

ABSTRACT
Myosin heavy chain-embryonic (MyHC-emb) is a skeletal muscle-specific contractile protein expressed during muscle development. Mutations in MYH3, the gene encoding MyHC-emb, lead to Freeman–Sheldon and Sheldon–Hall congenital contracture syndromes. Here, we characterize the role of MyHC-emb during mammalian development using targeted mouse alleles. Germline loss of MyHC-emb leads to neonatal and postnatal alterations in muscle fiber size, fiber number, fiber type and misregulation of genes involved in muscle differentiation. Deletion of Myh3 during embryonic myogenesis leads to the depletion of the myogenic progenitor cell pool and an increase in the myoblast pool, whereas fetal myogenesis-specific deletion of Myh3 causes the depletion of both myogenic progenitor and myoblast pools. We reveal that the non-cell-autonomous effect of MyHC-emb on myogenic progenitors and myoblasts is mediated by the fibroblast growth factor (FGF) signaling pathway, and exogenous FGF rescues the myogenic differentiation defects upon loss of MyHC-emb function in vitro. Adult Myh3 null mice exhibit scoliosis, a characteristic phenotype exhibited by individuals with Freeman–Sheldon and Sheldon–Hall congenital contracture syndrome. Thus, we have identified MyHC-emb as a crucial myogenic regulator during development, performing dual cell-autonomous and non-cell-autonomous functions.
This article has an associated ‘The people behind the papers’ interview.
KEY WORDS: Skeletal muscle, Myosin heavy chain-embryonic, Myogenesis, Development, Mice, Muscle progenitors, FGF, Signaling
Highlighted Article: Myh3 mouse alleles reveal that MyHC-embryonic is crucial for skeletal muscle development and adult Myh3 null mice exhibit scoliosis, a phenotype seen in congenital contracture syndromes.
INTRODUCTION
The vertebrate skeletal muscle develops through a complex series of steps involving cell fate determination, cell migration and differentiation. The early myogenic progenitors originate in the somites, expressing Pax3 followed by Pax7 paired box transcription factors, and migrate to their target tissues such as limbs, the diaphragm or the tongue. Commitment to the myogenic lineage occurs as a result of four basic helix-loop-helix transcription factors called myogenic regulatory factors (MRFs) – Myf5, MyoD, MRF4 and myogenin. Once committed, the progenitors become myoblasts that fuse with each other and with progenitors to generate myofibers, which are tubular multinucleate fibers that contain the contractile machinery essential for muscle contraction. Myofibers are composed of sarcomeres, the functional contractile unit of the skeletal muscle, made up of alternating arrays of thick and thin filaments. MRFs are crucial for the transcriptional regulation of the structural and contractile proteins that form the mature muscle fibers.
During mammalian development, myofibers are generated in two distinct stages, namely the embryonic and fetal phases of myogenesis. During the embryonic phase [embryonic day (E) 9.5-13.5 in mice], a small proportion of the embryonic myogenic progenitors fuse with each other to give rise to primary myofibers, whereas in the fetal phase (E14.5-17.5), fetal myogenic progenitors fuse with each other and with primary myofibers to give rise to secondary myofibers (Biressi et al., 2007a,b; Zammit et al., 2008). Although embryonic and fetal myogenic progenitors differ from each other with respect to their morphology, proliferation rates and response to growth factors, the myofibers derived from them, namely primary and secondary myofibers, respectively, also exhibit distinct characteristics (Biressi et al., 2007b; Stockdale, 1992; Zammit et al., 2008).
The sarcomeric thick filament is primarily composed of myosin, a heterohexamer comprising a pair each of myosin heavy chains (MyHCs), regulatory light chains and essential light chains. MyHCs have an N-terminal globular head region with actin-binding and ATPase activity, a neck region where the light chains bind, and a C-terminal coiled-coil tail region (Schiaffino and Reggiani, 1996). The contractile velocity of muscles is directly correlated with their myosin ATPase activity, varying from muscle to muscle and classified broadly as fast or slow (Barany, 1967). Of the seven mammalian skeletal muscle MyHCs, five are expressed during adult life: MyHC-IIa (encoded by Myh2), MyHC-IIx (encoded by Myh1) and MyHC-IIb (encoded by Myh4) are adult ‘fast’ isoforms; MyHC-slow (encoded by Myh7) is the ‘slow’ isoform; and MyHC-extraocular (encoded by Myh13) is a unique ‘fast’ isoform expressed in the extraocular and laryngeal muscles (Narusawa et al., 1987; Parker-Thornburg et al., 1992; Weydert et al., 1983; Wieczorek et al., 1985). Mice lacking MyHC-IIb or -IIx are significantly smaller, weigh less than the controls, and exhibit reduced grip strength, muscle weakness and increased interstitial fibrosis (Acakpo-Satchivi et al., 1997). In addition, two developmental isoforms, MyHC-embryonic (encoded by Myh3) and MyHC-perinatal (encoded by Myh8), are expressed during embryonic, fetal and neonatal development (Condon et al., 1990; Periasamy et al., 1984, 1985; Schiaffino et al., 2015; Whalen et al., 1981). Although MyHC-slow is expressed in the adult muscle, it is also expressed during developmental stages in the myofibers. MyHC-embryonic and -perinatal (MyHC-emb and -peri) are not normally expressed in adult muscle, except during skeletal muscle regeneration following injury or disease when they are transiently re-expressed (Sartore et al., 1982; Schiaffino et al., 1986).
Although several transcriptional regulators and their functions in the distinct phases of myogenesis have been well studied, little is known about the roles of downstream effectors such as MyHCs in myogenic differentiation. This is especially interesting with respect to developmental MyHCs because their expression correlates with specific developmental time points and myogenic phases, and mutations in these MyHCs lead to congenital syndromes. Mutations in the MyHC-emb encoding MYH3 gene lead to Freeman–Sheldon (FSS), Sheldon–Hall (SHS) and Multiple-Pterygium congenital contracture syndromes, indicating that developmental MyHCs have vital, yet unidentified, roles during development (Toydemir et al., 2006b,c).
In this study, we characterize the role of MyHC-emb in myogenic differentiation during embryonic, fetal, neonatal and postnatal stages of development using targeted mouse alleles. We find that MyHC-emb performs novel cell-autonomous and non-cell-autonomous functions during myogenesis. At the cell-autonomous level, MyHC-emb regulates muscle fiber size, fiber number and fiber type, whereas at the non-cell-autonomous level it regulates myogenic progenitor and myoblast differentiation. MyHC-emb expressed in myofibers, mediates non-cell-autonomous effects on myogenic progenitors and myoblasts via secreted fibroblast growth factor (FGF) signals. Supplementation with FGF in the culture media rescues the myogenic differentiation defects caused by loss of MyHC-emb function in vitro. Adult Myh3 null mice exhibit scoliosis, one of the phenotypes exhibited by individuals with FSS and SHS. Our results demonstrate that MyHC-emb is a crucial regulator of mammalian myogenesis.
RESULTS
Loss of MyHC-embryonic leads to alterations in myofiber number, area and fiber type
MyHC-emb is expressed during the embryonic and fetal stages of muscle development, and mutations in the MYH3 gene coding for MyHC-emb protein cause congenital contracture syndromes (Chong et al., 2015; Tajsharghi et al., 2008; Toydemir et al., 2006c). Because few studies have been undertaken to investigate the molecular functions of MyHC-emb, we generated Myh3 targeted mouse alleles. We generated a conditional Myh3fl3-7 allele, where exons 3-7 of Myh3 were flanked by LoxP sites (Fig. 1A), and a Myh3Δ allele, where exons 3-7 were deleted (Fig. 1B). Both Myh3fl3-7 and Myh3Δ alleles were verified by PCR (Fig. S1A-C). In crosses between Myh3Δ/+ heterozygous mice, Myh3Δ/Δ homozygous pups were not obtained at the expected 25% frequency but at about 14% (17 out of 126), suggesting that approximately half of Myh3Δ/Δ genotype animals die in utero during embryonic or fetal stages, and that MyHC-emb might have crucial functions during developmental stages. We find that MyHC-emb is expressed in the wild-type fetal heart and is absent in the Myh3Δ/Δ heart at E16.5, where it might have essential functions (Fig. S4D) (Rutland et al., 2011). We validated that Myh3Δ/Δ animals are null for MyHC-emb using immunofluorescence and qPCR (Fig. S2A-C).
Fig. 1.

Loss of MyHC-embryonic leads to neonatal myogenic differentiation defects. (A,B) Schematics depicting the Myh3fl3-7 allele, where LoxP sites (red arrowheads) flank exons 3-7 (A) and the Myh3Δ allele lacking exons 3-7 of Myh3 (B). (C-D″) Cross-sections through the hind limbs of P0 Myh3+/+ (C,C′) and Myh3Δ/Δ (D,D′) mice labeled by immunofluorescence for MyHC-slow (red), laminin (green) and DAPI (blue). ‘E’ and ‘S’ denote EDL and soleus muscles, respectively, and ‘Tib’ and ‘Fib’ denote tibia and fibula bones, respectively, in C′ and D′; C″ and D″ are magnifications of boxed areas of the flexor digitorum longus (FDL) muscle from C′ and D′, respectively. (E,F) Laminin (green) labeling on sections through the soleus muscles of P0 Myh3+/+ (E) and Myh3Δ/Δ (F) mice. (G,H) Quantification of myofiber number through the EDL (G) and soleus (H) muscles of P0 Myh3+/+ and Myh3Δ/Δ mice. (I,J) Quantification of myofiber area through the EDL (I) and soleus (J) muscles of P0 Myh3+/+ and Myh3Δ/Δ mice, grouped into 0-50 µm2, 50-150 µm2 and above 150 µm2. (K) Quantification of MyHC-slow+ fibers from P0 Myh3+/+ and Myh3Δ/Δ shank whole cross-section normalized to total area. (L-N) Western blots for MyHC-slow, MyHC-IIa and β-actin from P0 Myh3+/+ and Myh3Δ/Δ mice hind limb protein lysates (L) and densitometric quantification (M,N). Data are mean±s.e.m. of a minimum of three independent experiments. Scale bars: 200 μm (D′); 40 μm (D″); 20 μm (F).
Using Myh3Δ/Δ animals that survive, we investigated whether loss of MyHC-emb causes myofiber differentiation defects. We started by quantifying the number of myofibers per unit area in two representative muscles, the extensor digitorum longus (EDL), which is rich in fast fibers, and the soleus, which is rich in slow fibers, at postnatal day 0 (P0). There was a significant increase in the total fiber number in both the EDL and soleus upon loss of MyHC-emb (Fig. 1G,H). We also observed that myofiber size was reduced in the soleus in Myh3Δ/Δ animals, whereas it was not affected in the EDL (Fig. 1E,F; Fig. S2D,E). Upon quantification of the myofiber area of representative slow (soleus) and fast (EDL) muscles by grouping myofibers in 0-50 µm2, 50-150 µm2 and above 150 µm2 categories, we observed a significant increase in myofibers with the smallest area (0-50 µm2) in the soleus upon loss of MyHC-emb, whereas there was no effect on the EDL (Fig. 1I,J). Thus, MyHC-emb regulates two crucial muscle fiber properties in neonatal mice: myofiber number in fast and slow muscles, and myofiber cross-sectional area in slow muscles such as the soleus.
We next tested whether the loss of MyHC-emb affects muscle fiber type, by labeling Myh3+/+ and Myh3Δ/Δ P0 mouse hind limb cross-sections with MyHC-slow and laminin (basal lamina marker) (Fig. 1C-D″). We found a striking, almost twofold increase in the number of MyHC-slow+ fibers over the whole shank cross-section per unit area in Myh3Δ/Δ animals (Fig. 1C-D″,K). We validated the increase in MyHC-slow protein levels using western blot and found a dramatic sixfold increase in MyHC-slow protein levels in Myh3Δ/Δ animals (Fig. 1L,M). In addition, a fivefold increase in MyHC-IIa protein levels also occurred upon the loss of MyHC-emb, signifying that loss of MyHC-emb leads to changes in levels of other MyHC isoforms and fiber types (Fig. 1L,N). Thus, our findings indicate that MyHC-emb function is crucial for regulating myofiber number, size and type at neonatal stages.
At birth, Myh3Δ/Δ pups weighed significantly less than Myh3+/+ siblings (Fig. 2A). To test whether the myofiber defects observed at P0 persisted into postnatal stages, we documented the whole-muscle weights of the tibialis anterior (TA), gastrocnemius and quadriceps muscles of Myh3+/+ and Myh3Δ/Δ mice at P15 and P30 postnatal time points, and found that all three muscles weighed significantly less in Myh3Δ/Δ mice at both time points (Fig. 2B,C). Upon quantification of the myofiber area of P15 (Fig. 2H) and P30 (Fig. 2L) TA muscles, we found a significant increase in the proportion of myofibers with the smallest area (100-500 µm2) and a significant decrease in the proportion of myofibers with a larger area (1000-1500 µm2, 1500-2000 µm2 at P15 and 1500-2000 µm2, 2000-2500 µm2 at P30) in Myh3Δ/Δ mice. In the fast fiber-rich EDL muscle, we found a significant increase in the proportion of myofibers with the smallest area (100-500 µm2) and a significant decrease in the proportion of myofibers with a larger area (1000-1500 µm2) at P15 (Fig. 2I), whereas no significant differences were observed at P30 (Fig. 2M) in Myh3Δ/Δ mice. In the slow fiber-rich soleus muscle, we found a significant increase in the proportion of myofibers with the smallest area (100-500 µm2) and a significant decrease in the proportion of myofibers with a comparatively larger area (500-1000 µm2) at P15 (Fig. 2D,E,J) in Myh3Δ/Δ mice. The significant increase in the proportion of myofibers in the soleus with the smallest area (100-500 µm2) was maintained, although the decrease in the proportion of myofibers with a larger area was not significant at P30 (Fig. 2F,G,N) in Myh3Δ/Δ mice. Interestingly, the number of MyHC-slow+ fibers per unit area was significantly increased in the TA and soleus but not the EDL at P15 (Fig. 2D,E,K), whereas this increase was not observed at P30 in Myh3Δ/Δ mice (Fig. 2F,G,O).
Fig. 2.

Loss of MyHC-embryonic leads to postnatal myogenic differentiation defects. (A) Graph showing body weight of P0 Myh3+/+ and Myh3Δ/Δ pups. (B,C) Graphs showing TA, gastrocnemius and quadriceps muscle weights of Myh3+/+ and Myh3Δ/Δ animals at P15 (B) and P30 (C). (D-G) Representative images from cross-sections through the soleus of P15 and P30 Myh3+/+ mice (D,F), and Myh3Δ/Δ (E,G) mice labeled by immunofluorescence for MyHC-slow (red), laminin (green) and DAPI (blue). (H-J) Quantification of myofiber area through the TA (H), EDL (I) and soleus (J) muscles of P15 Myh3+/+ and Myh3Δ/Δ mice, grouped into 100-500 µm2, 500-1000 µm2, 1000-1500 µm2, above 1500 µm2 (for EDL and soleus), 1500-2000 µm2 (for TA), 2000-2500 µm2 (for TA) and above 2500 µm2 (for TA). (K) Quantification of MyHC-slow+ fibers in cross-sections of the TA, EDL and soleus muscles of P15 Myh3+/+ and Myh3Δ/Δ mice normalized to total area. (L-N) Quantification of myofiber area through the TA (L), EDL (M) and soleus (N) muscles of P30 Myh3+/+ and Myh3Δ/Δ mice, grouped into 100-500 µm2, 500-1000 µm2, 1000-1500 µm2, above 1500 µm2 (for EDL and soleus), 1500-2000 µm2 (for TA), 2000-2500 µm2 (for TA) and above 2500 µm2 (for TA). (O) Quantification of MyHC-slow+ fibers in cross-sections of the TA, EDL and soleus muscles of P30 Myh3+/+ and Myh3Δ/Δ mice normalized to total area. Data are mean±s.e.m. of a minimum of three independent experiments. Scale bars: 25 μm (E); 50 μm (G).
In summary, the observed reduced whole-body weight at P0 persists through the postnatal stages where individual muscles weigh significantly less in Myh3Δ/Δ mice. However, although the increase in the number of smaller myofibers and decrease in larger myofibers is apparent in all three muscles at P15 in Myh3Δ/Δ mice, the differences are seen in only two muscles at P30. Similarly, although the number of MyHC-slow+ fibers increases significantly in two muscles at P15 in Myh3Δ/Δ mice, it is not observed in any muscle tested at P30. Overall, these results indicate that several myofiber defects seen at neonatal stages in Myh3Δ/Δ mice persist into the postnatal stages, which are at least partially compensated for by P30.
Distinct muscles respond differently to loss of MyHC-embryonic
As loss of MyHC-emb led to alterations in crucial myofiber characteristics, we performed a whole-transcriptome RNA-Seq experiment to compare the gene expression profiles of P0 Myh3+/+ and Myh3Δ/Δ animals, in order to identify specific affected genes and pathways. For this, we used four muscles, the TA, quadriceps, gastrocnemius and diaphragm muscles, which express varying amounts of different MyHC isoforms (Fig. 3A-D). We found 218 genes that were significantly misregulated in Myh3 null muscles (Table S3). Only two genes, Nfil3 and Btg2, were misregulated across all four muscles, whereas 18 genes were misregulated across any three muscles, and 42 genes were misregulated across two muscles (Fig. 3H; Table S3).
Fig. 3.

Loss of MyHC-embryonic leads to global misregulation of genes involved in myogenic differentiation. (A-D) Volcano plots depicting results from the RNA-Seq experiment comparing P0 Myh3+/+ and Myh3Δ/Δ samples for quadriceps (A), TA (B), gastrocnemius (C) and diaphragm (D) muscles. The adjusted P-values are on a log10 scale, and significantly up- or down regulated candidates are marked as dark spots on the volcano plot. (E,F) Selected candidate genes from the RNA-Seq were validated by qPCR for quadriceps (E) and diaphragm (F). (G) Candidates from the RNA-Seq were tested on Myh3 or control siRNA-treated C2C12 cells at day 5. (H) Venn diagram depicting the number of candidate genes obtained and the degree of overlap in the RNA-Seq results comparing P0 Myh3+/+ and Myh3Δ/Δ muscles. (I,J) Quantification of Myh isoform transcript levels by qPCR on P0 quadriceps (I) and diaphragm (J) muscles of Myh3+/+ and Myh3Δ/Δ mice. The graphical data represent the mean±s.e.m. of a minimum of three independent experiments.
We found that the genes misregulated in two or more muscles were mostly involved in myogenic differentiation. For example, Btg2, a transcriptional coregulator misregulated in all four muscles, is involved in muscle development and differentiation (Feng et al., 2007), and Adamts15, a metalloproteinase misregulated in three muscles, is essential for myoblast fusion (Stupka et al., 2013). Transcripts for genes that encode proteins essential for muscle contractility, such as Tpm3, Myl3 and Myl2, were misregulated in multiple muscles (Table S3). Interestingly, 12 genes misregulated upon loss of Myh3 were identified in a study to discover transcriptional regulators involved in early muscle differentiation using C2C12 myoblasts (Rajan et al., 2012) (Table S4). Of these 12 candidates, eight genes were misregulated in more than one muscle in our study (Table S4). Six of these candidate genes were tested for their role in early C2C12 differentiation by knockdown assays and were found to be vital for differentiation (Rajan et al., 2012) (Table S4). All of this confirms that MyHC-emb is necessary for normal myogenic differentiation and its absence leads to the misregulation of muscle differentiation-related genes.
The diaphragm has the fewest misregulated genes in common with the three limb muscles, possibly because the diaphragm is a specialized muscle that is anatomically and functionally distinct from the limb (Fig. 3H). Another difference between these muscles is that the diaphragm is more mature at birth, as it starts working immediately with the onset of respiration, unlike limb muscles, which start working gradually (Kelly et al., 1991). One hundred and fifty-six genes were uniquely misregulated in a single muscle type, which could be genes that have distinctive roles in the specific muscles, depending on fiber type characteristics, functional properties and anatomical position (Fig. 3H). Next, we validated misregulated candidate genes in the quadriceps and the diaphragm using qPCR (Fig. 3E,F). Certain candidate genes identified by the RNA-Seq experiment were also misregulated upon Myh3 knockdown during C2C12 myogenic differentiation, indicating that MyHC-emb affects similar processes and pathways in vivo and in vitro (Fig. 3G).
In summary, genes related to differentiation were misregulated upon loss of MyHC-emb function, indicating that MyHC-emb is crucial for proper myogenic differentiation. Although MyHC-emb is expressed uniformly across muscles, the misregulated genes are not the same in different muscles, possibly because of inherent differences between muscles, which reflects their fiber type and metabolic diversity.
Loss of MyHC-embryonic leads to the misregulation of other MyHC isoforms
Myh3 is tightly linked with other fast Myh isoforms, and, therefore, we tested whether loss of MyHC-emb affects the expression of other MyHCs (Allen and Leinwand, 2001; Sartorius et al., 1998) (Fig. S2F). We found that Myh2 transcript levels were significantly upregulated in Myh3Δ/Δ animals (Fig. 3I,J; Fig. S2G,H). Myh1 transcript levels were significantly upregulated in the quadriceps and diaphragm of Myh3Δ/Δ mice (Fig. 3I,J), whereas Myh8 was significantly upregulated in the quadriceps and gastrocnemius (Fig. 3I; Fig. S2H). Remarkably, Myh2, which is upregulated in all of the muscles, is located adjacent to Myh3 in the Myh gene cluster on chromosome 11, whereas Myh1 and Myh8 are located downstream of Myh2 (Vikstrom et al., 1997; Weiss et al., 1999) (Fig. S2F). Thus, loss of MyHC-emb leads to compensatory upregulation of other Myh genes in the fast Myh gene cluster, with the genes located physically closest to Myh3 upregulated in more muscles. Transcript levels of Myh7, which is not part of the MyHC chromosome 11 cluster, were significantly downregulated in Myh3Δ/Δ animals in the TA and quadriceps (Fig. S2G and Fig. 3I).
MyHC-embryonic regulates myogenic progenitor differentiation in a non-cell-autonomous manner during development
As loss of MyHC-embryonic led to changes in myofiber size, number and type, as well as the misregulation of genes involved in muscle differentiation at neonatal stages, we hypothesized that the differentiation defects are most likely a result of embryonic- or fetal-specific requirements of MyHC-emb. To decipher the embryonic- and fetal-specific roles played by MyHC-emb, we made use of two Cre drivers, Pax3CreKI/+, which causes Cre-mediated recombination in embryonic and fetal myogenic lineages, and Pax7iCre/+, which recombines only in the fetal myogenic lineage (Engleka et al., 2005; Hutcheson et al., 2009; Keller et al., 2004). At E13.5, during embryonic myogenesis, deletion of Myh3 in Pax3CreKI/+;Myh3Δ/fl3-7 led to a significant reduction in the protein levels of the myogenic progenitor marker Pax7 to half that of the control (Fig. 4A,B), whereas levels of the committed myoblast markers MyoD and myogenin were significantly increased (Fig. 4A,B). This indicates that loss of MyHC-emb accelerates the differentiation of myogenic progenitors leading to the depletion of the progenitors, as seen by a reduction in Pax7 levels, and a concomitant increase in differentiated myoblasts, as indicated by the increased levels of the myoblast markers MyoD and myogenin (Fig. 4A,B). MyHC-slow protein levels were upregulated in Pax3CreKI/+;Myh3Δ/fl3-7 E13.5 embryos (Fig. 4A).
Fig. 4.

MyHC-embryonic non-cell-autonomously regulates myogenic progenitor differentiation during embryonic and fetal myogenesis. (A-G) Western blots for Pax7, MyoD, myogenin, MyHC-slow, caspase 3 and β-actin on protein lysates from hind limbs of E13.5 Pax3CreKI/+;Myh3Δ/fl3-7, E16.5 Pax7iCre/+; Myh3Δ/fl3-7 and P0 Myh3Δ/Δ, and controls (A); and their densitometric quantification (B-G). (H-I″) Immunofluorescence for Pax7 (green) and DAPI (blue) on cross-sections from E16.5 control (H-H″) and Pax7iCre/+;Myh3Δ/fl3-7 (I-I″) embryo hind limbs; H″ and I″ are magnifications of boxed areas from H′ and I′, respectively. (J,K) Immunofluorescence for MyoD (green) and DAPI (blue) on cross-sections from E16.5 control (J) and Pax7iCre/+;Myh3Δ/fl3-7 (K) embryo hind limbs. (L,M) Quantification of Pax7+ myogenic progenitors (L) and MyoD+ myoblast numbers (M) normalized to total area from E16.5 Pax7iCre/+;Myh3Δ/fl3-7 and the control embryo. Data are mean±s.e.m. of a minimum of three independent experiments. Scale bars: 200 μm (I′); 20 μm (I″ and K).
At E16.5, during fetal myogenesis, the deletion of Myh3 in Pax7iCre/+;Myh3Δ/fl3-7 also resulted in a significant reduction in Pax7 protein levels (Fig. 4A,C). Interestingly, unlike E13.5, the levels of the committed myoblast markers MyoD and myogenin were significantly decreased in Pax7iCre/+;Myh3Δ/fl3-7 E16.5 embryos (Fig. 4A,C). MyHC-slow protein levels were upregulated approximately sevenfold in Pax7iCre/+;Myh3Δ/fl3-7 E16.5 embryos (Fig. 4A,E), which suggests that the elevated MyHC-slow protein levels and MyHC-slow+ fibers seen at P0 (Fig. 1C-D″,K; Fig. 4A) are due to increased MyHC-slow protein levels during embryonic and fetal myogenesis. We investigated this further at the neonatal stage by comparing Pax7 protein levels in Myh3+/+ and Myh3Δ/Δ P0 mouse muscles. This comparison indicated that there was no difference in Pax7, MyoD or myogenin levels between the control and MyHC-emb null animals (Fig. 4A,D). Levels of cleaved caspase 3, a cell death marker, did not change in Pax7iCre/+;Myh3Δ/fl3-7 E16.5 embryos, indicating that decreased levels of the myogenic progenitor and myoblast markers were not due to elevated cell death (Fig. 4A,F). However, increased cell death was evident at P0 in Myh3Δ/Δ muscles (Fig. 4A,G and Fig. S5A-B′), which fits well with the increased Trim63 (MuRF1) transcript levels, a muscle-specific ubiquitin ligase that targets proteins for degradation, in P0 Myh3Δ/Δ muscles by RNA-Seq (Witt et al., 2005).
As we observed reduced Pax7 levels during embryonic and fetal myogenesis, we next examined whether the number of Pax7+ muscle progenitors varied between control and Pax7iCre/+;Myh3Δ/fl3-7 embryos (Fig. 4H-I″). We found a drastic ∼50% reduction in the number of Pax7+ myogenic progenitors in Pax7iCre/+;Myh3Δ/fl3-7 embryos (Fig. 4H″,I″,L). Similarly, we also observed a ∼60% decline in MyoD+ myoblasts in Pax7iCre/+;Myh3Δ/fl3-7 embryos (Fig. 4J,K,M). To validate this, we used germline Myh3 null E16.5 embryo (Myh3Δ/Δ) hind limb muscles. We found that both Pax7+ progenitors and levels of MyoD were significantly reduced in Myh3Δ/Δ embryos, corroborating the fetal-specific loss of MyHC-emb results (Fig. S5F,G,H). There were no changes observed in the number of dividing, phospho-histone H3+ (PHH3) nuclei between control and Pax7iCre/+;Myh3Δ/fl3-7 embryos, indicating that the rate of cell division is not responsible for the observed decrease in Pax7+ progenitors and MyoD+ myoblasts (Fig. S3A). This confirms that loss of MyHC-emb accelerates the differentiation of myogenic progenitors during fetal myogenesis, and causes the depletion of Pax7+ progenitors and MyoD+ myoblasts. We did not observe any difference in the number of Pax7+ muscle progenitors in Myh3Δ/Δ mice at P0, suggesting that the effect of MyHC-emb on Pax7+ myogenic progenitors is restricted to embryonic and fetal stages of development (Fig. S3B-D). We carried out semi-quantitative RT-PCR for Myh3, comparing cDNA derived from neonatal muscle progenitors (isolated by fluorescence-activated cell sorting) with whole-muscle cDNA, and found that Myh3 is not expressed in muscle progenitors (Fig. S4B). As MyHC-emb is not expressed in myogenic progenitors or myoblasts and its expression is limited to myofibers (Yoshida et al., 1998), the depletion of progenitor and myoblast pools that we observed must be mediated by secreted signals arising from the myofibers. Thus, MyHC-emb is essential for embryonic and fetal myogenesis by non-cell-autonomously regulating the differentiation rate of myogenic progenitors and myoblasts.
MyHC-embryonic knockdown causes reserve cell depletion and decreased fusion index
To confirm our in vivo results, we carried out siRNA-mediated knockdown of Myh3 during C2C12 myogenic differentiation in vitro. We observed a knockdown efficiency of ∼80% or higher (Fig. 5A,B). As observed in vivo, MyHC-slow protein levels exhibited an approximate sevenfold increase upon Myh3 knockdown at days 5 and 7 of differentiation (Fig. 5A,C). Next, we tested whether the depletion of Myh3 leads to an altered rate of differentiation of myogenic progenitors. To achieve this, we labeled the reserve cell pool, i.e. the population of cycling undifferentiated cells in the differentiated C2C12 culture, using F-actin (Burattini et al., 2004) (Fig. 5D-E′). We found that the number of reserve cells was dramatically reduced by ∼85% upon Myh3 knockdown (Fig. 5D-F). We hypothesized that any of three possibilities could explain the reduction in reserve cells: reduced cell proliferation, increased cell death or increased rate of differentiation. We did not observe any change in the number of proliferative PHH3+ cells between the control and Myh3 siRNA-treated cells (Fig. S3E,F). Although cell death, as measured by levels of cleaved caspase 3 protein, was elevated on day 1 of Myh3 knockdown, it was significantly less compared with control samples on days 5, 7 and 9 of differentiation (Fig. 5G,J). Thus, we ruled out decreased rate of cell division or increased cell death as being responsible for the reduction in reserve cell number, which must therefore occur as a result of the alteration in the rate of myogenic differentiation. To test this, we analyzed the protein levels of MyoD and myogenin following Myh3 knockdown, and observed an initial upregulation on days 3-5 (except for myogenin at day 3, where we observed a downregulation), followed by a reduction in expression of both at later time points (days 7-9) of differentiation (Fig. 5G-I). This indicates an initial increased rate of differentiation, which depletes the reserve cell pool, which in turn causes a decline in differentiation rate. Again, this effect must be non-cell-autonomous as MyHC-emb expression is restricted to myofibers and is not expressed by reserve cells or myoblasts. We found that differentiation was compromised upon Myh3 knockdown, with a significantly lower fusion index in Myh3 siRNA-treated cells, which could be caused by cell-autonomous and non-cell-autonomous signals (Fig. S5E). A significant reduction in the fusion index was also observed with differentiating primary myoblasts from Myh3Δ/Δ neonatal mice compared with controls (Fig. 5K; Fig. S5C,D). The results from these experiments have striking similarities to the developmental loss of MyHC-emb, with similar effects on the myogenic progenitor and myoblast pool, fiber type, and differentiation.
Fig. 5.

Myh3 depletion causes reduction in reserve cell number and decreased fusion index. (A-C) Western blots for MyHC-emb, MyHC-slow and β-actin on control and Myh3 siRNA-treated C2C12 cells over 9 days of differentiation (A), and densitometric quantification (B,C); ‘C’ and ‘S’ denote control and Myh3 siRNA along with the specific day of differentiation (A). (D-E′) MyHC (red), phalloidin (green) and DAPI (blue) immunofluorescence on control (D,D′) and Myh3 (E,E′) siRNA-treated C2C12 cells at day 5 of differentiation (D′ and E′ are magnifications from D and E, respectively, with white arrows marking reserve cells). (F) Quantification of reserve cell number per unit area (mm2) from control and Myh3 siRNA-treated C2C12 cells. (G-J) Western blots for MyoD, myogenin, caspase 3 and β-actin on control and Myh3 siRNA-treated C2C12 cells during differentiation (G) and densitometric quantification (H-J); ‘C’ and ‘S’ denote control and Myh3 siRNA along with the specific day of differentiation (G). (K) Fusion index of myofibers formed by differentiating myoblasts for 7 days, isolated from Myh3+/+ and Myh3Δ/Δ P0 mice, for which representative images are shown in Fig. S5C,D. Data are mean±s.e.m. of a minimum of three independent experiments. Scale bars: 100 μm (E); 25 μm (E′).
The non-cell-autonomous effects of MyHC-embryonic on myogenic differentiation are mediated by FGF signaling
To validate whether non-cell-autonomous signals mediated by MyHC-emb are crucial for normal myogenic differentiation, we differentiated C2C12 cells in conditioned media derived from Myh3 or control siRNA-treated C2C12 cells. Interestingly, we observed that C2C12 cells treated with Myh3 siRNA-conditioned media had larger and increased numbers of myofibers (Fig. 6A,B). The fusion index was significantly increased in cells differentiated in the presence of Myh3 siRNA-conditioned media (Fig. 6C), confirming that non-cell-autonomous, secreted signals mediated by MyHC-emb (expressed in myofibers) are crucial for regulating myogenic differentiation. The distinction between cell-autonomous and non-cell-autonomous effects are apparent in the fusion index experiments where cells were allowed to differentiate following Myh3 knockdown (both cell-autonomous and non-cell-autonomous effects) (Fig. S5E), compared with differentiating cells in conditioned media from Myh3-silenced cells (only non-cell-autonomous effects) (Fig. 6C).
Fig. 6.

The non-cell-autonomous effect of MyHC-embryonic on myogenesis is mediated by FGF signaling. (A-C) Immunofluorescence labeling for MyHC (red) and DAPI (blue) on C2C12 cells differentiated for 4 days in conditioned media from control and Myh3 siRNA-treated cells (A,B) and quantification of the fusion index (C). (D) Western blots for p-FGFR4, Spry2, p-Akt, p-Stat3 and β-actin on protein lysates from E16.5 Pax7iCre/+;Myh3Δ/fl3-7 and P0 Myh3Δ/Δ compared with controls. (E) Mass spectrometric analysis of secretome showing an abundance of FGF ligands in control and Myh3 siRNA-treated C2C12 cells. The graphical data represent the mean±s.e.m. of a minimum of three independent experiments. Scale bar: 25 μm.
Next, to identify signaling pathways that are misregulated upon loss of MyHC-emb, we carried out a pathway analysis using the RNA-Seq results comparing neonatal Myh3+/+ and Myh3Δ/Δ muscles. Among the top five misregulated pathways, four, namely IGF1 (insulin-like growth factor 1), ILK (integrin-like kinase), PTEN (phosphatase and tensin homolog) and STAT3 (signal transducer and activator of transcription 3), were related to mitogen-activated protein kinase (MAP kinase) signaling (Table 1).
Table 1.
RNA-Seq data analysis
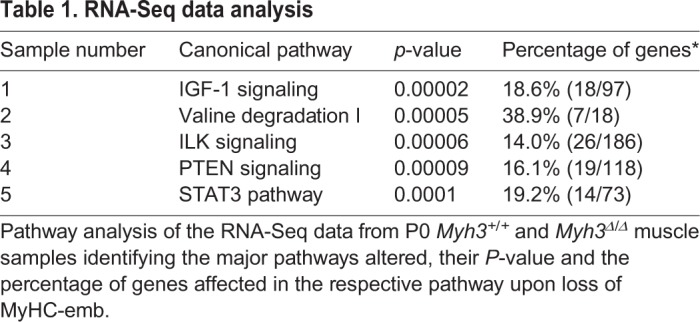
The FGF pathway, in which signaling is mediated through MAP kinase activation, has been reported to mediate differentiation and maintenance of the stem cell pool (Goetz and Mohammadi, 2013; Pawlikowski et al., 2017; Tsang and Dawid, 2004). FGFR4, a FGF family receptor, is central to myogenic differentiation during development, playing a key role in regulating the rate of differentiation of myogenic progenitors and myoblasts (Lagha et al., 2008; Marics et al., 2002). Interestingly, we found that levels of activated forms of known FGF pathway members, p-FGFR4, p-Akt and p-Stat3, were decreased during fetal myogenesis-specific (E16.5) loss of MyHC-emb (Fig. 6D). A similar decrease in the FGF target Spry2 was also observed, indicating that FGF pathway activation is compromised upon loss of MyHC-emb during fetal myogenesis (Fig. 6D). By P0, p-FGFR4 and Spry2 levels were similar to the control, whereas p-Akt and p-Stat3 levels continued to be reduced upon loss of MyHC-emb (Fig. 6D), possibly as a result of the compensatory effect of adult MyHCs, which are expressed by P0. To further confirm the effect of MyHC-emb on the FGF pathway, we carried out a mass spectrometric analysis to identify secreted FGF levels comparing the secretome from control and Myh3 siRNA-treated C2C12 cells. We found that levels of FGFs classically known to activate FGFR4 (especially FGF4, FGF6, FGF8 and FGF17) (Ornitz and Itoh, 2015) were reduced upon Myh3 knockdown (Fig. 6E). Furthermore, we quantified the transcript levels of FGFs known to bind FGFR4 (FGF1, FGF2, FGF4, FGF5, FGF6 and FGF8) using qPCR in E16.5 limb muscles of Pax7iCre/+;Myh3+/+ and Pax7iCre/+;Myh3Δ/fl embryos to identify the specific FGFs that are misregulated. We found that FGF1 and FGF2 are significantly downregulated, and FGF4 is significantly upregulated upon loss of MyHC-emb function (Fig. S4A). These results suggest that FGF signaling mediates the effect of MyHC-emb on myogenic differentiation.
Next, we tested whether FGF modulates the myogenic differentiation defects observed upon Myh3 knockdown. Knockdown of Myh3 led to a drastic reduction in the reserve cell number (Fig. 5D-F) and, therefore, we quantified the reserve cell number in Myh3 siRNA-treated cells grown in FGF-supplemented media. We found reserve cell numbers were restored to wild-type levels upon FGF supplementation, indicating that exogenous FGF can rescue the effect of loss of MyHC-emb function (compare Fig. 7A-C with Fig. 5D-F). A significant increase in the fusion index was observed upon FGF supplementation of Myh3 siRNA-treated C2C12 cells (Fig. 7D). To validate the effect of FGF signaling upon Myh3 knockdown, we also analyzed the levels of the myogenic differentiation markers MyoD and MyoG at days 3, 5 and 7 of culture (Fig. 7E). Interestingly, we found that both MyoD and MyoG levels decreased at days 3 and 5, whereas they increased at day 7, in the presence of exogenous FGF (Fig. 7E-G). This is in contrast to MyoD and MyoG levels increasing initially and decreasing at day 7 upon Myh3 knockdown (Fig. 5G-I). Levels of MyHC-slow, a marker of myogenic differentiation, increased upon FGF supplementation at days 5 and 7 (Fig. S4C). Thus, our results clearly demonstrate that FGF rescues the differentiation defects observed as a result of loss of MyHC-emb function.
Fig. 7.

Supplementation of FGF rescues the effect of loss of MyHC-embryonic on myogenesis. (A-B‴) Immunofluorescence labeling for MyHC (red), phalloidin (green) and DAPI (blue) on C2C12 cells at day 5, where Myh3 has been knocked down, treated with control media without FGF (A-A‴) or with FGF-supplemented media (B-B‴); A‴ and B‴ are magnifications from A″ and B″, respectively. (C,D) Quantification of reserve cell number per unit area (mm2) (C) and fusion index (D) from Myh3 siRNA-treated C2C12 cells at day 5, treated with FGF-supplemented media compared with the control. (E-G) Western blots for MyoD, myogenin and β-actin on Myh3 siRNA-treated C2C12 cells at days 3, 5 and 7 of differentiation, grown in the presence or absence of FGF (E), and densitometric quantification (F-G). The symbols, ‘−’ and ‘+’ denote absence or presence of FGF in the media (E). (H,I) Six-week old Myh3Δ/Δ mice exhibit scoliosis (I), compared with control Myh3Δ/+ animals (H). (J) Model summarizing the cell-autonomous and non-cell-autonomous roles of MyHC-embryonic during embryonic, fetal and neonatal myogenesis, where it regulates FGF levels, which control the rate of myogenic differentiation. Data are mean±s.e.m. of a minimum of three independent experiments. Scale bars: 100 μm (B″); 33 μm (B‴).
Although surviving Myh3Δ/Δ mice looked relatively normal at birth, other than decreased body weight, we observed that by 4-6 weeks of age, all Myh3Δ/Δ mice exhibit severe scoliosis, one of the phenotypes seen in individuals with FSS and SHS who have mutations in MYH3 (Fig. 7H,I).
DISCUSSION
We have characterized the role of MyHC-emb, one of two MyHCs expressed in the skeletal muscle during mammalian development. Although not absolutely required for muscle differentiation, we find that MyHC-emb underpins novel cell-autonomous and non-cell-autonomous functions during myogenesis.
Loss of MyHC-embryonic leads to cell-autonomous effects during muscle development
Loss of MyHC-emb function caused diverse cell-autonomous effects on neonatal muscle fibers, including alterations in fiber number, size and type. Intriguingly, although the effects on fiber number, type and size were not uniform across all muscles, they persisted into postnatal stages and were partially compensated for by ∼4 weeks of age, emphasizing the importance of MyHC-emb in myogenesis during development and neonatal stages. Although MyHC-emb is uniformly expressed across muscles, it might have distinct functions in different muscles based on muscle contractile and metabolic properties. Previous studies on adult MyHCs indicate that loss of Myh1 leads to an increase in the proportion of very small and large fibers (Acakpo-Satchivi et al., 1997), and specific adult muscles were affected to varying degrees with respect to fiber size and interstitial fibrosis in Myh4 and Myh1 null mice (Allen et al., 2000, 2001). The RNA-Seq experiment identified genes that are misregulated upon loss of Myh3, where 156 genes were uniquely misregulated in any of four muscles, confirming that loss of Myh3 has distinct effects on different muscles. Broadly, the misregulated genes were related to myogenic differentiation or muscle structure, indicating that MyHC-emb has essential roles during development.
MyHC-embryonic regulates embryonic and fetal myogenesis through non-cell-autonomous signals mediated by the FGF pathway
In addition to its cell-autonomous effects, loss of MyHC-emb function led to non-cell-autonomous effects on C2C12 reserve cells, the pool of undifferentiated cells that exhibit characteristics of satellite cells (Alli et al., 2013; Stuelsatz et al., 2010; Yoshida et al., 1998). We also find that MyHC-emb mediates non-cell-autonomous effects on myogenic progenitors and myoblasts during embryonic and fetal myogenesis in vivo. Previous studies show that the Pax7+ muscle stem cell pool and their differentiation are regulated by signals from differentiated myofibers, including the mechanical force of muscle contraction (Esteves de Lima et al., 2016). Although the myogenic progenitor pool depletion upon loss of MyHC-emb was consistent across embryonic and fetal myogenesis, the myoblast pool behaved differently, possibly as a result of inherent differences between embryonic and fetal myogenic progenitors, as previously described (Biressi et al., 2007a,b; Stockdale, 1992).
Both positive and negative signals regulate the rate of differentiation of myogenic progenitors and myoblasts. We and others have previously shown that extrinsic signals from the muscle connective tissue fibroblasts regulate muscle differentiation and fiber type (Joe et al., 2010; Mathew et al., 2011). The FGF pathway is crucial to myogenesis. FGF signaling has been reported to regulate Pax3, Pax7, MyoD and myogenin expression, as well as terminal differentiation (Edom-Vovard et al., 2001; Groves et al., 2005; Lagha et al., 2008). FGF signaling is also known to regulate the choice between proliferation and differentiation of myogenic precursors (Ben-Yair and Kalcheim, 2005; Marics et al., 2002). Our results indicate that MyHC-emb regulates the differentiation of myogenic progenitors and myoblasts via the FGF signaling pathway, potentially by regulating FGF secretion from the myofibers. Exogenous FGF was able to rescue the myogenic differentiation defects caused by Myh3 knockdown in C2C12 cells, demonstrating that FGF signaling mediates the non-cell-autonomous effects of MyHC-emb on myogenesis.
Recently, we and others described the first animal models for FSS using Drosophila, which has only one skeletal muscle MyHC isoform (Das et al., 2019; Rao et al., 2019). In the current work, we have generated the first mouse model for studying FSS. Individuals with FSS exhibit several clinical symptoms including muscle contractures, dental crowding, scoliosis, cryptorchidism, strabismus and hearing loss (Stevenson et al., 2006). Adult mice null for Myh3 exhibit scoliosis (Fig. 7H,I), a phenotype seen in most individuals with FSS (Stevenson et al., 2006). Detailed characterization of the Myh3 null mouse is required to verify whether they exhibit other symptoms seen in FSS patients. Whereas individuals with FSS have missense mutations in the MYH3 gene, which are thought to impede the ATPase activity of the protein, our mouse model is a loss of function of Myh3. Strikingly, missense mutations in human FGFR3 have been reported to cause camptodactyly-tall stature-scoliosis-hearing loss (CATSHL) syndrome, where individuals exhibit camptodactyly and scoliosis, phenotypes that are also observed in FSS and SHS caused by MYH3 mutations, indicating that MyHC-emb and FGF signaling might have common roles during development (Toydemir et al., 2006a,c).
Model and conclusions
Based on our findings, we propose that MyHC-embryonic has crucial cell-autonomous and non-cell-autonomous functions during embryonic, fetal, neonatal and postnatal stages of myogenesis. MyHC-emb within myofibers during embryonic and fetal stages is required to regulate FGF levels, which control the rate of differentiation of myogenic progenitors and myoblasts into myofibers (Fig. 7J). Reductions in FGF levels cause accelerated differentiation of progenitors and myoblasts, which depletes the pool of progenitors and myoblasts (a non-cell-autonomous effect of MyHC-emb) (Fig. 7J). In addition, MyHC-emb also regulates levels of other MyHCs, as well as fiber type, number and size (cell-autonomous effects of MyHC-emb) (Fig. 7J).
Thus, our study shows for the first time that developmental MyHCs are important regulators that play crucial cell-autonomous and non-cell-autonomous roles during skeletal muscle development. Future work on how the observed phenotypic changes contribute to scoliosis should shed light on the role of MyHC-embryonic in congenital contracture syndromes.
MATERIALS AND METHODS
Mice
Myh3fl3-7/+ mice were generated by flanking exons 3-7 of Myh3 with LoxP sites, according to published protocols (Wu et al., 2008). Briefly, an 11.3 kb genomic fragment of mouse Myh3 was recombineered into the pStartK vector from bacterial artificial chromosome clone RP23-67L23 (Children's Hospital Oakland Research Institute – CHORI, California, USA). By a series of recombineering and cloning steps, LoxP sites, FRT-PGKNeo-FRT and restriction enzyme sites were added to the construct and validated (Wu et al., 2008). The targeting construct was transferred to the pWS-TK2 vector, linearized and used for integration into embryonic stem cells (ESC) using positive and negative selection (Wu et al., 2008). Genomic DNA from ∼200 ESC clones was screened for 5′ and 3′ LoxP integration by PCR, and 24 clones were identified as positive for both 5′ and 3′ targeting events. Two clones were chosen for microinjection into blastocysts. The resulting chimeric animals from these were crossed with C57Bl/6J wild-type mice and the offspring tested for targeting. Once targeted animals were identified, they were bred for 5 or 6 generations to bring them into the C57Bl/6J background, and the neo cassette was removed by crossing with the R26RFlpe mice (Farley et al., 2000). Myh3Δ/+ mice were generated by crossing the Myh3fl3-7/+ mice with the ubiquitous Cre-expressing HprtCre mice (Tang et al., 2002). The Myh3fl3-7/+ mice were generated at the Transgenic and Gene Targeting Core (University of Utah, Salt Lake City, UT, USA).
Other Cre-drivers used were Pax3Cre (Engleka et al., 2005) and Pax7iCre (Keller et al., 2004). C57Bl/6J wild-type mice were used in this study. All of the animal maintenance and experiments were performed according to Institutional Animal Care and Use Committee (IACUC) approved protocols of the University of Utah and the Regional Centre for Biotechnology Institutional Animal Ethics Committee.
Genotyping
Genotyping was carried out by PCR using genomic DNA extracts prepared from mouse ear clips. Primer sequences used for genotyping Myh3 wild-type, floxed and delta alleles are listed in Table S1, under ‘Primers used for genotyping'. Briefly, mouse genomic DNA preparation was carried out using the HotSHOT lysis method (Truett et al., 2000). A PCR mastermix was prepared as follows: 7.5 µl GoTaq G2 Hot Start Green Master Mix (Promega, M7423); 8 µl of a 5 µM solution of primers for Myh3 (one forward and two reverse) (0.26 µM final concentration); 2.1 µl DNAse free water; and 3 µl HotSHOT DNA. The PCR cycling conditions were as follows: 95°C for 5 min; 34 cycles of 95°C for 30 s, 60°C for 20 s and 72°C for 40 s; 72°C for 10 min; and 10°C for 10 min. The PCR products were separated on 2% agarose gel, and Myh3 genotypes were determined based on band sizes (delta, 179 bp; wild type, 231 bp; floxed, 279 bp).
Cell culture
C2C12 mouse myoblasts (ATCC, CRL-1722) were cultured and maintained according to ATCC guidelines in growth medium containing DMEM-Dulbecco's Modified Eagle Medium (Gibco, 11995065) supplemented with 10% (v/v) fetal bovine serum (Sigma-Aldrich, F2442) and 2% penicillin-streptomycin (Gibco, 15140122). C2C12 cells were differentiated in differentiation media containing DMEM, 2% (v/v) horse serum (BioAbChem, 72-0460) and 2% penicillin-streptomycin.
For knocking down Myh3 expression in C2C12 myoblasts, reverse transfection was performed. Briefly, ∼30,000 cells were seeded on gelatin-coated coverslips (Neuvitro, GG-12) in each well of a 24-well plate (Nunc, 142485) layered with the transfection mix. The transfection mix comprised 100 µl Opti-MEM (Gibco, 31985070), 50 nM of Myh3 or control siRNA (Ambion, s70258 and 4390847, respectively) and 2 µl Lipofectamine RNAiMAX (Invitrogen, 13778150). Cells were cultured in growth medium for 48 h to allow growth to ∼80% confluence and efficient transfection. Subsequently, differentiation was induced by replacing the growth medium with differentiation medium. For RNA and protein lysate preparation, the same protocol was used, except that cells were seeded directly in the wells. All treatments were carried out uniformly between control and Myh3 siRNA-transfected cells.
For the conditioned media experiments, C2C12 cells were cultured in a 24-well dish after treatment with control and Myh3 siRNA, respectively, and the conditioned media collected at day 5 of plating. Next, ∼30,000 C2C12 cells were plated on gelatin-coated coverslips in growth media. After 48 h, the media were replaced with conditioned media from control and Myh3 siRNA-treated cells, and cultured for 4 days, following which coverslips were processed for immunofluorescence analysis.
For the FGF rescue experiments, C2C12 cells cultured in 24-well dishes were treated with Myh3 siRNA with (for immunofluorescence) or without (for protein lysates) gelatin-coated coverslips. For immunofluorescence analysis, 10 µg/ml FGF2 (Sigma-Aldrich, SRP4038) was added 48 h after plating to half of the wells and the other half were used as untreated controls. For protein lysate preparation, 10 µg/ml FGF2 was added to half of the wells at 48, 72 and 96 h after plating for 3-, 5- and 7-day samples, respectively. The other half were used as untreated controls for each time point.
For primary myoblast culture, hind limb muscles were isolated from Myh3+/+ and Myh3Δ/Δ mice at postnatal day 0 (P0), minced with surgical blades and then processed for pre-plating as described previously (Goetsch et al., 2015). After 3 h of pre-plating, the nonadherent cells in the supernatant (enriched for myoblasts) were transferred to gelatin-coated coverslips in individual wells of a 24-well dish, where they were grown in growth medium. After 3 days in growth medium, the cells were shifted to differentiation medium and grown for an additional 7 days, following which the coverslips were processed for immunofluorescence.
RNA isolation, cDNA synthesis and quantitative PCR (qPCR)
RNA was isolated from the TA, quadriceps, gastrocnemius and diaphragm muscles of Myh3+/+ and Myh3Δ/Δ mice at postnatal day 0 (P0) using the RNeasy Lipid Tissue Mini Kit (Qiagen, 74804). RNA was isolated from C2C12 cells using the RNeasy Mini Kit (Qiagen, 74106), cDNA was prepared using SuperScript III Reverse Transcriptase (Invitrogen, 18080-044) and oligo (dT) (Invitrogen, 58862) according to the manufacturer's protocol. Quantitative PCR was performed using SYBR Green (Applied Biosystems, 4367659) on the ABI 7500 Fast Real Time PCR System (Applied Biosystems) and normalized to Gapdh transcript levels. The genes studied and primers used for qPCR are listed in Table S1. Fgf qPCR primers are from a previous study (Du et al., 2016). The expression of target genes in the mutant muscles and Myh3 siRNA-transfected cells were normalized to that of wild-type muscles and control siRNA-transfected cells, respectively (Livak and Schmittgen, 2001). A minimum of four biological replicates of mutant and knockout mouse muscle samples and three biological replicates of Myh3 and control siRNA transfected samples were used for the expression analysis.
RNA-sequencing
Quadriceps, TA, gastrocnemius and diaphragm muscles from six Myh3+/+ and Myh3Δ/Δ animals, respectively, were harvested at postnatal day 0 (P0) and RNA extracted using the RNeasy Lipid Tissue Mini Kit (Qiagen, 74804) according to the manufacturer's protocol. The integrity and concentration of the isolated RNA was verified using the 2200 TapeStation (Agilent). Library preparation was performed using the TruSeq Stranded mRNA sample preparation kit (Illumina) with oligo dT selection according to the manufacturer's protocol and single-end 50 bp reads were generated using a HiSeq 2000 instrument (Illumina). Transcript annotations for mm10 (M_musculus, Dec 2011) were used from Ensembl. Reads were aligned using Pysano and annotated splice junctions generated using USeq (www.sourceforge.net/projects/USeq). Splice junction reads were mapped to genomic coordinates using the SamTranscriptomeParser application in USeq. Differential gene expression was identified using the Defined Region Differential Seq (DRDS) application in USeq, following which paired-sample differential gene expression analysis was performed using DESeq2 (Love et al., 2014). RNA-Seq metrics were generated using Picard's CollectRnaSeqMetrics, samples clustered using custom R scripts (Analysis/Plots), and significant genes were run in IPA to generate pathway analyses. A Venn diagram was generated to represent the differentially expressed genes identified by RNA-Seq, common between muscles or unique to specific muscles in Myh3Δ/Δ knockout animals compared with Myh3+/+ animals.
Western blots
For protein isolation, cells were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, R0278-500 ml) containing protease inhibitor (Sigma-Aldrich, P8340-5 ml). Protein was isolated from E13.5 Pax3Cre/+;Myh3+/+ and Pax3Cre/+;Myh3Δ/fl3-7, and E16.5 Pax7iCre/+;Myh3+/+ and Pax7iCre/+;Myh3Δ/fl3-7 embryo hind limbs using Qproteome FFPE Tissue Kit (Qiagen, 37623). Total protein was isolated from Myh3+/+ and Myh3Δ/Δ E16.5 embryo hearts or P0 hind limb muscles by homogenization using the Precellys 24 homogenizer (Bertin Technologies). Quantification of the protein samples was performed using a Pierce BCA Protein Assay Kit (Thermo Scientific, 23225) as per the manufacturer's protocol. Protein samples were separated on a 10% SDS-PAGE and transferred onto a PVDF membrane (Millipore, iPVH00010) at 4°C for 2 h. Western blot was performed using standard procedures: blocking in 5% skimmed milk (Himedia, RM1254-500GM) for 3 h; washes in PBS containing 0.1% Tween 20 (Sigma-Aldrich, P7949-100ML); incubation overnight with primary antibody at 4°C; 2 h incubation with HRP-conjugated secondary antibody at room temperature; and signal detection using the HRP substrate (Millipore, WBLUF0100). Blots were imaged using ImageQuant LAS 4000 (GE). Blots were stripped using standard procedures and reprobed for β-actin. Densitometry was performed to quantify the amount of protein normalized to beta-actin levels using the ImageQuant software. Antibodies used in these experiments are listed in Table S2.
Immunofluorescence and microscopy
Embryos at the appropriate stages were harvested from timed matings (E13.5 Pax3Cre/+;Myh3+/+ and Pax3Cre/+;Myh3Δ/fl3-7 embryos, and E16.5 Pax7iCre/+;Myh3+/+ and Pax7iCre/+;Myh3Δ/fl3-7), fixed and embedded in Optimal Cutting Temperature (OCT) (Tissue-Tek). Hind limbs from Myh3Δ/Δ and Myh3+/+ neonates at P0 were embedded in OCT and flash frozen in 2-methyl butane cooled in liquid nitrogen. Specific hind limb muscles from Myh3Δ/Δ and Myh3+/+ mice at P15 and P30 were also embedded in OCT and flash frozen in 2-methyl butane cooled in liquid nitrogen. Samples were sectioned at 10 µm using a cryomicrotome (Thermo Scientific; Microm HM 550) and adjacent sections collected on coated glass slides (VWR, VWRU48311-703). Sections from the middle of the limb (P0) or the muscle (P15 and P30) were processed as described below with adjacent sections used for detection of MyHC-emb, MyHC-slow, Pax7, MyoD, Laminin and PHH3. For immunofluorescence, tissue sections on slides or C2C12/primary cells on coverslips were fixed in 4% paraformaldehyde (PFA) for 20 min and washed with PBS. For tissue sections, the antigen was retrieved wherever required (specified in Table S2) by heating samples to 120°C for 5 min in citrate buffer (1.8 mM citric acid and 8.2 mM sodium citrate in water) in a 2100 PickCell Retriever (Aptum Biologics). Tissue sections and cells were blocked with 5% goat serum (BioAbChem, 72-0480) in PBS containing 0.1% Triton-X-100 (MP Biochemicals, 194854) for 1 h at room temperature, incubated overnight at 4°C in an appropriate concentration of primary antibody (Table S2), rinsed three times with PBS, incubated with secondary antibody for 2 h at RT, and rinsed with PBS. Where amplification was required, the samples were incubated with a biotin-conjugated secondary antibody for 2 h at room temperature and then with Strep-coupled fluorophore for 1 h at room temperature, and washed three times with PBS. Samples were postfixed in 4% PFA, rinsed in distilled water and mounted using DAPI Fluoromount-G (Southern Biotech, 0100-20). Fluorescence microscopy was performed using a Leica TCS SP5 II or Nikon A1R confocal microscope.
Preparation of secretome proteins and mass spectrometric analysis
Cells were cultured for 48 h in complete medium initially, following which it was replaced with serum-free medium. All traces of serum were removed by rinsing the cells three times with PBS before adding the serum-free medium. Medium for the secretome analysis was collected after 48 h of culture. Samples were centrifuged at 150 g for 5 min at 4°C to remove intact cells and debris. Protease inhibitor was added and the media were concentrated by ultrafiltration using the Amicon Ultra-15 Centrifugal Filter Unit (Millipore, UF C905024). Proteins were precipitated with cold acetone at −20°C overnight, and collected by centrifugation at 19,000 g for 1 h at 4°C. Pellets were air-dried at 37°C, solubilized in 4× Laemmli buffer and boiled for 10 min to denature proteins. Proteins were separated by 10% SDS-PAGE, stained with Coomassie Brilliant Blue R-250 and protein bands excised using sterile blades. The gel slices were destained, dehydrated and digested with mass spectrometry grade trypsin (Pierce Trypsin protease; Thermo Scientific, 90057) at 37°C overnight. Peptides were extracted by ultrasonication in extraction solution (50% Acetonitrile/5% trifluoroacetic acid) and the extract was dried in a vacuum concentrator at RT. The samples were desalted with modified Pierce C18 Zip-Tips (Thermo Scientific, 87 782) and loaded on a LC-MS/MS mass spectrometer (Triple TOF 5600 Sciex). The raw data of MS-MS (wiff and mgf files) were processed and subjected to database searches using Search GUI. Search results were processed, combined and interpreted in the form of emPAI (exponentially modified Protein Abundance Index) using Peptide Shaker, run with a false discovery rate of 1%.
Cell counts, fiber counts and statistics
For the fusion index analysis, five randomly selected nonoverlapping fields of view (2×3 tiles) per coverslip were imaged using identical settings. Myotubes were labeled for myosin heavy chain using a mixture of My32 and MyHC-slow antibodies and nuclei by DAPI. Counts for total nuclei, number of myotubes and nuclei within the myotubes per unit area, were performed using ImageJ (Schindelin et al., 2012; Schneider et al., 2012). The fusion index was calculated as the percentage of nuclei within myotubes (myotubes with at least two nuclei), compared with the total number of nuclei. For reserve cell counts, Myh3 and control siRNA-treated cells were cultured, stained using phalloidin, myosin heavy chain antibodies (mixture of My32 and MyHC-slow antibodies) and DAPI (to identify nuclei), after which images were captured. The total number of DAPI+ nuclei and phalloidin+ reserve cells were counted using spot and annotation functions in Imaris software (www.bitplane.com/), and normalized to unit area. PHH3+ nuclei were counted manually and the total DAPI+ nuclei were counted using the particle analyzer function in ImageJ (Schindelin et al., 2012; Schneider et al., 2012).
Pax7+, MyoD+ and PHH3+ cell counts from the entire P0 or E16.5 hind limb cross-section was performed using the spot function, and total P0 or E16.5 hind limb muscle cross-sectional area was quantified using the surface function in Imaris software. MyHC-slow fiber count was performed using the fiber typing function, and fiber area using the fiber properties function with the semiautomatic muscle analysis using segmentation of histology software (SMASH) (Smith and Barton, 2014).
Data from all of the experiments were analyzed with parametric unpaired t-tests using the GraphPad Prism software. Data are mean±s.e.m. The P-value is indicated on each graph and *P-value ≤0.05 is considered significant.
Supplementary Material
Acknowledgements
We acknowledge valuable suggestions and help from Dr Kirk Thomas, Dr Sen Wu and Dr Charles Murtaugh at the University of Utah with the Myh3 targeting design. Myh3 targeting was carried out at the transgenic gene-targeting mouse core facility at the University of Utah, and we are thankful to Susan Tamowski and staff for their services. We acknowledge the Cell Imaging core facility and Chris Rodesch at the University of Utah, the small animal facility (SAF) and imaging facilities at RCB, the mouse facility and Penny Noel at the University of Utah for help with mouse colony maintenance, and the genomics core facility at the University of Utah for RNA-sequencing. We thank Jennifer Lawson for important suggestions and help with the mouse work, Eric Bogenschutz and Dr Santhosh Karanth for suggestions on the RNA-Seq analysis, and Dr Tushar Maiti and Sandhini Saha for help with the mass spectrometry experiment design and analysis. We are grateful to Prof. Leslie Leinwand for helpful suggestions. We thank Dr Suchitra Gopinath from THSTI for providing some of the antibodies used in this work. We also acknowledge past and present members of the G.K. and S.J.M. labs for valuable suggestions and inputs. The authors acknowledge the support of DBT e-Library Consortium (DeLCON) for providing access to e-resources.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions metadata
Conceptualization: M.A., G.K., S.J.M.; Methodology: M.A., A.S., P.K., A.K., A.B., M.S., G.K., S.J.M.; Validation: M.A., A.S., P.K., A.K., A.B., M.S., S.J.M.; Formal analysis: M.A., A.S., P.K., A.K., A.B., M.S., S.J.M.; Investigation: M.A., A.S., P.K., A.K., A.B., M.S., G.K., S.J.M.; Resources: S.J.M.; Data curation: M.A., A.S., P.K., A.K., A.B., M.S., S.J.M.; Writing - original draft: M.A., A.S., P.K., A.K., A.B., M.S., S.J.M.; Writing - review & editing: M.A., P.K., M.S., S.J.M.; Supervision: G.K., S.J.M.; Project administration: G.K., S.J.M.; Funding acquisition: G.K., S.J.M.
Funding
This work was funded by an Intermediate Fellowship from the Wellcome Trust DBT India Alliance (IA/I/13/1/500872 to S.J.M.) and the National Institutes of Health (R01HD053728 to G.K.). We also acknowledge funding from the Department of Biotechnology (DBT), and the Regional Centre for Biotechnology (RCB). M.A. is funded by a senior research fellowship from the Indian Council of Medical Research, A.S. and P.K. by senior research fellowships from the Council of Scientific and Industrial Research, A.B. by senior research fellowship from the University Grants Commission, and M.S. by a Young Investigator Award from RCB initially and by a Wellcome Trust DBT India Alliance Early Career Fellowship later. Deposited in PMC for release after 6 months.
Data availability
RNA-Seq datasets have been deposited in GEO under accession number GSE100331.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.184507.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.184507.reviewer-comments.pdf
References
- Acakpo-Satchivi L. J. R., Edelmann W., Sartorius C., Lu B. D., Wahr P. A., Watkins S. C., Metzger J. M., Leinwand L. and Kucherlapati R. (1997). Growth and muscle defects in mice lacking adult myosin heavy chain genes. J. Cell Biol. 139, 1219-1229. 10.1083/jcb.139.5.1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D. L. and Leinwand L. A. (2001). Postnatal myosin heavy chain isoform expression in normal mice and mice null for IIb or IId myosin heavy chains. Dev. Biol. 229, 383-395. 10.1006/dbio.2000.9974 [DOI] [PubMed] [Google Scholar]
- Allen D. L., Harrison B. C. and Leinwand L. A. (2000). Inactivation of myosin heavy chain genes in the mouse: diverse and unexpected phenotypes. Microsc. Res. Tech. 50, 492-499. [DOI] [PubMed] [Google Scholar]
- Allen D. L., Harrison B. C., Sartorius C., Byrnes W. C. and Leinwand L. A. (2001). Mutation of the IIB myosin heavy chain gene results in muscle fiber loss and compensatory hypertrophy. Am. J. Physiol. Cell Physiol. 280, C637-C645. 10.1152/ajpcell.2001.280.3.C637 [DOI] [PubMed] [Google Scholar]
- Alli N. S., Yang E. C., Miyake T., Aziz A., Collins-Hooper H., Patel K. and McDermott J. C. (2013). Signal-dependent fra-2 regulation in skeletal muscle reserve and satellite cells. Cell Death Dis. 4, e692 10.1038/cddis.2013.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barany M. (1967). ATPase activity of myosin correlated with speed of muscle shortening. J. Gen. Physiol. 50 Suppl., 197-218. 10.1085/jgp.50.6.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yair R. and Kalcheim C. (2005). Lineage analysis of the avian dermomyotome sheet reveals the existence of single cells with both dermal and muscle progenitor fates. Development 132, 689-701. 10.1242/dev.01617 [DOI] [PubMed] [Google Scholar]
- Biressi S., Molinaro M. and Cossu G. (2007a). Cellular heterogeneity during vertebrate skeletal muscle development. Dev. Biol. 308, 281-293. 10.1016/j.ydbio.2007.06.006 [DOI] [PubMed] [Google Scholar]
- Biressi S., Tagliafico E., Lamorte G., Monteverde S., Tenedini E., Roncaglia E., Ferrari S., Ferrari S., Cusella-De Angelis M. G., Tajbakhsh S. et al. (2007b). Intrinsic phenotypic diversity of embryonic and fetal myoblasts is revealed by genome-wide gene expression analysis on purified cells. Dev. Biol. 304, 633-651. 10.1016/j.ydbio.2007.01.016 [DOI] [PubMed] [Google Scholar]
- Burattini S., Ferri P., Battistelli M., Curci R., Luchetti F. and Falcieri E. (2004). C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur. J. Histochem. 48, 223-233. [PubMed] [Google Scholar]
- Chong J. X., Burrage L. C., Beck A. E., Marvin C. T., McMillin M. J., Shively K. M., Harrell T. M., Buckingham K. J., Bacino C. A., Jain M. et al. (2015). Autosomal-dominant multiple Pterygium syndrome is caused by mutations in MYH3. Am. J. Hum. Genet. 96, 841-849. 10.1016/j.ajhg.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon K., Silberstein L., Blau H. M. and Thompson W. J. (1990). Development of muscle fiber types in the prenatal rat hindlimb. Dev. Biol. 138, 256-274. 10.1016/0012-1606(90)90196-P [DOI] [PubMed] [Google Scholar]
- Das S., Kumar P., Verma A., Maiti T. K. and Mathew S. J. (2019). Myosin heavy chain mutations that cause Freeman-Sheldon syndrome lead to muscle structural and functional defects in Drosophila. Dev. Biol. 449, 90-98. 10.1016/j.ydbio.2019.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W., Prochazka J., Prochazkova M. and Klein O. D. (2016). Expression of FGFs during early mouse tongue development. Gene Expr. Patterns 20, 81-87. 10.1016/j.gep.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edom-Vovard F., Bonnin M.-A. and Duprez D. (2001). Misexpression of Fgf-4 in the chick limb inhibits myogenesis by down-regulating Frek expression. Dev. Biol. 233, 56-71. 10.1006/dbio.2001.0221 [DOI] [PubMed] [Google Scholar]
- Engleka K. A., Gitler A. D., Zhang M., Zhou D. D., High F. A. and Epstein J. A. (2005). Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev. Biol. 280, 396-406. 10.1016/j.ydbio.2005.02.002 [DOI] [PubMed] [Google Scholar]
- Esteves de Lima J., Bonnin M.-A., Birchmeier C. and Duprez D. (2016). Muscle contraction is required to maintain the pool of muscle progenitors via YAP and NOTCH during fetal myogenesis. eLife 5, e15593 10.7554/eLife.15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley F. W., Soriano P., Steffen L. S. and Dymecki S. M. (2000). Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28, 106-110. [DOI] [PubMed] [Google Scholar]
- Feng Z., Tang Z.-L., Li K., Liu B., Yu M. and Zhao S.-H. (2007). Molecular characterization of the BTG2 and BTG3 genes in fetal muscle development of pigs. Gene 403, 170-177. 10.1016/j.gene.2007.08.009 [DOI] [PubMed] [Google Scholar]
- Goetsch K. P., Snyman C., Myburgh K. H. and Niesler C. U. (2015). Simultaneous isolation of enriched myoblasts and fibroblasts for migration analysis within a novel co-culture assay. BioTechniques 58, 25-32. 10.2144/000114246 [DOI] [PubMed] [Google Scholar]
- Goetz R. and Mohammadi M. (2013). Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 14, 166-180. 10.1038/nrm3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. A., Hammond C. L. and Hughes S. M. (2005). Fgf8 drives myogenic progression of a novel lateral fast muscle fibre population in zebrafish. Development 132, 4211-4222. 10.1242/dev.01958 [DOI] [PubMed] [Google Scholar]
- Hutcheson D. A., Zhao J., Merrell A., Haldar M. and Kardon G. (2009). Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for beta-catenin. Genes Dev. 23, 997-1013. 10.1101/gad.1769009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe A. W. B., Yi L., Natarajan A., Le Grand F., So L., Wang J., Rudnicki M. A. and Rossi F. M. V. (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153-163. 10.1038/ncb2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C., Hansen M. S., Coffin C. M. and Capecchi M. R. (2004). Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 18, 2608-2613. 10.1101/gad.1243904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A. M., Rosser B. W., Hoffman R., Panettieri R. A., Schiaffino S., Rubinstein N. A. and Nemeth P. M. (1991). Metabolic and contractile protein expression in developing rat diaphragm muscle. J. Neurosci. 11, 1231-1242. 10.1523/JNEUROSCI.11-05-01231.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagha M., Kormish J. D., Rocancourt D., Manceau M., Epstein J. A., Zaret K. S., Relaix F. and Buckingham M. E. (2008). Pax3 regulation of FGF signaling affects the progression of embryonic progenitor cells into the myogenic program. Genes Dev. 22, 1828-1837. 10.1101/gad.477908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J. and Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta C(T)) Method. Methods 25, 402-408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marics I., Padilla F., Guillemot J. F., Scaal M. and Marcelle C. (2002). FGFR4 signaling is a necessary step in limb muscle differentiation. Development 129, 4559-4569. [DOI] [PubMed] [Google Scholar]
- Mathew S. J., Hansen J. M., Merrell A. J., Murphy M. M., Lawson J. A., Hutcheson D. A., Hansen M. S., Angus-Hill M. and Kardon G. (2011). Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 138, 371-384. 10.1242/dev.057463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narusawa M., Fitzsimons R. B., Izumo S., Nadal-Ginard B., Rubinstein N. A. and Kelly A. M. (1987). Slow myosin in developing rat skeletal muscle. J. Cell Biol. 104, 447-459. 10.1083/jcb.104.3.447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M. and Itoh N. (2015). The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215-266. 10.1002/wdev.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker-Thornburg J., Bauer B., Palermo J. and Robbins J. (1992). Structural and developmental analysis of two linked myosin heavy chain genes. Dev. Biol. 150, 99-107. 10.1016/0012-1606(92)90010-E [DOI] [PubMed] [Google Scholar]
- Pawlikowski B., Vogler T. O., Gadek K. and Olwin B. B. (2017). Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev. Dyn. 246, 359-367. 10.1002/dvdy.24495 [DOI] [PubMed] [Google Scholar]
- Periasamy M., Wieczorek D. F. and Nadal-Ginard B. (1984). Characterization of a developmentally regulated perinatal myosin heavy-chain gene expressed in skeletal muscle. J. Biol. Chem. 259, 13573-13578. [PubMed] [Google Scholar]
- Periasamy M., Wydro R. M., Strehler-Page M. A., Strehler E. E. and Nadal-Ginard B. (1985). Characterization of cDNA and genomic sequences corresponding to an embryonic myosin heavy chain. J. Biol. Chem. 260, 15856-15862. [PubMed] [Google Scholar]
- Rajan S., Dang H. C. P., Djambazian H., Zuzan H., Fedyshyn Y., Ketela T., Moffat J., Hudson T. J. and Sladek R. (2012). Analysis of early C2C12 myogenesis identifies stably and differentially expressed transcriptional regulators whose knock-down inhibits myoblast differentiation. Physiol. Genomics 44, 183-197. 10.1152/physiolgenomics.00093.2011 [DOI] [PubMed] [Google Scholar]
- Rao D. S., Kronert W. A., Guo Y., Hsu K. H., Sarsoza F. and Bernstein S. I. (2019). Reductions in ATPase activity, actin sliding velocity, and myofibril stability yield muscle dysfunction in Drosophila models of myosin-based Freeman-Sheldon syndrome. Mol. Biol. Cell 30, 30-41. 10.1091/mbc.E18-08-0526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutland C. S., Polo-Parada L., Ehler E., Alibhai A., Thorpe A., Suren S., Emes R. D., Patel B. and Loughna S. (2011). Knockdown of embryonic myosin heavy chain reveals an essential role in the morphology and function of the developing heart. Development 138, 3955-3966. 10.1242/dev.059063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore S., Gorza L. and Schiaffino S. (1982). Fetal myosin heavy chains in regenerating muscle. Nature 298, 294-296. 10.1038/298294a0 [DOI] [PubMed] [Google Scholar]
- Sartorius C. A., Lu B. D., Acakpo-Satchivi L., Jacobsen R. P., Byrnes W. C. and Leinwand L. A. (1998). Myosin heavy chains IIa and IId are functionally distinct in the mouse. J. Cell Biol. 141, 943-953. 10.1083/jcb.141.4.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S. and Reggiani C. (1996). Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol. Rev. 76, 371-423. 10.1152/physrev.1996.76.2.371 [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Gorza L., Dones I., Cornelio F. and Sartore S. (1986). Fetal myosin immunoreactivity in human dystrophic muscle. Muscle Nerve 9, 51-58. 10.1002/mus.880090108 [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Rossi A. C., Smerdu V., Leinwand L. A. and Reggiani C. (2015). Developmental myosins: expression patterns and functional significance. Skelet. Muscle 5, 22 10.1186/s13395-015-0046-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S. and Eliceiri K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671-675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. R. and Barton E. R. (2014). SMASH - semi-automatic muscle analysis using segmentation of histology: a MATLAB application. Skelet. Muscle 4, 21 10.1186/2044-5040-4-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson D. A., Carey J. C., Palumbos J., Rutherford A., Dolcourt J. and Bamshad M. J. (2006). Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 117, 754-762. 10.1542/peds.2005-1219 [DOI] [PubMed] [Google Scholar]
- Stockdale F. E. (1992). Myogenic cell lineages. Dev. Biol. 154, 284-298. 10.1016/0012-1606(92)90068-R [DOI] [PubMed] [Google Scholar]
- Stuelsatz P., Pouzoulet F., Lamarre Y., Dargelos E., Poussard S., Leibovitch S., Cottin P. and Veschambre P. (2010). Down-regulation of MyoD by calpain 3 promotes generation of reserve cells in C2C12 myoblasts. J. Biol. Chem. 285, 12670-12683. 10.1074/jbc.M109.063966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupka N., Kintakas C., White J. D., Fraser F. W., Hanciu M., Aramaki-Hattori N., Martin S., Coles C., Collier F., Ward A. C. et al. (2013). Versican processing by a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats proteinases-5 and −15 facilitates myoblast fusion. J. Biol. Chem. 288, 1907-1917. 10.1074/jbc.M112.429647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajsharghi H., Kimber E., Kroksmark A.-K., Jerre R., Tulinius M. and Oldfors A. (2008). Embryonic myosin heavy-chain mutations cause distal arthrogryposis and developmental myosin myopathy that persists postnatally. Arch. Neurol. 65, 1083-1090. 10.1001/archneur.65.8.1083 [DOI] [PubMed] [Google Scholar]
- Tang S.-H. E., Silva F. J., Tsark W. M. K. and Mann J. R. (2002). A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis 32, 199-202. 10.1002/gene.10030 [DOI] [PubMed] [Google Scholar]
- Toydemir R. M., Brassington A. E., Bayrak-Toydemir P., Krakowiak P. A., Jorde L. B., Whitby F. G., Longo N., Viskochil D. H., Carey J. C. and Bamshad M. J. (2006a). A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am. J. Hum. Genet. 79, 935-941. 10.1086/508433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toydemir R. M., Chen H., Proud V. K., Martin R., van Bokhoven H., Hamel B. C. J., Tuerlings J. H., Stratakis C. A., Jorde L. B. and Bamshad M. J. (2006b). Trismus-pseudocamptodactyly syndrome is caused by recurrent mutation of MYH8. Am. J. Med. Genet. A 140, 2387-2393. 10.1002/ajmg.a.31495 [DOI] [PubMed] [Google Scholar]
- Toydemir R. M., Rutherford A., Whitby F. G., Jorde L. B., Carey J. C. and Bamshad M. J. (2006c). Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 38, 561-565. 10.1038/ng1775 [DOI] [PubMed] [Google Scholar]
- Truett G. E., Heeger P., Mynatt R. L., Truett A. A., Walker J. S. and Warman M. L. (2000). Preparation of PCR quality mouse genomic DNA with hot sodium hydroxide and tria (HotSHOT). BioTechniques 29, 52-54. 10.2144/00291bm09 [DOI] [PubMed] [Google Scholar]
- Tsang M. and Dawid I. B. (2004). Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci. STKE 2004, pe17 10.1126/stke.2282004pe17 [DOI] [PubMed] [Google Scholar]
- Vikstrom K. L., Seiler S. H., Sohn R. L., Strauss M., Weiss A., Welikson R. E. and Leinwand L. A. (1997). The vertebrate myosin heavy chain: genetics and assembly properties. Cell Struct. Funct. 22, 123-129. 10.1247/csf.22.123 [DOI] [PubMed] [Google Scholar]
- Weiss A., McDonough D., Wertman B., Acakpo-Satchivi L., Montgomery K., Kucherlapati R., Leinwand L. and Krauter K. (1999). Organization of human and mouse skeletal myosin heavy chain gene clusters is highly conserved. Proc. Natl. Acad. Sci. USA 96, 2958-2963. 10.1073/pnas.96.6.2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydert A., Daubas P., Caravatti M., Minty A., Bugaisky G., Cohen A., Robert B. and Buckingham M. (1983). Sequential accumulation of mRNAs encoding different myosin heavy chain isoforms during skeletal muscle development in vivo detected with a recombinant plasmid identified as coding for an adult fast myosin heavy chain from mouse skeletal muscle. J. Biol. Chem. 258, 13867-13874. [PubMed] [Google Scholar]
- Whalen R. G., Sell S. M., Butler-Browne G. S., Schwartz K., Bouveret P. and Pinset-Härstöm I. (1981). Three myosin heavy-chain isozymes appear sequentially in rat muscle development. Nature 292, 805-809. 10.1038/292805a0 [DOI] [PubMed] [Google Scholar]
- Wieczorek D. F., Periasamy M., Butler-Browne G. S., Whalen R. G. and Nadal-Ginard B. (1985). Co-expression of multiple myosin heavy chain genes, in addition to a tissue-specific one, in extraocular musculature. J. Cell Biol. 101, 618-629. 10.1083/jcb.101.2.618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt S. H., Granzier H., Witt C. C. and Labeit S. (2005). MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 350, 713-722. 10.1016/j.jmb.2005.05.021 [DOI] [PubMed] [Google Scholar]
- Wu S., Ying G., Wu Q. and Capecchi M. R. (2008). A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat. Protoc. 3, 1056-1076. 10.1038/nprot.2008.70 [DOI] [PubMed] [Google Scholar]
- Yoshida N., Yoshida S., Koishi K., Masuda K. and Nabeshima Y. (1998). Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J. Cell Sci. 111, 769-779. [DOI] [PubMed] [Google Scholar]
- Zammit P. S., Cohen A., Buckingham M. E. and Kelly R. G. (2008). Integration of embryonic and fetal skeletal myogenic programs at the myosin light chain 1f/3f locus. Dev. Biol. 313, 420-433. 10.1016/j.ydbio.2007.10.044 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.