

Abstract
The roles of cancer‐associated fibroblasts (CAF) in the progression of various types of cancers are well established. CAF promote cancer progression through pleiotropic mechanisms, including the secretion of soluble factors and extracellular matrix, physical interactions with cancer cells, and the regulation of angiogenesis, immunity and metabolism. Their contribution to therapeutic resistance is also well appreciated. Therefore, CAF have been considered as a therapeutic target in cancer. However, recent studies in autochthonous pancreatic cancer models suggest that specific subset(s) of CAF exhibit cancer‐restraining roles, indicating that CAF are functionally and molecularly heterogeneous, which is supported by recent single‐cell transcriptome analyses. While cancer‐promoting CAF (pCAF) have been extensively studied, the nature and specific marker(s) of cancer‐restraining CAF (rCAF) have remained uncharacterized. Interestingly, a recent study provided insight into the nature of rCAF and suggested that they may share molecular properties with pancreatic stellate cells (PSC) and mesenchymal stem/stromal cells (MSC). Complicating this finding is that PSC and MSC have been shown to promote the formation of a tumor‐permissive and tumor‐promoting environment in xenograft tumor models. However, these cells undergo significant transcriptional and epigenetic changes during ex vivo culture, which confounds the interpretation of experimental results based on the use of cultured cells. In this short review, we describe recent studies and hypotheses on the identity of rCAF and discuss their analogy to fibroblasts that suppress fibrosis in fibrotic diseases. Finally, we discuss how these findings can be exploited to develop novel anticancer therapies in the future.
Keywords: cancer‐restraining cancer‐associated fibroblasts, fibrosis, Meflin, mesenchymal stem/stromal cells, tumor microenvironment
Meflin marks CAF that first emerge around metaplastic or transformed cells, which behave as rCAF and later give rise to α‐SMA+ CAF with low Meflin expression, in pancreatic cancer. This results in CAF heterogeneity in an advanced stage of pancreatic cancer.

1. INTRODUCTION
In routine pathological practice, pathologists often observe that the stroma of any type of human solid cancer contains a large number of fibroblasts and a large quantity of extracellular matrix (ECM) produced by these cells, the volume of which may exceed that of the cancer cells themselves. Notably, fibroblast proliferation is conspicuous in aggressive and refractory cancers, such as pancreatic ductal adenocarcinoma (PDAC), and poorly differentiated tumors with extensive desmoplastic stromal reaction that arise in various organs (Figure 1A). These fibroblasts are termed tumor‐ or cancer‐associated fibroblasts (CAF) and, together with lymphoid cells and myeloid cells, adipocytes and tumor vessels, constitute a major compartment of the tumor microenvironment (TME).1, 2, 3, 4, 5, 6, 7 Numerous studies have revealed that CAF promote cancer progression through multiple mechanisms, including ECM remodeling and the production of growth factors, cytokines and chemokines, which promote cancer cell proliferation and metabolism as well as tumor angiogenesis.1, 2, 3, 4, 5, 6, 7 Studies on cancer immunotherapy have shed light on the involvement of CAF in the tumor immune microenvironment and have revealed that CAF expressing fibroblast activation protein‐α (FAPα) or α‐smooth muscle actin (α‐SMA) suppress antitumor immunity through various mechanisms, contributing to the formation of a tumor‐permissive microenvironment.8, 9, 10, 11, 12 Another intriguing feature of CAF is that they physically interact with cancer cells through trans‐heterophilic interaction between N‐cadherin expressed on CAF and E‐cadherin expressed on cancer cells, functioning to lead cancer cell cohorts and cause them to collectively invade the stroma.13, 14, 15 In this context, CAF produce various proteases that degrade the ECM deposited in the stroma to create paths or tunnels for the collective invasion of cancer cells. Interestingly, CAF and the ECM contribute to changes in the mechanical properties of the tumor stroma, making cancer cells more aggressive and resistant to chemotherapies.16, 17 For example, ECM deposited and remodeled by CAF makes the stroma less elastic (stiffer) and provides a scaffold that supports the growth, migration and invasion of cancer cells. In addition, the ECM induces the tumor vessel collapse, thereby hampering the penetration of therapeutic reagents and antibodies.18 Thus, CAF have been considered to be promoters of tumorigenesis with incredibly intricate and complex functions. Interested readers can refer to other comprehensive review articles on the functions and roles of CAF in many types of cancer.1, 2, 3, 4, 5, 6, 7
Figure 1.
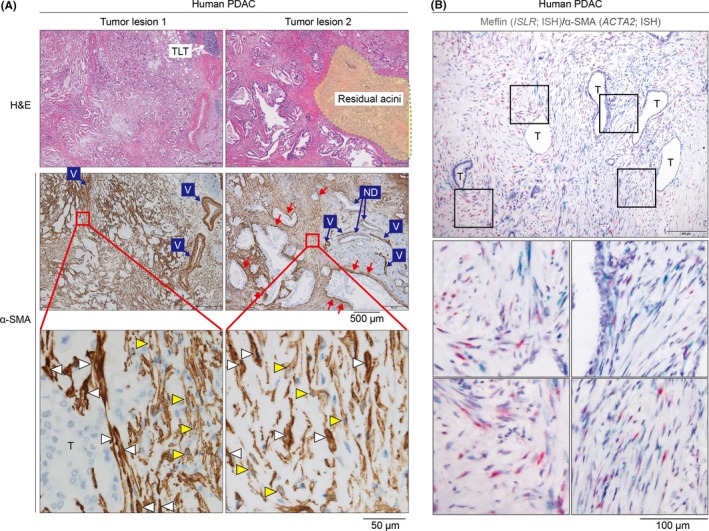
Cancer‐associated fibroblast (CAF) distribution and heterogeneity in human pancreatic ductal adenocarcinoma (PDAC). A, Representative histological images of human PDAC. Serial sections from two tumor lesions from a patient with PDAC stained with H&E are shown (upper panel). Residual acini are indicated in a dotted yellow‐colored area. In middle and lower panels, serial sections were stained for the CAF marker α‐smooth muscle actin (α‐SMA) (brown). Note that smooth muscle cells in the vessel walls (V) are also positive for α‐SMA. Tumor cells (T) as well as normal ducts (ND) are negative for α‐SMA. Magnified images of the boxed areas (lower panel) suggest that α‐SMA expression is heterogeneous: some CAF are strongly positive for α‐SMA (white arrowheads), whereas others are moderately or weekly positive for α‐SMA (yellow arrowheads). Note that some CAF that are predominantly positive for α‐SMA preferentially localize immediately adjacent to the tumor glands (red arrows in the middle panels). B, Meflin+ cancer‐restraining cancer‐associated fibroblasts (rCAF) and α‐SMA+ CAF in human PDAC. CAF in human PDAC were double stained for Meflin (ISLR; red) and α‐SMA (ACTA2; green) mRNA by in‐situ hybridization (ISH), revealing CAF heterogeneity in the tumor stroma. Boxed areas are magnified in lower panels. T, tumor glands
Recent single‐cell transcriptome studies have suggested that CAF are diverse in terms of their gene expression and spatial distribution in the tumor stroma.19, 20, 21, 22 The heterogeneity of CAF has also been observed by conventional immunohistochemistry for the CAF marker α‐SMA in PDAC tissue samples, where CAF exhibit varying levels of α‐SMA expression (Figure 1A). Numerous other CAF markers, including FAPα, podoplanin, C‐X‐C motif chemokine ligand 12 (CXCL12/stromal cell‐derived factor‐1), fibroblast‐specific protein‐1 (FSP1/S100A4), platelet‐derived growth factor receptors and periostin have been used to identify CAF subsets, with certain subsets expressing overlapping markers.1, 2, 3, 4, 5, 6, 7 Details on these CAF markers and the clinical relevance of individual CAF subsets have been extensively reviewed previously and, therefore, will not be discussed further here.1, 2, 3
The prevailing paradigm that CAF promote cancer progression has led to several attempts to develop novel therapeutics that specifically target CAF, some of which have progressed to clinical evaluation, as summarized and discussed elsewhere.23, 24 One caveat in targeting CAF, however, is described in three separate studies published approximately 5 years ago: selective genetic depletion of a proliferative α‐SMA+ CAF population or pharmacological blockade of Sonic Hedgehog (Shh) signaling, which is essential for desmoplastic reaction in the tumor stroma, resulted in tumor progression in PDAC mouse models.25, 26, 27 Interestingly, CAF depletion was accompanied by poor tumor differentiation in the PDAC mouse models, suggesting that tumor differentiation is partly regulated by the TME, and that a high degree of inherent plasticity of differentiation status exists in cancer cells within tumors.25, 26 Furthermore, CAF depletion led to escape from immune surveillance with increased regulatory T cell infiltration and changes in vascularity, suggesting that some CAF population(s), if not all, promote tumor immunity and regulate tumor angiogenesis, at least in the context of PDAC. These findings have led researchers to speculate that there are functionally and molecularly heterogeneous populations of CAF in the PDAC stroma, including pCAF, which have been extensively characterized, and cancer‐restraining cancer‐associated fibroblasts (rCAF) of yet unknown identity and origin.1, 2, 3, 5 Following these studies, it has been suggested that it may be prudent to deplete selected subset(s) of CAF, or modulate and fine‐tune pCAF functions rather than depleting the whole CAF population in the PDAC stroma. Importantly, the hypothesis that rCAF are present has not been limited to the context of PDAC;25, 26, 27 tumor‐suppressive roles of CAF have also been described in other cancers, including bladder, intestinal and colon cancers,28, 29, 30 suggesting the universal presence of rCAF across various types of cancer.
The complexity of CAF functions in cancer progression was appreciated by not only experimental data from mouse models but also clinical studies with Shh inhibitors such as saridegib (IPI‐926, Infinity) that have failed to show any obvious benefit in PDAC patients.3 The interpretation of these studies is, however, rendered difficult by conflicting data on the role of Shh signaling in cancer progression. Although Shh signaling has been described as a promoter of cancer progression based on arguments for the use of Shh inhibitors,31, 32, 33 other studies have shown that Shh signaling restrains the progression of bladder and colon cancers, in part by shaping TME, which involves bone morphogenetic protein (BMP) signaling to maintain the differentiation state of epithelial cells.28, 29 These observations have raised the possibility that the roles of Shh signaling are context‐dependent and individual pCAF or rCAF could respond selectively to Shh ligands, the details of which are currently elusive.
The aim of this short review is to shed light on the complex nature of CAF; however, the primary focus is on the identity and function of rCAF and their significance in cancer biology.
2. THE CONCEPT OF FIBROBLASTS BEING INNATELY TUMOR‐SUPPRESSIVE
In discussing the presence of rCAF in tumor stroma and their functions, it is important to understand that in‐depth debates have taken place regarding the inherent functions of fibroblasts in tumorigenesis. For instance, more than 50 years ago, Michael Stoker’s laboratory demonstrated that static normal fibroblasts cultured to confluent monolayers on plastic suppress the growth of polyoma virus‐transformed tumor cells.34 This inhibitory effect of fibroblasts on cancer cell proliferation has also been observed by other investigators.35 Such findings have led to the notion that the innate function of fibroblasts, which reside in every tissue of the body, is to suppress and protect against tumorigenesis.36, 37 In contrast, a pioneering study by Mina Bissell’s group published in the 1980s showed that the skin wound‐healing reaction promoted tumor formation in Rous sarcoma virus‐infected chickens.38 This study has significantly contributed to our understanding of the roles of the TME in initiation and promotion of tumorigenesis. Furthermore, the present study was among the first to suggest the possible involvement of fibroblasts in tumorigenesis based on the finding that the wound lesions contained activated fibroblasts and ECM produced by these cells. The involvement of fibroblasts in tumorigenesis has been corroborated by many subsequent studies, as described above. The fact that there seem to exist pCAF as well as rCAF in tumor stroma may indicate that we are seeing different aspects of the same fibroblasts depending on experimental conditions and observational contexts. Alternatively, pCAF and rCAF may have distinct origins, distribution and gene expression profiles. In any case, the molecular mechanisms by which fibroblasts suppress tumor formation and their specific marker(s) have not been adequately addressed to date.
The collective phenotypic changes in the components of the tumor stroma have been termed “stromagenic switch,” detailed descriptions of which have been provided elsewhere.39, 40 These include changes in the functions of fibroblasts that occur during cancer progression as well as other components of the TME, including the ECM, immune and myeloid cells, and tumor vessels, all of which are affected by each other and by signals derived from cancer cells. Given that CAF are a heterogeneous population in the tumor stroma, it is plausible to speculate that the stromagenic switch involves the conversion of rCAF to pCAF, contributing to cancer progression. An important question in view of this hypothesis is whether this conversion is irreversible or whether pCAF can become reprogrammed back into rCAF. If pCAF and rCAF represent distinct CAF populations with different origins, they may proliferate at different stages of tumor development.
Recent studies have begun to reveal several signaling pathways that differentially induce various CAF subsets in vitro and in vivo. For example, ECM constituents and substratum stiffness are crucial for the regulation of FAPα expression, a representative pCAF marker, in primary cultured lung fibroblasts.41 A recent study on PDAC revealed that interleukin (IL)‐1 and transforming growth factor (TGF)‐β, possibly derived from tumor cells, induce the differentiation of quiescent pancreatic stellate cells (PSC), which are a source of CAF, into IL‐6+ CAF (iCAF) and α‐SMA+ CAF (myCAF), respectively, yielding heterogeneous CAF subsets.19, 42 The presence of diverse CAF subsets was also shown in a mouse model of breast cancer, where CAF were classified into four groups: vascular CAF (vCAF), matrix CAF (mCAF), cycling CAF (cCAF) and developmental CAF (dCAF).21 These studies have led to the speculation that the induction of different CAF subsets during cancer progression leads to a decrease in, or loss of, the tumor‐suppressive microenvironment, which may differ depending on the cancer type and pathophysiological setting. Further spatio‐temporal single‐cell transcriptome and proteome analyses may reveal a more complete picture of CAF diversity and differentiation in various types of human cancer, as well as elucidate how different gene mutations in cancer cells result in varying degrees of CAF heterogeneity.
3. MEFLIN: A CANDIDATE MARKER OF CANCER‐RESTRAINING CANCER‐ASSOCIATED FIBROBLASTS IN PANCREATIC DUCTAL ADENOCARCINOMA
α‐smooth muscle actin was identified as a candidate marker of rCAF based on the observation that genetic depletion of proliferating α‐SMA+ cells led to an increase in tumor growth in a PDAC mouse model.25 Furthermore, blockade of Shh production by cancer cells inhibited CAF proliferation yet accelerated PDAC progression in mice, leading to the speculation that α‐SMA+ rCAF may proliferate in response to Shh signaling.25, 26 However, multiple studies have revealed a correlation between the number of α‐SMA+ CAF and poor outcomes in various types of human solid cancers, leading to the opposing hypothesis that states that α‐SMA+ CAF are a type of pCAF that shape a tumor‐permissive and tumor‐promoting environment.43, 44, 45, 46, 47, 48 In addition, there is ongoing discussion regarding the roles of α‐SMA+ CAF in tumor immunity. As stated previously, depletion of proliferating α‐SMA+ cells has been shown to lead to an increase in regulatory T cells, suggesting that α‐SMA+ CAF may promote antitumor immunity via direct or indirect mechanisms.25 However, another recent study showed that α‐SMA+ and FAPα+ CAF are critical for the retention and differentiation of regulatory T cells and, thus, have important roles in the suppression of antitumor immunity in a breast cancer mouse model.8 A possible, yet unvalidated, explanation for this inconsistency is that α‐SMA+ CAF functions differ depending on species and cancer types. Given that CAF are not uniformly positive for α‐SMA, and that α‐SMA expression levels are heterogeneous among CAF (Figure 1A), another attractive possibility is that there is functional or gene expression variability even within the α‐SMA+ subset of CAF. Furthermore, the use of α‐SMA as an rCAF marker poses a limitation as it is expressed by pericytes, smooth muscle cells that constitute vessel walls and smooth muscle layers, as well as myoepithelial cells in nearly all tissues.
A recent study by our group revealed that a CAF subset expressing Meflin, a glycosylphosphatidylinositol‐anchored protein,49 acts to suppress tumor progression in a PDAC mouse model and human PDAC.50 Examination of α‐SMA and Meflin expression by in‐situ hybridization (ISH) revealed that both genes are seemingly mutually exclusive (Figure 1B).50 However, quantification of the ISH signals and single‐cell transcriptome data analysis19 indicated that Meflin+ CAF were weakly positive for α‐SMA, while α‐SMA+ CAF were weakly positive for Meflin (Figure 2A).50 Interestingly, the notion that Meflin+ CAF function to inhibit PDAC progression has been supported by ISH findings in human PDAC tissue sections and comparison with patient outcomes, together with results from a study that knocked out the immunoglobulin superfamily containing leucine‐rich repeat (Islr) gene encoding Meflin in mice causing genetic depletion of Meflin+ CAF, and overexpression of Meflin in CAF in xenografted tumors in mice.50 As mentioned above, Meflin+ CAF weakly express α‐SMA, suggesting that in the previous study,25 in which all proliferating α‐SMA+ CAF were genetically depleted, a portion of Meflin+ CAF were also eradicated (Figure 2A). Together, the data, including data not described here,50 suggest that Meflin+ α‐SMAlow/− CAF represent rCAF in PDAC. Furthermore, these CAF were found to also be positive for Gli1, a transcription factor critical for Shh signaling, consistent with previous findings that describe the cancer‐promoting effect of Shh deletion in cancer cells.26, 50 We have also shown that Meflin is specifically expressed in PSC within the normal pancreas.50 Hence, it is feasible to hypothesize that Meflin+ PSC proliferate to become rCAF during tumorigenesis of PDAC (Figure 2A). Although the precise relationships among PSC, myCAF, iCAF and Meflin+ CAF have not been fully elucidated (Figure 2B), a transcriptome analysis showed that Meflin is expressed in PSC (base mean value 2623.5), iCAF (base mean value 665.7) and myCAF (base mean value 125.0),42 suggesting a mechanism whereby Meflin becomes downregulated during CAF differentiation.
Figure 2.
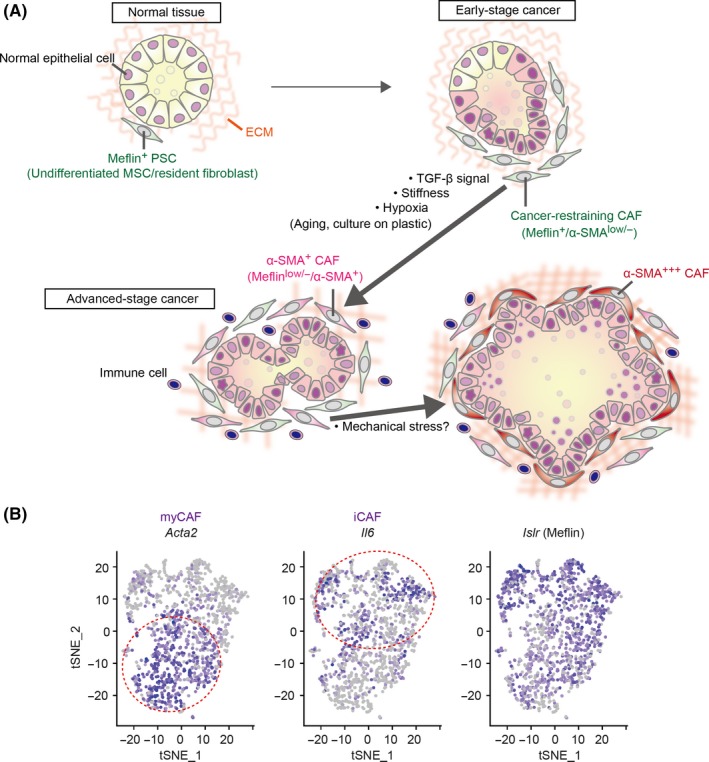
A possible mechanism underlying cancer‐associated fibroblast (CAF) heterogeneity. A, In the pancreas, Meflin marks pancreatic stellate cells (PSC), undifferentiated mesenchymal stem/stromal cells (MSC) and resident fibroblasts that localize around the acini, ducts and the islets of Langerhans. In the early stages of cancer development, including ADM and preinvasive (PanIN) stages, Meflin+ CAF or PSC that express low levels of α‐SMA start to proliferate around neoplastic cells, which we termed cancer‐restraining cancer‐associated fibroblasts (rCAF). Our recent studies showed that Meflin expression is downregulated by TGF‐β, stiff substrate and hypoxia, which we speculate are major factors that drive the differentiation of rCAF into α‐SMA+ and Meflin−/low CAF in advanced‐stage cancer. Aging and ex vivo culture of cells also cause downregulation of Meflin expression, although the relevance of these factors in cancer development and progression has not been demonstrated. In pancreatic ductal adenocarcinoma (PDAC), some CAF that are predominantly positive for α‐SMA preferentially localize immediately adjacent to the tumor glands, although there is currently no evidence that these CAF are derived from Meflin+ cells. The effect of mechanical stress imposed by the expanded tumor glands on α‐SMA expression in the CAF should also be considered. B, t‐distributed stochastic neighbor embedding (t‐SNE) plot showing the myCAF, iCAF and Meflin+ CAF subpopulations identified by scRNA‐seq of all cells isolated from tumors of a PDAC mouse model. Each dot is a CAF, and the intensity of the purple represents the expression level of the indicated genes. Reprinted from ref. 50, Copyright (2019), with permission from the American Association for Cancer Research
Interestingly, Meflin was previously identified as a marker of undifferentiated mesenchymal stem/stromal cells (MSC), which are distributed throughout every tissue in the body.49 As opposed to the other known MSC markers such as CD90, CD105, CD73, CD44 and CXCL12, Meflin is a marker of undifferentiated MSC, and is, therefore, not expressed by differentiated cell lineages, including mature adipocytes, chondrocytes and osteocytes.49 It is also expressed by interstitial fibroblast‐like cells found in the smooth muscle layer of the gastrointestinal tract as well as certain neuronal cells in the dentate gyrus and cerebral cortex, the significance of which has not yet been described. The finding that PSC express Meflin indicates shared features between these two cell types, details of which should be investigated in the future. Moreover, a previous study has shown that CD271 (also known as nerve growth factor receptor), a marker of PSC and MSC, is expressed by stromal cells in PDAC and the number of CD271+ cells is associated with a better prognosis, further supporting a view that PSC are closely related to MSC.51 However, it remains unclear whether Meflin+ CAF and CD271+ stromal cells are the same or represent distinct cell populations.
4. CANCER‐RESTRAINING CANCER‐ASSOCIATED FIBROBLASTS: SENTINEL CELLS THAT MONITOR CANCER CELL INITIATION?
We recently detected Meflin+ CAF in the vicinity of precancerous acinar‐to‐ductal metaplasia (ADM) lesions52, 53 in a PDAC mouse model.50 This mouse model, known as the KPC model,54 harbors oncogenic mutations in the genes encoding K‐Ras and p53 in the pancreatic epithelium and develops multiple ADM lesions around the interface between acinar, centroacinar, and ductal cells at 10‐12 weeks after birth (Figure 3). Notably, Meflin+ CAF appeared in the ADM lesions before α‐SMA expression was observed, suggesting that Meflin+ CAF have an elaborate response system for the detection of metaplastic or transformed cells or inflammation associated with tumor initiation (Figures 2A and 3). However, specific questions remain: for instance, it is unclear how Meflin+ CAF become mobilized and are recruited to metaplastic and transformed cells. Furthermore, the upstream regulators that control the activation of quiescent Meflin+ PSC or undifferentiated MSC have not yet been defined.
Figure 3.
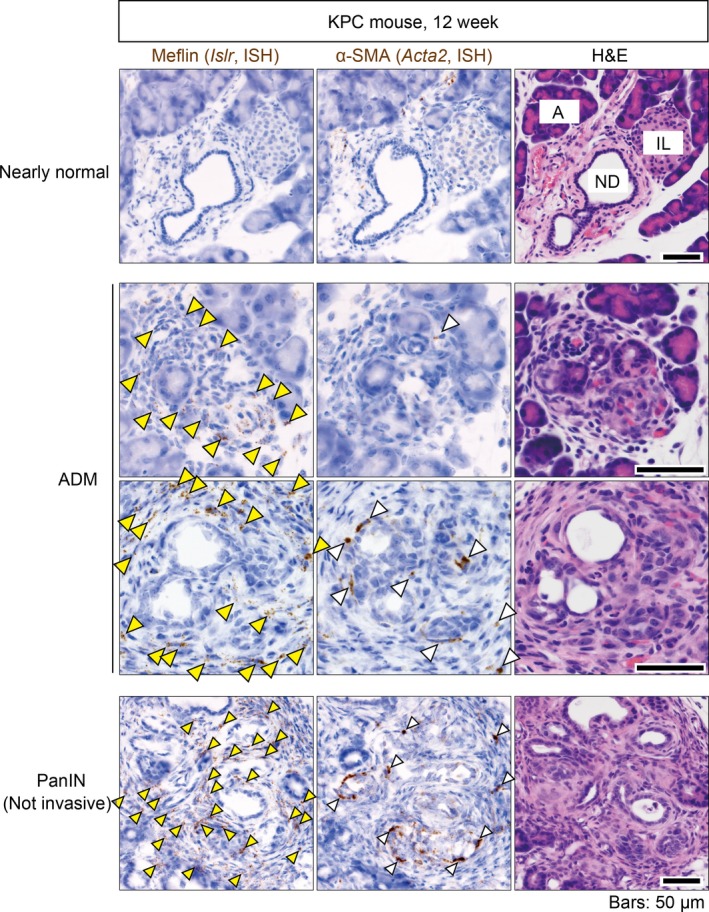
Meflin+ cancer‐associated fibroblasts (CAF) first emerge around metaplastic or tumor cells, followed by appearance of α‐SMA‐positive CAF in early stages of pancreatic cancer. Serial sections prepared from the pancreas of a 12‐week‐old KPC mouse were stained for Meflin and α‐SMA mRNA (left and middle panels) and with H&E (right panels). Different regions of the stained sections, including a nearly normal region (top panels), ADM lesions (middle panels) and a PanIN lesion (bottom panels) are shown. Yellow arrowheads indicate Meflin+ CAF or pancreatic stellate cells (PSC). White arrowheads denote α‐SMA+ CAF that preferentially emerge around metaplastic or tumor cells. A, normal acini; IL, islet of Langerhans; ND, normal duct
Meflin+ and α‐SMA+ CAF are differentially distributed in tumor tissues from the early to late stages of PDAC (Figures 1B, 2A and 3), suggesting that they are functionally mutually exclusive and develop from distinct cell lineages. Alternatively, they share the same lineage and change their gene expression patterns in response to cues derived from neoplastic cells and the TME. A lineage trace experiment showed that Meflin+ CAF gave rise to α‐SMA+ CAF in a xenograft tumor mouse model, which was accompanied by a decrease in Meflin expression.50 This phenotypic conversion was recapitulated in cultured MSC, where α‐SMA expression was upregulated while Meflin expression was significantly downregulated in response to transforming growth factor‐β (TGF‐β) signaling, substrate stiffness and hypoxia (Figure 2A).49, 50, 55 Given that TGF‐β is abundant in the TME and that tumors are stiffer and more hypoxic than normal tissues, it has been hypothesized that Meflin+ CAF first appear in close proximity to transformed cells as sentinel cells, and later differentiate into α‐SMA+ CAF in which Meflin expression is downregulated (Figure 2A). This phenotypic conversion of CAF may represent one aspect of the stromagenic switch that accompanies, and promotes, tumor progression.
A recently introduced concept for CAF diversification is that CAF may become “educated” by cancer cells through multiple direct and indirect mechanisms.24, 56, 57 A recent study that made use of an organoid culture system and PDAC mouse models revealed that IL‐1 signaling derived from cancer cells induces CAF differentiation into IL‐6+ iCAF, whereas TGF‐β blocks this pathway, causing a skewing toward the α‐SMA+ myCAF lineage; this suggested that complex mechanisms underlie CAF heterogeneity.19 Intriguingly, α‐SMA+ myCAF preferentially localize in the immediate proximity of tumor glands in PDAC (Figures 1A, 2A and 3),19, 42 suggesting that myCAF also serve as sentinel cells that recognize certain signals from proliferating cancer cells. However, given that α‐SMA expression is highly upregulated in response to various mechanical stresses acting on cells,58, 59 the high α‐SMA expression in myCAF may be induced by mechanical forces generated by the dilation of tumor glands (Figure 2A). Indeed, several studies have begun exploring the roles that changes in mechanical properties of the tumor stroma have in the regulation of cancer malignancy.16, 60, 61 CAF may serve as a central player in coupling mechanical to biochemical signals involved in increasing cancer malignancy.
5. MESENCHYMAL STEM/STROMAL CELLS: FOE OR FRIEND OF CANCER?
Our working hypothesis is that proliferating PSC, which share Meflin expression with undifferentiated MSC, represent rCAF at least in the context of PDAC (Figure 2A). This implies that MSC generally have a cancer‐restraining role in other types of cancer. However, many previous studies have examined the role of MSC and related stem cells in cancer progression with differing conclusions depending on the experimental design and model used.62, 63 Nevertheless, the common finding among these studies, although not conclusive, was that the net effect of MSC is the promotion of cancer through multiple mechanisms.64, 65, 66, 67, 68 One of the pioneering studies in the field showed that MSC are recruited from the bone marrow to the stroma of gastric cancer in mice, where they differentiate into pCAF in response to signaling induced by TGF‐β and CXCL12 derived from cancer cells and the TME.69 Another study revealed that co–implantation of MSC with a breast cancer cell line promoted cancer cell migration and metastasis through secretion of a chemokine C‐C motif chemokine ligand 5 in immunocompromised mice.64 These studies were followed by numerous studies that reached similar conclusions, which have been extensively reviewed elsewhere.62, 63, 65, 66 The mechanisms of the protumor function of MSC are similar to those of CAF, including the secretion of a wide repertoire of soluble and insoluble factors that directly or indirectly impact cancer cell proliferation and migration. This supports the hypothesis that MSC, whether resident in tissues or recruited from the bone marrow, are one of the major sources of pCAF in many types of cancer. One limitation of these studies on the protumor effects of MSC, however, was that the conclusions were largely based on syngeneic or allogeneic transplantation of MSC expanded on Petri dishes. Recent observations indicate that ex vivo culture of isolated MSC or tumor‐derived fibroblasts for a period of only 14 days induces the activation of DNA methyltransferases and DNA methylation changes in the promoter regions of various genes.70, 71 For example, the Islr promoter was methylated in MSC isolated from the umbilical cord and expanded in vitro, which was consistent with our finding that ex vivo culture on plastic induces a significant downregulation of Meflin expression in bone marrow‐derived MSC.49, 71 Further analysis revealed that culture substrate stiffness is key for downregulating Meflin expression.49, 55 The elastic modulus of normal body fluids and tissues is 102‐2 × 104 Pa, whereas that of culture plastic is 2‐3 GPa, indicating that MSC cultured on Petri dishes experience significant changes in mechanical stress, gene expression and the epigenome over time.16, 72 Thus, we believe that MSC in the body and those expanded on culture plastics exhibit unique gene expression profiles, functions, and properties, indicating that the findings from ex vivo‐expanded MSC do not necessarily accurately reflect the in vivo relevance of MSC. In support of this notion, a recent study revealed that PSC cultured in a soft 3D matrix immediately following isolation from the pancreas exhibited significant differences in gene expression compared to PSC cultured on Petri dishes.42 Similar to MSC, Meflin expression was significantly decreased in PSC cultured on Petri dishes, suggesting that care should be taken when interpreting data from studies employing cultured PSC.42
6. CANCER‐RESTRAINING CANCER‐ASSOCIATED FIBROBLASTS IN CANCER AND ANTIFIBROTIC FIBROBLASTS IN ORGAN FIBROSIS: THE SAME ON THE INSIDE?
The proliferation of fibroblasts has been clearly shown to serve as the primary etiology for many types of fibrotic diseases. The function of fibroblasts that appear in injured tissues is to produce the ECM as well as many types of soluble factors to promote tissue repair and suppress inflammation.73, 74 However, unfortunately, these fibroblasts gradually differentiate into α‐SMA+ myofibroblasts, leading to excessive and uncontrolled fibrosis and stiffening of the tissues. One important yet unaddressed question is how fibroblasts engaged in tissue repair can be discriminated from myofibroblasts that promote fibrosis. Our recent study revealed that Meflin+ MSC or fibroblasts rarely reside within the epicardium, endocardium and perivascular area of capillaries in the interstitium of the heart. Alternatively, they proliferate extensively and are recruited from other tissues immediately after acute myocardial infarction.55 Interestingly, a large proportion of the Meflin‐deficient mice died immediately after infarction due to cardiac rupture, suggesting that Meflin expression in fibroblasts or MSC is essential for acute repair of the injured cardiac tissue (Figure 4). Meflin deficiency also resulted in increased fibrosis and tissue stiffening in hearts with increased α‐SMA expression and fibrosis in a chronic heart failure condition, suggesting that Meflin may inhibit fibroblast differentiation into myofibroblasts (Figure 4).55 Mechanistically, Meflin binds to BMP‐7, a member of the BMP family, which counteracts the action of TGF‐β to prevent fibrosis.75 Studies on cultured cells have revealed that Meflin augments BMP‐7 signaling and inhibits TGF‐β signaling, suggesting that the primary function of Meflin is to inhibit profibrotic signaling in cardiac fibroblasts.55 Consistent herewith, failing hearts of Meflin‐deficient mice were stiffer than those of control mice and recapitulated the characteristics of an entity of diastolic heart failure in humans called “heart failure with preserved ejection fraction (HFpEF).”76 It is intriguing to note that Meflin+ fibroblasts and undifferentiated MSC exert a protective effect against the development of heart failure, and an antitumor function in PDAC development (Figure 4). One possible interpretation of these findings is that the primary function of these cells is the suppression of fibrosis in various types of diseases and the preservation of tissue homeostasis.
Figure 4.
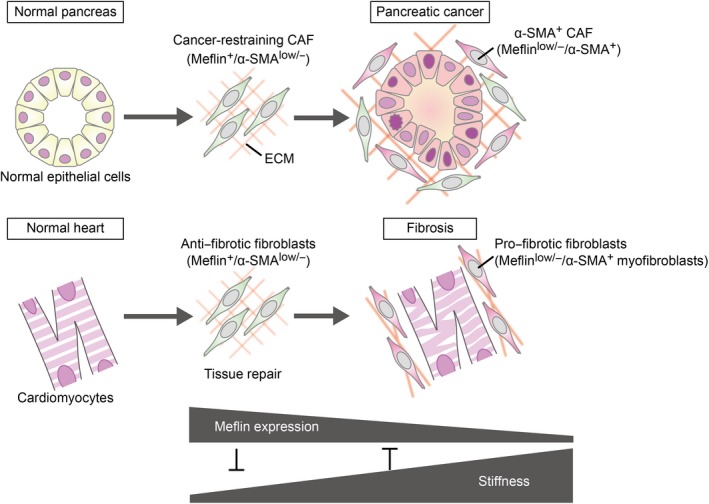
Analogy of cancer‐restraining cancer‐associated fibroblasts (rCAF) in pancreatic ductal adenocarcinoma (PDAC) to antifibrotic fibroblasts in cardiac fibrosis. In pancreatic ductal adenocarcinoma (PDAC), Meflin marks CAF that first emerge around metaplastic or transformed cells, which we showed behave as rCAF and later give rise to α‐SMA+ CAF with low Meflin expression, resulting in CAF heterogeneity in an advanced stage of cancer (upper panel). In cardiac infarction and fibrosis, we speculate that Meflin+ fibroblasts first proliferate in the injured area, and later yield α‐SMA+ myofibroblasts in the fibrotic phase (lower panel). The primary function of Meflin+ fibroblasts is to promote tissue repair and inhibit fibrosis, whereas loss of Meflin expression results in the differentiation of fibroblasts to myofibroblasts that contribute to the stiffening of cardiac tissue
7. PERSPECTIVE AND QUESTIONS
This review has focused on the emerging hypothesis that rCAF are present in the stroma of solid cancers. We have described our own data showing that Meflin, a marker of MSC, identifies and functionally contributes to rCAF that counteract pCAF in PDAC and fibroblasts with an antifibrotic function in chronic heart failure (Figure 4). These findings suggest that MSC, which reside in all tissues, are not prostrate cells that simply wait to differentiate into other cell types but rather act to protect against disease development and progression. Currently, literature on the presence of rCAF is scarce25, 26, 27, 50 and further investigations are essential to definitively prove the hypothesis.
There remain many questions to be answered in terms of the biology of Meflin+ CAF. For instance, what are the physiological roles of Meflin+ PSC in the normal pancreas and Meflin+ MSC in multiple organs? Which factors induce the proliferation of Meflin+ PSC? Are proliferating PSC identical to rCAF? Where are they recruited from to tumor lesions? Are there rCAF in cancers other than PDAC, and if so, do Meflin+ CAF represent rCAF in those cancers? How is CAF diversity, which is partly determined by the presence or absence of Meflin, involved in the immune response to cancer cells and tumor metabolism? Given that Meflin marks a subset of rCAF, how many types of rCAF exist in the tumor stroma, and does rCAF heterogeneity depend on the cancer type?
There are also many unanswered questions with respect to the functions of the Meflin protein. Meflin is a glycosylphosphatidylinositol‐anchored extracellular protein with leucine‐rich repeats that interacts with BMP7.49, 55 It is possible that Meflin has other binding partners, as do the matricellular proteins decorin and lumican, which are structurally similar to Meflin.77 In fact, Zhang et al (2018) identified Dvl2, a cytosolic component of the Wnt signaling pathway, as a new Meflin‐interacting protein and revealed that Meflin protects Dvl2 from autophagy‐dependent degradation, which contributes to the regenerative capacity of skeletal muscle stem cells.78
Finally, a remaining challenge in the CAF research field is to reprogram pCAF educated by cancer cells and the TME to rCAF. Previous studies have revealed that the administration of calcipotriol, a vitamin D analog, or all‐trans‐retinoic acid, a vitamin A derivative, may be capable of reprograming activated PSC to a more quiescent state, which sensitized PDAC to chemotherapy and inhibited PDAC progression in mice.79, 80 Consistent herewith, we found that calcipotriol treatment significantly increased Meflin expression in cultured fibroblasts isolated from PDAC tissues and mouse hearts,50, 55 indicating that calcipotriol may prove useful to revert pCAF and profibrotic fibroblasts into rCAF and antifibrotic fibroblasts, respectively. These findings may provide a rationale for the use of vitamin D analogs in the treatment of cancer patients. Indeed, several vitamin D analogs are currently in clinical trials in combination with chemotherapeutics such as gemcitabine and immune checkpoint inhibitors in patients with PDAC (https://clinicaltrials.gov/).
In conclusion, we discussed here the heterogeneous feature of CAF and proposed that there may be an rCAF subpopulation that restrains cancer progression. The shared properties of rCAF with tissue‐resident MSC and antifibrotic fibroblasts were also discussed. We believe that future investigations should seek to investigate this hypothesis, while working to develop novel therapies that increase the number of rCAF in any type of cancer.
DISCLOSURE
The authors declare no competing financial interests.
ACKNOWLEDGMENTS
We thank all the colleagues involved in the current research projects in our laboratory. We are grateful to Genichiro Ishii (National Cancer Center), Akira Orimo (Juntendo University) and Kenichiro Ishii (Mie University) for helpful discussions. This work was supported by: a Grant‐in‐Aid for Scientific Research (S) (26221304 to MT) and a Grant‐in‐Aid for Scientific Research (B) (18H02638 to AE) commissioned by the Ministry of Education, Culture, Sports, Science and Technology of Japan; AMED‐CREST (Japan Agency for Medical Research and Development, Core Research for Evolutional Science and Technology; 19gm0810007h0104 and 19gm1210008s0101 to AE); and the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) from AMED (19cm0106332h0002 to AE).
Miyai Y, Esaki N, Takahashi M, Enomoto A. Cancer‐associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020;111:1047–1057. 10.1111/cas.14346
Contributor Information
Masahide Takahashi, Email: mtakaha@med.nagoya-u.ac.jp.
Atsushi Enomoto, Email: enomoto@iar.nagoya-u.ac.jp.
REFERENCES
- 1. Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer‐associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2019;16:282‐295. [DOI] [PubMed] [Google Scholar]
- 2. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582‐598. [DOI] [PubMed] [Google Scholar]
- 3. Neesse A, Bauer CA, Öhlund D, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut. 2019;68:159‐171. [DOI] [PubMed] [Google Scholar]
- 4. Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gieniec KA, Butler LM, Worthley DL, Woods SL. Cancer‐associated fibroblasts—heroes or villains? Br J Cancer. 2019;121:293‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer‐associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99(Pt B):186‐196. [DOI] [PubMed] [Google Scholar]
- 7. Mezawa Y, Orimo A. The roles of tumor‐and metastasis‐promoting carcinoma‐associated fibroblasts in human carcinomas. Cell Tissue Res. 2016;365:675‐689. [DOI] [PubMed] [Google Scholar]
- 8. Costa A, Kieffer Y, Scholer‐Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463‐479. [DOI] [PubMed] [Google Scholar]
- 9. Kato T, Noma K, Ohara T, et al. Cancer‐associated fibroblasts affect intratumoral CD8+ and FoxP3+ T cells via IL6 in the tumor microenvironment. Clin Cancer Res. 2018;24:4820‐4833. [DOI] [PubMed] [Google Scholar]
- 10. Jiang H, Hegde S, DeNardo DG. Tumor‐associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother. 2017;66:1037‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer‐associated fibroblasts induce antigen‐specific deletion of CD8+ T cells to protect tumour cells. Nat Commun. 2018;9:948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti–PD‐L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212‐20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaggioli C, Hooper S, Hidalgo‐Carcedo C, et al. Fibroblast‐led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392‐1400. [DOI] [PubMed] [Google Scholar]
- 14. Labernadie A, Kato T, Brugués A, et al. A mechanically active heterotypic E‐cadherin/N‐cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19:224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neri S, Ishii G, Hashimoto H, et al. Podoplanin‐expressing cancer‐associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int J Cancer. 2015;137:784‐796. [DOI] [PubMed] [Google Scholar]
- 16. Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conklin MW, Keely PJ. Why the stroma matters in breast cancer: insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adh Migr. 2012;6:249‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583‐592. [DOI] [PubMed] [Google Scholar]
- 19. Biffi G, Oni TE, Spielman B, et al. IL1‐induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elyada E, Bolisetty M, Laise P, et al. Cross‐species single‐cell analysis of pancreatic ductal adenocarcinoma reveals antigen‐presenting cancer‐associated fibroblasts. Cancer Discov. 2019;9:1102‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bartoschek M, Oskolkov N, Bocci M, et al. Spatially and functionally distinct subclasses of breast cancer‐associated fibroblasts revealed by single cell RNA sequencing. Nat Commun. 2018;9:5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277‐1289. [DOI] [PubMed] [Google Scholar]
- 23. Whittle MC, Hingorani SR. Fibroblasts in pancreatic ductal adenocarcinoma: biological mechanisms and therapeutic targets. Gastroenterology. 2019;156:2085‐2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X, Song E. Turning foes to friends: targeting cancer‐associated fibroblasts. Nat Rev Drug Discov. 2019;18:99‐115. [DOI] [PubMed] [Google Scholar]
- 25. Özdemir BC, Pentcheva‐Hoang T, Carstens JL, et al. Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee JJ, Perera RM, Wang H, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci USA. 2014;111:E3091‐E3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin K, Lim A, Zhao C, et al. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell. 2014;26:521‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerling M, Büller NVJA, Kirn LM, et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat Commun. 2015;7:12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pallangyo CK, Ziegler PK, Greten FR. IKKβ acts as a tumor suppressor in cancer‐associated fibroblasts during intestinal tumorigenesis. J Exp Med. 2015;212:2253‐2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406‐410. [DOI] [PubMed] [Google Scholar]
- 32. Valenti G, Quinn HM, Heynen GJJE, et al. Cancer stem cells regulate cancer‐associated fibroblasts via activation of Hedgehog signaling in mammary gland tumors. Cancer Res. 2017;77:2134‐2147. [DOI] [PubMed] [Google Scholar]
- 33. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stoker MG, Shearer M, O’Niell C. Growth inhibition of polyoma‐transformed cells by contact with static normal fibroblasts. J Cell Sci. 1966;1:297‐310. [DOI] [PubMed] [Google Scholar]
- 35. Flaberg E, Markasz L, Petranyi G, et al. High‐throughput live‐cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int J Cancer. 2011;128:2793‐2802. [DOI] [PubMed] [Google Scholar]
- 36. Klein G. Evolutionary aspects of cancer resistance. Semin Cancer Biol. 2014;25:10‐14. [DOI] [PubMed] [Google Scholar]
- 37. Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV‐mediated tumor formation. Science. 1985;230:676‐678. [DOI] [PubMed] [Google Scholar]
- 39. Puré E, Lo A. Can targeting stroma pave the way to enhanced antitumor immunity and immunotherapy of solid tumors? Cancer Immunol Res. 2016;4:269‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lieberman A, Barrett R, Kim J, et al. Deletion of calcineurin promotes a pro‐tumorigenic fibroblast phenotype. Cancer Res. 2019;79:3928‐3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Avery D, Govindaraju P, Jacob M, Todd L, Monslow J, Puré E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018;67:90‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Öhlund D, Handly‐Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Underwood TJ, Hayden AL, Derouet M, et al. Cancer‐associated fibroblasts predict poor outcome and promote periostin‐dependent invasion in oesophageal adenocarcinoma. J Pathol. 2015;235:466‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alcaraz J, Carrasco JL, Millares L, et al. Stromal markers of activated tumor associated fibroblasts predict poor survival and are associated with necrosis in non‐small cell lung cancer. Lung Cancer. 2019;135:151‐160. [DOI] [PubMed] [Google Scholar]
- 45. Fujita H, Ohuchida K, Mizumoto K, et al. α‐Smooth muscle actin expressing stroma promotes an aggressive tumor biology in pancreatic ductal adenocarcinoma. Pancreas. 2010;39:1254‐1262. [DOI] [PubMed] [Google Scholar]
- 46. Sinn M, Denkert C, Striefler J, et al. α‐Smooth muscle actin expression and desmoplastic stromal reaction in pancreatic cancer: results from the CONKO‐001 study. Br J Cancer. 2014;111:1917‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valach J, Fík Z, Strnad H, et al. Smooth muscle actin‐expressing stromal fibroblasts in head and neck squamous cell carcinoma: Increased expression of galectin‐1 and induction of poor prognosis factors. Int J Cancer. 2012;131:2499‐2508. [DOI] [PubMed] [Google Scholar]
- 48. Yamashita M, Ogawa T, Zhang X, et al. Role of stromal myofibroblasts in invasive breast cancer: stromal expression of alpha‐smooth muscle actin correlates with worse clinical outcome. Breast cancer. 2012;19:170‐176. [DOI] [PubMed] [Google Scholar]
- 49. Maeda K, Enomoto A, Hara A, et al. Identification of Meflin as a potential marker for mesenchymal stromal cells. Sci Rep. 2016;6:22288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mizutani Y, Kobayashi H, Iida T, et al. Meflin‐positive cancer‐associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019;79:5367‐5381. [DOI] [PubMed] [Google Scholar]
- 51. Fujiwara K, Ohuchida K, Mizumoto K, et al. CD271⁺ subpopulation of pancreatic stellate cells correlates with prognosis of pancreatic cancer and is regulated by interaction with cancer cells. PLoS ONE. 2012;7:e52682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guerra C, Schuhmacher AJ, Cañamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K‐Ras oncogenes in adult mice. Cancer Cell. 2007;11:291‐302. [DOI] [PubMed] [Google Scholar]
- 53. Aichler M, Seiler C, Tost M, et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J Pathol. 2012;226:723‐734. [DOI] [PubMed] [Google Scholar]
- 54. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469‐483. [DOI] [PubMed] [Google Scholar]
- 55. Hara A, Kobayashi H, Asai N, et al. Roles of the mesenchymal stromal/stem cell marker Meflin in cardiac tissue repair and the development of diastolic dysfunction. Circ Res. 2019;125:414‐430. [DOI] [PubMed] [Google Scholar]
- 56. Erez N, Truitt M, Olson P, Hanahan D. Cancer‐associated fibroblasts are activated in incipient neoplasia to orchestrate tumor‐promoting inflammation in an NF‐κB‐dependent manner. Cancer Cell. 2010;17:135‐147. [DOI] [PubMed] [Google Scholar]
- 57. Ishii G, Hashimoto H, Asada K, et al. Fibroblasts associated with cancer cells keep enhanced migration activity after separation from cancer cells: a novel character of tumor educated fibroblasts. Int J Oncol. 2010;37:317‐325. [DOI] [PubMed] [Google Scholar]
- 58. Kobayashi N, Yasu T, Ueba H, et al. Mechanical stress promotes the expression of smooth muscle‐like properties in marrow stromal cells. Exp Hematol. 2004;32:1238‐1245. [DOI] [PubMed] [Google Scholar]
- 59. Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146‐155. [DOI] [PubMed] [Google Scholar]
- 60. Northey JJ, Przybyla L, Weaver VM. Tissue force programs cell fate and tumor aggression. Cancer Discov. 2017;7:1224‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leight JL, Drain AP, Weaver VM. Extracellular matrix remodeling and stiffening modulate tumor phenotype and treatment response. Annu Rev Cancer Biol. 2017;1:313‐334. [Google Scholar]
- 62. Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F III. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29:11‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Galland S, Stamenkovic I. Mesenchymal stromal cells in cancer: a review of their immunomodulatory functions and dual effects on tumor progression. J Pathol. 2019; 10.1002/path.5357 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557‐563. [DOI] [PubMed] [Google Scholar]
- 65. Melzer C, von der Ohe J, Hass R. Concise review: crosstalk of mesenchymal stroma/stem‐like cells with cancer cells provides therapeutic potential. Stem Cells. 2018;36:951‐968. [DOI] [PubMed] [Google Scholar]
- 66. Barcellos‐de‐Souza P, Gori V, Bambi F, Chiarugi P. Tumor microenvironment: bone marrow‐mesenchymal stem cells as key players. Biochim Biophys Acta. 2013;1836:321‐335. [DOI] [PubMed] [Google Scholar]
- 67. Mathew E, Brannon AL, Del Vecchio A, et al. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia. 2016;18:142‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Waghray M, Yalamanchili M, Dziubinski M, et al. GM‐CSF mediates mesenchymal–epithelial cross‐talk in pancreatic cancer. Cancer Discov. 2016;6:886‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Quante M, Tu SP, Tomita H, et al. Bone marrow‐derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Albrengues J, Bertero T, Grasset E, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer‐associated fibroblasts. Nat Commun. 2015;6:10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. De Witte SF, Peters FS, Merino A, et al. Epigenetic changes in umbilical cord mesenchymal stromal cells upon stimulation and culture expansion. Cytotherapy. 2018;20:919‐929. [DOI] [PubMed] [Google Scholar]
- 72. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kramann R, DiRocco DP, Humphreys BD. Understanding the origin, activation and regulation of matrix‐producing myofibroblasts for treatment of fibrotic disease. J Pathol. 2013;231:273‐289. [DOI] [PubMed] [Google Scholar]
- 74. Kendall RT, Feghali‐Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zeisberg M, Hanai J‐I, Sugimoto H, et al. BMP‐7 counteracts TGF‐β1–induced epithelial‐to‐mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964‐968. [DOI] [PubMed] [Google Scholar]
- 76. Redfield MM. Heart failure with preserved ejection fraction. N Engl J Med. 2016;375:1868‐1877. [DOI] [PubMed] [Google Scholar]
- 77. Appunni S, Anand V, Khandelwal M, Gupta N, Rubens M, Sharma A. Small leucine rich proteoglycans (decorin, biglycan and lumican) in cancer. Clin Chim Acta. 2019;491:1‐7. [DOI] [PubMed] [Google Scholar]
- 78. Zhang K, Zhang Y, Gu L, et al. Islr regulates canonical Wnt signaling‐mediated skeletal muscle regeneration by stabilizing Dishevelled‐2 and preventing autophagy. Nat Commun. 2018;9:5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sherman MH, Ruth TY, Engle DD, et al. Vitamin D receptor‐mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chronopoulos A, Robinson B, Sarper M, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630. [DOI] [PMC free article] [PubMed] [Google Scholar]