

Abstract
The production and secretion of matrix proteins upon stimulation of fibroblasts by transforming growth factor‐beta (TGFβ) play a critical role in wound healing. How TGFβ supports the bioenergetic cost of matrix protein synthesis is not fully understood. Here, we show that TGFβ promotes protein translation at least in part by increasing the mitochondrial oxidation of glucose and glutamine carbons to support the bioenergetic demand of translation. Surprisingly, we found that in addition to stimulating the entry of glucose and glutamine carbon into the TCA cycle, TGFβ induced the biosynthesis of proline from glutamine in a Smad4‐dependent fashion. Metabolic manipulations that increased mitochondrial redox generation promoted proline biosynthesis, while reducing mitochondrial redox potential and/or ATP synthesis impaired proline biosynthesis. Thus, proline biosynthesis acts as a redox vent, preventing the TGFβ‐induced increase in mitochondrial glucose and glutamine catabolism from generating damaging reactive oxygen species (ROS) when TCA cycle activity exceeds the ability of oxidative phosphorylation to convert mitochondrial redox potential into ATP. In turn, the enhanced synthesis of proline supports TGFβ‐induced production of matrix proteins.
Keywords: collagen, fibrosis, metabolism, proline, TGFβ
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Metabolism; Signal Transduction
TGFβ stimulates glutamine metabolism to fuel proline and collagen synthesis, acting as a sink to prevent excessive ROS generation.
Introduction
Mammalian cells depend on extracellular signals for growth, proliferation, and survival. In the absence of adequate extrinsic signals, cells cannot maintain viability due to an inability to take up nutrients (Rathmell et al, 2000). Growth factor‐directed nutrient uptake is critical to sustain cellular bioenergetics as well as the biosynthesis of macromolecules required for a cell to grow and proliferate. Glucose and glutamine are the major carbon sources for ATP production and biosynthesis, and glutamine provides the nitrogen required for biosynthesis (Altman et al, 2016; Hosios et al, 2016). Binding of receptor tyrosine kinases (RTKs) by extracellular growth factors promotes both the uptake and utilization of glucose and glutamine. For example, stimulation of RTKs can activate PI3K/AKT/mTOR signaling to increase surface localization of transporters for glucose and amino acids, and to promote utilization of these nutrients for glycolysis, anabolic cell growth, and survival (Lien et al, 2016).
Growth factor‐induced uptake of nutrients often exceeds a cell's need for maintenance of bioenergetics and biosynthesis (Bauer et al, 2004). Oxidation of carbon substrates in the tricarboxylic acid (TCA) cycle creates electrons that are transferred to nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD), yielding NADH and FADH2. NADH and FADH2 then donate electrons to the electron transport chain (ETC) at complexes I and II, creating an electron flow to complexes III and IV, and finally, to molecular oxygen, generating water. However, increased carbon oxidation in the TCA cycle in response to growth factor stimulation can yield NADH levels that exceed the capacity of the ETC to transfer electrons to molecular oxygen or the ability of complex V to dissipate the ETC‐produced proton gradient through ATP production (Murphy, 2009; Wellen & Thompson, 2010). This increases the chance of mitochondrial hyperpolarization, resulting in production of reactive oxygen species (ROS) which can damage the cell (Murphy, 2009; Schieber & Chandel, 2014). Certain cell types including liver, muscle, or adipose cells can store excess glucose carbon in the form of glycogen and fat. However, most cells in the human body are unable to store extra glucose. Therefore, to protect themselves from excessive mitochondrial ROS generation, these cells limit oxidation of glucose‐derived carbon (Thompson, 2011). Proliferating cells adapt by secreting excess glycolytic carbon in the form of lactate, often mediated by ROS‐induced HIF‐1α signaling (Lum et al, 2007; Vander Heiden et al, 2009). There are also efflux pathways for glycolytic pyruvate that enters the TCA cycle. Citrate secreted from the mitochondria supports lipid biosynthesis required for proliferation (Bauer et al, 2005); aspartate, generated from the TCA cycle intermediate oxaloacetate, is exported from the mitochondria to support nucleotide biosynthesis and protein translation (Birsoy et al, 2015; Sullivan et al, 2015). Thus, cell growth is directly coupled to the utilization of these metabolic “vents”, conferring proliferating cells with a mechanism to prevent ROS accumulation under growth factor stimulation. Another major nutrient reported to be under growth factor control is glutamine. Glutamine uptake in most transformed cells has been reported to be driven by myc‐dependent increase in glutamine uptake and mitochondrial deamination to produce glutamate and alpha‐ketoglutarate which support transamination reactions and TCA cycle anaplerosis, respectively (Wise et al, 2008; Gao et al, 2009). However, it is less clear how cells adapt to the accumulation of glutamine‐derived metabolites and prevent them from further increasing mitochondrial ROS as they enter the TCA cycle.
Unlike most extracellular growth factors, transforming growth factor‐beta‐1 (TGFβ‐1, hereafter TGFβ) does not bind to an RTK but activates a heterodimeric receptor with serine/threonine kinase activity, resulting in phosphorylation of Smad2 or Smad3 proteins which then bind to Smad4. Smad2/3‐Smad4 complexes bind to DNA and are responsible for the vast majority of TGFβ‐induced transcriptional output (Siegel & Massagué, 2003). The cellular responses to TGFβ are often context‐dependent; for example, TGFβ is known for its growth‐constraining and immunosuppressive effects, but it is also a potent inducer of epithelial–mesenchymal transition and fibrosis (Massagué, 2012). Recent evidence indicates that activation of TGFβ signaling supports these processes at least in part by reprogramming cellular metabolism. For instance, TGFβ‐mediated activation and differentiation of fibroblasts into extracellular matrix (ECM)‐producing myofibroblasts are accompanied by an increase in glucose uptake, glycolytic metabolism, and activation of the serine/glycine biosynthetic pathway (Andrianifahanana et al, 2016; Nigdelioglu et al, 2016; Selvarajah et al, 2019). In addition, glutaminolysis is required for TGFβ‐induced collagen production (Bernard et al, 2018; Hamanaka et al, 2019).
Here, we report that, unlike growth factors such as PDGF or IGF that primarily stimulate glucose metabolism, TGFβ stimulates both glucose and glutamine metabolism. Metabolic tracing experiments demonstrate that in TGFβ‐stimulated cells, glutamine that enters the mitochondria is preferentially used to support proline biosynthesis rather than TCA cycle anaplerosis. However, we observed that a rise in cellular proline levels in response to TGFβ was preceded by increased mitochondrial oxidative activity. We show that the TGFβ‐induced increase in proline occurs when TCA cycle oxidation exceeds the electron assimilation capacity of the electron transport chain. We propose that Smad4‐dependent induction of proline biosynthesis protects cells from the damaging effects of TGFβ‐induced increase in TCA cycle activity by diverting excess mitochondrial redox potential into the production of proline and supporting enhanced translation of collagens.
Results
TGFβ selectively promotes translation in serum‐stimulated fibroblasts
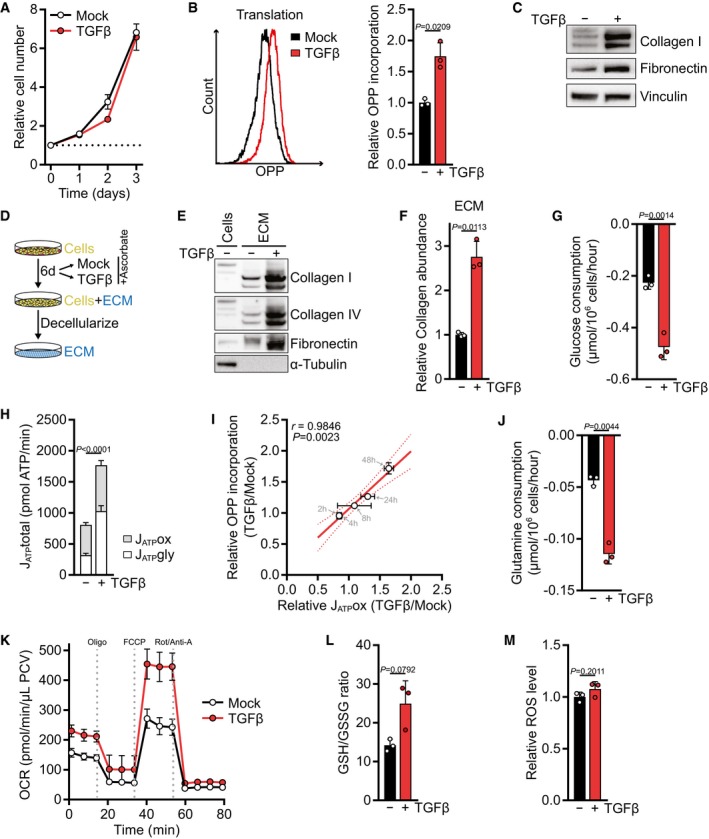
To study the primary response of fibroblasts to TGFβ stimulation in the presence of other growth factors, a condition that occurs during wound healing (Barrientos et al, 2008), we treated NIH‐3T3 fibroblasts for up to 72 h with TGFβ in the presence of 10% serum. During this period, TGFβ did not stimulate cell proliferation (Fig 1A). Nevertheless, TGFβ potently elevated the translation rate as measured by the incorporation of O‐propargyl‐puromycin, a tRNA‐mimetic compound, into nascent polypeptides (Figs 1B and EV1A). TGFβ treatment increased the expression of matrix proteins as reported by others (Fig 1C; Ignotz & Massagué, 1986), and we measured a higher deposition of collagens and fibronectin in cell‐derived ECM (Fig 1D–F). Similar observations were made in human lung fibroblasts (Fig EV1B and C). To sustain an increased translation rate, cells require high levels of ATP, both for charging of tRNAs with their cognate amino acid and for the regeneration of GTP which is consumed during elongation of the polypeptide chain. Consistent with a higher demand for ATP to meet the bioenergetic demand of increased translation, TGFβ treatment resulted in elevated consumption of glucose (Fig 1G) and increased the rate of ATP synthesis (Fig 1H). Both ATP production from glycolysis and oxidative phosphorylation were increased (Fig 1H).
Figure 1. TGFβ selectively promotes translation in serum‐stimulated fibroblasts.
-
AGrowth curve of NIH‐3T3 cells treated with TGFβ (2 ng/ml for all experiments) or vehicle alone (mock) for the indicated time. The cell number at the indicated days relative to the number at the start of the treatment (d0) is shown. The dotted line represents the cell number at d0.
-
BNIH‐3T3 cells were treated with TGFβ or mock for 48 h and incubated with O‐propargyl‐puromycin (OPP) for the last 60 min. The translation rate was determined by flow cytometry for OPP incorporation into proteins. A representative plot is shown on the left, and the quantification is shown on the right. Values are relative to mock‐treated cells.
-
CWestern blot of NIH‐3T3 cells treated with TGFβ or mock for 48 h.
-
DSchematic of extracellular matrix (ECM) production in vitro.
-
EWestern blot of ECM from NIH‐3T3 cells grown in the presence or absence of TGFβ and of cell lysates from NIH‐3T3 cells grown on a parallel plate. Note the absence of tubulin and the electromobility shift of collagens in ECM extracts.
-
FECM was produced from NIH‐3T3 cells according to (D), and collagen abundance was measured by picrosirius red staining, normalized to the packed cell volume before ECM extraction, and expressed relative to mock‐treated cells.
-
GNIH‐3T3 cells were treated with TGFβ or mock for 48 h. The medium was replaced in the last 12 h of the treatment (with the same treatments as before), and glucose consumption from the fresh media was measured using the YSI bioanalyzer.
-
HATP production rate from glycolysis (JATPgly) and mitochondrial oxidative phosphorylation (JATPox) from NIH‐3T3 cells treated with TGFβ or mock for 48 h, calculated using Seahorse data shown in (K) and Fig EV1F.
-
IPearson's correlation of mitochondrial ATP production rate (JATPox) and OPP incorporation of NIH‐3T3 cells treated with TGFβ or mock for 2, 4, 8, 24 and 48 h, relative to mock‐treated cells. Also see Fig EV1K and L. Note that the data points of the 2‐ and 4‐h measurements overlap.
-
JGlutamine consumption measured as in (G).
-
KNIH‐3T3 cells were treated with TGFβ or mock for 48 h, and the oxygen consumption rate (OCR) before and after treatment with mitochondrial inhibitors was measured using the Seahorse bioanalyzer. Oligo, oligomycin; Rot/Anti‐A, rotenone/antimycin; PCV, packed cell volume.
-
LNIH‐3T3 cells were treated with TGFβ or mock for 48 h, and reduced (GSH) and oxidized (GSSG) glutathione were measured by LC‐MS/MS. Shown is the GSH/GSSG ratio.
-
MNIH‐3T3 cells were treated with TGFβ for 48 h or mock and incubated with CM‐H2DCFDA for the last 30 min. ROS were measured by flow cytometry for CM‐H2DCFDA. Values are relative to mock‐treated cells.
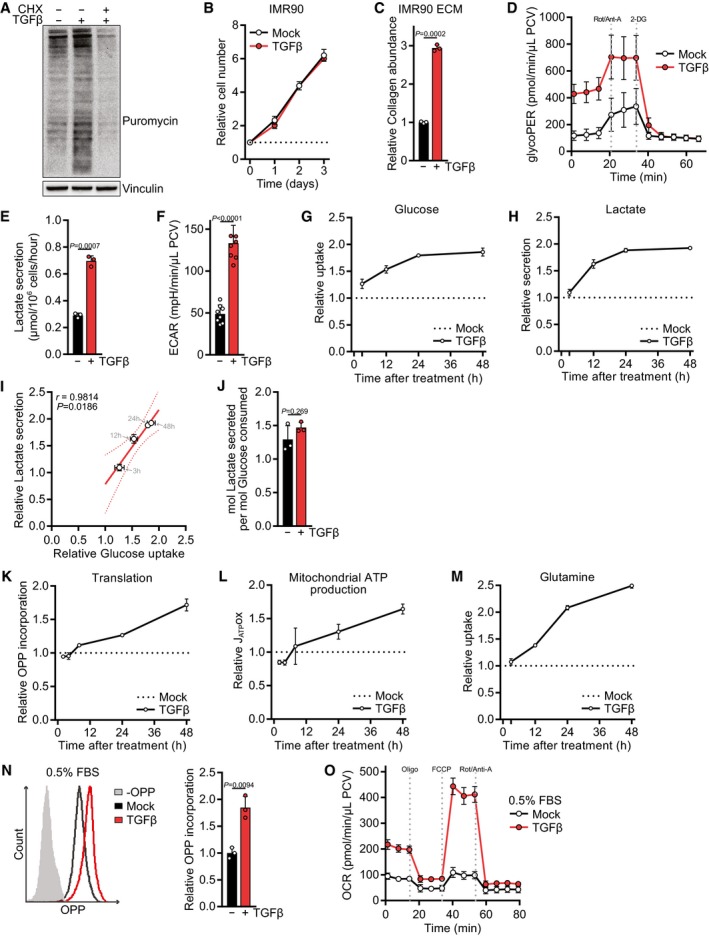
Figure EV1. TGFβ's effect on nutrient uptake and secretion, on glycolytic and oxidative metabolism, and on translation.
-
AWestern blot of NIH‐3T3 cells treated with TGFβ or mock for 24 h and incubated with puromycin for the last 10 min. The translation inhibitor cycloheximide (CHX) was added during puromycin incubation as a control.
-
BGrowth curve of IMR90 cells treated with TGFβ or mock for the indicated time. The cell number at the indicated days relative to the number at the start of the treatment (d0) is shown. The dotted line represents the cell number at d0.
-
CECM was produced from IMR90 cells grown in the presence or absence of TGFβ, and collagen abundance was measured by picrosirius red staining, normalized to the packed cell volume of cells grown on a separate plate under identical experimental conditions, and expressed relative to mock‐treated cells.
-
DNIH‐3T3 cells were treated with TGFβ or mock for 48 h, and the glycolytic rate (glycoPER) was measured at baseline and after subsequent injection of inhibitors using the Seahorse bioanalyzer. Rot/Anti‐A, rotenone/antimycin; 2‐DG, 2‐deoxyglucose; PCV, packed cell volume.
-
ENIH‐3T3 cells were treated with TGFβ or mock for 48 h, and lactate secretion into the medium was measured for the last 12 h of the experiment.
-
FNIH‐3T3 cells were treated with TGFβ or mock for 48 h, and the extracellular acidification rate (ECAR) was measured at baseline. Data were normalized to the packed cell volume.
-
G–INIH‐3T3 cells were treated with TGFβ or mock for 3, 12, 24 or 48 h, and the consumption of glucose from the media (G) or the secretion of lactate into the media (H) was measured in the last 12 h of the experiment. For each time point, values of TGFβ‐treated cells were normalized to the respective mock‐treated controls. Glucose uptake and lactate secretion of TGFβ‐treated relative to mock‐treated cells for each time point were analyzed by Pearson's correlation (I).
-
JRespiratory coupling, calculated by dividing the lactate secretion values from (E) through the glucose consumption values from Fig 1G.
-
KNIH‐3T3 cells were treated with TGFβ or mock for 2, 4, 8, 24, or 48 h and incubated with O‐propargyl‐puromycin (OPP) for the last 60 min. The translation rate was determined by flow cytometry for OPP incorporation into proteins. For each time point, values of TGFβ‐treated cells were normalized to the respective mock‐treated controls.
-
LNIH‐3T3 cells were treated with TGFβ or mock for 2, 4, 8, 24 or 48 h, and the oxygen consumption rate (OCR) before and after treatment with mitochondrial inhibitors was measured using the Seahorse bioanalyzer. From these data, the mitochondrial ATP production rate (JATPox) was calculated. For each time point, values of TGFβ‐treated cells were normalized to the respective mock‐treated controls.
-
MNIH‐3T3 cells were treated with TGFβ or mock for 3, 12, 24 or 48 h, and the consumption of glutamine from the media was measured in the last 12 h of the experiment. For each time point, values of TGFβ‐treated cells were normalized to the respective mock‐treated controls.
-
N, ONIH‐3T3 cells were serum‐deprived (0.5% FBS) and (N) treated with TGFβ or mock for 24 h and incubated with O‐propargyl‐puromycin (OPP) for the last 60 min followed by flow cytometry for OPP incorporation to measure the translation rate, or (O) treated with TGFβ for 48 h, and the oxygen consumption rate (OCR) before and after treatment with mitochondrial inhibitors was measured using the Seahorse bioanalyzer. Oligo, oligomycin. Values in (N) are relative to mock‐treated cells.
As reported by others (Nigdelioglu et al, 2016; Selvarajah et al, 2019), TGFβ‐stimulated cells displayed a higher glycolytic activity (Fig EV1D–F). We found that the increase in glucose uptake is an early event in response to TGFβ stimulation and plateaus after 24 h (Fig EV1G). Lactate secretion following TGFβ treatment showed similar kinetics (Fig EV1H) and is in proportion to the glucose taken up by TGFβ‐stimulated cells (Fig EV1I). Thus, lactate production per molecule glucose remained similar before and after TGFβ stimulation (Fig EV1J) and the amount of glucose utilized and not secreted as lactate also increases. Thus, TGFβ‐stimulated cells retain increased levels of glucose carbon to support biosynthesis and mitochondrial ATP production.
Using a kinetic analysis, we find that TGFβ‐induced translation and mitochondrial ATP production significantly correlated with each other (Figs 1I and EV1K and L), suggesting that an increase in oxidative phosphorylation is coupled to protein synthesis. In addition to glucose, TGFβ stimulated consumption of glutamine (Figs 1J and EV1M), which can act as anaplerotic substrate to support the TCA cycle (Deberardinis et al, 2007). To confirm that TGFβ stimulated oxidative phosphorylation, we measured the oxygen consumption rate on a Seahorse Bioanalyzer. This analysis demonstrated that TGFβ potently stimulated mitochondrial respiration and increased substrate availability to the electron transport chain (maximal respiration) (Fig 1K). An increased rate of protein synthesis and mitochondrial oxygen consumption in response to TGFβ treatment was also observed under serum‐deprived conditions (Fig EV1N and O).
Despite the increase in oxidative metabolism, cellular ROS production was not elevated in cells grown in TGFβ‐containing medium, as measured by the ratio of reduced/oxidized glutathione and the general ROS indicator DCFDA (Fig 1L and M). Taken together, these results indicate that TGFβ promotes nutrient uptake and mitochondrial oxidation primarily to support cellular bioenergetics and translation of ECM proteins, without increasing either cell proliferation or oxidative stress.
TGFβ promotes proline synthesis from glutamine for collagen production
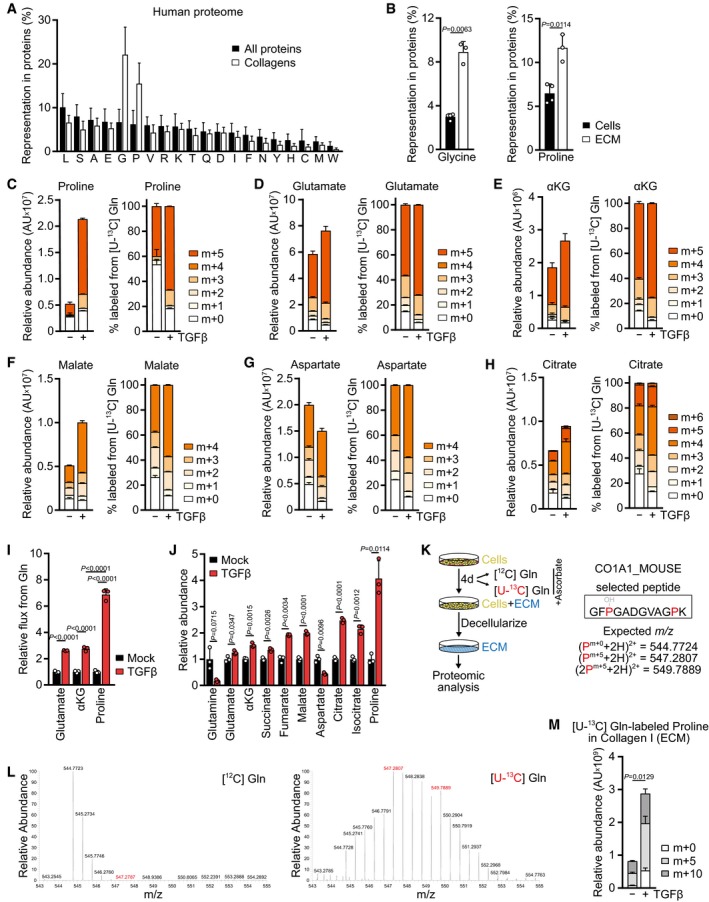
In addition to ATP, translation requires a continuous supply of amino acids for tRNA charging. Since TGFβ stimulated the synthesis of matrix proteins, but not growth, we hypothesized that TGFβ treatment induces the accumulation of intracellular amino acids that are particularly enriched in matrix proteins relative to other proteins. The amino acid composition of collagens, the most abundant proteins in the human body and in the ECM, is significantly different from that of other proteins (Fig 2A); compared to other proteins, collagens are particularly enriched in glycine (Fig 2A). To verify this in living cells, we performed acid hydrolysis of proteins isolated from NIH‐3T3 cells and their secreted ECM and confirmed a significant increase in the representation of glycine in cell‐derived ECM compared to total cellular protein (Fig 2B).
Figure 2. TGFβ promotes proline synthesis from glutamine for collagen production.
-
AComparison of the amino acid representation in all human proteins and all collagens. The x‐axis denotes individual amino acids shown by single‐letter code.
-
BGlycine and proline representation in protein acid hydrolysates of untreated NIH‐3T3 cells and their secreted ECM, produced as in Fig 1D. Amino acids were measured by GC‐MS.
-
C–HTracing of [U‐13C] l‐glutamine ([U‐13C] Gln) into indicated metabolites. NIH‐3T3 cells were treated with TGFβ (2 ng/ml for all experiments) or mock for 48 h, and the medium was replaced (including all treatments) with DMEM lacking l‐glutamine and supplemented with [U‐13C] Gln for the last 8 h. Metabolic steady state was reached at this time point (see kinetic labeling curves in Fig EV3B). Metabolites were measured by LC‐MS. The left graph shows the total pool size normalized to the packed cell volume; the right side shows the percent labeling. αKG, alpha‐ketoglutarate; AU, arbitrary units.
-
IGlutamine flux into glutamate, αKG, and proline in NIH‐3T3 cells treated with TGFβ or mock for 48 h, relative to mock‐treated cells.
-
JAbundance of indicated metabolites in NIH‐3T3 cells treated with TGFβ or mock for 48 h, relative to mock‐treated cells. Metabolites were measured by GC‐MS.
-
KSchematic of [U‐13C] Gln tracing into proline in collagen I‐α1 (CO1A1) in NIH‐3T3 cell‐derived ECM. Expected m/z of unlabeled (m+0) and fully labeled proline (m+5 for one proline, m+10 for two prolines) for the analytical peptide are shown.
-
LRepresentative MS2 spectra of the CO1A1 peptide in ECM generated with unlabeled ([12C] Gln, left) and fully labeled glutamine ([U‐13C] Gln, right). Peaks representing fully labeled (m+5, m+10) proline are highlighted in red. Peptides were measured by LC‐MS.
-
MRelative abundance of the analytical peptide containing unlabeled (m+0) or one/two fully labeled prolines (m+5/m+10) from the CO1A1 peptide in ECM derived from NIH‐3T3 cells treated with TGFβ or mock in the presence of fully labeled l‐glutamine ([U‐13C] Gln). Peptides were measured by LC‐MS. The amount of ECM analyzed was normalized to the packed cell volume of cells grown on a parallel plate under identical conditions.
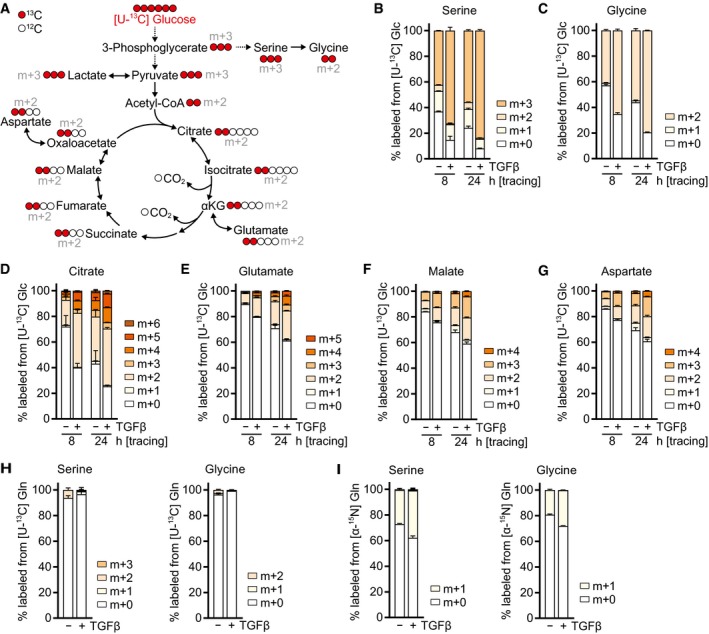
It has been reported that in addition to stimulating ATP production, glucose catabolism promotes glycine synthesis in the glycolytic pathway via serine from the glycolytic intermediate 3‐phosphoglycerate (Fig EV2A). To test whether TGFβ diverts glycolytic carbons into glycine biosynthesis, we traced the fate of glucose uniformly labeled with [13C] at all six carbons ([U‐13C] Glc). This analysis demonstrated that TGFβ stimulated the synthesis of both serine and glycine from glucose (Fig EV2B and C). In addition, TGFβ also increased labeling of citrate and downstream TCA cycle metabolites from glucose (Fig EV2D–G). These data are in line with a higher availability of glucose for both biosynthesis and mitochondrial oxidation upon TGFβ stimulation. We also assessed the contribution of glutamine to glycine biosynthesis. While glutamine carbon did not contribute to serine and glycine (Fig EV2H), both serine and glycine were substantially labeled by the glutamine alpha‐nitrogen (Fig EV2I). TGFβ treatment further increased alpha‐nitrogen labeling of serine and glycine (Fig EV2I), consistent with TGFβ‐induced expression of PSAT1 (Hamanaka et al, 2019) which catalyzes the transamination reaction in the serine biosynthetic pathway.
Figure EV2. TGFβ promotes serine and glycine biosynthesis and glucose oxidation.
-
ASchematic of stable isotope tracing using d‐glucose labeled with [13C] at all six carbons ([U‐13C] glucose, shown in red).
-
B–GTracing of [U‐13C] glucose ([U‐13C] Glc) into indicated metabolites. NIH‐3T3 cells were treated with TGFβ or mock for 48 h in DMEM lacking l‐serine and glycine in the presence of 0.5% dialyzed FBS, and the medium was replaced (including all treatments) with DMEM lacking d‐glucose, l‐serine, and glycine and supplemented with [U‐13C] Glc for the last 24 or 8 h in the presence of 0.5% dialyzed FBS. Metabolites were measured by LC‐MS.
-
H, ITracing of (H) [U‐13C] l‐glutamine ([U‐13C] Gln) or (I) [α‐15N] l‐glutamine ([α‐15N] Gln) into serine and glycine. NIH‐3T3 cells were treated with TGFβ or mock for 48 h in DMEM lacking l‐serine and glycine in the presence of 0.5% dialyzed FBS, and the medium was replaced (including all treatments) with DMEM lacking l‐glutamine, l‐serine, and glycine and supplemented with [U‐13C] Gln (H) or [α‐15N] Gln (I) for the last 8 h in the presence of 0.5% dialyzed FBS. Metabolites were measured by GC‐MS.
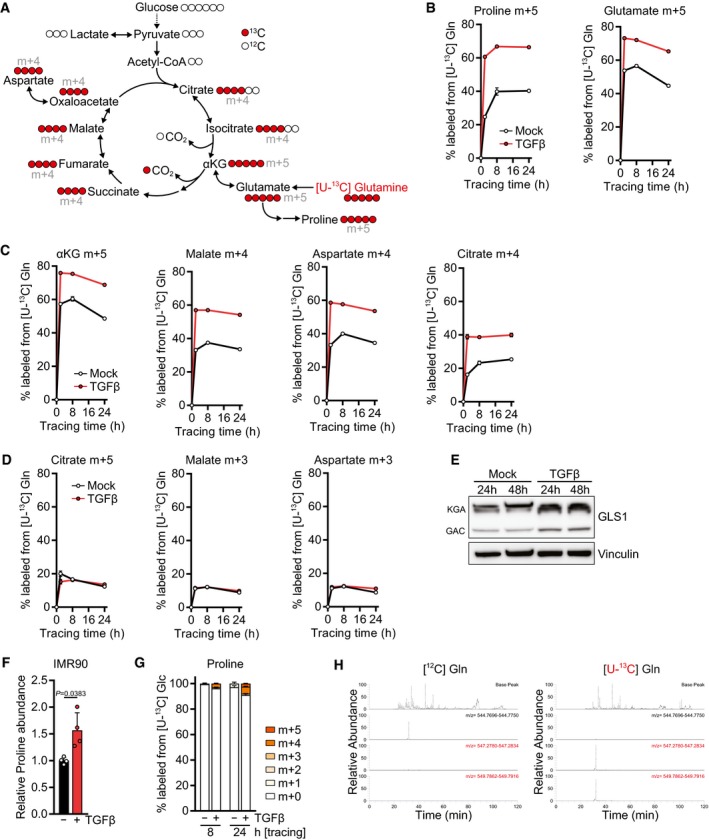
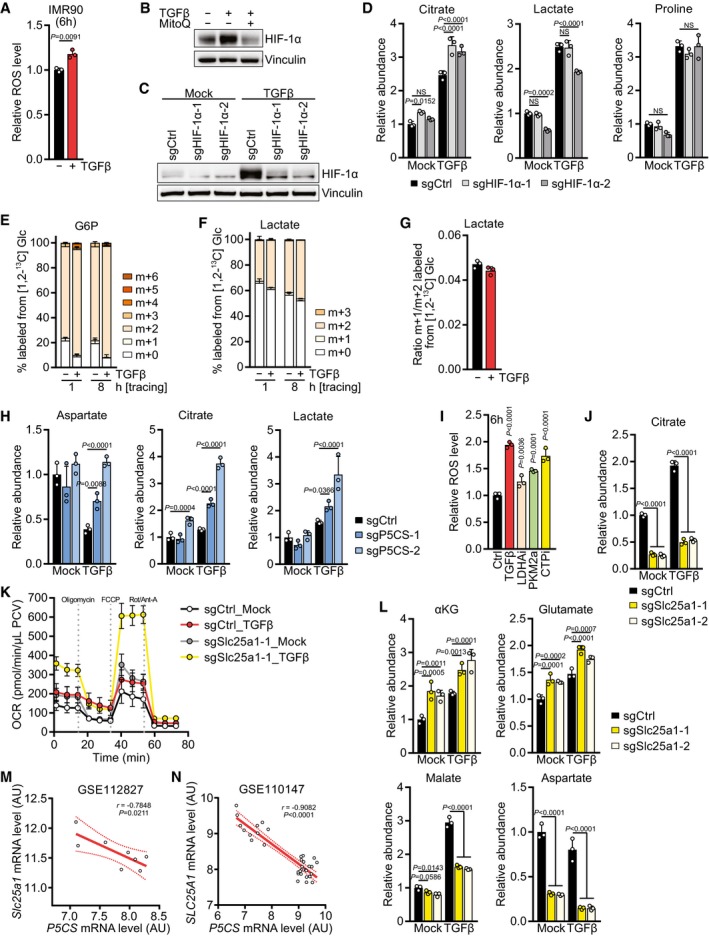
The second major amino acid enriched in collagens and the ECM is proline and its modified derivative hydroxyproline (Fig 2A and B). Since proline is a non‐essential amino acid produced from glutamine (Fig EV3A), we tested whether TGFβ promotes diversion of glutamine carbons into proline biosynthesis by tracing the fate of glutamine uniformly labeled with [13C] at all five carbons ([U‐13C] Gln). Indeed, we found that TGFβ treatment resulted in a substantial increase in the fraction and abundance of proline labeled from glutamine (m+5) (Figs 2C and EV3B). In addition, TGFβ‐treated cells displayed an increase in the contribution of glutamine to glutamate (m+5) (Figs 2D and EV3B) and to all TCA cycle metabolites we measured via oxidative metabolism (m+4) (Figs 2E–H and EV3C). Based on the low percentage of labeling of later TCA cycle intermediates such as malate from glucose (Fig EV2F), these data suggest that TGFβ‐treated cells increase glutamine anaplerosis. In contrast, reductive synthesis of citrate (m+5), malate and aspartate (m+3) from glutamine was unchanged upon TGFβ stimulation (Figs 2F–H and EV3D). Consistent with higher utilization of glutamine for proline biosynthesis and TCA cycle anaplerosis, glutaminase (GLS1) was upregulated in TGFβ‐treated cells (Fig EV3E).
Figure EV3. TGFβ promotes proline biosynthesis and glutamine oxidation.
-
ASchematic of stable isotope tracing using l‐glutamine labeled with [13C] at all five carbons ([U‐13C] glutamine, shown in red).
-
B–DKinetic labeling curves of the indicated isotopomers after tracing with [U‐13C] glutamine ([U‐13C] Gln). NIH‐3T3 cells were treated with TGFβ or mock for 48 h, and the medium was replaced (including all treatments) with DMEM lacking l‐glutamine and supplemented with [U‐13C] Gln for the last 2, 8, or 24 h. Metabolites were measured by LC‐MS. For other isotopomers, see Fig 2C–H.
-
EWestern blot of NIH‐3T3 cells treated with TGFβ or mock for the indicated time in the presence of 0.5% FBS. KGA (kidney type) and GAC (glutaminase C) denote the two isoforms of GLS1.
-
FIMR90 cells were treated with TGFβ or mock for 48 h, and abundance of proline was measured by GC‐MS. Values are relative to mock‐treated cells.
-
GTracing of [U‐13C] glucose ([U‐13C] Glc) into proline. NIH‐3T3 cells were treated with TGFβ or mock for 48 h in DMEM lacking l‐serine and glycine in the presence of 0.5% dialyzed FBS, and the medium was replaced (including all treatments) with DMEM lacking d‐glucose, l‐serine, and glycine and supplemented with [U‐13C] Glc for the last 8 or 24 h in the presence of 0.5% dialyzed FBS. Proline was measured by LC‐MS.
-
HRepresentative MS1 spectra of the CO1A1 peptide in ECM generated with unlabeled ([12C] Gln, left) and fully labeled glutamine ([U‐13C] Gln, right). Also see Fig 2K–M.
Quantification of metabolic fluxes revealed that the flux of glutamine carbons into alpha‐ketoglutarate was increased more than twofold in TGFβ‐treated cells and matched the elevated flux through glutaminase (Fig 2I). However, the flux of glutamine carbons into proline increased by sevenfold (Fig 2I). This was also reflected by a fourfold increase in free proline in cells upon treatment with TGFβ, while the maximal increase in the abundance of glutamate and TCA cycle intermediates was 2.5‐fold or less (Fig 2J). Proline levels were also increased when human lung fibroblasts were treated with TGFβ (Fig EV3F). Since a significant amount of glutamate carbons were labeled from glucose (Fig EV2E), we also assessed whether glucose carbons contributed to proline. However, < 10% of proline carbon was labeled after 24 h of [U‐13C] Glc tracing (Fig EV3G), indicating that glycolytic carbon does not substantially contribute to proline biosynthesis.
Glucose‐derived glycine has been shown to be incorporated into collagen I (Nigdelioglu et al, 2016), but whether fibroblasts use glutamine‐derived proline for the biosynthesis of collagen I is not known. To test this, we cultured NIH‐3T3 fibroblasts in the presence of unlabeled ([12C]) or fully labeled ([U‐13C]) glutamine and extracted cell‐derived ECM for proteomic analysis (Fig 2K). Glutamine carbons were readily incorporated as proline into collagen I secreted into the extracellular space (Figs 2L and EV3H), and the abundance of the collagen I peptide containing glutamine‐derived proline increased with TGFβ treatment (Fig 2M). Taken together, these data demonstrate that TGFβ promotes the utilization of glucose and glutamine for both mitochondrial oxidation and biosynthesis of glycine and proline which are utilized for translation of collagens.
P5CS is required for proline biosynthesis and is upregulated in lung fibrosis patients
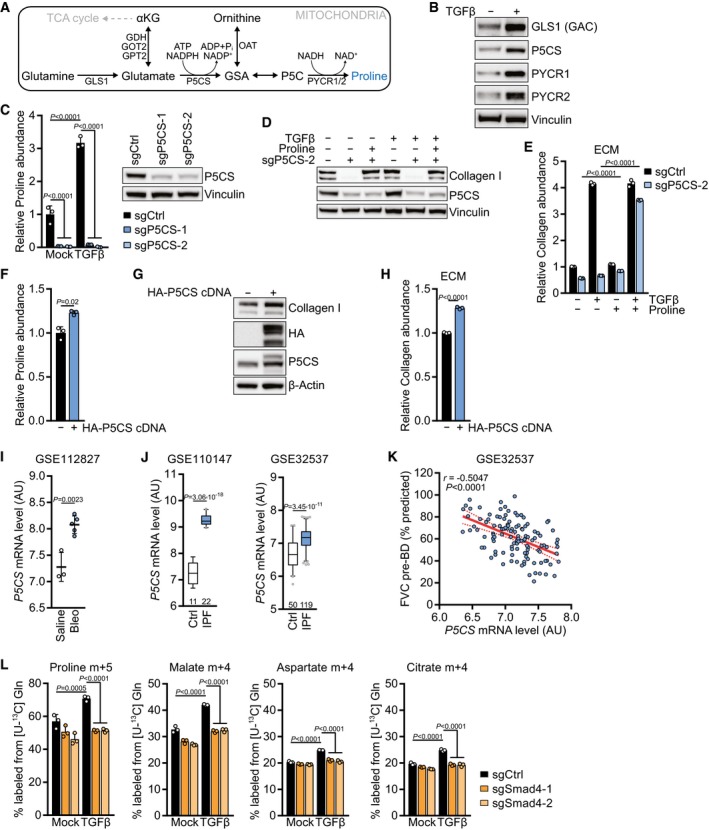
While the TGFβ‐mediated stimulation of glycine biosynthesis has been reported (Nigdelioglu et al, 2016; Selvarajah et al, 2019), it is less well understood how TGFβ promotes proline biosynthesis. In particular, how TGFβ directs the preferential flux of glutamine carbon into proline biosynthesis over TCA cycle anaplerosis is unknown. Proline is made in mitochondria from glutamine‐derived glutamate via two steps (Fig 3A): first, delta‐1‐pyrroline‐5‐carboxylate synthase (P5CS) catalyzes the ATP‐ and NADPH‐dependent phosphorylation and reduction conversion of glutamate to gamma‐glutamic semi‐aldehyde (GSA), which is in equilibrium with delta‐1‐pyrroline‐5‐carboxylate (P5C). Pyrroline‐5‐carboxylate reductase family members 1 and 2 (PYCR1/2) further reduce P5C to proline in a reaction that preferentially uses NADH (de Ingeniis et al, 2012). We observed that, in addition to upregulating GLS1, TGFβ treatment elevated the expression of all enzymes in the mitochondrial proline biosynthetic pathway (Fig 3B). Similar observations were made in human lung fibroblasts (Fig EV4A). To test whether the induction of these enzymes is required for proline biosynthesis, we genetically disrupted all enzymes in the pathway by CRISPR/Cas9‐mediated gene deletion. While deleting Pycr1 or Pycr2 individually did not affect cellular proline levels (Fig EV4B), proline was profoundly depleted in cells with deletion of P5CS encoded by aldehyde dehydrogenase 18 family member A1 (Aldh18a1, hereafter P5CS) (Fig 3C). Consistent with the high representation of proline in collagens (Fig 2A), P5CS deletion diminished the expression of collagen I protein both in untreated and TGFβ‐treated cells, which was restored by addition of proline to the culture medium (Fig 3D). Similar results were obtained when measuring collagen abundance in cell‐derived ECM (Fig 3E).
Figure 3. P5CS is required for proline synthesis and upregulated in lung fibrosis patients.
-
ASchematic of the mitochondrial proline biosynthetic pathway.
-
BWestern blot of NIH‐3T3 cells treated with TGFβ (2 ng/ml for all experiments) or mock for 48 h in the presence of 0.5% FBS.
-
CLeft: NIH‐3T3 cells expressing sgCtrl, sgP5CS‐1, or sgP5CS‐2 were treated with TGFβ or mock for 48 h, and proline abundance was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells. Right: Western blot of NIH‐3T3 cells expressing sgCtrl, sgP5CS‐1, or sgP5CS‐2.
-
DWestern blot of NIH‐3T3 cells expressing sgCtrl or sgP5CS‐2 and treated with TGFβ or mock for 48 h in the presence or absence of 0.15 mM proline.
-
ECollagen abundance in ECM produced by NIH‐3T3 cells expressing sgCtrl or sgP5CS‐2 grown in the presence or absence of TGFβ and 0.15 mM proline, measured by picrosirius red staining, and normalized to the packed cell volume of cells grown on a parallel plate under identical conditions. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
FProline abundance in NIH‐3T3 cells expressing empty vector or HA‐P5CS cDNA, measured by GC‐MS. Values are relative to mock‐treated empty vector‐expressing cells.
-
GWestern blot of NIH‐3T3 cells expressing empty vector or HA‐P5CS cDNA.
-
HCollagen abundance in ECM produced by NIH‐3T3 cells expressing empty vector or HA‐P5CS cDNA, measured by picrosirius red staining, and normalized to the packed cell volume of cells grown on a parallel plate under identical conditions. Values are relative to mock‐treated empty vector‐expressing cells. P < 0.0001 (sgP5CS ± proline in mock and TGFβ‐treated cells).
-
I, JAnalysis of the indicated gene expression datasets for mRNA levels of P5CS: (I) lung tissue from mice with pulmonary fibrosis induced by bleomycin (Bleo) treatment compared to saline treatment (GSE112827); (J) two datasets (GSE110147, GSE32537) from lungs of patients with idiopathic pulmonary fibrosis (IPF) compared to normal controls (Ctrl). AU, arbitrary units. The number of patients per group is indicated.
-
KPearson's correlation of P5CS mRNA level and forced vital capacity (FVC) before bronchodilator (pre‐BD) as percentage of what was predicted for each patient, from clinical data of GSE32537.
-
LTracing of [U‐13C] l‐glutamine ([U‐13C] Gln) into indicated metabolites. NIH‐3T3 cells expressing sgCtrl, sgSmad4‐1, or sgSmad4‐2 were treated with TGFβ or mock for 48 h, and the medium was replaced (including all treatments) with DMEM without l‐glutamine and supplemented with [U‐13C] Gln for the last 8 h. Metabolites were measured by GC‐MS.
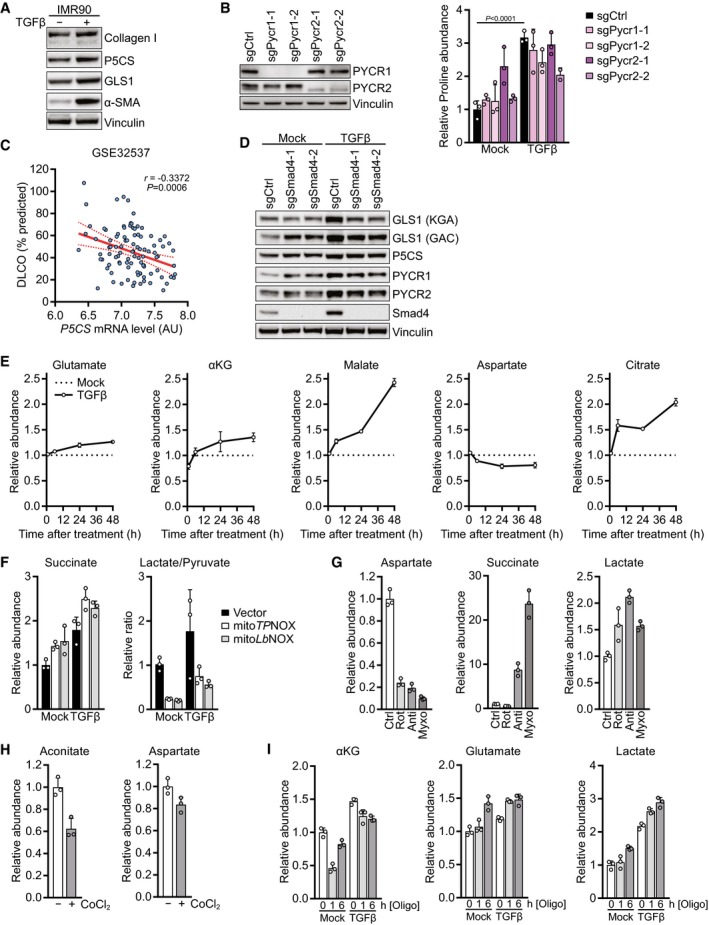
Figure EV4. Regulation of proline biosynthesis and controls for metabolic manipulations.
-
AWestern blot of IMR90 cells treated with TGFβ or mock for 48 h in the presence of 0.5% FBS.
-
BLeft: Western blot of NIH‐3T3 cells expressing sgCtrl, sgPycr1‐1, sgPycr1‐2, sgPycr2‐1, or sgPycr2‐2. Right: NIH‐3T3 cells expressing sgCtrl, sgPycr1‐1, sgPycr1‐2, sgPycr2‐1, or sgPycr2‐2 were treated with TGFβ or mock for 48 h, and proline abundance was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
CPearson's correlation of P5CS mRNA level and diffusing capacity for carbon monoxide (DLCO) in IPF patients from GSE32537 as a percentage of what was predicted for each patient. AU, arbitrary units.
-
DWestern blot of NIH‐3T3 cells expressing sgCtrl, sgSmad4‐1, or sgSmad4‐2 and treated with TGFβ or mock for 48 h in the presence of 0.5% FBS.
-
ENIH‐3T3 cells were treated with TGFβ or mock for 1, 6, 24 or 48 h, and abundance of the indicated metabolites was measured by GC‐MS. Values are relative to mock‐treated cells for each time point.
-
FNIH‐3T3 cells expressing empty vector, mitoTPNOX, or mitoLbNOX were treated with TGFβ or mock for 48 h, and abundance of succinate, lactate, and pyruvate was measured by GC‐MS. The lactate/pyruvate ratio was calculated as a surrogate for the cytosolic NADH/NAD+ ratio. Values are normalized to mock‐treated empty vector‐expressing cells.
-
GNIH‐3T3 cells were treated with rotenone, antimycin, myxothiazol, or vehicle control for 1 h, and abundance of the indicated metabolites was measured by GC‐MS. Values are normalized to control‐treated cells.
-
HNIH‐3T3 cells were treated with cobalt chloride (CoCl2) or vehicle control for 6 h, and abundance of the indicated metabolites was measured by GC‐MS. Values are normalized to control‐treated cells.
-
INIH‐3T3 cells were treated with TGFβ or mock for 48 h, and oligomycin or vehicle control was added for the last 1 or 6 h of the treatment. Abundance of the indicated metabolites was measured by GC‐MS. Values are normalized to mock and control‐treated cells.
We genetically engineered cells to overexpress P5CS to test whether the upregulation of P5CS by TGFβ contributes to increased proline and collagen biosynthesis. Indeed, ectopic expression of P5CS increased the abundance of proline in cells (Fig 3F) and elevated levels of collagen I in cells and the ECM (Fig 3G and H), although not to the same extent as did TGFβ stimulation. These data demonstrate that expression of P5CS is required and can be sufficient for proline and collagen biosynthesis in serum‐stimulated cells growing in complete medium, and that collagen levels depend on mitochondrial proline biosynthesis.
Based on these findings, we hypothesized that P5CS expression could also be relevant for fibrotic diseases which are characterized by excessive deposition of collagen by chronically activated myofibroblasts. Focusing on idiopathic pulmonary fibrosis (IPF), the most severe and lethal fibrotic disease (Coultas et al, 1994), we analyzed P5CS expression in publicly available gene expression datasets from lungs of mice treated with bleomycin to induce pulmonary fibrosis or from lungs of IPF patients (Yang et al, 2013; Cecchini et al, 2018). We found that P5CS was significantly upregulated in the bleomycin mouse model of pulmonary fibrosis (Fig 3I) and in IPF patients compared to normal controls in two independent datasets (Fig 3J). Moreover, the forced vital capacity (FVC) as well as the diffusing capacity for carbon monoxide (DLCO), two independent parameters of lung function, inversely correlated with expression levels of P5CS in IPF patients (Figs 3K and EV4C). Taken together, these data show that P5CS expression is critical for proline and collagen biosynthesis and correlates with disease‐relevant parameters.
Next, we investigated the regulation of proline biosynthesis by TGFβ using cells in which Smad4 was deleted via CRISPR/Cas9 to abrogate Smad4‐dependent transcription. While the increase in protein levels of proline biosynthetic enzymes in response to TGFβ was only partly regulated by Smad4, Smad4 deletion abolished the induction of glutaminase upon TGFβ treatment (Fig EV4D). The increase in labeling of proline and TCA cycle intermediates from glutamine in response to TGFβ treatment was completely dependent on Smad4 (Fig 3L), suggesting that both the TGFβ‐mediated increase in TCA cycle anaplerosis and proline biosynthesis were regulated by Smad4‐dependent glutaminase induction.
TGFβ‐induced stimulation of mitochondrial bioenergetics is required for increased proline biosynthesis
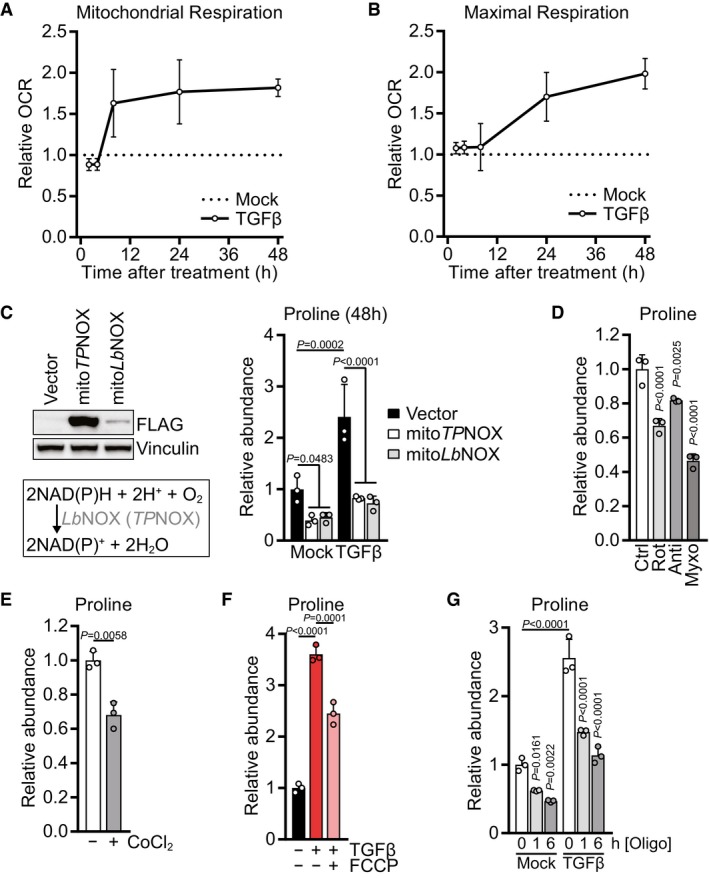
We next investigated the kinetics with which TGFβ stimulates mitochondrial TCA cycle activity and oxidative phosphorylation. TGFβ treatment increased mitochondrial respiration as soon as 6 h after TGFβ addition (Fig 4A). While respiration reached a relative plateau after 24 h of treatment, the maximal respiratory capacity continued to increase beyond this time point (Fig 4B). Similarly, levels of glutamate and TCA cycle intermediates steadily increased from 6 to 48 h after TGFβ treatment (Fig EV4E), indicating a progressive increase in mitochondrial TCA cycle flux after TGFβ stimulation. These data indicate that TGFβ continuously increases substrate availability to the ETC starting as soon as 6 h after the treatment, resulting in higher mitochondrial respiration and accumulation of mitochondrial redox potential.
Figure 4. TGFβ‐induced stimulation of mitochondrial bioenergetics is required for increased proline biosynthesis.
-
A, BMitochondrial basal respiration (A) and FCCP‐induced maximal respiration (B) of NIH‐3T3 cells treated with TGFβ (2 ng/ml for all experiments) or mock for 2, 4, 8, 24 or 48 h, based on Seahorse OCR measurements. The relative values were calculated by normalizing the respective values of TGFβ‐treated cells to those of mock‐treated cells for each time point.
-
CLeft: Western blot of NIH‐3T3 cells expressing empty vector, mitoTPNOX, or mitoLbNOX. MitoTPNOX or mitoLbNOX expression is detected by probing for their FLAG‐tag. Right: NIH‐3T3 cells expressing empty vector, mitoTPNOX, or mitoLbNOX were treated with TGFβ or mock for 48 h, and proline abundance was measured by GC‐MS. Values are normalized to mock‐treated empty vector‐expressing cells.
-
DNIH‐3T3 cells were treated with rotenone, antimycin, myxothiazol, or vehicle control for 1 h, and proline abundance was measured by GC‐MS. Values are normalized to control‐treated cells.
-
ENIH‐3T3 cells were treated with cobalt chloride (CoCl2) or vehicle control for 6 h, and proline abundance was measured by GC‐MS. Values are normalized to control‐treated cells.
-
FNIH‐3T3 cells were treated with TGFβ or mock for 48 h, and FCCP or vehicle control was added for the last 6 h of the treatment. Proline abundance was measured by GC‐MS. Values are normalized to mock‐ and control‐treated cells.
-
GNIH‐3T3 cells were treated with TGFβ or mock for 48 h, and oligomycin or vehicle control was added for the last 1 or 6 h of the treatment. Proline abundance was measured by GC‐MS. Values are normalized to mock‐ and control‐treated cells.
Given that mitochondrial redox equivalents in the form of NADH and NADPH act as coenzymes for proline biosynthesis (Fig 3A), we hypothesized that an accumulation of mitochondrial redox potential drives proline biosynthesis in cells stimulated with TGFβ. To test this idea, we genetically engineered NIH‐3T3 cells to express a mitochondria‐targeted version of the water‐forming NADH oxidase from Lactobacillus brevis (LbNOX) or to express a mitochondria‐targeted version of its NADPH‐specific mutant triphosphopyridine nucleotide oxidase (TPNOX) (Titov et al, 2016; Cracan et al, 2017). As reported previously (Titov et al, 2016; Cracan et al, 2017), these cells displayed succinate accumulation and a reduced lactate/pyruvate ratio (Fig EV4F), a surrogate for the cytosolic NADH/NAD+ ratio (Williamson et al, 1967). We observed that expression of either mitochondrial LbNOX or mitochondrial TPNOX reduced proline levels at baseline (Fig 4C). Notably, mitochondrial LbNOX or mitochondrial TPNOX abrogated proline accumulation in response to TGFβ stimulation (Fig 4C). These data suggest that an excess of mitochondrial redox potential beyond the electron assimilation capacity of the ETC can preferentially divert glutamine‐derived glutamate into proline biosynthesis rather than into the TCA in response to TGFβ (Fig 2I).
If mitochondrial redox potential is the sole regulator of proline biosynthesis, then inhibition of mitochondrial electron transport at complexes I and III might increase proline levels, as it results in an accumulation of NAD(P)H (Sullivan et al, 2015). As reported before (Mullen et al, 2014; Sullivan et al, 2015), treatment with rotenone, antimycin, or myxothiazol reduced aspartate levels and induced succinate and lactate accumulation (Fig EV4G), but decreased cellular proline levels (Fig 4D). Treatment with cobalt chloride (CoCl2), which can contribute to mitochondrial damage by disrupting iron–sulfur clusters (Ranquet et al, 2007; Dai et al, 2008; Macomber & Imlay, 2009) that are present within aconitase and multiple ETC components, also reduced levels of proline, aconitate, and aspartate (Figs 4E and EV4H). Together, these results suggest that intact mitochondrial electron transport is critical to maintain proline levels.
Electron transport within the ETC generates a proton motive force across the mitochondrial inner membrane that drives ATP synthesis. To uncouple electron transport from the proton gradient, we applied carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP) to cells treated with TGFβ for 48 h. Addition of FCCP in the last 6 h of the treatment was sufficient to suppress proline accumulation (Fig 4F). As FCCP uncouples mitochondrial electron transport from ATP synthesis, these data suggest that TGFβ‐induced proline biosynthesis also depends on coupled electron transport to maintain mitochondrial ATP production. To test this directly, we applied the ATP synthase inhibitor oligomycin to cells that had been stimulated with TGFβ for 48 h, resulting in reduced levels of alpha‐ketoglutarate but increased levels of glutamate and lactate (Fig EV4I). Notably, oligomycin treatment led to a progressive decrease in cellular proline levels both in untreated and TGFβ‐stimulated cells (Fig 4G). Thus, mitochondrial ATP synthesis is required for proline biosynthesis, likely for the reaction catalyzed by P5CS which consumes ATP (Fig 3A).
Proline biosynthesis is a vent for TGFβ‐induced mitochondrial redox stress
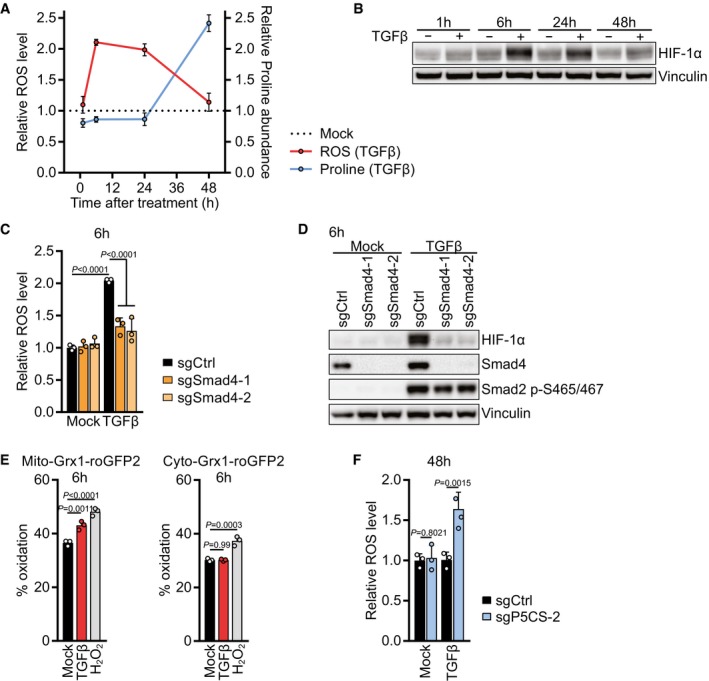
The above data indicate that TGFβ promotes mitochondrial oxidative activity, resulting in an increased rate of ATP synthesis and the accumulation of NAD(P)H, and that both ATP and NAD(P)H accumulation are required to convert mitochondrial glutamine into proline. Consistent with this, we found that unlike other TGFβ‐induced mitochondrial metabolites (Fig EV4E), proline did not increase until after mitochondrial respiration reached a plateau at 24 h after TGFβ treatment (Figs 4A and 5A). In fact, at earlier time points we found that TGFβ‐induced stimulation of mitochondrial metabolism was associated with increased ROS generation (Fig 5A). An excess of mitochondrial redox potential in response to increased mitochondrial oxidative metabolism can result in the generation of ROS (Murphy, 2009; Schieber & Chandel, 2014). We observed that ROS levels were increased 6 h after TGFβ stimulation but declined over time and went back to baseline after 48 h despite continued treatment with TGFβ (Fig 5A, see also Fig 1M). ROS were also transiently elevated in human lung fibroblasts 6 h after addition of TGFβ to the culture medium (Fig EV5A). The timing of ROS increase correlated with the early increase in TCA cycle intermediates and mitochondrial respiration in response to TGFβ stimulation (Figs 4A and EV4E), suggesting that TGFβ‐induced mitochondrial oxidative activity underlies the elevated ROS.
Figure 5. Proline biosynthesis is a vent for TGFβ‐induced mitochondrial redox stress.
-
ANIH‐3T3 cells were treated with TGFβ or mock for 1, 6, 24 or 48 h. ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment (Red line). Proline abundance was measured by GC‐MS (Blue line). The relative levels were calculated by normalizing the respective values of TGFβ‐treated cells to those of mock‐treated cells for each time point.
-
BWestern blot of NIH‐3T3 cells treated with TGFβ or mock for 1, 6, 24 or 48 h.
-
CNIH‐3T3 cells expressing sgCtrl, sgSmad4‐1, or sgSmad4‐2 were treated with TGFβ or mock for 6 h, and ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment. Values are normalized to mock‐treated sgCtrl‐expressing cells.
-
DWestern blot of NIH‐3T3 cells expressing sgCtrl, sgSmad4‐1, or sgSmad4‐2 and treated with TGFβ or mock for 6 h in the presence of 0.5% FBS.
-
ENIH‐3T3 cells expressing mito‐Grx1‐roGFP2 or cyto‐Grx1‐roGFP2 were treated with TGFβ or mock for 6 h or with 100 μM H2O2 as a control. Emission was measured with a 520/10‐nm filter after excitation with 405 nm and 488 nm lasers using flow cytometry. Oxidized roGFP2 gains excitability at 405 nm and loses excitability at 488 nm. Oxidation status is expressed as percentage of maximal oxidation which was determined by treating cells with 5 mM H2O2 for 5 min before harvest.
-
FNIH‐3T3 cells expressing sgCtrl or sgP5CS‐2 were treated with TGFβ or mock for 48 h in the presence of 0.15 mM proline. ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment.
Figure EV5. Deletion of HIF‐1α, P5CS, and Slc25a1, and tracing PPP activity.
-
AIMR90 cells were treated with TGFβ or mock for 6 h, and ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment. Values are relative to mock‐treated cells.
-
BWestern blot of NIH‐3T3 cells treated with TGFβ or mock for 6 h in the presence or absence of mitoquinol (MitoQ).
-
CWestern blot of NIH‐3T3 cells expressing sgCtrl, sgHIF‐1α‐1, or sgHIF‐1α‐2 and treated with TGFβ or mock for 6 h in the presence of 0.5% FBS.
-
DNIH‐3T3 cells expressing sgCtrl, sgHIF‐1α‐1, or sgHIF‐1α‐2 were treated with TGFβ or mock for 48 h, and abundance of the indicated metabolites was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
E–GTracing of [1,2‐13C] D‐glucose ([1,2‐13C] Glc) into (E) G6P and (F) lactate. NIH‐3T3 cells were treated with TGFβ or mock for 48 h in DMEM lacking l‐serine and glycine in the presence of 0.5% dialyzed FBS, and the medium was replaced (including all treatments) with DMEM lacking d‐glucose, l‐serine, and glycine and supplemented with [1,2‐13C] Glc for the last 1 or 8 h in the presence of 0.5% dialyzed FBS. Metabolites were measured by LC‐MS. (G) The ratio of m+1‐ versus m+2‐labeled lactate was calculated as a measure of PPP activity.
-
HNIH‐3T3 cells expressing sgCtrl, sgP5CS‐1, or sgP5CS‐2 were treated with TGFβ or mock for 48 h, and abundance of the indicated metabolites was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
INIH‐3T3 cells were treated with TGFβ, LDHAi (GSK 2837808A), PKM2a (DASA), CTPi, or vehicle control for 6 h, and ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment. Values are relative to control‐treated cells. P‐values represent comparison of individual samples to the control.
-
JNIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2 were treated with TGFβ or mock for 48 h, and abundance of citrate was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
KNIH‐3T3 cells expressing sgCtrl or sgSlc25a1‐1 were treated with TGFβ or mock for 48 h, and the oxygen consumption rate (OCR) before and after treatment with mitochondrial inhibitors was measured using the Seahorse bioanalyzer. Oligo, oligomycin; Rot/Anti‐A, rotenone/antimycin; PCV, packed cell volume.
-
LNIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2 were treated with TGFβ or mock for 48 h, and abundance of the indicated metabolites was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
M, NPearson's correlation of Slc25a1 and P5CS mRNA levels in mice (M) or patients (N) from the indicated datasets, as described in Fig 3. AU, arbitrary units.
HIF‐1α is stabilized by mitochondrial ROS which are generated by excess substrate availability to the ETC (Chandel et al, 2000; Brunelle et al, 2005; Guzy et al, 2005; Bell et al, 2007; Lum et al, 2007). In agreement with higher mitochondrial substrate availability, TGFβ also induced stabilization of HIF‐1α with similar kinetics as it induced ROS (Fig 5B). In line with Smad4 regulating glutaminolysis (Figs 3L and EV4D), ROS accumulation and HIF‐1α stabilization after 6 h TGFβ treatment were dependent on Smad4 (Fig 5C and D), suggesting that the Smad4‐dependent transcriptional program activated by TGFβ induces glutaminolysis, ROS accumulation, and HIF‐1α stabilization. TGFβ‐induced HIF‐1α stabilization depended on mitochondrial ROS, as it was prevented by mitoquinol (MitoQ), a mitochondria‐targeted antioxidant (Fig EV5B). To confirm whether TGFβ‐induced ROS were mitochondrial, we used either a mitochondria‐targeted or a cytosol‐targeted roGFP2 construct coupled to glutaredoxin (Grx1), allowing us to sensitively measure mitochondrial and cytosolic glutathione oxidation (Gutscher et al, 2008). Indeed, TGFβ induced glutathione oxidation only in mitochondria, not in the cytosol after 6 h of treatment (Fig 5E). Taken together, these data suggest that TGFβ activates glutaminolysis via Smad4, supporting TCA cycle anaplerosis, which increases substrate availability to the mitochondrial ETC, resulting in increased mitochondrial respiration and the generation of mitochondrial ROS that stabilize HIF‐1α. To test whether the TGFβ‐induced transient stabilization of HIF‐1α plays a role in proline biosynthesis, we expressed sgRNAs targeting HIF‐1α, which prevented HIF‐1α stabilization after 6 h of TGFβ stimulation (Fig EV5C). While cells expressing sgHIF‐1α displayed an increase in citrate and a decrease in lactate, proline levels were unchanged (Fig EV5D), indicating that HIF‐1α is not directly involved in proline upregulation in response to TGFβ stimulation.
We next investigated how the decline of ROS levels observed after TGFβ stimulation is regulated, despite ongoing mitochondrial oxidation and substrate accumulation (Fig 4A and B). We first tested whether TGFβ promoted diversion of the first glycolytic intermediate glucose 6‐phosphate (G6P) into the pentose phosphate pathway (PPP), which is a major producer of NADPH and is thus critical for antioxidant defense (Mullarky & Cantley, 2015). To analyze flux through the PPP, we traced the fate of glucose labeled at carbons one and two ([1,2‐13C] Glc) and calculated the ratio of m+1 lactate (derived from PPP) to m+2 lactate (derived from glycolysis) (Lee et al, 1998). In line with elevated glucose uptake (Fig 1G), TGFβ‐treated cells displayed increased m+2 labeling of G6P which was diluted out in lactate (Fig EV5E and F). However, PPP activity was low and was not enhanced in response to TGFβ treatment (Fig EV5G). These data suggest that TGFβ does not stimulate flux through the PPP relative to glycolysis.
Under hypoxia, HIF‐1α‐mediated metabolic reprogramming allows cells to divert glycolytic carbons away from mitochondrial oxidation to minimize ROS generation in the absence of a terminal electron acceptor (Lum et al, 2007). Similarly, proline biosynthesis could divert glutamine carbons away from mitochondrial oxidation and, in addition, uses up mitochondrial electrons that would otherwise be donated to the ETC (Fig 3A). This raised the intriguing possibility that TGFβ‐stimulated cells were using proline as a “vent” to prevent ROS generation due to the buildup of NADH in excess of the ability of oxidative phosphorylation to assimilate the extra electrons. To test whether activation of proline biosynthesis is important to resolve mitochondrial redox stress induced by TGFβ, ROS levels were assessed in cells unable to engage in proline biosynthesis due to deletion of P5CS (Fig 3C), and grown in medium supplemented with proline to avoid effects of proline loss on protein translation and growth (Loayza‐Puch et al, 2016; Sahu et al, 2016). While cells expressing sgCtrl did not display ROS after 48 h of TGFβ stimulation, as described above (Figs 1M and 5A), cells expressing sgP5CS were unable to resolve TGFβ‐induced ROS (Fig 5F), indicating that the decline in ROS after 48 h of TGFβ treatment depends on utilization of the proline biosynthetic pathway, not on proline itself. Taken together, these data indicate that proline biosynthesis functions in TGFβ‐induced mitochondrial oxidative metabolism to reduce ROS stress.
Mitochondrial citrate export limits proline biosynthesis and collagen production
If proline acts as a vent for redox stress imposed by increased TCA cycle activity, then we predicted that cells unable to engage in proline biosynthesis would utilize other vents to prevent excessive mitochondrial carbon oxidation and ROS accumulation in response to TGFβ treatment. Consistent with this idea, we observed that P5CS‐deleted cells displayed elevated levels of aspartate, citrate, and lactate when cultured in TGFβ‐containing medium (Fig EV5H), the alternative vents used by proliferating cells. In turn, we predicted that inhibition of mitochondrial citrate export in wild‐type cells should result in elevated ROS levels and increased utilization of the proline biosynthetic pathway. Using 4‐chloro‐3‐[[(3‐nitrophenyl)amino]sulfonyl]‐benzoic acid (CTPi), a competitive inhibitor of the mitochondrial citrate transport protein (CTP) (Aluvila et al, 2010), we found that cellular ROS levels increased in a dose‐dependent fashion after 6 h of treatment to a similar extent as upon TGFβ stimulation (Fig 6A). Similarly, inhibition of lactate dehydrogenase A (LDHAi) or activation of pyruvate kinase M2 (PKM2a) increased cellular ROS levels, although not to the same extent as did CTP inhibition (Fig EV5I).
Figure 6. Mitochondrial citrate export limits proline biosynthesis and collagen production.
-
ANIH‐3T3 cells were treated with vehicle control or CTPi at increasing concentrations (0.1, 0.2, 0.5 mM) or TGFβ (2 ng/ml for all experiments) for 6 h, and ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment. Values are relative to control‐treated cells. P‐values represent comparison of individual samples to the control.
-
BLeft: NIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2 were treated with TGFβ or mock for 6 h, and ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment. Values are relative to mock‐treated sgCtrl‐expressing cells. Right: Western blot of NIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2.
-
CNIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2 were treated with TGFβ or mock for 48 h, and proline abundance was measured by GC‐MS. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
DWestern blot of NIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2 and treated with TGFβ or mock for 48 h.
-
ECollagen abundance in ECM produced by NIH‐3T3 cells expressing sgCtrl, sgSlc25a1‐1, or sgSlc25a1‐2 grown in the presence or absence of TGFβ, measured by picrosirius red staining, and normalized to the packed cell volume of cells grown on a parallel plate under identical conditions. Values are relative to mock‐treated sgCtrl‐expressing cells.
-
FNIH‐3T3 cells expressing empty vector or Slc25a1 cDNA were treated with TGFβ or mock for 6 h, and ROS levels were measured by flow cytometry for CM‐H2DCFDA after incubation with CM‐H2DCFDA for the last 30 min of the treatment. Values are relative to mock‐treated empty vector‐expressing cells.
-
GNIH‐3T3 cells expressing empty vector or Slc25a1 cDNA were treated with TGFβ or mock for 48 h, and proline abundance was measured by GC‐MS. Values are relative to mock‐treated empty vector‐expressing cells.
-
HWestern blot of NIH‐3T3 cells expressing empty vector or Slc25a1 cDNA and treated with TGFβ or mock for 48 h.
-
ICollagen abundance in ECM produced by NIH‐3T3 cells expressing empty vector or Slc25a1 cDNA grown in the presence or absence of TGFβ, measured by picrosirius red staining, and normalized to the packed cell volume of cells grown on a parallel plate under identical conditions. Values are relative to mock‐treated empty vector‐expressing cells.
-
J, KAnalysis of the indicated gene expression datasets for mRNA levels of Slc25a1, as described in Fig 3: (J) lung tissue from mice with pulmonary fibrosis induced by bleomycin (Bleo) treatment compared to saline treatment (GSE112827); (K) two datasets (GSE110147, GSE32537) from lungs of patients with idiopathic pulmonary fibrosis (IPF) compared to normal controls (Ctrl). AU, arbitrary units. The number of patients per group is indicated.
-
LPearson's correlation of SLC25A1 and P5CS mRNA levels in IPF patients from GSE32537.
-
MIPF patients from GSE32537 were assigned into P5CS low /SLC25A1 high and P5CS high /SLC25A1 low groups based on the expression level of P5CS and SLC25A1. “High” represents patients with P5CS or SLC25A1 expression values being above the 75% percentile of the respective gene expression; “low” represents patients with expression values being below the 25% percentile of gene expression. The forced vital capacity (FVC) before bronchodilator (pre‐BD) as percentage of what was predicted for each patient was compared between the groups.
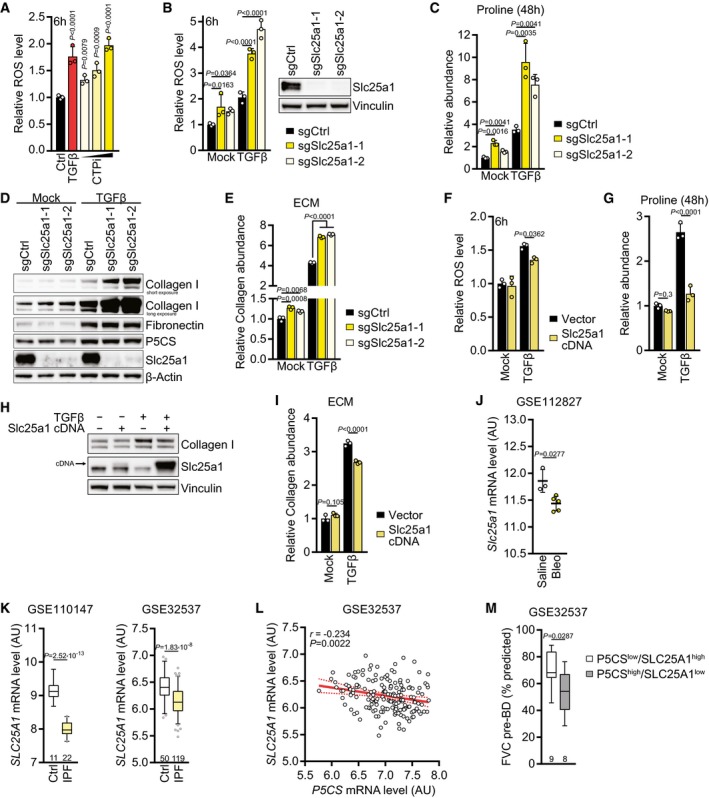
To confirm the above results with a genetic approach, we deleted Slc25a1 encoding CTP in NIH‐3T3 cells (Fig 6B). Like the CTP inhibitor, Slc25a1‐deleted cells displayed increased ROS levels which were further augmented upon 6‐h TGFβ treatment (Fig 6B). We hypothesized that blocking citrate export from mitochondria would increase mitochondria carbon load and, thus, TCA cycle activity, forcing cells to utilize other pathways to limit excessive mitochondrial oxidation. Slc25a1 deletion resulted in a reduction in the cellular citrate pool (Fig EV5J) and a higher level of mitochondrial respiration and substrate availability, in particular in response to TGFβ treatment (Fig EV5K). This indicated that citrate was oxidized in the TCA cycle, and consistent with that, levels of its downstream metabolites alpha‐ketoglutarate and glutamate were increased when Slc25a1 was deleted, both in mock‐ and TGFβ‐treated cells (Fig EV5L). However, this increase was not observed in later TCA cycle intermediates such as malate and oxaloacetate‐derived aspartate (Fig EV5L), possibly because CTP acts as a citrate–malate shuttle (Robinson et al, 1971), and thus, Slc25a1 deletion might prevent import of malate into mitochondria. Notably, Slc25a1 deletion caused an increase in the abundance of proline and synergized with TGFβ treatment to elevate cellular proline levels more than ninefold (Fig 6C). These data suggest that when citrate export from mitochondria is blocked, cells adapt to reduce carbon oxidation in the TCA cycle by further increasing the preference of glutamine‐derived carbons to enter the proline biosynthetic pathway.
Since proline biosynthesis is critical for collagen production (Fig 3D), we next asked whether fibroblasts handle proline accumulation induced by Slc25a1 deletion by diverting it into collagen, which can then be secreted into the ECM. Consistent with the high abundance of proline when Slc25a1 is deleted, these cells displayed elevated levels of collagen I but not fibronectin, an ECM protein which is not enriched in proline (Fig 6D). P5CS expression levels were similar in Slc25a1‐deleted cells (Fig 6D), indicating that the increase in proline and collagen I cannot simply be explained by differential expression of the rate‐limiting enzyme for proline biosynthesis. A higher abundance of collagen was also found in the ECM secreted by Slc25a1‐deleted cells (Fig 6E). Conversely, ectopic expression of Slc25a1 reduced cellular ROS levels, diminished proline accumulation, and led to a decline in collagen I levels in TGFβ‐treated cells and their secreted ECM (Fig 6F–I). These results indicate that mitochondrial citrate export can limit proline biosynthesis and collagen production.
Based on these findings, we asked whether expression of Slc25a1 was dysregulated in fibrotic disease. Consistent with Slc25a1 deletion being sufficient to augment collagen levels, Slc25a1 expression was downregulated in the bleomycin mouse model of pulmonary fibrosis (Fig 6J) as well as in IPF patients compared to normal controls in two independent datasets (Fig 6K). In all datasets analyzed, Slc25a1 expression inversely correlated with expression of P5CS (Figs 6L and EV5M and N). Since ectopic expression of P5CS and deletion of Slc25a1 both increased collagen levels in the ECM (Figs 3H and 6E), we predicted that IPF patients concomitantly displaying elevated P5CS expression and downregulation of SLC25A1 (P5CS high /SLC25A1 low) would present impaired lung function due to excessive collagen deposition. Consistent with this idea, lung function was significantly compromised in such patients compared to patients with a P5CS low /SLC25A1 high expression profile (Fig 6M). Taken together, these data demonstrate that fibroblasts engage in proline biosynthesis and collagen production when mitochondrial oxidative metabolism is elevated, and that this process correlates with the impairment in lung function exhibited by patients with idiopathic pulmonary fibrosis.
Discussion
In this study, we provide evidence that in fibroblasts, TGFβ is a potent stimulator of protein translation and nutrient uptake, reprogramming intermediate metabolism to promote both the production of the amino acids and the bioenergetic support for enhanced synthesis of matrix proteins. A major matrix component is collagens, triple‐helical proteins whose individual building blocks are enriched in proline residues. This suggests that intracellular proline availability could limit the translation of proline‐rich proteins such as collagen. To understand how TGFβ selectively stimulated matrix production, we examined whether TGFβ might exert its effects in part through stimulation of proline biosynthesis. First, we observed that TGFβ induced proline biosynthesis in a Smad4‐dependent manner. Second, we demonstrated that glutamine is preferentially used to support proline biosynthesis via P5CS, which requires an excess of mitochondrial NAD(P)H beyond which can be utilized for ATP synthesis. The subsequent incorporation of glutamine‐derived proline into collagen then provided an adaptive sink for excess mitochondrial carbon when NAD(P)H and ATP are high. Therefore, increasing collagen production and secretion provides fibroblasts with an effective means to deal with the high mitochondrial redox potential generated by TGFβ‐mediated stimulation of glucose and glutamine metabolism.
We provide evidence that proline biosynthesis allows fibroblasts to prevent excessive ROS accumulation in response to a TGFβ‐induced mitochondrial redox potential. Proline accumulation in response to redox stress is a well‐documented phenomenon in plants (Hayat et al, 2012). Stress‐induced increase in proline levels in plants is a result of enhanced biosynthesis from glutamate (Hare & Cress, 1997), and knockout mutants of plant P5CS display a reduction of stress‐induced proline biosynthesis, resulting in the accumulation of ROS (Székely et al, 2008). There is evidence that proline contributes to oxidative stress defense also in mammalian cells. The present data suggest that in the context of TGFβ‐stimulated fibroblasts, it is the consumption of mitochondrial NAD(P)H and ATP through proline biosynthesis from glutamine which contribute to ROS defense. In addition, other studies have demonstrated that proline itself may protect from oxidative damage by directly acting as ROS quencher (Kaul et al, 2008; Krishnan et al, 2008; Natarajan et al, 2012). Redox shuttling between proline and P5C via proline dehydrogenase (PRODH) and PYCR has also been proposed to facilitate the transfer of NAD(P)H between mitochondria and the cytosol (Phang, 1985; Liu et al, 2015).
What regulates the preferential diversion of glutamate into proline biosynthesis, rather than into the TCA cycle? Our data suggest that the accumulation of both mitochondrial ATP and reducing equivalents contributes to the regulation of this process. This is consistent with earlier reports of reduced proline levels in cell lines with impaired mitochondrial function due to mutations in fumarase or mitochondrial DNA‐encoded ETC components (Mullen et al, 2014; Chen et al, 2018). The oxidation of NADH in the ETC drives an electrochemical gradient that is used to synthesize ATP, and we found that uncoupling this gradient from ATP synthesis or direct inhibition of mitochondrial ATP synthase significantly reduces proline levels. ATP is, in addition to NADPH, required for the phosphorylation and reduction conversion of glutamate to P5C catalyzed by P5CS.
Consistent with the proposed role of TGFβ in idiopathic pulmonary fibrosis, we find that expression of P5CS is upregulated in lungs of IPF patients, while SLC25A1 is downregulated. We report that both P5CS and Slc25a1 are regulators of proline biosynthesis and collagen production, and IPF patients with a P5CS high /SLC25A1 low expression profile display significantly impaired lung function. The present results also suggest that inhibition of mitochondrial electron transport might limit proline biosynthesis and collagen production in such patients. Notably, metformin, which targets complex I (Cameron et al, 2018), was shown to attenuate hepatic and pulmonary fibrosis in rodents (Gamad et al, 2018; Rangarajan et al, 2018), raising the possibility that the therapeutic effects could—at least in part—be due to inhibition of proline biosynthesis.
In conclusion, TGFβ‐induced proline biosynthesis is another example of how cells can handle an excess of intracellular metabolites when these are not used to support cell proliferation. Fibroblasts deal with the accumulating proline induced by TGFβ by incorporating it into collagen, which is secreted into the ECM. This TGFβ‐induced conversion of available glucose and amino acids into enhanced extracellular matrix production would promote the regeneration of a damaged tissue during wound healing but, if stimulated in non‐physiological ways, could lead to an excessive fibrotic reaction.
Materials and Methods
Cell culture
NIH‐3T3 cells and IMR90 cells were obtained from ATCC and were cultured at 37°C in 5% CO2 and 20% O2. Cells were maintained in DMEM (25 mM d‐glucose, 2 mM l‐glutamine) supplemented with 10% FBS (Gemini), 100 units/ml penicillin, and 100 μg/ml streptomycin. The following supplements were added to the culture medium for maintenance of cells expressing the indicated guide RNAs: 0.15 mM l‐proline (sgP5CS‐expressing cells) and 1 mM sodium acetate (sgSlc25a1‐expressing cells). All experiments with sgP5CS‐expressing cells and all metabolite profiling experiments were done in DMEM supplemented with 10% dialyzed FBS (Gemini). For serum starvation experiments, the medium was changed to DMEM supplemented with 0.5% FBS 5–6 h after plating of cells, and treatment was started 16–20 h later. All media were prepared by the media preparation facility at MSKCC. Cells were verified as mycoplasma‐free by the MycoAlert Mycoplasma Detection Kit (Lonza).
Growth experiments
0.5 × 105 cells/well were plated in 12‐well culture plates, and treatment was started 5–6 h later. Cell numbers at the start of treatment (d0) and 1–3 days later were counted in triplicates using the Multisizer 3 Coulter Counter (Beckman) and normalized to the cell number at d0.
Chemicals
Antimycin (used at 1 μM), CoCl2 (100 μM), CTPi (0.1–0.5 mM), DASA (PKM2a, 20 μM), H2O2 (100 μM), and myxothiazol (1 μM) were purchased from Sigma; FCCP (2 μM), mitoquinol (MitoQ, 1 μM), oligomycin (1 μM), and rotenone (0.5 μM) were purchased from Cayman Chemical; GSK 2837808A (LDHAi, 10 μM) was purchased from Tocris Bioscience; and TGFβ‐1 (2 ng/ml) was purchased from PeproTech.
Ectopic gene expression and CRISPR/Cas9‐mediated gene deletion
P5CS cDNA, mitoLbNOX, and mitoTPNOX plasmids were obtained from Addgene and subcloned into a modified version of pTRE‐Tight (Clontech) with addition of a C‐ or N‐terminal HA‐tag (P5CS) or FLAG‐tag (mitoLbNOX, mitoTPNOX). Slc25a1 cDNA plasmid was obtained from DNASU. Cyto‐ and mito‐Grx1‐roGFP2 plasmids were obtained from Addgene. Guide RNAs targeting murine HIF‐1α, P5CS, Pycr1, Pycr2, Smad4, and Slc25a1 (Table 1) were designed using GuideScan (http://www.guidescan.com/) and cloned into pLentiCRISPRv2 (Addgene). Rosa26‐targeting guides were used as a control. Polyclonal cell populations were used for the experiments. Lentiviral particles were produced in 293T cells by using psPAX2 and pCMV‐VSV‐G packaging plasmids (Addgene). Retroviral particles were produced in 293T cells by using pCG‐gag‐pol and pCMV‐VSV‐G packaging plasmids (Addgene). Viral supernatant was passed through a 0.45‐μm nylon filter and used to transduce NIH‐3T3 cells in the presence of 8 μg/ml polybrene (Sigma) overnight. Cells were subjected to puromycin (2 μg/ml, Sigma), hygromycin (250 μg/ml, Millipore), or blasticidin (10 μg/ml, InvivoGen) antibiotic selection the following day. Inducible expression of HA‐P5CS cDNA, mitoLbNOX, and mitoTPNOX vectors was achieved with 100 ng/ml doxycycline (Sigma) and started when cells were plated for the experiment.
Table 1.
Sequences of sgRNAs used in this study
| Gene | Guide 1 sequence | Guide 2 sequence |
|---|---|---|
| Ctrl (Rosa26) | GAAGATGGGCGGGAGTCTTC | – |
| HIF‐1α | TCGTTAGGCCCAGTGAGAAA | CAAGATGTGAGCTCACATTG |
| P5CS (Aldh18a1) | CTTGCCGTGGGCACGACTGA | GCCGCACGCTCTGAGACAGA |
| Pycr1 | ACCCCGGTCGTGGTACGGGA | ACGGTCTCGGCCCTCCGGGT |
| Pycr2 | GCTGACCACGCGCTCACCCG | CGTGGGCAGGTCCATATCCG |
| Smad4 | AGACAGGCATCGTTACTTGT | AACTCTGTACAAAGACCGCG |
| Slc25a1 | CTTCACGTATTCGGTCGGGA | AGTGGTAGTCGTGTGCCCTA |
Western blot
Cells were lysed in 1× RIPA buffer (Millipore) supplemented with protease and phosphatase inhibitors (Thermo Fisher) on ice. Lysates were cleared by centrifugation for 10 min at 18,000 g and 4°C, and protein concentration was determined by BCA Protein Assay (Thermo Scientific). Equal amounts of protein were mixed with sample buffer and reducing agent (Thermo Scientific) and were separated on NuPAGE 4–12% Bis‐Tris gels (Thermo Scientific). Proteins were transferred to nitrocellulose membranes, blocked for 1 h with 5% milk in TBS‐T, and incubated in primary antibodies at 4°C overnight. Membranes were washed in TBS‐T and incubated in HRP‐coupled secondary antibodies at room temperature. Proteins were detected by chemiluminescence using ECL (Thermo Scientific) in a Bio‐Rad ChemiDoc Imager. The following primary antibodies were used: vinculin (Sigma, V9131), β‐actin (Sigma, A5441), α‐tubulin (Sigma, T9026), collagen I (Abcam, ab21286), collagen IV (Proteintech, 55131‐1‐AP), fibronectin (Abcam, ab2413), puromycin (EMD Millipore, MABE343), GLS1 (Abcam, ab156876), P5CS (Sigma, HPA008333), PYCR1 (Proteintech, 20962‐1‐AP), PYCR2 (Proteintech, 17146‐1‐AP), HA‐tag (Sigma, SAB4300603), FLAG‐tag (Sigma, F1804), Smad4 (Santa Cruz, sc‐7966), HIF‐1α (Cayman Chemical, 10006421), Smad2 phospho‐S465/467 (Cell Signaling, 3108S), and Slc25a1 (Proteintech, 15235‐1‐AP). The following secondary antibodies were used: anti‐rabbit HRP (GE, NA934V) and anti‐mouse HRP (Sigma, NA931).
Translation assays
For the assay involving Western blotting, cells were treated as indicated, and 90 μM puromycin was added in the last 10 min of the experiment with or without 10 μg/ml cycloheximide (CHX, Millipore) as control. Cells were lysed, and proteins were subjected to immunoblotting using an anti‐puromycin antibody as described above. For the assay involving flow cytometry, cells were treated as indicated and incubated with 20 μM O‐propargyl‐puromycin (OPP, Thermo Scientific) for the last 1 h of the experiment. Cells were harvested by trypsinization and fixed with methanol at −20°C, followed by permeabilization with 0.5% Triton X‐100 in PBS. Cells were stained using the Click‐iT Plus Alexa Fluor 647 Picolyl Azide Toolkit (Thermo Scientific) according to the manufacturer's instructions and analyzed by flow cytometry.
ECM extraction and collagen staining
Confluent NIH‐3T3 or IMR90 cells were grown for 6 days on plates coated with 0.1% gelatin (Sigma) in the presence of 50 μM ascorbate (Sigma) and treated as indicated, and the medium was replaced every other day. Plates were decellularized with 20 mM ammonium hydroxide/0.5% Triton X‐100 for 5 min on a rotating platform. Three times the volume of PBS was added and ECM was equilibrated overnight at 4°C, followed by four additional PBS washes. Sample buffer supplemented with 1 mM DTT was added, ECM was scraped off, and proteins were separated by SDS–PAGE followed by immunoblotting. To measure collagen abundance, extracted ECM was stained with the Picro Sirius Red Stain Kit (Abcam) according to the manufacturer's instructions. The stain was extracted with 0.1 M NaOH, and optical density was measured at 550 nm using a microplate reader. Values were normalized to the packed cell volume (number × volume) of cells grown on a separate plate under the same experimental conditions.
Measurement of glucose and glutamine consumption and lactate secretion
Cells were plated in 6‐well cell culture plates at a concentration aimed to reach 0.5–1 × 106 cells at the time of harvest and treated with TGFβ (2 ng/ml) for 48 h. Media were exchanged for an assay period of 12 h (+/− TGFβ) in the last 12 h of the treatment period, then collected, centrifuged, and analyzed using a 2950 Biochemistry Analyzer (YSI Life Sciences) to determine glucose, glutamine, and lactate concentration. Absolute rates of consumption/secretion of these metabolites were calculated by subtracting the concentration in medium incubated for the same amount of time without cells and then normalizing to the cell number at the time of harvest, media volume, and hours of incubation. For time course experiments, treatments were started in a staggered fashion. For the 48‐ and 24‐h treatments, TGFβ was added before the 12‐h assay period; for the 12‐h treatment, TGFβ was added when starting the assay period; for the 6‐ and 3‐h treatments, TGFβ was added during the assay period, such that the media for all conditions were harvested at the same time.
Measurement of oxygen consumption rate, extracellular acidification rate, ATP production rate, and glycolytic rate
Oxygen consumption rate (OCR), extracellular acidification rate (ECAR), ATP production rate (JATP), and glycolytic rate (glycoPER) were measured using a XFe96 Extracellular Flux Analyzer (Agilent). Cells were plated into Seahorse microplates (Agilent) at a density of 1.5 × 103 cells/well (10% FBS) or 5 × 103 cells/well (0.5% FBS), allowed to adhere overnight, and treated with TGFβ (2 ng/ml) for 48 h. In the time course experiment, TGFβ was added 2, 4, 8, 24 or 48 h before the assay. Treatment media were then removed and replaced with Seahorse media (DMEM containing 10 mM glucose, 2 mM glutamine, and 1 mM sodium pyruvate). OCR analysis was performed at basal level and after subsequent injections of oligomycin (1 μM), FCCP (2 μM), and rotenone/antimycin mix (0.5 μM) according to the manufacturer's instructions. ECAR was analyzed at basal level without any injection. Mitochondrial respiration was defined as the last measurement before oligomycin injection subtracted by the minimum rate after rotenone/antimycin injection (non‐mitochondrial respiration rate). Maximal respiration was calculated by subtracting the non‐mitochondrial respiration rate from the maximum rate measurement after FCCP injection. Glycolytic (JATPgly) and oxidative (JATPox) ATP production rates were calculated from Seahorse data according to Mookerjee et al (2017) using the bioenergetic spreadsheet downloaded from https://russelljoneslab.vai.org/tools/. For glycolytic rate analysis, treatment media were removed, cells were washed, and media were replaced with Seahorse media buffered with 5 mM HEPES. The media were replaced with fresh assay media immediately before the assay. Glycolytic rate analysis was performed at basal level and after subsequent injections of rotenone/antimycin mix (0.5 μM) and 2‐DG (5 mM) according to the manufacturer's instructions, and glycoPER was calculated from the resulting OCR and ECAR data using WAVE software (Agilent) under default settings.
ROS measurement
Intracellular ROS levels were measured by the CM‐H2DCFDA general oxidative stress indicator (Thermo Scientific). Cells were incubated with 1 μM CM‐H2DCFDA at 37°C for 30 min, harvested, and analyzed by flow cytometry. The mean fluorescence intensity of DAPI‐negative cells was measured in the FITC channel.
Measurement of cytosolic and mitochondrial glutathione oxidation
Cells expressing cyto‐ or mito‐Grx1‐roGFP2 were treated with TGFβ (2 ng/ml) for 6 h. Cells were washed and incubated with 20 mM N‐ethylmaleimide (NEM, Sigma) for 5 min to prevent further probe oxidation. Cells were harvested, fixed with 4% formaldehyde, and analyzed by flow cytometry using a 520/10‐nm filter. The ratio of emission after excitation at 405 and 488 nm was calculated as a measure of glutathione oxidation. The maximal oxidized and reduced form of the probe was determined for each experiment by incubating cells in extra wells with 5 mM H2O2 (Sigma) or 10 mM DTT (Thermo Scientific) for 5 min before adding NEM.
Stable isotope labeling and metabolite extraction
Cells were plated in 6‐well cell culture plates at a concentration aimed to reach 0.5‐1x106 cells at the time of harvest. For quantification of relative metabolite abundance, cells were treated with TGFβ (2 ng/ml) for 48 h in DMEM supplemented with 10% dialyzed FBS (Gemini). For [U‐13C] glutamine tracing experiments, cells were treated with TGFβ for 48 h in DMEM supplemented with 10% FBS. In the last 2, 8 or 24 h, medium was replaced with DMEM without l‐glutamine supplemented with 2 mM [U‐13C] l‐glutamine (Cambridge Isotope Laboratories) and 10% dialyzed FBS. For [U‐13C] glucose and [1,2‐13C] glucose tracing experiments, cells were treated with TGFβ for 48 h in DMEM without l‐serine and glycine supplemented with 0.5% dialyzed FBS. In the last 1, 8 or 24 h, medium was replaced with DMEM without d‐glucose, l‐serine, and glycine supplemented with 25 mM [U‐13C] d‐glucose (Cambridge Isotope Laboratories) or 25 mM [1,2‐13C] d‐glucose (Sigma) and 0.5% dialyzed FBS. For tracing of glutamine into serine and glycine, cells were treated with TGFβ for 48 h in DMEM without l‐serine and glycine supplemented with 0.5% dialyzed FBS. In the last 8 h, medium was replaced with DMEM without l‐glutamine, l‐serine, and glycine supplemented with 2 mM [U‐13C] or [α‐15N] l‐glutamine (Cambridge Isotope Laboratories) and 0.5% dialyzed FBS. For relative quantification of metabolites by GC‐MS, cells were washed briefly with PBS, which was then fully aspirated, and metabolism was quenched by immediately adding 1 ml of 80:20 methanol:water stored at −80°C containing 20 μM deuterated 2‐hydroxyglutarate (d5‐2HG) as an internal standard. For relative quantification of GSH and GSSG by LC‐MS/MS, metabolism was quenched without the PBS washing step by adding 1 ml of 80:20 methanol:water stored at −80°C containing 1 μg/ml deuterated SAM (d3‐SAM, (RS)‐S‐adenosyl‐L‐methionine‐d3 (S‐methyl‐d3) Tetra(p‐toluenesulfonate); CDN Isotopes) and 2 μg/ml deuterated SAH (d4‐SAH, S‐adenosylhomocysteine‐d4; Cayman Chemical). For stable isotope tracing experiments, metabolism was quenched without the PBS washing step by adding 1 ml of 80:20 methanol:water stored at −80°C. After overnight incubation at −80°C, the resulting mixtures were scraped on dry ice, transferred into a 1.5‐ml centrifuge tube, and centrifuged at 20,000 g for 20 min at 4°C. The supernatants were collected in clean tubes and dried in a vacuum evaporator (Genevac EZ‐2 Elite) for 2 h.
GC‐MS measurement for TCA cycle metabolites and amino acids
Dried metabolite extracts were dissolved in 40 mg/ml methoxyamine hydrochloride (Sigma) in pyridine (Thermo Scientific) for 90 min at 30°C and derivatized with MSTFA with 1% TMCS (Thermo Scientific) for 30 min at 37°C. Samples were analyzed using an Agilent 7890A GC connected to an Agilent 5975C Mass Selective Detector with electron impact ionization. The GC was operated in splitless mode with constant helium gas flow at 1 ml/min. 1 μl of trimethylsilyl‐derivatized metabolites was injected onto an HP‐5MS column, and the GC oven temperature ramped from 60 to 290°C over 25 min. Peak ion chromatograms for metabolites of interest were recorded and extracted at their specific m/z with MassHunter Quantitative Analysis software v10.0 (Agilent Technologies). Ions used for quantification of metabolite levels are as follows: d5‐2HG m/z 354; glycine m/z 174; serine m/z 204; aconitate m/z 375; citrate m/z 465; isocitrate m/z 245; alpha‐ketoglutarate m/z 304; succinate m/z 247; fumarate m/z 245; malate m/z 335; aspartate m/z 232; proline m/z 216; glutamate m/z 246; glutamine m/z 245; lactate m/z 219; and pyruvate m/z 174. All peaks were manually inspected and verified relative to known spectra for each metabolite. For relative quantification, integrated peak areas were normalized to the internal standard d5‐2HG and to the packed cell volume of each sample. Natural isotope abundance correction was performed with IsoCor software (Millard et al, 2012).
LC‐MS measurement for TCA cycle metabolites and amino acids
Metabolite extracts were resuspended in 100 μl of 60:40 acetonitrile:water, vortexed well, and incubated on ice for 20 min. Samples were further clarified by centrifugation at 20,000 g for 20 min at 4°C, and the supernatant was used for LC‐MS analysis. LC‐MS measurement for TCA cycle metabolites and amino acids isotopic labeling was performed with a 6545 Q‐TOF mass spectrometer with dual JetStream source (Agilent) operating in either positive or negative ion mode and coupled to hydrophilic interaction chromatography via electrospray ionization. For positive ionization mode, liquid chromatography separation was achieved on a ACQUITY UPLC BEH Amide column (150 mm × 2.1 mm, 1.7 μm particle size, Waters) using a gradient of solvent A (10 mM ammonium acetate in 10:90 acetonitrile:water with 0.2% acetic acid, pH 4) and solvent B (10 mM ammonium acetate in 90:10 acetonitrile:water with 0.2% acetic acid, pH 4). The gradient was 0 min, 95% B; 9 min, 70% B; 13 min, 30% B; 14 min, 30% B; 14.5 min, 95% B; 15 min, 95% B; and 20 min, 95% B. Other LC parameters were as follows: flow rate 400 μl/min, column temperature 40°C, and injection volume 5 μl. Other MS parameters were as follows: gas temp: 300°C; gas flow: 10 l/min; nebulizer pressure: 35 psig; sheath gas temp: 350°C; sheath gas flow: 12 l/min; VCap: 4,000 V; and fragmentor: 125 V. For negative ionization mode, liquid chromatography separation was achieved on a iHILIC‐(P) Classic column (100 mm × 2.1 mm, 5 μm particle size, HILICON) using a gradient of solvent A (10 mM ammonium bicarbonate in 10:90 acetonitrile:water with 5 μM medronic acid, pH 9.4) and solvent B (10 mM ammonium bicarbonate in 90:10 acetonitrile:water with 5 μM medronic acid, pH 9.4). The gradient was 0 min, 95% B; 15 min, 50% B; 18 min, 50% B; 19 min, 95% B; 19.10 min, 95% B; and 23 min, 95% B. Other LC parameters were as follows: flow rate 200 μl/min, column temperature 40°C, and injection volume 2 μl. Other MS parameters were as follows: gas temp: 300°C; gas flow: 10 l/min; nebulizer pressure: 40 psig; sheath gas temp: 350°C; sheath gas flow: 12 l/min; VCap: 3,000 V; and fragmentor: 125 V. Data were acquired from m/z 50 to 1,700 with active reference mass correction (m/z: 121.050873 and 922.009798 for acquisition in positive mode, and m/z: 119.0363 and 980.0163 for acquisition in negative mode) infused through a second nebulizer according to the manufacturer's instructions. Peak identification and integration were done based on exact mass and retention time match to commercial standards. Data analysis and natural isotope abundance correction were performed with MassHunter Profinder software v10.0 (Agilent Technologies). Flux from glutamine was calculated as the product of the first‐order rate constant of the kinetic labeling curve and relative metabolite pool size (Yuan et al, 2008).
GSH and GSSG measurements with LC‐MS/MS
The LC‐MS/MS method for relative quantification of GSH and GSSG involved hydrophilic interaction chromatography (HILIC) coupled to the triple‐quadrupole API 4000 mass spectrometer (Sciex). The LC separation was performed on a XBridge Amide column (50 mm × 2.1 mm, 3.5 μm particle size, Waters, Milford, MA). Solvent A was 10:90 acetonitrile:water with 10 mM ammonium acetate and 0.2% acetic acid, and solvent B was 90:10 acetonitrile:water with 10 mM ammonium acetate and 0.2% acetic acid. The gradient was as follows: 0 min, 92% B; 6 min, 55% B; 9.5 min, 30% B; and 11 min, 92% B. Other LC parameters were as follows: flow rate 400 μl/min, column temperature 40°C, and injection volume 5 μl. The mass spectrometer was operated in positive MRM mode. Other MS parameters were as follows: curtain gas: 20 psi; nebulizer gas: 65 psi; heater gas: 60 psi; probe voltage: 4,000 V; probe temperature: 500°C; and collision gas: 12 psi. Individual reactions that were monitored and collision energies (CE) were as follows: d3‐SAM m/z 402.2 → 301.1 (CE: 20 V)*, 250.2 (CE: 20 V); d4‐SAH m/z 389.1 → 253.9 (CE: 20 V)*, 140.4 (CE: 20 V); GSH m/z 308.1 → 179.0 (CE: 20 V)*, 233.0 (CE: 20V); GSSG m/z 613.4 → 484.0 (CE: 20 V)*, 355.0 (CE: 20 V), with * indicating the primary transition used to quantify each metabolite. Identities of metabolites were confirmed by presence of signal at both MRM transitions and by comparing to the retention times of pure standards. Data were analyzed using Analyst software v1.6.2 (SCIEX). Relative metabolite levels were quantified from raw peak areas corrected by d3‐SAM or d4‐SAH peak area factors and normalized to the packed cell volume.
Measurement of amino acid representation in proteins
Cellular lysates and ECM extracts were generated as described above. Acid hydrolysis was performed by incubating 200 μg cellular and ECM proteins in 1 ml 18% HCl containing 100 μM [U‐13C, U‐15N] labeled amino acid mix (Cambridge Isotope Laboratories) for 16 h at 100°C. Samples were cooled to room temperature and centrifuged at 20,000 g for 20 min. 500 μl supernatant was dried in a vacuum evaporator (Genevac EZ‐2 Elite) for 2 h. Samples were prepared for GC‐MS analysis as described above. Integrated peak areas of the unlabeled and fully labeled form of each amino acid were extracted, and their ratio was calculated. The sum of these ratios was calculated, and individual ratios were normalized to this sum to determine the percent representation of amino acids in proteins. Cysteine and tryptophan are acid‐labile and were not detected. We also did not detect methionine in acid hydrolysates.
Proteomic analysis of stable isotope‐labeled ECM
Confluent NIH‐3T3 cells were cultured for 4 days on gelatin‐coated plates in the presence of 50 μM ascorbate in DMEM without l‐glutamine supplemented with 2 mM unlabeled [12C] l‐glutamine or 2 mM [U‐13C] l‐glutamine and 10% dialyzed FBS, and medium was replaced after 2 days. ECM was extracted and subjected to SDS–PAGE as described above. Proteins were visualized by SimplyBlue SafeStain (Thermo Scientific) according to the manufacturer's instructions, and bands corresponding to collagen I were excised, enzymatically digested in situ with trypsin (Shevchenko et al, 2006), and desalted (Rappsilber et al, 2007). The purified peptides were diluted to 0.1% formic acid and analyzed by high‐resolution LC‐MS/MS in data‐dependent mode. We used a Thermo Q Exactive Plus Orbitrap mass spectrometer coupled to a Waters nanoACQUITY system (with a 100‐μm‐inner diameter × 10‐cm‐long C18 column (1.7 μm BEH130; Waters)) configured with a 180 μm × 2 cm trap column. Trapping was performed at 15 μl/min buffer A for 1 min and peptide elution with a 50% linear acetonitrile gradient over 120 min. MS data were collected in data‐dependent acquisition mode. Full scan MS1 spectra were acquired over 400–1,600 m/z at a resolution of 70,000 (m/z 400) with automatic gain control (AGC) at 1 × 106 ions. The top 10 most intense precursor ions were selected for HCD fragmentation performed at normalized collision energy (NCE) 27 with target ion accumulation value of 5 × 10(4). MS/MS spectra were collected with a resolution of 17,500. Protein/peptide identifications from the LC‐MS/MS data were performed using the Mascot search engine (Matrix Science, version 2.6.1.100; http://www.matrixscience.com) with the SwissProt mouse database (downloaded 20170705). The search parameters were as follows: (i) two missed cleavage tryptic sites were allowed; (ii) precursor ion mass tolerance = 10 ppm; (iii) fragment ion mass tolerance = 0.08 Da; and (iv) carbamidomethyl of cysteine was specified in Mascot as a fixed modification; and (v) deamidation of asparagine and glutamine, oxidation of methionine, acetyl of the N‐terminus, and S,T,Y phosphor were specified in Mascot as variable modifications. Two peptides minimum and protein FDR threshold at 1%. Peptides corresponding to collagen I α‐1 chain (CO1A1) were evaluated for their incorporation of the [13C] isotope from the samples cultured in the presence of [12C] or [U‐13C] l‐glutamine. Thermo Xcalibur version 2.2 and ICIS integration algorithm were used to detect and integrate the area of each peak in the isotopomer.
Analysis of gene expression datasets and patient data
Processed gene expression datasets GSE112827, GSE110147, and GSE32537 were downloaded from Gene Expression Omnibus (GEO). Samples were assigned to groups and compared using GEOquery and limma packages from the Bioconductor project in R studio (http://www.r-project.org). P‐values were adjusted for multiple comparison testing with the Benjamini and Hochberg false discovery rate method. Available clinical data for GSE32537 were correlated to individual gene expression profiles using Pearson's correlation analysis. Patients were grouped into low or high expressers according to the gene expression level being within the first or fourth quartile of the gene expression range.
Statistics
A Student t‐test with Welch's correction was applied to compare one variable between two groups. One‐way ANOVA was applied to compare one variable between three or more groups and corrected for multiple comparisons using the Holm–Sidak method. Two‐way ANOVA was applied to compare two independent variables between two groups and corrected for multiple comparisons using the Holm–Sidak method. Pearson's correlation was applied to analyze correlation between data from two groups. P‐values in gene expression datasets were calculated by moderate t‐statistics and corrected for multiple comparisons with the Benjamini and Hochberg false discovery rate method with FDR < 1%. Statistical analysis was done using GraphPad Prism 7 software and R (v3.5.3). Graphs show the mean + SD with individual datapoints, unless indicated otherwise. Results are representative of at least two independent experiments.
Author contributions
SS conceived the project, performed most experiments, analyzed the data, interpreted the results, and wrote the manuscript. MB and SV performed LC‐MS measurements and analysis, and MB provided support for Seahorse experiments. WQ performed YSI measurements and assisted with GSH/GSSG measurements. JZ subcloned P5CS, LbNOX, and TPNOX constructs and provided support for some of the methods used in this study. RCH performed proteomic measurements and analysis. JRC supervised LC‐MS and YSI experiments. CBT conceived the project, interpreted results, and wrote the manuscript.
Conflict of interest
CBT is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. He is also a former member of the board of directors and stockholder of Merck and Charles River Laboratories. He holds patents related to cellular metabolism.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank the members of the Thompson laboratory for critical discussions during manuscript preparation. The authors thank B. King for cloning of sgSmad4 constructs and T. Lindsten for critical reading of the manuscript. SS is supported by postdoctoral fellowships from the Human Frontier Science Program and the European Molecular Biology Organization. JZ is supported by the Leukemia and Lymphoma Society postdoctoral fellowship. JRC is supported by grants from the NIAID (R25 training grant AI140472‐01A1) and the Donald B. and Catherine C. Marron Cancer Metabolism Center. This work is supported by grants from the NCI (CBT) and by the Cancer Center Support Grant (P30 CA008748) to MSKCC.
The EMBO Journal (2020) 39: e103334
References
- Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16: 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluvila S, Sun J, Harrison DHT, Walters DE, Kaplan RS (2010) Inhibitors of the mitochondrial citrate transport protein: validation of the role of substrate binding residues and discovery of the first purely competitive inhibitor. Mol Pharmacol 77: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianifahanana M, Hernandez DM, Yin X, Kang JH, Jung MY, Wang Y, Yi ES, Roden AC, Limper AH, Leof EB (2016) Profibrotic up‐regulation of glucose transporter 1 by TGF‐β involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J 30: 3733–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic‐Canic M (2008) Growth factors and cytokines in wound healing. Wound Repair Regen 16: 585–601 [DOI] [PubMed] [Google Scholar]
- Bauer DE, Harris MH, Plas DR, Lum JJ, Hammerman PS, Rathmell JC, Riley JL, Thompson CB (2004) Cytokine stimulation of aerobic glycolysis in hematopoietic cells exceeds proliferative demand. FASEB J 18: 1303–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24: 6314–6322 [DOI] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GRS, Chandel NS (2007) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177: 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard K, Logsdon NJ, Benavides GA, Sanders Y, Zhang J, Darley‐Usmar VM, Thannickal VJ (2018) Glutaminolysis is required for transforming growth factor‐β1‐induced myofibroblast differentiation and activation. J Biol Chem 293: 1218–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu‐Remaileh M, Sabatini DM (2015) An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162: 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1: 409–414 [DOI] [PubMed] [Google Scholar]
- Cameron AR, Logie L, Patel K, Erhardt S, Bacon S, Middleton P, Harthill J, Forteath C, Coats JT, Kerr C et al (2018) Metformin selectively targets redox control of complex I energy transduction. Redox Biol 14: 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini MJ, Hosein K, Howlett CJ, Joseph M, Mura M (2018) Comprehensive gene expression profiling identifies distinct and overlapping transcriptional profiles in non‐specific interstitial pneumonia and idiopathic pulmonary fibrosis. Respir Res 19: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT (2000) Reactive oxygen species generated at mitochondrial Complex III stabilize hypoxia‐inducible factor‐1α during hypoxia: a mechanism of O2 sensing. J Biol Chem 275: 25130–25138 [DOI] [PubMed] [Google Scholar]
- Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, Valsecchi F, Primiano G, Calder EL, Carelli V et al (2018) Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab 27: 1007–1025.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas DB, Zumwalt RE, Black WC, Sobonya RE (1994) The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 150: 967–972 [DOI] [PubMed] [Google Scholar]
- Cracan V, Titov DV, Shen H, Grabarek Z, Mootha VK (2017) A genetically encoded tool for manipulation of NADP+/NADPH in living cells. Nat Chem Biol 13: 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai RP, Yu FX, Goh SR, Chng HW, Tan YL, Fu JL, Zheng L, Luo Y (2008) Histone 2B (H2B) expression is confined to a proper NAD+/NADH redox status. J Biol Chem 283: 26894–26901 [DOI] [PubMed] [Google Scholar]
- Deberardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104: 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamad N, Malik S, Suchal K, Vasisht S, Tomar A, Arava S, Arya DS, Bhatia J (2018) Metformin alleviates bleomycin‐induced pulmonary fibrosis in rats: pharmacological effects and molecular mechanisms. Biomed Pharmacother 97: 1544–1553 [DOI] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT et al (2009) C‐Myc suppression of miR‐23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutscher M, Pauleau A‐L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP (2008) Real‐time imaging of the intracellular glutathione redox potential. Nat Methods 5: 553–559 [DOI] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT (2005) Mitochondrial complex III is required for hypoxia‐induced ROS production and cellular oxygen sensing. Cell Metab 1: 401–408 [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, O'Leary EM, Witt LJ, Tian Y, Gökalp GA, Meliton AY, Dulin NO, Mutlu GM (2019) Glutamine metabolism is required for collagen protein synthesis in lung fibroblasts. Am J Respir Cell Mol Biol 0711: 1–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare PD, Cress WA (1997) Metabolic implications of stress‐induced proline accumulation in plants. Plant Growth Regul 21: 79–102 [Google Scholar]
- Hayat S, Hayat Q, Alyemeni MN, Wani AS, Pichtel J, Ahmad A (2012) Role of proline under changing environments: a review. Plant Signal Behav 7: 1456–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, Vander Heiden MG (2016) Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell 36: 540–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignotz RA, Massagué J (1986) Transforming growth factor‐beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 261: 4337–4345 [PubMed] [Google Scholar]
- de Ingeniis J, Ratnikov B, Richardson AD, Scott DA, Aza‐Blanc P, De SK, Kazanov M, Pellecchia M, Ronai Z, Osterman AL et al (2012) Functional specialization in proline biosynthesis of melanoma. PLoS ONE 7: e45190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul S, Sharma SS, Mehta IK (2008) Free radical scavenging potential of L‐proline: evidence from in vitro assays. Amino Acids 34: 315–320 [DOI] [PubMed] [Google Scholar]
- Krishnan N, Dickman MB, Becker DF (2008) Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic Biol Med 44: 671–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WNP, Boros LG, Puigjaner J, Bassilian S, Lim S, Cascante M (1998) Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2‐13C2]glucose. Am J Physiol Endocrinol Metab 274: 843–851 [DOI] [PubMed] [Google Scholar]
- Lien EC, Lyssiotis CA, Juvekar A, Hu H, Asara JM, Cantley LC, Toker A (2016) Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt‐driven breast cancer. Nat Cell Biol 18: 572–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Hancock CN, Fischer JW, Harman M, Phang JM (2015) Proline biosynthesis augments tumor cell growth and aerobic glycolysis: involvement of pyridine nucleotides. Sci Rep 5: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza‐Puch F, Rooijers K, Buil LCM, Zijlstra J, Oude Vrielink JF, Lopes R, Ugalde AP, Van Breugel P, Hofland I, Wesseling J et al (2016) Tumour‐specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530: 490–494 [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB (2007) The transcription factor HIF‐1alpha plays a critical role in the growth factor‐dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev 21: 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macomber L, Imlay JA (2009) The iron‐sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci USA 106: 8344–8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13: 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard P, Letisse F, Sokol S, Portais J‐C (2012) IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 28: 1294–1296 [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 292: 7189–7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E, Cantley LC (2015) Diverting glycolysis to combat oxidative stress In Innovative Medicine, Nakao K, Minato N, Uemoto S. (eds), Tokyo: Springer; [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM et al (2014) Oxidation of alpha‐ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 7: 1679–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan SK, Zhu W, Liang X, Zhang L, Demers AJ, Zimmerman MC, Simpson MA, Becker DF (2012) Proline dehydrogenase is essential for proline protection against hydrogen peroxide‐induced cell death. Free Radic Biol Med 53: 1181–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigdelioglu R, Hamanaka RB, Meliton AY, O'Leary E, Witt LJ, Cho T, Sun K, Bonham C, Wu D, Woods PS et al (2016) Transforming growth factor (TGF)‐β promotes de novo serine synthesis for collagen production. J Biol Chem 291: 27239–27251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang JM (1985) The regulatory functions of proline and pyrroline‐5‐carboxylic acid. Curr Top Cell Regul 25: 91–132 [DOI] [PubMed] [Google Scholar]
- Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, Locy ML, Ravi S, Deshane J, Mannon RB et al (2018) Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 24: 1121–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranquet C, Ollagnier‐de‐Choudens S, Loiseau L, Barras F, Fontecave M (2007) Cobalt stress in Escherichia coli: the effect on the iron‐sulfur proteins. J Biol Chem 282: 30442–30451 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906 [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Heiden MGV, Harris MH, Frauwirth KA, Thompson CB (2000) In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell 6: 683–692 [DOI] [PubMed] [Google Scholar]
- Robinson BH, Williams GR, Halperin ML, Leznoff CC (1971) Factors affecting the kinetics and equilibrium of exchange reactions of the citrate‐transporting system of rat liver mitochondria. J Biol Chem 246: 5280–5286 [PubMed] [Google Scholar]
- Sahu N, Dela Cruz D, Gao M, Sandoval W, Haverty PM, Liu J, Stephan JP, Haley B, Classon M, Hatzivassiliou G et al (2016) Proline starvation induces unresolved ER stress and hinders mTORC1‐dependent tumorigenesis. Cell Metab 24: 753–761 [DOI] [PubMed] [Google Scholar]
- Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–R462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarajah B, Azuelos I, Platé M, Guillotin D, Forty EJ, Contento G, Woodcock H V, Redding M, Taylor A, Brunori G et al (2019) mTORC1 amplifies the ATF4‐dependent de novo serine‐glycine pathway to supply glycine during TGF‐β 1 –induced collagen biosynthesis. Sci Signal 12: eaav3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havli J, Olsen JV, Mann M (2006) In‐gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1: 2856–2860 [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massagué J (2003) Cytostatic and apoptotic actions of TGF‐β in homeostasis and cancer. Nat Rev Cancer 3: 807–820 [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG (2015) Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162: 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Székely G, Abrahám E, Cséplo A, Rigó G, Zsigmond L, Csiszár J, Ayaydin F, Strizhov N, Jásik J, Schmelzer E et al (2008) Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J 53: 11–28 [DOI] [PubMed] [Google Scholar]
- Thompson CB (2011) Rethinking the regulation of cellular metabolism. Cold Spring Harb Symp Quant Biol 76: 23–29 [DOI] [PubMed] [Google Scholar]
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, Mootha VK (2016) Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352: 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB (2010) Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DH, Lund P, Krebs HA (1967) The redox state of free nicotinamide‐adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103: 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X‐Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105: 18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, Rosen R, Neidermyer AJ, McKean DF, Groshong SD et al (2013) Expression of cilium‐associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 68: 1114–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Bennett BD, Rabinowitz JD (2008) Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc 3: 1328–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File