

Key Readings Index
Structure and Function, 806
Dysfunction/Responses to Injury, 815
Portals of Entry/Pathways of Spread, 827
Defense Mechanisms/Barrier Systems, 828
Disorders of Domestic Animals, 830
Disorders of Horses, 876
Disorders of Ruminants (Cattle, Sheep, and Goats), 881
Disorders of Pigs, 888
Disorders of Dogs, 890
Disorders of Cats, 896
Structure and Function, 898
Dysfunction/Responses to Injury, 899
Responses of the Axon to Injury, 899
Portals of Entry/Pathways of Spread, 899
Defense Mechanisms/Barrier Systems, 899
Disorders of Domestic Animals, 899
Disorders of Horses, 904
Disorders of Dogs, 906
E-Glossary 14-1 Glossary of Abbreviations and Terms
Astrocytosis—Increased numbers of astrocytes.
Astrogliosis—Reactive astrocytic response with increased number (variable), length, and complexity of cell processes. In the CNS, reparative processes after injury, such as inflammation and necrosis, are facilitated by astrogliosis.
Axonopathy, distal of the CNS and PNS—Degeneration of axons involving distal portions of peripheral nerves and distal portions of long axons in the CNS (spinal cord).
Axonopathy, distal, of the PNS—A neuropathy with degeneration of the terminal and preterminal axon of peripheral nerves.
Axonotmesis—Axonal injury of a peripheral nerve in which there is degeneration of the part distal to the site of trauma, leaving the supporting framework intact and allowing for improved potential for regeneration and effective reinnervation.
Blood-brain barrier of the CNS—A barrier to free movement of certain substances from cerebral capillaries into CNS tissue. Relies on tight junctions between capillary endothelial cells and selective transport systems in these cells. Endothelial cell basement membrane and foot processes of astrocytes abutting the basement membrane may play a role in barrier function.
Blood-CSF barrier of the CNS—A barrier that consists of tight junctions located between epithelial cells of the choroid plexus and the cells of the arachnoid membrane that respectively separate fenestrated blood vessels of the choroid plexus stroma and dura mater from the CSF.
Blood-nerve barrier—A barrier to free movement of certain substances from the blood to the endoneurium of peripheral nerves. Barrier properties are conferred by tight junctions between capillary endothelial cells of the endoneurium and between perineurial cells and selective transport systems in the endothelial cells.
Brain edema—Increase in tissue water within the brain that results in an increase in brain volume. The fluid may be present in the intracellular or extracellular compartments or both. The term also is used to include the accumulation of plasma, especially in association with severe injury to the vasculature.
Brain swelling—Marked, rapidly developing, sometimes unexplained increase in cerebral blood volume and brain volume because of relaxation (dilation) of the arterioles that occurs after brain injury.
Büngner, cell bands of—A column of proliferating Schwann cells that forms within the space previously occupied by an axon following Wallerian degeneration. The proliferating column of cells is surrounded by the persisting basement membrane of the original Schwann cells.
CAE—Caprine arthritis encephalitis.
CCD—Canine cognitive dysfunction.
Central chromatolysis—Dissolution of cytoplasmic Nissl substance (arrays of rough endoplasmic reticulum and polysomes) in the central part of the neuronal cell body that results from injury to the neuron (often involving the axon). The cell body is swollen, and the nucleus frequently is displaced peripherally to the cell membrane. These structural changes functionally represent a response to injury that can be found (if the cell survives) by axonal regeneration with protein synthesis to produce components of the axon required for fast and slow axonal transport.
CNS—Central nervous system.
Cranium bifidum—A dorsal midline cranial defect through which meninges alone or meninges and brain tissue may protrude into a sac (-cele), covered by skin.
CSF—Cerebrospinal fluid.
Demyelination—A disease process in which demyelination (destruction of the myelin sheath) is the primary lesion, although some degree of axonal injury may occur. Primary demyelination is caused by injury to myelin sheaths and/or myelinating cells and their cell processes. Secondary demyelination occurs with axonal injury, as in Wallerian degeneration.
Dysraphism—Dysraphia, which literally means an abnormal seam, refers to a defective closure of the neural tube during development. This defect, which may occur at any point along the neural tube, is exemplified by anencephaly, prosencephalic hypoplasia, cranium bifidum, spina bifida, and myeloschisis.
Encephalitis—Inflammation of the brain.
Encephalo-—A combining form that refers to the brain.
Encephalopathy—A degenerative disease process of the brain.
Ganglionitis—Inflammation of peripheral (sensory or autonomic or both) ganglia.
Gemistocyte—Reactive, hypertrophied astrocyte that develops in response to injury of the CNS. The cell body and processes of gemistocytes become visible with conventional staining (e.g., H&E stain). The cell bodies and processes of normal astrocytes are not visible with H&E staining.
Gitter cell—Macrophage that accumulates in areas of necrosis of CNS tissue. The cytoplasm is typically distended, with abundant lipid-containing material derived from the lipid-rich nervous tissue. Gitter cell nuclei are often displaced peripherally to the cell membrane. These cells are often referred to as “foamy” macrophages.
H&E stain—Hematoxylin and eosin stain.
Hydranencephaly—A large, fluid-filled cavity in the area normally occupied by CNS tissue of the cerebral hemispheres resulting from abnormal development. The nervous tissue may be so reduced in thickness that the meninges form the outer part of a thin-walled sac. The lateral ventricles are variably enlarged because of their expansion into the area normally occupied by tissue.
Hydrocephalus—Accumulation of excess CSF resulting from obstruction within the ventricular system (noncommunicating form) associated with enlargement of any or all of the following: lateral ventricles, third ventricle, mesencephalic aqueduct, and fourth ventricle. Hydrocephalus can also occur with communication of the CSF between the ventricular system and the subarachnoid space (communicating form). Hydrocephalus ex vacuo (compensatory hydrocephalus) is characterized by an expansion of the lateral ventricle (or ventricles) that follows loss of brain tissue.
Joest-Degen bodies—Intranuclear inclusion bodies (rarely intracytoplasmic) found in neurons in the brains of animals with Borna disease.
Leuko-—Combining form referring to white matter of the brain or spinal cord.
Leukoencephalitis—Inflammation of the white matter of the brain.
Macroglia—A collective term referring to astrocytes and oligodendrocytes. Has also been variously used to refer solely to astrocytes or to astrocytes, oligodendrocytes, and ependymal cells of the CNS, and Müller cells of the retina.
Malacia—Grossly detectable (macroscopic lesion) softening of CNS tissue, usually the result of necrosis.
Meningo-—Combining form referring to meninges.
Meningomyelocele—A form of spina bifida in which meninges and spinal cord herniate through a defect in the vertebral column into a sac (-cele) covered by skin.
Microglia—Resident macrophages of the CNS that arise from mesodermal stem cells of the yolk sac.
Motor neuron, lower—Large multipolar neurons in the brainstem and ventral horns of the spinal cord with axons extending into the PNS.
Motor neuron, upper—Motor neurons with axons residing solely in the CNS that control lower motor neurons.
MVV—Maedi-visna virus.
Myelitis—Inflammation of the spinal cord.
Myelo-—Combining form referring to spinal cord.
Myelopathy—A degenerative disease process of the spinal cord.
Myeloschisis—Similar to spina bifida, except in its severe form is characterized by complete failure of the spinal neural tube to close, therefore a lack of development of the entire dorsal vertebral column.
Nageotte nodule—An aggregate of proliferating satellite cells (see later) in a ganglion in which there has been degeneration of neuronal cell bodies.
Neuroglial cells—Astrocytes, oligodendroglia, ependymal cells, and microglia of the CNS.
Neuronophagia—Accumulation of microglial cells around a dead neuron.
Neuropil—The gray matter feltwork that consists of intermingled and interconnected processes of neurons (axons and dendrites) and their synaptic junctions, plus processes of oligodendroglia, astrocytes, and microglia.
Neurapraxia—Traumatic injury to a peripheral nerve with temporary conduction block but with no permanent axonal damage.
Neurotmesis—Complete transection of a nerve and supporting framework with little potential for normal reinnervation.
Onion bulb—Concentric arrays of Schwann cell cytoplasm around an axon signifying multiple episodes of demyelination and remyelination.
PNS—Peripheral nervous system.
Polio—Combining form referring to gray matter of the CNS.
Polioencephalomalacia—Softening (usually the result of necrosis) of the gray matter of the brain.
Polioencephalomyelitis—Inflammation of the gray matter of the brain and spinal cord.
Poliomyelomalacia—Softening (usually the result of necrosis) of the gray matter of the spinal cord.
Porencephaly—A cleft or cystic defect in the cerebral hemisphere that communicates with the subarachnoid space and also may communicate with the ventricular system. The defect may contain CSF.
Radiculoneuritis (polyradiculoneuritis)—Inflammation of a spinal nerve rootlet (or rootlets).
Rarefaction—Reduction in density of CNS tissue that may result from edema, necrosis, and the like. This lesion is usually observed microscopically.
Satellite cells—Glial cells that cover the surface of nerve cell bodies in sensorimotor, autonomic, and enteric ganglia.
Satellitosis—An accumulation of oligodendroglia around neuronal cell bodies. Although this feature can be seen in normal brains, some consider that it may also be associated with neuronal injury.
Sclerosis—Literally means induration or hardening and, when used in describing lesions of the CNS, often refers to induration or hardening of the brain or spinal cord resulting from astrogliosis (astrocytic scar formation).
Spina bifida—A dorsal midline defect involving one to several vertebrae of the spinal column caused by failure of the neural tube to close, permitting exposure of the underlying meninges and spinal cord. The lesion may be associated with herniation of meninges alone or meninges and spinal cord tissue into a sac (-cele) covered by skin, or there may be no herniation (spina bifida occulta).
Status spongiosus—An encompassing term meaning the presence of small focal, ovoid to round “clear (unstained or poorly stained [H&E stain])” spaces in the CNS. The lesion can result from several different tissue alterations, which include splitting of the myelin sheath, accumulation of extracellular fluid, swelling of cellular (e.g., astrocytic and neuronal) processes, and axonal injury (Wallerian degeneration) when swollen axons are no longer detectable within distended spaces.
Syringomyelia—A tubular cavitation (syrinx) in the spinal cord that is not lined by ependyma and may extend over several segments.
Wallerian degeneration—Degeneration of the distal component of an injured (compressed or severed) axon. Although the term originally referred to injury of axons in the PNS, current usage also includes the CNS. This process also results in functional and structural alterations in the cell body (central chromatolysis) and proximal internode segment of the axon, and in secondary demyelination.
Nervous System
Development of the Adult Nervous System
The vertebrate embryo is formed by three layers of cells—the ectoderm (outermost layer), the mesoderm (middle layer), and the endoderm (innermost layer). Nervous tissues are derived from the ectoderm and eventually form all central nervous system (CNS) and peripheral nervous system (PNS) tissues of the adult animal. In the developing embryo, neurogenesis begins with a locally extensive, elongate proliferation of cells known as the neural plate, located along the cranial surface of the neuroectoderm (E-Fig. 14-1). The neural plate is bordered on either side by neural folds, which eventually fuse dorsally to form the neural tube (i.e., the eventual brain, spinal cord, and ventricular system). Immature neuroepithelial cells that line the neural tube ultimately become the source of neurons, astrocytes, ependymal cells, and oligodendrocytes. Resident microglial cells arise from mesodermal stem cells in the yolk sac that migrate to the neural tube during development, whereas the development and maturation of the brain and spinal cord proceed through a series of coordinated mechanisms characterized by cellular proliferation and subsequent remodeling (e.g., apoptosis) to produce the final morphologic features of the adult brain and spinal cord.
E-Figure 14-1.
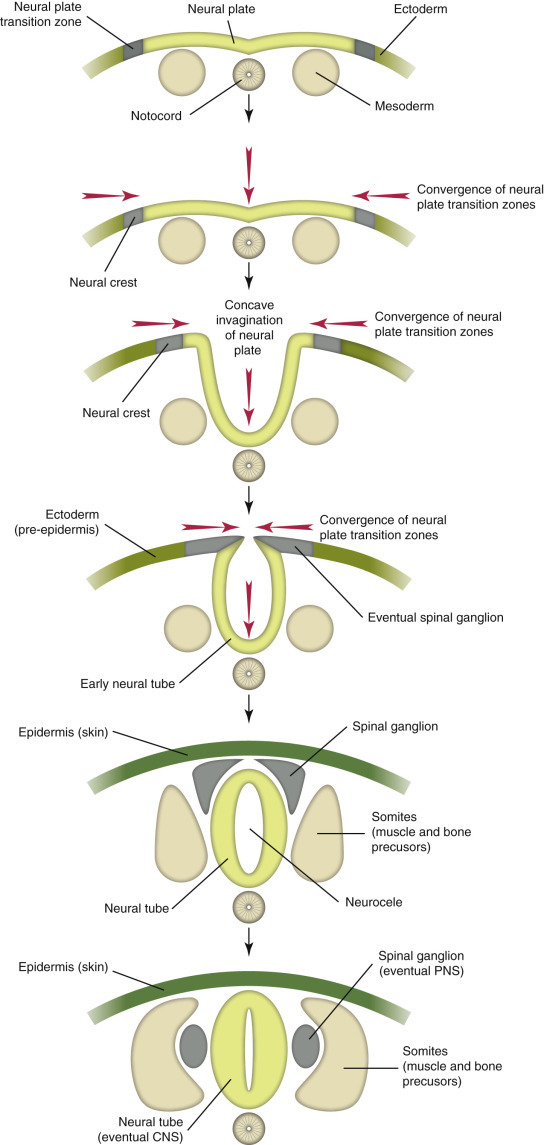
Neurogenesis of the Central Nervous System (CNS) and Peripheral Nervous System (PNS) in the Developing Embryo.
Neuroectoderm differentiates from ectoderm and forms the neural plate. The neural plate gives rise to neural crest cells and the neural tube, which in turn differentiate into PNS (spinal ganglion, peripheral nerves) and CNS (brain and spinal cord), respectively. The notochord eventually regresses, and portions remain as the nucleus pulposus of intervertebral disks.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Neuroepithelial cells, which form the spinal cord, reorganize during neurogenesis to produce centrally located gray matter (shaped like a butterfly with paired dorsal and ventral horns) and peripherally enveloping white matter (also see later section on gray and white matter). The white matter tracts of the spinal cord are further subdivided into funiculi, which contain variable numbers of ascending axons (i.e., action potential travels in the direction of the brain) and descending axons (i.e., action potential travels in the direction of the cauda equina). Segmentation (i.e., formation of the general regions of the brain and spinal cord) and stratification (i.e., formation of the lamina of the cerebral cortex) of the neural tube during embryologic development require a great deal more remodeling. Initial expansion of the neural tube results in a developing brain that is segmented into sections called the prosencephalon (forebrain), mesencephalon (midbrain), and rhombencephalon (hindbrain) (E-Box 14-1; E-Fig. 14-2). With further development the brain divides into the five segments: telencephalon, diencephalon (both derived from the prosencephalon), mesencephalon, and the metencephalon and myelencephalon (both derived from the rhombencephalon) (see E-Box 14-1). The telencephalon in the adult animal becomes paired cerebral hemispheres. The diencephalon becomes the thalamus and its associated structures. The mesencephalon gives rise to the corpora quadrigemina (superior and inferior colliculi) and the cerebral peduncles. The pons and cerebellum arise from the metencephalon, and lastly the medulla oblongata arises from the myelencephalon (see E-Fig 14-1). Concurrently, the spinal cord is segmented into cervical, thoracic, lumbar, and sacral sections, which are further subdivided into individual spinal nerves.
E-Box 14-1. Embryologic Development and Maturation of the Neural Tube.
| Neural tube | Prosencephalon (forebrain) | Telencephalon | Paired cerebral hemispheres |
| Diencephalon | Thalamus and related structures | ||
| Mesencephalon (midbrain) | Mesencephalon | Corpora quadrigemina (superior and inferior colliculi) | |
| Cerebral peduncles | |||
| Rhombencephalon (hindbrain) | Metencephalon | Pons and cerebellum | |
| Myelencephalon | Medulla oblongata | ||
| Spinal cord | Spinal cord | Cervical, thoracic, lumbar, and sacral spinal nerve levels |
E-Figure 14-2.
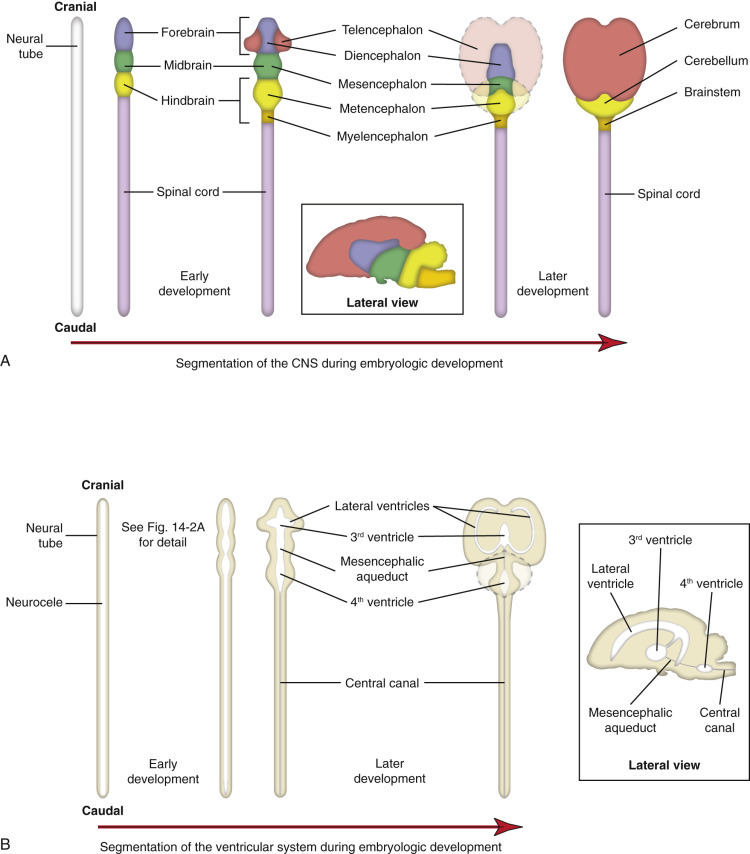
Segmentation and Stratification of the Neural Tube.
A, Formation of the central nervous system (CNS). B, Formation of the ventricular system.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The ventricular system develops in parallel with the brain and spinal cord. It originates from the space formed by the closing of the neural folds to form the neural tube and thus gives rise to the lateral ventricles, the third ventricle, the mesencephalic (cerebral) aqueduct, the fourth ventricle, and the central canal of the spinal cord. The dispersal of agents in the cerebrospinal fluid (CSF) to seemingly disparate regions of the CNS is explained by the interconnectedness of the ventricular system.
Following the development and differentiation of the brain into the five segments listed earlier, there is differential growth (i.e., occurs at different extents, rates, and times) in each of these parts of the developing brain. For example, the brainstem (pons and medulla) undergoes marked reorganization to form numerous specific nuclei that are the source of not only the majority of the cranial nerves, but also a neural relay network for the majority of neural impulses that arise in the prosencephalon (e.g., cerebral hemispheres and thalamus). It is also during this period of embryologic differentiation that stratification occurs within the cerebral cortex resulting in the formation of cerebral lamina. Lamina are distinct topographic layers of neuron cell bodies that have similar functions and innervate specific areas of the body as designated by these similar functions. They serve as topographic maps for specific activities within the CNS such as sensory, motor, and associative functions. Additionally, these activities are spread out into distinct “functional” lamina within lobes within the cerebral cortex, such as the frontal lobe (cognitive functions), parietal lobe (motor and sensory functions), occipital lobe (vision), and the temporal lobe (auditory functions). Lastly, the cerebellum undergoes significant reorganization with the development of multiple integrated layers of neurons, including the granule cell layer, the molecular cell layer, and the Purkinje cell layer.
The adult CNS is arranged to form two basic parts: the gray and white matter (Figs. 14-1 and 14-2 ). In the CNS, gray matter is found in the cerebral cortex, in the cerebellar cortex and cerebellar nuclei, around the base of the cerebral hemispheres (basal nuclei [often called basal ganglia]: caudate nucleus, lentiform nucleus [putamen, globus pallidus], amygdaloid nucleus, claustrum), and throughout the brainstem, often in nuclei. The gray matter is typified by numerous neuronal cell bodies, plus a feltwork of intermingled thinly myelinated axons and dendrites, their synaptic junctions, and processes of oligodendroglia, astrocytes, and microglia. This network of processes and synapses in the gray matter is referred to as the neuropil. The white matter consists of well-myelinated axons that arise from neuronal cell bodies in the gray matter and terminate distally in synapses or myoneural junctions, plus oligodendroglia, astrocytes, and microglia. In the cerebral hemispheres, white matter is located centrally, whereas in the brainstem, white matter is intermingled with gray matter (nuclei). In the spinal cord, white matter is located peripherally surrounding the gray matter.
Figure 14-1.
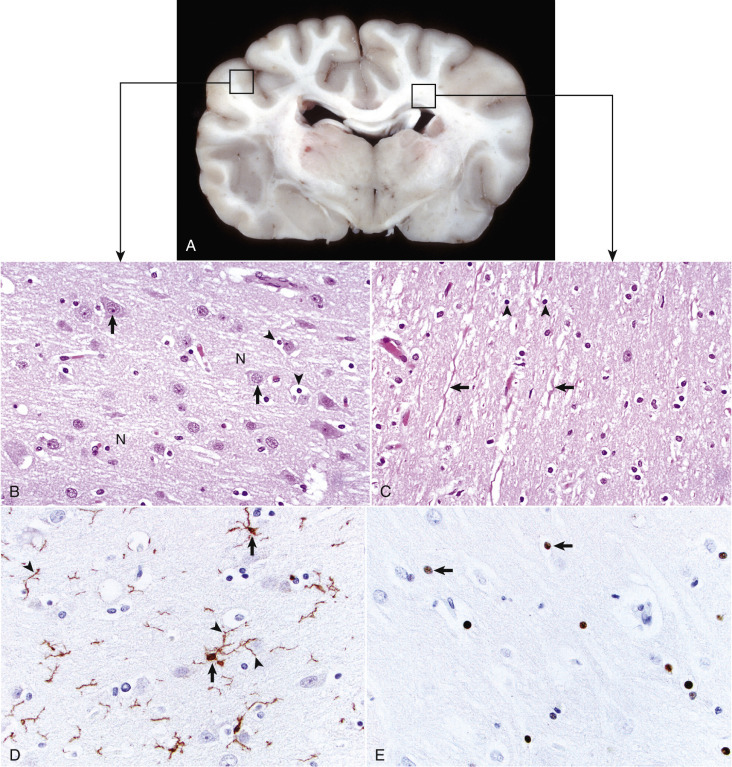
Organization of the Brain, Gray Matter, and White Matter.
A, Transverse section at the level of the thalamus, dog. Gray matter (darker areas) of the cerebral cortex lies beneath the leptomeninges on the external surface of the brain, whereas in the thalamus there is a mixture of gray and white matter. Major white matter areas (light areas) include corona radiata, centrum semiovale, and corpus callosum of the cerebrum, and internal capsule and optic tracts bordering the lateral and ventral surfaces of the thalamus, respectively. B, Gray matter consists primarily of the cell bodies of neurons (arrows) and a network of intermingled thinly myelinated axons, dendrites, and glial cell processes. This network is referred to as the neuropil (N). Other components include oligodendroglia (arrowheads), astrocytes, and microglia. H&E stain. C, White matter primarily consists of well-myelinated axons (arrows) plus oligodendroglia (arrowheads) and astrocytes. The clear spaces surrounding large axons are artifacts formed when the lipid components of myelin lamellae are dissolved away by solvents in the process of embedding tissue in paraffin for sectioning. H&E stain. D, Immunohistochemical (IHC) stain for ionized calcium binding adapter molecule 1 (Iba1). This IHC stain identifies microglia within a section of brain (arrows). Their ramified processes are similarly highlighted (arrowheads). DAB IHC stain. E, IHC stain for Olig2, a transcription factor that is expressed in the nucleus of oligodendrocytes (arrows). DAB IHC stain.
(A, B, and C courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. D and E courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
Figure 14-2.
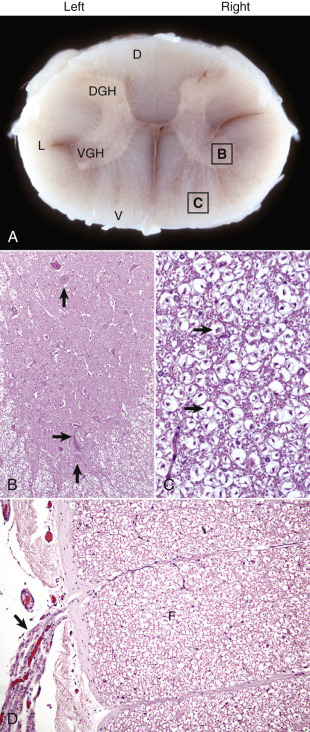
Organization of the Spinal Cord, Gray Matter, and White Matter.
A, White matter in the spinal cord is located peripherally and divided into dorsal, lateral, and ventral funiculi. As a general rule, dorsal funiculi (D) consist of ascending sensory axons, lateral funiculi (L) have a mixture of sensory and motor axons, and ventral funiculi consist of descending motor axons (V). Histologically, the right side is a mirror image of the left side. The areas labeled B and C and contained within the boxes correspond to the areas illustrated in B and C. B, Transverse section of spinal cord, ventral gray horn, horse. The cell bodies of large motor neurons (arrows) are those of lower motor neurons, and their axons extend in peripheral nerves to myoneural junctions that innervate skeletal muscle. H&E stain. C, Transverse section of spinal cord, ventral funiculus, horse. Because most axons course up and down the length of the spinal cord, in a transverse section, axons (arrows) are cut in cross section. They are surrounded by myelin sheaths whose lipid components are dissolved out during the preparation of paraffin-embedded sections, resulting in clear spaces that are an artifact. H&E stain. D, Efferent spinal nerve (longitudinal section shown here), transverse section of spinal cord, ventral funiculus, dog. Axons of lower motor neurons leave funiculi (F) and assemble as nerve rootlets (arrow) eventually forming peripheral nerves that innervate skeletal muscle. H&E stain. DGH, Dorsal gray horn; VGH, ventral gray horn.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The embryologic development of the PNS is as complex as the CNS and is also dependent on the normal development of the neural tube. A population of neural crest cells that form bilaterally in the dorsal regions of the neural tube is the origin of the majority of cells that populate the PNS, such as neurons and Schwann cells. These neural crest cells also migrate peripherally in developing tissues and organ systems within the mesoderm and endoderm to form structures such as spinal ganglia, enteric plexuses, and the adrenal medullas. Neurons of the PNS are divided into afferent and efferent types based on whether they conduct impulses to or from the CNS, respectively. Cell bodies for somatic efferent neurons, such as those in cranial or spinal nerves, are located in nuclei of the brain or the ventral horns of the gray matter of the spinal cord and project ventrolaterally long distances to innervate peripheral tissues, such as skeletal muscle. The cell bodies for somatic afferent neurons are located bilaterally within spinal ganglia (dorsal root ganglia) that are developmentally associated with specific spinal cord segments. The neurogenic control of visceral tissues and organ systems such as the alimentary system is complex; involves at a minimum two neurons, a preganglionic neuron and a postganglionic neuron; and is facilitated by the sympathetic, parasympathetic, and enteric nervous systems (i.e., visceral nervous systems). As a basic rule, for all three visceral nervous systems the preganglionic neuron cell body is located in the intermediate gray matter between the dorsal and ventral horns of the spinal cord or brain nucleus.
As noted earlier, during the development of the CNS a variety of differing cell types populate the brain and spinal cord, including the neurons, glia, ependyma, endothelial cells, pericytes and smooth muscle cells of blood vessels, and various cells in the meninges (Fig. 14-3 ; Box 14-1 ). Neurons vary in size, shape, and function, and their cell bodies are organized into functional groups such as nuclei, horns of gray matter in the spinal cord, and cerebral lamina. Neuronal processes called axons and dendrites traverse through the brain and spinal cord, the former often as organized bundles (tracts, fasciculi) forming synapses on cell bodies, dendrites, and axons of other functionally related neurons. It is estimated that there are 1 × 1011 neurons in the human brain. Each neuron makes approximately 10,000 synapses with other neurons; therefore there are approximately 1 × 1015 synapses in the human brain. The neurons maintain a close association with various glial cells, including microglia, astrocytes, and oligodendrocytes. The glia are responsible for helping to maintain CNS homeostasis and play an important role in the immune response and healing. Astrocytes, oligodendrocytes, and ependymal cells are derived from neuroectoderm, whereas microglia, part of the monocyte-macrophage system, are derived from progenitors in the embryonic yolk sac that populate the CNS during development. In the mammalian CNS, glia outnumber neurons 10 to 1. Ependymal cells line the ventricular system, whereas choroid plexus epithelial cells form the outer covering of the choroid plexuses. Lastly, the exterior of the CNS is covered by the meninges. The meninges consist of three layers named, from outermost to innermost layers, the dura mater, arachnoid, and pia mater. The arachnoid and pia enclose the subarachnoid space.
Figure 14-3.
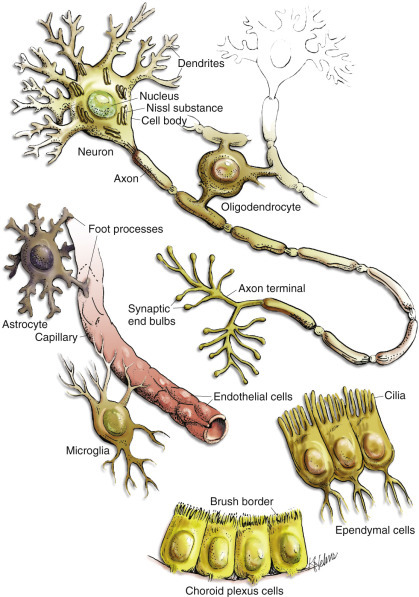
Cell Types in the Central Nervous System Include Neurons, Astrocytes, Oligodendroglia, Microglia, Ependymal Cells, Choroid Plexus Epithelial Cells, and Vascular Endothelial Cells.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Box 14-1. Cells of the Central Nervous System and Their Primary Functions.
Neurons
Transmission of electric and chemical impulses
Spatial and temporal interpretation of impulses
Inhibitory and stimulatory regulation of impulses
Astroglia (Protoplasmic [Type I] and Fibrous [Type II])
Regulation of extracellular neurotransmitter concentrations and fluid/electrolyte imbalances
Repair of injury by proliferation of astrocytic cellular processes
Support and bundling of functionally related axons traversing through the CNS
Participation in barrier systems
Glia limitans
Blood-brain barrier
Oligodendroglia
Myelination of axons within the CNS
Proposed neuronal cell body homeostasis within the CNS
Ependyma
Movement of CSF through the ventricular system
Choroid Plexus Epithelial Cells
Secretion of CSF
Barrier function (blood-CSF barrier)
Microglia
Immunosurveillance, immunoregulation, phagocytosis
Monocyte-macrophage system
Meninges
Arachnoid-CSF barrier
Subarachnoid CSF cushioning of head trauma
Endothelia
Barrier function (blood-brain barrier)
Selective molecule transport systems
CNS, Central nervous system; CSF, cerebrospinal fluid.
Central Nervous System
Structure2 and Function
Cells of the Central Nervous System
Neurons.
The structure and basic cellular biology of neurons is similar to that of other cells (Fig. 14-4 ); however, there are, as discussed later, some notable differences. The neuron consists of three structural components: dendrites, a cell body, and a single axon. The length of the axon varies, depending on the function of the neuron. The length of axons of motor or sensory neurons can be 10,000 to 15,000 times the diameter of the neuronal cell body, which results in these axons being several meters in length. The axon terminates in synaptic processes or neuromuscular junctions.
Figure 14-4.
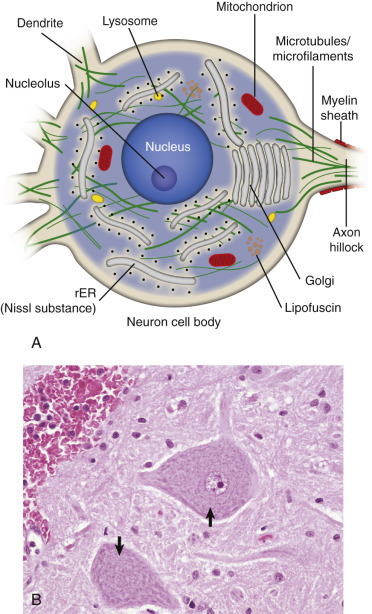
Neuron Structure.
A, Basic cell biology and structure of neurons are similar to other cells in the body. Additionally, neurons have dendritic arborizations and an axon, specializations for the initiation, propagation, and transmission of impulses that underlie the basic function of these cells. B, The cytoplasm of the neuronal cell body has blue (basophilic [H&E stain]) granular material (rough endoplasmic reticulum) called Nissl substance (arrows). Nissl substance synthesizes proteins, including precursor neurotransmitter proteins and the structural proteins (neurofilaments), active in maintaining the integrity (length and diameter) of the axon. H&E stain. rER, Rough endoplasmic reticulum.
(A courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Neuronal cell bodies vary considerably in size and shape, from the large neurons of the lateral vestibular nucleus, Purkinje cell layer of the cerebellum, and the ventral gray matter of the spinal cord to the very small lymphocyte-like granule cells of the cerebellar cortex (Fig. 14-5 ). Neuronal nuclei tend to be vesicular to spherical in shape, are usually centrally located, and often, particularly in large neurons, tend to contain a prominent central nucleolus. Neurons contain focal arrays of rough endoplasmic reticulum and polysomes, termed Nissl substance, that are responsible for the synthesis of proteins involved in many of the neuron's vital cellular processes such as axonal transport. Nissl substance is present in all neurons, regardless of the size of the cell body, but tends to be more prominent in those cells with voluminous cytoplasm such as motor neurons.
Figure 14-5.

Variations in Neuronal Morphologic Features, Cerebellum, Granule Cells, and Purkinje Neurons, Normal Animal.
The granule cell neurons of the cerebellar cortex (arrowheads) are very small basophilic cells that have relatively little demonstrable Nissl substance when compared with Purkinje neurons (arrows) and large motor neurons (depicted in Fig. 14-4, B). H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Axonal Transport (Axoplasmic Transport).
Axonal transport is a cellular mechanism used to move synaptic vesicles, proteins such as neurotransmitters, mitochondria, lipids, and other cell organelles from the neuron cell body through the axon to the synapses and then bring their degradation products back to the cell body.
As a result of structural differences between neurons and other cells, neurons have developed axonal transport systems to efficiently move molecules and cellular organelles from the cell body through the axon to the synapses and bring their degradation products back to the cell body (E-Fig. 14-3). Axons can be longer than a meter in length, especially in an animal such as a giraffe. Lower motor neurons, whose cell bodies lie in the ventral gray horn of the spinal cord, and lumbar dorsal root ganglia, whose axons extend both from the distal limb and the caudal medulla, have the longest axons in the body. The neuron expends considerable energy and materials to move biologic materials up and down the axon. Alterations in the function of these transport systems can lead to neuronal dysfunction.
E-Figure 14-3.
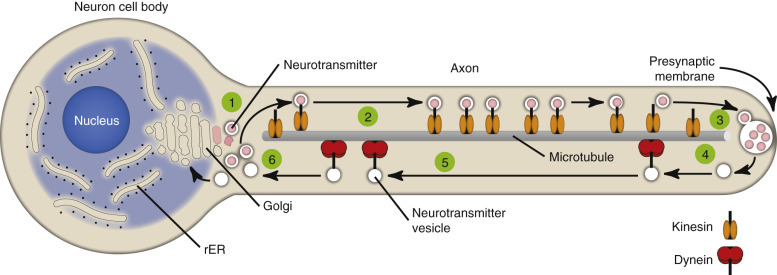
Axonal Transport Systems.
Neurotransmitter vesicles and neurofilament proteins, synthesized in the rough endoplasmic reticulum (rER) and packaged in the Golgi apparatus (1) are transported through the length of the axon (2) and to synapses by kinesin (3). Kinesin is a microtubule motor protein that uses chemical energy from adenosine triphosphate hydrolysis to generate mechanical force and thus bind to and move attached to microtubules. Used vesicles and effete neurofilament proteins (4) are returned along a microtubule (recycled) (5) to the neuron cell body (6) by cytoplasmic dynein, another microtubule motor protein. These transport systems are used by some pathogens (rabies virus, Listeria monocytogenes) to enter and spread within the central nervous system.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
These transport systems are divided into “fast axonal transport” and “slow axonal transport.” The fast axonal transport system has an anterograde component (toward the synapse) and a retrograde component (toward the cell body). The slow axonal transport system has only an anterograde component (toward the synapse).
Fast anterograde axonal transport (up to 400 mm per day) moves materials not intended for use in the cytoplasm of the neuron cell body. These materials formed from the Golgi apparatus are principally membrane-bound vesicles. They include mitochondria and membranous vesicles that contain peptide neurotransmitters, small transmitter molecules, and the enzymes necessary for their activation. These materials are moved down the axon on microtubules by specialized protein motors composed of kinesin and kinesin-related proteins using adenosine triphosphate (ATP) as an energy source.
Fast retrograde axonal transport (200 to 300 mm per day) returns endosomes, mitochondria, and catabolized proteins to the cell body of the neuron for degradation in lysosomes and reuse. This transported material is returned on microtubules, by dynein and microtubule-associated adenosine triphosphatase (ATPase) in the axon. This system will also transport certain toxins, such as tetanus toxin, and viruses, such as rabies virus, from the periphery via the PNS into the CNS.
Slow anterograde axonal transport (0.2 to 5 mm per day) transfers throughout the axon via microtubules, the major cytoskeletal proteins, such as microtubule and neurofilament proteins, which are necessary to maintain the structural integrity and transport systems within the axon.
Diseases of the axon that result directly or indirectly from alterations in axonal transport systems are discussed later. The character of the histologic lesions affecting injured nerve fibers can often be related to alterations in specific transport systems. Neurofilament proteins are synthesized in the neuronal cell body and are assembled and transported into axons. If neurofilaments accumulate in neuronal cell bodies and proximal axons, this lesion is called an axonopathy and is characterized by alterations in slow transport systems, which result in axonal swelling or atrophy and perikaryal neurofibrillary accumulations. Axonal injury and alterations in neurofilament transport can also cause secondary demyelination.
Membrane Potentials and Transmitter/Receptor Systems.
A fundamental activity of neurons is to modulate and effectively transmit chemical and electric signals from one neuron to another via synapses in the CNS or from one neuron to a muscle cell via junctional complexes, myoneural junctions, or motor end plates in the PNS. The process of nerve impulse conduction is made possible by the establishment and maintenance of an electric potential across the cell membrane of the neuron/axon.
A fundamental activity of neurons is to modulate and effectively transmit chemical and electric signals from one neuron to another via synapses in the CNS or from one neuron to a muscle cell via junctional complexes, myoneural junctions, or motor end plates in the PNS. The process of nerve impulse conduction is made possible by the establishment and maintenance of an electric potential across the cell membrane of the neuron/axon. Membrane potential is the difference in voltage between the inside and outside of the neuronal/axonal cell membrane and is called the resting potential. This potential is established and maintained by a membrane sodium ion (Na+)/potassium ion (K+)-ATPase (Na+/K+-ATPase) pump. The pump keeps the concentration of sodium ions outside the cell approximately 10 times greater than inside the cell, and the concentration of potassium ions inside the cell 20 times greater than outside the cell. The differences in concentrations of sodium ions outside and potassium ions inside of the cell membrane keep the membrane resting potential at approximately −70 mV. Thus the inside of the neuron/axon is 70 mV less than the outside. Sodium and potassium ions will leak across the cell membrane, and therefore concentration gradients are maintained by the Na+/K+-ATPase pump in the cell membrane. This established equilibrium and the membrane potential places the neuron in a “resting” condition, ready to generate an action potential.
An action potential arises when a neuron transmits information down an axon, away from the neuronal cell body. An action potential is initiated by an event that depolarizes the cell membrane and causes the resting potential to move toward 0 mV. When depolarization reaches a threshold level of approximately −50 mV, an action potential will occur. Once initiated, the strength of an action potential is always the same because the action potential is an intrinsic property of the neuron cell body and its axon.
Action potentials are caused by the movement of sodium and potassium ions across the neuron cell body/axon cell membrane. With an initiating event, sodium channels are first to open, and large concentrations of sodium ions enter the intracellular microenvironment (E-Fig. 14-4). Because sodium ions are positively charged, the polarity becomes more positive (−70 mV to −50 mV), and the neuron/axon becomes depolarized. Potassium channels open later in the depolarization process, concurrently with the closing of sodium channels. Potassium ions leave the cell and enter the extracellular fluid. These events cause repolarization of the neuron/axon and a return to a resting potential (−70 mV) via the membrane Na+/K+-ATPase pump. Alterations in these ion channels have been correlated with epilepsy in human beings and are starting to be discovered in animals.
E-Figure 14-4.
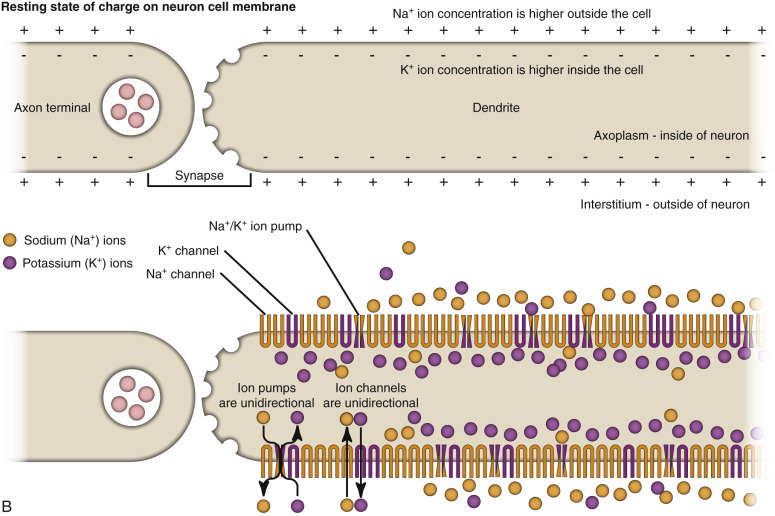
Resting and Action Potentials.
A, An action potential most commonly arises via neurotransmitter-induced chemical messages (depolarization) at synapses between presynaptic and postsynaptic neurons. It travels through dendrites, the cell body, and then down the axon to axon terminals of the postsynaptic neuron. B, Nerve impulse conduction occurs because of an electric potential sustained across the cell membrane of the dendrite/neuron cell body/axon. A resting membrane potential is maintained by differences in concentrations of potassium ions inside and sodium ions outside the cell membrane. When a neurotransmitter or other stimulus depolarizes the cell membrane to a threshold of approximately −50 mV, an action potential will occur. Sodium and potassium ions leak across the cell membrane, and therefore concentration gradients and thus the electric potential are maintained by sodium-potassium pumps in the cell membrane. C, When sodium channels are opened, an action potential occurs locally in the cell membrane. It is propagated down the cell membrane by the successive opening of voltage-gated sodium channels in adjacent sections of the membrane. Depolarized segments repolarize as sodium channels close and potassium ions move out of the cell.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Action potentials are most commonly initiated by neurotransmitters, such as acetylcholine, acting through synapses, but they also occur as a result of mechanical stimuli, such as stretching and sound waves. There are two main classes of synapses: inhibitory and excitatory. Stimulation of inhibitory synapses results in inhibitory postsynaptic potentials that cause hyperpolarization of dendrites and cell bodies. Hyperpolarization decreases the membrane potential (more negative, −80 mV), thus making the neuron less likely to reach the threshold for an action potential. Inhibitory neurotransmitters include γ-aminobutyric acid (GABA), glycine, dopamine, serotonin, norepinephrine (in the CNS), and acetylcholine (in heart muscle).
Stimulation of excitatory synapses results in excitatory postsynaptic potentials that cause depolarization of the dendrites and cell bodies. Depolarization increases the membrane potential (more positive, −50 mV), thus making the neuron more likely to reach the threshold for an action potential. Excitatory neurotransmitters include glutamate, norepinephrine in the PNS, and acetylcholine in skeletal muscle.
The generation of an action potential is a complicated process requiring depolarization of the cell membrane (−50 mV). Inhibitory and excitatory synapses and their inhibitory and excitatory postsynaptic potentials, respectively, are “summed” through processes, termed spatial and temporal summation, occurring in the dendritic network of the neuron. Spatial summation reflects additive input from different parts of the dendritic network, whereas temporal summation reflects additive input from stimuli that occur closely in time. This summation process is a graded potential and ultimately determines if the threshold for an action potential will occur.
The action potential is a flow of depolarization that travels down the axon to synapses at the distal axon. When the axon lacks myelin, the flow of depolarization down the axon is called continuous conduction. When the axon is myelinated, the speed of conduction is determined by the degree of myelination of the axon and is called saltatory conduction. The diameter of unmyelinated axons can range from 0.2 to 1 mm with action potential velocities ranging from 0.2 to 2 m/sec, whereas the diameter of myelinated axons can range from 2 to 20 mm with action potential velocities ranging from 12 to 120 m/sec. The greater the degree of myelination, the faster the speed of impulse conduction down the axon. In unmyelinated axons, action potentials are conducted at a relatively “slower” velocity by the process of ion exchange (continuous conduction). In myelinated axons, action potentials are conducted at a relatively “faster” velocity by a mechanism called saltatory conduction. In this process, action potentials move down the myelinated axon using cable properties, like electric current flow in insulated copper wires. This method is fast, efficient, and requires less energy than ion exchange. However, the action potential would decay if axons were myelinated continuously along their length and likely would not reach synapses at full strength or at all. This decay is caused by loss of current across the cell membrane and capacitance properties of the cell membrane as the action potential travels down the axon. To minimize the decay of action potentials, axons are myelinated in segments called internodes. A gap, called the node of Ranvier, is formed between consecutive internodes and measures between 0.2 and 2 mm in length. At this gap the action potential is restored to full strength by ion exchange. The node of Ranvier is highly enriched in sodium channels, and these channels are essential for impulse propagation via rapid action potential current restoration. Disease processes that disrupt myelination of axons will interfere with saltatory conduction, slow the action potential, and result in clinical dysfunction of the nervous system (see Fig. 14-21).
Figure 14-21.
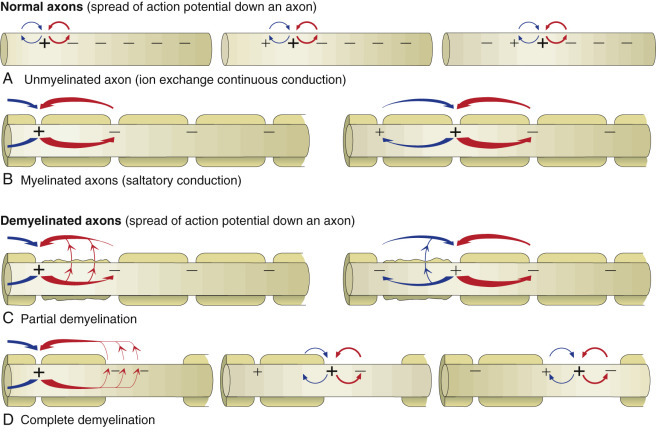
Axonal Action Potential Conduction and the Effect of Demyelination.
The speed of the conduction process is determined by the diameter of the axon and the degree of myelination. As axons increase in diameter, the resistance to ion flow decreases, allowing the action potential to flow faster. In addition, the degree of myelination is directly proportional to the diameter of the axon. Thus the concept that the more myelin the faster the speed of the impulse is true up to the point in which the myelin is normal in thickness. For an axon whose myelin is reduced, conduction of the action potential is slower. Under normal conditions, locomotion is a well-coordinated event that requires precise timing (speed) of impulse conduction to get coordinated movements. If the speed of the action potential is altered by disease, especially demyelination, then the conduction of the action potential will be delayed, and what are normally coordinated movements become uncoordinated. A, In unmyelinated axons, action potentials are conducted at a relatively “slower” velocity by the process of ion exchange continuous conduction (see E-Fig. 14-4). B, In myelinated axons, action potentials are conducted at a relatively “faster” velocity by a mechanism called saltatory conduction. Optimal function of saltatory conduction is dependent on having the proper degree of myelination of the axon (as determined by axonal diameter) throughout the full length of the axon. C, In axons that have lost some but not all of their myelin lamellae from one or more internodes so that there is a “thinner” covering of myelin, the speed of saltatory conduction is reduced because of leakage of the action potential across this thinner myelin sheath, resulting in clinical dysfunction of the nervous system. D, In axons that have lost all of their myelin from one or more internodes (complete primary demyelination of the internode), the speed of saltatory conduction is reduced because of the conversion from saltatory conduction to ion exchange continuous conduction in the areas where internodes have lost their myelin. Thus the speed and timing of the action potential is substantially reduced, leading to clinical dysfunction of the nervous system.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
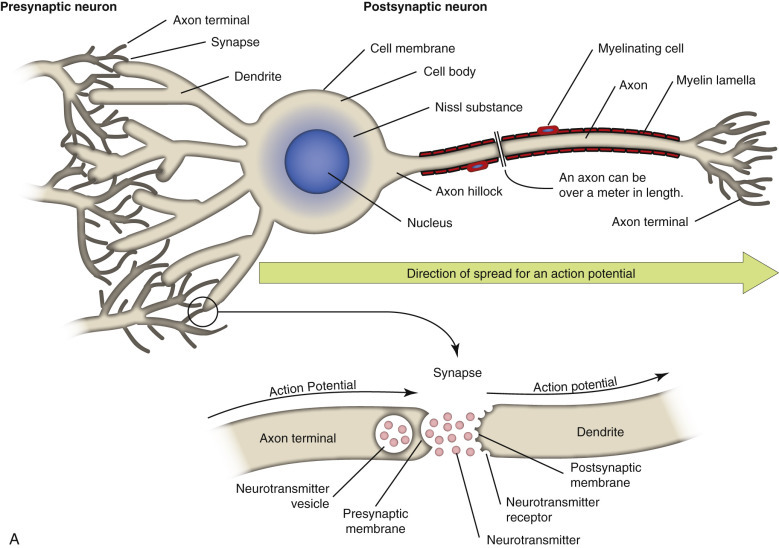
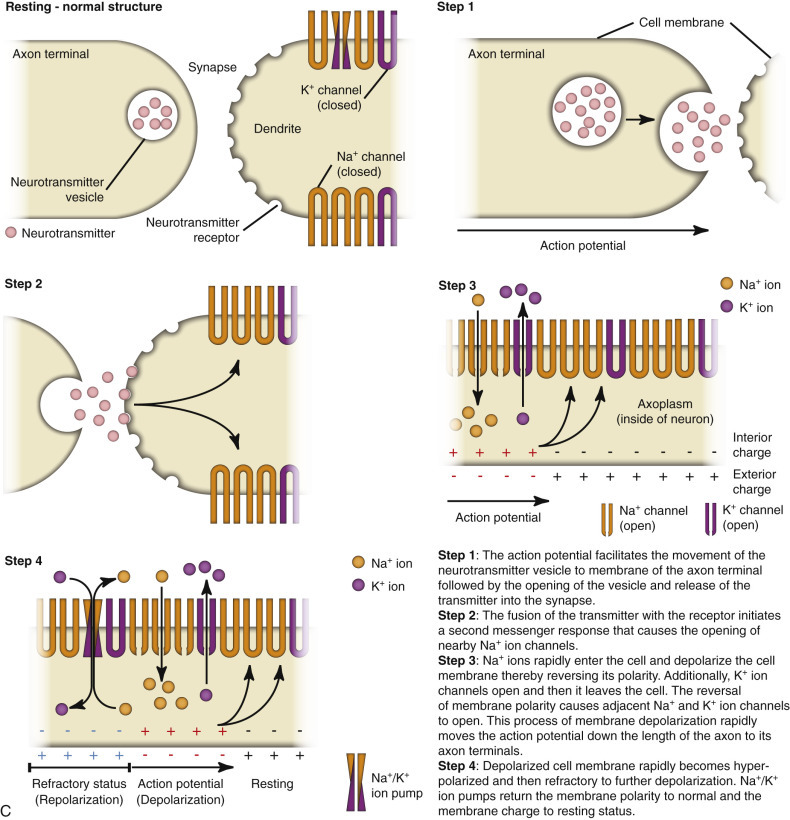
The axon can be a very long extension of the neuron cell body extending, for example, up to 2 m from the lumbar dorsal root ganglion in a giraffe. At its distal end the axon splits into several branches that end as specialized structures called axon terminals/terminal buttons/synaptic bulbs. Synapses present at these axon terminals are functional, and structural points of contact between “networked” neurons and these synapses convert the action potential into chemical signals that stimulate the next neuron in the conduction pathway. The cell membrane that releases chemical neurotransmitters is called the presynaptic membrane, and the cell membrane that has neurotransmitter receptors for the chemical neurotransmitters is called the postsynaptic membrane. These membranes are found on dendrites and cell bodies of the next neuron in the neural conduction pathway. The gap between the presynaptic and postsynaptic membranes that chemical neurotransmitters must cross is called the synaptic cleft. The mechanism of diseases, such as tetanus and botulism, is manifested through presynaptic and postsynaptic membrane receptors.
When an action potential reaches the axon terminal, it causes the release of chemical neurotransmitters from the presynaptic membrane by opening voltage-gated calcium channels, leading to membrane depolarization. The amount of chemical neurotransmitter released into the synaptic cleft is determined by the number of action potentials that reach the axon terminal over time. Chemical neurotransmitters traverse the synaptic cleft and bind to neurotransmitter receptors on dendrites and cell bodies of a new neuron in the neural conduction pathway.
There are two types of chemical neurotransmitter receptors, ionotropic and metabotropic, on the membrane of postsynaptic neurons. Functionally these receptor types differ in latency and duration of action. Ionotropic receptors have a fast response and short duration of effect, whereas metabotropic receptors have a slower response and a longer duration of effect. In addition, ionotropic receptors are localized to specific sites on the postsynaptic membrane, whereas metabotropic receptors are distributed diffusely and at random.
Chemical neurotransmitter stimulation of ionotropic receptors results in the opening of ion gates or channels, resulting in depolarization of the postsynaptic membrane. Excitatory neurotransmitters, such as glutamate, open postsynaptic membrane sodium channels. Inhibitory neurotransmitters, such as GABA, open postsynaptic membrane chloride channels.
Chemical neurotransmitter stimulation of metabotropic receptors results in the generation of a second messenger such as in the cyclic adenosine monophosphate (cAMP) pathway, which initiates a sequence of metabolic changes in the neuron. Metabotropic receptors are composed of protein subunits that span the postsynaptic cell membrane. An extracellular component of this protein has a high affinity for neurotransmitters and functions as a binding site. After binding the neurotransmitter, the receptor undergoes a configurational change that directly or indirectly activates a cell membrane enzyme, such as intracellular G proteins, leading to the formation of the second messenger. cAMP can activate protein kinase A–induced phosphorylation, leading to functional changes in ion channels and protein transcription. Dopamine is an example of a chemical neurotransmitter that uses metabotropic receptor pathways.
Astrocytes.
The functions of astrocytes in the CNS are regulation, repair, and support, as depicted in Figure 14-6 . All regions of the CNS contain astrocytes, and they are derived from pluripotential neuroepithelial progenitor cells during the development of the CNS. Astrocytes are the most numerous cell type in the CNS and have traditionally been classified into two types based on morphologic features. Protoplasmic astrocytes are located primarily in gray matter, whereas fibrous astrocytes occur chiefly in white matter. Microscopically, astrocytes have relatively large vesicular nuclei, indistinct or inapparent nucleoli, and no discernible cytoplasm with routine hematoxylin and eosin (H&E) staining (Fig. 14-7 ). With suitable histochemical stains, silver impregnation, or immunohistochemical staining for glial fibrillary acidic protein (GFAP [the major intermediate filament in astrocytes]), the cell body and the extensive arborization and interconnections of astrocytic processes can be demonstrated. Processes vary from short and brushlike to long branching processes in protoplasmic and fibrous astrocytes, respectively (Fig. 14-8 ). Expression of GFAP is the standard immunohistochemical marker for tumors of astrocyte origin and can also be used to qualitatively or quantitatively characterize disorders in which astrocytes are proliferative or reactive. However, caution should be taken when assessing astrocyte numbers and/or the extent of ramification of their processes because GFAP immunoreactivity can be diminished in terminal processes and/or cell bodies, and therefore the total GFAP immunoreactivity in any given section of brain may not be representative of the overall astrocytic response in the disorder.
Figure 14-6.
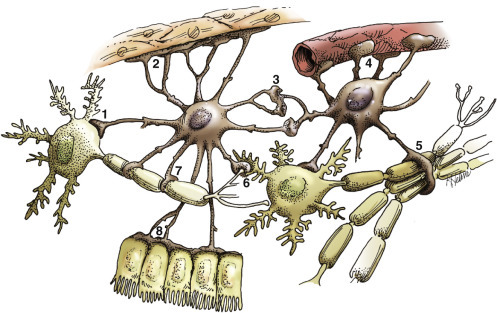
Functions of Astrocytes.
Astrocytes provide structural integrity and regulatory oversight, as depicted in this diagram. They: 1, monitor and regulate fluid and electrolyte balances within neurons and surrounding extracellular space; 2, form the glial limitans at the base of the pia mater; 3, interconnect with other astrocytes to provide a system to monitor and regulate fluid and electrolyte balances throughout the central nervous system (CNS); 4, participate in the formation and functions of the blood-brain barrier; 5, participate in the support of axon tracts of functionally related neurons; 6, monitor for and remove excessive release of neurotransmitters in synapses; 7, protect and insulate nodes of Ranvier; and 8, participate in the cerebrospinal fluid–brain barrier. In addition, astrocytes are a reparative (healing) cell after CNS injury with loss of tissue because nervous tissue, per se, is devoid of fibroblasts. Fibroblasts exist in the meninges and around blood vessels. Everywhere else, healing depends on the astrocyte, which responds by increased length, branching, and complexity of cellular processes (astrogliosis). The astrocyte has many functions in the nervous system; one of them is to act in healing to produce a scar in attempts to isolate cavities and abscesses. Fibroblasts may also contribute to the formation of a scar, if this cell type is present, as it is in the leptomeninges.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 14-7.
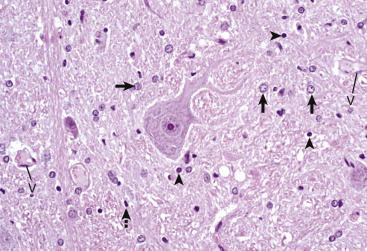
Histologic Features of Glial Cells, Ventral Gray Horn, Spinal Cord, Horse.
A neuronal cell body and its processes are in the center of the illustration. To the inexperienced, identifying specific types of glial cells in H&E-stained histologic sections can be challenging. Astrocytes (arrows) have larger vesicular nuclei (dispersed chromatin), and the cell membrane and cytoplasm are rarely seen in nondiseased conditions. Thus these nuclei just seem to “sit” in the midst of the neuropil. The majority of nuclei in the neuropil here are astrocytic. Oligodendroglial cells (arrowheads) have smaller and dense round nuclei (condensed chromatin) often surrounded by a clear zone indicative of cell cytoplasm and a cell membrane. Oligodendroglial cells in gray matter are called perineuronal satellite cells; those in white matter are called interfascicular oligodendrocytes. Microglial cells can be difficult to identify in H&E-stained sections of the central nervous system (CNS) but are often identified by their small, dense elongated nuclei (dashed arrow). The light pink homogeneous tissue distributed in large quantities between these cell types is the neuropil. V, Blood vessels. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 14-8.
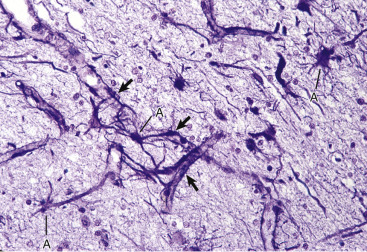
Astrocytic Processes, Brain, Cerebral Cortex, Normal Animal.
Processes of astrocytes arborize extensively throughout the central nervous system (structures stained purple). Note that some of the processes are on the outside of blood capillaries (end feet) (arrows). Holzer's stain. A, Cell body of astrocyte.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Functions of Astrocytes
Regulation of the Microenvironment.
The microenvironment of the CNS must be under strict control to maintain normal function. Astrocytes are involved in homeostasis of the CNS and regulate ionic and water balance, antioxidant concentrations, uptake and metabolism of neurotransmitters, and metabolism or sequestration of potential neurotoxins, including ammonia, heavy metals, and excitatory amino acid neurotransmitters such as glutamate and aspartate. Structurally, the homeostatic role of astrocytes is illustrated by the morphologic characteristics of the neuropil, where astrocytic processes surround synapses and maintain a microenvironment that is adequate for normal synaptic transmission.
Additionally, interactions between astrocytes, microglia, and neurons orchestrate immune reactions in the brain. In this regard, astrocytes can express major histocompatibility complex (MHC) class I and II antigens, a variety of cytokines and chemokines, and adhesion molecules that modulate inflammatory events in the CNS. Astrocytes also secrete growth factors and extracellular matrix molecules that play a role not only in embryonic development but also in repair of the CNS following injury. In this latter role, astrocytes can fuse with adjacent astrocytes via a variety of gap junctions, and the coupling of multiple astrocytes together can play an important role in normal CNS function and repair (see next section). The gap junctions between various astrocytes are mediated by connexins. Astrocytes also play an integral role in CNS metabolism and can accumulate glycogen that can later be used to sustain neurons, especially during periods of hypoglycemia.
Repair of Injured Nervous Tissue.
In the CNS, reparative processes that occur after injury, such as inflammation and necrosis, are chiefly the responsibility of astrocytes. In these reparative processes, astrocytes are analogous to fibroblasts in the rest of the body. Astrocytes do not synthesize collagen fibers, as do fibroblasts. Instead, repair is accomplished by astrocytic swelling and division, and abundant proliferation of astrocytic cell processes containing intermediate filaments composed of GFAP, a process called astrogliosis. As an example, neuronal necrosis occurs in some viral diseases of the CNS. When neurons die, the spaces left by the loss of the neuronal cell bodies are filled, and such spaces (<1 mm in diameter) are filled by processes of astrocytes. Larger spaces that form after injury, such as an infarct, are often too large to be filled and therefore exist in the CNS as fluid-filled spaces (cysts) surrounded by a capsule of astrocytic processes. Astrocytes will also attempt to wall off abscesses, but they are not as effective as fibroblasts, and the capsule can be incomplete or weak (Fig. 14-9 ). In the case of direct extension of bacteria from the meninges or meningeal blood vessels, which contain or are surrounded by fibroblasts, respectively, fibroblasts play a larger role in isolating the inflammatory process.
Figure 14-9.
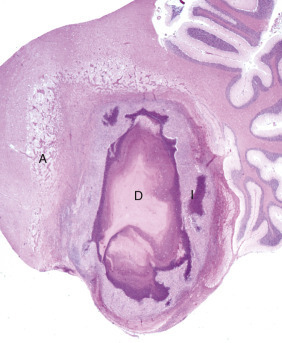
Astrocytic Repair, Bacterial Abscess, Brainstem, Sheep.
The abscess has a central core of necrotic debris (D) surrounded by a layer of inflammatory cells (I) and a less dense pink-staining zone representing an attempt by astrocytes and fibroblasts to form a capsule (A). This capsule is formed by fibrous tissue on the ventral and right sides, those sides closest to the pia, which contains fibroblasts. A fibrous capsule is absent from the dorsal and left sides of the abscess, adjacent to brain parenchyma. Here, there is no population of resident fibroblasts, and the capsule is formed by astrocytes and their processes, which are often delicate and do not form an effective capsule (A). H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Structural Support of the Central Nervous System.
Structurally, astrocytic processes provide support for other cellular elements and ensheathe and insulate synapses. Astrocytes also provide guidance and support of neuronal migration during development; thus tracts and fasciculi of axons with similar functions are arranged and structurally supported by astrocytic processes. Processes of astrocytes (foot processes) also terminate on blood vessels throughout the CNS, forming a component of the blood-brain barrier. Astrocytes influence the induction of tight junctions between endothelial cells that serve as the structural basis for the blood-brain barrier. A dense meshwork of astrocytic processes also forms the glia limitans beneath the pia mater and is variably prominent in subependymal areas. During CNS development, cells termed radial glia provide a scaffold and guidance for migrating neurons. When development is completed, radial glia mature into astrocytes. Some of these radial glia (also known as radial neural stem cells) remain active throughout life in the subventricular zone of the lateral ventricles, where they can repopulate lost populations of glial cells.
Oligodendroglia.
There are two types of oligodendroglia: (1) interfascicular oligodendrocytes and (2) satellite oligodendrocytes (satellite cells). The function of interfascicular oligodendroglia is myelination of axons, whereas the function of satellite oligodendroglia is thought to be regulation of the perineuronal microenvironment. Oligodendroglia have been compared with neurons with regard to their total cell size in that their processes occupy much more space than the cell body. Neurons have very long axons, which account for their size; oligodendroglia have extensive myelin sheaths, which account for their size. In H&E-stained sections, oligodendroglia are often confused with lymphocytes because of the similarity of the morphologic features of their nuclei and cytoplasmic volume. Interfascicular oligodendroglia and perineuronal satellite oligodendroglia are located primarily in white and gray matter of the CNS, respectively (Fig. 14-10 ); however, interfascicular oligodendroglia can also be found along axons that traverse through the gray matter. The mature, small oligodendrocyte has a spherical, hyperchromatic nucleus (see Figs. 14-7 and 14-10). As with astrocytes, the cell body and processes of this cell do not stain with conventional H&E staining methods and can only be demonstrated following special procedures that include metallic (silver) impregnation and immunohistochemical methods, including CNPase and Olig2.
Figure 14-10.
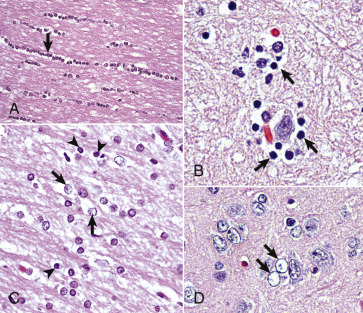
Responses of Glial Cells to Injury in H&E-Stained Central Nervous System (CNS) Sections.
A, White matter. In nondiseased states, oligodendroglia in white matter are often arranged linearly (interfascicular oligodendroglia) (arrow) and are responsible for the formation of myelin around axons. In gray matter (not shown; see Fig. 14-17), oligodendroglia are dispersed as individual cells around neuronal cell bodies as perineuronal satellite cells (B). H&E stain. B, Gray matter. When neurons are injured or there exists some perturbation of the perineuronal microenvironment, oligodendroglia around neurons can hypertrophy and proliferate in a process referred to as satellitosis. Perineuronal satellite oligodendroglia (arrows) surround a small degenerate neuron with condensed chromatin and little cytoplasm. H&E stain. C, White matter. Astrocytes (arrows) and oligodendroglia (arrowheads) have a limited repertoire of responses to injury in the CNS. Astrocytic proliferation can occur but is very difficult to determine in sections stained with H&E. Here, astrocyte nuclei are somewhat enlarged and appear more numerous than expected. H&E stain. D, Gray matter. Astrocytes respond to injury in hyperammonemia, such as occurs with hepatic encephalopathy, by forming astrocytes with enlarged, markedly vesicular (“watery”), often elongated nuclei called Alzheimer's type II astrocytes (arrows). This type of astrocyte may occur in pairs that are surrounded by a clear space indicative of cellular swelling. H&E stain.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B to D courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Most interfascicular oligodendroglia (see Fig. 14-10) are aligned in rows parallel to myelinated axons and are responsible for the formation and maintenance of segments (internodes) of myelin sheaths. One oligodendroglial cell can form as many as 50 different internodes of myelin, each of which can be located on many different axons (Fig. 14-11 ). Altered function of oligodendroglial cells, as occurs in infectious canine distemper virus (CDV) infection, can cause primary demyelination of these segments, resulting in severe neurologic dysfunction. Oligodendroglia also influence maturation and maintenance of axons and inhibit regeneration of established myelinated axons.
Figure 14-11.
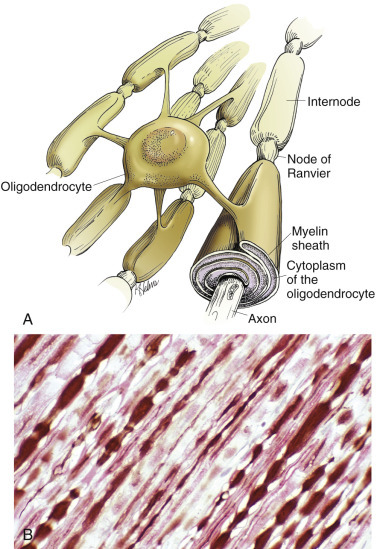
Central Nervous System (CNS) Myelin.
Oligodendroglia myelinate axons within the CNS (also see Fig. 14-3). A, As depicted in this illustration, each oligodendrocyte sends out numerous cytoplasmic processes that repetitively encircle (myelinate) the portion of an axon between two nodes of Ranvier (internode) on the same and several different axons. Direct or indirect injury to an oligodendrocyte can result in “demyelination” of those internodes myelinated by that oligodendrocyte. This injury will slow the rate of conduction of an action potential and depending on the site of the lesion, may lead to clinical signs of neural dysfunction (ataxia, proprioception deficits). B, CNS nerves, longitudinal section. Axons and their neurofilaments (brown stain) and myelin (red stain) are demonstrated by this immunohistochemical stain for neurofilament and myelin basic protein.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Perineuronal satellite oligodendroglia (see Fig. 14-10) are adjacent to neuronal cell bodies and are also located around blood vessels in the gray matter. They are thought by some investigators to regulate the perineuronal microenvironment and respond to perturbation by proliferation. When the perineuronal microenvironment is altered or neuron cell bodies are injured, perineuronal satellite oligodendroglia, in an attempt to regulate the environmental perturbation, hypertrophy and proliferate in a process referred to as satellitosis. However, this term is imprecise as other glial cells can also contribute to sattelitosis. Similarly, alterations in the microenvironment of gray and white matter away from areas surrounding neuron cell bodies result in hypertrophy of oligodendroglia (see Fig. 14-10). It should be noted that in some sections of the normal CNS there are increased numbers of oligodendrocytes that surround neurons, giving a false impression of pathologic satellitosis. This arrangement is especially true for interstitial white matter neurons found in the cerebral cortices.
Microglia.
The basic functions of microglia are immunosurveillance, immunoregulation, and reparative (phagocytic) activities after neural cell injury and death. Resident microglia originate from mesodermal stem cells in the yolk sac and enter and populate the CNS during embryonic development and early postnatal life, analogous to the formation of the monocyte-macrophage system in other organs. Microglia can become amoeboid by phagocytosing dead cells and cellular debris during remodeling and maturation of the CNS. Amoeboid cells then enter a quiescent stage and transform into ramified microglia. Ramified microglia constitute up to 20% of the glial cells and are present throughout the mature CNS, serving as sentinels of brain injury. Ramified microglia, also called resting cells, are most numerous in perineuronal and perivascular areas and in interfascicular locations in white matter. Evidence of pinocytosis in ramified cells suggests some role in maintaining the neural microenvironment. The principal function of microglia is phagocytosis, the initiation of and participation in the innate and adaptive immune responses, and in degenerative and inflammatory diseases of the CNS.
Microscopically, ramified microglia have small, hyperchromatic ovoid-, rod-, or comma-shaped nuclei and no appreciable cytoplasm with routine H&E staining; thus the term rod cell is sometimes used to describe them (see Fig. 14-7). With special labeling techniques or metallic impregnation, ramified cells have delicate branching processes. The small hyperchromatic nuclei and nuclear shape distinguish microglia from astrocytes and oligodendroglia. However, microglia are often difficult to identify in H&E-stained sections without some expertise in neuropathology.
Activated microglial cells are not the major source of active macrophages in inflammation of the CNS. Blood monocytes recruited from the circulation account for up to 70% of the macrophages in inflammatory and degenerative diseases of the CNS. These macrophages differentiate from blood monocytes involved in normal “leukocytic trafficking” through the CNS and can be involved in immunologic and phagocytic responses (gitter cells) to disease processes and infectious microbes. These macrophage populations are found mainly in the leptomeninges, choroid plexus, and perivascular areas.
Ependyma (Including Choroid Plexus Epithelial Cells).
The basic functions of ependymal cells, which line the ventricular system, are to help move CSF through the ventricular system via movement of their cilia and to regulate the flow of materials between the CNS and the CSF. The ependyma is a single-layered, cuboidal to columnar epithelium that lines the ventricles and mesencephalic aqueduct of the brain and central canal of the spinal cord (Fig. 14-12 ). This layer of cells is therefore situated between the CSF and nervous tissue. Ependymal cells have cilia that project into the CSF and beat in a coordinated manner in the direction of CSF flow. Other structures, referred to as circumventricular organs, which include the choroid plexuses, are covered by highly specialized ependymal cells. The surface of ependymal cells that form the choroid plexus have microvilli (microvillus border) and cilia that occur singly or more often in groups of three or more. The choroid plexus epithelial cells also have specialized tight junctions (zonulae occludens) that are a functional part of the blood-CSF barrier. In contrast to the choroid plexus, junctions between the conventional ependymal cells include gap junctions (transmembrane proteins form a pore, allowing communication between adjacent cells) and zonulae and fasciae adherentes, which permit movement of materials, such as proteins from the CSF, into the extracellular space of the brain. This cellular lining, however, is not a static membrane in that it regulates several processes that involve interaction between the CSF and brain. The functions include regulation of fluid homeostasis between the ventricular cavities and the brain, secretion and absorption of CSF, endocytosis, phagocytosis, and metabolism of substances such as iron resulting from the lysis of erythrocytes after hemorrhage into the ventricular system. Finally, ependymal cells have the structural and enzymatic characteristics necessary for scavenging and detoxifying a wide variety of substances in the CSF.
Figure 14-12.
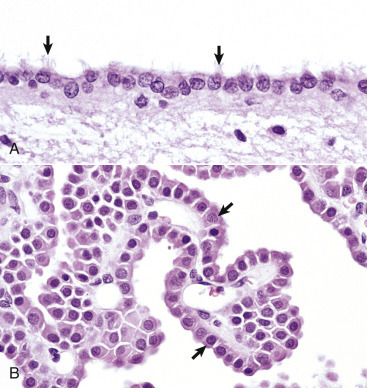
Ependymal and Choroid Plexus Epithelial Cells.
A, Ependymal cells are ciliated (arrows) and assist with the flow of cerebrospinal fluid (CSF) through the ventricular system. H&E stain. B, Choroid plexus epithelial cells (arrows) produce CSF from a brush border (microvilli) on the luminal surface. The surface of the choroid plexus also has cilia that occur singly or more often in groups of three or more on a single cell. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
During embryonic development the medial wall of the lateral ventricle (choroid fissure), the roof of the third ventricle, and the rostral part of the roof of the fourth ventricle consist of a single layer of neuroectoderm that is adherent on its outer surface to the pia mater. This neuroectoderm-pia union forms the tela choroidea, providing an anchor for the choroid plexuses, which is formed by an invagination of this bilayer membrane into the ventricular spaces.
Choroid plexus epithelial cells are modified ependymal cells. The choroid plexus epithelium is a single-layered, cuboidal to columnar epithelium with a microvillus border (see Fig. 14-12). CSF is secreted from the microvillus border. Choroid plexus epithelial cells, along with capillaries and the pia mater, form the choroid plexuses that project into the lateral, third, and fourth ventricles. The basic function of choroid plexuses is to produce the CSF that fills the ventricular system and the subarachnoid space. CSF has two important functions: (1) to act as a “shock absorber” to mitigate the effects of trauma to the brain and spinal cord and (2) to deliver nutrients to and remove wastes from the CNS.
The normal flow pattern of CSF is regulated by an intraventricular biologic pressure gradient in which the pressure created by secretion of CSF exceeds the pressure created by its absorption in arachnoid villi (arachnoid granulations). Arachnoid villi are focal extensions of the arachnoid and subarachnoid space that extend into the dorsal sagittal venous sinus of the brain. CSF is secreted by the choroid plexuses in the lateral, third, and fourth ventricles. It should be noted, however, that fluid from other sources, such as secretion by the ependyma, interstitial fluid of the brain, and ultrafiltrate of the blood, has also been reported to contribute to the formation of CSF. It moves from the lateral ventricles into the third ventricle, from the third ventricle through the mesencephalic aqueduct (aqueduct of Sylvius in human beings), and then to the fourth ventricle. Once in the fourth ventricle, the CSF exits through the two lateral apertures of the fourth ventricle to enter the subarachnoid space. Lateral apertures are the two openings in the caudal medullary velum that forms the roof of the fourth ventricle into the subarachnoid space, one at each side of the cerebellopontine angle. Although the central canal of the spinal cord is connected to the ventricular system at the caudal end of the fourth ventricle, there apparently is little active movement of CSF within the central canal. CSF in the subarachnoid space is reabsorbed by the arachnoid villi in the meninges. Recent evidence indicates that other routes of CSF drainage, in addition to arachnoid granulations, also exist and vary in different species. Venous sinuses, lymphatic drainage, and the cribriform plate appear to play important roles in CSF drainage and the maintenance of normal interventricular CSF pressure. In fact, experimental evidence suggests that the cribriform plate route may be the most important of the four. In human beings the entire volume of CSF is circulated approximately four times a day; however, with aging, the entire volume of CSF circulates less than two times a day.
Meninges.
The meninges, which enclose the CNS, consist of three layers: the dura mater (outermost layer), the arachnoid membrane, and the pia mater (innermost layer) (Fig. 14-13 ). Together, the arachnoid membrane and pia mater are frequently referred to as the leptomeninges, pia-arachnoid layer, or pia-arachnoid. The arachnoid membrane and pia mater are held together by bands of fibrous tissue called arachnoid trabeculae. This arrangement forms a compartment called the subarachnoid space in which CSF flows and which also contains blood vessels and nerves. The leptomeninges form a protective covering for the CNS and provide an external envelope filled with CSF that provides additional protection.
Figure 14-13.
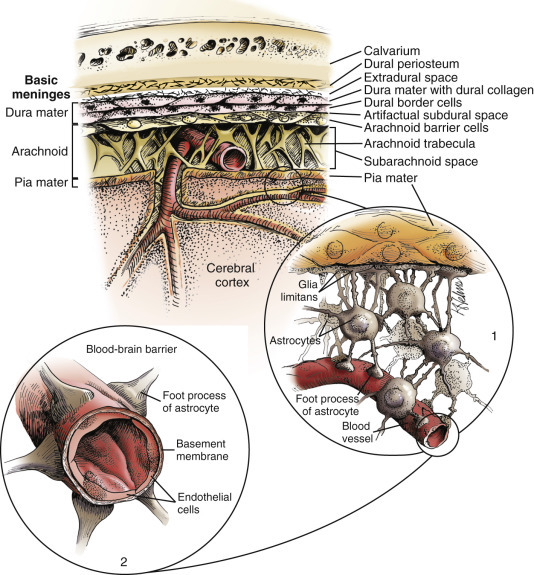
Organization of the Meninges.
The meninges, from outside to inside, are the dura mater, arachnoid mater, and pia mater as illustrated in the diagram. The arachnoid mater and the pia mater form the leptomeninges. These two layers of the leptomeninges also enclose the subarachnoid space, which contains the arteries, veins, and nerves and is filled with cerebrospinal fluid. The pia mater is attached to the surface of the central nervous system (CNS). Astrocytes and their foot processes underlie the pia mater and form the glia limitans (inset 1) and surround the endothelial cells that form the blood-brain barrier. As arterioles penetrate the cortex to supply the tissue with blood, they carry the pia and glia limitans with them for 1 to 3 mm until the arteriole structurally becomes a capillary. At this transition site within the cortex, the capillary penetrates the pia and is surrounded by the glia limitans, and the end feet of the astrocytes become part of the blood-brain barrier (inset 2). Components of the blood-brain barrier are capillary endothelial cells, basement membrane, and astrocytic foot processes, but the barrier is formed structurally by tight junctions between endothelial cells and functionally by specialized transport systems in these cells.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The dura mater, once referred to as the pachymeninx (thick meninges), is a strong and dense collagenous membrane (Fig. 14-14 ). In the cranium the dura consists of two layers that are fused with each other. The outer layer serves as the periosteum of the cranial bone, except in the areas of the venous sinuses (surrounded by dura) and falx cerebri, which is the longitudinal layer that extends ventrally between the two cerebral hemispheres. At the level of the foramen magnum, the two layers become separated; the outer layer continues to function as the periosteum of the vertebral (spinal) canal, and the inner layer forms the free dural membrane that surrounds the spinal cord. The inner aspect of dura mater is lined by elongated, flattened mesothelial-like cells. Except in neonates, there is no epidural (extradural) space in the cranial vault as there is in the spinal cord. There can be a “potential” epidural or extradural space in mature animals from hemorrhage caused by trauma.
Figure 14-14.
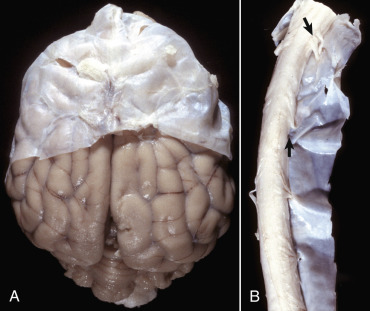
Layers of the Meninges.
A, Brain, dog. The dura matter is a thick opaque layer. Here it covers the rostral (cranial) half of the brain and has been dissected away from the caudal half of the brain to expose the underlying leptomeninges. In old animals the dura mater often fuses with the periosteum of the calvaria, and at necropsy to expose the brain, it is usually removed attached to the calvaria. The leptomeninges are present, but because they are so transparent, they are barely visible on the surface of the caudal half of the brain between gyri. B, Spinal cord, horse. The dura mater is the thick opaque layer dissected from and lying to the right of the spinal cord. The leptomeninges (pia-arachnoid layer) are present (but not readily visible in this photograph) on the exposed surface of the spinal cord. Arrows indicate spinal nerve roots.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The arachnoid consists of both the multilayered membrane composed of cells that overlap one another and the trabeculae that join it to the pia. The arachnoid has tight junctions between its cells, although other junctions have also been described. It contains no blood vessels and has an outer smooth surface formed by mesothelial-like cells that abut similar cells in the dura mater. The mesothelium-like surfaces of the dura and arachnoid oppose and slide over each other, analogous to the parietal and visceral surfaces of other serous membranes.
The pia mater is closely adherent to the surface of the brain and spinal cord and is penetrated by a large number of blood vessels that supply the underlying nervous tissue (Fig. 14-15 ). The pia mater consists of flat, thin, overlapping connective tissue cells (fibroblasts) that are separated from the underlying neural tissue by variable amounts of loose collagen fibers and the glia limitans. In many areas the pia, which lacks a basal lamina, is only one-cell-layer thick and has fenestrations, so that the glia limitans is exposed directly to the subarachnoid space. Pial and arachnoid cells also ensheathe blood vessels, collagen bundles, and nerves that are within or cross the subarachnoid space and also are around arteries that penetrate into the CNS up to 1 to 2 mm in depth. Macrophages including dendritic cells also are present throughout the leptomeninges.
Figure 14-15.
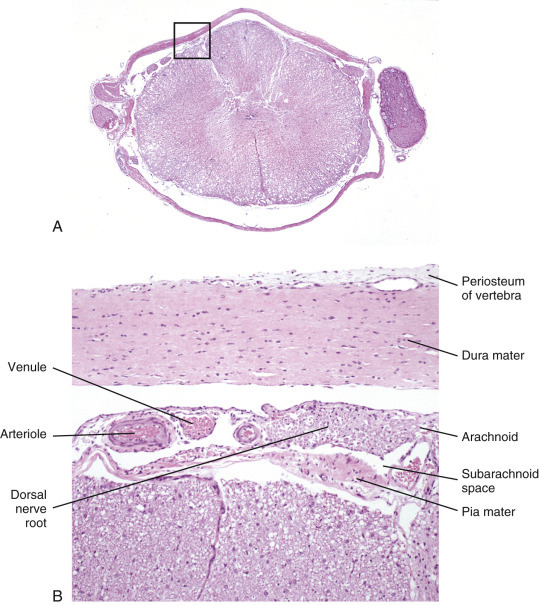
Histologic Section of Spinal Cord and Meninges.
A, Low magnification of a cross-section of the spinal cord and meninges with spinal nerve rootlets and a dorsal root ganglion from which B was selected (box). H&E stain. B, The inner surface of the dura mater and the outer surface of the arachnoid mater are covered with mesothelial cells, and the space between them is the subdural space. Blood vessels and nerves of the dorsal and ventral roots traverse in the subarachnoid space. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Endothelium.
The basic functions of endothelium in the CNS are to line luminal surfaces of blood vessels; form the blood-brain barrier; regulate thrombosis, thrombolysis, and platelet adherence; and maintain a nonthrombogenic boundary between coagulation cascade molecules and luminal surfaces of endothelial cells. Additionally, endothelial cells function as regulatory barriers to small and large molecules crossing the endothelium, and they control the adherence of leukocytes to their luminal surfaces. The endothelial cells of the blood-brain barrier actively transport those molecules that the brain consumes rapidly and in large quantities such as glucose, amino acids, lactate, and ribonucleosides.
Dysfunction/Responses to Injury
Ground Rules for Understanding Injury in the Central Nervous System
Before the responses of the CNS to injury are discussed, some fundamental concepts are reviewed in Box 14-2 .
Box 14-2. Concepts in Understanding Responses of the Central Nervous System to Injury.
The cells of the CNS vary in their susceptibility to injury (neurons > oligodendroglia > astrocytes > microglia > blood vessels). Neurons are the most sensitive to injury, whereas glial and other cells are more resistant to injury.
-
1.
Neurons have only small energy stores; therefore they depend on an intact blood flow to supply oxygen and nutrients, particularly glucose. Neurons with the highest metabolic rate, such as some neurons in the cerebral cortex, will die 6 to 10 minutes after the cessation of blood flow after cardiac arrest.
-
2.
There is no regeneration of neurons. The neurons you have now are the ones you were born with; however, their metabolism is dynamic, and metabolites are continually turned over and replaced.
-
3.
If nerve fibers in the CNS are cut by transection of the cord, no or little regeneration of nerve fibers results. Therefore if sufficient motor nerve fibers are cut, there is paralysis; if not, there is a neurologic deficit.
-
4.
If fibers in the PNS are cut, they can regenerate under certain circumstances. This outcome depends on axoplasmic flow, alignment of the proximal and distal portions of the nerve, and the preservation and alignment of the proximal and distal portions of the endoneurial tube (the structure in which the axon lies).
-
5.
Healing in the CNS is different than in the rest of the body. There are few fibroblasts in the CNS, and they are principally found only in the leptomeninges and in the outer few millimeters of the CNS, where they are pulled into the cerebral cortex with blood vessels. Therefore wounds deep in the CNS heal by proliferation of astrocyte processes. Astrocytic processes fill small dead spaces of less than a few millimeters and encapsulate large dead spaces and abscesses. Superficial wounds or wounds that extend through the leptomeninges heal by synthesis and deposition of collagen by fibroblasts (fibrous connective tissue) and by proliferation of astrocytic processes. However, in contrast to the fibroblast, astrocytic processes produce a very poor capsule, which can break down easily.
-
6.
The cranial cavity is nearly filled by the brain, its coverings, and fluids. Therefore many lesions, such as tumors, abscesses, hemorrhages, and hydrocephalus in the brain, produce clinical signs because they are space-occupying lesions, which in neuropathology implies that they cause atrophy or displacement of portions of the brain or cord, depending on the duration of the injury.
-
7.
The blood-brain barrier can exert control over drugs and antibodies and prevent them from entering the intact brain. It is also a barrier to infection and is formed by the tight junctions of the endothelial cells, aided by basement membrane, and the end feet of the astrocytes, which lie on the outside of the capillary.
-
8.
Although the CNS has the ability to resist infection and injury, once the CNS is infected, it has a low degree of resistance when compared with other tissues of the body. Microbes, such as Cryptococcus neoformans, which normally would be relatively nonpathogenic in other organs, may produce death if the CNS is infected. This outcome in part is attributable to the complexity of the CNS and the fact that it is the most vital organ in the body. Any disease process will often cause catastrophic results in the CNS, as opposed to tissues such as the lung, liver, and kidney.
CNS, Central nervous system; PNS, peripheral nervous system.
Neurons
Neurons are the most vulnerable cells in the nervous system and probably within the body. They have large requirements for energy to maintain normal metabolism, transport systems, and the formation of cytoskeleton proteins in the axon, which can extend over long distances (>1 m). Because neurons lack adequate intracellular glucose reserves, they are completely dependent for survival on an adequate blood supply to provide glucose. Additionally, neurons are vulnerable to free radical oxidative stresses and have a limited ability to buffer shifts of calcium ions into the cell, which can interfere with oxidative phosphorylation and ATP production, such as occurs with ischemia.
Neurons are especially sensitive to excessive stimulation with excitatory amino acid neurotransmitters called excitotoxins (e.g., glutamate and aspartate). These neurotransmitters are also released in a wide variety of neuronal injuries, especially ischemia. Under normal conditions, astrocytic processes surrounding synapses have efficient uptake systems to remove excitotoxins, and neurons are not injured. In excessive quantities, persistent binding of excitotoxins to receptors can lead to neuronal degeneration and death.
The microscopic appearance of the neuronal cell body can vary according to the injury. Characteristic changes of the neuronal cell body are reviewed in Box 14-3 .
Box 14-3. Microscopic Changes That Can Occur in the Neuronal Cell Body.
-
1.
Central chromatolysis after axonal injury, degenerative conditions, viral infection, or inherited conditions
-
2.
Ischemic cell change
-
3.
Enlargement of the cell body in lysosomal storage diseases
-
4.
Accumulation of lipofuscin pigment in aging
-
5.
Accumulation of neurofilaments in certain neuronal degenerative diseases
-
6.
Inclusion body formation in certain viral diseases
-
7.
Cytoplasmic vacuolation in spongiform encephalopathies
Neuronal Cell Death.
Neurons can die after injury as a result of one of two mechanisms: apoptotic cell death and necrotic cell death. These mechanisms are summarized next and discussed in greater detail in Chapter 1. Both apoptotic and necrotic neuronal cell death can occur concurrently or in temporal or spatial sequences within the nervous system. Although apoptotic and necrotic neuronal death represent different responses of neurons to injury, a network of receptors, messenger systems, and mechanisms of cytotoxicity are involved in both apoptotic and necrotic cell death. Factors that determine whether the apoptotic or necrotic pathway is activated include the character on the initiating ligand or injury, type of cell membrane receptors activated, and caspases expressed in response to injury.
Apoptotic Cell Death (Programmed Cell Death).
Apoptosis is a single cell-initiated, gene-directed, and self-destructive regulatory mechanism that leads to “programmed” cell death. This mechanism is used (1) during the development of the nervous system to ensure proper migration and orientation of cell layers and removal of excess embryonic cells, (2) to remove “aged” cells (i.e., cell turnover) in organs, and (3) to maintain cell number homeostasis in organ systems that have regenerative capacity (endocrine glands).
Apoptotic neuronal death is characterized by a sequence of cellular degenerative steps that can be identified biochemically and morphologically. After appropriate signals are recognized and interpreted by cell membrane receptors (Fas, tumor necrosis factor [TNF] receptor-1, TNF-related apoptosis-inducing ligand receptors), a family of proteins known as caspases are activated. Caspases cleave cellular substrates that are required for cellular function and include cytoskeleton proteins and nuclear proteins such as DNA repair enzymes. Caspases also activate other degradative enzymes, such as deoxyribonucleases, which cleave nuclear DNA.
The role of apoptotic neuronal death in specific neurologic diseases is discussed in greater detail in subsequent sections. As examples, some viral infections, such as feline panleukopenia virus infection, that occur in utero produce developmental anomalies by initiating apoptosis that leads to faulty differentiation of embryonic granule and Purkinje cell layers. Mild ischemia, excitotoxins, hormones, corticosteroids, and proinflammatory cytokines can similarly induce apoptotic cell death. Rabies virus and Borna disease virus have been linked experimentally to apoptotic neuronal death.
Apoptosis results in characteristic morphologic changes in cells such as shrinkage, cytoplasmic condensation and blebbing, and chromatin clumping and fragmentation (see Figs. 1-13, 1-22, 1-23Fig. 1-13Fig. 1-22Fig. 1-23, and E-Fig. 1-11). As cells continue to shrink, nuclear chromatin is cleaved into smaller units and along with condensed cytoplasm is packaged for removal by macrophages. Inflammation is not induced by apoptotic cell death.
Necrotic Cell Death.
Necrosis is a process that usually affects groups of cells in contrast to single isolated cells as observed in apoptosis. It is characterized by the following sequence: hydropic degeneration, swelling of mitochondria, loss of ionic gradient control across the cell membrane, activation of numerous cytoplasmic and lysosomal enzymes, pyknosis and fragmentation of the nucleus, and eventual cell lysis (see Figs 1-12, 1-13, 1-16Fig 1-12Fig 1-13Fig 1-16, and E-Fig. 1-11). Cellular debris associated with necrotic neuronal death elicits an inflammatory response in contrast to apoptotic neuronal death.
Acute Neuronal Necrosis.
Acute neuronal necrosis (also sometimes referred to as acidophilic or ischemic necrosis) is a common response to a variety of CNS injuries, such as cerebral ischemia caused by blood loss and hypovolemic shock, vascular thrombosis, and cardiac failure; inflammatory mediators; bacterial toxins; thermal injury; heavy metals; nutritional deficiencies, such as thiamine deficiency; and trauma. Additionally, conditions that reduce ATP generation through oxidative phosphorylation also lead to neuronal degeneration and death. Such conditions include (1) interference with cytochrome oxidase activity in mitochondria caused by cyanide poisoning, (2) competitive inhibition of oxygen uptake in carbon monoxide poisoning, and (3) inadequate availability of glucose for neuronal metabolism in hypoglycemia.
The susceptibility of cells and tissue structures of the CNS to ischemia in decreasing order of susceptibility are neurons, oligodendroglia, astrocytes, microglia, and blood vessels. However, within groups of neurons, some neurons are more sensitive to injury than others. This phenomenon is called selective neuronal vulnerability. Purkinje cells; some striatal neurons; neurons of the third, fifth, and sixth cerebral cortical lamina; and hippocampal pyramidal cells have the highest vulnerability. A regional vulnerability of neurons has also been reported (cerebral cortex and striatum > thalamus > brainstem > spinal cord). It is hypothesized that the most vulnerable neurons likely produce the most excitotoxins, such as glutamate, and are the most sensitive to them. Because of the microanatomic arrangement of the cerebral cortex, ischemic neurons often occur in a laminar pattern within the cerebrocortical gray matter. This microanatomic pattern accounts for the laminar lesions observed in thiamine deficiency–induced polioencephalomalacia in ruminants and in other diseases such as salt poisoning in pigs and lead poisoning in ruminants. Immunohistochemical stains for neuronal specific markers (e.g., neuronal nuclei) often delineate a linear pattern of neuronal necrosis better than that observed with an H&E stain and histologic examination.
After the various types of CNS injury, there is an early increase in ATP-dependent release of normally sequestered intracellular calcium ions from altered mitochondria and endoplasmic reticulum. Also during this time, neuronal depolarization potentiates the release of the neuroexcitatory neurotransmitter glutamate. Persistent activation of glutamate receptors of target cells results in a disturbance referred to as excitotoxicity. This altered activity leads to a notable influx of extracellular calcium into cells, causing further impairment of mitochondrial function and the generation of reactive oxygen species, such as superoxide, hydrogen peroxide, hydroxyl radicals, and nitric oxide. These reactive oxygen species, exerting their effects especially on lipid-rich cell membranes, can enhance the existing excitotoxicity, cause further influx of calcium into cells as a result of membrane damage, and ultimately result in neuronal dysfunction and death. Additionally, reperfusion of ischemic tissue after the initial ischemic injury can enhance the generation of reactive oxygen metabolites, thus amplifying the tissue damage. Other influencing factors include the temperature of the brain at the time of ischemia, with lower temperatures (as little as 2° C decrease) having a sparing effect and elevated temperatures having an enhanced effect on neuronal injury following ischemia.
Neurons depend on a continuous supply of oxygen to remain viable, and if the supply is interrupted for several minutes, the vulnerable neurons as described previously degenerate. Ischemic cell change can also result from metabolic disturbances other than ischemia, such as in thiamine deficiency and cyanide toxicosis, which interferes with oxygen use. In H&E-stained sections the cytoplasm of the neuronal cell body is shrunken, deeply eosinophilic, and frequently sharply angular to triangular in shape (Fig. 14-16 ). The nucleus is reduced in size, often triangular, frequently assumes a central position in the cell, and is pyknotic. The nucleolus and Nissl substance are usually not detectable. Ischemia neurons die and are removed either by a process called neuronophagia, which is phagocytosis by resident microglial cells and recruited macrophages or by lysis (see Fig. 14-16). After neuronal necrosis there is swelling of perineuronal and perivascular astrocytic processes and eventual replacement of the space left by loss of the neuron cell body by astrocytes and their processes.
Figure 14-16.
Neuronal Necrosis (Acute), So-Called Ischemic Cell Change, Cerebrum, Dog.
A, Neuronal ischemia. Neuronal cell bodies of cerebral cortical laminae are red, angular, and shrunken (arrows), and their nuclei are contracted and dense. This lesion can be caused by neuronal ischemia. H&E stain. B, Neuronophagia. This necrotic neuron cell body (center of figure) is surrounded and infiltrated by macrophages that will phagocytose the cell debris. H&E stain.
(A courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Chronic Neuronal Loss (Brain Atrophy).
Neuronal death and loss of neurons can occur as a result of progressive disease processes of long duration in the CNS. This loss, termed simple neuronal atrophy, is seen with slowly progressive neurologic diseases, such as cerebral cortical atrophy of aging, ceroid-lipofuscinosis, and various manifestations of selective or multisystem neuronal degeneration. Gross lesions are usually not visible, but when cerebrocortical neurons die, there can be atrophy of cerebral gyri, which results in widening of the sulci (Fig. 14-17 ). Microscopic lesions indicative of an earlier loss of neurons include diminished numbers of neurons, astrogliosis, and atrophy and loss of neurons in functionally related systems. Loss of neurons over time results in progressively worsening neurologic dysfunction as the afferent and efferent neurons that directly interact with the now dead neuron are similarly affected and eventually degenerate.
Figure 14-17.
Cerebral Cortical Atrophy, Horse.
Atrophy is seen with a variety of slowly progressive neurologic diseases in which there is a progressive loss of neurons. These diseases include cerebral cortical atrophy of aging and ceroid-lipofuscinosis. The characteristic gross lesions are narrowing of the cerebral gyri with a consequent widening of the sulci.
(Courtesy the Department of Veterinary Biosciences, The Ohio State University.)
Wallerian Degeneration and Central Chromatolysis.
Injury to axons of the CNS and PNS can result from a variety of causes such as (1) traumatic transection leading to Wallerian degeneration, (2) compression and crushing, (3) therapeutic neurectomies, (4) nerve stretching injury, and (5) intoxication.
Wallerian Degeneration.
In 1850 Dr. Augustus Volney Waller described the pattern of microscopic lesions (necrosis) in axons and myelin sheaths after transection. These changes became what we now refer to as Wallerian degeneration. Although Waller described this process in peripheral nerves, the term Wallerian degeneration is also used to describe necrosis that occurs in nerve fibers in the CNS after axons are injured (compressed or severed). Focal damage to a nerve fiber results in decreased or halted axonal transport, which manifests most prominently as segmental swellings in the axon called spheroids (Fig. 14-18 ). Eventually the axon's myelin degenerates, forming areas of vacuolation into which macrophages infiltrate and digest the now necrotic axonal and myelin debris (forming digestion chambers). In the neuronal cell body of the damaged axon, lesions include swelling of the neuronal cell body, peripheral displacement of the nucleus, and dispersion of centrally located Nissl substance (central chromatolysis) (Fig. 14-19 ). It must be stressed that this is only one of several ways in which chromatolysis can develop. Chromatolytic neurons can also be found in a wide variety of neurologic diseases including viral infection and degenerative diseases like equine motor neuron disease. The development of Wallerian degeneration is directly related to the diameter of the axon, with a larger axon undergoing a faster rate of Wallerian degeneration.
Figure 14-18.
Wallerian Degeneration, Transverse Section of Spinal Cord, Dog.
A, Longitudinal section. Arrows illustrate swollen axons. H&E stain. B, Transverse section. Laceration and/or severe compression of myelinated nerves cause a specific sequence of structural and functional changes in the axon and the myelin (distal from the point of injury), referred to as Wallerian degeneration (see E-Fig. 14-5). Axons are initially swollen (arrows) and eventually removed by phagocytosis to leave clear spaces, which were once the sites of nerve fibers. The cell bodies of affected neurons usually have central chromatolysis, but are metabolically active in an attempt to regenerate the lost portion of the axon (not shown; see Fig. 14-19). H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 14-19.
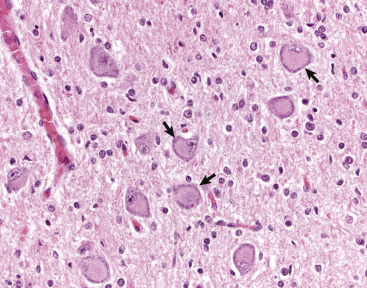
Central Chromatolysis, Neuron Cell Body, Dog.
Compare with Figs. 14-4, B, and 14-7. Affected neurons have eccentric nuclei and pale central cytoplasm with peripherally dispersed Nissl substance (arrows). H&E stain.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
Wallerian degeneration in the CNS follows the same sequence of events as in peripheral nerve fibers, but the speed of degeneration and phagocytosis is slower, and the capacity of the axon to fully regenerate is limited (E-Fig. 14-5). In addition, an axon of the PNS has the advantage of (1) efficient phagocytosis with removal of debris, (2) Schwann cells to remyelinate the regenerated axon, and (3) an endoneurial tube to guide the axon as it extends into the distal segment. On the other hand, an axon of the CNS has (1) few microglia (sparse in the white matter) to remove myelin debris and (2) oligodendrocytes with a more limited capability to remyelinate axons. In the CNS, severed axons have a very limited ability to regenerate and successfully reinnervate their appropriate sensory or motor structures. In the PNS, if the severed nerves have a large distance between the cut ends, fibrous tissue scarring can prevent the axons from the proximal segment from entering the distal endoneurial tubes and thus prevent the reparative response, resulting in the formation of a neuroma. If the severed ends of nerves are sewn together, but the endoneurial tubes are misaligned, which usually occurs, the sensory and motor nerves regenerate but reinnervate inappropriate sensory and motor structures.
E-Figure 14-5.
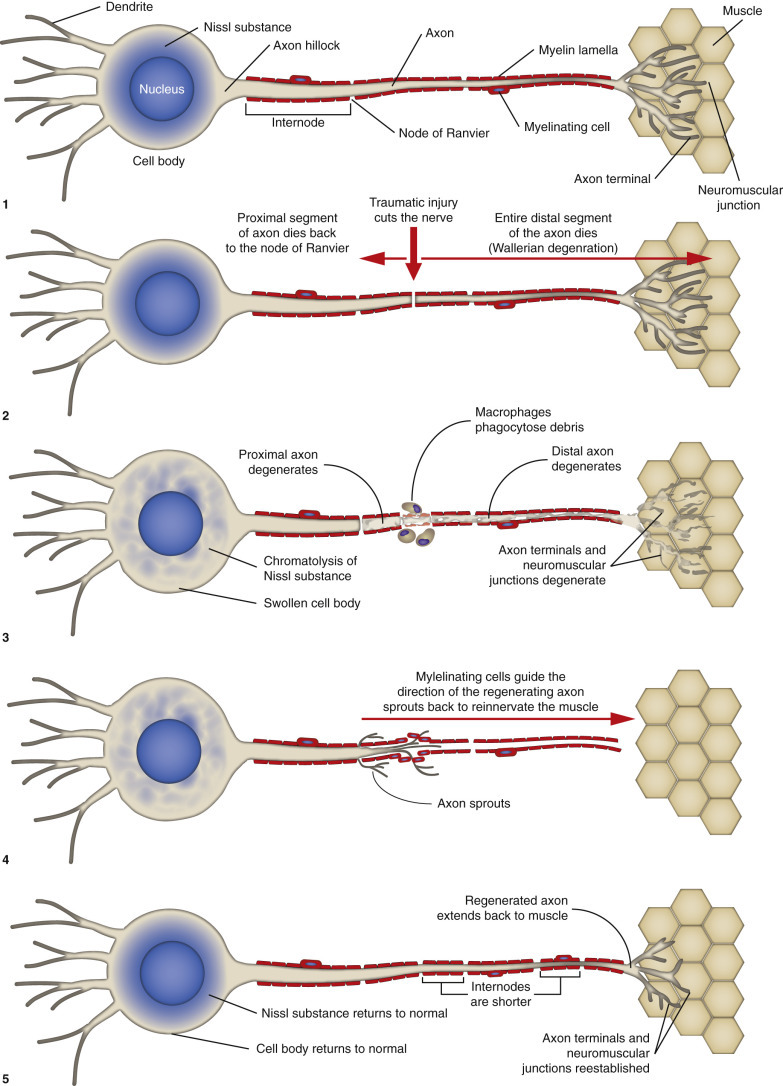
Peripheral Nerve Degeneration and Regeneration (Also Applicable to Neurons Within the Central Nervous System).
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The sequence of Wallerian degeneration in the PNS (similar in CNS except for significant regeneration; see previously) includes the following:
-
1.
Degeneration and fragmentation of axon and myelin within several days. Proximal segment degenerates back to the next node of Ranvier, but all the distal segment dies.
-
2.
Removal of axonal and myelin debris by phagocytosis. Some phagocytes are from the blood, and some phagocytosis is by Schwann cells. All of the debris is cleared out of the endoneurial tube within a few weeks.
-
3.
Regeneration of the axon if the endoneurium is intact to allow the axon of the proximal segment to enter and slide down the tube.
-
4.
Remyelinization by Schwann cells, which often arrange themselves in rows along the endoneurial tube and are termed Büngner's bands.
Microscopically, another early lesion in Wallerian degeneration is central chromatolysis, characterized by swelling of the neuronal cell body, dispersion of centrally located Nissl substance, and peripheral displacement of the nucleus (see Fig. 14-19). The time of onset depends on how much of the axon has been lost, and thus on how close to the neuronal cell body the axon has been transected. Onset can begin within 24 to 48 hours and reach its maximum in approximately 18 days after axonal injury. Chromatolysis is indicative of enhanced synthesis of transport and structural proteins required for regeneration of the axon and reestablishment of fast and slow axonal transport systems. The extent to which chromatolysis develops is related to the degree and location of axonal injury. It is more prominent and can even be followed by neuronal death, the more severe the loss of the volume of the axon and the closer the axonal injury is to the cell body. The time required for recovery of cell bodies can be several months and in most cases will vary from 3 to 6 months, depending on the severity of the axonal injury and the length of axon regenerated.
The change in the axon distal to the point of injury is first evident within 24 hours of injury. Wallerian degeneration is initially characterized by irregularity of the axonal diameter, which is followed after 48 to 72 hours by fragmentation of the axon and myelin along its length (see Fig. 14-19). This axonal alteration is followed by disintegration, and usually there is no evidence of the axon remaining by the second week after the injury.
Changes in the myelin sheath surrounding myelinated axons are evident by 28 to 96 hours after injury when axonal disintegration is well advanced. Initially there are irregularities in the sheath accompanied by folding, lamellar splitting, fracturing, and fragmentation (secondary demyelination). The fragmented myelin can form droplets (termed ellipsoids), which surround and enclose isolated fragments and debris of the former axon. Both axonal and myelin debris are then removed by macrophages through phagocytosis (forming digestion chambers in regions of necrosis). Degeneration of myelin is usually completed by the end of the second week, although evidence of myelin debris can be detected up to 1 to 3 months after axonal injury (the time required for macrophages in the PNS to phagocytose and clear the debris). Myelin debris can be detected in the CNS (when compared with the PNS) for a much longer time after injury.
If the neuronal cell body survives the injury to its axon, regeneration from the proximal stump in the PNS can occur. The degree of axonal regeneration depends on the status of the endoneurial tube (sheath) distal to the original point of injury (see E-Fig. 14-5). The normal endoneurial tube (sheath) and its contents consist (from outside inward) of (1) a connective tissue investment referred to as the endoneurium, (2) the basement membrane that surrounds the Schwann cells plus the Schwann cell cytoplasm, (3) the myelin sheath of myelinated axons, and (4) the axon. Approximately 24 to 72 hours after axonal injury, the endoneurial tube (sheath), formed by persisting basement membrane and endoneurium, contains degenerating remnants of the previously existing axon along with Schwann cells. Schwann cells begin to proliferate and eventually form Büngner's bands.
If the endoneurial tube (sheath) remains intact, as can occur following a compression injury to a peripheral nerve, neural regeneration through the formation of axonal sprouts can occur. A regenerating sprout from the proximal axonal stump can enter the column of Schwann cells and regenerate uninterrupted along its original pathway to the periphery, reestablishing innervation with an end-organ (skeletal muscle). Such axons then become remyelinated and regain their physiologic function of impulse transmission. Because of axoplasmic flow, a regenerating neuron lengthens at a rate of approximately 1 to 4 mm/day. Although the time required for axonal regeneration can vary, depending on the length of axon to be regenerated, examples of times required for morphologic and functional recovery after crush injury of a peripheral nerve are 250 to 300 and 456 to 486 days, respectively. If, however, the integrity of the endoneurial tube (sheath) is destroyed, as would occur after complete severance of a peripheral nerve, regeneration might not occur, because the proximal axonal stump might be prevented from reaching the distal endoneurial tube (sheath) by proliferated fibrous connective tissue (scar formation) at the site of axonal severance. Regenerating axons may also enter inappropriate endoneurial tubes (sheaths), resulting in improper impulse transmission, such as a sensory neuron axonal sprout entering an endoneurial tube intended for a nerve innervating a muscle.
The microscopic lesions for Wallerian degeneration within the CNS are similar to those described for the PNS, except that in the CNS, degenerated axons and myelin sheaths can remain for months to years before complete removal. With injury to the CNS, some cell bodies have central chromatolysis; others initially have central chromatolysis followed by atrophy and death. Affected axons and their myelin sheaths undergo a rather characteristic series of changes as they degenerate. Initially, axons form linear and bulbous swellings at and some distance from the site of injury. These enlargements are termed axonal spheroids. Spheroids consist of neurofilaments, microtubules, and cellular organelles. Because injury to the axon results in dysfunction of axoplasmic flow, any disruption of anterograde and retrograde axonal transport results in the accumulation of neurofilaments, microtubules, cellular organelles, and recycled molecules at or near the point of injury. This process occurs in peripheral nerves as well.
The axonal enlargements can be seen as early as a few hours at the site of injury and remain prominent, particularly for the first week or so (see Fig. 14-19). The surrounding myelin sheath is usually distended to create a space between the sheath and the axonal swelling. Progressively, such affected axons and myelin sheaths fragment along their length, forming ellipsoids as in the PNS. Eventually ellipsoids are removed through degeneration and phagocytosis, leaving an empty space, or one still containing myelin debris and macrophages. The latter lesion is termed a digestion chamber. With time and continued lysis and phagocytosis of the debris, most of the lesion will consist of enlarged empty spaces. The absence of swollen axons in such dilated spaces, especially in the early stages after CNS trauma, does not necessarily mean that the entire axon has degenerated and been removed, which can require several months. It might instead be the site of separation of an enlarged axon from the adjacent axon at the level of the section being examined.
Macroglia
Astrocytes.
Common astrocytic reactions in CNS injury are swelling, hypertrophy, division, and the laying down of intermediate filaments in cell processes. The term astrocytosis means that astrocytes have increased in size and number in response to injury, whereas the term astrogliosis (somewhat synonymous with hypertrophy) implies synthesis of intermediate filaments and an increased length, complexity, and branching of the astrocytic processes. The recognition of these differences is based on histopathologic and immunohistochemical evaluation.
Swelling is an acute response and is reversible, or it may progress with time to hypertrophy. Swollen astrocytes have clear-staining or vacuolated cytoplasm. Astrocytes swell after ischemia because of the increased uptake of sodium, chloride, and potassium ions and water in an effort to maintain homeostasis in the extracellular microenvironment. It is important to remember that such swelling depends on the astrocyte being viable and still having a semipermeable plasma membrane, even though its function may be altered. With progression and if the degree and duration of ischemia are sufficiently severe to result in cell death, the plasma membrane becomes fully permeable, and the cell does not swell but becomes shriveled or shrunken and undergoes disintegration, as described for the ischemic cell change of neurons.
If injury is severe, astrocytic processes fragment and disappear followed by lysis of the cell body. Hypertrophied astrocytes, often referred to as reactive, represent a response to a milder and more protracted injury to the CNS. Because of increases in intermediate filaments, mainly GFAP, the cytoplasm becomes apparent along with increased length and branching of the processes with H&E staining. The increase of intermediate filaments and consequently the intensity of GFAP immunohistochemical staining in these cells are so dramatic that some have defined reactive astrocytes on the basis of this change. In protracted degenerative or reparative conditions, astrocytes termed gemistocytes can be observed (Fig. 14-20 ). These cells have eccentric nuclei and abundant pink homogeneous cytoplasm, in contrast to the lack of visible cytoplasm in normal astrocytes, with routine H&E staining. Animals with hepatic and, less commonly, renal encephalopathy can have a unique microscopic lesion in the brain affecting astrocytes of the cerebral cortices. In these types of encephalopathies, astrocytic nuclei tend to be in pairs, triplets, quartets, or more occasionally with prominent central nucleoli and are surrounded by a clear space, which is the edematous cytoplasm. They are called Alzheimer's type II astrocytes (see Fig. 14-10, D).
Figure 14-20.
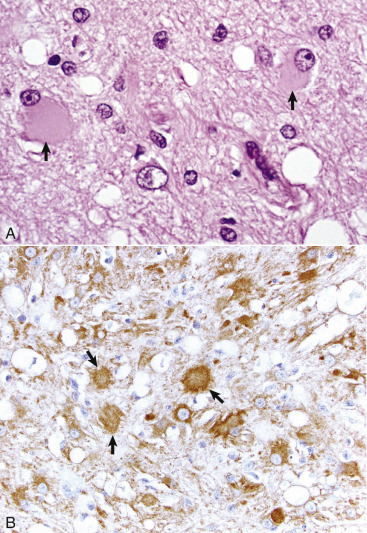
Gemistocytes (Gemistocytic Astrocytes), Cerebrum, Dog.
A, When astrocytes react to injury, initially by hypertrophy and later by the synthesis of increased glial filaments (astrogliosis), the nuclei enlarge and often the cell body, which is not normally visible in H&E-stained sections, will become visible. This type of reactive astrocyte is called a gemistocyte (plump astrocyte) (arrows). They occur in diseases in which there is alteration of intracellular and extracellular fluid balances or injury to the parenchyma, where healing will be by glial scarring (astrogliosis, e.g., to encapsulate a deep abscess or fill in a small area of dead space). H&E stain. B, Gemistocytes (arrows) are identified by immunohistochemical staining (brown color) with antibody to glial fibrillary acid protein. IHC DAB stain.
(A courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
Astrocytic proliferation can occur in CNS injury, but in most instances, proliferative capacity is limited. When it occurs, the most dramatic examples are associated with attempts by reactive astrocytes (astrogliosis) to “wall off” abscesses and neoplasms or to fill in cavitated areas that result after lysis of necrotic neurons with the processes of astrocytes. The astrocytes that form regions of astrogliosis are more commonly fibrillary in appearance. In large numbers they may form a glial scar, which is a network of interlaced astrocytic processes and provides a loose barrier that separates the injured brain from the more normal adjacent tissue. In this respect the astroglia act to reform a glia limitans around the injured region of the CNS in an effort to restore the blood-brain barrier and reestablish fluid and electrolyte balances.
Oligodendrocytes.
Oligodendroglia react to injury by cell swelling, hypertrophy, and degeneration. Both perineuronal and interfascicular oligodendroglia can swell, hypertrophy, and degenerate; however, only oligodendroglia precursor cells can proliferate to replace degenerate cells. The role that perineuronal or satellite oligodendroglia play in normal neuronal function and neuronal injury has not been definitively clarified. Microscopically, these cells swell and hypertrophy around injured neurons; this response to injury has been called satellitosis, although other glial cells can also contribute to satellitosis (see Fig. 14-10, B).
Degeneration of interfascicular oligodendroglia caused by ischemia, certain viruses, lead toxicity, and autoimmunity can result in selective degeneration of myelin sheaths referred to as primary demyelination. Primary demyelination is the loss of myelin around an intact axon and results in the alteration of the conduction velocity of an action potential down the axon, leading to clinical dysfunction (Fig. 14-21 ). Mechanisms of primary demyelination are summarized in Box 14-4 . Protracted or repetitive injury to myelinating cells and their myelin sheaths can lead to irreversible neuronal atrophy. Oligodendroglia precursor cells located in the subventricular zone of the CNS can mature into interfascicular oligodendroglia and can also proliferate in response to noncytocidal injury and become involved in remyelination after primary demyelination.
Box 14-4. Mechanisms of Primary Demyelination.
-
1.Inherited enzyme defects resulting in formation of abnormal myelin
- Leukodystrophies in human beings and animals
-
2.Impairment of myelin synthesis and maintenance
- Infection
- Mouse hepatitis virus in mice and progressive multifocal leukoencephalopathy in human beings; in both cases oligodendrocytes are selectively destroyed by viral agents, and myelin cannot be maintained.
- Nutritional
- Lack of maintenance of myelin is due to copper deficiency, malnutrition, vitamin B12 deficiency.
- Toxins
- Cyanide poisoning
- Cuprizone toxicity
-
3.Loss of myelin as a consequence of cytotoxic edema (status spongiosus)
- Hexachlorophene poisoning, usually prolonged edema
-
4.Destruction of myelin by detergent-like metabolites
- Lysolecithin, a metabolite of phospholipase A (normally present in the nervous system) may destroy myelin.
-
5.Immunologic destruction of myelin
- Cell mediated
- Experimental allergic encephalitis
- Landry-Guillain-Barré (human beings)
- Coonhound paralysis
- Marek's disease (chickens)
- Various stages of multiple sclerosis in human beings
- Various stages of canine distemper
CNS or PNS injury can also lead to loss of myelin secondary to injury of the axon and its cell body or to death of the neuron. When axons are injured, myelin lamellae forming the internodes are retracted and removed by phagocytosis. In some instances, oligodendroglia or Schwann cells, the myelin-forming cells in the PNS, also degenerate. This form of myelin degeneration is termed secondary demyelination and is secondary to axon degeneration or loss (resembles Wallerian degeneration).
Ependymal Cells.
Ependymal and choroid plexus epithelial cell responses to injury include atrophy, degeneration, and necrosis. Compression of ependymal cells lining the ventricles followed by atrophy usually occurs in response to enlargement of the ventricles as occurs with hydrocephalus. The cilia and microvilli of affected cells are reduced in number, and there is also a reduction in their cellular organelles such as endoplasmic reticulum and mitochondria. An additional lesion that accompanies ventricular enlargement is stretching and tearing of the ependymal lining. In such instances the resulting areas of ependymal discontinuity result in the subependymal CNS being directly exposed to the CSF. Unfortunately, mammalian ependymal cells do not regenerate and therefore do not repair the denuded areas. After 1 to 2 weeks, astrogliosis, which varies greatly in degree and uniformity, occurs in the exposed areas. Astrogliosis can extend into the ventricular space or be minimal in extent and confined to the periventricular area. Periventricular interstitial edema, myelin loss, and axon loss can ensue.
Inflammation of the ependyma, called ependymitis, can also occur following dissemination of microbes, especially bacteria, into the CSF. Microbes most commonly gain entrance to the ependyma via the circulation by lodging in the choroid plexuses, by direct contamination from a rupture of a cerebral abscess into the ventricular system, and by retrograde reflux through the lateral apertures of infected CSF from the subarachnoid space in cases of leptomeningitis. In the case of bacterial infection the suppurative exudate that forms in the CSF can cause obstructive hydrocephalus, although the development of hydrocephalus cannot always be explained solely on the basis of obstruction.
Microglia
Microglia are often the first cells in the CNS to react to injury, and the magnitude of the response is graded to correlate with the severity of damage. The responses of microglia to injury include hypertrophy, hyperplasia, phagocytosis of cellular and myelin debris, and neuronophagia, which is the removal of dead neuronal cell bodies. After injury, microglia progress through a stage of activation, becoming fully immunocompetent reactive cells. These reactive cells readily proliferate, either focally, forming glial nodules (Fig. 14-22 ), or more diffusely, depending on the nature of the injury. As mentioned, in concert with astrocytes and neurons, microglia help coordinate inflammatory events in the CNS. Resident microglia and blood-derived macrophages express major histocompatibility complex class I and II antigens, serve as antigen-presenting cells, and possess a broad armament of adhesion molecules, cytokines, and chemokines. Once activated, these cells can also produce nitric oxide, reactive oxygen intermediates, and other chemical mediators of inflammation that can damage the CNS if not under strict control. When tissue necrosis occurs, macrophages derived from blood monocytes phagocytose the lipid-laden debris of dead neurons, parenchyma, and glial remnants and accumulate in the damaged CNS. These cells are called gitter cells (Fig. 14-23 ).
Figure 14-22.
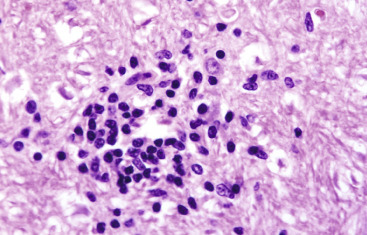
Glial Nodule, Brainstem, Dog.
These nodules (center of figure), formed by reactive microglial cells and infiltrating macrophages, occur most frequently in viral and protozoal encephalitides. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 14-23.
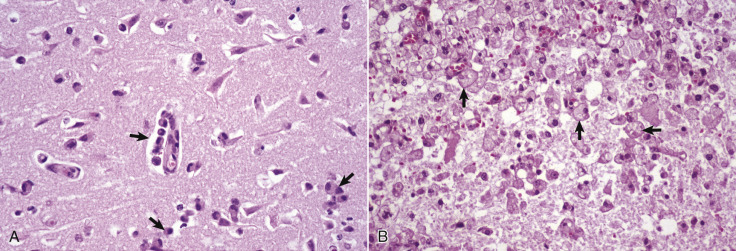
Gitter Cells, Cerebrum.
A, Early polioencephalomalacia, cow. Note the angular, eosinophilic neurons with pyknotic nuclei (ischemic cell change). Macrophages (arrows) in the perivascular space have been recruited from the circulating monocytes. These cells phagocytose cellular debris from the necrotic neurons and the myelin from the nerve fibers undergoing degeneration after the death of their neurons. Microglia also participate in this phagocytic response. Macrophages that have ingested degenerate myelin or other cellular debris have foamy cytoplasm and are termed gitter cells. H&E stain. B, Previous region of necrosis, dog. The normal brain parenchyma has liquefied, and the debris has been ingested by macrophages (arrows), which has resulted in the cytoplasm of these cells becoming foamy. They are now designated as gitter cells or, simply, foamy macrophages. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Meninges
Pathologic processes that initially involve the meninges, most commonly the leptomeninges, can secondarily invade the CNS because of the close apposition between the two tissues. Conversely, processes that primarily affect the CNS can secondarily affect the meninges, most commonly the leptomeninges.
Meningitis refers to inflammation of the meninges. In common usage the term generally refers to inflammation of the leptomeninges in contrast to inflammation of the dura mater, which is referred to as pachymeningitis. Leptomeningitis can be acute, subacute, or chronic and depending on the cause, suppurative, nonsuppurative, or granulomatous, and the exudate and inflammatory cells are chiefly in the subarachnoid space. Besides retrograde axonal transport, as occurs with, for example, Listeria monocytogenes, infectious microbes spread to the meninges hematogenously by direct extension or by leukocytic trafficking.
Other meningeal lesions include (1) inflammation of the external periosteal dura after osteomyelitis, formation of extradural abscesses, and skull fracture and involve the inner dura as an extension of leptomeningitis and (2) proliferation of the inner dural mesothelial cells, arachnoid cells, fibroblasts, and cells of the pia mater in response to irritation. Additional lesions likely related to aging or degeneration include formation of cellular nests of meningial cells on the outer surface of the arachnoid membrane, mineralization of the arachnoid membrane, and mineralization plus ossification of the dura mater of the spinal cord. Dural ossification in older dogs, which tends to affect the ventral, cervical, and lumbar dura mater, is most commonly encountered in large breeds, although smaller breeds can be affected. These lesions are of little clinical significance.
Circulatory System
Endothelial Cell (and Blood Vessel) Responses to Injury.
Because many of the infectious and neoplastic disease processes demonstrated in this book are spread through the body via the circulatory system, endothelial cells lining blood vessels, especially capillaries, are subject to a variety of injuries. Bacterial hematogenous CNS diseases occur at the interface between the white and gray matter in the cerebral hemispheres. This phenomenon is thought to result from abrupt changes in vascular flow or luminal diameter of vessels at the interface. These changes may make endothelial cells more susceptible to injury, vasculitis, and thrombosis or predispose the vessels to entrapment of tumor or bacterial emboli.
Endothelial injury can be reversible or nonreversible, resulting in necrosis. Injury resulting in endothelial dysfunction can include the activation and release of vasoactive mediators, such as histamine, leading to local and/or systemic changes in vascular flow, pressure, and permeability. Bacterial products and elicited inflammatory cytokines can directly or indirectly cause vascular inflammation (vasculitis) leading to thrombosis and disseminated intravascular coagulation. Thrombotic meningoencephalitis of cattle caused by the bacterium Histophilus somni is an example of this type of injury (see Fig. 14-89). Certain herpesviruses and protozoa can also infect endothelial cells and cause endothelial necrosis with vasculitis, hemorrhage, and thrombosis. Finally, some pathogens, such as angioinvasive fungi, directly invade blood vessels, resulting in necrosis of the endothelium. Vasculitis resulting in thrombosis can cause tissue ischemia, infarction, and vasogenic edema of the affected area of the CNS. A review of endothelial injury can be found in Chapter 2. Angioinvasive fungi are discussed in Chapter 4.
Figure 14-89.
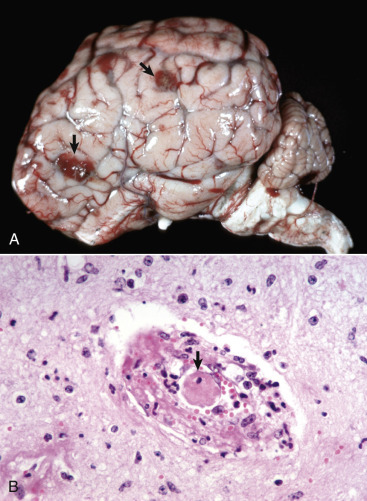
Thrombotic Meningoencephalitis, Cerebrum, Steer.
A, On the surface of the cerebral cortex (arrows) are several red-brown lesions. These lesions are areas of necrosis, hemorrhage, and inflammation secondary to vasculitis and thrombosis caused by Histophilus somni. Such septic infarcts are distributed randomly (hematogenous portal of entry) throughout the central nervous system, including the spinal cord. The lesions depicted here are unusually severe. B, A thrombus (arrow) is present in the vascular lumen. Note the acute inflammatory response, edema, fibrinogenesis, and hemorrhage in the vessel wall. H&E stain.
(A courtesy Dr. H. Leipold, College of Veterinary Medicine, Kansas State University. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Infarction.
Infarction means necrosis of a tissue after obstruction (ischemia) of its arterial blood supply. The rate at which ischemia occurs in the CNS determines the degree of injury that follows. The more rapid the onset of ischemia, the more severe the lesion. However, if the obstruction is sudden, as caused by an embolus, many of the neurons can die within minutes and other components within hours (Fig. 14-24 ). This outcome also applies to compressive injuries to the CNS that produce a sudden reduction in blood flow, such as can happen with sudden compression in rapidly occurring Hansen type I disk herniation in the dog. If the blood flow through an artery is gradually reduced, for example, because of atherosclerosis, there is often sufficient time for anastomotic vessels to dilate and compensate. Anastomoses of the arteries that penetrate from the ventral and cortical surfaces of the brain are insufficient to prevent infarction after sudden occlusion of one or more of these arteries. If the compression is slow—such as is caused by a slowly developing Hansen type II disk herniation in a dog or by a slowly growing neoplasm from the exterior, such as meningioma in a cat—adjacent neural tissue will atrophy to accommodate the mass.
Figure 14-24.
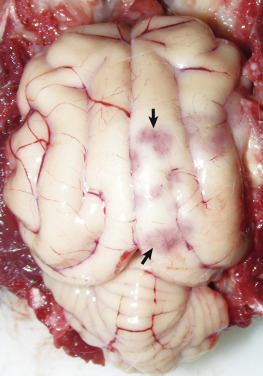
Malacia, Vascular Occlusion, Ischemia, Infarction, Cerebrum, Cat.
Several red-pink foci (arrows) are areas of ischemic necrosis secondary to vascular occlusion caused by cerebral metastasis of a bronchoalveolar carcinoma.
(Courtesy Drs. C.A. Lichtensteiger and R.A. Doty, College of Veterinary Medicine, University of Illinois.)
Cerebral necrosis, comparable to infarction after vascular occlusion, can also result from other causes, including cessation of cerebral circulation caused by cardiac arrest, sudden hypotension caused by reduced cardiac output, and reduced or absent oxygen in inspired air. Additional causes include altered function of hemoglobin as a result of carbon monoxide poisoning, inhibition of tissue respiration after cyanide poisoning, ingesting toxic substances and poisons, and nutritional deficiencies.
When an artery supplying the CNS is suddenly occluded, blood supply to cells at the center of the infarcted area is rapidly stopped, and if blocked for a sufficient period, all cells die. Neurons at the border of this area continue to receive some blood from unobstructed vessels. It is proposed that the axonal terminals of degenerated ischemic neurons in the center of the infarct release excessive amounts of the neurotransmitter glutamate, causing injury to still-viable neurons in the borders, which increases the extent of the infarct. This process begins after the binding of the neurotransmitter glutamate to receptors on viable neurons in the borders, inducing an abnormal movement of calcium ions into the recipient cells followed by an increase in intracellular calcium ion concentration. This buildup of calcium ions contributes to a multifunctional cascade that leads to neuronal death. When there is hemorrhage with the infarct, the mechanical injury from the pressure, plus tissue displacement by the hemorrhage, can cause additional damage. See Table 14-1 for the reparative responses associated with the resolution of infarcts.
Table 14-1.
Chronologic Sequence of Changes within Infarcted Tissue (in the Living Animal) after an Ischemic Event
| Time Following Ischemic Event | Tissue Change |
|---|---|
| Immediate (seconds) | Cessation of blood flow (ischemia) and accumulation of waste products |
| Few minutes | Cellular injury and death; necrosis and edema; hemorrhage (especially in gray matter) |
| 20 minutes | First microscopic evidence of neuronal injury (perfusion-fixation) |
| 1-2 hours | First microscopic evidence of neuronal injury (immersion-fixation) |
| 2 hours | Pale staining of infarct microscopically (white matter); swelling of capillary endothelium; increase in size of astrocytic nuclei |
| 3-5 hours | Ischemic cell change in most neurons; swelling of oligodendroglia and astroglia; beginning clasmatodendrosis of astrocytes |
| 6-24 hours | Beginning neutrophilic infiltration; alteration of myelin (pale staining), 8-24 hours; degeneration and decrease of oligodendroglia, 8-24 hours; astrocytic swelling and retraction and fragmentation of processes (clasmatodendrosis), and degeneration*; cytoplasm of astrocytes visible, 8-24 hours*; vascular degeneration and fibrin deposition, 8-24 hours; thrombosis,† 6-24 hours; beginning endothelial proliferation at margin of infarct, 9 hours |
| 8-24 (up to 48) hours | Initial gross detection of infarct unless hemorrhagic; infarct edematous (swollen), soft, pale, or hemorrhagic and demarcated |
| 1-2 days | Swelling of axons and myelin sheaths; prominent neutrophilic infiltration |
| 2 days | Prominent loss of neuroectodermal cells; continued proliferation of endothelial cells; reduced number of neutrophils; beginning increase in mononuclear cells (gitter cells) |
| 3-5 days | Prominent number of mononuclear cells (gitter cells); disappearance of neutrophils; continued endothelial cell proliferation; number of capillaries appear increased; beginning of astrocytic proliferation (often at margin of infarct) |
| 5-7 days | Grossly, swelling of infarct reaches maximum |
| 8-10 days | Reduction in gross swelling of infarct; liquefaction necrosis; prominent number of mononuclear cells (gitter cells); continued endothelial cell proliferation; beginning fibroblastic activity with collagen formation, variable but most prominent in CNS tissue adjacent to the meninges; beginning increase of astroglial fiber production, 5-13 days |
| 3 weeks-6 months | Mononuclear cells decreased; astroglial fiber density increased (especially at margin); astrocytic proliferation reduced; astrocytes return to original appearance; cystic stage of infarct, 2-4 months; vascular network may be present within cyst; endothelial cell proliferation reduced |
CNS, Central nervous system.
The degree of astrocytic injury depends on location (e.g., central or peripheral) of the cells within the infarct.
Obviously, thrombosis may occur earlier than 6 hours. This is the time when it may initially be prominent.
Although they occur through the same mechanisms, areas of cerebral infarction differ somewhat in gross appearance from infarcts in other tissues (Fig. 14-25 ). The abundance of lipids and enzymes, plus the relative lack of fibrous connective tissue stroma in the brain and spinal cord, results in the affected areas eventually becoming soft because of liquefaction necrosis. The gross appearance of infarction may also differ according to location. Lesions affecting the gray matter tend to be hemorrhagic, whereas infarction of the white matter is often pale. This difference is probably due in part to the less-dense capillary meshwork in the white matter because vessels supplying the white matter have fewer anastomoses than those of the gray matter. Infarcted tissue goes through a characteristic sequence of changes that can permit a relatively accurate determination of the age of the infarct. An outline of the chronologic events that occur after an ischemic episode that lasts more than 5 to 6 minutes and is followed by resuscitation of an animal is given in Table 14-1. As can be seen, the tissue changes listed in Table 14-1 take different periods of time to develop in the living resuscitated animal after ischemia occurs. Variation in the times that specific lesions occur depends on the extent and duration of the initial ischemic event. Following removal of cellular and myelin debris, the infarct is repaired by astrocytes. If the infarct is small (<1 mm), it is filled via astrogliosis; if the infarct is larger, it is encapsulated to form a cyst.
Figure 14-25.
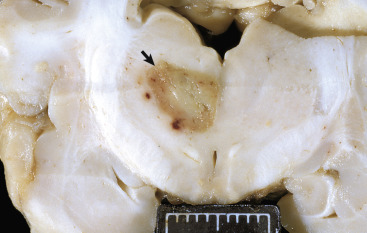
Central Nervous System Infarct, Brain, Thalamus, Dog.
The pattern of a focal, sharply demarcated region of yellow discoloration and malacia (softening) (arrow) in the left central thalamus indicates an infarct. Scale bar = 2 cm.
(Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Central Nervous System Swelling and Edema
Congestive Brain Swelling.
Congestive brain swelling, as distinguished from cerebral edema, partially represents unregulated vasodilation after trauma, and it can cause serious brain damage (even more severe than the primary injury) if not properly controlled. This lesion therefore represents an enlargement of the brain resulting in elevated intracranial pressure caused by the increased diameter of the blood-containing vasculature, whereas edema results in an increased pressure following accumulation of fluid in the interstitium or intracellularly outside the circulation. Acute brain swelling can be localized (usually of lesser significance) when associated with focal lesions or generalized (often serious) when caused by diffuse brain injury. Although rarely seen in domestic animals, one exception to the importance of focal lesions is extracerebral hemorrhage (acute subdural hematoma in human beings), which—though principally involving the surface of one hemisphere—can cause more mass effect (brain swelling) in the underlying cerebral hemisphere than the hematoma itself. In subdural hematomas, blood accumulates between the dura mater and the meninges. Hematomas also occur in the epidural region; however, the subdural variety are usually poorly circumscribed. If the hematoma is removed, the acute brain swelling can progress so rapidly that the brain protrudes (herniates) through the site of the craniotomy.
The more serious forms of diffuse brain injury are associated with generalized acute brain swelling. It is sometimes difficult to determine the relative importance of swelling in affected individuals because initial acute swelling (detectable as soon as 30 minutes after injury in human beings) can be followed after several hours to days by true cerebral edema (as a result of increased vascular permeability), which can be the actual deleterious lesion. Peroxidative injury to blood vessels has been one proposed cause of pathologic vasodilation in the posttraumatic CNS.
Cerebral Edema.
The basis of our current understanding of cerebral edema was advanced by Klatzo in 1967 when he proposed two distinct types: (1) cytotoxic edema, or cell swelling, caused by increased intracellular fluid with normal vascular permeability and (2) vasogenic edema, or tissue swelling, caused by increased extracellular fluid resulting from increased vascular permeability (Fig. 14-26 ). Other types of cerebral edema have been identified as hydrostatic (or interstitial) edema associated with increased hydrostatic pressure of the CSF (resulting from obstructive internal hydrocephalus) and hypo-osmotic edema, which is dependent on development of an abnormal osmotic gradient between the blood and nervous tissue. The types of edema in the CNS are summarized in Table 14-2 . It should be emphasized that, depending on the nature of the injury, multiple mechanisms can contribute to edema in the CNS and these distinctions are not always clearly defined or distinct. For continuity, spongiform change and status spongiosus will also be discussed in this section.
Figure 14-26.
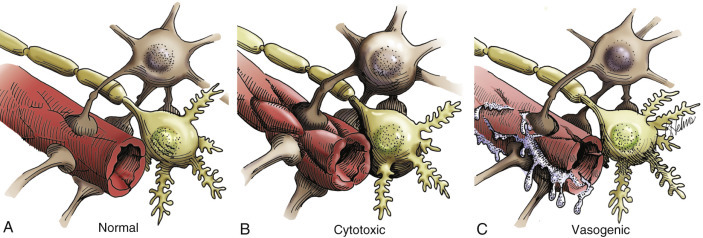
Types of Cerebral Edema.
A, Normal blood-brain barrier. Endothelial cells are red; astrocytes are beige; neurons are light yellow. B, Cytotoxic edema. Cytotoxic edema is characterized by the accumulation of fluid intracellularly (in neurons, astrocytes, oligodendroglia, and endothelial cells) as a result of altered cellular metabolism, often caused by ischemia. The gray and white matter are both affected. The fluid taken up by swollen cells is primarily derived from the extracellular space, which becomes reduced in size and has an increased concentration of extracellular solutes. C, Vasogenic edema. This type of edema is seen in acute inflammation, and its basic mechanism is an increase in vascular permeability from the breakdown of the blood-brain barrier. This breakdown allows movement of plasma constituents such as water, ions, and plasma proteins into the extracellular space, particularly that of the white matter.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. Redrawn and modified from an illustration from Leech RW, Shuman RM: Neuropathology: a summary for students, Philadelphia, 1982, Harper & Row.)
Table 14-2.
Types of Edema in the Central Nervous System
| Type of Edema | Cause | Outcome |
|---|---|---|
| Cytotoxic | Altered cellular metabolism (often due to ischemia) | Intracellular accumulation of fluid (neurons, glial cells, endothelial cells) |
| Vasogenic | Vascular injury with breakdown of the blood-brain barrier | Extracellular accumulation of fluid (cerebrocortical white matter) |
| Hydrostatic (interstitial) | Elevated ventricular hydrostatic pressure (hydrocephalus) | Extracellular accumulation of fluid (periventricular white matter) |
| Hypo-osmotic | Osmotic imbalances (blood plasma versus extracellular and intracellular microenvironments of the CNS) | Extracellular and intracellular accumulation of fluid (cerebrocortical gray and white matter) |
CNS, Central nervous system.
Vasogenic Edema.
In animals, vasogenic edema is the most common type of edema in the CNS. It occurs following vascular injury often adjacent to inflammatory foci, hematomas, contusions, infarcts, cerebral hypertension, and neoplasms. The underlying mechanism of vasogenic cerebral edema is a breakdown of the blood-brain barrier that results in movement of plasma constituents, such as water, ions, and organic osmolytes, and proteins into the perivascular extracellular space, particularly that of the white matter (Fig. 14-27 ). In addition to extracellular accumulation of fluid, vasogenic edema can also be accompanied by some cellular swelling involving astrocytes. Vasogenic edema with the resulting accumulation of extracellular fluid can cause an increase in intracranial pressure within the CNS. This pressure can also be so severe as to cause neurologic dysfunction and caudal displacement of brain structures such as the parahippocampal gyri and cerebellar vermis (see Figs. 14-59 and 14-60).
Figure 14-27.
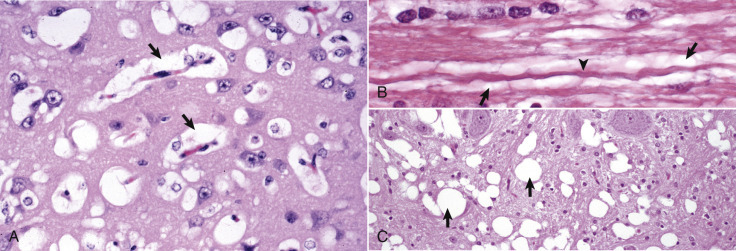
Edema.
A, Vasogenic edema. The perivascular spaces are wide as a result of fluid leakage through the blood-brain barrier (arrows) (see Fig. 14-26). A similar change can be seen around neurons. These fluid-filled spaces are often very difficult to differentiate from artifactual spaces caused by shrinkage from fixation and dehydration in the preparation of the paraffin-embedded sections. H&E stain. B, Intramyelinic edema. Note the accumulation of edema fluid (arrows) between myelin lamellae that surrounds the axon (arrowhead). This lesion was caused by hexachlorophene added to a medicated shampoo. Such products are no longer available for use in veterinary practice. H&E stain. C, Edema (spongy change, status spongiosus), hepatic encephalopathy, dog. This lesion is characterized by variably sized fluid-filled spaces within the white matter (arrows). It can develop by several different mechanisms, which include splitting of myelin sheaths, accumulation of extracellular fluid, and swelling of astrocytic and neuronal cellular processes. Such changes can reflect osmotic imbalances as well as a direct toxic effect on cells (cytotoxic edema). H&E stain.
(A and C courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 14-59.
Gyral Herniation, Parahippocampal Gyri, Brain, Transverse Section, Caudal Face, at Level of the Rostral Colliculi and Crus Cerebri, Horse.
The caudal displacement of the parahippocampal gyri (arrows) was caused by a sudden swelling of the brain (increase in intracranial pressure) from severe cerebral blunt force trauma to the head. The other cerebral gyri are swollen and flattened, and sulci are indistinct (cerebral edema).
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 14-60.
Coning of the Cerebellar Vermis, Brain, Cat.
A, Sagittal section. Coning of the cerebellum. The caudal cerebellar vermis has been displaced caudally through the foramen magnum; note the notch on the dorsal surface (arrow). This result has compressed the medulla oblongata (MO), which can cause death from compression of the respiratory center. Note the elevation of the corpus callosum (CC) and focal compression of the rostral cerebellar vermis by the tectum (quadrigeminal plate) (QP). B, Coning of the cerebellum through the foramen magnum, caudal view through the foramen magnum. Note that in this case not only has the cerebellar vermis (arrow) been displaced caudally, but also the medulla. The caudal cerebellar peduncles have been displaced caudally as far as the foramen magnum.
(A courtesy Dr. D. Cho, College of Veterinary Medicine, Louisiana State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy College of Veterinary Medicine, University of Illinois.)
Cytotoxic Edema.
Cytotoxic edema is characterized by the accumulation of fluid intracellularly in neurons, astrocytes, oligodendroglia, and endothelial cells (called hydropic degeneration in other cells of the body) as a result of altered cellular metabolism, often caused by ischemia. Although not all of the cells listed previously may be involved in all cases of cytotoxic edema, affected cells swell within seconds of injury. The mechanism is thought to involve an energy deficit that interferes with normal function of the cell's Na+/K+-ATPase pump. Thus the cell cannot maintain homeostasis, which requires the secretion of intracellular sodium, and the elevated concentration of intracellular sodium and presumably other ions, as well as organic osmolytes, is followed by an increased influx of water. The gray and white matter of the brain are both affected, the brain swells, and the sulci and gyri become indistinct and flattened (Fig. 14-28 ), respectively. The fluid taken up by the swollen cells is primarily derived from the extracellular space, which becomes reduced in size and has an increased concentration of extracellular solutes.
Figure 14-28.
Cerebral Edema, Dog.
On the dorsal surface, gyri are swollen and flattened and sulci have become less distinct. Accumulation of extracellular fluid has caused the brain to swell, and because space within the cranial vault is limited, the brain has been pressed against the calvaria. In extreme cases, notable brain swelling can cause caudal displacement of the parahippocampal gyri and vermis of the cerebellum (see Figs. 14-59 and 14-60).
(Courtesy Drs. C.A. Lichtensteiger and A. Gal, College of Veterinary Medicine, University of Illinois.)
However, for this lesion to be described accurately as cerebral edema, there must be additional fluid movement into the brain and not merely a change of existing fluid from extracellular to intracellular compartments. In practical terms, this is not always the case and can be difficult to determine. The term cytotoxic edema has been used rather loosely to refer simply to cellular swelling in many cases. Additional fluid may originate from the circulation by way of the transcapillary fluid exchange or possibly from the CSF, which has extensive diffusional communication with the extracellular fluid of the brain. The blood-brain barrier remains intact during development of this type of edema, so fluid does not enter the brain by a disturbance in vascular permeability. Specific causes of this lesion include hypoxia-ischemia, particularly in the early stages; intoxication with metabolic inhibitors, such as 2,4,dinitrophenol, 6-aminonicotinamide, and ouabain; and severe hypothermia.
Interstitial (Hydrostatic) Edema.
Interstitial edema is characterized by the accumulation of fluid in the extracellular space of the brain because of elevated ventricular hydrostatic pressure that accompanies hydrocephalus. Fluid moves across the ependyma of the ventricular wall and accumulates extracellularly in the periventricular white matter. Unlike the other forms of cerebral edema that cause swelling of affected CNS tissue, hydrostatic edema causes variable degeneration and loss of the periventricular white matter mostly through primary demyelination accompanied by loss of axons. As the periventricular white matter is reduced in volume, the ventricle expands to fill the void by displacement, thereby exacerbating the hydrocephalus. The blood-brain barrier remains intact in hydrostatic edema.
Hypo-osmotic Edema.
Hypo-osmotic edema occurs after overconsumption of water (water intoxication), leading to dilution of the osmolality of the plasma. Under normal conditions the osmolality of CSF and extracellular fluid in the CNS is slightly greater than that of plasma. When the osmolality of plasma is further decreased, water moves from the vasculature into the brain after the osmotic gradient, resulting in osmotic edema. This form of edema accounts for the clinical signs and lesions in osmotic demyelination syndrome and salt poisoning, as discussed later.
Spongiform Change and Status Spongiosus.
Spongiform change is a term whose exact meaning varies in different scientific disciplines and experimental situations. In this chapter, wherever possible, spongiform change is used to describe morphologic changes in H&E-stained sections that occur primarily in gray matter. These changes are characterized by small clear vacuoles of varied sizes that form in the cytoplasm of neuron cell bodies and proximal dendrites in diseases such as the transmissible spongiform encephalopathies (TSEs) and rabies encephalitis and in the processes of astrocytes that are spatially related to the affected neurons.
Status spongiosus (spongy degeneration) is also a phrase whose exact meaning varies. It is defined as multiple fluid-filled clear spaces in the white matter of H&E-stained sections of the CNS and may be extracellular or intracellular (see Fig. 14-27, C). This lesion results from the accumulation of edema fluid in the white matter secondary to a variety of causes, including cytotoxic edema, vasogenic edema, intramyelinic edema, Wallerian degeneration, and other hypoxic, toxic, and metabolic diseases.
In some instances the term spongy degeneration or spongy change has been used in the veterinary literature to describe microscopic lesions in a group of diseases of young dogs, cats, and cows characterized by fluid accumulation in white matter. These diseases are discussed in later sections.
Portals of Entry/Pathways of Spread
Disease processes of the CNS enter the brain and spinal cord through one of four principal portals (Box 14-5 ). These portals are (1) direct extension, (2) hematogenous entry, (3) leukocytic trafficking, and (4) retrograde axonal transport.
Box 14-5. Portals of Entry into the Central Nervous System.
Direct Extension
- Penetrating trauma through the calvaria or vertebral bodies
- Extension of middle and/or inner ear infection
- Extension of a nasal cavity/sinus infection through the cribriform plate or calvaria
- Extension from osteomyelitis
Hematogenous
- Localization within capillary beds of the meninges and parenchyma of the CNS
- Localization within capillary beds of the choroid plexuses and extension into the cerebrospinal fluid
Leukocyte Trafficking
Macrophages or lymphocytes (containing microbes) during their migration through the CNS
Retrograde Axonal Transport
Transported from the periphery into the CNS by neuronal retrograde axoplasmic flow
CNS, Central nervous system.
Direct Extension
Direct extension is a common portal of entry and includes a wide range of disease processes. Penetrating trauma through the calvaria or vertebrae as a result of a gunshot wound or other forms of trauma can provide a direct portal into the CNS. Disease processes can also extend into the brain and/or spinal cord as a result of (1) a middle and/or inner ear infection (Fig. 14-29 ), (2) nasal cavity/sinus infection or neoplasia through the cribriform plate or calvaria (Fig. 14-30 ), or (3) bacterial osteomyelitis or neoplasia of vertebral bodies with extension through the vertebrae into the vertebral canal.
Figure 14-29.
Chronic Bacterial Abscess, Leptomeninges of the Cerebellum, Sheep.
The abscess (arrow), which resulted from direct extension from an inner ear infection, compresses and distorts the cerebellum. It is walled off from the adjacent cerebellum by a distinct fibrous capsule synthesized by fibroblasts from the adjacent leptomeninges. This abscess likely arose in the leptomeninges of the cerebellum and grew to compress adjacent cerebellum.
(Courtesy College of Veterinary Medicine, University of Illinois.)
Figure 14-30.
Osteochondrosarcoma, Calvaria, Dog.
The neoplasm has destroyed and penetrated the calvaria and compressed the cerebral hemispheres (arrows). There is also invasion of frontal sinuses and the nasal cavity.
(Courtesy Dr. K. Bailey, College of Veterinary Medicine, University of Illinois.)
Benign growths of the calvaria and vertebrae, such as osteomas, chondromas, and osteochondromas, often extend into and compress the brain and spinal cord. One specific neoplasm, the multilobular osteochondrosarcoma, has been described as originating from the periosteum of the canine skull and can cause marked compression of the brain. Also, malignant neoplasms adjacent to the cranium or spinal column can cause injury by direct invasion. Some examples include osteosarcoma and fibrosarcoma in the dog. Also, malignant melanoma of the soft palate in the dog and melanoma involving the paravertebral lymph nodes in the horse can compress and invade adjacent CNS tissue. Other examples of direct extension include spinal cord lymphosarcoma in cattle, dogs, horses, and cats; nasal carcinomas in dogs and cats; and peripheral nerve sheath tumors in dogs.
Hematogenous Entry
The most common portal of entry into the CNS is the bloodstream. In neonates, infectious microbes, such as Escherichia coli, can enter the blood through the umbilical vein or through the venous system after surgical procedures such as castration. A CNS disease in cows called thrombotic meningoencephalitis is caused by H. somni bacteremia with localization of bacteria in blood vessels of the brain, which leads to vasculitis, hemorrhage, and thrombosis (see Fig. 14-89). In adult animals, sites of chronic inflammation, such as abscesses, bacterial skin disease, and ear infections, can also serve as sustained sources of bacteria, which can enter the venous system and spread to distant sites through the bloodstream hematogenously.
Capillary beds of the meninges, neuropil, and choroid plexuses are common sites for localization of specific infectious microbes. Such localization patterns may be attributable to receptor-mediated phenomena or vascular flow patterns related to the size of the infectious pathogen. The bloodstream is also a portal of entry into the CNS for metastasizing tumors such as hemangiosarcoma and a variety of carcinomas.
Leukocyte Trafficking
As part of the systemic immunologic surveillance system, macrophages (monocytes) and lymphoid cells continually move in and out of capillary beds in the CNS and thus serve as sentinel cells monitoring for the presence of disease processes within the brain and spinal cord (see Fig. 4-11). As examples, retroviruses, such as feline leukemia virus, and mycotic agents, such as B. dermatitidis, have stages of their life cycles within the cytoplasm of lymphocytes or macrophages. During the movement of lymphocytes and macrophages in and out of the CNS, cells infected with such agents are activated to release their infectious contents and infect cells of the CNS.
Retrograde Axonal Transport
Retrograde axonal transport provides a unique portal of entry for viruses such as rabies and the bacterium L. monocytogenes. These pathogens replicate in tissues richly innervated with receptors and motor end plates from sensory and motor neurons, respectively, which provide a connection between peripheral infection and the CNS. Retrograde axoplasmic flow is then used to gain entry into the CNS (see E-Fig. 14-3).
Defense Mechanisms/Barrier Systems
Barrier Systems
The CNS has several unique structural and functional barrier systems that serve to protect it from diseases affecting the vascular and ventricular systems and to actively facilitate transfer of necessary molecules, such as glucose, to cells within the CNS.
Blood-Brain Barrier.
The blood-brain barrier, formed by vascular endothelial cells, endothelial-derived basement membrane, and foot processes of astrocytes, exists in the capillaries of the CNS (see Fig. 14-13). The most important structural component of the blood-brain barrier is the tight junctions between endothelial cells of cerebral capillaries. By means of the blood-brain barrier the CNS can selectively regulate its extracellular compartment and isolate itself from sudden biochemical changes that may occur in the systemic circulation. In the majority of the CNS, endothelial cells are nonfenestrated and are held together by intercellular tight junctions. These tight junctions actively prevent the movement of protein, hydrophilic molecules, and ions from capillary lumina into the intercellular compartment of the CNS. Endothelial cells also have a transmembrane lipophilic pathway for the diffusion of small lipid molecules and numerous highly selective polarized receptor-mediated transport systems for molecules such as insulin, transferrin, glucose, purines, and amino acids. Finally, endothelial cells express a net negative charge on their abluminal side and at the basement membrane, providing an additional selective mechanism that impedes movement of anionic molecules such as chloride ions across the barrier. Foot processes of astrocytes cover more than 90% percent of the abluminal surface of capillary endothelial cells. Experimental evidence suggests that secretion of growth factors from astrocytes promotes the formation and maintenance of the blood-brain barrier.
Capillaries in the area postrema, median eminence, neurohypophysis, pineal body, subfornical organ, commissural organ, and supraoptic crest lack tight junctions and are fenestrated; thus the blood-brain barrier is absent at these sites.
Glia Limitans.
The CNS is separated from the subarachnoid CSF by the pia mater and the glia limitans (see Fig. 14-13). The glia limitans, which covers the outer surface of the brain and spinal cord and is situated immediately subjacent to the pia mater, consists of astrocytic fibers with many foot processes that form a distinct layer that lies subjacent to the pia mater. In many areas the pia is only one-cell-layer thick and has fenestrations, so that the glia limitans is exposed directly to the subarachnoid space. As arterioles penetrate the cerebral cortex to supply the CNS with blood, they carry the pia mater and surrounding glia limitans with them until the arteriole structurally and functionally becomes a capillary. At the capillary level the pia mater disappears, but the layer of pericapillary astrocytic foot processes remains and serves as a component of the blood-brain barrier. This transition zone occurs at a depth approximately 1 to 3 mm within the cerebral cortex and explains why in cases of meningitis the infiltrate can be observed to track along blood vessels a short distance into the parenchyma without causing actual parenchymal disease.
Blood–Cerebrospinal Fluid Barrier.
The blood-CSF barrier is formed by the choroid plexus and the arachnoid. This barrier is formed by tight junctions between apposing surfaces of choroid plexus epithelial cells that cover the choroid plexuses. As noted previously, blood vessels of the choroid plexus are fenestrated. The barrier formed by tight junctions between choroid plexus epithelial cells restricts the movement of molecules that leak from fenestrated capillaries into the extracellular compartment of the choroid plexus and then into the CSF. Similarly, the arachnoid membrane also has tight junctions that prevent the movement of molecules from the blood into the CSF. The arachnoid membrane is generally impermeable to hydrophilic molecules but lacks specialized transport systems, and its role in forming the blood-CSF barrier is largely passive.
Cerebrospinal Fluid–Brain Barrier (Ependymal Barrier).
The CNS is separated from ventricular CSF by ependymal epithelial cells and foot processes of astrocytes. Although the ependymal lining does form a cellular barrier of sorts, materials within the ventricular system can without too much difficulty penetrate into the brain. The CSF-brain barrier is far more permeable than the blood-brain barrier.
Innate and Adaptive Immune Responses
Although infectious microbes have developed unique approaches to gain entry into the CNS, the body has also evolved a strong suite of defense mechanisms to protect the CNS against infectious pathogens and disease processes. The skin and mucous membranes of the alimentary, respiratory, and urinary systems provide structural and functional barriers against disease. The inflammatory response, immune system, and monocyte-macrophage system provide a strong local and systemic defense against pathogen replication and disease spread. Finally, barrier systems in the CNS, reviewed in an earlier section, represent structural and functional protection against a wide range of pathogens and toxic injuries. These defense mechanisms are summarized in Box 14-6 .
Box 14-6. Defense Mechanisms against Injury and Infectious Microbes in the Central Nervous System.
Skin
Structural and functional (secretions) barrier.
Calvaria, Vertebrae
Structural barrier.
Meninges, Cerebrospinal Fluid
Structural and functional (continuous flow of CSF) barrier.
Barrier Systems
Blood-Brain Barrier
Structural and functional barrier formed by vascular endothelium, basement membrane, and astrocytic foot processes. This barrier regulates the movement of agents from the blood to the CNS.
Blood-CSF Barrier
Structural and functional barrier formed by choroid plexuses cells and the arachnoid membrane. This barrier regulates movement of agents from the blood to the CSF.
Glia Limitans
Formed by astrocytic foot processes immediately subjacent to the pia mater. This structure may have some barrier function in preventing movement of microbes from CSF into the CNS through the pia mater.
Microglia, Trafficking Macrophages
Resident and migrating cells that are part of the monocyte-macrophage system.
Immunologic Responses
Innate and adaptive immunologic responses that form the body's overall immune system.
CNS, Central nervous system; CSF, cerebrospinal fluid.
Inflammation of the Central Nervous System.
Inflammation of the CNS is different from inflammation in other organs because of the presence of the blood-brain barrier. Under normal conditions this barrier provides limited isolation of the CNS from circulating cellular and humoral elements of the immune system. Macrophages (monocytes) and T lymphocytes can penetrate an intact blood-brain barrier and enter the perivascular and subarachnoid spaces, transit these spaces, and return to the circulation in a role of protective immunologic surveillance of the CNS.
It is important to remember that inflammation in the CNS is regulated by a complex system of recognition and adhesion molecules, cytokines, chemokines, and their corresponding receptors (see Chapters 3 and 5). In particular within the CNS, chemokines and their receptors regulate physiologic and pathologic leukocyte trafficking and cellular migration events.
When pathogens use one of the four portals of entry to gain access to the CNS, the inflammatory process that ensues disrupts the blood-brain barrier. Thus, in addition to inflammation, edema and hemorrhage can result. Selectins and integrins in cooperation with chemokines are active in initiating and regulating the acute inflammatory response and the movement of neutrophils across the blood-brain barrier in response to a variety of pathogens. Migration of inflammatory cells within the CNS is poorly understood. Chemotactic gradients are likely established by chemokines that diffuse from sites of production within foci of inflammation. Activated glial cells, including astrocytes and resident microglia, form chemokine networks in areas of inflammation in response to cytokines produced by T lymphocytes that recognize foreign antigens.
Depending on the type of antigen and the pathogenicity of the infectious agent, the inflammatory response resolves (heals) or progresses to a chronic or granulomatous phase with attempts at resolution and clearance of the infectious agent. In the CNS the type of inflammatory response can vary with the cause. A rather simplistic guideline, to which there are always exceptions, that compares the type of inflammation with different causal agents is as follows:
-
1.
Serous to suppurative or purulent responses can be the result of several species of bacteria.
-
2.
Eosinophil responses occur in salt poisoning of pigs and with parasitic larval migration.
-
3.
Lymphocytic, monocytic/macrophage, nonsuppurative, lymphomonocytic, and lymphohistiocytic responses can be caused by viruses and certain protozoa.
-
4.
Granulomatous inflammation can be the result of fungi, certain protozoa, and some higher-order bacteria such as the Mycobacterium spp.
Disorders of Domestic Animals
Disorders that occur in many or all animal species are discussed in this section. Disorders of individual animal species are discussed in later sections covering disorders unique to that species.
Malformations3
Neural Tube Closure Defects (Dysraphia).
Dysraphia means an abnormal seam, and these anomalies result from defective interaction of neuroepithelium with adjacent notochordal and mesenchymal cells during closure of the neural tube in the early stages of development. Neuroepithelium is the progenitor cell for neurons and astrocytes, oligodendrocytes, and ependymal cells.
Experimental studies of closure of the neural tube show that it occurs at four distinct locations called closure initiation sites in the embryo, and disruption of this process at these sites leads to site-specific dysraphic anomalies. Closure site I contributes to the posterior neuropore (the opening at the posterior end of the embryonic neural canal), whereas closure sites II to IV contribute to the anterior neuropore (the opening at the anterior end of the embryonic neural canal). Anencephaly is caused by a failure of closure sites II or IV; spina bifida is caused by a failure of closure site I. Genes possibly involved in neural tube closure defects include those involved in folate metabolism and transport.
Dysraphic anomalies, also called neural tube closure defects, in animals are typified by anencephaly and prosencephalic hypoplasia, cranium bifidum, spina bifida, abnormal spinal cord development (duplication or abnormal cell migration), and syringomyelia.
Anencephaly and Prosencephalic Hypoplasia.
Anencephaly means an absence of the brain, but in many instances of so-called anencephaly only the rostral part of the brain (cerebral hemispheres) is absent, or very rudimentary, and to varying degrees the brainstem is preserved. Thus this abnormality is best designated prosencephalic hypoplasia. Such anomalies result from an abnormal development of the rostral aspect of the neural tube and failure of their fusion. Although the cause for these anomalies is largely unknown, anencephaly is most commonly reported in calves, where it is accompanied by other defects. It is an extremely rare event in other domestic species. Additionally, anencephaly—after initial cranium bifidum and exencephaly (protrusion of brain not covered by skin or meninges)—has been reported to occur in rat fetuses after exposure of the pregnant dam to excessive concentrations of vitamin A and cyclophosphamide.
Meningoencephalocele and Cranium Bifidum.
Cranium bifidum is characterized by a dorsal midline cranial defect through which meningeal and brain tissue can protrude. The protruded material, which forms a sac (-cele), is covered by skin and can be lined by meninges (meningocele) or meninges accompanied by a part of the brain (meningoencephalocele) (Fig. 14-31 ). Although the sac is readily apparent grossly, diagnosis of the presence or absence of brain tissue typically requires histologic examination. These malformations are hereditary in pigs and cats and are also caused by griseofulvin treatment in pregnant cats during the first week of gestation. They are uncommon and sporadic in other domestic species.
Figure 14-31.
Meningocele (M), Brain, Calf.
A defect in the caudodorsal portion of the skull has allowed the meninges to herniate into a large external pouch covered by skin. The pouch contains fluid and is lined by arachnoid and dura, which are continuous with those surrounding the brain. The cerebellum is small, and the occipital cortex truncated. Scale bar = 5 cm.
(Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Meningomyelocele and Spina Bifida.
Spina bifida is the vertebral counterpart of cranium bifidum. This lesion, which frequently tends to affect the caudal spine, is characterized by a dorsal defect in the closure of one to several vertebral arches that form the dorsal portions of the vertebral canal encasing the spinal cord. The lesion results from a failure of the neural tube and developing vertebral arches to close properly, which may result in herniation of either meninges (meningocele) or meninges and spinal cord (meningomyelocele) through the defect, forming a sac covered with skin. In some cases there is no herniation of the meninges or spinal cord through the defect, and this variation is termed spina bifida occulta (Fig. 14-32 ). In this variation there is an absence of skin over the affected vertebral arches, vertebral musculature is visible, and the dura mater and spinal cord can be seen in the spinal canal.
Figure 14-32.
Spina Bifida Occulta, Calf.
There is a cleft in several vertebrae of the dorsal spinal column resulting from defective closure of the neural tube. Although not always the case, note the lack of herniation of the meninges or spinal cord through the defect. The spinal cord is not visible (i.e., occulta) because it is located in the vertebral canal at the deepest ventral extent of the cleft and is covered by edematous muscle. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Spina bifida has been reported in several species, including horses, calves, sheep, dogs (especially English bulldogs), and cats, particularly the Manx breed, in which it is inherited as an autosomal dominant trait. An additional lesion, myeloschisis, also refers to failure of the neural tube to close and is therefore similar to spina bifida, except in its severe form it results from failure of the entire spinal neural tube to close. This lesion is therefore characterized by lack of development of the entire dorsal vertebral column, because the developing neural tube remained open, unable to fuse and develop normally.
Hydromyelia.
Congenital hydromyelia is an abnormal dilation of the central canal of the spinal cord (Fig. 14-33 ) that leads to the formation of a cavity in which CSF may accumulate. In animals this disorder likely results from infectious or genetic injury that results in damage to ependymal cells lining the canal and the subsequent disruption of the normal flow of CSF and the formation of abnormal CSF pressure gradients within the central canal. As CSF accumulates in the enlarging space, the increased pericanalicular pressure placed on the spinal cord compresses the white and gray matter, leading to loss of white matter and possibly neurons in gray matter. Acquired hydromyelia is rare and is caused by obstruction of the central canal CSF flow. Causes of obstruction include infection, inflammation, and neoplasia.
Figure 14-33.
End-Stage Hydromyelia, Spinal Cord, Dog.
The white and gray matter of the spinal cord are missing as a result of compression atrophy from a space-occupying, fluid-filled central canal. The only recognizable remnants of nervous tissue is the dura (arrows). In less severely affected animals, there would be variable dilation of the central canal of the spinal cord with much less severe compression atrophy.
(Courtesy College of Veterinary Medicine, University of Tennessee.)
Clinical signs in young animals with congenital hydromyelia vary, depending on the location and size of the dilation of the central canal in the spinal cord. Signs may include ataxia, urinary incontinence, respiratory difficulty, muscle weakness in front and/or hind limbs, and abnormal proprioceptive reflexes. It can accompany other defects in the spinal cord and has been seen as an abnormality associated with spinal dysraphism in Weimaraner dogs.
Neuronal Migration Disorders
Lissencephaly.
Lissencephaly (agyria) and a similar change called pachygyria (large, broad gyri) are developmental anomalies that result in part of or the entire cerebrum having smooth surfaces lacking normal gyri and sulci (Fig. 14-34 ). The cortex is thicker than normal on a transverse section, and the normal laminar pattern of neurons is disrupted. It has been reported most commonly in the Lhasa apso dog, but scattered reports also exist in kittens and lambs.
Figure 14-34.
Lissencephaly, Brain, Dog.
A, Note the smooth surfaces of the cerebral hemispheres, which are without gyri and sulci. Gyri and sulci fail to form, possibly from failure of neuronal development and migration. Lissencephaly is an abnormality in domestic animals but is a normal feature in some species, including mice, rats, rabbits, and birds. B, The development and migration of neurons have been disrupted such that the cortical gray matter lacks normal lamina formed by neuronal cell bodies. H&E stain.
(A courtesy Dr. L. Roth, College of Veterinary Medicine, Cornell University. B courtesy Dr. R. Mantene, College of Veterinary Medicine, University of Illinois.)
This lesion is thought to have a genetic basis and results from an arrest of or defect in neuronal migration during development. Recent experimental studies suggest that this migrational disorder is linked to mutations and/or deletions in the doublecortin, filamin-1, LIS1, and reelin genes. These genes control the spatial and temporal expression of proteins in the extracellular microenvironment that subsequently bind to receptors on migrating cells. Patterns of cell membrane binding signals are interpreted by migrating cells and are reflected in their movements by changes in intracellular cytoskeletal reorganization. This process allows cells to migrate to their final destinations within the CNS. Thus alterations in signaling pathways lead to abnormal neuronal migration and CNS anomalies.
The brains of many species, including birds and some laboratory animals, such as rabbits, rats, and mice, lack gyri and sulci; therefore agyria is normal in these species and has no functional significance.
Encephaloclastic Defects
Porencephaly and Hydranencephaly.
The formation of fluid-filled cavities in the brain, termed porencephaly (small cavities) and hydranencephaly (large cavities), usually occurs in utero during gestation. Porencephaly refers to a cleft or cyst in the wall of the cerebral hemisphere that typically communicates with the subarachnoid space, but it can also communicate with a lateral ventricle. The cavitation results from destruction of immature neuroblasts whose loss prevents normal development as a result of faulty or aberrant neuroblast migration. Hydranencephaly is considered a severe form of porencephaly and is characterized by cavitation in areas normally occupied by the white matter of the cerebral hemispheres and results from improper development of this part of the cerebrum. Hydranencephaly is often quite severe, with very little tissue present between the dilated lateral ventricles and the leptomeninges.
Type I and type II porencephaly have been described in human infants, and cases of porencephaly reported in animals can also be categorized using this scheme. Type I porencephaly is caused by vascular injury or vasculitis. Injury, resulting in infarction in the area of the subependymal germinal matrix, results in the formation of a cyst within the focus of dead cells and effete erythrocytes. The germinal matrix is very sensitive to ischemia because of sparse stroma, delicate vasculature, and high metabolism. The initial focus of hemorrhage can grow by centripetal expansion, depending on the severity of hemorrhage and hypoxia, into a cyst of considerable size. Type II porencephaly is caused by injury of neuroblasts in the germinal matrix and the failure of these neuroblasts to migrate within the matrix to form the cerebral cortex. A cyst results from the expansion of the subarachnoid space into the void left by the absence of the cortex.
Type II porencephaly appears to be the form of porencephaly that occurs in domestic animals. Viruses, such as those that cause Akabane, bovine viral diarrhea, blue tongue, border disease, Rift Valley fever, Schmallenberg disease, and Wesselsbron disease, infect and destroy differentiating neuroblasts and neuroglial cells in the developing fetus in utero. Although neuroblasts appear to be the primary target for viral infection in these diseases, additional experimental studies need to be conducted to clarify whether endothelial cells are also infected.
Grossly, porencephaly/hydranencephaly appears as thin-walled fluid-filled cysts of varied sizes in the cerebral hemispheres. Because of the lack of brain substance, the ventricles expand into this space (hydrocephalus ex vacuo), and the ependymal lining remains relatively preserved or may have scattered defects characterized by absent ependyma. The cranium and meninges are generally unaltered. In some cases cerebellar hypoplasia (all or part of the cerebellum) and hypoplasia of the spinal cord may also occur. Microscopically, necrosis of undifferentiated cells, including potential neuroblasts and neuroglia, surrounding a fluid-filled cavity is present in the subventricular zone of the cerebral hemispheres. Degeneration and loss of motor neurons of the ventral horns of the spinal cord may also be observed. This lesion may result in denervation atrophy of limb muscles with a resultant lack of joint movement and arthrogryposis, a persistent congenital flexure or contraction of a joint. Nonsuppurative encephalitis, typified by the accumulation of macrophages, lymphocytes, and plasma cells, also occurs.
Malformations of the Cerebellum
Cerebellar Hypoplasia.
In animals the most common causes of cerebellar hypoplasia are parvoviruses (kittens: panleukopenia virus [Fig. 14-35 ]) and pestiviruses (calves: bovine viral diarrhea virus [Fig. 14-36 ; E-Fig. 14-6] and piglets: classical swine fever virus). These viruses infect and destroy mitotic cells, primarily the cells of the external granule layer of the cerebellum that are still dividing during the late gestational and early neonatal periods. Necrosis of these cells means they are not available to form the granule layer, and thus the cerebellum is hypoplastic. In calves the cerebellar lesion (cerebellar hypoplasia/atrophy), which follows infection at 150 days of gestation (midtrimester), is considered to involve two processes. One process is typified by early necrosis of the undifferentiated cells in the external granule layer. A second process involves viral-induced vasculitis and ischemia of cerebellar folial white matter.
Figure 14-35.
Cerebellar Hypoplasia, Cerebellum, Cat.
In the cat, cerebellar hypoplasia (cerebellar hypoplasia, top specimen; normal cat, bottom specimen) most commonly is the result of in utero infection with feline panleukopenia virus (parvovirus). The virus infects and causes lysis of dividing cells in the external granule layer (on the outside of the cerebellum in the fetus). Because these cells are no longer available to migrate to form the granule layer, the cerebellum remains small.
(Courtesy Dr. Y. Niyo, College of Veterinary Medicine, Iowa State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Figure 14-36.
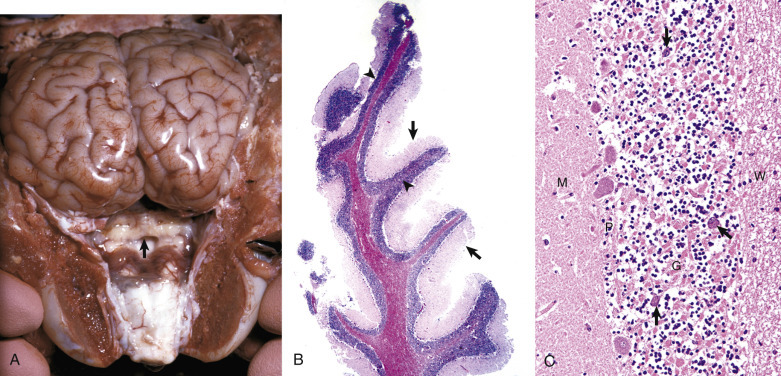
Cerebellar Hypoplasia, Cerebellum, Calf.
In the normal neonatal calf, cells of the external granule layer of the cerebellum migrate to form the granule layer (not shown). Bovine viral diarrhea virus infects and kills mitotic cells of the granule layer of the cerebellum. These cells are still dividing during the late gestational and early neonatal periods in the cat and between 100 to 180 days of gestation in the calf. Necrosis of these cells means they are not available to migrate to form the granule layer, and thus the cerebellum does not obtain full size. Depending on the stage of gestation, injury can also alter development of cells in others ways, including altered patterns of migration, resulting in various other lesions termed dysplasia. A, Cerebellar hypoplasia (arrow). In utero infection with bovine viral diarrhea virus (pestivirus) results in cytolysis of dividing germinal cells of the granule layer and vascular impairment secondary to vasculitis of the cerebellum during organogenesis. The severity of the lesion involving the granule cells is at its greatest if dividing cells are infected during the earliest stages of cellular differentiation, and occurs between 100 to 180 days of gestation. B, Note the folia of the cerebellum are hypoplastic and dysplastic with a reduced thickness of the molecular layer (arrows) and haphazardly organized and thinned granule cell layer (arrowheads). H&E stain. C, The molecular layer (M) of the cerebellum is reduced in thickness and lacks the normal number of neuronal nuclei. The Purkinje cell layer (P) has large gaps between adjacent cells as the result of the loss of neuron cell bodies or the failure of neurons to migrate properly to form this layer. Note the retention of Purkinje cells (arrows) in the granule cell layer (G). The granule cell layer has significantly reduced numbers of neurons as shown by the lack of nuclei. H&E stain. W, White matter.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B and C courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
E-Figure 14-6.
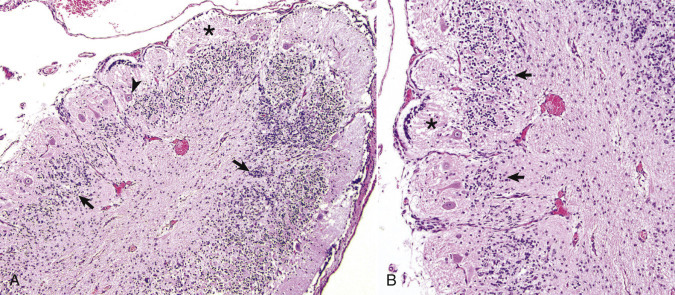
Cerebellar Hypoplasia, Cerebellum, Calf.
A, The cerebellar folia are disorganized with a discontinuous granule cell layer (arrows), sparsely populated Purkinje cell layer (arrowhead), and thin molecular layer (asterisk). H&E stain. B, Higher magnification of A illustrating the highly disorganized granule cell layer (arrows) and thinned molecular layer (asterisk). H&E stain.
(A and B courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
Grossly, the size of the cerebellum is reduced; the reduction in size varies in severity, depending on the age and developmental stage of the brain when the fetus or neonate is infected.
Microscopically, there is necrosis and loss of the external granule layer and degeneration and loss of Purkinje cells that are postmitotic but immature. Reasons for degeneration of Purkinje cells might include infection by the virus or lack of normal development of the cerebellar cortex. The Purkinje cells can also be malpositioned and located in the molecular layer as a result of the viral-induced alteration in development of the cerebellar cortex. In calves, edema of the folial white matter with focal hemorrhage in the cortex, followed by focal cavitation of the white matter and atrophy, may also be present. These latter lesions are due to ischemia resulting from vasculitis. Leptomeningitis, characterized by accumulation of lymphocytes and plasma cells and occasionally fibroplasia, may cause adhesions between adjacent cerebellar folia and focal obliteration of the subarachnoid space.
Malformations of the Spinal Cord
Syringomyelia.
Syringomyelia (congenital and acquired forms) is a disorder in which a cavity forms in the spinal cord. The cavity, called a syrinx, is not lined by ependyma and is separate from the central canal. The syrinx can extend over several spinal cord segments. The lesion is well known in human beings and has also been described most commonly in calves and also in many breeds of dogs. The syrinx can communicate with the central canal but should not be confused with hydromyelia, which means dilation of the central canal. In instances of communication, the term syringohydromyelia should be used. The cavity contains fluid and is unlined, except for varying degrees of mural astrocytosis. Proposed causes include the presence of an anomalous vascular pattern that results in low-grade ischemia, leading to infarction or failure of cells destined for this area to develop in utero trauma in human beings or an infection that causes degeneration and cavitation. An acquired form of syringomyelia is similar to congenital syringomyelia; however, it occurs in older animals. Proposed causes include injury after trauma to the central canal or its vascular supply caused by trauma, infection, or neoplasia that result in degeneration and cavitation of the spinal cord. The presence of syringomyelia and hydromyelia in some animals with vertebral malformations also suggests that local perturbations in CSF flow and pressure may allow for these dilations and cavities to develop.
Although the central canal of the spinal cord is connected to the ventricular system via the fourth ventricle, there apparently is little active movement of CSF within the central canal. Recently it has been hypothesized that there may be alteration of “normal” CSF flow (see the discussion on ependyma in the section on Cells of the Central Nervous System) with redirection of the flow along a pressure gradient into the central canal and into the syrinx. It has also been suggested that pressure differences in the vertebral column cause CSF to continually move into the cyst, resulting in enlargement of the syrinx and additional compressive damage to the spinal cord.
Clinical signs in young dogs and calves with syringomyelia vary, depending on the location and size of the spinal cord lesion. Signs may include ataxia, urinary incontinence, respiratory difficulty, muscle weakness in front and/or hind limbs, and abnormal proprioceptive reflexes.
Hydrocephalus.
By far the most common congenital CNS abnormality identified in domestic animals is hydrocephalus. It has a variety of causes, including in utero viral infection, developmental abnormalities in the ependyma or ventricular system, infection and subsequent blockage of the ventricular system, or periventricular parenchymal loss. There appears to be a genetic predisposition in some dog breeds (toy and brachycephalic), but the mechanism of injury has not been as clearly established in domestic animals as it has in human beings.
In laboratory animals, several neonatal in utero experimental viral infections, including mumps virus, reovirus type 1, and parainfluenza virus types 1 and 2, can induce congenital hydrocephalus. In utero infection with panleukopenia virus in cats and parainfluenza virus in dogs can also cause congenital hydrocephalus in affected offspring. Although there are some differences among the different viral infections, the basic lesion is stenosis of the mesencephalic aqueduct that results in the development of noncommunicating hydrocephalus. In the dog, closure of the mesencephalic duct can be incomplete. The virus grows in and causes destruction of ependymal cells lining the ventricular system. The infection is initially accompanied by an inflammation that resolves within 2 weeks.
The notable lesion resulting from this injury to the ependyma of the mesencephalic duct is its occlusion. This end-stage lesion is not the result of an astroglial response or due to the presence of viral antigen. Instead, the original ependyma-lined aqueduct is replaced by focal aggregates of remaining ependymal cells that have separated from the adjacent tissue, which appears normal. The appearance of the final lesion is therefore more suggestive of an agenesis than a viral infection. Infection of adult laboratory animals (mice with influenza viral infection) also can induce mesencephalic duct stenosis resulting in hydrocephalus, but in contrast to neonatal infection, there is a persistent astroglial response in the area of stenosis.
Congenital Hydrocephalus.
CSF can accumulate in the ventricular system, the subarachnoid space, or both. The type of hydrocephalus that develops depends on the site of blockage that disrupts normal flow of CSF.
Exactly which portions of the ventricular system will be dilated in hydrocephalus depends on the site of the blockage:
-
1.
Blockage of the interventricular foramen between a lateral and third ventricle leads to unilateral dilation of that lateral ventricle.
-
2.
Blockage of both interventricular foramina leads to bilateral dilation of both lateral ventricles.
-
3.
Blockage of the mesencephalic duct leads to bilateral dilation of the lateral ventricles, the third ventricle, and the segment of the mesencephalic duct proximal to the blockage.
-
4.
Blockage of the lateral apertures of the fourth ventricle leads to bilateral dilation of lateral ventricles, the third ventricle, the mesencephalic duct, and the fourth ventricle.
-
5.
Blockage of reabsorption leads to bilateral dilation of lateral ventricles, the third ventricle, the mesencephalic duct, the fourth ventricle, and the subarachnoid space.
As an example, following blockage of the interventricular foramina, the pressure in the lateral ventricles increases; the ventricles dilate; the ependyma becomes atrophied and focally discontinuous; and because of the pressure gradient, CSF is forced into the periventricular white matter, leading to hydrostatic edema. Hydrostatic edema results in degeneration and atrophy of myelin and axons, and this loss of tissue results in further expansion of the ventricles.
The forms of hydrocephalus are communicating and noncommunicating hydrocephalus. Communicating hydrocephalus, the least common of the two forms, occurs when there is communication of ventricular CSF with the subarachnoid space where the CSF can be in excess. Noncommunicating hydrocephalus results from obstruction within the ventricular system at, or rostral to, the lateral apertures of the fourth ventricle. An area of great vulnerability for obstruction is the mesencephalic aqueduct. Noncommunicating hydrocephalus can also occur without any evidence of obstruction to CSF flow as a result of failure of the reabsorption of CSF.
Another type of hydrocephalus, referred to as hydrocephalus ex vacuo (or compensating hydrocephalus), is not usually a congenital abnormality but occurs secondary to absence or loss of cerebral tissue. This type of hydrocephalus can occur in utero from destruction and loss of cerebral tissue surrounding the lateral ventricles (e.g., in hydranencephaly). Hydrocephalus ex vacuo is discussed further in the next section on acquired hydrocephalus.
Gross lesions associated with communicating and noncommunicating congenital hydrocephalus include enlargement (doming) of the cranium if obstruction occurs before the sutures have fused (Fig. 14-37 ). The bones of the calvaria are extremely thin, and the fontanelles are prominent (Fig. 14-38 ). In the brain there is prominent enlargement of the ventricular system proximal to the point of obstruction (Fig. 14-39 ). White matter adjacent to the dilated lateral ventricles is reduced in thickness, although the gray matter can retain a relatively normal appearance. As the hydrocephalus progresses, atrophy with fenestration and cavity formation of the interventricular septum (septum pellucidum), atrophy of the hippocampus in the floor of the lateral ventricles, and flattening of cortical gyri can occur. If the obstruction is abrupt and pressure builds rapidly, the cerebral hemispheres can be displaced caudally, causing herniation of the parahippocampal gyri under the tentorium cerebelli and of the vermis of the cerebellum through the foramen magnum. The resulting coning of the cerebellum can be accompanied by hemorrhage and necrosis of cells in the cerebellar folia as a result of ischemia and infarction. Microscopically the ependyma can become atrophied and focally discontinuous, and there is loss of cells and cell processes in adjacent white matter and variably in the gray matter.
Figure 14-37.
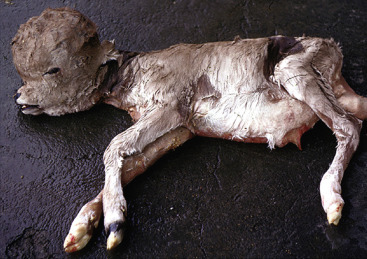
Congenital Hydrocephalus, Brain, Calf.
Note the symmetrically enlarged and dome-shaped calvaria. The bone of the calvaria is thinned and distorted from pressure from the expanding brain during gestation.
(Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
Figure 14-38.
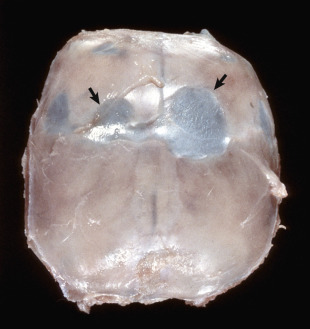
Calvaria, View of the Dorsal Surface, Congenital Hydrocephalus, Dog.
The bone of the calvaria is thin and the fontanelles (arrows) are enlarged. The translucent membrane covering the fontanelles is periosteum.
(Courtesy Drs. J. Wright and D. Duncan, College of Veterinary Medicine, North Carolina State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Figure 14-39.
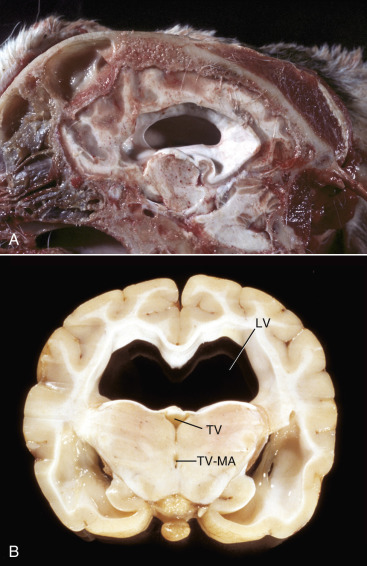
Hydrocephalus, Brain, Dog.
A, Midsagittal section of the head, third ventricle. Note the dilated third and lateral ventricles and the absence of most of the septum pellucidum between the left and right lateral ventricles. B, Junction between parietal and occipital lobes, level of thalamus. Bilateral dilation of lateral ventricles (LV) dorsally, and ventrolaterally. The fornix has separated and lies on the flattened floor of the ventricle. Note that the third ventricle (TV) and junctional area between the third ventricle and mesencephalic aqueduct (TV-MA) are not enlarged and are possibly even reduced in size, suggesting that the obstruction may be at, or rostral to, this plane of section.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Clinically, congenital hydrocephalus occurs most frequently in brachycephalic or toy breeds such as the Chihuahua, Lhasa apso, and toy poodle. Clinical signs occur within the first year of life, often before 3 months of age. Behavioral changes are the most common and include poor motor skill development; delay in learned behavior, such as house training; somnolence; dullness; episodic confusion; circling; periodic aggression; and seizures.
Acquired Hydrocephalus.
Noncommunicating acquired hydrocephalus has been associated with injury of the ependyma, resulting in obstruction of any of the following: the lateral apertures of the fourth ventricle, the cerebral aqueduct, or the interventricular foramen. Causes of obstruction include compression by cerebral abscesses and neoplasms, and blockages by infectious/inflammatory disease resulting in a ventriculitis and, uncommonly, by cholesteatomas in the choroid plexus of the lateral ventricles of the horse. Because the calvaria has now ceased to grow, unlike congenital hydrocephalus, it is of normal size and shape, and its bone is of normal thickness.
A second type of acquired hydrocephalus, referred to as hydrocephalus ex vacuo (or compensating hydrocephalus), usually occurs in the cerebral hemispheres secondary to loss of neural tissue. If there is loss of neurons in the cerebral cortex, as in bovine polioencephalomalacia or other types of laminar cortical necrosis, the axons of these neurons, which normally traverse the white matter of the cerebral hemispheres, will disappear, and there will be atrophy of the cortex from the loss of neuronal cell bodies and of the white matter from the loss of axons. The lateral ventricles will expand into the space once occupied by white matter. This dilation of the lateral ventricles may be bilateral when there has been a loss of white and gray matter from both cerebral hemispheres, or it may be unilateral. If the loss of cortex is localized, as in an infarct, then dilation of the lateral ventricle will not uniformly involve the whole lateral ventricle. Examples of disorders in which hydrocephalus ex vacuo occur include some storage diseases (ceroid-lipofuscinosis in sheep), aging, and postradiation exposure, all of which are associated with cerebral atrophy. There is no evidence of obstruction of the normal flow of CSF in this type of hydrocephalus.
Diseases Caused by Microbes
Bacteria
Brain Abscesses.
Cerebral abscesses in animals are relatively uncommon but arise after entry of bacteria into the CNS. This may occur either from direct extension or hematogenously. With direct extension, abscesses occur following penetrating wounds, such as calvarial fractures, or from spread of infection from adjacent tissues, such as the meninges, paranasal sinuses, and internal ear, and through the cribriform plate of the ethmoid (see Fig. 14-29). Diseases that cause bacteremia or septicemia result in infectious microbes being trapped in vascular beds within the CNS and meninges. Abscesses usually arise within gray matter because it receives a disproportionate share of blood flow in the CNS, usually at the gray-white (cortex–subcortical white matter) junction. They exert effects in the CNS by disruption and destruction of tissue and by displacement as space-occupying lesions. If the abscess grows quickly, tissue is more likely to be disrupted and destroyed and in the worst case penetrate the wall of the lateral ventricle and cause a ventriculitis. Bacteria in the CSF may be carried into the subarachnoid space and cause a leptomeningitis. On the other hand, if growth is slow, tissue is more likely to be displaced. Chronic abscesses become encapsulated by either fibrous tissue if they are close to the leptomeninges or by astrocytes away from the meninges. The mechanism of tissue injury is likely a secondary bystander effect related to the actions of the mediators of inflammation and the toxins and other products elaborated from bacteria. Bacteria appear to localize in specific areas of the CNS based on receptor-mediated attachment or because of vascular flow patterns unique to the gray matter–white matter interface of the CNS that allow bacteria to attach to and move through the blood-brain barrier. This latter flow mechanism likely occurs because small blood vessels supplying the cerebrum fail to continue into the white matter and end with their horizontal branches running parallel to the surface of the gyrus within the gray matter at the interface with the white matter. Once within the CNS or meninges, bacteria replicate and elicit an inflammatory response. Lytic enzymes released from lysosomes of neutrophils and other inflammatory cytokines secreted by lymphocytes and macrophages destroy neurons and their processes and disrupt synapses, thus affecting neurotransmission.
Grossly, brain abscesses can be single or multiple, be discrete or coalescing, and have varied sizes (Fig. 14-40 ). Early in the process, abscesses consist of a white to gray to yellow, thick to granular exudate. The color of the exudate can be influenced by the exuberance of the pyogenic response elicited by the inciting bacteria and by any pigments produced by the bacteria. Streptococcus spp., Staphylococcus spp., and Corynebacterium spp. may produce a pale-yellow to yellow, watery to creamy exudate. Coliforms, such as E. coli and Klebsiella spp., may produce a white to gray, watery to creamy exudate. Pseudomonas spp. may produce a green to bluish-green exudate. The borders of abscesses are often surrounded by a red zone of active hyperemia induced by inflammatory mediators acting on capillary beds.
Figure 14-40.
Chronic Cerebral Abscesses, Sheep.
Abscesses with caseous centers (arrow) have replaced most of the right cerebral hemisphere, enlarged it, and displaced the midline to the left. The abscesses are encapsulated by a thick fibrous capsule generated by fibroblasts of the pia and perivascular spaces of the outer cortex.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Brain abscesses can arise in some food animal species from an extension of otitis interna (see Fig. 14-29). These animals often display evidence of facial nerve paralysis, such as a drooping ear. The cerebellopontine angle and adjacent structures are the common locations for such abscesses. In horses, Streptococcus equi subsp. equi (strangles) can cause brain abscesses via hematogenous spread (Fig. 14-41 ). Direct penetration may also occur in small ruminants that lack frontal sinuses because of improper dehorning procedures. Brain abscesses are space-occupying lesions and as such can have a devastating effect on brain function. Depending on size and location, compression via mass effect (increased intracranial pressure) of vital structures (nuclei that regulate cardiac and respiratory rhythms) and brain displacements (cerebellar vermis, parahippocampal gyri) are two common sequelae to acute abscesses. Abscesses can occur in the spinal cord as a result of direct extension of bacterial vertebral osteomyelitis through the dura (Fig. 14-42 ), after tail docking in lambs, and occasionally from hematogenous spread.
Figure 14-41.
Abscess, Right Cerebral Hemisphere, Horse.
A, The cerebral cortex contains an abscess (arrow) caused by Streptococcus equi subsp. equi entering the central nervous system via the blood. A fibrous capsule is present on the lateral, medial, and dorsal sides of the abscess (most obvious on the lateral side as a gray band). There is no obvious capsule present on the ventral side (i.e., toward the right lateral ventricle). Microscopically, there is a thin glial capsule (astrogliosis). Note also the increased size of the right hemisphere with blurring of the distinction between gray and white matter, an indication of edema. B, Chains of Gram-positive (blue staining) cocci in the inflammatory exudate from an abscess caused by Streptococcus equi subsp. zooepidemicus. Gram stain.
(A courtesy Dr. K. Read, College of Veterinary Medicine, Texas A&M University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 14-42.
Diskospondylitis, Thoracic Spinal Cord, Pig.
This type of abscess (arrows) is commonly caused by bacterial emboli that lodge in intervertebral disks or in the body of vertebrae causing osteomyelitis, which can extend into intervertebral disks. Large intervertebral abscesses can compress the spinal cord and cause Wallerian degeneration of nerves, mainly in the ventral funiculi but also in other funiculi. In this case, remodeling and proliferation of the vertebral bone secondary to the infection also contributed to the narrowing of the spinal canal and compression of the spinal cord.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Clinically, animals with brain abscesses can show abnormal mental behaviors, ataxia, head tilt, circling, and loss of vision.
Diffuse Encephalitis.
Common bacteria have the potential to produce disease in the CNS by hematogenous spread and vasculitis (see the section on Neonatal Septicemia).
Ependymitis and Choroid Plexitis.
Infectious microbes, especially pus-forming bacteria, such as the coliform and Streptococcus spp., can enter the CNS hematogenously or via direct extension, invade the choroid plexuses, and be released into the CSF, gaining access to ependymal cells lining the ventricular system. Inflammation of the ependyma is called ependymitis, whereas inflammation of the choroid plexus is called choroid plexitis. Gross lesions usually consist of gray-white to yellow-green thick to gelatinous CSF within the ventricular system and choroid plexuses that are granular and gray-white, with areas of active hyperemia and hemorrhage. If the bacteria traverse through the lateral apertures of the fourth ventricle, they can enter and spread throughout the subarachnoid space, possibly inducing suppurative bacterial leptomeningitis. The exudate can also obstruct CSF flow, leading to noncommunicating hydrocephalus. Although most cases are caused by bacteria, cats infected with feline infectious peritonitis (FIP) can develop protein-rich fluid and exudate within the ventricular system that can lead to plugging of the mesencephalic aqueduct with subsequent hydrocephalus. Microscopically, inflammatory cells, especially neutrophils, mixed with fibrin, hemorrhage, and bacteria, can be seen in the exudate.
Meningitis.
Meningitis refers to inflammation of the meninges (Fig. 14-43 ). In animals, meningitis is most commonly caused by bacteria such as E. coli and Streptococcus spp. that traverse to the leptomeninges and subarachnoid space hematogenously. Bacteria can also spread to the meninges by direct extension and leukocytic trafficking. In common usage the term meningitis generally refers to inflammation of the leptomeninges (the pia mater, subarachnoid space, and adjacent arachnoid mater) in contrast to inflammation of the dura mater, which is referred to as pachymeningitis. Leptomeningitis can be acute, subacute, or chronic and, depending on the cause, suppurative, eosinophilic, nonsuppurative, or granulomatous. Inflammation of specific parts of the dura mater of the cranial cavity can occur in the external periosteal dura after osteomyelitis, formation of extradural abscesses and pituitary abscesses, and skull fracture and involve the inner dura in association with leptomeningitis. Abscesses of the pituitary fossa occur with some frequency in cattle, including in bulls with nose rings. Bacteria isolated from the cases include Pasteurella multocida and Trueperella pyogenes. The abscess can result from spread of infection arising in the caudal nasal cavity or sinuses, possibly through direct extension or through the venous circulation. Incision of the pituitary fossa releases a thick, viscous, opaque tan to yellow exudate, which can elevate the dura mater surrounding the fossa. Infection can extend via the infundibular recess of the third ventricle into the ventricular system, resulting in ventriculitis, ependymitis, and empyema. Systemic bacterial infections in neonates are a common cause of acute meningitis (leptomeningitis), which are suppurative and fibrinous. In animals, leptomeningitis secondary to a selective viral infection only of the leptomeninges is very rare and is usually seen in combination with viral-induced encephalitides.
Figure 14-43.
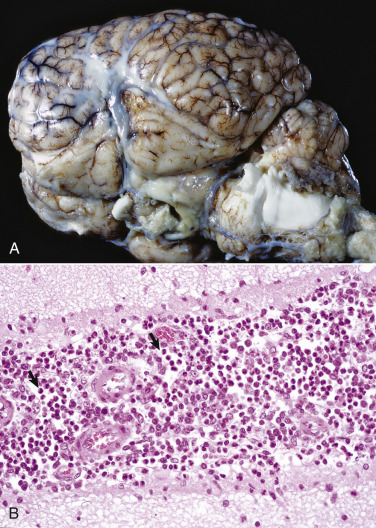
Suppurative Bacterial Meningitis, Cerebral Hemispheres, Horse.
A, Pale yellow-white thick exudate consistent with an infiltrate of neutrophils admixed with bacteria, cellular debris, edema fluid, and fibrin is present in the subarachnoid space on the lateral surface and also in the sulci. Overall the gyri are flattened, indicating brain swelling and compression. B, The arachnoid space of the leptomeninges in this sulcus contains a mixture of neutrophils (arrows), other mononuclear inflammatory cells, cellular debris, edema fluid, and fibrin. H&E stain.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Neonatal Septicemia.
Neonatal septicemia typically involves E. coli, Streptococcus spp., Salmonella spp., Pasteurella spp., and Haemophilus spp. The release of endotoxins and bacterial cell wall components, such as lipopolysaccharide, teichoic acid, and proteoglycans, in the CNS vasculature leads to the secretion of cytokines (TNF, interleukin, platelet-activating factor, prostaglandins, thromboxane, and leukotrienes) from the endothelium and trafficking CNS macrophages, followed by adhesion of neutrophils, injury to the endothelium and blood-brain barrier, and vasculitis resulting initially in brain swelling and brain edema and increased intracranial pressure.
Although there are differences in the diseases caused by these microbes, they tend to produce fibrinopurulent inflammation of membranous tissues (serosal surfaces) of the body. Leptomeninges, choroid plexus, and ependyma of the CNS—as well as sites often preferentially involved in hematogenous spread of bacteria like the synovium, uvea, and the serosal lining of body cavities—can be affected in various combinations. Infections are often acquired perinatally, and onset is usually within a few days of birth up to 2 weeks (Box 14-7 ). The initial portal of entry can be oral; intrauterine; umbilical; or surgical, via postsurgical procedures such as castration and ear notching; or via the respiratory system, but the bacteria eventually spread to the CNS hematogenously.
Box 14-7. Central Nervous System Bacterial Infections in Young Animals.
Calf
Escherichia coli: Leptomeningitis, choroiditis, ependymitis and ventriculitis, synovitis, ophthalmitis, and perioptic neuritis
Pasteurella/Mannheimia spp.: Leptomeningitis, ependymitis and ventriculitis
Streptococcus spp.: Leptomeningitis, synovitis, ophthalmitis
Foal
Escherichia coli: Leptomeningitis, ventriculitis, polyserositis, synovitis
Streptococcus spp.: Leptomeningitis, polyserositis, synovitis
Salmonella typhimurium: Leptomeningitis, ependymitis and ventriculitis, choroiditis, synovitis
Lamb
Escherichia coli: Leptomeningitis, ependymitis and ventriculitis, peritonitis, synovitis
Pasteurella/Mannheimia spp.: Leptomeningitis
Pig
Escherichia coli: Leptomeningitis, ophthalmitis
Haemophilus parasuis: Leptomeningitis, polyserositis, synovitis
Streptococcus suis type I and II: Leptomeningitis, choroiditis, ependymitis, cranial neuritis, myelitis
Salmonella choleraesuis: Leptomeningitis, ophthalmitis
Gross CNS lesions are commonly present and include congestion, hemorrhage, and diffuse to focal cloudiness or opacity in the leptomeninges, resulting in a leptomeningitis caused by accumulation of exudates (see Fig. 14-43). The ventricles contain fibrin, usually as a thin layer on the ependymal surface or as a pale coagulum in the CSF of the ventricular lumen, secondary to a choroid plexitis and/or ependymitis.
Microscopic lesions vary according to the microbe. With the exception of Salmonella spp., the lesions consist of deposits of fibrin and an infiltration of mainly neutrophils in and around the blood vessels and capillaries of the leptomeninges, choroid plexus, and ependymal or subependymal areas of the brain. The epithelium of the choroid plexus and ependymal lining of the ventricles can be disrupted by cellular degeneration, disorganization, and necrosis, and this inflammation can extend into the adjacent CNS. A vasculitis with thrombosis and hemorrhage can be associated with lesions caused by E. coli. Lesions caused by Salmonella spp. are not limited to the perinatal period. CNS involvement in salmonellosis is generally limited to foals, calves, and pigs, and in contrast to the infections mentioned earlier, the leukocytic response tends to have a greater proportion of macrophages and lymphocytes, often to the extent that the inflammation is designated histiocytic or granulomatous. This difference presumably reflects the fact that Salmonella spp. can be facultative pathogens of the monocyte-macrophage system. As is true in other tissues, vasculitis, thrombosis, necrosis, and hemorrhage often accompany Salmonella infections of the CNS. Haemophilus parasuis, which causes Glasser's disease, is also a frequent cause of leptomeningitis, polyserositis, and polyarthritis in 8- to 16-week-old pigs. Again, lesions are as previously noted with fibrinopurulent inflammation involving the leptomeninges, serosal linings of body cavities, and joints.
Bacterial infection with CNS and visceral involvement occurs in neonatal pigs and through the weaning period. These diseases are deserving of special mention because of the incidence and stereotyped nature of the infections. Several strains of Streptococcus suis are capable of causing disease. Type I strains generally cause disease in suckling pigs ranging in age from 1 to 6 weeks, whereas type II strains affect older pigs 6 to 14 weeks old. Type II strains are recognized as one of the more important serotypes, causing meningitis not only in pigs but also in human beings, particularly those working with pigs or handling porcine tissues. Other serotypes and untyped strains can also cause systemic disease that results in leptomeningitis, choroid plexitis, and ependymitis. Extension to involve cranial nerve roots or the central canal of the cervical spinal cord also occurs. The character of the inflammation is fibrinopurulent, and necrotic foci can be found in brainstem, cerebellum, and anterior spinal cord.
Clinically, affected animals are initially ataxic and then become laterally recumbent with rhythmic paddling of the limbs. As the disease progresses, they may become comatose and die.
Viruses.
The viruses causing CNS disease in domestic animals are listed in Table 14-3 .
Table 14-3.
Viruses Causing Central Nervous System Disease in Domestic Animals
| Virus Genus | Disease | Type of Injury |
|---|---|---|
| Arbovirus | Equine encephalomyelitis | Encephalitis/myelitis/meningitis/vasculitis |
| Japanese encephalitis | Encephalitis/myelitis/meningitis | |
| Louping ill | Encephalitis/myelitis/meningitis | |
| West Nile viral encephalomyelitis | Encephalitis/myelitis | |
| Wesselsbron virus | Malformations | |
| Bornavirus | Borna disease | Encephalitis/myelitis |
| Bunyavirus | Akabane disease | Malformations/encephalitis |
| Arthrogryposis hydranencephaly complex (Cache Valley fever) | Malformations/encephalitis | |
| Rift Valley fever, Schmallenberg virus | Malformations/encephalitis | |
| Coronavirus | Feline infectious peritonitis | Vasculitis/encephalitis/myelitis/meningitis |
| Hemagglutinating encephalomyelitis | Encephalitis/myelitis/meningitis/ganglioneuritis | |
| Enterovirus | Enterovirus-induced porcine polioencephalomyelitis | Encephalitis/myelitis |
| Herpesvirus | Equine herpesvirus 1 myeloencephalopathy | Encephalitis/myelitis/meningitis/vasculitis |
| Bovine malignant catarrhal fever | Encephalitis/myelitis/meningitis/vasculitis | |
| Infectious bovine rhinotracheitis | Encephalitis | |
| Pseudorabies | Encephalitis/myelitis/meningitis | |
| Lentivirus | Visna | Encephalitis/myelitis/demyelination |
| Caprine leukoencephalomyelitis-arthritis | Encephalitis/myelitis/demyelination | |
| Orbivirus | Bluetongue | Malformations/encephalitis |
| Paramyxovirus | Canine distemper | Demyelination/encephalitis/myelitis |
| Old-dog encephalitis | Encephalitis/demyelination/meningitis/vasculitis | |
| Parvovirus | Feline panleukopenia virus | Malformations/meningitis |
| Pestivirus | Classical swine fever | Malformations/hypomyelination/encephalitis/meningitis/vasculitis |
| Bovine viral diarrhea | Malformations/meningitis/dysmyelination | |
| Border disease | Malformations/hypomyelination | |
| Polyomavirus | Progressive multifocal leukoencephalopathy | Demyelination |
| Rhabdovirus | Rabies | Encephalitis/myelitis/meningitis/vasculitis/ganglioneuritis |
Arboviruses
Japanese Encephalitis.
See E-Appendix 14-2.
Louping Ill.
See E-Appendix 14-2.
Herpesviruses.
Encephalitic herpesviruses, members of the subfamily Alphaherpesvirinae, cause cell injury through (1) necrosis of infected neurons and glial cells, (2) necrosis of infected endothelial cells, and (3) secondary effects of inflammation, cytokines, and chemokines. Although necrosis appears to be the principal mechanism for cell injury, recent studies indicate that apoptotic cell death also plays a role.
Neurotropic herpesviruses enter the CNS principally by retrograde axonal transport; however, entry by hematogenous spread via viremia and leukocytic trafficking may occur. These viruses also have a unique survival mechanism that allows them to hide in a latent form in nervous tissue, for example, in trigeminal ganglion of pigs infected with pseudorabies virus. Stress or other factors can activate latent virus, resulting in encephalitis.
Rhabdoviruses
Rabies Encephalitis.
Rabies virus (family Rhabdoviridae) is one of the most neurotropic of all viruses infecting mammals. It is generally transmitted by a bite from an infected animal; however, respiratory infection has also been uncommonly reported after exposure to virus in bat caves, accidental human laboratory exposure, and corneal transplants.
The mechanism for spread of rabies virus from the inoculation site to the CNS is illustrated in Figure 14-44 . Rabies virus may first replicate locally at the site of inoculation. Infection of and replication in local skeletal muscle myocytes is an important initiating event. The virus then enters peripheral nerve terminals by binding to nicotinic acetylcholine receptors at the neuromuscular junction. The probability is greater that the virus will be taken up by both axon terminals and myocytes after a large inoculation dose. If the virus directly enters peripheral nerve terminals, the incubation period will more likely be short, regardless of whether muscle cells are infected. With progressively lower doses of virus, however, there is a greater possibility that the virus will enter either nerve terminals or myocytes but not both. This situation can result in a short incubation period if the virus directly enters nerve terminals as described previously or could result in a more prolonged incubation period if there was initial infection and retention of virus in myocytes before its release and uptake by nerve terminals.
Figure 14-44.
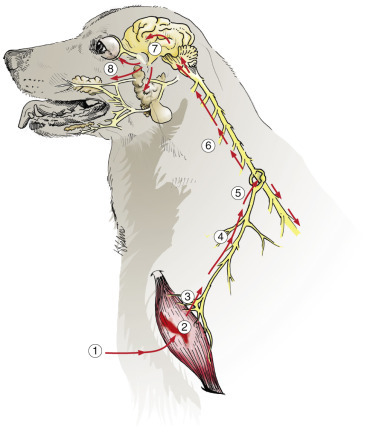
Pathogenesis of Rabies.
After a bite wound, 1, the rabies virus initially replicates in muscle (can enter peripheral nerves directly), 2, enters, 3, and ascends (retrograde axonal transport) the peripheral nerve, 4, to the dorsal root ganglion, 5, enters the spinal cord, 6, and ascends, 7, to the brain via ascending and descending nerve fiber tracts, infects brain cells, spreads to salivary glands, 8, and the eye and is excreted in saliva.
The virus moves from the periphery to the CNS by fast retrograde axoplasmic transport, apparently via sensory or motor nerves, at a rate of 12 to 100 mm per day. Experimental data suggest that rabies virus phosphoprotein interacts with dynein LC8, a microtubule motor protein used in retrograde axonal transport. With sensory axons the first cell bodies to be encountered after inoculation of a rear leg would be those of spinal ganglia, whose neuronal processes extend to the dorsal horn of the spinal cord. For motor axons the cell bodies of the lower motor neurons in ventral horn gray matter or neuronal cell bodies of the autonomic ganglia are the ones initially infected. It is not known whether viral infection and replication in neurons of dorsal root ganglia are essential for infection of the CNS. The virus then moves into the spinal cord and ascends to the brain using both anterograde and retrograde axoplasmic flow. During the spread of the virus between neurons within the CNS, there is also simultaneous centrifugal movement via anterograde axonal transport of the virus peripherally from the CNS to axons of cranial nerves. This process results in infection of various tissues, including the oral cavity and salivary glands, permitting transmission of the disease in saliva. An additionally important feature of rabies is that infection of nervous and nonnervous tissue, such as the salivary glands, occur at the same time, which permits affected animals to have the required aggressive behavior plus passage of the virus into the saliva to facilitate the transmission of the disease.
The results of recent experimental studies have helped clarify the mechanism by which the virus spreads within the CNS. After axoplasmic spread of the virus from an inoculated rear leg to neurons of the associated segments of the spinal cord, rapid spread of infection to the brain occurs via long ascending and descending fiber tracts, bypassing the gray matter of the rostral spinal cord. This early spread of the virus has been suggested to explain how induction of behavioral changes occurs before there is sufficiently severe injury to cause paralysis and allows dissemination of infection before there is time for a notable immune response. Spread of infection within neurons of the CNS occurs via both anterograde and retrograde axoplasmic flow, with corresponding neuron-to-neuron spread by axosomatic-axodendritic and somatoaxonal-dendroaxonal transfer of virus. Transsynaptic spread can occur by budding of developing virions from the neuronal cytoplasm (cell body or dendrite) into a synapsing axon or in the form of bare viral nucleocapsid (ribonucleoprotein-transcriptase complexes) in the absence of a complete virion.
In vivo experimental studies using a laboratory strain of rabies virus showed that the virus caused a downregulation of approximately 90% of genes in the brain at more than fourfold lower levels. Affected genes were those involved in regulation of cell metabolism, protein synthesis, growth, and differentiation. Other experimental studies have shown increased quantities of nitric oxide in brains of rabies-infected animals, suggesting that nitric oxide neurotoxicity may mediate neuronal dysfunction. Finally, the rabies virus has been shown to induce apoptotic cell death of brain neurons in mouse models. The exact mechanism of rabies virus–induced neuronal injury in domestic and wildlife species remains to be fully determined.
Gross lesions of the infected central nervous tissue are often absent but can include hemorrhage, especially in the spinal cord gray matter. Microscopic lesions of the CNS are typically lymphocytic and include a variable leptomeningitis and perivascular cuffing with lymphocytes, macrophages, and plasma cells; microgliosis, which sometimes is prominent; variable, but often not severe, neuronal degeneration; and ganglioneuritis. Emphasis should be given to the fact that infected neurons often are minimally altered morphologically, and in some cases the only lesion noted is intracytoplasmic, acidophilic inclusion bodies, called Negri bodies (Fig. 14-45 ). Also, dogs are reported to have a tendency to develop a more severe inflammatory reaction than other species, such as the cow, in which little if any inflammation might occur. Nonneural lesions include variable nonsuppurative sialitis accompanied by necrosis and presence of Negri bodies in salivary epithelial cells.
Figure 14-45.
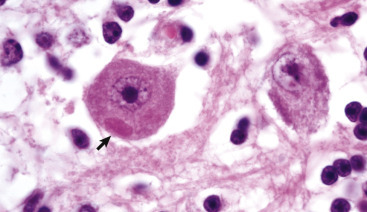
Rabies, Negri Body, Cerebellum, Purkinje Cell, Cow.
A large pale red (eosinophilic) inclusion (Negri body) is present in the cytoplasm of the neuron cell body (arrow). In the cow, Negri bodies are commonly seen in Purkinje cells and in other neurons, such as those of the red nucleus and cerebral cortex. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Negri bodies, formed within neurons of the CNS and even in the cranial trigeminal, spinal, and autonomic ganglia, have long been the hallmark of rabies infection, although they are not present in all cases. The inclusions are intracytoplasmic and initially develop as an aggregation of strands of viral nucleocapsid, which rather quickly transforms into an ill-defined granular matrix. Mature rabies virions, which bud from the nearby endoplasmic reticulum, can also be located around the periphery of the matrix. With time the Negri body becomes larger and detectable by light microscopy. Classically in H&E-stained sections the Negri body, which is eosinophilic, has one or more small, light clear areas called inner bodies that form as a result of invagination of cytoplasmic components (that include virions) in the matrix of the inclusion. Inclusions that do not possess “inner bodies” have been referred to as Lyssa bodies, but they are actually Negri bodies without cytoplasmic indentation. It should also be noted that both fixed viruses (adapted to the CNS by passage) and street viruses (that produce the naturally occurring disease) produce the same ultrastructural features, but fixed viral strains generally cause severe neuronal degeneration that precludes the development and thus the detection of Negri bodies. Negri bodies also tend to occur more frequently in large neurons such as the pyramidal neurons of the hippocampus (most common in carnivores, like the dog), neurons of the medulla oblongata, and Purkinje cells of the cerebellum (most common in herbivores, like cattle). The preferred tissues for rabies examination by light microscopy and by florescent antibody technique for virus include hippocampus, cerebellum, medulla, and the trigeminal ganglion. The typical samples submitted for fluorescent antibody staining are the medulla and cerebellum.
A spongiform lesion, indistinguishable qualitatively from the lesion characteristic for several of the spongiform encephalopathies, was described for the first time in 1984 by Charlton. This lesion was initially detected in experimental rabies in skunks and foxes and later in the naturally occurring disease in the skunk, fox, horse, cow, cat, and sheep. The lesion occurs in the neuropil of the thalamus and cerebral cortex most prominently, initially as intracytoplasmic membrane-bound vacuoles in neuronal dendrites and less commonly in axons and astrocytes. The vacuoles enlarge, compress surrounding tissue, and ultimately rupture, forming a tissue space. Although the mechanism responsible for the development of this lesion has not been determined, it is thought to result from an indirect effect of the rabies virus on neural tissue (possibly involving an alteration of neurotransmitter metabolism).
The clinical signs in domestic animals are similar with some differences between species. The clinical disease in the dog has been divided into three phases: prodromal, excitatory, and paralytic. In the prodromal phase, which lasts 2 to 3 days, the animal can have a subtle change in temperament. Furious rabies refers to animals in which the excitatory phase is predominant, and dumb rabies refers to animals in which the excitatory phase is extremely short or absent and the disease progresses quickly to the paralytic phase. Cattle and carnivores generally have the furious form of rabies, and affected animals are restless and aggressive. Other somewhat unique signs of cattle with rabies include bellowing, general straining, tenesmus, and signs of sexual excitement followed by paralysis and death. Mules, sheep, and pigs usually have the excitatory form of rabies. Horses can have early signs that are atypical for a neurologic disease but terminally tend to have the excitatory form.
When conducting a necropsy on an animal suspected of having rabies, it is important to remember (1) to provide additional protection (double gloves, mask, eye protection, and proper ventilation) for the prosector above those used for routine postmortem examination and (2) to collect the appropriate CNS tissues (hippocampus, cerebellum, and medulla and optionally the spinal cord) for examination by immunofluorescence and sometimes mouse inoculation. The remainder of the brain should be fixed by immersion in 10% neutral buffered formalin for histopathologic examination.
Bornaviruses
Borna Disease.
See E-Appendix 14-2.
Fungi and Algae.
Infection of the CNS by a variety of fungi and algae has been reported in domestic animals. Most reported cases are typically isolated occurrences and often represent opportunistic infection in immunocompromised individuals. Infections have involved genera such as Aspergillus, Candida, and Mucor; dematiaceous fungi; and the blue-green algae, Prototheca. These infections do not have a predilection for the nervous system. Of the systemic fungi, CNS infections have occurred with Coccidioides immitis, Blastomyces dermatitidis, Histoplasma capsulatum, and Cryptococcus neoformans, but only C. neoformans has a particular affinity for the CNS. These agents reach the CNS by leukocytic trafficking and hematogenous spread from primary sites of infection located in other areas (lung and skin commonly) of the body.
This group of pathogens usually elicits, as characterized by B. dermatitidis, a granulomatous to pyogranulomatous inflammatory response (Fig. 14-46 ). This response can be locally extensive, or distinct granulomas can form in the CNS and meninges. Grossly, CNS lesions consist of moderately well demarcated expansile yellow-brown foci that displace and disrupt normal tissue (Fig. 14-47 ). Microscopically, the exudate consists of neutrophils, macrophages (epithelioid type), and multinucleated giant cells. The latter two cell types may contain microbes in their cytoplasm. B. dermatitidis microbes are broad-based, budding, spherical yeastlike microbes 8 to 25 mm in diameter (Fig. 14-48 ). The inflammatory response, including cells (granulomatous inflammatory cells) and cytokines, leads to the axonal, neuronal, and myelin disruption observed in these mycotic diseases.
Figure 14-46.
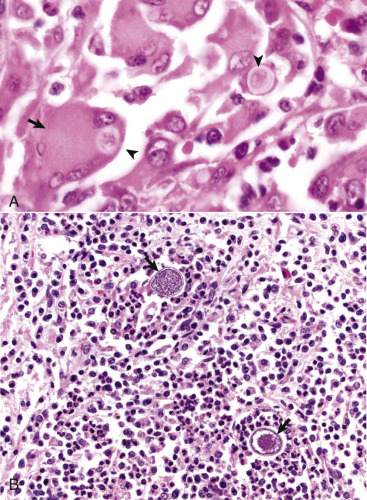
Granulomatous Encephalitis, Brain.
A, Dog. This inflammatory response, consisting of a mixture of macrophages, multinucleated giant cells (arrow), lymphocytes, varying numbers of neutrophils, and occasional plasma cells, is typical of central nervous system infections by fungi and algae. Blastomyces dermatitidis microbes are present in the exudate and within macrophages and giant cells (arrowheads). H&E stain. B, Alpaca. Coccidioides immitis–associated encephalitis. Large numbers of neutrophils and macrophages (pyogranulomatous inflammation) surround sporangia of C. immitis (arrows). H&E stain.
(A courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
Figure 14-47.
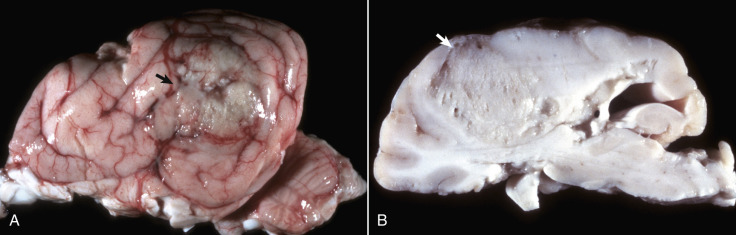
Blastomycosis, Cerebrum, Dog.
A, The subarachnoid space (leptomeninges) of the left cerebral hemisphere (parietal-temporal lobes) contains a locally extensive focus granuloma caused by Blastomyces dermatitidis (arrow) with extension into subjacent cortex. B, A parasagittal section of a similar lesion from another dog shows a moderately well demarcated granuloma in the white matter of the frontoparietal cortex (arrow).
(Courtesy College of Veterinary Medicine, University of Illinois.)
Figure 14-48.
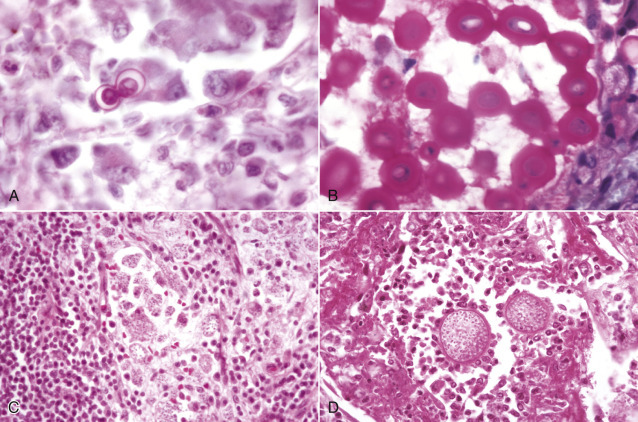
Morphologic Features of Fungi That May Infect the Central Nervous System.
A,Blastomyces dermatitidis, 8 to 25 mm in diameter, broad-based budding spherical yeast-like microbes, intracellular or extracellular location. H&E stain. B,Cryptococcus neoformans. In this illustration the microbe is surrounded by a mucinous capsule that is stained with Mayer's mucicarmine. The capsule varies in width but can be so thick as to give the microbe an overall diameter of 30 mm. The microbe without its capsule is 5 to 20 mm in diameter. The capsule does not stain with H&E, thus causing the microbe to appear to be surrounded by a clear halo (see Fig. 14-50, A). The microbes are oval to spherical but may be crescentic or cup shaped in routine mucicarmine- and H&E-stained sections. Dehydration that occurs during processing of the tissue to embed it in paraffin causes this shrinkage and distortion. Mayer's mucicarmine stain, aqueous wet mount. C,Histoplasma capsulatum, located intracellularly, is spherical to elongated, 5 to 6 mm in diameter. H&E stain. D,Coccidioides immitis, spherules (20 to 30 mm in diameter) containing endospores (<5 mm in diameter), can be intracellular or extracellular. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
CNS infections with C. immitis or H. capsulatum elicit an inflammatory response similar to that which occurs in B. dermatitidis. In coccidioidomycosis the microbes are extracellular and/or intracellular spherules (20 to 30 mm in diameter) containing endospores (<5 µm in diameter), whereas in histoplasmosis the microbe principally is located intracellularly and is 5 to 6 mm in diameter. The microscopic features of these fungi are compared in Figure 14-48.
Cryptococcus spp.
Cryptococcosis, most commonly occurring in cats, dogs, and occasionally horses, is caused by two species of Cryptococcus, C. neoformans and Cryptococcus gattii. These pathogens enter the leptomeninges and subarachnoid space by direct extension through the cribriform plate after a nasal or sinus infection or hematogenously by leukocytic trafficking usually from a pulmonary infection. Leptomeningeal inflammation can also extend along the roots of cranial nerves. Cryptococcus spp. secrete a thick mucopolysaccharide capsule that protects the microbe from host defenses. The accumulation of the microbe and its mucopolysaccharide capsule gives the leptomeninges a cloudy to viscous appearance. The leukocytic response can vary from sparse to granulomatous. In some infected cats Cryptococcus spp. may be present in large numbers without an inflammatory response. It is unclear whether this absence of inflammation is the result of suppression of the immune response by the microbe or a defect in the cat's immune and/or inflammatory responses to the pathogen.
Two virulence factors have been documented. First, a thick mucopolysaccharide capsule protects the microbe from host defenses. Second, virulent microbes possess a biochemical pathway that can use catecholamines and that consists of a specific transport pathway and the enzyme phenoloxidase with production of a melanin or melanin-like compound through a series of oxidation-reduction reactions. This pathway can help protect the microbe from oxidative damage in the brain. Both virulence factors are important for survival of the microbe in the host. In addition, CSF lacks alternative pathway complement components that bind to the microbe's carbohydrate capsule and facilitate phagocytosis and killing by neutrophils.
Grossly, in CNS tissue and the leptomeninges, multiple small “cysts” with a viscous, gelatinous appearance can be seen (Fig. 14-49 ). Microscopically, leptomeningeal lesions have a loosely organized, lacy appearance with often myriad cryptococcal microbes and little or no inflammation. This is especially true in the cat as opposed to the dog, where the inflammatory response is often more intense. The leptomeningeal reaction can extend along the roots of cranial nerves. Spread of the CNS infection results in ventriculitis and choroiditis. In CNS tissue, in addition to the presence of the microbe and its capsule, the response can vary from being sparse to a granulomatous inflammation.
Figure 14-49.
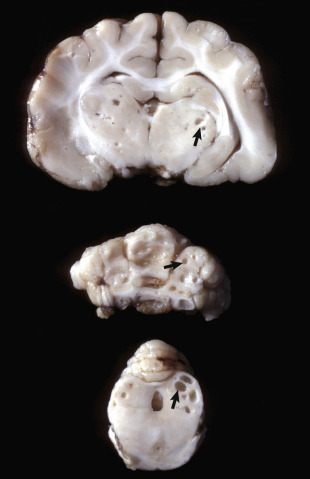
Cryptococcosis, Thalamus, Cerebellum, and Mesencephalon, Transverse Sections, Cat.
Note the “cavitational” lesions caused by Cryptococcus neoformans (arrows). Although the lesions look like cavities, they are filled with microbes, and the faint gray appearance is caused by the mucinous capsules of numerous cryptococci. Cryptococcus neoformans usually induces a granulomatous inflammation in most domestic animals, but in some animals, especially the cat, inflammation is minimal or absent.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The leukocytic response consists of neutrophils, eosinophils, macrophages, giant cells, and small mononuclear cells, depending on the immune status of the host. Animals with normal immune responses usually clear the infection from nasal cavities, sinuses, and the pulmonary system before its spreads systemically. Resistance to infection is provided by cell-mediated immunity. Immunosuppression of cell-mediated immunity caused by feline immunodeficiency virus and feline leukemia virus in cats and by Ehrlichia canis or long-term glucocorticoid therapy in dogs appears to increase susceptibility to cryptococcosis.
The yeast is spherical (2 to 10 mm in diameter), crescentic, or “cup shaped,” usually surrounded by a thick nonstaining (H&E stain) capsule (1 to 30 mm in diameter), and reproduces by narrow-based buds (Fig. 14-50, A ). Special stains, such as periodic acid–Schiff (PAS) and Gomori's methenamine silver, demonstrate the microbes readily, and the capsule can be stained with mucicarmine and Alcian blue (see Fig. 14-50, B).
Figure 14-50.
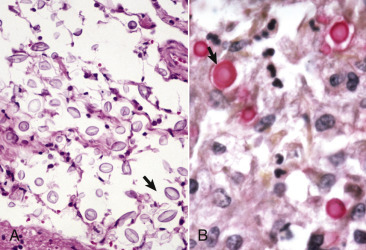
Leptomeningeal Cryptococcosis.
A, The thick unstained mucinous capsule surrounding the microbe results in the formation of a clear space (halo) in H&E-stained sections (arrow). This feature is useful in identifying the microbe in cytologic preparations and tissue sections. Also see Fig. 14-48, B. H&E stain. B, The mucinous capsule surrounding the microbe also stains with mucicarmine, providing a simple method to identify the microbe (arrow). Mayer's mucicarmine stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Clinically, the character of neurologic signs varies with the location of the lesions but can include depression, ataxia, seizures, paresis, and blindness.
Opportunistic Fungi.
Opportunistic fungi, including those fungi in the Zygomycetes group, such as Absidia corymbifera, Mucor spp., Rhizomucor pusillus, and Rhizopus arrhizus, and those fungi in the genus Aspergillus, such as Aspergillus niger, can invade blood vessels (angiotropic) and cause vascular thrombosis and infarcts in the CNS (Fig. 14-51 ). It must be noted that the term opportunistic implies that some form of tissue damage precedes fungal invasion. As an example, necrotizing enterocolitis caused by Salmonella spp. in the horse can provide an “open” vascular bed in the lamina propria of the intestinal mucosa that may be invaded by such fungi. Affected animals are often immunocompromised.
Figure 14-51.
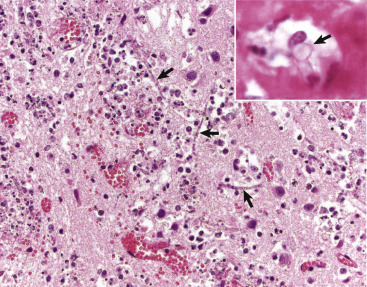
Opportunistic Angioinvasive Fungi.
Fungi such as Absidia corymbifera, Rhizomucor pusillus, and Rhizopus arrhizus and fungi in the genus Aspergillus, such as Aspergillus niger, can invade blood vessels (angiotropic) and cause vascular necrosis and infarcts in the central nervous system. Note the vasculitis, hemorrhage, and disruption of the vessel and the fungal hyphae in the lumen (arrows). H&E stain. Inset, Fungal hyphae in the lumen of a blood vessel (arrow). H&E stain.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University. Inset courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Protozoa
Neosporosis.
Neosporosis, caused by Neospora caninum, has been recognized in a variety of animals, including dogs, cats, cattle, sheep, and horses, as well as laboratory rodents. In horses, neosporosis can also be caused by Neospora hughesi. First described in 1988 as a multisystemic infection in the dog, the microbe has an affinity for the nervous system. The dog and its wild relatives, including coyotes and wolves, are the definitive hosts for the microbe, whereas herbivores like cattle are the typical intermediate hosts. Some of the features of the microbe are similar to those of Toxoplasma gondii, including division of tachyzoites by endodyogeny and having both proliferative (tachyzoites) and tissue cyst phases. However, N. caninum does not develop within a parasitophorous vacuole of a host cell, as does T. gondii. This latter feature is evident only with the use of transmission electron microscopy.
Although there are morphologic differences between the microbes (N. caninum has a thicker cyst wall), differentiation by light microscopy is unreliable, and electron microscopic examination or immunohistochemical analysis is required. Transmission occurs when the definitive host ingests tissue from an intermediate host that contains Neospora cysts. Such tissues include, but are not limited to, fetal membranes and aborted fetal tissues. Transplacental infection can also occur. N. caninum can infect a variety of cell types, but outside of the CNS, it appears to have an affinity for cells of the monocyte-macrophage system. The most likely method of spread to the CNS is via leukocytic trafficking.
Neurons and ependymal cells in the CNS, mononuclear cells in the CSF, and cells of blood vessels, including endothelium, intimal connective tissue, and tunica media smooth muscle cells, can harbor microbes. Microbes have also been detected in spinal nerves. Overall the morphologic pattern and character of lesions caused by Neospora spp. in the CNS are most consistent with multifocal necrotizing lesions with glial nodules and mixed inflammatory infiltrates. In dogs, lesions caused by Neospora caninum appear most frequently in the cerebellum.
Neurologic disease can be divided into two categories: that occurring during postnatal life and that associated with midterm to late-term abortions, the latter a notable problem in dairy cattle. Postnatal syndromes have been observed mainly in young and adult dogs, but horses are also affected. In young dogs, clinical signs are due to an ascending polyradiculoneuritis and polymyositis. In adult dogs, clinical signs are more referable to CNS lesions complicated by polymyositis, myocarditis, and dermatitis.
In horses the pathogen causing neosporosis is N. hughesi. Clinical signs resemble those of protozoal myeloencephalitis caused by Sarcocystis neurona. Lesions in horses include meningoencephalomyelitis; variable vasculitis and necrosis with microgliosis; and perivascular cuffing by macrophages, multinucleated giant cells, lymphocytes, plasma cells, or neutrophils, most commonly in the gray and white matter of the spinal cord but also in the pons and medulla.
Gross lesions can involve the white and/or gray matter. Peracute gross lesions may include foci of hemorrhage and necrosis distributed in a vascular pattern. Acute lesions have the same pattern of distribution but are on cut surface granular in texture and yellow-brown to gray. In some cases the periventricular white matter may be more affected. Chronic lesions have larger areas of granular yellow-brown to gray discoloration, which often makes white matter indistinguishable from gray matter. Microscopically the lesions and their temporal occurrence are similar to those described for T. gondii, including brain lesions that occur in aborted animals. Neospora spp. can be identified in tissue sections by H&E stain and immunohistochemical staining methods. Clinical signs are similar to those described for encephalitides induced by T. gondii.
Toxoplasmosis.
Toxoplasmosis is a disease in cats and other mammalian species caused by the obligate intracellular protozoan, T. gondii. Domestic, feral, and wild cats are the definitive hosts of T. gondii. Cats acquire T. gondii by ingesting infective cysts, oocysts, or tachyzoites when eating infected prey, such as rodents or birds. Ingestion of one of these stages initiates the intraintestinal life cycle, which occurs only in members of the cat family. T. gondii replicates and multiplies within epithelial cells of the small intestine and produces oocysts. Oocysts are released into the feces in large numbers for 2 to 3 weeks following initial ingestion of cysts, oocysts, or tachyzoites. When oocysts sporulate, usually within 5 days after passage in the feces, they become infectious for intermediate hosts. Sporulated oocysts are highly resistant and can survive in moist shaded soil or sand for months. Cats are unique in the biology of the microbe, serving as both definitive (intraintestinal life cycle) and intermediate (extraintestinal life cycle) hosts.
T. gondii can infect a wide variety of animals as intermediate hosts (extraintestinal life cycle), including fish, amphibians, reptiles, birds, human beings, and many other mammals. New World monkeys and Australian marsupials are the most susceptible, whereas Old World monkeys, rats, cattle, and horses seem highly resistant.
T. gondii can also parasitize a wide variety of cell types in the intermediate host and can cause lesions in such tissues as the lungs, lymphoid system, liver, heart, skeletal muscle, pancreas, intestine, eyes, and nervous system. After ingestion, bradyzoites from tissue cysts or sporozoites from oocysts enter intestinal epithelia and multiply. There is active penetration of plasma membranes by microbe-secreted lytic products, allowing a portal of entry rather than by uptake via phagocytosis. T. gondii can then spread locally, free in lymph or intracellularly in lymphocytes, macrophages, or granulocytes to Peyer's patches and regional lymph nodes. Intracellularly, microbes multiply as tachyzoites within a parasitophorous vacuole by repeated cycles of endodyogeny during the early acute stages of infection. Dissemination to distant organs is via lymph and blood, either as free microbes or intracellularly in lymphocytes, macrophages, or granulocytes via leukocytic trafficking.
With chronicity and an increasing antibody response by the host, tachyzoites of T. gondii transform into slow-growing bradyzoites that replicate in cysts within muscle. Infection of the CNS occurs hematogenously; neurons and astrocytes are the eventual target cells. The typical sequence of events in the pathogenesis of the characteristic lesion is similar to that in S. neurona infection. In utero infections in animals and human beings can result in CNS infection. In fetal brains, foci of necrosis are most common in the brainstem and induce the formation of microglial nodules. Additionally, foci of necrosis and mineralization occur in the cerebrocortical white matter and are caused by fetal hypoxia and ischemia resulting from severe placentitis, fetal myocardial damage, or initiation of a systemic inflammatory reaction in the fetus. In older more mature individuals, T. gondii infections have been associated with immunosuppression such as occurs in concurrent CDV infection and toxoplasmosis. In some cases this could represent activation of latent inactive T. gondii cysts (bradyzoites) in neural tissues.
Lysis of infected cells by primed cytotoxic, CD8+ T lymphocytes also could potentially contribute to the tissue damage through the production of cytokines, such as interferon-γ, which can activate microglia and astrocytes to inhibit parasite replication and induce cytotoxic T lymphocytes to kill infected cells. This exuberant inflammatory response and the cytokine cascade that ensues to kill the microbe also causes severe damage to cells in the area of inflammation, especially axons and neurons. Intracellular growth of tachyzoites also has been advanced as a cause of cellular necrosis. The microbe does not produce a cytotoxin.
The blood-brain barrier of the CNS is breached when free microbes or those located intracellularly (leukocytic trafficking) infect endothelial cells of the CNS vasculature, especially capillaries. Gross lesions can involve any area of the CNS without predilection for gray or white matter, may also involve nerve rootlets, and may initially include foci of hemorrhage and necrosis and later, granular, yellow-brown to gray foci. Peracute lesions initially include endothelial cell swelling as the result of infection by tachyzoites and vasculitis with hemorrhagic infarcts followed by vasogenic edema. If the edema is sufficiently severe to cause increased brain volume, the edema can lead to brain displacement and herniation.
Early microscopic lesions include infection of and proliferation within endothelial cells by T. gondii tachyzoites. Endothelial injury results in endothelial cell swelling, endothelial cell degeneration, hemorrhage, capillary occlusion, ischemic necrosis, and edema of adjacent tissue. Subsequently tachyzoites invade the CNS, inducing a prominent acute inflammatory response leading to necrosis and hemorrhage often striking in severity. With time the inflammatory response consists of perivascular cuffing of blood vessels within the CNS and leptomeninges by lymphocytes and macrophages. CNS responses to injury consist of microgliosis and astrogliosis; however, these responses are often insufficient to replace the loss of tissue in the cerebral hemispheres, resulting in dilation of the lateral ventricles (hydrocephalus ex vacuo) and the formation of persistent cysts in the tissue. With chronicity and increasing inflammatory and immunologic responses by the host, tachyzoites change to slow-growing bradyzoites that replicate in and form tissue cysts. Polyradiculoneuritis (i.e., inflammation of peripheral nerves, spinal nerve roots, and spinal cord) is also a potential sequela to infection with the microbe. Microbes in lesions can often be identified with an H&E stain, but immunohistochemical evaluation facilitates their detection and identification. Because the infection is systemic, lesions can occur in several other tissues.
Occasionally cysts (bradyzoites) can be observed in “normal” CNS tissue without an inflammatory or tissue lesion. These cysts are likely the result of a previous infection with T. gondii that was successfully resolved. Experimental studies have confirmed that administration of corticosteroids and thus immunosuppression increases susceptibility or exacerbates the infection with T. gondii or both or may contribute to the reactivation of tissue cysts.
Clinical signs can vary, depending on the age of the animal, species infected, and areas of the CNS involved and may include depression, weakness, incoordination, tremors, circling, paresis, and blindness.
Parasites.
As a general concept, lesions resulting from parasitic infestation of the CNS vary in degree of severity and distribution, depending on the parasite and the host response to infection. Gross lesions of hemorrhage and malacia in parasite migratory tracts or space-occupying cysts occur with the various parasitic stages. Microscopically, there is necrosis, hemorrhage, and a leukocytic response, typically with a significant infiltrate of eosinophils. The extent of the host response is often dictated by the degree of trauma and disruption created by the parasite and the level of sensitivity of the host for parasite antigens. This section is not intended to be an extensive review of veterinary parasitology but will cover those parasites most commonly seen in veterinary practice.
Insect Larvae.
Among the most common larvae are those of Oestrus ovis and Hypoderma bovis. The larvae of O. ovis develop in the nasal cavity of sheep but can penetrate into the cranial vault through the ethmoid bone. Larvae of H. bovis can enter the spinal canal during their migration in the subcutis from the hoof to the dorsal midline in cattle and rarely as an aberrant parasite in the brain of horses. Damage in the CNS caused by H. bovis in cattle is typically the result of inflammation directed at the degenerating parasites after anthelmintic treatment. Larvae of Cuterebra spp., usually a parasite of rabbits and rodents, can invade the CNS of dogs and cats and cause extensive meningeal or parenchymal lesions depending on the location through which it migrates (see the section on Feline Ischemic Encephalopathy).
Cestodes.
Coenurus cerebralis, the larval form of the dog tapeworm Taenia multiceps, most commonly infests sheep and occasionally other ruminants. The larval form reaches the CNS hematogenously and then cause damage during migration and encystation, forming space-occupying lesions. In this disease the initial larval migration is associated with severe necrosis, and inflammation with larvae is often present. Another form of the parasite, in which there is an encysted “bladder,” causes extensive compression of the parenchyma and associated atrophy. The parasite produces a neurologic disease known as gid. Another parasite for which human beings are the definitive host is Taenia solium, with pigs being the intermediate host. The larval stage, Cysticercus cellulosae, generally develops in muscle of the pig but can also occur in the meninges and brain, resulting in a disease called “cysticercosis.” Involvement of the CNS has been termed “neurocysticercosis.”
Initially, viable cysticerci become “trapped” within capillaries of the CNS, but they do not apparently elicit an inflammatory response. At some point the host responds immunologically and the cyst becomes denser, collapses inwardly, and disintegrates to eventually become calcified debris in a focus of inflammation. The inflammatory response has humoral and cellular components. Antibodies of the immunoglobulin G family are directed against the cyst; however, cysts are likely killed by mediators released from eosinophils, which are attracted to the site by mediators released from lymphoid cells in the inflammatory exudate. For undetermined reasons, in “susceptible” animals viable cysts can become established and grow slowly for years. Viable cysticerci can cause asymptomatic infection by actively evading and suppressing the immune response of the host. These cysts cause vasogenic edema and increased intracranial pressure related to behavior as “space-occupying” masses.
Gross lesions are usually seen in the cerebral hemispheres, commonly at the interface of gray and white matter in a hematogenous pattern. Cysts can also be found in the cerebellum, medulla, ventricles, subarachnoid space, and the spinal cord. There are usually no gross changes in the CNS surrounding the cysts. Cysts are round to oval and of varied sizes and number, many of which can be large and visible up to centimeters in diameter. They have a translucent cyst wall and contain a thick, clear fluid. Within the fluid is a scolex, visible as a small 2- to 3-mm nodule. Microscopically, there is little or no inflammation or tissue injury surrounding the cysts, except for compression and edema.
Nematodes (Cerebrospinal Nematodiasis).
Aberrant migration of larval stages of nematode parasites into and through the CNS is called cerebrospinal nematodiasis. Important causes of cerebrospinal nematodiasis include Parelaphostrongylus tenuis (ruminants, camelids), Strongylus vulgaris (horse), Elaphostrongylus rangiferi (small ruminants), Toxocara canis (dogs), and Baylisascaris procyonis (many species including dogs, primates, rabbits, and birds). They gain access to the CNS hematogenously and actively enter the CNS by crossing the blood vessel wall through their locomotive processes. Nematodes cause damage in the cerebromedullary area of the brain and/or spinal cord either from aberrant migration in the definitive host or migration in an aberrant host (Table 14-4 ; Fig. 14-52 ). Greater CNS damage is often created by the migration of the parasites in an aberrant host.
Table 14-4.
Nematodes Causing Central Nervous System Disease in Domestic Animals
| Parasite | Normal Host | Aberrant Host |
|---|---|---|
| NEMATODE MIGRATION IN ABERRANT HOST | ||
| Angiostrongylus cantonensis | Rat | Dog |
| Baylisascaris procyonis | Raccoon | Dog |
| Elaphostrongylus rangiferi | Reindeer | Sheep, goat |
| Parelaphostrongylus tenuis | Deer | Sheep, goat |
| Setaria digitate | Cattle | Sheep, goat, horse |
| ABERRANT NEMATODE MIGRATION IN NORMAL HOST | ||
| Angiostrongylus vasorum | Dog (coyote) | |
| Dirofilaria immitis | Dog (cat) | |
| Stephanurus dentatus | Pig | |
| Strongylus spp. | Horse | |
Figure 14-52.
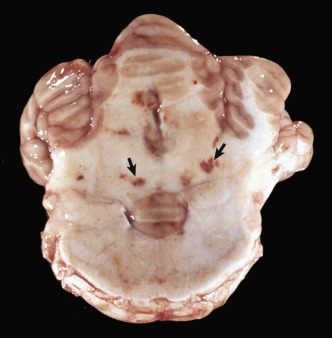
Cerebrospinal Nematodiasis, Brain, Cerebellum, and Medulla at the Level of the Pons, Horse.
Strongylus vulgaris migration. Several small foci of hemorrhage and necrosis in the cerebellar white matter are sites of larval migration (arrows).
(Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Macroscopic lesions of nematode larval migration often appear as linear or serpentine tracts of necrosis and/or hemorrhage in the tissue. Migration results in endothelial injury, vasculitis, and thrombosis, which may result in vascular occlusion and infarction. Larvae can often be found in histologic sections, and they induce a mononuclear cell inflammatory exudate, including abundant eosinophils (Fig. 14-53 ).
Figure 14-53.
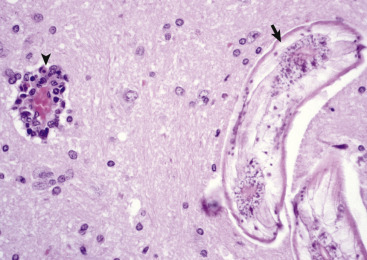
Cerebrospinal Nematodiasis, Central Nervous System (CNS), Rabbit.
Migration of Baylisascaris procyonis (arrow) in the CNS elicits a perivascular lymphomonocytic inflammatory response mixed with eosinophils (arrowhead) and results in direct injury to blood vessels, axons, and dendrites. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Halicephalobus gingivalis is a free-living rhabditiform nematode that can infest the nasal cavity, CNS, and kidneys of the horse. The life cycle, pathogenesis, and route of infection of Halicephalobus gingivalis are poorly understood. It has been proposed that the CNS is infected hematogenously in a manner similar to that described for cerebrospinal nematodiasis and that larvae penetrate skin and mucous membranes in recumbent horses with subsequent invasion of sinuses and/or blood vessels. In the CNS, microscopic lesions are prominently associated with blood vessels along which the parasite apparently migrates.
Prions
Transmissible Spongiform Encephalopathies.
Ovine spongiform encephalopathy (scrapie), bovine spongiform encephalopathy (BSE), and human spongiform encephalopathies are classified within a group of diseases called transmissible spongiform encephalopathies. Table 14-5 lists the known transmissible spongiform encephalopathies in animals and human beings. These diseases are caused by proteinaceous infectious particles (prions) that (1) are composed of an abnormal isoform of a normal cellular protein, the prion protein (PrPC [a 27- to 30-kD polypeptide]), designated PrPSc and (2) resist inactivation by procedures that degrade nucleic acids and proteins (i.e., heat, ultraviolet irradiation, and strong enzymes). PrPC is expressed throughout the body and is the product of a highly conserved gene found in microbes as diverse as fruit flies and human beings. The “Sc” superscript is derived from the word “scrapie” because scrapie is the prototype prion disease.
Table 14-5.
Transmissible Spongiform Encephalopathies (i.e., Prion Diseases)
| Disease | Natural Host(s) | Prion | Pathogenic PrP Isoform |
|---|---|---|---|
| ANIMALS | |||
| Ovine spongiform encephalopathy (scrapie) | Sheep, goats | Scrapie prion | OvPrPSc |
| Bovine spongiform encephalopathy (BSE) | Cows | BSE prion | BoPrPSc |
| Feline spongiform encephalopathy (FSE) | Cats | FSE prion | FePrPSc |
| Chronic wasting disease (CWD) Transmissible mink encephalopathy (TME) |
Mule deer, elk, black-tailed deer, white-tailed deer, mink | CWD prion TME prion |
MdePrPSc MkPrPSc |
| Exotic ungulate encephalopathy (EUE) | Nyala, greater kudu, oryx | EUE prion | UngPrPSc |
| HUMAN BEINGS | |||
| Kuru | Human beings | Kuru prion | HuPrPSc |
| Creutzfeldt-Jakob disease (CJD) | Human beings | CJD prion | HuPrPSc |
| Variant Creutzfeldt-Jakob disease (VCJD) | Human beings | VCJD prion | HuPrPSc |
| Gerstmann-Sträussler-Scheinker syndrome (GSS) | Human beings | GSS prion | HuPrPSc |
| Fatal familial insomnia (FFI) | Human beings | FFI prion | HuPrPSc |
Although the mechanism by which PrPSc forms has not been completely explained, a posttranslational modification of PrPC has been proposed (Fig. 14-54 ). This mechanism proposes that PrPSc acts as a template on which PrPC undergoes a conformational change (is refolded) by a process facilitated by another protein (referred to as protein X), whereby the α-helical content of PrPC diminishes and the amount of β-sheet increases, resulting in the formation of PrPSc. The features of a specific PrPSc are determined by the animal in which it is formed. When PrPSc of one species is inoculated into a different species, the recipient is less readily infected and generally has a prolonged incubation period. This resistance to infection is referred to as the species barrier.
Figure 14-54.
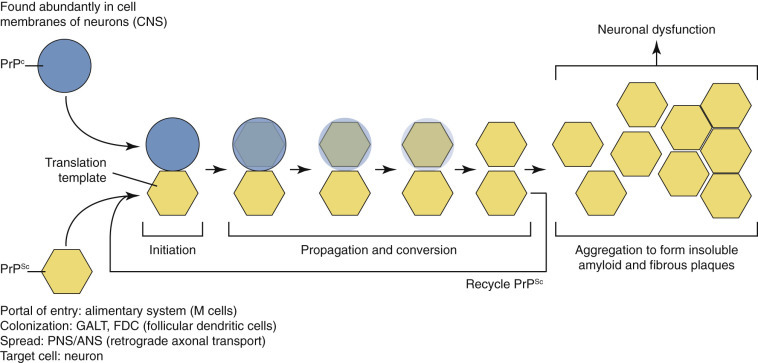
Prion Protein.
In prion diseases (spongiform encephalopathies), PrP (PrPC), a normal neuronal protein, is converted to an abnormal β-pleated sheet isoform (PrPSc) through the interaction of PrPSc with PrPC. ANS, Autonomic nervous system; CNS, central nervous system; GALT, gut-associated lymphoid tissue; PNS, peripheral nervous system.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Spongiform encephalopathies occur through horizontal transmission (feeding rendered cattle CNS tissue to cattle) or through an inherited mutation of the normal human prion gene. In animals the primary route of infection appears to be through horizontal transmission. For chronic wasting disease, horizontal transmission is surprisingly effective, and prions can be readily transmitted in saliva, blood, and urine. It has been proposed that prions ingested in infective feedstuffs enter the body through the intestine. Prions cross the intestinal wall at Peyer's patches and are phagocytosed and transported to other lymph nodes by leukocytic trafficking. Prions replicate in lymphocytes and macrophages of the lymphoid system before gaining access to the blood. The autonomic nervous system is important in delivering the prions to the CNS and neurons; however, the exact mechanisms of spread remain to be elucidated. Eventually neurons accumulate sufficient PrPSc to alter the normal function (it can take years), and neurologic signs are observed.
Prion diseases are fatal. The adaptive immune system does not recognize prions as foreign; therefore no immunologic protection develops. How the accumulation of PrPSc causes neurodegeneration and neuron loss in prion diseases is not clear; however, astrocyte and microglial cell activation and apoptosis appear to be likely components of the pathway leading to neuronal injury.
No gross lesions of the nervous system are detectable in animals with spongiform encephalopathies. Microscopic lesions in scrapie-infected sheep and goats are limited to the CNS and are most commonly present in the diencephalon, brainstem, and cerebellum (cortex and deep nuclei), with variable lesions in the corpus striatum and spinal cord. Except for some minor changes, the cerebral cortex is essentially unaffected.
The type of neuronal degeneration can vary and commonly is characterized by shrinkage with increased basophilia and cytoplasmic vacuolation (Fig. 14-55, A ), although other changes, such as central chromatolysis and ischemic cell change, variably occur. Astrocytosis in affected areas of the brain, including the cerebellar cortex, can be severe (see Fig. 14-55, B). There has been speculation whether the astrocytic reaction is a primary or a secondary response. An abnormal protein (prion amyloid protein) first accumulates in astroglial cells in the brain during scrapie infection, which could mean that this cell is the primary site of replication. The spongiform change tends to affect the gray matter, and greater severity of this lesion has been associated with long incubation periods. The lesion in the gray matter is the result of dilation of neuronal processes, but vacuolation of neuronal and astroglial perikarya, swelling of astrocytic processes, dilation of the periaxonal space, and splitting of myelin sheaths have also been reported. Finally, the disease is not accompanied by any notable inflammation within the CNS.
Figure 14-55.
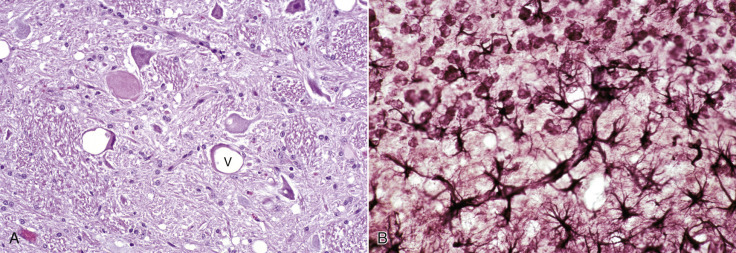
Spongiform Encephalopathy (Scrapie), Brain, Motor Neurons, Sheep.
A, Neuronal cell bodies contain one or more discrete and/or coalescing clear vacuoles (V). There are no inflammatory cells in this disease. Similar spongiosis is evident in the neuropil. H&E stain. B, Scrapie, experimental, brain, cerebellum, mouse. The cerebellar granule cells are at the top of the figure. There is notable hypertrophy and proliferation (astrocytosis) of astrocytes and their fibers (astrogliosis) (black branching fibers). Some of the processes (running diagonally across the illustration) end, as is normal for astrocytes, on the walls of capillaries. Cajal's gold sublimate stain for astrocytes.
(A courtesy Dr. D. Gould, College of Veterinary Medicine and Biomedical Sciences, Colorado State University; and Dr. M. McAllister, College of Veterinary Medicine, University of Illinois. B courtesy Dr. W.J. Hadlow.)
Degenerative Diseases
Metabolic
Aminoacidopathies.
Two diseases characterized by errors of amino acid metabolism have been described in neonatal calves. One disease, maple syrup urine disease (MSUD), occurs in young polled Hereford and Hereford calves. The second disease, bovine citrullinemia, which was originally described in Australia, occurs in neonatal Friesian calves.
Maple syrup urine disease is caused by an inherited defect in the branched-chain α-ketoacid enzyme decarboxylase and results in a deficiency of this enzyme, which is necessary to metabolize the branched-chain amino acids leucine, isoleucine, and valine. These amino acids are essential and must be obtained from protein in the diet. After consumption, proteins are digested, and the amino acids are released to be used to generate energy and for other metabolic processes. In maple syrup urine disease there is a mutation in one or more of the genes that regulate this degradation process; therefore abnormal metabolites and ketoacids accumulate to toxic levels and cause disease. Urine has a sweet odor attributable to a derivative of isoleucine resembling the smell of maple syrup. In human infants the disease is confirmed biochemically by finding elevated concentrations of leucine, isoleucine, and valine in the blood.
Gross lesions are not typically present. Microscopically, marked spongiosis, caused by vacuolation of myelin sheaths, is present throughout the neuraxis. The spongiosis affects both gray and white matter. Lesions are often most notable in areas, such as the brainstem, in which there is an intermingling of gray and white matter.
Affected calves may be normal at birth. Within a few days, depression, dullness, and weakness progress to recumbency and opisthotonus.
Bovine citrullinemia is a rare inborn error of metabolism of the urea cycle that results in a pronounced accumulation of citrulline and ammonia in the body fluids because of a failure of the normal synthesis of arginosuccinic acid by the enzyme arginosuccinate synthetase. In human infants the disease is confirmed biochemically by finding elevated concentrations of citrulline in the blood. The cerebral lesions have also been suspected to result from the hyperammonemia or possibly some defect in excitatory neurotransmitter metabolism. However, the pathogenesis of the disease in calves remains unsettled.
Grossly, brains are normal and have normal weights. Livers are pale yellow. Microscopically, there is fatty change in the liver. Lesions in the brain are characterized by mild to moderate diffuse astroglial swelling in the cerebral cortex. Perivascular, perineuronal, and periglial spaces are typically expanded by edema.
Calves are normal at birth. Within a few days a severe generalized CNS disorder develops characterized by apparent blindness, depression, and tremors that rapidly progress to seizures, coma, and death within a few hours.
Cerebral Cortical Atrophy.
Brain atrophy caused by the loss of neurons in the cerebral cortex can occur in all animal species and is most commonly associated with chronic degenerative conditions of the CNS in which neuronal dysfunction and eventual loss occurs. An example of cortical atrophy is seen in cerebral ceroid-lipofuscinosis, a storage disorder that occurs in many different species. Atrophy most frequently involves the cerebral hemispheres, especially the cortex. The cerebral hemispheres are increased in firmness and often have a tan color (lipofuscin), gyri are thinned, and the sulci widened. Microscopically, there is loss of neuron cell bodies in cortical laminae without inflammation. Astrogliosis in response to neuronal loss is also observed, as is an increased prominence of the adventitial layer of blood vessels.
Channelopathies.
Channelopathies are a newly emerging group of inherited neuromuscular diseases of human beings that affect the excitability of membranes of neurons and skeletal myocytes. These diseases result from mutations in genes encoding ion channel proteins that regulate calcium, sodium, and chloride channels and acetylcholine receptors. In human beings, neurologic diseases, such as epilepsy and migraine headaches, have been attributed to channelopathies. In veterinary neurology, channelopathies will likely be shown in the future to be the underlying mechanism for epilepsy and other primary neuronal degenerations; however, investigation into this type of neurologic disorder is still in its infancy in veterinary medicine. Examples of this type of disorder are the recent demonstration of mutated cyclic nucleotide-gated ion channels in several dog breeds with vision disorders.
Degenerative Leukomyelopathies.
Degenerative leukomyelopathies are a heterogeneous group of familial, likely inherited, and acquired diseases that have been described in dogs, cows, and horses. Although there is no universal agreement, the degenerative leukomyelopathies described here are best characterized as axonal degenerations with spheroid formation predominantly within spinal cord white matter and secondary changes in myelin sheaths and myelin loss. In dogs, familial or inherited diseases include degenerative axonopathy of Ibizan hounds, axonopathy in Labrador retrievers, and axonopathy in Jack Russell and smooth fox terriers. A disease in Rottweilers (see the section on Primary Cerebellar Neuronal Degeneration in the section on Dogs and the section on Cats) is another disorder with spinal cord white matter involvement that is familial and inherited. An acquired disease, hound ataxia, has been described in the United Kingdom and Ireland in harriers, beagles, and foxhounds. Hound ataxia may represent a nutritional disorder in hunting dogs fed paunch (tripe). Degenerative leukomyelopathies in cattle can be inherited as an autosomal recessive trait or have a familial predisposition. Leukomyelopathies have been reported in Murray Grey, Holstein-Friesian, and in certain lines of Brown Swiss cattle.
Gross lesions are usually not observed. Microscopically, lesions in the white matter of the spinal cord are bilaterally symmetric and consist of axonal degeneration with formation of spheroids, loss of axons, and secondary myelin degradation. Depending on the species or breed affected, lesions can involve any of the funiculi. Spinocerebellar tracts in the dorsolateral aspects of the lateral and septomarginal areas of ventral funiculi are commonly affected, as is the fasciculus gracilis in the dorsal funiculus. Severe involvement of the dorsal spinocerebellar tracts can extend into the caudal brainstem and via caudal cerebellar peduncles to the cerebellar cortex and Purkinje cells. In some species and breeds, there can also be involvement of additional specific brainstem structures.
Age of onset varies with the familial or inherited disorders. Paresis, ataxia, and dysmetria are the predominant clinical signs.
Epileptic Brain Damage.
Brain damage caused by prolonged (usually > 30 minutes) convulsive seizures (status epilepticus) is not widely recognized; however, it has been described secondary to either malformative lesions or tumors in the CNS. In human beings and experimental animals, brain damage resulting from status epilepticus is well documented. One study reported a relatively high incidence of brain lesions in dogs caused by status epilepticus. In this study, acute brain damage was widespread and corresponded well with areas of the brain prone to hypoxic-ischemic injury such as the cerebral cortex, pyriform cortex, basal nuclei, and hippocampus.
The cause of neuronal injury with prolonged convulsive seizures is debatable. It remains unclear if necrosis, apoptosis, or a combination of these two mechanisms causes neuronal injury in status epilepticus. During seizures there is an increased metabolic demand for glucose and oxygen by neurons; however, cerebral blood flow increases during seizures so that the amount of glucose and oxygen available for neurons to generate energy remains adequate, at least during earlier stages. Acute neuronal necrosis still occurs even when cerebral blood flow, oxygenation, body temperature, and other metabolic parameters are maintained within normal limits in experimental animals with status epilepticus.
Excitotoxic injury caused by accumulation of neurotoxic amino acid neurotransmitters, such as glutamate, during the extreme neuronal activity occurring in status epilepticus, is an attractive explanation for neuronal necrosis. Excitotoxicity would account for both the selective vulnerability of certain brain areas and the character of the lesions. Status epilepticus induced experimentally in rats with kainic acid, an excitatory amino acid receptor agonist, has been shown to cause primarily neuronal necrosis and some characteristics of apoptosis. Other experimental studies have suggested that astrocytes produce clusterin during status epilepticus. Clusterin (dimeric acidic glycoprotein), a sulfated glycoprotein, initiates apoptosis when expressed in cells in elevated concentrations. It is proposed that clusterin secreted by astrocytes during status epilepticus is actively endocytosed by hippocampal neurons, and these neurons die by an apoptotic mechanism. The exact mechanism of neuronal injury remains to be proved. There is some evidence that the mature brain is more prone to injury induced by status epilepticus than the immature brain.
Gross lesions, if present, usually consist of widened and flattened gyri and narrow indistinct sulci caused by cerebral edema. Acute neuronal ischemic cell change and astrocytic swelling are observed microscopically. In experimental animals with status epilepticus, neuronal degeneration is observed within 30 minutes and neuronal necrosis within 60 minutes.
Hepatic Encephalopathy.
Acute and chronic liver failure, as well as hepatic atrophy associated with congenital or acquired vascular shunts, often results in hepatic encephalopathy and disordered neurotransmission because of the accumulation of toxic substances, principally ammonia, in the systemic circulation and thus the CNS. Ammonia is formed in the gastrointestinal tract by bacterial degradation of amines, amino acids, purines, and urea from proteins consumed in the diet. In healthy animals, ammonia is detoxified in the liver by conversion to urea by the ornithine citrulline arginine urea cycle. Urea is far less toxic than ammonia and is excreted in the urine. Ammonia has several neurotoxic effects such as (1) changing the transit of amino acids, water, and electrolytes across the neuronal cell membranes and (2) inhibiting the generation of both excitatory and inhibitory postsynaptic potentials in neurons. Ammonia and other toxic metabolites also (1) cause increased permeability of the blood-brain barrier, leading to vasogenic edema, and (2) alter osmoregulation within the CNS. These mechanisms likely led to the spongy change (status spongiosus) characteristic of the disease microscopically. Because astrocytes play an important role in regulating fluid and electrolyte balances in the CNS and are the primary cell type having lesions (Alzheimer's type II astrocytes) in hepatic encephalopathy, it is not surprising that alterations in osmoregulation are a component of the pathogenesis of the disease. Astrocytes also contain high concentrations of glutamine synthetase, an enzyme that breaks ammonia into glutamine. Recent research indicates that glutamine, although a normal breakdown product, may actually have deleterious effects on astrocytes and may be one of the main contributors to astrocyte dysfunction noted in hyperammonemic states. Ammonia and other toxic metabolites likely also affect oligodendroglia. Finally, it has been proposed that alterations in the blood-brain barrier may facilitate passage of neurotoxins such as short-chain fatty acids, mercaptans, false (pseudo-) neurotransmitters (tyramine, octopamine, and β-phenylethanolamine), ammonia, and GABA into the CNS, leading to neuronal dysfunction. A similar condition, termed renal encephalopathy, has been described in dogs, ruminants, and horses. It is likely related to high concentrations of ammonia or ammonia-metabolites in the circulation because of inadequate renal clearance caused by severe glomerular or tubular injury.
In all species except the horse, lesions of hepatic encephalopathy are of two types: spongy change and formation of Alzheimer's type II astrocytes (see Fig. 14-10, D). Spongy change can be present throughout the neuraxis but typically involves areas of confluence or intermingling of gray and white. It is bilateral and symmetric in distribution. These areas include the deep cerebrocortical gray-white matter interface where the peripheral fibers that radiate from the corona radiata are found, basal nuclei and adjacent internal capsule, reticular areas throughout the brainstem, and deep cerebellar nuclei. The spongy change is due to intramyelinic edema, causing splitting and vacuolation of myelin sheaths. Spongy change can be produced experimentally by ammonia infusion and is reversible. Alzheimer's type II astrocytic change is a subtle alteration that has been reported in all domestic animals and is the only CNS change observed in horses with hepatic failure. Alzheimer's type II astrocytes are found in gray matter and have enlarged vesicular nuclei with peripheral chromatin, glycogen deposits, and demonstrable nucleoli or nucleolar-type bodies. They often occur in diads, triplets, or clusters of even high numbers. Immunohistochemical staining for GFAP is typically weak or absent, possibly indicating a toxic effect on astrocytes.
Clinically, affected animals show CNS signs such as seizures, ataxia, depressed mentation, walking aimlessly, and head pressing.
Mitochondrial Encephalopathies.
In human beings, various encephalopathic and myopathic syndromes caused by point mutations in mitochondrial DNA affecting tRNA genes are grouped under the acronyms MELAS (mitochondrial encephalopathy, lactic acidosis, strokelike episodes) and MERRF (myoclonic epilepsy with ragged red fibers). The various human syndromes include Leigh's disease (subacute necrotizing encephalomyelopathy), Kearns-Sayre syndrome, and Leber's hereditary optic atrophy.
Diseases that might be classified as mitochondrial encephalopathies are not well characterized in animals. Despite this caveat, the diseases reported in the Australian cattle dogs, English springer spaniel dogs, and a Jack Russell terrier, as well as in Limousin and Simmental cattle and New Zealand South Hampshire sheep, could represent mitochondrial disorders. Table 14-6 summarizes the salient features of the diseases. Characteristics of these diseases in both human beings and animals are symmetric bilateral involvement of the neuraxis and lesions typified by status spongiosus (edema of cerebral white matter) with variable progression to cavitation or necrosis. The CNS is highly dependent on oxidative metabolism and is therefore the most severely affected organ in mitochondrial disorders. Mitochondria isolated from affected human patients have impaired oxygen consumption and reduced respiratory chain enzyme complex activity.
Table 14-6.
Possible Mitochondrial Encephalopathies in Animals
| Animal | Age of Onset (months) | Clinical Signs | Primary Lesions |
|---|---|---|---|
| Cattle dog | 5 to 12 | Seizures, behavioral abnormalities followed by locomotor signs | Spongiosis and cavitation in cerebellum, brainstem nuclei, and spinal gray matter |
| English springer spaniel | 15 to 16 | Ataxia, disorientation, visual deficits | Status spongiosus in accessory olivary nucleus; axon loss and gliosis in optic nerve and tracts |
| Jack Russell terrier | 2.5 | Ataxia, hypermetria, and deafness | Neuronal degeneration and mineralization of the medulla oblongata, vestibulocochlear nerve, choroid plexus, and granule cell layer of the cerebellum |
| Limousin cattle | 1 to 4 | Locomotor signs, aggressive behavior, blindness | Spongiosis, cavitation in cerebral and cerebellar white matter, brainstem nuclei, optic chiasm |
| Simmental cattle | 5 to 12 | Ataxia, behavioral changes | Spongiosis and necrosis in internal capsule, caudate nucleus, putamen, brainstem nuclei, spinal gray matter |
Notably excluded from this list is the Alaskan husky, a breed that has a well-characterized encephalopathy that is morphologically similar to Leigh's disease in human beings. However, recent research has failed to reveal mutations in mitochondrial genes but rather has found an association between a mutation in the thiamine transporter 2 gene (SLC19A3) and the development of the disease. In this disease, bilaterally symmetric foci of encephalomalacia are often found in the thalamus, caudate, pons, medulla oblongata, and along the gray-white matter border of the cerebral cortices. The histologic lesions are typified by an abundance of astrocytes, some of which can be highly bizarre and vacuolated. The disease is included in this section because it can be considered a secondary mitochondrial disease, rather than a primary mitochondrial dysfunction.
Primary Neuronal Degeneration.
Primary neuronal degeneration that occurs in many or all animal species is discussed in this section. Disorders of individual animal species are discussed in sections covering disorders unique to that species.
The term primary neuronal degeneration encompasses three groups of diseases affecting specific regions of the CNS in a temporally and spatially stereotyped manner and are characterized by degeneration, necrosis, and loss of specific populations of functionally linked neurons. Box 14-8 gives an overview of these diseases. The first group includes the multisystem neuronal degenerations, which are diseases that affect populations of functionally related neurons in the basal ganglia, brainstem, and cerebellum. The second group includes the primary cerebellar neuronal degenerations, which are diseases that affect populations of neurons restricted to the cerebellum and cerebellar roof nuclei. The third group includes primary spinal cord degenerations, which are diseases associated with axonal swellings (axonal spheroids) in the neuraxis termed neuroaxonal dystrophies. Another term used to denote some of these diseases in the biomedical literature and veterinary textbooks is abiotrophy; this term was introduced by Gowers in 1902. The term literally means lack of (“a”) a vital (“bios”) nutrition (“trophy”) required to sustain the life of a tissue. For discussion of the primary neuronal degeneration affecting individual animal species, see sections covering disorders specific for the species.
Box 14-8. Multisystem Neuronal Degenerations and Brainstem/Spinal Syndromes in Domestic Animals.
Multisystem Neuronal Degenerations
Canine: Kerry blue terrier, red-haired cocker spaniel, Cairn terrier
Primary Cerebellar Degeneration
Neonatal Syndromes
Canine: Beagle, Samoyed, Irish setter
Ovine: Welsh mountain, Corriedale
Bovine: Hereford, Hereford cross, Ayrshire
Postnatal Syndromes
Canine: Airedale, German shepherd, Gordon setter, rough-coated collie, border collie, Finnish terrier, Bernese mountain dog, Bern running dog, Labrador retriever, golden retriever, cocker spaniel, Cairn terrier, Great Dane
Bovine: Holstein-Friesian, Hereford cross, Angus
Equine: Arabian, Arabian cross, Gotland pony
Ovine: Merino
Porcine: Yorkshire
Mitochondrial Encephalopathy (Encephalomyopathy)
Canine: English springer spaniel, Alaskan husky, Australian cattle dog, English setter dog, Jack Russell terrier
Bovine: Simmental, Limousin
Sheep: New Zealand South Hampshire
Spongy Degeneration
Canine: Labrador retriever, Saluki, silky terrier, Samoyed
Bovine: Jersey, shorthorn, Angus-shorthorn, Hereford
Feline: Egyptian Mau
Brainstem and Spinal Syndromes
Neuroaxonal Dystrophy
Canine: Border collie, Chihuahua, Rottweiler
Feline: Domestic
Equine: Morgan
Ovine: Suffolk
Motor Neuron Disease—Spinal Cord
Canine: Brittany spaniel, Swedish Lapland, English pointer, Rottweiler, German shepherd, sheepdog, collie, pug, dachshund, fox terrier
Feline: Siamese
Bovine: Brown Swiss, Hereford (shaker calf syndrome)
Equine: Various breeds (not believed hereditary)
Porcine: Hampshire, Yorkshire
Degenerative Leukomyelopathies (Spinal Cord—White Matter)
Canine: German shepherd, Afghan hound, Kooikerhondje, Labrador retriever, Ibizan hound, harrier, beagle, foxhound, Rottweiler, smooth fox terrier, Jack Russell terrier
Bovine: Brown Swiss, Holstein-Friesian, Murray Grey
Equine: Various breeds (see vitamin E deficiency)
Multisystem Neuronal Degeneration.
Multisystem neuronal degeneration is discussed in sections covering disorders unique to individual animal species.
Primary Cerebellar Neuronal Degeneration.
Depending on the degree of maturation of the cerebellum and related systems at the time of birth in the various species, clinical signs in animals with the neonatal syndromes can be manifest in the immediate postnatal period (bovine, ovine) or can be delayed until the time of ambulation (canine). Hereditary transmission is known or suspected in some instances. Lesions vary among affected species and breeds but overall include degeneration or absence of Purkinje cells, proximal swelling of Purkinje cell axons, variable loss of granule cells, cortical astrogliosis, and degeneration of nuclei in the cerebellar medulla.
Animals with postnatal cerebellar syndromes are normal at birth or at the time of ambulation. Onset of ataxia with various other clinical signs referable to cerebellar disease begins weeks, months, or even years after a period of apparently normal development. Initial clinical signs are often subtle. Progression of the signs can be slow or rapid, relentless, or with static periods. Some individuals reach a stage without further progression of signs, but this is not typical of the syndrome in most animals.
Grossly, the cerebellum can be normal or reduced in size and atrophic. Microscopically, lesions are analogous to those that occur in the neonatal syndromes with loss of Purkinje cells, variable neuronal depletion in the granule layer, and astrogliosis in the molecular layer. Fusiform swellings of proximal Purkinje cell axons are found in the rough-coated collie and the Yorkshire pig. In the rough-coated collie and Merino sheep, lesions occur in other areas of the neuraxis. In these syndromes, degeneration and loss of neurons in the deep cerebellar and other nuclei are accompanied by axonal degeneration in the cerebellum, brainstem, and spinal cord. Loss of spinal ventral horn motor neurons has been noted in the rough-coated collie. An autosomal recessive mode of inheritance is suspected or documented in several of the diseases. For individual disorders, see sections covering disorders of individual animal species.
For discussion of the primary cerebellar neuronal degeneration affecting individual animal species, see sections covering disorders specific for the species.
Neuroaxonal Dystrophy.
Diseases associated with axonal swellings (axonal spheroids) have been termed neuroaxonal dystrophy. Diseases include those putatively associated with vitamin E deficiency or that have been interpreted as an aging change (see the section on Degenerative Diseases). Included here are diseases with a species and breed association and onset relatively early in life, generally before 1 year of age but varying between 4 weeks and 3 years. A hereditary basis is often suspected or proved. Neuroaxonal dystrophies have been described in many animal species; however, they seem to be most common in dogs and horses. Neuroaxonal dystrophies of the horse are discussed in the section covering disorders unique to that species.
Dystrophy is defined as a disorder arising from defective or faulty nutrition of a cell, tissue, or organ, and the term is most commonly applied to muscle diseases. In this usage it applies to neurons and their axons (neuroaxonal). Lesions differ in severity and distribution, but are characterized by prominent axonal swellings in various nuclei (often sensory) in the brainstem, cerebellum, and spinal cord. Loss of cerebellar Purkinje and granule cells have been reported in Rottweilers and cats and the loss of brainstem neurons in cats. In the Morgan horse the axonal swellings are associated with vacuolation.
Neuroaxonal dystrophies are often characterized clinically by severe and often profound muscular weakness and widespread muscle atrophy. Sporadic cases in older adult animals also occur of unknown cause or suspected extraneous influence. Clinical signs vary but include gait abnormalities, dysmetria or hypermetria, proprioceptive disturbance, ataxia, or other cerebellar signs.
Motor Neuron Diseases.
Motor neuron diseases have been described predominately in dogs, cats, cows, horses, and pigs. Degeneration and loss of motor neurons in the ventral horns of the spinal cord and variable axonal degeneration in the ventral spinal nerve rootlets and peripheral nerves characterize the lesions in motor neuron diseases. In some of the motor neuron diseases there is prominent swelling of ventral horn neuronal cell bodies or axons, or both, associated with marked accumulation of neurofilaments. This accumulation is presumably caused by posttranslational protein modification and impairment of neurofilament protein transport. Degeneration in some diseases is not strictly limited to motor neurons of the spinal cord or to motor neurons in general. Other sites of involvement are motor or sensory nuclei, or both, in the brainstem and white matter tracts in the spinal cord.
In horses, lesions in motor neurons are analogous to those already described. The disease affects various breeds, no definitive familial association or age predilections are known, and an inherited basis is not suspected. Generalized weakness, muscle atrophy, and weight loss progress over 1 to several months.
In calves a disease known as shaker calf syndrome in horned Hereford calves can be only loosely termed a motor neuron disease. There is marked accumulation of neurofilaments within neurons of the central, peripheral, and autonomic nervous systems. All segments of the spinal cord are severely affected. Neurons and neuronal processes in ventral horns, intermediolateral nucleus, Clarke's column, and substantia gelatinosa are swollen and distended. Wallerian degeneration occurs in ventral nerve rootlets and white matter of the spinal cord. Brainstem lesions are less prominent. Swollen cerebellar Purkinje cells and neuronal degeneration in the lateral geniculate body and frontal cortex are reported. The disorder occurs in newborn calves and is characterized clinically by tremulous shaking of the head, body, and tail.
Nutritional
Vitamin B1 (Thiamine) Deficiency.
Thiamine pyrodiphosphate is the active form of thiamine. It is a critical cofactor for several thiamine-dependent enzymes involved in carbohydrate metabolism, and brain damage is thought to be related to a decline in thiamine-dependent enzymes, energy deprivation, and oxidative stress with the abnormal metabolism of free radicals in neurons. These enzymes are also important in the synthesis of several cell constituents, including neurotransmitters. Thiamine deficiency has been associated with neurologic disease in carnivores (Chastek's paralysis), human beings (Wernicke's encephalopathy), and ruminants. For discussion of nutritional diseases affecting individual animal species, see sections covering disorders specific for the species.
Vitamin A Deficiency.
See the section on the PNS and Chapter 21.
Toxicoses.
Space constraints do not allow a comprehensive discussion of all toxicities affecting the nervous system. Box 14-9 is a partial listing of poisons with the potential to cause CNS injury and neurologic illness. Some of these, such as mercury, have in the past caused high morbidity and mortality in isolated outbreaks. An example is the 1956 Minamata Bay incident in Japan of human beings eating fish containing high concentrations of methylmercury. Methylmercury accumulates in the aquatic food chain, and thus the highest concentrations exist in predatory fish at the top of the food chain. In human beings and animals with methylmercury toxicosis, neuronal cell bodies of the cerebral cortex and cerebellum die through a mechanism suggested to be apoptosis; however, microtubule dysfunction, oxidative stress, alterations of calcium homeostasis, and the potentiation of glutaminergic excitotoxicity may be involved. The potential remains for serious neurologic illnesses and death from these intoxications, and the interested reader is referred to more comprehensive reference sources. In this chapter, discussion of toxicities is limited to those conditions most likely to be encountered in veterinary practice.
Box 14-9. Other Toxicities Involving the Nervous System.
Chemicals
Heavy metals: Cadmium, manganese, mercury, tin (trimethyltin), zinc
Hexacarbons: n-Hexane, others
Pesticides: Carbaryl, bromethalin, chlorinated hydrocarbons
Drugs: Nitrofurazones, ivermectin, levamisole, metronidazole
Plants
Cycads, Chrysocoma tenuifolia, Helichrysum spp., Solanum spp. (dimidiatum, fastigiatum, kwebense), sorghum, Stypandra spp.
Mycotoxins
Acremonium, Aspergillus, Claviceps, fumonisin, Penicillium
Chemicals.
Chemically induced distal axonopathies have been classified by functional alterations affecting motor or sensory neurons, location of injury within the nerve (distal, proximal), or by the type of nerve affected (cranial or spinal). Because of the large number and wide use of chemicals in commerce, there exists an extensive list of experimental studies describing toxic axonopathies and neuropathies. Their complete discussion is outside the scope of this chapter.
Chemicals used in agricultural, industrial, and pharmaceutical commerce can injure nerves by interfering with axoplasmic flow. Such chemicals include acrylamide (polymerizing agent to strengthen paper), carbon disulfide (fat solvent, used for extraction of oil from oil-bearing fruit such as olives), triorthocresyl phosphate (high-performance lubricants in airplane engines), halomethane (refrigerants), methylene chloride (extraction agents, paint solvents, and degreasing agents), carbon tetrachloride (solvents), and butane (fuel source).
Acrylamide causes a unique distal axonopathy (dying-back axonopathy) primarily affecting axons of the PNS (less commonly the CNS) in which there is accumulation of neurofilaments within affected axons. Axonal spheroids are thought to be related to alteration of axonal transport resulting from phosphorylation of neurofilaments and their abnormal rearrangement within the axon. This “dying-back” axonopathy is microscopically characterized by degeneration of axons starting at or near synapses and proceeding toward the neuronal cell body. The most distal axonal projections are furthest from the cell body and thus cannot be maintained. Thus they are most vulnerable to functional alterations; however, it is unclear whether this degeneration is caused by energy deficits, lack of antioxidants, or physical obstruction of axoplasmic flow. Axonal degeneration is followed by secondary demyelination.
Certain types of toxic and biochemical injury to axons result in a stereotypic pattern of morphologic change that affects either distal or proximal segments of the axon and results in the formation of segmental axonal spheroids. Based on the location of the spheroids, such diseases are divided into one of two groups, either diseases affecting axons a distance away from their cell bodies (distal axonopathies) or diseases affecting axons near their cell bodies (proximal axonopathies).
Axonal spheroid formation and subsequent axonal degeneration are caused by alterations in axoplasmic flow and by alterations of anterograde or retrograde flow, depending on the nature of the injury resulting in the accumulation and/or rearrangement of cytoskeletal proteins. The histologic lesion common to these two types of axonopathies is the formation of axonal spheroids with subsequent degeneration of the axon and secondary demyelination, which is a process that in many ways resembles the lesions described for Wallerian degeneration. Axonal spheroids are common to a variety of neuronal derangements; therefore distal and proximal axonopathies must be differentiated from other diseases that cause spheroids such as the compressive axonopathies.
Distal and proximal axonopathies have been further subdivided by some scientific disciplines into groups based on whether initial axonal lesions progress in an anterograde or retrograde direction. The terminology and classification schemes, although useful to some, are outside the scope of this chapter and are often confusing. The occurrence of secondary anterograde or retrograde lesions is discussed in the context of some of the diseases presented next.
Organophosphates.
Organophosphates are divided into two groups according to their use, mode of action, and type of toxicity. The first group, organophosphate esters, used as pesticides (parathion, malathion, diazinon, carbaryl, or aldicarb), fungicides, herbicides, or rodenticides, cause acute toxicity by inhibiting cholinesterase either directly or indirectly and allowing acetylcholine to accumulate at synaptic (nerve-nerve junctions) or myoneural junctions (nerve-muscle junctions), resulting in persistent depolarization. In acute organophosphate toxicosis, clinical effects vary but are manifested in the following:
-
1.
Parasympathetic nervous system, leading to salivation, lacrimation, urination, defecation, bradycardia, and pupillary constriction
-
2.
Skeletal muscular system, resulting in muscle fasciculations followed by weakness and muscle paralysis (i.e., death is due primarily to respiratory failure)
-
3.
CNS, leading to anxiety, restlessness, hyperactivity, anorexia, and generalized seizures (i.e., observed in dogs and cats but uncommon in cattle)
Gross and microscopic lesions in the nervous system are absent, and those in other tissues are nonspecific.
The second group causes chronic toxicosis and is the most common cause of chemically induced distal axonopathy in veterinary medicine. This group of organophosphates includes the cresyl and related compounds such as triorthocresyl phosphate used in hydraulic fluids, lubricants, flame retardants, and plasticizers. The triaryl phosphate group of compounds used as high-temperature lubricants is toxic for several species of animals and human beings.
Chronic exposure (delayed neuropathy) to certain organophosphate pesticides and herbicides (trichlorphon, merphos, triorthocresyl phosphate, leptophos, parathion, malathion, and diazinon) causes delayed neurotoxicity unrelated to cholinesterase inhibition as seen in acute organophosphate toxicosis. The type of axonal injury caused by these chemicals follows the stereotypic process of morphologic changes described previously and occurs approximately 10 to 14 days after exposure. Organophosphorus compounds causing delayed neurotoxicity inhibit the activity of an enzyme referred to as neuropathy target esterase. The function of the enzyme in the PNS and CNS is not fully understood.
Phosphorylation of the enzyme by the toxic compound is proposed to interfere with its normal function, resulting in axonal injury. Other studies have shown that organophosphates causing delayed neurotoxicity interact with Ca2+ or calmodulin kinase II, an enzyme responsible for phosphorylation of cytoskeletal proteins, such as microtubules, neurofilaments, and microtubule-associated protein-2, resulting in disassembly and accumulation of these proteins in the distal portions of axons, producing axonal swelling and degeneration.
No specific gross lesions are present in chemically induced distal axonopathies. Microscopically, there is retrograde degeneration beginning in the distal part of axons, especially those with a larger diameter. Affected areas in the spinal cord include dorsal funiculi, spinocerebellar tracts in lateral funiculi, and ventromedial aspects of the ventral funiculi. Central chromatolysis of cell bodies of affected nerves has occurred.
Clinically, signs of toxicity are usually delayed 1 to 2 weeks after exposure. Young animals, because of their ability to compensate for the neurologic deficits, tend to be less seriously affected, whereas recovery is slow and incomplete in adults. Susceptible animals include cats, domestic and exotic ruminants, chickens, pheasants, and ducks. Small laboratory animals, dogs, and some nonhuman primates are less sensitive. Clinical signs are those of combined sensory and motor neuropathy and spinal cord damage, such as proprioceptive deficits expected by damage to the spinocerebellar nucleus and tract, as well as the fasciculus gracilis.
Selenium.
An acute paralytic syndrome termed bilateral poliomyelomalacia has been observed in feeder pigs associated with the inadvertent inclusion of toxic amounts of selenium (selenium-enriched yeast, sodium selenite, or sodium selenate) in pig rations. The pathogenesis of the lesions is not proved but could involve an induced nicotinamide or niacin deficiency. Experimentally, 6-aminonicotinamide, a gliotoxin and antagonist of the vitamin, causes lesions analogous to those seen in the natural porcine disease.
Grossly, bilateral (symmetric) areas of softening and yellow discoloration occur in the ventral spinal gray matter of the cervical and lumbar intumescences. Microscopically, acute lesions consist progressively of neuronal chromatolysis, neuronal necrosis, neuronal loss, microcavitation, and glial necrosis. As would be expected, these changes are subsequently followed by astrogliosis and the accumulation of gitter cells. Prominent capillaries are typical. Wallerian degeneration occurs in ventral spinal nerve rootlets of those cord segments whose ventral gray horn motor neurons have been destroyed. Identical lesions have been observed in the brainstem.
Clinically, affected pigs are alert, rest in sternal recumbency, and squeal loudly when disturbed. They eventually progress to quadriplegia with flaccid paralysis of the rear limbs. Cutaneous manifestations of the toxicity also occur and include rough hair coats, partial alopecia, and separation and sloughing of the hoofs. Historically, a similar ovine bilateral symmetric poliomyelomalacia has been reported from Africa, but an association with selenium toxicity was not made.
Sodium Chloride.
Sodium chloride toxicity, also known as sodium ion toxicosis, water deprivation syndrome, or salt poisoning, occurs primarily in pigs, poultry, and occasionally in ruminants, dogs, horses, nonhuman primates, and sheep. The disease occurs after overconsumption of sodium chloride in rations or supplements and can be complicated by limited availability of drinking water, resulting in severe dehydration. A similar sequence of events can occur with simple water restriction of sufficient duration to allow compensation by the brain's adaptive response to chronic hypernatremia (hyperosmolarity). Sodium chloride toxicity is due to hyperosmolarity (hypernatremia) caused by excessive intake of sodium salts or severe dehydration followed by rehydration and a “rapid” hypernatremic to normonatremic or hyponatremic shift.
During the initial hypernatremic phase the brain “shrinks” because of the osmotic loss of water. An influx of sodium, potassium, and chloride ions into the brain, beginning within minutes after the osmotic loss of water, is an acute adaptive response to equalize the sodium imbalance. Maintenance of a normal ionic balance in the brain is critical, however, for normal function, and although a new ionic equilibrium is established, this acute response alone cannot compensate for severe or prolonged hypernatremia.
A second, more delayed adaptive response of the brain is an influx or endogenous production of organic osmolytes, such as certain amino acids, polyols, and methylamines, to equalize the osmotic imbalance created by hypernatremia. This response requires hours or days to establish a new osmotic equilibrium. When animals are given free access to fresh water, an acute hypernatremic to hyponatremic shift occurs. Within minutes the brain attempts to offset this osmotic imbalance by eliminating sodium, potassium, and chloride ions by actively transporting these ions into the vasculature. This early response cannot, however, offset the osmotic stress created by the increased organic osmolytes in the brain. As a result of the osmotic gradient created by the elevated organic osmolytes, water enters the brain with subsequent brain swelling.
Grossly, lesions are inconsistent, but include cerebral and leptomeningeal congestion and edema. Zones of cerebrocortical laminar necrosis can be detected in transverse slices of fixed brain. Microscopically, cerebrocortical neuronal necrosis, often laminar, is accompanied by astrocytic swelling. In pigs, leptomeninges and perivascular spaces can have an infiltrate of eosinophils, and with longer survival, an influx of macrophages occurs, depending on the extent of necrosis (Fig. 14-56 ). The leptomeningeal and perivascular infiltrate of eosinophils is an inconsistent finding and can be a nonspecific finding in various porcine encephalitic disorders. However, when it occurs in conjunction with laminar cortical necrosis, it is indicative of salt toxicity in this species. Pallor of subcortical white matter is indicative of edema, and prominence of small cortical blood vessels is due to congestion and swelling of endothelial cell nuclei. In ruminants, arteriolar degeneration with a transmural neutrophilic infiltrate, cerebellar Purkinje cell necrosis, and edema of basal nuclei, thalamus, and midbrain have been observed.
Figure 14-56.
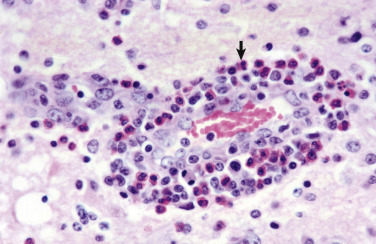
Eosinophilic Meningoencephalitis, Cerebral Cortex, Gray Matter, Pig.
Note the accumulation of eosinophils (arrow) in the perivascular space. This response is characteristic of the lesions of hypo-osmotic edema caused by water deprivation or excessive consumption of sodium salts. The surrounding neuropil is edematous. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Clinical signs include inappetence and dehydration early followed by heading pressing, incoordination, blindness, circling, paddling, and convulsions. Animals are often found dead in their pasture or pen.
Metals
Arsenic.
Toxicity caused by ingestion or cutaneous absorption can occur with inorganic and organic arsenicals and can affect multiple organs, including the nervous system. Inorganic compounds are predominantly herbicides or pesticides, whereas organic arsenicals (e.g., arsanilic acid) have been used as feed additives in the pig and poultry industries as growth promoters and to control enteric diseases.
Poisoning with inorganic arsenicals is an acute enteric disease with hepatic and renal manifestations, but neurologic signs can occur. Probably because of the nature of the organic compounds and the manner of their use, there is greater potential for neurotoxicity. Arsanilic acid has a greater tendency to cause peripheral and optic nerve and tract damage, whereas 3-nitro compounds tend to affect the spinal cord more severely.
Gross lesions are not present. Microscopically, lesions in cranial and peripheral nerves and spinal cord consist of axonal degeneration and fragmentation of myelin sheaths. In the spinal cord after 3-nitro poisoning, lesions are found chronologically in the cervical and thoracic cord followed by lesions in the lumbar cord. Spinocerebellar tracts and dorsal funiculi are predominantly affected. The distribution of lesions suggests that the distal segments of long ascending fiber tracts can be preferentially injured. Inorganic arsenicals inhibit sulfhydryl enzyme systems and disrupt cellular metabolism. The exact mode of action of organic arsenicals is unknown.
In pigs clinical signs include blindness resulting from damage to optic nerves and tracts and incoordination, paresis, and paralysis related to spinal cord and peripheral nerve lesions.
Lead.
Lead poisoning has occurred in a variety of animals, but with the increased awareness of the potential for toxicity and environmental contamination and current regulations, such as reduced concentrations in paint and unleaded gasoline, poisoning is uncommon and if it occurs is most common in cattle. Potential sources include discarded car batteries and old flaking or peeling lead paint in barns and farm buildings.
Depending on the quantity absorbed, poisoning can be peracute with no gross or microscopic lesions, acute, subacute, or chronic. In peracute or acute cases, contents of the upper digestive tract, such as fragments of battery plates or flakes of paint, could indicate the possibility of lead poisoning. Lead poisoning can affect many tissues and organs, including CNS, PNS, liver, kidneys, gastrointestinal tract, bone marrow, blood vessels, and organs of the reproductive and endocrine systems. In horses grazing lead-contaminated pastures, a cranial neuropathy with laryngeal and facial paralysis has been described.
Lead poisoning in cattle and other species is via the oral route or less commonly via the respiratory system or skin (inorganic lead). Lead can damage the brain through a variety of mechanisms. Direct toxic effects on neurons, astrocytes, and cerebral endothelial cells occur by disrupting metabolic pathways and altering the function of dopaminergic, cholinergic, and glutaminergic neurotransmitter systems. Lead crosses the blood-brain barrier rapidly using a cationic transporter, concentrates in the brain because of its ability to substitute for calcium ions in the pump, and enters astrocytes and neurons by voltage-sensitive cell membrane calcium channels. Lead disrupts calcium homeostasis, causing the accumulation of calcium in lead-exposed cells, and induces mitochondrial release of calcium, leading to apoptotic cell death. Astrocytes contain metallothionein and can sequester potentially toxic metals in the CNS, thus protecting more vulnerable neurons from the toxic effects of lead. However, astrocytes may also be sensitive to the toxic effects of lead, leading to functional deficits such as in the uptake, transport, and metabolism of neurotransmitters. Transplacental (human beings and sheep) and neonatal lead exposure can result in delayed brain maturation and biochemical abnormalities.
Gross lesions in the CNS are usually absent. When present, they can resemble those present in polioencephalomalacia of cattle, but this is uncommon. In general, gross lesions, if present, are distributed in a laminar pattern and include meningeal and cerebrovascular congestion, brain swelling with flattening of gyri, or hemorrhage. With longer survival times, there may be foci of cerebrocortical malacia (softening), cavitation, and laminar necrosis followed by cerebral cortical atrophy, widened sulci, narrowed gyri, and loss of white matter.
Microscopically, lesions in peracute cases are absent. In acute cases, congestion, astrocytic swelling, status spongiosus, and microvascular prominence caused by endothelial hypertrophy are present, and often ischemic neuronal cell change is characteristically confined to the tips of cerebrocortical gyri. For most cases in cattle, only a few necrotic neurons at gyral tips and minimal astrocytic swelling, vascular prominence, and congestion can be found. With longer survival times, cerebrocortical lesions progress to laminar necrosis, accumulations of macrophages, or liquefactive necrosis, although the last is rare. Because of their similarities, lesions of lead encephalopathy in ruminants must be differentiated from those of thiamine deficiency–associated polioencephalomalacia and sulfur-related polioencephalomalacia.
Lesions in dogs resemble those in cattle, but vascular damage is more obvious and consistent. Vascular lesions can progress to mural hyalinization, necrosis, and thrombosis. Other lesions include neuronal necrosis in the cerebral cortex, hippocampus, and cerebellum (Purkinje cells), myelin destruction in cerebrocortical white matter, and a peripheral neuropathy.
Clinically, affected cows are often found down or dead in the pasture. If clinical signs are present, they range initially from depression, inappetence, and diarrhea to teeth grinding (bruxism), circling, head pressing, incoordination, and blindness later. In small animals, especially dogs, clinical signs include ataxia, tremors, clonic-tonic seizures, blindness, and deafness.
Bromethalin.
Bromethalin, a highly lipophilic (i.e., high fat concentration in the brain) nonanticoagulant rodenticide, kills rodents, such as moles and voles, and domestic animals (predominantly dogs and cats secondary to accidental exposure) by uncoupling oxidative phosphorylation in mitochondria of neural cells. As a result, there is a decrease in ATP synthesis and dysfunction of ATP-dependent Na+ and K+ ion channel pumps. This outcome results in the accumulation of Na+ (and water) in neural cells and thus cytotoxic cerebral edema (see the section on Cerebral Edema [Permeability Changes]), leading to increased intracranial pressure and injury of neurons and their myelinated axons. Lesions, in addition to cerebral edema, include intramyelinic edema, myelin splitting, and axonal swelling, which all manifest as white matter spongiosis. Poisoned animals are usually depressed, have convulsions, varying degrees of ataxia, paresis, and paralysis, and may die.
Organotins.
Excessive exposure to organotins, such as triethyltin (stabilizer, catalyst, wood and textile preservative, fungicide, bactericide, and insecticide), causes cytotoxic edema principally affecting myelin sheaths of oligodendroglial cells in the white matter. Experimental studies have shown that triethyltin selectively damages myelin sheaths and causes a decrease in potassium concentrations in the white matter with a concurrent increase in intracellular water content. The blood-brain barrier is not affected. The mechanism of injury is thought to be uncoupling of oxidative phosphorylation and inhibition of mitochondrial ATPase activity within cell membranes. Loss of Na+/K+-dependent ATPase activity in cell membranes of myelin lamellae leads to the formation of intramyelinic edema.
Gross lesions, if present, consist of an enlarged brain and spinal cord. Because of compression against the cranium, an affected brain has flattened gyri and shallow indistinct sulci. Microscopically, fluid accumulates between myelin layers and leads to splitting of the myelin lamellae and the formation of intramyelinic spaces.
Microbial Toxins
Botulism.
See the section on Peripheral Nervous System.
Tetanus.
Tetanus is a spastic paralytic disease caused by the neurotoxin called tetanospasmin produced by Clostridium tetani. Similar to Clostridium botulinum, the bacterium is a ubiquitous Gram-positive spore-forming anaerobe commonly found in soil. Tetanospasmin is synthesized in anaerobic wounds and first binds at myoneural junctions and/or sensory receptors. It is transported via retrograde axoplasmic flow within the axon and across synaptic junctions until it reaches the CNS (see Fig. 4-28). In the CNS the toxin is transferred across synapses until it becomes fixed to gangliosides in the presynaptic inhibitory motor neuron. Tetanospasmin blocks the release of inhibitory neurotransmitters such as glycine and GABA. Inhibitory neurotransmitters act to dampen the actions of excitatory nerve impulses from upper motor neurons that are imposed on lower motor neurons. If these impulses cannot be dampened by normal inhibitory mechanisms, the generalized muscular spasms characteristic of tetanus ensue. Tetanospasmin appears to act by selective cleavage of a protein component of synaptic vesicles, thus preventing the release of neurotransmitters by the cells. Once toxin is bound to synapses, the administration of antitoxin is useless.
This disease is most common in horses but may also occur in lambs castrated in areas contaminated with spores of C. tetani. Tetanus also has been reported in cows, pigs, dogs, and cats. Except for the anaerobic wound, there are no macroscopic and microscopic tissue lesions in tetanus. Infected horses initially show signs of colic and muscle stiffness involving muscle groups of the lips, nostrils, ears, jaw (lockjaw), and tail. Horses are hyperesthetic and rapidly have a spastic and tetanic paralytic syndrome develop.
Plant Toxins
Astragalus, Oxytropis, and Swainsona Poisoning.
Astragalus, Oxytropis, and Swainsona represent three genera of plants with species that are toxic to livestock. As many as 300 species of Astragalus grow in North America, and the genus is the largest of any legume family in this part of the world. Three categories of toxicity can be observed with Astragalus, depending on the mechanism or manner of toxicity: nitro-containing, selenium-accumulating, and poisoning of the locoweed type. Only the last is discussed here. Locoweed poisoning, or locoism, is associated with ingestion of certain species of Astragalus and Oxytropis in North America and Swainsona in Australia. The toxic principles have been termed locoine and swainsonine, respectively.
The mechanism of toxicity has been clarified by the isolation of the indolizidine alkaloids swainsonine and swainsonine N-oxide from Astragalus lentiginosus. Recent discoveries have pinpointed the fungal endophyte Undifilum oxytropis as the ultimate source of swainsonine. The swainsonine compounds inhibit lysosomal α-mannosidase, thus inducing an acquired α-mannosidosis that mimics the inherited storage disease mannosidosis. Mannosidases are glycoside-hydrolyzing enzymes that are found in the Golgi, lysosomes, and cytoplasm of all mammalian cells. Analyses of tissue from animals poisoned with swainsonine have shown that swainsonine is present in all tissue; however, neurons; epithelial cells in organ systems, such as the liver; and macrophages of the monocyte-macrophage system of the spleen and lymph nodes are commonly affected. Therefore, as occurs in the inherited storage disease (mannosidosis), acquired swainsonine-induced storage diseases affect similar cells throughout the body. Additionally, swainsonine interferes with normal synthesis of glycoproteins containing asparagine-linked complex oligosaccharides. Swainsonine also inhibits Golgi mannosidase II, an effect not recognized in the inherited disorder.
There are no specific gross lesions in acquired swainsonine-induced storage diseases. Microscopically, lesions involve neuronal cell bodies throughout the neuraxis and autonomic ganglia and are analogous to the inherited lysosomal storage diseases.
Microscopically, neuron cell bodies are swollen, and nuclei are sometimes displaced to the periphery of the cell body. The cytoplasm appears foamy or finely vacuolated. The material that accumulates in the cytoplasm does not stain for lipid. Irregular fusiform enlargements, called meganeurites, occur in the proximal axon segment, and aberrant synapses form. With time, lesions include distal axonal degeneration and neuronal necrosis with mineralization. The presence of cytoplasmic lesions in other cells of the CNS, such as astrocytes, depends on the degree to which α-mannosidase is expressed in individual cell populations. Astrocytes are hydropic or swollen, but their appearance is less dramatic and diagnostic when compared with changes in neurons. Macrophages recruited from the bloodstream to phagocytose debris and mannose released from dead neurons also are affected by swainsonine. Microgliosis and neuronophagia are present but inconspicuous.
Similar to the swelling and vacuolation of neurons, this process also occurs in cells throughout the body, including hepatocytes, exocrine pancreatic cells, renal tubular epithelium, endocrine organs (thyroid, parathyroid, and adrenal glands), circulating leukocytes, and cells of the monocyte-macrophage system in liver, spleen, and lymph nodes. Ingestion of the plants of these species by females during gestation can also result in abortion or birth of weak neonates that have similar lesions.
Cattle, sheep, and horses are generally affected. Toxicity is usually insidious, and clinical signs are not observed until after the plants have been grazed on for 14 to 60 days. Clinical signs include poor condition, depression, head pressing, incoordination, stumbling, circling, blindness, recumbency, and paddling.
Miscellaneous Conditions
Meningeal Melanosis (Congenital).
The leptomeninges of animals and human beings with heavily pigmented skin, especially black-faced sheep and black-skinned pigs, can have melanin (Fig. 14-57 ). The extent and degree of pigment deposition varies dramatically from animal to animal. Similar pigment deposits can be found in other areas of the body, including the pleurae, uterine caruncles, liver, and respiratory and alimentary systems' mucous membranes. Congenital meningeal melanosis produces no clinical impairment in affected animals and is an expected, normal finding.
Figure 14-57.
Melanosis, Leptomeninges (Pia-Arachnoid Mater), Sheep.
Note the black pigmentation of the leptomeninges overlying the olfactory poles and dorsal aspect of the frontal lobe. Meningeal melanosis is a normal finding in black-faced sheep and other animals with heavily pigmented skin.
(Courtesy Dr. D. Morton, College of Veterinary Medicine, University of Illinois.)
Circulatory Disturbances
Many diseases of the CNS in veterinary medicine result from injury to the circulatory system and vascular endothelium. Vascular diseases of the CNS can result from inflammation/infection, either as a component of a systemic disease process or from extension of inflammatory meningeal or cerebral disease. The incidence of cerebrovascular diseases analogous to those in human beings, including trauma, is low in animals, and neurologic manifestations associated with these diseases are uncommon. Arteriosclerosis (“hardening” of arteries) can be categorized as lipid (atherosclerosis) or nonlipid arteriosclerosis with the latter including arterial fibrosis, mineralization, and amyloid deposition (see Chapter 10).
Atherosclerosis.
Atherosclerosis is reported in a variety of animals, including nonhuman primates, pigs, dogs, and several avian species. Older pigs are most commonly and severely affected. Occasional older dogs with chronic hypothyroidism or diabetes mellitus can have severe atherosclerosis. The pathogenesis of atherosclerosis and atherosclerotic plaque formation is most well understood in human beings, and the results of experimental studies may have some application to understanding atherosclerosis in domestic animals.
Atherosclerotic plaques arise from a complex and partially understood interaction among endothelium, smooth muscle cells, platelets, T lymphocytes, and monocytes. Endothelial injury induced by oxidized low-density lipoprotein (LDL) cholesterol results in vascular inflammation of the tunica intima. Monocytes migrate into the intima of the vessel wall to phagocytose LDL cholesterol. This process results in the formation of foam cells characteristic of early atherosclerosis (fatty streak). In addition, activated macrophages produce factors that also injure the endothelium. LDL cholesterol concentrations in foam cells and smooth muscle cells often exceed the antioxidant properties of normal endothelium. Oxidized LDL leads to additional metabolic changes that foster a procoagulant microenvironment and enhanced platelet-mediated thrombus formation, and initiates a cascade of events leading to the lesions associated with the development of mature atherosclerotic plaques (fibrous plaques with a cap [lipid-laden macrophages walled off by connective tissue]). The location of atherosclerotic plaques in the circulatory system depends on fluid shear stresses and their interaction with injured vascular endothelium. Atherosclerotic plaques characteristically occur in areas of vessel branching or areas where blood undergoes a sudden change in velocity and/or direction of flow.
Although atherosclerotic plaques can reach sizes large enough to significantly reduce blood flow to regions of the brain, the stability of the plaques determines the seriousness of the disease. A stable plaque is characterized by an excess of smooth muscle cells with few lipid-containing macrophages. An unstable plaque is characterized by a large lipid-rich core with abundant lipid-containing macrophages, thin fibrous cap, and inflammation. Rupture of unstable plaques can lead to vascular thrombosis or thromboembolism and infarction of areas supplied by these vessels in the CNS.
Grossly, vessels that can be involved include the aorta and its major branches, extramural coronary arteries, renal arteries, and cerebral arteries. Affected arteries are rigid, irregularly thickened, and white to yellow-white (atheromatous plaques) (Fig. 14-58, A ). Arterial lumina are narrowed or almost obliterated, but there is usually no appreciable ulceration, thrombosis, or hemorrhage (see Fig. 14-58, B). Intimal thickening in intracranial arteries often contains less lipid, and these arteries have a greater tendency for fibrosclerosis than other vessels. In arteries within the brain there is collagenous adventitial or transmural thickening. The arterial lesions can be associated with hemorrhage or infarcts involving basal nuclei, fornix, internal and external capsules, hippocampus, and thalamus.
Figure 14-58.
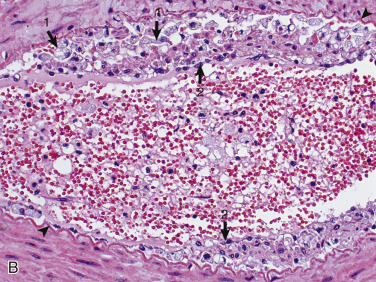
Atherosclerosis, Ventral Spinal Artery, Spinal Cord, Ventral Surface, Dog.
A, The ventral spinal artery is segmentally yellow, thickened, and beaded in appearance from atheroma (arrows). This dog had long-standing hypothyroidism. B, The tunica intima contains numerous foamy (lipid-laden) macrophages (arrows 1). Arrowheads, Internal elastic lamina; arrows 2, endothelium. H&E stain.
(A courtesy Dr. J. Hammond, Pieper Memorial Veterinary Center. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
In the dog, lesions most commonly involve cerebral, coronary, and renal arteries and are most severe in the intima and media. Hemorrhage, ischemia, and infarction of the cerebral cortex are uncommon but can occur. Infarctive changes associated with presumptive hypothyroidism-associated atherosclerosis have been identified in dogs by magnetic resonance imaging examinations. Many of these lesions resolve with treatment of the underlying endocrinopathy with minimal residual CNS lesions.
Cerebral Edema (Permeability Changes).
The causes and mechanisms of cerebral edema are presented in the section on vasogenic, cytotoxic, and interstitial edema. Cerebral edema has also been associated with “water intoxication,” which can result from an increased body hydration caused by (1) excessive, faulty intravenous hydration; (2) compulsive drinking caused by abnormal mental function; or (3) altered antidiuretic hormone secretion. The increased body hydration produces a hypotonic (hypo-osmolar) plasma, with subsequent development of an osmotic gradient between the hypotonic plasma and the relatively hypertonic state of the normal cerebral tissue. Fluid moves from the plasma into the brain. In this type of edema the blood-brain barrier remains intact. If it did not, the change in plasma osmolarity would soon be transmitted to the brain tissue (through vascular leakage) and would abolish the necessary osmotic gradient. Fluid accumulation occurs primarily intracellularly but can also be present extracellularly. In addition, typically a pronounced increase occurs in the rate of formation of CSF originating from the choroid plexus and the extracellular fluid of the brain.
The gross lesions that accompany cerebral edema are the result of enlargement of an organ in an enclosed, limited space; the degree of swelling obviously determines the type and extent of lesions that develop. In evaluating lesions it is particularly important initially to examine the brain and spinal cord in the fresh state and in situ.
Microscopically, in contrast to some other tissues, such as the lungs, the extracellular fluid associated with vasogenic edema fluid is often not detectable, except in instances of marked vascular injury. When the extracellular space-occupying fluid cannot be identified, only its effects (separation of the cells and their processes causing reduced staining intensity) can be recognized. Additionally, after prolonged vasogenic edema the lesions include hypertrophy and hyperplasia of astrocytes, activation of microglia, and demyelination. Cytotoxic edema is characterized by cellular swelling, including swelling of astrocytes.
Because of compression against the cranium, an affected brain has flattened gyri and shallow sulci, and it can shift in position. If the edema is confined to one side, the displacement is unilateral, which can be associated with herniation of the cingulated gyrus under the falx cerebri; the extent of the unilateral intracerebral enlargement can be best appreciated after the examination of transverse sections. Diffuse swelling usually causes a caudal shifting that can result in herniation of the brain (parahippocampal gyri of temporal lobes) beneath the tentorium cerebelli (Fig. 14-59 ) or herniation of the cerebellar vermis through the foramen magnum, resulting in “coning” of the vermis (Fig. 14-60 ). On a cut surface the white matter is most often affected (frequently with the vasogenic type of edema, which is the most common). It is swollen and soft, has a damp appearance, and is light yellow in the fresh, unfixed state.
Ischemic Myelopathy (Fibrocartilaginous Embolic Myelopathy).
Fibrocartilaginous embolic myelopathy has been described in almost all domestic species but it most commonly occurs in the dog. Herniation of fibrocartilage from the intervertebral disk into the vasculature, forming occlusive emboli, is the known cause, but the route taken by fibrocartilaginous material into the vessels of the spinal cord is uncertain. It has been suggested that trauma to the nucleus pulposus causes it to fragment and that the pressure of trauma forces small fragments into damaged veins, venous plexuses, or small arterioles. The ventral spinal artery and vein and their tributaries are commonly affected, presumably due to their proximity to the extruded disk material. Larger breed dogs are more frequently affected than chondrodystrophic breeds.
The gross lesion is an acute focal infarct involving cervical or lumbar spinal cord most commonly, but any portion can be affected (Fig. 14-61 ). Microscopically, emboli histochemically identical to the fibrocartilage of the nucleus pulposus of intervertebral disks occlude meningeal or CNS arteries or veins, or both, in affected areas (Fig. 14-62 ). Clinically, there is a sudden onset of spinal cord deficits, sometimes with cerebral involvement, in certain species. In dogs, larger breeds are more commonly affected. The disease occurs in young and old animals. One study reported that 60% of confirmed cases of canine ischemic myelopathy had a history of trauma or exercise.
Figure 14-61.
Spinal Cord Infarction (Ischemic Necrosis), Dog.
The yellow-brown region of necrosis (arrows) in the right lateral and ventral funiculi was the result of fibrocartilaginous emboli that occluded branches of the ventral spinal artery and obstructed blood flow.
(Courtesy Dr. J. Edwards, College of Veterinary Medicine, Texas A&M University; and Dr. J. King, College of Veterinary Medicine, Cornell University.)
Figure 14-62.
Fibrocartilaginous Embolus, Spinal Cord, Dog.
A, Vascular occlusion and infarction. Fibrocartilaginous emboli have obstructed the dorsolateral artery (top left) and branches of the ventral spinal artery to the right ventral gray horn and adjacent white matter, causing infarction (arrows). H&E stain. B, Fibrocartilaginous emboli in arterioles (arrows). C, Central canal. H&E stain.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. J. Van Vleet, College of Veterinary Medicine, Purdue University.)
Nonlipid Vascular Changes.
Arterial fibrosis occurs more frequently in older animals and has been described in dogs and horses. In the dog, fibrosis of the intima, media, or adventitia occurs with some frequency in cerebrospinal vessels of all types and caliber. Fibrous thickening of the adventitia of small meningeal and CNS arteries can be accompanied by variable degrees of extension of fibrosis into other layers of the vessel wall. A preferential site is the choroid plexus, where hyalinization and perivascular/vascular thickening is a common finding in older animals. In old horses a similar pattern of fibrosis occurs in vessels, and the adventitia may be preferentially affected. Amyloid deposits in meningeal and cerebral vessels are reported in older dogs and other animals. Mineralization (deposition of calcium or iron salts) of cerebral blood vessels occurs in the brains of several species but is especially common in adult horses. Vessels of the internal capsule, globus pallidus, cerebellar dentate nucleus, and infrequently the hippocampus are preferentially affected in horses, cattle, and less commonly, dogs. Meningeal vessels in old cats, old horses, and cattle and vessels of the choroid plexus in old cats are other sites of vascular mineralization. Almost always these areas of vascular mineralization should be considered incidental findings and are unlikely to be associated with clinical signs. Overt ischemic damage is rarely associated with these nonlipomatous vascular lesions in any species; therefore clinical signs are not seen with this lesion.
Lysosomal Storage Diseases
Dysfunction of lysosome-mediated degradation of products (substrates) of normal cellular metabolism results in diseases referred to as lysosomal storage diseases. These substrates cannot be degraded by lysosomes, and the accumulated substrate eventually results in death of the affected cells.
Cell death is the end point of a chronic and progressive process of substrate accumulation that interferes with cellular biochemical processes and transport systems. When neurons or myelinating cells die, they release their accumulated substrate into adjacent tissue. Macrophages are recruited from the bloodstream as monocytes, and they phagocytose cellular debris and unprocessed substrate released from dead cells. Macrophages, however, have the same genetic defect and thus also accumulate substrate in their lysosomes. Although less vulnerable to the effects of substrate accumulation, macrophages eventually die, and their released substrate is phagocytosed by additional macrophages recruited from the blood.
Lipid storage diseases, such as globoid cell leukodystrophy, are covered in more detail later. Features of some selected lysosomal storage diseases of animals are provided in Table 14-7 .
Table 14-7.
Classification of Selected Lysosomal Storage Diseases That Involve the Central Nervous System of Animals
| Disease | Storage Product | Deficient Enzyme | Species | Breed |
|---|---|---|---|---|
| GM1 gangliosidosis | GM1 ganglioside | β-Galactosidase | Bovine | Holstein-Friesian |
| Canine | Beagle, English springer spaniel, Portuguese water dog, Alaskan huskies | |||
| Feline | Siamese, domestic shorthair | |||
| Ovine | Suffolk, Coopworth-Romney | |||
| GM2 gangliosidosis | GM2 ganglioside | β-Hexosaminidase | Canine | German shorthair pointer, Japanese spaniel |
| Feline | Domestic shorthair, Korat | |||
| Porcine | Yorkshire | |||
| Globoid cell leukodystrophy (Krabbe's-like disease) | Galactosylceramide (galactocerebroside) and galactosylsphingosine (psychosine) | Galactosylceramidase (galactocerebroside β-galactosidase) | Canine | West Highland terrier |
| Cairn terrier, miniature poodle, bluetick hound, beagle, Pomeranian | ||||
| Feline | Domestic shorthair, domestic longhair | |||
| Ovine | Polled Dorset | |||
| α-Mannosidosis | Mannose-containing oligosaccharide | α-Mannosidase | Bovine | Angus, Murray Grey, Galloway |
| Feline | Persian, domestic shorthair | |||
| β-Mannosidosis | Mannose-containing oligosaccharide | β-Mannosidase | Caprine | Nubian |
| Bovine | Salers | |||
| Mucopolysaccharidosis | Different glycosaminoglycans | Several different enzyme deficiencies | Canine | Plott hound (type I, Hurler's disease), miniature pinscher (type VI, Maroteaux-Lamy disease) |
| German shepherd (type VII, Sly disease) | ||||
| Feline | Domestic shorthair (type I, Hurler's disease) | |||
| Domestic shorthair, Siamese (type VI, Maroteaux-Lamy disease) | ||||
| Domestic shorthair (type VII, Sly disease) | ||||
| Caprine | Nubian (type III, Sanfilippo's disease) | |||
| Ceroid-lipofuscinosis | Subunit c of mitochondrial ATPase | Prelysosomal defect? | Canine | English setter, border collie, Tibetan terrier |
| Ovine | South Hampshire | |||
| Bovine | Devon | |||
| Sphingolipid Activating proteins A and D |
Palmitoyl protein thioesterase | Canine | Miniature schnauzer | |
| Ovine | Swedish Landrace | |||
| Unknown | Unknown | Canine | Chihuahua, cocker spaniel, Saluki, terrier-cross, blue heeler, Yugoslavian shepherd, Dalmatian, Australian cattle dog, golden retriever, dachshund, corgi | |
| Ovine | Rambouillet | |||
| Bovine | Beefmaster | |||
| Feline | Siamese, domestic shorthair | |||
| Niemann-Pick type c disease | Primarily ganglioside in neurons | Unknown | Feline Canine |
Domestic shorthair Boxer |
ATPase, Adenosine triphosphatase.
Lysosomal storage diseases were originally thought to develop exclusively because of mutations that result in a reduction in lysosomal enzyme synthesis. More recently, however, it has become clear that there are other defects such as the following:
-
1.
Synthesis of catalytically inactive proteins that resemble normal active enzymes
-
2.
Defects in posttranslational processing (glycosylation, phosphorylation, addition of fatty acids in the Golgi) of the enzyme, which results in it being misdirected to sites (extracellular) other than to lysosomes
-
3.
Lack of enzyme activator (an enzyme that normally increases the rate of an enzyme-catalyzed reaction) or protector protein (facilitates repair and refolding of stress-damaged proteins)
-
4.
Lack of substrate activator protein required to assist with the hydrolysis of substrate
-
5.
Lack of transport protein required for elimination of digested material from lysosomes
Characterization of lysosomal disorders has therefore been broadened to include involvement of any protein that is essential for normal lysosomal function
The best-known diseases are characterized by accumulation of the substrate or substrate precursors and sometimes even by the absence of a critical metabolic product for normal lysosomal function. As a general principle, cell swelling and cytoplasmic vacuolation occur because of the accumulation of unprocessed substrate in the lysosomes; therefore differences in the size and appearance of cells (neurons versus hepatocytes) depend on the availability of the substrate (carbohydrate or lipid) in the organ system. Many lipids and glycolipids are unique to the nervous system; thus when there is a lysosomal defect, neural cells often accumulate in the substrate.
Examples of lysosomal storage diseases that affect human beings and animals are the gangliosidoses. With few exceptions, these diseases are inherited in an autosomal recessive pattern. They are also often gene-dose dependent and correspondingly, recessive homozygotes manifest the disease, whereas heterozygotes are phenotypically and functionally normal, but the affected enzyme's activity is reduced by approximately 50% of normal. The age of onset of clinical signs and the severity of the disease process can vary among the different diseases because the deficiency of the involved enzyme is not always the same. If the gene defect is such that the mutant enzyme is not synthesized at all, there is an early onset of a severe disease. Conversely, if there is some residual synthesis of the deficient enzyme, later onset and a milder form of the disease result because partial catabolism of the accumulated substrate permits a longer period of time before the lysosomes are so distended with substrate that they cause loss of cell function.
Gross lesions of the CNS vary among the different types of lysosomal storage diseases. Brain atrophy occurs with globoid cell leukodystrophy in latter stages of the diseases because of the loss of myelin. Brain atrophy is also seen with ceroid-lipofuscinosis but is not prominent in other lysosomal storage diseases, although brains of animals with gangliosidoses can have a firm, rubbery consistency. Microscopically, affected neurons often have a foamy, finely vacuolated, or granular cytoplasm, which is a reflection of the degree to which the stored material is removed during histologic processing (Fig. 14-63 ). The specific features of the stored material can be best appreciated by ultrastructural examination.
Figure 14-63.
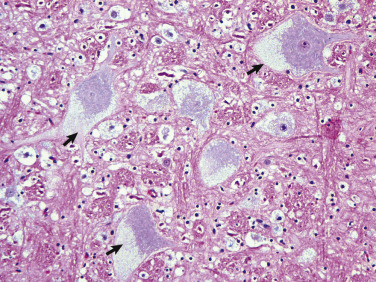
Glycogen/Carbohydrate (Lysosomal) Storage Disease, Brainstem, Neuron Cell Bodies, Cat.
Note the enlargement of the neuron cell bodies, displacement of nuclei, and accumulation of unprocessed substrate in the cytoplasm of the neuronal cell bodies (arrows) giving the appearance of “foamy” cytoplasm. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Ceroid-lipofuscinosis is a lysosomal storage disease characterized by abnormal sphingolipid (lipopigments) metabolism that occurs in cats, dogs, cattle, and sheep. Its lysosomal dysfunction has not been clearly identified, but experimental studies have shown alterations in the activity of palmitoyl-protein thioesterase and concentration of acid protease. The disease resembles other lysosomal storage diseases in that it can have a recessive mode of inheritance, but it is dissimilar in that it has no gene-dose effect. Brain atrophy occurs with ceroid-lipofuscinosis in later stages of the diseases (in sheep) (see Fig. 14-17). The atrophy, which most frequently involves the cerebral cortex but also sometimes the cerebellum, can result in a 50% reduction in brain weight. The cerebral hemispheres are increased in firmness and often have a tan color, whereas the gyri are thinned and the sulci widened, a clear indication of cerebrocortical atrophy. Microscopically, the cytoplasm of affected neurons has an eosinophilic granular material (with H&E staining) and a decrease in the number of neurons. Reactive astrogliosis is prominent, and microgliosis may also be observed.
Globoid Cell Leukodystrophy.
As discussed previously, lysosomal storage generally refers to a cellular alteration in which an increased amount of substrate material, which normally is degraded, accumulates within lysosomes, often eventually resulting in cell death. These diseases have a hereditary basis, occur in young animals, and are transmitted in an autosomal recessive pattern. Features of some selected lysosomal storage diseases of animals are given in Table 14-7.
Globoid cell leukodystrophy, a sphingolipidosis, is a lysosomal storage disease; its principal lesion is primary demyelination involving oligodendrocytes of the CNS and Schwann cells of the PNS. The disease, which has an autosomal recessive inheritance in the Cairn and West Highland white terrier, is generally seen in younger animals, often younger than 1 year old. It has also been described in beagles, miniature poodles, basset hounds, Pomeranians, bluetick hounds, and domestic short- and long-haired cats.
Mechanistically, the proposed sequence of events in this disease includes (1) early “normal” myelination that progresses up to a certain stage; (2) disruption of normal myelin turnover because of deficient galactosylceramidase activity; (3) degeneration and necrosis of myelinating cells because of the accumulation of psychosine; (4) primary demyelination; (5) recruitment of phagocytes, both resident microglia and trafficking blood monocytes; and (6) infiltration of macrophages, which become globoid cells after phagocytosing myelin byproducts into the nervous tissue. The last-named changes occur in response to the demyelination and unmetabolized galactocerebroside.
Affected oligodendroglia and Schwann cells are deficient in the lysosomal hydrolase, galactosylceramide β-galactosidase (GALC), which is responsible for degradation of galactosylsphingosine (psychosine) and galactosylceramide (galactocerebroside). Psychosine is highly toxic, and because it is not degraded, it has been hypothesized that it accumulates during the disease and causes direct injury to oligodendrocytes and Schwann cells, possibly through an apoptotic mechanism of cell death, in part, mediated by psychosine-induced production of cytokines and inducible nitric oxide synthase.
In globoid cell leukodystrophy the composition of myelin is not qualitatively abnormal. Galactosylceramide is highly concentrated in myelin but is nearly absent in systemic organs except for the kidney. Peak synthesis and turnover of galactosylceramide coincides with the peak period of myelin formation and turnover during the first year of life. GALC activity also increases in relation to the galactosylceramide peak. Myelination continues at a slower rate as animals mature, and in an adult myelin formation is stable with minimal turnover.
A deficiency in GALC activity results in the accumulation of galactosylceramide, especially during the early phase of myelin maturation and turnover, and in the formation of globoid cells discussed later. Psychosine also accumulates, leading to rapid and massive degeneration of oligodendroglia and Schwann cells, extensive myelinolysis, and reduction in myelination.
Gross lesions of the CNS are characterized by a gray discoloration of the white matter, especially of the centrum semiovale of the cerebral hemispheres and white matter of the spinal cord (Fig. 14-64 ). The lesion in the spinal cord tends to start in the peripheral white matter and spread inward. Microscopically, such areas have pronounced loss of myelin (Fig. 14-65, A ) and prominent globoid cells containing galactocerebroside that can be demonstrated with a periodic acid–Schiff stain (Fig. 14-65, B; E-Fig. 14-7). Peripheral nerves are also affected, and lesions are typified by primary demyelination and secondary axonal degeneration. Small sensory branches of peripheral nerves are useful sites to take biopsies to make diagnoses (see Fig. 14-112).
Figure 14-64.
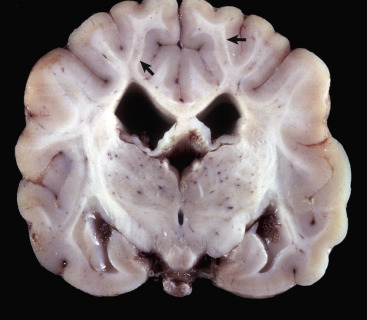
Lipid Storage Disease, Globoid Cell Leukodystrophy, Brain, Transverse Section at the Level of the Mammillary Body, Dog.
The white matter, especially of the gyri, has an off-white to light gray appearance (arrows). Macrophages (globoid cells) derived from blood monocytes (also enzymatically deficient in β-galactocerebrosidase) accumulate in white matter to phagocytose galactocerebroside and oligodendroglial debris secondary to the toxic effects of galactosylsphingosine (psychosine) on oligodendroglia (and in the peripheral nervous system Schwann cells). There is also bilateral hydrocephalus of the lateral ventricles, presumably hydrocephalus ex vacuo, as a result of the loss of neurons and their axons.
(Courtesy Dr. H.B. Gelberg, College of Veterinary Medicine, Oregon State University.)
Figure 14-65.
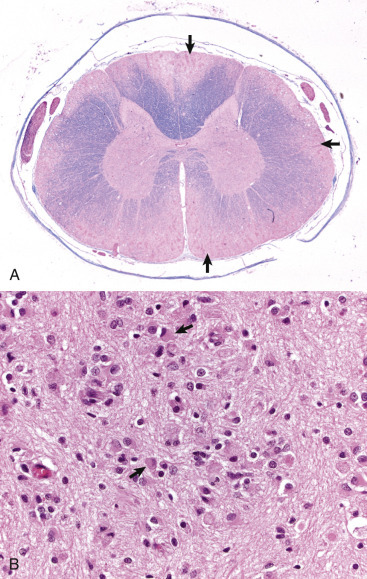
Globoid Cell Leukodystrophy, Dog.
A, Spinal cord. This section of spinal cord has been stained with Luxol fast blue, a histochemical reaction that stains myelin blue. Note the loss of myelin from the periphery of the cord, where axons are heavily myelinated (arrows), the first area to be affected. Luxol fast blue stain with a nuclear fast red counterstain. B, Early stage of the disease. The white matter contains globoid cells (macrophages) that are characterized by abundant eosinophilic cytoplasm and an eccentric nucleus (arrows). The number and size of macrophages increase over the time course of the disease due to progressive loss of myelin. H&E stain.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee; B courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
E-Figure 14-7.
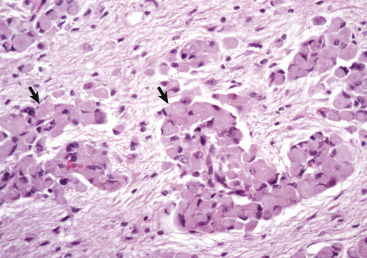
Globoid Cell Leukodystrophy, Cerebrum. Dog.
Late stage of the disease. Globoid cells deficient in β-galactocerebrosidase (arrows), usually located in the perivascular space, increase in size and number because they are unable to degrade galactocerebroside to a less complex substrate. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 14-112.
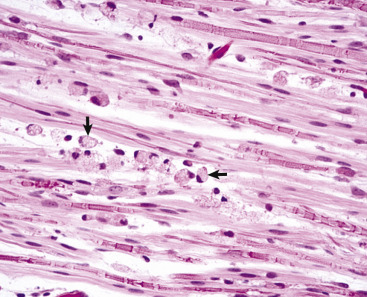
Globoid Cell Leukodystrophy, Small Branch of a Peripheral Sensory Nerve, Dog.
Primary demyelination, secondary axonal degeneration, and globoid cells (arrows) between the nerve fibers (also see Figs. 14-64 to 14-65). H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Clinically, affected animals are ataxic and have limb weakness and tremors that progress to paralysis and muscular atrophy. Poor vision or blindness may also occur.
Disease Processes Affecting Myelin Formation and Maintenance
Hypomyelination and Dysmyelination.
Disorders of myelin formation include hypomyelinogenesis (hypomyelination) and dysmyelination. Hypomyelinogenesis is a process in which there is underdevelopment of myelin. Dysmyelination refers to the formation of biochemically defective myelin. Hypomyelinogenesis and dysmyelination most often occur in the early postnatal period and have similar clinical and pathologic features. There are some differences in the lesions and the mechanisms by which they develop. Some of these diseases in domestic animals are outlined in Table 14-8 .
Table 14-8.
Hypomyelinogenesis and Dysmyelination in Animals
| Species | Breed | Disease Designation | Genetic Cause | Infectious Cause | Metabolic Cause |
|---|---|---|---|---|---|
| Bovine | All breeds | Bovine virus diarrhea (dysmyelination) | Bovine virus diarrhea (pestivirus) | ||
| Charolais | Progressive ataxia | Suspected | |||
| Ovine | All breeds | Border disease (hypomyelinogenesis-dysmyelination) | Border disease virus (pestivirus) | ||
| Porcine | Landrace | Congenital tremor (myelin agenesia) | Sex-linked recessive | ||
| Saddleback | Congenital tremor | Autosomal recessive | |||
| Chester-white | Myoclonia congenita | Autosomal recessive | Suspected | ||
| All breeds | Congenital tremor (dysmyelinogenesis and cerebellar hypoplasia) | Classic swine fever (pestivirus) | |||
| All breeds | Congenital tremor (dysmyelinogenesis) | Unknown virus suspected | |||
| All breeds | Congenital ataxia and tremor (hypomyelinogenesis and cerebellar hypoplasia) | Trichlorfon (acaricide) | |||
| Canine | Dalmatian | Hypomyelinogenesis | |||
| Chow Chow | Dysmyelination | Suspected | |||
| Springer spaniel | Shaking pups (hypomyelination) | Sex-linked recessive | |||
| Samoyed | Tremor (hypomyelination) | Suspected | |||
| Lurcher | Tremor syndrome (hypomyelination) | ||||
| Weimaraner | Hypomyelination | Suspected |
Hypomyelinogenesis
Diseases Caused by Viruses.
Classical swine fever (hog cholera) virus, a pestivirus, can be teratogenic in the porcine fetus. The best-known neural defects resulting from fetal infection are hypomyelinogenesis and cerebellar hypoplasia, although other lesions of the CNS, such as microencephaly and nonneural tissue, have been reported. The mechanism of lesion development has not been definitively determined, but a persistent infection that results in inhibition of cell division and function of selected tissues has been proposed.
Border disease viral infection (also a pestiviral infection) is capable of inducing maldevelopment in the CNS and nonneural tissues (skeleton) of lambs and goats after natural infection of the dam during pregnancy. One of the characteristic lesions in the CNS is hypomyelinogenesis, primarily affecting the white matter of the cerebrum and cerebellum. Grossly, it may be difficult to distinguish between white and gray matter in transverse sections of cerebrum and cerebellum. The brain and spinal cords from affected lambs may be smaller when compared with unaffected lambs. The PNS is unaffected. Hypomyelinogenesis may be related to a viral-induced decrease of myelin-associated glycoprotein, myelin basic protein, and activity of nucleotide phosphodiesterase in oligodendroglia. Other lesions detected in lambs include early inflammation, porencephaly-hydranencephaly, cerebellar malformation including hypoplasia, microencephaly, and reduction in diameter of the spinal cord.
Globoid Cell Leukodystrophy.
See the earlier section on Lysosomal Storage Diseases.
Spongy Degeneration (Status Spongiosus).
Spongy degeneration is a group of diseases of young animals characterized by a moth-eaten appearance (referred to here as status spongiosus) that primarily occurs in the white matter of the CNS but also extends into the gray matter. Status spongiosus is a somewhat nonspecific term and can develop by several different mechanisms. It includes a variety of lesions, such as splitting of lamella forming myelin sheaths (characteristic of the diseases discussed here), accumulation of extracellular fluid (extracellular cerebral edema; see section on Central Nervous System Swelling and Edema) swelling of cellular processes (astrocytic, neuronal), and Wallerian degeneration at a later stage when the necrotic myelin and axons have been phagocytosed and the spaces once occupied by these structures are empty.
Gross brain lesions reported for spongy degeneration range from no gross lesions to swelling, edema, and pallor of the white matter, and dilation of the ventricles. Microscopically, the lesion is characterized by variably sized empty spaces within the white matter. Ultrastructurally, with spongy degeneration and some other disease processes characterized by status spongiosus, there is splitting or separation of the myelin sheath at the intraperiod line with the formation of large intramyelinic spaces. In some cases, myelin formation is deficient.
Some species and breeds affected with spongy degeneration include the canine (Labrador retriever, Saluki, silky terrier, Samoyed), feline (Egyptian Mau), and bovine (Jersey, shorthorn, Angus shorthorn, Hereford), and an autosomal recessive mode of transmission has been proposed for some forms of this disorder. A unique form of the spongy degeneration also occurs in a group of metabolic inherited diseases called aminoacidopathies.
The term spongiform change should not be confused with spongy degeneration. Spongiform change is characterized by small clear vacuoles of varied sizes that form in the cytoplasm of neuron cell bodies and proximal dendrites in diseases, such as the transmissible spongiform encephalopathies and rabies encephalitis, and in the processes of astrocytes that are spatially related to the affected neurons.
Demyelination.
Demyelination, which means degeneration and loss of myelin already formed, can be divided into primary and secondary types. Primary demyelination refers to a disease process in which the myelin sheath is selectively affected, with the axon remaining essentially intact. Secondary demyelination, a designation criticized by some, refers to “secondary” degeneration of myelin after “primary” injury to and loss of the axon, as in Wallerian degeneration, and is not a selective injury of the myelin sheath.
Injury to oligodendroglia that results in myelin sheath breakdown or direct injury to myelin sheaths causes the release of lipids and other myelin components into the extracellular space. These materials readily activate microglial cells and attract blood monocytes, which phagocytose myelin debris.
Metabolic Causes
Osmotic Demyelination Syndrome.
In human beings, osmotic demyelination syndrome is termed central or extrapontine myelinolysis. The disorder was first reported in 1959, and the majority of cases were in severely malnourished alcoholics. Since then the disorder has been observed in a variety of clinical disease states. A major risk factor is chronic hyponatremia treated in hospitals by the administration of intravenous saline solution. The disease has been experimentally reproduced in dogs and laboratory rodents by inducing hyponatremia, allowing a period of stabilization (3 to 4 days is sufficient), and then administering saline-containing fluids.
Cases of osmotic demyelination syndrome are rarely reported in the veterinary literature. Several of the cases had Addison's disease with treatment consisting of intravenous administration of fluids containing normal saline solution to correct the hyponatremia typical of hypoadrenocorticism. The reported rates of correction were 22 mmol/L in 24 hours and 16.4 mmol/L in 24 hours. All of these rates of correction exceed the limits established for human beings and are consistent with human cases of the syndrome. Osmotic demyelination syndrome has also been reported in a cat and may have been due to poor nutrition.
The pathogenesis of osmotic demyelination syndrome is thought to be opposite to that occurring with salt poisoning (i.e., a hyponatremic to hypernatremic shift after saline administration). Lesions occur in areas of the brain in which there is a confluence or intermingling of gray and white matter. Rapid (within 24 to 48 hours) correction of chronic hyponatremia from an established equilibrium exceeds the adaptive responses of the brain, resulting in myelin destruction. The exact mechanism of demyelination is not known. It is proposed that osmotic imbalance and water shifts induce osmotic stress in the CNS that results in myelin destruction.
In contrast to human beings, gross lesions in dogs are either not apparent or subtle. Slight softening and discoloration has been observed in affected brain regions. Microscopically, lesions can be limited to the reticular formation at the level of the pons or can be extensive, affecting cerebellar folia, midbrain, thalamus, basal nuclei, and at the interface of the corona radiata and cerebrocortical gray matter. In affected areas the white matter is pale in routine H&E-stained sections and is heavily infiltrated with foamy macrophages. Special stains (Luxol fast blue for myelin) confirm acute myelin destruction and accumulation of myelin debris in macrophages. As is typical of strictly demyelinating lesions, axons are well preserved.
Circulatory and Physical Disturbances.
Physical compression of CNS tissue that results from various causes, usually chronic, can also induce demyelination. Some possible mechanisms include compression of myelin sheaths and oligodendroglia, interference with circulation resulting in CNS ischemia, and disease processes resulting in the accumulation of extracellular fluid.
It is well known that vasogenic and hydrostatic edema caused by inflammation, neoplasia, trauma, and obstructive hydrocephalus can cause degeneration of myelin sheaths. The underlying mechanisms for this injury are multiple and include creation of a hypoxic-anoxic environment, degeneration of oligodendroglia, and alteration in stability of the myelin sheath, permitting entrance of injurious proteolytic enzymes from the surrounding environment.
Diseases Caused by Microbes.
Progressive Multifocal Leukoencephalopathy.
See Table 14-3.
Immune-Mediated Diseases.
In veterinary medicine, naturally occurring immune-mediated demyelination in domestic animals is rare, and other than canine polyradiculoneuritis (coonhound paralysis), it is usually only suspected rather than proved. These diseases are mostly known to occur in human beings as sequelae to postinfectious and postvaccinal events. They result in primary demyelination of the CNS. Autoimmune diseases of the CNS are mechanistically either a type II hypersensitivity (antibody-mediated) or a type IV (cell-mediated) hypersensitivity, and T lymphocytes and macrophage-derived cytokines play contributory roles.
Autoimmune injury to oligodendroglia in the CNS arising from aberrant cellular and/or humoral immune responses can result from one of the following four proposed mechanisms:
-
1.
Molecular mimicry: The CNS has antigens that are similar or identical to those expressed by certain pathogens (virus or bacterium). The normal inflammatory and immunologic responses to these pathogens result in the expression of antibodies that cross-react with “antigens” normally expressed by CNS cells.
-
2.
Abrogation of immune tolerance: The CNS is an “immune privileged” organ (like the eye). The immune system therefore does not recognize CNS antigens as innate antigens, and if they are exposed to the immune system after inflammation or trauma, an autoimmune response can ensue. Injury, physical or otherwise, to blood vessels within the CNS can cause the release of “sequestered antigens” into the bloodstream, leading to an autoimmune response.
-
3.
Genetic factors: Functions of the immune system that are strictly regulated genetically may be under the control of abnormal inherited genes or altered normal genes that regulate immune responses to CNS antigens and thus increase the susceptibility to autoimmune diseases.
-
4.
Stress factors: Environmental stresses mediated through the CNS can depress the functions of the immune system, leading to the formation of autoantibodies.
The mechanism of myelin breakdown in immune-mediated demyelination is not clearly understood. The initial step is thought to be exposure of antigens in myelin basic protein of the major dense line of myelin lamellae after injury. Myelin proteins are substrates for calpain, a calcium-activated neutral proteinase. Calpain has been implicated in several autoimmune diseases and may play an important role in CNS demyelination. Exposed antigens are then recognized by the immune system, and experimental studies suggest that lesions result from a complex interaction between inflammatory cells and their mediators and lamellae of myelinating cells. T lymphocytes, some B lymphocytes, and activated macrophages (recruited monocytes) and microglia cells, adhesion molecules, cytokines, chemokines, and their receptors have been demonstrated in the lesions. This interaction results in primary demyelination.
Gross lesions are usually not present but can include a gray-to-yellow discoloration of white matter. Microscopically, lesions are best observed in white matter and are characterized by vacuolation of myelin and the presence of lymphocytes, macrophages, and plasma cells. Myelin sheaths degenerate from injury caused by (1) inflammatory mediators and (2) direct actions of macrophages on lamellae. Myelin lamellae separate as a result of intramyelinic edema, fragment, and are phagocytosed by macrophages.
The best-known model of immune-mediated demyelination is an experimental model referred to as experimental allergic encephalomyelitis (EAE). Experimental allergic encephalomyelitis is produced by inducing a hypersensitivity to myelin or more specifically, to myelin basic protein. If appropriate laboratory animals are inoculated with white matter or myelin basic protein (suspended in complete Freund's adjuvant), they become paralyzed after 2 to 3 weeks. Lesions are characterized by perivascular (perivenular) demyelination accompanied by accumulation of lymphocytes and macrophages.
A similar process, referred to as postvaccinal encephalomyelitis, occurred occasionally in human beings when human rabies vaccine contained CNS tissue. The incidence decreased after 1957 when duck embryo rabies vaccine came into use. In some cases with mild clinical signs, recovery was complete, and axons were remyelinated after immune-mediated demyelination.
A third situation in which this type of demyelination occurs follows infection with certain viruses in human beings (rubeola virus) and animals (e.g., influenza virus). These diseases, which are rare and designated as postinfectious encephalomyelitis, are also characterized by development of lesions in the CNS comparable to those of experimental allergic encephalomyelitis.
Traumatic Injury
Traumatic injury of the CNS is caused by physical insults such as compression, stretching, and/or laceration of neurons/axons. When the brain and spinal cord collide with the bony ridges lining the cranial vault and the bony wall of the vertebral canal, respectively, or when axial, rotational, and angular forces (Fig. 14-66 ) are applied to neurons and axons during trauma, the force of impact and sudden acceleration of neurons and axons, both within the CNS and in the adjacent cranial and spinal nerves, can cause them to compress, twist, stretch, and tear. Concurrently, the same type of forces can injure blood vessels in the CNS and leptomeninges and may result in small hemorrhages or hematomas within the parenchyma of the brain and in the leptomeninges (subarachnoid space) (Fig. 14-67 ).
Figure 14-66.
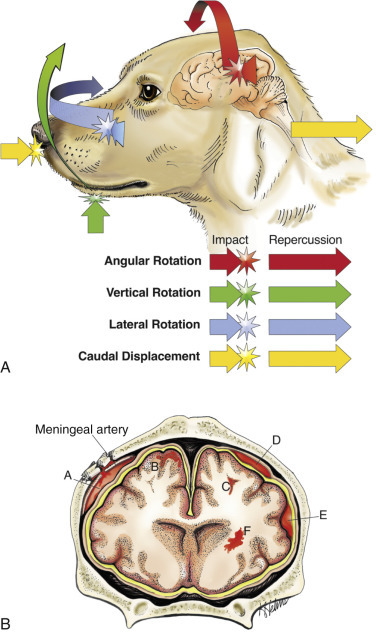
Traumatic Central Nervous System Injury and Hemorrhage.
A, Axial, rotational, and angular energy applied to the brain during trauma determines the severity of shear, tensile, and compressive forces that cause neuronal and vascular injury. B, Locations of hemorrhage, dog, brain. Epidural hemorrhage with laceration of meningeal artery (A); cortical hemorrhage (B); hemorrhage in subcortical white matter (C); subdural hemorrhage secondary to laceration of a bridging vein (D); subarachnoid hemorrhage (E); deep intracerebral hemorrhage (F).
(A courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. Redrawn and modified from an illustration in Leech RW, Shuman RM: Neuropathology: a summary for students, Philadelphia, 1982, Harper & Row.)
Figure 14-67.
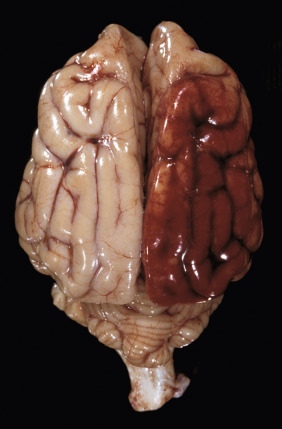
Leptomeningeal (Subarachnoid) Hemorrhage, Brain, Right Cerebral Hemisphere, Dog.
(Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Diffuse brain injury (often present in concussion) is caused by acceleration/deceleration forces applied to many areas of the CNS rather than in one specific location. Diffuse brain injury involves neuronal processes, cell bodies, transmitter mechanisms, and macroglial cells and blood vessels. The most severe injury to axons appears to be at the gray matter–white matter junction. A variant of diffuse brain injury is diffuse axonal injury in which axons of large myelinated nerve fibers are injured by shearing forces. In diffuse brain and axonal injury, mechanical deformation results in physical disruption of cell membranes and cytoskeleton and increased membrane permeability, resulting in major ionic fluxes in and out of the cell. Such changes can lead to excessive release of glutamate, excitotoxicity and cell death, free radical formation, apoptosis, and delayed inflammatory responses. The end result can be Wallerian degeneration.
In general, trauma of the CNS in animals occurs less frequently than in human beings. Animals are not exposed as frequently to potentially trauma-causing situations (e.g., automobile travel) as are human beings, and there are anatomic differences (see later), including a quadruped posture, which increases stability and helps protect the brains of animals.
Among animals, trauma to the brain is probably most frequently caused in dogs as a result of automobile-induced injury and in cats from falls from significant heights (high-rise apartment buildings, balconies, and roofs). Even after falling from considerable heights, cats often have remarkably minor injury to the CNS. Other examples include fracture of the spinal column or cranium of jumping horses and fractious animals, such as horses and ruminants, during excitement and restraint.
Predisposition to cerebral trauma is also influenced by anatomic differences. The percentage of brain mass in relation to skull size is much less in domestic animals than in primates, and in the bovine and porcine species the cranial cavity is additionally protected dorsally by prominent frontal sinuses. Birth trauma, which can be important in human beings, is essentially insignificant in animals because in the latter, the shoulders and particularly the pelvis, rather than the head, are likely to be compressed in the birth canal. Exceptions to this generalization include brachycephalic breeds of dogs. Several factors also influence the susceptibility of the spinal cord to trauma. The amount of space between the spinal cord and the wall of the vertebral canal is very important in determining the degree of injury after edema or compression with disk herniation. This space is greater in the cervical area of the dog than at the thoracolumbar level. Thus disk herniation at the latter area is more likely to result in severe spinal cord injury.
Functional factors also play an important role in brain injury. The brain of a freely movable head is much more susceptible to injury than one that is fixed in place. The increased susceptibility of the former has been attributed to the ability of the cranium (the bone) and its contents (the brain) to impact each other after nonpenetrating trauma. This interaction occurs because the brain does not completely fill the cranial cavity, thus resulting in a very short distance (or space) between brain and bone.
The type and location of the lesion (contusion and/or hemorrhage) depends on the location of the point of contact and the direction of the blow, relative to the head. If the blow is directly on the back or front of the head, the head and brain will move straight forward or backward, respectively (see Fig. 14-66, axial force). If the blow is horizontal to the top of the head or horizontal to the rostral portion, the head will rotate on the atlantooccipital axis (angularly and rotationally, respectively). In the case of an axial blow to the back of the head (an animal falling onto the back of its head), the head and thus the cranial vault will accelerate faster than the brain, which will lag behind, and the caudal aspect of the vault may move forward and contact the caudal aspect of the cerebral hemispheres, usually the occipital cortex. In the case of an animal falling onto the back of its head, the head will accelerate abruptly and the momentum will carry the brain caudally, where it may strike the inside of the caudal brain in the cranial vault. A vertical blow delivered directly down onto the dorsal surface of the head will have the same type of result on the dorsal aspect of the cerebral hemispheres as the blow to the back of the head. It is more common, from the same blows described previously, to see hemorrhage, usually subarachnoid, on the opposite side of the point of impact with the brain. For example, a vertical blow to the top of the head causes hemorrhage of the ventral surface of the medulla and cerebral hemispheres. Thus after an impact on a stationary, freely movable head, the bone of the cranial vault will move on the stationary brain and injure it (coup injury) and on the opposite side the nerves and blood vessels will be stretched, possibly resulting in nerve damage and hemorrhage (contrecoup injury). In addition, the mass and velocity of the object striking the head are important. Trauma after impact of a relatively large blunt object can create notable head movement and a large-impact injury, whereas a small object, such as a bullet moving at a high rate of speed, can cause less head movement and a smaller but deeper area of direct tissue damage. In summary, the basic concept is the transfer of kinetic energy by the striking object to the head. A large blunt object will cause the head to accelerate without deforming it; a smaller object, such as a bullet, will penetrate.
Factors involved in the protection of the brain include the rigidity of the cranium (depending on age), the round shape of the dorsum of the skull, the structure of the parietal, occipital, and temporal cranial bones (two layers of compact bone separated by spongy bone referred to as diploë), cranial sutures, sinuses, ridges in the floor of the cranial cavity, meninges, and CSF. The spinal cord is enclosed and protected by the vertebral column, which is surrounded by soft adipose tissue and muscle. Other structures that help protect the spinal cord by absorbing shock are the intervertebral disks and the cancellous bone of the vertebrae. Vertebral ligaments maintain the alignment of the vertebral column; denticulate ligaments support the spinal cord in the middle of the vertebral canal, and the meninges, particularly the CSF, cushions trauma.
Clinically, animals with CNS trauma have signs referable to the area injured, brain, or spinal cord. With brain trauma, signs can vary widely and range from unconsciousness lasting a few seconds followed by complete recovery and return to normal function to depression, abnormal behaviors such as disorientation and irritability, semiconsciousness with responsiveness only to noxious stimuli, and unconsciousness with no response to any stimulus. With spinal cord trauma, signs vary, depending on the severity of the injury and the rate of onset. Paralysis results from severance of the cord or ruptured disks. Paresis and ataxia result from less severe injury.
Concussion.
Concussion is often thought of as a clinical designation of temporary loss of consciousness with recovery after head injury. As in human beings, a movable head is much more susceptible to trauma than a fixed, supported one. Application of an appropriate concussive trauma to the mobile head of an animal results in a reversible cerebral dysfunction that lasts for a matter of seconds or at most a few minutes and is usually reversible, with stronger blows causing more severe injury and even death.
Concussive injuries of the diffuse type also occur in animals, but there are some differences between animals and human beings. For example, it is difficult to produce severe concussion in animals because the margin between the force of a stunning blow and one causing fatal injury is very small. The smaller the brain, the less vulnerable it is to rotational forces and the larger are the forces necessary to cause concussion. It should be noted, however, that concussion, particularly when there is rapid recovery from unconsciousness, can occur more frequently than appreciated in animals because the clinical signs may not be recognized.
Diffuse brain injury does not usually cause gross lesions. Microscopic lesions detected in animals include diffuse axonal injury characterized by axonal degeneration, and this may be followed by Wallerian degeneration. Damage to neurons ranges from central chromatolysis to death and neuronal loss. The more severe forms of diffuse brain injury can also have generalized acute brain swelling caused by unregulated vasodilation, which can be followed after some time by cerebral edema.
Spinal concussion is the term applied to the immediate and temporary loss of function that sometimes follows severe direct blows to the spinal column. Loss of function usually affects the long tracts/bundles of nerve fibers (funiculi), but usually there is no demonstrable external change in the vertebrae or spinal cord. As with cerebral concussion, there is often only a temporary functional disability of the cord after injury, but if the trauma is more severe, permanent neurologic deficits can result.
Contusion.
Contusion means bruising, which is generally associated with rupture of blood vessels, and in the cerebrum, this injury results in grossly detectable lesions, such as hemorrhage, which can, like concussion, result in unconsciousness and even death. The factors that cause concussion and contusion can occur together in the same animal. Lesions can be superficial (cerebral gyri) or more central (brainstem), and there can be concurrent skull fractures.
Although hemorrhage is the most common lesion, contusion of the brain can also result in tearing of CNS tissue. Tearing results in tissue necrosis and neuronal loss. Two designations are used to identify the location of contusive injury. A coup contusion is located at the impact site, and a contrecoup contusion at a location on the opposite side of the brain. When the two lesions occur together (coup-contrecoup or contrecoup-coup), the first term indicates the site of most severe injury. Box 14-10 summarizes the pathogenesis of coup-contrecoup contusion.
Box 14-10. Pathogenesis of Coup-Contrecoup Contusion.
Conditions Involved in Coup-Contrecoup Contusion
-
1.Head freely movable.
-
2.Head accelerated rapidly (by being struck by a broad object, such as an automobile) or decelerated rapidly (head strikes pavement after a fall from a standing position).
-
3.Because the brain does not fill the cranial vault, it may lag behind the movement of the cranium when the head is accelerated or decelerated rapidly.
-
4.As a result, the inside of the cranial vault may strike the stationary brain at the point of impact (coup injury), or the lesion may occur on the opposite side (contrecoup), either from the stretching and tearing of vessels at that site or by the brain being struck by the inside of the cranial vault on the opposite side when there is reduced amount of cerebrospinal fluid buffer present.
-
2.
Many investigations have been made to determine the mechanisms involved in the development of contusive lesions, and the kinetics are complicated and still not completely resolved. Factors considered to be significant include the ability of the head to move freely, the occurrence of a rotational movement of the brain over rough surfaces on the inside of the cranial vault, and the development within the cranial cavity of positive and negative pressures and gravitational forces. The basic results of the different types of blows to the heads of animals have been discussed previously, and it is interesting to compare these with the lesions in human beings. Several neuropathologic principles have been generally accepted regarding craniocerebral contusive trauma in human beings, as follows:
-
1.
A blow to the stationary (but freely movable) head produces a cerebrocortical coup contusion beneath the point of cranial impact, but with rare exceptions causes no cerebrocortical contrecoup contusion opposite the point of cranial impact. This outcome is not always true of animals, in which a blow to the dorsum of the head from a flat object, such as a spade, causes marked subarachnoid hemorrhage on the ventral surface (contrecoup).
-
2.
An impact of a moving head (moving before impact, as in a fall from a standing position) against a firm or unyielding surface causes a cerebrocortical contrecoup contusion opposite the point of cranial collision (often at the poles and inferior surfaces of the frontal and temporal lobes), but with rare exceptions there is no contusion beneath the point of impact. In contrast, horses that fall backward and land on their backs and strike the occiput often have subarachnoid hemorrhage over the occipital poles of the cerebral hemispheres.
-
3.
Falls from great heights and crushing of the head between a strong external force and unyielding surface are generally not associated with the occurrence of contrecoup lesions.
Dawson and coworkers proposed a mechanism for both contrecoup and coup injury of the human brain that also addressed the specific deficiencies of mechanisms that have been advanced by others. An example explaining the mechanism of contrecoup injury states that when a person falls backward from a standing position because of loss of balance, the gravitational torque acting on the body causes downward acceleration of the head in excess of the acceleration as a result of gravity. Under these circumstances the brain lags toward the trailing anterior surface of the cranium before impact (causing displacement of the protective CSF layer between the brain and skull) and permits compressive stress to develop at this site, although impact occurs at the opposite side of the head. Because dissipation of the CSF at the anterior (contrecoup) site allows compressive stress to be focal at this contrecoup site and because of the shearing stress generated, injury occurs. In addition, a relative rotational gliding motion between the brain and skull is produced when the impact suddenly stops the skull's motion and rotation, thus creating an additive shearing stress because the fluid lubrication necessary to facilitate gliding of the brain over the cranial surface is reduced. The concentration of this rotational shearing stress is likely to occur beneath the frontal and temporal lobes because of the rough surface of the skull that exists in this location. In contrast to contrecoup injury, coup contusions occur infrequently in this type of fall. In such situations the brain lags away from the impact site, which results in a thickening of the protective CSF layer between the brain and skull immediately beneath the point of impact, which helps explain the absence of coup injury in primates and human beings in typical moving head trauma.
Coup injury in human beings can occur when a stationary but freely movable head is impacted. In this type of trauma there is neither brain lag nor disproportionate distribution of CSF before impact, which accounts for the typical absence of contrecoup contusions. With regard to falls from great heights, the dynamics involving rotation of the body about a fixed point of ground contact associated with a fall from a standing position do not occur. Because gravity produces no torque on a freely falling object, no angular acceleration of the body is produced; therefore such a fall is a true free-fall state that is associated with an absence of brain lag. For this reason, contrecoup lesions occur infrequently with this type of trauma. One point should be emphasized with regard to the evaluation of coup and contrecoup cortical contusions just described. Displacement of bone associated with skull fracture can contuse the subjacent brain, regardless of the resting or moving status of the head, and such fracture-contusions have nothing to do with the coup-contrecoup mechanisms described. Also, even though the basic mechanisms discussed earlier apply to human beings, they should also be considered when evaluating cerebral contusions of domestic animals, but the situation in human brains is made far more complex because of the numerous wide bony ridges that project into the cranial vault.
Evaluation of spinal cord trauma should include examination not only of the spinal cord but also of the vertebral column and spinal nerve roots. Injuries to the spinal cord can involve concussion, contusion, hemorrhage, laceration, transection, and compression secondary to vertebral trauma and fracture. Contusion in the spinal cord is characterized by vascular tears, hemorrhage, and necrosis. Tears are generally focal and grossly visible. Contusion can occur without fracture of the vertebral column, with fracture, and with fracture plus dislocation of the spinal column. The latter combination can result in tearing and transection of the spinal cord.
In the acute phase of spinal cord trauma, contusions are microscopically identified by regions of perivascular hemorrhage. This lesion occurs rapidly and over time progresses to include blood diffusing into adjacent cortical tissue, where, if enough time lapses, reactive astrocytes and infiltration of macrophages occur. If allowed to heal, a region of gliosis and hemosiderin pigment is commonly found in these regions.
Central Nervous System Hemorrhage.
Although hemorrhage and hematomas in animal brains can be caused by a wide variety of injuries, trauma to the head is the most common cause. (Box 14-11 lists common causes of brain hemorrhage.)
Box 14-11. Common Causes of Hemorrhage in Animal Brains.
Vasculitis (such as Histophilus somni infection; angioinvasive fungal infections)
Damage to endothelium lining blood vessels (by the virus of canine infectious hepatitis, by septicemia or endotoxemia, by immune complexes, or by parasite larval migration)
Trauma
- Contusion
-
•Coup lesion
-
•Contrecoup lesion
-
•
Penetrating wounds
After trauma to the head, hemorrhages can develop in the epidural, subdural, and subarachnoid locations, under the pia mater (subpial), and in the brain (see Fig. 14-66, B). Hemorrhage can be diffuse (see Fig. 14-67) or focal (e.g., hematomas) (Fig. 14-68 ). Such hemorrhages can result from sliding of the brain over bony ridges within the cranium, with resultant stretching and tearing of blood vessels and tissue, after the cutting and penetration of bone fragments from skull fracture. Cerebral epidural hemorrhage, which is not commonly described in animals, has been reported in the horse, especially jumpers, resulting from falls while working. Epidural hemorrhage does not usually occur because the dura is tightly adhered to the inner surface of the calvaria and there is no epidural space. In trauma causing skull fractures, bleeding from local blood vessels can separate the dura from the calvaria, forming a hematoma in the dural space.
Figure 14-68.
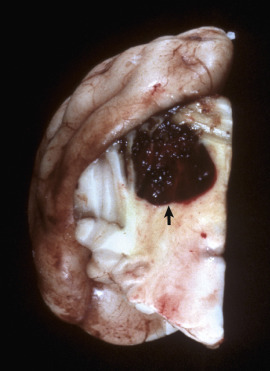
Hematoma, Cerebellum, Transverse Section, Dog.
Traumatic injury to the head resulted in hemorrhage and the formation of a hematoma (arrow) from shear, tensile, compressive axial, rotational, and angular forces.
(Courtesy Dr. H.B. Gelberg, College of Veterinary Medicine, Oregon State University.)
Subdural hemorrhage, which is an extravasation of blood between the dura mater and the cortex, occurs in dogs and cats. It is rare and usually diffuse and does not commonly organize into focal hematomas as seen in human beings, where they can be life threatening from compression and herniation of the brain. Subarachnoid and intracerebral hemorrhages are most common in all species after head injury (Fig. 14-69 ). Hemorrhage can result from injury to the brain with or without a fractured cranium by the mechanisms given previously and also from penetrating objects (bullets and stab wounds).
Figure 14-69.
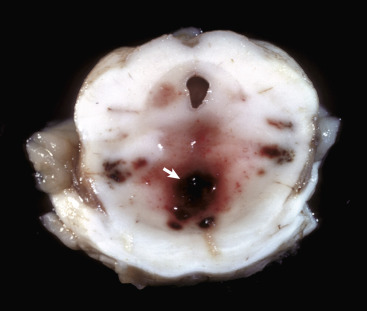
“Duret” Hemorrhages, Brainstem, Transverse Section at the Level of the Mesencephalon.
Note the multiple hemorrhages in the ventral mesencephalon (arrow). These hemorrhages are the result of twisting of the brainstem on a longitudinal axis from rotational and axial forces.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The same types of hemorrhage that affect the brain (epidural [rare], subdural [rare], and parenchymal) also occur in the spinal cord and its meninges. Causes are similar to those for the brain.
Hematomyelia (Hemorrhagic Myelomalacia).
Traumatic injury to the spinal cord can cause stretching and tearing of blood vessels, usually arterioles, within the gray matter, resulting in hematomyelia. Hematomyelia is also particularly associated with severe type I disk herniation. If larger vessels are torn, blood pressure can force blood into the gray matter. This outcome results in the formation of a dissecting blood-filled cavity ascending and/or descending initially within the gray matter of the spinal cord. This lesion, which is characterized by a softening to semiliquefaction (myelomalacia) and hemorrhage of the tissue, can develop within 12 to 24 hours after injury and can progress both cranially and caudally from the original site of trauma. As the cavity extends cranially, the hemorrhage at the original site also extends into the white matter and can transect the spinal cord. If this lesion extends to the fifth cervical cord segment, the phrenic nerves to the diaphragm will be denervated and respiratory paralysis will result. Bleeding continues until pressure in the blood-filled cavity is equal to the vascular pressure or until bleeding ceases because of hemostasis in the vessel. Hemorrhage can also result from bleeding in arteriovenous malformations within the spinal cord. Hematomyelia is characterized by neurologic deficits consistent with a sudden onset of ascending or descending flaccid paralysis and sensory abnormalities.
Compressive Injury.
Diseases resulting in compressive injury can affect the brain, spinal cord, or both concurrently. In the brain, diseases, such as neoplasia, reticulosis, canine granulomatous meningoencephalitis, and chronic cerebral abscesses, can compress adjacent nervous tissue. In the spinal cord, compression can be intramedullary (within the spinal cord) or extramedullary (outside the spinal cord). Causes of intramedullary compression include hemorrhages, neoplasms such as nephroblastoma of the young dog, and chronic expansile inflammatory diseases. Extramedullary compression can be caused by intervertebral disk herniation in the dog; cervical stenotic myelopathy (wobbler syndrome) in the horse and dog; vertebral fracture and dislocation; neoplasms of the meninges, such as the meningioma; nerve rootlets, such as nerve sheath tumors; or tumor metastasis, such as lymphosarcoma. Finally, developmental anomalies of bone, such as atlantooccipital malformation, and vertebral deformities with hemivertebrae, such as scoliosis, lordosis, and kyphosis (Fig. 14-70 ), can result in compression of the spinal cord.
Figure 14-70.
Vertebral Abnormalities, Vertebral Column.
A, Scoliosis, thoracic vertebrae, ventrodorsal view, sheep. Lateral deviation of the spinal column. B, Kyphosis, thoracolumbar vertebrae, lateral view, sheep. Dorsal deviation of several vertebrae and a wedge-shaped vertebra (hemivertebra; C) have resulted in compression of the spinal cord. C, Hemivertebra, lumbar vertebrae, dog. Note that the craniodorsal portion of the body of the hemivertebra has protruded into the vertebral canal.
(A and B courtesy College of Veterinary Medicine, University of Illinois. C courtesy Department of Veterinary Biosciences, The Ohio State University.)
Compression of CNS tissue causes neuronal dysfunction by impeding normal anterograde and retrograde axoplasmic flow in axons (see E-Fig. 14-3). In addition, compression of nerves may result in reduced blood flow to nerves and thus also contribute to neuronal dysfunction. Mild compression can result in partial blockage of slow axoplasmic flow and gradual accumulation of neurofilaments and microtubules, which results in mild enlargement of the axon proximal to the compression site and atrophy of the axon distal to the compression. Eventually, with a long period of time of complete blockage, the distal axon is lost.
Brain Displacements.
See the discussion of cerebral edema (permeability changes) in the section on Central Nervous System Swelling and Edema.
Cervical Stenotic Myelopathy.
Cervical stenotic myelopathy, or wobbler syndrome, is characterized by stenosis of the cervical vertebral canal, which causes compressive trauma to the cervical spinal cord (Fig. 14-71 ). This disease occurs primarily in young rapidly growing large breeds of horses and dogs. Reports indicate that the disease is not caused by a straightforward mechanism but apparently involves several factors (multifactorial disease). For example, stallions that have a genetic predisposition for rapid growth and large body size are reported to be at greater risk for developing the disease. Oversupplementation with protein, vitamins, and minerals used to promote rapid growth may also be another environmental factor in developing cervical stenotic myelopathy.
Figure 14-71.
Cervical Stenotic Myelopathy.
A, Cervical static stenosis, vertebral column, sagittal section, sixth cervical vertebra, horse. The vertebra on the bottom is stenotic (arrows) and will compress the spinal cord. The vertebra on the top is normal (arrows). B, Cervical vertebral instability, spinal cord, fifth cervical segment, dog. Note the narrowing of the spinal cord at the site of compression (arrow).
(Courtesy College of Veterinary Medicine, University of Illinois.)
Gross and microscopic lesions of the CNS in cervical stenotic myelopathy are similar to the lesions in intervertebral disk herniation. The severity depends on the speed and degree to which the compression is applied and the specific area of the cord that is involved. Central nervous tissue can tolerate a considerable degree of compression if it is applied slowly. Rapid compression can lead to quickly developing hypoxia-ischemia, necrosis, and direct damage to compressed axons. Lesions detected include a spectrum of changes characterized by axonal injury and disruption of myelin sheaths, resulting in Wallerian degeneration and necrosis of the gray or white matter or of both. Lesions can be visible grossly from the exterior but are more commonly seen on cross section of the spinal cord.
Microscopically, particularly at the site of injury, there is initial swelling of axons followed after several days by loss of architecture of the CNS as a result of necrosis and a beginning accumulation of gitter cells that have phagocytosed the lipid-rich tissue debris. Eventually the necrotic area is cleared and a cystic space is formed, which is surrounded by varying degrees of astrocytosis and astrogliosis, although not usually prominent unless there is severe destruction. Rostral and caudal to this location, the lesion is primarily one of Wallerian degeneration in the white matter, and the pattern of lesion development seen depends on the level of the spinal cord that is examined relative to the site of compression. At the site of injury, all parts of the white and gray matter of the spinal cord are affected and often necrotic, if the compressive force is sufficient. Rostral to this site, white matter degeneration is generally limited to the ascending tracts in the dorsal funiculi and the superficial portions of the dorsolateral part of the lateral funiculi. Caudal to the area of injury, degeneration is limited to the descending tracts in the ventral funiculi, and the more central portions of the lateral funiculi. It should also be noted that (1) a lesion may occur at the point of compression because of ischemia, but a lesion can also occur on the opposite side of the compression also because of ischemia that results from compression of the tissue against bone on that side, and (2) Wallerian degeneration can be observed in distal segments of affected axons far from the point of compression within the spinal cord. In the former case, compressive forces can be transferred through the spinal cord to axons and blood vessels away from the contact point, whereas in the latter example, degeneration of the distal axon segments can extend for a length of centimeters to meters away from the point of contact.
Cervical stenotic myelopathy has been known for many years to affect horses and more recently has been recognized in the large dog breeds. The disease in the horse has been referred to by several designations: the wobbler syndrome, wobbles, equine incoordination, and more recently, cervical stenotic myelopathy. The disease has been described in many horse breeds.
Cervical stenotic myelopathy in the horse has been divided into two syndromes: cervical static stenosis and cervical vertebral instability (dynamic stenosis). Cervical static stenosis commonly affects horses 1 to 4 years of age. The spinal cord is compressed at C5 through C7 as the result of an acquired dorsal or dorsolateral narrowing of the spinal canal (see Fig. 14-71). The stenosis is due to formation of bone that requires time to develop. The compressive effect with this type of stenosis is present regardless of head position. The second form of cervical stenotic myelopathy (cervical vertebral instability [dynamic stenosis]) occurs in horses ranging in age from 8 to 18 months and is characterized by a narrowing of the spinal canal during flexion of the neck, primarily at C3 through C5 vertebrae.
A disease process with many similarities to that in the horse also affects the dog. It has been known as wobbler syndrome, vertebral instability, vertebral subluxation, and cervical spondylolisthesis. The disease has been most frequently described in the Great Dane and Doberman pinscher breeds but has also been reported in the Saint Bernard, Irish setter, fox terrier, basset hound, Rhodesian ridgeback, and Old English sheepdog. Dogs can have signs develop between 8 months and 1 year of age, with a range of 1 month to 9 years. Great Danes tend to have lesions develop at a young age (8 months to 1 year), whereas Doberman pinschers are generally older, often more than 1 year of age. The vertebral and associated spinal cord lesions in the dog have been reported to most often involve the caudal cervical area from C5 through C7 vertebrae. An exception is the basset hound, in which C3 is affected.
Tumors.
In the brain, diseases, such as neoplasia, cause compression of adjacent nervous tissue. Compression of CNS tissue causes neuronal dysfunction by impeding normal anterograde and retrograde axoplasmic flow in axons (see the later discussion of specific types of CNS tumors).
Leptomeningeal Hemorrhage.
Physical trauma to the CNS compresses, twists, and stretches blood vessels until they are torn. Such injury results in bleeding and leptomeningeal (subarachnoid) hemorrhage (see Fig. 14-67). In the spinal cord, laceration of a large artery or vein can result in prolonged bleeding that ascends or descends the leptomeninges, causing neurologic dysfunction on examination compatible with ascending or descending neurologic deficits in spinal nerve roots.
Tumors
Neoplasms of the CNS in domestic animals occur most frequently in dogs, and at frequency and variety similar to human beings. These tumors share many gross and histologic features with their human counterparts. In human neuropathology, brain tumors are typically graded according the World Health Organization classification of tumors of the CNS (2007). This human grading scheme is being increasingly applied to brain tumors of domestic animals, most notably the dog; however, its usefulness and applicability in animal species needs to be established. The majority of neoplasms described have been in dogs and cats, and a large portion of these tumors occur in the older population. The intention in discussing CNS neoplasms in this chapter is to present a brief overview of the more common or better-known neoplasms that occur in animals and is not meant to be all-inclusive. The location and characteristics of the primary and common secondary (metastatic) tumors of the nervous system are summarized in Table 14-9 . Clinical signs in animals with intracranial tumors (e.g., gliomas, choroid plexus tumors) vary, depending on the location of the tumor in the CNS, but can include behavioral changes, ataxia, tetraparesis, seizures, circling, and abnormal cranial nerve and proprioceptive reflexes. The average age of affected dogs ranges from 6 to 12 years, and certain dog breeds are predisposed including brachycephalic breeds (e.g., boxer, Boston terrier) and golden retrievers.
Table 14-9.
Primary Tumors of the Nervous System
| Cell of Origin | Tumor Type | Location | Species Affected | Gross Appearance |
|---|---|---|---|---|
| CNS | ||||
| Embryonal cell | Medulloblastoma | Cerebellum (WM&GM) | Dog, cow, cat, pig (young animals) | Well-circumscribed, soft, gray to pink expansile mass (usually does not have hemorrhage, cysts, or necrosis). |
| Oligodendroglia | Oligodendroglioma | Cerebrum, brainstem, interventricular septum (WM&GM) | Dog, cat, cow | Well-demarcated, gray to pink-red, soft or gelatinous expansile mass, usually with areas of hemorrhage. |
| Astroglia | Astrocytoma | Pyriform lobe, cerebral hemispheres, thalamus-hypothalamus, midbrain, cerebellum, spinal cord (WM&GM) | Dog, cat, cow | Poorly demarcated, firm, gray-white when well differentiated; anaplastic neoplasms are moderately to well-demarcated, soft and friable (usually with areas of necrosis, hemorrhage, edema, and cavitation). |
| Ependyma | Ependymoma | Ventricular system (lateral; less commonly, third and fourth), central canal of the spinal cord | Dog, cat, cow, horse | Poorly to moderately to well-demarcated, soft and gray-white, invasive, destructive gelatinous expansile mass (usually with areas of hemorrhage and cavitation). |
| Choroid plexus epithelium | Choroid plexus tumor | Ventricular system (fourth; less commonly, third and lateral) | Dog, horse, cow | Well-demarcated, granular to papillary, gray-white to red, expansile mass. |
| Microglia | Microgliomatosis | Cerebrum, brainstem | Dog | Poorly demarcated, gray-white, infiltrative mass (may have perivascular pattern). |
| Endothelium | Hemangiosarcoma | Cerebrum, brainstem | Dog | Well-demarcated, invasive red to dark red, expansile mass. |
| Arachnoid cells | Meningioma | Meningeal surface of the CNS (convexity and lateral surfaces of the cerebral hemispheres, tentorium cerebelli, flax cerebri, ventral brainstem, spinal cord) | Cat, dog, horse, cow, sheep | Well-demarcated, variable shapes, firm, encapsulated, and gray-white to soft, red-brown or gray, expansile masses (usually with areas of hemorrhage and necrosis). |
| PNS | ||||
| Schwann cells (other supportive cells) | Peripheral nerve sheath tumor* | Cranial nerves, spinal nerves (brachial plexus) | Dog, cow, cat | Firm or soft (gelatinous), white or gray, nodular masses. |
CNS, Central nervous system; PNS, peripheral nervous system; WM&GM, white matter and gray matter.
Other names for this tumor type include schwannoma, neurofibroma, and neurilemmoma.
Embryonal or Primitive Neoplasms.
Considering the complexities of brain development and the fact that astrocytes and oligodendrocytes arise from common stem cells early in development, it should not be surprising that some neoplasms arising in the CNS have multiple lines of differentiation as indicated by immunohistochemical analysis. One such neoplasm is the rare primitive neuroectodermal tumor. These tumors have been described sporadically in cattle, horses, and dogs. They are found most commonly in young animals and are considered aggressive biologically. Histologic features include poorly differentiated embryonal cells with Homer Wright rosettes and a relatively high mitotic rate. Immunohistochemical staining for specific markers confirms multiple lines of differentiation such as neuronal, astrocytic, and oligodendroglial. Typically, one cell type dominates, most commonly neuronal.
Other uncommon embryonal tumors include medulloblastoma, ependymoblastoma, and malignant rhabdoid tumor. Like primitive neuroectodermal tumors, medulloblastomas are most common in young animals. They have been reported in horses, cattle, pigs, dogs, and cats and arise in the cerebellum, usually in close proximity to the vermis, and invade into adjacent structures. The cell of origin for the medulloblastoma has not been definitely determined. It has been proposed that the neoplasm arises from primitive cells originating in the neuroepithelial roof of the fourth ventricle that give rise to the external granule cell layer. Medulloblastomas are well circumscribed, soft, gray to pink masses and usually do not cause hemorrhage and/or necrosis or form cysts. Their expansile growth can compress the fourth ventricle and cause obstructive hydrocephalus and can also infiltrate adjacent structures, including the leptomeninges, and metastasize throughout the ventricular system. Histologic features, including Homer Wright rosettes and a high mitotic rate, are similar to those occurring in other embryonal tumors described in the veterinary literature; however, the neoplastic cells are typically small and round with variably elongate nuclei and ill-defined cytoplasm. Ependymoblastomas and rhabdoid tumors are extremely rare embryonal tumors that have been only sporadically reported and have not been adequately described in the veterinary literature.
Astrocytomas.
Astrocytomas have been morphologically classified based on their degree of differentiation (histologic features in H&E-stained sections) and include the following three types: diffuse astrocytomas, anaplastic astrocytomas, and glioblastoma multiforme. The degree of differentiation refers to how closely astrocytes forming the tumor resemble normal astrocytes within the CNS. Diffuse astrocytomas tend to have the most well differentiated astrocytes, whereas glioblastoma multiforme has the most poorly differentiated astrocytes. All of these tumors are malignant; however, the degree of malignancy is often inversely related to the degree of differentiation.
Astrocytomas have been reported most commonly in dogs (10% to 15% incidence) and cats, and rarely in horses, cattle, and pigs. Brachycephalic breeds, such as Boston terriers and boxers, and dogs 5 to 11 years of age are most commonly affected. Common sites include the cerebral hemispheres, especially the temporal and pyriform lobes, thalamus-hypothalamus, midbrain, and less frequently, the cerebellum and spinal cord.
Astrocytomas often displace normal tissue; however, their gross appearance often depends on their rate of growth and degree of differentiation (Fig. 14-72 ). Slow-growing, well-differentiated astrocytomas (less malignant) are usually difficult to distinguish from normal tissue and are rather solid or firm and gray-white. Rapidly growing, poorly differentiated astrocytomas are more malignant and easier to discern because they have areas of necrosis, hemorrhage, cavitation, and edema.
Figure 14-72.
Astrocytoma, Brain, Transverse Section at the Level of the Thalamus, Dog.
The deep ventromedial area (thalamus/hypothalamus) of the right hemisphere (arrows) contains a poorly demarcated, nonencapsulated, expansile mass, which is a space-occupying lesion that has displaced the midline to the left and compressed the right lateral ventricle. The left lateral ventricle is mildly dilated, most likely from compression of the interventricular foramen.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Microscopically, the more well-differentiated diffuse astrocytomas consist of a uniform cell type that is typically loosely organized. Cell size varies, and distinct ramifying cytoplasmic processes can be observed. Nuclei vary in size and shape and contain more chromatin than normal astrocytes. Cells tend to be arranged around and along blood vessels. The boundary between neoplastic and normal tissue is indistinct. Well-differentiated diffuse astrocytomas occur as pilocytic and gemistocytic variants, both of which recapitulate the histologic features of the astrocyte phenotype described earlier.
Conversely, anaplastic astrocytomas and glioblastoma multiforme have increased cellular pleomorphism, a high mitotic rate, and areas of necrosis. Glioblastoma multiforme is characterized by glomeruloid-like vascular proliferation and serpiginous tracts of necrosis that are often lined by neoplastic cells (pseudopalisading). Foci of oligodendroglial differentiation can be observed in these tumors. Grossly, hemorrhage is commonly observed in glioblastoma multiforme and can be a useful diagnostic feature.
Increasing use of immunohistochemical markers for glial cells and indicators of cellular proliferation can undoubtedly enhance the specificity and prognostic significance of the diagnosis of different glial cell neoplasms in animals. The most reliable marker for astrocytomas is GFAP, although vimentin has proved useful in some cases. Recent studies have also indicated that immunoreactivity for insulin-like growth factor–binding protein 2, epidermal growth factor receptor, and platelet-derived growth factor receptor-α may eventually become reliable markers for canine glioblastomas.
Oligodendrogliomas.
Neoplasms composed of oligodendrocytes occur most commonly in the dog, but cases have been reported in cats, horses, and cattle. The reported incidence varies; some reports indicate oligodendrogliomas as the most common neuroectodermal neoplasm (5% to 12% incidence), whereas others place it second to astroglial neoplasms. As with astrocytomas in dogs, there is a predilection for brachycephalic breeds (Boston terriers, boxers, and bulldogs), and the age range is the same as for astrocytomas (5 to 11 years of age). Neoplasms occur in all areas of the cerebrum and brainstem, especially in close proximity to the lateral ventricles (Fig. 14-73 ; E-Fig. 14-8). Neoplasms tend to extend to meningeal and ventricular surfaces, and dissemination via the CSF and the ventricular system to distant brain sites can be a common finding.
Figure 14-73.
Oligodendroglioma, Brain, Cerebrum, Transverse Section.
The tumor (arrows) is arising in the ventral cerebral cortex. It is well demarcated, gray, and gelatinous.
(Courtesy Drs. A. de Lahunta and A.D. Miller, Cornell University College of Veterinary Medicine, Cornell University.)
E-Figure 14-8.
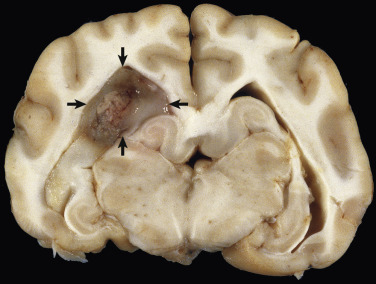
Oligodendroglioma, Brain, Cerebrum, Transverse Section Through the Lateral Ventricles, Dog.
The tumor (arrows) is arising in a periventricular location and is gray, soft, and gelatinous.
(Courtesy Drs. A. de Lahunta and A.D. Miller, Cornell University College of Veterinary Medicine, Cornell University.)
Grossly, the typical oligodendroglioma is a well-demarcated mass of variable size that is characteristically gray to pink-red and soft to gelatinous with areas of hemorrhage. In larger tumors the central area may be cystic. Microscopically, the neoplasms are composed of densely packed cells. Nuclei are round, centrally located, and hyperchromatic. The cells are often surrounded by a halo (so-called honeycomb effect), which is an artifactual retraction of the cytoplasm caused by delayed fixation. Other patterns of growth include cells arranged in rows, especially peripherally in the mass, or in semicircles. Mitoses are generally infrequent. Other changes include mucoid degeneration (accounting for the gelatinous appearance seen grossly), edema, cavitation, and rarely mineralization. Extensive necrosis is uncommon, except in higher-grade variants (i.e., more anaplastic morphologic changes) of the tumor, where serpiginous tracts of necrosis, nuclear atypia, glomeruloid-like vascular proliferation, and high mitotic rate are commonly encountered. Immunohistochemical identification of neoplastic oligodendrocytes can be aided by the use of CNPase and Olig1/Olig2; however, these markers are not 100% specific for the identification of neoplastic oligodendrocytes, and routine histologic examination remains the standard by which to diagnose these tumors.
Oligoastrocytomas.
Mixed gliomas (i.e., oligoastrocytomas) are characterized by atypical populations of neoplastic oligodendroglia and astrocytes. These tumors are uncommon in veterinary medicine with most cases reported in the dog. Although foci of oligodendroglioma and astrocytoma differentiation can be seen in astrocytomas and oligodendrogliomas, respectively, to be diagnosed as a mixed glioma, the proportion of each cell population in the tumor needs to be greater than 30%. Histologic features are similar to those described earlier in the sections covering astrocytomas and oligodendrogliomas. The degree of necrosis, mitotic activity, and microvascular proliferation vary based on the anaplastic grade of the tumor.
Gliomatosis Cerebri.
Gliomatosis cerebri is a widely infiltrating glioma that is uncommonly encountered in veterinary medicine with most reports confined to the dog. Gross lesions are typically not present, unless the diffusely infiltrating cells coalesce to form a distinct “mass lesion.” The cells, which infiltrate the CNS, are elongate with a prominent nuclear chromatin pattern and scant cytoplasm. They do not have any topographic perivascular orientation. Mitoses can be common and typically increase in number directly correlated to the anaplastic grade of the tumor. An astrocytic origin for the majority of these tumors is suspected, and neoplastic cells can stain positive by immunohistochemical reaction for GFAP. However, some of these tumors are negative for GFAP, suggesting either a different cell of origin (i.e., microglia) or the possibility that the mass consists of dedifferentiated astrocytes that are not expressing GFAP. Rarely oligodendroglial tumors can have a similar pattern of spread.
Ependymomas.
Ependymomas are one of the less frequently occurring neoplasms in dogs, cats, cattle, and horses. Some reports on the dog indicate a higher frequency in brachycephalic breeds. Ependymomas usually involve the lateral or less commonly, mesencephalic aqueduct, third, and fourth ventricles. They also occur in the central canal of the spinal cord. The neoplasm can be observed within the ventricular system and anywhere along the subarachnoid space, likely attributable to local metastasis via the CSF. Noncommunicating hydrocephalus may result from obstruction of CSF flow within the ventricular system.
Grossly, ependymomas are usually large expansile intraventricular masses with generally well-demarcated margins (Fig. 14-74 ). The neoplasm is soft and gray-white to red, depending on blood content, and has a smooth cut surface in dogs. In cats the cut surface can have a granular texture. In some ependymomas the cut surface may have gelatinous consistency and be cavitated. More aggressive tumors show invasion into the normal tissue at its margins. Microscopically, ependymomas are highly cellular and well vascularized. Cells have hyperchromic, round-to-oval nuclei with scant or undetectable cytoplasm. Cells form perivascular rosettes (pseudorosettes) with nuclear polarity away from the vessel wall. Cells are also arranged in sheets and bands. The mitotic rate is variable. Hemorrhage, cystic degeneration, and capillary proliferation occur. Higher-grade tumors display invasive growth, frequent mitoses, and anaplasia. Currently no reliable specific immunohistochemical markers for ependymomas exist; however, GFAP immunoreactivity can be observed, especially in the pseudorosettes where the anuclear areas are GFAP positive. Cytokeratin immunoreactivity has a patchy pattern of distribution.
Figure 14-74.
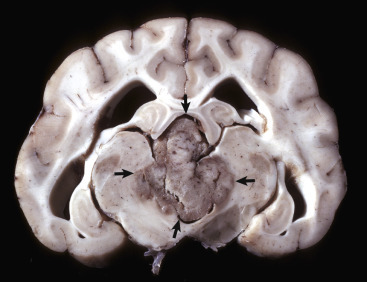
Ependymoma, Brain, Transverse Section at the Level of the Hippocampus, Dog.
The third ventricle contains a moderately well demarcated expansile mass (arrows) that has invaded normal tissue ventral to it. Moderate hydrocephalus is present in both lateral ventricles from blockage of the third ventricle.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Choroid Plexus Tumors.
Choroid plexus tumors (papillomas and carcinomas) occur most commonly in the dog but have been reported in horses and cattle. In the dog they are reported to be 5% to 10% of all primary brain tumors, and there is a breed predilection reported for golden retrievers. The age at clinical presentation varies widely, but it is usually seen in middle-aged to older dogs. In the dog the neoplasm occurs most frequently in the fourth ventricle, but it also can be located in the third and lateral ventricles.
Grossly, the neoplasm is a well-defined, expansive, granular to papillary growth located within the ventricular system that is gray-white to red and compresses the adjacent nervous tissue (Fig. 14-75 ). Noncommunicating hydrocephalus may result from obstruction of CSF flow within the ventricular system. Microscopically, these neoplasms generally resemble the choroid plexus and are characterized by an arborizing vascular connective tissue stroma that is covered with a cuboidal to columnar epithelial layer. Mitoses are not typically present in the benign form. A more malignant variety, choroid plexus carcinoma, is characterized by invasiveness, increased mitoses, additional occurrence of solid tumor growth, and a tendency to metastasize within the ventricular system or into the subarachnoid space (carcinomatosis), where implantation in the ependyma or meninges, respectively, occurs. Immunohistochemical identification with cytokeratin and E-cadherin can be useful. Patchy GFAP immunoreactivity is also reported.
Figure 14-75.
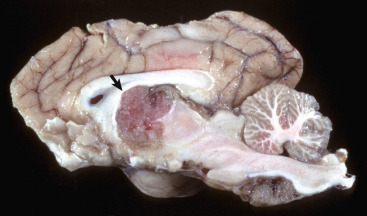
Choroid Plexus Tumor (Carcinoma), Brain, Sagittal Section, Dog.
The third ventricle contains an expansile mass (arrow) that has invaded the normal tissue ventral to it. The mass ventral to the medulla (right) may be a metastasis arising from tumor cells that entered the third ventricle and then spread in the cerebrospinal fluid caudally, through the mesencephalic duct, into the fourth ventricle, and out through a lateral aperture into the subarachnoid space.
(Courtesy Dr. Y. Niyo, College of Veterinary Medicine, Iowa State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Meningiomas.
Meningiomas are the most common neoplasm of the CNS in dogs and cats, and there are rare reports in horses and ruminants. The majority of meningiomas occur in dogs between 7 and 14 years of age and in cats 10 years old or older. Sites of occurrence in the dog include the basal area of the brain, the area over the convexity of the cerebral hemispheres, the cerebellum-tentorium area, the lateral surface of the brain, the falx cerebri, and the surface of the spinal cord. Retrobulbar involvement (originating from the optic nerve sheath) also occurs. In the cat the neoplasm uniquely occurs in the tela choroidea of the third ventricle (E-Fig. 14-9) but also occurs over the cerebral hemispheres, along the falx cerebri, over the cerebellum and tentorium, and rarely at the base of the brain. Occurrence in the meninges of the spinal cord is not common. Meningiomas arise from the arachnoid cell layer, which is on the external surface of the arachnoid membrane. These cells cover the surface of the arachnoid layer that opposes the surface layer of the dura mater, and thus these tumors project into the subdural space and often compress or invade the underlying CNS parenchyma.
E-Figure 14-9.
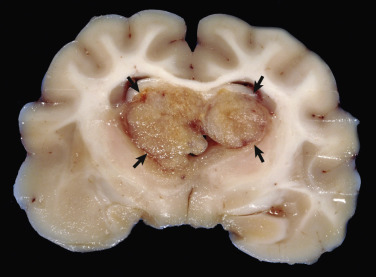
Ventricular Meningioma, Brain, Transverse Section at the Level of the Hippocampus, Cat.
The third ventricle is expanded by a multilobulated, tan neoplasm (arrows). Meningiomas occur with some regularity in this location in the cat. H&E stain.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University.)
Grossly, neoplasms in the dog are solitary and vary in size. The neoplasms are well defined, spherical, lobulated, lenticular, or plaquelike in shape; firm; encapsulated; and gray-white (Fig. 14-76 ). Sometimes on the cut surface there are soft, red, brown, or gray areas of hemorrhage and necrosis. Because these neoplasms grow slowly, they cause pressure atrophy of the adjacent nervous tissue. Meningiomas can be invasive, and sometimes there is hyperostosis of the overlying bone. In the cat, meningiomas vary in size from barely detectable to 2 cm in diameter. Cats can develop more than one neoplasm over the cerebrum, and meningiomas can also be incidental, old-age findings. Other characteristics are comparable with those described in the dog.
Figure 14-76.
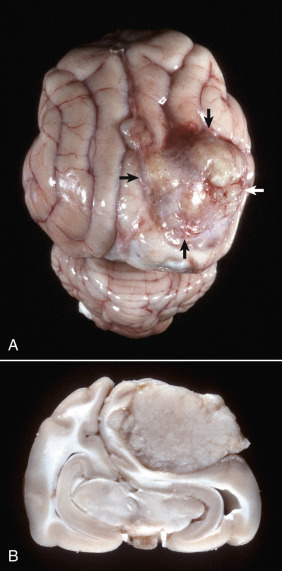
Meningioma, Brain, Cat.
A, On the surface of the right parietal cortex is a mass (arrows) that has compressed and distorted the adjacent parenchyma. It is a space-occupying lesion that has displaced the midline (cerebral longitudinal fissure) to the left. B, Transverse section at the level of the hippocampus of the brain depicted in A. The tumor has compressed the right cerebral hemisphere, and this has resulted in the midline being displaced to the left with compression of the left cerebral hemisphere. The meningioma does not invade the brain and can be “shelled” out at necropsy or surgery.
(Courtesy College of Veterinary Medicine, University of Illinois.)
Microscopically, several patterns of neoplastic cells can occur, and more than one pattern can be present in a given neoplasm. Based on their cytomorphologic features, the majority of these tumors can be subtyped as follows: (1) transitional; (2) meningothelial; (3) psammomatous; (4) fibrous; (5) atypical; or (6) malignant. The transitional, meningothelial, psammomatous, fibrous, and atypical subtypes appear to be the most common in the dog. In the cat, almost all meningiomas have features that are more typical of the meningothelial or transitional type with psammoma bodies being a common feature. Other less common subtypes include angiomatous, chordoid, microcystic, malignant, and papillary. Although cytomorphologic features of all subtypes are beyond the scope of this chapter, general features of the most common subtypes are mentioned here. The transitional subtype is composed of nests of polygonal to spindle cells arranged in whorls. The meningothelial variant is composed of cellular sheets lacking distinct morphologic characteristics, whereas the fibrous variant is composed of interlacing bundles of spindle cells. The psammomatous variant is characterized by a predominance of mineral concretions (psammoma bodies). Typically in these variants the neoplastic cells have large cell bodies with abundant cytoplasm, ill-defined cell boundaries, and elongated, oval, open-faced nuclei with peripherally located chromatin. The number of cells making up a whorl can vary from a few to many. Additional microscopic lesions include hemorrhage, foci of necrosis, and infiltrates of neutrophils. Whereas necrosis is an indication of a poorer prognosis in human beings, it is common in canine variants of meningioma, and its significance in tumor progression is unknown. Invasive growth occurs but is less common than growth by expansion. Currently, immunohistochemical staining is not as advanced in veterinary medicine as it is in the diagnosis of meningiomas in human beings; however, vimentin can be useful.
Hemangiosarcoma.
Primary hemangiosarcoma of the CNS is a rare neoplasm arising from endothelial cells. The disease is most common in dogs but can occur in all domestic species. The neoplasm is a solitary expansile red to dark red mass within the cerebral cortex. Its color and bloody consistency are helpful in differentiating it from a primary or metastatic melanoma. Most hemangiosarcomas found in the CNS are from metastases.
Hematopoietic Tumors
Lymphoma.
A variety of hematopoietic tumors affect the CNS, both as primary and as metastatic neoplasms. The most common of these tumors is lymphoma. Primary CNS lymphoma occurs in all domestic animal species but is most common in the dog and the cat. In the cow it can occur as part of a manifestation of bovine leukosis virus–associated lymphoma; however, this tumor more commonly presents as an extradural white to yellow lobulated compressive mass in the spinal canal. Primary lymphoma in the dog and the cat has no sites of predilection and can involve adjacent organs, including the pineal gland and the pituitary gland. They are composed of dense sheets of neoplastic lymphocytes that typically have a high mitotic rate and many regions of single cell to confluent necrosis. The primary cell type may be a T or B lymphocyte. Identification of the cell type requires immunohistochemical analysis; CD3 (T lymphocyte) and CD79a/CD20/Pax5 (B lymphocyte) are the markers most commonly used to classify these tumors.
An uncommon variant of CNS lymphoma is intravascular lymphoma. This variant occurs almost exclusively in the dog. Histologically, it presents as dense aggregates of neoplastic lymphocytes that fill meningeal and parenchymal blood vessels. Thus these tumors are often associated with the formation of thrombi and infarction of local tissues. Neoplastic infiltration into the parenchyma apparently only occurs with rupture of the affected vessels. Presumably, these neoplastic lymphocytes lack appropriate migration signals or receptors needed to cross the blood-brain barrier. These tumors typically have a B lymphocyte origin in dogs, which contrasts to the more typical T lymphocyte phenotype in human beings.
Histiocytic Sarcoma.
Histiocytic sarcoma is another hematopoietic neoplasm that can present either as a primary CNS tumor or as part of disseminated disease. The primary tumor typically presents as a meningeal mass that compresses and less commonly invades the underlying neuroparenchyma. Although certain breed predilections are reported for this (including Bernese mountain dog and Pembroke Welsh corgi), many breeds have been reported to develop this disease. Histologically, these tumors are composed of neoplastic round cells that often have reniform nuclei. Multinucleated cells are common, and the mitotic index is often quite high. These tumors can be further identified through a variety of immunohistochemical stains, including CD18 and Iba1.
Other Hematopoietic Tumors.
Other hematopoietic tumors, including extramedullary plasmacytomas, have been reported rarely in the CNS; however, these tumors represent a very small fraction of the primary hematopoietic tumors of the CNS.
Metastatic Tumors.
Hematogenously metastasizing neoplasms occur and affect the brain more often than the spinal cord. The species in which metastasis has been most commonly reported is the dog; the next most frequent is the cat. Of the metastasizing carcinomas, mammary gland carcinoma in the dog has been reported to occur most frequently, although others, especially nasal carcinoma due to local metastasis, have been described. Hemangiosarcoma is one of the most common metastasizing sarcomas in the dog and the most common metastasizing tumor to the brain. Metastases of many other sarcomas, including lymphosarcoma, fibrosarcoma, and malignant melanoma, have also been reported. In the CNS, metastatic hemangiosarcomas appear to have predilection for the gray matter–white matter interface (Fig. 14-77 ). In the cat the neoplasms that metastasize to the CNS include the mammary gland carcinoma (Fig. 14-78 ) and lymphosarcoma.
Figure 14-77.
Hemangiosarcoma, Metastatic, Brain, Formalin-Fixed, Transverse Section at the Level of the Thalamus, Dog.
Note the prominent hematogenous metastases, which appear as black nodules of various sizes distributed throughout the brain, sometimes at the gray matter–white matter interface. In an unfixed (fresh) specimen the nodules would be red to dark red. Black nodules in a fresh specimen would be consistent with metastatic melanoma.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 14-78.
Mammary Carcinoma, Metastatic, Brain, Transverse Section at the Level of the Hippocampus, Dog.
The right cerebral hemisphere contains a well-demarcated mass (arrow), which has caused enlargement of the right cerebral hemisphere and compression of the right lateral ventricle. The left lateral ventricle is slightly dilated, probably because of pressure on the interventricular foramen.
(Courtesy Drs. F. Moore and J. Carpenter, Angell Memorial Animal Hospital; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Aging Changes
Throughout this chapter, specific disorders have been correlated with aging because they occur in older animals and include, as examples, cerebral cortical atrophy of aging, hydrocephalus ex vacuo, the accumulation of lipofuscin pigment in neurons, and some forms of neuroaxonal dystrophy. Their relationship to behavioral changes is unclear. More recently, suspected aging changes in the nervous system of dogs and cats (i.e., senility of advanced age or “dementia”) have been correlated with behavioral changes such as deficiencies in learning and memory, as well as loss of other “normal routine” and long-established behavioral patterns. This collection of clinical signs in dogs has been termed canine cognitive dysfunction (CCD) and has been compared, in some respects, to Alzheimer's disease in human beings. Senile plaques and cerebrovascular amyloidosis have been described in the brains of aged dogs and are thought to result in structural and functional alterations in chemical pathways leading to cognitive dysfunction.
Disorders of Horses
Disorders that occur in many or all animal species are discussed in the section on Disorders of Domestic Animals.
Diseases Caused by Microbes
Viruses
Arboviruses
Equine Encephalomyelitis.
Eastern equine encephalomyelitis (EEE), western equine encephalomyelitis (WEE), and Venezuelan equine encephalomyelitis (VEE) viruses are members of the family Togaviridae, genus Alphavirus. The primary target cell for infection and injury is the neuron; however, these viruses can cause vasculitis followed by thrombosis. After inoculation (by mosquito) the hematogenously circulating virus initially infects several tissues, including endothelial cells of local blood vessels, tissues of the lymphoreticular system (also known as monocyte-macrophage system), muscle, and connective tissue. In lymphoid tissue and bone marrow this infection may cause cellular depletion, necrosis, or both. A second viremia results in hematogenous infection of the CNS. Experimental evidence suggests that the virus replicates in endothelial cells before entering the nervous system and infecting neurons and to a lesser extent glia. There is also evidence that viruses of this group (notably Venezuelan equine encephalomyelitis) can cause alterations in the metabolism of neurotransmitters in the CNS and that these alterations are responsible for some of the clinical signs.
Recent experimental evidence from in vivo and in vitro models of Venezuelan equine encephalomyelitis suggests that the virus causes upregulation of multiple proinflammatory genes, including inducible nitric oxide synthase and TNF-α. This upregulation, occurring principally in astrocytes, affected other glial cells and influenced neuronal survival. In addition to these mediators of innate immune responses, apoptotic cell death also contributes to neurodegeneration after virus infection.
In the CNS, all three viruses induce a polioencephalomyelitis that has similar characteristics, but there are some differences. Overall, gross lesions are uncommon and nonspecific. They can include cerebral hyperemia, edema, petechiation, focal necrosis, and increased CSF in the subarachnoid space. Gross lesions, although not often present, are usually found in gray matter, which is appreciated best in the spinal cord (Fig. 14-79 ). Microscopic lesions are most prominent in the gray matter of the brain and spinal cord and are characterized by perivascular cuffing with lymphocytes, macrophages, and neutrophils; variable neutrophilic infiltration of the gray matter; microgliosis; neuronal degeneration; focal cerebrocortical necrosis; perivascular edema and hemorrhage; necrotizing vasculitis; thrombosis; choroiditis; and leptomeningitis. Neutrophils are detectable during the early stages (2 days) of clinical eastern equine encephalomyelitis and Venezuelan equine encephalomyelitis. Vasculitis, thrombosis, and cerebrocortical necrosis are particularly evident in Venezuelan equine encephalomyelitis, but also in eastern equine encephalomyelitis. No lesions are detected in the trigeminal ganglion.
Figure 14-79.
Hemorrhagic Encephalitis and Myelitis, Eastern Equine Polioencephalomyelitis, Brainstem and Spinal Cord, Horse.
A, Brain, transverse section at the level of the hippocampus, horse. The gray matter of the brainstem has dark red to black discoloration as a result of congestion and hemorrhage. The lesion is the result of viral infection, which has an affinity for neurons; this virus also causes vascular necrosis followed by thrombosis, but this is not common. B, Spinal cord, horse. Note the red to brown discoloration of the gray matter in the dorsal and ventral horns (caused by congestion and hemorrhage). The lesion is the result of viral infection that has an affinity for neurons; however, this virus can also cause vascular necrosis followed by thrombosis.
(Courtesy College of Veterinary Medicine, University of Florida; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Infection of horses with eastern equine encephalomyelitis, western equine encephalomyelitis, and Venezuelan equine encephalomyelitis viruses produces a range of progressive clinical maladies, including fever, rapid heart rate, anorexia, depression, muscle weakness, and behavioral changes, such as dementia, aggression, head pressing, wall leaning, circling, blindness, and paralysis of facial muscles. Eastern equine encephalomyelitis has also been reported in cattle, sheep, camelids, and pigs.
West Nile Viral Encephalomyelitis.
West Nile virus (WNV), a mosquito-borne virus (family Flaviviridae, genus Flavivirus) that causes acute polioencephalomyelitis primarily in human beings, birds, and horses, is most commonly transmitted via a bird-mosquito cycle. In 2002, West Nile virus infection was diagnosed in 47,000 horses in 40 states in the United States, and it was estimated that more than 4500 horses died after infection. It remains an important cause of viral encephalitis in horses, and the disease is ubiquitous in the lower 48 states of the United States.
The virus passes freely through a variety of wild-songbird populations with members of the Corvidae (crow family) and American robins playing important roles in the propagation and dissemination of the virus. Additionally, a variety of Culex mosquitoes disseminates the virus among animal populations. Birds develop a fulminate viremia with viral spread to all organs, whereas horses, representing a dead-end species for the virus, only have infections of the CNS.
The pathogenesis of West Nile virus encephalomyelitis remains to be fully elucidated; however, it is likely to be similar to the mechanism described previously for equine encephalomyelitis viruses. The primary target cell for infection and injury is the neuron; microglial cells are also affected. Gross lesions of West Nile viral infection in horses usually involve the gray matter and include hyperemia and petechiation to prominent hemorrhage with prevalent involvement of the lower brainstem and ventral horns of the thoracolumbar spinal cord. Gross lesions of hemorrhage are more likely to occur with West Nile virus than with the three equine encephalitides (eastern equine encephalomyelitis, western equine encephalomyelitis, Venezuelan equine encephalomyelitis). Microscopic lesions in birds and horses that have died of the disease are characterized by a polioencephalomyelitis and hemorrhage of the CNS that can vary in degree of severity. The inflammation is predominately mononuclear; however, neutrophils can be detected in some cases. Lesions outside of the nervous system in affected horses do not occur. Clinical signs of the equine West Nile viral infection include variable fever, depression, ataxia, weakness to paralysis of the hind limbs, tetraplegia, convulsions, coma, and death.
Herpesviruses
Equine Herpesvirus 1 Myeloencephalopathy.
Equine herpesvirus 1 (EHV-1) (an alphaherpesvirus) is an important cause of equine abortion and perinatal foal infection and death, in addition to myeloencephalitis. EHV-1 can also cause rhinopneumonitis. EHV-1 does not appear to be neurotropic, which is in contrast to some herpesvirus encephalitides of other species in which the virus replicates in neurons (herpes simplex viral infection in the human, infectious bovine rhinotracheitis viral infection in calves, and pseudorabies viral infection in pigs). In addition to vasculitis being the principal lesion, the infection in the horse also differs somewhat from most other herpetic infections of the CNS in being primarily a disease of the adult, although young animals can be affected.
Equine herpesvirus myeloencephalopathy begins with inhalation of the EHV-1. The virus infects epithelial cells of the nasopharynx and spreads to local lymphoreticular tissue, where it infects lymphocytes and macrophages (monocytes). Through trafficking by monocytes, EHV-1 is transferred to endothelial cells of the CNS. The virus, which is endotheliotropic, even though infection of neurons and astrocytes can occur, localizes in small arteries and capillaries of the CNS and some other tissues after direct spread from the circulating infected cells. Inflammation of endothelial cells then results in vasculitis, leading to thrombosis and infarction of the neural tissue supplied by the thrombosed vessel. Latent infection of the trigeminal ganglion and lymphoid tissues can also occur. Although viral infection can be identified in neurons, the virus is not neurovirulent and has no effect on neural cells; thus the gross and histologic lesions noted in this disease are entirely related to vasculitis.
The characteristic lesion in the CNS caused by EHV-1 infection is a vasculitis affecting endothelial cells of small blood vessels with thrombosis and resulting in focal CNS necrosis (infarction). Lesions occur in both the gray and white matter of the spinal cord, medulla oblongata, mesencephalon, diencephalon, and cerebral cortex (Fig. 14-80, A ). The endothelium appears to be the initial site of involvement (see Fig. 14-80, B), with the subsequent intimal and medial degeneration resulting in hemorrhage, thrombosis, extravasation of plasma proteins into the perivascular space, axonal swelling with ballooning of the myelin sheath and degeneration of the cell body, and variable mononuclear cell cuffing. Other lesions include cerebrospinal ganglioneuritis and vasculitis in nonneural tissues, including the endometrium, nasal cavity, lungs, uvea of the eye, hypophysis, and skeletal muscle. Inclusion bodies are not observed in CNS lesions.
Figure 14-80.
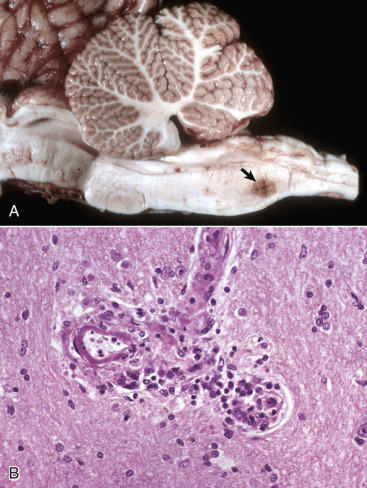
Equine Herpesvirus 1 Myeloencephalopathy, Brain, Midsagittal Section.
A, Hemorrhage, brainstem, horse. Focal or multifocal areas of hemorrhage and/or necrosis (arrow) are characteristic of equine herpesvirus encephalitis but also occur in equine arteritis virus encephalitis, cerebrospinal nematodiasis, and equine protozoal encephalomyelitis (Sarcocystis neurona). B, Vasculitis and hemorrhage, brainstem, horse. Vasculitis is the primary lesion. The virus localizes in small arteries, venules, and capillaries of the central nervous system, resulting in vasculitis and fibrinoid necrosis, which at times leads to thrombosis and focal infarction of the brain and spinal cord. H&E stain.
(A courtesy College of Veterinary Medicine, University of Illinois. B courtesy Dr. J. Simon, College of Veterinary Medicine, University of Illinois.)
The neurologic form of EHV-1 infection has a worldwide distribution and affects other Equidae, including zebras in addition to the horse, but appears to be relatively uncommon when compared with the incidence of abortion and upper respiratory tract disease caused by EHV-1. The neurologic disease may accompany or follow outbreaks of respiratory disease or abortion. An outbreak of epizootic acute encephalitis in Thomson's gazelle (Gazella thomsoni) was reported in 1997 from a zoologic garden in Japan. That disease resembled equine herpesvirus encephalitis, and the virus, named gazelle herpesvirus 1, was serologically related to EHV-1 and had a strong tropism for endothelium.
Protozoa
Equine Protozoal Encephalomyelitis (Sarcocystosis).
Equine protozoal encephalomyelitis is a disease in horses caused by Sarcocystis neurona. The microbe enters the body through ingestion of sporocysts, but how the parasite enters the CNS is unclear. Experimental studies suggest that ingested sporocysts multiply in visceral tissue, perhaps the intestine, and then are transported to the CNS probably via leukocytic trafficking. In the CNS the typical sequence of events in the pathogenesis of the characteristic lesion is thought to be (1) leukocytic trafficking with focal parasitic activation and replication, (2) marked inflammation, (3) edema, and (4) pronounced tissue destruction affecting both white and gray matter.
Gross lesions can occur throughout the neuraxis but are more common in the spinal cord, particularly the cervical and lumbar intumescences, than in the brain. In the brain, lesions are most commonly seen in the brainstem. When gross lesions are present, they consist of discolored necrotic foci, often with hemorrhages of varied sizes that are located randomly throughout the white or gray matter (Fig. 14-81 ).
Figure 14-81.
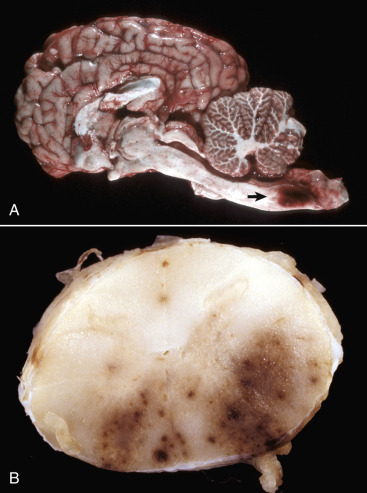
Protozoal Encephalomyelitis, Brain, Sagittal Section, Horse.
A, Note the large focus of hemorrhage and necrosis (arrow) in the caudal medulla caused by Sarcocystis neurona. B, Lumbar spinal cord, transverse section. Myelitis due to S. neurona infection. Prominent focal hemorrhage and necrosis are present in the right lateral funiculus and in the right and left ventral funiculi.
(A courtesy College of Veterinary Medicine, University of Illinois. B courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Microscopic lesions occur in both the white and gray matter and include necrosis, hemorrhage, and accumulations of lymphocytes, macrophages, neutrophils, eosinophils, and variable numbers of multinucleated giant cells in perivascular areas and the neuropil, or less commonly, the leptomeninges and the axonal swelling. Gemistocytic astrocytosis can be prominent. In lesions, S. neurona is small and crescent shaped to round, has a well-defined nucleus, and is often arranged in aggregates or rosettes (Fig. 14-82 ). Microbes can be difficult to detect but occur intracellularly in neurons, giant cells, neutrophils, or macrophages or occur extracellularly in cysts within the neuropil. S. neurona is not typically seen in vascular endothelial cells, a common site for other members of this genus.
Figure 14-82.
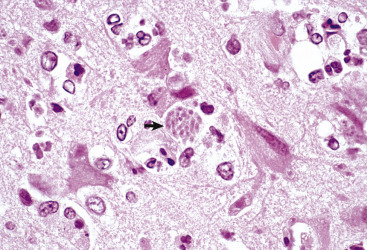
Sarcocystis Neurona, Brain, Horse.
S. neurona is a small, crescent-shaped to round protozoan microbe found in neurons, endothelial cells, and microglial cells. The microbes can be arranged in nonencysted aggregates (arrow) or rosettes intracellularly and in central nervous system tissue often with a mixed leukocytic cellular inflammatory response of varied severity. Microbes can be difficult to detect in histologic sections. H&E stain.
(Courtesy Dr. J. Simon, College of Veterinary Medicine, University of Illinois.)
S. neurona can be propagated in tissue culture where it develops in the host cell cytoplasm. It divides by endopolygeny, with development of schizonts containing merozoites arranged in rosettes around a prominent residual body. The schizont stage of the microbe differs from those of the genera Toxoplasma, Isospora, Eimeria, Besnoitia, Hammondia, and Neospora because the merozoites lack rhoptries but resemble the schizont stage of other members of Sarcocystis spp. and of the Frenkelia genus. Studies have shown that the opossum represents the definitive host and avian species represent the intermediate host, providing a reservoir for infection of the horse.
Sarcocystis spp. infection in nonequine species involves microbes similar to S. neurona. Such microbes have been associated with encephalomyelitis in cattle, sheep, cats, and dogs, as well as raccoons, but such infections are sporadic. Lesions and microbes have additionally been seen in the CNS of infected bovine fetuses and consist of multifocal glial nodules with or without microbes. Rare cases of equine protozoal myelitis have occurred concurrently with Neospora hughesi or N. caninum.
Clinical infection typically occurs in young adult horses, and signs depend on the area of the CNS parasitized. Signs can include depression, behavioral changes, seizures, gait abnormalities, ataxia, facial nerve paralysis, head tilt, paralysis of the tongue, urinary incontinence, dysphagia, atrophy of masseter and/or temporal muscles, and atrophy of the quadriceps and/or gluteal muscles.
Degenerative Diseases
Metabolic
Equine Degenerative Myeloencephalopathy and Neuroaxonal Dystrophy.
Equine degenerative myeloencephalopathy has been reported in a variety of purebred and mixed-breed horses, and a similar disease exists in zebras. To date the exact mechanism by which the disease develops is unknown; however, a genetic component likely underlies many of the cases. For cases in which a genetic basis cannot be determined, and for cases in which a genetic basis is known, the primary predisposing factor is dietary insufficiency of vitamin E. In some horses with low plasma concentrations of vitamin E, supplementation with vitamin E has resulted in clinical improvement of the horse. Additionally, the use of vitamin E supplementation in rations on farms with a high incidence of degenerative myeloencephalopathy has reduced the occurrence of the disease. A hereditary predisposition for equine degenerative myeloencephalopathy has not been excluded, but identifying a specific mechanism has so far been elusive.
In horses with degenerative myeloencephalopathy, lesions are microscopic. Axonal degeneration in the spinal cord is the most notable lesion, is bilateral, and can affect all funiculi, although lesions predominate in the sensory axons. Dorsal spinocerebellar tracts in lateral funiculi and septomarginal areas of the ventral funiculus are often severely affected. Myelin loss occurs secondary to the axonal degeneration. Depending on duration of clinical illness, astrogliosis occurs and is often most prominent in the region adjacent to the glia limitans. Another less dramatic lesion in affected horses is the formation of eosinophilic spheroids (Fig. 14-83 ). In horses, spheroids have been described in the nucleus gracilis, medial and lateral cuneate nuclei of the terminal brainstem, and thoracic nucleus of the spinal cord. Spheroids represent a focal eosinophilic swelling in the course of an axon that can be somewhat homogeneous, laminated, or granular. The swellings are filled with amorphous debris, membranous profiles, and effete organelles.
Figure 14-83.
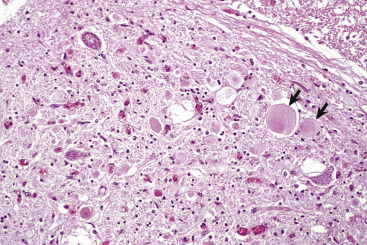
Axonal Spheroids and Dystrophic Axons, Degenerative Myeloencephalopathy, Spinal Cord, Dorsal Gray Horn, Horse.
Axons of the thoracic nucleus are swollen, rounded, and pale pink (axonal spheroids, which are accumulations of neurofilaments and effete organelles) (arrows). Attributed to vitamin E deficiency, through which vitamin E functions as an antioxidant, protecting cells from free radical–mediated injury. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Neuroaxonal dystrophy has been reported in several horse breeds, most commonly the Morgan and Haflinger breeds. Lesions consist of spheroids in brainstem nuclei as previously noted, but the severe axonal degeneration does not occur in the spinal cord. The cause of axonal spheroids and dystrophic axons in equine degenerative myeloencephalopathy and neuroaxonal dystrophy is unclear; however, alteration of axoplasmic flow is suspected. Immunohistochemical analyses have shown that spheroids and dystrophic axons contain elevated quantities of proteins involved in movement, docking, and fusion of synaptic vesicles to plasma membranes. These findings suggest that disruption of axoplasmic flow plays a role in the pathogenesis of dystrophic axons in equine degenerative myeloencephalopathy and neuroaxonal dystrophy.
Clinically, equine degenerative myeloencephalopathy and neuroaxonal dystrophies typically occur in young horses. The onset is insidious, and clinical signs are symmetric with spasticity, ataxia, and paresis of the limbs.
Primary Neuronal Degeneration
Primary Cerebellar Neuronal Degeneration.
Equine cerebellar abiotrophy, a primary cerebellar neuronal degeneration, occurs in Arabian or part-Arabian foals and Swedish Gotland ponies. The disease is inherited in an autosomal recessive pattern; however, the underlying pathologic mechanism for Purkinje cell loss remains elusive. Altered expression of MUTYH, a DNA-repair enzyme, has been shown in some affected horses. Grossly, the cerebellum may be slightly reduced in size and retracted from the overlying calvaria. Microscopically, early in the disease course Purkinje cells and their proximal axons are swollen (Fig. 14-84 ). With time there is loss of Purkinje cells and neurons of the granule layer. Clinical signs appear between the time of birth and 9 months of age and include head tremors, ataxia, and spasticity.
Figure 14-84.
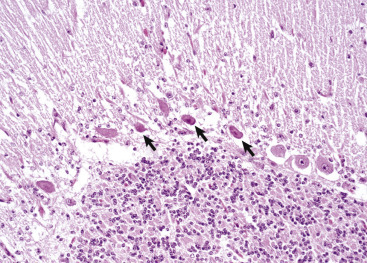
Equine Cerebellar Degeneration, Cerebellum, Horse.
Purkinje cells in the cerebellum are undergoing necrosis with shrunken cell bodies and nuclear pyknosis (arrows). H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Nutritional
Vitamin E Deficiency.
See the previous section on Equine Degenerative Myeloencephalopathy and Neuroaxonal Dystrophy.
Toxicoses
Microbial Toxins
Leukoencephalomalacia.
Ingestion of moldy feed composed of corn or corn by-products contaminated with the fungus Fusarium moniliforme causes an acute fatal neurologic disease in horses called leukoencephalomalacia. The primary toxin isolated from F. moniliforme has been named fumonisin B1, although other fumonisins have been extracted. Based on the character and progression of lesions, vascular damage has been inferred as the primary injury; however, fumonisins can also disrupt sphingolipid metabolism via the inhibition of ceramide synthase. Sphingolipids are bioactive compounds that participate in the regulation of cell growth, differentiation, metabolic functions, and apoptotic cell death. In addition, fumonisins are associated with lipid peroxidation of cells and cellular membranes, inhibit synthesis of macromolecules and DNA, and may enhance production of TNF-α by macrophages.
Gross lesions involve the white matter of the frontal and parietal lobes of the cerebral hemispheres most commonly, but cases with involvement of major white matter tracts in the brainstem and deep cerebellar white matter have occurred (Fig. 14-85 ). As a result of the white matter damage, including edema, brain swelling is marked with flattening of cerebrocortical gyri. The lesions are often bilateral but not symmetric, are unequal in severity, and can be quite extensive. The characteristic gross lesion at the time of death is malacia and liquefaction of the affected white matter due predominantly to the breakdown of lipids accompanied by hemorrhage. The reason white matter, including subcortical white matter, is principally involved—whereas the cerebral cortical gray matter is spared—is thought to be related to a unique vulnerability of the blood vessels in the white matter. The mechanism for this selective vulnerability is unknown.
Figure 14-85.
Leukoencephalomalacia, Brain, Horse.
A, Sagittal section. The white matter of the frontal and parietal lobes is necrotic (malacia). The gray matter is not affected. This disease is caused by the toxin fumonisin B1 produced by the fungus Fusarium moniliforme, which grows in damaged feed grains. Note that this case demonstrates the extent and distribution of liquefactive necrosis in the white matter in this disease. A more typical presentation is shown in B. B, Transverse section. The white matter of the three cerebral gyri located at the top of the illustration has areas of yellow gelatinous softening (arrows) and hemorrhage. Because of the absence of cavitation (liquefactive necrosis), the age of this lesion is likely less than the lesion depicted in C. C, Note the severe injury of the white matter. Myelinated axons are fragmented, and myelin debris is abundant. Numerous macrophages are present in the space previously occupied by myelinated axons, and they are phagocytosing the cellular debris. H&E stain.
(A courtesy Dr. J. Simon, College of Veterinary Medicine, University of Illinois. B courtesy Dr. W. Crowell, College of Veterinary Medicine, University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. C courtesy Dr. W. Haschek-Hock, College of Veterinary Medicine, University of Illinois.)
Microscopically, the affected white matter is liquefied, and the parenchyma is disrupted by accumulation of pink-staining proteinaceous fluid with scattered neutrophils, lymphocytes, macrophages, and rarely eosinophils (see Fig. 14-85, C). The border of the lesion is surrounded by diffuse or perivascular edema, perivascular hemorrhage, and blood vessels with small leukocytic cuffs. In the acute phase especially, blood vessel walls are degenerate or necrotic, and some are infiltrated with neutrophils, plasma cells, and eosinophils. Although not often detected, thrombosis occurs. Less characteristic changes include edema and perivascular cuffing in the leptomeninges and neuronal necrosis in deeper layers of the overlying gray matter near the affected white matter.
Leukoencephalomalacia can also be associated with hepatotoxicity, or hepatotoxicity can be the sole manifestation. Additionally, other animals, including pigs, are susceptible, but clinical disease and lesions generally reflect pulmonary, hepatic, or renal injury. Clinical signs can include depression, somnolence, head pressing, aimless wandering, blindness, or seizures. Rapid progression of these clinical signs followed by death is typical, ranging from 1 to 10 days after onset.
Plant Toxins
Centaurea spp. Poisoning.
Horses grazing Centaurea solstitialis (yellow star thistle) or Centaurea repens (Russian knapweed) develop a disorder known as nigropallidal encephalomalacia. This disease is similar to Parkinson's disease in human beings and has been proposed as a model for experimental studies; however, the equine disease is not associated with abnormal cytoplasmic accumulation of α-synuclein as seen in Parkinson's disease. The specific cause of the syndrome is not proved. A sesquiterpene lactone isolated from C. repens termed repin could provide a basis for the neurotoxicity. Cytotoxicity in cell culture was associated with depletion of glutathione (a major antioxidant), an increase in reactive oxygen species, and evidence of membrane damage in PC12 cells (a pheochromocytoma cell line) and mouse astrocytes. High concentrations of monoamine oxidase involved in dopamine metabolism normally found in the dopaminergic strionigral tract (i.e., striatum and substantia nigra [regulate balance and movement]) could render these areas of the brain more susceptible to oxidative damage caused by repin. The mechanism is unknown. Repin is also reported to inhibit dopamine release in the rat striatum, potentially contributing to the clinical manifestations of the disorder.
Gross brain lesions are sharply demarcated foci of yellow discoloration and malacia in the globus pallidus (pallidal) and substantia nigra (nigro) (Fig. 14-86 ). Lesions are usually bilateral and vary in severity; however, unilateral lesions also occur. Microscopically, necrosis with loss of neurons is the primary lesion; however, axons, glia, and blood vessels also are necrotic. The debris is phagocytosed by macrophages recruited into the lesion from the bloodstream.
Figure 14-86.
Equine Nigropallidal Encephalomalacia, Brain, Transverse Section Through the Midbrain at the Level of the Rostral Colliculi, Horse.
This lesion is caused by yellow star thistle poisoning. Note the symmetrically cavitated (malacia) lesions in the substantia nigra (arrows), resulting from necrosis and phagocytosis by gitter cells.
(Courtesy Dr. L. Lowenstine, School of Veterinary Medicine, University of California-Davis; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Grazing the plants for 1 month or longer during the hot summer months when other forage is dried and unpalatable can cause clinical disease. Affected horses are somnolent, have persistent chewing movements, and have difficulty in prehension of feed and drinking water. With clinical evaluation, lip and tongue paralysis with reduced jaw tone are commonly recognized early in the disorder. Death generally is due to emaciation and starvation.
Miscellaneous Conditions.
For discussion of miscellaneous conditions affecting individual animal species, see sections covering diseases specific for the species.
Cholesteatomas.
Cholesteatomas, also called cholesterol granulomas, form in choroid plexuses of the ventricles in horses typically as an aging change, although rarely these granulomas can form in young horses. These masses are usually incidental, but if they grow large enough and occlude the flow of CSF, acquired hydrocephalus can result. Cholesteatomas are tan to yellow-brown firm masses with a smooth often glistening surface (Fig. 14-87 ). Occasionally the masses are mineralized. This lesion is thought to result from edema and minor, but repeated hemorrhages within the choroid plexuses, which result in cholesterol deposits. These deposits elicit a foreign body inflammatory reaction (foreign body granuloma) in the choroid plexus.
Figure 14-87.
Cholesterol Granuloma (Cholesteatoma), Brain, Sagittal Section, Horse.
The choroid plexus of the lateral ventricle contains an expansile mass consisting chiefly of cholesterol and a granulomatous inflammatory response (arrow).
(Courtesy College of Veterinary Medicine, University of Illinois.)
Circulatory Disturbances
Peripartum Asphyxia Syndrome.
Peripartum asphyxia syndrome (dummy foal, neonatal maladjustment syndrome, or barker foal) is attributed to impairment of normal umbilical blood flow between the mare and foal during parturition, resulting in decreased vascular flow to the brain. Causes usually are those related to interruption of umbilical blood flow, such as a twisted or pinched umbilical cord as occurs in a dystocia or premature separation of the placenta, possibly caused by endophyte fescue toxicity.
In the early stages of the syndrome, hemorrhages are often widespread in nervous tissue, and cerebral gyri are edematous and swollen. If the foal survives for several days or longer, laminar cortical necrosis may occur. Microscopically, initial lesions consist of laminar cortical edema and neuronal necrosis, followed by gitter cell accumulation and phagocytosis of cellular debris. Ischemic neuronal injury can be found throughout the neuraxis.
Clinical signs in foals with peripartum asphyxia syndrome include barking like dogs, seizures, aimless wandering, absence of a suckling reflex, and loss of affinity for the mare. Affected foals are usually normal for the first 12 to 24 hours and then decline rapidly as neuronal necrosis ensues.
Postanesthetic Myelopathy.
A hemorrhagic myelopathy has been reported in young horses secondary to general anesthesia in which the animals are placed in dorsal recumbency. Following surgery, they are typically unable to stand. Grossly, there is marked hemorrhage throughout the gray matter of the spinal cord and acute neuronal necrosis, histologically. Presumably the lesion develops secondary to the weight of the animal blocking normal venous drainage from the vertebral sinuses leading to an infarctive lesion in the spinal cord.
Disorders of Ruminants (Cattle, Sheep, and Goats)
Disorders that occur in many or all animal species are discussed in the section on Disorders of Domestic Animals.
Diseases Caused by Microbes
Bacteria
Listeriosis.
Listeriosis, a bacterial disease with particular affinity for the CNS, is seen mainly in domestic ruminants. L. monocytogenes, a facultative intracellular Gram-positive bacterium, invades through the mucosa of the oral cavity and into sensory and motor branches of the trigeminal nerve. Other cranial nerve branches that innervate the oral cavity and pharynx may also be involved. The invasion through the mucosa is facilitated by trauma to the mucosa, thereby allowing the pathogen to gain access to the underlying tissues and nerve endings. The bacteria migrate via sensory axons using retrograde axonal transport to the trigeminal ganglion and then into the brainstem or via motor axons directly to the midbrain and medulla (motor neurons—nucleus of cranial nerve V). The infection can then spread rostrally and caudally to other areas of the CNS. These sites are likely the result of direct extension of infection because L. monocytogenes is a motile bacterium that spreads from cell to cell in its replicative phase.
The mechanism of tissue injury is not completely defined; however, injury to neurons and axons is likely a secondary bystander effect related to inflammation. A correlation between the degree of cell-mediated immunity and severity of brain damage suggests that immunologic injury may also occur. The microbe produces multiple virulence factors, including a hemolysin (listeriolysin), which is required for intracellular multiplication, and internalin, which internalizes E-cadherin, thereby allowing the infection to spread beyond localized regions.
Once bacteria enter the CNS, recent experimental studies suggest that L. monocytogenes can directly infect neurons, glia cells, and macrophages recruited in the inflammatory exudate. The bacteria spread from cell to cell using a secreted phospholipase that cleaves a variety of phospholipids, including sphingomyelin (a component of myelin) and phospholipids in cell membranes (see Fig. 4-30). Axonal injury and neuronal death are likely attributable to inflammatory processes, especially to the action of listeriolysin and lipases.
Gross lesions are usually absent, but leptomeningeal opacity, foci of yellow-brown discoloration (0.1 to 0.2 mm in diameter in the area of the nuclei of cranial nerves V and VIII), hemorrhage, necrosis in the terminal brainstem, and cloudy CSF can all be observed. Microscopically, a meningoencephalitis centered on the pons and medulla and involving both gray and white matter is characteristic (Fig. 14-88 ). The lesions, however, can extend from the diencephalon to the caudal medulla or cranial cervical spinal cord. Small, early lesions consist of loose clusters of microglial cells. With time, these lesions enlarge and contain variable numbers of neutrophils (see Fig. 14-88, B). Later in the process, microabscesses form, and neutrophils are the principal inflammatory cell. Although uncommon, some microabscesses contain macrophages as the principal cell type. Necrosis and accumulation of gitter cells can be prominent in some cases. Numerous Gram-positive bacilli can be detected in some lesions (see Fig. 14-88, C). Leptomeningitis is regularly present and is often severe, and the exudate is composed predominantly of mononuclear cells (macrophages, lymphocytes, plasma cells) with fewer neutrophils. Cranial ganglioneuritis involving the trigeminal nerve and ganglion is often present. Vasculitis may occur in cases of cerebral listeriosis.
Figure 14-88.
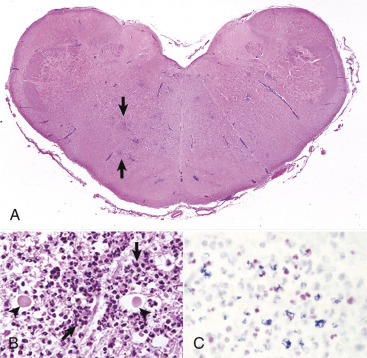
Listeriosis, Medulla, Cow.
A, Microabscesses. Note the areas of faint blue discoloration in this subgross magnification of the medulla (arrows). The less well defined blue areas are aggregates of neutrophils (microabscesses), and the blue linear lesions are perivascular cuffs. Listeria monocytogenes, the causative agent, uses retrograde axonal transport via the cranial nerves to enter the central nervous system and localize in the medulla (brainstem) and proximal cervical spinal cord. The lesion is rarely visible on gross observation. H&E stain. B, Early microabscesses (arrows) and inflammation are the result of inflammatory mediators that have injured axons (arrowheads) and will lead to Wallerian degeneration, seen here at the stage of swollen eosinophilic axons. H&E stain. C,L. monocytogenes, which is Gram-positive (blue coccobacilli), can sometimes be detected in microabscesses in a histologic section stained with a Gram stain. Gram stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Listeriosis presents in three disease forms: meningoencephalitis, abortion and stillbirth, and septicemia. The last commonly develops in young animals, possibly from an in utero infection. The encephalitic and reproductive forms of the disease rarely occur together in an individual animal or in the same flock or herd. Infections in human beings also occur. Clinical signs in meningoencephalitic listeriosis are related to lesions in the brainstem and include dullness, torticollis, circling, unilateral facial paralysis, and drooling caused by pharyngeal paralysis. Signs of cranial nerve dysfunction occur because of inflammation in the brainstem. Death usually occurs within a few days after the initial signs and is preceded by recumbency and paddling of the limbs. Silage is the most common source of infection. If silage is contaminated with soil containing L. monocytogenes and is improperly prepared and stored (pH > 5.4), the microbe can multiply.
Thrombotic Meningoencephalitis.
H. somni, a small Gram-negative bacillus, causes septicemia in cattle with variable clinical presentations, including pneumonia, polyarthritis, myocarditis, abortion, and meningoencephalitis. The disease is most prevalent in feedlot cattle but can occur in other situations. All manifestations, particularly meningoencephalitis, tend to be sporadic with single to multiple animals in a herd affected. The CNS form of the disease has been termed thrombotic meningoencephalitis, previously referred to as thromboembolic meningoencephalitis. Mural thrombi from local vascular injury rather than thromboemboli from distal sites of vascular injury, such as the lungs, are the major type of thrombus in this disease.
The pathogenesis of H. somni infection is not completely understood. Many cattle harbor the microbe in the upper digestive tract without evidence of disease, but under some circumstances it invades to cause severe clinical infection. The mechanism(s) of invasion into the bloodstream is not definitely known, but the respiratory tract is the initial site of bacterial replication followed by hematogenous spread to the CNS. The bacterium produces profound damage to endothelial cells, from which the majority of its thrombotic lesions develop. One of the primary bacterial virulence factors, lipooligosaccharide, has been shown in experimental studies to cause apoptosis of endothelial cells. This mechanism likely contributes to the profound damage of endothelial cells occurring with this disease. Once endothelial cells are damaged, thrombosis occurs, and tissues dependent on the obstructed blood vessel undergo infarction and necrosis.
Gross lesions in the CNS are irregularly sized foci of hemorrhage and necrosis scattered randomly and visible both externally and on cut surfaces (Fig. 14-89 ). Lesions are most frequent in the cerebrum, commonly at the cortical gray matter–white matter interface. The location of the lesion may reflect a change in the diameter and flow patterns of blood vessels, allowing bacteria to preferentially damage these areas. The spinal cord can also be affected. Other lesions include brain swelling caused by edema and leptomeningitis with cloudiness of the CSF.
Microscopic lesions in all organs, including the CNS, consist of a marked vasculitis and vascular necrosis, which are followed by thrombosis and infarction. The vasculitis is associated with regional edema and infiltration of neutrophils and macrophages in and around the affected blood vessel. Colonies of small Gram-negative bacilli can be found in the thrombi, affected blood vessels, and in the infarcted tissue.
Clinically, affected cows are initially ataxic and circle, head press, and appear blind. As the disease progresses, they may have convulsions, become comatose, and die.
Viruses
Herpesviruses
Bovine Malignant Catarrhal Fever.
Malignant catarrhal fever is usually a sporadic highly fatal disease of cattle and other ruminants, including deer, buffalo, and antelope, and can involve several animals in a herd. The disease has a worldwide distribution, and the clinicopathologic features do not differ significantly from one part of the world to another. The primary target tissues are the vasculature, lymphoid organs, and epithelium (particularly of the respiratory and gastrointestinal tracts), but the kidneys, liver, eyes, joints, and CNS can also be affected. The virus appears to be transferred between lymphoid tissue/cells and endothelial cells via leukocytic trafficking in T lymphocytes. Two general types of the disease occur, the sheep-associated and wildebeest-derived forms, although several other less common causes of the disease have also been described. The causative agents involved belong to the herpesvirus subfamily Gammaherpesvirinae. The disease occurring outside Africa, caused by ovine herpesvirus 2, often involves close contact of presumed “carrier” sheep with susceptible ruminants. The disease has recently been reported in musk ox (Ovibos moschatus), Nubian ibex (Capra nubiana), and gemsbok (Oryx gazella). In Africa and occasionally in wildlife facilities outside the continent, the source of the infection (designated Alcelaphine herpesvirus 1) is the wildebeest. Two other antigenically related viruses, which apparently do not cause natural disease, include Alcelaphine herpesvirus 2 and Hippotraginae herpesvirus 1, which have been isolated from African hartebeest and roan antelope, respectively. It is generally accepted that cattle and other susceptible ruminants contract the disease in nature after respiratory or oral infection during association with carrier sheep (presumed) and wildebeest, particularly at the time of parturition. A cell-mediated and cytotoxic lymphocytic process has been proposed as involved in the development of the necrotizing vasculitis.
Gross lesions of the CNS include active hyperemia and cloudiness of the leptomeninges caused by nonsuppurative meningoencephalomyelitis and vasculitis. Lymphocytic perivascular cuffing and varying degrees of necrotizing vasculitis occur in the leptomeninges and in all parts of the brain and occasionally in the spinal cord, with the white matter most consistently involved. Other lesions in the affected CNS include variable neuronal degeneration, microgliosis, choroiditis, necrosis of ependymal cells, and ganglioneuritis. Clinical signs referable to CNS infection may include trembling, shivering, ataxia, and nystagmus.
Bovine Alphaherpesvirus Meningoencephalitis.
Although bovine herpesvirus 1 (BHV-1) occasionally causes a nonsuppurative meningoencephalitis, primarily in young cattle, two variants of BHV-1 isolated in Argentina and Australia (referred to as BHV-1, subtypes 3a and 3b, respectively) and more recently BHV-5 (isolated in South America, mainly Argentina and Brazil) have a particular tropism for the CNS. Recent evidence suggests that BHV-5 uses an intranasal route to infect and replicate in the nasal mucosa and then enters the CNS by retrograde axonal transport predominantly through olfactory nerves.
Gross lesions in the CNS are nonspecific and include meningeal congestion, petechiation, hemorrhage, and malacia in the frontal and olfactory cortices. Microscopic lesions consist of dense perivascular infiltrates of lymphocytes, plasma cells, and macrophages accompanied by neuronal degeneration, vasculitis, necrosis, and presence of intranuclear acidophilic inclusions in neurons and astrocytes. Trigeminal ganglia, where the virus remains latent, often have some degree of ganglioneuritis, satellitosis, and Nageotte nodule formation.
Clinically, outbreaks of disease occur in young cattle ranging in age from 5 to 18 months. Lesions can also involve the eyes (conjunctivitis) and tissues of the reproductive, alimentary, and integumentary systems in addition to the nervous system.
Bunyaviruses
Schmallenberg Virus.
Schmallenberg virus causes fetal malformations in ruminants. It is an emergent orthobunyavirus that was first detected in Germany in 2011 before spreading widely throughout Europe. This virus appears to be a novel viral infection whose origin continues to be elusive. Evidence for a reemergent virus as a cause was not found when this novel virus was compared to older, stored samples that had similar pathologic findings. The virus is in the Bunyaviridae family and the Orthobunyavirus genus. The life cycle of the virus remains poorly characterized; however, it is believed to be similar to other members of the Bunyaviridae family. The virus is likely transmitted by a variety of Culicoides spp. (i.e., biting midges) vectors.
Adult sheep, cattle, and goats can develop fever, diarrhea, and reduced milk production associated with viral infection; however, the most important lesions are observed in fetuses secondary to circulating viremia and viral replication in the developing fetus. Similar to other members of the Bunyaviridae family like Akabane virus and Aino virus, fetal malformations are the primary manifestation of in utero infection with the virus. Gross lesions include congenital malformations of the fetus such as torticollis, ankyloses, arthrogryposis, brachygnathia inferior, hydranencephaly, porencephaly, and cerebral/cerebellar hypoplasia. The skull can be domed. Microscopically, affected CNS tissue has lymphocytic and histiocytic perivascular inflammation in the gray and white matter as well as overlying meninges. Inflammation occurs in animals that survive early gestational infection. Sporadic glial nodules can be found, most commonly in the mesencephalon and hippocampus. Diffuse gliosis consisting of increased numbers of astrocytes and microglia is also a consistent finding. Clinical signs in animals born alive include ataxia and behavioral abnormalities.
Other Bunyavirus Diseases.
Akabane disease, arthrogryposis hydranencephaly complex (Cache Valley fever), and Rift Valley fever are additional diseases caused by members of the Bunyaviridae family. These insect vector–transmitted viruses have a shared tropism for fetal tissues and cause congenital malformations and encephalitis.
Lentiviruses
Visna.
Visna, which means wasting in Icelandic, is a slowly progressive, transmissible disease of sheep. The disease is caused by a distinct viral strain of the ovine maedi-visna virus (MVV) complex, a lentivirus within the family Retroviridae. A different strain of the same virus causes a lymphocytic interstitial pneumonia referred to as maedi.
Visna is a persistent viral disease in which the elimination of the virus does not occur. Similar to other lentiviruses, the virus is cell associated, can undergo antigenic drift (see Chapter 4), and replicates slowly (hence lentivirus, from the Latin “lente,” meaning slow). Although the visna virus and other lentiviruses can infect promonocytes and monocytes in the bone marrow and blood, viral replication is restricted in these cells, where it remains as proviral DNA until their maturation and differentiation to macrophages. Primary viral replication occurs in cells of monocyte-macrophage-microglial lineage. Thus visna virus enters the CNS by way of leukocytic trafficking of virus-infected macrophages. Visna virus can also be present in oligodendrocytes and astrocytes located in foci of demyelination. Recent studies suggest the virus can also infect and productively replicate in endothelial cells. This mechanism may provide an additional route for viral entry into the CNS and the potential for alterations of the blood-brain barrier.
Although gross lesions are not frequently observed, they may occur in areas of prominent inflammation, where they appear as yellow-tan areas. Early microscopic lesions primarily affect the gray and white matter subjacent to the ependyma of the ventricular system of the brain and central canal of the spinal cord. These lesions are characterized by a nonsuppurative encephalomyelitis accompanied by pleocytosis, variable edema, CNS necrosis, astrocytosis, choroiditis, and nonsuppurative leptomeningitis. Degeneration of myelin sheaths also occurs at this time but is often accompanied by axonal degeneration, suggesting that it could be a secondary lesion, such as in Wallerian degeneration. Oligodendroglial and neuron injury and necrotic cell death are attributable to cytokines and other toxic factors secreted by inflammatory cells and glial cells. Recent in vitro studies suggest that caspase activation leading to apoptotic cell death may play a role in visna.
The primary demyelination that occurs during the late stages of the disease process (6 months to 8 years after infection) has been proposed to result from oligodendroglial cell infection. The main sources of excreted virus are the udder and lungs (as cell-associated virus), and transmission occurs most readily between the dam and lamb via the milk and between confined individuals, probably via respiratory secretions. MVV has also been detected in semen of infected rams. It is important to emphasize that no profound immune deficiency occurs with MVV infections, as is the case for immunodeficiency lentiviral infections that infect CD4+ T lymphocytes. Nonetheless, secondary infections can still significantly accompany MVV infection.
In addition to pneumonia, MVVs can also cause mastitis, arthritis (uncommonly of the carpus or hind limb joints), and a mesangial glomerulitis. Irrespective of the target organ, the lesions are to be regarded as chronic and lymphoproliferative, in contrast to the well-known immunodeficiency infections caused by human immunodeficiency virus 1, simian immunodeficiency virus, and feline immunodeficiency virus.
Viral strain differences and breed of sheep can influence the lesions that develop. For example, the visna form of the disease does not commonly occur in ovine breeds of North America, although some degree of visna-like lesions can be seen with maedi. Visna originally was described in Iceland, but sheep with similar lesions of the CNS have been detected in the Netherlands, Kenya, the United States, and Canada. CNS signs include an abnormal hind limb gait that progresses to incoordination and rear limb paresis over a period of weeks or months.
Caprine Leukoencephalomyelitis.
Caprine leukoencephalomyelitis was first described in the United States and has since been recognized in other parts of the world. The infection can be readily transmitted by colostrum and milk after birth, or by direct contact. A close relationship exists between the MVV of sheep and the causative agent of caprine leukoencephalomyelitis, caprine arthritis encephalitis (CAE) virus. As with MVV infection of sheep, caprine arthritis encephalitis viral infection is also a lymphoproliferative disease with a tropism for the macrophage, which acts as a carrier for the virus.
Gross lesions in the nervous system include tan- to salmon-colored foci of necrosis and inflammation that can occasionally be detected in the brain (Fig. 14-90 ) and most prominently in the spinal cord. Microscopic lesions of the nervous system are similar to those of visna (nonsuppurative encephalomyelitis) but can be more severe with caprine arthritis encephalitis. Nonneural lesions include interstitial pneumonia of moderate severity in some affected kids. Such lungs fail to collapse completely and are mottled red or blue. As discussed under visna, the mechanism of infection and cell death appear similar.
Figure 14-90.
Leukoencephalitis, Caprine Retrovirus-Induced Encephalitis, Brainstem, Goat.
A focal area of the brainstem (arrow) is yellow-brown and was found, microscopically, to be infiltrated by lymphocytes, plasma cells, and histiocytes. A similar lesion commonly occurs in the spinal cord.
(Courtesy Dr. H.E. Whiteley, College of Veterinary Medicine, University of Illinois.)
The pattern of disease in caprine arthritis encephalitis is age dependent. Neurologic manifestations are usually seen in young kids 2 to 4 months of age, but unlike visna in sheep, there is a more rapid progression and signs progressing to quadriplegia develop within weeks to months. As with visna, affected goats have pleocytosis. In adult goats the primary target tissue is the synovium of the joints, and animals that survive the initial infection have lymphoproliferative synovitis and arthritis develop. Pneumonia, lymphocytic mastitis, and encephalomyelitis also occur in adult animals.
Chlamydia
Sporadic Bovine Encephalomyelitis.
See E-Appendix 14-2.
Prions.
See the section on Disorders of Domestic Animals for additional discussion of prions.
Bovine Spongiform Encephalopathy.
Bovine spongiform encephalopathy was originally identified in the United Kingdom in 1986 but likely existed there as early as April 1985. Through the end of 2003, more than 183,000 cows from more than 35,000 herds were reported with BSE. With regard to the origins of BSE, epidemiologic evidence indicates that the disease was caused initially (during the early 1980s) by the feeding of rations containing meat and bone meal supplements that were contaminated with the scrapie agent. Countries that have reported cases of BSE or are considered to have a substantial risk to have animals with BSE include Albania, Austria, Belgium, Bosnia-Herzegovina, Bulgaria, Canada, Croatia, Czech Republic, Denmark, Yugoslavia, Finland, France, Germany, Greece, Hungary, Ireland, Israel, Italy, Japan, Liechtenstein, Luxembourg, Macedonia, the Netherlands, Norway, Oman, Poland, Portugal, Romania, Slovak Republic, Slovenia, Spain, Sweden, Switzerland, the United States, and the United Kingdom (Great Britain, including Northern Ireland and the Falkland Islands). Since the number of affected animals peaked in the 1990s and early 2000s, the number of animals affected yearly has diminished to only small numbers (<6/year).
In December 2003, BSE was confirmed in a single dairy cow from a herd in Washington State. This cow was apparently imported into the United States 2 to 3 years earlier from a farm in northern Alberta, Canada, which was the source of the positive BSE case in Canada confirmed in May 2003. In 2004, additional cases of BSE likely linked to a common source were reported in Washington State, Oregon, and Canada, and in June 2005 a cow tested positive for BSE in Texas.
Signs that accompany BSE include changes in behavior such as nervousness or aggressiveness, abnormal posture, abnormal gait, incoordination, difficulty in rising, decreased milk production, and loss of body weight despite continued appetite. Clinically, affected cattle deteriorate until they either die or require euthanasia. This clinical period usually ranges from 2 weeks to 6 months. All cases of the disease in cattle have occurred in adult animals, with an age range of 3 to 11 years, but most animals have clinical signs develop between 3 and 5 years of age.
The National Veterinary Services Laboratory (Animal and Plant Health Inspection Service [APHIS]) of the U.S. Department of Agriculture has developed and implemented through local and regional veterinary diagnostic laboratories procedures for sampling, preparing, and submitting brains for BSE analysis. Space limitations prevent describing these procedures. They can be obtained through APHIS or a diagnostic laboratory. Dead or sick animals that display neurologic signs and are suspected of being potential BSE candidates are tested using immunohistochemical analysis, Western blot analysis, or enzyme-linked immunosorbent assay (ELISA) to identify PrPSc in brain tissue. Genetically valuable animals that may be exported or have their embryos or DNA saved for future use and are from transmissible spongiform encephalopathy risk groups can be tested for PrPSc. Immunohistochemical analysis for PrPSc of samples obtained (i.e., under anesthesia) from biopsies of tonsil, rectal lymphoid tissue, and lymphoid tissue of the third eyelid can be performed ante mortem.
Ovine Spongiform Encephalopathy (Scrapie).
Scrapie is best known as a degenerative disease that affects the CNS of sheep and was first recognized in Great Britain and other countries of western Europe more than 250 years ago. The disease, which is currently reported throughout the world except for Australia and New Zealand, also occurs naturally in the domestic goat. The name is derived from the characteristic clinical signs of pruritus, which often results in loss of wool in sheep. The disease progresses inexorably with early signs of subtle change in behavior or temperament followed by scratching and rubbing against fixed objects because of the pruritus. Additional signs include incoordination, weight loss (despite retention of appetite), biting of the feet and limbs, lip smacking, gait abnormalities, trembling (when suddenly stressed), recumbency, and eventually death after 1 to 6 months or longer.
Much of our current understanding of the pathogenesis of the natural infection of scrapie in Suffolk sheep has been advanced by Dr. William Hadlow and coworkers. It should be noted that Dr. Hadlow is a veterinary pathologist and diplomate of the American College of Veterinary Pathologists, who first recognized and reported the similarity between scrapie and kuru of human beings. This important contribution led to the current understanding of the transmissible spongiform encephalopathies and a Nobel Prize for the medical scientist (Dr. D. Gajdusek) who originally investigated kuru in the South Pacific.
Degenerative Diseases
Metabolic
Primary Neuronal Degeneration
Primary Cerebellar Neuronal Degeneration.
Primary cerebellar neuronal degeneration has been reported to occur in Merino and Charolais lambs, Holstein-Friesian calves, and Angus calves. An autosomal recessive mode of inheritance is suspected or documented in several of the diseases, but the mechanism of injury is unclear. Grossly, the cerebellum can be normal or reduced in size and atrophic. Microscopically, lesions may include loss of Purkinje cells, variable neuronal depletion in the granule layer, fusiform swellings of proximal Purkinje cell axons, and astrogliosis in the molecular layer.
Animals with postnatal primary cerebellar neuronal degeneration are normal at birth or at the time of ambulation. Onset of ataxia with various other clinical signs referable to cerebellar disease begins weeks or months after a period of apparently normal development. Initial clinical signs are often subtle. Progression of the signs can be slow or rapid, relentless, or with static periods. Some individuals reach a stage without further progression of signs, but this is not typical of the syndrome in most animals.
Nutritional
Vitamin B1 (Thiamine) Deficiency
Thiamine Deficiency in Ruminants.
Thiamine deficiency in cattle, sheep, and less commonly, goats, has been termed polioencephalomalacia. Rumen microbes are able to synthesize thiamine, and therefore only very young ruminants, whose rumens have not yet been populated by thiamine-producing microbes, should be susceptible to thiamine deficiency. Hence conclusive evidence of an absolute thiamine deficiency as the sole cause of polioencephalomalacia in ruminants has been elusive. Evidence or theories linking thiamine with the ruminant disorder include the following:
-
1.
Clinical response to thiamine injection in some individuals.
-
2.
Decreases in ruminal thiamine or overgrowth of thiaminase-producing microbes such as Bacillus thiaminolyticus.
-
3.
Ingestion of thiaminase-containing plants such as bracken fern.
-
4.
Production of inactive thiamine analogues.
-
5.
Decreased absorption or increased fecal excretion of thiamine.
-
6.
Sulfur toxicosis can result in identical lesions through the process of sulfite cleaving thiamine.
Gross lesions, if present, are limited primarily to the cerebral cortex. Initially, 2 days after onset, the surface of the brain can be swollen (cerebral edema) as indicated by flattening of cerebrocortical gyri and narrow sulci. In rare cases with more severe brain swelling, brain displacement with herniation of the parahippocampal gyri beneath the tentorium cerebelli and the vermis of the cerebellum into the foramen magnum can occur. By 4 days after onset, yellow discoloration of the cerebrocortical gray matter occurs (Fig. 14-91 ), and it is at this time that autofluorescence (see later for more detail) is seen when the brain is examined under 365-nm ultraviolet light (Fig. 14-92 ). Eight to 10 days after onset, edematous separation and necrosis involving the middle to deep lamina or gray matter–white matter interface may be appreciable (Fig. 14-93 ). In advanced cases with prolonged survival, areas of marked atrophy of cerebral gyri with an attenuated or absent gray matter zone are covered by meninges (Fig. 14-94 ).
Figure 14-91.
Acute Polioencephalomalacia, Cerebral Cortex, Cross-Sectional View, Cow.
Gyri are yellow and swollen (arrows). The cause of this yellow color is unknown but has been shown experimentally not to be caused by ceroid-lipofuscin pigments. Changes involving the sulci and gyri in acute polioencephalomalacia are shown in Figure 14-93.
(Courtesy Dr. L. Roth, College of Veterinary Medicine, Cornell University.)
Figure 14-92.
Acute Polioencephalomalacia, Brain, Cerebral Hemispheres and Midbrain, Steer.
A, Compare the relative lack of gross lesions with those revealed by ultraviolet (UV) light in B. B, Bilaterally symmetric laminar pattern of apple-green autofluorescence (from mitochondrial derivatives) involving the full thickness of the cortex indicates areas of necrosis in the gray matter. Although not shown here, the colliculi were also autofluorescent. The cerebrum is exposed to 365-nm UV light from a Wood's lamp. Similar results can be obtained with fixed (preserved) brain.
(Courtesy Dr. P.N. Bochsler, School of Veterinary Medicine, University of Wisconsin-Madison.)
Figure 14-93.
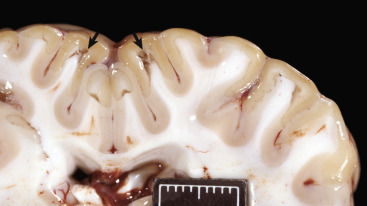
Acute cerebrocortical polioencephalomalacia, thiamine deficiency, brain, parietal lobe, level of thalamus, goat.
Note the liquefactive necrosis with varying degrees of tissue separation (arrows) in the deep cortex. Scale bar = 2 cm.
(Courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Figure 14-94.
Chronic Cerebral Cortical Atrophy, Brain, Cow.
Gyri are atrophic and narrow with widened sulci. In this case the loss of cerebral cortex was caused by thiamine deficiency several years previously.
(Courtesy College of Veterinary Medicine, University of Illinois.)
Microscopically, the earliest lesions are laminar cortical necrosis and astrocytic swelling. Laminar cortical necrosis is characterized by neuronal necrosis (ischemic change), with a laminar pattern of edema in the cerebral cortex. Neurons in the middle to deep lamina of the parietal and occipital lobes of the cerebral cortex are preferentially affected (Fig. 14-95, A ). In the early stages or in mild cases, lesions can be limited to the depths of cerebrocortical sulci, but generally there is involvement of entire gyri that may be confluent over extensive areas of the cortex. After 4 to 5 days, neuronal necrosis and edema are more severe, and there is an early influx of blood monocytes that mature into tissue macrophages and become gitter cells as they phagocytose necrotic debris. Macrophages and gitter cells are observed most commonly in perivascular and perineuronal spaces and in the pia arachnoid (see Fig. 14-23, A). After 8 to 10 days, necrosis and edema have resulted in laminar separation (at the gray matter–white matter interface) in which there are prominent accumulations of macrophages (see Fig. 14-95, B). Lesions that accompany the necrosis include vascular prominence caused by endothelial cell and perithelial cell hypertrophy and hyperplasia, congestion, and a minimal, if any, influx of neutrophils. Bilaterally symmetric focal lesions, similar to those seen in carnivores, occur in the thalamus and midbrain or colliculi and rarely in other brainstem structures. Animals that survive can have cerebral atrophy and develop hydrocephalus ex vacuo and have been known to live 1 to 2 years. It should be noted that laminar cortical necrosis can be caused by a variety of metabolic abnormalities. In ruminants, in addition to thiamine deficiency, water deprivation–sodium ion toxicosis, and lead poisoning can cause polioencephalomalacia and laminar cortical necrosis.
Figure 14-95.
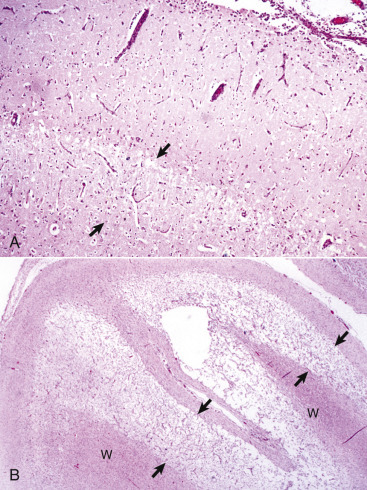
Polioencephalomalacia, Cerebral Cortex, Cross-Sectional View, Cow.
A, Acute stage. Note the zone of edema and acute neuronal necrosis affecting lamina 4-6 (area between arrows) of the cerebral cortex. Monocytes can be seen in the pia-arachnoid layer and subarachnoid space (upper right) in response to neuronal injury and the need to phagocytose cellular debris. Monocytes will also rapidly appear in perivascular spaces of blood vessels in the area of laminar edema and neuronal necrosis. H&E stain. B, Chronic stage. Areas of microcavitation in the deep cortical laminae next to the subcortical white matter are poorly stained (area between arrows) when compared with those of the normal superficial cortex (left). W, White matter. H&E stain.
(A courtesy Dr. W. Haschek-Hock, College of Veterinary Medicine, University of Illinois. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
When exposed to ultraviolet light (365-nm wavelength), the brains of affected ruminants may have autofluorescent bands of necrotic cerebral cortex. Autofluorescence is likely secondary to the accumulation of autofluorescent substances in degenerating neurons that seem to be localized to mitochondria. The belief that ceroid-lipofuscin pigments are the cause of the autofluorescence is still unproven. An association between mitochondria and autofluorescence has also been shown in the neuronal ceroid lipofuscinoses (Batten's disease, ceroid-lipofuscin storage diseases). These diseases are characterized by the accumulation of an autofluorescent (365-nm ultraviolet light), intracytoplasmic storage material in CNS neurons composed mostly of subunit c of mitochondrial ATP synthase.
Clinically, the disease is seen most commonly in cattle 6 to 18 months of age fed concentrated rations. In sheep, most cases occur in younger age groups (2 to 7 months). Clinical signs in cattle and small ruminants may include depression, stupor, ataxia, head pressing, apparent cortical blindness, opisthotonos, convulsions, and recumbency with paddling and then death. If animals survive or respond to therapy, clinical signs can persist.
Polioencephalomalacia commonly occurs in cattle fed rations rich in carbohydrates with little roughage and is also associated with clinical or subclinical acidosis that may precipitate changes in rumen microflora. The disorder also occurs in association with other dietary factors, including cobalt deficiency, molasses- and urea-based diets, and diets high in elemental sulfur and sulfates and sulfides, some of which are not specifically associated with thiamine deficiency.
There has been considerable interest in the relationship of high dietary sulfur to polioencephalomalacia. All sources of sulfur, including formulated rations, plants rich in sulfur (Kochia scoparia), and high concentrations of sulfur in the drinking water, are additive. Total dietary intake should not exceed 0.3% to 0.4%. The exact mechanism to explain sulfur-induced polioencephalomalacia in ruminants has not been proved but most likely involves interference with cytochrome oxidases thereby distrupting the mitochondrial respiratory chain.
Copper Deficiency.
Swayback and enzootic ataxia are descriptive terms used to describe the disease related to copper deficiency in lambs and kid goats. Swayback refers to the congenital form of the disease, whereas in enzootic ataxia, onset is delayed for up to 6 months after birth. Although copper deficiency is involved, the pathogenesis is likely complex and involves interplay with other metals, including zinc, iron, and molybdenum. Lesions occur in the cerebrum, brainstem, and spinal cord in the congenital form, but only in the brainstem and spinal cord in cases with a postnatal onset.
Additionally, deficiency of copper can affect wool, hair growth, and pigmentation; musculoskeletal development; and integrity of connective tissue. Copper deficiency can be primarily the result of copper-deficient soils and inadequate intake from forage or secondary from defective absorption because of interactions between copper, molybdenum, zinc, cadmium, or inorganic sulfates. Copper is a component of several enzyme systems, including cytochrome and lysyl oxidases, such as mitochondrial cytochrome-c oxidase; dopamine β-monooxygenase; peptidylglycine α-amidating monooxygenase; tyrosinase; and superoxide dismutase and the protein ceruloplasmin. These enzyme systems are essential for energy generation by mitochondria in the brain, regulating oxidative stress, catecholamine synthesis, and the modification of peptide neurotransmitters.
The pathogenesis of the lesions that occur in swayback and enzootic ataxia is poorly understood. It has been suggested that cerebral lesions result from loss of embryonic cells at the same stage of brain development as occurs in porencephaly and hydranencephaly after in utero viral infection or dysgenesis caused by a biochemical disturbance. Biochemical disturbances could also account for the axonal/neuronal degeneration in the brainstem and spinal cord. Altered function of the mitochondrial enzyme cytochrome oxidase leading to energy failure could play a role in cerebral dysgenesis and axonal and ultimately neuronal degeneration.
More intriguing is the possible involvement of the enzyme copper-zinc superoxide dismutase. A mutation in this enzyme is present in approximately 20% of human beings with familial amyotrophic lateral sclerosis and in some individuals with the sporadic form of the disease. This human disorder, which is classified as a motor neuron disease, has a much later onset, and the mutation results in a “gain of function” of the enzyme rather than lack of function as might be present in copper deficiency. Neurofilament accumulation in brainstem and spinal ventral horn neurons and fiber tract degeneration (corticospinal in human beings versus spinocerebellar in lambs and kids with copper deficiency) are similar. Drawing a relevant association between these diseases of human beings and abnormal function of superoxide dismutase in swayback and enzootic ataxia in animals is premature at this time, however. It is also possible that the cortical white matter degeneration and brainstem or spinal lesions arise from different mechanisms.
Grossly, approximately 50% of congenitally affected lambs and rarely kids have bilateral cerebrocortical lesions. Externally the cerebral cortex can be focally soft and fluctuant or collapsed. These foci correspond to areas of rarefaction, which have either a gelatinous consistency or cystic cavities filled with clear serous fluid, in the white matter of the corona radiata and centrum semiovale. Microscopically, a variable astrogliosis is associated with the degeneration of white matter, but the cavities lack a capsule of glial fibers. Myelin degradation and an influx of macrophages are minimal. Delicate neuronal and astroglial processes traverse the cavities. Neuronal necrosis in cortical gray matter overlying these white matter lesions is sometimes observed.
Microscopic lesions in the brainstem and spinal cord in both the congenital (swayback) and delayed-onset (enzootic ataxia) forms of copper deficiency are similar in lambs and kids, and they affect both gray and white matter. Large multipolar neurons of the brainstem reticular formation, certain brainstem nuclei—such as the red and vestibular nuclei—and the ventral, lateral, and less commonly, dorsal horns of the spinal cord are affected. Neuronal cell bodies lack stainable Nissl substance (chromatolysis). The cytoplasm is variably dense, pink, and homogeneous to fibrillar as a result of the accumulation of neurofilaments, and nuclei are often displaced to an eccentric position against the cell membrane. The extent of neuronal necrosis varies. Lesions in the white matter of the spinal cord consist of bilateral areas of pallor in the dorsolateral aspects of the lateral funiculi (corresponding roughly to the spinocerebellar tracts) and also in the ventral funiculi adjacent to the ventral median fissure. The pallor of the white matter is due to degeneration of myelinated axons. Involvement of the medial (septomarginal) aspect of the dorsal funiculi is infrequent. In the terminal brainstem, lesions are similar but have a somewhat scattered distribution. The spinocerebellar tracts extending into the middle cerebellar peduncles are affected. Astrogliosis is usually mild. Definitive microscopic lesions in the cerebellum and ventral spinal nerve rootlets and peripheral nerves are usually lacking in lambs but can be frequent in kids. Changes in Purkinje cells are analogous to those already noted in neurons in other areas. Additionally, ectopic Purkinje cells and thinning of the granule cell layer occur. Bergmann glial cell processes in the molecular layer undergo hypertrophy. Lesions in ventral spinal nerve rootlets and peripheral nerve are caused by axonal degeneration secondary to injury of motor neurons in the ventral gray horns.
Clinically, CNS disease as a result of copper deficiency in animals is mainly a disorder of sheep and goats and can be present at birth (swayback in lambs, rarely kids), or onset can be delayed up to 6 months (enzootic ataxia in lambs and kids). Swayback occurs in newborn lambs from ewes with inadequate dietary copper intake. Affected lambs can be born dead, weak, or unable to stand. If mobile, they are ataxic. Enzootic ataxia is characterized by ataxia.
Toxicoses
Microbial Toxins
Clostridium perfringens Type D Encephalopathy (Pulpy Kidney Disease, Overeating Disease).
Clostridium perfringens type D enterotoxemia, associated with epsilon toxin production, is a disease of sheep, goats, and cattle, but only sheep commonly exhibit the neurologic manifestations of the disease. Brain damage is due to vascular injury and breakdown of the blood-brain barrier. Binding of epsilon toxin to endothelial cell surface receptors results in opening of tight junctions, disturbed transport processes, increased vascular permeability that results in vasogenic edema, swelling of astrocytic foot processes, and ultimately necrosis caused by hypoxic-ischemic mechanisms. Some of the effects of epsilon toxin can be mediated by an adenyl cyclase–cyclic adenosine monophosphate (cAMP) system.
Gross lesions are absent in some peracute cases but when present, consist initially of bilaterally symmetric foci of malacia, leading to yellow-gray to red foci with malacia and cavitation (Fig. 14-96, A ). Lesions can be found in the internal capsule, basal nuclei, thalamus, hippocampus, rostral colliculus and substantia nigra, pons, corona radiata of frontal cortex, and cerebellar peduncles, especially the middle peduncle. Lesions in other tissue consist of pulmonary congestion and edema, serous pericardial effusion, petechiation, and soft (pulpy) kidneys (see Fig. 11-42).
Figure 14-96.
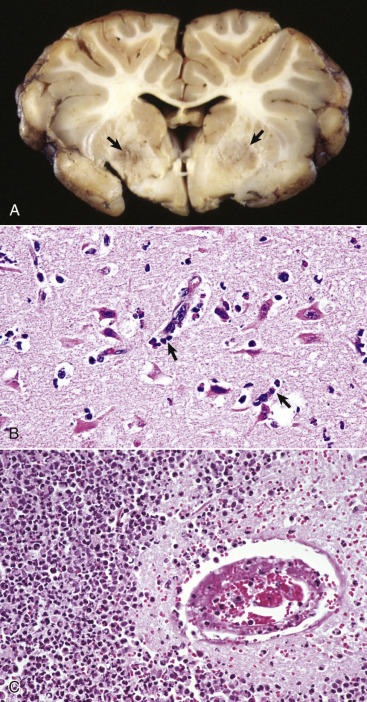
Focal Symmetric Encephalomalacia, Brain, Transverse Section at the Level of the Basal Nuclei and Rostral Thalamus, Sheep.
A, There are bilateral discoloration and malacia (arrows) in a portion of the basal nuclei. These lesions are caused by an enterotoxin produced by Clostridium perfringens type D. B, Early stage. Note the acute neuronal necrosis (red neurons) with perivascular and perineuronal edema. Inflammatory cells (arrows), including neutrophils and macrophages, are beginning to appear in the perivascular space and will migrate to the necrotic neurons and phagocytose the debris. H&E stain. C, Later stage. Microscopically, the walls of arterioles can be hyalinized and endothelial cell nuclei swollen and vesicular (not shown here) with perivascular leakage of proteinaceous fluid. Pericapillary hemorrhage and acute necrosis of neurons and macroglia can occur. With longer survival, as seen here, lesions include destruction of neuropil, accumulation of neutrophils and foamy macrophages, and lymphocytic perivascular cuffing. In this case the inflammatory response is pronounced and would not be considered typical of this disease. H&E stain.
(A courtesy Dr. D. Cho, College of Veterinary Medicine, Louisiana State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. B.E. Walling, College of Veterinary Medicine, University of Illinois. C courtesy Dr. J. Simon, College of Veterinary Medicine, University of Illinois.)
Microscopically, the CNS lesion in acute cases is vasogenic edema secondary to vascular injury. The fluid in perivascular spaces is frequently protein rich and eosinophilic. Walls of arterioles can be hyalinized, and endothelial cell nuclei swollen and vesicular (see Fig. 14-96, B). Vasogenic edema, which is interstitial in location, gives a light or pink-staining, spongy appearance to the CNS parenchyma. Both gray matter and white matter are affected. Pericapillary hemorrhage and acute necrosis of neurons and macroglia occur. Other changes occur with longer survival and include axonal swelling, accumulation of neutrophils and foamy macrophages (see Fig. 14-96, B and C), vascular prominence caused by perithelial-endothelial nuclear enlargement or swelling, lymphocytic perivascular cuffing, and liquefactive necrosis.
Sheep of all ages, except newborns, are susceptible; incidence peaks between 3 and 10 weeks of age and in feeder lambs shortly after arrival at a feedlot. Resistance of newborns can be related to lack in the gut of pancreatic proteolytic enzymes necessary for activation of the epsilon toxin, and to trypsin inhibitors in colostrum. Lambs are typically in good condition and are found dead.
Disorders of Pigs
Disorders that occur in many or all animal species are discussed in the section on Disorders of Domestic Animals.
Diseases Caused by Microbes
Viruses
Enteroviruses
Enterovirus-Induced Porcine Polioencephalomyelitis.
See E-Appendix 14-2.
Coronaviruses
Hemagglutinating Encephalomyelitis.
See E-Appendix 14-2.
Herpesviruses
Pseudorabies.
Pseudorabies virus (suid herpesvirus 1), an alphaherpesvirus, causes encephalitis primarily in pigs; although a wide variety of domestic and wild animals are also susceptible. The ingestion of infected pig meat is the traditional source of infection in domestic dogs and cats. The disease is also known as Aujeszky's disease and “mad itch” and is not zoonotic. Pseudorabies is not related to rabies but was named because its clinical signs sometimes resemble those seen with rabies. The disease in susceptible species other than pigs is generally fatal. Although pigs—particularly young, suckling piglets—can die from infection, most mature pigs remain persistently infected and act as latent carriers.
The route of natural infection in pigs is intranasal, pharyngeal, tonsillar, or pulmonary by direct contact or aerosolization, followed by reproduction of virus in epithelial cells of the upper respiratory tract. The virus then travels to the tonsil and local lymph nodes by way of the lymph vessels. After replication in the nasopharynx, virus invades sensory nerve endings and is then transported in axoplasm via the trigeminal ganglion and olfactory bulb to the brain. The virus has also been reported to be capable of spreading transsynaptically. Recent studies have additionally shown that some strains produce lesions in the gastrointestinal tract and myenteric plexuses, suggesting that infection might spread from the intestinal mucosa to the CNS via autonomic nerves. In latently infected pigs the oronasal epithelium can be recurrently infected by virus spreading from the nervous system, followed by its excretion in oronasal fluid. The virus can also spread hematogenously, although in low titer, to other tissues of the body. As an example, it can cause placenta disease in pregnant sows and is an important cause of fetal maceration and mummification in the pig. Cellular attachment, entry, and cell-to-cell spread of the virus are mediated by glycoprotein projections that extend from the surface of the viral particle.
Gross lesions in pigs occur in several nonneural tissues, including organs of the respiratory system, lymphoid system, digestive tract, and reproductive tract. Focal tissue necrosis also occurs in the liver, spleen, and adrenal glands, particularly in young suckling pigs, and mortality can be high. Evidence of facial pruritus is a common sequela to infection, although the actual incidence varies considerably. The CNS is free of gross lesions except for nonspecific leptomeningeal congestion. Microscopic lesions in pigs are characterized by a nonsuppurative meningoencephalomyelitis with trigeminal ganglioneuritis. Injury to CNS tissue can be marked, with neuronal degeneration and necrosis. Intranuclear amphophilic inclusion bodies are not commonly detected in pigs but can be present in neurons and astrocytes. In cattle, sheep, dogs, and cats, the pathogenesis involving axonal spread to the CNS is comparable to that of pigs, with lesions that include nonsuppurative encephalomyelitis accompanied by ganglioneuritis and intraneuronal inclusion bodies
Pestiviruses
Classical Swine Fever (Hog Cholera).
See E-Appendix 14-2 and Chapters 4 and 10.
Degenerative Diseases
Toxicoses
Microbial Toxins
Edema Disease (Enterotoxemic Colibacillosis).
Edema disease is a disorder of rapidly growing, healthy feeder pigs being fed a high-energy ration. The disease is caused by strains of E. coli producing a Shiga-like toxin, which is similar to toxins produced by Shigella dysenteriae and is designated Shiga-like toxin type IIe. Serotypes O138, O139, and O140 seem to be most commonly reported. This toxin causes necrosis of smooth muscle cells in small arteries and arterioles and a reduction focally in the degree of circulation to the CNS parenchyma, leading to infarction manifested grossly as malacia in the CNS. Glycolipid cell surface receptors on endothelial cells, globotriaosylceramide or globotetraosylceramide, are binding sites for the toxin, and their presence confers susceptibility to the disease. Binding of the toxin to these receptors can initiate a chain of inflammatory and immunologic reactions that lead to vascular damage.
The basic lesion is an angiopathy that leads to edematous and hypoxic-ischemic injury in a variety of tissue, including the brain. Grossly, edema is present in the subcutis often prominent in the palpebrae, cardiac region of the gastric submucosa, gallbladder, colonic mesentery, mesenteric lymph nodes, larynx, and lungs, and serous effusions occur in the thoracic cavity and pericardial sac (see Figs. 7-169 and 7-170). Congestion and sometimes hemorrhage also occur. The characteristic gross lesions in the brain are usually bilaterally symmetric foci of necrosis in the caudal medulla, but they can extend rostrally as far as the basal nuclei. The lesions are yellow-gray, soft, and slightly depressed.
The primary microscopic lesion, a degenerative angiopathy/vascular necrosis, is noted most frequently and is most severe in the caudal medulla to the diencephalon and in cerebral and cerebellar meninges (see Fig. 10-69). Cerebral, cerebellar, and spinal blood vessels are also affected. Initially, perivascular edema results from early vascular injury. Edema is followed by necrosis of medial smooth muscle cells, deposition of fibrinoid material, and accumulation of macrophages and lymphocytes in the adventitia; however, inflammation is not a primary process in this disease. Although endothelial cells and their nuclei become swollen and vesicular, this layer generally remains intact, and consequently thrombosis is not a feature. Lesions associated with the angiopathy include pallor and spongiosis of the CNS parenchyma caused by vasogenic edema and necrosis of neurons and glia. An influx of macrophages into the necrotic lesions can be observed in chronic lesions.
Pigs are usually 4 to 8 weeks of age, but younger and older pigs can be affected. Clinically, affected animals are initially ataxic and then become laterally recumbent with rhythmic paddling of the limbs. As the disease progresses, they may become comatose and die. Most pigs die within 24 hours; however, pigs that survive for several days typically develop CNS lesions (see Table 14-1).
Disorders of Dogs
Disorders that occur in many or all animal species are discussed in the section on Disorders of Domestic Animals.
Diseases Caused by Microbes
Viruses
Morbillivirus
Canine Distemper.
Canine distemper is one of the most important viral diseases of the canine species. It is caused by a Morbillivirus (family Paramyxoviridae) and has a worldwide distribution. Morbilliviruses other than CDV include measles virus, rinderpest virus, peste-des-petits-ruminants virus, phocine distemper virus of seals, and dolphin and porpoise morbilliviruses. The virus is pantropic and has a particular affinity for lymphoid and epithelial tissues (lung, gastrointestinal tract, urinary tract, skin) and the CNS (including the optic nerve). In the CNS there is demyelination without any substantial amount of inflammation.
Dr. Brian Summers (formerly of Cornell University, College of Veterinary Medicine) has commented on the sequence of events in CDV infection based on his studies of its pathogenesis. CDV is spread between dogs by aerosol transmission. The virus is trapped in the mucosa of the nasal turbinates (centrifugal turbulence), infects local macrophages, and is spread by macrophages (leukocytic trafficking) to regional lymph nodes (retropharyngeal). CDV replicates in these regional lymph nodes, and replication is followed by a primary viremia that infects systemic lymph nodes, spleen, and the thymus approximately 48 hours after exposure. With infection of the lymphoid system, immunosuppression can occur, resulting in secondary bacterial infections, such as conjunctivitis, rhinitis, and bronchopneumonia, which are commonly seen in CDV infections.
Four to 6 days after the primary viremia, a secondary viremia occurs largely via leukocytic trafficking. CDV spreads from cells of the lymphoid system to infect the CNS and epithelial cells of the respiratory mucosa, urinary bladder mucosa, and gastrointestinal tract. In the CNS, trafficking leukocytes form perivascular cuffs, and from these cells CDV is disseminated throughout the CNS. It should be noted that the degree of inflammation in the CNS at this stage is minimal.
Under laboratory conditions the severity of the disease and the cell populations and areas infected in the CNS depend on the (1) age of the dog, (2) strain of CDV, and (3) kinetics of the antiviral immune response. Virtually all cells of the CNS, including those in the meninges, choroid plexus, neurons, and glia, are susceptible to infection, but oligodendrocytes are novel in that the infection in these cells is usually defective (incomplete). In dogs infected experimentally with the A75-17 strain of CDV, isolated from a dog with CDV in 1975, approximately one-third of the dogs died from encephalomyelitis and the effects attributable to severe immunosuppression. One-third of infected dogs developed a timely systemic immune response, and the CNS disease was quickly resolved and they recovered. Finally, one-third of the dogs infected with CDV developed a subacute to chronic inflammatory/demyelinating disease of the white matter with some gray matter involvement because of a delayed and deficient immune response.
The encephalomyelitis of canine distemper is initiated after viral entry into the CNS, perhaps 1 week after exposure to CDV. Leukocytic trafficking spreads CDV to the gray and white matter of the CNS and to epithelial cells and macrophages of the choroid plexuses. The virus is shed from choroid plexus epithelial and ependymal cells into the CSF in infected macrophages. It is disseminated throughout the ventricular system, infects ependymal cells lining the ventricular system, and then spreads locally to infect astrocytes and microglia. Periventricular white matter lesions, especially those surrounding the fourth ventricle, are the result of this sequence of events.
By 25 days after exposure, CDV-infected leukocytes in the perivascular cuffs have disappeared; however, lesions in the white matter consist of periventricular foci of myelin degeneration and swollen astrocytes. CDV infects astrocytes, microglia, and other cells. Microglia and recruited blood monocytes phagocytose myelin fragments.
Inflammatory mediators released from lymphocytes, microglial cells, and trafficking macrophages result in expansion of the initial lesions. These inflammatory mediators may cause necrosis of cells and cell processes in the focus but do not result in a “selective” demyelinating process affecting axons. The characteristic white matter vacuolation (intramyelinic edema) seen in H&E-stained sections of CDV-infected CNS is apparently caused by a direct effect of the virus on oligodendrocytes, because it appears in the earliest white matter lesions before they have acquired an “inflammatory” character—a dense infiltrate of lymphocytes, monocytes, and plasma cells.
Gross lesions characteristically occur in the cerebellum (medullary area, folial white matter, and subpial white matter) and cerebellar peduncles (with both white matter and sometimes gray matter involvement of the pons). Lesions also occur in medulla oblongata (particularly in the subependymal area of the fourth ventricle), rostral medullary vellum, cerebrum (both white matter and gray matter), optic nerves, optic tracts, spinal cord, and meninges.
Microscopically, in addition to demyelination there is status spongiosus, astrocytic hypertrophy and hyperplasia with focal and variable syncytial cell formation, reduced numbers of oligodendroglia, and variable neuronal degeneration (Fig. 14-97, A ). Inclusion bodies (cytoplasmic, nuclear, or both) are detectable, particularly in astrocytes, which are important target cells for the distemper virus, but also in ependymal cells and occasionally in neurons (Fig. 14-97, B). The earliest evidence of myelin injury is a ballooning change resulting from a split in the myelin sheath or more degenerative changes, including axonal swelling. This lesion is also variably associated with astroglial and microglial proliferation. This initial injury of the myelin sheath, which has been suggested to be a result of perturbed astrocytic function after viral infection, is followed by a progressive removal of compact myelin sheaths by phagocytic microglial cells that infiltrate the myelin lamellae and variable axonal necrosis.
Figure 14-97.
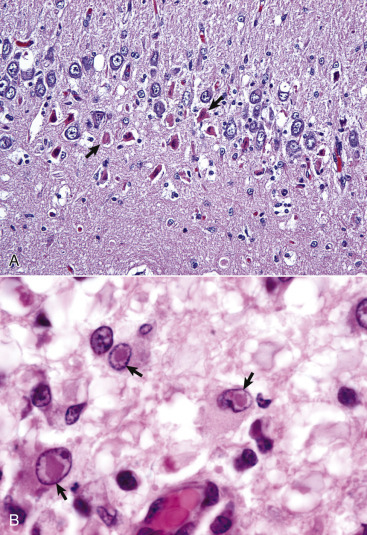
Canine Distemper, Dog.
A, Acute polioencephalomyelitis. Hippocampus. Note the necrotic neurons (arrows) and edema of the dentate gyrus. Low numbers of mononuclear inflammatory cells are present. H&E stain. B, Inclusion bodies, brain, midbrain periventricular white matter, dog. Distinct acidophilic (red) intranuclear inclusion bodies (arrows) are present in astrocytes and some gemistocytes. Similar inclusions can be observed in the cytoplasm of epithelial cells throughout the body (bladder epithelium, respiratory epithelium, gastric epithelium). H&E stain.
(A courtesy Dr. W. Haschek-Hock, College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
A late stage of demyelination, which is a reflection of an affected animal's improved immune status, is more pronounced and is characterized by nonsuppurative inflammation (perivascular cuffing, leptomeningitis, and choroiditis) and also can be accompanied by tissue degeneration and accumulation of gitter cells.
In addition to the dog, animals of the families Ailuridae (red panda), Canidae (fox, wolf), Hyaenidae (hyena), Mustelidae (ferret, mink), Procyonidae (raccoon, panda), Ursidae (bear), Viverridae (civet, mongoose), and Felidae (exotic felids including lions, tigers, and leopards) are susceptible to canine distemper viral infection. Additionally, canine distemper has recently been reported in javelinas (collared peccaries) of the family Tayassuidae in the United States.
Neurologic signs in all affected species include convulsions, myoclonus, tremor, disturbances in voluntary movement, circling, hyperesthesia, paralysis, and blindness.
Old-Dog Encephalitis.
Old-dog encephalitis is thought to arise from long-term persistent infection of the CNS with a defective form of CDV. This pathogenesis has been demonstrated in experimental infections with the CDV. Although the virus has the same general polypeptide composition and contains all of the major viral proteins as the one causing conventional distemper, some differences among peptides have been reported. The mechanisms involved in development of lesions are not known; however, they result in a proliferation of nonsuppurative inflammatory cells.
Lesions are primarily in the cerebral hemispheres and brainstem. Microscopic lesions are characterized primarily by demyelination with disseminated, nonsuppurative encephalitis with variable, sometimes prominent, lymphoplasmacytic perivascular cuffing, microgliosis, astrogliosis, and variable leptomeningitis and neuronal degeneration. Nuclear and cytoplasmic inclusions, positive for distemper viral antigen, have been detected in neurons and astrocytes in the cerebral cortex, thalamus, and brainstem but in contrast to distemper, not in the cerebellum.
Old-dog encephalitis is a rare condition occurring in mature adult dogs. Clinical signs include depression, circling, head pressing, visual deficits, seizures, and muscle fasciculations.
Degenerative Diseases
Metabolic
Aging-Related Degenerative Myelopathy (German Shepherd Myelopathy).
A degenerative myelopathy is most commonly seen in the German shepherd, but a similar condition has been described in other predominatly large canine breeds (Belgium shepherd, Old English sheepdog, Rhodesian ridgeback, Weimaraner, Pembroke Welsh corgi, and Great Pyrenees). Based on its prevalence in German shepherds, it has been suggested that there is a genetic “aging” predisposition in this breed. Altered suppressor lymphocyte activity has been noted in affected dogs, but the relevance to the CNS disease is unknown. Some investigators have reported low vitamin E concentrations and suggested oxidative stress injury; others have found elevated concentrations of acetylcholinesterase in CSF. The cause of this disorder remains to be discovered.
Gross lesions in the CNS are not present in dogs with age-related degenerative myelopathy; however, atrophy of caudal axial and appendicular muscles occurs. Microscopic lesions are most notable in the thoracic spinal cord and can be diffuse or multifocal. Dorsal aspects of the lateral and ventromedial areas of ventral funiculi can be more severely affected, but lesions can be diffuse in all funiculi. Lesions consist predominantly of ballooning and degeneration of myelin sheaths accompanied by axonal degeneration and loss. Degeneration of dorsal nerve rootlets and peripheral nerves, loss of neuron cell bodies in spinal gray matter, and involvement of brainstem nuclei are also described.
Clinically, affected dogs are usually older than 8 years, but the disease has occurred as early as 5 years of age. They have progressive ataxia referable to the thoracolumbar spinal cord and muscle weakness.
Primary Neuronal Degeneration
Multisystem Neuronal Degeneration
Progressive Neuronal Abiotrophy of Kerry Blue Terriers.
Progressive neuronal abiotrophy of Kerry blue terriers (hereditary striatonigral and cerebello-olivary degeneration of Kerry blue terriers) is a well-characterized example of a disease with multisystem neuronal degeneration. The disease has an autosomal recessive inheritance pattern and affects connected neural systems, including basal nuclei and the substantia nigra (i.e., striatonigral) and the cerebellar cortex and caudal olivary nucleus (cerebello-olivary).
The pathogenesis is unknown, but an excitotoxic mechanism associated with abnormalities in the glutaminergic corticostriatal and in the granule–Purkinje cell neurotransmitter systems is proposed. The caudate nucleus and cerebellar cortex are believed to be the primary sites of involvement, whereas lesions in the olivary nucleus and substantia nigra represent transsynaptic degeneration.
Gross lesions are a slight reduction in size of the cerebellum and narrowing of cerebellar folia (Fig. 14-98 ). In more advanced stages, small foci of softening and discoloration occur in the caudal olivary nucleus and substantia nigra (Fig. 14-99 ). Lesions in the caudate nucleus initially consist of vague areas of pallor that progress to marked malacia and cavitation. There can be similar involvement of the putamen at this advanced stage. Microscopic lesions in chronologic order are degeneration and loss of cerebellar Purkinje and granule cells, followed by neuronal loss in the caudal olivary nucleus, caudate nucleus, putamen, and substantia nigra. Astrogliosis occurs in later stages and is especially prominent in the cerebellar molecular layer. The caudate nucleus and putamen are eventually reduced to microcystic cavities bisected by a few nerve and glial fibers.
Figure 14-98.
Striatonigral and Cerebello-Olivary Degeneration, Brain, Cerebellum, Kerry Blue Terrier.
Marked atrophy and thinning of folia of dorsal cerebellum (arrows) has resulted in increased width of sulci.
(Courtesy Drs. D. Montgomery and R. Storts, College of Veterinary Medicine, Texas A&M University.)
Figure 14-99.
Striatonigral and Cerebello-Olivary Degeneration, Brain, Rostral Parietal Lobe, at Level of Optic Chiasm, Kerry Blue Terrier.
Note malacia (softening), microscopically due to microcavitation and loss of neurons, in caudate nuclei (arrows) and putamen (arrowheads).
(Courtesy Drs. D. Montgomery and R. Storts, College of Veterinary Medicine, Texas A&M University; and Vet Pathol 20:143-159, 1983.)
Affected dogs typically start to develop clinical signs in the first 2 to 5 months of age. Clinical signs include rear limb ataxia, intention tremors, hypermetria of front and rear limbs, and atrophy of appendicular and epaxial muscles, presumably from disuse.
Multisystem Neuronal Degeneration of the Red-Coated English Cocker Spaniel.
Multisystem neuronal degeneration of the red-coated English cocker spaniel is suspected of being inherited. The pathogenesis is unknown. Bilaterally symmetric diffuse nerve cell loss, astrogliosis, and axonal swellings occur in several nuclei, including septal nuclei, globus pallidus, subthalamic nuclei, substantia nigra, tectum, medial geniculate bodies, and cerebellar and vestibular nuclei. Central cerebellar white matter, corpus callosum, thalamic striae, and subcortical (particularly subcallosal gyri) white matter involvement also are noted. White matter lesions consist of axonal spheroids, intense astrogliosis, subtle myelin loss, and perivascular accumulation of macrophages. Clinical signs occur during the first year of life and consist of progressive ataxia and behavioral changes.
Multisystemic Neuronal Degeneration of the Cairn Terrier.
Multisystemic neuronal degeneration of the Cairn terrier has features of an inherited disorder. The pathogenesis is unknown. Widespread neuronal chromatolysis affecting multiple neuronal systems is observed in the CNS and PNS. Affected areas include brainstem sensory and motor nuclei, cerebellar nuclei, ventral and dorsal gray columns of the spinal cord, and various ganglia. Other lesions include degeneration in lateral and ventral spinal funiculi, necrosis of substantia gelatinosa and adjacent white matter most notable in caudal thoracic and cranial lumbar segments, and degeneration in dorsal and ventral spinal nerve rootlets and peripheral nerves. Clinical signs occur usually by 5 months of age, and there is onset of progressive cerebellar ataxia, spastic paresis, and collapse.
Primary Cerebellar Neuronal Degeneration.
Primary cerebellar neuronal degenerations (referred to as abiotrophies) occur in many breeds of dogs, including American Staffordshire terriers, Australian kelpie, Italian hound, border collie, Brittany spaniel, beagle, Portuguese podengo, and Scottish terriers. This list is not inclusive and serves only to provide a few examples. Although most common in young animals of the breeds listed, cerebellar degeneration in the American Staffordshire terrier and Brittany spaniel occurs later in life. An autosomal recessive mode of inheritance is suspected or documented in several of the diseases. Grossly, the cerebellum can be normal or reduced in size and atrophic. Microscopically, the distribution and characteristics of lesions vary, depending on the breed and species of animal affected. A detailed discussion of the microscopic lesions for each breed and species is outside the scope of this chapter; however, lesions may include a combination of the following changes: loss of Purkinje cells and neurons of the granule layer, astrogliosis, fusiform swelling of proximal Purkinje cell axons, and axonal degeneration in the cerebellum, brainstem, and spinal cord.
Neuronal Vacuolation and Spinocerebellar Degeneration.
A syndrome causing neuronal vacuolation and spinocerebellar degeneration has been reported most commonly in Rottweiler dogs as well as sporadically in boxers and mixed-breed dogs. The cause of this disease has not been determined but appears to have a hereditary basis. Apoptotic cell death is apparently not involved in neuronal degeneration. No gross lesions are observed in the brain, but atrophy of the dorsal cricoarytenoid muscles of the larynx has been seen. Microscopic lesions are characterized by spongiform change affecting neuron cell bodies and the neuropil. The cytoplasm of neurons of the cerebellar nuclei and nuclei of the extrapyramidal system contain one or more clear vacuoles (1 to 45 µm in diameter). Similar vacuoles are found in neurons in dorsal nerve root ganglia, myenteric plexuses, and other ganglia of the autonomic nervous system. Purkinje cells are also vacuolated, and in terminal stages of the disease there is degeneration with segmental Purkinje cell loss.
Clinical signs, seen as early as 6 weeks (commonly between 3 and 8 months of age in both sexes), include generalized weakness, ataxia, proprioceptive deficits, and paresis that progress in severity over the course of the disease.
Nutritional
Vitamin B1 (Thiamine) Deficiency
Thiamine Deficiency in Carnivores.
In monogastric carnivores and human beings the relationship between neurologic disease and thiamine deficiency is firmly established, and lesions in these species are similar. In carnivores (dog, cat, mink, and fox), there is an absolute dietary requirement for vitamin B1. Dietary factors, such as the ingestion of fish containing thiaminase, deficient diets, or diets in which the vitamin has been destroyed by other means such as heating, can all lead to thiamine deficiency.
Gross and microscopic lesions are bilaterally symmetric and commonly involve the midlaminae of the cerebral cortex, with the occipital and temporal cortices most consistently affected. Additionally, a variety of brainstem nuclei are affected, with the caudal colliculus being the most common (Fig. 14-100 ). Lesions consist of neuronal vacuolation accompanied by neuronal degeneration and necrosis with variable hemorrhage, vascular proliferation, and astrogliosis, the latter of which is time dependent. If the degree of necrosis is severe enough, accumulation of gitter cells is a common sequela. Clinical signs in carnivores may include a combination of the following: anorexia, vomiting, depression, wide-based stance, ataxia, spastic paresis, circling, seizures, muscle weakness, recumbency, opisthotonus, coma, or death.
Figure 14-100.
Thiamine Deficiency Encephalopathy, Midbrain, Caudal Colliculi, Dog.
In the dog, lesions of thiamine deficiency encephalopathy are generally restricted to the brainstem. Note the symmetrically cavitated (malacic) lesions in the caudal colliculi (arrows) resulting from neuronal necrosis.
(Courtesy Dr. J. Edwards, College of Veterinary Medicine, Texas A&M University; and Dr. J. King, College of Veterinary Medicine, Cornell University.)
Miscellaneous Conditions
Dural Ossification.
Dural ossification (ossifying pachymeningitis and dural osseous metaplasia) is a metaplastic aging change of predominantly large breed dogs. Grossly, the dura of the cervical and lumbar enlargements of the spinal cord has reddish-brown plaques that histologically are composed of bone, adipose tissue, and hematopoietic cells. These plaques typically recapitulate the normal bone marrow (Fig. 14-101 ). Thoracolumbar pain in affected dogs is thought to arise after flexion/extension of the spinal column as a result of compression of either the spinal cord or the spinal nerve roots; however, they are generally viewed as incidental, age-associated lesions.
Figure 14-101.
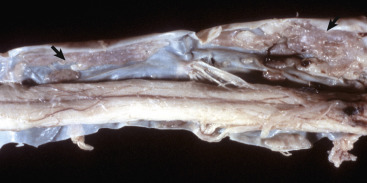
Osseous Metaplasia, Dura Mater, Dog.
Also called ossifying pachymeningitis and dural ossification, the dura mater contains well-differentiated bone and bone marrow (arrows). With movement of the vertebrae, the metaplastic bone can impinge on nerve roots and cause pain in large breed dogs.
(Courtesy College of Veterinary Medicine, University of Illinois.)
Inherited Necrotizing Myelopathy of Afghan Hounds.
Inherited necrotizing myelopathy of Afghan hounds has an autosomal recessive inheritance. A similar disease has been reported in Dutch Kooikerhondje dogs, but the relative degrees of axonal degeneration versus demyelination have not been adequately described.
Topographically, gross lesions are consistently observed in the midthoracic segments, with extension to the midcervical and midlumbar segments in the most severe cases. Gross lesions include softening and cavitation of the affected segments. Lesions are bilateral and symmetric regardless of the region affected. In the thoracic segments the ventral funiculi are most severely affected, although lesions can be found circumferentially around the spinal cord white matter. In the cervical and lumbar segments, the lesions tend to be focused either in the dorsal or ventral horns. Histologically, in severe lesions there is destruction of white matter, with an influx of macrophages accompanied by myelin degradation, glial cell loss, and vascular proliferation. The gray matter and the fasciculus proprius are typically spared in this disease. Neuronal cell bodies in spinal cord gray matter and ventral spinal nerve rootlets are similarly unaffected.
Clinical signs begin between 3 and 13 months of age and progress rapidly to paraplegia or tetraplegia within 1 to 3 weeks.
Leptomeningeal Fibrosis.
Aging dogs have varying degrees of leptomeningeal fibrosis involving the recesses of the cerebral sulci (Fig. 14-102 ). This lesion is not present in the leptomeninges covering the outermost surfaces of the gyri. This latter feature can be useful in differentiating meningeal fibrosis from suppurative meningitis. In cases of suppurative meningitis, exudate accumulates over the entire surface of the gyri.
Figure 14-102.
Meningeal Fibrosis, Leptomeninges (Pia-Arachnoid Mater), Dog.
In old dogs the leptomeninges can have areas of fibrosis (white areas around blood vessels in sulci), particularly in the sulci. This lesion must not be confused with acute leptomeningitis and accumulation of exudate in the leptomeninges and subarachnoid space. In the latter the exudate extends into the sulci and also covers the gyri (see Fig. 14-43, A).
(Courtesy College of Veterinary Medicine, University of Illinois.)
Granulomatous Meningoencephalitis.
Granulomatous meningoencephalitis (GME) is one of two important idiopathic encephalitides of the dog, the second one being necrotizing encephalitis, of which there are two variants (see later). Granulomatous meningoencephalitis occurs most commonly in young to middle-aged small breed dogs, including poodles and various terrier breeds. No sex predilection is known, although some studies are skewed slightly toward female. Clinical signs in cases with brain lesions are variable and include behavioral changes and circling. Spinal cord lesions can result in paresis and ataxia.
When present, gross lesions consist of gray-white to red, expansive areas within the white matter of the brain and brainstem (Fig. 14-103, A ). Lesions can have irregular, well-defined margins and a gelatinous or rubbery consistency or appear granular. There are two different distribution patterns of the disease, a disseminated form and a focal form. The focal form is most commonly seen in the thalamus and brainstem. Histologically, this disease causes distinct perivascular cuffs composed almost entirely of lymphocytes and macrophages with scattered numbers of plasma cells and neutrophils. These cuffs are almost entirely restricted to the white matter. The proportion of lymphocytes and macrophages can vary in affected foci. Macrophages are commonly epithelioid, and in chronic cases there is abundant deposition of reticulin and collagen in the affected perivascular regions (see Fig. 14-103, B). The inflammatory lesion in granulomatous meningoencephalitis is predominantly CD3+ lymphocytes and perivascular accumulations of CD163+ macrophages. This observation has led to the suggestion that the underlying mechanism of the disease process is a T lymphocyte–mediated delayed-type hypersensitivity of an organ-specific autoimmune disease. Regardless, the cause of granulomatous meningoencephalitis has remained enigmatic, and various attempts to consistently isolate pathogens from affected dogs have proved fruitless.
Figure 14-103.
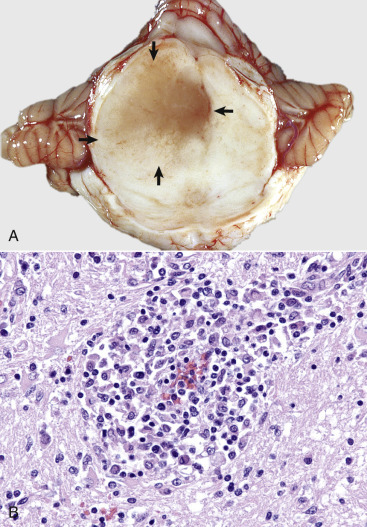
Granulomatous Meningoencephalitis, Transverse Section of Midbrain Just Rostral to the Pons, Dog.
A, The mesencephalon is swollen, discolored, markedly distorted, and soft as the result of extensive granulomatous inflammation (arrows), which has displaced the midline to the right. The mesencephalic aqueduct is also compressed and distorted. B, Note the accumulation of granulomatous inflammatory cells in the perivascular space. Such layers of cells expand over time and compress adjacent neural tissue, resulting in Wallerian-like degeneration of affected myelinated axons and atrophy of affected neuron cell bodies. H&E.
(A courtesy Dr. J. Edwards, College of Veterinary Medicine, Texas A&M University; and Dr. J. King, College of Veterinary Medicine, Cornell University. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Necrotizing Encephalitis.
Necrotizing encephalitis, a second important idiopathic encephalitis of the dog, has two variants, necrotizing meningoencephalitis (NME) and necrotizing leukoencephalitis (NLE). Necrotizing meningoencephalitis is a disease that is almost always restricted to small breed dogs like the pug, shih tzu, and Maltese terrier. It was once referred to as pug-dog encephalitis before it was recognized in a wide variety of small breed dogs. Grossly, this disease appears as bilateral foci of malacia and discoloration of the cerebral hemispheres. Histologically, this gross lesion corresponds to regions of meningoencephalitis in which the inflammation is typically robust, nonsuppurative, and associated with marked gliosis. Unlike in cases of granulomatous meningoencephalitis, spinal cord involvement is uncommon. The second disease is known as necrotizing leukoencephalitis. This disease predominates in the white matter. It is most commonly seen in Yorkshire terriers, although sporadic reports exist in other small breed dogs. Gross lesions consist of prominent bilateral cavitation and necrosis of the cerebral white matter. Histologically, there is robust nonsuppurative inflammation accompanied by gliosis, cavitation, edema, and gitter cell infiltration. The etiopathogenesis of both diseases is unknown, and whether they represent a continuum of the same disease or distinct diseases is still debated. Populations of lymphocytes and macrophages as determined by experimental analysis are similar in the two variants. Clinical signs in both variants are typically nonspecific and can involve a variety of prosencephalic processes such as behavior, processing of sensory and associative information, and the initiation of voluntary movement.
Traumatic Injury
Intervertebral Disk Disease.
Although the anatomy of vertebrae and intervertebral disks is similar in dogs and human beings, there are notable differences in the anatomy of the spinal cord and spinal nerve roots that result in differences between the two species in clinical signs caused by herniated disks. Disk herniations in human beings commonly occur laterally, rather than dorsally as in dogs, contributing to the differences in clinical presentations (i.e., lateral herniations compress spinal nerve rootlets, whereas dorsal compression in dogs impinges on the spinal cord directly). In human beings the spinal cord terminates at the level of the second lumbar vertebra. Spinal nerve roots forming the cauda equina traverse the remaining lumbar and sacral vertebrae before they exit the spinal canal to innervate structures. In dogs the spinal cord terminates at the level of the sixth lumbar vertebra, and nerve roots forming the cauda equina traverse the remaining lumbar, sacral, and coccygeal vertebrae before they exit the spinal canal to innervate structures. Therefore in human beings, disk disease involving the caudal lumbar vertebrae (caudal to L2) results in compression of spinal nerve roots that innervate limbs and are reflected clinically in a condition known as sciatica that is defined as referred pain in the sciatic nerve. In dogs, herniated disks in the lumbar vertebra primarily compress the spinal cord and under certain circumstances spinal nerves. Dogs thus present clinically with different neurologic signs.
Differences in clinical signs between dogs and human beings caused by herniated disks arise not only from anatomic differences discussed previously, but also from postural differences and the application of shear and stress forces applied to the vertebrae and intervertebral disks. Human beings, with bipedal locomotion and erect posture, dissipate the forces of walking (also running) by transferring these forces from the legs up the vertebral column to the lumbar vertebrae (i.e., first in line to absorb and dissipate forces). In addition, lumbar vertebrae are not stabilized by the rib cage, and thus lumbar vertebrae must also absorb rotational forces (i.e., twisting forces) of motion. Therefore lumbar vertebrae are the primary sites for disk herniation in human beings; however, because of the anatomy discussed earlier, herniated disks compress nerve roots and not the spinal cord. This arrangement results in pain but rarely leg paralysis.
Dogs, having quadrupedal locomotion and horizontal posture, normally dissipate the forces of walking by transferring these forces up the limbs at right angles to the vertebral column and spinal cord. However, when a dog jumps with downward motion, as an example, from a chair to the floor, the force is directed down the vertebral column and results in greater “end on” compression of disks and increased likelihood of herniation. Additionally, thoracic vertebrae are fixed in place by the ribs, and lumbar vertebrae can rotate freely around the axial skeleton. This arrangement directs the impact of stress and shear forces to the thoracolumbar vertebrae and is a primary site of disk herniation and spinal cord compression. In dogs, because the spinal cord is compressed, paralysis of the rear limbs results.
Intervertebral disk disease occurs in the canine species, particularly dogs of the chondrodystrophic breeds typified by the dachshund and Pekingese (Fig. 14-104 ). Contiguous vertebrae are held together by the annulus fibrosus of intervertebral disks and dorsal and ventral longitudinal ligaments. This anatomic arrangement results in the spinal canal being properly aligned in an axial plane, so the spinal cord can traverse through the space without compression. Extradural space surrounding the cervical, cranial thoracic, and caudal lumbar spinal cord is sufficient to allow for the accumulation of herniated disk material without substantial compression of the spinal cord. In contrast, there is little extradural space surrounding the thoracolumbar spinal cord; this is the site most likely to have clinically significant disk herniation. As a result in dogs, thoracolumbar disk herniation is often more debilitating than cervical disk herniation.
Figure 14-104.
Intervertebral Disk Disease, Dog.
A, Disk rupture (herniated intervertebral disk), spinal cord compression. Disk material compresses the spinal cord (arrow) resulting in Wallerian degeneration. B, Vertebral column, lumbar vertebrae. Herniated intervertebral disk (arrow) protrudes into the vertebral canal. C, Herniated intervertebral disk, spinal cord. The disk material (arrows) lies in the epidural space, touches the dura mater, and compresses the overlying spinal cord. An area of necrosis, possibly caused by infarction, is present in the ventral area of the left lateral funiculus (arrowhead). The multiple small holes in all funiculi are the sites of lost nerves as the result of spinal cord compression, which caused Wallerian degeneration. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Degeneration of intervertebral disks in chondrodystrophic breeds of dogs is a genetically programmed metaplastic change of the nucleus pulposus, resulting in peripheral to central replacement of the nucleus pulposus with cartilage. It begins as early as 6 months of age, progresses rapidly, and results in the loss of elasticity of the nucleus pulposus. The loss of elasticity places additional mechanical stress forces on the annulus fibrosus, which itself is experiencing degenerative changes similar to those occurring in the nucleus pulposus. The annulus fibrosus is thinnest and thus weakest at its point of contact with the spinal canal. If the annulus fibrosus is ruptured after stress forces placed on the disk by movement, such as jumping downward from a chair, fragments of the nucleus pulposus can be released into the spinal canal (Hansen type I herniation). If the annulus fibrosus ruptures dorsally, fragments compress the ventral funiculi of the spinal cord. If the annulus fibrosus ruptures dorsolaterally to laterally, fragments compress the ventral funiculi, lateral funiculi, and/or spinal nerve roots. Degeneration of intervertebral disks in nonchondrodystrophic breeds of dogs is an aging change of the nucleus pulposus resulting from fibrous metaplasia. This change causes a gradual loss of elasticity of the nucleus pulposus, which may be noticed clinically by 8 to 10 years of age. The gradual loss of elasticity places mechanical stress forces on the annulus fibrosus and results in its protrusion into and compression of the spinal canal (Hansen type II herniation).
Disk herniation causes injury to the CNS by several mechanisms. As discussed previously, the primary injury is caused by the physical trauma of compression and the resulting Wallerian degeneration of affected axons. Additionally, disk material can compress the vascular supply to a spinal cord segment, resulting in ischemia, neuronal excitotoxicity, and necrosis. Type I herniation causes the most severe spinal cord damage because there is insufficient time for the spinal cord to compensate and for collateral circulation to develop, as may occur in type II herniation.
Aging Changes
See Disorders of Domestic Animals, Aging Changes.
Canine cognitive dysfunction has been compared to Alzheimer's disease in human beings. Senile plaques and cerebrovascular amyloidosis have been described in the brains of aged dogs and are thought to result in structural and functional alterations in chemical pathways, leading to cognitive dysfunction.
Disorders of Cats
Disorders that occur in many or all animal species are discussed in the section on Disorders of Domestic Animals.
Diseases Caused by Microbes
Viruses
Coronaviruses
Feline Infectious Peritonitis.
Feline infectious peritonitis, which is caused by a coronavirus and has a worldwide distribution, is mainly a disease of domestic cats, although wild felids can be affected. A similar virus has recently been recognized in ferrets and can cause similar lesions in the CNS. There are two recognized feline coronaviruses: feline enteric coronavirus (FECV) and feline infectious peritonitis virus (FIPV), which cause FECV and feline infectious peritonitis infections, respectively. The viruses for each infection are antigenically and morphologically indistinguishable and are currently considered to represent avirulent (FECV) and virulent (FIPV) strains of the same basic feline coronavirus. After ingestion, FECV infects and replicates in epithelial cells of the intestine and usually is an insignificant infection, although severe intestinal disease can occur. Recent research suggests that a mutation in the gene for the furin cleavage site of a spike protein (a structural glycoprotein) in the FECV viral envelope gives the virus the ability to mutate to FIPV and replicate in monocytes/macrophages, use leukocyte trafficking (see Chapter 4) by these cells to spread systemically, and cause lesions characteristic of feline infectious peritonitis in other organ systems.
FIPV enters the susceptible cat primarily by ingestion of contaminated saliva or feces, although transmission by direct inoculation (e.g., cat bites, licking open wounds) and in utero (rarely) have been reported. After infection the virus replicates in macrophages that spread the virus to the liver, serosal surfaces, uvea, the meninges, and ependyma of the brain and spinal cord.
After dissemination of the virus in the body, the development of disease depends on the type and degree of immunity that develops. Virus containment with resistance to disease occurs following development of a strong cell-mediated immunity. Humoral immunity by itself is not protective and can actually enhance development of the effusive form of feline infectious peritonitis (wet form) by two proposed mechanisms. The first involves the development of virus-antibody-complement complexes that particularly accumulate in the same areas as infected macrophages around small blood vessels, resulting in inflammation and subsequent vascular injury (type III hypersensitivity) accompanied by effusion of large amounts of fluid. The second mechanism involves a process referred to as antibody-dependent enhancement (demonstrated to occur experimentally), which involves uptake of virus-antibody-complement complexes by macrophages followed by significant viral replication. The heavily infected macrophages, frequently perivascularly oriented, release cytokines that result in alteration of endothelial junctional complexes that leads to leakage of substantial amounts of fluid.
Noneffusive feline infectious peritonitis (i.e., dry form), in comparison, is thought to occur when partial cell-mediated immunity (type IV hypersensitivity) develops and represents an intermediate stage between nonprotective humoral immunity alone and protective cellular immunity. Support for this mechanism is the fact that cats that develop the noneffusive form of feline infectious peritonitis after experimental infection usually have a preceding and transient bout of effusive-type disease. In addition, there is evidence to support the theory that cats recovered from feline infectious peritonitis are immune by a process of “infection immunity” or “premunition.” Once these cats no longer retain such infections, they seem to also lose protective (cell-mediated) immunity and are in fact more sensitive to a subsequent challenge exposure because of the presence of humoral antibody.
The basic lesion in effusive and noneffusive feline infectious peritonitis is a pyogranulomatous inflammation, leading to vasculitis followed by an inconsistent vascular necrosis resulting in infarction. Unlike the majority of immune-mediated vasculitides in human medicine, feline infectious peritonitis shows preferential involvement of small- and medium-sized veins. The phlebitis that occurs is associated with activated, virally infected monocytes as they egress from the blood vessel to become macrophages. The effusive form is typified by serositis, accumulation of fluid in the abdominal and thoracic cavities, with varying degrees of severity of inflammation. Lesions of the noneffusive form more frequently result in leptomeningitis, chorioependymitis, focal encephalomyelitis, and ophthalmitis, although involvement of the kidneys, hepatic and mesenteric lymph nodes, and less frequently, serosa and other abdominal viscera can occur. In the CNS, pyogranulomatous vasculitis tends to affect blood vessels of (1) the leptomeninges, especially in sulci and near their entrance into subjacent CNS tissue and around the circle of Willis (Fig. 14-105 ), and (2) the periventricular white matter, especially around the fourth ventricle (Fig. 14-106 ). The uvea, retina, and optic nerve sheath are also commonly involved in feline infectious peritonitis.
Figure 14-105.
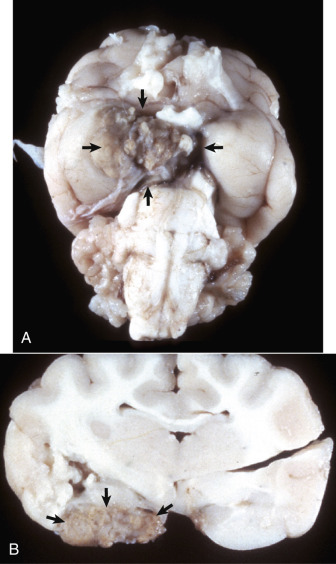
Pyogranulomatous Vasculitis, Feline Infectious Peritonitis, Cat.
A, Ventral brain, cerebral vasculature of the circle of Willis. A white-yellow pyogranulomatous inflammation distorts and obscures the blood vessels. Lesions are attributed to viral induced inflammation that targets vessel walls (arrows). The character of the inflammatory response can vary from an exudate with accumulation of serous fluid and fibrin mixed with neutrophils and histiocytes to a reaction that is more pyogranulomatous, and in which commonly there are lymphocytes and plasma cells. The severity and magnitude of the lesion depicted here is much more dramatic than usual. B, A cross-sectional view of A. The pyogranuloma (arrows) is principally in the subarachnoid space and has compressed the adjacent cerebral cortex.
(A and B courtesy Dr. J. Sundberg, College of Veterinary Medicine, University of Illinois.)
Figure 14-106.
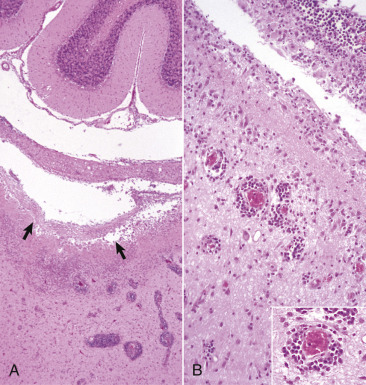
Pyogranulomatous Vasculitis, Feline Infectious Peritonitis (FIP), Cat.
A, Periventricular white matter (arrows) beneath the fourth ventricle (between the medullary vellum and medulla). The pyogranulomatous inflammation that occurs with FIP causes vascular and perivascular injury, vasogenic edema, and parenchymal disruption. H&E stain. B, A higher magnification of A. Ventriculitis and ependymitis are evident. Note the prominent perivascular cuffs of small mononuclear cells and macrophages. H&E stain. Inset, Higher magnification of B. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Feline infectious peritonitis generally occurs sporadically in cats of all ages, but it is most common in younger cats between the ages of 3 months and 3 years and can be clinically significant because it can result in death. The disease manifests itself in effusive (wet) or noneffusive (dry) forms. Clinical signs caused by involvement of blood vessels in the CNS can include behavioral changes, dullness, coma, paresis, ataxia, paralysis, and seizures.
Degenerative Diseases
Primary Neuronal Degeneration
Primary Cerebellar Neuronal Degeneration.
A discussion of primary cerebellar neuronal degeneration in dogs and cats is in the previous section on Disorders of Dogs.
Circulatory Disturbances
Feline Ischemic Encephalopathy.
Feline ischemic encephalopathy is associated with aberrant cerebrospinal migration of Cuterebra fly larvae that enter the brain via the nasal cavity. A vascular-mediated vasospasm of the middle cerebral artery, resulting from hemorrhage or a toxin elaborated by the parasite, is the most likely mechanism behind the vascular lesions. Acute vascular lesions are uncommonly noted; however, the chronic inflammatory lesions are typical for the disease.
The gross lesions of acute disease are predominantly unilateral and occur in the white and gray matter of the cerebral hemispheres, usually in the area supplied by the middle cerebral artery (Fig. 14-107 ). The necrosis can be multifocal or involve up to two-thirds of one hemisphere. Hemorrhages can occur in the CNS or leptomeninges. In chronic cases, cerebral atrophy, most severe adjacent to the middle cerebral artery of the affected hemisphere, is most typical. Microscopically, lesions include vasculitis, thrombosis, ischemia, and infarction, and the cerebral cortical lesions follow the sequence of changes for infarction listed in Table 14-1.
Figure 14-107.
Feline Ischemic Encephalopathy, Brain.
A, Lateral view of a collapsed area of the cerebral cortex. Note the torturous pattern of the vascular supply, likely a component of the reparative response to ischemic injury. B, Transverse section at the junction between the left parietal and occipital lobes, level of thalamus, cat. Chronic feline ischemic encephalopathy with unilateral cerebral degeneration-atrophy. The dorsolateral aspect of the left cerebral hemisphere has undergone necrosis, followed by cyst formation and collapse after phagocytic removal of the necrotic debris. Cystic spaces (arrows) have placed the previously existing parenchyma and the left lateral ventricle (LV) has expanded into the area of lost tissue (hydrocephalus ex vacuo).
(A courtesy Drs. V. Hsiao and A. Gillen, College of Veterinary Medicine, University of Illinois. B courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University.)
Feline ischemic encephalopathy has a peracute to acute onset and affects cats of any age. Clinical signs usually reflect unilateral cerebral involvement. The disease most often occurs in the summer months and is accompanied by signs that can include depression, mild ataxia, seizures, behavioral changes, and blindness.
Peripheral Nervous System
Structure and Function
The PNS is typically divided into three parts: the sensorimotor division, the autonomic division, and the enteric division. The sensorimotor division is formed by sensory neurons (afferent components of cranial and spinal nerves, sensory receptors, and cranial and spinal ganglia) and motor neurons (efferent components of cranial and spinal nerves and lower motor neurons) that innervate skeletal muscle via myoneural junctions (Fig. 14-108 ). The autonomic and enteric divisions consist of networks of afferent and efferent nerves and their ganglia (Meissner's [submucosal] and Auerbach's [myenteric] plexuses) that regulate, as examples, the contractility and relaxation of smooth muscle of the vascular and alimentary (peristalsis) systems and glandular secretions via sympathetic and parasympathetic fibers. Afferent and efferent nerve fibers of the autonomic and enteric divisions are carried in the afferent and efferent branches of the sensorimotor division (cranial and spinal nerves).
Figure 14-108.
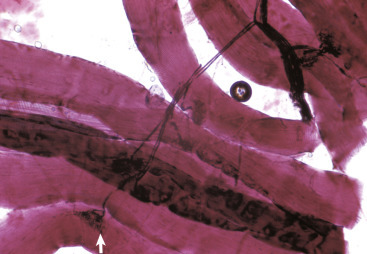
Myoneural Junctions.
Peripheral nerve with terminal axons ending at myoneural junctions on muscle fibers (arrow). Dissected and glycerol-mounted muscle fibers.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Peripheral nerves are composed of groups of axons, both myelinated and nonmyelinated, and of varying caliber (Fig. 14-109 ). As with the CNS, conspicuous components of axons are neurofilaments and microtubules. Neurofilaments provide structural support; microtubules are intimately involved in bidirectional axoplasmic flow of structural components, nutrients, and trophic factors to and from the cell body required for maintenance of the axons and neuronal integrity. Transport from the neuron cell body to the distal axon (anterograde flow) occurs at fast (400 mm per day or approximately 0.25 mm per minute) and slow (1 to 4 mm per day) rates. Retrograde transport from the distal axon to the cell body progresses at a rate of 200 mm per day (approximately 0.125 mm per minute) (see E-Fig. 14-3).
Figure 14-109.
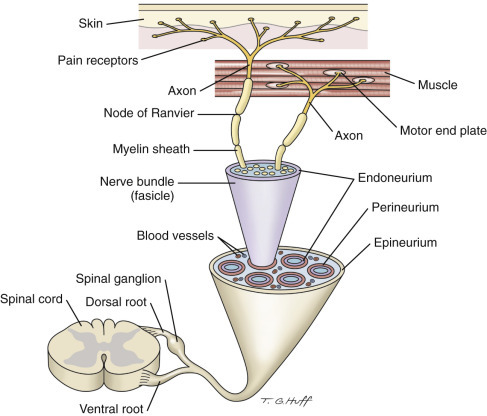
Organization of a Peripheral Nerve, and Sensory and Motor Branches and Their Coverings.
Supporting cells in the PNS include Schwann cells and fibroblasts of the endoneurium and the satellite cells (Schwann cell–like cells) of the dorsal root ganglion. Schwann cells surround both myelinated and nonmyelinated axons and are responsible for formation of the myelin sheaths (see Fig. 14-109). In contrast to the CNS, where one oligodendroglial cell can send out numerous processes to myelinate many different axonal internodes of several different axons, one Schwann cell myelinates one internode of one axon. As a result, the entire length of an axon in the PNS is myelinated by many individual Schwann cells.
Although Schwann cells do not appear to play a role in axon guidance during formation of the PNS, these cells are necessary for maintenance of axons and secrete neurotrophic factors that play a role in regeneration. Axons are grouped into fascicles with surrounding loosely organized tissue fibrils and specialized endoneurial fibroblastic cells with phagocytic capabilities (see Fig. 14-109). When an axon is damaged so badly as to cause Wallerian degeneration, removal of the debris is by these putative endogenous phagocytic cells, augmented by an influx of blood monocytes. Mast cells and small blood vessels also are present among the nerve fibers.
Depending on species and anatomic location, endothelial cells of endoneurial blood vessels can be joined by tight junctions, preventing free passage of some macromolecules and providing an incomplete blood-nerve barrier. Collagen bundles and modified fibroblastic cells, termed perineurial cells, form the perineurium that ensheathes individual nerve fascicles. The perineurium contributes some barrier properties by preventing the free diffusion of macromolecules into the nerve fascicles. The fibrous epineurium is continuous with the dura mater as a peripheral nerve joins the CNS and encloses groups of nerve fascicles. The epineurium contains fibroblasts, mast cells, and adipocytes, the latter probably providing some protection to the nerve. Satellite cells are found in dorsal root ganglia within the matrix formed by the endoneurium that envelops cell bodies of peripheral nerves. They function in a supportive, nonmyelinating role, much like perineuronal oligodendroglia in the CNS.
The autonomic and enteric divisions of the PNS function primarily to transmit impulses from the CNS to peripheral organs (efferent nerves) that regulate (involuntary control) the function of these organ systems (heart, vascular system, visceral smooth muscle, and exocrine and endocrine glands). These effects include but are not limited to the rate and force of contraction and relaxation in smooth (visceral organs and blood vessels) and striated muscle (heart). Afferent nerves, which transmit from the periphery to the CNS, mediate visceral sensation and vasomotor and respiratory reflexes through baroreceptors and chemoreceptors in the carotid sinus and aortic arch. Autonomic and enteric functions are regulated in the medulla, pons, and hypothalamus of the CNS.
The autonomic division has two structural and functional components: the sympathetic and parasympathetic systems. These systems usually have opposing effects on innervated organ systems. The parasympathetic system acts, for example, to lessen the effects of increased vasoconstriction (smooth muscle) and contractility (heart rate) exerted by the sympathetic system.
The enteric division of the PNS system exerts effects on digestive processes, such as motility, secretion, and absorption, and blood flow. The main components of the enteric nervous system are myenteric plexuses (Auerbach's plexuses), located between the longitudinal and circular layers of muscle, and submucosal plexuses (Meissner's plexuses) that innervate esophageal and intestinal smooth muscle. Injury to these plexuses can lead to dysautonomias, which are discussed in a later section.
Dysfunction/Responses to Injury
See Central Nervous System, Dysfunction/Responses to Injury
Responses of the Axon to Injury
See a discussion on Wallerian degeneration and central chromatolysis in the section on Responses of Neurons to Injury in the section on the CNS.
Portals of Entry/Pathways of Spread
See Central Nervous System, Portals of Entry/Pathways of Spread
Defense Mechanisms/Barrier Systems
Blood-Nerve Barrier
The blood-nerve barrier regulates the free movement of certain substances from the blood to the endoneurium of peripheral nerves. Barrier properties are conferred by tight junctions between endothelial cells of the capillaries of the endoneurium and perineurium and by selective transport systems in the endothelial cells.
Disorders of Domestic Animals
Peripheral Neuronopathies and Myelinopathies
Many disorders affecting the CNS are also manifested in lesions in the PNS, either (1) because of damage to neuron cell bodies of lower motor neurons residing in the CNS or (2) because the PNS is equally vulnerable to the disease. An example in the first case is lysosomal storage diseases, in which substrate accumulates in cell bodies of lower motor neurons. Cell death and axonal degeneration of the PNS are the end points of a chronic and progressive process of substrate accumulation that interferes with cellular biochemical processes and transport systems. In the second case, substrate also accumulates in cell bodies of sensory neurons located in the dorsal root ganglion of the PNS, resulting in cell death and axonal degeneration. Despite this caveat, there are certain diseases that primarily affect the PNS. Depending on whether the lesion is in a sensory or motor nerve or both, diseases of the PNS can manifest clinically as motor disturbance, sensory deprivation, or a combination of motor and sensory alterations. Space constraints do not allow an exhaustive coverage of disorders of the PNS. Many of the reported disorders seem to represent isolated occurrences in a specific breed. This section covers the major types of PNS diseases with reference to specific disorders for illustrative purposes.
Congenital/Hereditary/Familial Diseases
Primary Sensory Neuropathies.
Primary sensory neuropathies included here are the hereditary, familial, and breed- or species-associated syndromes reported in a variety of domestic animals that result in degeneration of PNS sensory neurons (dorsal root ganglion) or axons innervating the limbs. Two examples of primary sensory neuropathies have been described in English pointers and long-haired dachshunds. In pointer dogs the onset is 2 to 12 months of age with signs of self-mutilation and insensitivity to pain resulting in neuro-pododermatitis or acral mutilation syndrome (Fig. 14-110 ). Additional signs can include ataxia, loss of conscious proprioception, and patellar hyporeflexia. In dachshunds a sensory neuropathy is manifested shortly after birth by ataxia and alterations in the function of the autonomic division of the PNS, such as urinary incontinence and digestive disturbances. Lesions in pointer dogs consist of small dorsal root ganglia with neuronal loss and replacement by satellite cells (Nageotte nodules) and mild reduction in size of dorsal nerve rootlets because of degeneration and loss of myelinated and nonmyelinated axons with the presence of cell bands of Büngner (indicative of attempts at remyelination). In dachshunds, lesions are a distal axonopathy with loss of large myelinated and unmyelinated axons. Lesions can occur in the vagus nerve.
Figure 14-110.
Neuro-Pododermatitis (Acral Mutilation Syndrome), Dog.
This disorder, a primary sensory peripheral neuronopathy with self-mutilation and insensitivity to pain, is caused by the absence of (or small) dorsal root ganglia, reduction in size of dorsal nerve rootlets, and degeneration and loss of myelinated and nonmyelinated sensory axons. This dog wore off its footpads when placed on a concrete run. Satellite cell proliferation is commonly present in other large autonomic ganglia (i.e., celiac ganglion).
(Courtesy College of Veterinary Medicine, University of Illinois.)
Other degenerative distal axonopathies of the PNS have also been reported in Birman cats and in dog breeds, including Bouvier des Flandres, Siberian huskies and crossbreeds, boxer dogs (sensory axonopathy), Rottweilers (sensory axonopathy), dachshunds (sensory axonopathy), Dobermans (dancing Doberman disease is thought to be a primary myopathy), German shepherds (giant axonal neuropathy), and Dalmatians. They may have a genetic basis and be inherited.
A noninherited form of sensory neuropathy also occurs in dogs and is associated with ganglioradiculitis. Although many viruses, including pseudorabies virus and rabies virus, can cause inflammation in the spinal ganglia, this specific sensory neuropathy appears not to be associated with a viral infection. Gross lesions in affected animals often reveal a zone of pallor in the dorsal funiculus corresponding to Wallerian degeneration secondary to severe inflammation within the dorsal root ganglia. The inflammation is often associated with Nageotte nodule formation, neuronal loss and degeneration, and some degree of nonspecific gliosis. The inflammation is also typically mononuclear (i.e., the character of the inflammatory cells involved), raising the possibility that this form of ganglioradiculitis has an autoimmune basis.
In all forms of sensory neuropathy, animals are usually young (birth to 15 months of age) and show signs of ataxia, muscle weakness (paresis and tetraparesis) followed by muscle atrophy, proprioceptive deficits, urinary incontinence, and digestive disturbances (enteric division involvement).
Dysautonomias
Hereditary Dysautonomias.
Dysautonomia is a degeneration of neurons in the ganglia of the enteric division of the PNS that has been reported in dogs, cats (Key-Gaskell syndrome), a llama, sheep, cattle, and horses. The cause is unknown, and a hereditary basis is suspected in some cases. A toxic cause has been postulated in the cat. Lesions recently reported in sheep with abomasal emptying defect resemble those reported in dysautonomic diseases of human beings and other animals.
Lesions are observed in peripheral and enteric (autonomic) ganglia and vary from neuronal chromatolysis and nuclear pyknosis in more acute cases to loss of neurons and proliferation of satellite cells in cases with longer duration (Fig. 14-111 ). Minimal to mild leukocytic infiltrates occur, but the lesions are not overtly inflammatory. In cats and dogs, clinical signs are varied and include gastrointestinal disturbances, urinary incontinence, mydriasis, unresponsive pupils, bradycardia, and other signs associated with autonomic dysfunction.
Figure 14-111.
Dysautonomia, Submucosal (Auerbach's) Plexus, Dog.
Neuronal central chromatolysis, nuclear pyknosis, and loss of neurons are the characteristic histologic features of enteric dysautonomia. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Acquired Dysautonomias
Equine Grass Sickness (Equine Dysautonomia).
Equine grass sickness (equine dysautonomia) is discussed in the later section on Disorders of Horses.
Peritonitis-Induced Dysautonomias.
Degeneration of autonomic neurons in the myenteric and submucosal ganglion (plexuses) can occur in animals with peritonitis. The degree of neuronal degeneration appears to be related to the severity and type of inflammatory response in the peritoneal cavity and the ability of inflammatory mediators and other potentially toxic molecules to reach the ganglia by diffusion or hematogenously. Degeneration does not appear to progress to neuronal cell death if the peritonitis is resolved. Affected neurons have vesicular nuclei that are two to three times normal size (see Fig. 14-111). Nissl substance is also displaced (central chromatolysis). Nerve fiber bundles are edematous, and supporting cells can be remarkably hyperplastic and compress adjacent supporting stroma.
This lesion is thought to arise from inflammatory cytokine-mediated injury to autonomic neurons, and this may cause alterations of intestinal motility. It appears that the morphologic changes observed in autonomic and enteric neuron cell bodies are reversible with resolution of the peritonitis. Whether the neuronal lesion results from diffusion or hematogenous or retrograde axonal transport of cytokines to the ganglion from the site of inflammation remains to be proved.
This lesion is likely the cause of paralytic ileus seen with peritonitis.
Hypomyelination/Dysmyelination Diseases.
In contrast to the CNS, congenital and postnatal disorders of myelin formation are rare in the PNS but have been described in dogs, calves, and a cat. These disorders are thought to have a genetic predisposition and to be inherited. In dogs, hypomyelination has been described in golden retrievers with onset at approximately 7 weeks of age. Clinical signs include a peculiar hopping gait, depressed spinal reflexes, and circumduction of the limbs while walking. Lesions in peripheral nerves include thin myelin sheaths, increased numbers of Schwann cells, neurolemma cells with abnormally increased cytoplasmic volume, and no evidence of active demyelination or effective remyelination. The lesions are believed to involve a defect in Schwann cells or an abnormal axon–Schwann cell interaction.
In calves a myelinopathic peripheral neuropathy has been described in Santa Gertrudis–Brahman crossbreeds. Microscopically, lesions were present in the vagus nerves, somatic peripheral nerves of the brachial plexuses, and the sciatic nerves. Dorsal and ventral spinal nerve roots also had similar lesions. There was “sausage-shaped” thickening of the myelin sheaths as a result of excess myelin arranged about the axons or as irregularly folded myelin sheaths not surrounding axons. Onset of clinical signs was at 6 to 10 months of age. Clinical signs were dysphagia, abnormal rumination with bloat, and a weak shuffling gait. Congenital hypomyelination has also been described in a 2-month-old Dorset lamb with tremors and incoordination.
Demyelination Diseases.
A variety of injuries similar to those described in the CNS can cause primary demyelination in the PNS. Specific demyelinating diseases are covered in the next sections. In response to injury, Schwann cells can proliferate to restore the myelin sheaths, often forming longitudinal columns along the course of a degenerated axon termed Büngner's bands. Remyelination results in internodes that are shorter than the internodes of adjacent normal myelinated axons, and this change is used microscopically to detect areas of remyelination in peripheral nerves. Another lesion that occurs with repeated episodes of demyelination is proliferation of Schwann cell processes forming concentric whirls, called onion bulbs that surround the axon.
Coyotillosis.
Another cause of primary demyelination is the shrub coyotillo (Karwinskia humboldtiana) affecting mainly small ruminants in the semidesert areas of the southwestern United States. Seeds in the fruit contain polyphenolic compounds that are toxic when ingested. Four toxic compounds have been isolated, including a substance called karwinol A that induces primary demyelination of peripheral nerves. Based on electron microscopic evaluation of lesions, the damage appears to be due directly to axonal injury caused by consolidation of neurofilaments and margination of microtubules.
Endocrine Diseases.
Endocrine disorders, such as hypothyroidism, hyperadrenocorticism, and diabetes mellitus, can affect the PNS. The lesions of these neuropathies are not well characterized and can include evidence of primary demyelination, remyelination, and axonal degeneration. Distal portions of the axon are commonly affected. The extent to which demyelination or axonal degeneration is the primary lesion remains to be determined. From a clinical standpoint, it may be difficult to distinguish neurologic signs from those signs attributed to hormonal-influenced injury of myofibers. Clinical signs can be caused by sensory and motor deficits.
Nutritional Diseases
Vitamin A, Vitamin D, and Riboflavin Deficiencies.
Nutritional axonopathies are relatively uncommon and are chiefly caused by vitamin A and some of the B vitamin deficiencies. Vitamin A deficiency results indirectly in peripheral neuropathy by affecting bone growth and remodeling. In neonatal calves the neuropathy is due to narrowing of the optic foramina caused by continued bone deposition with decreased resorption resulting in compression of the optic nerves, Wallerian degeneration, and blindness. B vitamin deficiencies are primarily diseases of pigs and poultry. In pigs, deficiency of pantothenic acid (B-complex vitamin, vitamin B5) causes a sensory neuropathy with axonal degeneration, demyelination, and chromatolysis and neuron loss in the dorsal root ganglia, resulting in proprioceptive deficits, goose-stepping, and dysmetria. The exact sequence of events in pantothenic acid–deficiency neuropathy is controversial because one study in pigs described initial lesions in the axon, whereas a second study described initial lesions in the cell body. Riboflavin deficiency in poultry, named curled-toe paralysis, is primarily a demyelinating neuropathy. Peripheral nerves are swollen because of endoneurial edema, and there is subsequent demyelination with mild axonal degeneration.
Vitamin E Deficiency
Equine Motor Neuron Disease.
Equine motor neuron disease is discussed in the later section on Disorders of Horses.
Toxic Diseases
Chemicals.
Toxic diseases resulting from chemicals are discussed in greater detail in the section on the CNS. Examples of chemical toxins causing distal axonal degeneration are the vinca alkaloids, vincristine and colchicine, both causing disassembly of microtubules and inhibiting axoplasmic flow. Paclitaxel (Taxol), an alkaloid from the western yew (Taxus brevifolia), promotes the assembly of and stabilizes microtubules but also causes an axonopathy. An outbreak of distal polyneuropathy has been reported in cats fed commercial diets contaminated with the ionophore salinomycin, which is used as a coccidiostatic drug in poultry and growth promoter in cows. There was acute onset of lameness and paralysis affecting the hind limbs that progressed to the forelimbs. Demyelination of peripheral nerves followed the axonal degeneration. Some toxins seem to cause different patterns of injury in the CNS and PNS. For example, in the CNS, lead is noted to cause neuronal necrosis, whereas in the PNS, demyelination, preferentially affecting Schwann cells, is prominent in some species.
Other Toxic Neuropathies.
A number of toxins can affect the PNS, with or without damage in the CNS. The initial toxic effects can be at the level of the neuron cell body, the axon, or the myelin sheaths. Examples of toxins targeting neuronal cell bodies are organomercurial compounds such as methylmercury and the cancer chemotherapeutic agent doxorubicin. Methylmercury is particularly toxic because it directly alters biochemical reactions. Although methylmercury poisoning can result from ingestion of water or forage contaminated with industrial discharge, in animals the consumption of fish containing excessive concentrations of methylmercury is the most likely source of the toxin. Fish accumulate methylmercury in their muscle as a result of a “normal” environmental process called biomethylation. Biomethylation converts elemental mercury to methylmercury, which is ingested in the diet of fish. In mercury poisoning, sensory neuron cell bodies of the dorsal root ganglion are preferentially involved, and the motor neurons are spared, whereas with doxorubicin, both dorsal root ganglion and autonomic cell bodies are affected. Experimental studies suggest that neuronal cell death is caused by apoptosis of neuronal cell bodies resulting in axonal and Wallerian degeneration.
Autoimmune Diseases
Neuromuscular Junction Diseases
Myasthenia Gravis.
Myasthenia gravis is a disorder of neuromuscular impulse transmission at myoneural junctions and results in flaccid paralysis of skeletal muscle. The disease can be caused by an autoimmune mechanism (acquired) or result from inherited genetic abnormalities (congenital). In autoimmune myasthenia gravis the antibody binds to acetylcholine receptors (type II hypersensitivity) on postsynaptic muscle membranes. This interaction results in distortion of receptors and blocks binding of the receptors with acetylcholine. Acquired myasthenia gravis often occurs concurrently with thymic abnormalities such as thymoma and thymic hyperplasia. Because the thymus is responsible for immunologic self-tolerance, thymic abnormalities leading to induced alterations in tolerance have been suggested as the mechanism for developing an antibody response against acetylcholine receptors.
Congenital myasthenia gravis is caused by a genetically determined deficiency in the number of acetylcholine receptors expressed in motor end plates.
There are no gross or microscopic lesions in the PNS or CNS caused by myasthenia gravis. Clinical signs and lesions are the result of impairment of skeletal and esophageal muscles and muscle weakness followed by muscle atrophy.
Diseases Caused by Microbes
Bacteria
Botulism.
Botulism is characterized by a flaccid paralysis caused by the neurotoxin of Clostridium botulinum type A, B, or C in North America and type D in South Africa. The bacterium is a ubiquitous Gram-positive spore-forming anaerobe commonly found in soil. This disease most commonly occurs in horses in North America and cows in South Africa. In North America, type B is most prevalent along the Mid-Atlantic states to Kentucky, whereas type A often predominates in the western United States. However, recent literature suggests that type A may be more prevalent along the Mid-Atlantic states that previously recognized. Different forms of botulism affect foals and adult horses. Toxicoinfectious botulism occurs in foals. Foals contract the disease by ingesting soil contaminated with clostridial spores.
In adult horses, poisoning after ingestion of toxin-contaminated forage and less commonly wound botulism occur. Adult horses contract the disease principally through the ingestion of preformed toxin in contaminated feeds, usually haylage that is prepared and stored improperly. Less commonly, adult horses contract the disease through tissue injury and an anaerobic environment, such as hoof abscesses and skin wounds. Spores of C. botulinum are either carried into wounds by nails or other contaminated foreign objects or into gastric ulcers by ingestion of contaminated soil. In either case the spores germinate only in necrotic tissue that has an anaerobic environment. The bacteria replicate and produce exotoxin, which is absorbed through the capillary endothelium and enters the bloodstream. In adult horses that ingest contaminated feeds, the toxin is absorbed from the alimentary system and enters the bloodstream.
Other than the wound where the bacteria replicates, the toxin of C. botulinum causes no macroscopic and microscopic tissue lesions. Once botulinum toxin is in the bloodstream, it enters myoneural junctions and binds to receptors on presynaptic terminals of peripheral cholinergic synapses (see Fig. 4-28). The toxin is then internalized into vesicles, translocated to the cytosol, and then mediates the proteolysis of components of the calcium-induced exocytosis apparatus, thus interfering with acetylcholine release. Inhibition (blockage) of the release of acetylcholine results in flaccid paralysis of muscles innervated by cholinergic cranial and spinal nerves, but there is no impairment of adrenergic or sensory nerves.
Clinically, affected horses have progressive paralysis of the muscles of the limbs, mandible, larynx/pharynx, upper eyelid, tongue, and tail. Death is usually caused by flaccid paralysis of the diaphragm, resulting in respiratory failure. Blockage of acetylcholine release at presynaptic cholinergic terminals is permanent. Improvement occurs only when axons develop (sprout) new terminals to replace those damaged by botulinum toxin.
Viruses and Protozoa.
Inflammation of the PNS can occur in conjunction with viruses such as herpesvirus and rabies. Neuritis of the cauda equina in horses and dogs is primarily an inflammatory disorder with secondary demyelination. The cause of the neuritis in the horse is unknown, and attempts to consistently recover infectious microbes from affected animals have proven fruitless. These so-called cases of cauda equina neuritis (also known as polyneuritis equi) are often granulomatous with multinucleate giant cells and degenerative changes in the nerves and ganglia. Cranial nerve ganglia can be similarly affected. Polyradiculoneuritis and to a lesser extent ganglionitis occur in toxoplasmosis and neosporosis. The vomiting in pigs infected with hemagglutinating encephalomyelitis virus is presumed to result from altered function of the vagal nucleus and its ganglion and gastric intramural autonomic plexuses.
Lysosomal Storage Diseases
Globoid Cell Leukodystrophy.
Peripheral nerves are also affected in globoid cell leukodystrophy, and lesions are typified by primary demyelination followed by axonal degeneration. Small sensory branches of peripheral nerves are useful sites for biopsies to make the diagnoses (Fig. 14-112 ). Gross lesions are not evident; however, microscopically, such areas have pronounced loss of myelin and abundant globoid cells (activated blood monocytes).
Other lysosomal storage diseases known to affect the PNS include α-l-fucosidosis (nerve enlargement due to macrophage infiltration); α- and β-mannosidosis (vacuolated Schwann cells), and gangliosidoses (vacuoles in neurons with demyelination).
Traumatic Injury
Trauma to peripheral nerves (lower motor or sensory) is relatively common in animals and can result from lacerations, violent stretching and tearing, or compression or contusion. Reaction patterns after PNS injury are analogous to those in the CNS, but peripheral nerves have a greater capacity for repair. Three patterns of lesions in the PNS have been described. Mild injury that leaves the axon intact (neurapraxia) can result in temporary conduction block, but total recovery of function is possible. More severe damage that destroys the axon but leaves the connective tissue framework intact (axonotmesis) results in Wallerian degeneration distal to the point of injury, but the potential for regeneration and reinnervation is good. Finally, severance of the nerve with destruction of the supporting framework (neurotmesis) results in Wallerian degeneration distal to the injury with the potential for regeneration but little chance of normal reinnervation. Destruction of the supporting framework results in fibrosis between the proximal and distal ends of the nerve and this gap may be large depending on the severity of the injury. Fibrous tissue can obstruct the regenerating proximal axon from reaching the distal supporting framework of the axon. If the regenerative response is exuberant but unproductive, a “potentially” palpable bulbous-like growth can form at the severed stump of the proximal axon called a “neuroma.” The pattern of Wallerian degeneration and the reaction of the neuronal cell body to damage of its axon are described in an earlier section.
Recurrent Laryngeal Paralysis.
Recurrent laryngeal paralysis is discussed in the later section on Disorders of Horses.
Neurogenic Cardiomyopathy (Brain-Heart Syndrome).
Neurogenic cardiomyopathy, or brain-heart syndrome, is discussed in the later section on Disorders of Dogs.
Neurogenic Shock.
Neurogenic shock is caused by an alteration in the function of the autonomic nervous system and its regulation of muscle tone in systemic blood vascular beds (Fig. 14-113 ). The onset of neurogenic shock usually coincides with traumatic injury to the CNS; however, the factors that determine whether it occurs are poorly understood. It is thought to be caused by massive discharge of the autonomic nervous system. After trauma there is immediate vasoconstriction of vascular smooth muscle. Vasoconstriction is shortly followed by vasodilation, expanded circulatory volume, and a reduction in blood pressure leading to shock. Brain-heart syndrome in veterinary medicine is likely a manifestation of neurogenic shock and vasoconstriction of arterioles leading to myocardial necrosis.
Figure 14-113.
Mechanism of Neurogenic Shock.
(Courtesy Dr. A.D. Miller, College of Veterinary Medicine, Cornell University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Tumors
The objective of this section is not to be all encompassing regarding neoplasms of the PNS but to review one of the best-known examples of how neoplasia can involve this system. Although tumors of the PNS are often lumped into categories of either benign or malignant nerve sheath tumors, pathologists currently recognize three distinct types of nerve sheath tumors: Schwannomas, perineuriomas, and neurofibromas. Schwannomas and perineuriomas are derived from Schwann cells and perineurial cells respectively, whereas neurofibromas include elements derived from Schwann cells and fibroblasts. Malignant tumors show more anaplastic cytoarchitectural features and aggressive growth into adjacent normal tissue. Nerve sheath tumors occur in both cranial (Fig. 14-114 ) and spinal nerves (Fig. 14-115 ) of the PNS. Currently there are no reliable immunohistochemical markers for these tumors.
Figure 14-114.
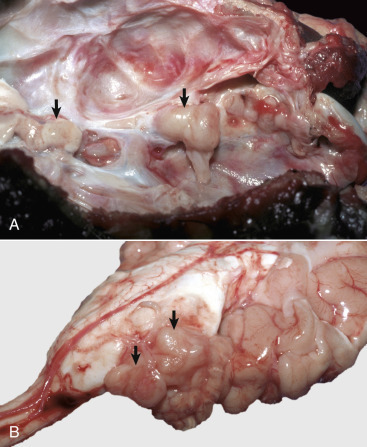
Nerve Sheath Tumors.
A, Inner surface of the cranial vault, cranial nerves, dog. These tumors are usually lobulated, well-defined, tan, solitary to multiple masses that arise from the coverings of a cranial or spinal nerve (arrows). In the central nervous system, the trigeminal nerve is usually affected, and the masseter and temporalis muscles innervated by it may atrophy. Tumors compress the nerves, causing wallerian degeneration. B, Brain from dog in A. Peripheral nerve sheath tumors (arrows).
(A and B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 14-115.
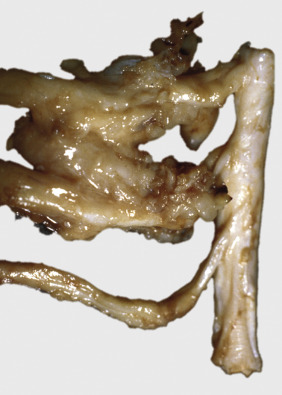
Nerve Sheath Tumor, Spinal Nerve, Cow.
These tumors are similar to those described in Figure 14-114 and occur most commonly in cows and dogs. Although schwannoma has been proposed as the best term to classify these tumors, the term nerve sheath tumor groups all morphologic diagnoses under a common umbrella.
(Courtesy College of Veterinary Medicine, University of Illinois.)
Schwannomas of animals have been most recognized in the dog and less commonly in the cat, horse, and cow. In the dog the neoplasm most commonly affects the cranial (fifth) or spinal nerve roots (posterior cervical–anterior thoracic roots of the brachial plexus and their extensions and roots at the thoracic and lumbar levels). Although true schwannomas of the dermal soft tissues have been reported, there should always be careful consideration of other soft tissue sarcomas that can have similar morphologic features.
Grossly, schwannomas are nodular or varicose thickenings along nerve trunks or nerve roots. They can be firm or soft (gelatinous) and white or gray. Schwannomas of the spinal cord nerve roots can remain inside the dura mater or extend through a vertebral foramen to the exterior.
Histologically, schwannomas are composed entirely of neoplastic Schwann cells and are typically composed of two morphologic patterns, known as Antoni type A and type B, that occur in variable proportions within the neoplasm. Antoni A tissue is cellular and consists of monomorphic sheets, fascicles, and whorls of spindle-shaped Schwann cells. These cells have poorly defined eosinophilic cytoplasm and pointed basophilic nuclei and are present in a collagenous stroma of variable extent. The nuclei of these cells are commonly arranged in rows, between which are parallel arrays (stacks) of their cytoplasmic processes, and this arrangement is called a Verocay body. Antoni B areas are also composed of Schwann cells, but their cytoplasm is inconspicuous, and their nuclei appear to be suspended in a copious myxoid, often microcystic, matrix. Other histologic findings include hyalinized stroma and the absence of nerve fibers. These tumors can reliably be stained immunohistochemically with antibodies to laminin or type IV collagen that highlight the basal lamina produced by the neoplastic Schwann cells. S-100 immunoreactivity is typically diffuse throughout the neoplasm.
Neurofibromas consist of Schwann cells, perineurial cells, and fibroblasts. This type of tumor is not common in domestic animals. However, a unique form occurs in the cow as part of a benign neurofibromatosis syndrome in which it occurs most commonly in mature animals, although the lesion has also been reported in young calves and involves the cranial eighth nerve, brachial plexus, and intercostal nerves. Additionally, autonomic nerves of the liver, heart, mediastinum, and thorax can be affected. The skin can be infrequently involved. Some microscopic features of the neurofibroma include elongated spindle cells with poorly defined pale eosinophilic, tapering wavy or buckled nuclei, and numerous small nerve fibers (which are not present in schwannomas). Presence of mast cells is also reported. These neoplastic components are situated in a variably prominent fibromyxoid to myxoid matrix (myxoid neurofibroma), although another variant of the neoplasm contains prominent collagen (collagenous neurofibroma). Neurofibromas have far less immunoreactivity for either laminin or S-100, with the latter staining the Schwann cell component, but not the other cell types.
The last nerve sheath tumor is the perineurioma. It usually behaves as a benign neoplasm. It is a very rare variant of nerve sheath tumors in domestic animals, with only rare reports in the dog. It consists of tight, concentric whorls of perineurial cells that surround a central axon. Immunohistochemical stains for neurofilament proteins often highlight the central axon, whereas stains like laminin highlight the spindle cell population.
All of the aforementioned neoplasms can undergo malignant transformation and are then referred to as malignant nerve sheath tumors. They typically invade surrounding structures or can, based on location, invade or surround the spinal cord.
Disorders of Horses
Peripheral Neuropathies
Congenital/Hereditary/Familial Diseases
Colonic Agangliosis.
Colonic agangliosis (lethal white foal syndrome) is a disorder involving development of the enteric division of the PNS and is analogous to Hirschsprung's disease in infants. This disease occurs most commonly in foals of American paint horses with overo markings. Affected foals have white or nearly white skin color. Specific information on these skin marking patterns can be obtained from the American Paint Horse Association. The gene, which results in the “colonic agangliosis” phenotype being expressed, is inherited as a homozygous dominant.
Mutations in the endothelin-B receptor gene have been detected in affected horses and in some patients with Hirschsprung's disease. Both glial-derived neurotrophic factor and endothelin-3 are required for normal development of the enteric nervous system and enteric ganglia. It is proposed that glial-derived neurotrophic factor is required for proliferation and differentiation of neuronal precursor cells destined to populate the gut. Endothelin-3 might modulate these effects by inhibiting differentiation, thus allowing sufficient time for precursor cells to migrate and populate the intestinal wall in a cranial to caudal progression before they differentiate to form enteric ganglia.
The lumen of the large intestine is often small or narrowed in affected animals. Microscopically, the myenteric and submucosal enteric ganglia are absent, and the areas affected vary but can extend anywhere between the ileum and distal large colon. Affected foals die within days after birth from functional blockage of the ileum and/or colon because of the lack of innervation and thus normal gut motility.
Toxic Diseases
Acquired Dysautonomias
Equine Grass Sickness (Equine Dysautonomia).
Equine dysautonomia is a disease that affects the postganglionic sympathetic and parasympathetic neurons and is most commonly reported in the United Kingdom and a variety of European countries. In addition, prevertebral and paravertebral ganglia are commonly affected, as are cranial nerve nuclei of the brainstem. The lesions consist of neuronal chromatolysis followed by degeneration and loss of lower motor neurons of the general visceral efferent nuclei of cranial nerves III and X and the general somatic efferent nuclei of cranial nerves III, V, VII, and XII. It has been suggested that equine dysautonomia should be classified as a multisystem disease. Clinically, injury of neurons results in dysphagia, reflux esophagitis, and gut stasis (colic).
Although the cause is unknown, oxidative stress, fungal toxins, changes in weather, and exposure to C. botulinum type C have been proposed. Pasture grasses stressed by rapid growth or sudden cold weather can have reduced concentrations of antioxidants and increased concentrations of glutamate and aspartate (excitotoxic amino acids) and the neurotoxin malonate. It has been proposed that ingestion of high concentrations of these compounds either directly (excitotoxicity-apoptotic cell death) or indirectly (nitric oxide toxicity) induces neuronal injury within the autonomic nervous system, resulting in alimentary system dysfunction. Because mycotoxins were suspected as a cause of equine grass sickness, studies conducted to investigate this hypothesis demonstrated high levels of Fusarium spp. fungi in pastures from confirmed cases of grass sickness. The significance of these fungi in the pathogenesis of grass sickness is unclear. Current research is focused on the role that C. botulinum type C plays in the development of the disease. Higher levels of immunoglobulin A against C. botulinum type C1 neurotoxin have been reported in acute cases. Historical evidence also suggests that use of C. botulinum vaccination may prevent some aspects of the disease.
There are no gross lesions in the PNS, except potentially for lesions related to paralytic ileus (i.e., distended gastrointestinal tract, impacted large intestine, esophagitis due to reflux); however, microscopically and principally in the small intestine (ileum), the cell bodies of neurons in ganglia of the autonomic and enteric divisions of the PNS are chromatolytic, have displaced and pyknotic nuclei, are swollen and vacuolated, and with time there is neuronal loss and satellite cell proliferation in affected ganglia. Interstitial cells of Cajal are also reduced in number. Equine dysautonomia affects horses, ponies, and donkeys primarily between the ages of 2 and 7 years old. The disorder occurs principally between the months of April and July. Injury to enteric neurons results clinically in acute to chronic dysphagia and gut stasis (colic). The only way to diagnose equine grass sickness ante mortem is by taking a biopsy of the small intestine during surgery.
Nutritional Diseases
Deficiencies
Equine Motor Neuron Disease.
Equine motor neuron disease resembles amyotrophic lateral sclerosis in human beings. Because vitamin E concentrations are very low in affected horses, this and other dietary antioxidant deficiencies have been suggested as a possible factor in the mechanism of equine motor neuron disease. Thus dietary factors, especially the long-term absence (>1 year) of green feeds with high vitamin E concentrations, have been implicated in the pathogenesis of the disease. Vitamin E supplementation may be useful in treating this disease if detected and treated early in its course.
Neural injury in equine motor neuron disease involves the cell bodies and axons of lower motor neurons (ventral horn cells, cranial nerves). Microscopically, cell bodies are swollen, have chromatolysis, and contain spheroids. As the disease progresses, the cell bodies become shrunken and degenerate and are removed by neuronophagia. When the cell bodies are lost, the resulting empty neuronal space can be replaced by astrogliosis. The axons of affected lower motor neurons have lesions consistent with Wallerian degeneration. Abundant amounts of lipofuscin are often found in neurons and endothelial cells.
The injury in lower motor neurons has been attributed to an oxidative stress mechanism because vitamin E is an antioxidant that offsets the harmful effects of free radicals and reactive oxygen species that can cause membrane lipid peroxidation. However, it is not linked to a mutation in the equine Cu/Zn superoxide dismutase gene. This gene regulates the production of the enzyme superoxide dismutase, whose function is to convert free radicals and reactive oxygen species (highly toxic to cells) to hydrogen peroxide (much less toxic to cells). The enzyme catalase is used to convert hydrogen peroxide to water and oxygen molecules. The muscle lesion in equine motor neuron disease is atrophy of type I myofibers secondary to loss of type 1 lower motor neurons.
Clinically, equine motor neuron disease is characterized by progressive degeneration and loss of lower motor neurons resulting in muscle atrophy, weight loss, difficulty standing, and muscle fasciculation
Vitamin E Deficiency
Equine Degenerative Myeloencephalopathy.
Equine degenerative myeloencephalopathy is discussed in the section on Disorders of Horses in the section on the CNS.
Traumatic Injury
Recurrent Laryngeal Paralysis.
Laryngeal paralysis (roarer syndrome) is caused by axonal injury to the left recurrent laryngeal nerve, which results in neurogenic atrophy of the left dorsal, lateral, and transverse cricoarytenoid muscles and consequently dysfunction of the larynx and laryngeal folds (see Fig. 15-16). The cricoarytenoid dorsalis muscle is the main abductor muscle of the larynx, which keeps the arytenoid cartilages in a lateral position. The possible causes of this axonopathy are multiple, and there may be different causes for different age groups of animals and different forms of the disease. Known causes include (1) transection of the axon by extension of inflammation from the guttural pouches because the nerve runs through the pouch within a connective tissue fold and (2) other trauma to the nerve. There is also some evidence that laryngeal paralysis may be inherited in younger horses. Currently a genetic age-onset abnormality of axoplasmic flow appears to be the most likely cause in horses in which trauma and inflammation can be excluded as causes.
Affected horses have disabilities of performance and a characteristic and diagnostic “roaring” sound with inspiration. Laryngeal hemiplegia can affect the right or left dorsal cricoarytenoid muscles; however, 95% of cases involve the left side. The cause of this specificity is unclear. Some have suggested it is related to the long course of the left recurrent laryngeal, which extends down into the chest and loops under the arch of the aorta to return to the larynx, but this hypothesis is weakened by the fact that the axonal injury is distal to where the nerve innervates the larynx. Curiously, studies have shown that other long nerves in the horse can also show degenerative changes, which may indicate that this disorder is a polyneuropathy, rather than a specific disease of the recurrent laryngeal nerve.
Gross lesions can vary from being recognizable to being inapparent. Microscopically, the lesion is Wallerian degeneration. Affected nerves are often shrunken with loss of axons and myelin, and Büngner's bands can be plentiful. Laryngeal hemiparesis is primarily a disease of large horse breeds between the ages of 2 and 7 years old.
Disorders of Dogs
Peripheral Neuropathies
Congenital/Hereditary/Familial Diseases
Dysautonomias.
A discussion on hereditary dysautonomias can be found in the section on Peripheral Nervous System, Disorders of Domestic Animals.
Miscellaneous Neuropathies
Canine Inherited Hypertrophic Polyneuropathy.
Canine inherited hypertrophic polyneuropathy is a familial disorder in Tibetan mastiffs. The primary defect is in the Schwann cells, but the pathogenesis is undetermined. There are no gross lesions in the PNS. Microscopic lesions consist of demyelination with onion bulb formation. The Schwann cell's cytoplasm is distended by accumulations of actin filaments. Axonal degeneration occurs but is mild. Clinical signs in canine inherited hypertrophic polyneuropathy begin at 7 to 10 weeks of age. They include pelvic limb muscle weakness, depressed spinal reflexes, and muscle atrophy that later progress to involve forelimbs and eventually cause recumbency. Neuropathies with primary developmental demyelination have also been reported in Alaskan malamutes and in beagle–basset hound crosses. A hypertrophic polyneuropathy has rarely been reported in unrelated domestic cats with onset at approximately 1 year of age.
Toxic Diseases
Dysautonomias
Acquired Dysautonomias
Peritonitis-Induced Dysautonomias.
Peritonitis-induced dysautonomias are discussed in the section on Peripheral Nervous System, Disorders of Domestic Animals.
Miscellaneous Neuropathies
Acute Idiopathic Polyneuritis.
Acute idiopathic polyneuritis (coonhound paralysis) is an acute, fulminating polyradiculoneuritis with ascending paralysis that occurs in dogs after the bite or scratch of a raccoon. By definition, polyradiculitis refers to disease or injury involving multiple cranial or spinal nerve roots, whereas polyradiculoneuritis refers to disease or injury involving multiple cranial or spinal nerve roots and their corresponding peripheral nerves.
Coonhound paralysis has been compared with Guillain-Barré syndrome. This human syndrome typically follows a viral illness, vaccination, or some other antecedent disease that results in an autoimmune response resulting in primary demyelination of cranial and spinal rootlets and nerves and delayed conduction of action potentials down the axon. Humoral and cell-mediated components are suspected to be involved in the autoimmune response.
Coonhound paralysis, like Guillain-Barré syndrome, is believed to represent an autoimmune primary demyelination. Despite the lack of close association of macrophages with the degenerating myelin and axons early in the development of the lesions, secretion of TNF-α by these cells could explain both the demyelination and axonal degeneration.
Acute idiopathic polyneuritis has been reported in dogs without an association with raccoons and also occurs rarely in cats, suggesting multiple factors might be involved in this type of nerve damage. Lesions in coonhound paralysis are most severe in ventral spinal nerve rootlets and progressively diminish distally in the peripheral nerve. Involvement of dorsal spinal nerve rootlets and ganglia is not constant and relatively minor. Lesions in the ventral nerve rootlets consist of segmental demyelination with a variable influx of neutrophils, depending on the acuteness and severity of clinical signs, along with lymphocytes, plasma cells, and macrophages (Fig. 14-116 ). Axonal degeneration is a common sequela. Evidence of remyelination with Büngner's bands and axonal sprouting occur during the recovery phase, but the effectiveness of the latter to establish continuity of the nerve rootlet and thus reinnervation of muscle is limited.
Figure 14-116.
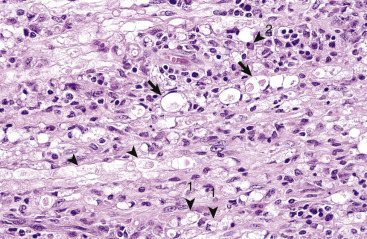
Polyradiculoneuritis, Coonhound Paralysis, Peripheral Nerve, Dog.
This disease results from an autoimmune response leading to primary demyelination of cranial and spinal rootlets and nerves. Myelin sheaths in this peripheral nerve are distended and fragmented along their length (arrowheads) and have been infiltrated by a mixed population of inflammatory cells consisting of lymphocytes, macrophages (1), and plasma cells (2). Enlarged spaces in the myelin sheath, termed digestion chambers (arrows), which form in response to inflammatory and degradative processes, contain myelin debris and macrophages (not shown in this example). Axonal degeneration can occur secondary to primary demyelination. H&E stain.
(Courtesy Drs. R.A. Doty, J.J. Andrews, and J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
A chronic polyradiculoneuritis with infiltrations of lymphocytes, plasma cells, or macrophages; demyelination; and variable axonal degeneration in cranial and spinal nerve rootlets and cranial nerves is also reported in dogs and cats. With repeated episodes of demyelination, onion bulbs can be apparent. Both sensory and motor nerves can be involved with sensory disturbances and muscle atrophy.
Clinically, affected dogs have signs of coonhound paralysis that develop 1 to 2 weeks after exposure to raccoon saliva. Initial signs of hyperesthesia, weakness, and ataxia are replaced in 1 to 2 days by tetraparesis and/or tetraparalysis that may last from weeks to months. Dogs can die from respiratory paralysis. Recovery is common, but the paralysis can be prolonged in dogs with extensive muscular atrophy.
Traumatic Injury
Neurogenic Cardiomyopathy (Brain-Heart Syndrome).
Neurogenic cardiomyopathy is a syndrome in dogs characterized by unexpected death 5 to 10 days after diffuse CNS injury (usually hit by car). Affected dogs die of cardiac arrhythmias caused by myocardial degeneration. Grossly, the myocardium has numerous discrete and coalescing pale white streaks and/or poorly defined areas of necrosis. Neurogenic cardiomyopathy is thought to be caused by overstimulation of the heart by autonomic neurotransmitters and systemic catecholamines released at the time of trauma. It is unknown why there is a 5- to 10-day delay in the development of myocardial necrosis.
E-Appendix 14-1 Necropsy
Difficulties in the Examination of the Central Nervous System
There are inherent difficulties in performing a satisfactory examination of the central nervous system (CNS) in all animal species, including rapid autolysis and the fact that removal of the brain and spinal cord is very hard work in older, large animals whose bones are very hard. The gross examination of the CNS requires time (30 to 60 minutes) and a reasonable degree of physical strength.
Necropsy Procedures
The brain can be removed from the calvaria without great difficulty, but experience gained in veterinary school (diagnostic necropsy rotations) does make the process more efficient and helps to preserve the anatomic integrity of the brain. In the cow and other mammals with large frontal sinuses (pig), the task is more difficult. The bone of the calvaria is cut and removed using a handsaw, hatchet, or Stryker saw (electrically powered saw) with a large blade in large animals or a small handsaw, Stryker saw with a small blade, bone rongeurs, or bone-cutting forceps in smaller animals. The equipment used depends on the thickness of the bone and the experience of the prosector.
The head is usually disarticulated from the body at the occipitoatlantal joint before examination. A midline incision should be cut through the skin over the calvaria from the occipitoatlantal joint to a point midway between the eyes. The skin is reflected laterally to expose the entire calvaria and the masseter, temporalis, and other muscles. These muscles should be dissected free from the calvaria. The bone of the calvaria is cut in a plane extending from the foramen magnum to a point just behind the eye on each lateral side of the calvaria. A single cut is then made in the calvarial bone behind the eyes at right angles to the initial two cuts. This approach results in connecting the planes of all these cuts through the foramen magnum. If the cuts are complete and connected, the calvaria can be pulled free from the head by pulling in a rostral to caudal direction. In animals with frontal sinuses, the same process is essentially repeated twice to expose the brain.
Once the calvaria is removed, the dura mater, if not removed with the calvaria, should be cut along the midline and reflected laterally to expose the surface of the brain. The falx cerebri and tentorium cerebelli must be cut and removed before attempting to remove the brain; otherwise the brain can be torn during removal. The head is turned upside down and at the foramen magnum, the cranial nerve roots severed in descending numeric order to release the brain from the cranial vault. The force of gravity pulls the brain out of the cavity onto the necropsy table.
The spinal cord can be removed from small and large animals, but the process is labor intensive; without experience and proper equipment, the process is prone to spinal cord damage and risk to the prosector. The vertebral column must be separated from the limbs, ribs, and visceral and thoracic organs. In large animals the vertebral column is cut transversely into cervical, thoracic, and lumbar segments. Each segment is then cut in a lateral to medial direction on a sagittal plane to the depth of the spinal cord on a heavy-duty band saw. When the vertebral canal is reached, the dura mater is longitudinally cut the length of the cord, the spinal nerves are severed, and the spinal cord removed. A similar method can be used in large dogs, but in smaller animals, a complete dorsal laminectomy can be performed and then this procedure is followed. The anatomic orientation of the spinal cord needs to be maintained for subsequent histopathologic evaluation.
Even light compression of the spinal cord during necropsy fractures the gray matter. Heavier pressure squeezes it out like toothpaste. The CNS is very delicate and must be handled carefully. Bone dust from saws can cause confusion in interpretation of the histologic sections.
Gross Examination before Fixation
Once the CNS has been removed from the calvaria and vertebral canal and before placing the brain and spinal cord in a fixative, they should be carefully examined for any gross abnormalities. The CNS is a symmetric organ with the right and left sides mirroring each other in structural and functional features across an imaginary midline. These sides should be compared for any alterations in size, symmetry, or color. The sulci, gyri, and the meninges should be examined for inflammation and for any changes suggestive of edema. It is not advisable to section an unfixed specimen of brain or spinal cord unless absolutely necessary.
It is also at this time that a decision must be made regarding the saving of specimens for ancillary testing such as for bacteriologic, virologic, and toxicologic testing. The necessity for these tests will be based on the history, findings of the postmortem examination, and probability of a specific disease occurring in the animal. Fresh CNS tissue should be provided for these ancillary tests in quantities sufficient and satisfactory for the requested analysis, but not in a manner that interferes with further macroscopic and microscopic evaluation of the CNS after fixation. Contact your regional diagnostic laboratory for specific information regarding proper sample selection for these ancillary tests and for evaluation of tissues for rabies virus.
Fixation Procedures
The CNS should be fixed by immersion in 10% neutral-buffered formalin for histopathologic evaluation. Once the CNS is fixed, it should be sliced like a “loaf of bread” from the olfactory bulbs to the cauda equina in approximately 1-cm-thick sections. The cut surfaces of each section should be carefully examined for changes in size, shape, color, and symmetry when compared with the opposite side. Specimens for microscopic evaluation should be taken from any areas that have visible changes and from areas of the CNS that likely would have lesions for diseases listed in the list of differential diagnoses. In addition, all routine microscopic evaluations of the CNS, in which gross lesions are or are not detected, should include specimens from the cerebral cortices (two areas), thalamus/hypothalamus, hippocampus, cerebellum and roof nuclei, brainstem (two areas), and the cervicothoracic and thoracolumbar enlargements of the spinal cord.
The spinal cord and peripheral nerve can be fixed by immersion in formalin. Fetal and neonatal brains should be fixed in 20% neutral-buffered formalin. It is best to fix the entire brain without prior sectioning by submerging it in 10% neutral-buffered formalin and then adding 37% formalin until the brain floats in the solution. The brain should not be randomly sliced in the hope of better tissue fixation. These procedures may vary based on the academic institution.
E-Appendix 14-2 Diseases Caused by Microbes
Viruses
Disorders of Domestic Animals
Borna Disease
Borna disease is an encephalomyelitis caused by an unusual enveloped RNA virus that replicates in the nucleus of neurons, has no cytopathic effect, and until 1997, its virions had not been visualized ultrastructurally. The disease has been recognized in central Europe for more than 250 years but likely exists worldwide. Borna disease virus has been classified as the prototype of a relatively new virus family, Bornaviridae. It most commonly infects horses and sheep, but cases have also been reported in dogs, cats, and cattle. Borna disease virus infection in horses was originally considered to result in a high mortality, up to 80% to 100% after 1 to 3 weeks of clinical illness. More recent evidence indicates that the majority of infected animals are either asymptomatic or have mild clinical disease that can be accompanied by behavioral changes, followed by recovery. The clinically severe disease in the horse is still largely confined to certain regions in Germany and Switzerland; the asymptomatic infection, which probably also occurs in other species, including human beings, is considered to have a worldwide distribution. Proposed peripheral spread of virus by axonal transport in the autonomic and enteric divisions of the peripheral nervous system (PNS) has led to the suggestion that infection of these neurons might cause lesions responsible for colic and other gastroenteric dysfunctions in horses.
Experimental evidence indicates that infection of the central nervous system (CNS) via olfactory nerves can follow intranasal viral exposure. The virus is highly neurotropic, similar to rabies virus, and is transported by retrograde axonal transport from the periphery to the CNS. Infection of astrocytes, oligodendroglia, ependyma, choroid plexus epithelial cells, and Schwann cells occurs by direct extension when these cells are in close proximity to virus-infected neurons. The virus can also infect the retina by retrograde axonal transport via the optic nerve and cause blindness under experimental conditions. After reaching the CNS, the virus spreads intraaxonally and transsynaptically (proposed) as described for rabies virus. The virus can then also spread centrifugally via the PNS, resulting in infection of various nonneural tissues, including the lacrimal and salivary glands, endocrine tissues, such as the pituitary and adrenal glands, and other tissues. It has also been determined that peripheral blood monocytes can be infected. Experimentally the specific lesions that develop in the CNS appear to depend on a viral-induced, cell-mediated immune mechanism. Antibody to Borna disease virus does not appear to play any significant role in the disease process.
There are no major gross lesions in the CNS. Microscopic lesions, which are limited to the nervous system, consist of a nonsuppurative encephalomyelitis with neuronal degeneration. Lesions are confined largely to the gray matter and are most severe in the midbrain, midbrain-diencephalon junction, hypothalamus, and hippocampus. Inflammation of the meninges and spinal cord is generally mild. Small, round to oval, eosinophilic intranuclear inclusions (Joest-Degen bodies) occur in neurons of the brainstem, hippocampus, and cerebrospinal ganglia. In the PNS, inflammation occurs in the cranial, spinal, and autonomic ganglia and in the peripheral nerves.
Louping Ill
Louping ill, a tick-transmitted sheep encephalomyelitis, has been recognized in the British Isles for at least 200 years. It is primarily a disease of sheep but also affects cattle, horses, pigs, goats, red deer, dogs, human beings, and the red grouse. It is caused by a flavivirus (family Flaviviridae) and occurs in the British Isles and Norway. A similar disease of sheep occurs in Turkey, Greece, Bulgaria, and Spain. Some cases have been determined to be caused by similar flaviviruses but are distinct from each other and from louping ill virus. The disease occurs in the spring and summer when ticks are alive.
After infection caused by the tick Ixodes ricinus, the virus replicates in lymph nodes and spleen and reenters the blood stream to cause a high-titered viremia, which is the probable route of infection of neurons. Excretion of the virus in milk of infected ewes and goats has also been reported, with the suggestion that transmission by ingestion is of possible significance in suckling kids.
No major gross lesions are present. Microscopic lesions are characterized by a meningoencephalomyelitis, which is primarily nonsuppurative, although occasional neutrophils can be present. Specific changes also include neuronal degeneration, necrosis, and neuronophagia, most consistently occurring in Purkinje cells of the cerebellar cortex but also affecting neurons of the medulla oblongata, pons, and spinal cord. Lesions in Purkinje cells have been proposed to be at least partially responsible for the unique clinical signs of a peculiar leaping gait displayed by affected animals. No inflammation of spinal ganglia occurs, but inflammation has been detected in sciatic nerves.
Japanese Encephalitis
Japanese encephalitis is a particularly important disease in human beings, but infection also occurs in horses, pigs, cattle, and sheep. The causative virus is classified as a member of the family Flaviviridae (closely related to St. Louis encephalitis and West Nile virus) and is transmitted by mosquitoes, mainly Culex tritaeniorhynchus. In nature, infection is maintained in a cycle involving vector mosquitoes, birds, and pigs. Although young susceptible pigs can have signs, detectable illness is not a feature of viral infection in adult or pregnant pigs. However, transplacental fetal infection during pregnancy can result in mummification and stillbirth of fetuses or the birth of weak live pigs with nervous signs accompanied by nonsuppurative encephalitis and neuronal degeneration.
Factors involved in the pathogenesis of this viral infection include the recent finding that in rats the ability of the virus to infect neurons is closely associated with neuronal immaturity. Such an age-dependent susceptibility of brain-to-viral infection has also been noted with other flaviviruses, including St. Louis encephalitis virus and yellow fever virus. The fact that fetal and neonatal pigs and young horses appear to be more susceptible than adult animals suggests that such a correlation could also exist in naturally occurring infections of animals.
Grossly, CNS lesions include mild leptomeningeal congestion and hyperemia and occasional hemorrhages within the brain and spinal cord. Microscopically, neurons in the CNS are the target cell. Lesions are characterized by an early leptomeningitis and encephalitis in which neutrophils predominate, followed by nonsuppurative encephalomyelitis. This virus also causes degeneration of neurons, especially Purkinje cells of the cerebellum, with necrosis, neuronophagia, microgliosis, and axonal degeneration. Neuronal necrosis is accompanied by lymphocytic perivascular cuffs. There are no inclusion bodies. The lesions are distributed diffusely throughout the nervous system but affect the gray matter more than the white matter. Well-documented outbreaks of meningoencephalomyelitis caused by Japanese encephalitis virus in horses have been reported. Young or immature horses are more susceptible to infection than older animals. Its geographic distribution includes India, China, and southeastern Asia.
Disorders of Pigs
Classical Swine Fever
Classical swine fever (hog cholera) of pigs is caused by a pestivirus. It has a worldwide distribution, except for several countries, including the United States, from which it has been successfully eradicated. Infection under natural conditions occurs by the oronasal route. The virus initially infects epithelial cells of the tonsillar crypts and surrounding lymphoid tissue and then spreads to submandibular and pharyngeal lymph nodes, where it replicates. The virus disseminates via leukocytic trafficking to the spleen, bone marrow, visceral lymph nodes, and lymphoid tissue of the intestine, where high titers of virus are attained. Target cells for virus replication include endothelial cells, lymphoid cells and macrophages, and epithelial cells. Hematogenous spread of the virus via leukocytic trafficking to endothelial cells throughout the infected pig is usually completed in 5 to 6 days. Infected animals die of disseminated intravascular coagulation.
Lesions of the acute disease, which primarily result from a tropism of the virus for vascular endothelium with subsequent hemorrhage, are present in many organs, including the kidneys, intestinal serosa, lymph nodes, spleen, liver, bone marrow, lungs, skin, heart, stomach, gallbladder, and CNS. Grossly, cerebral edema may be observed. Microscopic lesions of the CNS occur in both gray and white matter and tend to be most prominent in the medulla oblongata, pons, colliculi, and thalamus but also occur in the cerebrum, cerebellum, and spinal cord. Lesions are characterized by swelling, proliferation, and necrosis of endothelium; perivascular lymphocytic cuffing; hemorrhage and thrombosis; microgliosis; and neuronal degeneration. Choroiditis and leptomeningitis also occur. Special histochemical stains may be required to satisfactorily identify elementary bodies in mononuclear inflammatory cells. Clinical signs resulting from involvement of the CNS include ataxia, paresis, and convulsions.
Enterovirus-Induced Porcine Polioencephalomyelitis
Teschen disease and Talfan disease, the enterovirus encephalomyelitides of pigs caused by porcine enteroviruses (family Picornaviridae), are characterized by polioencephalomyelitis. Teschen disease is caused by porcine enterovirus 1. Natural infection occurs by the oral route and is followed by viral localization and replication in the tonsil, Peyer's patches, and the intestinal tract (primarily ileum, large intestine, and cervical and mesenteric lymph nodes). These viruses then enter the bloodstream and spread hematogenously through the blood-brain barrier to the CNS, where they target motor neurons.
No gross lesions are detectable. Cerebral, cerebellar, and spinal cord involvement vary with the different viruses causing the polioencephalomyelitis. All forms of the disease are characterized microscopically by a nonsuppurative polioencephalomyelitis, which targets motor neurons of the ventral gray horns and craniospinal ganglia. These viruses cause degeneration of neurons with acute swelling, central chromatolysis, necrosis, neuronophagia, microgliosis, and axonal degeneration. Neuronal necrosis is accompanied by lymphocytic perivascular cuffs, especially in the spinal cord. Astrocytosis and particularly astrogliosis also occur. Ganglioneuritis, particularly of dorsal root ganglia of the spinal cord, and variable leptomeningitis of varying severity also occur.
Clinical signs include ataxia, excessive squealing, altered or lost vocalization, irritability, muscular tremors/rigidity, grinding of the teeth, and convulsions. In the different diseases, severity varies from notable and associated with death of affected animals (Teschen disease occurring sporadically in Europe and Africa) to less severe disease with signs that include fever, diarrhea, and paralysis, sometimes most severe in the hind legs, in pigs in North America and some other regions of the world (Talfan disease).
Hemagglutinating Encephalomyelitis Viral Infection of Pigs
In 1958 a disease of nursing pigs characterized by high morbidity, vomiting, anorexia, constipation, and severe progressive emaciation was reported in Ontario, Canada. The causal agent was found to be a coronavirus. Infection by the oronasal route, which has been demonstrated experimentally, is followed by viral replication in epithelial cells of the nasal mucosa, tonsils, lungs, and small intestine. After local replication the virus spreads to the CNS by retrograde axonal transport in the peripheral nerves, which include the trigeminal and olfactory nerves, vagus, and extensions from intestinal plexuses to the spinal cord. Neurons of craniospinal ganglia also are infected. The vomiting associated with the disease is presumed to result from altered function of neurons (in the vagal nucleus and its ganglion, and gastric intramural plexuses in the enteric division of the PNS) secondary to viral infection.
Gross lesions of the CNS are not present. Microscopic lesions occur in the respiratory tract, stomach, and CNS and PNS. Similar to enterovirus encephalomyelitis, lesions in the CNS are most pronounced in the gray matter and are characterized by a nonsuppurative meningoencephalomyelitis with neuronal degeneration, lymphocytic perivascular cuffs, and microglial nodules. The caudal brainstem, particularly the medulla and pons, and spinal cord are affected. In the peripheral ganglia, the lesions are nonsuppurative inflammation and neuronal degeneration.
Clinically affected animals may show vomiting caused by neuronal lesions in enteric plexuses, depression, hyperesthesia, trembling, ataxia, convulsions, and paddling of the limbs if laterally recumbent.
Chlamydia-Induced Vasculopathies
Disorders of Ruminants
Sporadic Bovine Encephalomyelitis
Sporadic bovine encephalomyelitis (chlamydial encephalomyelitis) is uncommon and is caused by either Chlamydophila pecorum or psittaci (formally named Chlamydia psittaci). The disease was first described in the United States, but cases have since occurred in other countries. Chlamydiaceae are obligate intracellular microbes now classified as bacteria. C. psittaci is inhaled or ingested via an oronasal route and infects epithelial cells of oronasal mucous membranes and lungs, endothelial cells, and monocytes and lymphocytes. It appears to spread systemically to infect endothelial cells within the CNS via leukocytic trafficking in monocytes and lymphocytes. Lesions are nonsuppurative meningoencephalomyelitis and serofibrinous polyserositis, arthritis, and tenosynovitis. Gross changes in the CNS, when present, are limited to active hyperemia and edema of the leptomeninges. Microscopic lesions extend throughout the neuraxis and consist of leptomeningeal and perivascular infiltrates of macrophages, plasma cells, and a few neutrophils. The basilar leptomeninges are most severely affected. Leukocytes extend into the adventitia of blood vessels accompanied by endothelial swelling and necrosis leading to vasculitis, thrombosis, and ischemia. Additional lesions include neuronal degeneration and parenchymal necrosis with microgliosis. Immune-mediated mechanisms have been suggested as the cause of the vasculitis. Calves younger than 6 to 12 months of age are most susceptible. Affected animals initially are ataxic but terminally may become recumbent and opisthotonos develops.
Suggested Readings
Suggested Readings are available at www.expertconsult.com.
Footnotes
For a glossary of abbreviations and terms used in this chapter see E-Glossary 14-1.
Postmortem examination of the CNS is discussed in E-Appendix 14-1.
See E-Box 1-1 for a listing of potential, suspected, or known genetic disorders in the nervous system.
Suggested Readings
- Abbott NJ, Patabendige AA, Dolman DE. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony DC, Couch Y. The systemic response to CNS injury. Exp Neurol. 2014;258:105–111. doi: 10.1016/j.expneurol.2014.03.013. Review. [DOI] [PubMed] [Google Scholar]
- Arancibia-Carcamo IL, Attwell D. The node of Ranvier in CNS pathology. Acta Neuropathol. 2014;128:161–175. doi: 10.1007/s00401-014-1305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. Review. [DOI] [PubMed] [Google Scholar]
- Bolon B, Butt M. Wiley-Blackwell; Hoboken: 2011. Fundamental neuropathology for pathologists and toxicologists: principles and techniques. [Google Scholar]
- Brosius Lutz A, Barres BA. Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev Cell. 2014;28:7–17. doi: 10.1016/j.devcel.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantile C, Youssef S. The nervous system. In: Maxie MG, editor. ed 6. vol 1. Saunders; Philadelphia: 2015. (Jubb KVF, Kennedy PC, Palmer N: Pathology of domestic animals). [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Su JH, Cotman CW. Beta-amyloid accumulation in aged canine brain: a model of early plaque formation in Alzheimer's disease. Neurobiol Aging. 1993;14:547–560. doi: 10.1016/0197-4580(93)90038-d. [DOI] [PubMed] [Google Scholar]
- de Lahunta A, Glass EN. ed 4. Saunders; Philadelphia: 2014. Veterinary neuroanatomy and clinical neurology. [Google Scholar]
- Dowling AL, Head E. Antioxidants in the canine model of human aging. Biochim Biophys Acta. 2012;1822:685–689. doi: 10.1016/j.bbadis.2011.09.020. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP, Lindsay DS, Saville WJ. A review of sarcocystis neurona and equine protozoal myeloencephalitis (EPM) Vet Parasitol. 2001;95:89–131. doi: 10.1016/s0304-4017(00)00384-8. [DOI] [PubMed] [Google Scholar]
- Edgar JM, Nave KA. The role of CNS glia in preserving axon function. Curr Opin Neurobiol. 2009;19:498–504. doi: 10.1016/j.conb.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Elsheikha HM, Mansfield LS. Molecular typing of sarcocystis neurona: current status and future trends. Vet Parasitol. 2007;149:43–55. doi: 10.1016/j.vetpar.2007.06.039. Review. [DOI] [PubMed] [Google Scholar]
- Gallo V, Deneen B. Glial development: the crossroads of regeneration and repair in the CNS. Neuron. 2014;83:283–308. doi: 10.1016/j.neuron.2014.06.010. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci. 2010;11:87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- Goering R, Dockrell H, Zuckerman M. ed 5. Mosby; St. Louis: 2012. Mims' medical microbiology. [Google Scholar]
- Hadlow WJ, Kennedy RC, Race RE. Natural infection of Suffolk sheep with scrapie virus. J Infect Dis. 1982;146:657–664. doi: 10.1093/infdis/146.5.657. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Harkin KR, Andrews GA, Nietfeld JC. Dysautonomia in dogs: 65 cases (1993-2000) J Am Vet Med Assoc. 2002;220:633–639. doi: 10.2460/javma.2002.220.633. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Tilson HA. ed 3. CRC Press; Boca Raton: 2010. Neurotoxicology. (target organ toxicology series)). [Google Scholar]
- Head E. A canine model of human aging and Alzheimer's disease. Biochim Biophys Acta. 2013;1832:1384–1389. doi: 10.1016/j.bbadis.2013.03.016. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC. Rabies pathogenesis. J Neurovirol. 2002;8:267–269. doi: 10.1080/13550280290100725. [DOI] [PubMed] [Google Scholar]
- Jolly RD, Walkley SU. Lysosomal storage diseases of animals: an essay in comparative pathology. Vet Pathol. 1997;34:527–548. doi: 10.1177/030098589703400601. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, editors. Principles of neural science. ed 5. McGraw-Hill; New York: 2012. [Google Scholar]
- Katsumoto A, Lu H, Miranda AS. Ontogeny and functions of central nervous system macrophages. J Immunol. 2014;193:2615–2621. doi: 10.4049/jimmunol.1400716. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Han SJ, Yang I. Microglia and central nervous system immunity. Neurosurg Clin N Am. 2010;21:43–51. doi: 10.1016/j.nec.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Kierszenbaum AL, Tres L. ed 4. Saunders; Philadelphia: 2015. Histology and cell biology: an introduction to pathology. [Google Scholar]
- Kigerl KA, de Rivero Vaccari JP, Dietrich WD. Pattern recognition receptors and central nervous system repair. Exp Neurol. 2014;258:5–16. doi: 10.1016/j.expneurol.2014.01.001. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacbawan FL, Muenke M. Central nervous system embryogenesis and its failures. Pediatr Dev Pathol. 2002;5:425–447. doi: 10.1007/s10024-002-0003-3. [DOI] [PubMed] [Google Scholar]
- Lorenz MD, Coates J. ed 5. Saunders; Philadelphia: 2010. Handbook of veterinary neurology. [Google Scholar]
- Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- Maclachlan NJ, Dubovi EJ. ed 4. Academic Press; San Diego: 2010. Fenner's veterinary virology. [Google Scholar]
- Meuten DJ. ed 4. Iowa State Press; Ames: 2002. Tumors in domestic animals. [Google Scholar]
- Miron VE, Franklin RJ. Macrophages and CNS remyelination. J Neurochem. 2014;130:165–171. doi: 10.1111/jnc.12705. Review. [DOI] [PubMed] [Google Scholar]
- Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. doi: 10.1146/annurev-immunol-032713-120240. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Soma LA, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94:1077–1098. doi: 10.1152/physrev.00041.2013. Review. [DOI] [PubMed] [Google Scholar]
- Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–38. doi: 10.1016/j.neulet.2013.12.071. Review. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. Review. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. ed 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. Prion biology and diseases. [Google Scholar]
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–623. doi: 10.1146/annurev-genet-110711-155524. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rofina J, van Andel I, van Ederen AM. Canine counterpart of senile dementia of the Alzheimer type: amyloid plaques near capillaries but lack of spatial relationship with activated microglia and macrophages. Amyloid. 2003;10:86–96. doi: 10.3109/13506120309041730. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2013;20:160–172. doi: 10.1177/1073858413504466. Review. [DOI] [PubMed] [Google Scholar]
- Spencer PS, Schaumburg HH, Ludolph AC. ed 2. Oxford University Press; New York: 2000. Experimental and clinical neurotoxicology. [Google Scholar]
- Squire LR, Berg D, Bloom F. ed 3. Academic Press; San Diego: 2008. Fundamental neuroscience. [Google Scholar]
- Summers BA, Cummings JF, de Lahunta A. Mosby; St. Louis: 1995. Veterinary neuropathology. [Google Scholar]
- Thomson CE, Hahn C. Saunders; Philadelphia: 2012. Veterinary neuroanatomy: a clinical approach. [Google Scholar]
- Vandevelde M, Higgins R, Oevermann A. Wiley-Blackwell; Hoboken: 2012. Veterinary neuropathology: essentials of theory and practice. [Google Scholar]
- Vandevelde M, Zurbriggen A. Demyelination in canine distemper virus infection: a review. Acta Neuropathol. 2005;109:56–68. doi: 10.1007/s00401-004-0958-4. [DOI] [PubMed] [Google Scholar]
- van Noort JM, Bsibsi M. Toll-like receptors in the CNS: implications for neurodegeneration and repair. Prog Brain Res. 2009;175:139–148. doi: 10.1016/S0079-6123(09)17509-X. [DOI] [PubMed] [Google Scholar]
- Venneti S. Prion diseases. Clin Lab Med. 2010;30:293–309. doi: 10.1016/j.cll.2009.11.002. Review. [DOI] [PubMed] [Google Scholar]
- Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci. 2014;15:84–97. doi: 10.1038/nrn3638. Review. [DOI] [PubMed] [Google Scholar]
- Wang W, Bu B, Xie M. Neural cell cycle dysregulation and central nervous system diseases. Prog Neurobiol. 2009;89:1–17. doi: 10.1016/j.pneurobio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Young B, O'Dowd G, Woodford P. ed 6. Churchill Livingstone; Edinburgh: 2013. Wheater's functional histology: a text and colour atlas. [Google Scholar]
- Young W. Spinal cord regeneration. Cell Transplant. 2014;23:573–611. doi: 10.3727/096368914X678427. Review. [DOI] [PubMed] [Google Scholar]