

ABSTRACT
The Gsx2 homeodomain transcription factor promotes neural progenitor identity in the lateral ganglionic eminence (LGE), despite upregulating the neurogenic factor Ascl1. How this balance in maturation is maintained is unclear. Here, we show that Gsx2 and Ascl1 are co-expressed in subapical progenitors that have unique transcriptional signatures in LGE ventricular zone (VZ) cells. Moreover, whereas Ascl1 misexpression promotes neurogenesis in dorsal telencephalic progenitors, the co-expression of Gsx2 with Ascl1 inhibits neurogenesis. Using luciferase assays, we found that Gsx2 reduces the ability of Ascl1 to activate gene expression in a dose-dependent and DNA binding-independent manner. Furthermore, Gsx2 physically interacts with the basic helix-loop-helix (bHLH) domain of Ascl1, and DNA-binding assays demonstrated that this interaction interferes with the ability of Ascl1 to bind DNA. Finally, we modified a proximity ligation assay for tissue sections and found that Ascl1-Gsx2 interactions are enriched within LGE VZ progenitors, whereas Ascl1-Tcf3 (E-protein) interactions predominate in the subventricular zone. Thus, Gsx2 contributes to the balance between progenitor maintenance and neurogenesis by physically interacting with Ascl1, interfering with its DNA binding and limiting neurogenesis within LGE progenitors.
KEY WORDS: E-protein, Proximity ligation assay, Subapical progenitor, Tcf3, Telencephalon
Summary: This study demonstrates that Gsx2 physically interacts with the proneural factor Ascl1 in LGE progenitors and interferes with the DNA binding capacity of Ascl1, thus priming progenitors for neurogenesis while allowing for further expansion.
INTRODUCTION
Throughout evolution, the mammalian telencephalon, including both the cerebral cortex and the striatum (also known as the caudate-putamen), has enlarged dramatically, compared with more caudal central nervous system (CNS) regions. This expansion has been made possible, at least in part, through a major increase in the number of telencephalic progenitors during embryogenesis (reviewed by Wilsch-Bräuninger et al., 2016). Along the rostral-caudal axis of the CNS, most neural progenitors in the ventricular zone (VZ) divide at the ventricular (i.e. apical) surface and have thus been termed apical progenitors (APs) (Wilsch-Bräuninger et al., 2016; Fish et al., 2008). APs comprise radial glia that undergo a process called interkinetic nuclear migration, wherein the soma migrates to the apical surface to undergo M phase. The ventricular surface area thereby represents a limiting factor in this developmental process, and, thus, a secondary progenitor population has arisen in the telencephalon at the basal extent of the VZ, termed basal progenitors (BPs) (Smart, 1976; Fish et al., 2008). BPs comprise the subventricular zone (SVZ) and typically expand through one or two rounds of cellular divisions before undergoing a terminal symmetric division, thereby doubling neuronal output (Haubensak et al., 2004; Noctor et al., 2004). Recently, a new intermediate progenitor was identified in the VZ of the medial and lateral ganglionic eminences (MGE and LGE, respectively). These dividing cells, which possess aspects of radial glia, have been termed subapical progenitors (SAPs) and give rise to the BPs (Pilz et al., 2013). Hence, SAPs are akin to the neural progenitors of the outer SVZ of the cerebral cortex of higher mammals, including primates (Hansen et al., 2010; Fietz et al., 2010). Altogether, the APs and SAPs in the VZ and the BPs in the SVZ allow for significant neuronal expansion in the mammalian telencephalon from rodents to primates. However, the molecular mechanisms that regulate their generation and differentiation remain unclear.
The LGE gives rise to striatal projection neurons, which comprise the majority of neurons in the striatum, as well as olfactory bulb interneurons and a subset of amygdala interneurons called intercalated cells (ITCs) (Deacon et al., 1994; Olsson et al., 1995, 1998; Wichterle et al., 2001; Waclaw et al., 2010). These neuronal subtypes arise from two defined progenitor domains within the LGE; the dorsal (d)LGE produces olfactory bulb interneurons and ITCs, and the ventral (v)LGE generates striatal projection neurons (Yun et al., 2001; Stenman et al., 2003; Waclaw et al., 2010). The homeodomain protein Gsx2 (also known as Gsh2) is expressed by neural progenitors of the LGE (Szucsik et al., 1997; Toresson et al., 2000) with high levels in dLGE progenitors, and moderate levels defining those in the vLGE (Yun et al., 2001). Genetic lineage studies have shown that Gsx2-expressing LGE progenitors ultimately give rise to both neurons and glia (Kessaris et al., 2006; Fogarty et al., 2007; Qin et al., 2016). Accordingly, analysis of Gsx2 mutants revealed an essential role for Gsx2 in generating the above-mentioned LGE-derived neuronal subtypes (Toresson et al., 2000; Corbin et al., 2000; Yun et al., 2001; Waclaw et al., 2009, 2010; Kuerbitz et al., 2018). Gsx2 regulates the generation of these neuronal subtypes through the upregulation of the proneural basic helix-loop-helix (bHLH) factor Ascl1 (Toresson et al., 2000; Corbin et al., 2000; Yun et al., 2001; Waclaw et al., 2009; Wang et al., 2009). In fact, some Gsx2+ LGE progenitors co-express Ascl1 (Yun et al., 2003; Wang et al., 2013). However, previous studies have indicated that sustained Gsx2 expression limits the ability of LGE (Pei et al., 2011; Méndez-Gómez and Vicario-Abejón, 2012) and postnatal SVZ cells (López-Juárez et al., 2013) to differentiate. Furthermore, Gsx2 mutant mice, which exhibit reduced Ascl1 levels, have LGE progenitors that precociously generate oligodendrocyte precursor cells (Chapman et al., 2013). Thus, Gsx2 primes LGE progenitors for neurogenesis by upregulating Ascl1, but these cells remain as undifferentiated progenitors that undergo further expansion until Gsx2 expression is downregulated (Pei et al., 2011). In this study, we describe how molecular interactions between Gsx2 and Ascl1 within LGE progenitors impact the choice between progenitor expansion versus neurogenesis.
RESULTS
Gsx2 and Ascl1 define the progression of LGE progenitor maturation
Previous studies revealed that Gsx2 is upstream of the neurogenic factor Ascl1 in LGE VZ progenitors (Toresson et al., 2000; Corbin et al., 2000; Yun et al., 2001; Waclaw et al., 2009; Wang et al., 2009), thus specifying a neurogenic potential within this population. Moreover, a portion of Gsx2-expressing LGE progenitors co-express Ascl1 (Yun et al., 2003; Wang et al., 2013), which suggests that progenitors progress from Gsx2-only VZ cells to Ascl1-only VZ/SVZ cells via a transitional state in which both factors are co-expressed. To better define this progenitor progression, we examined Gsx2 and Ascl1 protein expression within LGE progenitors between embryonic day (E) 11.5 and E15.5, which represents the early neurogenic period when secondary progenitors (e.g. BPs) are established (Bhide, 1996; Pilz et al., 2013). Gsx2/Ascl1 double-labeled cells occurred in a ‘salt and pepper’ fashion throughout the LGE VZ but were generally absent in the most apical cells (Fig. 1A-C; Fig. S1). At early stages of neurogenesis, Ascl1+ cells comprise only about one-third of Gsx2+ LGE VZ cells; however, the vast majority of Ascl1+ cells co-express Gsx2 (Fig. 1A-C,H,I; Fig. S1A). The proportion of double-labeled cells in the vLGE, which contributes to striatal neurogenesis extensively between E11.5 and E15.5 (Waclaw et al., 2009; Kelly et al., 2018), was dynamic. Indeed, the number of Gsx2+Ascl1+ double-labeled cells as a percentage of Gsx2+ cells in the VZ peaked at E13.5 (Fig. 1H; Fig. S1A-C), which correlates with the establishment of the proliferative SVZ (Bhide, 1996). Moreover, by E15.5, the percentage of double-labeled vLGE cells as a proportion of Ascl1+ cells, fell to approximately half of that seen at E11.5 (Fig. 1I), resulting in more Ascl1+-only cells (likely BPs) during peak striatal neurogenesis. In contrast, the proportion of double-labeled cells as a ratio of either Gsx2+ or Ascl1+ did not change substantially over time in the dLGE (Fig. 1H,I; Fig. S1A-C), where robust neurogenesis occurs after E15.5 (Hinds, 1968; Tucker et al., 2006; Waclaw et al., 2006; López-Juárez et al., 2013).
Fig. 1.

Gsx2 and Ascl1 co-expression marks LGE subapical progenitors (SAPs). (A-G) Triple immunohistochemistry for Gsx2 (A,C,D,G), Ascl1 (B,C,E,G) and phosphohistone 3 (PH3) (C,F,G) in the E12.5 LGE. Box in C has been rotated 90° counterclockwise for the images in D-G. Note that most APs (i.e. PH3+ cells at the apical surface indicated by dotted lines in A-C or positioned at the top of D-G) express low or undetectable levels of either Gsx2 or Ascl1 (C,G). In contrast, PH3+ cells at abventricular positions (i.e. SAPs) within the VZ frequently colocalize Gsx2 and Ascl1 (D-G). (H,I) Quantification of Gsx2+Ascl1+ co-expressing cells in the dLGE (white bars) versus vLGE (black bars) as either a ratio of the Gsx2+ (H) or Ascl1+ (I) cells from the embryonic stages shown in Fig. S1. Data shown in H and I represent the mean±s.d. (n=3). Note, a small but significant (P<0.05) difference was detected in the Gsx2+Ascl1+/Ascl1+ cells of the dLGE at E13.5 (H). One-way ANOVA was performed between the dLGE or the vLGE data at each embryonic stage with a Tukey's HSD post-hoc. *P<0.05, **P<0.01 and ***P<0.001. Scale bars: 50 µm (A-C); 20 µm (D-G).
To examine how the dynamic expression of Gsx2 and Ascl1 correlates with LGE progenitor subtypes (i.e. APs, SAPs and BPs), we triple-labeled with phosphohistone 3 (PH3) to identify dividing progenitors in M phase. Most PH3-positive M-phase cells lined the ventricular (i.e. apical) surface, and thus represent APs (Fig. 1C). These apical dividing cells typically did not express, or only expressed low levels of, Gsx2 and, in rare instances, they expressed low levels of Ascl1 (Fig. 1D-G). In contrast, the vast majority of subapical PH3+ LGE cells were found to co-express Ascl1 and Gsx2 (Fig. 1D-G, arrows). The Ascl1+ cells that divide at abventricular positions within the VZ were previously described as a unique progenitor population within the VZ, i.e. SAPs (Pilz et al., 2013). SAPs are dependent on Ascl1 for both their appearance in the LGE VZ and the normal generation of proliferative progenitors in the SVZ (i.e. BPs) (Castro et al., 2011; Pilz et al., 2013). Thus, Gsx2 and Ascl1 co-expression appears to be a defining feature of LGE SAPs and together these factors might regulate the expansion potential of these intermediate progenitors.
To characterize gene expression in each progenitor subtype, we performed single cell RNA-seq (scRNA-seq) studies on the mouse E12.5 ventral telencephalon (including MGE, LGE and septum) using the 10x Genomics platform. A total of 29,873 cells (from two independent rounds with 15,376 and 14,497 cells) were recovered with a median of approximately 3000 genes/cell. Unsupervised cell clustering identified nine distinct cell groups, including neural progenitors and different neuronal subtypes derived from the ventral telencephalon (Fig. 2A; Table S1). To analyze Gsx2- and Ascl1-expressing progenitors in the LGE more closely, we focused on cell groups corresponding to VZ and SVZ progenitors (groups 0 and 2, respectively, in Fig. 2A and Table S1) after excluding cells expressing the MGE gene Nkx2.1 or the septal gene Zic1, leaving a total of 5364 cells. We classified these cells into four groups: Gsx2+ only (411 cells), Gsx2+Ascl1+ (1506 cells) double positive, Ascl1+ only (2182 cells) and a double-negative cell group (1265 cells). Although these groups were similar to each other at the overall transcriptome level, we observed gene enrichment that differed notably among the groups (Table 1; Table S2). The Gsx2+ group showed genes that characterize VZ progenitors, such as Fabp7 (Blbp) and Slc1a3 (Glast), whereas the double-expressing group showed genes that typically mark SVZ cells, such as Dlx1/2 (Table 1). Interestingly, the Ascl1+ cell group was the only group with enrichment of neuronal genes, such as Tubb3 (βΙΙΙ tubulin), Gad2 (Gad65) and Elavl3 (Table 1) despite the fact that they are still progenitors. Accordingly, feature plots show that Gsx2+-only cells are largely confined to the Slc1a3+ VZ (i.e. radial glia) group (cluster 0; Fig. 2B,E). By contrast, Gsx2+Ascl1+ and Ascl1+ cells are distributed through both VZ and SVZ clusters, correlating well with cells expressing the SVZ gene Dlx1 (Fig. 2C,F, clusters 0 and 2). Moreover, Ascl1+-only cells showed the highest correlation with progenitors expressing neurogenic genes, such as Dcx (Fig. 2D,G). These data support the progressive maturation of LGE progenitors from Gsx2+ radial glia to Gsx2+Ascl1+ SAPs and finally to Ascl1+ neurogenic BPs.
Fig. 2.

Single cell transcriptome analysis of Gsx2+ and Ascl1+ progenitors show progressive maturation of LGE progenitors. (A) UMAP plot of distinct cell types identified from E12.5 ventral telencephalon cells. Clusters with characteristics of VZ and SVZ progenitors (i.e. clusters 0 and 2, respectively) are outlined and labeled. (B-D) Feature plots showing cells expressing the VZ marker Slc1a3 (Glast) (B), the SVZ-enriched Dlx1 (C) and the neuronal marker Dcx (D). (E-G) Feature plots showing Gsx2+ only (E), Gsx2+Ascl1+ (F) and Ascl1+ only (G) LGE cells within the progenitor compartments. Note that Gsx2+-only cells correlate well with those expressing the VZ radial glial marker Slc1a3 (Glast) (B,E), whereas the double-labeled cells and Ascl1+-only cells correlate best with the SVZ marker Dlx1 (C,F,G). Moreover, the Ascl1+-only cells correlate best with the neurogenic (i.e. Dcx+) cells (D,G).
Table 1.
Genes enriched in Gsx2+ and/or Ascl1+ progenitors

Gsx2 expression overrides Ascl1-induced neurogenesis in the mouse telencephalon
To better understand the impact of Gsx2 and Ascl1 co-expression in telencephalic progenitors, we used a mouse Foxg1tTA transgenic system to misexpress each factor individually or together, in the dorsal telencephalon (Hanashima et al., 2002; Waclaw et al., 2009; Ueki et al., 2015). We previously used this system to show that Gsx2 misexpression ventralizes dorsal telencephalic progenitors and induces an LGE fate, including the upregulation of Ascl1 (Waclaw et al., 2009; Chapman et al., 2013). Moreover, Gsx2 misexpression maintained telencephalic cells in a neural progenitor state with a concomitant reduction in neurogenesis (Pei et al., 2011).
To determine the impact of Ascl1 misexpression on dorsal telencephalic progenitors, we analyzed E12.5 Foxg1tTA; tet-O-Ascl1 and Foxg1tTA control embryos for Gsx2 and Ascl1, as well as neuronal differentiation markers. As expected, Foxg1tTA; tet-O-Ascl1 embryos expressed Ascl1 throughout the dorsal-ventral extent of the telencephalon with no change in Gsx2 expression, which stops at the pallio-subpallial boundary (Fig. 3A,B, arrowheads). A previous study by Fode et al. (2000) showed that ectopic Ascl1 expression in dorsal telencephalic progenitors drives ventral (i.e. LGE) identity, similar to the effects of Gsx2 misexpression (Waclaw et al., 2009), along with the specification of GABAergic neuronal phenotypes. Using this misexpression system, we found that ectopic Ascl1 induced a significant increase in neurogenesis in the dorsal telencephalon as marked by β-III-tubulin (Tubb3) and doublecortin (Dcx) (Fig. 3F,I,J), compared with controls (Fig. 3E,I,J). Because the dense staining for these immature neuron markers made cellular analysis difficult, we measured the thickness of the pallial region expressing Tubb3 and Dcx (Fig. 3E-H, short bars) as a ratio of total pallial thickness (Fig. 3E-H, longer bars). Consistent with our previous publication (Pei et al., 2011), we found that Gsx2 misexpression, despite upregulating Ascl1 (Fig. 3D), resulted in reduced Tubb3 and Dcx expression in the dorsal telencephalon (Fig. 3H-J). Not only was the ratio of the Tubb3/Dcx-positive staining domain to total pallial thickness significantly reduced relative to Ascl1 misexpressors, the amount of staining for either neuronal marker was greatly diminished compared with control embryos (compare Fig. 3H with Fig. 3E,F). As the levels of Gsx2 are likely to be significantly higher than the amount of upregulated Ascl1 in Foxg1tTA; tet-O-Gsx2 embryos, the imbalance could favor neural progenitor maintenance over neurogenesis. To test this idea, we generated Foxg1tTA; tet-O-Ascl1; tet-O-Gsx2 embryos to simultaneously express both factors in dorsal telencephalic progenitors (Fig. 3C) and, similar to Gsx2 misexpressing embryos, neurogenesis was reduced in embryos misexpressing both Gsx2 and Ascl1 compared with the Ascl1 misexpressors (Fig. 3F,G-J). Thus, co-expression of Gsx2 and Ascl1 in dorsal progenitors limits Ascl1-driven neurogenesis, consistent with Gsx2/Ascl1 co-expression in LGE SAPs, allowing for progenitor expansion at the expense of neurogenesis.
Fig. 3.

Gsx2 inhibits Ascl1-driven neurogenesis in a transgenic misexpression assay. (A-H) Coronal sections through the telencephalon of E12.5 control (i.e. Foxg1tTA) (A,E), Ascl1-misexpressing (i.e. Foxg1tTA; tetO-Ascl1) (B,F), Gsx2- and Ascl1-misexpressing (i.e. Foxg1tTA; tetO-Ascl1; tetO-Gsx2) (C,G) and Gsx2-misexpressing (i.e. Foxg1tTA; tetO-Gsx2) (D,H) embryos. (B) Misexpression of Ascl1 throughout the telencephalon did not alter Gsx2 expression, stopping at the pallio-subpallial boundary (indicated by arrowheads) as in controls (A). Misexpression of Ascl1 within the dorsal telencephalon did, however, lead to an increase in Tubb3 and Dcx staining (compare F with E). Insets in C and D represent high power views of the dorsal telencephalon VZ showing broad co-expression of Gsx2 and Ascl1. E and F were counterstained with DAPI. Misexpression of Gsx2 alone upregulated Ascl1 throughout the telencephalon (D) and reduced neurogenesis (H), which was similar to misexpression of both Gsx2 and Ascl1 (C,G). (I,J) Quantification was carried out by measuring the Tubb3/Dcx-positive cortical staining (smaller white bar) and represented as a ratio of the total pallial wall (larger white bar). Data presented in I (Tubb3) and J (Dcx) represent mean±s.d. for each genotype (C, control, n=3; A, Ascl1 misexpression, n=3; D, Gsx2 and Ascl1 misexpression, n=3; G, Gsx2 misexpression, n=3). One-way ANOVA was performed between the data from C (Control), A (Ascl1-misexpression), D (double-misexpression) and G (Gsx2-misexpression) embryos with a Tukey's HSD post hoc. *P<0.05 as compared to C and †P<0.01 as compared with A misexpressing embryos. Scale bars: 200 µm [A-D; insets: 10 µm (C,D)]; 100 µm (E-H).
Gsx2 inhibits Ascl1-mediated gene expression in a dose-dependent manner
Ascl1, like other bHLH transcription factors, activates gene expression by binding as a dimer to palindromic DNA sequences referred to as E-boxes (consensus CANNTG sequence) (Johnson et al., 1992; Henke et al., 2009). Homodimeric or heterodimeric complexes can bind these targets, but for class II tissue-specific bHLHs such as Ascl1, heterodimerization with more widely expressed class I bHLHs, termed E-proteins, is often important for target activation (Massari and Murre, 2000; Henke et al., 2009). To examine whether Gsx2 interferes with Ascl1-mediated gene expression, we developed an in vitro luciferase assay in Drosophila S2 cells. We utilized Drosophila S2 cells to eliminate the confounding effects of Gsx2 upregulation of endogenous Ascl1 in mammalian cell lines. In this assay, we used a portion of the Drosophila achaete promoter (Jafar-Nejad et al., 2003), which contains three distinct E-box binding sites fused to a luciferase cassette, termed Ac-Luc (Fig. 4A). This construct shows low basal activity and, when Ascl1 alone is added, no significant increase in luciferase was detected (Fig. 4B). The addition of Daughterless (i.e. Drosophila E-protein) alone resulted in a modest increase (approximately fivefold) in luciferase activity, whereas co-expression of Ascl1 and E-protein resulted in a ∼33-fold increase in luciferase expression (Fig. 4B). Titrating in increasing levels of Gsx2 resulted in a stepwise reduction of luciferase expression (Fig. 4B), supporting the notion that Gsx2 limits the transcriptional activity of Ascl1. In contrast, Gsx2 failed to alter the ability of Daughterless homodimers to activate luciferase in this cell-based assay (Fig. 4C). However, as Gsx2 is known to have repressor functions (Winterbottom et al., 2010, 2011), it is possible that Gsx2 directly binds DNA to repress the achaete promoter. To test this idea, we mutated the asparagine (N) at amino acid position 253 to an alanine (A) in the Gsx2 homeodomain (Gsx2N253A), which abrogates DNA binding (Fig. 4D-F). Importantly, we found that titrating in increasing levels of Gsx2N253A also resulted in a significant reduction in luciferase activity, with the exception of the lowest concentration tested (Fig. 4B). Furthermore, we observed similar results when we repeated this analysis in mammalian mK4 cells (Valerius et al., 2002) using a reporter containing six multimerized E2-boxes (CAGCTG) (Fig. 4G). In this case, Ascl1 was sufficient to activate gene expression in the absence of exogenous E-protein, and titrating in increasing levels of either Gsx2 or Gsx2N253A resulted in a significant reduction of luciferase activity (Fig. 4G). These findings suggest that the ability of Gsx2 to reduce Ascl1-mediated gene expression is largely independent of Gsx2 DNA binding.
Fig. 4.

Gsx2 interferes with Ascl1-mediated reporter activation independent of its ability to bind DNA. (A) Schematic of the luciferase reporter construct used in B and C. The promoter of the Drosophila acheate gene contains three E-box sequences that can be bound by Ascl1 (Fig. S2). (B) Luciferase assay in S2 cells using the Ac-Luc reporter cotransfected with the indicated amounts of Ascl1, Drosophila E-protein (Daughterless) and Gsx2. Values represent fold activation over the Ac-Luc reporter added alone. Effects of co-transfecting an empty pAC5.1 expression vector, Gsx2 wild type, and Gsx2 DNA binding mutant (N253A) are shown. (C) 100 ng of Ac-Luc reporter was co-transfected with the indicated amount of Drosophila E-protein with 100 ng of wild-type Gsx2. Values represent fold activation over the Ac-Luc reporter added alone. (D) Schematic of the Gsx2 protein indicating the homeodomain and the position of the amino acid mutated to disrupt DNA binding. (E,F) Equimolar amounts of Gsx2 (E) and Gsx2N253A (F) were added to probes containing a predicted high affinity Gsx2 binding site. Note the complete loss of DNA binding with the Gsx2N253A protein. (G) Schematic of the Luciferase reporter construct containing six copies of an E-box with the sequence CAGCTG. This 6xE2box reporter (5 ng) was co-transfected into the mouse mK4 cell line with 25 ng Ascl1 and the indicated amount of Gsx2. Values represent fold activation over reporter alone. For luciferase assays, all conditions were performed in triplicate and normalized to a Renilla luciferase transfection control. Data represent means±s.d. In B and G, a one-way ANOVA was performed between vector, Gsx2 and Gsx2N253A with a Tukey's HSD post-hoc test. * indicates significant difference (P<0.01) between transfection of empty vector and Gsx2 WT. ‡ indicates significant difference (P<0.01) between transfection of empty vector and Gsx2N253A. In C, an unpaired, two-tailed Student's t-test was performed between the results from vector and Gsx2 for each condition.
Gsx2 interacts with Ascl1 in LGE progenitors
The mechanism by which Gsx2 limits the activity of Ascl1 is unclear. One possibility is that Gsx2 might bind Ascl1 at the protein level and interfere with its function. To test this hypothesis, we performed a yeast two-hybrid interaction assay, using either Ascl1 or Olig2, another bHLH factor expressed in subsets of LGE progenitors (Takebayashi et al., 2000; Chapman et al., 2013), as prey and Gsx2 as bait. In this system, we found that Gsx2 robustly interacts with Ascl1 but not Olig2 (Fig. 5A,B). Moreover, consistent with the results from the luciferase assay showing that Gsx2 did not alter E-protein homodimer-mediated gene activation (Fig. 4C), we did not observe interactions between Gsx2 and Tcf3 (i.e. mouse E-protein) in the yeast two-hybrid assay (Fig. 5C). Furthermore, co-immunoprecipitation (co-IP) assays on lysates from E12.5 telencephalons, using antibodies specific for either Gsx2 or Ascl1, revealed that Gsx2 co-immunoprecipitates with Ascl1 and vice versa (Fig. 5D). Thus, these data suggest that Gsx2 and Ascl1 physically interact within LGE progenitors of the mouse embryonic telencephalon.
Fig. 5.
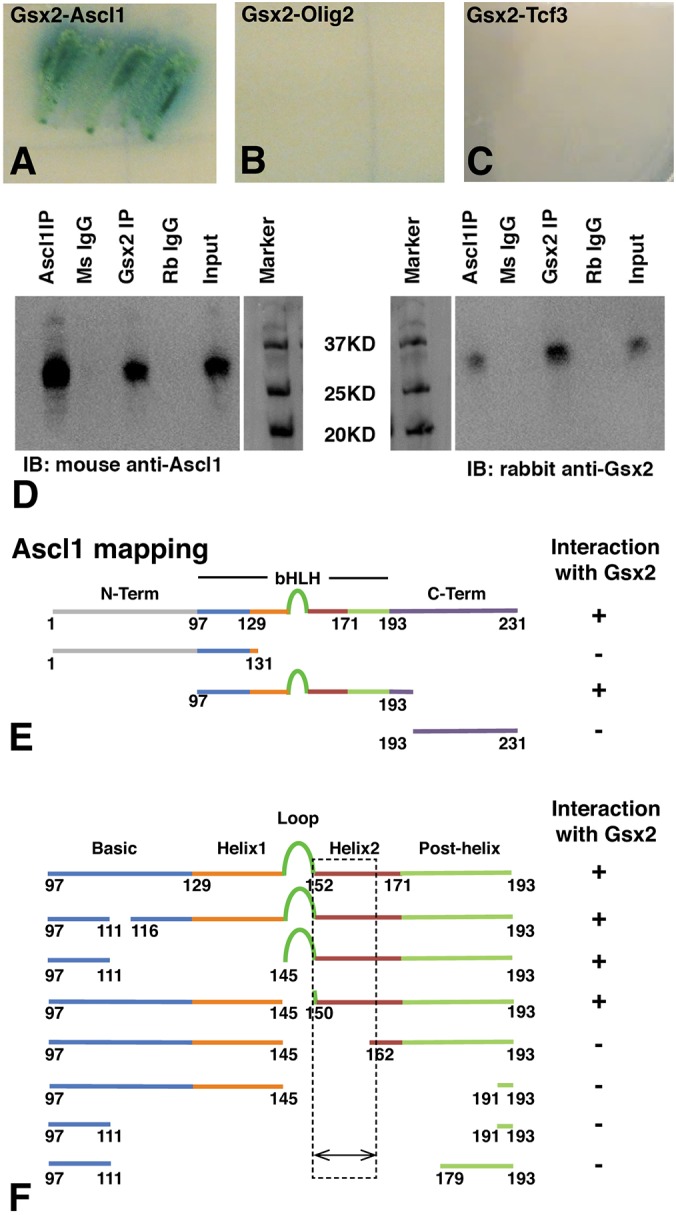
Gsx2 physically interacts with the bHLH domain of Ascl1 in the mouse telencephalon. (A-C) Yeast two-hybrid experiments using either Gsx2 as bait and Ascl1 (A), Olig2 (B), or Tcf3 as prey (C). Note robust reporter gene expression (i.e. α-gal) was only observed in the Gsx2-Ascl1 experiment (A). (D) Co-IP experiments using lysates from E12.5 mouse telencephalon. Pulling down Gsx2 with a rabbit antibody and blotting with a mouse Ascl1 antibody showed association of Ascl1 with Gsx2. Conversely, pulling down Ascl1 and blotting with a Gsx2 antibody showed association of these two proteins in the embryonic mouse telencephalon. Rabbit (Rb) IgG and mouse (Ms) IgG were used as controls for the Gsx2 and Ascl1 pull downs, respectively, and the input lane contained 10% input. (E) Using truncated portions of Ascl1 (e.g. N-Terminal, bHLH and C-Terminal) in a yeast two-hybrid assay, we found that only the bHLH domain of Ascl1 interacts with Gsx2. (F) Further deletion mapping studies using the yeast two-hybrid assay showed that the second helix (amino acids 150-162) of Ascl1 is required for interactions with Gsx2. See Fig. S2 for detailed amino acid deletions.
Gsx2 binds the second helix of the Ascl1 bHLH domain
As Gsx2 interferes with the neurogenic function of Ascl1, we hypothesized that Gsx2 might interact with a portion of Ascl1 necessary for DNA binding and/or forming transcription complexes. To test this possibility, we performed yeast two-hybrid assays with constructs that express the Ascl1 N-terminus (Ascl11-131), the bHLH domain (Ascl197-193), or the C terminus (Ascl1193-231), and found that only the bHLH domain interacted with Gsx2 (Fig. 5E). To further map the subdomain of Ascl1 that interacts with Gsx2, we used site-directed mutagenesis to create a series of small deletions (Fig. 5F). Within the bHLH, the basic region and first helix are necessary for DNA binding, whereas the second helix is involved in protein dimerization with other bHLH proteins, including itself (homodimers) and E-proteins, such as Tcf3 (heterodimers) (Johnson et al., 1992; Massari and Murre, 2000; Nakada et al., 2004; Henke et al., 2009). Interestingly, only mutations inside or spanning the second helix of Ascl1's bHLH domain disrupted Gsx2 binding (Fig. 5F). This result suggests that Gsx2 might compete with Ascl1 and/or E-proteins (e.g. Tcf3) in the formation of homodimers and heterodimers.
Gsx2:Ascl1 interactions inhibit Ascl1 homodimer and heterodimer complexes from binding DNA target E-boxes
As Gsx2 physically interacts with the same portion of Ascl1 that mediates dimer formation with other bHLH proteins, we hypothesized that Gsx2 interferes with the ability of Ascl1 to form homodimers or heterodimers with E-proteins on DNA. To test this idea, we used purified Gsx2, Ascl1 and Tcf3 (i.e. E-protein) in electromobility shift assays (EMSAs). To select an appropriate DNA probe, we tested the three E-box sequences from the Ac-Luc (Fig. 4A) for Ascl1-Ascl1 homodimer, Ascl1-Tcf3 heterodimer and Tcf3-Tcf3 homodimer binding (Fig. S3). From these studies, we found that the E2-box (Fig. 6A), which most closely matches the consensus Ascl1 binding motif (Castro et al., 2011), mediated the most robust formation of both Ascl1-Ascl1 and Ascl1-Tcf3 complexes (Fig. S3A-I). To examine the effect of Gsx2 on homodimer and heterodimer formation on the E2 site, we performed EMSAs with increasing concentrations of Gsx2. Initial experiments were performed with the Gsx2N253A protein, which is unable to bind DNA, but capable of inhibiting Ascl1-induced gene expression (Fig. 4B,F,G). Titrating in increasing levels of Gsx2N253A efficiently disrupted Ascl1 homodimer formation on the E2 sequence (Fig. 6B,C). As it is technically difficult to determine the relative concentrations of Ascl1 or Tcf3 within LGE progenitors, we performed EMSAs with Ascl1:Tcf3 ratios of 32:1, 1:2 and 1:12 (Fig. 6D,F,H). In EMSAs performed with a high ratio of Ascl1 relative to Tcf3 (32:1), only Ascl1-Ascl1 homodimers and Ascl1-Tcf3 heterodimers were detected and increasing the amount of Gsx2N253A preferentially diminished Ascl1 homodimer formation (Fig. 6D,E). At a 1:2 Ascl1:Tcf3 ratio, we predominantly detected Ascl1-Tcf3 heterodimers, with only weak Tcf3-Tcf3 homodimers observed (Fig. 6F, lane 16). Under these conditions, adding increasing amounts of Gsx2N253A did not significantly alter Ascl1-Tcf3 heterodimer formation (Fig. 6F,G). However, a slight increase in Tcf3 homodimer binding was observed (Fig. 6F,G), probably as a result of Gsx2 binding Ascl1, and thereby freeing increasing amounts of Tcf3 that can form homodimer complexes on DNA. EMSAs performed with a 1:12 Ascl1 to Tcf3 ratio and increasing amounts of Gsx2N253A, showed no effect on Tcf3 homodimer formation but a significant reduction of Ascl1-Tcf3 heterodimers (Fig. 6H,I). Finally, no effect was observed by increasing levels of Gsx2N253A on the formation of Tcf3 homodimers in the absence of Ascl1 (Fig. 6J,K), consistent with Gsx2 neither physically interacting with Tcf3 (Fig. 5C) nor inhibiting Tcf3 homodimer-induced luciferase activation (Fig. 4C). Notably, we observed similar disruption of Ascl1 DNA binding using wild-type Gsx2 protein (Fig. S4). Altogether, these data support a model in which Gsx2 physically interacts with the Ascl1 bHLH domain, and thereby interferes with the formation of Ascl1 homodimers and Ascl1-Tcf3 heterodimers on DNA. Hence, the inhibition of Ascl1-mediated gene expression by Gsx2 in luciferase assays (Fig. 4B,G) is likely to result from its ability to interfere with Ascl1 binding to E-box DNA sequences.
Fig. 6.

Gsx2 interferes with Ascl1 homodimer and heterodimer binding to an E-box DNA sequence. (A) Sequence of the E-box (highlighted in red) probe used in lanes 1-33 of EMSAs shown in B-K. (B) Gsx2 DNA binding mutant, Gsx2N253A, is titrated in increasing amounts from 0 to 80 pmoles in samples containing a constant 2.5 pmoles of Ascl1. (C) Percentage of probe bound by Ascl1-Ascl1 homodimers (red bars) in lanes 2-5. (D) Ascl1 (2.5 pmoles) and E-protein (0.08 pmoles) were added in a 32:1 ratio in each lane (9-13) with increasing levels of Gsx2N253A from 0 to 80 pmoles. (E) Percentage of probe bound by Ascl1-Ascl1 homodimers (red) and Ascl1-E47 heterodimers (yellow) in lanes 9-13. (F) Ascl1 (0.15 pmoles) and E-protein (0.3 pmoles) were added in a 1:2 ratio in each lane (16-20) with increasing levels of Gsx2N253A from 0 to 80 pmoles. (G) Percentage of probe bound by Ascl1-E47 heterodimers (yellow) and E47-E47 homodimers (green) in lanes 15-20. (H) Ascl1 (0.026 pmoles) and E-protein (0.31 pmoles) were added in a 1:12 ratio in each lane (25-28) with increasing levels of Gsx2N253A from 0 to 80 pmoles. (I) Percentage of probe bound by Ascl1-E47 heterodimers (yellow), and E47-E47 homodimers (green) in lanes 24-28. (J) E-protein (0.3 pmoles) was added to each lane (30-33) with increasing levels of Gsx2N253A from 0 to 80 pmoles. (K) Percentage of probe bound by E47-E47 homodimers (green) in lanes 30-33. Each EMSA was performed in triplicate, and data in C,E,G,I,K represent mean±s.d. with the intensity of bands representing each complex normalized to total probe intensity. Note, the y axes in C,E,G,I,K are different scales in order to accentuate the relative changes. An unpaired, two-tailed Student's t-test was performed between the no Gsx2N253A condition and the maximum Gsx2N253A condition, *P<0.05.
Gsx2 competes with the E-protein Tcf3 to bind to Ascl1
Ascl1 can homodimerize and heterodimerize with Tcf3 isoforms (e.g. E-proteins E12/E47) via interactions that require the second helix (Johnson et al., 1992; Nakada et al., 2004; Henke et al., 2009). This helix also interacts with Gsx2, so we assessed whether Gsx2 competes with Tcf3 to interact with Ascl1. Note that we did not observe interactions between Tcf3 and Gsx2 in a yeast two-hybrid assay (Fig. 5C). To investigate if Gsx2 competes with E-proteins to interact with Ascl1, we used a yeast three-hybrid system in which a third interfering protein is turned on or off using a methionine (met) inducible switch (Tirode et al., 1997). We made use of the ‘met off’ system to test the impact of either Tcf3 on Gsx2-Ascl1 interactions or Gsx2 on Ascl1-Tcf3 interactions. In the absence of Tcf3 (+met), Ascl1 (prey) interacted with Gsx2 (bait) (Fig. 7A). In the presence of Tcf3 (−met), however, the Ascl1-Gsx2 interaction was largely abrogated (Fig. 7B). Thus, a typical Ascl1-interacting partner, Tcf3, can interfere with Gsx2-Ascl1 interactions. Interestingly, the converse experiment using Tcf3 as bait and Ascl1 as prey, showed that Gsx2 is also capable of abrogating Ascl1-Tcf3 interactions (Fig. 7C,D). Hence, these data are consistent with Ascl1 interacting with either Gsx2 or Tcf3 but not both at the same time.
Fig. 7.

Gsx2 and Tcf3 compete for molecular interactions with Ascl1 in a yeast three-hybrid assay. The yeast three-hybrid assay utilizes an interfering protein that is capable of interacting with the bait or the prey to disrupt their interaction. (A) Using Gsx2 as bait and Ascl1 as prey, with no interfering protein, results in reporter (α-gal) expression, whereas the addition of Tcf3 (E-protein) as an interfering protein disrupts the interaction as shown in B. (C) Likewise, with Tcf3 as bait and Ascl1 as prey, and no interfering protein, α-gal expression is activated, which is disrupted by the addition of Gsx2 as the interfering protein as seen in D.
Spatial localization of Gsx2-Ascl1 and Ascl1-Tcf3 interactions within the LGE
To further characterize Gsx2-Ascl1 protein interactions in vivo, we adapted a proximity ligation assay (PLA) protocol associated with cultured cells (Söderberg et al., 2006; Bagchi et al., 2015), for use with fixed mouse embryonic forebrain sections. PLA uses two secondary antibodies conjugated to short oligonucleotides, to which mutually complementary oligonucleotides are ligated in situ. If two proteins are adjacent to or bound to each other, and the antibodies are within 40 nm (Bagchi et al., 2015), the oligonucleotides prime a fluorescence-based polymerization reaction that results in repetitive loops using a rolling circle amplification model. We performed a PLA to detect Gsx2-Ascl1 interactions in E12.5 telencephalon and found that there is widespread signal throughout the LGE VZ (Fig. 8A), which appears as single fluorescent dots that are largely confined to the nucleus (Fig. 8B). Indeed, the spatial pattern of the PLA signal for Gsx2-Ascl1 interactions fits well with the double immunofluorescent staining for Gsx2 and Ascl1 (see Fig. 1C). To demonstrate PLA signal specificity, we performed a PLA for Gsx2 and Ascl1 in Gsx2 knockout mice and found no detectable signal (Fig. 8C). In addition, we utilized sections from embryos that misexpress either Gsx2 (Foxg1tTA; tetO-Gsx2) or Ascl1 (Foxg1tTA; tet-O-Ascl1). In embryos misexpressing Gsx2, we detected PLA signal in VZ progenitors throughout the dorsal-ventral aspect of the telencephalon (Fig. 8D). This finding is in accordance with the fact that Gsx2 upregulates Ascl1 in dorsal telencephalic progenitors (Waclaw et al., 2009) (see also Fig. 3D). In contrast, Foxg1tTA; tet-O-Ascl1 embryos only showed PLA signal in the ventral telencephalon (Fig. 8E), consistent with Gsx2 not being upregulated in the pallium of these embryos (Fig. 3B). Finally, we performed a PLA for Gsx2 and Ascl1 in sections from the Foxg1tTA; tet-O-Ascl1; tet-O-Gsx2 embryos and observed robust PLA signal within VZ progenitors throughout the dorsal-ventral aspect of the telencephalon (Fig. 8F). Taken together, our data demonstrate that Gsx2 and Ascl1 physically interact within the LGE VZ progenitors that co-express these two factors.
Fig. 8.

Proximity ligation assay (PLA) shows Ascl1-Gsx2 interactions and Ascl1-Tcf3 interactions in a distinct portion of the LGE germinal zone. (A) When PLA was performed using rabbit anti-Gsx2 and guinea pig anti-Ascl1 antibodies, a strong signal (magenta) was detected in the LGE and septal VZ. (B) High power magnification of the VZ region shows a punctate signal associated with the DAPI-stained nuclei. (C) The PLA signal between Gsx2 and Ascl1 antibodies was specific as no signal was detected in Gsx2 knockout (KO) tissue sections. (D) In Gsx2-misexpressing embryos, PLA signal is expanded throughout the telencephalon as is the case for Gsx2 and Ascl1 expression. (E) PLA signal is not expanded throughout the telencephalon in the Ascl1-misexpressing embryos (pallio-subpallial boundary indicated by arrow in E as Gsx2 is not upregulated outside of the ventral telencephalon (Fig. 2B). However, the PLA signal is intensified in the ventral telencephalon. (F) Misexpression of both Gsx2 and Ascl1 leads to increased PLA signal throughout the telencephalon. (G) Immunostaining for Tcf3 protein in the E12.5 telencephalon shows staining throughout the germinal zones including both the VZ and SVZ. (H) PLA using the goat anti-Tcf3 and guinea pig anti-Ascl1 antibodies shows signal in both the LGE VZ as well as the SVZ, with stronger signal in the latter region. The boundary between the VZ and SVZ is indicated by the dashed line in G and H. (I) Schematic model showing LGE progenitor subtypes, with SAPs co-expressing Gsx2 and Ascl1 thus limiting Ascl1's neurogenic function and allowing for progenitor expansion. Gsx2 expression is lost in BPs, allowing Ascl1:Tcf3 heterodimers to drive direct neurogenesis. Note that Gsx2 was observed in some APs (indicated by blue hatching) but not together with Ascl1. In this model, both APs and SAPs could undergo direct neurogenesis if Gsx2 was downregulated (indicated by thin arrows). Scale bars:100 µm (A); 10 µm (B); 200 µm (C); 100 µm (D-H). N, neuron.
Our results from the yeast three-hybrid assay raise the possibility that competition between Gsx2 and Tcf3 to interact with Ascl1 might occur in LGE cells. Indeed, immunostaining for Tcf3 in E12.5 telencephalon showed broad staining, with the highest signal confined to the germinal regions including the VZ, where Gsx2 and Ascl1 are co-expressed (Fig. 8G). Thus, there is extensive overlap between these transcription factors in the LGE, particularly in the VZ. To investigate Ascl1-Tcf3 interactions in the LGE, we performed PLA using Tcf3 and Ascl1 antibodies. Interestingly, a strong PLA signal was predominantly detected within the SVZ of the LGE, whereas a weaker signal was observed in the VZ (Fig. 8H). As Gsx2 is largely confined to the VZ, the reduced PLA signal for Ascl1-Tcf3 interactions suggests that Gsx2 competes with Tcf3 for Ascl1 interactions in the VZ. Taken together, our results suggest that Gsx2-Ascl1 interactions predominate in LGE VZ cells (i.e. SAPs) to maintain cells in a progenitor state, whereas proneurogenic Ascl1-Tcf3 interactions characterize SVZ cells (i.e. BPs) (Fig. 8I). This hypothesis is supported by our scRNA-seq results (Fig. 2 and Table 1), which show that neuronal gene expression (i.e. Dcx, Tubb3, Gad2 and Elavl3) is only enriched in the Ascl1+ cell group and not in the Gsx2+ or Gsx2+Ascl1+ groups.
DISCUSSION
In this study, we show that a subset of LGE VZ progenitors, namely the SAPs, co-express Gsx2 and Ascl1. Moreover, our scRNA-seq transcriptome analysis suggests that SAPs are positioned between the Gsx2+ APs (i.e. radial glia) and the neurogenic Ascl1+ BPs. Furthermore, we demonstrate that Gsx2 limits Ascl1-driven neurogenesis, at least in part, through an interaction with its bHLH domain, thereby limiting the ability of Ascl1 to form the homodimers and heterodimers that bind DNA and activate gene expression. Finally, using a PLA, we found that Gsx2-Ascl1 interactions occur predominantly in LGE VZ cells (presumably SAPs), whereas Ascl1-Tcf3 interactions largely occur in the BPs of the SVZ. Thus, interactions between Gsx2 and Ascl1 probably contribute to the expansion of LGE progenitors, particularly the SAPs, by delaying neurogenesis until Gsx2 is downregulated in the LGE SVZ BPs (Fig. 8I). Indeed, Ascl1+-only cells (i.e. BPs) uniquely showed enrichment for neuronal genes (e.g. Tubb3 and Gad2) that have been shown to have Ascl1 ChIP peaks nearby (Table 1; Castro et al., 2011). In this way, Gsx2 primes the neuronal potential of LGE progenitors by upregulating Ascl1 expression but also allows for progenitor (e.g. SAP) expansion to generate the proper number of neurons.
Gsx2 and Ascl1 co-expression within LGE SAPs is consistent with several previous findings in Gsx2 and Ascl1 mutants. First, Gsx2 null mutants lack a proliferative SVZ (i.e. BPs) in the LGE at early stages of neurogenesis, which recovers partially after the Gsx1 family member is upregulated in Gsx2 mutants (Toresson and Campbell, 2001). A similar loss of BPs was also reported in the septal region when Gsx2 was conditionally inactivated (Qin et al., 2017), which supports the notion that Gsx2 is required to generate secondary progenitors (e.g. SAPs and BPs) in the ventral telencephalon. Second, Ascl1 not only marks the LGE SAPs but is required for their generation as well as the production of BPs (Pilz et al., 2013). As Gsx2 is necessary for normal Ascl1 expression in LGE progenitors (Toresson et al., 2000; Corbin et al., 2000; Yun et al., 2001), both BPs (Toresson and Campbell, 2001) and SAPs would be predicted to be reduced in the Gsx2 mutant LGE. Altogether, these data show that co-expression of Gsx2 and Ascl1 marks the LGE SAPs and regulates their development into BPs (see Fig. 8I).
The vLGE gives rise to striatal projection neurons (Yun et al., 2001; Stenman et al., 2003) that are organized into two neurochemically and anatomically distinct compartments, termed the patch (also known as the striosome) and matrix (Graybiel and Ragsdale, 1978; Gerfen, 1992). The patch, which occupies only 15% of the striatum, is generated at early stages in the rodent (e.g. E10-E12 in the mouse), whereas the matrix occupies approximately 85% of the striatum and is populated at later stages (e.g. E13 and onward) (van der Kooy and Fishell, 1987; Johnston et al., 1990). This difference in timing and the larger size contribution of matrix projection neurons to the striatum correlates well with the appearance of Ascl1+ SAPs in the mouse vLGE. For example, SAPs appear in the mouse forebrain between E12 and E14 and represent nearly half of all dividing LGE VZ progenitors by E16 (Pilz et al., 2013). Moreover, consistent with the Ascl1 lineage undergoing an abrupt transition around E13, Ascl1-expressing progenitors exhibit an expanded capacity for proliferation at this time, an occurrence that can be predicted if they progress through the SAP to BP expansion route (Kelly et al., 2018). Thus, this change in the Ascl1 lineage correlates with the switch from generating the patch to matrix compartments of the striatum and suggests that to generate the large numbers of matrix neurons, expansion through SAPs and BPs is probably necessary.
The co-expression of Gsx2 and the neurogenic factor Ascl1 in LGE SAPs presents a challenge for these cells to maintain progenitor status. We previously showed that Gsx2 plays a role in maintaining progenitor identity within the LGE lineage (Pei et al., 2011). Hence, despite upregulating Ascl1, Gsx2-expressing progenitors do not undergo rapid neuronal differentiation. In this study, we confirmed this finding, and compared and contrasted the ability of Ascl1 to induce neurogenesis in cells that do not express Gsx2 (i.e. the dorsal telencephalon) versus those that co-express Ascl1 and Gsx2 (i.e. double misexpressing embryos). Our findings reveal that even when expressing high levels of both Gsx2 and Ascl1, dorsal telencephalic progenitors show a pronounced decrease in neurogenesis to a level similar to those misexpressing Gsx2 only. As with Ascl1, the Dlx genes require Gsx2 for their correct expression in LGE progenitors (Toresson et al., 2000; Corbin et al., 2000; Yun et al., 2001; Wang et al., 2013). Both Ascl1 and Dlx factors promote the differentiation of GABAergic phenotypes typical of LGE-derived neurons (Yun et al., 2002; Long et al., 2009a,b; Pla et al., 2018; Lindtner et al., 2019). Furthermore, loss of Gsx2 leads to the upregulation of the oligodendrocyte precursor marker Pdgfrα (Corbin et al., 2003) and concomitant precocious oligodendrocyte differentiation (Chapman et al., 2013, 2018). Thus, it appears that Gsx2 specifies a neurogenic potential in LGE progenitors by virtue of upregulating Ascl1 (and Dlx factors) but maintains the co-expressing cells as progenitors capable of expanding as SAPs and subsequently generating BPs.
Our data suggest that the mechanism by which Gsx2 limits Ascl1-driven neurogenesis in LGE progenitors is disruption of Ascl1 homodimer and heterodimer formation, and thereby decreasing DNA binding to E-box sequences. Formation of Ascl1-Ascl1 and Ascl1-E-protein dimers occurs via interactions between amino acids in the second helix of the bHLH, and is independent of DNA binding (Massari and Murre, 2000; Nakada et al., 2004). Remarkably, Gsx2 interacts with this portion of the bHLH in Ascl1. Thus, in cells that express Gsx2, Ascl1 and E-protein, such as the LGE SAPs, Gsx2 might limit the neurogenic capacity of Ascl1. Indeed, our three-hybrid experiments support this notion. It is important to mention, however, that in addition to inhibiting Ascl1-Ascl1 and Ascl1-E-protein dimer binding to target DNA, it is possible that Gsx2-Ascl1 complexes bind novel DNA sequences that contribute to progenitor maintenance. Unlike the case for Ascl1, Gsx2 target DNA sequence binding was not disrupted by titrating in higher amounts of Ascl1 (Fig. S5). This finding suggests that the region of Gsx2 that interacts with Ascl1 is outside of the homeodomain. So far, we do not know the specific residues of Gsx2 that interact with Ascl1, nor do we know whether Ascl1 binding to Gsx2 affects other potential Gsx2 partners or leads to unique target gene selection (e.g. progenitor genes over neuronal genes). Further studies will be needed to address these questions.
Although Ascl1 is a well-known neurogenic factor, it has also been implicated in progenitor maintenance and proliferation. In fact, Imayoshi et al. (2013) showed that Ascl1 mRNA and protein levels oscillate at moderate levels in neural progenitors, and only when Ascl1 expression levels increase and become sustained does it drive neuronal differentiation. Thus, it might be that the interaction with Gsx2 limits the ability of the moderate oscillatory Ascl1 levels from regulating genes involved in neuronal differentiation. However, once Gsx2 expression is downregulated (e.g. in BPs), Ascl1 expression might stabilize and reach a sufficient level to activate gene expression required for neurogenesis. Alternatively, Ascl1 might utilize a Gsx2-independent mechanism to reach a sustained high level of expression in LGE progenitors. Upon reaching such a threshold, Ascl1 could overcome the inhibitory interactions with Gsx2. It also should be noted that Castro et al. (2011) identified two sets of Ascl1 gene targets: one group that contributes to neurogenesis and another that regulates progenitor proliferation. Our EMSA data (Fig. 6) using the ‘E2’ E-box sequence suggests that Ascl1-Ascl1 homodimers might be more sensitive to Gsx2 disruption than are Ascl1-Tcf3 heterodimers. Although it is unclear whether the homodimer versus heterodimer complexes have distinct preferred DNA-binding sites, and by extension, regulate genes differentially, it could be that Gsx2 co-expression favors Ascl1-Tcf3 over Ascl1-Ascl1 complexes in progenitor cells, aiding in the maintenance of progenitor identity.
Finally, we modified the PLA technique (Söderberg et al., 2006; Bagchi et al., 2015) to examine the LGE cells in which Gsx2-Ascl1 and/or Ascl1-E-protein interactions occur in tissue sections. This technology allows for the identification of cells in which two proteins are within 40 nm of each other and, thus, are probably in direct contact (Bagchi et al., 2015). The great advantage of this technology is that we can test for the regional localization of different partner proteins within complex tissues. Indeed, using this technique, we were able to detect Gsx2-Ascl1 interactions in LGE VZ cells, whereas Ascl1-E-protein interactions predominate in the LGE SVZ. These findings support the progenitor lineage model proposed in Fig. 8I, wherein Gsx2-Ascl1 interactions enriched in LGE SAPs limit the neurogenic potential of Ascl1 and thereby permit further expansion of neuronally specified progenitors, either as SAPs or as BPs. In contrast, because BPs lack Gsx2, Ascl1-E-protein or Ascl1-Ascl1 interactions could promote symmetric neurogenic divisions to generate neurons. Thus, the differential protein-protein interactions between Gsx2 and Ascl1, and between Ascl1 and Tcf3, within tissues provide a novel mechanism in which the choice of transcription factor partner ultimately dictates distinct cellular responses: Gsx2-Ascl1 interactions allow for continued progenitor expansion, whereas Ascl1-Tcf3 interactions promote cell cycle exit and subsequent neurogenesis.
MATERIALS AND METHODS
Animals
All experiments using mice were approved by the Institutional Animal Care and Use Committee (IACUC) of the Cincinnati Children's Hospital Research Foundation and were conducted in accordance with US National Institutes of Health guidelines. The previously described transgenic mice used included Foxg1tTA (Hanashima et al., 2002), tet-O-Gsx2 (Waclaw et al., 2009), tet-O-Ascl1 (Ueki et al., 2015) and Gsx2RA/+ (Waclaw et al., 2009). Maintenance and genotyping of animals and embryos was performed as described previously (Waclaw et al., 2009; Ueki et al., 2015). For misexpression studies, Foxg1tTA males were crossed with either tet-O-Gsx2, tet-O-Ascl1 or tet-O-Gsx2; tet-O-Ascl1 transgenic females. Additionally, Gsx2RA/+ mice were intercrossed to generate Gsx2RA/RA null embryos. The day of vaginal plug detection was deemed E0.5.
Immunohistochemistry
Embryos were fixed for 3 h (E11.5), 6 h (E12.5) or overnight (E15.5) at 4°C in 4% paraformaldehyde (PFA) then washed three times in PBS and cryopreserved in 30% sucrose. Embryos were sectioned coronally at 12 µm thickness on a cryostat and slides were stored at −20°C.
Immunohistochemistry on embryonic brain sections was performed as described previously (Waclaw et al., 2009). Primary antibodies were used at the following concentrations: guinea pig anti-Ascl1, 1:10,000 [provided by Jane Johnson, University of Texas Southwestern, Dallas, TX, USA (Kim et al., 2008)]; mouse anti-Ascl1, 1:500 (BD Pharmingen, 556604); guinea pig anti-Dcx, 1:3000 (Millipore, AB2253); rabbit anti-Gsx2, 1:3000 (Toresson et al., 2000); mouse anti-phosphohistone 3 (PH3), 1:500 (Cell Signaling, 9706S); goat anti-Tcf3, 1:500 (Abcam, ab59117); and rabbit anti-Tubb3, 1:1000 (Covance, PRB-435P). Secondary antibodies used were: donkey anti-rabbit conjugated to Alexa 594 or Alexa 647, 1:200 (Jackson ImmunoResearch, 711-586-152 or 711-606-152); donkey anti-guinea pig conjugated to Alexa 594 or Alexa 647, 1:200 (Jackson ImmunoResearch, 706-586-148 or 706-606-148); donkey anti-goat IgG conjugated to Alexa 594, 1:200 (Jackson ImmunoResearch, 705-586-147); and goat anti-mouse IgG1 conjugated to Alexa 568, 1:500 (Invitrogen, A21124). Because each tet-O transgene has an IRES-EGFP (Waclaw et al., 2009; Ueki et al., 2015), no Alexa 488-conjugated secondary antibodies were used on these sections. Slides were counterstained with DAPI for 10 min, coverslip-mounted with Fluoromount-G (SouthernBiotech, 0100-01) and dried overnight at room temperature. In certain cases, slides were coverslipped with Fluoromount-G containing DAPI (SouthernBiotech, 0100-20). Stained slides were imaged using confocal microscopy using a Nikon A1 LSM system with a GsAsP solid state laser.
Quantification of immunostainings
For quantification of Gsx2 and Ascl1 cellular expression in the embryonic LGE, a box spanning the Gsx2-expressing VZ was drawn in the dLGE and vLGE, respectively, and Gsx2+ single-, Ascl1+ single- and Gsx2+Ascl1+ double-labeled cells were manually counted. Four LGEs per embryo and three embryos at each time point were quantified. Statistical significance was determined by one-way ANOVA with the Tukey's honest significant difference (HSD) post-hoc test.
To quantify the effects of misexpressing Ascl1 alone, Gsx2 alone or Ascl1 and Gsx2 together on Dcx and Tubb3 expression in the dorsal telencephalon, immunostaining thickness (indicated by the smaller white bar in Fig. 3) was measured at ten locations spanning the dorsal pallium for each hemisphere as a ratio of the total mean pallial wall thickness at the same ten locations (indicated by the larger white bar in Fig. 3). Three embryos for each genotype were analyzed. Statistical significance was determined by one-way ANOVA with the Tukey's HSD post-hoc test.
scRNA-seq: barcoding, cDNA amplification, library construction and data matrix generation
Two separate rounds of dissections were performed on E12.5 CD1 embryos in which the ventral telencephalon (including the MGE, LGE and septum) was removed and dissociated into single cells, as described previously (Nagao et al., 2008). The single cell suspension was adjusted to 1000 cells/µl and ∼16,000 cells were loaded into a well on a 10x Chromium Single Cell instrument (10x Genomics). Barcoding, cDNA amplification and library construction were performed using the Chromium Single Cell 3′ Reagent Kits v. 3 according to manufacturer's instructions. Post cDNA amplification reaction and cleanup were performed using SPRIselect reagent (Beckman Coulter, B23318). Post cDNA amplification and library construction quality were analyzed using the Bioanalyzer High Sensitivity Kit (Agilent, 5067–4626). The final single cell 3′ library contains standard Illumina paired-end constructs (begin and end with P5 and P7 primer sequences, and 16 bp 10× Barcode, 10 bp UMI, Read one primer sequence, Read two primer sequence, and the 8 bp i7 sample index). Libraries were sequenced using an Illumina HiSeq 2500 and the paired-end 75 bp sequencing flow cell. Sequencing parameters used were: Read 1, 27 cycles; i7 index, 8 cycles; Read 2, 147 cycles, according to manufacturer's recommendations, which produced ∼300 million reads.
Initial analysis of the two data sets produced very similar results so they were merged as a single dataset, and potential doublets were removed using DoubletDecon (DePasquale et al., 2019). Initial cell filtering selected cells that expressed >1000 genes. Cells containing high percentages of mitochondrial (>20%) and hemoglobin genes (>0.025%) were also filtered out. Genes included in the analysis were expressed in a minimum of three cells. Only one read per cell was needed for a gene to be counted as expressed per cell. The resulting gene expression matrix was normalized to 10,000 molecules per cell and log transformed (Macosko et al., 2015). Cell-type clusters and marker genes for each cluster were identified using the R v. 3.6.1 library Seurat v. 3.1.0 (Butler et al., 2018; Stuart et al., 2019). All clustering was unsupervised, without driver genes. The influence of the number of unique molecular identifiers was minimized by regression within the ScaleData function. The top 1730 genes with highest variability among cells were used for principal components analysis. Cell clusters were determined by the Louvain algorithm. Dimension reduction was performed using the Python implementation of UMAP (Uniform Manifold Approximation and Projection) using the top 18 significant principal components determined by JackStraw plot. Marker genes were determined for each cluster using the Wilcoxon Rank Sum test within the FindAllMarkers function using genes expressed in a minimum of 10% of cells and a fold change threshold of 1.3. FeaturePlots were generated using all cells, but only showing expression of the gene of interest in cells that were in clusters 0 and 2 and not expressing Nkx2.1 and Zic1.
Plasmids, molecular cloning and site-directed mutagenesis
Details describing all plasmids generated for this study can be found in Table S3, and all primer sequences used for PCR-based cloning can be found in Table S4. The following plasmids were purchased and used in this study: the pGBKT7, pGADT7 and pBRIDGE yeast vectors (Clontech); the pET14b bacterial protein expression vector (Novagen); the pAc5.1 Drosophila expression vector (Invitrogen); and the pCDNA6 mammalian expression vector (Thermo Fisher Scientific). The pGL3-basic vector (Promega) was modified by destroying the existing BamHI site, then cloning a new BamHI site between KpnI and XhoI followed by cloning of the minimal promoter from the Drosophila Ac gene between BamHI and XhoI sites. The Ac-Luciferase and Da-pAc5.1 used for luciferase assays (Jafar-Nejad et al., 2003), and the E47 (isoform of Tcf3) amino acids 430-648 in pET14b used for bacterial protein expression (Zhang et al., 2019) were described previously. PCR reactions were performed using either CloneAmp Hi-fidelity polymerase mix (Clontech) or ACCUZYME DNA polymerase. Ligation reactions were performed using T4 DNA ligase or In-Fusion HD (Clontech). Site-directed mutagenesis was performed using inverse PCR followed by In-Fusion ligation. All DNA clones and mutations were verified by DNA sequencing.
Yeast two- and three-hybrid assays
Yeast two-hybrid assays were performed using the Matchmaker Gold yeast two-hybrid system (Clontech 630489), following the manufacturer's protocols with minor modifications. For Gsx2-Ascl1, Gsx2-Tcf3 and Gsx2-Olig2 interaction assays, a ‘Mate and Plate’ (Letteboer and Roepman, 2008) approach was used. Briefly, a Y2H-Gold strain of Saccharomyces cerevisiae (Sc) was freshly made competent by the lithium acetate method using the manufacturer's protocols, and the bait construct (Gsx2 in the pGBKT7 vector) was transformed by PEG/DMSO and heat shock, as described by the manufacturer's protocols. The Y187 strain of Sc was similarly made competent and transformed with prey constructs (Ascl1, Olig2 or Tcf3 in the pGADT7 vector). Positive colonies were obtained using single-dropout nutritional auxotrophy on SD-Trp and SD-Leu plates, respectively. Bait and prey colonies were mixed and co-cultured overnight in 2×YPDA, mating confirmed by microscopy, and two hybrids were selected on double dropout (DDO) plates (SD-Leu,-Trp). Bait-prey interactions were determined on DDO+Aerobasidin A+X-α-Gal plates. The prresence of blue colonies indicated positive interaction. The interactions were confirmed by patching the colonies in SD-Leu,−Trp, −His, −Ade, +ABA and +X-α−Gal plates.
To study interactions between full-length Gsx2 and Ascl1 deletion mutants, a ‘co-transformation’ approach was taken, as described previously (Chien et al., 1991). Briefly, both bait and prey constructs were co-transformed into the Y187 strain and two hybrids were selected on DDO plates. The rest of the assay was performed as described above.
Yeast three-hybrid assays were performed essentially as described previously (Tirode et al., 1997). A fresh competent Y187 strain of Sc was cotransformed with Ascl1 in the pGADT7 vector and either Gsx2-MCS1; E47 MCS2, E47 MCS1; Gsx2 MCS2, Gsx2 MCS1; or Tcf3 MCS1 constructs, as described above. Three hybrids were selected on DDO plates, bait-prey interaction determined on DDO/X/A plates and interference determined on SD-Trp, −Leu, −Met, XaGal, ABA or the same plates+Met. Interference was confirmed by patching colonies on SD-Trp, −Leu, −His, −Ade, X-α−Gal and ABA with or without Met.
Immunoprecipitation and western blots
Immunoprecipitations (IPs) were performed as described previously (Lin and Lai et al., 2017) with minor variations. Briefly, telencephalons were dissected from E12.5 mouse embryos in ice-cold PBS. Each telencephalon was homogenized on ice with a plastic pestle in a microcentrifuge tube in 400 µl of modified radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris (pH 7.4), 150 mM NaCl and 1% Triton X-100] supplemented with protease inhibitor cocktail (PIC) (Sigma-Aldrich, P8340). Lysates were centrifuged at 15,000 g for 10 min at 4°C, and the supernatant was collected in a fresh, prechilled tube. Next, 2 µl of rabbit anti-Gsx2 antibody (1:200, Toresson et al., 2000) or 5 µl of mouse anti-Ascl1 antibody (1:80, BD Pharmingen, 556604) was added and the tube was rotated end over end at 4°C overnight. A 10 µl slurry of Protein G agarose (Sigma-Aldrich, P7700) in ice-cold RIPA buffer was prepared, added to the lysate and incubated for a further 2 h. The beads were separated from the liquid by centrifugation at 5000 g for 5 min at 4°C and washed five times in ice cold RIPA buffer with PIC. The immunoprecipitated proteins were eluted in Glycine-HCl buffer (pH 3) and denatured for 10 min at 70°C in SDS loading buffer containing β-mercaptoethanol.
For western blot analysis, samples were separated on a 12% acrylamide gel using Tris-Glycine buffer. Gels were transferred to PVDF membranes with Tris/Glycine/Methanol transfer buffer for 2 h at 4°C in a Bio-Rad Protean wet transfer apparatus. Membranes were washed three times in TBS and blocked with 0.5% Casein (Sigma-Aldrich, C7087) for 1 h at room temperature. The membranes were then probed with primary antibodies [rabbit anti-Gsx2 (1:5000, Toresson et al., 2000) or mouse anti-Ascl1 (1:1000, BD Pharmingen, 556604)] in TBS+0.1% Tween 20 (TBST) overnight at 4°C with gentle rocking, washed three times in TBST, and subsequently incubated with 1:20,000 goat anti-rabbit (Thermo Fisher Scientific, 31460) or 1:10,000 goat anti-mouse (Bio-Rad, STAR207P) secondary antibodies at room temperature for 1 h. The membranes were then washed three times in TBST, once with TBS and overlaid with hydrogen peroxide+ECL (Pierce). The membranes were then imaged in a Bio-Rad imager.
Luciferase reporter assays
Drosophila S2 cells (Schneider, 1972) were ordered from the Drosophila Genomics Resource Center (S2-DGRC stock number: 6). For the S2 luciferase assays, 6×105 cells were cultured in 12-well plates in HyClone media (Fisher Scientific) for 24 h prior to transfection. Each well was transfected with a total of 0.3 μg of DNA (100 ng Ac-luciferase reporter, 2 ng copia Renilla and the indicated amount of expression constructs) using Effectene Transfection Reagent (Qiagen). Empty pAc5.1 was added to bring the total DNA to 0.3 μg per well. Cells were harvested 36 h after transfection. For mammalian mK4 cell (Valerius et al., 2002) luciferase assays, 4×104 cells were cultured in 48-well plates in DMEM with 10% fetal bovine serum for 24 h prior to transfection. Each well was transfected with 85 ng total DNA (5 ng 6xE2-Luciferase, 5 ng Renilla-Luciferase, and the indicated amount of expression constructs or empty pCDNA6 expression vector) using Effectene Transfection Reagent. Cells were harvested 48 h after transfection. All lysates were isolated and luciferase activity was measured via the Promega Dual Luciferase Assay kit. All firefly luciferase values were normalized to Renilla luciferase values to normalize for transfection efficiency, and each experiment was performed in triplicate. Results are reported as fold expression over the luciferase reporter alone.
Protein purification and DNA probe design
His-tagged proteins were purified from BL21 cells via nickel chromatography as described previously (Uhl et al., 2010). Fragments of the following Mus musculus proteins were used: Ascl1 amino acids 92-231 containing the bHLH domain, E47 (isoform of Tcf3 gene) amino acids 430-648 containing the bHLH domain, Gsx2 amino acids 167-305 containing the homeodomain, and Gsx2N253A amino acids 167-305 containing the mutated homeodomain. Protein concentrations were determined using the Bradford assay and confirmed by SDS-PAGE and GelCode Blue stain (Thermo Fisher Scientific). Probes were generated as described previously by annealing a 5′-IRDye 700-labeled oligo (IDT) to the oligos listed in the section below and filling in with Klenow (Uhl et al., 2016).
Electrophoretic mobility shift assays (EMSAs)
EMSAs were performed as previously described using native PAGE (Uhl et al., 2010, 2016). For EMSAs in Fig. S3, 0.04 pmoles of the indicated probe was added to 20 µl binding reactions. For EMSAs in Fig. 6 and Fig. S4, 0.68 pmoles of probe was added to each 20 µl binding reaction. To facilitate exchange between bHLH partners in experiments where Ascl1 and E47 were used, all binding reactions were incubated at 37°C for 30 min before addition of the probe. For binding reactions testing the ability of GsxN253A to alter Ascl1-Ascl1 or Ascl1-E47 binding, the Ascl1 and Gsx2N253A proteins were mixed first, incubated at 37°C for 30 min, and then E47 was added and samples were incubated at 37°C for an additional 30 min. Probes were added and samples incubated at room temperature for 10 min. For EMSAs in Fig. 6, the number of pmoles of protein added in each lane is indicated in Table S5. Quantification of EMSAs was performed using LI-COR Biosciences’ Image Studio software by calculating the total probe in each lane and the percentage of probe bound by either homodimer or heterodimer complexes. For EMSAs performed in Fig. S4B, 0.68 pmoles of Dll1 E1box probe was added to 20 µl binding reactions. Wild-type Gsx2 and Ascl1 were mixed first, then E47 was added, and the indicated probes were added last, followed by room temperature incubation for 10 min. For EMSAs performed in Fig. S4C, 0.04 pmoles of Ac-Luc E2 box probe was added to 20 µL binding reactions. All binding reactions were incubated at 37°C for 30 min before addition of the probe. Then samples were incubated at room temperature for 10 min. For EMSAs shown in Fig. S4, the amount of Ascl1, E47 and Gsx2 added is indicated in the figure legend. The following oligos were used for EMSAs (bold text indicates the E-box binding site): Ac-Luc E1: GTCACGCAGGTGGGATCCTAGTGCGGGCGTGGCT; Ac-Luc E2: CACCAACAGCTGCGTTTTTAGTGCGGGCGTGGCT; Ac-Luc E3: GACAGGCAGCTGAAAATGTAGTGCGGGCGTGGCT; Dll1 E1: AGAGAGCAGGTGCTGTAGTGCGGGCGTGGCT; Gsx2_site: CAGAGTGTAATTAACATTCAGTAGTGCGGGCGTGGCT.
Proximity ligation assay (PLA)
Duolink PLA kit components (MilliporeSigma, DUO92105) were used for performing PLA with a modified protocol. Positive (+ve) and negative (−ve) rabbit and goat PLA-conjugated antibodies were purchased from Sigma-Aldrich. For the guinea pig PLA antibody, the −ve PLA DNA tag was covalently attached to 100 µl of 1 mg/ml unconjugated donkey anti-guinea pig IgG Fab (Jackson ImmunoResearch, 706-005-148) using the Duolink in situ Probemaker MINUS kit (MilliporeSigma, DU092010) as per manufacturer's instructions. PFA-fixed cryostat sections (four per slide, one of each genotype) were dried immediately after cryosectioning and rehydrated in 1×PBS for 5 min. The tissue sections were then incubated for 20 min in 10% donkey serum (NDS) in PBS containing 0.1% Triton X-100, and incubated overnight with 1:2500 rabbit anti-Gsx2 and 1:5000 guinea pig anti-Ascl1 (for the Gsx2:Ascl1 PLA), or 1:500 goat anti-Tcf3 and 1:5000 guinea pig anti-Ascl1 (for the Ascl1:Tcf3 PLA), at room temperature in a humidified chamber. The slides were then washed twice in PBS containing 0.1% Triton X-100, once with PBS without detergent and once with 20 mM Tris (pH 8), 150 mM NaCl and 0.05% Tween 20. Individual sections were then encircled using a hydrophobic pen before adding 100 µl of secondary antibody mix containing 20 µl of anti-rabbit +ve conjugated antibody (Sigma-Aldrich, DUO92002) together with 5 µl −ve DNA conjugated anti-guinea pig antibody in dilution buffer containing 1% NDS, and incubated at room temperature for 2 h. Slides were washed twice in Olink buffer A (Millipore-Sigma) and probe-ligation was initiated by the addition of 0.5 µl ligase in 39.5 µl 1× ligase buffer, and incubation at 37°C for 30 min. The slides were then washed three times in Olink buffer A and the amplification reaction was initiated by adding 100 µl of polymerase to each section in 1× polymerase stock buffer followed by overnight incubation at 37°C in a humidified chamber. The following morning, the slides were washed three times in TBS and dipped in water. They were then coverslipped in Hoechst 33342 stain containing wet mounting medium and sealed with nail polish. PLA slides were imaged using an Olympus BX51 microscope with epifluorescence.
Supplementary Material
Acknowledgements
We thank Ron Waclaw and Jeff Kuerbitz for critical reading of the manuscript. We also thank Jane Johnson (UT Southwestern, Dallas, TX, USA) for providing the guinea pig anti-Ascl1 antibody and Hugo Bellen (Baylor College of Medicine, Houston, TX, USA) for providing the Ac-Luciferase and Daughterless expression plasmids.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.N., B.G., K.C.; Methodology: K.R., J.S., S.Q., B.C., M.A., S.S.P.; Formal analysis: K.R., J.S., S.Q., M.A., S.S.P.; Investigation: K.R., J.S., S.Q., B.C., M.A., S.S.P.; Resources: M.N.; Data curation: K.R., J.S., S.Q., M.A., S.S.P.; Writing - original draft: K.R., J.S., S.Q., B.G., K.C.; Writing - review & editing: K.R., J.S., S.Q., B.C., M.A., S.S.P., M.N., B.G., K.C.; Supervision: B.G., K.C.; Project administration: M.N., B.G., K.C.; Funding acquisition: M.N., B.G., K.C.
Funding
This work was supported by National Institutes of Health (R01 NS044080 to K.C. and B.G.; NS069893 to M.N. and K.C.) and a Chan Zuckerberg Initiative grant (2017-173968 to S.S.P.). Deposited in PMC for release after 12 months.
Data availability
scRNA-seq data have been submitted to GEO under accession number GSE142768.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.185348.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.185348.reviewer-comments.pdf
References
- Bagchi S., Fredriksson R. and Wallén-Mackenzie Å. (2015). In situ proximity ligation assay (PLA). Methods Mol. Biol. 1318, 149-159. 10.1007/978-1-4939-2742-5_15 [DOI] [PubMed] [Google Scholar]
- Berger M. F., Badis G., Gehrke A. R., Talukder S., Philippakis A. A., Peña-Castillo L., Alleyne T. M., Mnaimenh S., Botvinnik O. B., Chan E. T. et al. (2008). Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133, 1266-1276. 10.1016/j.cell.2008.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide P. G. (1996). Cell cycle kinetics in the embryonic mouse corpus striatum. J. Comp. Neurol. 374, 506-522. [DOI] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E. and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411-420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro D. S., Martynoga B., Parras C., Ramesh V., Pacary E., Johnston C., Drechsel D., Lebel-Potter M., Garcia L. G., Hunt C. et al. (2011). A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes. Dev. 25, 930-945. 10.1101/gad.627811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H., Waclaw R. R., Pei Z., Nakafuku M. and Campbell K. (2013). The homeobox gene Gsx2 controls the timing of oligodendroglial fate specification in mouse lateral ganglionic eminence progenitors. Development 140, 2289-2298. 10.1242/dev.091090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H., Riesenberg A., Ehrman L. A., Kohli V., Nardini D., Nakafuku M., Campbell K. and Waclaw R. R. (2018). Gsx transcription factors control neuronal versus glial specification in ventricular zone progenitors of the mouse lateral ganglionic eminence. Dev. Biol. 442, 115-126. 10.1016/j.ydbio.2018.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien C. T., Bartel P. L., Sternglanz R. and Fields S. (1991). The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci USA 88, 9578-9582. 10.1073/pnas.88.21.9578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin J. G., Gaiano N., Machold R. P., Langston A. and Fishell G. (2000). The Gsh2 homeodomain gene controls multiple aspects of telencephalic development. Development 127, 5007-5020. [DOI] [PubMed] [Google Scholar]
- Corbin J. G., Rutlin M., Gaiano N. and Fishell G. (2003). Combinatorial function of the homeodomain proteins Nkx2.1 and Gsh2 in ventral telencephalic patterning. Development 130, 4895-4906. 10.1242/dev.00717 [DOI] [PubMed] [Google Scholar]
- Deacon T. W., Pakzaban P. and Isacson O. (1994). The lateral ganglionic eminence is the origin of cells committed to striatal phenotypes: neural transplantation and developmental evidence. Brain Res. 668, 211-219. 10.1016/0006-8993(94)90526-6 [DOI] [PubMed] [Google Scholar]
- DePasquale E. A. K., Schnell D. J., Van Camp P.-J., Valiente-Alandí Í., Blaxall B. C., Grimes H. L., Singh H. and Salomonis N. (2019). DoubletDecon: deconvoluting doublets from single-cell RNA-sequencing data. Cell Rep. 29, 1718-1727.e8. 10.1016/j.celrep.2019.09.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fietz S. A., Kelava I., Vogt J., Wilsch-Bräuninger M., Stenzel D., Fish J. L., Corbeil D., Riehn A., Distler W., Nitsch R. et al. (2010). OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 13, 690-699. 10.1038/nn.2553 [DOI] [PubMed] [Google Scholar]
- Fish J. L., Dehay C., Kennedy H. and Huttner W. B. (2008). Making bigger brains-the evolution of neural-progenitor-cell division. J. Cell. Sci. 121, 2783-2793. 10.1242/jcs.023465 [DOI] [PubMed] [Google Scholar]
- Fode C., Ma Q., Casarosa S., Ang S. L., Anderson D. J. and Guillemot F. (2000). A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes. Dev. 14, 67-80. [PMC free article] [PubMed] [Google Scholar]
- Fogarty M., Grist M., Gelman D., Marín O., Pachnis V. and Kessaris N. (2007). Spatial genetic patterning of the embryonic neuroepithelium generates GABAergic interneuron diversity in the adult cortex. J. Neurosci. 27, 10935-10946. 10.1523/JNEUROSCI.1629-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen C. R. (1992). The neostriatal mosaic: multiple levels of compartmentalization in the basal ganglia. Annu. Rev. Neurosci. 15, 285-320. 10.1146/annurev.ne.15.030192.001441 [DOI] [PubMed] [Google Scholar]
- Graybiel A. M. and Ragsdale C. W. Jr. (1978). Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylcholinesterase staining. Proc. Natl. Acad. Sci. USA 75, 5723-5726. 10.1073/pnas.75.11.5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashima C., Shen L., Li S. C. and Lai E. (2002). Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J. Neurosci. 22, 6526-6536. 10.1523/JNEUROSCI.22-15-06526.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. V., Lui J. H., Parker P. R. and Kriegstein A. R. (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554-561. 10.1038/nature08845 [DOI] [PubMed] [Google Scholar]
- Haubensak W., Attardo A., Denk W. and Huttner W. B. (2004). Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc. Natl. Acad. Sci USA 101, 3196-3201. 10.1073/pnas.0308600100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke R. M., Meredith D. M., Borromeo M. D., Savage T. K. and Johnson J. E. (2009). Ascl1 and Neurog2 form novel complexes and regulate Delta-like3 (Dll3) expression in the neural tube. Dev. Biol. 328, 529-540. 10.1016/j.ydbio.2009.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds J. W. (1968). Autoradiographic study of histogenesis in the mouse olfactory bulb. I. Time of origin of neurons and neuroglia. J. Comp. Neurol. 134, 287-304. 10.1002/cne.901340304 [DOI] [PubMed] [Google Scholar]
- Imayoshi I., Isomura A., Harima Y., Kawaguchi K., Kori H., Miyachi H., Fujiwara T., Ishidate F. and Kageyama R. (2013). Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342, 1203-1208. 10.1126/science.1242366 [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H., Acar M., Nolo R., Lacin H., Pan H., Parkhurst S. M. and Bellen H. J. (2003). Senseless acts as a binary switch during sensory organ precursor selection. Genes. Dev. 17, 2966-2978. 10.1101/gad.1122403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. G., Gerfen C. R., Haber S. N. and van der Kooy D. (1990). Mechanisms of striatal pattern formation: conservation of mammalian compartmentalization. Brain Res. Dev. Brain Res. 57, 93-103. 10.1016/0165-3806(90)90189-6 [DOI] [PubMed] [Google Scholar]
- Johnson J. E., Birren S. J., Saito T. and Anderson D. J. (1992). DNA binding and transcriptional regulatory activity of mammalian achaete-scute homologous (MASH) proteins revealed by interaction with a muscle-specific enhancer. Proc. Natl. Acad. Sci. USA 89, 3596-3600. 10.1073/pnas.89.8.3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S. M., Raudales R., He M., Lee J. H., Kim Y., Gibb L. G., Wu P., Matho K., Osten P., Graybiel A. M. et al. (2018). Radial glial lineage progression and differential intermediate progenitor amplification underlie striatal compartments and circuit organization. Neuron 99, 345-361. 10.1016/j.neuron.2018.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N., Fogarty M., Iannarelli P., Grist M., Wegner M. and Richardson W. D. (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 9, 173-179. 10.1038/nn1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. J., Battiste J., Nakagawa Y. and Johnson J. E. (2008). Ascl1 (Mash1) lineage cells contribute to discrete cell populations in CNS architecture. Mol. Cell. Neurosci. 38, 595-606. 10.1016/j.mcn.2008.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuerbitz J., Arnett M., Ehrman S., Williams M. T., Vorhees C. V., Fosher S. E., Garratt A. N., Muglia L. J., Waclaw R. R. and Campbell K. (2018). Loss of intercalated cells (ITCs) in the mouse amygdala of Tshz1 mutants correlates with fear, depression, and social interaction phenotypes. J. Neurosci. 38, 1160-1177. 10.1523/JNEUROSCI.1412-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letteboer S. J. and Roepman R. (2008). Versatile screening for binary protein-protein interactions by yeast two-hybrid mating. Methods. Mol. Biol. 484, 145-159. 10.1007/978-1-59745-398-1_10 [DOI] [PubMed] [Google Scholar]
- Lin J.-S. and Lai E.-M. (2017). Protein-protein interactions: co-immunoprecipitation. Methods Mol. Biol. 1615, 211-219. 10.1007/978-1-4939-7033-9_17 [DOI] [PubMed] [Google Scholar]
- Lindtner S., Catta-Preta R., Tian H., Su-Feher L., Price D. J., Dickel D. E., Greiner V., Silberberg S. N., McKinsey G. L., McManus M. T. et al. (2019). Genomic resolution of DLX-orchestrated transcriptional circuits driving development of forebrain GABAergic neurons. Cell Rep. 28, 2048-2063. 10.1016/j.celrep.2019.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. E., Cobos I., Potter G. B. and Rubenstein J. L. R. (2009a). Dlx1&2 and Mash1 transcription factors control MGE and CGE patterning and differentiation through parallel and overlapping pathways. Cereb. Cortex 19 Suppl. 1, i96-i106. 10.1093/cercor/bhp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. E., Swan C., Liang W. S., Cobos I., Potter G. B. and Rubenstein J. L. R. (2009b). Dlx1&2 and Mash1 transcription factors control striatal patterning and differentiation through parallel and overlapping pathways. J. Comp. Neurol. 512, 556-572. 10.1002/cne.21854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Juárez A., Howard J., Ullom K., Howard L., Grande A., Pardo A., Waclaw R., Sun Y. Y., Yang D., Kuan C. Y. et al. (2013). Gsx2 controls region-specific activation of neural stem cells and injury-induced neurogenesis in the adult subventricular zone. Genes. Dev. 27, 1272-1287. 10.1101/gad.217539.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M. et al. (2015). highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202-1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari M. E. and Murre C. (2000). Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 20, 429-440. 10.1128/MCB.20.2.429-440.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Gómez H. R. and Vicario-Abejón C. (2012). The homeobox gene Gsx2 regulates the self-renewal and differentiation of neural stem cells and the cell fate of postnatal progenitors. PLoS ONE 7, e29799 10.1371/journal.pone.0029799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao M., Campbell K., Burns K., Kuan C.-Y., Trumpp A. and Nakafuku M. (2008). Coordinated control of self-renewal and differentiation of neural stem cells by Myc and the p19ARF-p53 pathway. J. Cell Biol. 183, 1243-1257. 10.1083/jcb.200807130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada Y., Hunsaker T. L., Henke R. M. and Johnson J. E. (2004). Distinct domains within Mash1 and Math1 are required for function in neuronal differentiation versus neuronal cell-type specification. Development 131, 1319-1330. 10.1242/dev.01008 [DOI] [PubMed] [Google Scholar]
- Noctor S. C., Martínez-Cerdeño V., Ivic L. and Kriegstein A. R. (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136-144. 10.1038/nn1172 [DOI] [PubMed] [Google Scholar]
- Olsson M., Campbell K., Wictorin K. and Björklund A. (1995). Projection neurons in fetal striatal transplants are predominantly derived from the lateral ganglionic eminence. Neuroscience 69, 1169-1182. 10.1016/0306-4522(95)00325-D [DOI] [PubMed] [Google Scholar]
- Olsson M., Björklund A. and Campbell K. (1998). Early specification of striatal projection neurons and interneuronal subtypes in the lateral and medial ganglionic eminence. Neuroscience 84, 867-876. 10.1016/S0306-4522(97)00532-0 [DOI] [PubMed] [Google Scholar]
- Pei Z., Wang B., Chen G., Nagao M., Nakafuku M. and Campbell K. (2011). Homeobox genes Gsx1 and Gsx2 differentially regulate telencephalic progenitor maturation. Proc. Natl. Acad. Sci USA 108, 1675-1680. 10.1073/pnas.1008824108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilz G.-A., Shitamukai A., Reillo I., Pacary E., Schwausch J., Stahl R., Ninkovic J., Snippert H. J., Clevers H., Godinho L. et al. (2013). Amplification of progenitors in the mammalian telencephalon includes a new radial glial cell type. Nat. Commun. 4, 2125 10.1038/ncomms3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pla R., Stanco A., Howard M. A. A., Rubin A. N., Vogt D., Mortimer N., Cobos I., Potter G. B., Lindtner S., Price J. D. et al. (2018). Dlx1 and Dlx2 promote interneuron GABA synthesis, synaptogenesis, and dendritogenesis. Cereb. Cortex. 28, 3797-3815. 10.1093/cercor/bhx241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S., Madhavan M., Waclaw R. R., Nakafuku M. and Campbell K. (2016). Characterization of a new Gsx2-cre line in the developing mouse telencephalon. Genesis 54, 542-549. 10.1002/dvg.22980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S., Ware S. M., Waclaw R. R. and Campbell K. (2017). Septal contributions to olfactory bulb interneuron diversity in the embryonic mouse telencephalon: role of the homeobox gene Gsx2. Neural. Dev. 12, 13 10.1186/s13064-017-0090-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider I. (1972). Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 27, 353-365. [PubMed] [Google Scholar]
- Smart I. H. (1976). A pilot study of cell production by the ganglionic eminences of the developing mouse brain. J. Anat. 121, 71-84. [PMC free article] [PubMed] [Google Scholar]
- Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.-J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.-G. et al. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995-1000. 10.1038/nmeth947 [DOI] [PubMed] [Google Scholar]
- Stenman J., Toresson H. and Campbell K. (2003). Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J. Neurosci. 23, 167-174. 10.1523/JNEUROSCI.23-01-00167.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M., Hao Y., Stoeckius M., Smibert P. and Satija R. (2019). Comprehensive integration of single-cell data. Cell 177, 1888-1902.e21. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucsik J. C., Witte D. P., Li H., Pixley S. K., Small K. M. and Potter S. S. (1997). Altered forebrain and hindbrain development in mice mutant for the Gsh-2 homeobox gene. Dev. Biol. 191, 230-242. 10.1006/dbio.1997.8733 [DOI] [PubMed] [Google Scholar]
- Takebayashi H., Yoshida S., Sugimori M., Kosako H., Kominami R., Nakafuku M. and Nabeshima Y. (2000). Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech. Dev. 99, 143-148. 10.1016/S0925-4773(00)00466-4 [DOI] [PubMed] [Google Scholar]
- Tirode F., Malaguti C., Romero F., Attar R., Camonis J. and Egly J. M. (1997). A conditionally expressed third partner stabilizes or prevents the formation of a transcriptional activator in a three-hybrid system. J. Biol. Chem. 272, 22995-22999. 10.1074/jbc.272.37.22995 [DOI] [PubMed] [Google Scholar]
- Toresson H. and Campbell K. (2001). A role for Gsh1 in the developing striatum and olfactory bulb of Gsh2 mutant mice. Development 128, 4769-4780. [DOI] [PubMed] [Google Scholar]
- Toresson H., Potter S. S. and Campbell K. (2000). Genetic control of dorsal-ventral identity in the telencephalon: opposing roles for Pax6 and Gsh2. Development 127, 4361-4371. [DOI] [PubMed] [Google Scholar]
- Tucker E. S., Polleux F. and LaMantia A.-S. (2006). Position and time specify the migration of a pioneering population of olfactory bulb interneurons. Dev. Biol. 297, 387-401. 10.1016/j.ydbio.2006.05.009 [DOI] [PubMed] [Google Scholar]
- Ueki Y., Wilken M. S., Cox K. E., Chipman L., Jorstad N., Sternhagen K., Simic M., Ullom K., Nakafuku M. and Reh T. A. (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci USA 112, 13717-13722. 10.1073/pnas.1510595112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl J. D., Cook T. A. and Gebelein B. (2010). Comparing anterior and posterior Hox complex formation reveals guidelines for predicting cis-regulatory elements. Dev. Biol. 343, 154-166. 10.1016/j.ydbio.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl J. D., Zandvakili A. and Gebelein B. (2016). A Hox transcription factor collective binds a highly conserved Distal-less cis-regulatory module to generate robust transcriptional outcomes. PLoS Genet. 12, e1005981 10.1371/journal.pgen.1005981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerius M. T., Patterson L. T., Witte D. P. and Potter S. S. (2002). Microarray analysis of novel cell lines representing two stages of metanephric mesenchyme differentiation. Mech. Dev. 110, 151-164. 10.1016/S0925-4773(01)00581-0 [DOI] [PubMed] [Google Scholar]
- van der Kooy D. and Fishell G. (1987). Neuronal birthdate underlies the development of striatal compartments. Brain Res. 401, 155-161. 10.1016/0006-8993(87)91176-0 [DOI] [PubMed] [Google Scholar]
- Waclaw R. R., Allen Z. J. II, Bell S. M., Erdélyi F., Szabó G., Potter S. S. and Campbell K. (2006). The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron 49, 503-516. 10.1016/j.neuron.2006.01.018 [DOI] [PubMed] [Google Scholar]
- Waclaw R. R., Wang B., Pei Z., Ehrman L. A. and Campbell K. (2009). Distinct temporal requirements for the homeobox gene Gsx2 in specifying striatal and olfactory bulb neuronal fates. Neuron 63, 451-465. 10.1016/j.neuron.2009.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclaw R. R., Ehrman L. A., Pierani A. and Campbell K. (2010). Developmental origin of the neuronal subtypes that comprise the amygdalar fear circuit in the mouse. J. Neurosci. 30, 6944-6953. 10.1523/JNEUROSCI.5772-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Waclaw R. R., Allen Z. J. II, Guillemot F. and Campbell K. (2009). Ascl1 is a required downstream effector of Gsx gene function in the embryonic mouse telencephalon. Neural. Dev. 4, 5 10.1186/1749-8104-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Long L. E., Flandin P., Pla R., Waclaw R. R., Campbell L. and Rubenstein J. L. R. (2013). Loss of Gsx1 and Gsx2 function rescues distinct phenotypes in Dlx1/2 mutants. J. Comp. Neurol. 521, 1561-1584. 10.1002/cne.23242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H., Turnbull D. H., Nery S., Fishell G. and Alvarez-Buylla A. (2001). In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 128, 3759-3771. [DOI] [PubMed] [Google Scholar]
- Wilsch-Bräuninger M., Florio M. and Huttner W. B. (2016). Neocortex expansion in development and evolution - from cell biology to single genes. Curr. Opin. Neurobiol. 39, 122-132. 10.1016/j.conb.2016.05.004 [DOI] [PubMed] [Google Scholar]
- Winterbottom E. F., Illes J. C., Faas L. and Isaacs H. V. (2010). Conserved and novel roles for the Gsh2 transcription factor in primary neurogenesis. Development 137, 2623-2631. 10.1242/dev.047159 [DOI] [PubMed] [Google Scholar]
- Winterbottom E. F., Ramsbottom S. A. and Isaacs H. V. (2011). Gsx transcription factors repress Iroquois gene expression. Dev. Dyn. 240, 1422-1429. 10.1002/dvdy.22648 [DOI] [PubMed] [Google Scholar]
- Yun K., Potter S. and Rubenstein J. L. (2001). Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development 128, 193-205. [DOI] [PubMed] [Google Scholar]
- Yun K., Fischman S., Johnson J., Hrabe de Angelis M., Weinmaster G. and Rubenstein J. L. (2002). Modulation of the notch signaling by Mash1 and Dlx1/2 regulates sequential specification and differentiation of progenitor cell types in the subcortical telencephalon. Development 129, 5029-5040. [DOI] [PubMed] [Google Scholar]
- Yun K., Garel S., Fischman S. and Rubenstein J. L. R. (2003). Patterning of the lateral ganglionic eminence by the Gsh1 and Gsh2 homeobox genes regulates striatal and olfactory bulb histogenesis and the growth of axons through the basal ganglia. J. Comp. Neurol. 461, 151-165. 10.1002/cne.10685 [DOI] [PubMed] [Google Scholar]
- Zhang X., McGrath P. S., Salomone J., Rahal M., McCauley H. A., Schweitzer J., Kovall R., Gebelein B. and Wells J. M. (2019). A comprehensive structure-function study of neurogenin3 disease-causing alleles during human pancreas and intestinal organoid development. Dev. Cell 50, 367-380. 10.1016/j.devcel.2019.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.