

Abstract
Dysregulation or mutation of DNA binding proteins such as transcription factors (TFs) is associated with the onset and progression of various types of disease, including cancer. Alteration of TF activity occurs in numerous cancer tissues due to gene amplification, deletion, and point mutations, and epigenetic modification. Although cancer‐associated TFs are promising targets for cancer therapy, development of drugs targeting these TFs has historically been difficult due to the lack of high‐throughput screening methods. Recent advances in technology for identification and selective inhibition of DNA binding proteins enable cancer researchers to develop novel therapeutics targeting cancer‐associated TFs. In the present review, we summarize known cancer‐associated TFs according to cancer type and introduce recently developed high‐throughput approaches to identify selective inhibitors of cancer‐associated TFs.
Keywords: DNA binding protein, drug discovery, high‐throughput screening, telomere, transcription factor
Alteration of transcription factor (TF) activity occurs in numerous cancer tissues due to gene amplification, deletion, and point mutations, and epigenetic modification. The development of drugs targeting these TFs has historically been difficult due to the lack of high‐throughput screening methods. Recent advances in technology for identification and selective inhibition of DNA binding proteins enable cancer researchers to develop novel therapeutics targeting cancer‐associated TFs.

Abbreviations
- AEP
AF4 family/ENL family/P‐TEFb
- ARNT
aryl hydrocarbon receptor nuclear translocator
- BRD4
bromodomain‐containing protein 4
- CBP
CREB‐binding protein
- DUX4
double homeobox protein 4
- ETO
eight‐twenty one
- EWS
Ewing's sarcoma
- FLI1
friend leukemia virus integration 1
- HIF
hypoxia‐inducible factor
- MLL
mixed‐lineage leukemia
- SOX2
Sry‐related high‐mobility box 2
- STAT3
signal transducer and activator of transcription 3
- TF
transcription factor
- TRF
telomere‐repeat binding factor
1. INTRODUCTION
A number of DNA binding proteins, such as TFs, are critical regulators of gene expression. TFs generally regulate expression of their target genes through specific binding to the promoter or enhancer region of their targets and can either activate or suppress transcription of target genes. Transcription factors play important roles in various biological process such as stem cell maintenance and differentiation. 1 Therefore, the expression profiles of TFs differ between tissues and organs, and dysregulation of TFs occurs not only in cancer, but also in other diseases.
Recent progress in clinical sequencing studies has revealed that somatic mutations cause and promote tumor initiation and progression. Multiple studies reported that these alternations can affect transcriptional activity, resulting in cancer development. 2 Chromosomal abnormality also affects tumor malignancy, as these abnormalities frequently result in formation of fusion genes with TFs that activate oncogenic signaling pathways. 3
Because TF dysregulation is associated with tumor development in both blood tumors and solid tumors, TFs are a promising putative approach for development of anticancer agents that selectively inhibit activity of dysregulated TFs. Indeed, recent advances in technologies such as high‐throughput screening and protein knockdown methods have enabled identification and development of small molecule inhibitors against cancer‐associated TFs. In the present review, we summarize the roles of TFs in tumor development and introduce the recent advances in methodology for identification and development of small molecule inhibitors targeting cancer‐associated TFs.
2. CANCER‐ASSOCIATED TFS ARE A PROMISING TARGET FOR CANCER THERAPY
The first identified cancer‐associated TFs were fusion proteins in leukemia cells (Table 1). Accumulating lines of evidence have revealed that these fusion proteins function as drivers of disease onset and progression by inhibiting cell differentiation and maintaining a more stem cell‐like state. 4 , 5 Recently, cancer‐associated TFs that promote invasiveness and metastasis of solid tumors have also been identified (Table 1). In the subsequent section, we introduce the cancer‐associated TFs known to play important roles in blood cancers and solid tumors.
Table 1.
Cancer‐associated DNA binding proteins
| Cancer | DNA binding protein | Function | Reference |
|---|---|---|---|
| AML | MLL‐AEP | Immortalization of hematopoietic progenitors | 8 |
| AML | AML1‐ETO | G1 cell cycle progression | 34 |
| CML | Fox3a | Acquisition of resistance to imatinib | 59 |
| Breast cancer | c‐Myc | Upregulation of mitotic‐associated genes | 36 |
| Breast cancer | SOX2 | Induction of tamoxifen resistance | 37 |
| Melanoma | DUX4 | Immune invasion through the downregulation of MHC class I | 38 |
| Glioma | TRF1 | Telomeric damage and inhibition of CSC properties | 60 |
| Colon | TRF2 | Induction of endothelial cell differentiation and angiogenesis | 30 |
Each DNA binding protein is associated with tumor malignancies.
AEP, AF4 family/ENL family/P‐TEFb; CSC, cancer stem cell; DUX4, double homeobox protein 4; ETO, eight‐twenty one; MLL, mixed‐lineage leukemia; SOX2, Sry‐related high‐mobility box 2; TRF1, telomere‐repeat binding factor 1.
2.1. Cancer‐associated TFs in leukemia
Leukemia is classified into acute myeloid leukemia (AML), acute lymphocytic leukemia, Chronic myelogenous leukemia (CML), and chronic lymphocytic leukemia. Chromosomal rearrangements at 11q23 are observed in both pediatric and adult leukemias, which led to the identification of the mixed‐lineage leukemia gene (MLL). 6
Approximately 10% of acute leukemia patients harbor MLL translocations. 7 MLL belongs to a family of histone methyltransferases and specifically methylates histone H3 on lysine 4. MLL frequently fuses with a component of the AEP coactivator complex in acute leukemia, resulting in immortalization of hematopoietic progenitors through the aberrant activation of genes associated the hematopoietic stem cell program, such as HOXA9 and MEIS1. 8 , 9 Okuda et al 10 reported that AEP and MLL‐AEP fusion proteins induce transcriptional activation through physical interaction with selectivity factor 1, which is a core component of the RNA polymerase I preinitiation complex. Acute myeloid leukemia 1 (AML1/RUNX1) is a TF and forms a fusion protein with the ETO gene, which is involved in transcriptional repression. 11 The AML1‐ETO fusion protein is associated with development of acute leukemia by upregulating a number of genes such as SOX4, IL‐17BR, CD200, and γ‐catenin. 12 , 13 A recent study reported that AML1‐ETO cooperates with AP‐1 to drive cyclin D2 expression, resulting in G1 cell cycle progression and leukemic propagation. 4
Chronic myelogenous leukemia is (CML) caused by a genetic abnormality that leads to generation of BCR‐ABL, a constitutively active tyrosine kinase. Therefore, the tyrosine kinase inhibitor imatinib was a breakthrough CML therapy. However, Naka et al 14 reported that initiating cells in CML are resistant to imatinib. In a mouse model used in this study, treatment of leukemia‐initiating cells with transforming growth factor‐β induced nuclear localization of Foxo3a through Akt activation, leading to development of imatinib resistance.
2.2. Cancer‐associated TFs in solid tumors
Recent studies reported that TF are also dysregulated in solid cancers, which results in acquisition of tumor malignancies such as cell proliferation, drug resistance, invasiveness, immune evasion, and metastasis (Table 1). c‐Myc is an important cancer‐associated TF and promotes tumor cell growth and proliferation by regulating numerous target genes such as Cdc25A, hTERT, and glutamine synthetase. 15 , 16 , 17 Santoro et al 18 reported that p53 loss induces c‐Myc activation in breast tumors. Although c‐Myc is important for symmetric cell division of mammary stem cells and induces reprogramming of progenitor cells to stem cells in a mouse model, concomitant p53 loss and c‐Myc activation upregulates expression of 189 mitosis‐associated genes, resulting in the expansion of cancer stem cells in human breast cancer cells.
Sry‐related high‐mobility box 2 is a pluripotency‐associated TF and is essential for the maintenance of stemness. Therefore, SOX2 plays important roles in regulating developmental processes. 1 Aberrant SOX2 expression is present in many cancers and promotes tumor seeding ability and drug resistance. For example, in breast cancer cells, SOX2 suppression induces sensitization to tamoxifen by activating Wnt‐signaling pathway‐related genes such as DKK1 and AXIN2. 19 , 20
Immune checkpoint blockade against T‐cell inhibitory receptors such as CTL‐associated protein‐4 (CTLA‐4) and PD‐1 is considered to be one of the most effective approaches across diverse cancers. 21 However, it is difficult to predict patient response to these approaches during cancer treatment. Therefore, it is critical to identify genes that modulate antigen presentation and tumor‐immune interaction. Chew et al 22 reported that the early embryonic TF DUX4, which is silenced in somatic tissues, is reexpressed in diverse solid cancers, resulting in suppression of MHC class I. Therefore, DUX4 inhibits T cell recognition of cancer cells. Consistent with these findings, low DUX4 expression is correlated with progression‐free and overall survival in response to anti‐CTLA‐4 therapy in metastatic melanoma patients.
Nuclear receptors are also important TFs and known to function as a critical regulator in cancer biology. 23 Estrogen receptor (ER) is one of the nuclear receptors and associated with luminal type breast cancer. 24 Therefore, ER is important to determine the subtypes of breast cancer and therapeutic approaches. As the expression level of ER is transcriptionally regulated by ER factor‐1, 25 ER factor‐1 is also considered to regulate the gene expression that is characteristic of the luminal type breast cancer phenotype.
2.3. Telomeric DNA binding protein
In addition to cancer‐associated TFs, telomere binding proteins are considered to be viable therapeutic targets, as these proteins are associated with tumor malignancies such as tumorigenesis and proliferation. 26 Mammalian telomeres are comprised of a double‐stranded TTAGGG‐repeat tract terminating in a 3′ single‐stranded overhang that forms a T‐loop with a specialized protein complex known as the shelterin complex. The shelterin complex is comprised of six proteins, including TRF1, TRF2, POT1, RAP1, TIN2, and TPP1. Among them, TRF1, TRF2, and POT1 are telomeric DNA binding proteins. The telomere‐repeat binding factors TRF1 and TRF2 bind duplex TTAGGG repeats to stabilize telomeric DNA.
Bejarano et al 26 reported that TRF1 is highly expressed in mouse and human glioblastoma multiforme (GBM). In GBM mouse models, brain‐specific Trf1 genetic deletion efficiently inhibits GBM initiation and progression, improving survival rate. Importantly, TRF1 small molecule inhibitors have similar effects in human GBM cell lines and xenografts generated from patient‐derived primary glioblastoma CSCs .
Because TRF2 expression is elevated in several types of cancers, including breast, liver, lung, and colon cancer, TRF2 is considered to be associated with tumorigenesis. 27 , 28 , 29 , 30 Blanco et al 31 used telomerase‐deficient mice that also expressed TRF2 under the keratin 5 promoter to demonstrate that telomerase deficiency promotes TRF2‐mediated epithelial carcinogenesis. These findings suggest that TRF2 inhibition could be an effective therapeutic approach for treatment of telomerase‐deficient cancers.
The 3′ overhang of human telomeres forms tetra‐stranded DNA structures known as G‐quadruplexes, which are important for elongation of telomeric DNA by telomerase. 32 Because stabilization of G‐quadruplexes has the potential to inhibit telomere replication by interfering with the telomerase‐mediated elongation step, small molecules that stabilize G‐quadruplexes are promising agents for cancer therapy. Consistent with these findings, the G‐quadruplex stabilizer telomestatin induces tumor suppression by promoting dissociation of TRF2 from the telomeres. 33
3. CLINICAL EVALUATION OF SMALL MOLECULE INHIBITORS TARGETING DNA BINDING PROTEINS
Several inhibitors of DNA binding proteins have been evaluated in clinical trials (Table 2). 34 , 35 , 36 , 37 , 38 Signal transducer and activator of transcription 3 plays key roles in multiple cancer‐related signaling pathways and is aberrantly expressed in various human cancers. 39 Activation of STAT3 is associated with cancer stem cell properties such as tumor seeding ability and drug resistance. 40 , 41 The small molecule napabucasin (BBI608) was identified as a STAT3 inhibitor that targets cancer stem cells. 35 Combination treatment with napabucasin and conventional chemotherapy is under evaluation in clinical trials, particularly for advanced cancer patients 42 (Table 2).
Table 2.
Clinical evaluation of cancer‐associated transcription factor small molecule inhibitors
| Target DNA binding protein | Inhibitor name | Company | Mode of action | Clinical trial no. | Reference |
|---|---|---|---|---|---|
| STAT3 | Napabucasin | Boston Biomedical | Inhibition of target genes driven by STAT3 | NCT02753127 | 35 |
| CBP/β‐catenin | E7386 | Eisai | Inhibition of Wnt target genes through modulation of β‐catenin/CBP interaction | NCT04008797 | 34 |
| HIF2α | PT2385 | Peloton Therapeutics | Allosteric inhibition of the dimerization of HIF2α with ARNT | NCT02293980 | 59 |
| NF‐κB and GATA3 | MLN9708 | Millennium Pharmaceuticals | Proteasome inhibitor targeting NF‐κB and GATA3 | NCT02181413 | 36 |
| BRD4 | AZD5153 | AstraZeneca | Disruption of the chromatin binding activity of bromodomain‐containing protein 4 | NCT03205176 | 37 |
| EWS‐FLI1 | TK216 | Oncternal Therapeutics | Blocking of the physical interaction of E26 transformation‐specific transcription factors with RNA helicases | NCT02657005 | 38 |
| MDM2 | BI‐907828 | Boehringer Ingelheim | Inhibition of physical interaction between MDM2 and p53 | NCT03449381 | 60 |
DNA binding proteins, including fusion genes, are promising targets for cancer therapy and are currently being evaluated in clinical trials.
ARNT, aryl hydrocarbon receptor nuclear translocator; BRD4, bromodomain‐containing protein 4; CBP, CREB‐binding protein; EWS, Ewing's sarcoma; FLI1, friend leukemia virus integration 1; HIF‐2α, hypoxia‐inducible factor‐2α; MDM2, murine double minute homolog 2; NF‐κB, nuclear factor‐κB; STAT3, signal transducer and activator of transcription 3.
Hypoxia‐inducible factors are a family of TFs consisting of HIF1α, HIF2α, and HIF3α. 43 , 44 , 45 The HIF proteins are stabilized and localized to the nucleus under hypoxic conditions. In the nucleus, HIF proteins heterodimerize with ARNT (also known as HIFβ). The heterodimer binds to hypoxia‐responsive elements of target genes involved in redox homeostasis, metabolism, angiogenesis, tumorigenesis, and inflammation. In more than 90% of clear cell renal cell carcinoma (ccRCC), HIF2α is aberrantly stabilized and is associated with malignant phenotypes. 46 Wallace et al 47 developed the small molecule PT2385 as a specific antagonist of HIF2α, which allosterically inhibits dimerization of HIF2α with ARNT. This study indicated that PT2385 induces tumor regression in an animal model, and that HIF2α functions as a pivotal oncogenic driver in ccRCC. PT2385 is now under evaluation in clinical trials (Table 2). Therefore, targeting HIF2α is a promising approach for treatment of ccRCC.
4. DEVELOPMENT OF HIGH‐THROUGHPUT SCREENING METHODS
Although DNA binding proteins are attractive therapeutic targets for cancer therapy, efficiently identifying small molecules that selectively target cancer‐associated DNA binding proteins remains problematic. Conventional methods used to evaluate interactions between DNA and protein, such as EMSA and ELISA, are not suitable for high‐throughput screening. Therefore, a number of studies sought to develop high‐throughput screening methods based on reporter assays, EMSA, fluorescence polarization assays, and luminescence‐based binding assay, enabling rapid and high‐throughput identification of small molecule inhibitors of cancer‐associated DNA binding proteins. 48 , 49 , 50 , 51 , 52 , 53
4.1. Microfluidic‐based EMSA (QPID) assay
Electrophoretic mobility‐shift assay is considered a gold standard method to analyze and measure the binding affinity between a DNA binding protein and its target sequence. 54 Combining the EMSA assay with microfluidics technology facilitated more rapid and quantitative performance of the EMSA. Glick et al 51 succeeded in developing a high‐throughput microfluidic platform termed quantitative protein interaction with DNA (QPID). This is an integrated microfluidic‐based assay with a DNA microarray that enables analysis of 4096 samples and calculation of the binding affinity of DNA binding proteins to their target sequences in a single run (Figure 1). The interaction of cyclic AMP‐dependent transcription factor 1 (ATF1) and ATF3 with 128 genomic cAMP response elements was examined using this platform. The study revealed that the difference in DNA binding affinity between ATF1 and ATF3 was due to a minor groove width of genomic DNA.
Figure 1.
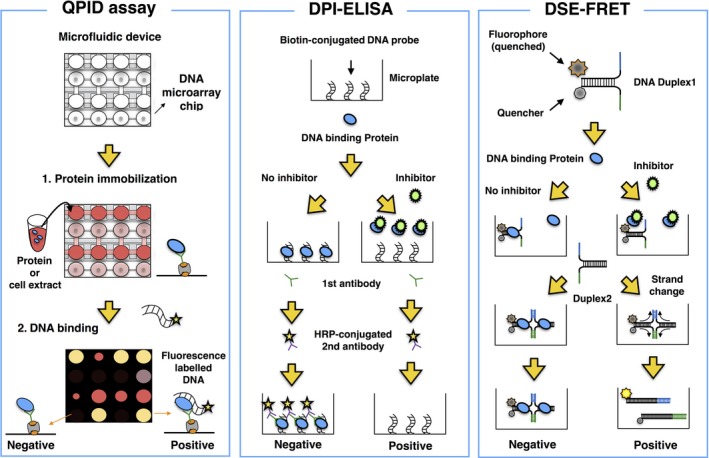
High‐throughput screening methods for the identification of small inhibitors targeting DNA binding proteins. Left panel (quantitative protein interaction with DNA [QPID] assay): integrated microfluidic‐based assay with a DNA microarray to investigate the physical interactions of DNA binding proteins with their target sequences in high‐throughput screening. Middle panel (DNA‐protein interaction [DPI]‐ELISA): high‐throughput ELISA for identification of small molecules that inhibit the binding of DNA binding proteins to DNA sequences. Right panel (DNA strand exchange‐fluorescence resonance energy transfer [DSE‐FRET]): FRET‐based assay for identification of small compounds that inhibit the interaction of DNA binding proteins with DNA oligos, which leads to DNA strand exchange
4.2. DNA‐protein interaction‐ELISA
The ELISA is a well‐known and useful method for detection of target proteins such as antigens and Abs, quantifying relative amounts of the target protein based on an enzymatic reaction. Brand et al 48 developed a novel method based on the ELISA termed DPI‐ELISA, in which biotin‐labelled oligonucleotides are conjugated to streptavidin‐coated microplates. The DPI‐ELISA enables analysis of DNA‐protein interactions in multiple samples in a single run (Figure 1). Using the DPI‐ELISA method, Alonso et al 49 identified netropsin as a specific inhibitor of mammalian high‐mobility group protein AT‐hook 2 (HMGA2), which is associated with metastasis in several types of cancers. 55 , 56 , 57
4.3. DNA strand exchange‐fluorescence resonance energy transfer
To investigate the interactions between DNA binding proteins and their target sequences more simply and rapidly, Miyagi et al 50 developed a novel method based on 2 phenomena: DNA binding protein‐dependent inhibition of spontaneous DSE between partially double‐stranded DNA probes, and FRET (Figure 1). For the DSE‐FRET assay, 2 types of oligonucleotides are used. One is a quencher‐conjugated oligo, and the other is a fluorescence‐labelled oligo. During the interaction of the DNA binding protein with these 2 oligonucleotides, strand exchange is inhibited. Therefore, after inhibition of the interaction of the DNA binding protein with the oligonucleotides, fluorescence is detected.
4.4. Amplified luminescent proximity homogeneous assay
An amplified luminescent proximity homogeneous assay is based on a luminescent oxygen‐channeling chemistry. 58 In this assay, an analyte is sandwiched by a biotinylated Ab conjugated to streptavidin‐coated donor beads and a second Ab conjugated to acceptor beads. Donor beads are located in proximity to acceptor beads through the binding of these Abs to the analyte. After the excitation of donor beads at 680 nm, singlet oxygen is transferred from donor beads to acceptor beads, which results in the chemiluminescent emission at 615 nm. Using this system, Nomura et al 53 established a high‐throughput screening system and identified pyrrothiogatain as a novel inhibitor of GATA3 DNA‐binding activity, which is important for T helper 2 cell differentiation.
5. CONCLUDING REMARKS
Recent progress in technologies such as DPI‐ELISA and DSE‐FRET has enabled high‐throughput identification of small molecules that selectively target disease‐associated DNA binding proteins using chemical libraries. Therefore, cancer‐associated TFs are no longer considered undruggable targets. Although numerous challenges for pharmaceutical targeting of cancer‐associated TFs remain, this is a promising approach for treatment of diverse cancers. These approaches are particularly well‐suited to identifying cancer‐associated TF inhibitors because of sequence specificity and affinity of TFs, which could improve the selective inhibition of the target proteins. Using these technologies, a novel platform could also be developed for identification of small molecule inhibitors of cancer‐associated RNA‐binding proteins, which also play important roles in cancer development and progression. Furthermore, combination treatment of these inhibitors with conventional cancer treatments will contribute positively to the clinical outcomes of cancer patients.
DISCLOSURE
Professor Hidetoshi Tahara is a founder, stock owner, and board director of MiRTeL Co. Ltd. HT owns stock in MiRTeL. Family member Kanoko Tahara is an employee of MiRTeL Co. Ltd. The other authors have no conflicts of interest.
ACKNOWLEDGMENTS
This study was undertaken as part of the Project for Development of Innovative Research on Cancer Therapeutics (P‐Direct), Ministry of Education, Culture, Sports, Science and Technology of Japan.
Shiroma Y, Takahashi R‐U, Yamamoto Y, Tahara H. Targeting DNA binding proteins for cancer therapy. Cancer Sci. 2020;111:1058–1064. 10.1111/cas.14355
Yoshitomo Shiroma and Ryou‐u Takahashi contributed equally to this work.
REFERENCES
- 1. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 2. Dang CV. MYC on the path to cancer. Cell. 2012;149:22‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yokoyama A. Transcriptional activation by MLL fusion proteins in leukemogenesis. Exp Hematol. 2017;46:21‐30. [DOI] [PubMed] [Google Scholar]
- 4. Martinez‐Soria N, McKenzie L, Draper J, et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell. 2018;34(626–642):e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu B, Klingbeil O, Tarumoto Y, et al. A transcription factor addiction in leukemia imposed by the MLL promoter sequence. Cancer Cell. 2018;34(970–981):e978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Djabali M, Selleri L, Parry P, Bower M, Young BD, Evans GA. A trithorax‐like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet. 1992;2:113‐118. [DOI] [PubMed] [Google Scholar]
- 7. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem‐cell development. Nat Rev Cancer. 2007;7:823‐833. [DOI] [PubMed] [Google Scholar]
- 8. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher‐order complex containing AF4 and ENL family proteins with P‐TEFb facilitates oncogenic and physiologic MLL‐dependent transcription. Cancer Cell. 2010;17:198‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL‐AF9. Nature. 2006;442:818‐822. [DOI] [PubMed] [Google Scholar]
- 10. Okuda H, Kanai A, Ito S, Matsui H, Yokoyama A. AF4 uses the SL1 components of RNAP1 machinery to initiate MLL fusion‐ and AEP‐dependent transcription. Nat Commun. 2015;6:8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ajore R, Dhanda RS, Gullberg U, Olsson I. The leukemia associated ETO nuclear repressor gene is regulated by the GATA‐1 transcription factor in erythroid/megakaryocytic cells. BMC Mol Biol. 2010;11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci U S A. 1991;88:10431‐10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tonks A, Pearn L, Musson M, et al. Transcriptional dysregulation mediated by RUNX1‐RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia. 2007;21:2495‐2505. [DOI] [PubMed] [Google Scholar]
- 14. Naka K, Hoshii T, Muraguchi T, et al. TGF‐beta‐FOXO signalling maintains leukaemia‐initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676‐680. [DOI] [PubMed] [Google Scholar]
- 15. Galaktionov K, Chen X, Beach D. Cdc25 cell‐cycle phosphatase as a target of c‐myc. Nature. 1996;382:511‐517. [DOI] [PubMed] [Google Scholar]
- 16. Wu KJ, Grandori C, Amacker M, et al. Direct activation of TERT transcription by c‐MYC. Nat Genet. 1999;21:220‐224. [DOI] [PubMed] [Google Scholar]
- 17. Bott AJ, Peng IC, Fan Y, et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22:1068‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santoro A, Vlachou T, Luzi L, et al. p53 loss in breast cancer leads to Myc activation, increased cell plasticity, and expression of a mitotic signature with prognostic value. Cell Rep. 2019;26(624–638):e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yin Y, Xie CM, Li H, et al. The FBXW2‐MSX2‐SOX2 axis regulates stem cell property and drug resistance of cancer cells. Proc Natl Acad Sci U S A. 2019;116:20528‐20538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piva M, Domenici G, Iriondo O, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chew GL, Campbell AE, De Neef E, et al. DUX4 suppresses MHC Class I to promote cancer immune evasion and resistance to checkpoint blockade. Dev Cell. 2019;50(658–671):e657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bushweller JH. Targeting transcription factors in cancer ‐ from undruggable to reality. Nat Rev Cancer. 2019;19:611‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747‐752. [DOI] [PubMed] [Google Scholar]
- 25. deConinck EC, McPherson LA, Weigel RJ. Transcriptional regulation of estrogen receptor in breast carcinomas. Mol Cell Biol. 1995;15:2191‐2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bejarano L, Schuhmacher AJ, Mendez M, et al. Inhibition of TRF1 telomere protein impairs tumor initiation and progression in glioblastoma mouse models and patient‐derived xenografts. Cancer Cell. 2017;32(590–607):e594. [DOI] [PubMed] [Google Scholar]
- 27. Nijjar T, Bassett E, Garbe J, et al. Accumulation and altered localization of telomere‐associated protein TRF2 in immortally transformed and tumor‐derived human breast cells. Oncogene. 2005;24:3369‐3376. [DOI] [PubMed] [Google Scholar]
- 28. Wu M, Lin Z, Li X, et al. HULC cooperates with MALAT1 to aggravate liver cancer stem cells growth through telomere repeat‐binding factor 2. Sci Rep. 2016;6:36045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lantuejoul S, Raynaud C, Salameire D, et al. Telomere maintenance and DNA damage responses during lung carcinogenesis. Clin Cancer Res. 2010;16:2979‐2988. [DOI] [PubMed] [Google Scholar]
- 30. Zizza P, Dinami R, Porru M, et al. TRF2 positively regulates SULF2 expression increasing VEGF‐A release and activity in tumor microenvironment. Nucleic Acids Res. 2019;47:3365‐3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blanco R, Munoz P, Flores JM, Klatt P, Blasco MA. Telomerase abrogation dramatically accelerates TRF2‐induced epithelial carcinogenesis. Genes Dev. 2007;21:206‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han H, Hurley LH. G‐quadruplex DNA: a potential target for anti‐cancer drug design. Trends Pharmacol Sci. 2000;21:136‐142. [DOI] [PubMed] [Google Scholar]
- 33. Tahara H, Shin‐Ya K, Seimiya H, Yamada H, Tsuruo T, Ide T. G‐Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3' telomeric overhang in cancer cells. Oncogene. 2006;25:1955‐1966. [DOI] [PubMed] [Google Scholar]
- 34. Katoh M. Multilayered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/betacatenin signaling activation (Review). Int J Mol Med. 2018;42:713‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Y, Rogoff HA, Keates S, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A. 2015;112:1839‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boonstra PS, Polk A, Brown N, et al. A single center phase II study of ixazomib in patients with relapsed or refractory cutaneous or peripheral T‐cell lymphomas. Am J Hematol. 2017;92:1287‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bradbury RH, Callis R, Carr GR, et al. Optimization of a Series of Bivalent Triazolopyridazine Based Bromodomain and Extraterminal Inhibitors: The Discovery of (3R)‐4‐[2‐[4‐[1‐(3‐Methoxy‐[1,2,4]triazolo[4,3‐b]pyridazin‐6‐yl)‐4‐piperidyl]phen oxy]ethyl]‐1,3‐dimethyl‐piperazin‐2‐one (AZD5153). J Med Chem. 2016;59:7801‐7817. [DOI] [PubMed] [Google Scholar]
- 38. Spriano F, Chung EYL, Gaudio E, et al. The ETS Inhibitors YK‐4‐279 and TK‐216 are novel antilymphoma agents. Clin Cancer Res. 2019;25:5167‐5176. [DOI] [PubMed] [Google Scholar]
- 39. Bai L, Zhou H, Xu R, et al. A potent and selective small‐molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36(498–511):e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang W, Hu H, Zhang Q, et al. Regulatory networks in mechanotransduction reveal key genes in promoting cancer cell stemness and proliferation. Oncogene. 2019;38:6818‐6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Niu J, Sun Y, Chen B, et al. Fatty acids and cancer‐amplified ZDHHC19 promote STAT3 activation through S‐palmitoylation. Nature. 2019;573:139‐143. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Shitara K, Yodo Y, Iino S. A Phase I study of napabucasin plus paclitaxel for Japanese patients with advanced/recurrent gastric cancer. Vivo. 2019;33:933‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72‐82. [DOI] [PubMed] [Google Scholar]
- 44. Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N. Expression and characterization of hypoxia‐inducible factor (HIF)‐3alpha in human kidney: suppression of HIF‐mediated gene expression by HIF‐3alpha. Biochem Biophys Res Commun. 2001;287:808‐813. [DOI] [PubMed] [Google Scholar]
- 45. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447‐5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear‐cell renal cell carcinoma. Nat Genet. 2013;45:860‐867. [DOI] [PubMed] [Google Scholar]
- 47. Wallace EM, Rizzi JP, Han G, et al. A small‐molecule antagonist of HIF2alpha is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 2016;76:5491‐5500. [DOI] [PubMed] [Google Scholar]
- 48. Brand LH, Kirchler T, Hummel S, Chaban C, Wanke D. DPI‐ELISA: a fast and versatile method to specify the binding of plant transcription factors to DNA in vitro. Plant Methods. 2010;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alonso N, Guillen R, Chambers JW, Leng F. A rapid and sensitive high‐throughput screening method to identify compounds targeting protein‐nucleic acids interactions. Nucleic Acids Res. 2015;43:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miyagi T, Shiotani B, Miyoshi R, et al. DSE‐FRET: a new anticancer drug screening assay for DNA binding proteins. Cancer Sci. 2014;105:870‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Glick Y, Orenstein Y, Chen D, et al. Integrated microfluidic approach for quantitative high‐throughput measurements of transcription factor binding affinities. Nucleic Acids Res. 2016;44:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Salcher S, Spoden G, Hagenbuchner J, et al. A drug library screen identifies Carbenoxolone as novel FOXO inhibitor that overcomes FOXO3‐mediated chemoprotection in high‐stage neuroblastoma. Oncogene. 2019;39:1080‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nomura S, Takahashi H, Suzuki J, Kuwahara M, Yamashita M, Sawasaki T. Pyrrothiogatain acts as an inhibitor of GATA family proteins and inhibits Th2 cell differentiation in vitro. Sci Rep. 2019;9:17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Miki T, Matsumoto T, Zhao Z, Lee CC. p53 regulates Period2 expression and the circadian clock. Nat Commun. 2013;4:2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dong J, Wang R, Ren G, et al. HMGA2‐FOXL2 axis regulates metastases and epithelial‐to‐mesenchymal transition of chemoresistant gastric cancer. Clin Cancer Res. 2017;23:3461‐3473. [DOI] [PubMed] [Google Scholar]
- 56. Chiou SH, Dorsch M, Kusch E, et al. Hmga2 is dispensable for pancreatic cancer development, metastasis, and therapy resistance. Sci Rep. 2018;8:14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gao X, Dai M, Li Q, Wang Z, Lu Y, Song Z. HMGA2 regulates lung cancer proliferation and metastasis. Thorac Cancer. 2017;8:501‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ullman EF, Kirakossian H, Switchenko AC, et al. Luminescent oxygen channeling assay (LOCI): sensitive, broadly applicable homogeneous immunoassay method. Clin Chem. 1996;42:1518‐1526. [PubMed] [Google Scholar]
- 59. Courtney KD, Ma Y, Diaz de Leon A, et al. HIF‐2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first‐in‐class HIF‐2 inhibitor in clear cell renal cell carcinoma patients. Clin Cancer Res. 2020;26:793‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cornillie J, Wozniak A, Li H, et al. Anti‐tumor activity of the MDM2‐TP53 inhibitor BI‐907828 in dedifferentiated liposarcoma patient‐derived xenograft models harboring MDM2 amplification. Clin Transl Oncol. 2019. 10.1007/s12094-019-02158-z [DOI] [PubMed] [Google Scholar]