

Abstract
Protein prenylation is essential for many cellular processes including signal transduction, cytoskeletal reorganization, and membrane trafficking. Here, we identify a novel type of protein prenyltransferase, which we named geranylgeranyltransferase type‐III (GGTase‐III). GGTase‐III consists of prenyltransferase alpha subunit repeat containing 1 (PTAR1) and the β subunit of RabGGTase. Using a biotinylated geranylgeranyl analogue, we identified the Golgi SNARE protein Ykt6 as a substrate of GGTase‐III. GGTase‐III transfers a geranylgeranyl group to mono‐farnesylated Ykt6, generating doubly prenylated Ykt6. The crystal structure of GGTase‐III in complex with Ykt6 provides structural basis for Ykt6 double prenylation. In GGTase‐III‐deficient cells, Ykt6 remained in a singly prenylated form, and the Golgi SNARE complex assembly was severely impaired. Consequently, the Golgi apparatus was structurally disorganized, and intra‐Golgi protein trafficking was delayed. Our findings reveal a fourth type of protein prenyltransferase that generates geranylgeranyl‐farnesyl Ykt6. Double prenylation of Ykt6 is essential for the structural and functional organization of the Golgi apparatus.
Keywords: Golgi, protein prenylation, PTAR1, SNARE, Ykt6
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics;
A new type of protein prenyltransferase formed by PTAR1 and RabGGTβ heterodimers drives double prenylation of Golgi SNARE Ykt6, which is critical for Golgi complex maintenance and function in mammalian cells.

Introduction
Prenylation is one of the most common post‐translational lipid modifications found in eukaryotic cells (Wang & Casey, 2016). Bioinformatics and chemical biology studies predict that approximately 1% of mammalian proteins are modified with prenyl groups (Maurer‐Stroh et al, 2007; Nguyen et al, 2009). Protein prenylation involves the attachment of a 15‐carbon farnesyl group or a 20‐carbon geranylgeranyl group to a cysteine residue of target proteins through a thioether linkage. These prenyl groups are synthesized as farnesyl pyrophosphate (FPP) or geranylgeranyl pyrophosphate (GGPP) in the cholesterol synthesis pathway. Protein prenylation is essential for the membrane localization of target proteins and their proper biological functions.
Studies in the early 1990s have identified three types of protein prenyltransferases: farnesyltransferase (FTase), geranylgeranyltransferase type‐I (GGTase‐I), and Rab geranylgeranyltransferase (RabGGTase, also called GGTase‐II) (Reiss et al, 1990; Seabra et al, 1991, 1992a,b). FTase and GGTase‐I transfer a farnesyl group and a geranylgeranyl group, respectively, to a cysteine residue at the fourth position from the C‐terminus of protein substrates (Lane & Beese, 2006). This sequence is called the CAAX motif, where C is cysteine, A is usually an aliphatic residue, and X is any amino acid. FTase prefers Ser, Met, or Gln at the X position, whereas GGTase‐I prefers Leu or Phe. After prenylation, the CAAX proteins are further processed by two enzymes that reside in the endoplasmic reticulum (ER) membrane. Ras converting enzyme 1 (RCE1) cleaves the AAX tripeptide, and then, isoprenylcysteine methyltransferase (ICMT) methylates the exposed carboxyl group of the prenylcysteine using S‐adenosyl methionine (SAM) as a methyl donor. These two post‐prenylation steps make the C‐terminus of CAAX proteins more hydrophobic and facilitate their stable membrane attachment. Proteins farnesylated by FTase include Ras proteins, nuclear lamins, several protein kinases and phosphatases, and a few centromere proteins. The Golgi SNARE protein Ykt6 is also known to be farnesylated by FTase (Fukasawa et al, 2004). Proteins geranylgeranylated by GGTase‐I include the γ subunits of trimeric G proteins, members of the Rho family of small GTPases, and many other regulatory proteins.
RabGGTase, the third identified enzyme, exclusively modifies members of the Rab family of small GTPases (Leung et al, 2006), which have diverse C‐terminal sequences that typically contain closely located two cysteines (e.g., CC, CCXX, CXC). In contrast to the CAAX prenyltransferases, RabGGTase cannot recognize Rab proteins directly. Instead, RabGGTase recognizes them through an accessory molecule called Rab escort protein (REP) that binds unmodified Rab and presents it to the enzyme (Seabra et al, 1992a; Andres et al, 1993; Alexandrov et al, 1994). This indirect mode of substrate recognition enables RabGGTase to modify the various C‐terminal cysteine motifs of Rab proteins. RabGGTase can doubly geranylgeranylate Rab proteins, and REP delivers the di‐geranylgeranylated Rab to its destination membrane. In mammals, there are two closely related REP isoforms, REP1 and REP2. Mutations in the CHM gene, which encodes REP1, lead to hereditary retinal degeneration known as choroideremia due to under‐prenylation of Rab proteins including Rab27A (Seabra et al, 1993, 1995; Rak et al, 2004).
All of the protein prenyltransferases exist as heterodimers composed of tightly bound α and β subunits (Lane & Beese, 2006; Leung et al, 2006). FTase and GGTase‐I share a common α subunit but have distinct β subunits. RabGGTase consists of its own α and β subunits. REP interacts specifically with the α subunit of RabGGTase (Pylypenko et al, 2003). Structural studies have revealed that the three enzymes have highly similar structures (Park et al, 1997; Zhang et al, 2000; Long et al, 2002; Taylor et al, 2003; Guo et al, 2008), despite relatively low amino acid sequence conservation (15–30%). The α subunits adopt crescent‐shaped helical hairpin structures. The β subunits form α‐α barrel structures with a deep central cavity that harbors the catalytic site. All protein prenyltransferases are metalloenzymes that contain a zinc ion in the catalytic site.
SNAREs constitute a family of membrane proteins that acts as the minimal protein machinery for intracellular membrane fusion (Malsam & Sollner, 2011). Vesicular membrane‐ and target membrane‐localized SNAREs assemble into four‐helix bundles, called the trans‐SNARE complexes, to drive fusion of the two membranes (Weber et al, 1998). Most SNARE proteins have a transmembrane domain at the C‐terminus and localize to distinct membrane compartments via the C‐terminal anchor. Ykt6 is a unique SNARE that has no transmembrane domain but instead has a conserved double cysteine motif at the C‐terminus (C194C195AIM) (McNew et al, 1997). It has been shown that farnesylation of Cys195, at the fourth position from the C‐terminus, is required for Ykt6 localization to the Golgi membrane (Fukasawa et al, 2004).
Interestingly, the human genome encodes a poorly characterized protein with significant homology to the known prenyltransferase α subunits. This protein, designated as prenyltransferase alpha subunit repeat containing 1 (PTAR1), is highly conserved from fruit fly to mammals. A recent structural study suggested that PTAR1 can act as a prenyltransferase α subunit (Kuchay et al, 2019); however, the molecular function of PTAR1 remains unclear. In this study, we show that PTAR1 exists as a heterodimer with the β subunit of RabGGTase, which we term geranylgeranyltransferase type‐III (GGTase‐III). Using an unbiased approach, we identify the Golgi SNARE protein Ykt6 as a substrate of GGTase‐III. Our biochemical and structural analyses reveal that GGTase‐III recognizes Cys195‐farnesylated Ykt6 and attaches a geranylgeranyl group to Cys194, generating doubly prenylated Ykt6. Double prenylation is essential for Ykt6 to function as a Golgi SNARE, and the loss of doubly prenylated Ykt6 leads to disorganization of the Golgi apparatus.
Results
PTAR1 exists as a heterodimeric complex with the β subunit of RabGGTase
Human PTAR1 is a 429 amino acid protein with a predicted size of 49 kDa. PTAR1 is highly conserved across species; the mouse, zebrafish, and fruit fly orthologues show 88, 62, and 32% amino acid sequence identity to human PTAR1, respectively (Appendix Fig S1). Immunoblot analysis showed that PTAR1 was ubiquitously expressed in rat tissues with highest expression in the kidney (Fig 1A). The homology of PTAR1 to the known prenyltransferase α subunits suggested that PTAR1 also functions as a heterodimer. We purified FLAG‐PTAR1 binding proteins from HeLa S3 cell cytosol and found that the β subunit of RabGGTase bound to FLAG‐PTAR1 (Fig 1B). We confirmed that endogenous PTAR1 was complexed with RabGGTβ in rat brain and thymus cytosol, as shown by co‐immunoprecipitation assays (Fig 1C). To further characterize this complex, we fractionated rat brain cytosol by anion exchange chromatography and analyzed the column fractions with antibodies against RabGGTα, RabGGTβ, and PTAR1. Interestingly, RabGGTβ was eluted in two peaks; the earlier minor peak (peak A, Fig 1D) contained PTAR1 but not RabGGTα, whereas the later major peak (peak B, Fig 1D) contained RabGGTα but not PTAR1. When PTAR1 was immunodepleted from the peak A fraction, RabGGTβ was concomitantly depleted (Fig 1E). These results indicate that RabGGTβ is shared not only by RabGGTα but also by PTAR1. Quantification of the band intensities of RabGGTβ in the peak A and B fractions indicates that ~ 18% of RabGGTβ is complexed to PTAR1 in rat brain cytosol. In gel filtration chromatography, the brain PTAR1–RabGGTβ complex eluted in the same position (~ 90 kDa) as the recombinant complex purified from Sf9 insect cells (Fig 1F and G), suggesting that the endogenous complex contains no other accessory proteins. Collectively, these results demonstrate the existence of a fourth protein prenyltransferase. We term this complex geranylgeranyltransferase type‐III (GGTase‐III). As summarized in Fig 1H, the prenyltransferase family comprises three α subunit isoforms and three β subunit isoforms. Their combination makes four distinct prenyltransferase complexes.
Figure 1. PTAR1 exists as a heterodimeric complex with the β subunit of RabGGTase.
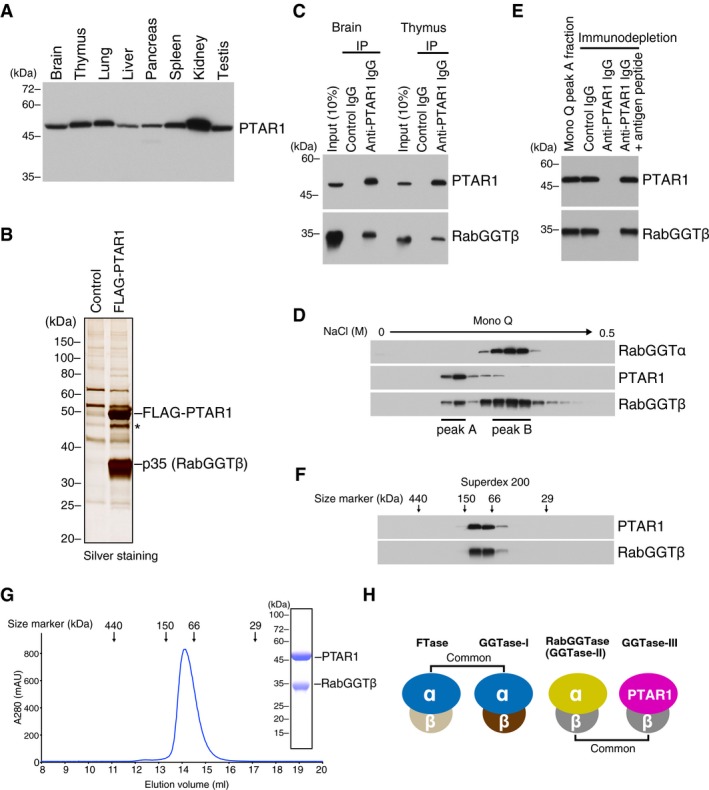
-
APTAR1 protein expression in rat tissues. Proteins extracted from various rat tissues (15 μg each) were analyzed by immunoblotting with antibodies against PTAR1.
-
BIdentification of RabGGTβ as a binding protein of PTAR1. Anti‐FLAG immunoprecipitates from control HeLa S3 cells or FLAG‐PTAR1‐expressing HeLa S3 cells were analyzed by SDS–PAGE and silver staining. The 35 kDa protein was identified as RabGGTβ by mass spectrometry. The asterisk denotes a degradation product of FLAG‐PTAR1.
-
CCo‐immunoprecipitation of endogenous PTAR1 and RabGGTβ from rat brain and thymus cytosol.
-
DFractionation of rat brain cytosol by Mono Q anion exchange column chromatography. Column fractions were analyzed by immunoblotting with antibodies against RabGGTα, PTAR1, and RabGGTβ. RabGGTβ eluted in two peaks (peak A and peak B).
-
ECo‐immunodepletion of PTAR1 and RabGGTβ by anti‐PTAR1 IgG. PTAR1 was immunodepleted from the Mono Q peak A fraction using anti‐PTAR1 IgG‐bound protein A beads, and the supernatant was analyzed by immunoblotting with antibodies against PTAR1 and RabGGTβ. Normal rabbit IgG and anti‐PTAR1 IgG preabsorbed with the antigen peptide were used as controls.
-
FFractionation of the Mono Q peak A fraction by Superdex 200 gel filtration chromatography. Column fractions were analyzed by immunoblotting with antibodies against PTAR1 and RabGGTβ.
-
GGel filtration chromatography of the recombinant PTAR1–RabGGTβ complex purified from Sf9 insect cells on a Superdex 200 column. Inset shows SDS–PAGE and Coomassie staining analysis of the purified complex.
-
HSchematic diagram of the protein prenyltransferase family. FTase and GGTase‐I share the same α subunit. RabGGTase and the PTAR1–RabGGTβ complex (GGTase‐III) share the same β subunit.
Source data are available online for this figure.
GGTase‐III has no prenyltransferase activity on known prenyltransferase substrates
We first examined the possible prenyltransferase activity of GGTase‐III using known prenyltransferase substrates, Ras, Rho, and Rab. The results demonstrated that GGTase‐III had no detectable CAAX prenyltransferase activity on H‐Ras (Fig EV1A) or RhoA (Fig EV1B). Despite sharing the same β subunit with RabGGTase, GGTase‐III failed to geranylgeranylate Rab proteins including Rab1A, Rab5A, and Rab11A even in the presence of REP1 or REP2 (Fig EV1C and D). We could not detect an interaction between GGTase‐III and REPs (Fig EV1E). Thus, GGTase‐III appeared to have no activity on substrates of known prenyltransferases. These results led us to expect that GGTase‐III has its own substrates.
Figure EV1. GGTase‐III has no prenyltransferase activity on known prenyltransferase substrates.
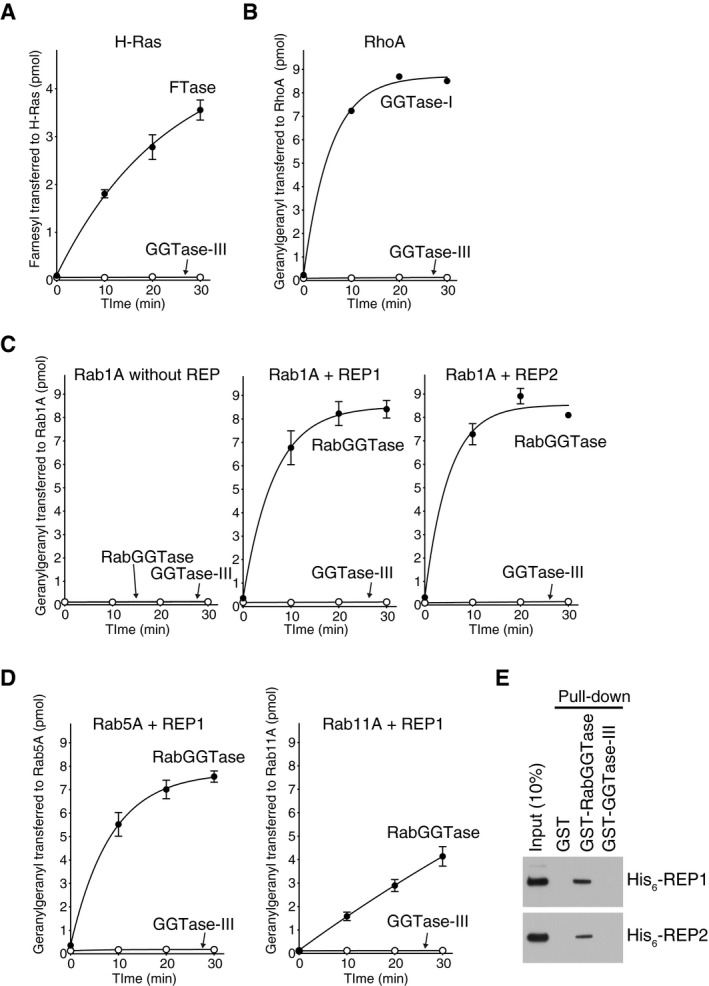
-
A–DPrenylation activity of GGTase‐III on Ras, Rho, and Rab proteins. (A) His6‐H‐Ras (5 μM) was incubated with FTase (50 nM) or GGTase‐III (50 nM) and 3H‐FPP (1 μM) at 37°C. Reactions were stopped at the indicated time points, and the amount of 3H‐farnesyl transferred to H‐Ras was quantified by scintillation counting (mean ± SEM, n = 3). (B) His6‐RhoA (5 μM) was incubated with GGTase‐I (50 nM) or GGTase‐III (50 nM) and 3H‐GGPP (1 μM), and the amount of 3H‐geranylgeranyl transferred to RhoA was quantified (mean ± SEM, n = 3). (C) His6‐Rab1A (5 μM) was incubated with RabGGTase (50 nM) or GGTase‐III (50 nM) and 3H‐GGPP (1 μM) in the absence or presence of either His6‐REP1 (100 nM) or His6‐REP2 (100 nM), and the amount of 3H‐geranylgeranyl transferred to Rab1A was quantified (mean ± SEM, n = 3). (D) His6‐Rab5A and His6‐Rab11A were incubated with RabGGTase (50 nM) or GGTase‐III (50 nM) and 3H‐GGPP (1 μM) in the presence of His6‐REP1 (100 nM), and the amount of 3H‐geranylgeranyl transferred to Rab5A and Rab11A was quantified (mean ± SEM, n = 3).
-
EREP pull‐down assay. Recombinant His6‐REP1 or His6‐REP2 was incubated with glutathione Sepharose beads coated with GST, GST‐RabGGTase, or GST‐GGTase‐III. Bound His6‐REP proteins were analyzed by immunoblotting with anti‐His6 antibody.
Source data are available online for this figure.
Identification of Ykt6 as a GGTase‐III substrate
To identify substrate proteins of GGTase‐III, we used biotin‐geranyl pyrophosphate (BGPP), a biotinylated analogue of GGPP (Fig 2A). It has been shown that RabGGTase can use BGPP as a prenyl donor and transfer the biotinylated geranyl moiety to Rab proteins (Nguyen et al, 2009). We reasoned that if GGTase‐III also can use BGPP as a prenyl donor, substrate proteins would be labeled with the biotin tag and can easily be detected. Figure 2B shows our strategy. First, we treated HeLa S3 cells with simvastatin, an inhibitor of the cholesterol synthesis pathway, to deplete prenyl pyrophosphates in the cells. This treatment would keep the putative substrate proteins in unprenylated states. Then, a cytosolic fraction was prepared from the statin‐treated cells and applied to an affinity column containing GST‐GGTase‐III (consisting of GST‐PTAR1 and RabGGTβ) to concentrate candidate substrate proteins. Bound proteins were eluted with a high salt buffer, dialyzed, and incubated with recombinant GGTase‐III and BGPP. The reaction products were purified by avidin beads and analyzed by protein blotting using horseradish peroxidase (HRP)‐conjugated avidin to detect biotinylated proteins. We detected a single, specific band at 25 kDa (Fig 2C). Mass spectrometry identified the protein as Ykt6 (Table EV1). Bacterially purified recombinant Ykt6 was indeed biotin‐geranylated by GGTase‐III, but not by RabGGTase, in the presence of BGPP (Fig 2D).
Figure 2. Identification of Ykt6 as a substrate of GGTase‐III .
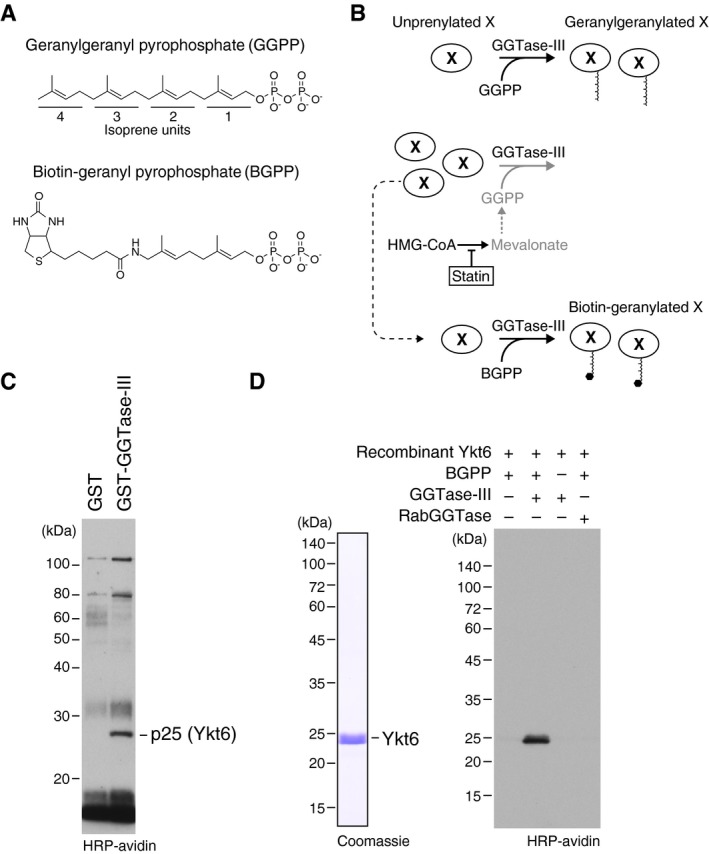
-
AMolecular structures of geranylgeranyl pyrophosphate (GGPP) and its biotinylated analogue biotin‐geranyl pyrophosphate (BGPP). Geranylgeranyl moiety consists of four repeats of 5‐carbon isoprene unit.
-
BPurification strategy for GGTase‐III substrate proteins using statin and BGPP. “X” represents a putative substrate protein of GGTase‐III. HMG‐CoA, hydroxymethylglutaryl‐CoA.
-
CIdentification of Ykt6 as a protein biotin‐geranylated by GGTase‐III. Cytosolic proteins extracted from statin‐treated HeLa S3 cells were applied to GST or GST‐GGTase‐III affinity columns. Bound proteins were eluted and incubated with recombinant GGTase‐III and BGPP. Reaction products were separated by SDS–PAGE, transferred to a nitrocellulose membrane, and probed with horseradish peroxidase (HRP)‐labeled avidin to detect biotinylated proteins. Mass spectrometry identified the 25 kDa protein as Ykt6.
-
DBiotin‐geranylation of recombinant Ykt6 by GGTase‐III. Bacterially produced recombinant untagged Ykt6 (left) was incubated with buffer, GGTase‐III, or RabGGTase in the absence or presence of BGPP for 30 min at 37°C. Biotin‐geranylated Ykt6 was detected as in (C).
Source data are available online for this figure.
GGTase‐III transfers a geranylgeranyl group to Cys194 of Cys195‐farnesyl Ykt6
Ykt6 is a SNARE protein regulating intra‐Golgi membrane trafficking (Parlati et al, 2002; Xu et al, 2002; Volchuk et al, 2004) and other membrane transport pathways including autophagosome‐vacuole/lysosome fusion (Bas et al, 2018; Gao et al, 2018; Matsui et al, 2018; Takats et al, 2018). Human Ykt6 is a 198 amino acid protein consisting of an N‐terminal longin domain and a C‐terminal SNARE domain (Fig 3A). Unlike most of the SNARE proteins, Ykt6 lacks a transmembrane domain, but instead has a prenylation motif containing two cysteines at its C‐terminus (C194C195AIM, Fig 3A). These two cysteines are universally conserved from yeast to mammals (Fig 3B). The second cysteine (Cys195), which conforms to the consensus CAAX sequence, has been shown to be farnesylated by FTase (Fukasawa et al, 2004). Although the first cysteine (Cys194) has been thought to be modified with a palmitoyl group (Fukasawa et al, 2004), the possibility of its prenylation has not been explored.
Figure 3. GGTase‐III geranylgeranylates Cys194 of Cys195‐farnesyl Ykt6.
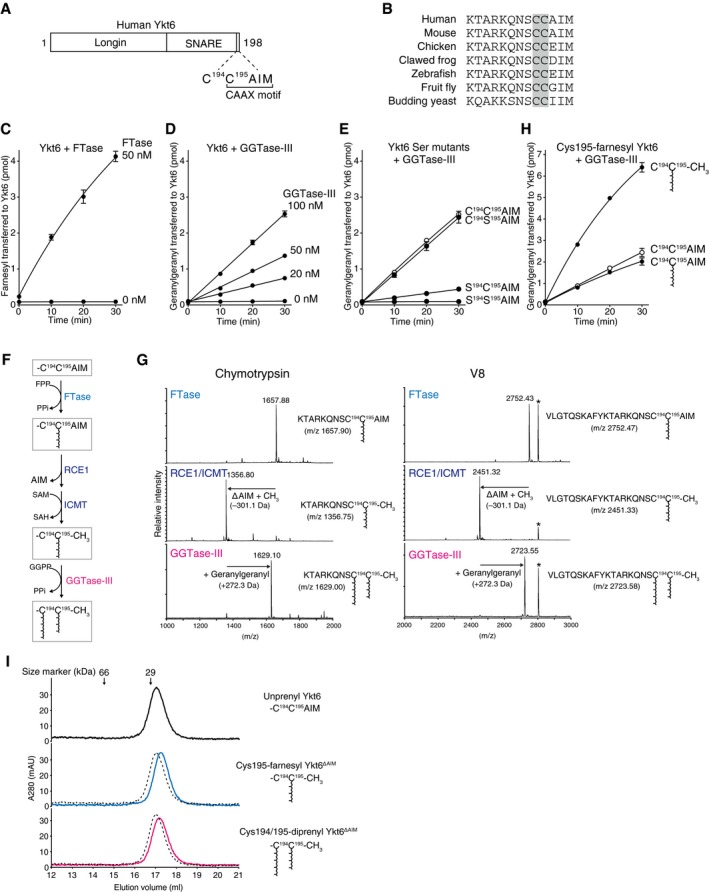
-
ASchematic diagram showing the domain structure of human Ykt6 protein. Ykt6 has two cysteine residues, Cys194 and Cys195, at the fifth and fourth positions from the C‐terminus.
-
BAlignment of the C‐terminal amino acid sequences of Ykt6 from various species. The conserved tandem cysteines are highlighted in gray.
-
CFarnesylation of recombinant Ykt6 by FTase. Recombinant Ykt6 (5 μM) was incubated with FTase (0 or 50 nM) and 3H‐FPP (1 μM; ˜ 3,500 dpm/pmol) at 37°C. Reactions were stopped at the indicated time points, and the amount of 3H‐farnesyl transferred to Ykt6 was quantified by scintillation counting (mean ± SEM, n = 3).
-
DGeranylgeranylation of recombinant Ykt6 by GGTase‐III. Recombinant Ykt6 (5 μM) was incubated with increasing concentrations of GGTase‐III (0–100 nM) and 3H‐GGPP (1 μM; ˜ 3,000 dpm/pmol) for the indicated times, and the amount of 3H‐geranylgeranyl transferred to Ykt6 was quantified (mean ± SEM, n = 3).
-
EGeranylgeranylation of Cys to Ser mutants of Ykt6 by GGTase‐III. WT and mutant Ykt6 proteins (5 μM each) were incubated with GGTase‐III (100 nM) and 3H‐GGPP (1 μM) for the indicated times, and the amount of 3H‐geranylgeranyl transferred to Ykt6 was quantified (mean ± SEM, n = 3).
-
FSequential prenylation process of Ykt6. SAM, S‐adenosylmethionine; SAH, S‐adenosylhomocysteine; PPi, inorganic pyrophosphate.
-
GMass spectra of prenylated Ykt6 peptides. Recombinant Ykt6 was farnesylated by FTase (FTase), further processed by RCE1 and ICMT (RCE1/ICMT), and finally geranylgeranylated by GGTase‐III (GGTase‐III). After purification to homogeneity, the prenylated Ykt6 proteins were digested with chymotrypsin (left) or V8 protease (right) and analyzed by MALDI‐TOF mass spectrometry. The calculated m/z values for the C‐terminal peptide ions are shown in parentheses. The peaks labeled with an asterisk correspond to amino acids 14–38 of Ykt6 (AKVVLLKAAYDVSSFSFFQRSSVQE; m/z 2,806.48).
-
HEffects of C‐terminal processing and Cys195 farnesylation on Cys194 geranylgeranylation by GGTase‐III. Purified recombinant unprenyl Ykt6, Cys195‐farnesyl Ykt6, and Cys195‐farnesyl Ykt6ΔAIM (5 μM each) were incubated with GGTase‐III (100 nM) and 3H‐GGPP (1 μM) for the indicated times, and the amount of 3H‐geranylgeranyl transferred to Ykt6 was quantified (mean ± SEM, n = 3).
-
IGel filtration chromatography of purified unprenyl Ykt6, Cys195‐farnesyl Ykt6ΔAIM, and Cys194/195‐diprenyl Ykt6ΔAIM (135 μg each) on a Superdex 200 column. Dotted lines indicate the elution profile of unprenyl Ykt6 for comparison. The prenylated Ykt6 proteins were eluted slightly later than the unprenylated Ykt6, suggesting more compact and stable folds.
We first confirmed that FTase efficiently farnesylated Ykt6 (Fig 3C). To test whether GGTase‐III can geranylgeranylate Ykt6, we incubated Ykt6 with 3H‐GGPP and increasing concentrations of GGTase‐III. We found that GGTase‐III was able to transfer 3H‐geranylgeranyl to Ykt6 (Fig 3D), albeit less efficiently than FTase. To determine which cysteine is geranylgeranylated by GGTase‐III, we prepared Ykt6 mutants in which either or both cysteines were mutated to serine. Interestingly, GGTase‐III geranylgeranylated the C194S195AIM mutant to the same extent as wild‐type (WT) Ykt6, but showed very low activity for the S194C195AIM mutant (Fig 3E), indicating that GGTase‐III has a preference for Cys194. Since Cys195 is farnesylated by FTase, this result raised the intriguing possibility that Ykt6 could be doubly prenylated in both cysteines by FTase and GGTase‐III. Because FTase cannot add a second prenyl group to a mono‐prenylated peptide (Lane & Beese, 2006), we considered the possibility that FTase first farnesylates Cys195 and then GGTase‐III geranylgeranylates Cys194 (Fig 3F).
To test this possibility, we enzymatically prepared recombinant Cys195‐farnesylated Ykt6 by incubating unmodified Ykt6 with FTase and an excess amount of FPP. Matrix‐assisted laser desorption ionization time‐of‐flight (MALDI‐TOF) mass spectrometry analysis of chymotrypsin‐ or V8 protease‐digested peptides showed uniform modification of the C‐terminal peptides with a farnesyl group (Fig 3G, FTase). Since prenylated CAAX proteins are sequentially processed by RCE1 and ICMT in vivo, we further incubated Cys195‐farnesyl Ykt6 with insect cell membranes containing recombinant RCE1 and ICMT in the presence of S‐adenosylmethionine (SAM). MALDI‐TOF analysis confirmed cleavage and methylation of the farnesylated peptides after incubation with RCE1 and ICMT (Fig 3G, RCE1/ICMT). The Cys195‐farnesylated, C‐terminally cleaved, and methylated form of Ykt6 (hereafter referred to as Cys195‐farnesyl Ykt6ΔAIM) was purified again to homogeneity and tested in the geranylgeranylation assay. Remarkably, Cys195‐farnesyl Ykt6ΔAIM was geranylgeranylated much more efficiently than the unprenylated form (Fig 3H). We also tested Cys195‐farnesylated Ykt6 with uncleaved C‐terminus and found that the cleavage of the AIM sequence was required for efficient Cys194 geranylgeranylation (Fig 3H). These results indicate that GGTase‐III preferentially recognizes the farnesylated ΔAIM form of Ykt6. Importantly, MALDI‐TOF analysis of fully geranylgeranylated Cys195‐farnesyl Ykt6ΔAIM showed a +272.3 Da mass shift (Fig 3G, GGTase‐III), which corresponds to the addition of a geranylgeranyl group. These results unambiguously demonstrate that GGTase‐III can attach a geranylgeranyl group to Cys194 of Cys195‐farnesyl Ykt6, thereby generating doubly prenylated Ykt6. Apart from Rab proteins, this is the first example of a protein modified with double prenyl groups.
Although Ykt6 is lipid‐modified, it mainly localizes to the cytosol (Fukasawa et al, 2004). Structural studies of rat Ykt6 in complex with a fatty acid dodecylphosphocholine (DPC) have shown that Ykt6 exists in the cytosol by adopting a closed, autoinhibited conformation with the SNARE motif wrapping around the longin domain (Wen et al, 2010). In the structure, the lipid molecule was bound in a hydrophobic groove formed between the longin domain and the SNARE motif, predicting that the Cys195‐linked farnesyl group could be sequestered in the hydrophobic groove. Surprisingly, the doubly prenylated recombinant Ykt6 was still water‐soluble and showed similar properties to the mono‐farnesylated form of Ykt6 in gel filtration chromatography (Fig 3I). This suggests that the second prenyl group could also be sequestered in the hydrophobic groove and shielded from the aqueous environment.
Crystal structures of GGTase‐III in apo form and in complex with Ykt6
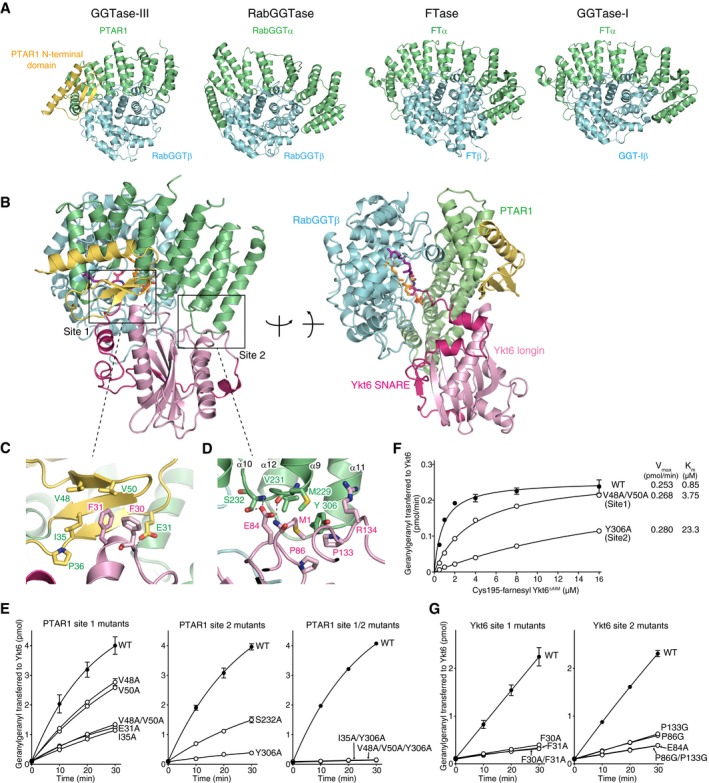
To gain insight into the structural features of GGTase‐III, we determined the crystal structure of GGTase‐III. As full‐length GGTase‐III produced no crystals in our initial crystallization trials, we subjected it to limited trypsin digestion to identify unstable regions and found that the complex containing C‐terminally truncated PTAR1 (residues 1–366) yielded diffraction‐quality crystals. PTAR1 exhibits a crescent‐like structure composed of α helices, which is similar to the α subunits of other prenyltransferases (Fig 4A and Appendix Table S1). Additionally, PTAR1 has a characteristic N‐terminal domain (residues 1–57) composed of an α helix and a three‐stranded β sheet (Fig 4A, yellow). This N‐terminal domain is unique among the prenyltransferase α subunits and highly conserved in PTAR1 orthologues (Appendix Fig S1). The β subunit adopts an α‐α barrel structure with a funnel‐shaped central cavity and binds to the concave surface of PTAR1. The structure of the β subunit is essentially identical to that of RabGGTase (Zhang et al, 2000; Guo et al, 2008).
Figure 4. Crystal structure of apo‐GGTase‐III and its complex with Cys195‐farnesyl Ykt6Δ AIM and GGPP .
-
AStructural comparison of GGTase‐III and other prenyltransferases. The α and β subunits of the prenyltransferases are shown in green and cyan, respectively. The unique N‐terminal domain of PTAR1 is shown in yellow.
-
BOverall structure of GGTase‐III complexed with Cys195‐farnesyl Ykt6ΔAIM and GGPP in two orientations. GGTase‐III is colored as in (A). The longin domain of Ykt6 is in pink, and SNARE domain is in magenta.
-
C, DMagnified views of site 1 (C) and site 2 (D) depicting the interaction between PTAR1 and Ykt6. Dashed lines indicate hydrogen bonds.
-
EGeranylgeranylation activity of WT GGTase‐III and the indicated mutants (100 nM each). Cys195‐farnesyl Ykt6ΔAIM (1 μM) and 3H‐GGPP (1 μM) were used as substrates (mean ± SEM, n = 3).
-
FKinetic analysis of WT GGTase‐III and the site 1 or site 2 mutant (100 nM each) using increasing concentrations of Cys195‐farnesyl Ykt6ΔAIM (mean ± SEM, n = 3 for WT and Y306A, n = 1 for V48A/V50A).
-
GGeranylgeranylation of unprenyl WT Ykt6 and the site 1 or site 2 mutants by GGTase‐III. WT and mutant Ykt6 proteins (5 μM each) were incubated with GGTase‐III (100 nM) and 3H‐GGPP (1 μM), and the amount of 3H‐geranylgeranyl transferred to Ykt6 was quantified (mean ± SEM, n = 3).
To elucidate the catalytic mechanism, we next sought to determine the structure of GGTase‐III in complex with Ykt6. To facilitate crystallization, we further truncated the C‐terminus of PTAR1 to Asn327. The truncated GGTase‐III was fully active compared to the full‐length enzyme produced in insect cells (Appendix Fig S2A). We first determined the crystal structure of GGTase‐III in complex with unprenyl Ykt6 and then in complex with Cys195‐farnesyl Ykt6 and GGPP. However, in these structures, we could not observe the electron density corresponding to the C‐terminus of Ykt6, possibly due to the presence of uncleaved AIM residues. Finally, using the ΔAIM form of Cys195‐farnesyl Ykt6 and GGPP, we successfully determined the complex structure including the terminal two cysteines of Ykt6 bound in the active site of the enzyme. Unexpectedly, the geranylgeranyl moiety of GGPP was transferred to Cys194 in the obtained structure (described below). Thus, the present structure likely represents a post‐catalytic state.
In the complex, Ykt6 binds primarily to PTAR1 in a closed conformation, where the SNARE domain forms three well‐separated helices and wraps around the longin domain as observed in the DPC‐bound Ykt6 structure (Fig 4B; Wen et al, 2010). Two binding interfaces were found between PTAR1 and Ykt6 (site 1 and site 2, Fig 4B). In site 1, the unique N‐terminal domain of PTAR1 forms a hydrophobic pocket consisting of Ile35, Pro36, Val48, and Val50, which accommodates Phe30 and Phe31 of Ykt6 (Fig 4C). The side chain of PTAR1 Glu31 forms a hydrogen bond with the main chain NH group of Ykt6 Phe30. In site 2, the Ykt6‐binding interface of PTAR1 is formed by the loops between α9 and α10 and between α11 and α12 (Fig 4D). The main chain CO group of PTAR1 Met229 forms a hydrogen bond with the terminal amino group of Ykt6 Met1. Either the main chain NH group or side chain OH group of PTAR1 Ser232 forms a hydrogen bond with the side chain carboxyl group of Ykt6 Glu84. Furthermore, Tyr306 of PTAR1 is accommodated in a hydrophobic pocket formed by Met1, Pro86, Pro133, and the aliphatic portion of Arg134 of Ykt6. Val231 of PTAR1 hydrophobically interacts with Met1 and the aliphatic portion of Glu84 of Ykt6.
To verify these interactions, we prepared a series of GGTase‐III mutants consisting of mutant PTAR1 and WT RabGGTβ (Appendix Fig S2B). We then tested their activities using Cys195‐farnesyl Ykt6ΔAIM and 3H‐GGPP as substrates. All of the site 1 mutants (E31A, I35A, V48A, V50A, and V48A/V50A) displayed significantly lower activity than WT enzyme (Fig 4E). The site 2 mutants (S232A and Y306A) also showed markedly reduced activity (Fig 4E). Simultaneous mutations in both sites (I35A/Y306A and V48A/V50A/Y306A) nearly completely abolished the enzyme activity (Fig 4E). Kinetic analysis showed that K m values for the site 1 mutant V48A/V50A and the site 2 mutant Y306A increased 4.4‐ and 27.4‐fold, respectively (Fig 4F). We also prepared Ykt6 site 1 and site 2 mutants (Appendix Fig S2C). When tested with WT GGTase‐III, all of the Ykt6 site 1 mutants (F30A, F31A, and F30A/F31A) and site 2 mutants (E84A, P86G, P133G, and P86G/P133G) showed substantially reduced geranylgeranyl acceptor activity (Fig 4G). Collectively, these results support the view that the two binding sites mediate the interaction between GGTase‐III and Ykt6.
Structural basis of Ykt6 double prenylation by GGTase‐III
The C‐terminal tail of Ykt6 (Thr187–Cys195) is stretched and inserted into the central cavity of the β subunit (Fig 5A). The electron densities of the farnesylated Cys195 and geranylgeranylated Cys194 are clearly visible (Fig 5B; simulated‐annealing F o–F c omit map contoured at 3σ level), although the density of the region between Thr187 and Ser193 is not observed, likely owing to its conformational flexibility. The geranylgeranyl moiety is accommodated in a lipid substrate binding pocket of the β subunit in the same conformation as observed in the GGPP‐bound GGTase‐III structure, except that the first two isoprene units (C1–C10) are shifted toward the catalytic site by rotating through the second isoprene unit (Figs 5C and D, and EV2A). This movement allows the formation of a covalent bond between the C1 carbon and the sulfur atom of Cys194. The geranylgeranylated Cys194 is located at the catalytic site formed by the conserved zinc‐coordinating residues of the β subunit, Asp238, Cys240, and His290 (Fig EV2B). This arrangement of Cys194 suggests that GGTase‐III has the same catalytic mechanism as FTase and GGTase‐I (Lane & Beese, 2006). Unexpectedly, however, the catalytic zinc ion is not coordinated in the GGTase‐III–product structure probably because it was chelated and removed by tartaric acid included in the crystallization condition.
Figure 5. Structural basis of Ykt6 double prenylation by GGTase‐III .
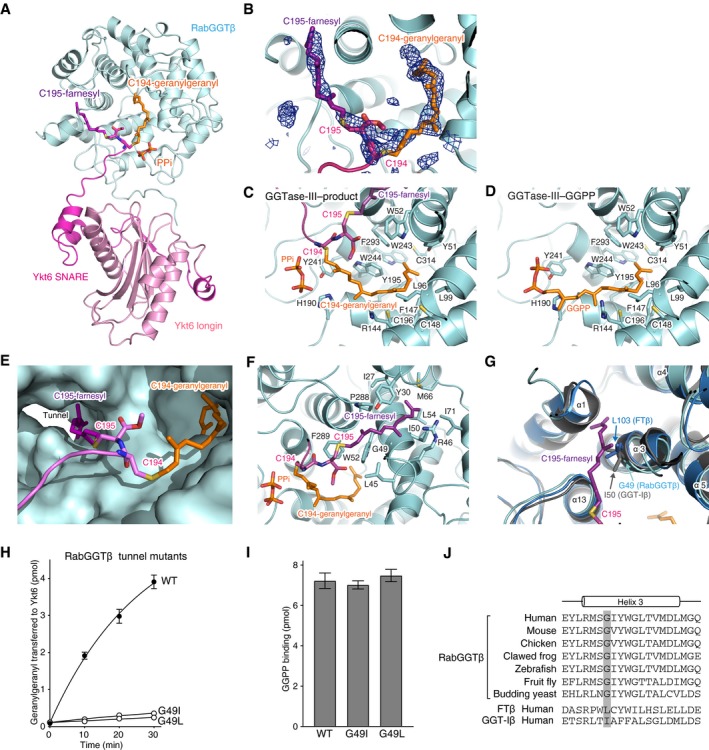
-
AOverall view of the central cavity of the β subunit showing insertion of the C‐terminal tail of Ykt6. PTAR1 is omitted for clarity. The Cys194‐linked geranylgeranyl and Cys195‐linked farnesyl moieties are shown in orange and purple, respectively. PPi, inorganic pyrophosphate.
-
BElectron density of the Cys194‐linked geranylgeranyl and Cys195‐linked farnesyl moieties (blue mesh; simulated‐annealing F o–F c omit map contoured at 3σ level).
-
C, DDetailed views of the geranylgeranyl moiety in the GGTase‐III–product complex (C) and GGPP in the GGTase‐III–GGPP complex (D). Residues of the β subunit that form the lipid substrate binding pocket are shown.
-
ESurface representation of the central cavity of the β subunit showing a tunnel formed near the active site. The Cys195‐linked farnesyl moiety is anchored into the hydrophobic tunnel.
-
FDetailed view of the Cys195‐linked farnesyl group bound in the hydrophobic tunnel. Residues of the β subunit that line the hydrophobic tunnel are shown.
-
GSuperposition of the β subunits of FTase (blue), GGTase‐I (gray), and GGTase‐III (cyan). In FTase and GGTase‐I, the hydrophobic tunnel is blocked by the side chain of Leu103 and Ile50, respectively. The corresponding position is replaced by Gly49 in GGTase‐III.
-
HGeranylgeranylation activity of WT GGTase‐III and tunnel mutants (100 nM each). Cys195‐farnesyl Ykt6ΔAIM (1 μM) and 3H‐GGPP (1 μM) were used as substrates (mean ± SEM, n = 3).
-
IGGPP binding to GGTase‐III. WT GGTase‐III and tunnel mutants (1 μM each) were incubated with 3H‐GGPP (2 μM) at 4°C for 10 min. After desalting, enzyme‐bound 3H‐GGPP was quantified by scintillation counting (mean ± SEM, n = 3).
-
JAlignment of the helix 3 sequences of RabGGTβ orthologues and human FTβ and GGT‐Iβ. The position of the conserved glycine residue (Gly49 in human) is highlighted in gray.
Figure EV2. Active site of GGTase‐III .
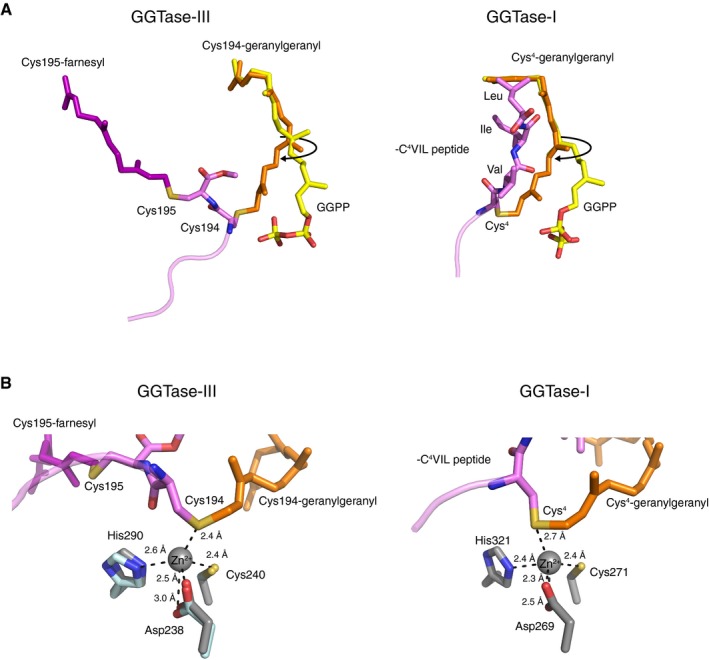
-
ALeft, comparison of the Cys194‐linked geranylgeranyl moiety (orange) in the GGTase‐III–product complex and GGPP (yellow) in the GGTase‐III–GGPP complex. In the product complex, the first and second isoprene units of GGPP are rotated to form a covalent bond with Cys194. The Cys195‐linked farnesyl moiety (purple) is anchored into the hydrophobic tunnel. Right, comparison of the geranylgeranyl moiety (orange) linked to Cys4 of C4VIL peptide in the GGTase‐I–product complex (PDB 1N4R) and GGPP (yellow) in the GGTase‐I–GGPP complex (PDB 1N4P). GGTase‐III and GGTase‐I show similar isoprene movements during catalysis.
-
BLeft, the catalytic zinc ion and zinc‐coordinating residues of RabGGTase (gray; PDB 3DST) superposed on the GGTase‐III–product complex (cyan). The sulfur atom of geranylgeranylated Cys194 is located in the vicinity of the superposed catalytic zinc ion. Right, the catalytic site of the GGTase‐I–product complex (PDB 1N4R). GGTase‐III has a similar active site arrangement as GGTase‐I.
Remarkably, the Cys195‐linked farnesyl moiety is deeply anchored into a hydrophobic tunnel formed near the active site (Fig 5E and F). This tunnel originates from a shallow hydrophobic groove corresponding to the “exit groove” of FTase and GGTase‐I and extends through the β subunit (Long et al, 2002; Taylor et al, 2003). In FTase and GGTase‐I, this tunnel is blocked by the branched side chain of Leu103 and Ile50, respectively, of their β subunits (Fig 5G). In GGTase‐III, these residues are substituted by Gly49, making a path that can accommodate a prenyl group. To test the importance of this farnesyl anchoring, we occluded the hydrophobic tunnel by replacing Gly49 of the β subunit with leucine or isoleucine, mimicking the β subunits of FTase and GGTase‐I. The mutant enzymes could no longer transfer the second prenyl group to the mono‐farnesylated Ykt6 (Fig 5H and Appendix Fig S2D). These mutations did not appear to affect the active site as GGPP bound normally to the mutants (Fig 5I). Thus, the unique hydrophobic tunnel of RabGGTβ plays an essential role in the second prenyl transfer most likely by anchoring the Cys195‐linked farnesyl moiety. This anchoring would enable proper positioning of the unmodified Cys194 in the active site. These structural findings are consistent with the results that GGTase‐III preferentially modifies Cys195‐farnesylated Ykt6 (Fig 3H). Gly49 of the β subunit is strictly conserved from yeast to mammals (Fig 5J), suggesting that the hydrophobic tunnel is a common structural feature of the RabGGTβ orthologues.
Native Ykt6 is doubly prenylated
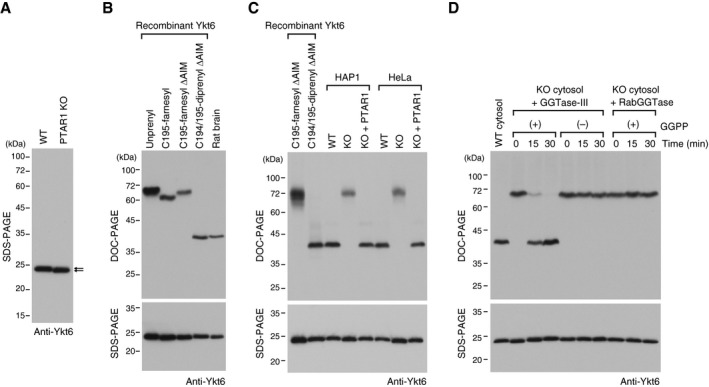
Although our biochemical and structural analyses indicate that recombinant Ykt6 can be doubly prenylated, the C‐terminal lipid modification status of native Ykt6 protein is unknown. To determine whether native Ykt6 is doubly prenylated, we generated PTAR1 knockout (KO) HAP1 and HeLa cell lines using CRISPR‐Cas9 genome editing and examined the prenylation status of Ykt6 in the cells. The established cell lines have frameshift mutations in the exon 2 of PTAR1 that introduce premature stop codons (Fig EV3A and B). Immunoblot analysis confirmed the absence of PTAR1 in these cells (Fig EV3C). In PTAR1 KO cells, Ykt6 was mainly localized to the cytosol as in WT cells (Fig EV3D). Interestingly, however, we noticed a slight difference in the electrophoretic mobility of Ykt6 between WT cells and PTAR1 KO cells (Fig 6A). Using this observation as a guide, we found that the prenylation status of Ykt6 can be clearly distinguished by a modified gel electrophoresis using deoxycholate (DOC) instead of sodium dodecyl sulfate (SDS). This method, termed DOC‐PAGE, revealed that the di‐prenylated form of recombinant Ykt6 migrated much faster than the unprenylated or mono‐farnesylated forms of Ykt6 (Fig 6B). This is presumably due to binding of the anionic detergent to the diprenyl moiety of Ykt6 during electrophoresis. Importantly, we found that native Ykt6 in rat brain cytosol migrated at exactly the same position as diprenyl Ykt6ΔAIM (Fig 6B). Endogenous Ykt6 in HAP1 and HeLa cells also migrated at the diprenyl position (Fig 6C). In contrast, Ykt6 in PTAR1 KO cells migrated at the monofarnesyl position, which was rescued by exogenous expression of PTAR1 (Fig 6C). The PTAR1 mutants unable to geranylgeranylate Ykt6 could not rescue this mobility shift (Fig EV3E).
Figure EV3. Generation of PTAR1 KO cells by CRISPR‐Cas9 genome editing.
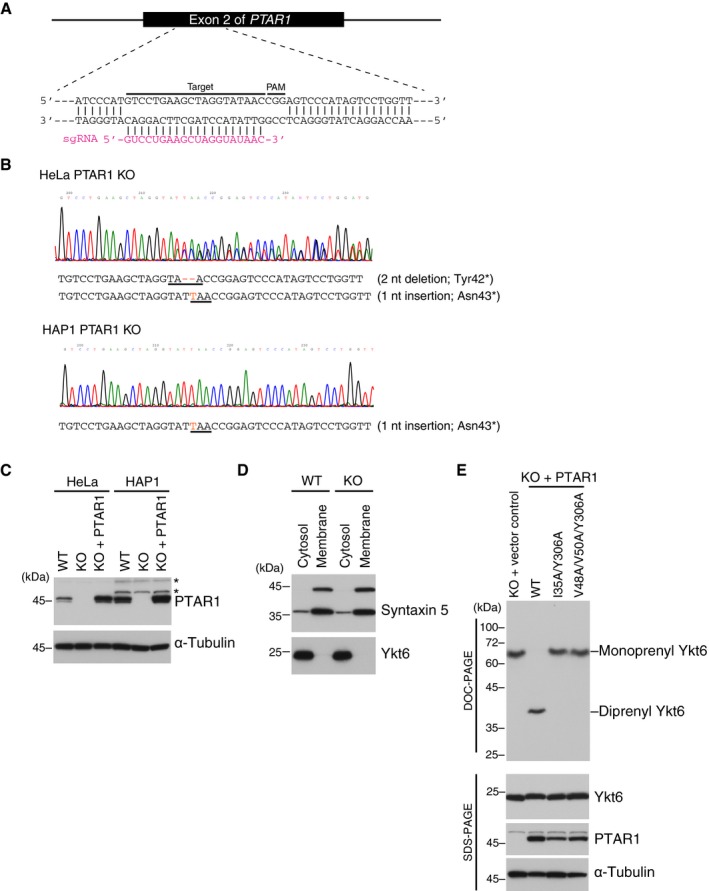
-
AGeneration of PTAR1 KO cell lines by CRISPR‐Cas9 using single‐guide RNA (sgRNA) targeting exon 2 of the human PTAR1 gene. The target sequence and PAM sequence are indicated.
-
BSequence data of the genomic region containing the exon 2 of PTAR1. Insertion and deletion mutations are shown in red, and the resulting premature stop codons are underlined. These mutations introduce a stop codon at the amino acid position 42 or 43. HAP1 is a haploid cell line and contains a single allele for PTAR1. nt, nucleotide.
-
CImmunoblot analysis of WT cells, PTAR1 KO cells, and PTAR1 KO cells stably expressing PTAR1 (KO + PTAR1) using anti‐PTAR1 antibody. Asterisks denote proteins that cross‐react with the antibody.
-
DCytosolic localization of Ykt6 in HeLa cells. WT and PTAR1 KO HeLa cells were fractionated into cytosol and membrane fractions, and analyzed by immunoblotting with anti‐syntaxin 5 and anti‐Ykt6 antibody. Syntaxin 5 has two isoforms with different translation initiation sites.
-
EPrenylation status of Ykt6 in PTAR1 KO HAP1 cells and PTAR1 KO HAP1 cells stably expressing WT PTAR1 or Yk6‐binding defective mutants of PTAR1. The prenylation status of Ykt6 was analyzed by DOC‐PAGE followed by immunoblotting with anti‐Ykt6 antibody. Lower panels show conventional immunoblot analysis of the same samples using the indicated antibodies.
Source data are available online for this figure.
Figure 6. Native Ykt6 is doubly prenylated.
-
AImmunoblot of cell lysates from WT HAP1 cells and PTAR1 KO HAP1 cells using anti‐Ykt6 antibody. Note the slight difference in the gel mobility of Ykt6 (arrows).
-
BPrenylation status of rat brain Ykt6. Recombinant unprenyl Ykt6, Cys195‐farnesyl Ykt6, Cys194/195‐diprenyl Ykt6 samples (1 ng each), and rat brain cytosol were electrophoresed on a polyacrylamide gel using deoxycholate (DOC)‐containing buffer and analyzed by immunoblotting with anti‐Ykt6 antibody (DOC‐PAGE; upper panel). The same samples were analyzed by conventional SDS–PAGE and immunoblotting with anti‐Ykt6 antibody (SDS–PAGE; lower panel).
-
CPrenylation status of Ykt6 in WT and PTAR1 KO cells. Recombinant Ykt6 samples and cell lysates of WT cells, PTAR1 KO cells, and PTAR1 KO cells stably expressing PTAR1 (KO + PTAR1) were separated by DOC‐PAGE (upper) or SDS–PAGE (lower), and analyzed by immunoblotting with anti‐Ykt6 antibody.
-
DIn vitro reconstitution of Ykt6 double prenylation. Dialyzed cytosol prepared from PTAR1 KO HAP1 cells was incubated at 37°C for the indicated times with recombinant GGTase‐III (100 nM) or RabGGTase (100 nM) in the absence or presence of GGPP (10 μM). After incubation, reaction products were separated by DOC‐PAGE (upper) or SDS–PAGE (lower), and analyzed by immunoblotting with anti‐Ykt6 antibody.
Source data are available online for this figure.
To demonstrate that the observed mobility shift is due to the absence of a geranylgeranyl moiety, we reconstituted the second prenylation reaction with PTAR1 KO cytosol and recombinant GGTase‐III. Incubation of PTAR1 KO cytosol with GGTase‐III, but not with RabGGTase, shifted the band at the monofarnesyl position to the diprenyl position in a time‐dependent manner (Fig 6D). This conversion was observed only in the presence of exogenously added GGPP (Fig 6D). SDS–PAGE analysis of the same samples showed no obvious differences (Fig 6D, lower panel), indicating that the band shift is not caused by proteolysis. These results provide strong evidence that native Ykt6 exists in the doubly prenylated form and GGTase‐III is responsible for the double prenylation.
GGTase‐III‐deficient cells exhibit abnormal Golgi morphology and function
Ykt6 is implicated in COPI‐dependent intra‐Golgi transport (Xu et al, 2002; Volchuk et al, 2004). To test the functional significance of Ykt6 double prenylation, we examined morphology and function of the Golgi apparatus by co‐immunostaining of GM130 (a cis‐Golgi marker) and golgin‐97 (a trans‐Golgi marker). In PTAR1 KO cells, the cis compartments were abnormally dilated and disorganized, and the trans compartments were embedded in the dilated cis compartments (Fig 7A). Electron microscopy analysis of PTAR1 KO cells revealed that the Golgi cisternae were swollen and did not form a tightly stacked structure (Fig 7B). Accumulation of unfused vesicles was observed around the swollen cisternae (Fig 7B).
Figure 7. GGTase‐III‐deficient cells exhibit structural and functional Golgi defects.
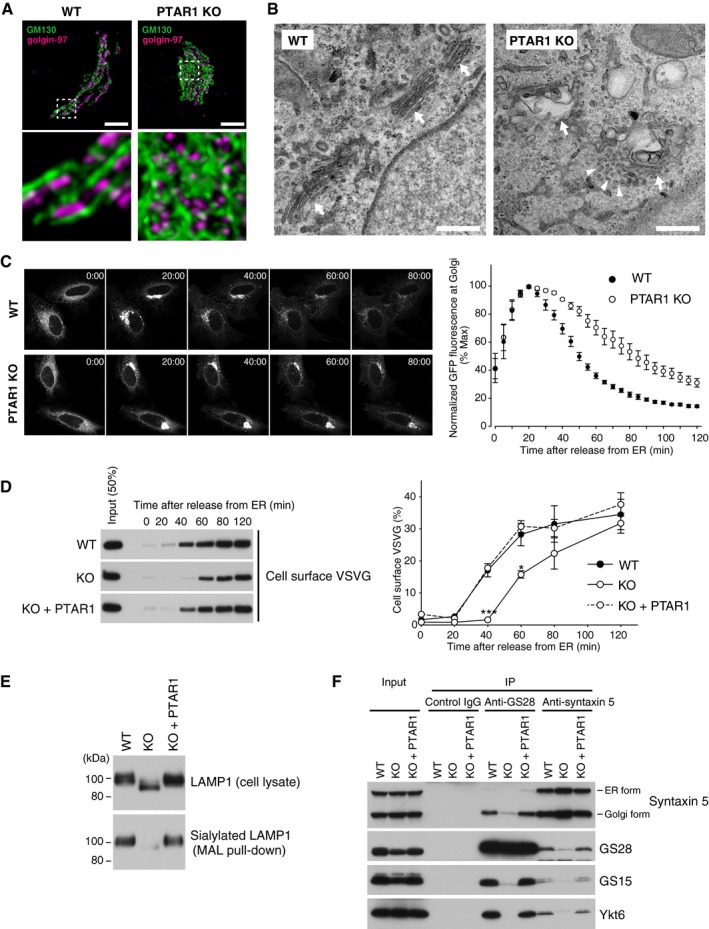
-
ARepresentative confocal images of the Golgi apparatus in WT or PTAR1 KO HeLa cells. Cells were co‐immunostained for GM130 (a cis‐Golgi marker) and golgin‐97 (a trans‐Golgi marker). Images were deconvoluted using Huygens software. The lower panels show magnified images of the boxed region of the upper panels. Scale bars, 5 μm.
-
BElectron micrographs of the Golgi apparatus in WT or PTAR1 KO HAP1 cells. Arrows indicate Golgi stacks. Arrowheads indicate examples of unfused vesicles accumulated around the swollen Golgi cisternae in PTAR1 KO cells. Scale bars, 500 nm.
-
C, DDefect in intra‐Golgi trafficking in PTAR1 KO cells. (C) VSVG‐GFP expressing WT and PTAR1 KO HeLa cells were cultured at 40°C and then shifted to 32°C. VSVG‐GFP fluorescence images taken at the indicated times after the temperature shift are shown. The right panel shows quantification of VSVG‐GFP fluorescence in the Golgi region after the temperature shift (mean ± SEM, n = 6). (D) VSVG‐GFP expressing WT HeLa cells, PTAR1 KO HeLa cells, and PTAR1 KO HeLa cells stably expressing PTAR1 (KO + PTAR1) were cultured at 40°C and then shifted to 32°C. At the indicated time points, cell surface proteins were biotinylated using sulfo‐NHS‐LC‐biotin. Biotinylated VSVG‐GFP was purified from cell lysates using avidin agarose and analyzed by immunoblotting with anti‐GFP antibody. The right panel shows quantification of the cell surface biotinylated VSVG‐GFP (means ± SEM, n = 3). Data were analyzed by one‐way ANOVA with Dunnett's post‐hoc test. *P < 0.05, ***P < 0.001.
-
EAnalysis of LAMP1 glycosylation. Cell lysates of WT HeLa cells, PTAR1 KO HeLa cells, and PTAR1 KO HeLa cells stably expressing PTAR1 (KO + PTAR1) were analyzed by immunoblotting with anti‐LAMP1 antibody (upper). Sialylated LAMP1 was precipitated from the cell lysates using Maackia amurensis leucoagglutinin (MAL) agarose and analyzed by immunoblotting (lower).
-
FDefect in the Golgi SNARE assembly in PTAR1 KO cells. The Golgi SNARE complex was immunoprecipitated from NEM‐treated WT HAP1 cells, PTAR1 KO HAP1 cells, and PTAR1 KO HAP1 cells stably expressing PTAR1 (KO + PTAR1) using control mouse IgG, anti‐GS28 IgG, or anti‐syntaxin 5 IgG. Immunoprecipitates were analyzed by immunoblotting with antibodies against syntaxin 5, GS28, GS15, and Ykt6. Syntaxin 5 has two isoforms with different translation initiation sites. Inputs were 20% (syntaxin 5, GS28, and GS15) and 2% (Ykt6). The data shown are representative of three independent experiments with similar results.
Source data are available online for this figure.
To investigate intra‐Golgi trafficking, we used a temperature‐sensitive mutant of vesicular stomatitis virus glycoprotein tagged with green fluorescent protein (VSVG‐GFP). The mutant VSVG protein is misfolded and retained in the ER at 40°C and released upon temperature shift to 32°C. After temperature reduction, VSVG‐GFP fluorescence in PTAR1 KO cells accumulated in the Golgi region with peak intensity at 20 min as observed in WT cells. However, the Golgi fluorescence signal decreased much more slowly in PTAR1 KO cells than in WT cells (Fig 7C), suggesting a defect in intra‐Golgi trafficking. Consistent with this, VSVG‐GFP arrival to the plasma membrane was significantly delayed in PTAR1 KO cells, as shown by cell surface biotinylation assays (Fig 7D). This delay was rescued by restoration of PTAR1 expression (Fig 7D). These observations indicate that PTAR1 KO cells have a functional defect in intra‐Golgi transport.
Next, we examined protein glycosylation in PTAR1 KO cells. Immunoblot analysis of LAMP1, a lysosomal membrane protein that is heavily sialylated in the Golgi, showed that LAMP1 in PTAR1 KO cells migrated faster than that in WT cells (Fig 7E). To examine sialylation of LAMP1, we used Maackia amurensis leucoagglutinin (MAL), an α2,3‐linked sialic acid‐binding lectin. We found that the amount of LAMP1 precipitated by MAL agarose was markedly reduced in PTAR1 KO cells (Fig 7E), indicating that sialylation of LAMP1 is greatly decreased in PTAR1 KO cells.
Doubly prenylated Ykt6 is required for the Golgi SNARE complex formation
Ykt6 forms a fusion‐competent trans‐SNARE complex with three Golgi‐resident SNAREs, syntaxin 5, GS28 (also known as GOS‐28), and GS15 (Parlati et al, 2002; Xu et al, 2002). We performed co‐immunoprecipitation assays to examine the functional importance of Ykt6 double prenylation in the Golgi SNARE complex assembly. Cells were pretreated with N‐ethylmaleimide (NEM) to prevent disassembly of once formed SNARE complexes. In WT cells, Ykt6 was stably co‐precipitated with syntaxin 5, GS28, and GS15 (Fig 7F). In contrast, the amounts of Ykt6 complexed with these partner SNAREs were severely decreased in PTAR1 KO cells (Fig 7F). Thus, double prenylation is essential for Ykt6 to participate in the Golgi SNARE complex assembly. This result suggests that the failure of the Golgi SNARE complex formation may underlie the structural and functional abnormality of the Golgi apparatus in PTAR1 KO cells.
Discussion
In this study, we have revealed that the Golgi SNARE protein Ykt6 undergoes sequential prenylation by FTase and the newly characterized enzyme GGTase‐III, which generates doubly prenylated Ykt6. This previously unrecognized SNARE double prenylation is essential for the maintenance of Golgi structure and function. GGTase‐III prenylates its substrate Ykt6 in a fundamentally different mode from other prenyltransferases. First, GGTase‐III directly recognizes Ykt6 through the α subunit PTAR1 (Fig 4B), whereas FTase and GGTase‐I recognize the last four amino acids of protein substrates mainly through the β subunits (Lane & Beese, 2006), and RabGGTase indirectly recognizes Rab proteins through REP (Leung et al, 2006). Second, GGTase‐III preferentially recognizes an already mono‐farnesylated form of Ykt6 and adds a second prenyl group to generate doubly prenylated Ykt6. Although di‐geranylgeranylation of Rab proteins is well‐established, this is the first example of a protein modified with both farnesyl and geranylgeranyl groups. This double prenylation would be catalyzed first by FTase, and then by GGTase‐III, because (i) Cys195 farnesylation of unmodified Ykt6 by FTase is much faster than Cys194 geranylgeranylation by GGTase‐III (Fig 3A and B) and (ii) GGTase‐III preferentially recognizes the farnesylated ΔAIM form of Ykt6 over the unprenylated form (Fig 3H). In GGTase‐III‐deficient cells, Ykt6 remains in a singly prenylated form (Fig 6C) and cannot participate in the Golgi SNARE complex assembly (Fig 7F), revealing that double prenylation is essential for the function of Ykt6.
Although our biochemical and structural analyses provide clear evidence that Ykt6 is doubly prenylated, Fukasawa et al (2004) have shown that Cys194 of human Ykt6 could be palmitoylated. However, this seemingly contradictory observation can be reconciled with our findings by considering the following experimental results. In the previous study, Cys194 palmitoylation was observed only in the N‐terminally tagged, F42E mutant of Ykt6. Our structural analysis reveals that Met1 of Ykt6 directly contacts with PTAR1 (Fig 4D), indicating that an N‐terminal tag on Ykt6 sterically interferes with PTAR1 binding. Indeed, GGTase‐III cannot geranylgeranylate Ykt6 with an N‐terminal His6 tag (Appendix Fig S2E). Furthermore, the F42E mutant is forced to adopt an open, activated conformation and cannot form a closed conformation (Wen et al, 2010). As shown in Fig 4B, GGTase‐III recognizes the closed form of Ykt6 and thus shows markedly reduced activity for the open form mutant (Appendix Fig S2E). These results indicate that, if an N‐terminally tagged, open form mutant of Ykt6 is expressed in cells, it will not be recognized by GGTase‐III and remain in a singly prenylated form. Considering that protein palmitoyltransferases have relatively relaxed substrate specificities (Roth et al, 2006) and are enriched in the Golgi membrane (Ernst et al, 2018), the observed Cys194 palmitoylation of Ykt6 could be a non‐physiological modification that occurred under conditions where GGTase‐III cannot function. Our results suggest that studies using tagged versions of Ykt6 and/or the open form mutant of Ykt6 may need to be carefully reconsidered as such constructs may not reflect the function of doubly prenylated Ykt6.
The finding that Ykt6 is doubly prenylated raises an important question as to how cytosolic Ykt6 is activated and recruited to the Golgi membrane. Structural studies and molecular dynamics simulations suggest that unprenylated Ykt6 exists in both open and closed conformations and farnesylation of Cys195 stabilizes it in a closed, autoinhibited state (Dai et al, 2016; Pylypenko et al, 2008; Wen et al, 2010; Weng et al, 2015; Fig EV4A). In the previous palmitoylation model, the open/closed transition of Ykt6 was explained by a palmitoylation/depalmitoylation cycle of Cys194. Upon palmitoylation of Cys194, Ykt6 becomes an open conformation and is inserted into the Golgi membrane. Conversely, depalmitoylation of Cys194 makes Ykt6 adopt the closed conformation, releasing it into the cytosol. However, since prenylation is irreversible, this model is incompatible with the finding that Ykt6 constitutively exists in the doubly prenylated form. Our biochemical analysis indicates that doubly prenylated Ykt6 is still in a soluble state (Fig 3I), suggesting that a different mechanism may regulate the open/closed transition of Ykt6 (Fig EV4A).
Figure EV4. Scheme of the double prenylation of Ykt6.
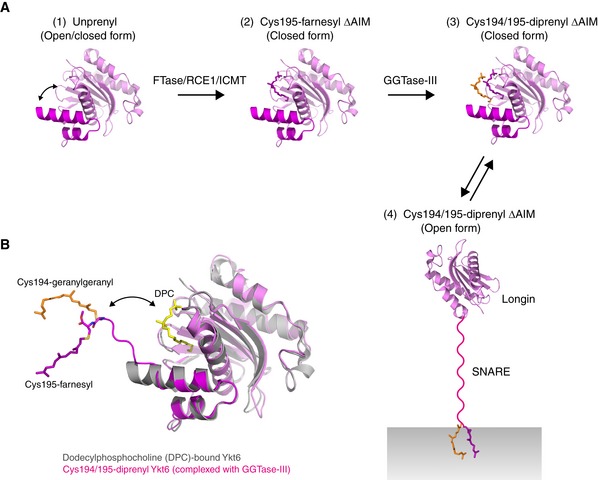
-
AModel for the sequential double prenylation of Ykt6. (1) Nascent unprenylated Ykt6 exists in both closed and open conformations. (2) Farnesylation of Cys195 stabilizes the closed conformation of Ykt6. (3) Doubly prenylated Ykt6 keeps the closed conformation by sequestering both farnesyl and geranylgeranyl groups into the putative prenyl binding groove. (4) Upon activation, the SNARE domain is unfolded and the C‐terminal two prenyl groups are inserted into the membrane.
-
BSuperposition of the dodecylphosphocholine (DPC)‐bound, closed form of Ykt6 (gray; PDB 3KYQ) and Cys194/195‐diprenyl Ykt6ΔAIM complexed to GGTase‐III (longin, pink; SNARE, magenta). GGTase‐III is omitted for clarity. The putative prenyl binding groove of Ykt6, occupied by DPC (yellow), is located closed to the active site of the enzyme. Upon binding to GGTase‐III, the Cys195‐linked farnesyl moiety accommodated in the putative prenyl binding groove of Ykt6 is translocated into the hydrophobic tunnel of the enzyme, allowing the transfer of geranylgeranyl moiety to Cys194. The attached two prenyl groups may easily translocate back to the prenyl binding groove of Ykt6.
Our biochemical and structural analyses provide a mechanistic model for Ykt6 double prenylation. As discussed above, nascent Ykt6 would be first processed by FTase and RCE1/ICMT and then recognized by GGTase‐III. The crystal structure of the GGTase‐III–Ykt6 complex shows that Ykt6 interacts with PTAR1 in a closed conformation (Fig 4B). In this binding mode, the C‐terminal helix of the SNARE domain is directed toward the central cavity of the enzyme. Since the putative prenyl binding groove of Ykt6 is positioned close to the active site (Fig EV4B), the Cys195‐linked farnesyl group of Ykt6 may easily translocate into the hydrophobic tunnel of the enzyme. The C‐terminal AIM sequence may sterically interfere with this translocation as it is inhibitory to the second prenyl transfer compared with the ΔAIM form (Fig 3H). The farnesyl anchoring into the hydrophobic tunnel would ensure stable attachment of the otherwise unstable C‐terminus of Ykt6 to the active site. After the second prenyl transfer, the two prenyl groups would detach from the enzyme, likely accompanied by binding of a new GGPP molecule to the lipid substrate binding site, and translocate back to the prenyl binding groove of Ykt6 (Fig EV4B). This is analogous to the function of REP that sequesters two geranylgeranyl groups of Rab proteins into its prenyl binding pocket following the second prenyl transfer (Rak et al, 2004).
This study reveals the first structure of RabGGTβ complexed with a prenylated peptide product, providing a possible explanation for the double prenylation mechanism of RabGGTase. RabGGTase can processively add two geranylgeranyl groups to Rab proteins in a manner that proceeds without dissociation of the monoprenyl intermediate (Thoma et al, 2001). However, it is unclear whether the first prenyl moiety is located in a defined binding site during the second prenyl transfer (Guo et al, 2008). Our structural results suggest that the first geranylgeranyl moiety of Rab proteins could be translocated into the hydrophobic tunnel of RabGGTβ. This anchoring may be particularly important for RabGGTase to accomplish double prenylation because the C‐terminal sequences of Rab proteins are various and cannot make specific contacts with the active site. Further, by varying the insertion lengths of the first geranylgeranyl moiety, the unmodified second cysteine at various positions within the C‐termini of Rab proteins may be positioned properly in the active site (Long et al, 2002). Our mutational analysis demonstrates a critical requirement for the invariant glycine residue (Gly49) located in helix 3 of RabGGTβ (Fig 5G). In the β subunits of FTase and GGTase‐I, the corresponding residue is always a larger amino acid across many species (leucine or cysteine in FTβ and isoleucine in GGT‐Iβ), which would block the entry of a prenyl group into the tunnel. RabGGTβ may have evolved to allow protein double prenylation by acquiring the hydrophobic tunnel structure.
Interestingly, PTAR1 has been identified as a host factor required for Lassa virus infection in a haploid genetic screen (Jae et al, 2014). After internalization by endocytosis, Lassa virus invades into the host cell cytoplasm through binding to an α2,3‐linked sialic acid on the lysosomal membrane protein LAMP1. We found that α2,3‐sialylation of LAMP1 is severely impaired in PTAR1 KO cells (Fig 7E), suggesting that the resistance of PTAR1‐deficient cells to Lassa virus infection is due to the sialylation defect of LAMP1. PTAR1 is also identified as an essential host factor for Rift Valley fever virus infection (Riblett et al, 2016), whose cellular receptors are heparan sulfate proteoglycans. Since glycosylation is sequentially processed in the Golgi apparatus by multiple enzymes that are arrayed in an ordered manner from cis to trans, the observed glycosylation defects may be due to the disordered structure of the Golgi apparatus caused by the loss of doubly prenylated Ykt6.
Our unbiased approach led to the discovery of a single protein substrate for GGTase‐III, but we cannot exclude the possibility that GGTase‐III has other unknown protein substrates. A recent structural study by Kuchay et al (2019) has shown that PTAR1 heterodimerizes with RabGGTβ and interacts with FBXL2, a regulatory subunit of a membrane‐localized ubiquitin ligase complex. They further showed that GGTase‐III can geranylgeranylate FBXL2, which terminates in the sequence CVIL. However, since CVIL is a typical substrate sequence for GGTase‐I (Taylor et al, 2003), it remains uncertain whether FBXL2 is a physiological substrate for GGTase‐III. In our analysis, FBXL2 was still able to localize to the plasma membrane in PTAR1 KO HeLa cells (Fig EV5A), indicating that GGTase‐III is dispensable for the membrane localization of FBXL2. Furthermore, we found that GGTase‐III could not geranylgeranylate FBXL2 under conditions where GGTase‐I robustly geranylgeranylated it (Fig EV5B and C). These results contradict the findings of the previous study regarding FBXL2 as a GGTase‐III substrate. However, FBXL2 has additional cysteines adjacent to the CAAX motif (CRCCVIL), raising the possibility that these cysteines could be geranylgeranylated by GGTase‐III once the CAAX cysteine is geranylgeranylated by GGTase‐I. Further investigation is needed to determine whether FBXL2 is a physiological substrate for GGTase‐III.
Figure EV5. GGTase‐III cannot geranylgeranylate FBXL2.
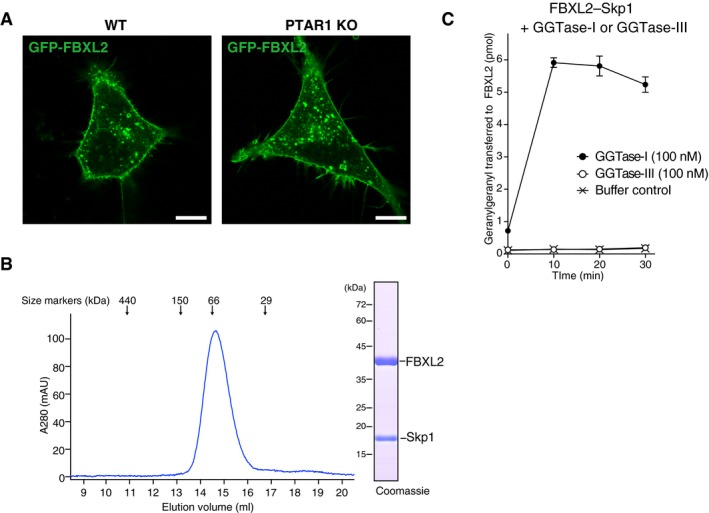
-
ALocalization of FBXL2 in HeLa cells. WT HeLa cells and PTAR1 KO HeLa cells transiently expressing GFP‐FBXL2 and myc‐Skp1 were observed for GFP fluorescence. In PTAR1 KO cells, GFP‐FBXL2 was still able to localize to the plasma membrane as observed in WT cells. Scale bars, 10 μm.
-
BPurification of the recombinant FBXL2–Skp1 complex by Superdex 200 gel filtration chromatography. The peak fraction was analyzed by SDS–PAGE and Coomassie staining.
-
CGeranylgeranylation assay showing that GGTase‐III cannot geranylgeranylate FBXL2. The purified recombinant FBXL2–Skp1 complex (5 μM) was incubated with GGTase‐I (100 nM) or GGTase‐III (100 nM) and 3H‐GGPP (1 μM) at 37°C. Reactions were stopped at the indicated time points, and the amount of 3H‐geranylgeranyl transferred to FBXL2 was quantified by scintillation counting (mean ± SEM, n = 3). GGTase‐III failed to geranylgeranylate FBXL2, whereas GGTase‐I efficiently geranylgeranylated FBXL2 under the same conditions.
Source data are available online for this figure.
In summary, we have discovered a fourth type of protein prenyltransferase, GGTase‐III. In cooperation with FTase, GGTase‐III catalyzes the double prenylation of the Golgi SNARE protein Ykt6. Our findings demonstrate that doubly prenylated Ykt6 is essential for the Golgi SNARE assembly and the organization of the Golgi apparatus. Why double prenylation is required for the function of Ykt6 and how doubly prenylated Ykt6 works to organize the Golgi apparatus remain important questions.
Materials and Methods
Reagents
Geranylgeranyl pyrophosphate (GGPP) was purchased from Cayman. Farnesyl pyrophosphate (FPP) was from Isoprenoids. [1‐3H] GGPP and [1‐3H] FPP were from American Radiolabeled Chemicals. Biotin‐labeled geranyl pyrophosphate (BGPP) was from Jena Bioscience. Protein A agarose, EDTA‐free cOmplete protease inhibitor cocktail, chymotrypsin, and V8 protease were from Roche. Leupeptin and pepstatin A were from Peptide Institute. LB broth was from BD Biosciences. Glutathione Sepharose 4B, NHS‐activated Sepharose, PreScission protease, and streptavidin Mag Sepharose were from GE Healthcare. Ni‐NTA agarose was from Qiagen. SulfoLink coupling resin, maleimide‐activated keyhole limpet hemocyanin, sulfo‐NHS‐LC‐biotin, bissulfosuccinimidyl suberate (BS3), NeutrAvidin agarose, high‐sensitivity NeutrAvidin‐HRP, fetal bovine serum (FBS), goat serum, and Grace's insect medium were from Thermo Fisher Scientific. Dulbecco's modified Eagle's medium (DMEM), Iscove's modified Dulbecco's medium (IMDM), penicillin–streptomycin solution, blasticidin S hydrochloride, puromycin dihydrochloride, amphotericin B, simvastatin, sodium ampicillin, cycloheximide, isopropyl‐β‐d‐thiogalactopyranoside (IPTG), deoxycholate (DOC), sodium dodecyl sulfate (SDS), and dithiothreitol (DTT) were from Fujifilm Wako Chemicals. Thrombin restriction grade was from Merck Millipore. Angiotensin II, ACTH fragment 18–39, α‐cyano‐4‐hydroxycinnamic acid (CHCA), poly‐l‐lysine, fibronectin, imidazole, phenylmethylsulfonyl fluoride (PMSF), N‐ethylmaleimide (NEM), l‐glutathione, FLAG peptide, S‐adenosylmethionine (SAM), CHAPS, and Triton X‐100 were from Sigma‐Aldrich. Ethylenediaminetetraacetic acid (EDTA), ethylene glycol tetraacetic acid (EGTA), and n‐octyl‐β‐d‐glucoside were from Dojindo Laboratories. FuGENE 6 was from Promega. Maackia amurensis leucoagglutinin (MAL) agarose was from J‐Oil Mills. Restriction enzymes were from New England Biolabs. Other general reagents were purchased from Fujifilm Wako Chemicals or Sigma‐Aldrich.
Antibodies
Anti‐PTAR1 rabbit polyclonal antibodies were raised against a synthetic peptide corresponding to residues 371–384 of human PTAR1 (SSKQGYSQETKRLK; synthesized by Eurofins Genomics) coupled to keyhole limpet hemocyanin and affinity purified using SulfoLink coupling resin. Anti‐Ykt6 rabbit polyclonal antibodies were raised against recombinant Ykt6 and affinity purified using NHS‐activated Sepharose. Commercial antibodies used were as follows: mouse anti‐Ykt6 (E‐2), mouse anti‐syntaxin 5 (B‐8), mouse anti‐RabGGTβ (E‐8), and mouse anti‐LAMP1 (H4A3) from Santa Cruz Biotechnology; mouse anti‐GM130 (Clone 35), mouse anti‐GS28 (Clone 1), and mouse anti‐GS15 (Clone 19) from BD Biosciences; rabbit anti‐golgin‐97 (D8P2K) from Cell Signaling Technology; mouse anti‐α‐Tubulin (B512), mouse anti‐hexahistidine (His6) tag (HIS‐1), normal rabbit IgG, and anti‐FLAG M2 affinity gel from Sigma; rabbit polyclonal anti‐RabGGTα from Abcam; rabbit polyclonal anti‐GFP from MBL; mouse isotype control IgG2A (Clone 20102) from R&D systems; rat anti‐PA tag (NZ‐1) from Fujifilm Wako Chemicals; horseradish peroxidase (HRP)‐labeled secondary antibodies from Jackson ImmunoResearch; and Alexa‐labeled fluorescent secondary antibodies from Molecular Probes.
DNA constructs
The coding sequence of PTAR1 was amplified by PCR from human brain Marathon‐Ready cDNA (Clontech) using primers 5′‐AATTGGATCCATGGCCGAGACCAGCGAGGAGGTG‐3′ and 5′‐AATTCTCGAGTCATTGACTCAAAGTAACCAGCCATTTCC‐3′. The PCR product was digested with BamHI and XhoI and ligated into a modified lentiviral expression vector containing an N‐terminal FLAG epitope tag (pBIB‐FLAG). Five independent clones were sequenced, and identical sequences were obtained. The cloned sequence was identical to the NCBI reference sequence NM_001366936 (protein prenyltransferase alpha subunit repeat containing 1 isoform 3). The plasmids containing human RhoA (Higashi et al, 2008) and canine Rab5A (Shirakawa et al, 2004) were previously described. Other cDNAs used in this study were amplified by PCR using the brain cDNA as a template and cloned into respective expression vectors. Site‐directed mutagenesis was performed by PCR. All constructs were verified by sequencing. pEGFP‐VSVG was a gift from Jennifer Lippincott‐Schwartz (Addgene 11912). pSpCas9(BB)‐2A‐Puro (PX459) was from Feng Zhang (Addgene 48139). DH5α chemically competent cells (Nippon Gene) were used for molecular cloning.
Recombinant protein expression and purification
Recombinant FTase was produced in Escherichia coli using a modified pETDuet‐1 bicistronic expression vector (Merck Millipore) containing an N‐terminal His6 tag followed by a thrombin cleavage sequence in the multiple cloning site 1 (MCS1) (pETDuet‐His). The human FTase α subunit (FNTA) and FTase β subunit (FNTB) coding sequences were cloned into MCS1 and MCS2, respectively, of pETDuet‐His. BL21 (DE3) cells (Nippon Gene) were transformed with the plasmid and grown in LB broth containing 100 μg/ml ampicillin to optical density at 600 nm (OD600) of ~ 0.6. Protein expression was induced with 0.1 mM IPTG for 16 h at 20°C. Cells were harvested by centrifugation and lysed by sonication in buffer A (50 mM HEPES‐KOH pH7.4, 50 mM NaCl, 5 mM MgCl2, and 1 mM DTT) containing 1 mM PMSF and 10 mM imidazole. Cell lysate was centrifuged at 100,000 × g for 1 h, and the supernatant was incubated with Ni‐NTA agarose beads for 2 h at 4°C. The beads were transferred to a Poly‐Prep column (Bio‐Rad) and washed with 20 column volumes of buffer A containing 20 mM imidazole. Bound proteins were eluted with buffer A containing 200 mM imidazole and subjected to anion exchange chromatography using a HiTrap Q HP column connected to an AKTA purifier chromatography system (GE Healthcare). Fractions containing FTase were pooled and applied to a Superdex 200 gel filtration column equilibrated with buffer A. Peak fractions were pooled, aliquoted, and stored at –80°C until use. Recombinant GGTase‐I was purified in a similar manner using the pETDuet‐His vector containing the GGTase‐I β subunit (PGGT1B) in MCS2.
Recombinant GGTase‐III was produced in Sf9 insect cells (Thermo Fisher Scientific) co‐infected with recombinant baculoviruses encoding GST‐tagged PTAR1 and untagged RabGGTβ. The PTAR1 coding sequence was subcloned into a modified pFastBac1 vector (Thermo Fisher Scientific) containing an N‐terminal GST tag followed by a thrombin cleavage sequence (pFastBac‐GST). The human RabGGTase β subunit (RABGGTB) coding sequence was cloned into pFastBac1. Sf9 cells were grown in Grace's insect medium supplemented with 5% (v/v) FBS, 50 units/ml penicillin, 50 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. Recombinant baculoviruses were produced according to the manufacturer's instructions. Sixty hours after co‐infection, cells were harvested and lysed in buffer A containing 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 1 mM PMSF. Cell lysate was centrifuged at 100,000 × g for 1 h, and the supernatant was incubated with glutathione Sepharose beads for 2 h at 4°C. The beads were washed with buffer A, and bound proteins were eluted with buffer A containing 10 mM glutathione. After cleavage of the GST tag with thrombin, the protein was further purified by Mono Q anion exchange chromatography followed by Superdex 200 gel filtration chromatography in buffer A. For RabGGTase expression, the human RabGGTase α subunit (RTABGGTA) coding sequence was cloned into pFastBac‐GST. RabGGTase was expressed and purified in the same manner as described above. For GST pull‐down assays and affinity column purification experiments, GGTase‐III and RabGGTase were purified without thrombin digestion.
For mutant enzyme assays in Figs 4 and 5, wild‐type (WT) and mutant GGTase‐III was produced in E. coli using a modified pETDuet‐1 vector containing an N‐terminal GST sequence followed by an HRV3C protease cleavage sequence in MCS1 (pETDuet‐GST). The RabGGTβ and full‐length PTAR1 coding sequences were cloned into MCS1 and MCS2, respectively, of pETDuet‐GST. Protein expression was induced in BL21 (DE3) cells with 0.1 mM IPTG for 16 h at 20°C, and proteins were purified using glutathione Sepharose as described above. The GST tag was cleaved with PreScission protease overnight at 4°C. After digestion, the proteins were further purified by Mono Q chromatography and Superdex 200 chromatography in buffer A. Purified proteins were aliquoted and stored at −80°C until use.
For purification of REP proteins, the human REP1 and REP2 coding sequences were cloned into pFastBac HT (Thermo Fisher Scientific). His6‐REP1 and His6‐REP2 were expressed in Sf9 cells and purified using Ni‐NTA agarose as described above. Purified His6‐REP1 and His6‐REP2 were dialyzed against buffer A and stored at −80°C until use.
For the preparation of human RCE1 and ICMT containing membrane fractions, the human RCE1 and ICMT coding sequences were cloned into a modified pFastBac1 vector containing a C‐terminal PA epitope tag (pFastBac‐PA), and proteins were expressed in Sf9 cells as described above. Cells were harvested and disrupted by sonication in buffer A containing 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 1 mM PMSF. Cell debris was removed by centrifugation at 1,000 × g for 10 min, and crude membrane fractions were prepared by centrifugation at 100,000 × g for 1 h. The membrane pellets were resuspended in buffer A containing 1.5 M NaCl to wash out peripherally membrane‐bound proteins and pelleted again by centrifugation at 100,000 × g for 1 h. The membrane pellets were resuspended in buffer A at ~ 10 mg total protein/ml, aliquoted, and stored at −80°C until use. The expression levels of RCE1 and ICMT were examined by immunoblotting with anti‐PA tag antibody.
All protein substrates were produced in E. coli. The H‐Ras, RhoA, Rab1A, and Rab11A coding sequences were cloned into pRSET A (Thermo Fisher Scientific). The Rab5 coding sequence was cloned into pDEST17 (Thermo Fisher Scientific). Protein expression was induced in BL21 (DE3) cells with 0.1 mM IPTG for 6 h at 30°C. Proteins were purified using Ni‐NTA agarose, dialyzed against buffer A, and stored at −80°C until use.
For purification of untagged Ykt6, the human Ykt6 coding sequence was cloned into the NdeI and XhoI sites of pRSET A. Untagged Ykt6 expression was induced in BL21 (DE3) cells with 0.1 mM IPTG for 6 h at 30°C. Cells were harvested and lysed by sonication in buffer A containing 1 mM PMSF. After centrifugation at 100,000 × g for 1 h, the supernatant was treated with ammonium sulfate to 60% saturation and left on ice for 30 min with stirring. The suspension was centrifuged at 10,000 × g for 15 min, and the precipitated material was dissolved and dialyzed overnight against buffer A containing 1 M ammonium sulfate. The dialyzed material was applied to a HiTrap Phenyl HP column equilibrated with buffer A containing 1 M ammonium sulfate. The column was eluted with a decreasing gradient of 1–0 M ammonium sulfate in buffer A. Fractions containing Ykt6 were pooled, concentrated, and applied to a Superdex 200 column equilibrated with buffer A. Peak fractions were pooled, aliquoted, and stored at −80°C. Mutant Ykt6 proteins were purified in the same manner as the WT protein.
Generation and purification of prenylated Ykt6
To generate Cys195‐farnesyl Ykt6, approximately 10 mg of purified unprenylated Ykt6 was mixed with 4 mg of FTase and 2 μmol of FPP in buffer A containing 20 μM ZnCl2 and incubated for 2 h at 37°C. After incubation, the solution was adjusted to 1 M ammonium sulfate and applied to a HiTrap Phenyl HP column equilibrated with buffer A containing 1 M ammonium sulfate. The column was eluted with a decreasing gradient of 1–0 M ammonium sulfate in buffer A. Fractions containing Cys195‐farnesyl Ykt6 were pooled, concentrated, and applied to a Superdex 200 column equilibrated with buffer A. The ΔAIM form of Cys195‐farnesyl Ykt6 was prepared in a similar manner except that farnesylation reaction was performed with Sf9 membranes containing recombinant RCE1 and ICMT (30 and 60 mg total protein, respectively). Membranes were removed by centrifugation before HiTrap Phenyl chromatography. To generate the ΔAIM form of Cys194/195‐diprenyl Ykt6, 4 mg of purified Cys195‐farnesyl Ykt6ΔAIM was mixed with 2 mg of GGTase‐III and 1 μmol of GGPP in buffer A containing 20 μM ZnCl2 and incubated for 2 h at 37°C. Cys194/195‐diprenyl Ykt6ΔAIM was purified to homogeneity by HiTrap Phenyl chromatography followed by Superdex 200 gel filtration chromatography as described above. The prenylation status of purified prenylated Ykt6 proteins was examined by DOC‐PAGE and MALDI‐TOF mass spectrometry.
MALDI‐TOF mass spectrometry
Two micrograms of purified recombinant Ykt6 samples were digested with 0.1 μg of chymotrypsin or 0.2 μg of V8 protease for 3 h at 37°C. Digested peptides were purified using ZipTip C18 columns (Merck Millipore), mixed with 5 mg/ml CHCA in 80% (v/v) acetonitrile and 0.1% (v/v) trifluoroacetic acid, and analyzed using an AXIMA Performance MALDI‐TOF mass spectrometer (Shimadzu). Angiotensin II and ACTH fragment 18–39 were used as calibration standards.
DOC‐PAGE
Protein samples were electrophoresed on a 12.5% polyacrylamide gel in a buffer containing 25 mM Tris, 192 mM glycine, and 0.1% (w/v) sodium deoxycholate. After electrophoresis, the gel was incubated for 5 min in a buffer containing 25 mM Tris, 192 mM glycine, and 0.1% (w/v) SDS, transferred to a nitrocellulose membrane (Protran BA85; GE Healthcare), and processed for immunoblotting with anti‐Ykt6 antibody.
Protein purification for crystallization
For crystallization, RabGGTβ and PTAR1 (residues 1–366 or 1–327) were co‐expressed in BL21 (DE3) cells using the pETDuet‐GST vector. Protein expression and purification were performed in the same manner as the full‐length complex, except that the final gel filtration step was performed with 20 mM HEPES‐KOH pH 7.4, 50 mM NaCl, and 1 mM DTT. Cys195‐farnesyl Ykt6 and Cys195‐farnesyl Ykt6ΔAIM were prepared as described above using 17 mg of unprenylated Ykt6 as starting material and purified by HiTrap Phenyl chromatography followed by Superdex 200 gel filtration chromatography in 20 mM HEPES‐KOH pH 7.4, 50 mM NaCl, and 1 mM DTT.
Crystallization
All samples were crystallized by the sitting drop vapor diffusion method at 20°C. The apo‐GGTase‐III complex (PTAR1 residues 1–366 and full‐length RabGGTβ) was concentrated to 11 g/l and mixed with an equal volume of the reservoir solution (40 mM MgCl2, 50 mM Na cacodylate pH 6.0, 5% 2‐methyl‐2,4‐pentanediol (MPD), and 8 mM CHAPS). The crystal of apo‐GGTase‐III was cryoprotected by the addition of MPD (35%) to the reservoir solution and flash‐cooled by liquid nitrogen. To form the complex of GGTase‐III (PTAR1 residues 1–327 and full‐length RabGGTβ) and unprenyl Ykt6, GGTase‐III and Ykt6 were mixed in an equimolar ratio, concentrated to 15 g/l, and mixed with an equal volume of the reservoir solution (1.18 M K2HPO4 and 0.82 M NaH2PO4). The crystal of the complex was cryoprotected by the addition of ethylene glycol (35%) and flash‐cooled by liquid nitrogen. To form the complex of GGPP‐bound GGTase‐III (PTAR1 residues 1–327 and full‐length RabGGTβ) and Cys195‐farnesyl Ykt6, GGTase‐III was co‐purified with GGPP and mixed with Cys195‐farnesyl Ykt6 in an equimolar ratio. The mixture was concentrated to 9 g/l and mixed with an equal volume of the reservoir solution (0.1 M Na formate pH 7.0 and 11% (w/v) PEG3350). The complex of GGPP‐bound GGTase‐III with the ΔAIM form of Cys195‐farnesyl Ykt6 was formed in the same manner and mixed with an equal volume of the reservoir solution (1.0 M ammonium tartrate dibasic pH 7.0). The crystals were cryoprotected by the addition of ethylene glycol (30%) and flash‐cooled by liquid nitrogen.
Data collection and structure determination
All diffraction data sets were collected at 100 K at beamline BL41XU of SPring‐8 (Hyogo, Japan) and processed with HKL2000 (Otwinowski & Minor, 1997) and CCP4 program suite (Winn et al, 2011). The structure of apo‐GGTase‐III was determined by the molecular replacement method using the program MOLREP (Vagin & Teplyakov, 2010). The structure of RabGGTase (PDB 3DSS) was used as the search model. The structure of the GGTase‐III–unprenyl Ykt6 complex was determined by the molecular replacement method using the structures of the apo‐GGTase‐III and the Ykt6 longin domain (PDB 3KYQ) as the search models. The structure of GGTase‐III in complex with Cys195‐farnesyl Ykt6 and GGPP was determined by the molecular replacement method using the GGTase‐III structure in the GGTase‐III–unprenyl Ykt6 complex as the search model for the first trial with the program MOLREP. Subsequently, the unprenyl Ykt6 structure in the same complex was used as the search model for the second trial with the program Phaser (McCoy et al, 2007). The structure of GGTase‐III complexed with Cys195‐farnesyl Ykt6ΔAIM and GGPP was determined by the molecular replacement method using the program Phaser with the GGTase‐III–Cys195‐farnesyl Ykt6–GGPP structure as the search model. The initial models were improved by the repetitive cycles of manual model building using Coot (Emsley & Cowtan, 2004) and refinement using Phenix (Adams et al, 2010). Data collection and refinement statistics are shown in Appendix Table S1. All structural figures were prepared using the program PyMOL (Schrödinger, LLC).
In the apo‐GGTase‐III crystal, cacodylate ions contained in the crystallization reagent covalently bound to Cys60 and Cys92 of PTAR1 to form dimethyl arsenic cysteine residues via a two‐step reaction utilizing DTT in the sample solution, as proposed previously (Maignan et al, 1998). The attached arsenic atoms were confirmed by anomalous difference Fourier maps.
Limited proteolytic analysis
Full‐length GGTase‐III (6 g/l) was incubated with 0.04% (w/w, protease/sample) trypsin for 1 h at 20°C. The reaction was stopped by the addition of SDS sample buffer and subjected to SDS–PAGE analysis followed by blotting to a membrane (Immobilon; Merck Millipore). The blotted bands of the digested samples were subjected to N‐terminal sequencing using an ABI Procise Model 492 peptide sequencer.
Fractionation of rat brain cytosol
Freshly obtained brains from male Wister rats (5 weeks old) were homogenized in buffer A containing protease inhibitor cocktail using a Potter‐Elvehjem homogenizer. The homogenate was centrifuged at 2,000 × g for 10 min, and the supernatant was further centrifuged at 200,000 × g for 20 min to obtain a cytosolic fraction. The cytosolic fraction was applied to a Mono Q anion exchange column equilibrated with buffer A without NaCl. The column was eluted with a linear gradient of 0–0.5 M NaCl in buffer A, and an aliquot of each fraction was analyzed by immunoblotting with the indicated antibodies. The fractions containing PTAR1 (peak A, Fig 1D) were pooled and applied to a Superdex 200 column equilibrated with buffer A, and the column fractions were analyzed by immunoblotting with the indicated antibodies. The gel filtration column was calibrated with ferritin (440 kDa), alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa), and carbonic anhydrase (29 kDa).
Immunoprecipitation and immunodepletion assays
For immunoprecipitation of endogenous PTAR1, protein A agarose beads were crosslinked with 2 μg of anti‐PTAR1 IgG or control rabbit IgG using the chemical crosslinker BS3. Cytosolic fractions were prepared from rat brain and thymus tissues as described above and incubated with the beads for 6 h at 4°C. After washing the beads with buffer A containing 0.1% (w/v) Triton X‐100, bound proteins were eluted with glycine–HCl buffer (pH 2.5) and analyzed by immunoblotting with the indicated antibodies. For immunodepletion assays, the Mono Q peak A fractions (Fig 1D) were pooled and incubated with protein A agarose beads precoated with 5 μg of anti‐PTAR1 IgG, control IgG, or anti‐PTAR1 IgG preabsorbed with the antigen peptide for 6 h at 4°C. Beads were sedimented by brief centrifugation, and the supernatant was analyzed by immunoblotting with the indicated antibodies.
Cell culture
HAP1 cells (Horizon) were maintained in IMDM supplemented with 10% (v/v) FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. HeLa cells and HeLa S3 cells (JCRB Cell Bank) were maintained in DMEM supplemented with 10% (v/v) FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Lentivirus‐infected cells were selected and maintained in the culture medium supplemented with 5 μg/ml blasticidin. HeLa S3 cells were grown in spinner flasks (Chemglass Life Sciences).
Identification of RabGGTβ as a PTAR1‐binding protein
HeLa S3 cells stably expressing FLAG‐tagged PTAR1 were established by lentivirus infection. Approximately 7 × 108 cells were harvested and lysed by sonication in buffer B (50 mM HEPES‐KOH pH 7.4, 78 mM KCl, 4 mM MgCl2, 2 mM EGTA, 0.2 mM CaCl2, and 1 mM DTT) containing protease inhibitor cocktail. Cell lysates were centrifuged at 100,000 × g for 1 h, and the supernatant was incubated with anti‐FLAG M2 agarose beads for 3 h at 4°C. The beads were washed three times with buffer B containing 0.1% (w/v) CHAPS, and bound proteins were eluted with buffer B containing 1 mg/ml FLAG peptide. The eluted proteins were analyzed by SDS–PAGE, followed by silver staining. The 35 kDa protein band was identified as RabGGTβ by mass spectrometry analysis.
Purification of GGTase‐III substrates
Three‐liter suspension culture of HeLa S3 cells (~ 3 × 109 cell) was treated with 10 μg/ml simvastatin for 24 h. The cells were harvested by centrifugation and disrupted by sonication in buffer A containing 2 mM EDTA, 2 mM EGTA, and protease inhibitor cocktail. The cell lysate was centrifuged at 100,000 × g for 1 h, and the supernatant was dialyzed against buffer A overnight at 4°C. The dialyzed material was applied to GST or GST‐GGTase‐III affinity columns (0.5 ml bed volume of glutathione Sepharose beads containing 0.8 mg of GST or 3.2 mg of GST‐GGTase‐III), and bound proteins were eluted with 3 ml of buffer A containing 0.5 M NaCl. After dialysis and concentration, the eluted proteins were incubated with 100 nM GGTase‐III and 20 μM BGPP in buffer A for 2 h at 30°C. Biotinylated proteins were purified using NeutrAvidin agarose and boiled in SDS sample buffer. The samples were resolved by SDS–PAGE, transferred to a nitrocellulose membrane, and probed with NeutrAvidin–HRP. Aliquots of the samples were parallelly analyzed by SDS–PAGE and silver staining, and the 25 kDa band was identified as Ykt6 by mass spectrometry.
Prenylation assays
Prenylation assays were performed in a 20‐μl reaction mixture containing 50 mM HEPES‐KOH pH 7.4, 50 mM NaCl, 5 mM MgCl2, 10 μM ZnCl2, 1 mM DTT, 0.2% (w/v) octylglucoside, 1 μM 3H‐GGPP (~ 3,000 dpm/pmol) or 1 μM 3H‐FPP (~ 3,500 dpm/pmol), and indicated amounts of recombinant prenyltransferases and protein substrates. RabGGTase assays were performed in the presence of 100 nM His6‐REP1 or His6‐REP2. Reactions were incubated at 37°C for the indicated times and terminated by adding 0.5 ml of cold ethanol containing 1 M HCl. After incubation on ice for 10 min, reaction products were filtered through glass microfiber filters (GF/C; GE Healthcare) prewashed with 5 ml of cold ethanol. After washing twice with 5 ml of cold ethanol, filter‐bound radioactivity was quantified using a liquid scintillation counter (Aloka).
GGPP binding assay
GGTase‐III (1 μM) was incubated in 50 mM HEPES‐KOH pH 7.4, 50 mM NaCl, 5 mM MgCl2, 10 μM ZnCl2, 1 mM DTT, 0.2% (w/v) octylglucoside, and 2 μM 3H‐GGPP (~ 5,000 dpm/pmol) for 10 min on ice and then desalted using PD MiniTrap G‐25 columns (GE Healthcare) equilibrated with buffer A containing 0.1% (w/v) octylglucoside. Radioactivity eluted in fractions from 0.6 to 0.9 ml was quantified by scintillation counting.
REP pull‐down assay
GST‐RabGGTase or GST‐GGTase‐III (10 μg each) was immobilized on glutathione Sepharose beads and incubated with 1 μg of recombinant His6‐REP1 or His6‐REP2 in buffer A containing 0.01% (w/v) Triton X‐100 for 1 h at 4°C. After washing the beads with the same buffer, bound proteins were eluted with buffer A containing 10 mM glutathione and analyzed by immunoblotting with anti‐His6 antibody.
Genome editing using CRISPR‐Cas9
PTAR1 KO cell lines were generated using the CRISPR‐Cas9 system as described (Ran et al, 2013). Briefly, the guide sequence targeting the exon 2 of PTAR1 (5′‐GTCCTGAAGCTAGGTATAAC‐3′) was ligated into the pSpCas9(BB)‐2A‐Puro vector using BbsI sites. HAP1 and HeLa cells were transfected with the plasmid using FuGENE 6 and cultured for 48 h with 0.5 μg/ml puromycin. Puromycin‐resistant cells were seeded in 96‐well plates, and clonal cell lines were established. Insertion and deletion mutations of the cell lines were confirmed by direct genomic sequencing. The absence of PTAR1 protein was confirmed by immunoblotting with anti‐PTAR1 antibody. For rescue experiments, PTAR1 KO cells stably expressing exogenous PTAR1 or PTAR1 mutants were generated by lentiviral infection using a lentiviral expression vector containing a thymidine kinase promoter (pTIB).
Immunofluorescence analysis
HeLa cells growing on a poly‐l‐lysine‐coated cover glass chamber (Matsunami Glass) were fixed with 4% formaldehyde in phosphate‐buffered saline (PBS) for 15 min, permeabilized with 0.1% (w/v) Triton X‐100 for 5 min, and blocked with 10% (v/v) goat serum in PBS for 30 min at room temperature. Cells were stained with primary antibodies diluted in the blocking buffer for 1 h at room temperature and then stained with Alexa‐labeled secondary antibodies for 1 h. After mounting with 80% glycerol, the cells were observed with a Leica TCS SP8 confocal microscopy using a 63 × Apochromat oil‐immersion objective. Images were deconvoluted with Huygens software (Scientific Volume Imaging).
Electron microscopy
HAP1 cells growing on fibronectin‐coated cover glasses were fixed with a mixture of 2.5% glutaraldehyde and 4% formaldehyde in 0.1 M HEPES‐KOH buffer (pH 7.4) containing 1 mM CaCl2 at 4°C overnight, and then post‐fixed by a mixture of 1% osmium tetroxide and 0.1% potassium ferrocyanide in 0.1 M sodium cacodylate buffer (pH 7.4) at room temperature for 1 h. The samples were dehydrated and embedded in epoxy resin. Ultrathin sections stained with lead citrate were observed using a JEOL JEM1011 electron microscope operated at 100 kV.
Live cell imaging of VSVG‐GFP
HeLa cells growing on poly‐l‐lysine‐coated 35‐mm glass bottom dishes (Matsunami Glass) were transfected with pEGFP‐VSVG using FuGENE 6. Twelve hours after transfection, cells were transferred to 40°C and incubated for 6 h. After the addition of cycloheximide (20 μg/ml), cells were imaged in a temperature‐controlled chamber set at 32°C using a Leica TCS SP8 confocal microscope. The intensity of GFP fluorescence in the Golgi region was quantified using ImageJ software.
Cell surface biotinylation assay
HeLa cells plated on 12‐well plates were transfected with pEGFP‐VSVG using FuGENE 6. Twelve hours after transfection, cells were transferred to 40°C and incubated for 6 h. After the addition of cycloheximide (20 μg/ml), cells were shifted to 32°C and incubated for the indicated times. After incubation, the cells were washed with cold PBS and incubated with 0.5 mM sulfo‐NHS‐LC‐biotin in PBS for 15 min on ice to biotinylate cell surface proteins. The cells were then incubated with 50 mM Tris–HCl pH 7.4 in PBS for 15 min on ice to inactivate unreacted NHS esters. The cells were lysed in 50 mM HEPES‐KOH pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1% (w/v) Triton X‐100, and protease inhibitor cocktail. Biotinylated VSVG‐GFP was purified from the cell lysates with streptavidin Mag Sepharose and analyzed by immunoblotting with anti‐GFP antibody.
MAL lectin pull‐down assay
HeLa cells growing on 6‐cm dishes were lysed in buffer A containing 1 mM CaCl2, 1% (w/v) Triton X‐100, and protease inhibitor cocktail. After centrifugation at 200,000 × g for 10 min, supernatants were incubated with MAL lectin agarose beads for 1 h at 4°C. After washing the beads with the same buffer, bound proteins were eluted with SDS sample buffer and analyzed by immunoblotting with anti‐LAMP1 antibody.
Immunoprecipitation of the Golgi SNARE complex
Subconfluent cultures of HAP1 cells in 15‐cm dishes were incubated with 1 mM NEM in IMDM for 15 min on ice, followed by incubation with 2 mM DTT in IMDM for additional 15 min to quench unreacted NEM. Cells were then incubated with prewarmed IMDM containing 10% (v/v) FBS and penicillin/streptomycin for 30 min at 37°C. After washing with cold PBS, the cells were lysed in buffer A containing 1% (w/v) Triton X‐100 and protease inhibitor cocktail, and centrifuged at 200,000 × g for 10 min. The supernatants were incubated with 0.5 μg of control mouse IgG, anti‐syntaxin 5 IgG, or anti‐GS28 IgG, and protein A agarose beads for 12 h at 4°C. After washing the beads with buffer A containing 1% (w/v) Triton X‐100, bound proteins were boiled in SDS sample buffer and analyzed by immunoblotting with the indicated antibodies.
Analysis of FBXL2
The coding sequences of human Skp1 and FBXL2 were synthesized (gBlocks Gene Fragments; Integrated DNA Technologies) and cloned into MCS1 and MSC2, respectively, of pETDuet‐GST. The GST‐Skp1–FBXL2 complex was purified with glutathione Sepharose as described above. After cleavage of the GST tag with PreScission protease, the complex was further purified by Mono Q and Superdex 200 chromatography in buffer A and used in prenylation assays. For live cell imaging of GFP‐FBXL2, the FBXL2 and Skp1 coding sequences were subcloned into pEGFP‐C1 (Clontech) and pBIB‐myc (a modified mammalian expression vector containing an N‐terminal myc tag sequence), respectively. HeLa cells growing on a poly‐l‐lysine‐coated cover glass chamber were co‐transfected with the pEGFP‐FBXL2 and pBIB‐myc‐Skp1 plasmids using FuGENE 6. Eighteen hours after co‐transfection, the cells were imaged using a Leica TCS SP8 confocal microscope.
Statistical analysis
Quantification of prenylation assays is represented as the mean of three independent experiments unless otherwise stated. Cell surface VSVG biotinylation assay was analyzed by one‐way ANOVA with Dunnett's post‐hoc test using Prism 8 software (GraphPad). Error bars represent the standard error of the mean (SEM).
Author contributions
RS and KG performed biochemical and cell biological experiments. SG‐I conducted crystallization and structural analysis with assistance from YS, AY, and SF. SW and HK contributed to the initial studies. NS and DAT analyzed data. HM performed preliminary mass spectrometry analysis. JC and TF performed electron microscopy. RS, SG‐I, and SF wrote the manuscript with input from all authors. SF and HH supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to Tomohito Higashi for comments on the manuscript. We thank the beamline staff of BL41XU of SPring‐8 for technical help during data collection. We thank the staff of Biomedical Research Core of Tohoku University Graduate School of Medicine for technical support. We also thank Jennifer Lippincott‐Schwartz and Feng Zhang for providing plasmids. This work was supported by JSPS Grants‐in‐Aid for Scientific Research 16K08574 (R.S.), 17K15072 (S.G.‐I.), and 16H05148 (H.H.), JST CREST JPMJCR12M5 (S.F.), and a grant from Takeda Science Foundation (R.S.).
The EMBO Journal (2020) 39: e104120
See also: https://doi.org/10.15252/embj.2020104744 (April 2020)
Contributor Information
Ryutaro Shirakawa, Email: ryutaro.shirakawa.b1@tohoku.ac.jp.
Shuya Fukai, Email: fukai@iam.u-tokyo.ac.jp.
Data availability
The coordinates and structure factors reported in this study have been deposited in the Protein Data Bank under the accession codes 6J6X (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J6X) (apo‐GGTase‐III), 6J74 (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J74) (GGTase‐III–Ykt6), 6J7X (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J7X) (GGTase‐III–Cys195‐farnesyl Ykt6–GGPP), and 6J7F (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J7F) (GGTase‐III–Cys195‐farnesyl Ykt6ΔAIM–GGPP).
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov K, Horiuchi H, Steele‐Mortimer O, Seabra MC, Zerial M (1994) Rab escort protein‐1 is a multifunctional protein that accompanies newly prenylated rab proteins to their target membranes. EMBO J 13: 5262–5273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres DA, Seabra MC, Brown MS, Armstrong SA, Smeland TE, Cremers FP, Goldstein JL (1993) cDNA cloning of component A of Rab geranylgeranyl transferase and demonstration of its role as a Rab escort protein. Cell 73: 1091–1099 [DOI] [PubMed] [Google Scholar]
- Bas L, Papinski D, Licheva M, Torggler R, Rohringer S, Schuschnig M, Kraft C (2018) Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome‐vacuole fusion. J Cell Biol 217: 3656–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Seeger M, Weng J, Song S, Wang W, Tan YW (2016) Lipid regulated intramolecular conformational dynamics of SNARE‐protein Ykt6. Sci Rep 6: 30282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Ernst AM, Syed SA, Zaki O, Bottanelli F, Zheng H, Hacke M, Xi Z, Rivera‐Molina F, Graham M, Rebane AA et al (2018) S‐palmitoylation sorts membrane cargo for anterograde transport in the Golgi. Dev Cell 47: 479–493.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa M, Varlamov O, Eng WS, Sollner TH, Rothman JE (2004) Localization and activity of the SNARE Ykt6 determined by its regulatory domain and palmitoylation. Proc Natl Acad Sci USA 101: 4815–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Reggiori F, Ungermann C (2018) A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J Cell Biol 217: 3670–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Wu YW, Das D, Delon C, Cramer J, Yu S, Thuns S, Lupilova N, Waldmann H, Brunsveld L et al (2008) Structures of RabGGTase‐substrate/product complexes provide insights into the evolution of protein prenylation. EMBO J 27: 2444–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Ikeda T, Shirakawa R, Kondo H, Kawato M, Horiguchi M, Okuda T, Okawa K, Fukai S, Nureki O et al (2008) Biochemical characterization of the Rho GTPase‐regulated actin assembly by diaphanous‐related formins, mDia1 and Daam1, in platelets. J Biol Chem 283: 8746–8755 [DOI] [PubMed] [Google Scholar]
- Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P et al (2014) Virus entry. Lassa virus entry requires a trigger‐induced receptor switch. Science 344: 1506–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchay S, Wang H, Marzio A, Jain K, Homer H, Fehrenbacher N, Philips MR, Zheng N, Pagano M (2019) GGTase3 is a newly identified geranylgeranyltransferase targeting a ubiquitin ligase. Nat Struct Mol Biol 26: 628–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane KT, Beese LS (2006) Thematic review series: lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J Lipid Res 47: 681–699 [DOI] [PubMed] [Google Scholar]
- Leung KF, Baron R, Seabra MC (2006) Thematic review series: lipid posttranslational modifications. geranylgeranylation of Rab GTPases. J Lipid Res 47: 467–475 [DOI] [PubMed] [Google Scholar]
- Long SB, Casey PJ, Beese LS (2002) Reaction path of protein farnesyltransferase at atomic resolution. Nature 419: 645–650 [DOI] [PubMed] [Google Scholar]
- Maignan S, Guilloteau JP, Zhou‐Liu Q, Clement‐Mella C, Mikol V (1998) Crystal structures of the catalytic domain of HIV‐1 integrase free and complexed with its metal cofactor: high level of similarity of the active site with other viral integrases. J Mol Biol 282: 359–368 [DOI] [PubMed] [Google Scholar]
- Malsam J, Sollner TH (2011) Organization of SNAREs within the Golgi stack. Cold Spring Harb Perspect Biol 3: a005249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, Mizushima N (2018) Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol 217: 2633–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer‐Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F (2007) Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput Biol 3: e66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew JA, Sogaard M, Lampen NM, Machida S, Ye RR, Lacomis L, Tempst P, Rothman JE, Sollner TH (1997) Ykt6p, a prenylated SNARE essential for endoplasmic reticulum‐Golgi transport. J Biol Chem 272: 17776–17783 [DOI] [PubMed] [Google Scholar]
- Nguyen UT, Guo Z, Delon C, Wu Y, Deraeve C, Franzel B, Bon RS, Blankenfeldt W, Goody RS, Waldmann H et al (2009) Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat Chem Biol 5: 227–235 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Park HW, Boduluri SR, Moomaw JF, Casey PJ, Beese LS (1997) Crystal structure of protein farnesyltransferase at 2.25 angstrom resolution. Science 275: 1800–1804 [DOI] [PubMed] [Google Scholar]
- Parlati F, Varlamov O, Paz K, McNew JA, Hurtado D, Sollner TH, Rothman JE (2002) Distinct SNARE complexes mediating membrane fusion in Golgi transport based on combinatorial specificity. Proc Natl Acad Sci USA 99: 5424–5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylypenko O, Rak A, Reents R, Niculae A, Sidorovitch V, Cioaca MD, Bessolitsyna E, Thoma NH, Waldmann H, Schlichting I et al (2003) Structure of Rab escort protein‐1 in complex with Rab geranylgeranyltransferase. Mol Cell 11: 483–494 [DOI] [PubMed] [Google Scholar]
- Pylypenko O, Schonichen A, Ludwig D, Ungermann C, Goody RS, Rak A, Geyer M (2008) Farnesylation of the SNARE protein Ykt6 increases its stability and helical folding. J Mol Biol 377: 1334–1345 [DOI] [PubMed] [Google Scholar]
- Rak A, Pylypenko O, Niculae A, Pyatkov K, Goody RS, Alexandrov K (2004) Structure of the Rab7:REP‐1 complex: insights into the mechanism of Rab prenylation and choroideremia disease. Cell 117: 749–760 [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS (1990) Inhibition of purified p21ras farnesyl:protein transferase by Cys‐AAX tetrapeptides. Cell 62: 81–88 [DOI] [PubMed] [Google Scholar]
- Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, Brummelkamp TR, Wojcechowskyj JA (2016) A haploid genetic screen identifies heparan sulfate proteoglycans supporting rift valley fever virus infection. J Virol 90: 1414–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR III, Davis NG (2006) Global analysis of protein palmitoylation in yeast. Cell 125: 1003–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabra MC, Reiss Y, Casey PJ, Brown MS, Goldstein JL (1991) Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell 65: 429–434 [DOI] [PubMed] [Google Scholar]
- Seabra MC, Brown MS, Slaughter CA, Sudhof TC, Goldstein JL (1992a) Purification of component A of Rab geranylgeranyl transferase: possible identity with the choroideremia gene product. Cell 70: 1049–1057 [DOI] [PubMed] [Google Scholar]
- Seabra MC, Goldstein JL, Sudhof TC, Brown MS (1992b) Rab geranylgeranyl transferase. A multisubunit enzyme that prenylates GTP‐binding proteins terminating in Cys‐X‐Cys or Cys‐Cys. J Biol Chem 267: 14497–14503 [PubMed] [Google Scholar]
- Seabra MC, Brown MS, Goldstein JL (1993) Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science 259: 377–381 [DOI] [PubMed] [Google Scholar]
- Seabra MC, Ho YK, Anant JS (1995) Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem 270: 24420–24427 [DOI] [PubMed] [Google Scholar]
- Shirakawa R, Higashi T, Tabuchi A, Yoshioka A, Nishioka H, Fukuda M, Kita T, Horiuchi H (2004) Munc13‐4 is a GTP‐Rab27‐binding protein regulating dense core granule secretion in platelets. J Biol Chem 279: 10730–10737 [DOI] [PubMed] [Google Scholar]
- Takats S, Glatz G, Szenci G, Boda A, Horvath GV, Hegedus K, Kovacs AL, Juhasz G (2018) Non‐canonical role of the SNARE protein Ykt6 in autophagosome‐lysosome fusion. PLoS Genet 14: e1007359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JS, Reid TS, Terry KL, Casey PJ, Beese LS (2003) Structure of mammalian protein geranylgeranyltransferase type‐I. EMBO J 22: 5963–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma NH, Niculae A, Goody RS, Alexandrov K (2001) Double prenylation by RabGGTase can proceed without dissociation of the mono‐prenylated intermediate. J Biol Chem 276: 48631–48636 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66: 22–25 [DOI] [PubMed] [Google Scholar]
- Volchuk A, Ravazzola M, Perrelet A, Eng WS, Di Liberto M, Varlamov O, Fukasawa M, Engel T, Sollner TH, Rothman JE et al (2004) Countercurrent distribution of two distinct SNARE complexes mediating transport within the Golgi stack. Mol Biol Cell 15: 1506–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Casey PJ (2016) Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol 17: 110–122 [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
- Wen W, Yu J, Pan L, Wei Z, Weng J, Wang W, Ong YS, Tran TH, Hong W, Zhang M (2010) Lipid‐induced conformational switch controls fusion activity of longin domain SNARE Ykt6. Mol Cell 37: 383–395 [DOI] [PubMed] [Google Scholar]
- Weng J, Yang Y, Wang W (2015) Lipid regulated conformational dynamics of the longin SNARE protein Ykt6 revealed by molecular dynamics simulations. J Phys Chem A 119: 1554–1562 [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Martin S, James DE, Hong W (2002) GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol Biol Cell 13: 3493–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Seabra MC, Deisenhofer J (2000) Crystal structure of Rab geranylgeranyltransferase at 2.0 A resolution. Structure 8: 241–251 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The coordinates and structure factors reported in this study have been deposited in the Protein Data Bank under the accession codes 6J6X (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J6X) (apo‐GGTase‐III), 6J74 (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J74) (GGTase‐III–Ykt6), 6J7X (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J7X) (GGTase‐III–Cys195‐farnesyl Ykt6–GGPP), and 6J7F (http://www.rcsb.org/pdb/explore/explore.do?structureId=6J7F) (GGTase‐III–Cys195‐farnesyl Ykt6ΔAIM–GGPP).