

Short abstract
Late-onset Alzheimer’s disease (LOAD) is a long-enduring neurodegenerative disease that progresses for decades before the symptoms of cognitive decline and loss of executive function are measurable. Amyloid deposits among other pathological changes, tau hyperphosphorylation, synapse loss, microglia and astroglia activation, and hippocampal atrophy are among the pathological hallmarks of the disease. These are present in the brain before memory complaints are reported and an AD diagnosis is made. The attempt to postpone or prevent the disease is becoming a more and more plausible goal because new early electrophysiological, cognitive, blood-based, and imaging-based diagnostics are being brought forward at the same time as the first anti-amyloid antibody is about to be approved. In view of known contributions of neuroinflammation to the pathology of LOAD, we should not focus solely on anti-amyloid therapies and ignore the interactive neuroinflammatory component of AD. Our belief is that it would be more rewarding to start clinical trials using combination therapies that are based on approved, safe, and efficacious anti-neuroinflammatory agents such as anti-interleukin-1 signaling agents in combination with the anti-amyloid antibodies that have been shown to be safe in multiyear trials. The proposal is that we should administer these two classes of safe biologicals to symptom-free individuals in midlife who are identified as having a high-risk-for-Alzheimer’s-disease using “precision medicine.”
Keywords: dementia and neurological disorders, microglia, neurodegenerative diseases, inflammation, neuroinflammatory disorders, pharmacology
Late-onset Alzheimer’s disease (LOAD) is a long-enduring neurodegenerative disease (Jack et al., 2018) that progresses for decades before the symptoms of cognitive decline and loss of executive function are measurable.
The societal cost of AD in all its manifestations is likely to continue to grow for the foreseeable future especially in an aging population. The growth of the incidence of AD is very likely to put a severe strain on families as well as medical and support services. The projected financial cost can be measured in trillions of dollars (Bartfai and Lees, 2013).
Many pharmaceutical companies were pursuing candidate drugs. However, the only medications approved for use in AD are a few cholinesterase inhibitors and memantine, an N-methyl-D-aspartate receptor antagonist that has been shown to improve memory in some cases. They are inadequate medicines to ameliorate the disease. Trials are of very long duration and difficult to design and manage in a population often suffering from multiple age-related conditions. In addition, the trials now cost billions of dollars to conduct. As such, the majority of Big Pharma companies have given up; there are easier ways to make money. A new approach was needed.
Among the pathological hallmarks of the disease are amyloid deposits, tau hyperphosphorylation (Orr et al., 2017), synapse disruption and loss (Mucke and Selkoe, 2012), microglia and astroglia activation, and hippocampal atrophy—processes most of which we can now image (Jack et al., 2018). These pathological markers are present in the brain before memory complaints are reported and an AD diagnosis is made. The attempt to postpone or prevent the disease (Selkoe, 2012) in individuals at high risk defined by diagnostic markers from genetics (Alzheimer’s Prevention Registry Gene Match) from cerebrospinal fluid (CSF) measurements, positron-emission tomography (PET) imaging, and, recently, from emerging blood-based markers (Hampel et al., 2018; Niculescu et al., 2019) is becoming a more and more plausible goal.
While multiple targets have been identified (Bartfai and Lees, 2006, 2013), both basic and therapeutic research has focused largely on reducing the amyloid load. A number of companies created several anti-amyloid biologicals. But, regrettably, the clinical trials showed that while the drugs “worked” in that amyloid load in the brain was reduced, no significant effect on cognition and cognitive decline could be demonstrated. This may be illustrated and exemplified most vividly by news of the development of aducanumab by Biogen and Eisai.
The announcement that aducanumab would move to Phase 3 clinical trials was greeted with guarded enthusiasm (Netzer, 2015).
The announcement that the Phase 3 trial was being suspended because of lack of efficacy was greeted as yet another disappointment (Fagan, 2019).
Biogen/Eisai continued to assemble and assess data and now conclude that aducanumab is effective (Schneider, 2020).
Why the change? The futility analysis that caused the trial to be stopped used data from approximately 1,750 subjects from 2 trials comprising 2,700 patients. The data met the pre-established negative criteria, and the announcement of ineffectiveness was made. Dennis Selkoe (Fagan, 2019) responded to the announcement by saying that
Perhaps “the trial patients were too symptomatic” and had a too advanced stage of AD.
“The ‘amyloid hypothesis’ that we have been working on for decades is incorrect.”
Aducanumab, in particular, is an antibody that does not sufficiently neutralize and clear the key form of amyloid—the soluble “Aβ oligomers.”
Earlier, Biogen had increased the original sample size to 3,300 patients, and more people had completed the trial by the end of 2018. Biogen/Eisai continued to analyze the data incorporating these new data. Patients carrying the Apoε4 gene had been given lower doses than noncarriers in the same group. Then, they were switched to the higher dose of both the low- and high-dose groups. The two trials, EMERGE and ENGAGE, had different outcomes with EMERGE showing improvement. The statistics are complex because of the dynamic nature of the trial, shifting doses, and post hoc decisions, some related to side effects. But, randomized data from both trials showed an improvement (Schneider, 2020). The fortunes of aducanumab in these trials were not smooth (Selkoe, 2019a, 2019b).
We are very likely to find ourselves in the situation where we will have aducanumab, an anti-Aβ 1-40/42 antibody (Sevigny et al., 2016) regarded as efficacious and approved by the Food and Drug Administration (FDA). The evidence that the data from the aducanumab trials were not immediately compelling may indicate that treatment of AD with this monotherapy is not sufficient to meet the societal need even if financially very successful for Biogen/Eisai.
The Case For and Against Anti-Amyloid Monotherapy
The disease-modifying property and slowing of cognitive decline of aducanumab may be shared by several similar antibodies from previously “failed” clinical trials. For example, gantenerumab, bapineuzumab, and solanezumab were all shown to reduce amyloid load by measuring amyloid concentrations in CSF and blood and/or by PET imaging (Rinne et al., 2010; Novakovic et al., 2013; Doody et al., 2014).Companies will be keen to revisit their data with the hope of reassessing efficacy. It is likely that these alternatives might be rapidly approved following any approval of Biogen’s aducanumab. Meanwhile, Roche’s gantenerumab continues in trials in both familial AD (Dominantly Inherited Alzheimer Network Trials Unit) and in sporadic AD (Graduate 1 and Graduate 2 trials) with read outs expected several years from now. The Banner Institute and several National Institutes of Health-supported studies focus on early onset familial AD using single-agent therapy with gantenerumab.
Obviously, not everyone gave up in March 2019. The market likes alternatives, not just one drug in a class.
The aducanumab data may be used to support the argument for using single anti-amyloid/tau therapeutic approaches. Even if the start of single-agent therapy moved toward midlife (see Gandy et al., 2017), we might still be preparing for new disappointments from modest improvements. However successful this approach might seem in terms of prescriptions filled, it represents a new instance of lost time and funds to find an effective therapy to postpone and prevent LOAD or to achieve slowing of disease progression.
The data are indeed an important gain for our designed therapy. However, the focus and concentration on a monotherapy would, we believe, represent a missed opportunity or even a grave mistake. We should not forget the neuroinflammatory component of AD.
Insights of the past 4 to 14 years show that microglia activation (Butovsky and Weiner, 2018; Dong et al., 2019) and neuroinflammatory processes (Sheng et al., 1996; Heneka et al., 2018) are locked in a vicious cycle with amyloidosis, and, hence, the new pharmacotherapies should target both processes. To prevent, postpone, or slow AD, such combination treatments should start in symptom-free patients and be closely monitored within new clinical trials.
These trials would address both arms of this self-enhancing “neuroinflammation–amyloidosis–neuroinflammation” process simultaneously (Figure 1).
Figure 1.
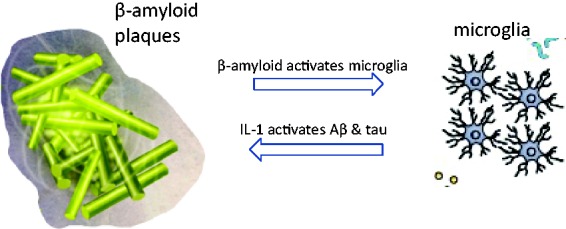
“Two Hit” Synergy to Break the Vicious Cycle: Anti-IL-1 Strategy Backed Up by Simultaneous Aβ Reduction. Amyloid oligomers and plaque increase IL-1 and NLRP3 and activate microglia. Anti-Aβ antibody reduces oligomer and plaque burden, relieving proinflammatory signaling. Anti-IL-1 biologicals, such as anakinra, canakinumab, or rilonacept, block IL-1 signaling, relieving proinflammatory activation of amyloidosis and tau phosphorylation (see text for details).
IL = interleukin.
The anti-neuroinflammatory biologicals we are suggesting (Table 1) are already approved as subcutaneously administered treatments. These anti-neuroinflammatory agents that reduce or block interleukin (IL)-1 action have been used as approved drugs in tens of thousands of patients for many years in several inflammatory diseases involving inflammation in the periphery—rheumatoid arthritis, Crohn’s disease, and psoriasis—but lately also in the brain in reducing effects of Mediterranean fever (Özçakar et al., 2016; De Benedetti et al., 2018), neurotrauma, and acting on certain forms of epilepsy in children (Kenney-Jung et al., 2016; Dilena et al., 2019). Thus, safe and efficacious subcutaneous anti-neuroinflammatory drugs can be easily deployed as part of a combination with subcutaneous or even intravenous anti-amyloid biologicals.
Table 1.
Available Anti-Neuroinflammatory and Anti-Amyloidosis Agents That May Be Combined for Postponement and/or Prevention of LOAD in High-Risk Individuals
| Anti-IL-1 signaling biologicals | Anti-Amyloidosis biologicals |
|---|---|
| IL-1Ra—anakinra (sc) | antibodies to monomers or oligomers of Aβ 1-40/42 |
| IL-1 Trap—rilonacept (sc) | gantenerumab (iv, sc), solanezumab (iv, sc), bapineuzumab (iv, sc) |
| anti-IL-1β antibody—canakinumab (sc) | aducanumab (iv), crenezumab (iv), BAN2401 (iv) |
Note. sc = subcutaneous; iv = intravenous.
Multipharmacy and Safety
Before any Phase 2 and 3 trials of anti-amyloid and anti-neuroinflammatory combinations are designed, it is only the safety as a fixed-dose combination (FDC) that needs to be established. However, there are many diseases treated with multipharmacy—pain, epilepsy, diabetes, and so forth—and among these are approved FDCs such as angiotensin-converting enzyme (ACE) inhibitors with diuretic thiazides, for example, Zestoretic (lisinopril/hydrochlorothiazide) and Stalevo (carbidopa/levodopa/entacapone) for Parkinson's disease (PD). Some drug combinations have been used in millions of patients. ACE inhibitor–diuretic and angiotensin type 2 receptor blocker–diuretic combinations have been around for decades.
When proposing combinations of drugs for a treatment, in particular as here, for chronic treatment in symptom-free patients, the safety of the combination is paramount. It should be noted, therefore, that we are suggesting combining biologicals—antibodies to amyloid peptide and oligomers, and a small protein recombinant IL-receptor antagonist, or a medium-sized recombinant protein IL-1 Trap, or a large protein antibody to IL-1b—and hence, they likely will not have drug–drug interaction issues as it often is the case when combining two or more low-molecular weight (MW) drugs. With low-MW drugs, their metabolism may proceed via the same cytochrome p450 isoenzyme and/or the same drug transporters, and thus, one needs to match their pharmacokinetics and pharmacodynamics. These concerns are very unlikely to present a problem when two biologicals are combined. It is also suggested that neither of the components of the combination of the two biologicals should be used in a higher dose than is known to be safe individually. We have data from years of administration in the large Phase 3 studies of the anti-amyloid antibodies—some of which were administered for 1 to 4 years duration in up to 14,000 patients—and we have data from the more than a decade of treatments for the approved anti-IL-1 agents that show that these are safe and also that they have positive effects on central nervous system diseases.
This suggests that safety will not be a major obstacle for the combined use of two suitable biologicals in larger clinical trials on postponement/prevention of conversion of mild cognitive impairment (MCI) to LOAD or clinical trials on postponement of onset of LOAD in high-risk individuals.
The Efficacy of Anti-Amyloid Biologicals: Can AD Be Ameliorated Once It Is Established?
The anti-amyloid antibodies bapineuzumab, solanezumab, gantenerumab, and aducanumab have all led to a reduction in amyloid load, but there has not been any large pivotal study to prove that the reduction in amyloid load or in phospho-tau was accompanied with a slowing of the cognitive decline and loss of the executive function. Except, that is, for the small Phase 2 study of aducanumab that led to the large Phase 3 study by Biogen that started aducanumab’s remarkable “roller-coaster ride.” Now, Biogen is urging the FDA to approve it as the first AD-disease-modifying biological. However, these up to 30% reductions of total amyloid burden did not reduce the rate of cognitive decline in patients already presenting with cognitive decline and massive amyloid loads.
It was surmised that perhaps the anti-amyloid or anti-tau treatment needs to start in midlife, some 15 to 20 years before memory complaints appear. This agreement on moving the start of the treatment earlier is almost universal now—see Gandy et al. (2017)—but it adds to another problem. Trials take 4 to 7 years, and patent life for the drugs that require these long trials has not been adjusted by governments. Therefore, it is a losing economical proposition to work on disease modification of slow neurodegenerative diseases, while some success of symptom treatment in other diseases may be shown much faster after just 12 to 24 months. Here, the emerging blood-based biomarkers that in small studies start to show predictive value of the conversion from no or mild memory complaint to mild AD within 12 to 24 months may offer means to reduce clinical trial duration (Hampel et al., 2018; Niculescu et al., 2019).
Anti-Neuroinflammatory Therapies
Why is this monotherapeutic anti-amyloid approach a so deeply rooted approach for such a complex and chronically progressive neurodegenerative disease? After all, we have used multipharmacy to treat pain for millennia and cancer for decades. There is definitely more than one risk factor for LOAD and hence more than one therapeutic target. Many studies in the past 4 years found that microglia play an important role in AD. It is hardly surprising that these “brain macrophages” that mediate the innate immune response within the brain should be activated when neurodegeneration is starting, ongoing, or becoming dominant. For example, over past years, triggering receptor expressed on myeloid cells 2-DNAX-activating protein of 12 kDa (TREM2-DAP12) articles are now in great abundance showing that microglial activity and their proteins play a role in AD and may also prevent the strong transgenically driven amyloidosis in transgenic AD models (see, e.g., Leyns et al., 2017).
The therapeutic targeting of neuroinflammation in the brain was accepted in the case of neurotrauma, when the blood–brain barrier integrity is compromised, and it has been well known now for decades that the endogenous pyrogen is the key proinflammatory cytokine IL-1 in the brain, acting on the neuronal IL-1 receptor (IL1R1, IL1RAP/IL1R3) complex. IL-1β is the major proinflammatory cytokine in excitable tissues such as heart, brain, and smooth muscle. It induces other proinflammatory cytokines, such as IL-6 and tumor necrosis factor, and the β-secretase pathway. The molecular mechanism of its action on amyloid precursor protein (APP) processing and on neuronal activity during the hyperactivity of cortical neurons in early AD involves signaling through the IL1R1-IL1RAP (IL1R accessory protein or IL1R3) heterodimer. The pathway that leads to both serine and tyrosine kinase activation is thus involved in protein phosphorylation that accelerates APP β-secretase processing, tau hyperphosphorylation, and changes in ion channel activity.There is now extensive literature showing that Aβ peptide and its oligomers stimulate microglia, which, after activation of the inflammasome, secrete IL-1β, IL-18, IL-1Ra, and other cytokines (Halle et al., 2008; Cai et al., 2014; Venegas et al., 2017; Hansen et al., 2018; Lučiūnaitė et al., 2019). Therefore, it is not such a surprise that we now see publications suggesting Alzheimer’s drug development should target microglia (Butovsky and Weiner, 2018; Dong et al., 2019).
Yet, the more than 2,250 studies listed in ClinicalTrials.gov under AD are sadly deficient in trials with combination therapies and even more so of combinations with a good scientific rational. Moreover, the changes in the fortune of aducanumab brought forth a monotherapeutic approach focusing on anti-amyloid–anti-tau when clearly we should try simultaneously to prevent amyloidosis and the neuroinflammation it causes.
Conclusions
Given the available data, we conclude that we should be conducting clinical trials with anti-amyloid antibodies combined with approved anti-neuroinflammatory agents such as the three classes of anti-IL-1 agents: anakinra, rilonacept, or canakinumab (Kahlenberg, 2016) and that these combinations should be administered early, in midlife, to symptom-free high-risk-for-AD individuals. We recommend safe biologicals from both classes of agents. This extreme focus on safety enables the treatment over many years of symptom free but high-risk-for-AD individuals. Some combinations can be administered in subcutaneous formulations. This would make such treatments self-administered and cause little interruption of normal life. For other combinations, only an intravenous formulation of anti-amyloid currently exists. These proposed biologicals can be administered once weekly to once monthly.
How do we identify individual at high-risk-for-AD while symptom-free? Today, we have more than 20 genome-wide association study (GWAS)-identified genes that very significantly increase the risk of LOAD: Apoε4 heterozygotes make up approximately 20% of the population and increase risk by 8- to 12-fold; IL1RAP G-mutation carriers have a 4 to 8 times higher risk; and there are approximately 20 more identified targets with lesser but very significant risk for AD.The genotyping company 23andMe reported that they have genotyped more than four million subjects for Apoε genotype. There are thousands of those who are Apoε4 heterozygotes and have one or several close relatives who suffer(ed) from dementia that in many cases was confirmed as being LOAD. Blood-based markers are advancing predictions of who will convert from a nonsymptomatic state or from MCI to mild AD (Hampel et al., 2018). Thus trial recruitment of such individuals will be possible, for whom an 18- to 36-month trial may clearly show whether postponement or prevention of LOAD is achieved. We thus believe that recruiting to a prevention/postponement study in LOAD will not be difficult if two approved safe biologicals are used in an FDC.
How might a multipharmaceutical approach lead to successful disease modification of LOAD? A major obstacle is that clinical trials for this long-lasting progressive disease are already astonishingly long and expensive, and combination drug trials are, in themselves, notoriously complex, large, and expensive. Nevertheless, we have to do the following to have any chance of therapeutic success in postponing, preventing or slowing AD.
Move the start of treatment to midlife, nonsymptomatic subjects at risk;
Address pathways (e.g., amyloidosis, tauopathy, neuroinflammation) that contain one or more of the major risk factors identified; and
Use precision medicine to select those for the preventive decade of therapy—by genotyping, by imaging, and by blood-based markers. Note that subjects voluntarily do this with 23andMe and other genotyping companies, or as they do by participating in the 350,000-people strong Alzheimer’s Prevention Registry GeneMatch Program.
To make starting trials with combinations somewhat easier, some biological therapeutics can all be given subcutaneously. For example, any combination of gantenerumab or bapineuzumab, with rilonacept, anakinra, or canakinumab could be administered subcutaneously by the subjects themselves (Table 1). The subjects of these trials should be annually tested for cognitive decline, blood AD biomarkers, imaged for amyloidosis and tested for markers of neuroinflammation by blood assays or imaging. The safety of anti-amyloid antibodies in causing amyloid-related imaging abnormalities (ARIA) needs to be tested by magnetic resonance imaging as already determined for these agents. It is a distinct possibility that with an early start of treatment, when less oligomeric and fibrillary amyloid is present and in the simultaneous presence of an anti-neuroinflammatory agent, ARIA will be reduced.
Multipharmacy holds promise in this chronic neurodegenerative disease as it has shown in the clinic in addressing pain, cardiovascular diseases, and other diseases. These combinations could be put into trials almost immediately and could pave the way for other combination therapies, each potentially more effective at different stages of this long neurodegenerative and neuroinflammatory disease. The doses would be determined before the trial, but the doses might be modulated during the trial according to the individual-specific monitoring data. If indeed such therapies are approved, the ratio of the components of the combination medicine might be adapted according to patients’ needs as determined by disease progression monitoring. Indeed, the particular biologicals might change as other biologicals might become more efficacious and available.
The vicious cycle that reigns under the neuroinflammation-stimulated progression of amyloidosis (Mrak and Griffin, 2001; Jiang et al., 2018) and amyloid-stimulated microglial activation (Butovsky and Weiner, 2018; Dong et al., 2019) has been known for many years but has not been addressed simultaneously using a drug combination. Indeed, it is likely that many patients who were in trials for reducing amyloidosis were also being treated with drugs blocking neuroinflammation, as both conditions last for decades and afflict many people. Instead of finding those who have been on anti-neuroinflammatory therapy while being enrolled in anti-amyloid trials and performing a highly difficult meta-analysis of individuals who had both classes of drugs, it would be more prudent to start an interventional double-blind large study at midlife, on high-risk-for-LOAD persons to investigate and hopefully demonstrate the benefit of combination therapy.
One could in the long term devise better patent laws and clinical trial regulations for developing drugs in diseases with such a long prodromal phase as LOAD has. But we already have many drugs that are safe, exhibiting pharmacodynamic results, which have been administered to tens of thousands of subjects, that is, amyloid-load-lowering anti-amyloid antibodies. They were administered far too late, and alone, without simultaneous anti-neuroinflammatory therapy, to succeed.
We propose here concrete combination therapies that will use the existing and already clinically tested drugs for both the amyloidosis pathway and the neuroinflammatory IL-1 signaling pathway where IL1RAP stands out among risk factors uncovered by GWAS.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD
Graham V. Lees https://orcid.org/0000-0002-1856-8732
References
- Bartfai T., Lees G. V. (2006). Drug discovery: From bedside to Wall Street. Elsevier Academic Press. [Google Scholar]
- Bartfai T., Lees G. V. (2013). The future of drug discovery: Who decides which diseases to treat. Academic Press. [Google Scholar]
- Butovsky O., Weiner H. L. (2018). Microglial signatures and their role in health and disease. Nature Rev Neurosci, 19, 622–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Hussain M. D., Yan L. J. (2014). Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci, 124, 307–321. 10.3109/00207454.2013.833510 [DOI] [PubMed] [Google Scholar]
- De Benedetti F., et al. Gattorno, M., Anton, J., Ben-Chetrit, E., Frenkel, J., Hoffman, H. M., Koné-Paut, I., Lachmann, H. J., Ozen, S., Simon, A., Zeft, A., Calvo Penades, I., Moutschen, M., Quartier, P., Kasapcopur, O., Shcherbina, A., Hofer, M., Hashkes, P. J., Van der Hilst, J., Hara, R., Bujan-Rivas, S., Constantin, T., Gul, A., Livneh, A., Brogan, P., Cattalini, M., Obici, L., Lheritier, K., Speziale, A., & Junge, G. (2018). Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med, 378, 1908–1919. 10.1056/NEJMoa1706314 [DOI] [PubMed] [Google Scholar]
- Dilena R., Mauri E., Aronica E., Bernasconi P., Bana C., Cappelletti C., Carrabba G., Ferrero S., Giorda R., Guez S., Scalia Catenacci S., Triulzi F., Barbieri S., Calderini E., Vezzani A. (2019). Therapeutic effect of anakinra in the relapsing chronic phase of febrile infection-related epilepsy syndrome. Epilepsia Open, 4, 344–350. 10.1002/epi4.12317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y., Li X., Cheng J., Hou L. (2019). Drug development for Alzheimer’s disease: Microglia induced neuroinflammation as a target? Int J Mol Sci, 20, 558–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody R. S., Thomas R. G., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., Raman R., Sun X., Aisen P. S., Siemers E., Liu-Seifert H., Mohs R., Alzheimer’s Disease Cooperative Study Steering Committee, & Solanezumab Study Group. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med, 370, 311–321. 10.1056/NEJMoa1312889 [DOI] [PubMed] [Google Scholar]
- Fagan T. (2019). Biogen/Eisai halt Phase 3 aducanumab trials. Alzforum https://www.alzforum.org/news/research-news/biogeneisai-halt-phase-3-aducanumab-trials
- Gandy S., Bartfai T., Lees G. V., Sano M. (2017). Midlife interventions are critical in prevention, delay, or improvement of Alzheimer’s disease and vascular cognitive impairment and dementia. F1000Res, 6, 413 10.12688/f1000research.11140.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A., Hornung V., Petzold G. C., Stewart C. R., Monks B. G., Reinheckel T., Fitzgerald K. A., Latz E., Moore K. J., Golenbock D. T. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol, 9, 857–865. 10.1038/ni.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H., O’Bryant S. E., Molinuevo J. L., Zetterberg H., Masters C. L., Lista S., Kiddle S. J., Batrla R., Blennow K. (2018). Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat Rev Neurol, 14, 639–652. 10.1038/s41582-018-0079-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. V., Hanson J. E., Sheng M. (2018). Microglia in Alzheimer’s disease. J Cell Biol, 217, 459–472. 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T., McManus R. M., Latz E. (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci, 19, 610–621. 10.1038/s41583-018-0055-7 [DOI] [PubMed] [Google Scholar]
- Jack C. R., Jr., Bennett D. A., Blennow K., Carrillo M. C., Dunn B., Haeberlein S. B., Holtzman D. M., Jagust W., Jessen F., Karlawish J., Liu E., Molinuevo J. L., Montine T., Phelps C. H., Rankin K. P., Rowe C. C., Scheltens P., Siemers E., Snyder H. M., Sperling R., Elliott, C., Masliah, E., Ryan, L. M., & Silverberg, N. (2018). NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement, 14, 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C., Zou X., Zhu R., Shi Y., Wu Z., Zhao F., Chen L. (2018). The correlation between accumulation of amyloid beta with enhanced neuroinflammation and cognitive impairment after intraventricular hemorrhage. J Neurosurg, 131, 54–63. 10.3171/2018.1.JNS172938 [DOI] [PubMed] [Google Scholar]
- Kahlenberg J. M. (2016). Anti-inflammatory panacea? The expanding therapeutics of IL-1 blockade. Curr Opin Rheumatol, 28, 197–203. 10.1097/BOR.0000000000000266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney-Jung D. L., Vezzani A., Kahoud R. J., LaFrance-Corey R. G., Ho M. L., Muskardin T. W., Wirrell E. C., Howe C. L., Payne E. T. (2016). Febrile infection-related epilepsy syndrome treated with anakinra. Ann Neurol, 80, 939–945. 10.1002/ana.24806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns C. E. G., Ulrich J. D., Finn M. B., Stewart F. R., Koscal L. J., Serrano J. R. (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci, 114, 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lučiūnaitė, A., McManus, R. M., Jankunec, M., Rácz, I., Dansokho, C., Dalgėdienė, I., Schwartz, S., Brosseron, F., & Heneka, M. T. (2019). Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem, e14945 10.1111/jnc.14945 [DOI] [PubMed] [Google Scholar]
- Mrak R. E., Griffin W. S. T. (2001). Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging, 22, 903–908. [DOI] [PubMed] [Google Scholar]
- Mucke L., Selkoe D. J. (2012). Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb Perspect Med, 2, a006338 10.1101/cshperspect.a006338PMID:22762015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer W. J. (2015). Experimental Alzheimer’s drug, aducanumab, slows cognitive decline in early trials. Alzinfo https://www.alzinfo.org/articles/diagnosis/experimental-alzheimers-drug-aducanumab-slows-cognitive-decline-in-early-trials/
- Niculescu A. B., Le-Niculescu H., Roseberry K., Wang S., Hart J., Kaur A., Robertson H., Jones T., Strasburger A., Williams A., Kurian S. M., Lamb B., Shekhar A., Lahiri D. K., Saykin A. J. (2019). Blood biomarkers for memory: Toward early detection of risk for Alzheimer disease, pharmacogenomics, and repurposed drugs. Mol Psychiatry. 10.1038/s41380-019-0602-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic D., Feligioni M., Scaccianoce S., Caruso A., Piccinin S., Schepisi C., Errico F., Mercuri N. B., Nicoletti F., Nisticò R. (2013). Profile of gantenerumab and its potential in the treatment of Alzheimer’s disease. Drug Des Devel Ther, 7, 1359–1364. 10.2147/DDDT.S53401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr M. E., Sullivan A. C., Frost B. A. (2017). Brief overview of tauopathy: Causes, consequences, and therapeutic strategies. Trends Pharmacol Sci, 38, 637–648. 10.1016/j.tips.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özçakar Z. B., Özdel S., Yılmaz S., Kurt-Şükür E. D., Ekim M., Yalçınkaya F. (2016). Anti-IL-1 treatment in familial Mediterranean fever and related amyloidosis. Clin Rheumatol, 35, 441–446. 10.1007/s10067-014-2772-2 [DOI] [PubMed] [Google Scholar]
- Rinne J. O., Brooks D. J., Rossor M. N., Fox N. C., Bullock R., Klunk W. E., Mathis C. A., Blennow K., Barakos J., Okello A. A., de LIano S. R. M., Liu E., Koller M., Gregg K. M., Schenk D., Black R., Grundman M. (2010). 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol, 9, 363–372. 10.1016/S1474-4422(10)70043-0 [DOI] [PubMed] [Google Scholar]
- Schneider L. (2020). A resurrection of aducanumab for Alzheimer’s disease, Lancet Neurol, 19, 111–112. 10.1016/S1474-4422(19)30480-6 [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2012). Preventing Alzheimer’s. Science, 337(6101), 1488–1492. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2019. a). Alzheimer disease and aducanumab: Adjusting our approach. Nat Rev Neurol, 15, 365–366. 10.1038/s41582-019-0205-1PMID:31138932 [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2019. b, October). Biogen’s good news on aducanumab could ‘open the floodgates’ for Alzheimer’s drugs. STAT.
- Sevigny J., et al. Chiao, P., Bussiére, T., Weinreb, P. H., Williams, L., Maier, M., Dunstan, R., Salloway, S., Chen, T., Ling, Y., O'Gorman, J., Qian, F., Arastu, M., Li, M., Chollate, S., Brennan, M. S., Quintero-Monzon, O., Scannevin, R. H., Arnold, H. M., Engber, T., Rhodes, K., Ferrero, J., Hang, Y., Mikulskis, A., Grimm, J., Hock, C., Nitsch, R. M., & Sandrock, A. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature, 537(7618), 50–56. 10.1038/nature19323 (update in: Nature 2017; 546: 564). [DOI] [PubMed] [Google Scholar]
- Sheng J. G., Ito K., Skinner R. D., Mrak R. E., Rovnaghi C. R., van Eldik L. J., Griffin W. S. T. (1996). In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging, 17, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C., Kumar S., Franklin B. S., Dierkes T., Brinkschulte R., Tejera D., Vieira-Saecker A., Schwartz S., Santarelli F., Kummer M. P., Griep A., Gelpi E., Beilharz M., Riedel D., Golenbock D. T., Geyer M., Walter J., Latz E., Heneka M. T. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature, 552(7685), 355–361. 10.1038/nature25158 [DOI] [PubMed] [Google Scholar]