

Abstract
Hepatocyte growth factor activator inhibitor‐1 (HAI‐1), encoded by the SPINT1 gene, is a membrane‐bound protease inhibitor expressed on the surface of epithelial cells. Hepatocyte growth factor activator inhibitor‐1 regulates type II transmembrane serine proteases that activate protease‐activated receptor‐2 (PAR‐2). We previously reported that deletion of Spint1 in Apc Min/+ mice resulted in accelerated formation of intestinal tumors, possibly through enhanced nuclear factor‐κB signaling. In this study, we examined the role of PAR‐2 in accelerating tumor formation in the Apc Min/+ model in the presence or absence of Spint1. We observed that knockout of the F2rl1 gene, encoding PAR‐2, not only eliminated the enhanced formation of intestinal tumors caused by Spint1 deletion, but also reduced tumor formation in the presence of Spint1. Exacerbation of anemia and weight loss associated with HAI‐1 deficiency was also normalized by compound deficiency of PAR‐2. Mechanistically, signaling triggered by deregulated protease activities increased nuclear translocation of RelA/p65, vascular endothelial growth factor expression, and vascular density in Apc Min/+‐induced intestinal tumors. These results suggest that serine proteases promote intestinal carcinogenesis through activation of PAR‐2, and that HAI‐1 plays a critical tumor suppressor role as an inhibitor of matriptase, kallikreins, and other PAR‐2 activating proteases.
Keywords: angiogenesis, colon cancer, HAI‐1, PAR‐2, Spint1
Hepatocyte growth factor activator inhibitor‐1 insufficiency induces protease‐activated receptor‐2 activation, resulting in tumor promotion through nuclear factor‐κB activation and tumor angiogenesis

1. INTRODUCTION
Hepatocyte growth factor activator inhibitor‐1 (HAI‐1)/serine peptidase inhibitor, Kunitz type 1 (SPINT1), encoded by the human SPINT1 gene, is a membrane‐associated Kunitz‐type serine protease inhibitor that is expressed by most epithelial cells and placental cytotrophoblasts.1 It was initially identified as a cellular inhibitor of serum hepatocyte growth factor activator and subsequent studies have indicated that HAI‐1 is also a critical regulator of epithelial type II transmembrane serine proteases (TTSPs), particularly matriptase.2, 3 Previously, we generated several Spint1 mutant mouse models and showed that HAI‐1 contributes to the integrity of epithelia, including the intestinal epithelium.2, 4 Intestine‐specific Spint1 knockout mice showed increased susceptibility to dextran sulfate sodium‐induced experimental colitis.4 Moreover, the Spint1 deletion led to accelerated tumor formation in the Apc Min/+ mouse model, indicating that HAI‐1 is a tumor suppressor.5 Indeed, the cell surface immunoreactivity of HAI‐1 was markedly reduced in carcinoma cells compared to adjacent normal enterocytes or adenoma cells in human colon cancers,6, 7 and this trend was confirmed in a murine Apc Min/+ model.5 Consequently, the ratio of HAI‐1 expression relative to its target epithelial protease, matriptase, was decreased along with the progression of colon cancers.8 Enhanced tumor formation observed in HAI‐1‐deficient Apc Min/+ mice was mediated, at least partly, by activation of nuclear factor (NF)‐κB signaling9; however, detailed mechanisms underlying NF‐κB activation in the absence of HAI‐1 remain unclear.
Protease‐activated receptor‐2 (PAR‐2) is a 7 transmembrane‐spanning domain G protein‐coupled receptor widely expressed by epithelial, endothelial, and smooth muscle cells, with diverse physiological and pathological functions.10, 11, 12, 13 Specific cleavage by trypsin‐like serine proteases frees an endogenous ligand for interaction with the core of the receptor, inducing a conformational change that triggers signal transduction.10, 14, 15 Type II transmembrane serine proteases are known to activate PAR‐216, 17, 18, 19, 20, 21 and PAR‐2 reportedly contributes to tumor progression through its promotion of invasive growth by cancer cells and by stimulating angiogenesis.11, 19, 22 Among TTSPs, matriptase is the most potent PAR‐2 activator known in epithelial and tumor tissues.22, 23 Transgenic expression of matriptase in murine keratinocytes induces skin carcinogenesis24 that is entirely dependent on PAR‐2 signaling.25 Activation of PAR‐2 by transgenic matriptase expression led to protumorigenic cytokine expression through activation of NF‐κB signaling.25 Previously, we reported that the loss of intestinal HAI‐1/Spint1 led to increased trypsin‐like serine protease activity in Apc Min/+ mouse intestine.9 We thus hypothesize that HAI‐1 insufficiency permits the unrestricted activity of pericellular trypsin‐like serine proteases, including matriptase, leading to activation of PAR‐2/NF‐κB signaling in colon cancer tissues.
To test this hypothesis, we generated Apc Min/+ mice with an intestine‐specific Spint1‐deletion with or without the superimposition of global PAR‐2/F2rl1 deletion to analyze the effects of PAR‐2 signaling on intestinal carcinogenesis and on the enhanced tumor susceptibility induced by HAI‐1 deficiency. We found that the deletion of F2rl1 reduced tumor formation both in control and Spint1‐deleted Apc Min/+ mice and decreased the activation of NF‐κB and angiogenesis in HAI‐1 deficient tumors.
2. MATERIALS AND METHODS
2.1. Mice
All animal experiments were carried out using protocols approved by the Institutional Animal Care and Use Committee of the University of Miyazaki. Apc Min/+ mice and Villin‐Cre mice were obtained from The Jackson Laboratory. Apc Min/+ Spint1LoxP/LoxP/Villin‐Cre mice4, 5 were crossed with F2rl1‐deficient mice26 to generate Apc Min/+ mice with an intestine‐specific Spint1 deletion and global PAR‐2/F2rl1 deletion (Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+). Mice were assessed daily, and body weights were recorded weekly. Blood samples were obtained from the right ventricle, and EDTA‐containing plasma samples were used for analyzing hemoglobin concentration.
At 15 weeks of age, all mice were killed to evaluate the number and sizes of intestinal tumors. The tumor size was scored as previously described.5 For histological analysis, intestinal tissues were fixed in 4% paraformaldehyde in PBS and embedded in paraffin. Four‐micrometer‐thick sections were stained with H&E or processed for immunohistochemical analysis.
2.2. Quantitative RT‐PCR
Total RNA was prepared with TRIzol (Life Technologies Japan), followed by DNase I (Takara Bio) treatment. For RT‐PCR, 3 µg total RNA was reverse‐transcribed with a mixture of Oligo (dT)12‐18 (Life Technologies Japan) and random primers (6 mers) (Takara Bio) using 200 units of ReverTra Ace (Toyobo), and 1/30 of the resulting cDNA was processed for quantitative RT‐PCR. Real‐time RT‐PCR was undertaken in a Thermal Cycler Dice Real Time System II (Takara Bio) using the SYBR Premix Ex Taq II (Takara Bio). For internal control, β‐actin mRNA was also measured. The following primers were used: β‐actin forward, 5′‐TGACAGGATGCAGAAGGAGA, and reverse, 5′‐GCTGGAAGGTGGACAGTGAG; Pecam1 (CD31) forward, 5′‐GGAAGCCAACAGCCATTACGG, and reverse, 5′‐GAGCCTTCCGTTCTCTTGGTGA; Vegfa forward, 5′‐CAGGCTGCTGTAACGATGAA, and reverse, 5′‐CTGCATTCACATCTGCTGTG; Bcl2 (Bcl‐2) forward, 5′‐ACCGTCGTGACTTCGCAGAG, and reverse, 5′‐GGTGTGCAGATGCCGGTTCA; and Bad forward, 5′‐GCCCTAGGCTTGAGGAAGTC, and reverse, 5′‐CAAACTCTGGGATCTGGAACA.
2.3. Immunohistochemical analyses
For immunohistochemistry, tissue sections were processed for antigen retrieval by microwaving for 10 minutes at 96°C in 10 mmol/L citrate buffer (pH 6.0), followed by treatment with 3% H2O2 in PBS for 10 minutes. After blocking in 5% normal goat serum (Dako) in PBS, the sections were incubated with anti‐NF‐κB p65 (RelA/p65) rabbit mAb (Cell Signaling Technology) or anti‐CD31 rabbit polyclonal Ab (Cell Signaling Technology), or antiphosphorylated MET (Y1235) rabbit polyclonal Ab,27 or anti‐β‐catenin rabbit polyclonal Ab (Sigma‐Aldrich) or anti‐CD45 rat mAb (Wuxi Biosciences) for 16 hours at 4°C and then incubated with Envision labeled polymer reagents (Dako) for 30 minutes at room temperature. The reactions were revealed by nickel and cobalt‐3,3′‐diaminobenzidine (Pierce) and counterstained with Mayer’s hematoxylin. To quantify RelA/p65 nuclear translocation and CD31+ vessels, stained sections with nonneoplastic mucosa and tumor tissues were selected and photographed at 200× magnification. Two independent investigators counted the RelA/p65+ nuclei and CD31+ vessels and the mean number per field was calculated. To evaluate the immunoreactivity of phosphorylated MET, we scored as described by Fukushima et al.27
2.4. Cell culture
The Caco2 cell line was obtained from the Riken BRC Cell Bank. Cells were cultured in DMEM containing 10% FBS. For transient silencing of F2RL1, 2 kinds of siRNA were used. One (PAR‐2 siRNA #1) was an siRNA pool (ON‐TARGETplus SMARTpool siRNA; Thermo Fisher Scientific) used with the siGENOME Non‐Targeting siRNA pool as a control. The other (PAR‐2 siRNA #2) was Stealth siRNA (Invitrogen) and the sequence was 5′‐UCAACCACUGUUAAGACCUCCUAUU‐3′. Transfection was carried out using Lipofectamine RNAiMax reagent (Invitrogen) followed by cultivation in DMEM supplemented with 10% FBS for 24 hours. The cells were treated with or without 10 µmol/L PAR‐2 agonist, Ser‐Leu‐Ile‐Gly‐Arg (SLIGR)‐NH2, or 10 µmol/L PAR‐2 selective antagonist, Phe‐Ser‐Leu‐Leu‐Arg‐Tyr (FSLLRY)‐NH2 (Peptides International).28
2.5. Immunoblot analysis
Mouse intestinal tumors were homogenized on ice in CelLytic MT (Sigma‐Aldrich) supplemented with protease inhibitor cocktail (Sigma‐Aldrich), 100 mmol/L NaF, and 1 mmol/L Na3VO4. The extracts were centrifuged at 20 000 g for 15 minutes at 4°C, and the resulting supernatants were used for experiments. Cellular proteins were extracted with CelLytic M (Sigma‐Aldrich) supplemented with protease inhibitor cocktail, 100 mmol/L NaF, and 1 mmol/L Na3VO4, centrifuged 15 000 g for 15 minutes, and the supernatants were collected. Equal amounts of total proteins were separated by SDS‐PAGE under reducing conditions using 4%‐12% gradient gels and transferred onto an Immobilon‐P membrane (Millipore). After blocking with 5% nonfat milk in 25 mmol/L TBS with 0.1% Tween‐20, pH 7.6 (TBS‐T), the membranes were incubated overnight at 4°C with primary Ab, followed by washing with TBS‐T and incubation with HRP‐conjugated secondary Ab diluted in TBS‐T with 1% BSA for 1 hour at room temperature. The labeled proteins were visualized with a chemiluminescence reagent (PerkinElmer Life Science). The following primary Abs were used: anti‐β‐actin mouse mAb (Sigma‐Aldrich), anti‐PAR‐2 rabbit mAb, anti‐phospho‐NF‐κB p65 (Ser536) rabbit mAb and anti‐NF‐κB p65 rabbit mAb (Cell Signaling Technology Japan).
2.6. Enzyme‐linked immunosorbent assay
Serum vascular endothelial growth factor (VEGF)‐A levels of mice and human VEGF‐A levels in culture supernatants of Caco2 were measured with a Quantikine VEGF ELISA kit (R&D Systems) according to the manufacturer’s instructions.
2.7. Statistical analysis
Statistical analysis was carried out using StatView 5.0 (SAS). Comparison between 2 unpaired groups was made with repeated‐measure of variance or the Mann‐Whitney U test. Significance was set at P < .05.
3. RESULTS
3.1. Deficiency in PAR‐2 alleviates increased tumor formation in Spint1‐deleted Apc Min/+ mice
Consistent with the previously reported increase in tumor burden,5 Spint1‐deleted Apc Min/+ (Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+) mice gained significantly less weight with age than Apc Min/+ control mice. Strikingly, this phenotype was completely reversed by concomitant deletion of F2rl1 (Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+; Figure 1A). Profound anemia of Spint1‐deleted Apc Min/+ mice was also normalized to Apc Min/+ control levels with compound PAR‐2 deficiency, although hematocrits remained lower than in naïve mice (Figure 1B). Fifteen weeks after birth, the number of intestinal tumors was significantly (P = .0001) increased in Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ (146.6 ± 4.5, n = 10) compared with control Spint1 LoxP/LoxP/Apc Min/+ mice (85.4 ± 5.7, n = 11) (Figure 2A,B). Again, concomitant deletion of F2rl1 in Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice significantly (P = .0003) reduced the number of intestinal tumors (92.9 ± 7.3, n = 11) (Figure 2B). Intriguingly, deletion of F2rl1 also reduced the number of tumors formed in Apc Min/+ mice in the presence of Spint1 (Spint1 LoxP/LoxP/F2rl1 −/−/Apc Min/+) (P = .0486). F2rl1 deletion also significantly decreased the size of tumors in both the control (P = .0046) and Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice (P = .0092) (Figures 2B and S1). However, tumor histology was not visibly altered by the deletion of F2rl1 (Figure 2C).
Figure 1.
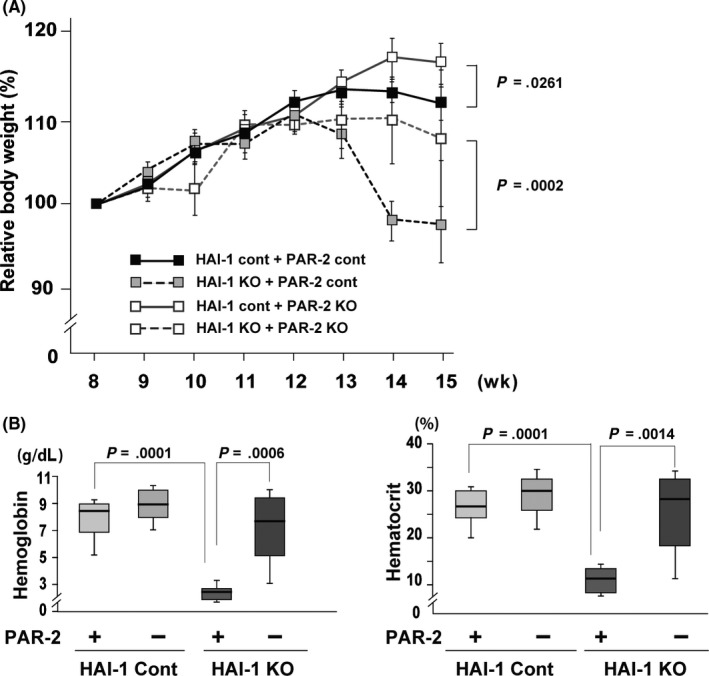
Protease‐activated receptor‐2 (PAR‐2) deficiency improved the health of Apc Min/+ mice deficient in intestinal hepatocyte growth factor activator inhibitor‐1 (HAI‐1). A, Relative body weight (mean ± SEM) of Spint1LoxP/LoxP/Apc Min/+ mice (HAI‐1 control [cont] + PAR‐2 cont) (n = 11), Spint1LoxP/LoxP/F2rl1 −/−/Apc Min/+ mice (HAI‐1 cont + PAR‐2 knockout [KO]) (n = 11), Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice (HAI‐1 KO + PAR‐2 cont) (n = 10), and Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ mice (HAI‐1 KO + PAR‐2 KO) (n = 11). B, Mean hemoglobin concentration (left panel) and hematocrit levels (right panel) of the mice. Box and whiskers indicate the interquartile range and sample maxima and minima, respectively. Median is indicated by a bold line
Figure 2.
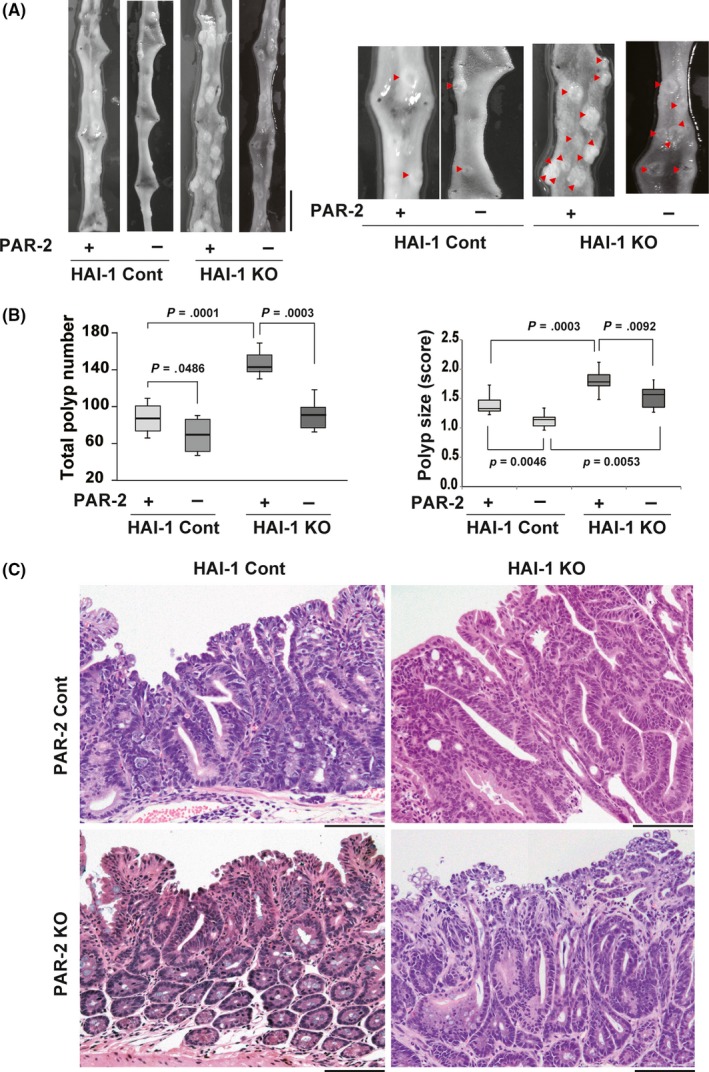
Effect of protease‐activated receptor‐2 (PAR‐2)/F2rl1 deletion on intestinal tumor formation in Apc Min/+ mice. A, Representative macroscopic appearance of the proximal small intestine from Spint1LoxP/LoxP/Apc Min/+ mice (hepatocyte growth factor activator inhibitor‐1 [HAI‐1] control [cont] + PAR‐2 cont) (n = 11), Spint1LoxP/LoxP/F2rl1 −/−/Apc Min/+ mice (HAI‐1 cont + PAR‐2 knockout [KO]) (n = 11), Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice (HAI‐1 KO + PAR‐2 cont) (n = 10), and Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ mice (HAI‐1 KO + PAR‐2 KO) (n = 11). Higher magnification photographs are also shown (right panel). Arrowheads indicate neoplastic polyps. Bar = 1 cm. B, Number of intestinal polyps (mean ± SEM; left panel) and polyp size scores of the mice (right panel). Box and whiskers indicate the interquartile range and sample maxima and minima, respectively. Median is indicated by a bold line. C, Histology of tumors (H&E stain). Bar = 100 μm
3.2. Protease‐activated receptor‐2 drives NF‐κB activation induced by Spint1 deletion
We previously reported that NF‐κB signaling is activated in HAI‐1‐deficient Apc Min/+ tumors. Moreover, an NF‐κB inhibitor suppressed the HAI‐1 loss‐mediated enhancement of tumorigenicity in Apc Min/+ mice.9 Protease‐activated receptor‐2 is known to activate NF‐κB signaling in various human cancers, including colon cancer,25, 29, 30, 31 and HAI‐1 regulates PAR‐2‐activating TTSPs.22 Thus, the activation of NF‐κB in HAI‐1‐deficient intestine might result from the excessive activation of PAR‐2. To test this hypothesis, we evaluated the effect of F2rl1 deletion on the nuclear translocation of the NF‐κB subunit RelA/p65. The ablation of PAR‐2/F2rl1 abrogated the enhanced nuclear translocation (Figure 3A,B) and phosphorylation of RelA/p65 resulting from the loss of HAI‐1 (Figure 3C) in intestinal tumors. The enhanced nuclear translocation of RelA/p65 was also observed in stroma cells adjacent to the tumor cells in HAI‐1‐deficient intestine, which was alleviated by the concomitant deletion of F2rl1 (Figure S2A). To support this, the number of CD45+ cells increased in the HAI‐1‐deficient intestine (Figure S2B). These results indicate that increased NF‐κB activation in Spint1‐deleted Apc Min/+ mice is driven by PAR‐2 signaling.
Figure 3.
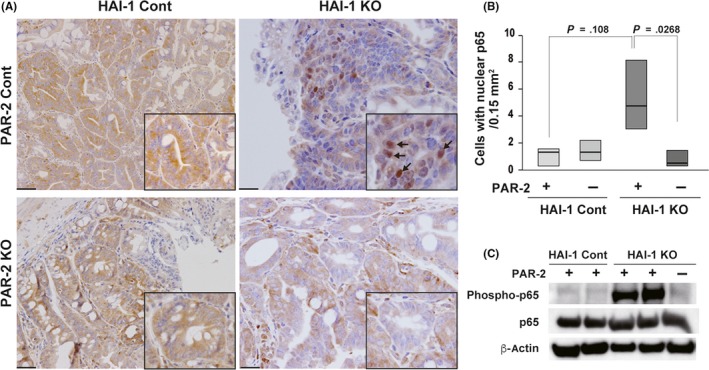
Nuclear factor‐κB (NF‐κB) activation was decreased by the absence of protease‐activated receptor‐2 (PAR‐2) in the intestines of Spint1‐deficient Apc Min/+ mice. A, Representative photographs of RelA/p65 nuclear translocation in intestinal tumor cells (arrows). Higher magnification images are also shown with indication of RelA/p65‐positive nuclei by arrows (inset). Bar = 50 μm. B, Quantitation of nuclear RelA/p65‐positive cells from Spint1LoxP/LoxP/Apc Min/+ mice (hepatocyte growth factor activator inhibitor‐1 [HAI‐1] control [cont] + PAR‐2 cont) (n = 4), Spint1LoxP/LoxP/F2rl1 −/−/Apc Min/+ mice (HAI‐1 cont + PAR‐2 knockout [KO]) (n = 4), Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice (HAI‐1 KO + PAR‐2 cont) (n = 4), and Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ mice (HAI‐1 KO + PAR‐2 KO) (n = 4) at 15 weeks of age. Box shows interquartile range. Median is indicated by a bold line. C, Immunoblot analysis for the effect of Spint1 deletion (HAI‐1 KO) or Spint1/F2rl1 double deletions (HAI‐1 KO/PAR2 ‐) on the phosphorylation (Phospho‐) of RelA/p65 in the intestinal tumor tissues
3.3. Protease‐activated receptor‐2 signaling increases VEGF‐A expression and vascular density
Next, we explored the mechanism by which PAR‐2/NF‐κB signaling increased mean intestinal tumor size and number in Apc Min/+ mice. Protease‐activated receptor‐2 deficiency did not affect the Ki‐67 labelling index or mRNA levels of Bcl2 and Bad genes in the intestinal tumors (Figure S3), indicating that cell proliferation rate and apoptotic signals were not altered. We then examined the expression of VEGF‐A, a key regulator of angiogenesis, in mouse tissues and serum. Intriguingly, there was a highly significant increase in serum VEGF‐A protein levels in Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice that was abolished with compound deficiency of PAR‐2 (Figure 4A). Accordingly, HAI‐1 deficiency was associated with a nonsignificant increase in Vegfa gene expression in the Apc Min/+ tumors that was significantly (P = .035) decreased with the compound deficiency of PAR‐2 (Figure 4B). Consistent with PAR‐2‐dependent VEGF expression, capillary density was significantly (P = .0282) decreased in tumors from Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ compared with those from Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice, although Spint1 deficiency did not by itself significantly increase vascular density in tumors or adjacent mucosa (Figure 5A). Quantitative RT‐PCR confirmed decreased mRNA levels for the Pecam1 gene, which encodes the endothelial marker CD31, after F2rl1 deletion in both tumors and adjacent mucosa (Figure 5B). These results suggest a PAR‐2‐dependent effect of local HAI‐1 deficiency on local and systemic VEGF‐A levels that in turn impacts vascular density and tumor growth.
Figure 4.
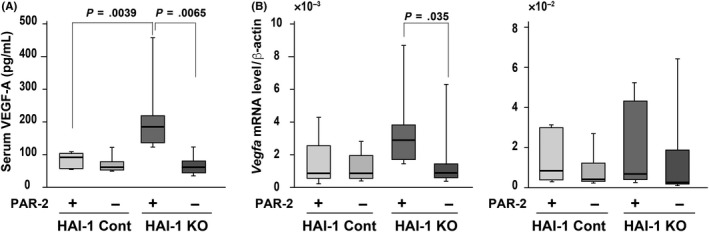
Vascular endothelial growth factor‐A (VEGF‐A) levels in the intestinal tissues and sera of mice. A, ELISA measurements of VEGF‐A in sera from each group. B, Quantitative RT‐PCR for Vegfa mRNA levels in tumor (left panel) and nontumor mucosa tissues (right panel) from Spint1LoxP/LoxP/Apc Min/+ mice (hepatocyte growth factor activator inhibitor‐1 [HAI‐1] control [cont] + PAR‐2 cont) (n = 7), Spint1LoxP/LoxP/F2rl1 −/−/Apc Min/+ mice (HAI‐1 cont + PAR‐2 knockout [KO]) (n = 7), Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice (HAI‐1 KO + PAR‐2 cont) (n = 7), and Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ mice (HAI‐1 KO + PAR‐2 KO) (n = 7). Box and whiskers indicate the interquartile range and sample maxima and minima, respectively. Median is indicated by a bold line
Figure 5.
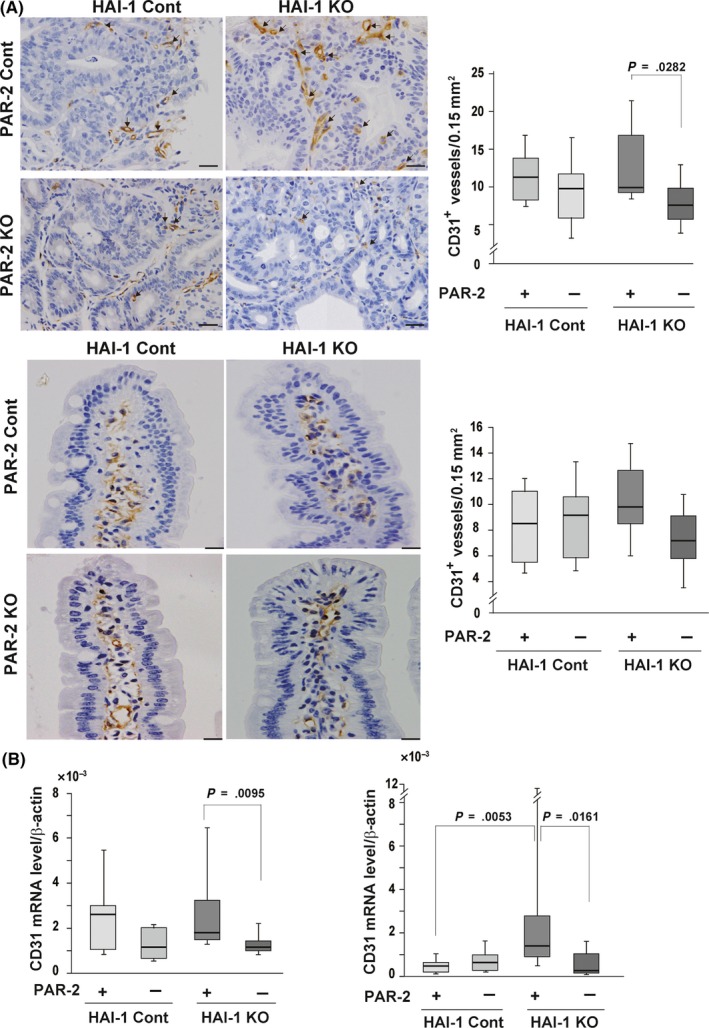
Effect of protease‐activated receptor‐2 (PAR‐2) deficiency on capillary density. A, Immunohistochemical analyses of CD31. Representative photographs of CD31+ vessels (arrows) (left panel), quantitation of CD31+ vessels (right panel) of tumor tissues (upper panel), and nontumor mucosa tissues (lower panel) from Spint1LoxP/LoxP/Apc Min/+ mice (hepatocyte growth factor activator inhibitor‐1 [HAI‐1] control [cont] + PAR‐2 cont) (n = 7), Spint1LoxP/LoxP/F2rl1 −/−/Apc Min/+ mice (HAI‐1 cont + PAR‐2 knockout [KO]) (n = 6), Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice (HAI‐1 KO + PAR‐2 cont) (n = 5), and Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ mice (HAI‐1 KO + PAR‐2 KO) (n = 5). Bar = 20 μm. B, Quantitative RT‐PCR for Pecam1 (CD31) mRNA expression in tumor (left panel) and nontumor mucosa (right panel) tissues. Box and whiskers indicate the interquartile range and sample maxima and minima, respectively. Median is indicated by a bold line
3.4. Protease‐activated receptor‐2 signaling activates NF‐κB signaling and enhances VEGF‐A expression in Caco2 human colon cancer cell line
Finally, to examine the role of PAR‐2 in NF‐κB activation and VEGF‐A expression of colon cancer cells, we analyzed the effects of F2RL1 silencing, PAR‐2 antagonist, or PAR‐2 agonist on the Caco2 human colon carcinoma cell line. Knockdown of F2RL1 or treatment of the cells with PAR‐2 antagonist suppressed the phosphorylation of RelA/p65 (Figure 6A). In contrast, PAR‐2 agonist enhanced the phosphorylation of RelA/p65 (Figure 6A). Next, we examined the effects of PAR‐2 agonist and antagonist on the VEGF‐A expression by Caco2 cells. As shown in Figure 6B, incubation of the cells with PAR‐2 agonist enhanced VEGF‐A mRNA levels. On the contrary, PAR‐2 antagonist suppressed the VEGF‐A expression. The released VEGF‐A proteins tended to be increased by the PAR‐2 agonist treatment, but the difference was not statistically significant (Figure 6B).
Figure 6.
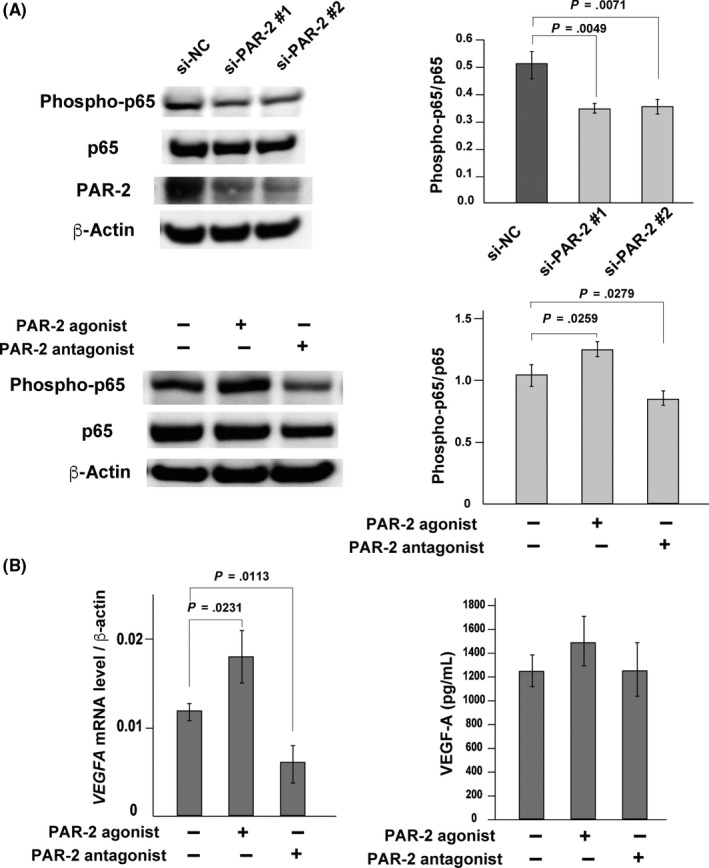
Protease‐activated receptor‐2 (PAR‐2) activated nuclear factor‐κB (NF‐κB) signaling and induced the expression of vascular endothelial growth factor‐A (VEGF‐A) in Caco2 colon cancer cell line. A, Immunoblot analysis of phosphorylated (Phospho‐)p65 and total p65. Caco2 cells were treated with 2 kinds of F2RL1 siRNAs: PAR‐2 siRNA #1 (si‐PAR‐2 #1) and PAR‐2 siRNA#2 (si‐PAR‐2 #2). Control siRNA (si‐NC)‐treated cells were also analyzed (upper panel). Effects of PAR‐2 agonist (10 µmol/L) and antagonist (10 µmol/L) on Caco2 cells (lower panel). Ratio of phospho‐p65 to p65 of each group is shown in the right panels. Error bar, SD of 3 independent experiments. B, Quantitative RT‐PCR for VEGF‐A mRNA levels in Caco2 cells treated with PAR‐2 agonist (10 µmol/L) or antagonist (10 µmol/L) (left panel) and ELISA measurements of VEGF‐A in the culture supernatant of Caco2 cells (right panel). Error bar, SD of 3 independent experiments
4. DISCUSSION
In this study, we show that ablation of PAR‐2/F2rl1 nearly eliminates the enhancement of NF‐κB signaling and tumor formation caused by the loss of HAI‐1/Spint1 in Apc Min/+ mice, providing evidence that PAR‐2 is critically involved in NF‐κB activation and the susceptibility to carcinogenesis caused by HAI‐1 insufficiency. The significance of PAR‐2 on NF‐κB signaling was also confirmed in the Caco2 human colon carcinoma cell line. Protease‐activated receptor‐2 deficiency also reduced tumor size and number, even in the presence of Spint1. These results suggest that protease signaling is also spontaneously deregulated in the Apc Min model, contributing significantly to tumor growth.
The detailed molecular mechanisms linking activated NF‐κB signaling to enhanced tumor formation remain unclear. Protease‐activated receptor‐2‐dependent VEGF‐A expression and angiogenesis induced by HAI‐1 deficiency could play a role. However, it remains to be determined whether the increased VEGF‐A level in Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice was a direct effect of HAI‐1 deficiency‐induced PAR‐2 activation or an epiphenomenon of the increased tumor burden. In this regard, the mean tumor size in Spint1LoxP/LoxP/Villin‐Cre/F2rl1 −/−/Apc Min/+ mice was larger than that of Spint1LoxP/LoxP/ F2rl1 −/−/Apc Min/+ mice, but the serum VEGF‐A levels and VEGFA mRNA levels were similar between these 2 groups. Thus, the increased serum VEGF‐A proteins did not simply rely on the secondary effect of increased tumor burden. As PAR‐2 activation in fact enhanced the VEGFA mRNA levels in Caco2 cells, we hypothesized that both factors (increased tumor burden and HAI‐1 deficiency‐induced PAR‐2 activation) account for the enhanced VEGF‐A production in Spint1LoxP/LoxP/Villin‐Cre/Apc Min/+ mice. Importantly, nuclear translocation of β‐catenin and activation of the hepatocyte growth factor‐MET axis, which were enhanced in Spint1‐deleted Apc Min/+ mice,5 were not significantly altered by compound PAR‐2/F2rl1 deletion (Figure S4).
Nuclear factor‐κB signaling has important roles in cancer progression, primarily through its stimulation of cell proliferation, survival, and angiogenesis.32, 33, 34 In this regard, activation of PAR‐2 by an extracellular trypsin‐like serine protease transduces NF‐κB signaling.22, 29, 35 Although several in vitro studies have reported that PAR‐2 promotes the proliferation of human colon cancer cell lines,36, 37, 38 the role of PAR‐2 in colorectal cancer has not been addressed in vivo. Here, we report for the first time that PAR‐2 promotes tumor formation and growth in mouse intestine harboring the Apc mutation. Protease‐activated receptor‐2 is expressed not only in epithelial cells but also leukocytes, endothelial cells, and smooth muscle cells.19, 39, 40 Although PAR‐2 is hardly detectable in normal fibroblasts, activated fibroblasts express PAR‐2, as do cancer‐associated fibroblasts.28, 41 Protease‐activated receptor‐2 is also expressed by most cancer cells of epithelial origin, including colorectal cancer cells.42 In this study, we did not address which cells were responsible for PAR‐2‐induced tumor promotion and VEGF‐A expression. In a mouse model of matriptase‐induced skin carcinogenesis, the excess activation of keratinocyte PAR‐2 was crucial for tumor promotion.25 As HAI‐1 and TTSP are both membrane anchored with mostly local actions, it is likely that epithelial PAR‐2 of the intestine is also responsible for the increased frequency of tumor formation in the current study. To clarify this question, further studies using mice with intestinal epithelium‐specific F2rl1‐deletion will be required.
This study did not identify the protease(s) responsible for the presumed excess activation of PAR‐2 in the Spint1‐deleted Apc Min/+ intestine. Protease‐activated receptor‐2 is activated by various trypsin‐like serine proteases.14, 15, 16, 17, 18, 19, 20, 21, 22 Among them, the main candidate protease is matriptase, a HAI‐1 regulated protease that is widely expressed in epithelial cells, including enterocytes.2, 43 The matriptase‐PAR‐2 axis has been implicated in development, inflammation, and cancer.23, 25, 44, 45, 46 The ST14 gene, encoding matriptase, was originally proposed as a suppressor of colon cancer.47 Indeed, ablation of St14/matriptase in the intestinal epithelium impaired the barrier function and induced mucosal inflammation, eventually resulting in the formation of colon adenocarcinoma in mice.48 Excess matriptase activity also leads to the disturbance of epithelial integrity in the intestine.49, 50 Moreover, matriptase is known to be upregulated in various human cancers and deregulated activities of matriptase contribute to tumor progression.51, 52, 53 In colorectal cancers, the ratio of matriptase / HAI‐1 mRNA is increased during the early stages of carcinogenesis.8 These lines of evidence indicate that normal, tightly regulated matriptase activity is critically required for the integrity of intestinal epithelium, and dysregulation of matriptase contributes to neoplastic progression of the intestinal epithelium. Protease‐activated receptor‐2 can also be activated by the tissue factor (TF)/factor VIIa (FVIIa) complex, coagulation factor Xa (FXa), and the ternary TF/FVIIa/FXa complex.19, 54, 55 Like matriptase and PAR‐2, TF is frequently expressed in various cancers, including colorectal cancer.54, 56 A recent study reported that TF‐dependent coagulation initiation on epithelia triggers matriptase activation, which in turn activates PAR‐2, suggesting that cancer‐associated loss of vascular integrity might trigger TTSP activation secondary to activation of the coagulation cascade.57 In a previous study of TF expression in human colorectal carcinoma cell lines, most lines expressed TF with augmented expression in a highly metastatic subline.58 In colorectal cancers, high TF expression in tumor cells also correlated with poor patient prognosis.59, 60 Kallikrein‐related peptidases (KLKs) are also candidate PAR‐2 activators in this context.61 Matriptase is an activator of KLK5, which is also directly inhibited by HAI‐1.62, 63, 64 Kallikrein‐related peptidase 5 is trypsin‐like serine proteinase that can efficiently activate PAR‐2 as well as KLK14, another PAR‐2 activating KLK.65 Kallikrein‐related peptidase 7 is known to be expressed by human colon cancer cell lines in vitro and colon cancer tissues in vivo.14, 61
In summary, we showed that a protease‐activated G‐protein‐coupled receptor, PAR‐2, contributes to intestinal carcinogenesis in the murine Apc Min model, and accounts, at least partly, for the increased tumor susceptibility associated with genetically induced deficiency of the TTSP inhibitor HAI‐1 in intestinal epithelial cells. Mechanistically, PAR2 signaling promotes NF‐κB activation and tumor angiogenesis. This study illustrates the importance of tight regulation of pericellular serine protease activities for epithelial homeostasis and points to serine proteases and protease‐activated receptors as therapeutic targets in epithelial carcinogenesis.
DISCLOSURE
The authors declare that there is no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We thank Ms Yukari Torisu and Junko Kurogi for their excellent technical assistance. This work was supported by Japan Society for the Promotion of Science KAKENHI 15K08311 (MK), 16H05175 (HK), and 17K08764 (TF) and by the French National Research Agency (ANR‐15‐CE14‐0009; EC).
Kawaguchi M, Yamamoto K, Kataoka H, et al. Protease‐activated receptor‐2 accelerates intestinal tumor formation through activation of nuclear factor‐κB signaling and tumor angiogenesis in Apc Min/+ mice. Cancer Sci. 2020;111:1193–1202. 10.1111/cas.14335
REFERENCES
- 1. Kataoka H, Suganuma T, Shimomura T, et al. Distribution of hepatocyte growth factor activator inhibitor type 1 (HAI‐1) in human tissues. Cellular surface localization of HAI‐1 in simple columnar epithelium and its modulated expression in injured and regenerative tissues. J Histochem Cytochem. 1999;47(5):673‐682. [DOI] [PubMed] [Google Scholar]
- 2. Kataoka H, Kawaguchi M, Fukushima T, Shimomura T. Hepatocyte growth factor activator inhibitors (HAI‐1 and HAI‐2): emerging key players in epithelial integrity and cancer. Pathol Int. 2018;68(3):145‐158. [DOI] [PubMed] [Google Scholar]
- 3. Antalis TM, Bugge TH, Wu Q. Membrane‐anchored serine proteases in health and disease. Prog Mol Biol Transl Sci. 2011;99:1‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawaguchi M, Takeda N, Hoshiko S, et al. Membrane‐bound serine protease inhibitor HAI‐1 is required for maintenance of intestinal epithelial integrity. Am J Pathol. 2011;179(4):1815‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoshiko S, Kawaguchi M, Fukushima T, et al. Hepatocyte growth factor activator inhibitor type 1 is a suppressor of intestinal tumorigenesis. Cancer Res. 2013;73(8):2659‐2670. [DOI] [PubMed] [Google Scholar]
- 6. Nagaike K, Kohama K, Uchiyama S, et al. Paradoxically enhanced immunoreactivity of hepatocyte growth factor activator inhibitor type 1 (HAI‐1) in cancer cells at the invasion front. Cancer Sci. 2004;95(9):728‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kataoka H, Hamasuna R, Itoh H, Kitamura N, Koono M. Activation of hepatocyte growth factor/scatter factor in colorectal carcinoma. Cancer Res. 2000;60(21):6148‐6159. [PubMed] [Google Scholar]
- 8. Vogel LK, Sæbø M, Skjelbred CF, et al. The ratio of Matriptase/HAI‐1 mRNA is higher in colorectal cancer adenomas and carcinomas than corresponding tissue from control individuals. BMC Cancer. 2006;6:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawaguchi M, Yamamoto K, Kanemaru A, et al. Inhibition of nuclear factor‐κB signaling suppresses Spint1‐deletion‐induced tumor susceptibility in the ApcMin/+ model. Oncotarget. 2016;7(42):68614‐68622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase‐activated receptors (PARs) ‐ focus on receptor‐receptor‐interactions and their physiological and pathophysiological impact. Cell Commun Signal. 2013;11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ungefroren H, Witte D, Rauch B, et al. Proteinase‐activated receptor 2 may drive cancer progression by facilitating TGF‐β signaling. Int J Mol Sci. 2017;18(11):2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lohman R‐J, Cotterell AJ, Suen J, et al. Antagonism of protease‐activated receptor 2 protects against experimental colitis. J Pharmacol Exp Ther. 2012;340(2):256‐265. [DOI] [PubMed] [Google Scholar]
- 13. Tahara T, Shibata T, Nakamura M, et al. Promoter methylation of protease‐activated receptor (PAR2) is associated with severe clinical phenotypes of ulcerative colitis (UC). Clin Exp Med. 2009;9(2):125‐130. [DOI] [PubMed] [Google Scholar]
- 14. Gratio V, Loriot C, Virca GD, et al. Kallikrein‐related peptidase 14 acts on proteinase‐activated receptor 2 to induce signaling pathway in colon cancer cells. Am J Pathol. 2011;179(5):2625‐2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coughlin SR. Thrombin signalling and protease‐activated receptors. Nature. 2000;407(6801):258‐264. [DOI] [PubMed] [Google Scholar]
- 16. Seitz I, Hess S, Schulz H, et al. Membrane‐type serine protease‐1/matriptase induces interleukin‐6 and ‐8 in endothelial cells by activation of protease‐activated receptor‐2. Arterioscler Thromb Vasc Biol. 2007;27(4):769‐775. [DOI] [PubMed] [Google Scholar]
- 17. Liu C, Li Q, Zhou X, Kolosov VP, Perelman JM. Human airway trypsin‐like protease induces mucin5AC hypersecretion via a protease‐activated receptor 2‐mediated pathway in human airway epithelial cells. Arch Biochem Biophys. 2013;535(2):234‐240. [DOI] [PubMed] [Google Scholar]
- 18. Wilson S, Greer B, Hooper J, et al. The membrane‐anchored serine protease, TMPRSS2, activates PAR‐2 in prostate cancer cells. Biochem J. 2005;388(Pt 3):967‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Protease‐activated receptors (PARs)‐biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015;34(4):775‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Driesbaugh KH, Buzza MS, Martin EW, Conway GD, Kao JPY, Antalis TM. Proteolytic activation of the Protease‐activated Receptor (PAR)‐2 by the glycosylphosphatidylinositol‐anchored serine protease testisin. J Biol Chem. 2015;290(6):3529‐3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane‐type serine protease 1 and identification of protease‐activated receptor‐2 and single‐chain urokinase‐type plasminogen activator as substrates. J Biol Chem. 2000;275(34):26333‐26342. [DOI] [PubMed] [Google Scholar]
- 22. Pawar NR, Buzza MS, Antalis TM. Membrane‐anchored serine proteases and protease‐activated receptor‐2–mediated signaling: co‐conspirators in cancer progression. Cancer Res. 2019;79(2):301‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Camerer E, Barker A, Duong DN, et al. Local protease signaling contributes to neural tube closure in the mouse embryo. Dev Cell. 2010;18(1):25‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. List K, Szabo R, Molinolo A, et al. Deregulated matriptase causes ras‐independent multistage carcinogenesis and promotes ras‐mediated malignant transformation. Genes Dev. 2005;19(16):1934‐1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sales KU, Friis S, Konkel JE, et al. Non‐hematopoietic PAR‐2 is essential for matriptase‐driven pre‐malignant progression and potentiation of ras‐mediated squamous cell carcinogenesis. Oncogene. 201434:346‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindner JR, Kahn ML, Coughlin SR, et al. Delayed onset of inflammation in protease‐activated receptor‐2‐deficient mice. J Immunol. 2000;165(11):6504‐6510. [DOI] [PubMed] [Google Scholar]
- 27. Fukushima T, Kawaguchi M, Yamamoto K, et al. Aberrant methylation and silencing of the SPINT2 gene in high‐grade gliomas. Cancer Sci. 2018;109(9):2970‐2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanemaru AI, Yamamoto K, Kawaguchi M, et al. Deregulated matriptase activity in oral squamous cell carcinoma promotes the infiltration of cancer‐associated fibroblasts by paracrine activation of protease‐activated receptor 2. Int J Cancer. 2017;140(1):130‐141. [DOI] [PubMed] [Google Scholar]
- 29. Johnson JJ, Miller DL, Jiang R, et al. Protease Activated Receptor‐2 (PAR‐2)‐mediated Nf‐κB activation suppresses inflammation‐associated tumor suppressor MicroRNAs in oral squamous cell carcinoma. J Biol Chem. 2016;291(13):6936‐6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo D, Zhou H, Wu Y, et al. Involvement of ERK1/2/NF‐κB signal transduction pathway in TF/FVIIa/PAR2‐induced proliferation and migration of colon cancer cell SW620. Tumor Biol. 2011;32(5):921‐930. [DOI] [PubMed] [Google Scholar]
- 31. Das K, Prasad R, Ansari SA, Roy A, Mukherjee A, Sen P. Matrix metalloproteinase‐2: A key regulator in coagulation proteases mediated human breast cancer progression through autocrine signaling. Biomed Pharmacother. 2018;105:395‐406. [DOI] [PubMed] [Google Scholar]
- 32. Perkins ND. The diverse and complex roles of NF‐κB subunits in cancer. Nat Rev Cancer. 2012;12(2):121‐132. [DOI] [PubMed] [Google Scholar]
- 33. Ben‐Neriah Y, Karin M. Inflammation meets cancer, with NF‐κB as the matchmaker. Nat Immunol. 2011;12(8):715‐723. [DOI] [PubMed] [Google Scholar]
- 34. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature. 2006;441(7092):431‐436. [DOI] [PubMed] [Google Scholar]
- 35. Kanke T, Macfarlane SR, Seatter MJ, et al. Proteinase‐activated receptor‐2‐mediated activation of stress‐activated protein kinases and inhibitory kappa B kinases in NCTC 2544 keratinocytes. J Biol Chem. 2001;276(34):31657‐31666. [DOI] [PubMed] [Google Scholar]
- 36. Wu Y, Wang J, Zhou H, et al. Effects of calcium signaling on coagulation factor VIIa‐induced proliferation and migration of the SW620 colon cancer cell line. Mol Med Rep. 2014;10(6):3021‐3026. [DOI] [PubMed] [Google Scholar]
- 37. Ma Y, Bao‐Han W, Lv X, et al. MicroRNA‐34a mediates the autocrine signaling of PAR2‐activating proteinase and its role in colonic cancer cell proliferation. PLoS ONE. 2013;8(8):e72383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu L, Xia L, Zhou H, et al. TF/FVIIa/PAR2 promotes cell proliferation and migration via PKCα and ERK‐dependent c‐Jun/AP‐1 pathway in colon cancer cell line SW620. Tumor Biol. 2013;34(5):2573‐2581. [DOI] [PubMed] [Google Scholar]
- 39. Allard B, Bara I, Gilbert G, et al. Protease activated receptor‐2 expression and function in asthmatic bronchial smooth muscle. PLoS ONE. 2014;9(2):e86945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johansson U, Lawson C, Dabare M, et al. Human peripheral blood monocytes express protease receptor‐2 and respond to receptor activation by production of IL‐6, IL‐8, and IL‐1β. J Leukoc Biol. 2005;78(4):967‐975. [DOI] [PubMed] [Google Scholar]
- 41. Gruber BL, Marchese MJ, Santiago‐Schwarz F, Martin CA, Zhang J, Kew RR. Protease‐activated receptor‐2 (PAR‐2) expression in human fibroblasts is regulated by growth factors and extracellular matrix. J Invest Dermatol. 2004;123(5):832‐839. [DOI] [PubMed] [Google Scholar]
- 42. Elste AP, Petersen I. Expression of proteinase‐activated receptor 1–4 (PAR 1–4) in human cancer. J Mol Histol. 2010;41(2–3):89‐99. [DOI] [PubMed] [Google Scholar]
- 43. Oberst MD, Singh B, Ozdemirli M, Dickson RB, Johnson MD, Lin C‐Y. Characterization of matriptase expression in normal human tissues. J Histochem Cytochem. 2003;51(8):1017‐1025. [DOI] [PubMed] [Google Scholar]
- 44. Bardou O, Menou A, François C, et al. Membrane‐anchored serine protease matriptase is a trigger of pulmonary fibrogenesis. Am J Respir Crit Care Med. 2016;193(8):847‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Szabo R, Peters DE, Kosa P, Camerer E, Bugge TH. Regulation of feto‐maternal barrier by matriptase‐ and PAR‐2‐mediated signaling is required for placental morphogenesis and mouse embryonic Survival. PLoS Genet. 2014;10(7):e1004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ye J, Kawaguchi M, Haruyama Y, et al. Loss of hepatocyte growth factor activator inhibitor type 1 participates in metastatic spreading of human pancreatic cancer cells in a mouse orthotopic transplantation model. Cancer Sci. 2014;105(1):44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y, Cai X, Schlegelberger B, Zheng S. Assignment of human putative tumor suppressor genes ST13 (alias SNC6) and ST14 (alias SNC19)to human chromosome bands 22q13 and 11q24→q25 by in situ hybridization. Cytogenet Cell Genet. 1998;83(1–2):56‐57. [DOI] [PubMed] [Google Scholar]
- 48. Kosa P, Szabo R, Molinolo AA, Bugge TH. Suppression of Tumorigenicity‐14, encoding matriptase, is a critical suppressor of colitis and colitis‐associated colon carcinogenesis. Oncogene. 2012;31(32):3679‐3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kawaguchi M, Yamamoto K, Takeda N, et al. Hepatocyte growth factor activator inhibitor‐2 stabilizes Epcam and maintains epithelial organization in the mouse intestine. Commun Biol. 2019;2(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Szabo R, Callies LLK, Bugge TH. Matriptase drives early‐onset intestinal failure in a mouse model of congenital tufting enteropathy. Development. 2019;146(22):dev183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tanabe LM, List K. The role of type II transmembrane serine protease‐mediated signaling in cancer. FEBS J. 2017;284(10):1421‐1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Webb SL, Sanders AJ, Mason MD, Jiang WG. Type II transmembrane serine protease (TTSP) deregulation in cancer. Front Biosci. 2011;16(2):539‐552. [DOI] [PubMed] [Google Scholar]
- 53. Martin CE, List K. Cell surface–anchored serine proteases in cancer progression and metastasis. Cancer Metastasis Rev. 2019;38(3):357‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kasthuri RS, Taubman MB, Mackman N. Role of tissue factor in cancer. J Clin Oncol. 2009;27(29):4834‐4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Camerer E, Huang W, Coughlin SR. Tissue factor‐ and factor X‐dependent activation of protease‐activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97(10):5255‐5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jia Z‐C, Wan Y‐L, Tang J‐Q, et al. Tissue factor/activated factor VIIa induces matrix metalloproteinase‐7 expression through activation of c‐Fos via ERK1/2 and p38 MAPK signaling pathways in human colon cancer cell. Int J Colorectal Dis. 2012;27(4):437‐445. [DOI] [PubMed] [Google Scholar]
- 57. Le Gall SM, Szabo R, Lee M, et al. Matriptase activation connects tissue factor‐dependent coagulation initiation to epithelial proteolysis and signaling. Blood. 2016;127(25):3260‐3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kataoka H, Uchino H, Asada Y, et al. Analysis of tissue factor and tissue factor pathway inhibitor expression in human colorectal carcinoma cell lines and metastatic sublines to the liver. Int J cancer. 1997;72(5):878‐884. [DOI] [PubMed] [Google Scholar]
- 59. Lykke J, Nielsen HJ. The role of tissue factor in colorectal cancer. Eur J Surg Oncol. 2003;29(5):417‐422. [DOI] [PubMed] [Google Scholar]
- 60. Seto S‐I, Onodera H, Kaido T, et al. Tissue factor expression in human colorectal carcinoma: correlation with hepatic metastasis and impact on prognosis. Cancer. 2000;88(2):295‐301. [DOI] [PubMed] [Google Scholar]
- 61. Chung H, Hamza M, Oikonomopoulou K, et al. Kallikrein‐related peptidase signaling in colon carcinoma cells: targeting proteinase‐activated receptors. Biol Chem. 2012;393(5):413‐420. [DOI] [PubMed] [Google Scholar]
- 62. Yoon H, Laxmikanthan G, Lee J, et al. Activation profiles and regulatory cascades of the human kallikrein‐related peptidases. J Biol Chem. 2007;282(44):31852‐31864. [DOI] [PubMed] [Google Scholar]
- 63. Emami N, Diamandis EP. Human kallikrein‐related peptidase 14 (KLK14) is a new activator component of the KLK proteolytic cascade. J Biol Chem. 2008;283(6):3031‐3041. [DOI] [PubMed] [Google Scholar]
- 64. Sales KU, Masedunskas A, Bey AL, et al. Matriptase initiates activation of epidermal pro‐kallikrein and disease onset in a mouse model of Netherton syndrome. Nat Genet. 2010;42(8):676‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Briot A, Deraison C, Lacroix M, et al. Kallikrein 5 induces atopic dermatitis‐like lesions through PAR2‐mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206(5):1135‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials