cells.
3.2. Characterization of extracellular vesicles
The secretion of sEV is an essential means by which tumors induce systemic changes; however, how they regulate the behavior of recipient cells remains largely unknown. RS4;11 cells were treated with bortezomib, and sEV were isolated through ultracentrifugation and determined by NTA. We observed that the concentration and size of sEV were increased in bortezomib‐treated cells (Figure 2A), indicating that PI treatment induced the release of sEV in MLL cells. Subsequently, sEV from the conditioned medium of naïve and bortezomib‐treated RS4;11 cells were isolated through ultracentrifugation and characterized comprehensively. The NTA results showed that the size of sEV was around 95 and 106 nm, in naïve and bortezomib‐treated RS4;11 cells, respectively (Figure 2B). Figure 2C demonstrates that the vesicles have a membrane structure and the size is almost 100 nm, visualized by TEM. Exosome representative markers, including endosomal TSG101 as well as tetraspanins CD9, CD63 and CD81, were specifically enriched in the isolated sEV (Figure 2D), whereas intracellular proteins, calnexin and GRP78, were negligible. The presence of these exosome marker proteins together with their size31 indicate that the sEV are from endosomal origin, and most are exosomes.
Figure 2.
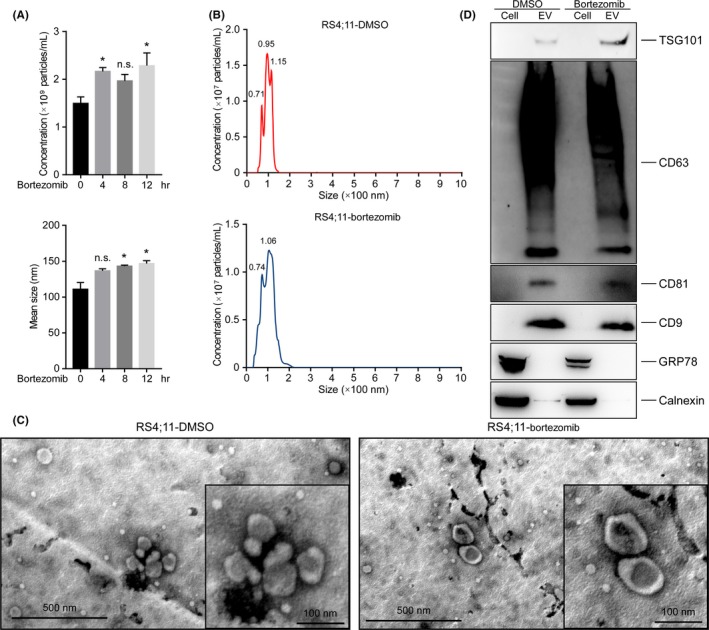
Characterization of extracellular vesicles. A, Nanoparticle tracking analysis of isolated small extracellular vesicles (sEV) from RS4;11 cells treated with 10 nM bortezomib. Ten frames per sample were analyzed. B, Nanoparticle tracking analysis of isolated sEV from DMSO‐treated or bortezomib‐treated RS4;11 cells. C, Transmission electron microscopy analysis of isolated sEV showed the presence of vesicles with the appropriate size and morphology. D, Immunoblot of sEV showed the absence of calnexin and GRP78 and presence of tetraspanins CD9, CD63 and CD81, as well as endosomal TSG101, which were enriched. n.s., not significant; *P < 0.05; two‐tailed t test. Data are represented as mean ± SD
3.3. Exosomes derived from proteasome inhibitor‐treated cells induce cell cycle arrest and proteasome inhibitor resistance
Exosomes have been previously reported to promote resistance of cancer cells to chemotherapies through different mechanisms.19, 23 Therefore, we reasoned that exosomes might contribute to tolerance transitions to facilitate the acquisition of PI resistance that underlies inevitable relapse under treatment stress. To confirm the critical role of exosomes in mediating tolerance transition, we exposed PI‐sensitive cells to bortezomib‐treated RS4;11‐released exosomes (RS4;11‐Rexo) and observed the aggregation of naïve RS4;11 cells (Figure 3A), consistent with the idea that cell adhesion and aggregation lead to enhanced cell viability.32 Furthermore, compared with naïve cells and cells treated with RS4;11‐naïve cell‐derived exosomes (RS4;11‐Nexo), cells treated with RS4;11‐Rexo showed significant resistance to bortezomib (Figure 3B,C, and Figure S2A). Moreover, cells pre–treated with very low concentrations of RS4;11‐Rexo were resistant to PI‐induced apoptosis (Figure 3C). These results indicate that exosomes released from cells under treatment stress change the microenvironment and transmit chemoresistance to drug‐sensitive naïve cells.
Figure 3.
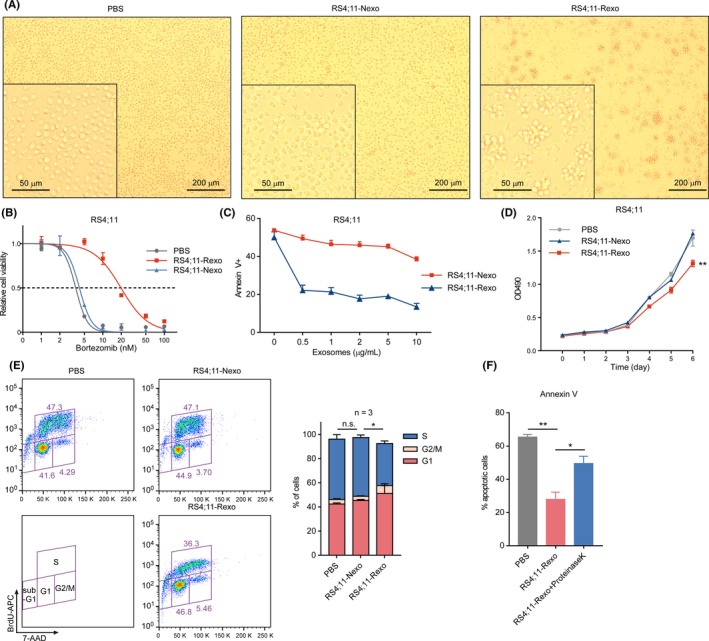
Exosomes derived from PI‐treated cells induce cell cycle arrest and proteasome inhibitor (PI) resistance. A, Representative images of RS4;11 cells treated either with PBS, 5 µg/mL RS4;11‐naïve exosomes (RS4;11‐Nexo) or PI‐treated RS4;11‐released exosomes (RS4;11‐Rexo) for 4 d. B, RS4;11 cells were treated with PBS, 5 µg/mL RS4;11‐Nexo or 5 µg/mL RS4;11‐Rexo for 4 d. Cell viability with bortezomib at the indicated concentrations for 24 h was detected. C, RS4;11 cells were treated with the indicated concentrations of exosomes for 4 d. The percentage of annexin V‐positive cells was determined after a 16 h treatment with 5 nmol/L bortezomib. D, The proliferation of RS4;11 cells treated with PBS or 5 µg/mL indicated exosomes for 6 d. E, Cell cycle profiling of RS4;11 cells treated with PBS or 5 µg/mL indicated exosomes for 4 d. Stacked barplot shows the fraction of cells viable in G1, S and G2/M phases of the indicated cells. F, RS4;11 cells were treated with PBS, 5 µg/mL RS4;11‐Rexo or exosomes treated with proteinase K and 1% Triton X‐100 for 30 min at 4°C. The percentage of Annexin V‐positive cells was determined after a 16‐h treatment with 5 nmol/L bortezomib. n.s, not significant; *P < 0.05; **P < 0.01; two‐tailed t test. Data are represented as mean ± SD
To further characterize these recipient cells, we evaluated cell proliferation, and observed that the growth of cells treated with RS4;11‐Rexo was notably decreased (Figures 3D and S2B). Subsequently, these recipient cells were analyzed by flow cytometry and the results showed that the S‐phase of the cell cycle was significantly reduced, accompanied by an increase in G1‐phase in RS4;11‐Rexo‐treated cells (Figures 3E and S2C). The above results revealed that exosomes derived from cells under therapy stress suppressed cell cycle processes of naïve cells to render these cells insensitive to drugs and make them resistant to current therapies.
We then sought to confirm whether exosomal proteins contribute to PI resistance. The membrane of sEV can be disrupted by the addition of detergent and allow the proteinase K to enter, which degrades exosomal proteins.20 We found that RS4;11‐Rexo‐induced PI resistance was abolished by protease treatment (Figure 3F), confirming that exosomal proteins were essential in transmitting PI resistance. These results suggest that exosomal proteins are a major source of intercellular communication factors, which play a critical role in regulating the drug‐resistant phenotype of recipient cells.
3.4. Tied Diffusion through Interacting Events integration reveals specific tolerant networks in mixed‐lineage leukemias
In MLL cell line RS4;11, the gene expression was notably changed under therapy stress (Figure S3A), and enrichment of genes involved in translation and cell cycle processes were observed (Figure S3B), which may also lead to changes in the secretion and components of exosomes. On uptake by recipient cells, exosomal proteins control many differentially expressed genes. Therefore, exosomal proteins are considered as “upstream” events that may influence and be statistically associated with activity of gene regulation in recipient cells.33 We first performed quantitative label‐free mass spectrometry using isolated exosomes from naïve and PI‐treated RS4;11 cells and analyzed the proteomic datasets. Within this dataset, 159 differentially expressed proteins were identified (FDR < 0.05, fold change > 2; Figure S3C, Table S1). Importantly, the large number of differentially expressed proteins enabled an integrated computational approach to identify pathways implicated from this proteomic dataset.
To identify and prioritize the causal protein drivers of the “downstream” gene sets specifically deregulated in recipient cells, we performed RNA sequencing using recipient cells treated with naïve or PI‐treated cell‐derived exosomes, and used the TieDIE algorithm for integrated pathway analysis (Figure 4A). Using TieDIE, we integrated differentially expressed proteins in exosomes and transcriptomic data of recipient cells to synthesize a robust signaling network, which consisted of 357 nodes (56 exosomal proteins, 48 genes and 253 linker proteins) connected by 2044 edges (Table S2). To simplify this network, interactions that were supported by the proteomic or transcriptomic data were retained, respectively. This resulted in a compact subnetwork with a high level of specificity (Figures 4B,C, and S4 and S5), which revealed the master regulator proteins in exosomes and genes in recipient cells, respectively.
Figure 4.
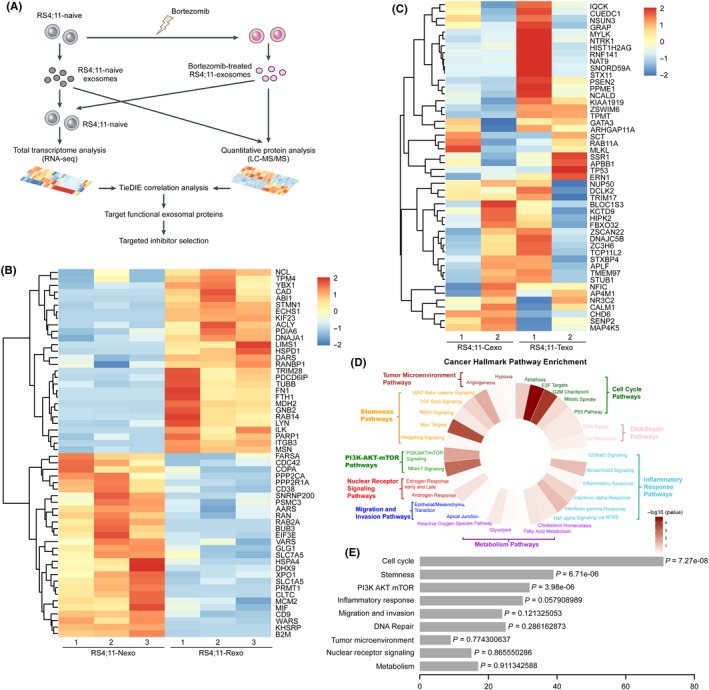
Tied Diffusion through Interacting Events (TieDIE) integration reveals specific tolerant networks in mixed‐lineage leukemias (MLL). A, Flow diagram depicting the integration of gene expression and proteome datasets for TieDIE analysis. B, TieDIE analysis identified 56 exosomes regulatory proteins as “upstream” of the integrated network. C, TieDIE analysis identified 48 differently expressed genes as “downstream” of the integrated network. D, Enriched cancer hallmarks generated by dataset integration using TieDIE after inclusion of the gene expression and proteomic data. E, Detailed P‐value of enriched cancer hallmark pathways
Given a complex integration network, one goal from these approaches was to select proteins corresponding to genes or pathways from PI resistance and then match targeted therapies based on these lesions. Therefore, we compared the 56 selected exosomal regulatory proteins (Figure 4B) with the DrugBank database and created a list of 31 proteins (31/56, 55.4%) as well as their corresponding drugs or compounds (Figure S6, Table S3). For instance, we identified an increased level of PRMT1 in exosomes released from PI‐treated RS4;11 cells, which has been previously reported to facilitate stem cell‐like properties and resistance to chemotherapy.4 The data‐induced network also revealed targets such as B2M, HSPA4 and ITGB3, which have been previously reported as targets that could potentially overcome drug resistance.2, 3, 34 Furthermore, the combination of PI with CD38 monoclonal antibody daratumumab or PARP1 inhibitors is effective in patients with multiple myeloma, and may prevent the emergence of resistant cells; the efficacy of this combination therapeutic strategy may also be promising in MLL.35, 36 Thus, the differences in exosomal regulatory protein levels could help explain these differences as well as offer new treatment options that could abrogate signaling upstream of these differentially expressed genes.
Next, we further analyzed the results of TieDIE analysis in the form of cancer‐related hallmark pathways (Figure 4D). The enrichment of genes notably involved in cell cycle pathways and stemness pathways was observed (Figure 4D,E). The enrichment of the cell cycle process is intriguing as our results above showed that PI‐treated cell‐derived exosomes suppressed the cell cycle processes of recipient cells, and the subnetwork revealed that TP53 and NUP50 genes were involved in the cell cycle deregulation (Figure S7). In addition, we found higher relative enrichment of proteins involved in the stemness pathways (Figures 4D,E, and S8), which have previously been able to connect a primary basal stem cell signature to cell resistance with this gene set.37
3.5. Inhibiting secretion of exosomes promotes the therapeutic effect of bortezomib in mice
Exosomes create a permissive microenvironment for the emergence of resistance to chemotherapies.19 Taking our results above, we hypothesized that the altered microenvironment, using GW4869 to inhibit the secretion of exosomes, might overcome PI resistance. To investigate the therapeutic effects of GW4869 in combination with bortezomib on leukemia progression, we transplanted RS4;11 cells into NOD‐SCID mice and evaluated the in vivo efficacy (Figure 5A). As expected, GW4869 single‐agent treatment showed no benefit for these xenograft mice, suggesting that these cells were insensitive to GW4869 treatment. In contrast, a notably decreased percentage of resistant cells and better responsiveness were observed with the combination treatment of bortezomib and GW4869 than with either of the two agents singly (Figure 5B,C). Moreover, the xenograft mice with combination treatment had significant improvement in overall survival (Figure 5D). These results strongly suggest that the inhibition of exosome secretion strengthens the anticancer effects of bortezomib in treating MLL by preventing the emergence of resistant cancer cells.
Figure 5.
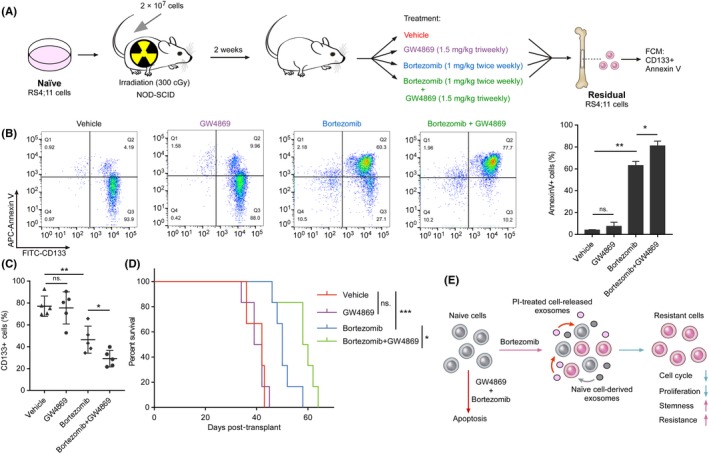
Inhibiting secretion of exosomes strengthens therapeutic effect of bortezomib in mice. A, Treatment schedule for the administration of bortezomib, GW4869 or vehicle. B, Bone marrow cells from the xenograft mice were obtained and dead cells were removed. Flow cytometric detection of CD133 expression and apoptosis was measured 16 h after the addition of bortezomib. Histogram of three independent biological replicates with three technical replicates (right panel). C, The percentage of leukemic cells in the bone marrow of the indicated groups of xenograft mice was monitored using CD133 antibody. D, Kaplan‐Meier survival curves for xenograft mice. E, The illustration depicts how proteasome inhibitor (PI)‐treated cell‐released exosomes induce PI tolerance in recipient cells and the therapeutic function of the combination therapy in mixed‐lineage leukemias (MLL). n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; by two‐tailed t test or log‐rank test for significance. Data represent means of triplicate reactions ± SD
4. DISCUSSION
One of the major causes of low efficacy of therapy in MLL is the rapid acquisition of drug resistance. Continuous interactions and exchanges between tumor cells and the TME are involved in the development of drug resistance phenotypes.38 Investigation of the mechanisms underlying microenvironment‐mediated drug resistance is important to identify novel pharmacological targets for this aggressive leukemia. This work demonstrates the involvement of exosomes in the acquisition of drug‐resistant phenotype in MLL cells (Figure 5E). In this study, we demonstrated that exosomes derived from cells under therapy stress could induce cell cycle arrest and growth suppression in recipient cells, thereby transmitting resistance to drug‐sensitive cell populations (Figure 5E). Therefore, exosomes conclusively play a critical role in PI resistance, and our findings implicate that inhibition of exosome secretion during treatment can lead to a systemic and durable therapeutic response and obviate drug resistance.
An expanding body of literature emphasizes the contribution of drug‐tolerant cancer cells in the development of inevitable drug resistance.9, 10 Tolerance is generally understood to be the ability, whether inherited or not, of a cell population to survive transient exposure to chemotherapy, which is often achieved by slowing down an essential cellular process.39 Drug‐resistant cancer cells can not only emerge from the treatment‐mediated selection of subpopulations that present at the start of therapy but also from a general adaptation process under therapy stress. Moreover, the microenvironment and pharmacokinetic conditions of solid tumors as well as hematological malignancies put cancer cells in different situations during therapy stress.40, 41 As chemotherapy drugs poison the cancer, neighboring cancer cells respond to the carnage, triggering apoptosis and releasing signals that encourage the survival of nearby sensitive cells. Our study demonstrated that exosomes might function as a driver in the adaptation process under therapy stress. This non–autonomous control of the recipient cell adaptation suggests that exosomes may mediate other systemic stresses.42 Our study also highlights the importance of preventing the emergence of drug‐tolerant cancer cells through inhibition of exosome secretion. Conceivably, attenuating the acquisition of drug tolerance along with chemotherapy would be an effective way to prevent treatment failure and relapse. To this end, combination therapy strategies, as opposed to sustained mono–therapy, should be used to prevent cancer cells from adapting to a drug‐tolerant state.39, 43
The critical role of exosomes in mediating cell‐cell communication with the TME has gained increasing attention. Functionally, exosomes have elaborate roles in cancer progression via transferring a variety of proteins, DNA and RNA.44, 45 Prior studies have described the role of exosomes in cancer progression and aggressiveness.46 The transport of exosomes is believed to be an effective means of modulating cell signaling and biological function between donor cells and recipient cells.47 Importantly, the ability of exosomes, derived from drug‐resistant cells, to transmit the resistant phenotype to drug‐sensitive cells has also been indicated as an important mechanism for transference of drug resistance, mainly through transferring of miRNA.48 However, the mechanisms underlying the contribution of exosomes to the development of transient drug tolerance remain poorly understood. Here, our data suggest that exosomes derived from cells under therapy stress transmit PI resistance to sensitive cells by facilitating cell cycle arrest and stemness pathway activation in MLL cells. We also found that the development of resistance conferred by exosomes could be inhibited by GW4869, possibly due to the blockade of exosome secretion by donor cells. GW4869 has already been used in vivo to inhibit exosome secretion in different disease models,49, 50 and combination treatment with bortezomib led to a significant effect on tumor load.51
In addition, our data showed that PI treatment induced the release of sEV in MLL cells, indicating a role of sEV in response to PI. Given the essential roles of sEV in the TME, we assumed that the induced sEV might change the microenvironment to favor tumor cell survival. We also observed that a very low concentration of PI‐treated cell‐derived exosomes was sufficient to resist drug‐induced apoptosis (Figure 3C). These results suggest that the cargos rather than the amount of exosomes play causal roles in the development of PI tolerance. Due to intratumor heterogeneity, whether the exosomes secreted by different cell subpopulations vary in amount and composition is largely unknown and requires further investigation.
Continued development of bioinformatic strategies allows for the integration of datasets, which will enable accurate analysis of the high‐throughput sequencing data and avoid perturbations in single data analysis as well. TieDIE analyses establish an integrative, pathway‐based reference for protein signaling regulation of aggressive MLL and are a valuable reference for further investigation. Specifically, our results revealed information potentially suitable for preclinical models to assess co–targeting or combination therapies. Future in vitro and in vivo experiments will be necessary for further validation of the therapeutic targets for overcoming aggressive acquisition of drug resistance that we present.
In conclusion, using integration analysis, our study demonstrated that exosomes derived from cells under therapy stress transmit PI tolerance to recipient cells through cell cycle arrest and enhanced stemness. Exosomes can act not only as a mediator of development of PI tolerance but also as a therapeutic target to overcome PI resistance, thereby enhancing the clinical benefits of PI therapy in MLL.
DISCLOSURE
The authors declare no competing financial interests.
Supporting information
FigS1‐S8
TableS1‐S3
ACKNOWLEDGMENTS
We thank Dr Shuhong Shen at Shanghai Jiao Tong University for provision of MLLr‐PDX. This work was supported by the National Key Research and Development Program of China (2018YFA0107802), the National Natural Science Foundation of China (81973996 and 81570119), the Shanghai Municipal Education Commission Gaofeng Clinical Medicine Grant (20161304), the Program of Shanghai Academic/Technology Research Leader (19XD1402500), the Shanghai Municipal Health Commission (2019CXJQ01), the Collaborative Innovation Center of Hematology and the Samuel Waxman Cancer Research Foundation.
Ge M, Qiao Z, Kong Y, Lu H, Liu H. Exosomes mediate intercellular transfer of non–autonomous tolerance to proteasome inhibitors in mixed‐lineage leukemia. Cancer Sci. 2020;111:1279–1290. 10.1111/cas.14351
Maolin Ge, Zhi Qiao and Yan Kong contributed equally to this work.
Contributor Information
Maolin Ge, Email: molin_ge@sjtu.edu.cn.
Han Liu, Email: liuhan68@sjtu.edu.cn.
DATA AVAILABILITY STATEMENT
RNA‐Seq data are available in the Gene Expression Omnibus (GEO) database under accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134879. The mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD014711.
REFERENCES
- 1. Muntean AG, Hess JL. The pathogenesis of mixed‐lineage leukemia. Annu Rev Pathol. 2012;7:283‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hong SK, Lee H, Kwon OS, et al. Large‐scale pharmacogenomics based drug discovery for ITGB3 dependent chemoresistance in mesenchymal lung cancer. Mol Cancer. 2018;17:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gettinger S, Choi J, Hastings K, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao Y, Lu Q, Li C, et al. PRMT1 regulates the tumour‐initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death Dis. 2019;10:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu H, Westergard TD, Cashen A, et al. Proteasome inhibitors evoke latent tumor suppression programs in pro–B MLL leukemias through MLL‐AF4. Cancer Cell. 2014;25:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neff T, Armstrong SA. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood. 2013;121:4847‐4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown R, Curry E, Magnani L, Wilhelm‐Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer. 2014;14:747‐753. [DOI] [PubMed] [Google Scholar]
- 8. Kartal‐Yandim M, Adan‐Gokbulut A, Baran Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol. 2016;36:716‐726. [DOI] [PubMed] [Google Scholar]
- 9. Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug‐induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee MC, Lopez‐Diaz FJ, Khan SY, et al. Single‐cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci USA. 2014;111:E4726‐E4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharma SV, Lee DY, Li B, et al. A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell. 2010;141:69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hrusovsky I, Emmerich B, von Rohr A, et al. Bortezomib retreatment in relapsed multiple myeloma ‐ results from a retrospective multicentre survey in Germany and Switzerland. Oncology. 2010;79:247‐254. [DOI] [PubMed] [Google Scholar]
- 13. Moreau P, Richardson PG, Cavo M, et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120:947‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petrucci MT, Giraldo P, Corradini P, et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br J Haematol. 2013;160:649‐659. [DOI] [PubMed] [Google Scholar]
- 15. Sun XX, Yu Q. Intra‐tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015;36:1219‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569‐579. [DOI] [PubMed] [Google Scholar]
- 17. Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome mediated communication within the tumor microenvironment. J Controlled Release. 2015;219:278‐294. [DOI] [PubMed] [Google Scholar]
- 18. Ludwig AK, Giebel B. Exosomes: small vesicles participating in intercellular communication. Int J Biochem Cell Biol. 2012;44:11‐15. [DOI] [PubMed] [Google Scholar]
- 19. Wang J, Hendrix A, Hernot S, et al. Bone marrow stromal cell‐derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014;124:555‐566. [DOI] [PubMed] [Google Scholar]
- 20. Qiao Z, Zhang Y, Ge M, et al. Cancer cell derived small extracellular vesicles contribute to recipient cell metastasis through promoting HGF/c‐met pathway. Mol Cell Proteomics. 2019;18:1619‐1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boelens MC, Wu TJ, Nabet BY, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shao H, Chung J, Lee K, et al. Chip‐based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat Commun. 2015;6:6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu H, Han H, Song S, et al. Exosome‐transmitted PSMA3 and PSMA3‐AS1 promote proteasome inhibitor resistance in multiple myeloma. Clin Cancer Res. 2019;25:1923‐1935. [DOI] [PubMed] [Google Scholar]
- 24. Drake JM, Paull EO, Graham NA, et al. Phosphoproteome integration reveals patient‐specific networks in prostate cancer. Cell. 2016;166:1041‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paull EO, Carlin DE, Niepel M, Sorger PK, Haussler D, Stuart JM. Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE). Bioinformatics. 2013;29:2757‐2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ge M, Luo Z, Qiao Z, et al. HERP binds TBK1 to activate innate immunity and repress virus replication in response to endoplasmic reticulum stress. J Immunol. 2017;199:3280‐3292. [DOI] [PubMed] [Google Scholar]
- 27. Khurana E, Fu Y, Chen J, Gerstein M. Interpretation of genomic variants using a unified biological network approach. PLoS Comput Biol. 2013;9:e1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li D, Hu Y, Jin Z, et al. TanCAR T cells targeting CD19 and CD133 efficiently eliminate MLL leukemic cells. Leukemia. 2018;32:2012‐2016. [DOI] [PubMed] [Google Scholar]
- 30. Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244‐1247. [DOI] [PubMed] [Google Scholar]
- 31. Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 2016;113:E968‐E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumbrink J, Kirsch KH. p130Cas acts as survival factor during PMA‐induced apoptosis in HL‐60 promyelocytic leukemia cells. Int J Biochem Cell Biol. 2013;45:531‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen JC, Alvarez MJ, Talos F, et al. Identification of causal genetic drivers of human disease through systems‐level analysis of regulatory networks. Cell. 2014;159:402‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adachi T, Sakurai T, Kashida H, et al. Involvement of heat shock protein a4/apg‐2 in refractory inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:31‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neri P, Ren L, Gratton K, et al. Bortezomib‐induced "BRCAness" sensitizes multiple myeloma cells to PARP inhibitors. Blood. 2011;118:6368‐6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palumbo A, Chanan‐Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754‐766. [DOI] [PubMed] [Google Scholar]
- 37. Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brauner A, Fridman O, Gefen O, Balaban NQ. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol. 2016;14:320‐330. [DOI] [PubMed] [Google Scholar]
- 39. Salgia R, Kulkarni P. The genetic/non–genetic duality of drug ‘resistance’ in cancer. Trends Cancer. 2018;4:110‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martin JD, Seano G, Jain RK. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu Rev Physiol. 2019;81:505‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burger JA, Gribben JG. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: insight into disease biology and new targeted therapies. Semin Cancer Biol. 2014;24:71‐81. [DOI] [PubMed] [Google Scholar]
- 42. Taylor RC, Berendzen KM, Dillin A. Systemic stress signalling: understanding the cell non–autonomous control of proteostasis. Nat Rev Mol Cell Biol. 2014;15:211‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldman A, Majumder B, Dhawan A, et al. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy‐induced phenotypic transition. Nat Commun. 2015;6:6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654‐659. [DOI] [PubMed] [Google Scholar]
- 45. Peinado H, Alec kovic M, Lavotshkin S, et al. Corrigendum: Melanoma exosomes educate bone marrow progenitor cells toward a pro–metastatic phenotype through MET. Nat Med. 2016;22:1502. [DOI] [PubMed] [Google Scholar]
- 46. Ela S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discovery. 2013;12:347‐357. [DOI] [PubMed] [Google Scholar]
- 47. Melo SA, Sugimoto H, O’Connell JT, et al. Cancer exosomes perform cell‐independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sousa D, Lima RT, Vasconcelos MH. Intercellular transfer of cancer drug resistance traits by extracellular vesicles. Trends Mol Med. 2015;21:595‐608. [DOI] [PubMed] [Google Scholar]
- 49. Nagashima S, Jirintai S, Takahashi M, et al. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J Gen Virol. 2014;95:2166‐2175. [DOI] [PubMed] [Google Scholar]
- 50. Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2014;35:1792‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Faict S, Muller J, De Veirman K, et al. Exosomes play a role in multiple myeloma bone disease and tumor development by targeting osteoclasts and osteoblasts. Blood Cancer J. 2018;8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigS1‐S8
TableS1‐S3
Data Availability Statement
RNA‐Seq data are available in the Gene Expression Omnibus (GEO) database under accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134879. The mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD014711.