

Introduction
Type 1 Brugada syndrome (BrS) is characterized by an electrocardiogram (ECG) pattern of ST-segment elevation with an upward convexity “coved-type” morphology seen in the right precordial leads. This condition is associated with a high risk of fatal ventricular arrhythmia and sudden cardiac death.1 BrS can develop secondary to drugs, electrolyte abnormalities, hyperthermia, and other conditions, such as mechanical compression of the right ventricular outflow tract (RVOT) or acute ischemia in the RVOT. However, the precise mechanisms underlying the acquired forms of BrS remain controversial.
We describe the case of a 66-year-old patient who presented with a spontaneous type 1 Brugada phenotype induced by cardiac iron overload after transfusion of a large volume of packed red blood cells (RBCs) in a short period of time. The patient’s ventricular tachycardia (VT) did not resolve with the administration of isoprenaline and actually worsened. The potential reason for such arrhythmia might be uncontrolled cardiac ion channel dysfunction in the context of iron overload. This case can increase awareness about the connection between cardiac ion channel dysfunction in patients with iron overload and BrS, leading to improved management and outcomes of patients with iron-overload cardiomyopathy and heart failure.
Case report
A 66-year-old man presented to our hospital with aggravation of dyspnea and intermittent palpitations after physical activity. The patient had a 2-year history of multiple myeloma, which had been treated with chemotherapy, including a total doxorubicin dose of 120 mg/m2. He also received approximately 7600 mL of packed RBCs to relieve his severe myelosuppression symptoms over the 6 months before his admission to our hospital. As a result of long-term intravenous chemotherapy and intermittent blood transfusion, the patient had been hospitalized 41 times from June 2016 to June 2018.
Upon the patient’s presentation to our hospital, blood test results showed an abnormal elevation in B-type natriuretic peptide levels, with a value of 1186 pg/mL (0–100 pg/mL). Compared with the patient’s ultrasound cardiography (UCG) 4 months prior, his recent UCG showed marked enlargement of the heart, with a left ventricular end-diastolic diameter of 61 mm, a right ventricular internal diameter of 43 mm, and reduced ejection fraction of 41% (Figure 1). The patient had extremely elevated levels of serum ferritin, with a value of 2497.80 ng/mL. Magnetic resonance imaging showed a significant decrease in signal intensity on T2-weighted images in the liver parenchyma (Figure 2). The ECG obtained on initial presentation revealed a J-point elevation of >2 mm, followed by T-wave inversion in the right precordial leads (V1 to V3) (Figure 3A). On the third day after admission, the patient started feeling severe dyspnea associated with intermittent palpitations. Urgent ECGs were performed and evaluated (Figure 3B), which showed frequent regular, wide QRS complex tachycardia with a rate of 100 beats per minute.
Figure 1.

Ultrasound cardiography (UCG) testing upon this presentation and about 4 months before this hospitalization. A: Apical 4-chamber view of UCG testing upon this presentation. B: Optimal parasternal long-axis view of UCG testing upon this presentation. C: Apical 4-chamber view of UCG testing about 4 months before this hospitalization. D: Optimal parasternal long-axis view of UCG testing about 4 months before this hospitalization.
Figure 2.
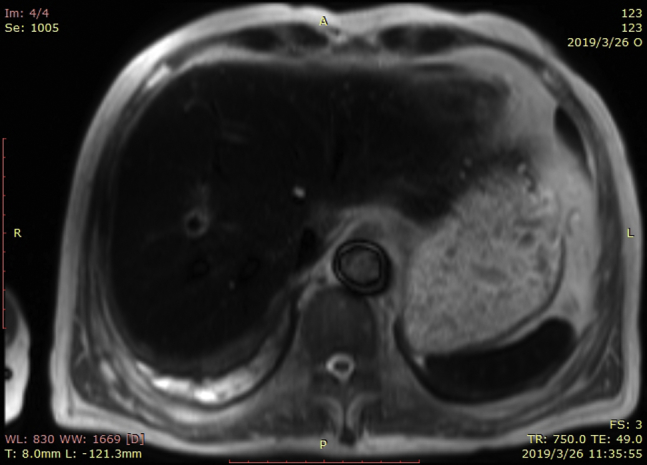
Abdominal magnetic resonance imaging.
Figure 3.

Electrocardiograph (ECG) recordings. A: ECG obtained on initial presentation. B: ECG obtained on the third day after admission. C: Three ECG segments recorded in the second, third, and fourth intercostal space, respectively, about 2 weeks later.
At first, lidocaine was administered at the rate of 2 mg/min, but the arrhythmias did not improve. Isoprenaline was then administered at the rate of 0.05 μg/kg/min, in an attempt to normalize J waves. Several minutes after the initiation of isoprenaline, however, the duration of nonsustained VT was prolonged, and the arrhythmia eventually converted to sustained VT. After we discontinued the infusion of isoprenaline, the patient’s vital signs remained stable.
After 1-week management of heart failure and iron chelation therapy, his nonsustained VT and frequent premature ventricular contractions were resolved. About 2 weeks later, the type 1 Brugada pattern also resolved (Figure 3C). However, the patient could not discontinue RBC transfusion and the level of serum ferritin could not be decreased effectively. Unfortunately, his heart failure was severe and progressed. Two months after his discharge from our hospital, he died of congestive heart failure.
Discussion
In our case, the patient presented a type 1 Brugada ECG pattern, which was characterized by a pronounced elevation of the J point followed by T-wave inversion in the right precordial leads (Figure 3A). He had never presented with similar ECG abnormalities during any of his previous 41 hospitalizations. Thus, this was a spontaneous type 1 Brugada phenotype. He also experienced frequent episodes of VT (Figure 3B) originating from the RVOT, which was thought to be the location of the arrhythmogenic substrate in patients with BrS. However, we did not think his VT was closely associated with BrS because his VT was slow and not polymorphic, which was much more likely to be an idioventricular rhythm owing to conduction and structural abnormality causing right ventricular delay.
The patient experienced progressive dyspnea with edema in both lower extremities. B-type natriuretic peptide levels were abnormally high and UCG showed that the whole heart was significantly enlarged, which confirmed that the patient had developed cardiomyopathy and congestive heart failure. Because this patient had received a large volume of RBCs to relieve his severe myelosuppression symptoms, he had extremely elevated levels of serum ferritin, with a value of 2497.80 ng/mL. Serum ferritin can reliably predict cardiac iron overload when levels are >2485 ng/mL.2 The manifestation of his abdominal magnetic resonance imaging, a significant decrease in signal intensity on T2-weighted images in the liver parenchyma, is consistent with iron overload. Cardiac iron deposition can result in cardiomyopathy, arrhythmia, and heart failure.3 We suspected that cardiac iron overload was the underlying cause of the patient’s ECG abnormalities and clinical manifestations. In addition, the management of heart failure and iron chelation therapy led to resolution of VT and Brugada pattern, which also supported our suspicions.
Some reliable evidence from previous studies was found. Reportedly, endocardial biopsies conducted in 4 patients with secondary hemochromatosis demonstrated that the subendocardial iron concentration was approximately half of that found in the epicardial layer.4 The heterogeneous patterns of myocardial iron distribution may be implicated in the arrhythmogenesis of human iron overload cardiomyopathy.5
In the presence of iron overload, transferrin becomes saturated, leading to an increase in non-transferrin-bound iron (NTBI), a free form of iron with a high toxic potential, in the serum.6 Excess NTBI infiltrates the cardiomyocytes and influences the functions of many cardiac ion channels, which can accentuate J-point and ST-segment elevations.1 Kuryshev and colleagues found that transient outward K+ currents [Ito] were increased and Na+ currents [INa] were decreased in iron-overload gerbils.5 Several studies have shown that L-type calcium currents [ICa,L] were also affected by iron overload.6,7 ICa,L is a major pathway for NTBI uptake into cardiomyocytes, which competes with cardiac calcium channels (Ca2+). Excessive cardiac iron accumulation generates highly toxic reactive oxygen species, then intracellular Ca2+ is accumulated and ICa,L is decreased by mediating oxidative stress.7 Thus, the combination of increased Ito and decreased INa and ICa,L might contribute to the development of the type 1 Brugada phenotype.
This patient received low-dose isoprenaline, which did not suppress, but rather exacerbated, his VT. Isoprenaline was previously proven effective in suppressing acute VT episodes associated with BrS as a result of its ability to increase ICaL.8 However, in this case we thought his VT was not associated with BrS. It might be the reason why isoprenaline was not effective. Furthermore, isoprenaline likely facilitates Fe2+ entry into the cardiomyocytes through ICa,L, which worsens the condition of cardiac iron overload.
Although some different secondary forms of BrS have been described, to our knowledge this is the first case report to show that cardiac iron overload can induce type 1 Brugada phenotype.
Conclusions
We presented a case of a spontaneous type 1 Brugada phenotype associated with iron overload and heart failure. This case illustrates the need for increased clinical recognition of cardiac iron overload as an underlying pathophysiologic mechanism of heart failure and arrhythmia, and a phenocopy of BrS.
Key Teaching Points.
-
•
Type 1 Brugada phenotype could be induced by cardiac iron overload as a result of cardiac ion channel dysfunction.
-
•
Cardiac iron overload might be life-threatening. Iron chelation therapy is critical to the management of heart failure and the resolution of Brugada electrocardiography pattern.
-
•
In the presence of cardiac iron overload and heart failure, isoprenaline should be recommended cautiously to suppress the acute ventricular tachycardia episodes.
Footnotes
Funding Support: This study is supported by the National Natural Science Foundation of China (Grant No. 81370293 to Dr Liu).
References
- 1.Mizusawa Y., Wilde A.A. Brugada syndrome. Circ Arrhythm Electrophysiol. 2012;5:606–616. doi: 10.1161/CIRCEP.111.964577. [DOI] [PubMed] [Google Scholar]
- 2.Krittayaphong R., Viprakasit V., Saiviroonporn P., Wangworatrakul W., Wood J.C. Serum ferritin in the diagnosis of cardiac and liver iron overload in thalassaemia patients real-world practice: a multicentre study. Br J Haematol. 2018;182:301–305. doi: 10.1111/bjh.14776. [DOI] [PubMed] [Google Scholar]
- 3.Siddique A., Kowdley K.V. Review article: the iron overload syndromes. Aliment Pharmacol Ther. 2012;35:876–893. doi: 10.1111/j.1365-2036.2012.05051.x. [DOI] [PubMed] [Google Scholar]
- 4.Fitchett D.H., Coltart D.J., Littler W.A. Cardiac involvement in secondary haemochromatosis: a catheter biopsy study and analysis of myocardium. Cardiovasc Res. 1980;14:719–724. doi: 10.1093/cvr/14.12.719. [DOI] [PubMed] [Google Scholar]
- 5.Kuryshev Y.A., Brittenham G.M., Fujioka H. Decreased sodium and increased transient outward potassium currents in iron-loaded cardiac myocytes. Implications for the arrhythmogenesis of human siderotic heart disease. Circulation. 1999;100:675–683. doi: 10.1161/01.cir.100.6.675. [DOI] [PubMed] [Google Scholar]
- 6.Oudit G.Y., Trivieri M.G., Khaper N., Liu P.P., Backx P.H. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J Mol Med (Berl) 2006;84:349–364. doi: 10.1007/s00109-005-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khamseekaew J., Kumfu S., Chattipakorn S.C., Chattipakorn N. Effects of iron overload on cardiac calcium regulation: translational insights into mechanisms and management of a global epidemic. Can J Cardiol. 2016;32:1009–1016. doi: 10.1016/j.cjca.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Ohgo T., Okamura H., Noda T. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm. 2007;4:695–700. doi: 10.1016/j.hrthm.2007.02.014. [DOI] [PubMed] [Google Scholar]