

Chapter 13.1. Introduction
Over the last century, much has been learned about the functions of eosinophils, eosinophilia, and human disease. Indeed, from its initial appearance as a casual, innocent bystander in disease to a major pathogenic effector cell causing the disease, the complexity of eosinophil activities at the molecular and cellular level is now being unraveled. An extension of this intense investigation is the recognition that targeting the eosinophil, its production, and its accumulation in target tissues, is an important and necessary area of therapeutic exploration.
In healthy individuals, eosinophils represent a minor leukocyte subpopulation, accounting for less than 5% of total circulating white blood cells. Tissue compartments with abundant resident populations of eosinophils include bone marrow, primary, and secondary lymphoid tissues, the uterus, and most of the gastrointestinal tract (with the exception of the esophagus under homeostatic conditions). These tissues share features of substantial cellular turnover and regenerative capacity. To a large extent, eosinophils serve as effector cells, capable of inducing significant tissue damage as a result of their release of preformed cytotoxic mediators, including the granule proteins, major basic protein, and eosinophilic cationic protein. These mediators lead to the production of reactive oxygen species and generate an array of lipid mediators. The role of eosinophils was previously considered to be defensive in the setting of parasitic infections or offensive in the development of an allergic response to an environmental allergen. This binary expression of the role or function of eosinophils, especially in the context of human disease, has recently undergone considerable evolution. Eosinophilia and eosinophil products are now centrally positioned in ongoing immune responses through production of pivotal cytokines and chemokines, expression of features of antigen-presenting cells, ligation of Toll-like receptors, and the elicitation of T-helper (Th2) immune responses. In these activities, eosinophils have been shown on the one hand to enhance local inflammatory responses, while on the other to dampen such responses. With this extensive array of activities, it is not surprising that a role for eosinophils has been demonstrated in normal tissue homeostasis and in many disease states. This chapter details the unique positions eosinophils play in a wide range of disease states, in both pathological and protective roles.
In Chapter 13.2, Per Venge begins by addressing the proteome of human eosinophils and the differences in molecular forms of many of the proteins in healthy and allergic subjects. Identification of the spectrum of proteins produced and the genetic polymorphisms of the major secretory molecules is fundamental to our understanding of the role of eosinophils in health and disease and provides the potential for targeted regulation of specific functions.
Eosinophilia is a hallmark of allergic disorders characterized by the activation of selective hematopoietic processes during the onset and maintenance of allergic inflammation. The appearance of eosinophils in the circulation and in tissue involves processes in the bone marrow that lead to accumulation, differentiation, and proliferation of eosinophil lineage-committed hematopoietic progenitors at tissue sites. In Chapter 13.6, Gavreau and Denburg summarize mechanisms that lead to the accumulation of eosinophils and their progenitors in the airways of allergic asthmatics. The role of eosinophils in asthma is further developed by Thomas and Busse. Recognizing the controversial relationship between airway eosinophilia and asthma severity, they focus on the dynamic contribution of eosinophils to asthma. What emerges are two major roles, one in which the eosinophil serves as an effector cell in airway remodeling, the other as a biomarker for asthma exacerbations. The link between eosinophils and asthma is strengthened with the recognition that viruses are a primary cause of asthma exacerbations. In Chapter 13.7, Bivins-Smith and Jacoby explore the association of eosinophils with virus-induced asthma, especially eosinophil contact with airway nerves, which become activated and release mediators that cause dysfunction. In releasing excess acetylcholine, the altered airway nerves modulate airway smooth muscle responses and induce the development of bronchoconstrictive responses.
Moving from the airways to the skin, in Chapter 13.3 Simon and Simon discuss eosinophil infiltration of the skin in a wide variety of disorders, both allergic and nonallergic. However, it remains somewhat unclear what mechanisms are responsible for eosinophil recruitment and activation in the skin, especially as conditions and pathogenic roles vary from disorder to disorder. A similar scenario is observed in the various primary eosinophilic gastrointestinal disorders. In Chapter 13.8, Davis and Rothenberg identify common and uncommon features of these disorders and the surprising and somewhat frightening increases in prevalence of these conditions, especially eosinophilic esophagitis.
Beyond the association of eosinophilic infiltration of the airways, skin, or gastrointestinal tract, the entity hypereosinophilic syndrome has emerged, in which there is significant peripheral blood eosinophilia in the absence of evidence for parasitic, allergic, infections, or other causes. In Chapter 13.9, Khoury and Klion define this rare group of disorders, taking advantage of advances in molecular diagnostics and the use of targeted therapies.
Importantly, eosinophils are now appreciated to be important players in newly recognized roles. In several cancers, eosinophilia is associated with the tumors. In Chapter, 13.10, Lofti and colleagues characterize tumor-associated eosinophils, with activities such as destructive effector functions potentially limiting tumor growth as well as immunomodulatory and remodeling activities, which may suppress immune responses. Eosinophils have been implicated in transplant rejection and have long been seen in graft-versus-host disease. In Chapter 13.13, Roufosse and colleagues review the friend or foe sides of eosinophil infiltration in solid organ and hematopoietic stem cell transplantation. In Chapter 13.14, Krahn and colleagues review the clinical and pathological features of the rare condition of eosinophil myositis. In the absence of known causes of eosinophilic myositis, such as parasite infections, systemic disorders or toxic causes, some cases of idiopathic eosinophilic myositis have been linked to calpain-3 mutations.
Over the last century, much has been learned about the functions of eosinophils, eosinophilia, and human disease. Indeed, from its initial appearance as a casual, innocent bystander in disease to its recognition as a major pathogenic effector cell causing the disease, the complexity of eosinophil activities at the molecular and cellular level is now being unraveled. An extension of this intense investigation is the recognition that targeting the eosinophil, its production and its accumulation in target tissues, is an important and necessary area of therapeutic exploration.
Chapter 13.2. Genomics and Proteomics of the Human Eosinophil
Per Venge
The human eosinophil contains some unique proteins shared only with primates. These are eosinophil cationic protein and eosinophil-derived neurotoxin/RNase2, two potent multifunctional secretory proteins likely to have a great impact on the biology of eosinophils in disease. This is illustrated by the close associations of genetic polymorphisms in these two genes with allergy and parasitic disease. The study of the proteome of human eosinophils has revealed intriguing differences in molecular forms of several proteins between healthy and allergic subjects. Some proteins, such as the heat shock cognate protein 70, were only detected in eosinophils of allergic individuals and other proteins, such as eosinophil peroxidase, showed dramatic molecular alterations in the allergic population. The study of the proteome of human eosinophils and of genetic polymorphisms of the major secretory molecules should, together with assaying eosinophil proteins in biological fluids, provide us with decisive knowledge on the role of eosinophils in health and disease.
Introduction
Almost 40 years ago, the eosinophil granule major basic protein (MBP) was purified by Gleich and colleagues from guinea pig cells1., 2. and the eosinophil cationic protein (ECP/RNase3) purified from human leukemia cells by our group.3., 4. These achievements were the starting points for the study of the proteome of the human eosinophil, with the subsequent purification of MBP from human eosinophils5 and the identification and purification of the two other major eosinophil granule proteins, eosinophil protein X/eosinophil-derived neurotoxin (EPX/EDN)6., 7. and eosinophil peroxidase (EPO).8., 9. Remarkably, these four highly basic proteins make up about 90% of the proteins contained in the secretory granules of the human eosinophil. The eosinophil is regarded to be a secretory cell and it was consequently assumed that the biological activities of the human eosinophil are governed to a large extent by the activities of these proteins. Thus, in attempts to understand the role of the human eosinophil in health and disease, a detailed study of the proteins and their genetics in relation to human disease therefore seemed logical. In this context, it should be emphasized that the granule content of human eosinophils is unique and shared only with other primates, since the duplicated gene products ECP and EPX/EDN are rapidly evolving and highly divergent orthologues are present in nonprimate mammalian species.10 Such findings indicate that the interpretation of activities of eosinophils of other species should be extrapolated to humans with care. In this subchapter, I will describe some of the key activities of the four major granule proteins and the experience of assaying these proteins in various biological materials in human disease. I will also describe recent attempts to map the protein content further using modern proteomics techniques. At the end of the subchapter, I will summarize the genetic findings of the proteins and the associations of single nucleotide polymorphisms (SNPs) of the genes encoding these proteins with disease. More details of the biological activities of the four proteins are found throughout this volume.
The Four Major Granule Proteins of Human Eosinophils
The four major granule proteins of human eosinophils will be considered in turn (Table 13.2.1 ).
TABLE 13.2.1.
Some Characteristics of the Four Major Granule Proteins
| Molecule | Chromosome | Protein Size (kDa) | pI | Major (Minor) Cellular Localization | Biological Activities |
|---|---|---|---|---|---|
| ECP (RNase3) | Chr 14q | 15.5–22 | 10.5–11 | Eosinophil (Neutrophil, Monocyte) | Cytotoxic, Noncytotoxic activities (RNase) |
| EPO | Chr 17q | ~66 | >11 | Eosinophil | Peroxidase, Nonperoxidase activities |
| EPX/EDN (RNase 2) | Chr 14q | 18.6 | ~9 | Eosinophil (Neutrophil, Liver) | RNase, Alarmin |
| MBP | Chr 11q | 13.8 | 10.8 | Eosinophil, Placental cells (Basophil) | Cytotoxic, Noncytotoxic activities |
ECP, eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; EPO, eosinophil peroxidase; EPX, eosinophil protein X; MBP, major basic protein; pI, isoelectric point; RNase 2, non-secretory ribonuclease.
Eosinophil Cationic Protein
ECP is a single chain, highly basic protein [isoelectric point (pI) ranging from 10.5 to 11] with apparent molecular masses ranging from 15.7 kDa to 22 kDa. The heterogeneity is largely due to glycosylation of the protein.11., 12. The gene encoding ECP comprises two exons and one intron and is located on the q arm of chromosome 14 (14q). Exon 2 is the coding DNA sequence for ECP. ECP is located in the secretory granules of human eosinophils and is unique to humans and primates. Minute amounts of ECP may be produced by monocytes and neutrophils under certain conditions.13., 14., 15. However, most of the ECP located in neutrophils probably derives from the active uptake of ECP from the environment.16 ECP belongs to the large family of RNases and is also named RNase3. In addition to being an RNase, ECP is a true multifunctional protein with both cytotoxic and noncytotoxic activities. The cytotoxic activities are determined by post-translational glycosylations and the majority, if not all, of ECP stored in the granules is richly glycosylated and noncytotoxic.12., 17., 18. Upon release from the eosinophil, the molecule is deglycosylated and acquires cytotoxic capabilities.19 Several SNPs have been found in the DNA sequence of ECP; however, only two are in the coding part of exon 2.20., 21. The most commonly found SNP is located at position 434, in which guanine (G) is replaced by cytosine (C). In Scandinavian populations 434G is most commonly found, whereas in African populations 434C is the most common.22 Thus, in Scandinavia about 60% of the population carry the 434GG genotype and about 8% the 434CC genotype, whereas the reverse is the case in a Ugandan population. The 434G>C SNP results in an amino acid shift from arginine at position 96 to threonine and a fundamental change in biological activity, since the cytotoxic activity is lost.18 Whether the loss in cytotoxic activity is due to the amino acid shift per se affecting the cytotoxic site of the molecule or due to the fact that the replacement of arginine with threonine potentially creates a new glycosylation site that might disguise another cytotoxic site is at present not entirely clear. Attempts to identify the bactericidal active sites were made by engineering recombinant protein and peptides.23 These experiments suggest a location for the activity at the N-terminal portion of ECP. The presence in the ECP molecule of several active sites, possibly with different targets, seems likely. The SNP 277C>T in the coding region of the ECP gene is much less common and gives rise to a replacement of arginine at position 47 with cysteine. The possible functional consequences of this amino acid shift are unknown, but it is predicted to have a great impact on the molecular structure. Other biological activities of ECP, such as the RNase activity and the ability of ECP to activate fibroblasts, are not affected by the amino acid shift from arginine to threonine, which shows that these capabilities of ECP are dissociated from each other.17 Other SNPs in the ECP gene are associated with protein expression. Thus, the 562G>C SNP in the 3′UTR region was found to closely correlate with the cellular content of ECP.24 The affected sequence is a binding site for the transcription factor, retinoic acid receptor (RXR), which in turn acts as a cofactor to the transcription factor, Sp1. Sp1 was shown to affect ECP synthesis by binding to the promoter region of the gene. Also, the intronic SNP c.-38A>C has been shown to relate to the ECP content of eosinophils. Thus, several parts of the ECP gene seem to affect ECP production. In a Japanese population, a promoter polymorphism -393C>T is common, but is not found at all in the Scandinavian population.25 This mutation is closely related to serum levels of ECP, thus suggesting an impact on ECP production. The activities of ECP are counteracted by heparin, but also by a protease-modified α2-macroglobulin.11
Eosinophil Protein X/Eosinophil-Derived Neurotoxin
EPX/EDN is a single chain protein of 18.6 kDa that shares 70% homology with ECP, since the formation of the two proteins is the result of a gene duplication 30–40 million years ago.26., 27. The ancestral gene was an RNase and this property has been conserved by the gene product EPX/EDN, but almost completely lost in the gene product ECP, which has instead acquired cytotoxic properties. The protein was independently described, purified, and named (EPX) by our group7 and the group of Gleich (named EDN)28 and both names are used in the literature. For the sake of clarity the name eosinophil derived neurotoxin (EDN) will be used throughout this volume, since this is the more commonly used name and also because the gene of eosinophil peroxidase (EPO) recently has been renamed EPX. The EDN gene is located close to the ECP gene at chromosome 14. Chromosome 14 is also the location of the genes of the RNase A superfamily to which ECP and EDN belong; hence, the alternative name of RNase2. EDN is also a highly basic protein (pI of about 9), although less so than ECP. EDN is produced in small amounts by macrophages and neutrophils, but also by liver cells. EDN is stored in the eosinophil within the secretory granules together with ECP, but is also stored in a separate compartment of easily mobilized secretory vesicles.29 The biological activities of EDN are related to its RNase activity and involve antiviral properties. However, recent studies indicate several other activities of great interest. Thus, EDN has been added to the growing list of alarmins,30 which are proteins that attract and enhance the activities of antigen-presenting cells, such as dendritic cells. Activation of cells through Toll-like receptor 2 (TLR2) further links the activity of EDN to components of innate immunity. The neurotoxic activity, which is the basis for the name EDN, suggests cytotoxic properties for EDN, although the cytotoxic activity of the molecule against any other cell is modest and mostly absent. In our previous studies, we could show some alterations in Purkinje cells in the cerebellum of rabbits following injection of EDN, thus resembling the Gordon phenomenon.31 However, the injection of 100 times lower amounts of ECP had much more detrimental consequences, with the disappearance of Purkinje cells and the rapid development of ataxia and other neurological disturbances. The neurotoxic activities of the eosinophil proteins and the development of the Gordon phenomenon may therefore be the combined actions of the potent RNase EDN and the cytotoxic ECP. Four SNPs were identified in the EDN gene in a Scandinavian population, none of which gives rise to an amino acid shift. One SNP, 405G>C, is located in the intron and is closely related to the cellular content of EDN. This locus is the binding site for several different transcription factors that may be involved in the expression of EDN.
Eosinophil Peroxidase
EPO is a two chain heme-binding protein with one heavy chain of about 52 kDa and one light chain of about 14 kDa.9 The gene is located on chromosome 17q31 and consists of 12 exons and 11 introns. The amino acid sequence shows an almost 70% homology with that of myeloperoxidase and also considerable homology with other members of the peroxidase family of proteins.32 EPO is a highly basic protein with a pI of >11. It is located in the matrix of the secretory granules and is probably specific to eosinophil granulocytes, since no other locations have been identified in mature cells. EPO is difficult to extract from mixed blood leukocytes, since it has a high affinity for neutrophil membrane structures.33 The biological activities of EPO are partly related to its peroxidase activity and partly to other properties of the molecule. The peroxidase catalyzes halidation reactions leading to the formation of long-acting hypohalides, such as hypobromous acid, oxidation of thiocyanate, and nitration of tyrosine.34., 35. Such radicals may act on cellular membranes and take part in defense reactions against a variety of microbes. Numerous mutations and polymorphisms have been found in the EPO gene, five of which result in amino acid shifts. The possible consequence to functional activities of these amino acid shifts is unknown.
Major Basic Protein
Eosinophil MBP was named from findings in guinea pig eosinophils, since it appeared to make up the majority of the proteins contained in the secretory granules.1., 36., 37., 38., 39., 40. In human eosinophils the content of MBP is in the range of the other three major proteins, i.e., 5–10 μg/106 eosinophils. The mass of MBP is 13.8 kDa and its pI is 11.4. The MBP gene is located on chromosome 11q12 and consists of six exons and five introns. MBP is apparently produced as a much larger preproprotein, and an acidic portion of proMBP is cleaved off upon storage in the eosinophil granules. This acidic portion of proMBP may serve to protect cellular structures from its cytotoxic activities during synthesis and packaging. The larger proMBP, however, has been identified in immature bone marrow cells. proMBP has also been found in placental cells in complex with the metalloproteinase pappalysin-1, or pregnancy associated protein A (PAPP-A), and shown to inhibit the activities of PAPP-A. The MBP molecule makes up the typical crystals seen in the specific granules of human eosinophils. An MBP homologue was identified, characterized, and named MBP2.40 This protein was purified from human eosinophils and has a molecular mass of about 13.5 kDa and a much lower isoelectric point of 8.7. The gene encoding hMBP2 is located in close proximity to the gene of MBP1 at chromosome 11q12 and has five exons. MBP1 is expressed in several cell types other than human eosinophils, such as basophils and placental cells, whereas hMBP2 seems to be located only in eosinophils. The biological activities of MBP are predominantly related to its cytotoxic capabilities, but numerous noncytotoxic activities have also been identified, many of which will be described throughout this volume. In the MBP genes, several mutations and polymorphisms have been identified, five of which may result in an amino acid shift. Consequences to the activities of MBP resulting from these amino acid shifts have not been described.
Proteomics Studies of Human Eosinophils
As discussed elsewhere in this volume, the human eosinophil is capable of producing and secreting a number of other proteins in addition to the major proteins described above. These include large numbers of adhesion molecules, chemokines, cytokines, and others. In an attempt to gain further insight into the biology of human eosinophils, modern proteomics techniques may be applied to map the major protein content of normal and diseased eosinophils. In this regard, several different approaches may be applied. One is the description of as many proteins as possible and another is the selected description of proteins based on criteria such as extraction procedures or detection methods, e.g., based on the identification of phosphorylated proteins only. One study incubated eosinophils with sonicates of mast cells and the cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor (TNF-α) and used [35S]methionine to monitor protein synthesis.41 Extracts of eosinophils were run on two-dimensional (2-D) gels and the number of protein spots increased dramatically following these stimuli compared to control cells. In addition, the position of the spots differed depending on the stimuli used, which suggests that eosinophils respond differently to these stimuli. Unfortunately, no attempts were made to identify the proteins in these spots. Another study showed differences in 51 spots between healthy subjects and those affected by atopic dermatitis.42 One such difference was downregulation of the Grb7 adaptor protein in cells from patients, which may relate to eosinophilia of the patients and antiapoptotic features of these cells. Overall, 1121 spots were identified in healthy subjects and 1310 spots in the eosinophils of atopic dermatitis patients, which emphasizes that circulating eosinophils of such patients are exposed to various stimuli that induce protein synthesis. One upregulated spot of particular interest in atopic dermatitis relates to increased expression of the low-affinity receptor for immunoglobulin E (IgE). A different approach involved the study of phosphoproteins in an acute myelogenous leukemia (AML) eosinophil cell line after exposure to dexamethasone or IL-5.43 Fourteen phosphoproteins showed significant changes, i.e., were either phosphorylated or dephosphorylated, after IL-5 and 12 after dexamethasone. Phosphorylation of the translation initiation factor elf-3 subunit was increased by IL-5 and was also found to be increased in patients with atopic dermatitis. Interestingly, phospho-apolipoprotein E (p-APOE) was induced in eosinophils by dexamethasone but was decreased by IL-5 treatment. p-APOE levels could therefore be used as an indicator of proliferation or apoptosis of eosinophils. A 2-D gel of a survey of proteins in whole eosinophil extracts and extracts of membrane fractions of eosinophils of healthy subjects and of eosinophils obtained from allergic subjects during a pollen season is shown (Fig. 13.2.1 ). Altogether more than 336 spots were identified by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), representing 98 different proteins.44 Among the proteins identified were the four major granule proteins described above and a number of other proteins hitherto not associated with human eosinophils. As expected, the proteins represent, to a large part, cytoskeleton-related proteins such as actin, but more than 11% of all proteins are of granular origin (Fig. 13.2.2 ). The study also showed large differences in the expected pIs of several proteins. Thus, the cytoskeleton-related proteins cofilin-1, profilin-1, adenylyl cyclase-associated protein 1 (CAP 1), and coronin-1A were all found to be significantly acidified, whereas EDN and MBP2 were much more basic than expected. The actual biological significance of alterations to cytoskeleton-related proteins is uncertain, but may relate to the well-documented migrating capacity of eosinophils. In eosinophils obtained from allergic subjects exposed to pollen, several intriguing changes were observed (Table 13.2.2 ). One such was a change of more than three units in the pI of two protein spots identified as the heavy chain of EPO due to heavy chain glycosylations. We speculate that such heavy glycosylation may interfere with the enzymatic activity of EPO. In support of such speculations are our previous findings that the peroxidase-dependent luminol-enhanced chemiluminescence reaction of blood eosinophils purified from allergic subjects during the pollen season is significantly reduced.45 Altogether 12 spots were significantly changed in eosinophils from pollen-exposed allergic subjects, five of which were identified by MALDI-TOF MS. The other three identified spots were heat shock cognate protein 70 (hsc70) and the α and β subunits of CAP 1. As indicated above, CAP 1 subunits are involved in cell motility. The upregulation of these proteins, therefore, indicates that eosinophils from allergic subjects have an increased potential to respond to chemoattractants, a capacity that is well documented, since eosinophils harvested from the blood of allergic subjects show increased migration toward several chemoattractants.46 The upregulation of hsc70 has a number of biological implications of interest, such as changes in protein folding, intracellular protein transportation, and antigen presentation. The latter may lend further support to the eosinophil being actively involved in antigen presentation.47
FIGURE 13.2.1.
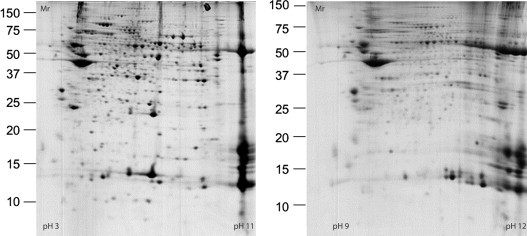
Two-dimensional separation of the proteins of eosinophils from healthy subjects.
The molecular weight is given on the vertical axis. The left panel represents separation of proteins using a pH gradient 3–11 and the right panel separation from pH 9 to pH 12. A large number of proteins are gathered at the end, i.e., at about pH 11, of the left panel. These highly basic proteins were further separated and identified as shown in the right panel. Due to poor solubility, some proteins were not possible to separate using 2-D electrophoresis and their isoelectric points could therefore not be estimated. Among the insoluble proteins were eosinophil lysophospholipase (Charcot–Leyden protein), part of eosinophil peroxidase (EPO) and major basic protein (MBP). Mr, relative molecular mass.
FIGURE 13.2.2.
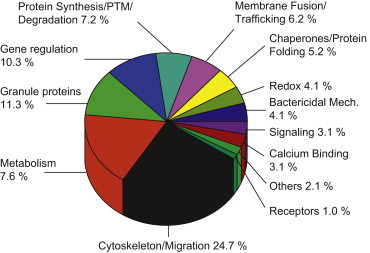
Distribution of the eosinophil proteins according to their biological functions.
The largest proportion of proteins is related to the cytoskeleton and metabolism of the cell. However, as much as 11% of total proteins identified are granule secretory components. PTM, post-translational modification.
TABLE 13.2.2.
Isoelectric Points and Molecular Weights (Mr) of the Four Major Granule Proteins of Healthy or Pollen-Allergic Subjects
| Molecule | Theoretical pI/Mr | Experimental pI/Mr (Healthy Subjects) | Experimental pI/Mr (Allergic Subjects) |
|---|---|---|---|
| ECP | 10.72/15.7 | 10.16–10.72/15.5–20.8 | Similar to healthy |
| EPO, heavy chain | 10.79/53.4 | 10.97/55.6 | 7.48/53.7 |
| EPO, heavy chain | 10.79/53.4 | 9.37/56.9 | 7.30/50.7 |
| EPX/EDN (RNase 2) | 9.2/15.5 | 9.67–10.65/17.2–25.3 | Similar to healthy |
| MBP | 10.8/13.8 | 12.6/ND | Similar to healthy |
ECP, eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; EPO, eosinophil peroxidase; EPX, eosinophil protein X; MBP, major basic protein; ND, not done; pI, isoelectric point; RNase 2, non-secretory ribonuclease.
Assaying Eosinophil Granule Proteins in Disease
The eosinophil marker that has become most widely used in the everyday clinical routine of the allergist is ECP, although several reports have shown that the measurement of EDN, EPO, or MBP may also be useful. The measurement of any of these eosinophil proteins may indicate the activity and turnover of the eosinophil granulocyte. Currently, ECP is measured in serum/plasma, but measurements in nasal lavage fluid, sputum, and possibly saliva are interesting alternatives, since ECP in these biological fluids more accurately reflects the local process. The advantages and disadvantages of measuring ECP in various biological fluids will be discussed below, and the current evidence that ECP may be a useful complement to the diagnostic armamentarium for monitoring and characterizing disease activity in the allergic patient. The emerging evidence of the clinical usefulness of urine measurement of EDN as alternative to serum ECP measurement to reflect eosinophil turnover and activity will also be considered.
Eosinophil Cationic Protein in Sputum and Other Secretions
Numerous reports show that assaying ECP in nasal lavage, saliva, and sputum has the potential to become a clinical instrument for characterizing and monitoring inflammatory processes in the airways.11., 48. This has been particularly shown in patients with asthma, chronic obstructive respiratory disease, and cystic fibrosis. In most cases, sputum has to be induced by hypertonic saline and cells in the sputum need to be separated from the supernatant in order to analyze mediators released from inflammatory cells. The relatively time consuming and complicated procedures required to achieve this are probably the main obstacles for a more widespread use of sputum measurement as a clinical tool. An alternative and much simpler procedure is the measurement of specific markers of various cells in whole sputum extracts. The numbers of eosinophil granulocytes in sputum have been estimated using ECP and several publications show that the numbers of eosinophils measured in this way correlate well with disease activity in asthma and are reduced as a consequence of corticosteroid treatment. An interesting alternative to sputum is saliva, since we showed recently that asthmatics have significantly raised levels of ECP in saliva that are reduced by corticosteroid treatment.49 Still, however, we do not know what the ECP levels in saliva actually reflect, as they may be indicative of either systemic or local eosinophil activity. In addition, the measurements of specific cell markers in nasal lavage fluids or ear secretions have been widely used, and the usefulness of such measurements in the understanding of cellular involvement has been clearly indicated.50 However, their clinical application is still not established.
Eosinophil Cationic Protein in Serum/Plasma
ECP may be measured in both serum and plasma.11 If plasma is chosen, the blood should be anticoagulated with ethylenediaminetetraacetic acid (EDTA) or citrate in order to prevent spontaneous extracellular release of ECP and subsequent interaction with heparin. The levels of ECP in serum are consistently higher than in plasma, due to the fact that eosinophils in the test tube continue their extracellular release of ECP ex vivo. This is an active process that is both time and temperature dependent, which means that higher extracellular levels are achieved with increasing time and ambient temperature, and vice versa.51 Thus, if ECP is measured in serum, strict standardization of the blood sampling procedure and handling of the blood sample are necessary in order to avoid unacceptable variations in ECP levels. Our recommendation is that blood should be taken in tubes with a gel separator and that coagulation is allowed for 1 h at room temperature (22°C) before centrifugation and separation of serum. Both plastic and glass tubes may used. However, differences in the material and the inclusion of coagulation activators in the tubes may affect measurements. This means that normal ranges of ECP have to be determined in each laboratory that does not follow the recommendations of the manufacturer. The levels of ECP in EDTA–plasma probably correctly reflect the circulating levels of ECP at the time of blood sampling. These levels are the consequences of production and elimination of ECP, i.e., local or systemic release of ECP to the circulation as well as variations in the turnover rate of ECP. Normally, turnover is quite rapid, with a half-life (t½) of about 45 min. For several reasons, we can assume that the turnover is more rapid in subjects with ongoing inflammation. This means that an increased release of ECP to the circulation in certain diseases does not always lead to the anticipated increase in plasma levels, since the increased release may be partly or fully counteracted by an increased elimination rate. The dynamics of such counteracting principles may be the main explanation of the fact that in most cases clinical information obtained by EDTA–plasma measurements of ECP is less clear and less useful than the information obtained by serum measurements. In addition to the circulating levels, serum levels of ECP also reflect the secretory activity of the eosinophil population in the blood and, since the levels in serum are often 5–10 times those in plasma, we may draw the conclusion that it is above all the secretory activity of the eosinophils that determines ECP levels. The propensity of blood eosinophils to release ECP is increased in subjects exposed to allergen.52 My own interpretation of serum ECP levels is that they reflect the propensity of the eosinophil population to release ECP in the local process, e.g., in the lung of asthmatics. The higher this propensity is, the more damage is inflicted on the patient. In order to eliminate the influence of eosinophil counts on the serum levels of ECP and thereby obtain a purer reflection of eosinophil activity, serum levels may be divided by the eosinophil count, thus forming an ECP/eosinophil ratio. In some studies, such a ratio was found to be more closely related to disease severity in asthma.
More than a thousand papers have been published dealing with the relation between ECP levels and allergic or other inflammatory diseases.11., 48., 53., 54. The majority of these publications indicate that ECP provides novel information about the process and that the information may be used in treatment stratification and monitoring of the disease, since ECP levels are closely related to exacerbation propensity and severity in diseases such as asthma and atopic dermatitis. Recent data also indicate that serum levels of ECP and EPO predict the further development of allergic disease.55 Thus, serum levels of the two proteins were significantly elevated in a group of patients with allergic rhinitis who developed asthma-like symptoms 6 years later. The prediction was not seen for blood eosinophil counts or nasal lavage findings. Several publications, though, have questioned and sometimes rejected ECP as a clinically useful marker. One reason for this may be the simplified view that asthma is one disease and that the disease is caused by one cell, the eosinophil, and that one marker such as ECP will solve all clinical problems. This is obviously not true, since we know today that the involvement of eosinophils in the asthmatic process is very variable between individuals. Another cause of variations in the results is probably the lack of awareness of the importance of correct sample handling. Still another possibility may be related to the recent discovery of several genetic variants of ECP and the fact that these variants are related to the expression of allergic symptoms and serum levels of ECP. The levels of ECP in blood or any other biological fluid are not disease specific, but provide us with information about the extent to which eosinophils are involved in the particular disease process.
Eosinophil Protein X/Eosinophil-Derived Neurotoxin in Urine
In a search for noninvasive means to monitor eosinophil involvement in inflammatory diseases, urine measurement of EDN has emerged as a promising candidate, particularly in children.56., 57., 58. In order to minimize the influence of differences in water dilution of the urine, EDN levels have to be adjusted to creatinine concentrations unless 24-hour samplings are carried out. It is also useful to bear in mind that all eosinophil markers, including the excretion of EDN, show circadian rhythms with the highest levels occurring at night.59 Thus, for the sake of standardization of blood or urine measurements of eosinophil markers, sampling should always be carried out at the same time of the day. Several studies have shown that EDN in urine is elevated in asthma and atopic dermatitis, is related to disease activity, and reduced by anti-inflammatory treatment, i.e., corticosteroid treatment. Elevated levels of EDN in urine in wheezy children also seem to predict the development of asthma.
Single Nucleotide Polymorphisms in the Major Granule Proteins and their Associations with Disease
As mentioned above, two major granule proteins of human eosinophils are unique to humans and primates. Thus, knowledge of the role of these proteins in human disease cannot be extrapolated from mouse gene knockout experiments. The alternative is therefore to search for human counterparts to such knockouts, i.e., SNPs or mutations that have an impact on the biological activities of these proteins either with regard to their functional alterations or altered production. As shown above, only one SNP is known to lead to a functional alteration in any of these proteins: the ECP434G>C gene polymorphism, in which the replacement of G with C in the DNA sequence results in the production of a noncytotoxic protein with the amino acid threonine at position 97 instead of arginine.18
ECP434G>C (rs2073342) Genotypes and Clinical Findings
In the first study examining the possible association of the ECP434G>C SNP with human disease, we found strong associations with the development of allergic symptoms, both in a group of 209 medical students and in a group of 79 subjects with allergic or nonallergic asthma.21 In the group of medical students, the diagnosis of allergy was based on a self-assessment questionnaire and in the asthma group the distinction was based on a clinical diagnosis. We found that the genotype ECP GG, giving rise to the production of the cytotoxic species, was more common in those experiencing allergic symptoms than in nonallergic subjects or those with nonallergic asthma. However, most notable was the absence of any allergic manifestations in the two cohorts carrying the ECP CC genotype. The ECP CC genotype therefore seemed to exclude the development of allergic symptoms and provided strong support for a key role for eosinophils in allergy. In a larger community-based study on 574 randomly selected subjects in Estonia and Sweden [The European Community Respiratory Health Survey (ECRHS)], symptoms and signs of allergy were based on a structured interview.60 The results of this study were much less clear, although the ECP434G>C genotypes (ECP434GG, ECP434GC, and ECP434CC) showed significant associations with various expressions of allergic symptoms. However, it also became clear that ethnicity, gender, and smoking habits are important confounders. One intriguing finding of the ECRHS study was the associations of the ECP434G>C genotypes with lung function with reduced lung functions found in both women and men carrying the ECP434G>C G-allele compared to those carrying the C-allele. If confirmed, these findings suggest a detrimental effect of the cytotoxic ECP on lung tissues. In a Norwegian–Dutch study, no associations between allergy and the ECP434G>C genotype were found.61 In contrast, this study showed an association with nonallergic asthma, which was also the case in our ECRHS study. An association with allergic rhinitis of the ECP434G>C genotype was also negated in a Korean study.62 The association between asthma/allergy and the ECP434G>C genotype is at present confusing and the seeming differences in findings not easily explained. Ethnicity and gender differences may have an impact, but the definition of the phenotypes studied is probably more important. It is important to clarify these relationships, since the cytotoxic activities of ECP could be targets for therapeutic interventions if such associations are confirmed and established.
ECP has the capacity to kill Schistosoma mansoni larvae. Knowing that the cytotoxic capacity is lost by an amino acid shift from arginine to threonine at position 97, we conducted a study on subjects living in Uganda in an endemic area of S. mansoni infections.22 The ECP434G>C genotype distribution in this population was dominated by subjects carrying the ECP434 CC genotype, i.e., the opposite of the distribution found in non-African populations. Thus, the majority of people living in these endemic areas have a genotype that gives rise to the production of a noncytotoxic ECP and possibly a poorer defense against S. mansoni infection. We examined parasite egg excretion in feces, to reflect the level of defense against the infection, and indeed found higher numbers in subjects carrying the C-allele, thus suggesting the involvement of ECP in this defense reaction. We also found that subjects carrying the G-allele are much more prone to develop liver fibrosis, one of the serious consequences of the infection that affects about 10% of those infected by S. mansoni. Thus, it seems that the capacity to produce the cytotoxic ECP has some effect on defense against S. mansoni, but that the host’s reaction to the infection is the major threat to the infected subject. In this regard, cytotoxic ECP may play a key role.
ECP 562G>C (rs2233860) Genotypes and Clinical Findings
As mentioned above, the ECP562G>C genotype is closely related to the cellular content of ECP, with the lowest levels found in those carrying the ECP CC genotype.24 We found few, if any, associations between this genotype and the expression of allergic symptoms or asthma, but close relations to gender, with a higher prevalence of the G-allele in women, and relations to smoking habits.60 Similar findings were seen for the 434G>C genotypes and may relate to the fact that these two genotypes are in strong disequilibrium. In the Korean study, the ECP562 G>C C-allele was found to be more prevalent in allergic rhinitis.62 In the Norwegian–Dutch study, no apparent relations to allergy, asthma, and ECP levels were found, whether evaluated alone or as part of a haplotype.61
ECP c.-38A>C (rs2233859) Genotypes and Clinical Findings
The intronic SNP ECP c.-38A>C is located in a part of the gene that may be involved in regulating ECP production. The Norwegian–Dutch study showed a higher proportion of elevated serum levels of ECP and IgE, as well as higher proportions of subjects with asthma and bronchial hyperreactivity, in those carrying the adenine (A)-allele.61 Most of these associations, though, were only seen in the Dutch population. In a Japanese study, no association between the intronic SNP and serum ECP levels were found.25 In our ECRHS study, we found an intriguing association between the ECP c.-38A>C genotypes and atopy.60 Among males, but not among women, atopy was associated with the ECP c.-38(A>C) genotypes, with a significantly higher frequency of the CC genotype. In a logistic regression analysis, the ECP c.-38CC genotype was independently associated with an increased risk of atopy with an odds ratio of 1.9 and confidence interval (CI) of 1.2–3.1 when adjusted for gender, ethnicity, and smoking habits.
ECP -393C>T (rs11575981) Genotypes and Clinical Findings
The ECP -393C>T SNP is located in the promoter region of the ECP gene. This SNP has only been described in the Japanese population.25 Interestingly, the -393C>T genotypes are related to serum levels of ECP, with undetectable levels in subjects carrying the TT genotype.
Eosinophil Protein X/Eosinophil-Derived Neurotoxin, Eosinophil Peroxidase, and Major Basic Protein Genotypes and Clinical Findings
One study examined a large number of EDN and ECP SNPs in a South Indian population with microfilaria infection and tropical pulmonary eosinophilia.63 No associations between either of these conditions and the SNPs examined were seen. However, the South Indian population seemed to have unique SNPs and haplotypes of the EDN and ECP genotypes compared to Asian and Scandinavian populations. In two Japanese reports, SNPs in the coding parts of the EPO gene were found to be associated with cedar pollinosis.64., 65. In particular, an SNP in exon 7, resulting in the amino acid shift from proline to leucine and which might affect the activity of the protein, showed a strong association. In the second study, an association with a silent SNP in exon 6 was shown in addition to the exon 7 SNP. In a recent Czech study on allergic rhinitis, the exon 6 SNP was also found to be associated, whereas the exon 7 SNP was not present in this population.66 These reports suggest the involvement of EPO in the allergic process, although whether it involves the peroxidase activity of EPO or reflects other mechanisms is unknown. One report from Germany studied patients with atopic dermatitis and possible associations between nine different SNPs located in the four major eosinophil granule protein genes.67 However, no associations with atopic dermatitis were found for any of the SNPs studied, despite the fact that eosinophils are regarded to be important effector cells in this disease. No other studies have investigated SNPs in the MBP gene in relation to human disease.
References
- 1.Gleich G.J., Loegering D.A., Maldonado J.E. Identification of a major basic protein in guinea pig eosinophil granules. J Exp Med. 1973;137:1459–1471. doi: 10.1084/jem.137.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gleich G.J., Loegering D.A., Kueppers F., Bajaj S.P., Mann K.G. Physiochemical and biological properties of the major basic protein from guinea pig eosinophil granules. J Exp Med. 1974;140:313–332. doi: 10.1084/jem.140.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olsson I., Venge P. Cationic proteins of human granulocytes. I. Isolation of the cationic proteins from the granules of leukaemic myeloid cells. Scand J Haematol. 1972;9:204–214. doi: 10.1111/j.1600-0609.1972.tb00932.x. [DOI] [PubMed] [Google Scholar]
- 4.Olsson I., Venge P. Cationic proteins of human granulocytes. II. Separation of the cationic proteins of the granules of leukemic myeloid cells. Blood. 1974;44 N0.2:235–246. [PubMed] [Google Scholar]
- 5.Gleich G.J., Loegering D.A., Mann K.G., Maldonado J.E. Comparative properties of the Charcot-Leyden crystal protein and the major basic protein from human eosinophils. J Clin Invest. 1976;57:633–640. doi: 10.1172/JCI108319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durack D.T., Ackerman S.J., Loegering D.A., Gleich G.J. Purification of human eosinophil-derived neurotoxin. Proc Natl Acad Sci U S A. 1981;78:5165–5169. doi: 10.1073/pnas.78.8.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson C.G.B., Venge P. Purification and characterization of a new cationic protein—eosinophil protein-x(EPX)—from granules of human eosinophils. Immunology. 1983;50:19–26. [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen R.L., Little C. Purification and some properties of myeloperoxidase and eosinophil peroxidase from human blood. Biochem J. 1983;209:781–787. doi: 10.1042/bj2090781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson MGCh, Peterson C.G.B., Venge P. Human eosinophil peroxidase: purification and characterization. J Immunol. 1985;134 N0.3:1875–1879. [PubMed] [Google Scholar]
- 10.Rosenberg H.F., Dyer K.D. Eosinophil cationic protein and eosinophil-derived neurotoxin. Evolution of novel function in a primate ribonuclease gene family. J Biol Chem. 1995;270:21539–21544. doi: 10.1074/jbc.270.37.21539. [DOI] [PubMed] [Google Scholar]
- 11.Venge P., Byström J., Carlson M., Håkansson L., Karawacjzyk M., Peterson C. Eosinophil cationic protein (ECP): molecular and biological properties and the use of ECP as a marker of eosinophil activation in disease. Clin Exp Allergy. 1999;29:1172–1186. doi: 10.1046/j.1365-2222.1999.00542.x. [DOI] [PubMed] [Google Scholar]
- 12.Eriksson J., Woschnagg C., Fernvik E., Venge P. A SELDI-TOF MS study of the genetic and post-translational molecular heterogeneity of eosinophil cationic protein. J Leukoc Biol. 2007;82:1491–1500. doi: 10.1189/jlb.0507272. [DOI] [PubMed] [Google Scholar]
- 13.Sur S., Glitz D.G., Kita H., Kujawa S.M., Peterson E.A., Weiler D.A. Localization of eosinophil-derived neurotoxin and eosinophil cationic protein in neutrophilic leukocytes. J Leukoc Biol. 1998;63:715–722. doi: 10.1002/jlb.63.6.715. [DOI] [PubMed] [Google Scholar]
- 14.Monteseirin J., Vega A., Chacon P., Camacho M.J., El B.R., Asturias J.A. Neutrophils as a novel source of eosinophil cationic protein in IgE-mediated processes. J Immunol. 2007;179:2634–2641. doi: 10.4049/jimmunol.179.4.2634. [DOI] [PubMed] [Google Scholar]
- 15.Byström J., Tenno T., Håkansson L., Amin K., Trulson A., Högbom E. Monocytes, but not macrophages, produce the eosinophil cationic protein. APMIS. 2001;109:507–516. doi: 10.1111/j.1600-0463.2001.apm090704.x. [DOI] [PubMed] [Google Scholar]
- 16.Byström J., Garcia R.C., Håkansson L., Karawajczyk M., Moberg L., Soukka J. Eosinophil cationic protein is stored in, but not produced by, peripheral blood neutrophils. Clin Exp Allergy. 2002;32:1082–1091. doi: 10.1046/j.1365-2222.2002.01408.x. [DOI] [PubMed] [Google Scholar]
- 17.Rubin J., Zagai U., Blom K., Trulson A., Engström A., Venge P. The coding ECP 434(G>C) gene polymorphism determines the cytotoxicity of ECP but has minor effects on fibroblast-mediated gel contraction and no effect on RNase activity. J Immunol. 2009;183:445–451. doi: 10.4049/jimmunol.0803912. [DOI] [PubMed] [Google Scholar]
- 18.Trulson A., Byström J., Engström A., Larsson R., Venge P. The functional heterogeneity of eosinophil cationic protein is determined by a gene polymorphism and post-translational modifications. Clin Exp Allergy. 2007;37:208–218. doi: 10.1111/j.1365-2222.2007.02644.x. [DOI] [PubMed] [Google Scholar]
- 19.Woschnagg C., Rubin J., Venge P. Eosinophil cationic protein (ECP) is processed during secretion. J Immunol. 2009;183:3949–3954. doi: 10.4049/jimmunol.0900509. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J., Rosenberg H.F. Sequence variation at two eosinophil-associated ribonuclease loci in humans. Genetics. 2000;156:1949–1958. doi: 10.1093/genetics/156.4.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jönsson U.B., Byström J., Stålenheim G., Venge P. Polymorphism of the eosinophil cationic protein-gene is related to the expression of allergic symptoms. Clin Exp Allergy. 2002;32:1092–1095. doi: 10.1046/j.1365-2222.2002.01410.x. [DOI] [PubMed] [Google Scholar]
- 22.Eriksson J., Reimert C.M., Kabatereine N.B., Kazibwe F., Ireri E., Kadzo H. The 434(G>C) polymorphism within the coding sequence of Eosinophil Cationic Protein (ECP) correlates with the natural course of Schistosoma mansoni infection. Int J Parasitol. 2007 doi: 10.1016/j.ijpara.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Torrent M., de la Torre B.G., Nogues V.M., Andreu D., Boix E. Bactericidal and membrane disruption activities of the eosinophil cationic protein are largely retained in an N-terminal fragment. Biochem J. 2009;421:425–434. doi: 10.1042/BJ20082330. [DOI] [PubMed] [Google Scholar]
- 24.Jönsson U.B., Byström J., Stålenheim G., Venge P. A (G->C) transversion in the 3′ UTR of the human ECP (eosinophil cationic protein) gene correlates to the cellular content of ECP. J Leukoc Biol. 2006;79:846–851. doi: 10.1189/jlb.0904517. [DOI] [PubMed] [Google Scholar]
- 25.Noguchi E., Iwama A., Takeda K., Takeda T., Kamioka M., Ichikawa K. The promoter polymorphism in the eosinophil cationic protein gene and its influence on the serum eosinophil cationic protein level. Am J Respir Crit Care Med. 2003;167:180–184. doi: 10.1164/rccm.200204-292OC. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg H.F., Domachowske J.B. Eosinophil-derived neurotoxin. Methods Enzymol. 2001;341:273–286. doi: 10.1016/s0076-6879(01)41158-x. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg H.F. Eosinophil-derived neurotoxin/RNase 2: connecting the past, the present and the future. Curr Pharm Biotechnol. 2008;9:135–140. doi: 10.2174/138920108784567236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durack D.T., Ackerman S.J., Loegering D.A., Gleich G.J. Purification of human eosinophil-derived neurotoxin. Proc Natl Acad Sci USA. 1981;78 N0.8:5165–5169. doi: 10.1073/pnas.78.8.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karawajczyk M., Sevéus L., Garcia R., Björnsson E., Peterson C.G., Roomans G.M. Piecemeal degranulation of peripheral blood eosinophils: a study of allergic subjects during and out of the pollen season. Am J Respir Cell Mol Biol. 2000;23:521–529. doi: 10.1165/ajrcmb.23.4.4025. [DOI] [PubMed] [Google Scholar]
- 30.Yang D., Chen Q., Su S.B., Zhang P., Kurosaka K., Caspi R.R. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J Exp Med. 2008;205:79–90. doi: 10.1084/jem.20062027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fredens K., Dahl R., Venge P. The Gordon phenomenon induced by the eosinophil cationic protein and eosnophil protein-X. J Allergy Clin Immunol. 1982;70 N0.5:361–366. doi: 10.1016/0091-6749(82)90025-2. [DOI] [PubMed] [Google Scholar]
- 32.Davies M.J., Hawkins C.L., Pattison D.I., Rees M.D. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 33.Zabucchi G., Menegazzi R., Soranzo M.R., Patriarca P. Uptake of human eosinophil peroxidase by human neutrophils. Am J Pathol. 1986;124:510–518. [PMC free article] [PubMed] [Google Scholar]
- 34.Aldridge R.E., Chan T., van Dalen C.J., Senthilmohan R., Winn M., Venge P. Eosinophil peroxidase produces hypobromous acid in the airways of stable asthmatics. Free Radic Biol Med. 2002;33:847–856. doi: 10.1016/s0891-5849(02)00976-0. [DOI] [PubMed] [Google Scholar]
- 35.Ulrich M., Petre A., Youhnovski N., Promm F., Schirle M., Schumm M. Post-translational tyrosine nitration of eosinophil granule toxins mediated by eosinophil peroxidase. J Biol Chem. 2008;283:28629–28640. doi: 10.1074/jbc.M801196200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gleich G.J., Loegering D.A., Frigas E., Filley W.V. The eosinophil granule major basic protein: biological activities and relationship to bronchial asthma. Monogr Allergy. 1983;18:277–283. [PubMed] [Google Scholar]
- 37.Hamann K.J., Barker R.L., Ten R.M., Gleich G.J. The molecular biology of eosinophil granule proteins. Int Arch Allergy Appl Immunol. 1991;94:202–209. doi: 10.1159/000235362. [DOI] [PubMed] [Google Scholar]
- 38.Popken-Harris P., Thomas L., Oxvig C., Sottrup-Jensen L., Kubo H., Klein J.S. Biochemical properties, activities, and presence in biologic fluids of eosinophil granule major basic protein. J Allergy Clin Immunol. 1994;94:1282–1289. doi: 10.1016/0091-6749(94)90343-3. [DOI] [PubMed] [Google Scholar]
- 39.Plager D.A., Stuart S., Gleich G.J. Human eosinophil granule major basic protein and its novel homolog. Allergy. 1998;53:33–40. doi: 10.1111/j.1398-9995.1998.tb04937.x. [DOI] [PubMed] [Google Scholar]
- 40.Plager D.A., Adolphson C.R., Gleich G.J. A novel human homolog of eosinophil major basic protein. Immunol Rev. 2001;179:192–202. doi: 10.1034/j.1600-065x.2001.790119.x. [DOI] [PubMed] [Google Scholar]
- 41.Levi-Schaffer F., Temkin V., Simon H.U., Kettman J.R., Frey J.R., Lefkovits I. Proteomic analysis of human eosinophil activation mediated by mast cells, granulocyte macrophage colony stimulating factor and tumor necrosis factor alpha. Proteomics. 2002;2:1616–1626. doi: 10.1002/1615-9861(200211)2:11<1616::AID-PROT1616>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 42.Yoon S.W., Kim T.Y., Sung M.H., Kim C.J., Poo H. Comparative proteomic analysis of peripheral blood eosinophils from healthy donors and atopic dermatitis patients with eosinophilia. Proteomics. 2005;5:1987–1995. doi: 10.1002/pmic.200401086. [DOI] [PubMed] [Google Scholar]
- 43.Ryu S.I., Kim W.K., Cho H.J., Lee P.Y., Jung H., Yoon T.S. Phosphoproteomic analysis of AML14.3D10 cell line as a model system of eosinophilia. J Biochem Mol Biol. 2007;40:765–772. doi: 10.5483/bmbrep.2007.40.5.765. [DOI] [PubMed] [Google Scholar]
- 44.Woschnagg C., Forsberg J., Engström A., Odreman F., Venge P., Garcia R.C. The human eosinophil proteome. Changes induced by birch pollen allergy. J Proteome Res. 2009;8:2720–2732. doi: 10.1021/pr800984e. [DOI] [PubMed] [Google Scholar]
- 45.Woschnagg C., Rak S., Venge P. Oxygen radical production by blood eosinophils is reduced during birch pollen season in allergic patients. Clin Exp Allergy. 1996;26:1064–1072. [PubMed] [Google Scholar]
- 46.Håkansson L., Carlson M., Stålenheim G., Venge P. Migratory responses of eosinophil and neutrophil granulocytes from patients with asthma. J Allergy Clin Immunol. 1990;85 N0.4:743–750. doi: 10.1016/0091-6749(90)90193-8. [DOI] [PubMed] [Google Scholar]
- 47.Wang H.B., Ghiran I., Matthaei K., Weller P.F. Airway eosinophils: allergic inflammation recruited professional antigen-presenting cells. J Immunol. 2007;179:7585–7592. doi: 10.4049/jimmunol.179.11.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venge P. Monitoring the allergic inflammation. Allergy. 2004;59:26–32. doi: 10.1046/j.1398-9995.2003.00386.x. [DOI] [PubMed] [Google Scholar]
- 49.Schmekel B., Ahlner J., Malmström M., Venge P. Eosinophil cationic protein (ECP) in saliva: a new marker of disease activity in bronchial asthma. Respir Med. 2001;95:670–675. doi: 10.1053/rmed.2001.1123. [DOI] [PubMed] [Google Scholar]
- 50.Hurst D.S., Venge P. Evidence of eosinophil, neutrophil, and mast-cell mediators in the effusion of OME patients with and without atopy. Allergy. 2000;55:435–441. doi: 10.1034/j.1398-9995.2000.00289.x. [DOI] [PubMed] [Google Scholar]
- 51.Björk A., Venge P., Peterson C.G. Measurements of ECP in serum and the impact of plasma coagulation. Allergy. 2000;55:442–448. [PubMed] [Google Scholar]
- 52.Carlson M., Håkansson L., Kämpe M., Stålenheim G., Peterson C., Venge P. Degranulation of eosinophils from pollen-atopic patients with asthma is increased during pollen season. J Allergy Clin-Immunol, 89 N0.1. Part. 1994;1:131–139. doi: 10.1016/s0091-6749(05)80050-8. [DOI] [PubMed] [Google Scholar]
- 53.Koh G.C., Shek L.P., Goh D.Y., Van B.H., Koh D.S. Eosinophil cationic protein: is it useful in asthma? A systematic review. Respir Med. 2007;101:696–705. doi: 10.1016/j.rmed.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 54.Wolthers O.D. Eosinophil granule proteins in the assessment of airway inflammation in pediatric bronchial asthma. Pediatr Allergy Immunol. 2003;14:248–254. doi: 10.1034/j.1399-3038.2003.00030.x. [DOI] [PubMed] [Google Scholar]
- 55.Nielsen L.P., Peterson C.G., Dahl R. Serum eosinophil granule proteins predict asthma risk in allergic rhinitis. Allergy. 2009;64:733–737. doi: 10.1111/j.1398-9995.2008.01869.x. [DOI] [PubMed] [Google Scholar]
- 56.Kristjánsson S., Strannegård I.L., Strannegård Q., Peterson C., Enander I., Wennergren G. Urinary eosinophil protein X in children with atopic asthma: A useful marker of antiinflammatory treatment. J Allergy Clin Immunol. 1996;97:1179–1187. doi: 10.1016/s0091-6749(96)70182-3. [DOI] [PubMed] [Google Scholar]
- 57.Labbe A., Aublet-Cuvelier B., Jouaville L., Beaugeon G., Fiani L., Petit I. Prospective longitudinal study of urinary eosinophil protein X in children with asthma and chronic cough. Pediatr Pulmonol. 2001;31:354–362. doi: 10.1002/ppul.1058. [DOI] [PubMed] [Google Scholar]
- 58.Gore C., Peterson C.G., Kissen P., Simpson B.M., Lowe L.A., Woodcock A. Urinary eosinophilic protein X, atopy, and symptoms suggestive of allergic disease at 3 years of age. J Allergy Clin Immunol. 2003;112:702–708. doi: 10.1016/s0091-6749(03)01886-4. [DOI] [PubMed] [Google Scholar]
- 59.Wolthers O.D., Heuck C. Circadian variations in serum eosinophil cationic protein, and serum and urine eosinophil protein X. Pediatr Allergy Immunol. 2003;14:130–133. doi: 10.1034/j.1399-3038.2003.02038.x. [DOI] [PubMed] [Google Scholar]
- 60.Jönsson U.B., Håkansson L.D., Jogi R., Janson C., Venge P. Associations of ECP (eosinophil cationic protein)-gene polymorphisms to allergy, asthma, smoke habits and lung function in two Estonian and Swedish sub cohorts of the ECRHS II study. BMC Pulm Med. 2010;10:36. doi: 10.1186/1471-2466-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munthe-Kaas M.C., Gerritsen J., Carlsen K.H., Undlien D., Egeland T., Skinningsrud B. Eosinophil cationic protein (ECP) polymorphisms and association with asthma, s-ECP levels and related phenotypes. Allergy. 2007;62:429–436. doi: 10.1111/j.1398-9995.2007.01327.x. [DOI] [PubMed] [Google Scholar]
- 62.Kang I., An X.H., Oh Y.K., Lee S.H., Jung H.M., Chae S.C. Identification of polymorphisms in the RNase3 gene and the association with allergic rhinitis. Eur Arch Otorhinolaryngol. 2010;267:391–395. doi: 10.1007/s00405-009-1103-8. [DOI] [PubMed] [Google Scholar]
- 63.Kim Y.J., Kumaraswami V., Choi E., Mu J., Follmann D.A., Zimmerman P. Genetic polymorphisms of eosinophil-derived neurotoxin and eosinophil cationic protein in tropical pulmonary eosinophilia. Am J Trop Med Hyg. 2005;73:125–130. [PubMed] [Google Scholar]
- 64.Nakamura H., Miyagawa K., Ogino K., Endo T., Imai T., Ozasa K. High contribution contrast between the genes of eosinophil peroxidase and IL-4 receptor alpha-chain in Japanese cedar pollinosis. J Allergy Clin Immunol. 2003;112:1127–1131. doi: 10.1016/j.jaci.2003.08.051. [DOI] [PubMed] [Google Scholar]
- 65.Nakamura H., Higashikawa F., Miyagawa K., Nobukuni Y., Endo T., Imai T. Association of single nucleotide polymorphisms in the eosinophil peroxidase gene with Japanese cedar pollinosis. Int Arch Allergy Immunol. 2004;135:40–43. doi: 10.1159/000080222. [DOI] [PubMed] [Google Scholar]
- 66.Hrdlickova B., Izakovicova-Holla L. Association of single nucleotide polymorphisms in the eosinophil peroxidase gene with allergic rhinitis in the Czech population. Int Arch Allergy Immunol. 2009;150:184–191. doi: 10.1159/000218122. [DOI] [PubMed] [Google Scholar]
- 67.Parwez Q., Stemmler S., Epplen J.T., Hoffjan S. Variation in genes encoding eosinophil granule proteins in atopic dermatitis patients from Germany. J Negat Results Biomed. 2008;7:9. doi: 10.1186/1477-5751-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chapter 13.3. Eosinophils and Skin Diseases
Eosinophil infiltration in the skin has been observed in allergic and reactive diseases, autoimmune diseases, and infectious diseases, as well as in lymphomas and solid tumors. Since eosinophils are not present under physiological conditions in the skin, cutaneous eosinophilia requires an increased production, recruitment, and/or survival of eosinophils, which may be due to intrinsic defects in the eosinophils, as in myeloproliferative forms of hypereosinophilic syndromes, or extrinsic stimulation by cytokines released by T cells or tumor cells. Here, we discuss the present knowledge on eosinophils in selected skin disorders. However, the exact mechanisms of how eosinophils are recruited and activated in the skin under each condition and their pathogenic role(s) are not fully understood.
Introduction
Tissue eosinophilia with or without associated blood eosinophilia is observed in a number of skin disorders, including allergic diseases, autoimmune diseases, bacterial or viral infections, hematologic diseases, parasitic infestations, as well as in association with tumors.1., 2. The presence or absence of eosinophils in skin specimens is often used for differential diagnoses by dermatopathologists. For instance, eosinophils in the upper dermis might be a clue for the diagnosis of early lesions of bullous pemphigoid (BP), even in the absence of blisters. In addition, the detection of eosinophils might indicate the differential diagnosis of drug reactions, which often cannot be distinguished from other inflammatory skin diseases.
In hematoxylin and eosin (H&E) stained skin specimens, eosinophils are seen as round shaped cells stuffed with coarse eosinophil granules (Fig. 13.3.1 A). Disrupted oval-shaped eosinophils may also be found, e.g., in subacute and chronic eczematous lesions (Fig. 13.3.1A, B).3 Depending on the disease, eosinophils are located among other inflammatory cells in the perivascular areas (e.g., in eczema), between collagen bundles in the dermis (e.g., in eosinophilic cellulitis), or in the epidermis (e.g., in pemphigus foliaceus; Fig. 13.3.1B–D). Moreover, in eosinophilic pustular folliculitis, eosinophils enter the hair follicle.4 Extracellular granular proteins can be detected in varying amounts either as separate deposits or as a thin coating on collagen bundles. The latter are called flame figures and are typically seen in eosinophilic cellulitis. Recently, deposition of eosinophil granule proteins in association with extracellular DNA traps was reported in several allergic, autoimmune, and infectious skin diseases.5 Immunofluorescence staining using antibodies directed against eosinophil cationic protein (ECP) or major basic protein (MBP) allows a more sensitive detection of eosinophils and extracellular granular protein depositions than H&E staining.6
FIGURE 13.3.1.
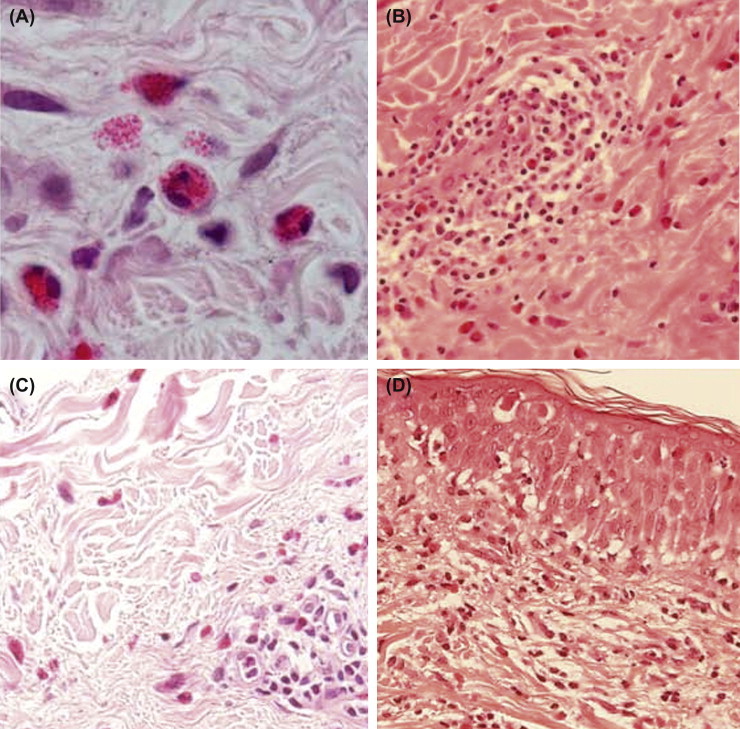
Eosinophil infiltration in the skin.
A, Round, oval shaped and disrupted eosinophils (magnification ×1000). B, Perivascular infiltrate containing eosinophils in allergic contact dermatitis. C, Eosinophils between collagen bundles in eosinophilic cellulitis (Wells' syndrome). D, Eosinophilic spongiosis in pemphigus foliaceus.
Under physiological conditions, eosinophils are usually not detectable in the skin. Therefore, primary causes (intrinsic) or stimuli (extrinsic) are required for initiating the increased production, recruitment, and/or survival of eosinophils under pathological conditions.1 Myeloproliferative forms of hypereosinophilic syndromes (HES) represent intrinsic eosinophilic disorders,7 in which mutations of multipotent or pluripotent hematopoietic stem cells occur, with subsequent increased eosinophil proliferation, often affect the skin (Table 13.3.1 ). Cutaneous manifestations vary from multiple erythematous papules, plaques, and nodules, to generalized erythematous maculopapular eruptions, often associated with pruritus.8., 9. Clonal eosinophilia can occur as a consequence of gene rearrangements that result in increased tyrosine kinase activity.10 As a consequence, patients with hypereosinophilia due to fusion of the platelet-derived growth factor receptor α (PDGFRA) and FIP1 like 1 (FIP1L1) genes respond to imatinib therapy.11
TABLE 13.3.1.
| Intrinsic Disorders | Extrinsic Disorders | |
|---|---|---|
| Mutations of Hematopoietic Stem Cells | Cytokines Released by | |
| T cells | Tumor cells | |
| Chronic eosinophilic leukemia | Allergic diseases: Atopic dermatitis, Urticaria, Drug reactions | Cutaneous T cell lymphoma |
| Acute myeloid leukemia | Langerhans cell histiocytosis | |
| Chronic myeloid leukemia | B-cell lymphomas | |
| Myelodysplastic syndromes | Hodgkin’s lymphomas | |
| Idiopathic hypereosinophilic syndromes | Autoimmune diseases: Bullous pemphigoid, Dermatitis herpetiformis, Pemphigus, Epidermolysis bullosa | Acute T-cell leukemia/lymphoma |
| Infectious diseases: HIV, Ectoparasitosis, Insect bites, Erythema chronicum migrans, Erythema toxicum neonatorum | ||
| Hyper-IgE syndrome (Job syndrome) | ||
| Eosinophilic pustular folliculitis | ||
| Granuloma annulare | ||
| Angiolymphoid hyperplasia with eosinophilia | ||
| Localized scleroderma | ||
| Eosinophilic fasciitis | ||
| Eosinophilic cellulitis (Wells syndrome) | ||
| Hypereosinophilic syndromes | ||
| Inflammatory clonal T-cell disease | ||
Extrinsic eosinophilic disorders due to cytokine release by either T cells or tumor cells are more common than intrinsic HES forms due to genetic abnormalities within hematopoietic stem cells (Table 13.3.1). Cytokines involved in the development of skin eosinophilia are interleukin-3 (IL-3), IL-5, and granulocyte/macrophage colony-stimulating factor (GM-CSF). The expression of IL-5 in association with eosinophilic skin disorders has been reported for atopic dermatitis (AD), BP, cutaneous T cell lymphoma, episodic angioedema with eosinophilia, eosinophilic cellulitis, eosinophilic fasciitis, eosinophilic folliculitis, exanthematous drug reactions, hypereosinophilic syndrome with skin involvement, and urticaria.2 IL-3 expression has been detected in blister fluids of BP and blood leukocytes of HES patients.12., 13., 14. In Langerhans cell histiocytosis, as well as in AD, atopy patch test reactions, and cutaneous late phase reactions, the expression of both GM-CSF and IL-3 has been shown.2 The chemokine eotaxin/C-C motif chemokine 11 (CCL11) is important for tissue recruitment and activation of eosinophils. Eotaxin expression has been observed in AD, autoimmune-blistering diseases like dermatitis herpetiformis and BP, drug reactions, eosinophilic folliculitis, and parasitic dermatoses, and also in lymphomas, e.g., cutaneous T-cell lymphoma and Hodgkin disease.2
The primary function of eosinophils was originally thought to be related to the protection against helminth parasites.15 Recently, a novel mechanism of eosinophil function in innate immunity has been reported. By releasing mitochondrial DNA and granule proteins, eosinophils form extracellular structures that can bind and kill bacteria invading the gastrointestinal tract.16 Such extracellular DNA structures generated by eosinophils have recently also been reported in inflammatory skin diseases.5 Furthermore, eosinophils are presumed to cause tissue damage.15., 17. In addition, eosinophils play an important role in repair and remodeling processes, as well as in immunomodulation.18., 19. The role of eosinophils under pathological conditions has mostly been studied in parasitic infections and bronchial asthma. In contrast, the role of eosinophils has not been explored in substantial depth in skin diseases. With regard to skin diseases, it can be assumed that eosinophils directly contribute to or amplify pruritus (itch) in the skin, by releasing neurotrophins (nerve growth factor and brain-derived neurotrophic factor) or indirectly by acting on mast cells.20 Pruritus is associated with most eosinophilic skin diseases, in particular with AD, BP, cutaneous T-cell lymphoma, and parasitic infections.
In the following sections, we summarize the current knowledge on the activation and function of eosinophils in selected eosinophilic skin disorders.
Atopic Dermatitis
Tissue eosinophilia is a typical feature of eczema, in particular of AD. The numbers of eosinophils in the skin are usually modest (2.8 cells/mm2; range 0–90.3) and correlate with disease severity, as well as with the degree of spongiosis in acute exacerbations and marked epidermal hyperplasia in chronic stages.21 Besides eosinophils, eosinophil-derived products, such as ECP, eosinophil-derived neurotoxin (EDN), and MBP, are present in increased amounts in the blood and the skin of AD patients.22 Immunostaining with antibodies to ECP and MBP, as well as electron-microscopic evaluation revealed eosinophil granule proteins inside eosinophils, but also in the extracellular spaces, and the near absence of intact eosinophils, suggesting eosinophil degranulation and degeneration.3., 23. In AD, eosinophil production, differentiation, recruitment, survival, and activation are under tight control of cytokines, particularly GM-CSF, IL-3, and IL-5, and chemokines, including eotaxin and RANTES (C-C motif chemokine 5; CCL5), as well as adhesion molecules, complement factors and leukotrienes.22 However, the pathogenic role(s) of eosinophils in AD have not yet been defined. The release of granule proteins suggests a role in host defense and/or tissue damage. Furthermore, eosinophils release a broad spectrum of mediators such as cytokines [GM-CSF, IL-1, IL-3, IL-4, IL-5, IL-8, IL-10, IL-13, and transforming growth factor (TNF)] and leukotrienes (in particular, the cysteinyl leukotrienes LTC4, LTD4, and LTE4) and thus they can regulate immune responses or initiate tissue repair processes.18 Improvement of AD upon both systemic and topical therapy is usually associated with a decrease in eosinophils and other inflammatory cells in the skin.22 However, the administration of an anti-IL-5 antibody showed only moderate effects on clinical symptoms, although blood eosinophils were almost completely depleted.24 Currently, it remains unclear whether anti-IL-5 antibody treatment reduces eosinophil tissue infiltration in lesional AD skin.
Urticaria
Although the development of pruritic wheals in urticaria is attributed to the release of histamine by mast cells and basophils, other cell types, including eosinophils, neutrophils, and macrophages, and T cells, are also present in urticarial lesions. Eosinophils and extracellular deposits of eosinophil granule proteins have been described in chronic idiopathic urticaria, delayed pressure urticaria, and solar urticaria.25 Extracellular deposits of MBP have been observed as granular deposits and coating tissue fibers in the dermis, as well as in small blood vessel walls.26 ECP may stimulate histamine release by mast cells and basophils.27 By generating eicosanoid mediators and secreting neuropeptides, such as substance P, eosinophils may contribute to vasodilation.25 Vascular endothelial growth factor, which is elevated in the plasma of chronic urticaria patients and correlates with disease severity, has been reported to be predominantly expressed by eosinophils.28 Recently, an involvement of the coagulation cascade in the pathogenesis of chronic urticaria has been suggested. Eosinophils that were shown to express tissue factor in urticarial lesions may activate the tissue factor pathway of coagulation, resulting in the generation of thrombin, which stimulates mast cells for histamine release.29
Eosinophilic Cellulitis (Wells' Syndrome)
Eosinophilic cellulitis is characterized by an intense infiltration of eosinophils, extracellular granule deposition, and flame figures in the dermis.30 Recently, high numbers of eosinophils generating extracellular DNA traps in association with ECP have also been observed in this eosinophilic disorder.5 Patients present with recurrent episodes of acute pruritic dermatitis and occasionally with blisters, painful edematous swellings, or persistent urticarial eruptions.30 The cause is unknown, but some patients develop eosinophilic cellulitis in association with hematological disorders, infections, or anti-TNF-α therapy. Corticosteroids are usually helpful in this disease. In 37% of patients with HES, the skin is affected.31 Skin manifestations of HES include blisters, eosinophilic cellulitis, erythematous macules, lichenoid eruptions or urticarial lesions, necrosis, papules or nodules, pruritus, purpura, and ulcerations. Cutaneous symptoms are usually present in a subgroup of patients with HES, in which IL-5-producing T cells exhibiting an abnormal immunophenotype have been identified.13 Anti-IL-5 antibody therapy has been shown to improve skin symptoms in HES patients.6
Eosinophilic Pustular Folliculitis
Eosinophilic Pustular Folliculitis (EPF) presents with recurring clusters of sterile follicular papules and pustules, predominantly on the face and trunk.4 EPF may affect immunocompetent subjects (Ofuji disease), but is most commonly seen together with immunosuppression. EPF has been reported in association with infections, in particular acquired immunodeficiency syndrome (AIDS), autoimmune diseases, and medications, as well as autologous peripheral blood stem cell and allogeneic bone marrow transplantation.4 The histology shows a dense follicular and perifollicular infiltrate of eosinophils and scattered lymphocytes, and sometimes follicle destruction. A pathogenic role for eosinophils in response to fungi (Malassezia), Demodex mites, and bacteria has been suggested.4
Bullous Pemphigoid
The histopathology of BP reveals eosinophil infiltration in and below blisters and along the basement membrane,32 as well as in nonblistering, urticarial, or eczematous lesions of BP.33 Patients with active BP exhibit increased eotaxin and IL-5 levels, as well as eosinophil numbers in the blood compared with patients in clinical remission and healthy controls,34., 35., 36. associated with significant eosinophil infiltration in the skin, as well as disease intensity.37., 38. Whether BP180 and/or BP230 autoantibodies of immunoglobulin G (IgG) or IgE types can activate eosinophils with or without preceding priming has not been investigated so far. Eosinophils exhibit CD16, the Fc-γ receptor, and degranulate upon stimulation with IgG immune complexes.39 Furthermore, IL-5-primed eosinophils from these patients release granule proteins upon stimulation with complement C5a.40 It has been hypothesized that in the presence of complement, eosinophils release enzymes and reactive oxygen onto the basement membrane, causing tissue destruction and blister formation in BP.41 Eosinophil granule protein depositions have been observed in both blistering and evolving lesions.42 Recently, extracellular DNA traps generated by eosinophils were also described in BP.5 MMP9 has been reported to be expressed by eosinophils in lesional skin, as well as in blister fluids of BP.43 Moreover, MMP9 cleaves the extracellular, collagenous domain of BP180 autoantigen in vitro.43
Other Autoimmune Bullous Diseases
Eosinophilic spongiosis can be observed in early pemphigus including pemphigus foliaceus.44 The presence of eosinophils may be due to IL-5 as part of the mixed T-helper 1/2 (Th1)/Th2 cytokine profile that has been found in pemphigus vulgaris.45 Complement-fixing antibodies were shown to induce eosinophil infiltration in pemphigus.46 Charcot–Leyden crystals have been observed in pemphigus vegetans.47
Eosinophils can be seen in the papillary dermis in dermatitis herpetiformis, although neutrophils and leukocytoclasis are more characteristic in this disorder.32 Furthermore, both neutrophils and eosinophils are the predominant infiltrating cells in linear IgA bullous dermatosis.48 Epidermolysis bullosa acquisita following GM-CSF therapy has been related to eosinophil infiltration and deposition of eosinophil peroxidase (EPO) and MBP at the dermal–epidermal junction.49
Cutaneous Drug Eruptions
In drug reactions, the presence of eosinophils in the skin is quite a striking finding, despite various clinical and histopathological presentations [e.g., acute generalized exanthematous pustulosis (AGEP), erythema multiforme, maculopapular rashes, and pseudolymphomatous and granulomatous drug reactions].50 Drug reaction with eosinophilia and systemic symptoms (DRESS) is a drug reaction with both blood and tissue eosinophilia and systemic symptoms. It presents with an acute, severe skin eruptions that may develop from a maculopapular rash into erythroderma, as well as with blood eosinophilia, fever, hepatitis, lymphadenopathy, and other organ involvement.51 Eosinophils accompanied by other inflammatory cells are found in the skin and lymph nodes. Severe hepatitis, in which eosinophilic infiltration or granulomas as well as hepatocyte necrosis and cholestasis are striking features, may result in liver failure, accounting for the high mortality rate of 10%.51 The treatment is based on high-dose corticosteroids. Drugs known to cause DRESS are anticonvulsants, anticancer drugs, antidiabetics, antimicrobial agents, nonsteroidal anti-inflammatory drugs, and sulfa drugs.1
Lymphomas and Tumors
In Langerhans cell histiocytosis (LCH), among the infiltrate of Langerhans cells (LC), scattered or clusters of eosinophils can be found in the papillary and deeper dermis, respectively.52 Since activated LC generate a broad spectrum of proinflammatory cytokines, e.g., chemokines and GM-CSF, they are able to recruit and activate eosinophils directly or via stimulation of other cell types. A predominant Th2 type cytokine production by cutaneous T-cell lymphoma (CTCL) results in eosinophilia, extracellular granule protein depositions, as well as increased IL-5 levels in the skin and/or peripheral blood.53 In angiolymphoid hyperplasia with eosinophilia, clonal T cell populations have been identified.54 Currently, it is not known whether the eosinophils inhibit or modulate proliferation of the malignant cells.
Scleroderma-Like Disorders
Eosinophilia is a common histological finding in a number of diseases characterized by marked tissue fibrosis, such as cutaneous forms of systemic sclerosis, drug induced scleroderma-like illness (e.g., bleomycin), eosinophilia–myalgia syndrome, eosinophilic fasciitis, localized scleroderma, and toxic oil syndrome.55 Elevated eosinophil granule protein levels have been detected in the serum of patients with diffuse cutaneous forms of systemic sclerosis, suggesting eosinophil activation and degranulation.56 As they produce fibrogenic cytokines, e.g., transforming growth factor β (TGF-β), eosinophils are thought to contribute to skin fibrosis.55
Conclusion
Together, these examples show that most observations on eosinophils in skin diseases are rather descriptive. The exact mechanisms whereby eosinophils are recruited and activated, as well as their pathogenic role(s), are not yet fully understood. Depending on the skin disease, a role in host defense, immunoregulation and/or remodeling, and fibrosis can be assumed. Further research is therefore required in order to understand the function of eosinophils in skin diseases and to develop new therapeutic strategies.
Acknowledgements
Our research is supported by the Stanley Thomas Johnson Foundation and Swiss National Science Foundation, Bern, Switzerland.
References
- 1.Simon D., Simon H.U. Eosinophilic disorders. J Allergy Clin Immunol. 2007;119:1291–1300. doi: 10.1016/j.jaci.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Simon D., Wardlaw A., Rothenberg M.E. Organ-specific eosinophilic disorders of the skin, lung, and gastrointestinal tract. J Allergy Clin Immunol. 2010;126:3–13. doi: 10.1016/j.jaci.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leiferman K.M., Ackerman S.J., Sampson H.A., Haugen H.S., Venencie P.Y., Gleich G.J. Dermal deposition of eosinophil-granule major basic protein in atopic dermatitis: comparison with onchocerciasis. N Engl J Med. 1985;313:282–285. doi: 10.1056/NEJM198508013130502. [DOI] [PubMed] [Google Scholar]
- 4.Nervi S.J., Schwartz R.A., Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285–289. doi: 10.1016/j.jaad.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 5.Simon D., Hoesli S., Roth N., Staedler S., Yousefi S., Simon H.U. Eosinophil extracellular DNA traps in skin diseases. J Allergy Clin Immunol. 2010;127(1):194–199. doi: 10.1016/j.jaci.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Plötz S.G., Simon H.U., Darsow U., Simon D., Vassina E., Yousefi S. Use of an anti-interleukin-5 antibody in the hypereosinophilic syndrome with eosinophilic dermatitis. N Engl J Med. 2003;349:2334–2339. doi: 10.1056/NEJMoa031261. [DOI] [PubMed] [Google Scholar]
- 7.Simon H.U., Rothenberg M.E., Bochner B.S., Weller P.F., Wardlaw A.J., Wechsler M.E. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol. 2010;126:45–49. doi: 10.1016/j.jaci.2010.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon H.U., Plötz S.G., Simon D., Dummer R., Blaser K. Clinical and immunological features of patients with interleukin-5-producing T cell clones and eosinophilia. Int Arch Allergy Immunol. 2001;124:242–245. doi: 10.1159/000053723. [DOI] [PubMed] [Google Scholar]
- 9.Kaddu S., Zenahlik P., Beham-Schmid C., Kerl H., Cerroni L. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966–978. doi: 10.1016/s0190-9622(99)70086-1. [DOI] [PubMed] [Google Scholar]
- 10.Gotlib J., Cross N.C., Gilliland D.G. Eosinophilic disorders: molecular pathogenesis, new classification, and modern therapy. Best Pract Res Clin Haematol. 2006;19:535–569. doi: 10.1016/j.beha.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 11.Cools J., DeAngelo D.J., Gotlib J., Stover E.H., Legare R.D., Cortes J. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 12.Simon H.U., Yousefi S., Dommann-Scherrer C.C., Zimmermann D.R., Bauer S., Barandun J. Expansion of cytokine-producing CD4-CD8-T cells associated with abnormal Fas expression and hypereosinophilia. J Exp Med. 1996;183:1071–1082. doi: 10.1084/jem.183.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon H.U., Plötz S.G., Dummer R., Blaser K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med. 1999;341:1112–1120. doi: 10.1056/NEJM199910073411503. [DOI] [PubMed] [Google Scholar]
- 14.Vassina E.M., Yousefi S., Simon D., Zwicky C., Conus S., Simon H.U. cIAP-2 and survivin contribute to cytokine-mediated delayed eosinophil apoptosis. Eur J Immunol. 2006;36:1975–1984. doi: 10.1002/eji.200635943. [DOI] [PubMed] [Google Scholar]
- 15.Klion A.D., Nutman T.B. The role of eosinophils in host defense against helminth parasites. J Allergy Clin Immunol. 2004;113:30–37. doi: 10.1016/j.jaci.2003.10.050. [DOI] [PubMed] [Google Scholar]
- 16.Yousefi S., Gold J.A., Andina N., Lee J.J., Kelly A.M., Kozlowski E. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–953. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- 17.Frigas E., Motojima S., Gleich G.J. The eosinophilic injury to the mucosa of the airways in the pathogenesis of bronchial asthma. Eur Respir J. 1991;13:S123–S135. [PubMed] [Google Scholar]
- 18.Jacobsen E.A., Taranova A.G., Lee N.A., Lee J.J. Eosinophils: singularly destructive effector cells or purveyors of immunoregulation. J Allergy Clin Immunol. 2007;119:1313–1320. doi: 10.1016/j.jaci.2007.03.043. [DOI] [PubMed] [Google Scholar]
- 19.Straumann A., Conus S., Gronzka P., Kita H., Kephart G., Bussmann C. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic esophagitis: A randomized, placebo-controlled, double-blind trial. Gut. 2010;59:21–30. doi: 10.1136/gut.2009.178558. [DOI] [PubMed] [Google Scholar]
- 20.Steinhoff M., Bienenstock J., Schmelz M., Maurer M., Wei E., Biro T. Neurophysiological, neuroimmunological, and neuroendocrine basis of pruritus. J Invest Dermatol. 2006;126:1705–1718. doi: 10.1038/sj.jid.5700231. [DOI] [PubMed] [Google Scholar]
- 21.Kiehl P., Falkenberg K., Vogelbruch M., Kapp A. Tissue eosinophilia in acute and chronic dermatitis: a morphometric approach using quantitative image analysis of immunostaining. Br J Dermatol. 2001;145:720–729. doi: 10.1046/j.1365-2133.2001.04456.x. [DOI] [PubMed] [Google Scholar]
- 22.Simon D., Braathen L.R., Simon H.U. Eosinophils and atopic dermatitis. Allergy. 2004;59:561–570. doi: 10.1111/j.1398-9995.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 23.Cheng J.F., Ott N.L., Peterson E.A., George T.J., Hunkee M.J., Gleich G.J. Dermal eosinophils in atopic dermatitis undergo cytolytic degeneration. J Allergy Clin Immunol. 1997;99:683–692. doi: 10.1016/s0091-6749(97)70031-9. [DOI] [PubMed] [Google Scholar]
- 24.Oldhoff J.M., Darsow U., Werfel T., Katzer K., Wulf A., Laifaoui J. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy. 2005;60:693–696. doi: 10.1111/j.1398-9995.2005.00791.x. [DOI] [PubMed] [Google Scholar]
- 25.Staumont-Salle D., Dombrowicz D., Capron M., Delaporte E. Eosinophils and urticaria. Clin Rev Allergy Immunol. 2006;30:13–18. doi: 10.1385/CRIAI:30:1:013. [DOI] [PubMed] [Google Scholar]
- 26.Peters M.S., Schroeter A.L., Kephart G.M., Gleich G.J. Localization of eosinophil granule major basic protein in chronic urticaria. J Invest Dermatol. 1983;81:39–43. doi: 10.1111/1523-1747.ep12538380. [DOI] [PubMed] [Google Scholar]
- 27.O’Donnell M.C., Ackerman S.J., Gleich G.J., Thomas L.L. Activation of basophil and mast cell histamine release by eosinophil granule major basic protein. J Exp Med. 1993;157:1981–1991. doi: 10.1084/jem.157.6.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tedeschi A., Asero R., Marzano A.V., Lorini M., Fanoni D., Berti E. Plasma levels and skin-eosinophil-expression of vascular endothelial growth factor in patients with chronic urticaria. Allergy. 2009;64:1616–1622. doi: 10.1111/j.1398-9995.2009.02069.x. [DOI] [PubMed] [Google Scholar]
- 29.Cugno M., Marzano A.V., Tedeschi A., Fanoni D., Venegoni L., Asero R. Expression of tissue factor by eosinophils in patients with chronic urticaria. Int Arch Allergy Immunol. 2009;148:170–174. doi: 10.1159/000155748. [DOI] [PubMed] [Google Scholar]
- 30.Moossavi M., Mehregan D.R. Wells’ syndrome: a clinical and histopathologic review of seven cases. Int J Dermatol. 2003;42:62–67. doi: 10.1046/j.1365-4362.2003.01705.x. [DOI] [PubMed] [Google Scholar]
- 31.Ogbogu P.U., Bochner B.S., Butterfield J.H., Gleich G.J., Huss-Marp J., Kahn J.E. Hypereosinophilic syndrome: A multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol. 2009;124:1319–1325. doi: 10.1016/j.jaci.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blenkinsopp W.K., Haffenden G.P., Fry L., Leonard J.N. Histology of linear IgA disease, dermatitis herpetiformis, and bullous pemphigoid. Am J Dermatopathol. 1983;5:547–554. doi: 10.1097/00000372-198312000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Strohal R., Rappersberger K., Pehamberger H., Wolff K. Nonbullous pemphigoid: prodrome of bullous pemphigoid or a distinct pemphigoid variant? J Am Acad Dermatol. 1993;29:2993–2999. doi: 10.1016/0190-9622(93)70179-w. [DOI] [PubMed] [Google Scholar]
- 34.Engineer L., Bhol K., Kumari S., Razzaque Ahmed A. Bullous pemphigoid: interaction of interleukin 5, anti-basement membrane zone antibodies and eosinophils. A preliminary observation. Cytokine. 2001;13:32–38. doi: 10.1006/cyto.2000.0791. [DOI] [PubMed] [Google Scholar]
- 35.Frezzolini A., Teofoli P., Cianchini G., Barduagni S., Ruffelli M., Ferranti G. Increased expression of eotaxin and its specific receptor CCR3 in bullous pemphigoid. Eur J Dermatol. 2002;12:27–31. [PubMed] [Google Scholar]
- 36.Nakashima H., Fujimoto M., Asashima N., Watanabe R., Kuwano Y., Yazawa N. Serum chemokine profile in patients with bullous pemphigoid. Br J Dermatol. 2007;156:454–459. doi: 10.1111/j.1365-2133.2006.07601.x. [DOI] [PubMed] [Google Scholar]
- 37.Ameglio F., D’Auria L., Bonifati C., Ferrano C., Mastroianni A., Giacalone B. Cytokine pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with disease intensity. Br J Dermatol. 1998;138:611–614. doi: 10.1046/j.1365-2133.1998.02169.x. [DOI] [PubMed] [Google Scholar]
- 38.Wakugawa M., Nakamura K., Hino H., Toyama K., Hattori N., Okochi H. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol. 2000;143:112–116. doi: 10.1046/j.1365-2133.2000.03599.x. [DOI] [PubMed] [Google Scholar]
- 39.Davoine F., Labonte I., Ferland C., Mazer B., Chakir J., Laviolette M. Role and modulation of CD16 expression on eosinophils by cytokines and immune complexes. Int Arch Allergy Immunol. 2004;134:165–172. doi: 10.1159/000078650. [DOI] [PubMed] [Google Scholar]
- 40.Simon H.U., Weber M., Becker E., Zilberman Y., Blaser K., Levi-Schaffer F. Eosinophils maintain their capacity to signal and release eosinophil cationic protein upon repetitive stimulation with the same agonist. J Immunol. 2000;165:4069–4075. doi: 10.4049/jimmunol.165.7.4069. [DOI] [PubMed] [Google Scholar]
- 41.Sams W.M., Jr., Gammon W.R. Mechanism of lesion production in pemphigus and pemphigoid. J Am Acad Dermatol. 1982;6:431–452. doi: 10.1016/s0190-9622(82)70036-2. [DOI] [PubMed] [Google Scholar]
- 42.Borrego L., Maynard B., Peterson E.A., George T., Iglesias L., Peters M.S. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. 1996;148:897–909. [PMC free article] [PubMed] [Google Scholar]
- 43.Ståhle-Bäckdahl M., Inoue M., Guidice G.J., Parks W.C. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest. 1994;93:2022–2030. doi: 10.1172/JCI117196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brodersen I., Frentz G., Thomsen K. Eosinophilic spongiosis in early pemphigus foliaceus. Acta Derm Venereol. 1978;58:368–369. [PubMed] [Google Scholar]
- 45.Rico M.J., Benning C., Weingart E.S., Streilein R.D., Hall R.P. Characterization of skin cytokines in bullous pemphigoid and pemphigus vulgaris. Br J Dermatol. 1999;140:1079–1086. doi: 10.1046/j.1365-2133.1999.02907.x. [DOI] [PubMed] [Google Scholar]
- 46.Iwatsuki K., Tagami H., Yamada M. Pemphigus antibodies mediate the development of an inflammatory change in the epidermis. A possible mechanism underlying the feature of eosinophilic spongiosis. Acta Derm Venereol. 1983;63:495–500. [PubMed] [Google Scholar]
- 47.Kanitakis J. Charcot-Leyden crystals in pemphigus vegetans. J Cutan Pathol. 1987;14:127. [PubMed] [Google Scholar]
- 48.Caproni M., Rolfo S., Bernacchi E., Bianchi B., Brazzini B., Fabbri P. The role of lymphocytes, granulocytes, mast cells and their related cytokines in lesional skin of linear IgA bullous dermatosis. Br J Dermatol. 1999;140:1072–1078. doi: 10.1046/j.1365-2133.1999.02904.x. [DOI] [PubMed] [Google Scholar]
- 49.Ward J.C., Gitlin J.B., Garry D.J., Jatoi A., Luikart S.D., Zelickson B.D. Epidermolysis bullosa acquisita induced by GM-CSF: a role for eosinophils in treatment-related toxicity. Br J Haematol. 1992;81:27–32. doi: 10.1111/j.1365-2141.1992.tb08166.x. [DOI] [PubMed] [Google Scholar]
- 50.Ramdial P.K., Naidoo D.K. Drug-induced cutaneous pathology. J Clin Pathol. 2009;62:493–504. doi: 10.1136/jcp.2008.058289. [DOI] [PubMed] [Google Scholar]
- 51.Bocquet H., Bagot M., Roujeau J.C. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (Drug Rash with Eosinophilia and Systemic Symptoms: DRESS) Semin Cutan Med Surg. 1996;15:250–257. doi: 10.1016/s1085-5629(96)80038-1. [DOI] [PubMed] [Google Scholar]
- 52.Newman B., Hu W., Nigro K., Gilliam A.C. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302–316. doi: 10.1016/j.jaad.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 53.Ionescu M.A., Rivet J., Daneshpouy M., Briere J., Morel P., Janin A. In situ eosinophil activation in 26 primary cutaneous T-cell lymphomas with blood eosinophilia. J Am Acad Dermatol. 2005;52:32–39. doi: 10.1016/j.jaad.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Kempf W., Haeffner A.C., Zepter K., Sander C.A., Flaig M.J., Mueller B. Angiolymphoid hyperplasia with eosinophilia: evidence for a T-cell lymphoproliferative origin. Hum Pathol. 2002;33:1023–1029. doi: 10.1053/hupa.2002.128247. [DOI] [PubMed] [Google Scholar]
- 55.Foti R., Leonardi R., Rondinone R., Di Gangi M., Leonetti C., Canova M. Scleroderma-like disorders. Autoimmun Rev. 2008;7:331–339. doi: 10.1016/j.autrev.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Cox D., Earle L., Jimenez S.A., Leiferman K.M., Gleich G.J., Varga J. Elevated levels of eosinophil major basic protein in the sera of patients with systemic sclerosis. Arthritis Rheum. 1995;38:939–945. doi: 10.1002/art.1780380709. [DOI] [PubMed] [Google Scholar]
Chapter 13.4. The Evolving Role of Eosinophils in Asthma
Since the discovery of the eosinophil in the late 1800s, this cell has been considered to be not only a characteristic feature of asthma, but possibly an essential component of its pathophysiology. This concept has been supported by many observations, including the demonstration of tissue eosinophilia in postmortem analyses of the lungs of status asthmaticus, as well as parallel relationships between blood eosinophilia and markers of asthma severity. The concept of the eosinophil’s central role in asthma was further strengthened by animal studies that showed that anti-interleukin-5 (anti-IL-5) reduced blood and airway eosinophils and in parallel decreased airway hyperresponsiveness and the development of the late-phase response to inhaled antigen. Surprisingly, when anti-IL-5 was given to patients with asthma, although bronchoalveolar lavage and peripheral blood eosinophils dramatically diminished, this treatment failed to show any significant improvement in airway hyperresponsiveness or airflow obstruction to inhaled antigen, or in clinical markers. What has emerged in the wake of anti-IL-5 studies are the two roles in which the eosinophil appears to play a significant role in asthma: that of an effector cell in airway remodeling and as a biomarker for asthma exacerbations.
Introduction: Why has the Eosinophil held a Position of Prominence in Asthma?
Asthma is characterized by variable airflow obstruction, bronchial hyperresponsiveness, and chronic airway inflammation, all of which are likely to be intertwined and interdependent. The immune processes involved in the development of these characteristics in asthma are complex, redundant, and interactive, making it difficult to specifically define which factor, or factors, are the principal contributors to these processes and the eventual pathophysiology of asthma. As has also become apparent, asthma is represented by multiple phenotypes in which the clinical profiles and patterns of inflammation have distinct, though overlapping, characteristics. To appreciate the mechanisms of disease in asthma and the role of eosinophils, it is helpful to explore the contribution of individual cells to the pathophysiology of asthma.
As early as the turn of the 20th century, the eosinophil was identified as a prominent cell associated with asthma.1., 2. For example, postmortem examinations of the lungs of patients who died from status asthmaticus showed, in many cases, sheets of eosinophils infiltrating the airways. These telling findings led to the long-held belief that eosinophils are an inherent characteristic and possibly an essential component of asthma, particularly in severe exacerbation of disease. As the role of airway inflammation became more fully defined, the assumption that eosinophils are a primary, if not the principal, contributor to asthma was integrated into concepts of asthma, at least until the last decade.3
This positioning into asthma was further supported by animal models, which strengthened the hypothesis that eosinophils are a principal contributor to inflammation, airflow obstruction, and airway hyperresponsiveness (AHR).4 From animal models, it was possible to separate individual components that contribute to inflammation, and the cytokine, interleukin-5 (IL-5), was found to be responsible not only for terminal differentiation of eosinophils, but by ablating IL-5 with a specific monoclonal antibody, many of asthma-like airway responses to inhaled antigen were inhibited. Based upon these findings, eosinophils and IL-5 became a major pathway in allergic inflammation and a target to regulate and more mechanistically control asthma.
These theories would subsequently be tested with the administration of anti-IL-5 to patients with asthma. As expected, there was a significant reduction in circulating and sputum eosinophils but, surprisingly, little to no impact on features of asthma—airflow obstruction or AHR—and only a 50% reduction in bronchial mucosa eosinophils. Based upon these data, the role of the eosinophil in and contribution to asthma appeared less apparent. What has evolved since these dramatic shifts from an early appreciation of the eosinophil to asthma has been a more accurate and informed picture of this cell’s involvement and contribution to asthma, which is the objective of our subchapter.
To most fully appreciate current views of eosinophils in asthma, we feel that it is helpful to trace the key observations that have attempted to define this cell’s role in asthma. Retracing these discoveries has led to a more well-defined elucidation of the eosinophil’s role in asthma.
What has been Learned from Investigations into the Eosinophil’s Contribution to Asthma?
Peripheral Blood Eosinophils
Clinical evidence to suggest that eosinophils are key players in asthma first emerged from examinations of peripheral blood samples, largely because of the ease and safety of such studies. Peripheral eosinophilia had been recognized as a feature of asthma for decades, with clinical correlations arising between airflow obstruction and the magnitude of peripheral blood eosinophilia.5 In 1975, a pivotal study by Horn et al.6 found that the total peripheral blood eosinophil count in a cohort of asthmatic patients was greater in those individuals with a more severe disease. This was an important early observation and provided a clue to what the eosinophil may contribute to asthma as an association arose between the degree of airflow obstruction and peripheral blood eosinophil counts. These and subsequent observations suggested that circulating eosinophils may be a key clinical feature of asthma and, because of these associations, contribute to airflow obstruction and hence disease severity.
In a subsequent study of 43 patients with chronic asthma, Bousquet et al.7 assessed clinical symptoms of patients in relationship to peripheral blood eosinophil counts. These investigators found a positive correlation between the eosinophil count and disease severity. To extend the value of peripheral blood eosinophils to features of asthma, Taylor et al.8 was also able to find correlations between peripheral blood eosinophilia and another key characteristic of asthma, bronchial hyperresponsiveness. Collectively, these studies were helpful in gaining an understanding of the role of eosinophils in asthma as they showed that the level of circulating eosinophils may serve as a biomarker not only for the presence of disease but also to indicate the degree of severity.
How Did the Eosinophil’s Biology Further Link This Cell Type to Asthma?
The eosinophil was named for the ability of eosin to stain its basic granules and thus impart the cell’s well-recognized red color.5 These eosin-staining granules, however, were found to be more than a marker for this cell type. Eosinophil-derived granule products have been found to have multiple actions, some of which can produce pathological and physiological features associated with asthma, including injury to respiratory epithelium (desquamation) and AHR.
The eosinophil granules consist of four major cytotoxic cationic proteins: major basic protein (MBP), eosinophil peroxidase (EPO), eosinophil-derived neurotoxin (EDN) or nonsecretory ribonuclease, and eosinophil cationic protein (ECP).9 MBP accounts for 55% of the eosinophil granule content, is toxic to parasites, and has similar cytotoxic effects on the respiratory epithelial cells of both animals and humans (Table 13.4.1 ).10., 11. In vitro studies have shown that the application of MBP to respiratory tissue, at concentrations consistent with values found in sputum and bronchoalveolar lavage (BAL) fluid of asthmatic patients (50–100 mg/mL), leads to almost complete erosion of tracheal epithelium and parallels the observed desquamation of respiratory epithelium in the bronchial mucosa of subjects with asthma. These effects of MBP suggest a potential mechanism by which eosinophils may contribute to airway injury, initiate repair (i.e., remodeling), and subsequently induce hyperresponsiveness.12 To extend these observations, Flavahan et al. incubated guinea pig tracheal rings with MBP and found bronchial smooth muscle tension to acetylcholine was significantly enhanced compared to tissues not exposed to eosinophils.13 In a dog model, the intraepithelial administration of MBP also increased the bronchoconstriction response.14
TABLE 13.4.1.
Eosinophil-Derived Inflammatory Mediators
| Cytotoxicity | Epithelial Damage | Airway Hyper-Responsiveness | Broncho-Constriction | Mucus Production, Vascular Leakage, Vasodilation | Eosinophil-Attraction Activation | Mast-Cell Proliferation | ||
|---|---|---|---|---|---|---|---|---|
| Granule proteins | MBP | + | + | + | ||||
| ECP | + | + | + | |||||
| EDN | + | + | + | |||||
| EPO | + | + | ||||||
| Lipid mediators | Leukotrienes C4, D4, E4 | + | + | |||||
| Prostaglandin E2, I2 | − | + | ||||||
| PAF | + | |||||||
| Cytokines | IL-3 | + | + | |||||
| GM-CSF | + | |||||||
| IL-5 | + | |||||||
GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; PAF, platelet-activating factor.
ECP also causes cytotoxic effects on respiratory tract cells. By adding small amounts of ECP to the respiratory tract, Dahl et al.15 found damage and denudation of bronchial and tracheal epithelial cells, which paralleled observations in asthma. Elevated ECP values are found in BAL fluid of asthmatic subjects during the late-phase response (LPR) to an allergen challenge, as well as in lavage fluid of patients with chronic, persistent asthma.16 In guinea pig models, the application of increasing concentrations of EPO to tracheal mucosa caused epithelial cell exfoliation, ciliostasis, and bleb formation.17 Serum EDN levels are elevated in asthma and return to normal levels following treatment with prednisolone and the achievement of disease control.18 Finally, when compared to normal subjects, EDN concentrations in urine are greater in asthmatic children.19
Eosinophils also produce other inflammatory products such as cysteinyl leukotrienes, platelet activating factor (PAF), reactive oxygen species (ROS), and substance P. In in vitro studies designed to assess eosinophil biology, PAF stimulated the release of granule products and led to superoxide anion generation.20 PAF was also chemotactic for the eosinophil, as reflected by the induction of an eosinophilic infiltrate of the airway after local or systemic delivery of PAF in guinea pigs.21., 22. These early findings in animals suggested that the release of PAF by eosinophils could potentially create a self-perpetuating cycle to further the development of eosinophilic inflammation and hence airway injury. PAF also has direct proinflammatory properties. When administered to humans, PAF causes rapid bronchoconstriction, which can last up to 2 h.23 Similar to changes that follow an airway allergen challenge, PAF causes a prolonged increase in bronchial responsiveness.24 Although a role for PAF in human asthma has not been established, the biology of this eosinophil product is associated with airway changes reflective of asthma and also implies an important role for eosinophils in this process.
Eosinophils also generate the cysteinyl leukotrienes, LTC4 and LTD4, which can cause both an increase in vascular permeability and bronchoconstriction.25 LTC4 is secreted by eosinophils and is increased in asthmatic children.26 Activated eosinophils can also release substance P, a neuropeptide, to increase vascular permeability and promote plasma extravasation.27 Eosinophils, like other phagocytes, generate ROS, which are produced in greater concentrations in asthma.28 In asthma, ROS can cause mucus secretion, which leads to increased vascular permeability and airway obstruction. When damaged by ROS, respiratory epithelium produces fewer bronchodilatory substances, such as prostaglandin E2 (PGE2) and nitric oxide, with the net result of increased airflow obstruction29 (Fig. 13.4.1 ).
FIGURE 13.4.1.
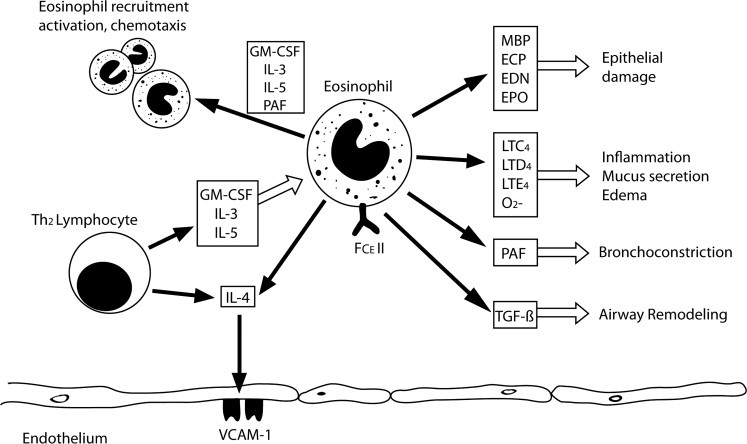
Eosinophil activities.
Activated T-helper type 2 (Th2) lymphocytes result in the generation of granulocyte macrophage colony stimulating factor (GM-CSF), interleukin-3 (IL-3), and IL-5. These cytokines have several proinflammatory properties, including activation, adhesion, chemotaxis, enhanced maturation, and prolonged survival. Antigen-induced stimulation of FCε receptor II also activates the eosinophil. IL-4, produced by Th2 cells and the eosinophil, upregulates the vascular cell adhesion molecule 1 (VCAM-1) adhesion molecule and promotes adherence and migration through the vascular endothelium. The activated eosinophil is capable of mediator release. Granule constituents eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), eosinophil peroxidase (EPO), and major basic protein (MBP) are capable of epithelial damage. Leukotrienes and reactive oxygen species (ROS) are capable of causing inflammation, edema and mucus secretion. Platelet activating factor (PAF) and transforming growth factor β (TGF-β) cause bronchoconstriction and airway remodeling, respectively. O2−, peroxide; FcEII, Fc component of IgE.
Eosinophils Generate Inflammatory Cytokines
Although T cell-derived cytokines may be the major source of factors that influence eosinophil development and function, eosinophils themselves can also produce several cytokines to perpetuate their own inflammatory biology and that associated with asthma. Transforming growth factor β (TGF-β) is a profibrotic cytokine produced by eosinophils and found in increased concentrations in asthma.30 TGF-β stimulation of fibroblasts leads to a thickening of the reticular lamina of the airways, and thus provides a conceptual link between eosinophilic inflammation and structural remodeling of the airway in asthma.30 Eosinophils also produce several other mediators/cytokines, such as IL-13, matrix metalloproteinase-9 (MMP-9), tissue inhibitor of metalloproteinases 1 (TIMP-1; or metalloproteinase inhibitor 1), and vascular endothelial growth factor (VEGF), which have also been implicated in matrix remodeling in asthma.31
IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, and IL-16 are all produced by eosinophils in varying concentrations. IL-1, initially observed in mice and later found in hypereosinophilic patients, is associated with HLA-DR expression and is thought to contribute to the eosinophil’s function as an antigen-presenting cell.32 IL-3 and granulocyte macrophage colony stimulating factor (GM-CSF), are also produced by eosinophils, and can function in an autocrine fashion to prolong the cell’s survival.33 IL-4, which upregulates vascular cell adhesion molecule (VCAM) receptors on endothelial cells and is a cofactor in immunoglobulin E (IgE) isotype switching, is produced by eosinophils from patients with atopic asthma.34 IL-5, which regulates terminal differentiation and survival of eosinophils, is found in eosinophils obtained from BAL fluid of asthmatic patients.35 IL-5 release also follows eosinophil stimulation by IgA, IgG, or IgE immune complexes.36
What has been Learned from the Study of Sputum and Bal Eosinophilia?
The presence of eosinophils in sputum has been a characteristic finding of asthma since the early 19th century.2 Bousquet et al. increased sputum and peripheral blood eosinophils in asthma patients.7 Sputum eosinophilia is also a feature of nonasthmatic eosinophilic bronchitis, a relatively nonspecific term. In contrast, sputum eosinophilia is not a feature of chronic obstructive pulmonary disease (COPD). In addition, previous studies have suggested a relationship between BAL fluid eosinophilia and the level of bronchial responsiveness.37 The concentrations of ECP and MBP in BAL fluid were also found to directly relate to the percentage of eosinophils, implying that eosinophils in asthmatic airways are activated and have undergone degranulation.38 To extend these observations, Uchida et al.4 evaluated the effect of MBP on airway responsiveness in an animal model. Within 1 h of direct instillation of MBP onto the trachea of rats, significant increases in airway responsiveness to methacholine occurred.
Finally, 22 subjects with asthma were identified, half of whom were treated with inhaled corticosteroids (ICS) and the other half given only bronchodilators, to be used as needed.39 ICS use led to a fall in serum and BAL ECP levels but, interestingly, the eosinophil count was unchanged in both the corticosteroid or bronchodilator treated groups. Collectively, eosinophil-derived products can have a significant influence on the function and pathohistology of the airway and mirror many features of asthma.
What has been Learned from the Study of Bronchial Mucosal Eosinophils?
In the 1950s, a further relationship of asthma to eosinophils was supported by the finding of a marked infiltration of eosinophils in the lung tissue of patients who died suddenly of status asthmaticus.40 In cases of death from acute, severe asthma, eosinophilic infiltrates were found in the lung parenchyma, bronchial lumen, and entire thickness of the bronchial wall.41 Bronchial biopsies obtained by bronchoscopy from patients with mild disease also showed eosinophils as a prominent cellular infiltrate.42 In addition to the presence of eosinophils and their granule products in airways of asthmatic patients, the airway histology showed epithelial desquamation, impaired ciliary function, basement membrane thickening and mucous plugs.9., 43. Expanding upon these findings, Ohashi et al.44 found the eosinophilic infiltration of mucosal tissue was associated with opening of tight junctions of bronchial epithelial cells. These histological analyses showed eosinophil infiltration of the airways was associated with epithelial damage and possibly linked to subsequent airway hyper-responsiveness.44
What has been Learned About Eosinophil Involvement in Airway Inflammation from Inhaled Antigen Challenges?
When subjects with allergic asthma inhale allergens to which they are sensitized, there is an immediate, or early, response, characterized by an acute fall in forced expiratory volume in one second (FEV1). Approximately 40% of subjects will go on to develop an LPR, which occurs 4–8 h after allergen exposure. In LPR, eosinophils are the prominent airway cellular infiltrate both in animal models and human subjects. To define the kinetics of eosinophil recruitment and eotaxin/C-C motif chemokine 11 (CCL11) generation, Humbles et al.45 found the chemokine eotaxin was increased 2–3 h postallergen challenge in sensitized guinea pigs. By 12–24 h postallergen challenge, there was a significant increase in eosinophil numbers in the BAL fluid, but no further rise in eotaxin. These findings in the guinea pig suggest that eosinophil recruitment to the airway following an inhalation of allergen is, in part, regulated by eotaxins. Findings of an early recruitment of eosinophils and the later development of a late-phase airflow obstruction to allergen suggested that these events reflect processes in asthma that could provide insight into how eosinophils may contribute to asthma.
Histopathological findings of the airway in LPR are also similar to events found in patients with chronic asthma and persistent airway obstruction. In guinea pigs sensitized to ovalbumin, 17 h after an inhalation challenge with ovalbumin the cellular composition in BAL fluid is predominantly neutrophils.46., 47. By 72 h post antigen exposure, 50% of BAL cells are eosinophils. In addition, the subsequent eosinophilic infiltration of the peribronchial smooth muscle and epithelium persists for up to 7 days. Eosinophils in the BAL of immunized guinea pigs with an LPR were found to be activated, suggesting that these cells are primed during recruitment to the airway.48
Similarly in humans, de Monchy et al.16 found significant eosinophilia only in patients who developed LPR to inhaled antigen. Moreover, the eosinophils in the BAL fluid had undergone degranulation, as evidenced by an elevated ECP:albumin ratio. There was also evidence that peripheral blood eosinophils and ECP concentrations are increased prior to antigen challenge in those subjects who eventually develop an LPR. Subsequently, Cockcroft et al.49 evaluated the LPR in a population of asthma patients who had been given a single dose of inhaled beclomethasone dipropionate, inhaled salbutamol or inhaled cromoglycate in a randomized, double-blind, placebo-controlled crossover trial. While beclomethasone had no effect on the allergen-induced early pulmonary obstructive response, there was a significant inhibition of the LPR at 7 h and 30 h later. Collectively, these data suggest that allergen provocation of allergic inflammation is likely to be regulated by eosinophils and can be controlled by corticosteroids, which block both the late-phase rise in recruited eosinophils and the subsequent reduction in lung function. Thus, a logical conclusion from work at this time was that a direct association between eosinophil recruitment and altered lung function existed in asthma, a finding substantiated by Kidney et al.50
In 2000, Gauvreau et al.51 extended these observations when she evaluated the development of the LPR and recruitment of eosinophils in asthma patients who were initially treated with inhaled budesonide for 1 week and then underwent an inhaled allergen challenge. Budesonide administration reduced the intensity of the LPR and also inhibited eosinophil recruitment to the airway, reflected by sputum eosinophils and the parallel increase in peripheral blood eosinophils 24 h after challenge. From these data, it was assumed that allergic activation of the airway and the subsequent development of the LPR were caused by eosinophils recruited to the lung.
What has been Learned about the Eosinophil and Asthma from Segmental Antigen Challenges?
To extend observations from whole-lung allergen challenges, a number of groups developed the technique of using a bronchoscope to deliver antigen into single segments of the airway. To perform these studies, a bronchoscope is introduced into the lung and then wedged into an isolated airway segment, where a dose of allergen is introduced and a lavage performed immediately; the cells and lavage fluid obtained at this time represent events associated with the acute or early response. Bronchoscopy is repeated 24–48 h later, the challenged segment of the airway identified, and lavage performed: the airway events analyzed at this time model the LPR. This approach, while not allowing for measures of pulmonary function, provides a direct measure of cells, mediators, and retrieval of cells for ex vivo study to compare early- and late-phase allergic reactions.
When antigen is introduced to airway segments in allergic patients, there is a strong eosinophilic response 24–48 h postchallenge.52 The analysis of BAL is characterized by large concentrations of granule proteins, ECP, EDN, EPO, and MBP, as well as LTC4, suggesting that the recruited eosinophils are activated when they appear in the airway. Additionally, when eosinophils are retrieved from BAL fluid, they are phenotypically distinct from circulating cells and have greater superoxide anion release, collagen adherence and cell surface adherence receptors compared to peripheral blood eosinophils. These findings suggested that LPR-associated eosinophils, which are recruited to the lung during the late phase and are terminally differentiated, have a greater capacity to generate inflammation. BAL levels of IL-5 were also increased and correlated with the eosinophils, suggesting a key role for this cytokine in these processes. From these studies, a more expanded picture of the allergic inflammatory response was made, along with the identification of key mediators and evidence for an enhancement of the eosinophil’s inflammatory potential.
Calhoun et al.53 used segmental antigen challenges in subjects with allergic rhinitis who had been inoculated with rhinovirus to further evaluate the interactions between viral upper respiratory infections and allergic reactions, and gain insight into mechanisms of asthma exacerbations. During an acute rhinovirus infection, BAL fluid obtained 48 h after antigen challenge contained increased numbers of eosinophils. In some subjects, the increase in eosinophils persisted for up to 1 month following the acute viral infection and initial antigen challenge. This augmented antigen-induced eosinophil recruitment was thought to be a possible mechanism for an intensified inflammatory airway response during viral infections and to account for greater asthma symptoms at that time. Alternatively, virus-induced epithelial damage, with a subsequent increase in mucosal permeability, was hypothesized to increase allergen contact with immune cells, thus creating a greater inflammatory response.54
What has been Learned about Eosinophils in Asthma by Evaluating the Effects of Treatment with Corticosteroids?
The presence of eosinophils is regulated by apoptosis and modulated by cytokines, growth factors, and lipid mediators, which are released during allergic inflammation but suppressed by glucocorticoids.55 Consequently, the persistence of airway eosinophilia in some patients with asthma was attributed to cells that had developed resistance to corticosteroids.56 The reduction in airway eosinophils with corticosteroids and improved asthma control further supported a central role for eosinophils in asthma.3 Under both circumstances, either a reduction or persistence of eosinophils, and their correlation with symptoms of asthma, supported a direct link of eosinophils to the pathobiology of asthma.
In 1991, Evans et al.57 measured eosinophil numbers in 10 asthmatic subjects 14 days following the initiation of inhaled budesonide treatment. Airway responsiveness to methacholine was improved and was associated with a fall in peripheral eosinophils. These findings also suggested that eosinophil production, maturation, and differentiation may take place in the lung, as well as the bone marrow. As corticosteroid treatment reduced serum, sputum, and tissue eosinophils, and these reductions were associated with improved asthma symptoms, the eosinophil’s place as a primary contributor to asthma appeared further substantiated. However, not all patients with asthma have eosinophilia, nor are all asthma patients responsive to corticosteroids. These observations, in the face of data already discussed, raised questions as to how essential the eosinophil is to all of the clinical features of asthma.
What has been Learned by Comparisons of Eosinophilic and Noneosinophilic Asthma?
While many studies of asthma confirm the presence of elevated eosinophils, circulating and BAL fluid eosinophilia are not always present in asthma.40., 58. Theories as to these differences included the possibility that eosinophils may be present only during an exacerbation and their absence may result from the actions of medication, particularly ICS.59., 60. These observations also led to an emerging theory that at least two asthma phenotypes exist, based on the presence or absence of tissue eosinophils.48 Persistent eosinophilic inflammation, despite treatment, was found to be more common in adult onset asthma and less common in classic allergic asthma in patients receiving ICS.61 While up to 40% of cases of severe asthma appear to start later in life, the presence of eosinophils in patients with late-onset asthma is also more variable. Interestingly and importantly, patients with asthma and existing eosinophilia had greater airway remodeling and more exacerbations, despite treatment.62
To begin to dissect and clarify these relationships, Woodruff et al.63 used a number of innovative approaches for study. Firstly, to determine the molecular basis for asthma heterogeneity and the involvement of eosinophils, the effect of an underlying T-helper type 2 (Th2)-mediated profile of inflammation was evaluated in a cohort of 42 patients with mild to moderate asthma and 28 healthy control subjects. The recruited subjects were stratified based on high or low expression of IL-13-inducible genes in samples of their airway epithelium. Using the response of their epithelium to stimulation with IL-13, investigators were able to classify the reaction as Th-2-high vs. Th-2-low. The Th2-high asthma group was indistinguishable from the Th2-low asthma group in relationship to demographics, lung function, and response to bronchodilators. However, the Th2-high group had significantly greater AHR, total IgE levels, and BAL and peripheral blood eosinophils. The presence of airway remodeling was also evaluated in this study population, and both the reticular basement membrane thickness and epithelial mucin stores were increased in subjects with a Th2-high profile. Following this classification, subjects were randomized and treated with either inhaled fluticasone or placebo to determine if there was a difference in clinical response to ICS based upon their Th2 profiles.
In the Th2-high group, ICS improved the FEV1, whereas no change occurred in the low Th2 group. This study further supported the concept of heterogeneity in the pathogenesis and pathophysiology of asthma that can be characterized by the presence of Th2-driven inflammation with eosinophilia and responsiveness to corticosteroids as markers of this profile. These findings also indicated that other phenotypes of asthma exist but their features were poorly understood.
What has been Learned about Asthma by Studying Interleukin-5?
Th2 cell-derived IL-5 has been identified as the major cytokine involved in terminal differentiation of eosinophils, activation of mature eosinophils, and prolongation of eosinophil cell survival (Fig. 13.4.2 ).64 IL-5 enhances eosinophil degranulation, chemotaxis, antibody-dependent cytotoxicity, and adhesion to endothelium.65 Van Der Veen et al.66 identified 22 patients with mild to moderate, dust mite-sensitive allergic asthma and found a significant correlation between the magnitude of the LPR, the allergen-specific proliferative response of peripheral T lymphocytes, and an increase in IL-5 in vivo following inhaled antigen. Extending beyond observational studies of an elevation of IL-5 in relationship to LPR, inhaled IL-5 was found to cause, or significantly contribute to, AHR.67 Shi et al.68 also demonstrated that inhaled IL-5 in asthma acted as an eosinophil chemoattractant and activator of the recruited eosinophils to the airway. In a subsequent, blinded, placebo-controlled crossover study of eight patients with allergic bronchial asthma, Shi et al.68 found a significantly enhanced methacholine PC20 (the dose of the inhaled antagonist that provokes a 20% drop in FEV1) responsiveness 24 h and 48 h after an inhalation of IL-5, as well as a significant increase in sputum eosinophils and ECP. Using a mouse model with IL-5 knocked out, inhaled allergen did not lead to eosinophilia or an increase in AHR.69 IL-5 was also decreased during treatment with corticosteroids that improved asthma control.70 From these and other data, IL-5 emerged as the dominant cytokine regulating the eosinophil’s involvement in allergic airway disease.
FIGURE 13.4.2.
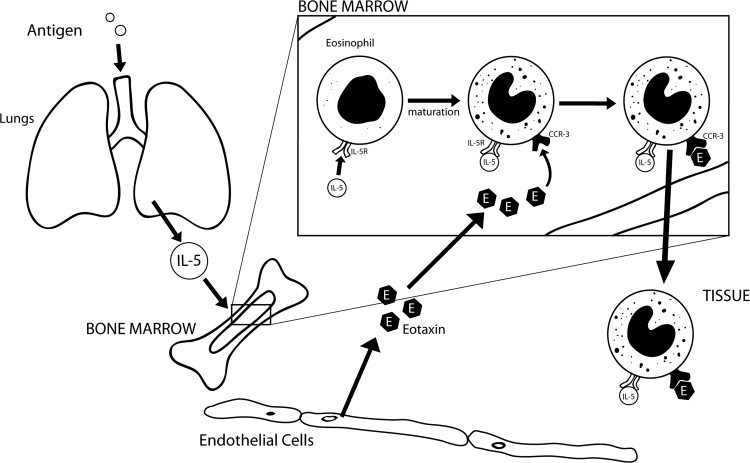
Interleukin-5.
Interleukin-5 (IL-5) is a signaling molecule that stimulates eosinophil proliferation, maturation, and activation. An antigen-stimulated immune response in tissues leads to the secretion of IL-5 by cells such as eosinophils, mast cells, and T-helper type 2 (Th2) cells. IL-5 then acts on the bone marrow to mobilize existing eosinophils and induce further eosinophil production. These maturing eosinophils become responsive to eotaxin/ C-C motif chemokine 11 (CCL11), produced by the endothelium, allowing for their exit from the bone marrow to tissue. CCR3, C-C chemokine receptor type 3; IL-5R, interleukin-5 receptor.
What has been Learned About Eosinophils in Asthma from Treatment with Anti-Interleukin-5?
The anti-IL-5 monoclonal antibody is an IgG antibody that binds with high affinity to free IL-5, thus preventing its binding to the IL-5 receptor on the surface of eosinophils and their progenitors. Van Oosterhouet et al.71 used ovalbumin to challenge sensitized guinea pigs and induce airway eosinophilia, neutrophilia, and tracheal hyperreactivity. When the sensitized guinea pigs were treated with anti-IL-5 and then challenged with ovalbumin, airway eosinophilia, not neutrophil recruitment, was suppressed as well as the allergen challenge-induced increase in AHR. When sensitized guinea pigs were treated with the anti-IL-5 monoclonal antibody by Akutsu et al.,72 there was a decrease in AHR, tracheal wall eosinophil infiltration, and the LPR. Animal studies in which anti-IL-5 decreased airway eosinophilia, AHR to allergen, and LPR provided further support for the hypothesis that eosinophils are central to many important aspects of the pathogenesis of asthma, and suggested that if eosinophil migration to the airway could be inhibited, signs and symptoms of asthma could be controlled or prevented.
What has been Learned about Eosinophil Involvement in Asthma with Anti-Interleukin-5 Treatment?
In 2000, Leckie et al.73 conducted a randomized, double-blind, placebo-controlled trial in which a single infusion of either of two doses of anti-IL-5 humanized monoclonal antibody or placebo was administered to 24 men with mild allergic asthma. The study goal was to evaluate the effect of anti-IL-5 on the LPR to inhaled antigen, on the premise that this treatment would ablate eosinophil recruitment and hence the airway responses to allergen, including the development of the LPR and an associated increase in AHR. The investigators found anti-IL-5 to decrease blood and sputum eosinophils following inhaled allergen challenge. Surprisingly, anti-IL-5 treatment had no effect on either the LPR or postallergen increase in airway responsiveness to histamine. Thus, in striking contrast to animal studies, there was no significant effect of anti-IL-5 on the development of an LPR, despite the absence of eosinophils in the blood or the airways following antigen exposure. This study, though conducted in a small numbers of patients, prompted a total reevaluation of the eosinophil’s role in asthma, including whether it had one at all.
Extending this study, Flood-Page et al.74 evaluated anti-IL-5 monoclonal antibody treatment in asthma patients by examining its multidose effect on blood and sputum, as well as bone marrow and airway tissue eosinophils. In a randomized, double-blind, placebo-controlled study, 24 patients with mild asthma received three intravenous doses of mepolizumab (i.e., IL-5 monoclonal antibody) or placebo for 20 weeks. Within 4 weeks of the first anti-IL-5 dose, there was a significant decrease in peripheral blood eosinophils. At weeks 4 and 10 of the study, there was nearly a 100% reduction of eosinophils in blood and sputum samples following anti-IL-5 treatment. In contrast, eosinophils were reduced by only 52% in bone marrow aspirates. Similarly, bronchial mucosa eosinophils were reduced by 55% from baseline. Despite this reduction of intact eosinophils, staining of the bronchial biopsy for intracellular MBP was unchanged by mepolizumab. Despite these reductions in eosinophils, there was no change in AHR, exacerbations, FEV1 values, peak flow measurements, or symptoms between the anti-IL-5 and placebo-treated groups.
While a reduction in blood and BAL fluid/sputum eosinophils occurred with anti-IL-5, there was only a 50% reduction in bone marrow and bronchial eosinophils.74 This finding confirmed that, despite anti-IL-5 therapy, residual airway eosinophils persisted and the total amount of MBP present in airway tissues was unchanged. From these observations, it was also hypothesized that the residual eosinophil population in the airway continued to exist and release, or retain, granule proteins, despite anti-IL-5 treatment. Furthermore, it was proposed that these persistent effects may be responsible for the lack of improved asthma control, FEV1, and AHR.
These findings with anti-IL-5 contrasted sharply with effects noted with oral corticosteroids, which caused an 80% decrease of bronchial mucosal eosinophilia and led to significant clinical improvements of asthma symptoms.75 Given the possibility that, despite anti-IL-5 treatment, residual airway eosinophils may be sufficient to continue to exert their influence on clinical outcomes, including lung function and symptoms, the eosinophil should not have been excluded as a target for asthma therapy, although the outcome may not be symptoms or airflow obstruction.74
What is the Role of Eosinophils in Exacerbations of Asthma?
In 2008, Rothenberg et al.76 reported the results of a randomized, double-blind, placebo-controlled trial of anti-IL-5 in 85 prednisone-dependent (20–60 mg/d) patients with hypereosinophilic syndrome. In addition to a significant lowering of peripheral blood eosinophilia, 84% of the mepolizumab-treated subjects were able to reduce their oral prednisone dose to ≤10 mg/d compared to only 43% in the placebo group. In addition, mepolizumab significantly reduced the likelihood of an exacerbation of their hypereosinophilic disease. While the effect of therapy on hypereosinophilic syndrome patients may not be extrapolated to asthma, this study did show that mepolizumab has the ability to decrease eosinophils, the prednisone requirement to maintain disease control, and exacerbations from the hypereosinophilic syndrome.
Expanding on this theme, Green et al.3 examined a strategy designed to determine the effects of treatment directed toward reducing sputum eosinophil counts rather than administering a dose of ICS based on symptoms alone, i.e., a guidelines approach. Seventy-four patients with moderate to severe asthma were identified and randomly assigned to either a management strategy based upon British Thoracic Society guidelines or one using a dose of ICS that reduced sputum eosinophils to <3%. The sputum management strategy group had an average eosinophil count that was 63% lower than that of the guideline-managed group during the study. As an apparent consequence of this reduction in eosinophils, patients in the sputum management group had significantly fewer asthma exacerbations and were admitted less frequently to the hospital for asthma (Fig. 13.4.3 ). However, total asthma quality of life scores, mean peak flow measurements, postbronchodilator FEV1, and the use of rescue bronchodilators were not different in the two management groups.3 While this study did not show a relationship between sputum eosinophils and variable airflow obstruction and other parameters, such as daily symptoms, it did support the notion that eosinophils play a central role in asthma by either serving as a marker for exacerbation risks or being possibly linked to an increased susceptibility for exacerbations.
FIGURE 13.4.3.
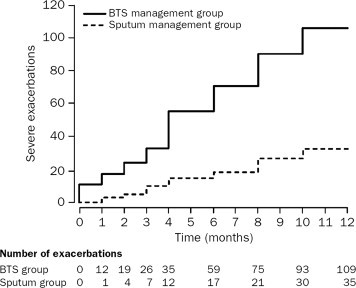
Cumulative asthma exacerbations in the British Thoracic Society (BTS) guideline management group and the sputum management group.
(Reproduced with permission from Green et al.3)
It can be argued that previous studies of anti-IL-5 had examined patients with only mild asthma and with outcome measures that were not specifically related to ongoing eosinophilic inflammation. To extend upon this hypothesis, Haldar et al.77 and Nair et al.78 published results of studies designed to evaluate mepolizumab treatment in patients with severe asthma, persistent eosinophilia in sputum, and frequent exacerbations. In an approach similar to that of Green et al.,3 Haldar et al.77 evaluated the effect of a reduction in sputum eosinophils with anti-IL-5. In this randomized, double-blind, placebo-controlled trial, 61 patients were enrolled with asthma and sputum eosinophilia that was refractory to treatment and a history of recurrent exacerbations. Patients were given anti-IL-5 monoclonal antibody or placebo monthly for 1 year. The mepolizumab group had 57 exacerbations requiring prednisone (a mean of 2.0 exacerbations per year per subject) compared to 109 exacerbations (3.4 exacerbations per subject per year) in the placebo group (Fig. 13.4.4 ).
FIGURE 13.4.4.

The cumulative number of severe exacerbations that occurred in each study group over the course of 50 weeks. The mean number of exacerbations per subject over the course of the 50-week treatment period was 2.0 in the mepolizumab group, compared with 3.4 in the placebo group (relative risk, 0.57; 95% confidence interval, 0.32 to 0.92; p = 0.02).
(Reproduced with permission from Haldar P, et al.77)
The mepolizumab treatment group also had a greater improvement in scores of the Asthma Quality of Life Questionnaire, with a mean improvement of 0.55 compared to 0.19 in the placebo group.77 Similar to findings of Flood-Page et al.,31., 74. the anti-IL-5 treatment group had significantly lower eosinophil counts in BAL, blood, and bronchial wash, but less of a decrease in bronchial mucosa eosinophils. As noted in previous studies,74 there were no significant changes from baseline in AHR, bronchodilator use, or FEV1 in the mepolizumab group. Interestingly, when prednisolone was given after the mepolizumab treatment, there was an improvement in exhaled nitric oxide or lung function, suggesting these symptoms and pathways may be dissociated from eosinophilic inflammation, and are possibly mediated through other mechanisms.
What Effects do Eosinophils have on Airway Remodeling?
Reticular basement membrane thickening in asthma is associated with the presence of bronchial mucosal eosinophils. Using allergen-sensitized mice, Humbles et al.79 found eosinophil-deficient mice were protected from the development of peribronchiolar collagen deposition and increased airway smooth muscle mass following allergen challenge. However, increases in AHR and mucus secretion occurred in the eosinophilic deficient mice, just like wild-type mice. The authors concluded that eosinophils contribute substantially to airway remodeling, but are not obligatory for allergen-induced lung dysfunction.79
As noted earlier by Flood-Page et al.,74 residual bronchial wall eosinophils persists despite treatment with anti-IL-5, even when blood and sputum eosinophils are nearly eliminated. In addition, anti-IL-5 has little effect on the MBP found in the airways, airflow obstruction, and AHR. These observations raise the possibility that bronchial mucosal eosinophils are a privileged cell or are in a privileged location when lodged in tissues, and their levels are not responsive to previously used methods of eosinophil depletion. Furthermore, these findings suggest that another important contribution of eosinophils to the pathophysiology of asthma is airway remodeling, which is supported by other studies.
In a 2-year study, Sont et al.80 evaluated an asthma treatment strategy aimed at reducing AHR compared to treatment that followed international guidelines and was based primarily on symptom and lung function assessments. In this randomized, prospective, parallel trial of 75 adults with mild to moderate asthma, 41 patients were placed in the reference strategy group, with a treatment based on current guidelines, while 34 patients were placed in the AHR group with a treatment strategy based on guidelines as well as a reduction in AHR. In addition to measuring bronchodilator use, FEV1, peak expiratory flow (PEF) and symptoms, AHR was also quantified following methacholine challenges. Patients in the AHR strategy group received higher doses of ICS to reduce AHR. This use of higher doses of ICS and reduction in AHR was associated with a lower incidence of exacerbations and a greater improvement in lung function.
Of the 75 patients in the study,80 55 also underwent bronchial biopsies before and after their individual treatment approaches. In AHR strategy group subjects, who received an additional 400 μg/d of ICS, there was a significantly greater decrease in subepithelial reticular layer airway thickness and bronchial mucosa eosinophils compared to the reference group (Fig. 13.4.5 ). The study also showed that the decrease in mucosal eosinophils related to an accompanying improvement in AHR. When the investigators evaluated the relationship between improvements in AHR and changes in the histology of the airway biopsies, they found a correlation with the reduction in tissue eosinophils (Fig. 13.4.6 ). These findings further support for a role of eosinophils in airway remodeling.
FIGURE 13.4.5.
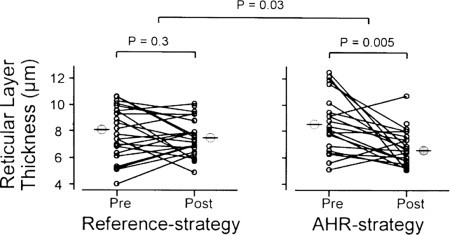
Individual changes in reticular layer thickness beneath the epithelium in bronchial biopsy specimens before and after treatment for 2 years according to the reference and AHR strategies. Bars indicate mean values at the visits for both strategies. There is a significant decrease in reticular layer thickness within the AHR strategy group, which is significantly greater than in the reference strategy group.
(Reproduced with permission from Sont JK, et al.80)
FIGURE 13.4.6.

Relationship between changes in EG2+ eosinophils and changes in methacholine PC20 (the dose of the inhaled antagonist that provokes a 20% drop in FEV1, the forced expiratory volume in one second) during 2 years of treatment according to the reference and airway hyperresponsiveness (AHR) strategies. The greater the decrease in number of EG2+ eosinophils, the greater the improvement in AHR to inhaled methacholine. EG2, antibody marker for activated esinophils.
(Reproduced with permission from Sont JK, et al.80)
The Haldar et al.77 study also evaluated the effects of anti-IL-5 on features of airway remodeling. Airway wall thickness and airway wall area were evaluated by chest x-ray computed tomography (CT). Changes in these assessments of airway structure were made following a year of treatment in both the mepolizumab and placebo groups. The investigators found a significant reduction in airway wall thickness and total wall area (TA and WA) in the mepolizumab-treated group (Fig. 13.4.7 ).
FIGURE 13.4.7.
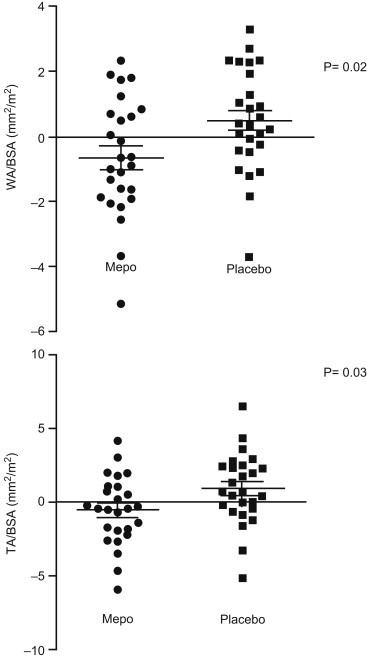
Mean change in CT measured wall area (WA) and total area (TA), corrected for body surface area (BSA) after 12 months of therapy with mepolizumab or placebo. Horizontal bars represent mean change from baseline and error bars +/−SEM.
(Reproduced with permission from Haldar P, et al.77)
Flood-Page et al.31 also hypothesized that a reduction in bronchial wall eosinophils with anti-IL-5 would reduce markers of airway remodeling. Twenty-four mild atopic asthma patients received three monthly infusions of mepolizumab and had bronchial biopsies taken before and after each infusion. In an attempt to determine if bronchial mucosa eosinophilic inflammation was associated with an increased deposition of extracellular matrix (ECM) proteins, researchers found a positive correlation between the thickness, density and expression of the ECM protein tenascin, and bronchial mucosal eosinophil numbers. As noted previously, mepolizumab caused a significant, though incomplete elimination of bronchial mucosal eosinophils. Associated with these changes in eosinophils was a decrease in the thickness and density of tenascin, as well as density of the ECM proteins lumican and procollagen III, in the reticular basement membrane. In addition, there was a decrease in expression of both tenascin and lumican. BAL fluid in the mepolizumab-treated group also had a significant decrease in TGF-β. Not only did this study confirm that the expression of ECM proteins is greater in asthma, it also showed that a reduction in eosinophil numbers is associated with a decrease in ECM protein deposition in the airway.
Thus, the above studies provide convincing evidence for a relationship between the presence of eosinophils and features of airway remodeling. Firstly, an increased ICS dose leads to a decrease in bronchial mucosal eosinophils and airway thickness, as well as a reduction in bronchial hyperresponsiveness.80 Anti-IL-5 studies built on such observations by showing that a decrease in bronchial mucosal eosinophils is associated with a reduction in airway thickness, as assessed by CT scans of the chest.77 Finally, evidence for a decrease in TGF-β and ECM proteins following the use of anti-IL-5 provides further support of the link between bronchial mucosa eosinophilic inflammation and airway remodeling in asthma.31
Conclusion
Since the discovery of the eosinophil in the late 1800s, this cell has been considered to be a characteristic feature and possibly an essential and etiological component of the pathophysiology of asthma. These assumptions were supported by demonstrating tissue eosinophilia in postmortem analyses of the lungs of patients who died in status asthmaticus, as well as by finding parallel relationships between blood eosinophilia and asthma severity. The belief in the central role of eosinophil in asthma was further strengthened by animal models that convincingly found anti-IL-5 strategies to reduce AHR, blood and lung eosinophils, and the LPR to inhaled antigen. These studies identified eosinophils as a central contributor to asthma. Surprisingly, when anti-IL-5 was evaluated in patients with asthma, airway and peripheral eosinophils dramatically diminished, but there were no significant improvements in daily symptoms of asthma or airflow obstruction. What has emerged in the wake of anti-IL-5 studies is perhaps a more informed opinion on two major roles for eosinophils in asthma: as effector cells of airway remodeling and exacerbations.
References
- 1.Wardlaw A.J., Brightling C., Green R., Woltmann G., Pavord I. Eosinophils in asthma and other allergic diseases. Br Med Bull. 2000;56:985–1003. doi: 10.1258/0007142001903490. [DOI] [PubMed] [Google Scholar]
- 2.Gibson P.G., Girgis-Gabardo A., Morris M.M., Mattoli S., Kay J.M., Dolovich J. Cellular characteristics of sputum from patients with asthma and chronic bronchitis. Thorax. 1989;44:693–699. doi: 10.1136/thx.44.9.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green R.H., Brightling C.E., McKenna S., Hargadon B., Parker D., Bradding P. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 4.Uchida D.A., Ackerman S.J., Coyle A.J., Larsen G.L., Weller P.F., Freed J. The effect of human eosinophil granule major basic protein on airway responsiveness in the rat in vivo. A comparison with polycations. Am Rev Respir Dis. 1993;147:982–988. doi: 10.1164/ajrccm/147.4.982. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch J.G., Hirsch B.I. Paul Ehrich and the discovery of the eosinphil. In: Mahmoud A.A.F., Austen K.F., editors. The Eosinophil in Health and Disease. Grune and Stratton; New York: 1980. pp. 2–23. [Google Scholar]
- 6.Horn B.R., Robin E.D., Theodore J., van Kessel A. Total eosinophil counts in the management of bronchial asthma. N Engl J Med. 1975;292:1152–1155. doi: 10.1056/NEJM197505292922204. [DOI] [PubMed] [Google Scholar]
- 7.Bousquet J., Chanez P., Lacoste J.Y., Barneon G., Ghavanian N., Enander I. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 8.Taylor K.J., Luksza A.R. Peripheral blood eosinophil counts and bronchial responsiveness. Thorax. 1987;42:452–456. doi: 10.1136/thx.42.6.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gleich G.J., Adolphson C.R. The eosinophilic leukocyte: structure and function. Adv Immunol. 1986;39:177–253. doi: 10.1016/s0065-2776(08)60351-x. [DOI] [PubMed] [Google Scholar]
- 10.Frigas E., Loegering D.A., Gleich G.J. Cytotoxic effects of the guinea pig eosinophil major basic protein on tracheal epithelium. Lab Invest. 1980;42:35–43. [PubMed] [Google Scholar]
- 11.Frigas E., Loegering D.A., Solley G.O., Farrow G.M., Gleich G.J. Elevated levels of the eosinophil granule major basic protein in the sputum of patients with bronchial asthma. Mayo Clin Proc. 1981;56:345–353. [PubMed] [Google Scholar]
- 12.Gleich G.J., Frigas E., Loegering D.A., Wassom D.L., Steinmuller D. Cytotoxic properties of the eosinophil major basic protein. J Immunol. 1979;123:2925–2927. [PubMed] [Google Scholar]
- 13.Flavahan N.A., Slifman N.R., Gleich G.J., Vanhoutte P.M. Human eosinophil major basic protein causes hyperreactivity of respiratory smooth muscle. Role of the epithelium. Am Rev Respir Dis. 1988;138:685–688. doi: 10.1164/ajrccm/138.3.685. [DOI] [PubMed] [Google Scholar]
- 14.Brofman J.D., White S.R., Blake J.S., Munoz N.M., Gleich G.J., Leff A.R. Epithelial augmentation of trachealis contraction caused by major basic protein of eosinophils. J Appl Physiol. 1989;66:1867–1873. doi: 10.1152/jappl.1989.66.4.1867. [DOI] [PubMed] [Google Scholar]
- 15.Dahl R., Venge P., Fredens K. The eosinophil. In: Barnes P.J., Rodger I., Thomson N., editors. Asthma: Basic Mechanisms and Clinical Management. Academic Press; London: 1988. pp. 115–130. [Google Scholar]
- 16.De Monchy J.G., Kauffman H.F., Venge P., Koeter G.H., Jansen H.M. Bronchoalveolar eosinophilia during allergen-induced late asthmatic reactions. Am Rev Respir Dis. 1985;131:373–376. doi: 10.1164/arrd.1985.131.3.373. [DOI] [PubMed] [Google Scholar]
- 17.Motojima S., Frigas E., Loegering D.A., Gleich G.J. Toxicity of eosinophil cationic proteins for guinea pig tracheal epithelium in vitro. Am Rev Respir Dis. 1989;139:801–805. doi: 10.1164/ajrccm/139.3.801. [DOI] [PubMed] [Google Scholar]
- 18.Robinson D.S., Assoufi B., Durham S.R., Kay A.B. Eosinophil cationic protein (ECP) and eosinophil protein X (EPX) concentrations in serum and bronchial lavage fluid in asthma. Effect of prednisolone treatment. Clin Exp Allergy. 1995;25:1118–1127. doi: 10.1111/j.1365-2222.1995.tb03259.x. [DOI] [PubMed] [Google Scholar]
- 19.Lugosi E., Halmerbauer G., Frischer T., Koller D.Y. Urinary eosinophil protein X in relation to disease activity in childhood asthma. Allergy. 1997;52:584–588. doi: 10.1111/j.1398-9995.1997.tb02605.x. [DOI] [PubMed] [Google Scholar]
- 20.Kroegel C., Yukawa T., Dent G., Venge P., Chung K.F., Barnes P.J. Stimulation of degranulation from human eosinophils by platelet-activating factor. J Immunol. 1989;142:3518–3526. [PubMed] [Google Scholar]
- 21.Lellouchtubiana A., Lefort J., Simon M.T., Pfister A., Vargaftig B.B. Eosinophil Recruitment into Guinea-Pig Lungs after Paf-Acether and Allergen Administration—Modulation by Prostacyclin, Platelet Depletion, and Selective Antagonists. American Review of Respiratory Disease. 1988;137:948–954. doi: 10.1164/ajrccm/137.4.948. [DOI] [PubMed] [Google Scholar]
- 22.Lellouchtubiana A., Lefort J., Pirotzky E., Vargaftig B.B., Pfister A. Ultrastructural Evidence for Extravascular Platelet Recruitment in the Lung Upon Intravenous-Injection of Platelet-Activating Factor (Paf-Acether) to Guinea-Pigs. Brit J Exp Pathol. 1985;66:345–355. [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin A.H.E., Smith L.J., Patterson R. The Bronchoconstrictor Properties of Platelet-Activating-Factor in Humans. American Review of Respiratory Disease. 1987;136:1145–1151. doi: 10.1164/ajrccm/136.5.1145. [DOI] [PubMed] [Google Scholar]
- 24.Cuss F.M., Dixon C.M., Barnes P.J. Effects of inhaled platelet activating factor on pulmonary function and bronchial responsiveness in man. Lancet. 1986;2:189–192. doi: 10.1016/s0140-6736(86)92489-x. [DOI] [PubMed] [Google Scholar]
- 25.Dahlen S.E., Hedqvist P., Hammarstrom S., Samuelsson B. Leukotrienes are potent constrictors of human bronchi. Nature. 1980;288:484–486. doi: 10.1038/288484a0. [DOI] [PubMed] [Google Scholar]
- 26.Isono T., Koshihara Y., Murota S., Fukuda Y., Furukawa S. Measurement of immunoreactive leukotriene C4 in blood of asthmatic children. Biochem Biophys Res Commun. 1985;130:486–492. doi: 10.1016/0006-291x(85)90443-7. [DOI] [PubMed] [Google Scholar]
- 27.Garland A., Necheles J., White S.R., Neeley S.P., Leff A.R., Carson S.S. Activated eosinophils elicit substance P release from cultured dorsal root ganglion neurons. Am J Physiol. 1997;273:L1096–L1102. doi: 10.1152/ajplung.1997.273.5.L1096. [DOI] [PubMed] [Google Scholar]
- 28.Chanez P., Dent G., Yukawa T., Barnes P.J., Chung K.F. Generation of oxygen free radicals from blood eosinophils from asthma patients after stimulation with PAF or phorbol ester. Eur Respir J. 1990;3:1002–1007. [PubMed] [Google Scholar]
- 29.Henricks P.A., Nijkamp F.P. Reactive oxygen species as mediators in asthma. Pulm Pharmacol Ther. 2001;14:409–420. doi: 10.1006/pupt.2001.0319. [DOI] [PubMed] [Google Scholar]
- 30.Minshall E.M., Leung D.Y., Martin R.J., Song Y.L., Cameron L., Ernst P. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1997;17:326–333. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- 31.Flood-Page P., Menzies-Gow A., Phipps S., Ying S., Wangoo A., Ludwig M.S. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weller P.F., Rand T.H., Barrett T., Elovic A., Wong D.T., Finberg R.W. Accessory cell function of human eosinophils. HLA-DR-dependent, MHC-restricted antigen-presentation and IL-1 alpha expression. J Immunol. 1993;150:2554–2562. [PubMed] [Google Scholar]
- 33.Kita H., Ohnishi T., Okubo Y., Weiler D., Abrams J.S., Gleich G.J. Granulocyte/macrophage colony-stimulating factor and interleukin 3 release from human peripheral blood eosinophils and neutrophils. J Exp Med. 1991;174:745–748. doi: 10.1084/jem.174.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moqbel R., Ying S., Barkans J., Newman T.M., Kimmitt P., Wakelin M. Identification of messenger RNA for IL-4 in human eosinophils with granule localization and release of the translated product. J Immunol. 1995;155:4939–4947. [PubMed] [Google Scholar]
- 35.Broide D.H., Paine M.M., Firestein G.S. Eosinophils express interleukin 5 and granulocyte macrophage-colony-stimulating factor mRNA at sites of allergic inflammation in asthmatics. J Clin Invest. 1992;90:1414–1424. doi: 10.1172/JCI116008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kay A.B., Barata L., Meng Q., Durham S.R., Ying S. Eosinophils and eosinophil-associated cytokines in allergic inflammation. Int Arch Allergy Immunol. 1997;113:196–199. doi: 10.1159/000237545. [DOI] [PubMed] [Google Scholar]
- 37.Kirby J.G., Hargreave F.E., Gleich G.J., O’Byrne P.M. Bronchoalveolar cell profiles of asthmatic and nonasthmatic subjects. Am Rev Respir Dis. 1987;136:379–383. doi: 10.1164/ajrccm/136.2.379. [DOI] [PubMed] [Google Scholar]
- 38.Wardlaw D.F., Dunnett S., Gleich G.J. Eosinophils and mast cells in bronchoalveolar lavage and mild asthma: relationship to bronchial hyperreactivity. Am Rev Respir Dis. 1988;137:62. doi: 10.1164/ajrccm/137.1.62. [DOI] [PubMed] [Google Scholar]
- 39.Adelroth E., Rosenhall L., Johansson S.A., Linden M., Venge P. Inflammatory cells and eosinophilic activity in asthmatics investigated by bronchoalveolar lavage. The effects of antiasthmatic treatment with budesonide or terbutaline. American Review of Respiratory Disease. 1990;142:91–99. doi: 10.1164/ajrccm/142.1.91. [DOI] [PubMed] [Google Scholar]
- 40.Earle B.V. Fatal bronchial asthma; a series of fifteen cases with a review of the literature. Thorax. 1953;8:195–206. doi: 10.1136/thx.8.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kroegel C. The role of eosinophils in asthma. Lung. 1990;168(Suppl):5–17. doi: 10.1007/BF02718107. [DOI] [PubMed] [Google Scholar]
- 42.Metzger W.J., Nugent K., Richerson H.B., Moseley P., Lakin R., Zavala D. Methods for Bronchoalveolar Lavage in Asthmatic-Patients Following Bronchoprovocation and Local Antigen Challenge. Chest. 1985;87:S16–S19. doi: 10.1378/chest.87.1_supplement.16s. [DOI] [PubMed] [Google Scholar]
- 43.Frigas E., Gleich G.J. The eosinophil and the pathophysiology of asthma. J Allergy Clin Immunol. 1986;77:527–537. doi: 10.1016/0091-6749(86)90341-6. [DOI] [PubMed] [Google Scholar]
- 44.Ohashi Y., Motojima S., Fukuda T., Makino S. Airway hyperresponsiveness, increased intracellular spaces of bronchial epithelium, and increased infiltration of eosinophils and lymphocytes in bronchial mucosa in asthma. American Review of Respiratory Disease. 1992;145:1469–1476. doi: 10.1164/ajrccm/145.6.1469. [DOI] [PubMed] [Google Scholar]
- 45.Humbles A.A., Conroy D.M., Marleau S., Rankin S.M., Palframan R.T., Proudfoot A.E. Kinetics of eotaxin generation and its relationship to eosinophil accumulation in allergic airways disease: analysis in a guinea pig model in vivo. J Exp Med. 1997;186:601–612. doi: 10.1084/jem.186.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunn C.J., Elliott G.A., Oostveen J.A., Richards I.M. Development of a prolonged eosinophil-rich inflammatory leukocyte infiltration in the guinea-pig asthmatic response to ovalbumin inhalation. Am Rev Respir Dis. 1988;137:541–547. doi: 10.1164/ajrccm/137.3.541. [DOI] [PubMed] [Google Scholar]
- 47.Gulbenkian A.R., Fernandez X., Kreutner W., Minnicozzi M., Watnick A.S., Kung T. Anaphylactic challenge causes eosinophil accumulation in bronchoalveolar lavage fluid of guinea pigs. Modulation by betamethasone, phenidone, indomethacin, WEB 2086, and a novel antiallergy agent, SCH 37224. Am Rev Respir Dis. 1990;142:680–685. doi: 10.1164/ajrccm/142.3.680. [DOI] [PubMed] [Google Scholar]
- 48.Wenzel S.E. Eosinophils in asthma—closing the loop or opening the door? N Engl J Med. 2009;360:1026–1028. doi: 10.1056/NEJMe0900334. [DOI] [PubMed] [Google Scholar]
- 49.Cockcroft D.W., Murdock K.Y. Comparative effects of inhaled salbutamol, sodium cromoglycate, and beclomethasone dipropionate on allergen-induced early asthmatic responses, late asthmatic responses, and increased bronchial responsiveness to histamine. J Allergy Clin Immunol. 1987;79:734–740. doi: 10.1016/0091-6749(87)90204-1. [DOI] [PubMed] [Google Scholar]
- 50.Kidney J.C., Boulet L.P., Hargreave F.E., Deschesnes F., Swystun V.A., O’Byrne P.M. Evaluation of single-dose inhaled corticosteroid activity with an allergen challenge model. J Allergy Clin Immunol. 1997;100:65–70. doi: 10.1016/s0091-6749(97)70196-9. [DOI] [PubMed] [Google Scholar]
- 51.Gauvreau G.M., Wood L.J., Sehmi R., Watson R.M., Dorman S.C., Schleimer R.P. The effects of inhaled budesonide on circulating eosinophil progenitors and their expression of cytokines after allergen challenge in subjects with atopic asthma. Am J Respir Crit Care Med. 2000;162:2139–2144. doi: 10.1164/ajrccm.162.6.2001120. [DOI] [PubMed] [Google Scholar]
- 52.Sedgwick J.B., Calhoun W.J., Gleich G.J., Kita H., Abrams J.S., Schwartz L.B. Immediate and late airway response of allergic rhinitis patients to segmental antigen challenge. Am Rev Respir Dis. 1991;144:1274–1281. doi: 10.1164/ajrccm/144.6.1274. [DOI] [PubMed] [Google Scholar]
- 53.Calhoun W.J., Dick E.C., Schwartz L.B., Busse W.W. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest. 1994;94:2200–2208. doi: 10.1172/JCI117581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gern J.E. Mechanisms of virus-induced asthma. J Pediatr. 2003;142:S9–13. doi: 10.1067/mpd.2003.20. discussion S13–14. [DOI] [PubMed] [Google Scholar]
- 55.Woolley K.L., Gibson P.G., Carty K., Wilson A.J., Twaddell S.H., Woolley M.J. Eosinophil apoptosis and the resolution of airway inflammation in asthma. Am J Respir Crit Care Med. 1996;154:237–243. doi: 10.1164/ajrccm.154.1.8680686. [DOI] [PubMed] [Google Scholar]
- 56.Saeed W., Badar A., Hussain M.M., Aslam M. Eosinophils and eosinophil products in asthma. J Ayub Med Coll Abbottabad. 2002;14:49–55. [PubMed] [Google Scholar]
- 57.Evans P.M., O’Connor B.J., Fuller R.W., Barnes P.J., Chung K.F. Effect of inhaled corticosteroids on peripheral blood eosinophil counts and density profiles in asthma. J Allergy Clin Immunol. 1993;91:643–650. doi: 10.1016/0091-6749(93)90270-p. [DOI] [PubMed] [Google Scholar]
- 58.Gleich G.J., Motojima S., Frigas E., Kephart G.M., Fujisawa T., Kravis L.P. The eosinophilic leukocyte and the pathology of fatal bronchial asthma: evidence for pathologic heterogeneity. J Allergy Clin Immunol. 1987;80:412–415. doi: 10.1016/0091-6749(87)90063-7. [DOI] [PubMed] [Google Scholar]
- 59.Koch-Weser J. Beta adrenergic blockade and circulating eosinophils. Arch Intern Med. 1968;121:255–258. [PubMed] [Google Scholar]
- 60.Ohman J.L., Jr., Lawrence M., Lowell F.C. Effect of propranolol on the isoproterenol, responses of cortisol, isoproterenol, and aminophylline. J Allergy Clin Immunol. 1972;50:151–156. doi: 10.1016/0091-6749(72)90046-2. [DOI] [PubMed] [Google Scholar]
- 61.Miranda C., Busacker A., Balzar S., Trudeau J., Wenzel S.E. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol. 2004;113:101–108. doi: 10.1016/j.jaci.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 62.Haldar P., Pavord I.D., Shaw D.E., Berry M.A., Thomas M., Brightling C.E. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178:218–224. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Woodruff P.G., Modrek B., Choy D.F., Jia G., Abbas A.R., Ellwanger A. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanderson C.J. Interleukin-5, eosinophils, and disease. Blood. 1992;79:3101–3109. [PubMed] [Google Scholar]
- 65.Walsh G.M., Hartnell A., Wardlaw A.J., Kurihara K., Sanderson C.J., Kay A.B. IL-5 enhances the in vitro adhesion of human eosinophils, but not neutrophils, in a leucocyte integrin (CD11/18)-dependent manner. Immunology. 1990;71:258–265. [PMC free article] [PubMed] [Google Scholar]
- 66.van der Veen M.J., Van Neerven R.J., De Jong E.C., Aalberse R.C., Jansen H.M., van der Zee J.S. The late asthmatic response is associated with baseline allergen-specific proliferative responsiveness of peripheral T lymphocytes in vitro and serum interleukin-5. Clin Exp Allergy. 1999;29:217–227. doi: 10.1046/j.1365-2222.1999.00466.x. [DOI] [PubMed] [Google Scholar]
- 67.Shi H.Z., Xiao C.Q., Zhong D., Quin S.M., Liu Y., Liang G.R. Effect of inhaled interleukin-5 on airway hyperreactivity and eosinophilia in asthmatics. Am J Respir Crit Care Med. 1998;157:204–209. doi: 10.1164/ajrccm.157.1.9703027. [DOI] [PubMed] [Google Scholar]
- 68.Shi H., Qin S., Huang G., Chen Y., Xiao C., Xu H. Infiltration of eosinophils into the asthmatic airways caused by interleukin 5. Am J Respir Cell Mol Biol. 1997;16:220–224. doi: 10.1165/ajrcmb.16.3.9070605. [DOI] [PubMed] [Google Scholar]
- 69.Foster P.S., Hogan S.P., Ramsay A.J., Matthaei K.I., Young I.G. IL-5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson D., Hamid Q., Ying S., Bentley A., Assoufi B., Durham S. Prednisolone treatment in asthma is associated with modulation of bronchoalveolar lavage cell interleukin-4, interleukin-5, and interferon-gamma cytokine gene expression. Am Rev Respir Dis. 1993;148:401–406. doi: 10.1164/ajrccm/148.2.401. [DOI] [PubMed] [Google Scholar]
- 71.Van Oosterhout A.J., Ladenius A.R., Savelkoul H.F., Van Ark I., Delsman K.C., Nijkamp F.P. Effect of anti-IL-5 and IL-5 on airway hyperreactivity and eosinophils in guinea pigs. Am Rev Respir Dis. 1993;147:548–552. doi: 10.1164/ajrccm/147.3.548. [DOI] [PubMed] [Google Scholar]
- 72.Akutsu I., Kojima T., Kariyone A., Fukuda T., Makino S., Takatsu K. Antibody against interleukin-5 prevents antigen-induced eosinophil infiltration and bronchial hyperreactivity in the guinea pig airways. Immunol Lett. 1995;45:109–116. doi: 10.1016/0165-2478(94)00241-i. [DOI] [PubMed] [Google Scholar]
- 73.Leckie M.J., ten Brinke A., Khan J., Diamant Z., O’Connor B.J., Walls C.M. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 74.Flood-Page P.T., Menzies-Gow A.N., Kay A.B., Robinson D.S. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 75.Bentley A.M., Hamid Q., Robinson D.S., Schotman E., Meng Q., Assoufi B. Prednisolone treatment in asthma. Reduction in the numbers of eosinophils, T cells, tryptase-only positive mast cells, and modulation of IL-4, IL-5, and interferon-gamma cytokine gene expression within the bronchial mucosa. Am J Respir Crit Care Med. 1996;153:551–556. doi: 10.1164/ajrccm.153.2.8564096. [DOI] [PubMed] [Google Scholar]
- 76.Rothenberg M.E., Klion A.D., Roufosse F.E., Kahn J.E., Weller P.F., Simon H.U. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med. 2008;358:1215–1228. doi: 10.1056/NEJMoa070812. [DOI] [PubMed] [Google Scholar]
- 77.Haldar P., Brightling C.E., Hargadon B., Gupta S., Monteiro W., Sousa A. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nair P., Pizzichini M.M., Kjarsgaard M., Inman M.D., Efthimiadis A., Pizzichini E. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 79.Humbles A.A., Lloyd C.M., McMillan S.J., Friend D.S., Xanthou G., McKenna E.E. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 80.Sont J.K., Willems L.N., Bel E.H., Van Krieken J.H., Vandenbroucke J.P., Sterk P.J. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med. 1999;159:1043–1051. doi: 10.1164/ajrccm.159.4.9806052. [DOI] [PubMed] [Google Scholar]
Chapter 13.5. Eosinophil-Targeted Treatment of Asthma
Bronchitis is a central component of airway diseases. However, this is not measured in routine clinical practice. Quantitative cell counts in sputum is currently the most reliable and comprehensive noninvasive assessment of cellular inflammation, and particularly of eosinophilic inflammation, in the airways. Treatment strategies that decrease eosinophil numbers in sputum, whether nonspecific using corticosteroids or specific using anti-interleukin-5 (anti-IL-5) monoclonal antibodies, decrease asthma exacerbations, improve asthma control and lung function, and improve quality of life for patients. Studies using anti-IL5 strategies have confirmed the importance of identifying an eosinophilic phenotype of asthma, the role of eosinophils in asthma pathophysiology, and the activity of luminal eosinophils in disease severity. There is an urgent need to translate these principles into clinical practice and to incorporate the use of sputum cell counts in the management of severe obstructive airway diseases.
Introduction
The role of the eosinophils as key players in the pathophysiology of asthma has been debated, despite evidence that the cells are present and activated in the airway lumen and tissue1 of patients with current asthma; are increased in number when asthma is uncontrolled2 or severe3 and decreased when asthma is controlled4; and treatment strategies that aim to control airway eosinophilia are significantly more effective and less expensive in improving asthma control5., 6. and decreasing asthma exacerbations compared to guideline-based clinical strategies.
Cynicism was fueled by observations that in murine models of allergic sensitization, airway hyperresponsiveness could be induced without eosinophils.7 Skepticism grew stronger when therapy using monoclonal antibodies against interleukin-5 (IL-5), which has no known clinically relevant biologic activity other than targeting eosinophils, failed to demonstrate improvement in asthma outcomes despite decreasing airway and blood eosinophil numbers.8 The molecule did not reduce allergen-induced airway constriction or hyperresponsiveness, airflow limitation, exacerbations, or symptoms. The likely explanations for this apparent paradox are inappropriate methodology, inadequate sample size,9 or an inadequate reduction in bronchial mucosal eosinophil numbers.10
This subchapter will describe the clinical studies that demonstrated an improvement in asthma control using treatment strategies that aimed to normalize sputum eosinophil count using corticosteroids; to evaluate critically the clinical trials that failed to demonstrate an improvement in asthma using monoclonal antibodies directed against IL-5; and to present evidence from a prospective audit of clinical outcomes of patients managed by normalizing sputum cell counts.
Eosinophil-Based Treatment Strategies Using Corticosteroids
Airway eosinophilia can be reliably and relatively noninvasively assessed in sputum.11 In clinical practice, approximately 30% of patients with asthma attending a tertiary clinic have eosinophilic bronchitis.12 More severe asthma and more severe airflow limitation are associated with more intense sputum eosinophilia.13 Two studies in adults and one study in children have evaluated the outcomes of titrating anti-inflammatory treatment with the intention of normalizing eosinophils in sputum. The first single center, 1-year trial that examined the effect of treating asthma to reduce eosinophils to 2% resulted in a significant reduction of severe exacerbations compared with a control group treated without sputum eosinophil counts.5 The large number of exacerbations and their severity was probably a result of the policy at the time to reduce corticosteroid use further if control was maintained for 2 months. The second trial6 was a multicenter trial conducted over 2 years, and differed in that the minimum dose of corticosteroid to maintain sputum eosinophils at 3% was determined first and then maintained for the duration of the study. Exacerbations were few and mild compared with the first study and were reduced by about 50% compared with the group treated with the same best-guideline approach to treatment without sputum cell counts. The active treatment reduced eosinophilic exacerbations but had no effect on neutrophilic exacerbations, which were regarded to be probably of viral cause. The benefits in both studies were achieved without any increase in corticosteroid dose over that required by the control group. In contrast, a similar study in children showed a nonstatistically significant effect on reducing exacerbations using a sputum strategy that aimed to keep eosinophil levels to below 2.5%.14 The modest benefit was most likely due to the inadequate control of eosinophils in the treatment arm that was probably related to the inadequate dose of inhaled corticosteroids allowed in the study. The effectiveness of using sputum eosinophils as a marker to decrease exacerbations in adults and children with moderate to severe asthma was recently confirmed in a systematic review and meta-analysis.15 A critical review of the recently published clinical trial literature reveals that the reduction in exacerbation reported in all the most recent large clinical trials for either asthma or chronic obstructive pulmonary disease (COPD) for any new medication compared to placebo is significantly less than those reported for strategies employing the judicious use of currently available medications guided by cell counts in sputum (Table 13.5.1 ). In addition to a significant reduction of exacerbations at a reduced cost,16 the adverse consequences of new therapies and suboptimal treatment may also be avoided.
TABLE 13.5.1.
Relative Risk Reduction of Exacerbations for Various Interventions for Asthma and for Chronic Obstructive Pulmonary Disease
| Asthma | COPD | ||
|---|---|---|---|
| Intervention | RRR | Intervention | RRR |
| ICS + LABA (AJRCCM 2004;170:836–44) |
10–15% | ICS + LABA (NEJM 2007;356:775–89) |
25% |
| Tiotropium (NEJM 2010;363:1715–26) |
~5% | Tiotropium (NEJM 2008;359:1543–54) |
14% |
| Omalizumab (Ann Intern Med 2011;154:573–82) |
25% | Tiotropium + ICS/LABA (NEJM 2011;364:1093–1103) |
27% |
| ICS + Montelukast (BMJ 2003;327:891–7) |
~1% | Roflumilast (Lancet 2009;374:695–703) |
16–21% |
| Thermoplasty (AJRCCM 2010;181:116–24) |
22% | Community-based multi-disciplinary programs (Thorax 2010;65:7–13) |
∼ |
| Sputum strategy (ERJ 2006;27:483–94) |
49% | Sputum strategy (ERJ 2007;29:906–13) |
62% |
COPD, chronic obstructive pulmonary disease; ICS, inhaled corticosteroid; LABA, long-acting β agonist; RRR, relative risk reduction.
Anti-Interleukin-5 Clinical Trials
The beneficial effects of corticosteroids are not limited to decreasing eosinophil numbers in the airways.17 They can also reduce the numbers of other cells, such as lymphocytes and mast cells, and decrease some markers of remodeling. Thus, it is not possible to conclude definitively the pathobiological role of eosinophils in asthma from those studies. Definitive proof would be obtained by reducing eosinophil numbers in the airway using treatments that directly target the eosinophils. Recently, the availability of monoclonal antibodies directed against IL-5 has provided us with the opportunity to examine this question. In two recently published randomized controlled trials (RCTs) on the effect of mepolizumab18., 19. and a clinical trial on the effect of reslizumab,20 these drugs reduced sputum eosinophils numbers to almost zero. This reduction was associated with a reduction of exacerbations compared with the placebo group in the first mepolizumab study18 and a prednisone-sparing effect and improvement in clinical outcomes in a small sample number in the second.19 In the larger reslizumab clinical trial,20 the reduction in sputum eosinophils was associated with an improvement in the forced expiratory volume in one second (FEV1) and in asthma controls over a 5-month period in patients with moderate to severe asthma. The results of these three studies contrasted with the negative results of five other trials, in which the effect of the antieosinophil drug was not examined in patients with asthma and current sputum eosinophilia. In two of the five studies that measured sputum eosinophils and in the three RCTs, the greater the certainty that an increase in eosinophils was persistent, the greater the success of the treatment (Table 13.5.2 ).
TABLE 13.5.2.
Response to Anti-Interleukin-5 and the Eosinophil Phenotype
| Study | Intervention | Sputum Eosinophils at Study Entry | Success |
|---|---|---|---|
| Flood-Page et al. (AJRCCM 2007; 176: 1062–71) | Mepolizumab | 5% of patients had >3% eosinophils | X |
| Kips et al. (AJRCCM 2003; 167: 1655–9) | Reslizumab | ~30% of patients had >3% eosinophils | X |
| Haldar et al. (NEJM 2009; 360: 973–84) | Mepolizumab | all had >3% on one occasion in 2 years | √ |
| Castro et al. (AJRCCM 2011; Aug 18 epub) | Reslizumab | all had >3% at randomization | √√ |
| Nair et al. (NEJM 2009; 360: 985–93) | Mepolizumab | All had >3% on ≥3 occasions | √√√ |
Observational Study of Normalizing Sputum Eosinophils
Clinical outcomes are significantly improved when patients who require daily prednisone are monitored using sputum cell counts. Sixty-three patients with asthma (36 men; mean age, 52 years; mean BMI, 29.1) were followed for a median period of 7 years (range, 0.25–26 year).21 Thirty-seven patients had associated chronic airflow limitation (postbronchodilator FEV1/vital capacity <70%). Twenty had never smoked. Forty-two percent were nonatopic. Significant comorbidities included gastroesophageal reflux disease (70%), nasal polyps and sinusitis (65%), and sensitivity to nonsteroidal anti-inflammatory drugs (28%). Ethmoid and sphenoid sinusitis were the most important predictors of persistent airway eosinophilia.22 At the time of their initial assessment, the majority of the patients were not on daily prednisone (median daily dose, 0 mg; sputum eosinophils: mean, 18.8%; median, 5.3%; minimum, 0%; maximum, 84%). Monitoring with the aim of keeping sputum eosinophils at <2% resulted in higher doses of corticosteroids (median daily dose of prednisone was 10 mg and for inhaled corticosteroids was 1000 μg of fluticasone equivalent), and this was associated with predictable significant adverse effects. Over the period of follow-up, despite decreasing the eosinophilic exacerbations to 0.2 years/patient, there were 22 noneosinophilic neutrophilic exacerbations. Overall, there was no significant loss of lung function over the period of follow-up (mean decrease in FEV1, 35 mL/year).
Other Antieosinophil Treatment Strategies
Corticosteroids are very effective in reducing eosinophil numbers and activation in the airways of most patients with asthma. Therefore novel treatment strategies such as anti-IL5 should probably be reserved for patients who require high doses of inhaled or regular ingested corticosteroids to control their airway eosinophilia and asthma. Although a larger number of antisense molecules, monoclonal antibodies, and small molecules are currently being evaluated to target a number of relevant cytokines or chemoattractants involved in eosinophil recruitment into the airway, such as eotaxin/ C-C motif chemokine 11 (CCL11), IL-4, and IL-13,23 none of them have yet been demonstrated to be effective in suppressing an airway eosinophilia that persists despite being treated with prednisone. These are currently being investigated.
Conclusions
These studies illustrate a number of important principles. Firstly, they confirm that eosinophils are important in the pathophysiology of asthma. Second, since the nature of airway inflammation can change over time in the same patient,24 antieosinophil treatment will be effective only when eosinophils are present in the airway. Third, luminal eosinophils represent biologically active cells. Fourth, clinical trials with small numbers of carefully characterized patients can demonstrate biologically relevant mechanisms, whereas large studies have not. Fifth, monitoring of exhaled nitric oxide is not effective in directing antieosinophil therapy in patients with severe asthma and airway eosinophilia.25 Finally, since sputum cell count-based treatment strategies are more effective than any other currently available medications for the treatment of asthma or COPD, we are doing our patients with severe asthma and COPD a disservice by delaying the introduction of sputum cell counts in clinical practice.
Acknowledgements
Dr. Nair is supported by a Canada Research Chair in Airway Inflammometry. This chapter is dedicated to the fond memory of Professor Freddy Hargreave who tirelessly argued for the implementation of measurement of bronchitis by sputum examination in clinical practice.
References
- 1.Bousquet J., Chanez P., Lacoste J.Y., Barnéon G., Ghavanian N., Enander I. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 2.Leuppi J.D., Salome C.M., Jenkins C.R., Anderson S.D., Xuan W., Marks G.B. Predictive markers of asthma exacerbation during stepwise dose reduction of inhaled corticosteroids. Am J Respir Crit Care Med. 2001;163:406–412. doi: 10.1164/ajrccm.163.2.9912091. [DOI] [PubMed] [Google Scholar]
- 3.van Veen I.H., ten Brinke A., Gauw S.A., Sterk P.J., Rabe K.F., Bel E.H. Consistency of sputum eosinophilia in difficult-to-treat asthma: a 5-year follow-up study. J Allergy Clin Immunol. 2009;124:615–617. doi: 10.1016/j.jaci.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 4.Lemière C., Ernst P., Olivenstein R., Yamauchi Y., Govindaraju K., Ludwig M.S. Airway inflammation assessed by invasive and noninvasive means in severe asthma: eosinophilic and noneosinophilic phenotypes. J Allergy Clin Immunol. 2006;118:1033–1039. doi: 10.1016/j.jaci.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Green R.H., Brightling C.E., McKenna S., Hargadon B., Parker D., Bradding P. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 6.Jayaram L., Pizzichini M.M., Cook R.J., Boulet L.P., Lemière C., Pizzichini E. Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur Respir J. 2006;27:483–494. doi: 10.1183/09031936.06.00137704. [DOI] [PubMed] [Google Scholar]
- 7.Wills-Karp M., Karp C.L. Eosinophils in asthma: remodeling a tangled tale. Science. 2004;305:1726–1729. doi: 10.1126/science.1104134. [DOI] [PubMed] [Google Scholar]
- 8.Leckie M.J., ten Brinke A., Khan J., Diamant Z., O’Connor B.J., Walls C.M. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 9.O’Byrne P.M., Inman M.D., Parameswaran K. The trials and tribulations of IL-5, eosinophils, and allergic asthma. J Allergy Clin Immunol. 2001;108:503–508. doi: 10.1067/mai.2001.119149. [DOI] [PubMed] [Google Scholar]
- 10.Flood-Page P.T., Menzies-Gow A.N., Kay A.B., Robinson D.S. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 11.Nair P., Hargreave F.E. Measuring bronchitis in airway diseases: clinical implementation and application. Chest. 2010;138(2 Suppl):38S–43S. doi: 10.1378/chest.10-0094. [DOI] [PubMed] [Google Scholar]
- 12.D’silva L., Hassan N., Wang H.Y., Kjarsgaard M., Efthimiadis A., Hargreave F.E. Heterogeneity of bronchitis in airway diseases in tertiary care clinical practice. Can Respir J. 2011;18:144–148. doi: 10.1155/2011/430317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ten Brinke A., Zwinderman A.H., Sterk P.J., Rabe K.F., Bel E.H. Factors associated with persistent airflow limitation in severe asthma. Am J Respir Crit Care Med. 2001;164:744–748. doi: 10.1164/ajrccm.164.5.2011026. [DOI] [PubMed] [Google Scholar]
- 14.Fleming L., Wilson N., Regamey N., Bush A. Use of sputum eosinophil counts to guide management in children with severe asthma. Thorax. 2011;67(3):193–198. doi: 10.1136/thx.2010.156836. [DOI] [PubMed] [Google Scholar]
- 15.Petsky H.L., Cates C.J., Lasserson T.J., Li A.M., Turner C., Kynaston J.A. A systematic review and meta-analysis: tailoring asthma treatment on eosinophilic markers (exhaled nitric oxide or sputum eosinophils) Thorax. 2012 Mar;67(3):199–208. doi: 10.1136/thx.2010.135574. [DOI] [PubMed] [Google Scholar]
- 16.D’silva L., Gafni A., Thabane L., Jayaram L., Hassack P., Hargreave F.E. Cost analysis of monitoring asthma treatment using sputum cell counts. Can Respir J. 2008;15:370–374. doi: 10.1155/2008/946735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakir J., Loubaki L., Laviolette M., Milot J., Biardel S., Jayaram L. Monitoring sputum eosinophils in mucosal inflammation and remodelling: a pilot study. Eur Respir J. 2010;35:48–53. doi: 10.1183/09031936.00130008. [DOI] [PubMed] [Google Scholar]
- 18.Haldar P., Brightling C.E., Hargadon B., Gupta S., Monteiro W., Sousa A. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair P., Pizzichini M.M., Kjarsgaard M., Inman M.D., Efthimiadis A., Pizzichini E. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 20.Castro M., Mathur S., Hargreave F., Boulet L.P., Xie F., Young J. for the Res-5–0010 Study Group. Reslizumab for Poorly Controlled, Eosinophilic Asthma: A Randomized, Placebo-Controlled Study. Am J Respir Crit Care Med. 2011 Nov 15;184(10):1125–1132. doi: 10.1164/rccm.201103-0396OC. [DOI] [PubMed] [Google Scholar]
- 21.Aziz-ur-Rehman A., Kjarsgaard M., Efthimiadis A., Hargreave F.E., Nair P. An audit of prednisone-dependent asthma [abstract] Am J Respir Crit Care Med. 2008;177(suppl):A107. [Google Scholar]
- 22.Lui B.T., Boylan C., Aziz-Ur-Rehman A., Sommer D.D., Nair P. Paranasal sinus disease and sputum eosinophilia in prednisone dependent asthma. J Otolaryngol Head Neck Surg. 2010;39:703–709. [PubMed] [Google Scholar]
- 23.Bochner B.S., Gleich G.J. What targeting eosinophils has taught us about their role in diseases. J Allergy Clin Immunol. 2010;126:16–25. doi: 10.1016/j.jaci.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’silva L., Cook R.J., Allen C.J., Hargreave F.E., Nair P. Changing pattern of sputum cell counts during successive exacerbations of airway disease. Respir Med. 2007;101:2217–2220. doi: 10.1016/j.rmed.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 25.Nair P., Kjarsgaard M., Armstrong S., Efthimiadis A., O’Byrne P.M., Hargreave F.E. Nitric oxide in exhaled breath is poorly correlated to sputum eosinophils in patients with prednisone-dependent asthma. J Allergy Clin Immunol. 2010;126:404–406. doi: 10.1016/j.jaci.2010.05.032. [DOI] [PubMed] [Google Scholar]
Chapter 13.6. Eosinophils and Hemopoietic Processes in Allergic Asthma
Gail M. Gauvreau, Judah A. Denburg
Eosinophilia is a hallmark of a number of allergic disorders, including asthma. Activation of selective hemopoietic processes is associated with the onset and maintenance of allergic inflammation in atopic adults.
Following allergen exposure in the airways, the subsequent appearance of eosinophils in the circulation involves processes in the bone marrow leading to efflux of both mature and immature eosinophil populations into the circulation, with differentiation and proliferation of eosinophil lineage-committed hemopoietic progenitors occurring at tissue sites. The accumulation of eosinophils within the airway tissue occurs as a response to locally generated signals that induce a number of events, including further initiation of production of eosinophils in the bone marrow, continued migration of eosinophils and their progenitors, and increased survival and effector functions of these cells. This subchapter will summarize mechanisms leading to the accumulation of eosinophils and their progenitors in the airways in allergic asthma, and review therapies that target these cells and hemopoietic processes, as measures designed to control allergen-induced eosinophilic airway inflammation.
Eosinophils in Asthma: An Overview
Eosinophils, a prominent feature of asthma, are found in increased numbers in the circulation and airways in relation to the severity of asthma.1., 2. Inhaled allergen challenge in asthmatic subjects results in the appearance and accumulation of mature and immature eosinophils in the bone marrow, blood,3 and airways.4., 5., 6. The kinetics of eosinophilia are compartment-specific (Fig. 13.6.1 ),4., 7., 8. and the number of eosinophils correlates with the severity of the late asthmatic reaction.9 This will be covered in detail elsewhere in this book.
FIGURE 13.6.1.
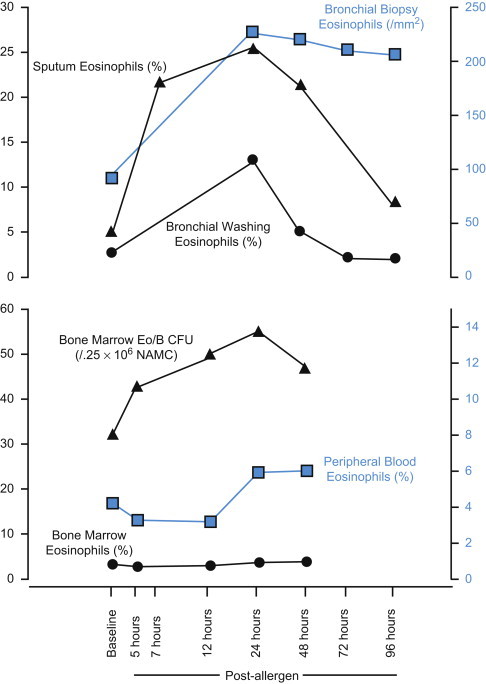
Kinetics of allergen-induced eosinophilia in airways, blood, and bone marrow.
A rapid increase in airway eosinophils postallergen is mirrored by a decrease in mature eosinophils from the bone marrow and blood as these cells migrate to the airways, and an increase in eosinophil progenitors to replace the mature cells.4., 7., 8. Eo/B CFU, eosinophil/basophil colony-forming units.
Eosinophils are terminally differentiated myeloid leukocytes that migrate to tissues as effector cells in a number of inflammatory processes, including allergic diseases and helminth infections.10 The migration and accumulation of eosinophils is highly regulated via signaling of cytokines and chemokines through cell-surface receptors, and by induction of adhesion molecule expression.11 Since allergic asthma is primarily a T-helper type 2 (Th2)-mediated disease, it is not surprising that cytokines driving eosinophilia are Th2 cell products: specifically, granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), and interleukin-5 (IL-5), which signal through specific high-affinity cell-surface receptors linked to a common β-chain—all of which can act as eosinophil growth factors that promote formation of eosinophil/basophil (Eo/B) colony-forming units (CFU) in functional assays.12 The common β-chain is especially important for Eo/B proliferation, production of cytokines from eosinophils, and eosinophil migration to effector sites.13 Of the three Eo/B differentiation-inducing cytokines, IL-5 is necessary for mobilization of eosinophil progenitors from the bone marrow and their terminal differentiation.14 Other Th2 cytokines, such as IL-4 and IL-13, are known to regulate transmigration of eosinophils from the vascular bed into the tissue compartments by augmenting expression of adhesion molecules on the endothelium15 and inducing expression of potent eosinophil chemokines, such as eotaxin/C-C motif chemokine 11 (CCL11) and RANTES/CCL5 in the airways.16., 17. IL-9 has also been shown to play a supportive role in both mast cell and Eo/B differentiation.18., 19. Once in the inflamed tissue, eosinophils contribute to the manifestation of symptoms through release of granule proteins and proinflammatory mediators.10
Hemopoietic Activity in Allergic Asthma
Hemopoietic progenitors, which are found in the circulation under steady-state and disease conditions, exist in the bone marrow at various stages of lineage commitment; in the latter compartment, these progenitors can be allowed or selected to differentiate in specific directions in response to various ambient stimuli, such as hemopoietic cytokines engaging specific cell-surface receptors. This hemopoietic activity occurs in the bone marrow proper under the influence of stromal cells and microvascular endothelial cells, and in tissues, also in response to epithelial cells. Each of these resident cell populations can respond to inflammatory stimuli by increasing transcription and translation of hemopoietic growth factors and/or cytokines. The bone marrow environment itself, as well as primed tissues, can each support and accelerate production of eosinophils for either release into the circulation or amplification of tissue eosinophilic inflammation, respectively.20., 21. The process of tissue amplification of eosinophilic inflammation through progenitor differentiation at the site has been termed in situ hemopoiesis (Figure 13.6.2 ).22., 23.
FIGURE 13.6.2.
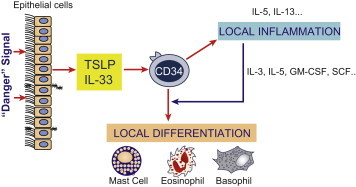
TSLP effects on progenitors: a new vista on in situ hemopoiesis.
Thymic stromal lymphopoietin (TSLP) can induce Th2 cytokines within cord blood CD34+ progenitors, effectively enhancing inflammatory effector function of these progenitors, as well as providing lineage differentiating stimuli (via IL-5 acting on CD34+ CD45+ IL-5RAhi+ cells in an autocrine fashion) for tissue-resident (bronchial or nasal mucosal) CD34+ cells. GM-CSF, granulocyte-macrophage colony-stimulating factor; IL-3, interleukin-3; IL-5, interleukin-5; IL-33, interleukin-33; SCF, stem cell factor.
(Delespesse and Allakhverdi, personal communication.)
CD34-Positive Eosinophil/Basophil Progenitors in Allergic Asthma
Hemopoietic progenitors express the cell stage-specific antigen, CD34, which is present at the highest levels on early hemopoietic myeloid progenitors and is progressively lost on terminally differentiating cells.24 These CD34+ hemopoietic progenitors contribute to the ongoing recruitment of eosinophils and basophils to sites of allergen challenge in allergic diseases, including asthma.25 Indeed, the number of CD34+ cells, as well as both mature and immature eosinophil cell numbers, are higher in blood and bone marrow of atopic subjects compared with nonatopic controls;26., 27. mild asthmatic subjects have fewer circulating CD34+ cells than severe asthmatic subjects,28 and CD34+ cell numbers have been shown to correlate with the level of airflow obstruction.28
Interleukin-5 Receptor Subunit Alpha
After airway allergen inhalation challenge in atopic asthmatic subjects, CD34+ cells from bone marrow synthesize mRNA and protein for membrane-bound IL-5 receptor subunit α (IL-5RA),29 permitting them to further respond to the eosinophilopoietic cytokine, IL-5. Progenitor cells from the bone marrow of atopic subjects show increased responsiveness to IL-5,26 probably due to increased levels of bone marrow CD34+IL-5RA+ cells, a unique feature of atopic disease.30 The phenotype of bone marrow CD34+ cells from mild asthmatic subjects has been carefully examined by flow cytometry, demonstrating a higher proportion of CD34+ IL-5RA+ after allergen challenge in those subjects who developed airway eosinophilia and increased methacholine airway responsiveness.29., 31. Taken together with in vitro experiments demonstrating IL-5-induced expression of IL-5RA on human progenitor cells,32., 33. these data demonstrate eosinophil lineage skewing of CD34+ cells in response to an allergic stimulus. Thus, increased production of Eo/B lineage-committed progenitors within the bone marrow, the subsequent development of blood and tissue eosinophilia,29 and the maintenance of an allergic inflammatory response26., 34. have been linked. Reports of antigen-induced increases in CD34+ IL-5RA+ cell numbers in murine bone marrow coincident with enhancement of IL-5-dependent eosinophilopoiesis provide further evidence that eosinophil production occurs as a result of expansion of the relevant eosinophil progenitor population rather than exclusively from demargination or release of sequestered mature eosinophils into the circulation.35., 36., 37., 38., 39. The rapid increase in expression of IL-5RA on CD34+ cells from the bone marrow of atopic asthmatics28., 40. clearly demonstrates that progenitor cells in these subjects are primed to respond to IL-5; indeed, CD34+ IL-5RA+ cell numbers circulate at higher numbers in subjects with asthma compared to controls, and correlate with asthma severity.28
Although controversial, chemokines such as eotaxin may also play a role not only in the migration41 but also in the differentiation of progenitor cells. Since CD34+ progenitor cells are found to express C-C chemokine receptor 3 (CCR3),42 which is upregulated in a Th2 environment, it is possible that signaling through this receptor also causes progenitor cells to differentiate into eosinophils independently of GM-CSF, IL-3, and IL-5.43
Colony-Forming Units
Colony assays are functional measures for the quantification of hemopoietic differentiation potential. Eo/B CFU are clusters of immature, nascent eosinophils and basophils derived from single progenitors44., 45., 46. present in nonadherent mononuclear or purified CD34+ cell populations seeded into a semisolid medium in the presence of hemopoietic growth factors. There are significantly greater numbers of Eo/B CFU in the peripheral blood of subjects with various allergic airway disorders, including asthma, nasal polyposis, and rhinitis.47., 48. Greater numbers of CD34+ cells are detected in blood and bone marrow of atopic than nonatopic subjects, correlating positively with higher numbers of Eo/B CFU grown from the blood and marrow of these subjects.26 The higher levels of CD34+ cells and IL-5-responsive Eo/B CFU in atopic subjects indicate a role for Eo/B progenitors in allergic diseases, such as asthma.26 The growth of Eo/B CFU is also influenced by exposure to specific antigen, which initiates a cascade of events with a stimulatory effect on the bone marrow to produce and release newly formed inflammatory cells. Importantly, allergic individuals can exhibit fluctuations in the numbers of circulating progenitors and tissue eosinophils during an allergen pollen season.49., 50. An initial increase in circulating Eo/B CFU at the beginning of seasonal allergen exposure is followed by a significant decline at the peak of the season, coincident with nasal symptoms and inflammation, probably reflecting the migration and differentiation in situ of progenitor cells from the blood to the inflamed tissue.49., 50., 51. This increase in Eo/B CFU has also been demonstrated in allergic asthma exacerbation.52., 53. Supporting the latter in vivo observation, higher numbers of bone marrow31 and circulating54 Eo/B CFU can be measured 24 h after allergen inhalation. Studies conducted in animal models of allergic asthma that carefully investigate the trafficking of cells from bone marrow confirm that allergen-induced airway inflammation is associated with increased numbers of newly formed cells in the blood and airways.34., 55. In mouse models of allergic rhinitis and/or asthma, changes in bone marrow eosinophilopoiesis, accompanied by appropriate peaks of hemopoietic cytokines and chemokines, such as eotaxin, GM-CSF and IL-5, and changes in cell surface receptors for these factors, were shown to occur as early as 2 h after allergen challenge,38., 39., 56., 57., 58. indicating that progenitor cell fluctuations and hemopoietic processes contributing to eosinophilic airways inflammation can occur quite rapidly in response to allergen challenge.
Studies to assess changes in cytokines levels within the bone marrow of sensitized mice or atopic asthmatics following allergen exposure have detected increases in levels of IL-5 consistent with the kinetics of eosinophil lineage commitment.37., 56., 57., 59. Using colony-forming assays, bone marrow progenitor cells from asthmatic subjects developing late-phase responses and airway eosinophilia postallergen challenge were shown to be more responsive to IL-5 than cells from subjects without these allergen-induced responses.29 A subsequent investigation demonstrated that allergen inhalation by allergic asthmatic subjects induces a time-dependent change in the levels of growth factors and cytokines in the bone marrow: subjects with elevated levels of airway and circulating eosinophils postchallenge had increased numbers of IL-5-responsive progenitors at 12 h and 24 h postallergen (Fig. 13.6.1), coincident with increased IL-5 protein levels in the bone marrow.4 As such, allergen-induced activation of an eosinophilopoietic process highlights the relationship between increased bone marrow IL-5 levels and the regulation of eosinophil production from bone marrow progenitor cells. Given the observed delayed interferon γ (INF-γ) increase in the marrow of these subjects,4 it could be postulated that activated T cells migrate from the airways to the bone marrow and release cytokines such as IL-5 and IL-9 that may locally orchestrate activation of hemopoietic events during an allergic inflammatory response.60., 61., 62., 63. To this end, investigations of the cell-associated cytokine production within the bone marrow have shown that CD34+ cells64., 65. and T cells61., 62., 63., 65. produce IL-5 during the course of the allergic inflammatory response. Nascent eosinophils and basophils picked from Eo/B CFU have also been shown to constitute autocrine sources of GM-CSF and IL-5,54., 61., 62., 63., 66., 67. which could further amplify the process of differentiation and proliferation. More recently, it has been shown that CD34+ cells from blood and bone marrow release more IL-5 following stimulation with calcium ionophore and phorbol-12-myristate-13-acetate than do the equivalent number of CD3+ T-lymphocytes.68
In Situ Hemopoiesis
Traditionally, the focal point of the differentiation and maturation process involving hemopoietic progenitors has been the bone marrow, under the regulation of its proper microenvironment. However, there is now an abundance of evidence demonstrating that eosinophil progenitors can traffic as fully or partly undifferentiated cells to allergic inflammatory tissues in the upper and lower airways, where they can and do differentiate into mature eosinophils under the control of local stimuli. Indeed, Eo/B CD34+ cells are found in tissues such as the bronchial mucosa,69 nasal polyps,70 and even in atopic dermatitis lesions.71 Despite the importance of systemic and local IL-5 production for the differentiation of CD34+ cells in the bone marrow, it is apparent that Eo/B can differentiate similarly at sites of tissue inflammation. This has been demonstrated functionally in several studies. Firstly, inflamed tissue from subjects with allergic rhinitis and nasal polyposis has been shown to produce hemopoietic cytokines that promote the differentiation and maturation of Eo/B CFU.23., 72., 73., 74., 75., 76., 77., 78. Next, mononuclear cells extracted from nasal polyp tissue have the potential to produce Eo/B CFU in response to growth factors such as IL-5.23., 70., 72. This supports the concept of in situ hemopoiesis, in which locally elaborated growth factors can drive the differentiation and maturation of hemopoietic progenitors into mature eosinophils (Fig. 13.6.2). Finally, phenotypic evaluations have identified CD34+ cells in various airway compartments, including the mucosa of the upper and lower airways of subjects with allergic rhinitis and asthma, respectively.60., 69., 70. The already elevated levels of CD34+ cells in induced sputum samples collected from mild asthmatic subjects are further increased after allergen inhalation challenges compared to healthy controls.3 With respect specifically to in situ eosinophilopoiesis, increased numbers of cells double positive for CD34 and IL5RA mRNA are found in nasal biopsies60 and lung biopsies of allergic asthmatic subjects compared to normal controls,60., 69. suggesting that IL-5-responsive CD34+ cells committed to the Eo/B lineage are present within the inflamed tissue. Furthermore, CD34+ cells appearing in sputum of allergic asthmatic subjects stain positive for IL-5 and IL-13 intracellularly, suggesting that they themselves may act as proinflammatory effector cells in the microenvironment of the inflamed tissue.64 Autocrine production of growth factors in Eo/B CFU grown from the blood of allergic asthmatic subjects has likewise been documented, which suggests that cytokine expression by differentiating progenitors may provide an additional stimulus to enhance differentiation in situ.54., 66. A similar autocrine upregulation of both eosinophilopoietic factors and their receptors has recently been demonstrated for TLR-ligated CD34+ cells in cord blood.79., 80. In situ IL-5-dependent differentiation of eosinophil progenitors can be triggered in the nasal mucosa of allergic rhinitic donors: ex vivo nasal mucosa cultures stimulated with allergen were shown to contain reduced numbers of CD34+ IL5RA mRNA+ cells and increased numbers of major basic protein (MBP) immunoreactive cells, thus shifting cells locally from an immature to a mature phenotype.60
In a mouse model of allergen-induced airway inflammation, newly produced CD34+ CCR3+ cells, isolated from lung and stimulated with eotaxin-2/CCL24 or IL-5, differentiated into significant numbers of CFU; cell cycle analysis showed significant increases in the number of CD34+ CCR3+ proliferating cells in allergen-exposed animals.81 These data support the notion of local Eo/B differentiation within tissues, suggesting that the CCR3/eotaxin pathway is also involved in the regulation of this allergen-driven in situ hemopoiesis, at least in mice. Recently, it has been shown that human airway smooth muscle of allergic asthmatics can also stimulate increased Eo/B differentiation in vitro,82 adding to the complexity of systemic and local hemopoietic processes in the generation of eosinophilic tissue inflammation.
Eosinophil Progenitors and Innate Immunity
The evidence described above for in situ hemopoiesis43 is also in keeping with a recent observation that IL-33/thymic stromal lymphopoietin (TSLP) is sufficient to induce the differentiation of CD34+ progenitors into eosinophils, in addition to activating Th2 and mast cells.64 A major recent discovery is that TSLP promotes Th2 pathways (via IL-25 and IL-33) that induce the development of a c-kitintSca-1+ multipotent progenitor (MMP; also known as ‘nuocytes’)83 population in gut-associated lymphoid tissue;84., 85. this provides further support for in situ hemopoiesis, and promotes the idea that TSLP provides a critical link between adaptive and innate immunity in the process of hemopoietic CD34+ cell differentiation within tissues (Fig. 13.6.2).
As mentioned above, recent data showing that CD34+ IL-5+ and CD34+ IL-13+ double-positive cells can be detected in sputum of asthmatic patients, with increases after allergen challenge,64 are consistent with the concept of allergen activation, via TSLP, of multipotent as well as lineage-committed progenitors, some of which can thus potentially function as inflammatory effector cells without further differentiation. Preliminary data show that recombinant TSLP, with or without IL-33 as a coligand, can also upregulate IL-5RA on CD34+ cells, rendering them more responsive to the effects of IL-5, with concomitant functional Eo/B CFU differentiation.86 Moreover, stimulation of CD34-enriched human cord blood cells with the Toll-like receptor (TLR) agonists, lipopolysaccharide (LPS) or CpG-oligodeoxynucleotides (CpG ODN), induces increased expression of TLR-2, TLR-4, and TLR-9 on CD34+ cells, as well as increases in GM-CSFRA, IL-3RA, and IL-5RA.79., 80. These observations demonstrate additional mechanisms through which innate immunity can regulate the responsiveness of Eo/B progenitors. CD34+ cells stimulated with a combination of TLR agonists and hemopoietic cytokines have been shown to give rise to more Eo/B CFU responsive to IL-3 and IL-5 than through hemopoietic cytokine stimulation alone.79., 80. Collectively, these data point to TSLP–TLR–TLR ligand interactions that can result in autocrine upregulation of Th2 cytokines (e.g., IL-5, IL-13) in CD34+ cells,61., 64., 65. and further support the idea that TSLP provides a critical link between adaptive and innate immunity in the process of hemopoietic CD34+ cell differentiation within tissues (Fig. 13.6.2).
The Role of Interleukin-5 in Eosinophil Progenitor Recruitment
IL-5 is central for upregulation of myeloid progenitors in the bone marrow after airway allergen challenge,38., 87. and for trafficking from the marrow to the airways in several animal models of either upper or lower airways inflammation.56., 88., 89. Airway, blood, and nasal eosinophilia are completely inhibited by either neutralizing the biologic effects of IL-590 or through deletion of the gene encoding IL-5.62 Although the asthmatic lung can release abundant amounts of IL-5, studies in animal models have led to debate regarding the distribution and sources of IL-5 required to drive airway eosinophilia. Studies in mice have shown that circulating, rather than local, IL-5 in the lung is critical for the development of allergic airways eosinophilia.91 This was investigated through systemic IL-5 gene transfer to IL-5-knockout mice, which effectively supported ovalbumin (OVA)-induced eosinophilia in the blood, bone marrow, and lung. This contrasts with IL-5 gene transfer into the airways, which did not induce eosinophilia following OVA challenge.91 Thus, systemic IL-5 seems to be necessary for the development of eosinophilia in the mouse. However, this is more controversial in humans.
Further studies have demonstrated that inhalation of IL-5 by allergic asthmatic subjects leads to the development of peripheral blood and sputum eosinophilia,92 and inhaled IL-5 induces airway eosinophilia accompanied by increased airway hyperresponsiveness (AHR).93 In contrast, three subsequent studies reported no effect of IL-5 inhalation on eosinophil levels in blood, airways, or AHR in allergic asthmatics.94., 95., 96. By inhalation, IL-5 was found to significantly decrease CD34+ IL5RA mRNA+ cells within the bronchial mucosa and decrease the percentage of CD34+ CCR3+ cells in the bone marrow of atopic asthmatics.96 It has been hypothesized that lung-derived IL-5 provides a signal to a population of cells within the bronchial mucosa that traffic to the bone marrow, where they locally induce the efflux of CD34+ CCR3+ cells.65., 96. Elegant experiments involving perfusion of the femoral bone marrow of guinea pigs confirmed that whereas IL-5 induces chemokinesis of bone marrow eosinophils, mobilization of mature eosinophils by IL-5 occurs synergistically with, and requires, the presence of eotaxin.41
Roles of Eotaxins
Eotaxin is a potent and eosinophil-specific chemoattractant.97., 98., 99. Eotaxin-1 is thought to be more important than eotaxin-2 for inducing mobilization of eosinophils and their progenitors from the bone marrow into the blood circulation.41., 100. A reduction of eosinophil numbers was observed in the lungs of mice treated with eotaxin-1 blocking antibodies101., 102. and in strain-specific eotaxin-1 knockout mice.103., 104. Eotaxin-1 is released from lung structural cells,105 and is released at increased levels following allergen challenge.106 Human endothelial progenitor cells also express eotaxin-1,107 and have been shown to be rapidly mobilized to the lung after allergen challenge in sensitized mice107 and atopic asthmatic subjects,108 where they also can contribute to the development of lung eosinophilia through the expression and secretion of eotaxin-1.
More recently, both eotaxin-1 and eotaxin-2 have been shown to induce migration of murine bone marrow and blood CD34+ CCR3+ cells using an in vitro transmigration assay.81 These data suggest that the CCR3/eotaxin pathway is involved in the regulation of allergen-driven accumulation/mobilization of eosinophil lineage-committed progenitor cells in the lung. The receptor for eotaxin, CCR3, is upregulated on CD34+ cells after allergen challenge, thereby facilitating eotaxin-mediated progenitor cell mobilization from the bone marrow to the peripheral circulation.42., 109.
Other Migration Signals
Studies in sensitized mice have indicated that T cells are the major gatekeepers regulating eotaxin and IL-5 levels, and thus eosinophilia. Reductions in airway, bone marrow, and peripheral blood eosinophil levels in CD4−/− and CD8−/− mice suggest that these T cell populations are critical regulators of allergen-induced eosinophilia. Furthermore, reduced serum IL-5 and bronchoalveolar lavage eotaxin-2 levels in CD4−/− mice suggest that CD4+ T cells are obligatory for the development of allergen-induced airway eosinophilia.110
In addition to eotaxin and IL-5, many other mediators have been shown to induce responses in both mature eosinophils and Eo/B progenitors. Cysteinyl leukotrienes, which are released in the airways following perturbation by allergen, are chemoattractants for mature eosinophils,111 but have also been shown to aid differentiation112 and induce chemotaxis and in vitro transendothelial migration of Eo/B progenitors.113 Preliminary work has shown that both IL-4 and IL-13 can prime hemopoietic progenitor cells in a transmigration assay,114 and studies are under way to investigate whether progenitors are also responsive to the eosinophil chemoattractants prostaglandin D2 receptor 2 (CRTH2; reviewed in115) and peroxisome proliferator-activated receptor γ (PPARγ).116., 117. In the allergen challenge model, there is attenuation of CXCR4 (SDF-1α receptor) expression on bone marrow CD34+ cells from mild asthmatic subjects, as well as a reduction in SDF-1α levels in the bone marrow. This demonstrates a mechanism whereby retention of progenitors in the bone marrow regulates their egress into the blood.109
Eosinophils and Progenitors as Targets of Asthma Therapy
The discoveries that IL-5 is a specific eosinophil growth factor in humans and that eotaxins can selectively induce eosinophil recruitment, as described above, were instrumental for the development of drugs targeting the eosinophil. This is reviewed in detail elsewhere.118
Interleukin-5 Blockade
Anti-IL-5 treatment in mild atopic asthmatic subjects has been shown to induce a reduction of airway eosinophils, arrest bone marrow eosinophil maturation, and decrease eosinophil progenitors in the bronchial mucosa.119., 120. Though initial clinical trials of IL-5 blockade in patients with asthma were unsuccessful in demonstrating clinical efficacy,119., 121., 122., 123., 124. a number of issues may have contributed to the failure of these studies, including lack of depletion of tissue eosinophils and their granule products; patient selection; and methodological problems.125 Subsequent studies, conducted in a subgroup of asthmatic patients selected on the basis of having persistent eosinophilic asthma, have demonstrated that blocking IL-5 with the humanized IL-5 antibody, mepolizumab, has a steroid-sparing effect, reduces exacerbations, and improves quality of life for asthma patients.126., 127. The anti-IL-5 approach is troubled by the inability to completely abolish eosinophilia, consistent with murine observations (reviewed in Matthaei et al.89), since it is hypothesized that if eosinophilia were solely controlled by IL-5 then a more complete suppression of eosinophils would ensue from therapeutic anti-IL-5 interventions. A propos this possibility, however, MEDI-563 is a humanized anti-IL-5RA monoclonal antibody that binds IL-5RA with high affinity and mediates cell lysis via antibody-directed cell-mediated cytotoxicity. As such, MEDI-563 kills all cells bearing IL-5RA, including eosinophil progenitors, and has been shown to eliminate eosinophils from the circulation of subjects with mild asthma.128., 129. This antibody is currently under investigation in other clinical models of asthma.
That eosinophils can remain in the lung tissue of asthmatic subjects despite IL-5 blockade suggests that additional signals promote eosinophil survival. Indeed in IL-5-deficient mice, responses to GM-CSF and IL-3 are normal, despite absence of eosinophilia in the nasal mucosa and the bone marrow, significantly lower numbers of IL-5-responsive Eo/B CFU and maturing CFU eosinophils, and reduced expression of IL-5RA on bone marrow-derived CD34+ CD45+ progenitor cells.56 These results indicate that redundant cytokine mechanisms can compensate for IL-5 deficiency and highlight the multifactorial nature of allergic inflammation, indicating that combined, as opposed to single-line, therapies may be more effective in the treatment of diseases such as asthma.
Antisense Therapy
One such therapy is currently being tested in clinical trials of allergic asthma. TPI-ASM8 is a combination of two antisense oligonucleotides, one blocking translation of the IL-3/IL-5/GM-CSF receptor common β chain, and the other blocking translation of CCR3. As such, TPI-ASM8 prevents expression of receptors for GM-CSF, IL-3, IL-5, eotaxin-1, and eotaxin-2, and will thus hypothetically inhibit many of the signals shown to be crucial for eosinophilopoiesis, as well as eosinophil migration, activation and survival. Following allergen challenge in mild atopic asthmatic subjects, inhaled TPI-ASM8 inhibited accumulation of mature eosinophils and CD34+ IL-5RA+ cells in the sputum, in addition to inhibiting the late asthmatic response.130., 131.
Steroid Therapy
Despite advances made in the development of eosinophil-specific therapies, corticosteroids remain the gold standard for the treatment of allergic inflammatory diseases like asthma.132., 133., 134. Local delivery using inhalers is intended for topical treatment of the affected tissue; however, the anti-inflammatory actions of corticosteroids have been shown to extend beyond the environment of the airways, probably due to a small amount of systemic availability. These systemic effects are beneficial for regulating hemopoietic mechanisms that originate in the bone marrow. Stepwise withdrawal of inhaled corticosteroids results in a rapid and substantial increase in Eo/B progenitors assayed in peripheral blood, which returns to baseline when treatment is reinstated.135 Only 1 week of treatment with inhaled corticosteroid is sufficient to significantly attenuate allergen-induced levels of circulating Eo/B CFU66 and reduce baseline numbers of bone marrow Eo/B CFU34 in mild allergic asthmatic subjects, further demonstrating the efficacy of corticosteroids on progenitors in peripheral blood. However, the inhaled steroid has no effect on the allergen-induced increase in the number of bone marrow CD34+ cells, the increase in IL-5RA expression on these cells, or the number of Eo/B CFU. These findings suggest that topical corticosteroids may exert indirect suppressive effects on the differentiation of eosinophil progenitors.
Conclusion
Eosinophil progenitors are now emerging as effector cells that can migrate to inflamed tissue where they rapidly proliferate in response to allergic stimuli. Understanding communication between eosinophil progenitors and the innate immune system will require further exploration.
Acknowledgements
The authors would like to thank Drs Guy Delespesse and Zoulfia Allakhverdi, Laboratory on Allergy, CHUM Research Center, Notre Dame Hospital, Montreal, Canada, for providing input from their research into TSLP effects on progenitor cells (Fig. 13.6.2). Thanks also to Lynne Larocque for her expert assistance with the preparation of the manuscript. Funding for this research was provided by the Canadian Institutes of Health Research (CIHR), and the Allergy, Genes and Environment Network of Centres of Excellence (AllerGen NCE Inc.).
References
- 1.Alfaro C., Sharma O.P., Navarro L., Glovsky M.M. Inverse correlation of expiratory lung flows and sputum eosinophils in status asthmaticus. Ann Allergy. 1989;63:251–254. [PubMed] [Google Scholar]
- 2.Walker C., Kaegi M.K., Braun P., Blaser K. Activated T cells and eosinophilia in bronchoalveolar lavages from subjects with asthma correlated with disease severity. J Allergy Clin Immunol. 1991;88:935–942. doi: 10.1016/0091-6749(91)90251-i. [DOI] [PubMed] [Google Scholar]
- 3.Dorman S.C., Efthimiadis A., Babirad I., Watson R.M., Denburg J.A., Hargreave F.E. Sputum CD34+IL-5Rα+ cells increase after allergen: evidence for in situ eosinophilopoiesis. Am J Respir Crit Care Med. 2004;169:573–577. doi: 10.1164/rccm.200307-1004OC. [DOI] [PubMed] [Google Scholar]
- 4.Dorman S.C., Sehmi R., Gauvreau G.M., Watson R.M., Foley R., Jones G.L. Kinetics of bone marrow eosinophilopoiesis and associated cytokines after allergen inhalation. Am J Respir Crit Care Med. 2004;169:565–572. doi: 10.1164/rccm.200307-1024OC. [DOI] [PubMed] [Google Scholar]
- 5.De Monchy J.G., Kauffman H.F., Venge P., Koeter G.H., Jansen H.M., Sluiter H.J. Bronchoalveolar eosinophilia during allergen-induced late asthmatic reactions. Am Rev Respir Dis. 1985;131:373–376. doi: 10.1164/arrd.1985.131.3.373. [DOI] [PubMed] [Google Scholar]
- 6.Gauvreau G.M., Evans M.Y. Allergen inhalation challenge: a human model of asthma exacerbation. Contrib Microbiol. 2007;14:21–32. doi: 10.1159/000107052. [DOI] [PubMed] [Google Scholar]
- 7.Gauvreau G.M., Newbold P., Perrett J., Craggs B., Foster M., Drong M. Kinetics of cellular infiltration post allergen challenge in asthma subjects: differences between bronchial washings and lung tissue (abst) Am J Respir Crit Care Med. 2007;175:A679. [Google Scholar]
- 8.Gauvreau G.M., Watson R.M., O’Byrne P.M. Kinetics of allergen-induced airway eosinophilic cytokine production and airway inflammation. Am J Respir Crit Care Med. 1999;160:640–647. doi: 10.1164/ajrccm.160.2.9809130. [DOI] [PubMed] [Google Scholar]
- 9.Bousquet J., Chanez P., Lacoste J.Y., Barneon G., Ghavanian N., Enander I. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 10.Weller P.F. Eosinophils: structure and functions. Curr Opin Immunol. 1994;6:85–90. doi: 10.1016/0952-7915(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg H.F., Phipps S., Foster P.S. Eosinophil trafficking in allergy and asthma. J Allergy Clin Immunol. 2007;119:1303–1310. doi: 10.1016/j.jaci.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 12.Clutterbuck E.J., Sanderson C.J. Regulation of human eosinophil precursor production by cytokines: a comparison of recombinant human interleukin-1 (rhIL-1), rhIL-3, rhIL-5, rhIL-6, and rh granulocyte-macrophage colony-stimulating factor. Blood. 1990;75:1774–1779. [PubMed] [Google Scholar]
- 13.Asquith K.L., Ramshaw H.S., Hansbro P.M., Beagley K.W., Lopez A.F., Foster P.S. The IL-3/IL-5/GM-CSF common receptor plays a pivotal role in the regulation of Th2 immunity and allergic airway inflammation. J Immunol. 2008;180:1199–1206. doi: 10.4049/jimmunol.180.2.1199. [DOI] [PubMed] [Google Scholar]
- 14.Shalit M., Sekhsaria S., Malech H.L. Modulation of growth and differentiation of eosinophils from human peripheral blood CD34+ cells by IL5 and other growth factors. Cell Immunol. 1995;160:50–57. doi: 10.1016/0008-8749(95)80008-7. [DOI] [PubMed] [Google Scholar]
- 15.Ying S., Meng Q., Barata L.T., Robinson D.S., Durham S.R., Kay A.B. Associations between IL-13 and IL-4 (mRNA and protein), vascular cell adhesion molecule-1 expression, and the infiltration of eosinophils, macrophages, and T cells in allergen-induced late-phase cutaneous reactions in atopic subjects. J Immunol. 1997;158:5050–5057. [PubMed] [Google Scholar]
- 16.Li L., Xia Y., Nguyen A., Lai Y.H., Feng L., Mosmann T.R. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J Immunol. 1999;162:2477–2487. [PubMed] [Google Scholar]
- 17.Zimmermann N., Hogan S.P., Mishra A., Brandt E.B., Bodette T.R., Pope S.M. Murine eotaxin-2: a constitutive eosinophil chemokine induced by allergen challenge and IL-4 overexpression. J Immunol. 2000;165:5839–5846. doi: 10.4049/jimmunol.165.10.5839. [DOI] [PubMed] [Google Scholar]
- 18.Gounni A.S., Gregory B., Nutku E., Aris F., Latifa K., Minshall E. Interleukin-9 enhances interleukin-5 receptor expression, differentiation, and survival of human eosinophils. Blood. 2000;96:2163–2171. [PubMed] [Google Scholar]
- 19.Matsuzawa S., Sakashita K., Kinoshita T., Ito S., Yamashita T., Koike K. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J Immunol. 2003;170:3461–3467. doi: 10.4049/jimmunol.170.7.3461. [DOI] [PubMed] [Google Scholar]
- 20.Mohle R., Salemi P., Moore M.A., Rafii S. Expression of interleukin-5 by human bone marrow microvascular endothelial cells: implications for the regulation of eosinophilopoiesis in vivo. Br J Haematol. 1997;99:732–738. doi: 10.1046/j.1365-2141.1997.4713279.x. [DOI] [PubMed] [Google Scholar]
- 21.Hogan M.B., Piktel D., Landreth K.S. IL-5 production by bone marrow stromal cells: implications for eosinophilia associated with asthma. J Allergy Clin Immunol. 2000;106:329–336. doi: 10.1067/mai.2000.108309. [DOI] [PubMed] [Google Scholar]
- 22.Denburg J.A. Hemopoietic progenitors and cytokines in allergic inflammation. Allergy. 1998;53(supp.145):22–26. doi: 10.1111/j.1398-9995.1998.tb04935.x. [DOI] [PubMed] [Google Scholar]
- 23.Denburg J.A. Bone marrow in atopy and asthma: hematopoietic mechanisms in allergic inflammation. Immunol Today. 1999;20:111–113. doi: 10.1016/s0167-5699(98)01423-6. [DOI] [PubMed] [Google Scholar]
- 24.Krause D.S., Fackler M.J., Civin C.I., May W.S. CD34: structure, biology, and clinical utility. Blood. 1996;87:1–13. [PubMed] [Google Scholar]
- 25.Sergejeva S., Johansson A.K., Malmhall C., Lotvall J. Allergen exposure-induced differences in CD34+ cell phenotype: relationship to eosinophilopoietic responses in different compartments. Blood. 2004;103:1270–1277. doi: 10.1182/blood-2003-05-1618. [DOI] [PubMed] [Google Scholar]
- 26.Sehmi R., Howie K., Sutherland D.R., Schragge W., O’Byrne P.M., Denburg J.A. Increased levels of CD34+ hemopoietic progenitor cells in atopic subjects. Am J Respir Cell Mol Biol. 1996;15:645–654. doi: 10.1165/ajrcmb.15.5.8918371. [DOI] [PubMed] [Google Scholar]
- 27.Zeibecoglou K., Ying S., Yamada T., North J., Burman J., Bungre J. Increased mature and immature CCR3 messenger RNA+ eosinophils in bone marrow from patients with atopic asthma compared with atopic and nonatopic control subjects. J Allergy Clin Immunol. 1999;103:99–106. doi: 10.1016/s0091-6749(99)70532-4. [DOI] [PubMed] [Google Scholar]
- 28.Makowska J.S., Grzegorczyk J., Cieslak M., Bienkiewicz B., Kowalski M.L. Recruitment of CD34+ progenitor cells into peripheral blood and asthma severity. Ann Allergy Asthma Immunol. 2008;101:402–406. doi: 10.1016/S1081-1206(10)60317-1. [DOI] [PubMed] [Google Scholar]
- 29.Sehmi R., Wood L.J., Watson R., Foley R., Hamid Q., O’Byrne P.M. Allergen-induced increases in IL-5 receptor α-subunit expression on bone marrow-derived CD34+ cells from asthmatic subjects. A novel marker of progenitor cell commitment toward eosinophilic differentiation. J Clin Invest. 1997;100:2466–2475. doi: 10.1172/JCI119789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sehmi R., Howie K., Rerecich T., Watson R.M., Foley R., O’Byrne P.M. Increased numbers of eosinophil progenitor cells (CD34+IL5Rα+) in the bone marrow of atopic asthmatic subjects (abst) J Allergy Clin Immunol. 2000;105:S172. [Google Scholar]
- 31.Wood L.J., Inman M.D., Watson R.M., Foley R., Denburg J.A., O’Byrne P.M. Changes in bone marrow inflammatory cell progenitors after inhaled allergen in asthmatic subjects. Am J Respir Crit Care Med. 1998;157:99–105. doi: 10.1164/ajrccm.157.1.9704125. [DOI] [PubMed] [Google Scholar]
- 32.Tavernier J., Van der Heyden J., Verhee A., Brusselle G., Van Ostade X., Vandekerckhove J. Interleukin 5 regulates the isoform expression of its own receptor alpha-subunit. Blood. 2000;95:1600–1607. [PubMed] [Google Scholar]
- 33.Denburg J.A., Sehmi R., Upham J. Regulation of IL-5R on eosinophil progenitors in allergic inflammation: role of retinoic acid. Int Arch Allergy Immunol. 2001;124:246–248. doi: 10.1159/000053724. [DOI] [PubMed] [Google Scholar]
- 34.Wood L.J., Sehmi R., Gauvreau G.M., Watson R.M., Foley R., Denburg J.A. An inhaled corticosteroid, budesonide, reduces baseline but not allergen-induced increases in bone marrow inflammatory cell progenitors in asthmatic subjects. Am J Respir Crit Care Med. 1999;159:1457–1463. doi: 10.1164/ajrccm.159.5.9808123. [DOI] [PubMed] [Google Scholar]
- 35.Ohkawara Y., Lei X.F., Stampfli M.R., Marshall J.S., Xing Z., Jordana M. Cytokine and eosinophil responses in the lung, peripheral blood, and bone marrow compartments in a murine model of allergen-induced airways inflammation. Am J Respir Cell Mol Biol. 1997;16:510–520. doi: 10.1165/ajrcmb.16.5.9160833. [DOI] [PubMed] [Google Scholar]
- 36.Gaspar Elsas M.I., Joseph D., Elsas P.X., Vargaftig B.B. Rapid increase in bone-marrow eosinophil production and responses to eosinopoietic interleukins triggered by intranasal allergen challenge. Am J Respir Cell Mol Biol. 1997;17:404–413. doi: 10.1165/ajrcmb.17.4.2691. [DOI] [PubMed] [Google Scholar]
- 37.Inman M.D., Ellis R., Wattie J., Denburg J.A., O’Byrne P.M. Allergen-induced increase in airway responsiveness, airway eosinophilia and bone-marrow eosinophil progenitors in mice. Am J Respir Cell Mol Biol. 1999;21:473–479. doi: 10.1165/ajrcmb.21.4.3622. [DOI] [PubMed] [Google Scholar]
- 38.Saito H., Howie K., Wattie J., Denburg A., Ellis R., Inman M.D. Allergen-induced murine upper airway inflammation: local and systemic changes in murine experimental allergic rhinitis. Immunology. 2001;104:226–234. doi: 10.1046/j.0019-2805.2001.01253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J., Saito H., Crawford L., Inman M.D., Cyr M.M., Denburg J.A. Hemopoietic mechanisms in murine allergic upper and lower airways inflammation. Immunology. 2005;114:386–396. doi: 10.1111/j.1365-2567.2005.02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou C.L., Wang C.H., Kuo H.P. Upregulation of IL-5 receptor expression on bone marrow-derived CD34+ cells from patients with asthma. Changgeng Yi Xue Za Zhi. 1999;22:416–422. [PubMed] [Google Scholar]
- 41.Palframan R.T., Collins P.D., Williams T.J., Rankin S.M. Eotaxin induces a rapid release of eosinophils and their progenitors from the bone marrow. Blood. 1998;91:2240–2248. [PubMed] [Google Scholar]
- 42.Sehmi R., Dorman S., Baatjes A., Watson R., Foley R., Ying S. Allergen-induced fluctuation in CC chemokine receptor 3 expression on bone marrow CD34+ cells from asthmatic subjects: significance for mobilization of haemopoietic progenitor cells in allergic inflammation. Immunology. 2003;109:536–546. doi: 10.1046/j.1365-2567.2003.01686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamkhioued B., Abdelilah S.G., Hamid Q., Mansour N., Delespesse G., Renzi P.M. The CCR3 receptor is involved in eosinophil differentiation and is up-regulated by Th2 cytokines in CD34+ progenitor cells. J Immunol. 2003;170:537–547. doi: 10.4049/jimmunol.170.1.537. [DOI] [PubMed] [Google Scholar]
- 44.Denburg J.A., Richardson M., Telizyn S., Bienenstock J. Basophil/mast cell precursors in human peripheral blood. Blood. 1983;61:775–780. [PubMed] [Google Scholar]
- 45.Denburg J.A., Silver J.E., Abrams J.S. Interleukin-5 is a human basophilopoietin: Induction of histamine content and basophilic differentiation of HL-60 cells and of peripheral blood basophil-eosinophil progenitors. Blood. 1991;77:1462–1468. [PubMed] [Google Scholar]
- 46.Denburg J.A., Telizyn S., Messner H., Lim B., Jamal N., Ackerman S.J. Heterogeneity of human peripheral blood eosinophil-type colonies: Evidence for a common basophil-eosinophil progenitor. Blood. 1985;66:312–318. [PubMed] [Google Scholar]
- 47.Denburg J.A., Telizyn S., Belda A., Dolovich J., Bienenstock J. Increased numbers of circulating basophil progenitors in atopic patients. J Allergy Clin Immunol. 1985;76:466–472. doi: 10.1016/0091-6749(85)90728-6. [DOI] [PubMed] [Google Scholar]
- 48.Sehmi R., Wood L.J., Inman M.D., Watson R.M., O’Byrne P.M., Lopez A.F. Increases in bone marrow derived CD34+ hemopoietic progenitor cells expressing the alpha-subunit of IL-3 receptors following allergen challenge in mild asthmatics (abst) Am J Respir Crit Care Med. 1996;153:A880. [Google Scholar]
- 49.Otsuka H., Dolovich J., Befus A.D., Telizyn S., Bienenstock J., Denburg J.A. Basophilic cell progenitors, nasal metachromatic cells, and peripheral blood basophils in ragweed-allergic patients. J Allergy Clin Immunol. 1986;78:365–371. doi: 10.1016/s0091-6749(86)80091-4. [DOI] [PubMed] [Google Scholar]
- 50.Linden M., Svensson C., Andersson M., Greiff L., Andersson E., Denburg J.A. Circulating eosinophil/basophil progenitors and nasal mucosal cytokines in seasonal allergic rhinitis. Allergy. 1999;54:212–219. doi: 10.1034/j.1398-9995.1999.00756.x. [DOI] [PubMed] [Google Scholar]
- 51.Cyr M.M., Baatjes A.J., Hayes L.M., Crawford L., Denburg J.A. The effect of desloratadine on eosinophil/basophil progenitors and other inflammatory markers in seasonal allergic rhinitis: a placebo-controlled randomized study (abst) J Allergy Clin Immunol. 2002;109:S117. doi: 10.1159/000088721. [DOI] [PubMed] [Google Scholar]
- 52.Gibson P.G., Dolovich J., Girgis-Gabardo A., Morris M.M., Anderson M., Hargreave F.E. The inflammatory response in asthma exacerbation: changes in circulating eosinophils, basophils and their progenitors. Clin Exp Allergy. 1990;20:661–668. doi: 10.1111/j.1365-2222.1990.tb02705.x. [DOI] [PubMed] [Google Scholar]
- 53.Gibson P.G., Manning P.J., O’Byrne P.M., Girgis-Gabardo A., Dolovich J., Denburg J.A. Allergen-induced asthmatic responses: Relationship between increases in airway responsiveness and increases in circulating eosinophils, basophils, and their progenitors. Am Rev Respir Dis. 1991;143:331–335. doi: 10.1164/ajrccm/143.2.331. [DOI] [PubMed] [Google Scholar]
- 54.Gauvreau G.M., O’Byrne P.M., Moqbel R., Velazquez J., Watson R.M., Howie K.J. Enhanced expression of GM-CSF in differentiating eosinophils of atopic and atopic asthmatic subjects. Am J Respir Cell Mol Biol. 1998;19:55–62. doi: 10.1165/ajrcmb.19.1.2871. [DOI] [PubMed] [Google Scholar]
- 55.Johansson A.K., Sergejeva S., Sjostrand M., Lee J.J., Lotvall J. Allergen-induced traffic of bone marrow eosinophils, neutrophils and lymphocytes to airways. Eur J Immunol. 2004;34:3135–3145. doi: 10.1002/eji.200425043. [DOI] [PubMed] [Google Scholar]
- 56.Saito H., Matsumoto K., Denburg A.E., Crawford L., Ellis R., Inman M.D. Pathogenesis of murine experimental allergic rhinitis: a study of local and systemic consequences of IL-5 deficiency. J Immunol. 2002;168:3017–3023. doi: 10.4049/jimmunol.168.6.3017. [DOI] [PubMed] [Google Scholar]
- 57.Saito H., Morikawa H., Howie K., Crawford L., Baatjes A.J., Denburg E. Effects of a cysteinyl leukotriene receptor antagonist on eosinophil recruitment in experimental allergic rhinitis. Immunology. 2004;113:246–252. doi: 10.1111/j.1365-2567.2004.01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Southam D.S., Widmer N., Ellis R., Hirota J.A., Inman M.D., Sehmi R. Increased eosinophil-lineage committed progenitors in the lung of allergen-challenged mice. J Allergy Clin Immunol. 2005;115:95–102. doi: 10.1016/j.jaci.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 59.Hart T.K., Cook R.M., Zia-Amirhosseini P., Minthorn E., Sellers T.S., Maleeff B.E. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Allergy Clin Immunol. 2001;108:250–257. doi: 10.1067/mai.2001.116576. [DOI] [PubMed] [Google Scholar]
- 60.Cameron L., Christodoulopoulos P., Lavigne F., Nakamura Y., Eidelman D., McEuen A. Evidence for local eosinophil differentiation within allergic nasal mucosa: inhibition with soluble IL-5 receptor. J Immunol. 2000;164:1538–1545. doi: 10.4049/jimmunol.164.3.1538. [DOI] [PubMed] [Google Scholar]
- 61.Minshall E.M., Schleimer R., Cameron L., Minnicozzi M., Egan R.W., Gutierrez-Ramos J.C. Interleukin-5 expression in the bone marrow of sensitized Balb/c mice after allergen challenge. Am J Respir Crit Care Med. 1998;158:951–957. doi: 10.1164/ajrccm.158.3.9709114. [DOI] [PubMed] [Google Scholar]
- 62.Sitkauskiene B., Johansson A.K., Sergejeva S., Lundin S., Sjostrand M., Lotvall J. Regulation of bone marrow and airway CD34+ eosinophils by interleukin-5. Am J Respir Cell Mol Biol. 2004;30:367–378. doi: 10.1165/rcmb.2002-0311OC. [DOI] [PubMed] [Google Scholar]
- 63.Sitkauskiene B., Radinger M., Bossios A., Johansson A.K., Sakalauskas R., Lotvall J. Airway allergen exposure stimulates bone marrow eosinophilia partly via IL-9. Respir Res. 2005;6:33. doi: 10.1186/1465-9921-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allakhverdi Z., Comeau M.R., Smith D.E., Toy D., Endam L.M., Desrosier M. CD34+ hemopoietic progenitor cells are potent effectors of allergic inflammation. J Allergy Clin Immunol. 2009;123:472–478. doi: 10.1016/j.jaci.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 65.Wood L.J., Sehmi R., Dorman S., Hamid Q., Tulic M.K., Watson R.M. Allergen-induced increases in bone marrow T lymphocytes and interleukin-5 expression in subjects with asthma. Am J Respir Crit Care Med. 2002;166:883–889. doi: 10.1164/rccm.2108015. [DOI] [PubMed] [Google Scholar]
- 66.Gauvreau G.M., Wood L.J., Sehmi R., Watson R.M., Dorman S.C., Schleimer R.P. The effects of inhaled budesonide on circulating eosinophil progenitors and their expression of cytokines after allergen challenge in subjects with atopic asthma. Am J Respir Crit Care Med. 2000;162:2139–2144. doi: 10.1164/ajrccm.162.6.2001120. [DOI] [PubMed] [Google Scholar]
- 67.Gauvreau G.M., Lee J.M., Watson R.M., Irani A.M., Schwartz L.B., O’Byrne P.M. Increased numbers of both airway basophils and mast cells in sputum after allergen inhalation challenge of atopic asthmatics. Am J Respir Crit Care Med. 2000;161:1473–1478. doi: 10.1164/ajrccm.161.5.9908090. [DOI] [PubMed] [Google Scholar]
- 68.Bossios A., Sjostrand M., Dahlborn A.K., Samitas K., Malmhall C., Gaga M. IL-5 expression and release from human CD34 cells in vitro; ex vivo evidence from cases of asthma and Churg-Strauss syndrome. Allergy. 2010;65:831–839. doi: 10.1111/j.1398-9995.2009.02271.x. [DOI] [PubMed] [Google Scholar]
- 69.Robinson D.S., Damia R., Zeibecoglou K., Molet S., North J., Yamada T. CD34+/interleukin-5Rα messenger RNA+ cells in the bronchial mucosa in asthma: potential airway eosinophil progenitors. Am J Respir Cell Mol Biol. 1999;20:9–13. doi: 10.1165/ajrcmb.20.1.3449. [DOI] [PubMed] [Google Scholar]
- 70.Kim Y.K., Uno M., Hamilos D.L., Beck L., Bochner B., Schleimer R. Immunolocalization of CD34 in nasal polyposis. Effect of topical corticosteroids. Am J Respir Cell Mol Biol. 1999;20:388–397. doi: 10.1165/ajrcmb.20.3.3060. [DOI] [PubMed] [Google Scholar]
- 71.Mastrandrea F., Cadario G., Nicotra M.R., Natali P.G. Hemopoietic progenitor cells in atopic dermatitis skin lesions. J Investig Allergol Clin Immunol. 1999;9:386–391. [PubMed] [Google Scholar]
- 72.Denburg J.A., Dolovich J., Ohtoshi T., Cox G., Gauldie J., Jordana M. The microenvironmental differentiation hypothesis of airway inflammation. Am J Rhinology. 1990;4:29–32. [Google Scholar]
- 73.Metcalf D. Clonal analysis of the response of HL60 human myeloid leukemia cells to biological regulators. Leuk Res. 1983;7:117–132. doi: 10.1016/0145-2126(83)90002-4. [DOI] [PubMed] [Google Scholar]
- 74.Ohnishi M., Ruhno J., Bienenstock J., Milner R., Dolovich J., Denburg J.A. Human nasal polyp epithelial basophil/mast cell and eosinophil colony-stimulating activity: the effect is T-cell-dependent. Am Rev Respir Dis. 1988;138:560–564. doi: 10.1164/ajrccm/138.3.560. [DOI] [PubMed] [Google Scholar]
- 75.Ohnishi M., Ruhno J., Dolovich J., Denburg J.A. Allergic rhinitis nasal mucosal conditioned medium stimulates growth and differentiation of basophil/mast cell and eosinophil progenitors from atopic blood. J Allergy Clin Immunol. 1988;81:1149–1154. doi: 10.1016/0091-6749(88)90883-4. [DOI] [PubMed] [Google Scholar]
- 76.Cox G., Ohtoshi T., Vancheri C., Denburg J.A., Dolovich J., Gauldie J. Promotion of eosinophil survival by human bronchial epithelial cells and its modulation by steroids. Am J Respir Cell Mol Biol. 1991;4:525–531. doi: 10.1165/ajrcmb/4.6.525. [DOI] [PubMed] [Google Scholar]
- 77.Vancheri C., Gauldie J., Bienenstock J., Cox G., Scicchitano R., Stanisz A. Human lung fibroblast-derived granulocyte-macrophage colony stimulating factor (GM-CSF) mediates eosinophil survival in vitro. Am J Respir Cell Mol Biol. 1989;1:289–295. doi: 10.1165/ajrcmb/1.4.289. [DOI] [PubMed] [Google Scholar]
- 78.Vancheri C., Ohtoshi T., Cox G., Xaubet A., Abrams J.S., Gauldie J. Neutrophilic differentiation induced by human upper airway fibroblast-derived granulocyte/macrophage colony-stimulating factor (GM-CSF) Am J Respir Cell Mol Biol. 1991;4:11–17. doi: 10.1165/ajrcmb/4.1.11. [DOI] [PubMed] [Google Scholar]
- 79.Reece P., Thanendran A., Crawford L., Tulic M.K., Thabane L., Prescott S.L. Maternal allergy modulates cord blood hematopoietic progenitor Toll-like receptor expression and function. J Allergy Clin Immunol. 2011;127:447–453. doi: 10.1016/j.jaci.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 80.Reece P., Crawford L., Baatjes A., Cyr M., Sehmi R., Denburg J.A. Cord blood hemopoietic progenitor cell Toll-like receptor expression and function: a mechanism underlying allergic inflammation in early life (abst) J Allergy Clin Immunol. 2010;125:AB105. [Google Scholar]
- 81.Radinger M., Bossios A., Sjostrand M., Lu Y., Malmhall C., Dahlborn A.K. Local proliferation and mobilization of CCR3(+) CD34(+) eosinophil-lineage-committed cells in the lung. Immunology. 2011;132:144–154. doi: 10.1111/j.1365-2567.2010.03349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fanat A.I., Thomson J.V., Radford K., Nair P., Sehmi R. Human airway smooth muscle promotes eosinophil differentiation. Clin Exp Allergy. 2009;39:1009–1017. doi: 10.1111/j.1365-2222.2009.03246.x. [DOI] [PubMed] [Google Scholar]
- 83.Neill D.R., Wong S.H., Bellosi A., Flynn R.J., Daly M., Langford T.K. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saenz S.A., Siracusa M.C., Perrigoue J.G., Spencer S.P., Urban J.F., Jr., Tocker J.E. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ziegler S.F., Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. 2010;11:289–293. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hui C.C.K., Murphy D.M., Thong B., Delespesse G., Denburg J.A., Larché M. Induction of thymic stromal lymphopoietin (TSLP) in airway epithelium by recombinant allergens (abst) J Allergy Clin Immunol. 2011;127:AB125. [Google Scholar]
- 87.Wood L.J., Inman M.D., Denburg J.A., O’Byrne P.M. Allergen challenge increases cell traffic between bone marrow and lung. Am J Respir Cell Mol Biol. 1998;18:759–767. doi: 10.1165/ajrcmb.18.6.3006. [DOI] [PubMed] [Google Scholar]
- 88.Egan R.W., Athwahl D., Chou C.C., Chapman R.W., Emtage S., Jehn C.H. Pulmonary biology of anti-interleukin 5 antibodies. Mem Inst Oswaldo Cruz. 1997;92(Suppl. 2):69–73. doi: 10.1590/s0074-02761997000800011. [DOI] [PubMed] [Google Scholar]
- 89.Matthaei K.I., Foster P.S., Young I.G. The role of interleukin-5 (IL-5) in vivo: studies with IL-5 deficient mice. Mem Inst Oswaldo Cruz. 1997;92(Suppl. 2):63–68. doi: 10.1590/s0074-02761997000800010. [DOI] [PubMed] [Google Scholar]
- 90.Kopf M., Brombacher F., Hodgkin P.D., Ramsay A.J., Milbourne E.A., Dai W.J. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity. 1996;4:15–24. doi: 10.1016/s1074-7613(00)80294-0. [DOI] [PubMed] [Google Scholar]
- 91.Wang J., Palmer K., Lotvall J., Milan S., Lei X.F., Matthaei K.I. Circulating, but not local lung, IL-5 is required for the development of antigen-induced airways eosinophilia. J Clin Invest. 1998;102:1132–1141. doi: 10.1172/JCI2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi H.Z., Li C.Q., Qin S.M., Xie Z.F., Liu Y. Effect of inhaled interleukin-5 on number and activity of eosinophils in circulation from asthmatics. Clin Immunol. 1999;91:163–169. doi: 10.1006/clim.1999.4699. [DOI] [PubMed] [Google Scholar]
- 93.Shi H.Z., Xiao C.Q., Zhong D., Qin S.M., Liu Y., Liang G.R. Effect of inhaled interleukin-5 on airway hyperreactivity and eosinophilia in asthmatics. Am J Respir Crit Care Med. 1998;157:204–209. doi: 10.1164/ajrccm.157.1.9703027. [DOI] [PubMed] [Google Scholar]
- 94.Stirling R.G., van Rensen E.L., Barnes P.J., Chung K.F. Interleukin-5 induces CD34(+) eosinophil progenitor mobilization and eosinophil CCR3 expression in asthma. Am J Respir Crit Care Med. 2001;164:1403–1409. doi: 10.1164/ajrccm.164.8.2010002. [DOI] [PubMed] [Google Scholar]
- 95.van Rensen E.L., Stirling R.G., Scheerens J., Staples K., Sterk P.J., Barnes P.J. Evidence for systemic rather than pulmonary effects of interleukin-5 administration in asthma. Thorax. 2001;56:935–940. doi: 10.1136/thorax.56.12.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Menzies-Gow A.N., Flood-Page P.T., Robinson D.S., Kay A.B. Effect of inhaled interleukin-5 on eosinophil progenitors in the bronchi and bone marrow of asthmatic and non-asthmatic volunteers. Clin Exp Allergy. 2007;37:1023–1032. doi: 10.1111/j.1365-2222.2007.02735.x. [DOI] [PubMed] [Google Scholar]
- 97.Jose P.J., Griffiths-Johnson D.A., Collins P.D., Walsh D.T., Moqbel R., Totty N.F. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J Exp Med. 1994;179:881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fulkerson P.C., Fischetti C.A., McBride M.L., Hassman L.M., Hogan S.P., Rothenberg M.E. A central regulatory role for eosinophils and the eotaxin/CCR3 axis in chronic experimental allergic airway inflammation. Proc Natl Acad Sci U S A. 2006;103:16418–16423. doi: 10.1073/pnas.0607863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garcia-Zepeda E.A., Rothenberg M.E., Ownbey R.T., Celestin J., Leder P., Luster A.D. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2:449–456. doi: 10.1038/nm0496-449. [DOI] [PubMed] [Google Scholar]
- 100.Mould A.W., Matthaei K.I., Young I.G., Foster P.S. Relationship between interleukin-5 and eotaxin in regulating blood and tissue eosinophilia in mice. J Clin Invest. 1997;99:1064–1071. doi: 10.1172/JCI119234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Campbell E.M., Kunkel S.L., Strieter R.M., Lukacs N.W. Temporal role of chemokines in a murine model of cockroach allergen-induced airway hyperreactivity and eosinophilia. J Immunol. 1998;161:7047–7053. [PubMed] [Google Scholar]
- 102.Gonzalo J.A., Lloyd C.M., Wen D., Albar J.P., Wells T.N., Proudfoot A. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J Exp Med. 1998;188:157–167. doi: 10.1084/jem.188.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rothenberg M.E., MacLean J.A., Pearlman E., Luster A.D., Leder P. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J Exp Med. 1997;185:785–790. doi: 10.1084/jem.185.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang Y., Loy J., Ryseck R.P., Carrasco D., Bravo R. Antigen-induced eosinophilic lung inflammation develops in mice deficient in chemokine eotaxin. Blood. 1998;92:3912–3923. [PubMed] [Google Scholar]
- 105.Smit J.J., Lukacs N.W. A closer look at chemokines and their role in asthmatic responses. Eur J Pharmacol. 2006;533:277–288. doi: 10.1016/j.ejphar.2005.12.064. [DOI] [PubMed] [Google Scholar]
- 106.Brown J.R., Kleimberg J., Marini M., Sun G., Bellini A., Mattoli S. Kinetics of eotaxin expression and its relationship to eosinophil accumulation and activation in bronchial biopsies and bronchoalveolar lavage (BAL) of asthmatic patients after allergen inhalation. Clin Exp Immunol. 1998;114:137–146. doi: 10.1046/j.1365-2249.1998.00688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Asosingh K., Hanson J.D., Cheng G., Aronica M.A., Erzurum S.C. Allergen-induced, eotaxin-rich, proangiogenic bone marrow progenitors: a blood-borne cellular envoy for lung eosinophilia. J Allergy Clin Immunol. 2010;125:918–925. doi: 10.1016/j.jaci.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Imaoka H., Babirad I., Watson R.M., Obminski G., Strinich T.X., Howie K. Increased lung-homing of vascular endothelial progenitor cells in asthmatic subjects (abst) Am J Respir Crit Care Med. 2010;181:A5624. [Google Scholar]
- 109.Dorman S.C., Babirad I., Post J., Watson R.M., Foley R., Jones G.L. Progenitor egress from the bone marrow after allergen challenge: role of stromal cell-derived factor 1alpha and eotaxin. J Allergy Clin Immunol. 2005;115:501–507. doi: 10.1016/j.jaci.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 110.Radinger M., Bossios A., Alm A.S., Jeurink P., Lu Y., Malmhall C. Regulation of allergen-induced bone marrow eosinophilopoiesis: role of CD4+ and CD8+ T cells. Allergy. 2007;62:1410–1418. doi: 10.1111/j.1398-9995.2007.01509.x. [DOI] [PubMed] [Google Scholar]
- 111.Fregonese L., Silvestri M., Sabatini F., Rossi G.A. Cysteinyl leukotrienes induce human eosinophil locomotion and adhesion molecule expression via a CysLT1 receptor-mediated mechanism. Clin Exp Allergy. 2002;32:745–750. doi: 10.1046/j.1365-2222.2002.01384.x. [DOI] [PubMed] [Google Scholar]
- 112.Braccioni F., Dorman S.C., O’Byrne P.M., Inman M.D., Denburg J.A., Parameswaran K. The effect of cysteinyl leukotrienes on growth of eosinophil progenitors from peripheral blood and bone marrow of atopic subjects. J Allergy Clin Immunol. 2002;110:96–101. doi: 10.1067/mai.2002.125000. [DOI] [PubMed] [Google Scholar]
- 113.Bautz F., Denzlinger C., Kanz L., Mohle R. Chemotaxis and transendothelial migration of CD34(+) hematopoietic progenitor cells induced by the inflammatory mediator leukotriene D4 are mediated by the 7-transmembrane receptor CysLT1. Blood. 2001;97:3433–3440. doi: 10.1182/blood.v97.11.3433. [DOI] [PubMed] [Google Scholar]
- 114.Punia N., Thomson J., Babirad I., Sehmi R. IL-4 and IL-13 prime the transmigrational responses of hemopoietic progenitor cells (abst) Am J Respir Crit Care Med. 2010;181:A4034. [Google Scholar]
- 115.Pettipher R. The roles of the prostaglandin D(2) receptors DP(1) and CRTH2 in promoting allergic responses. Br J Pharmacol. 2008;153(Suppl. 1):S191–S199. doi: 10.1038/sj.bjp.0707488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ueki S., Kato H., Kobayashi Y., Ito W., Adachi T., Nagase H. Anti- and proinflammatory effects of 15-deoxy-delta-prostaglandin J2(15d-PGJ2) on human eosinophil functions. Int Arch Allergy Immunol. 2007;143(Suppl. 1):15–22. doi: 10.1159/000101399. [DOI] [PubMed] [Google Scholar]
- 117.Smith S.G., Sehmi R., Howie K., Watson R.M., Campbell H., Obminski G. Effects of peroxisome proliferator-activated receptors (PPARs) on eosinophil migration (abst) Am J Respir Crit Care Med. 2010;181:A2787. [Google Scholar]
- 118.Bochner B.S., Gleich G.J. What targeting eosinophils has taught us about their role in diseases. J Allergy Clin Immunol. 2010;126:16–25. doi: 10.1016/j.jaci.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Flood-Page P., Menzies-Gow A., Phipps S., Ying S., Wangoo A., Ludwig M.S. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Menzies-Gow A., Flood-Page P., Sehmi R., Burman J., Hamid Q., Robinson D.S. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J Allergy Clin Immunol. 2003;111:714–719. doi: 10.1067/mai.2003.1382. [DOI] [PubMed] [Google Scholar]
- 121.Leckie M.J., ten Brinke A., Khan J., Diamant Z., O’Connor B.J., Walls C.M. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 122.Kips J.C., O’Connor B.J., Langley S.J., Woodcock A., Kerstjens H.A., Postma D.S. Effect of SCH55700, a humanized anti-human interleukin-5 antibody, in severe persistent asthma: a pilot study. Am J Respir Crit Care Med. 2003;167:1655–1659. doi: 10.1164/rccm.200206-525OC. [DOI] [PubMed] [Google Scholar]
- 123.Flood-Page P.T., Menzies-Gow A.N., Kay A.B., Robinson D.S. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 124.Flood-Page P., Swenson C., Faiferman I., Matthews J., Williams M., Brannick L. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062–1071. doi: 10.1164/rccm.200701-085OC. [DOI] [PubMed] [Google Scholar]
- 125.O’Byrne P.M., Inman M.D., Parameswaran K. The trials and tribulations of IL-5, eosinophils, and allergic asthma. J Allergy Clin Immunol. 2001;108:503–508. doi: 10.1067/mai.2001.119149. [DOI] [PubMed] [Google Scholar]
- 126.Nair P., Pizzichini M.M., Kjarsgaard M., Inman M.D., Efthimiadis A., Pizzichini E. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 127.Haldar P., Brightling C.E., Hargadon B., Gupta S., Monteiro W., Sousa A. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Busse W.W., Katial R., Gossage D., Sari S., Wang B., Kolbeck R. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol. 2010;125:1237–1244. doi: 10.1016/j.jaci.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 129.Kolbeck R., Kozhich A., Koike M., Peng L., Andersson C.K., Damschroder M.M. MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J Allergy Clin Immunol. 2010;125:1344–1353. doi: 10.1016/j.jaci.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 130.Gauvreau G.M., Boulet L.P., Cockcroft D.W., Baatjes A., Cote J., Deschesnes F. Antisense therapy against CCR3 and the common beta chain attenuates allergen-induced eosinophilic responses. Am J Respir Crit Care Med. 2008;177:952–958. doi: 10.1164/rccm.200708-1251OC. [DOI] [PubMed] [Google Scholar]
- 131.Gauvreau G.M., Pageau R., Seguin R., Carballo D., D’Anjou H., Campbell H. Efficacy of increasing doses of TPI ASM8 on allergen inhalation challenges in asthmatics (abst) Am J Respir Crit Care Med. 2010;181:A5669. [Google Scholar]
- 132.Bousquet J., Clark T.J., Hurd S., Khaltaev N., Lenfant C., O’Byrne P. GINA guidelines on asthma and beyond. Allergy. 2007;62:102–112. doi: 10.1111/j.1398-9995.2006.01305.x. [DOI] [PubMed] [Google Scholar]
- 133.Barnes P.J., Pedersen S., Busse W.W. Efficacy and safety of inhaled corticosteroids. New developments. Am J Respir Crit Care Med. 1998;157:S1–53. doi: 10.1164/ajrccm.157.3.157315. [DOI] [PubMed] [Google Scholar]
- 134.Hargreave F.E., Dolovich J., Newhouse M.T. The assessment and treatment of asthma: a conference report. J Allergy Clin Immunol. 1990;85:1098–1111. doi: 10.1016/0091-6749(90)90056-a. [DOI] [PubMed] [Google Scholar]
- 135.Gibson P.G., Wong B.J., Hepperle M.J., Kline P.A., Girgis-Gabardo A., Guyatt G. A research method to induce and examine a mild exacerbation of asthma by withdrawal of inhaled corticosteroid. Clin Exp Allergy. 1992;22:525–532. doi: 10.1111/j.1365-2222.1992.tb00161.x. [DOI] [PubMed] [Google Scholar]
Chapter 13.7. Eosinophil Activities and Virus-Induced Asthma
Elizabeth R. Bivins-Smith, David B. Jacoby
Virus infections are the primary cause of asthma exacerbations in both children and adults. During virus-induced asthma, eosinophils in contact with airway nerves become activated and release mediators that cause airway nerve dysfunction. Airway parasympathetic nerves, which provide the dominant control over airway smooth muscle, release excess acetylcholine, which causes bronchoconstriction. Soluble mediators released by activated eosinophils also participate in virus clearance in the host. Eosinophils can recognize viruses, become activated, present viral antigens, and release virucidal mediators. Once considered solely detrimental, these cells are now gaining recognition for their beneficial antiviral role in virus-induced asthma. Further investigation is warranted to elucidate the mechanisms of eosinophil-mediated virus clearance in patients with asthma.
Virus-Induced Asthma Attacks: An Overview
Asthma is a chronic pulmonary disease characterized by airway remodeling, airway inflammation, and bronchoconstriction. Patients with asthma cycle between periods of exacerbations, which result in significant morbidity, and recovery. Virus infections are the leading cause of asthma exacerbations in children and adults, yet specific treatment and prevention strategies for virus-induced asthma are limited. The tissue damage caused during virus-induced asthma is divided into two categories: damage caused by the virus itself and damage from the host immune response. Eosinophils play a significant role in both virus clearance and immunity-mediated tissue damage.
As many as 80% of childhood cases and 55% of adult cases of asthma attacks have an identifiable underlying virus infection.1., 2. Diagnosis of virus infection in acute asthma is often presumptive and based on patient history and physical examination, but other diagnostic laboratory techniques include serology, virus culture, and reverse-transcription polymerase chain reaction. Respiratory RNA viruses are the main types of virus that induce asthma attacks, with rhinovirus (the common cold virus) accounting for approximately two-thirds of viruses identified.3 Coronavirus, influenza, parainfluenza, and respiratory syncytial virus (RSV) comprise the remainder of respiratory RNA viruses that induce asthma attacks.1
The immune response to viral infections is T-helper type 1 (Th1)-driven and involves the production of proinflammatory cytokines such as interleukin-12 (IL-12) and interferon γ (IFN-γ). Studies show that Th1 responses in patients with asthma are impaired, which results in decreased Th1 cytokine production during viral infection.4 Asthma also skews the immune system away from a Th1 response toward a Th2 immune environment through increased production of Th2 cytokines, such as IL-4, IL-5, and IL-13. IL-5 is the central cytokine involved in eosinophil proliferation, maturation, and survival. When the immune environment of a virus-infected patient with asthma shifts from Th1 to a Th2 response, these patients experience more severe symptoms of infection and delayed pathogen clearance, which likely contribute to increased asthma exacerbations and hospital admissions.4., 5.
The association of eosinophils with virus-induced asthma is well documented. Eosinophil products are present in the sputum of patients with virus-induced asthma,6 and histology studies show that in patients who have died from severe asthma, eosinophils are clustered around airway nerves (Fig. 13.7.1 ).7 In the following sections, we will present the mechanisms of eosinophil pathophysiology in virus-induced asthma, focusing on virus-induced airway inflammation, viral activation of eosinophils, virus-induced eosinophil-mediated neural changes, and T cell–eosinophil interactions during virus infection. We then discuss the beneficial role of eosinophils in virus-induced asthma and potential targets for prevention and treatment of the disease.
FIGURE 13.7.1.

Eosinophils are physically associated with airway nerves in asthma.
Postmortem airway tissues from patients with fatal asthma were stained with antibodies to the nerve-specific protein PGP9.5 (black) and to eosinophil MBP (red). A, Eosinophils line up along nerve fibers in airway smooth muscle layer. B, Eosinophils surround an airway nerve bundle, seen longitudinally. C, Eosinophils surround a nerve bundle, cut in cross section. Note extracellular MBP (arrow). D, A nerve fiber is encrusted with extracellular MBP.7
Eosinophils in Virus-Induced Airway Inflammation
Respiratory virus infection induces airway inflammation in all individuals, but the inflammatory response is different in patients with asthma from in nonasthmatic individuals. Experimental rhinovirus infection of human subjects increases eosinophil numbers in the bronchial epithelium in both nonasthmatic and asthmatic volunteers. Eosinophils remained in airway tissues of virus-infected patients with asthma 6 weeks longer than in infected, nonasthmatic controls. These data suggest that virus-induced eosinophil influx into the airways occurs in individuals both with and without asthma and that the eosinophil response to viral infection is accentuated in patients with asthma.8
The airway epithelium is the primary site of respiratory virus infection. Virus-infected epithelial cells release a wide array of proinflammatory cytokines that recruit inflammatory cells into the airways. Among these proinflammatory cytokines are eotaxin/C-C motif chemokine 11 (CCL11), granulocyte-macrophage colony-stimulating factor (GM-CSF), MIP-1α/CCL3, and RANTES/CCL5, which are chemoattractants and activators of eosinophils.9., 10., 11., 12., 13., 14., 15., 16. Virus infection increased eosinophil inflammation in the airways of antigen-sensitized and antigen-challenged mice compared to sensitized-challenged, mock-infected animals. In addition, virus infection of epithelial macrophages induced the release of eotaxin, and the blockade of eotaxin bioactivity with a neutralizing antibody inhibited airway eosinophilia in sensitized-challenged, virus-infected but not sensitized-challenged, mock-infected mice. These data suggest that virus infection recruits eosinophils to the lungs of sensitized-challenged animals and that this is dependent on eotaxin.17
Virus infections of allergic individuals may lead to the development of eosinophilic inflammation as well as features of asthma. Calhoun et al. showed that patients with allergic rhinitis who were experimentally infected with rhinovirus had increased eosinophil influx into the airways following allergen challenge. These patients also had increased histamine release and edema from fluid leakage, which are consistent with asthma. These data suggest that virus infection may induce asthma in individuals with preexisting allergic respiratory diseases. The Th2 immune environment that allergic rhinitis elicits closely resembles the cytokine profiles of asthma, so a subsequent viral infection may tip airway physiology toward an asthma phenotype.18
Virus-Induced Eosinophil Activation
Eosinophils are activated during viral infection. Studies of patients with RSV bronchiolitis showed the presence of eosinophil cationic protein (ECP) and eosinophil-derived neurotoxin (EDN) in their lower airway secretions.19., 20. Other studies have shown that cell surface expression of CD11b, a marker of cellular activation, is upregulated in eosinophils of RSV-infected patients compared to uninfected volunteers.21 These data suggest that eosinophils are activated and degranulate following viral infection.
Toll-like receptors (TLRs) are pattern recognition receptors that recognize pathogen-associated molecular patterns (PAMPs). Many groups have shown that TLRs are present and functional in eosinophils. TLRs mediate both the innate and adaptive immune responses, and some believe they are involved in controlling sensitization. Studies suggest that polymorphisms in TLR2, TLR4, TLR9, and TLR10 are associated with an increased risk of developing asthma.22., 23., 24., 25. Human eosinophils express TLR1, TLR4, TLR7, TLR9, and TLR10 mRNA. TLR7 recognizes single-stranded RNA (ssRNA) found in respiratory RNA viruses. Nagase et al. showed that R848 (a synthetic ligand for TLR7 and TLR8) increases CD11b expression on human eosinophils, induces superoxide generation, and promotes cell survival.26 Phipps et al. showed that mouse eosinophils express TLR7 on their cell surface and TLR3, TLR4, and TLR7 in endosomes. Furthermore, ssRNA treatment of mouse eosinophils increases eosinophil peroxidase (EPO) release, increases CD11b expression, and induces degranulation.27
Eosinophil TLR responses to stimulation may differ between atopic and nonatopic individuals. Mansson et al. showed that eosinophils treated with IL-5 and then stimulated with polyI:C (a TLR3 agonist) had increased IL-8 release compared to IL-5 treatment alone.28 In addition, IL-5 pretreatment potentiates R837 (a TLR7-specific synthetic ligand)-induced IL-8 release. Moreover, eosinophils collected from atopic patients and stimulated with R837 release increased IL-8 compared to R837-stimulated eosinophils from nonallergic volunteers.29 These data suggest that atopy affects eosinophil TLR responses and that aberrant virus-induced eosinophil activation in patients with asthma may exacerbate their airway disease.
Eosinophils can be infected and activated by respiratory viruses. Eosinophils can be activated directly by virus binding to receptors on either the cell surface or in endosomes, depending on the virus’s mechanism of entry. RSV mediates entry into target cells by binding TLR4. Although it has not been demonstrated that RSV binds TLR4 on eosinophils, in vitro culture of RSV with human eosinophils prolongs cell survival.30 Transmission electron microscopy studies showed that RSV is internalized by human eosinophils and identified virions in phagocytic vacuoles near the cell surface. In addition, infected eosinophils underwent piecemeal degranulation, suggesting that these cells were activated upon infection.31
In some cases, virus-induced eosinophil activation requires priming prior to infection and, in other cases, viruses prime eosinophils to respond to treatment with additional stimuli. Handzel et al. showed that rhinovirus binds intracellular adhesion molecule-1 (ICAM-1) on human eosinophils primed with GM-CSF and that GM-CSF treatment upregulates ICAM-1 on eosinophils.32 Additional studies showed that RSV increased superoxide generation and CD11b in eosinophils that were first treated with platelet activating factor.33 RSV infection of eosinophils potentiates phorbol-12-myristate-13-acetate (PMA)-induced superoxide generation and leukotriene C4 release.34 These data suggest that virus-induced eosinophil activation in asthma may lead to an exaggerated host immune response, resulting in airway inflammation and constriction.
Virus-Induced Airway Hyperreactivity: The Role of Eosinophils
During virus-induced asthma, eosinophils cause dysfunction of parasympathetic nerve signaling. Airway parasympathetic nerves provide the dominant control over airway smooth muscle.35 These neurons release acetylcholine (ACh), which binds M3 muscarinic receptors (M3Rs) on smooth muscle to cause bronchoconstriction.36 ACh release is normally limited via negative feedback inhibition of ACh on neuronal M2 muscarinic receptors (M2Rs).37 When neuronal M2Rs are dysfunctional, ACh release by neurons is not inhibited, and increased levels of ACh binding to M3Rs on airway smooth muscle cause airway hyperresponsiveness (AHR).
In antigen-sensitized, antigen-challenged guinea pigs, eosinophils mediate AHR.38 Parasympathetic nerves recruit eosinophils to the nerves via eotaxin signaling.39 Cholinergic nerves express ICAM-1 and vascular cell adhesion protein (VCAM), which bind to eosinophils.40 Eosinophils bind to nerves and release major basic protein (MBP), which blocks M2Rs on nerves. This causes loss of M2R function, resulting in increased ACh release and bronchoconstriction.41., 42.
Viral infection of nonsensitized guinea pigs causes the loss of M2R function and AHR, but this loss is not eosinophil-mediated (not prevented by antibody to IL5 or antibody to MBP).43 In nonsensitized guinea pigs, the virus-induced loss of M2R function appears to be on the level of reduced M2R gene expression. This appears to be the result of production of TNF-α, which decreases M2R mRNA stability.44 In contrast, in sensitized guinea pigs, virus-induced loss of M2R function and AHR is mediated by eosinophils. Adamko et al. showed that depletion of eosinophils with an anti-IL-5 antibody prevents virus-induced AHR in sensitized guinea pigs, but not in nonsensitized, virus-infected animals. Additional studies showed that blockade of MBP bioactivity inhibits virus-induced AHR and M2R dysfunction in sensitized guinea pigs. Collectively, these data suggest that in the context of asthma, eosinophils are recruited to airway nerves, virus infection activates the eosinophils to release MBP, and MBP antagonizes M2Rs, resulting in bronchoconstriction (Fig. 13.7.2 ).45
FIGURE 13.7.2.

Viral infection activates airway eosinophils in a CD8+ T cell-dependent process.
Activated eosinophils release major basic protein, which binds to inhibitory M2 muscarinic receptors on parasympathetic nerves. Blocking these receptors increases acetylcholine (Ach) release, causing bronchoconstriction. M3, M3 muscarinic receptor.
Interactions Between T Lymphocytes and Eosinophils in Virus-Induced Asthma
In addition to eosinophil–nerve interactions, eosinophils communicate with T cells in virus-induced asthma. As mentioned above, eosinophils mediate virus-induced AHR and M2R dysfunction in sensitized guinea pigs but not in nonsensitized animals.45 Adamko et al. demonstrated that depletion of CD8+ T cells prevents virus-induced AHR and M2R dysfunction in sensitized but not in nonsensitized guinea pigs. Sensitization increases the number of eosinophils in the airways, and virus infection of both nonsensitized and sensitized guinea pigs reduces the total number of eosinophils in the airways and eosinophils associated with nerves, suggesting that virus infection induces eosinophil degranulation. CD8+ T cell depletion inhibits virus-induced eosinophil cytolysis, suggesting that CD8+ T cells promote virus-induced eosinophil degranulation.46 These data suggest that CD8+ T cells interact with eosinophils in the airways of virus-infected, sensitized animals to promote AHR and M2R dysfunction. Schwarze et al. showed that CD8+ T cells are also necessary for AHR and lung eosinophilia during RSV infection of mice.47
In vitro studies have shown that eosinophils directly interact with T cells to induce eosinophil degranulation and promote antiviral, T cell-mediated immunity. When human eosinophils are incubated with parainfluenza virus in the presence of T cells and antigen-presenting cells (APCs; macrophages or dendritic cells), they release EPO. UV-inactivated virus also induces EPO release from eosinophils, suggesting that this process occurs in the absence of viral replication. Additional studies showed that RSV is also able to induce EPO release from eosinophils in the presence of T cells and APCs.48 Handzel et al. showed that eosinophils present rhinovirus antigens to CD4+ T cells, inducing their clonal expansion and the release of IFN-γ.32 These data suggest that eosinophils directly interact with CD4+ and CD8+ T cells, and that their interactions result in eosinophil degranulation, which may augment symptoms of asthma as well as initiate antiviral adaptive immunity. Despite the detrimental role eosinophils play in the pathophysiology of asthma, the antiviral effect of eosinophils may also be beneficial in the context of virus infection and asthma.
Benefits of Eosinophils in Virus-Induced Asthma
The antibacterial and antiparasitic effects of eosinophils are well documented, yet their antiviral properties have been studied only recently. While eosinophils have historically been acknowledged for their pathophysiological role in asthma, recent studies suggest that these cells also play a beneficial antiviral role in virus-induced asthma. Adamko et al. showed that guinea pigs sensitized to ovalbumin had reduced parainfluenza titers in their lungs 4 days following infection compared to nonsensitized, virus-infected animals. Furthermore, depletion of eosinophils with an anti-IL-5 antibody increased viral levels in sensitized virus-infected animals to above those observed in nonsensitized infected animals.45 These data suggest that eosinophils serve an antiviral role in the context of virus infection and allergy.
Additional studies have investigated the mechanisms by which eosinophils promote virus clearance in vivo. Phipps et al. showed that IL-5 transgenic mice, which have increased levels of eosinophils, have improved RSV clearance compared to wild-type RSV-infected mice. MyD88 is the adaptor molecule for many TLR7 signaling pathways, and studies show that adoptive transfer of wild-type eosinophils, but not eosinophils deficient in MyD88, into the lungs of infected wild-type mice improves virus clearance and inhibits AHR. Blockade of nitric oxide synthase 2 (NOS-2; inducible nitric oxide synthase) activity inhibits RSV clearance in vivo. These data suggest that eosinophils contribute to RSV clearance in both a MyD88-dependent and NOS-dependent manner. Furthermore, these data suggest that eosinophils may aid the prevention of RSV-induced AHR.27
As mentioned above, eosinophils can be activated by viruses to degranulate. Upon degranulation, eosinophils release granule proteins and reactive oxygen species, which can cause damage to pathogen moieties. Several groups have shown that eosinophils increase superoxide generation in response to virus exposure.33., 34. Klebanoff et al. showed that human eosinophils stimulated with PMA are virucidal against human immunodeficiency virus (HIV) type 1 in vitro and that purified EPO, when incubated with hydrogen peroxide and a halide (its substrate), is also virucidal against HIV.49 ECP and EDN contain intrinsic ribonuclease activity that can destroy the genomes of RNA viruses. Domachowske et al. showed that eosinophils and human recombinant EDN have antiviral activity toward RSV in vitro.50 Others have demonstrated an antiviral effect of EDN against HIV in vitro.51., 52. Collectively, these data suggest that eosinophils inactivate RNA viruses by a number of different mechanisms involving granule proteins and reactive oxygen species.
While some studies have shown that eosinophils destroy virus, other groups have shown that eosinophils are productively infected by viruses. As mentioned previously, eosinophils can bind and internalize viruses. Kimpen et al. showed that RSV can bind to the eosinophil membrane and observed virus in the phagocytic vacuoles of eosinophils.31., 34. Dyer et al. showed that RSV and mouse pneumovirus productively infect human and mouse eosinophils, respectively, resulting in the release of infectious virions and proinflammatory cytokines in vitro.53 In contrast, our laboratory has presented evidence suggesting that parainfluenza virus abortively infects human eosinophils. Parainfluenza virus can enter eosinophils and replicate the viral RNA genome, but infectious virus particles are not released.54 A productive infection of eosinophils could potentially serve to induce cytokine release and attract other immune cells, while an abortive infection would remove extracellular virus from the local environment and/or promote the presentation of virus antigens to T cells. Collectively these data suggest that interactions between eosinophils and different types of viruses are varied and complex.
Therapeutic Targets and Prevention Strategies in Virus-Induced Asthma
Standard treatments for patients suffering from virus-induced asthma exacerbations consist of β-agonists in combination with anticholinergics and steroids. β-agonists relax smooth muscle by stimulating adenylate cyclase activity and closing calcium channels, while anticholinergics block the effects of the neurotransmitter ACh on airway smooth muscle constriction. Viruses and interferon downregulate inhibitory M2Rs on parasympathetic nerves, thereby increasing ACh release, and steroids can reverse these effects.55 Steroids can also reduce eosinophil influx into the lungs, ICAM-1 expression on nerves, and neuronal M2R dysfunction.56., 57.
While eosinophil in sensitized animals can prevent virus-induced hyperreactivity,45 the effects of eosinophil depletion in human virus-induced asthma attacks are less clear. While eosinophil depletion with anti-IL5 has limited effects on the response to antigen challenge in human subjects58 and a clinical study of anti-IL5 had unimpressive effects in unselected asthmatics, when asthmatics with sputum eosinophilia were treated with anti-IL5, steroids were withdrawn without exacerbation in the majority of cases.59 Whether eosinophil depletion can prevent virus-induced asthma exacerbation awaits further studies.
Another potential treatment for virus-induced asthma is the correction of deregulated cytokine production. As mentioned previously, the immune environment of patients with asthma is skewed toward a Th2 phenotype, with decreased interferon production.4 Treatments that promote a Th1 phenotype, the immune response typically observed in viral infection, may prove clinically valuable.
The most direct method of treatment or prevention of virus-induced asthma is targeting the viral infection itself. However, such treatments are at present limited.
Rhinovirus
Treatment and prevention of rhinovirus, the cause of two-thirds of virus-induced asthma exacerbations, presents a significant challenge because over 100 serotypes of the virus exist. The varying antigenicity among serotypes further hinders vaccine design. As mentioned previously, rhinovirus uses ICAM-1 binding for entry into cells, thus the blockade of cellular infection by blocking virus binding to target cells presents one promising mechanism for drug design. While ribavirin, which is believed to interfere with viral RNA synthesis, has activity against a range of viruses, including rhinovirus and RSV, its clinical efficacy is limited and it is not widely used in the treatment of rhinovirus.
Respiratory Syncytial Virus
RSV vaccine development has been problematic, with paradoxical worsening of the clinical response being an issue. Palivizumab is a monoclonal antibody that prevents RSV infection by interfering with the RSV fusion protein and, thus, viral entry into cells. In addition, studies show that type IV phosphodiesterase inhibitors inhibit RSV-induced AHR and eosinophilia in the lungs.60
Influenza
Effective vaccines for influenza are recommended for patients with airway disease, including asthma. Although it is difficult to prove that influenza-induced asthma attacks are reduced by vaccination, this may be due to the relatively small number of asthma attacks caused by influenza. Treatment for influenza viral infections consists of neuraminidase inhibitors. Again, the role of neuraminidase inhibitors in preventing asthma attacks is difficult to demonstrate.
Coronavirus and Parainfluenza Virus
Currently no treatments or vaccines exist for parainfluenza virus and coronavirus. Improved immunological understanding of these viruses and of virus interactions with the host, and eosinophils specifically, will aid in the advancement of future vaccines and treatments.
Conclusion
Respiratory virus infections are the primary cause of asthma exacerbations in children and adults. In asthma, eosinophils are recruited to the airways as a part of the Th2 immune response, where they contribute to asthma pathophysiology. However, eosinophils may play a dual role in virus-induced asthma; on the one hand recognizing viruses, releasing virucidal mediators, and presenting antigens, and on the other hand becoming activated and increasing airway reactivity. Current treatments for virus-induced asthma are not specific to virus infection and instead focus on smooth muscle relaxation and reduction of airway inflammation. Further investigation of the interactions between eosinophils and viruses is warranted and will likely lead to more targeted treatments and prevention.
References
- 1.Johnston S.L., Pattemore P.K., Sanderson G., Smith S., Lampe F., Josephs L. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atmar R.L., Guy E., Guntupalli K.K., Zimmerman J.L., Bandi V.D., Baxter B.D. Respiratory tract viral infections in inner-city asthmatic adults. Arch Intern Med. 1998;158:2453–2459. doi: 10.1001/archinte.158.22.2453. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson K.G., Kent J., Ireland D.C. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papadopoulos N.G., Stanciu L.A., Papi A., Holgate S.T., Johnston S.L. A defective type 1 response to rhinovirus in atopic asthma. Thorax. 2002;57:328–332. doi: 10.1136/thorax.57.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gern J.E., Vrtis R., Grindle K.A., Swenson C., Busse W.W. Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am J Respir Crit Care Med. 2000;162:2226–2231. doi: 10.1164/ajrccm.162.6.2003019. [DOI] [PubMed] [Google Scholar]
- 6.Grunberg K., Smits H.H., Timmers M.C., de Klerk E.P., Dolhain R.J., Dick E.C. Experimental rhinovirus 16 infection. Effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am J Respir Crit Care Med. 1997;156:609–616. doi: 10.1164/ajrccm.156.2.9610079. [DOI] [PubMed] [Google Scholar]
- 7.Costello R.W., Schofield B.H., Kephart G.M., Gleich G.J., Jacoby D.B., Fryer A.D. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am J Physiol. 1997;273:L93–103. doi: 10.1152/ajplung.1997.273.1.L93. [DOI] [PubMed] [Google Scholar]
- 8.Fraenkel D.J., Bardin P.G., Sanderson G., Lampe F., Johnston S.L., Holgate S.T. Lower airways inflammation during rhinovirus colds in normal and in asthmatic subjects. Am J Respir Crit Care Med. 1995;151:879–886. doi: 10.1164/ajrccm/151.3_Pt_1.879. [DOI] [PubMed] [Google Scholar]
- 9.Griego S.D., Weston C.B., Adams J.L., Tal-Singer R., Dillon S.B. Role of p38 mitogen-activated protein kinase in rhinovirus-induced cytokine production by bronchial epithelial cells. J Immunol. 2000;165:5211–5220. doi: 10.4049/jimmunol.165.9.5211. [DOI] [PubMed] [Google Scholar]
- 10.Subauste M.C., Jacoby D.B., Richards S.M., Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest. 1995;96:549–557. doi: 10.1172/JCI118067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsukura S., Kokubu F., Noda H., Tokunaga H., Adachi M. Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J Allergy Clin Immunol. 1996;98:1080–1087. doi: 10.1016/s0091-6749(96)80195-3. [DOI] [PubMed] [Google Scholar]
- 12.Noah T.L., Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol. 1993;265:L472–L478. doi: 10.1152/ajplung.1993.265.5.L472. [DOI] [PubMed] [Google Scholar]
- 13.Becker S., Reed W., Henderson F.W., Noah T.L. RSV infection of human airway epithelial cells causes production of the beta-chemokine RANTES. Am J Physiol. 1997;272:L512–L520. doi: 10.1152/ajplung.1997.272.3.L512. [DOI] [PubMed] [Google Scholar]
- 14.Olszewska-Pazdrak B., Casola A., Saito T., Alam R., Crowe S.E., Mei F. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol. 1998;72:4756–4764. doi: 10.1128/jvi.72.6.4756-4764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawaguchi M., Kokubu F., Kuga H., Tomita T., Matsukura S., Kadokura M. Expression of eotaxin by normal airway epithelial cells after influenza virus A infection. Int Arch Allergy Immunol. 2000;122(Suppl. 1):44–49. doi: 10.1159/000053632. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulos N.G., Papi A., Meyer J., Stanciu L.A., Salvi S., Holgate S.T. Rhinovirus infection up-regulates eotaxin and eotaxin-2 expression in bronchial epithelial cells. Clin Exp Allergy. 2001;31:1060–1066. doi: 10.1046/j.1365-2222.2001.01112.x. [DOI] [PubMed] [Google Scholar]
- 17.Nagarkar D.R., Bowman E.R., Schneider D., Wang Q., Shim J., Zhao Y. Rhinovirus infection of allergen-sensitized and -challenged mice induces eotaxin release from functionally polarized macrophages. J Immunol. 2010;185:2525–2535. doi: 10.4049/jimmunol.1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calhoun W.J., Dick E.C., Schwartz L.B., Busse W.W. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest. 1994;94:2200–2208. doi: 10.1172/JCI117581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison A.M., Bonville C.A., Rosenberg H.F., Domachowske J.B. Respiratory syncytical virus-induced chemokine expression in the lower airways: eosinophil recruitment and degranulation. Am J Respir Crit Care Med. 1999;159:1918–1924. doi: 10.1164/ajrccm.159.6.9805083. [DOI] [PubMed] [Google Scholar]
- 20.Garofalo R., Kimpen J.L., Welliver R.C., Ogra P.L. Eosinophil degranulation in the respiratory tract during naturally acquired respiratory syncytial virus infection. J Pediatr. 1992;120:28–32. doi: 10.1016/s0022-3476(05)80592-x. [DOI] [PubMed] [Google Scholar]
- 21.Lindemans C.A., Kimpen J.L., Luijk B., Heidema J., Kanters D., van der Ent C.K. Systemic eosinophil response induced by respiratory syncytial virus. Clin Exp Immunol. 2006;144:409–417. doi: 10.1111/j.1365-2249.2006.03084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eder W., Klimecki W., Yu L., von Mutius E., Riedler J., Braun-Fahrlander C. Toll-like receptor 2 as a major gene for asthma in children of European farmers. J Allergy Clin Immunol. 2004;113:482–488. doi: 10.1016/j.jaci.2003.12.374. [DOI] [PubMed] [Google Scholar]
- 23.Raby B.A., Klimecki W.T., Laprise C., Renaud Y., Faith J., Lemire M. Polymorphisms in toll-like receptor 4 are not associated with asthma or atopy-related phenotypes. Am J Respir Crit Care Med. 2002;166:1449–1456. doi: 10.1164/rccm.200207-634OC. [DOI] [PubMed] [Google Scholar]
- 24.Lazarus R., Raby B.A., Lange C., Silverman E.K., Kwiatkowski D.J., Vercelli D. TOLL-like receptor 10 genetic variation is associated with asthma in two independent samples. Am J Respir Crit Care Med. 2004;170:594–600. doi: 10.1164/rccm.200404-491OC. [DOI] [PubMed] [Google Scholar]
- 25.Lazarus R., Klimecki W.T., Raby B.A., Vercelli D., Palmer L.J., Kwiatkowski D.J. Single-nucleotide polymorphisms in the Toll-like receptor 9 gene (TLR9): frequencies, pairwise linkage disequilibrium, and haplotypes in three U.S. ethnic groups and exploratory case-control disease association studies. Genomics. 2003;81:85–91. doi: 10.1016/s0888-7543(02)00022-8. [DOI] [PubMed] [Google Scholar]
- 26.Nagase H., Okugawa S., Ota Y., Yamaguchi M., Tomizawa H., Matsushima K. Expression and function of Toll-like receptors in eosinophils: activation by Toll-like receptor 7 ligand. J Immunol. 2003;171:3977–3982. doi: 10.4049/jimmunol.171.8.3977. [DOI] [PubMed] [Google Scholar]
- 27.Phipps S., Lam C.E., Mahalingam S., Newhouse M., Ramirez R., Rosenberg H.F. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood. 2007;110:1578–1586. doi: 10.1182/blood-2007-01-071340. [DOI] [PubMed] [Google Scholar]
- 28.Mansson A., Fransson M., Adner M., Benson M., Uddman R., Bjornsson S. TLR3 in human eosinophils: functional effects and decreased expression during allergic rhinitis. Int Arch Allergy Immunol. 2010;151:118–128. doi: 10.1159/000236001. [DOI] [PubMed] [Google Scholar]
- 29.Mansson A., Cardell L.O. Role of atopic status in Toll-like receptor (TLR)7- and TLR9-mediated activation of human eosinophils. J Leukoc Biol. 2009;85:719–727. doi: 10.1189/jlb.0808494. [DOI] [PubMed] [Google Scholar]
- 30.Lindemans C.A., Coffer P.J., Schellens I.M., de Graaff P.M., Kimpen J.L., Koenderman L. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J Immunol. 2006;176:5529–5537. doi: 10.4049/jimmunol.176.9.5529. [DOI] [PubMed] [Google Scholar]
- 31.Kimpen J.L., Garofalo R., Welliver R.C., Fujihara K., Ogra P.L. An ultrastructural study of the interaction of human eosinophils with respiratory syncytial virus. Pediatr Allergy Immunol. 1996;7:48–53. doi: 10.1111/j.1399-3038.1996.tb00105.x. [DOI] [PubMed] [Google Scholar]
- 32.Handzel Z.T., Busse W.W., Sedgwick J.B., Vrtis R., Lee W.M., Kelly E.A. Eosinophils bind rhinovirus and activate virus-specific T cells. J Immunol. 1998;160:1279–1284. [PubMed] [Google Scholar]
- 33.Tachibana A., Kimura H., Kato M., Nako Y., Kozawa K., Morikawa A. Respiratory syncytial virus enhances the expression of CD11b molecules and the generation of superoxide anion by human eosinophils primed with platelet-activating factor. Intervirology. 2002;45:43–51. doi: 10.1159/000050086. [DOI] [PubMed] [Google Scholar]
- 34.Kimpen J.L., Garofalo R., Welliver R.C., Ogra P.L. Activation of human eosinophils in vitro by respiratory syncytial virus. Pediatr Res. 1992;32:160–164. doi: 10.1203/00006450-199208000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Nadel J.A., Barnes P.J. Autonomic regulation of the airways. Annu Rev Med. 1984;35:451–467. doi: 10.1146/annurev.me.35.020184.002315. [DOI] [PubMed] [Google Scholar]
- 36.Roffel A.F., Elzinga C.R., Zaagsma J. Muscarinic M3 receptors mediate contraction of human central and peripheral airway smooth muscle. Pulm Pharmacol. 1990;3:47–51. doi: 10.1016/0952-0600(90)90009-8. [DOI] [PubMed] [Google Scholar]
- 37.Fryer A.D., Maclagan J. Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol. 1984;83:973–978. doi: 10.1111/j.1476-5381.1984.tb16539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fryer A.D., Costello R.W., Yost B.L., Lobb R.R., Tedder T.F., Steeber D.A. Antibody to VLA-4, but not to L-selectin, protects neuronal M2 muscarinic receptors in antigen-challenged guinea pig airways. J Clin Invest. 1997;99:2036–2044. doi: 10.1172/JCI119372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fryer A.D., Stein L.H., Nie Z., Curtis D.E., Evans C.M., Hodgson S.T. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest. 2006;116:228–236. doi: 10.1172/JCI25423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawatzky D.A., Kingham P.J., Court E., Kumaravel B., Fryer A.D., Jacoby D.B. Eosinophil adhesion to cholinergic nerves via ICAM-1 and VCAM-1 and associated eosinophil degranulation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1279–L1288. doi: 10.1152/ajplung.00279.2001. [DOI] [PubMed] [Google Scholar]
- 41.Jacoby D.B., Gleich G.J., Fryer A.D. Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest. 1993;91:1314–1318. doi: 10.1172/JCI116331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans C.M., Fryer A.D., Jacoby D.B., Gleich G.J., Costello R.W. Pretreatment with antibody to eosinophil major basic protein prevents hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. J Clin Invest. 1997;100:2254–2262. doi: 10.1172/JCI119763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fryer A.D., Yarkony K.A., Jacoby D.B. The effect of leukocyte depletion on pulmonary M2 muscarinic receptor function in parainfluenza virus-infected guinea-pigs. Br J Pharmacol. 1994;112:588–594. doi: 10.1111/j.1476-5381.1994.tb13115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nie Z., Jacoby D.B., Fryer A.D. Etanercept prevents airway hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. Br J Pharmacol. 2009;156:201–210. doi: 10.1111/j.1476-5381.2008.00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adamko D.J., Yost B.L., Gleich G.J., Fryer A.D., Jacoby D.B. Ovalbumin sensitization changes the inflammatory response to subsequent parainfluenza infection. Eosinophils mediate airway hyperresponsiveness, m(2) muscarinic receptor dysfunction, and antiviral effects. J Exp Med. 1999;190:1465–1478. doi: 10.1084/jem.190.10.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adamko D.J., Fryer A.D., Bochner B.S., Jacoby D.B. CD8+ T lymphocytes in viral hyperreactivity and M2 muscarinic receptor dysfunction. Am J Respir Crit Care Med. 2003;167:550–556. doi: 10.1164/rccm.200206-506OC. [DOI] [PubMed] [Google Scholar]
- 47.Schwarze J., Cieslewicz G., Joetham A., Ikemura T., Hamelmann E., Gelfand E.W. CD8 T cells are essential in the development of respiratory syncytial virus-induced lung eosinophilia and airway hyperresponsiveness. J Immunol. 1999;162:4207–4211. [PubMed] [Google Scholar]
- 48.Davoine F., Cao M., Wu Y., Ajamian F., Ilarraza R., Kokaji A.I. Virus-induced eosinophil mediator release requires antigen-presenting and CD4+ T cells. J Allergy Clin Immunol. 2008;122:69–77. doi: 10.1016/j.jaci.2008.03.028. 77 e1–2. [DOI] [PubMed] [Google Scholar]
- 49.Klebanoff S.J., Coombs R.W. Virucidal effect of stimulated eosinophils on human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1996;12:25–29. doi: 10.1089/aid.1996.12.25. [DOI] [PubMed] [Google Scholar]
- 50.Domachowske J.B., Dyer K.D., Bonville C.A., Rosenberg H.F. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J Infect Dis. 1998;177:1458–1464. doi: 10.1086/515322. [DOI] [PubMed] [Google Scholar]
- 51.Lee-Huang S., Huang P.L., Sun Y., Kung H.F., Blithe D.L., Chen H.C. Lysozyme and RNases as anti-HIV components in beta-core preparations of human chorionic gonadotropin. Proc Natl Acad Sci U S A. 1999;96:2678–2681. doi: 10.1073/pnas.96.6.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rugeles M.T., Trubey C.M., Bedoya V.I., Pinto L.A., Oppenheim J.J., Rybak S.M. Ribonuclease is partly responsible for the HIV-1 inhibitory effect activated by HLA alloantigen recognition. AIDS. 2003;17:481–486. doi: 10.1097/00002030-200303070-00002. [DOI] [PubMed] [Google Scholar]
- 53.Dyer K.D., Percopo C.M., Fischer E.R., Gabryszewski S.J., Rosenberg H.F. Pneumoviruses infect eosinophils and elicit MyD88-dependent release of chemoattractant cytokines and interleukin-6. Blood. 2009;114:2649–2656. doi: 10.1182/blood-2009-01-199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bivins-Smith E. Eosinophils mediate antiviral effects in vivo and directly kill parainfluenza virus in vitro. 6th Biennial Symposium International Eosinophil Society. 2009 [Google Scholar]
- 55.Jacoby D.B., Xiao H.Q., Lee N.H., Chan-Li Y., Fryer A.D. Virus- and interferon-induced loss of inhibitory M2 muscarinic receptor function and gene expression in cultured airway parasympathetic neurons. J Clin Invest. 1998;102:242–248. doi: 10.1172/JCI1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans C.M., Jacoby D.B., Fryer A.D. Effects of dexamethasone on antigen-induced airway eosinophilia and M(2) receptor dysfunction. Am J Respir Crit Care Med. 2001;163:1484–1492. doi: 10.1164/ajrccm.163.6.2007047. [DOI] [PubMed] [Google Scholar]
- 57.Nie Z., Nelson C.S., Jacoby D.B., Fryer A.D. Expression and regulation of intercellular adhesion molecule-1 on airway parasympathetic nerves. J Allergy Clin Immunol. 2007;119:1415–1422. doi: 10.1016/j.jaci.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Leckie M.J., ten Brinke A., Khan J., Diamant Z., O’Connor B.J., Walls C.M. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 59.Nair P., Pizzichini M.M., Kjarsgaard M., Inman M.D., Efthimiadis A., Pizzichini E. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 60.Ikemura T., Schwarze J., Makela M., Kanehiro A., Joetham A., Ohmori K., Gelfand E.W. Type 4 phosphodiesterase inhibitors attenuate respiratory syncytial virus-induced airway hyper-responsiveness and lung eosinophilia. J Pharmacol Exp Ther. 2000;294:701–706. [PubMed] [Google Scholar]
Chapter 13.8. Eosinophils and Gastrointestinal Disease
Benjamin P. Davis, Marc E. Rothenberg
Primary eosinophilic gastrointestinal disorders (EGID) are diseases with eosinophilia in the absence of known causes. These disorders include eosinophilic colitis, eosinophilic enteritis, eosinophilic esophagitis, eosinophilic gastritis, and eosinophilic gastroenteritis, a term used when more than one gastrointestinal segment is involved. Common symptoms of EGID include abdominal pain, diarrhea, dysphagia, failure to thrive, food impaction, gastric dysmotility, irritability, and vomiting. Clues to the diagnosis of EGID include gastrointestinal problems refractory to conventional therapy, a history of atopy, peripheral blood eosinophilia, and a family history of EGID. Ruling out other secondary causes of tissue eosinophilia is important in the diagnosis of EGID. Diagnosis is dependent on the histopathology of an endoscopic biopsy.
Introduction
Primary eosinophilic gastrointestinal disorders (EGID) are diseases with eosinophilia in the absence of known causes (e.g., drug reactions, malignancy, and parasitic infections). These disorders include eosinophilic colitis, eosinophilic enteritis, eosinophilic esophagitis (EoE), eosinophilic gastritis, and eosinophilic gastroenteritis, a term used when more than one gastrointestinal (GI) segment is involved.1., 2. The symptoms of EGID include abdominal pain, diarrhea, dysphagia and food impaction, failure to thrive, gastric dysmotility, irritability, and vomiting. Both genetic and environmental factors have a role in EGID. Approximately 10% of patients with EGID have a first-degree relative that also has EGID. It appears that allergy may play a role. In fact, approximately 75% of EGID patients have atopy, allergen-free diets have been shown to be therapeutic, and tissue specimens show mast cell degranulation. Animal studies of EGID also provide evidence of allergic etiology.2 Although food-specific immunoglobulin E (IgE) is common in EGID patients, food-induced anaphylactic responses occur in only a minority of patients. Thus, EGID appears to fall between pure IgE-mediated food allergy and cellular-mediated hypersensitivity disorders (Fig. 13.8.1 ). In fact, a recent study shows a higher incidence of EoE in patients with celiac disease, supporting a cell-mediated hypersensitivity or T-helper type 1 (Th1)-mediated mechanism,3 although another recent study demonstrates a lack of celiac disease-associated alleles in EoE.4
FIGURE 13.8.1.

The spectrum of eosinophilic gastrointestinal disorders.
Eosinophilic gastrointestinal disorders (EGID) fall in the middle of the spectrum between immunoglobulin E (IgE)-mediated and non-IgE, cellular-mediated hypersensitivity disorders. EGID, eosinophilic gastrointestinal disorders; IBD, inflammatory bowel disease.
The chief differential diagnoses include gastroesophageal reflux disease (GERD), inflammatory bowel disease (IBD), and specific infections including parasites and Helicobacter pylori.5., 6. The prevalence of EGID has not been rigorously calculated, but they appear to be widespread and not uncommon.1 For example, EoE has been reported in Australia,7 Brazil,8 England, Italy, Japan,9 Spain, and Switzerland.10 In one study approximately 10% of pediatric patients with GERD-like symptoms who were unresponsive to acid blockade had EoE.11 Another study reported that 6% of children with chronic esophagitis have EoE.1 Prevalence estimates vary from 1:70,000 adults in Australia to 1:2000 children in Cincinnati, USA. The increased prevalence of the disease is primarily due to increased recognition, as the disease accounted for approximately 30% of refractory chronic esophagitis in the 1980s and 1990s; however, a bona fide increase in disease incidence is also occurring.12 It appears that EGID may be even more prevalent than pediatric IBD.
The blood eosinophil level in EoE patients is typically not dramatically elevated, although it averages twice the normal value.13 The relative normal value of blood eosinophilia compared with esophageal eosinophilia highlights the tissue-specific pathogenesis of the disease. However, some EGID patients have peripheral eosinophilia high enough to meet criteria for hypereosinophilic syndrome (HES), defined by sustained peripheral blood eosinophilia (>1500 cells/mm3) and end-organ involvement in the absence of known causes of eosinophilia.14., 15. While HES often involves the GI tract, the other organs typically affected in HES (heart and skin) are rarely involved in EGID.
Eosinophils and the Gastrointestinal Tract
In most tissues, eosinophils are present in minute amounts. Organs with substantial eosinophils include GI tract, lymph nodes, spleen, and thymus. Interestingly, in a large study of healthy patients, on autopsy eosinophil degranulation was only seen in the GI tract. Eosinophils are normally present in the lamina propria of the colon, small intestine, and stomach, but are not normally present in the epithelium or Peyer patches, although they infiltrate these regions in EGID.2 Eosinophil homing to the GI tract appears to occur independent of gut flora, as evidenced by prenatal, adult, and germ-free mice having eosinophils in similar locations and concentrations. Additionally, mice deficient in innate signaling responses (i.e., Myd88 deficient) have normal numbers of GI eosinophils. Together these data indicate that eosinophils respond to signals distinct from those of most other intestinal inflammatory cells that typically require gut flora-induced signaling. In fact, eotaxin/C-C motif chemokine 11 (CCL11) is the unique signal that induces localization of eosinophils to the GI tract.
In vitro, the granule components of eosinophils are toxic to many tissues, including intestinal epithelium. The eosinophil cationic proteins major basic protein (MBP), eosinophil peroxidase (EPO), and eosinophil cationic protein (ECP) are cytotoxic to epithelium at concentrations similar to those found in biological fluids from patients with eosinophilia. ECP can render cell membranes porous. MBP increases smooth muscle reactivity via vagal muscarinic M2 receptors and can trigger mast cell and basophil degranulation. In patients with eosinophilic gastroenteritis, MBP and ECP are deposited extracellularly in the small bowel, and ultrastructural changes in eosinophil secondary granules (indicating degranulation and mediator release) are found in duodenal biopsies. Furthermore, Charcot–Leyden crystals are commonly found in feces, and disease severity correlates with mucosal eosinophil numbers.
Eosinophils can secrete a number of different cytokines, suggesting that they may modulate multiple aspects of the immune response, as well as epithelial growth, fibrosis, and tissue remodeling. In addition, tissue eosinophils have distinct cytokine expression patterns under inflammatory versus noninflammatory conditions. For example, esophageal eosinophils from EoE patients express high levels of Th2 cytokines and transforming growth factor β (TGF-β).16 Other molecules secreted by eosinophils include halide acids, hydrogen peroxide, and leukotrienes, which increase vascular permeability and mucus secretion and stimulate smooth muscle contraction.
In IBD, eosinophils form only a small percentage of the infiltrating leukocytes, but their level has been proposed to be a negative prognostic indicator. Observations of eosinophilia in IBD suggest that eosinophils mediate axonal necrosis. Recently, it has been shown that eotaxin is upregulated in intestinal macrophages and epithelial cells in pediatric ulcerative colitis and thus may be a future target for therapy.17
Evaluation of Eosinophilic Gastrointestinal Disorders
Common symptoms of EGID patients include abdominal pain, diarrhea, dysphagia, failure to thrive, gastric dysmotility, hypoproteinemia, irritability, microcytic anemia, and vomiting. Patients with these refractory problems, especially individuals with a strong history of allergic diseases, peripheral blood eosinophilia, and/or a family history of EGID, should be evaluated for EGID. Evaluation for EGID starts with a comprehensive history and physical examination. Symptoms vary depending on the intestinal segment involved (e.g., abdominal pain and dysphagia are most common in eosinophilic gastroenteritis and EoE, respectively). In most EGID patients, peripheral eosinophilia is not present. Total IgE levels may help classify patients with atopy or those with occult parasite infection. In atopic EGID patients, food allergy is common and can be verified by skin prick and skin patch testing.18 There are no pathognomonic symptoms or blood tests for diagnosing EGID. Therefore, the diagnosis of EGID is based on endoscopic biopsy procurement and appropriate clinical information.
The diagnosis of EGID is dependent on histological evaluation of biopsy samples, contingent upon the quantity, location, and characteristics of the eosinophilic inflammation. EGID patients often have a variety of endoscopic findings,10 but it is not uncommon for the lumen to look normal endoscopically; thus, a microscopic evaluation of biopsy samples is required. EGID often has patchy involvement, requiring analysis of biopsies from multiple intestinal segments. While the normal esophagus is devoid of eosinophils, the rest of the GI tract contains readily detectable eosinophils. With the lack of diagnostic criteria, diagnosis of EGID depends on clinical and histopathological expertise. Diagnosis of EGID depends on several factors including the following: (1) eosinophil quantification, (2) the histological location of eosinophils, (3) associated histopathological abnormalities, and (4) the absence of pathological features suggestive of other primary disorders. Recently, gene expression profiles (transcriptomes) have been proposed to be helpful for the diagnosis of EoE; elevated levels of esophageal eotaxin-3/CCL26 in a single biopsy specimen has approximately 90% sensitivity.19 Other disease processes, such as drug hypersensitivity, collagen vascular disease, malignancy, or infection, should be ruled out (Table 13.8.1 ). Evaluation for intestinal parasites via stool samples, colonoscopy aspirates, or antibody titers should be performed, especially if patients have high-risk exposure (e.g., drinking well water, living on a farm, or travel to endemic area). In one study of eosinophilic enteritis, the dog hookworm Ancylostoma caninum was identified as the cause in 15% of cases.20 Infection with Strongyloides stercoralis should be excluded before treating for EGID, as immunosuppressants can be life threatening with this infection.21
TABLE 13.8.1.
Classification and Examples of Eosinophilic Gastrointestinal Disorders
| Primary | Secondary |
|---|---|
| Eosinophilic esophagitis | Parasitic infection |
| Eosinophilic gastritis | Inflammatory bowel disease |
| Eosinophilic enteritis | Gastroesophageal reflux disease |
| Eosinophilic colitis |
|
| Eosinophilic gastroenteritis |
|
Eosinophilic Esophagitis
The esophagus is normally devoid of eosinophils.1., 2. Disorders associated with esophageal eosinophil infiltration include allergic vasculitis, bullous pemphigoid, carcinomatosis, drug injury, EoE, eosinophilic gastroenteritis, esophageal leiomyomatosis, GERD, HES, IBD, myeloproliferative disorders, parasitic and fungal infections, periarteritis, pemphigus vegetans, recurrent vomiting, and scleroderma.6., 22. Eosinophil-associated esophageal disorders are classified as primary or secondary (i.e., due to another known disease process) (Table 13.8.1). The primary subtype includes the atopic, nonatopic, and familial variants, and the secondary subtype is subdivided into those with and without systemic eosinophilia. EoE is familial in about 10% of patients.
Etiology
The etiology of EoE is poorly understood, but food allergy has been implicated. Most EoE patients have specific IgE to foods and aeroallergens, but only a few have experienced anaphylaxis.1 Esophageal eosinophilia may also be linked to allergic airway disease. Several independent studies have linked EoE to allergic etiology. For example, repeated delivery of allergens or interleukin-13 (IL-13) to murine lungs (via direct delivery or transgenic overexpression) induces experimental EoE; patients with allergic rhinitis have increased esophageal eosinophils;23 intranasal delivery of indoor insect allergen induces EoE in mice;24 patients with EoE commonly report seasonal variations in symptoms; and there is a seasonal variation in EoE diagnosis.25., 26.
In addition to eosinophils, T cell and mast cell numbers are increased in esophageal biopsies, suggesting a chronic Th2-associated inflammation (Fig. 13.8.2 ).27 Mast cells have been shown to be elevated in the esophagus and to degranulate in EoE, and they appear to be stimulated by the kit ligand. Carboxypeptidase A3 and tryptase appear to be good surrogate markers for mast cell involvement.28 IL-13 is overproduced in the esophagus of EoE patients. In addition, in experimental systems, such as IL-13 lung transgenic systems, IL-13 induces eosinophilic esophagitis and tissue remodeling with features both dependent and independent of eosinophils.29 Consistent with Th2 activation, IL-5 overexpression induces EoE, and IL-5 neutralization completely blocks allergen- or IL-13-induced EoE in mice.30 However, anti-IL-5 therapy in humans has not yet been shown to be effective at ameliorating clinical aspects of the disease, although esophageal eosinophilia improves.31., 32. Interestingly, a cytokine with more of a Th1 skew, IL-15, appears to mediate CD4+ T cells and to be involved in the pathogenesis of EoE.33
FIGURE 13.8.2.
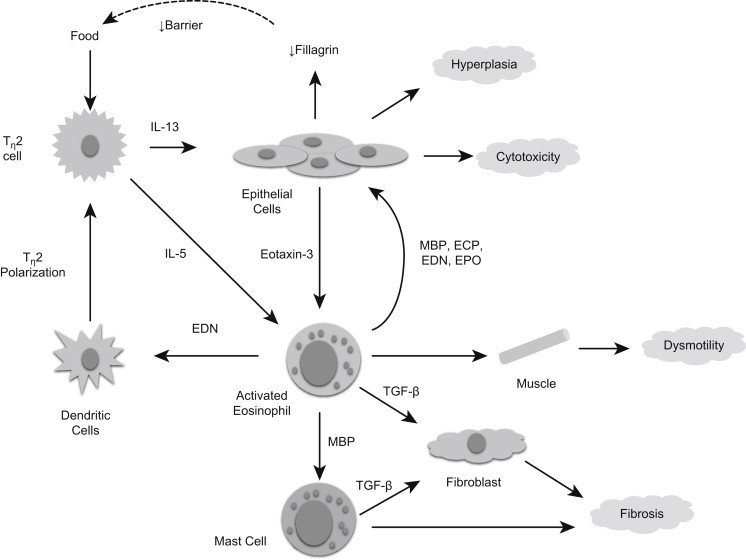
The inflammatory response in EoE.
Food antigens trigger T-helper type 2 (Th2) cells to release interleukin-5 (IL-5) and IL-13, which stimulate eosinophils and esophageal epithelial cells, respectively. IL-13 induces epithelial cells to produce eotaxin-3/C-C motif chemokine 26 (CCL26) and downregulate filaggrin. Reduced production of filaggrin might inhibit esophageal barrier function, which could perpetuate the inflammation by maintaining local food antigen uptake. IL-5 and eotaxin-3 activate eosinophils to release major basic protein (MBP) and eosinophil-derived neurotoxin (EDN), which activate mast cells and dendritic cells, respectively. Eosinophils and mast cells also produce TGF-β, which activates fibroblasts and muscle cells and contributes to dysmotility, fibrosis, and hyperplasia.24., 62. ECP, eosinophil cationic protein; EPO, eosinophil peroxidase; TGF-β, transforming growth factor β.
In a recent genome-wide microarray expression profile analysis of esophageal tissue19 from patients with EoE and normal controls, an EoE transcriptome was found to contain changed expression of approximately 1% of the entire human genome. Interestingly, eotaxin-3 was the most prominently overexpressed gene in EoE patients, and levels correlated with disease severity. Furthermore, a single nucleotide polymorphism (SNP) in eotaxin-3 was overrepresented in EoE patients. Conversely, mice lacking the eotaxin receptor (C-C chemokine receptor type 3; CCR3) were protected from developing experimental EoE. Notably, eotaxin-3 is induced by IL-13. Two recent studies have implicated genetic susceptibility for EoE with genetic variants of the TSLP gene; TSLP encodes thymic stromal lymphopoietin, an epithelial gene product that targets dendritic cells and promotes their Th2-polarizing ability. These studies included a genome-wide analysis study of common variants, as well as a large-scale candidate SNP approach. Notably, sex-associated TSLP receptor polymorphisms on Xp22.3/Yp11.3 may explain male predisposition to EoE.34
Clinical and Diagnostic Studies
Symptoms of EoE include dysphagia, epigastric or chest pain, GI obstructive problems, and vomiting.35 Patients are predominantly male35 and have robust esophageal eosinophilia,2 extensive epithelial hyperplasia, and are commonly atopic compared to GERD patients. In adults with EoE, dysphagia and food impaction are common complaints.36 One study presented a symptom scoring tool that can help identify patients with EoE and correlates with tissue inflammation.37 However, another study demonstrated dissociation between symptom scores and histology in treated EoE patients.38 At present, it is important to develop validated disease instruments so that these important parameters can be tracked, especially in clinical trials.
EoE is associated with esophageal dysmotility/dysphagia, which may be related to motor dysfunction of the esophagus rather than to physical narrowing.39 Esophageal ultrasound shows dysfunctional muscularis mucosa in EoE.40 Radiographic and endoscopic studies demonstrate furrowing, mucosal rings, polyps, strictures, ulcerations, and whitish papules.41 It appears that a small diameter on barium esophagography can help diagnosis of EoE,42 although many patients have normal barium assessments and need further evaluation by endoscopy.43 Recently, it has been demonstrated that esophageal distensibility, defined as cross-sectional area following intraluminal pressure, is significantly reduced in EoE patients.44
The number and location of eosinophils helps distinguish EoE from GERD. Greater than 15 eosinophils per high-power field (hpf) and a lack of response to proton-pump inhibitors suggest EoE. Proximal and distal esophageal eosinophilia suggests EoE, whereas eosinophilia confined to the distal esophagus suggests GERD.1 Histopathological changes in EoE include esophageal mucosa thickening with basal layer hyperplasia and papillary lengthening. Tissue eosinophil counts may underestimate eosinophil involvement, particularly with marked degranulation. Eosinophil-derived neurotoxin (EDN) staining of biopsy specimens may be useful for diagnosis and management.45
Clinical assessment of EoE includes analysis of food and aeroallergen sensitization and exclusion of GERD as well as other causes of eosinophils in the esophagus. However, EoE and GERD are not mutually exclusive and may coexist in the same patient (Table 13.8.2 ). Skin patch testing may facilitate identification of food allergy and lead to improved dietary therapy.46
TABLE 13.8.2.
Comparison of EoE and GERD
| Associated Features | EoE | GERD |
|---|---|---|
| Clinical | ||
|
|
|
| Procedural findings | ||
|
|
|
| Histopathology | ||
|
|
|
| Treatment effectiveness | ||
|
|
|
CCL26, C-C motif chemokine 26; hpf, high-power field.
Treatment
Specific food antigen and aeroallergen avoidance, identified by skin testing or history, is indicated for patients with atopic EoE. If feasible, an elemental diet is recommended, as it improves symptoms and reduces the number of eosinophils in the esophageal biopsies of patients with EoE (allergic or nonallergic subtypes). Elemental diet therapy frequently requires placement of a gastrostomy tube to achieve adequate caloric intake.
Glucocorticoids are also effective. Systemic steroids are used for acute exacerbations, and topical steroids provide day-to-day control.47 For topical steroid delivery, the patient is instructed to swallow the dose to promote deposition on the esophageal mucosa. Topical fluticasone lowers the level of eosinophils, CD3+ cells, and CD8+ cells in the proximal and distal esophagus48 and improves symptoms.36 Side effects of inhaled glucocorticoids are less likely with swallowed fluticasone, as this drug undergoes extensive first-pass metabolism in the liver. However, local esophageal candidiasis may occur.48 In addition to inhaled steroids, an oral suspension of budesonide can be used.49 Viscous budesonide improves symptoms and endoscopic/histological appearance and also appears to reach the distal esophagus.50 A recent study shows FK506 binding protein 5 (FKBP5) transcript levels increase with glucocorticoid exposure, thus helping to distinguish responders from nonresponders and treated from untreated. This also provides the best evidence that swallowed steroids induce local effects in the esophagus.51
Although food antigen avoidance and glucocorticoids are the mainstay of treatment, additional therapies may be beneficial. In EoE patients for whom impaction has become an issue, esophageal dilation appears to be safe and effective.52., 53. Even if GERD is not present, neutralizing gastric acidity (with proton-pump inhibitors) may improve symptoms and esophageal pathology. On the horizon, an anti-IL-13 antibody is a promising future therapy. In an animal model of IL-13-induced esophageal eosinophilia, an anti-IL-13 antibody was effective, and clinical studies are now under way.54 Additionally, anti-IL-5 antibodies prevent experimental EoE in mice55 56 and appear to decrease eosinophilia of the human esophagus in early-stage clinical trials.57 Human clinical trials are currently under way.
Prognosis
Although the natural history of EoE is not fully known, it is not uncommon for children with EoE to have a parent with a history of chronic esophageal strictures. Esophageal biopsies in some of these parents have revealed EoE.
The following symptoms occur in order of increasing age: feeding problems, vomiting, abdominal pain, dysphagia, and food impaction.13 Thus, if left untreated, EoE is likely to progress to esophageal scarring and dysfunction. Recent data show that pediatric EoE patients, diagnosed by retrospective biopsy analysis, are at increased risk of developing persistent disease characterized by dysphagia, food impaction, a need for esophageal dilation, and food allergy.12 58 Despite this, the development of Barrett esophagus in EoE has not been studied. However, it appears that EoE requires chronic treatment.
Having EoE increases the risk of developing other forms of EGID. Thus, routine surveillance guided by symptoms of the entire GI tract by endoscopy is recommended.
Eosinophilic Gastritis and Gastroenteritis
At baseline, the stomach and intestine have readily detectable eosinophils. Therefore, diagnosis of enteritis, eosinophilic gastritis, and gastroenteritis is more complex than diagnosis of EoE. These diseases are characterized by selective infiltration of eosinophils in the stomach and/or small intestine with variable involvement of the esophagus and/or large intestine. Eosinophilic enteritis, gastritis, and gastroenteritis can be divided into primary or secondary. The primary disorders have also been called idiopathic or allergic gastroenteropathy. Primary eosinophilic gastroenteritis is subdivided on the basis of the level of histological involvement into mucosal, muscularis, and serosal forms. Endoscopic biopsy can be normal in the latter two subtypes. Importantly, many other disorders feature eosinophil infiltration in the stomach, including allergic vasculitis, drug hypersensitivity, drug injury, HES, IBD, myeloproliferative disorders, parasitic and bacterial infections (e.g., H. pylori), periarteritis, and scleroderma.
Etiology
Because total IgE is elevated and food-specific IgEs are detectable in most EGID patients, an allergic mechanism is suspected. However, even though most patients have positive skin tests to a variety of food antigens, they do not have typical anaphylactic reactions, suggesting a delayed form of food hypersensitivity syndrome. In support of an allergic mechanism, mast cells are increased in EGID.59 Also, eosinophilic gastroenteritis can be induced by feeding enteric-coated allergen beads to sensitized mice.60 These mice develop eosinophil-associated GI dysfunction, including delayed food transit, gastromegaly, and weight loss.61 In addition, duodenal lamina propria T cells in EGID preferentially secrete Th2 cytokines (especially IL-13) when stimulated with milk proteins. Furthermore, elevated secretion of IL-4 and IL-5 by peripheral blood T cells has been observed in eosinophilic gastroenteritis. IgA deficiency can also be associated with eosinophilic gastroenteritis and may be related to the increased rate of atopy or to occult GI infection in these patients.
Clinical and Diagnostic Studies
In general, these disorders present with symptoms related to the degree and area of the GI tract affected. The mucosal form of eosinophilic gastroenteritis (the most common variant) is characterized by abdominal pain, bloody stools, diarrhea, failure to thrive, iron-deficiency anemia, malabsorption, protein-losing enteropathy, and vomiting. In the muscularis form, thickening of the bowel wall may result in GI obstructive symptoms. The serosal form is characterized by exudative ascites.
There are no standards for the diagnosis of eosinophilic gastritis or gastroenteritis, but the presence of increased eosinophils in biopsy specimens, infiltration of eosinophils within intestinal crypts and gastric glands, lack of involvement of other organs, and exclusion of secondary causes of eosinophilia support a diagnosis of eosinophilic gastroenteritis. Patients with eosinophilic gastritis may have micronodules (and/or polyposis), and these lesions often contain aggregates of lymphocytes and eosinophils. Food allergy and peripheral eosinophilia may be present but are not required for diagnosis.
Treatment
Eliminating foods implicated by skin prick or radioallergosorbent (RAST) testing has variable results, whereas complete resolution is generally achieved with amino acid-based elemental diets. Once remission has been achieved by dietary modification, specific food groups are reintroduced (at approximately 3-week intervals for each food group), and endoscopy is reperformed to identify sustained remission or disease flares.
Systemic or topical steroids are the main therapy when diet restriction has failed or is not feasible. For systemic steroid therapy, a course of 2–6 weeks of therapy with relatively low doses seems to work better than a 7-day course of glucocorticoid bursts. Various topical glucocorticoid preparations are designed to deliver drugs to specific segments of the GI tract [e.g., budesonide tablets (Entocort EC) targeted to the ileum and proximal colon]. As with asthma, topical steroids have a better risk–benefit ratio than systemic steroids. In cases refractory to or dependent on glucocorticoid therapy, parenteral nutrition or antimetabolite therapy (azathioprine or 6-mercaptopurine) may help.
Drugs such as cromoglycate, ketotifen, mycophenolate mofetil (an inosine monophosphate dehydrogenase inhibitor), suplatast tosilate, and various alternative therapies are not generally very useful, although successful long-term remission of eosinophilic gastroenteritis has been reported following montelukast treatment. Use of proton-pump inhibitors can improve symptoms and the degree of esophageal and gastric pathology, even if GERD is not present.
Prognosis
The natural history of eosinophilic gastritis, enteritis, and gastroenteritis is not well documented; however, they are often chronic, relapsing/remitting diseases. When the disease presents in infancy and specific food sensitization can be identified, remission is likely by late childhood. In food antigen-induced disease, abnormal levels of circulating IgE and eosinophils often serve as markers for tissue involvement/relapse. As noted earlier, due to concern for HES, routine surveillance of the cardiopulmonary system is recommended.
Eosinophilic Colitis
Colonic eosinophilia occurs in a variety of disorders, including allergic colitis of infancy, drug reactions, eosinophilic gastroenteritis, infections (e.g., helminths), IBD, and vasculitis (e.g., Churg–Strauss syndrome). Allergic colitis in infancy (or dietary protein-induced proctocolitis of infancy syndrome) is the most common cause of bloody stools in the first year of life. Similar to other EGID, these disorders are classified into primary and secondary.
Etiology
In contrast to other EGID, eosinophilic colitis is usually not IgE associated. Some studies point to a T cell-mediated process, though the exact mechanism is unclear. Allergic colitis of infancy may be an early expression of protein-induced enteropathy. Cow’s milk and soy proteins are the foods most frequently associated, but other food proteins can also induce this disorder. Interestingly, this condition may more commonly occur in infants who are exclusively breastfed and can occur in infants fed with protein hydrolysate formulas. An association between allergic colitis and the later development of IBD has been reported but remains controversial.
Clinical and Diagnostic Studies
Similar to eosinophilic gastroenteritis, the symptoms of eosinophilic colitis vary depending on the degree and location of tissue involvement. Although diarrhea is a classic symptom, symptoms that can occur independent of diarrhea commonly include abdominal pain, anorexia, and weight loss. In infants, bloody diarrhea precedes diagnosis by several weeks, and anemia due to blood loss is not uncommon. Most infants affected do not have constitutional symptoms and are otherwise healthy. There is a bimodal age distribution, with the infantile form presenting with a mean age at diagnosis of approximately 60 days, and the second group presenting during adolescence and early adulthood.
There is no single gold standard diagnostic test, but peripheral blood eosinophilia or eosinophils in the stool are suggestive of eosinophilic colitis. On endoscopic examination, patchy erythema, loss of vascularity and lymphonodular hyperplasia are seen; findings are mostly localized to the rectum but can affect the entire colon. Histological examination often reveals preservation of mucosal architecture, with focal aggregates of eosinophils in the crypt epithelium, lamina propria, and muscularis mucosa and occasionally, multinucleated giant cells in the submucosa.
Treatment
Treatment of eosinophilic colitis varies according to the disease subtype. Clinical symptoms of eosinophilic colitis of infancy resolve within 72 hours of withdrawal of the offending protein. Treatment of eosinophilic colitis in older patients, for which IgE-associated triggers are rarely identified, usually requires medical management. Anti-inflammatory drugs, including aminosalicylates and systemic or topical steroids, are commonly used and appear to be efficacious, but clinical trials have not been conducted. Several forms of rectally delivered glucocorticoids treat the distal colon, though eosinophilic colitis typically also involves the proximal colon. In cases refractory or dependent on systemic glucocorticoid therapy, parenteral nutrition or antimetabolite therapy (azathioprine or 6-mercaptopurine) are alternatives.
Prognosis
Eosinophilic colitis presenting in the first year of life has a very good prognosis with the vast majority of patients able to tolerate the culprit food(s) by 1–3 years of age. The prognosis for eosinophilic colitis developing in adolescence or adulthood is less favorable. As with eosinophilic gastroenteritis, the natural history has not been studied, and this disease is considered to be a chronic relapsing/remitting disorder. Because eosinophilic colitis can often be a manifestation of other disease processes, ruling out autoimmune disease and other secondary causes of eosinophilia is recommended.
Evaluation of Hypereosinophilic Syndrome in Eosinophilic Gastrointestinal Disorders Patients
The term HES describes patients with systemic symptoms due to marked eosinophilia. Diagnostic criteria for HES include persistent eosinophilia of at least 1500 cells/mm2 for a sustained duration; lack of known causes of eosinophilia; and symptoms and signs of organ system involvement. Patients with EGID and blood eosinophil counts greater than 1500/mm2 meet these diagnostic criteria but generally do not have the high risk of life-threatening complications associated with classic HES (i.e., cardiomyopathy or central nervous system involvement).
As is true in any patient with prolonged and marked eosinophilia, HES patients are prone to develop eosinophilic endomyocardial disease with embolization. Thus, routine echocardiograms are warranted in patients with EGID and peripheral blood eosinophilia. Additionally, the diagnosis of HES should be considered in patients with EGID who develop extra-GI manifestations. Additional testing may include bone marrow analysis (searching for myelodysplasia), serum tryptase and vitamin B12 levels (both moderately elevated in classic HES), and genetic analysis for the FIP1-like 1/platelet-derived growth factor receptor-α (FIP1L1–PDGFRA) fusion event.
A major advance in our understanding of HES has come about through treatment of HES with imatinib mesylate, a tyrosine kinase inhibitor. In many HES patients, treatment with imatinib mesylate dramatically reduces peripheral blood and bone marrow eosinophils, suggesting that certain HES patients express a kinase that is sensitive to imatinib mesylate. A fusion gene, the result of an 800-kb deletion in chromosome 4, yields a novel activated kinase (FIP1L1–PDGFR). FIP1L1–PDGFR is inhibited by imatinib in vitro, explaining the sensitivity of HES patients to imatinib. Those responsive to imatinib are typically 20–50-year-old males who present with marked peripheral eosinophilia (i.e., classic HES). These patients have been shown to meet minor criteria for systemic mastocytosis, having elevated levels of serum mast cell tryptase and high numbers of dysplastic mast cells in the bone marrow.
Conclusion
EGID are now being recognized more frequently. They have strong genetic and allergic components and share clinical and immunopathogenic features with asthma. EGID are associated with a variety of nonspecific common GI symptoms and laboratory findings, making their diagnosis dependent on microscopic examination biopsies and ruling out of other eosinophil-associated disease.
Eosinophils have potent proinflammatory effects mediated by their cytotoxic granule constituents and various lipid mediators and cytokines. In Th2-associated GI inflammatory conditions, eosinophilia in the lamina propria is dependent on IL-5 and eotaxin. Esophageal eosinophilia can be induced experimentally by pulmonary deposition of aeroallergens or the Th2 cytokine, IL-13.
Many new therapeutic approaches are now being developed for EGID, including eotaxin-3 receptor/CCR3 antagonists, eotaxin-3 blockers, humanized anti-IL-5, and IL-4/IL-13 inhibitors. However, although much progress has been made concerning GI eosinophils and EGID, there is still a paucity of knowledge compared with other cell types and GI diseases that may be even less common (e.g., IBD). A better understanding of the pathogenesis and treatment of EGID will emerge by combining comprehensive clinical and research approaches involving experts in the fields of allergy, gastroenterology, nutrition, and pathology.
References
- 1.Fox V.L., Nurko S., Furuta G.T. Eosinophilic esophagitis: it’s not just kid’s stuff. Gastrointest Endosc. 2002;56(2):260–270. doi: 10.1016/s0016-5107(02)70188-0. [DOI] [PubMed] [Google Scholar]
- 2.Rothenberg M.E. Pathogenesis and clinical features of eosinophilic esophagitis. J Allergy Clin Immunol. 2001;108(6):891–894. doi: 10.1067/mai.2001.120095. [DOI] [PubMed] [Google Scholar]
- 3.Leslie C., Mews C., Charles A., Ravikumara M. Celiac disease and eosinophilic esophagitis: a true association. J Pediatr Gastroenterol Nutr. 2010;50(4):397–399. doi: 10.1097/MPG.0b013e3181a70af4. [DOI] [PubMed] [Google Scholar]
- 4.Lucendo A.J., Arias Á, Pérez-Martínez I., López-Vázquez A., Ontañón-Rodríguez J., González-Castillo S. Adult Patients with Eosinophilic Esophagitis Do Not Show an Increased Frequency of the HLA-DQ2/DQ8 Genotypes Predisposing to Celiac Disease. Dig Dis Sci. 2011;56:1107–1111. doi: 10.1007/s10620-010-1383-2. [DOI] [PubMed] [Google Scholar]
- 5.Rothenberg M.E., Mishra Anil, Brandt Eric B., Simon P. Hogan. Gastrointestinal eosinophils. Immunol Rev. 2001;179:139–155. doi: 10.1034/j.1600-065x.2001.790114.x. [DOI] [PubMed] [Google Scholar]
- 6.Rothenberg M.E., Mishra Anil, Brandt Eric B., Simon P. Hogan. Gastrointestinal eosinophils in health and disease. Adv Immunol. 2001;78:291–328. doi: 10.1016/s0065-2776(01)78007-8. [DOI] [PubMed] [Google Scholar]
- 7.Croese J., Fairley S.K., Masson J.W., Chong A.K., Whitaker D.A., Kanowski P.A. Clinical and endoscopic features of eosinophilic esophagitis in adults. Gastroint Endosc. 2003;58(4):516–522. doi: 10.1067/s0016-5107(03)01870-4. [DOI] [PubMed] [Google Scholar]
- 8.Cury E.K., Schraibman V., Faintuch S. Eosinophilic infiltration of the esophagus: gastroesophageal reflux versus eosinophilic esophagitis in children—discussion on daily practice. J Pediatr Surg. 2004;39(2):e4–e7. doi: 10.1016/j.jpedsurg.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 9.Fujiwara H., Morita A., Kobayashi H., Hamano K., Fujiwara Y., Hirai K. Infiltrating eosinophils and eotaxin: their association with idiopathic eosinophilic esophagitis. Ann Allergy Asthma Immunol. 2002;89(4):429–432. doi: 10.1016/S1081-1206(10)62047-9. [DOI] [PubMed] [Google Scholar]
- 10.Straumann A., Spichtin H.P., Bucher K.A., Heer P., Simon H.U. Eosinophilic esophagitis: red on microscopy, white on endoscopy. Digestion. 2004;70(2):109–116. doi: 10.1159/000080934. [DOI] [PubMed] [Google Scholar]
- 11.Markowitz J.E., Liacouras C.A. Eosinophilic esophagitis. Gastroent Clin N Am. 2003;32(3):949–966. doi: 10.1016/s0889-8553(03)00047-5. [DOI] [PubMed] [Google Scholar]
- 12.DeBrosse C.W., Collins M.H., Buckmeier Butz B.K., Allen C.L., King E.C., Assa’ad A.H. Identification, epidemiology, and chronicity of pediatric esophageal eosinophilia, 1982–1999. J Allergy Clin Immunol. 2010;126(1):112–119. doi: 10.1016/j.jaci.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noel R.J., Putnam P.E., Rothenberg M.E. Eosinophilic esophagitis. N Engl J Med. 2004;351(9):940–941. doi: 10.1056/NEJM200408263510924. [DOI] [PubMed] [Google Scholar]
- 14.Assa’ad A.H., Spicer R.L., Nelson D.P., Zimmermann N., Rothenberg M.E. Hypereosinophilic syndromes. Chem Immunol. 2000;76:208–229. doi: 10.1159/000058787. [DOI] [PubMed] [Google Scholar]
- 15.Roufosse F., Cogan E., Goldman M. The hypereosinophilic syndrome revisited. Annu Rev Med. 2003;54:169–184. doi: 10.1146/annurev.med.54.101601.152431. [DOI] [PubMed] [Google Scholar]
- 16.Straumann A., Kristl J., Conus S., Vassina E., Spichtin H.P., Beglinger C. Cytokine expression in healthy and inflamed mucosa: probing the role of eosinophils in the digestive tract. Inflamm Bowel Dis. 2005;11(8):720–726. doi: 10.1097/01.mib.0000172557.39767.53. [DOI] [PubMed] [Google Scholar]
- 17.Ahrens R., Waddell A., Seidu L., Blanchard C., Carey R., Forbes E. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol. 2008;181(10):7390–7399. doi: 10.4049/jimmunol.181.10.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spergel J.M., Beausoleil J.L., Mascarenhas M., Liacouras C.A. The use of skin prick tests and patch tests to identify causative foods in eosinophilic esophagitis. J Allergy Clin Immunol. 2002;109(2):363–368. doi: 10.1067/mai.2002.121458. [DOI] [PubMed] [Google Scholar]
- 19.Blanchard C., Wang N., Stringer K.F., Mishra A., Fulkerson P.C., Abonia J.P. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest. 2006;116(2):536–547. doi: 10.1172/JCI26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker N.I., Croese J., Clouston A.D., Parry M., Loukas A., Prociv P. Eosinophilic enteritis in northeastern Australia. Pathology, association with Ancylostoma caninum, and implications. Am J Surg Pathol. 1995;19(3):328–337. doi: 10.1097/00000478-199503000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Liepman M. Disseminated Strongyloides stercoralis. A complication of immunosuppression. JAMA. 1975;231(4):387–388. [PubMed] [Google Scholar]
- 22.Ahmad M., Soetikno R.M., Ahmed A. The differential diagnosis of eosinophilic esophagitis. J Clin Gastroenterol. 2000;30(3):242–244. doi: 10.1097/00004836-200004000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Onbasi K., Sin A.Z., Doganavsargil B., Onder G.F., Bor S., Sebik F. Eosinophil infiltration of the oesophageal mucosa in patients with pollen allergy during the season. Clin Exp Allergy. 2005;35(11):1423–1431. doi: 10.1111/j.1365-2222.2005.02351.x. [DOI] [PubMed] [Google Scholar]
- 24.Rayapudi M., Mavi P., Zhu X., Pandey A.K., Abonia J.P., Rothenberg M.E. Indoor insect allergens are potent inducers of experimental eosinophilic esophagitis in mice. J Leukoc Biol. 2010;88(2):337–346. doi: 10.1189/jlb.0110025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almansa C., Krishna M., Buchner A.M., Ghabril M.S., Talley N., DeVault K.R. Seasonal distribution in newly diagnosed cases of eosinophilic esophagitis in adults. Am J Gastroenterol. 2009;104(4):828–833. doi: 10.1038/ajg.2008.169. [DOI] [PubMed] [Google Scholar]
- 26.Prasad G.A., Alexander J.A., Schleck C.D., Zinsmeister A.R., Smyrk T.C., Elias R.M. Epidemiology of eosinophilic esophagitis over three decades in Olmsted County, Minnesota. Clin Gastroenterol Hepatol. 2009;7(10):1055–1061. doi: 10.1016/j.cgh.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Straumann A., Bauer M., Fischer B., Blaser K., Simon H.U. Idiopathic eosinophilic esophagitis is associated with a TH2-type allergic inflammatory response. J Allergy Clin Immunol. 2001;108(6):954–961. doi: 10.1067/mai.2001.119917. [DOI] [PubMed] [Google Scholar]
- 28.Abonia J.P., Blanchard C., Butz B.B., Rainey H.F., Collins M.H., Stringer K. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1):140–149. doi: 10.1016/j.jaci.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuo L., Fulkerson P.C., Finkelman F.D., Mingler M., Fischetti C.A., Blanchard C. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13R alpha2-inhibited pathway. J Immunol. 2010;185(1):660–669. doi: 10.4049/jimmunol.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishra A., Rothenberg M.E. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology. 2003;125(5):1419–1427. doi: 10.1016/j.gastro.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Straumann A., Conus S., Grzonka P., Kita H., Kephart G., Bussmann C. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut. 2010;59(1):21–30. doi: 10.1136/gut.2009.178558. [DOI] [PubMed] [Google Scholar]
- 32.Conus S., Straumann A., Bettler E., Simon H.U. Mepolizumab does not alter levels of eosinophils, T cells, and mast cells in the duodenal mucosa in eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1):175–177. doi: 10.1016/j.jaci.2010.04.029. [DOI] [PubMed] [Google Scholar]
- 33.Zhu X., Wang M., Mavi P., Rayapudi M., Pandey A.K., Kaul A. Interleukin-15 expression is increased in human eosinophilic esophagitis and mediates pathogenesis in mice. Gastroenterology. 2010;139(1):182–193. doi: 10.1053/j.gastro.2010.03.057. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherrill J.D., Gao P.S., Stucke E.M., Blanchard C., Collins M.H., Putnam P.E. Variants of thymic stromal lymphopoietin and its receptor associate with eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126(1) doi: 10.1016/j.jaci.2010.04.037. p. 160–5.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orenstein S.R., Shalaby T.M., Di Lorenzo C., Putnam P.E., Sigurdsson L., Mousa H. The spectrum of pediatric eosinophilic esophagitis beyond infancy: a clinical series of 30 children. Am J Gastroenterol. 2000;95:1422–1430. doi: 10.1111/j.1572-0241.2000.02073.x. [DOI] [PubMed] [Google Scholar]
- 36.Konikoff M.R., Noel R.J., Blanchard C., Kirby C., Jameson S.C., Buckmeier B.K. A randomized double-blind-placebo controlled trial of fluticasone proprionate for pediatric eosinophilic esophagitis. Gastroenterology. 2006;131:1381–1391. doi: 10.1053/j.gastro.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 37.Aceves S.S., Newbury R.O., Dohil M.A., Bastian J.F., Dohil R. A symptom scoring tool for identifying pediatric patients with eosinophilic esophagitis and correlating symptoms with inflammation. Ann Allergy Asthma Immunol. 2009;103(5):401–406. doi: 10.1016/S1081-1206(10)60359-6. [DOI] [PubMed] [Google Scholar]
- 38.Pentiuk S., Putnam P.E., Collins M.H., Rothenberg M.E. Dissociation between symptoms and histological severity in pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2009;48(2):152–160. doi: 10.1097/MPG.0b013e31817f0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nurko S., Rosen R., Furuta G.T. Esophageal dysmotility in children with eosinophilic esophagitis: a study using prolonged esophageal manometry. Am J Gastroenterol. 2009;104(12):3050–3057. doi: 10.1038/ajg.2009.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fox V.L., Nurko S., Teitelbaum J.E., Badizadegan K., Furuta G.T. High-resolution EUS in children with eosinophilic allergic esophagitis. Gastrointest Endosc. 2003;57(1):30–36. doi: 10.1067/mge.2003.33. [DOI] [PubMed] [Google Scholar]
- 41.Fox V.L. Pediatric endoscopy. Gastrointest Endosc Clin N Am. 2000;10(1):175–194. viii. [PubMed] [Google Scholar]
- 42.White S.B., Levine M.S., Rubesin S.E., Spencer G.S., Katzka D.A., Laufer I. The small-caliber esophagus: radiographic sign of idiopathic eosinophilic esophagitis. Radiology. 2010;256(1):127–134. doi: 10.1148/radiol.10091060. [DOI] [PubMed] [Google Scholar]
- 43.Binkovitz L.A., Lorenz E.A., Di Lorenzo C., Kahwash S. Pediatric eosinophilic esophagitis: radiologic findings with pathologic correlation. Pediatr Radiol. 2010;40(5):714–719. doi: 10.1007/s00247-009-1484-2. [DOI] [PubMed] [Google Scholar]
- 44.Kwiatek M.A., Hirano I., Kahrilas P.J., Rothe J., Luger D., Pandolfino J.E. Mechanical properties of the esophagus in eosinophilic esophagitis. Gastroenterology. 2011;140(1):82–90. doi: 10.1053/j.gastro.2010.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kephart G.M., Alexander J.A., Arora A.S., Romero Y., Smyrk T.C., Talley N.J. Marked deposition of eosinophil-derived neurotoxin in adult patients with eosinophilic esophagitis. Am J Gastroenterol. 2010;105(2):298–307. doi: 10.1038/ajg.2009.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spergel J., Rothenberg M.E., Fogg M. Eliminating eosinophilic esophagitis. Clin Immunol. 2005;115(2):131–132. doi: 10.1016/j.clim.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Liacouras C.A., Spergel J.M., Ruchelli E., Verma R., Mascarenhas M., Semeao E. Eosinophilic esophagitis: A 10-year experience in 381 children. Clin Gastroenterol Hepatol. 2005;3(12):1198–1206. doi: 10.1016/s1542-3565(05)00885-2. [DOI] [PubMed] [Google Scholar]
- 48.Teitelbaum J.E., Fox V.L., Twarog F.J., Nurko S., Antonioli D., Gleich G. Eosinophilic esophagitis in children: immunopathological analysis and response to fluticasone propionate. Gastroenterology. 2002;122(5):1216–1225. doi: 10.1053/gast.2002.32998. [DOI] [PubMed] [Google Scholar]
- 49.Aceves S.S., Dohil R., Newbury R.O., Bastian J.F. Topical viscous budesonide suspension for treatment of eosinophilic esophagitis. J Allergy Clin Immunol. 2005;116(3):705–706. doi: 10.1016/j.jaci.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 50.Dohil R., Newbury R., Fox L., Bastian J., Aceves S. Oral viscous budesonide is effective in children with eosinophilic esophagitis in a randomized, placebo-controlled trial. Gastroenterology. 2010;139(2):418–429. doi: 10.1053/j.gastro.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Caldwell J.M., Blanchard C., Collins M.H., Putnam P.E., Kaul A., Aceves S.S. Glucocorticoid-regulated genes in eosinophilic esophagitis: a role for FKBP51. J Allergy Clin Immunol. 2010;125(4):879–888. doi: 10.1016/j.jaci.2010.01.038. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dellon E.S., Gibbs W.B., Rubinas T.C., Fritchie K.J., Madanick R.D., Woosley J.T. Esophageal dilation in eosinophilic esophagitis: safety and predictors of clinical response and complications. Gastrointest Endosc. 2010;71(4):706–712. doi: 10.1016/j.gie.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 53.Bohm M., Richter J.E., Kelsen S., Thomas R. Esophageal dilation: simple and effective treatment for adults with eosinophilic esophagitis and esophageal rings and narrowing. Dis Esophagus. 2010;23(5):377–385. doi: 10.1111/j.1442-2050.2010.01051.x. [DOI] [PubMed] [Google Scholar]
- 54.Blanchard C., Mishra A., Saito-Akei H., Monk P., Anderson I., Rothenberg M.E. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354) Clin Exp Allergy. 2005;35(8):1096–1103. doi: 10.1111/j.1365-2222.2005.02299.x. [DOI] [PubMed] [Google Scholar]
- 55.Mishra A., Hogan S.P., Brandt E.B., Rothenberg M.E. An etiological role for aeroallergens and eosinophils in experimental esophagitis. J Clin Invest. 2001;107(1):83–90. doi: 10.1172/JCI10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishra A., Hogan S.P., Brandt E.B., Rothenberg M.E. IL-5 promotes eosinophil trafficking to the esophagus. J Immunol. 2002;168(5):2464–2469. doi: 10.4049/jimmunol.168.5.2464. [DOI] [PubMed] [Google Scholar]
- 57.Stein M.L., Collins M.H., Villanueva J.M., Kushner J.P., Putnam P.E., Buckmeier B.K. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol. 2006;118(6):1312–1319. doi: 10.1016/j.jaci.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 58.DeBrosse C.W., Franciosi J.P., King E.C., Butz B.K., Greenberg A.B., Collins M.H. Long-term outcomes in pediatric-onset esophageal eosinophilia. J Allergy Clin Immunol. 2011;128(1):132–138. doi: 10.1016/j.jaci.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brandt E.B., Strait R.T., Hershko D., Wang Q., Muntel E.E., Scribner T.A. Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest. 2003;112(11):1666–1677. doi: 10.1172/JCI19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hogan S.P., Mishra A., Brandt E.B., Foster P.S., Rothenberg M.E. A critical role for eotaxin in experimental oral antigen-induced eosinophilic gastrointestinal allergy. Proc Nat Acad Sci USA. 2000;97:6681–6686. doi: 10.1073/pnas.97.12.6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aceves S.S., Newbury R.O., Dohil R., Bastian J.F., Broide D.H. A pathological function for eotaxin and eosinophils in eosinophilic gastrointestinal inflammation. Nat Immunol. 2001;2(4):353–360. doi: 10.1038/86365. [DOI] [PubMed] [Google Scholar]
- 62.Aceves S.S. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2007;119(1):206–212. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
Chapter 13.9. Rare Hypereosinophilic Syndromes
Hypereosinophilic syndrome is defined as blood eosinophilia of >1500/mm3 on at least two separate occasions or evidence of prominent tissue eosinophilia associated with symptoms and marked tissue eosinophilia in the absence of secondary causes. The workup for secondary causes of eosinophilia consists of determining exogenous triggers such as medications, parasitic diseases, and a comprehensive clinical and laboratory evaluation. With advances in molecular diagnostics, hypereosinophilic syndrome is increasingly being recognized as a heterogeneous group of rare disorders with differing pathogenesis. Knowledge of the pathogenesis of distinct variants of HES has improved approaches to therapy.
Introduction
Although case reports describing eosinophilic infiltration of tissues accompanied by pronounced peripheral eosinophilia have existed since the 1950s, the concept of a hypereosinophilic syndrome (HES) was not introduced until 1975 in a landmark paper by Chusid and colleagues.1 The original description of HES was based largely on clinical findings highlighting the heterogeneous presentations of HES and proposed the following diagnostic criteria:1 blood eosinophilia >1500/mm3 for at least 6 months (or death before 6 months with signs and symptoms of eosinophilic disease);2 lack of evidence for allergic, infectious, parasitic, or other known causes of eosinophilia; and3 presumptive signs of organ involvement. Since that time, the evolution of diagnostic testing has led to the identification of several HES variants with defined etiologies. Furthermore, the availability of novel targeted therapies precludes waiting 6 months for a diagnosis. As a result, several new conflicting classification schemes have been proposed.
The 2008 World Health Organization (WHO) classification specifically distinguishes between HES and eosinophilic disorders with proven clonal populations of myeloid or lymphoid origin.2 Although correct from a diagnostic standpoint, there are many issues that are not addressed, namely the clinical overlap between patients with a detectable clonal population and those with similar disease in whom the mutation is not detectable with current techniques, and the heterogeneity of patients with nonclonal HES. In contrast, an HES classification scheme proposed at a 2005 NIH-sponsored workshop,3 and subsequently refined by the same authors, groups clinically similar disorders with known and unknown etiologies into HES variants. Although useful from the standpoint of treatment approaches, this approach creates confusion with respect to diagnostic labels, since a patient with proven clonal eosinophilia is classified as having both HES and chronic eosinophilic leukemia (CEL).3
Despite these limitations, for the purposes of this subchapter, HES will be defined as (1) blood eosinophilia of >1500/mm3 on at least two separate occasions or evidence of prominent tissue eosinophilia associated with symptoms and marked tissue eosinophilia, and (2) exclusion of secondary causes of eosinophilia, such as drug hypersensitivity, hypoadrenalism, and parasitic infection.4 The approach to classification, diagnosis, pathophysiology, and treatment of the various forms of HES and other rare manifestations of eosinophilic disease will be discussed.
Epidemiology
Hypereosinophilic syndromes are rare diseases, and correct estimates of incidence and prevalence are unavailable. By extrapolation from the Surveillance, Epidemiology and End Results (SEER) database of cancer statistics in the United States, the incident rate of HES has been estimated to be between 0.018 and 0.036 per 100,000 person-years in the period from 2001 to 2005. The calculated prevalence was estimated at 0.36–6.3 per 100,000.5 These estimates are approximations and rely on coding of eosinophilia by individual physicians. In addition, the lack of specific codes for HES variants precludes a more specific characterization of incidence. Unified comprehensive patient databases would be useful in this regard. HES characteristically develops between the ages of 20 and 50 years; however, young children and the elderly can also be affected. A male predominance has been described, although this is due primarily to overrepresentation of males with the FIP1-like 1/platelet-derived growth factor receptor-α (FIP1L1/PDGFRA) mutation in most series.6
Diagnosing Rare Hypereosinophilic Syndromes
Hypereosinophilia may be attributable to secondary causes requiring specific treatment. Therefore, a careful history and physical examination are of paramount importance for making the correct diagnosis (Fig. 13.9.1 ). The history should include the degree and duration of eosinophilia, with documentation if available. Specific symptoms related to individual organ systems should be elicited, as patients can present with a multitude of findings ranging from the most common complaints of skin involvement to more rare presentations of connective tissue or ocular involvement. Pertinent medication, occupational, and travel histories should also be obtained to exclude drug allergy, hypersensitivity reactions, and helminth infections, respectively. A family history of eosinophilia should also be investigated.
FIGURE 13.9.1.

Approach to diagnosis and treatment.
B12, vitamin B12; CK, creatine kinase; CT, computerized tomography; EKG, electrocardiogram; ESR, erythrocyte sedimentation rate; HES, hypereosinophilic syndrome; HIV, human immunodeficiency virus; IgE, immunoglobulin E; PFTs, pulmonary function tests; US, United States.
The initial physical exam should be complete, with a focus on examination for organ involvement commonly seen in eosinophilic syndromes. Notably, careful examination of the skin, abdomen (to assess organomegaly), cardiovascular system (to assess evidence of heart failure and valvular disease), lungs, neurological system (to exclude neuropathy) should be performed.
Finally, laboratory and diagnostic testing for eosinophilia should be directed by the history and prior physical findings, but should include basic testing to assess end-organ involvement (Fig. 13.9.1). Initial laboratory evaluation should include, at a minimum, a complete blood count (CBC) with a differential, chemistry panel to assess creatine kinase, creatinine, electrolytes, erythrocyte sedimentation rate (ESR), liver enzymes, quantitative immunoglobulin E (IgE), levels serum B12, and serum tryptase. Assessment of cardiac status should include echocardiogram, electrocardiogram (EKG), and measurement of serum troponin. Spirometry with assessment of lung volumes and diffusion capacity should be performed to assess occult pulmonary involvement. Often the initial evaluation will determine the need for further testing. For example, a patient with asthma and sinusitis should be screened for Churg-Strauss syndrome (CSS) with serum anti-neutrophil cytoplasmic antibody (ANCA). Electrolyte abnormalities with eosinophilia might necessitate a work-up for hypoadrenalism. Findings of anemia, neutropenia, or thrombocytopenia would initiate a search for hematological disorders, including CEL. A pertinent drug history should prompt discontinuation of potentially offending agents to see if the eosinophilia resolves. Exceedingly uncommon in secondary eosinophilia, an elevated serum B12 or tryptase level should prompt evaluation for CEL and systemic mastocytosis (SM).
If no secondary or reactive cause is identified, one can reasonably proceed to further evaluation for HES and other rare eosinophilic disorders. Additional screening tests at this point should include computed tomography (CT) of the abdomen, chest, and pelvis, and bone marrow examination to assess cellularity, dysplastic changes of either eosinophils or mast cells, eosinophil precursors, mast cell numbers, and myelofibrosis. Cytogenetic analysis of dividing cells should be performed, as well as specific testing by real-time polymerase chain reaction (RT-PCR) or fluorescence in situ hybridization (FISH) for the most common genetic abnormalities associated with eosinophilia, including PDGFR-associated CELs. D816V KIT analysis should also be performed to exclude SM in patients with elevated serum tryptase and/or suggestive findings on bone marrow biopsy. T-cell clonality should be assessed by RT-PCR for T-cell receptor rearrangement and/or flow cytometry. Phenotypic assessment should also be performed to identify aberrant populations of T cells that may or may not be clonal and should include CD3, CD4, CD8, and if possible CD5 and CD7. Aberrant T-cell populations often exhibit elevated intracellular interleukin-4 (IL-4), IL-5, and IL-13 levels;7 however, intracellular cytokine analysis is not routinely available except at specialized academic centers. Various biomarkers have been clinically correlated with particular variants of HES, including serum B12 and tryptase elevation in myeloproliferative forms of HES and elevated serum IgE and thymus and activation-regulated chemokine (TARC)/C-C motif chemokine 17 (CCL17) levels in lymphocytic variant HES and are discussed further in the next section. It is important to recognize that the diagnosis of HES is an iterative process and may be revisited if new clinical findings develop and as new biomarkers and diagnostic tests become available.
Hypereosinophilic Syndrome Variants
As stated in the introduction, the current classification of HES variants is in evolution as specific etiologies become better defined. Despite the molecular uncertainties and current unknowns, it remains useful to subdivide patients into HES variants based on clinical characteristics and a combination of molecular and biologic data (Fig. 13.9.2 ), since this has important implications regarding prognosis, monitoring, and treatment. Unfortunately, the vast majority (60–70%) of patients with HES remain classified as idiopathic. In the following section, we discuss HES variants in further detail with a focus on clinical features, diagnosis, mechanisms, pathogenesis, and treatment. Areas with considerable controversy surrounding either diagnosis or treatment are also addressed.
FIGURE 13.9.2.
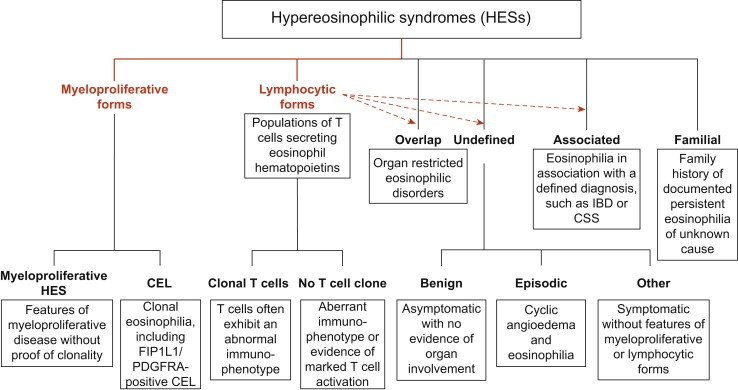
Classification of hypereosinophilic syndrome.
Dashed arrows identify variants of hypereosinophilic syndrome (HES) that are thought to be T cell-mediated.
(Reproduced with permission from Elsevier4).
Lymphocytic Variant Hypereosinophilic Syndrome
Mechanisms and Pathogenesis
Lymphocytic variant HES (L-HES) is defined by the expansion of an aberrant and/or clonal T-lymphocyte population with increased production of eosinophilopoietic cytokines in a patient who meets criteria for HES. Historically, the evidence for T-cell-mediated pathogenesis came to light when peripheral T-cell clones generated from a patient with HES were cultured with bone marrow progenitors, leading to outgrowth of eosinophilic colonies in culture.8 Further studies demonstrated that IL-5 was overproduced; however, other cytokines such as IL-2, interferon γ (IFN-γ), and additional T-helper type 2 (Th2) cytokines, IL-4 and IL-13, may also be increased in individual clones.9
Clinical Features and Diagnosis
L-HES affects males and females in equal proportions. Patients frequently have elevated serum IgE levels and skin involvement, ranging from urticaria and eczematous rashes to subcutaneous involvement with angioedema. Rare patients present with cyclic eosinophilia and angioedema in the setting of an abnormal lymphocyte population (Gleich’s syndrome). Lymphadenopathy and splenomegaly are uncommon except in the case of occult lymphoma, and bone marrow examination is generally normocellular with increased eosinophils. In contrast to the myeloproliferative variant, life-threatening end-organ involvement, including endomyocardial fibrosis, neurologic complications, and hypercoagulability are extremely rare in L-HES. Furthermore, L-HES is usually glucocorticoid-responsive, although moderate to high doses may be necessary to control symptoms.
Blood counts reflect elevated eosinophil levels; there may be lymphocytosis, but this is uncommon. There is often a polyclonal expansion of IgG or IgM, and markers of inflammation, such as ESR and C-reactive protein (CRP), and T-cell activation, such as CCL17, may be elevated. Thrombocytosis may also be present. The majority of aberrant T-cell populations in L-HES are detectable by flow cytometry using standard panels. Although CD3−CD4+ is the most common phenotype, CD3+ CD4− CD8− and CD4+ CD7dim/− have also been described in some patients.10 It is important to include appropriate markers to distinguish these cell populations from monocytes, especially if CD4 expression is decreased,9 and to exclude T-cell lymphomas that may present with eosinophilia, including cutaneous T-cell lymphoma11 and angioimmunoblastic T-cell lymphoma. Clonal populations of IL-5-producing cells with a normal surface phenotype have also been described in L-HES. Consequently, T-cell receptor (TCR) rearrangement analysis and/or assessment of clonality by flow cytometry using TCR-Vβ staining should be performed. Demonstration of increased Th2 cytokine production by the aberrant and/or clonal T-cell population by intracellular flow cytometry is also diagnostic, but is impractical at most centers.12
Therapy
The decision to treat L-HES patients depends on the nature and extent of disease. Patients with clinical manifestations attributable to the eosinophilia should be treated initially with corticosteroids (20–60 mg prednisone/d depending on the severity of the signs and symptoms), reducing slowly to the lowest dose that suppresses symptoms and eosinophilia. Steroid-sparing agents should be considered for patients with elevated eosinophil counts and symptoms requiring moderate to high dose (>10–15 mg prednisone equivalent daily) corticosteroids. Aims of therapy should be to reduce disease manifestations and prevent organ dysfunction. A number of steroid-sparing agents targeting T cells have been tried with variable success.6 Among currently available agents, IFN-α (at a dose range of 1–3 mU daily) has shown the greatest efficacy. Due to in vitro data demonstrating decreased apoptosis of the clonal population in the presence of IFN-α, concomitant low dose corticosteroid therapy is recommended in patients with L-HES.13 More recently, anti-IL-5 therapy with mepolizumab (available only for compassionate use through GlaxoSmithKline) has been shown to be safe and effective as a steroid-sparing agent in this subgroup of patients.14
It is important to monitor patients with a clonal T-cell population for progression to T-cell lymphoma throughout the course of their disease. This may be preceded by lymphadenopathy, expansion of the clone, and/or the development of cytogenetic abnormalities. Yearly bone marrow examination with karyotyping has been recommended,9 although the impact of this approach on prognosis is unknown.
Myeloproliferative Variant Hypereosinophilic Syndrome
Mechanisms and Pathogenesis
The most common cause of marked eosinophilia with myeloproliferative features is an interstitial deletion in chromosome 4q12 between FIP1L1 and the tyrosine kinase, PDGFRA, that results in the fusion gene product FIP1L1/PDGFRA.15 Other PDGFRA fusion partners have been identified, but are less common. Colony forming assays using an FIP1L1/PDGFRA reporter showed that expression of the fusion gene in human hematopoietic progenitors induces differentiation of erythrocytes and neutrophils in addition to eosinophils,16 and multiple lineages, including B- and T-lymphocytes, monocytes, mast cells, and neutrophils, can express the fusion gene in affected patients.17 Current data suggest, however, that the detrimental clinical effects are mediated primarily by eosinophils. In fact, the clinical presentation of FIP1L/PDGFRA-positive CEL is indistinguishable from that of a subset of idiopathic HES patients with myeloproliferative features.
A number of other myeloproliferative disorders (MPDs) can present with marked blood and bone marrow eosinophilia and have been defined at the molecular level. These include PDGFRB-associated chronic myelomonocytic leukemia (CMML), Janus kinase 2 (JAK2)-associated MPDs, 5q- syndrome and D816V KIT-associated SM. Although SM shares bone marrow features with FIP1L1/PDGFRA-positive CEL, the clinical presentation is often quite different from HES18 and the eosinophils are not usually implicated in disease pathogenesis.
Clinical Features and Diagnosis
Myeloproliferative variant HES (MHES) is the most aggressive form of HES, with fatality rates of up to 50% at 3 years prior to the availability of imatinib therapy. Patients are predominantly male with a near 100% male predominance in PDGFRA-associated disease. MHES typically presents between the ages of 25 and 50 years, although children and the elderly can be affected. The clinical presentation ranges from fatigue and malaise to endomyocardial fibrosis and stroke. Splenomegaly is common and characteristic laboratory findings include anemia and thrombocytopenia, occasional neutrophilia, and elevation of serum B12 and tryptase levels. Bone marrow examination reveals a hypercellular marrow with increased and dysplastic eosinophils and eosinophil precursors, fibrosis, and, in most cases, a concomitant increase in atypical mast cells. Eosinophilia is unresponsive to corticosteroids in most patients with MHES.
Although the existence of a myeloproliferative (leukemic) variant of HES had long been recognized, the availability of molecular testing has revolutionized diagnosis of this syndrome. The FIP1L1/PDGFRA fusion gene can be detected either by RT-PCR or FISH in peripheral blood. Additional testing, including cytogenetics, FISH, and/or quantitative PCR, should be performed, as indicated, to identify other MPDs associated with peripheral eosinophilia.
Therapy
The discovery that the tyrosine kinase inhibitor, imatinib, can decrease eosinophilia and improve symptoms in patients with HES facilitated the discovery of the FIP1L1/PDGFRA fusion gene15 and has dramatically improved prognosis in MHES. Some patients with FIP1L1–PDGFRA-negative MHES also respond to imatinib. Therefore, a trial of imatinib in such patients is reasonable if they fail low dose corticosteroids, particularly in the setting of myeloproliferative features. Most FIP1L1/PDGFRA-positive patients achieve clinical and molecular remission within 1–2 weeks of beginning imatinib therapy and can be maintained on low-dose therapy (100 mg daily) for many years without disease progression. Although FIP1L1/PDGFRA-negative patients may require longer to respond, a trial of imatinib 400 mg daily for 4 weeks is sufficient to assess response. Side effects of therapy are comparable to that seen in the treatment of chronic myeloid leukemia (CML), but rarely lead to discontinuation of therapy, and the development of resistance appears to be extremely rare. Unfortunately, imatinib is not curative in MHES.19
Since the clinical symptoms may be rapidly progressive, any new diagnosis of HES with myeloproliferative features should be evaluated without delay so that treatment can be initiated rapidly. EKG, troponin, and echocardiogram should be performed prior to initiating therapy. If cardiac dysfunction is present or serum troponin is elevated, corticosteroids should be initiated concurrent with imatinib therapy, due to reports of necrotizing myocarditis after initiation of therapy in patients with preexisting cardiac involvement. Initial monitoring on imatinib therapy should include weekly complete blood counts, liver function tests, and serum troponin levels. Bone marrow examination should be repeated at 4–8 weeks, even in the setting of hematologic response, to exclude occult leukemia or lymphoma that may have been masked by the marked eosinophilia. Long-term monitoring should include echocardiograms every 3–6 months to assess progression of disease, as well as to monitor for possible treatment-related cardiomyopathy.
For patients with FIP1L1/PDGFRA-positive MHES who fail or are intolerant to imatinib therapy, a trial of one of the newer tyrosine kinase inhibitors with activity against the most common resistance mutations is indicated. For FIP1L1/PDGFRA-negative MHES patients who fail imatinib, a step-wise approach of commonly used medications for reduction of eosinophils should be employed, balancing toxicities of the drugs and the individual patient’s comorbidities. Drugs that have been used successfully include hydroxyurea, IFN-α, anti-IL-5 therapy, and vincristine.20 Patients with rapidly progressive or aggressive disease unresponsive to standard therapies should be considered for nonmyeloablative allogeneic bone marrow transplant, a strategy that has proven curative in a number of cases.21
Idiopathic Hypereosinophilic Syndrome
Mechanisms and Pathogenesis
By definition, the mechanism of pathogenesis in idiopathic HES is unknown. Similar to other forms of HES, pathogenesis is due to tissue infiltration by eosinophils, with deposition of granule proteins and release of inflammatory mediators.
Clinical Features and Diagnosis
Patients with idiopathic HES may have relatively few or no symptoms (benign eosinophilia) or a wide variety of manifestations attributable to eosinophilic infiltration of target organs. The most commonly affected organs are the gastrointestinal tract, lungs, and skin (Fig. 13.9.3 ).6 Nonspecific symptoms, including arthralgias, fatigue, malaise, and myalgias, are also common. Cardiac manifestations, including eosinophilic myocarditis and endomyocardial fibrosis, and neurological involvement each occur in 15–20% of patients with idiopathic HES and are major causes of morbidity and mortality in this patient group.
FIGURE 13.9.3.
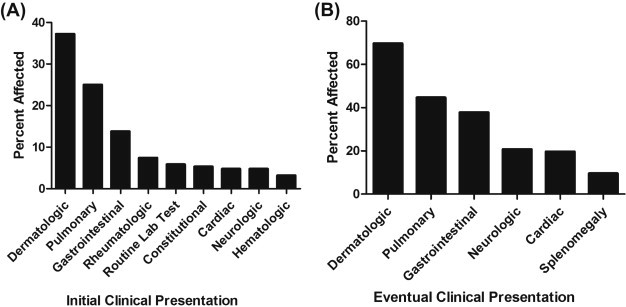
A, Presenting end-organ involvement. B, Eventual end-organ involvement.
Two areas of confusion with regards to secondary forms of eosinophilia may delay treatment for true HES. The first involves appropriate testing to rule out parasitic disease. Prompt referral to an infectious disease specialist can target helminth infection testing based on the patient’s travel history and likelihood of exposure. Whether to empirically treat for Strongyloides according to Centers for Disease Control and Prevention (CDC) guidelines is an area of controversy. Recommendations of the CDC are that all patients who are ‘at risk of disseminated strongyloidiasis should be treated.’ The drug of choice for treatment of uncomplicated strongyloidiasis is ivermectin. The second area of confusion is that of medications causing hypersensitivity syndromes such as drug reaction with eosinophilia and systemic symptoms (DRESS). Many drugs can cause eosinophilia starting from days to years after initiation. Simplification of drug regimens can vastly improve the ability to make the diagnosis of HES. Once the diagnosis of HES is made, the decision to treat is tailored to the likely etiology.
Therapy
Since patients with idiopathic HES are heterogeneous in their presentation, virtually all treatment must be individualized based on the presence of signs and symptoms and the likelihood of disease progression with end-organ involvement. Although the discovery of imatinib has dramatically altered prognosis in myeloproliferative HES, and in particular for patients with FIP1L1/PDGFRA-positive CEL, patients with idiopathic HES are much less likely to respond.
Corticosteroids remain first-line therapy for the majority of patients with HES, including L-HES, although many patients require moderate to high doses or develop significant steroid-related toxicity. Patients who fail to respond to corticosteroids or have significant side effects from prolonged high dose therapy should be considered for second-line therapy. A short (4–6 week) trial of imatinib, the only United States Food and Drug Administration (FDA)-approved drug for the treatment of HES, may be warranted in steroid-resistant patients.
Hydroxyurea is the most commonly used of the cytotoxic medications and has been used alone or in combination with IFN-α at doses ranging from 500 mg to 3 g daily. It can be associated with cytopenias or other adverse effects at high doses, thus limiting its use as a solitary agent. Furthermore, hydroxyurea may be associated with an increased risk of secondary malignancy with prolonged use. Of the immunomodulatory medications, IFN-α is most frequently used and at doses of 1–3 mU daily is effective in up to 30% of patients.6 Prolonged remission of clinical symptoms and eosinophilia has rarely been reported after prolonged IFN-α use or when INF-α is used in combination with cytotoxic medications.22 Other cytotoxic and immunomodulatory agents have been used with varied success and include cytosine arabinoside (ara-C),23 vincristine,20 alemtuzumab,24., 25. intravenous immunoglobulin,26 cyclosporine,27 cyclophosphamide,28 azathioprine, and methotrexate. However, bone marrow transplantation remains the only curative therapy.21
Familial Eosinophilia
Clinical Features
Familial hypereosinophilia is an autosomal dominant disorder discovered in one kindred family. Eosinophilia is present at birth in affected individuals and remains remarkably stable over time. Although the index case and his sister presented with eosinophilic end-organ involvement (endomyocardial fibrosis and peripheral neuropathy) that progressed despite therapy, most affected family members have followed a benign course despite eosinophil counts ranging from 2000–5000/mm3 over many years without treatment.29 There have been isolated reports of additional families with eosinophilia, but no clear genetic inheritance pattern has been found. Common environmental exposures, including helminth infection, must be excluded prior to attributing the eosinophilia to a familial origin.
Mechanisms and Pathogenesis
The genetic defect in familial hypereosinophilic syndrome is not known. The gene has been mapped to an area on chromosome 5q harboring the cytokine gene cluster;30 however, sequencing of a number of candidate genes in this region, including GM-CSF, IL-3, and IL-5, has failed to identify polymorphisms that could account for the affected phenotype. Additional sequencing is currently under way. Eosinophils from affected family members appear to be less activated than those from patients with HES, coincident with the general lack of eosinophil-mediated pathology.29 No further family members have developed organ involvement, thus precluding further studies of disease pathogenesis in this disorder.
Organ-Restricted Eosinophilia
Single organ eosinophilic tissue infiltration has been described for nearly all organ systems, although isolated involvement of the skin, lung or gastrointestinal tract is most common. The mechanism whereby certain tissues are targeted preferentially over others is not well understood. Some organ-restricted disorders, such as eosinophilic esophagitis, have distinct clinical presentations and rarely progress to multiorgan involvement even in the presence of marked peripheral eosinophilia. Other disorders, such as chronic eosinophilic pneumonia (CEP), may overlap considerably in presentation with, or be the initial manifestation of, systemic HES. Treatment varies depending on the specific clinical manifestations, but local or systemic corticosteroids are often effective. It is beyond the scope of this subchapter to discuss all organ-restricted eosinophilic syndromes, several of which are covered in other subchapters. Consequently, only a few examples are provided to illustrate the diversity of syndromes seen.
Pulmonary Eosinophilia
Eosinophilia restricted to the lung, with or without peripheral eosinophilia, is associated with a wide variety of allergic, infectious, inflammatory, and neoplastic disorders, including asthma, allergic bronchopulmonary aspergillosis, drug hypersensitivity reactions, fungal pneumonia, helminth infection, and sarcoidosis. Pulmonary eosinophilia can also be the first indicator of systemic eosinophilic diseases, including CSS. When no trigger is identified and disease remains restricted to the lung, pulmonary eosinophilia can be classified into two distinct syndromes: acute eosinophilic pneumonia (AEP) and CEP.
AEP typically occurs in healthy men between the ages of 20 and 40 years without a history of asthma. Symptoms include the abrupt onset of dyspnea, fever, malaise, night sweats, nonproductive cough, and pleuritic chest pain, and respiratory failure requiring mechanical ventilation is common. Physical examination may reveal bibasilar rales or rhonchi. Radiological findings include diffuse alveolar infiltrates or reticular opacities in the early stages and bronchoscopy reveals increased absolute eosinophil counts as well as an increased percentage of eosinophils (>25%) relative to the total inflammatory cell content.31 Whether an environmental exposure plays a role in the pathophysiology remains controversial. Patients typically respond well to high-dose corticosteroid therapy despite respiratory failure at presentation, and long-term sequelae are extremely uncommon.
CEP presents as a subacute illness, often mistaken for asthma in the early stages. It progresses to involve similar symptoms of cough and dyspnea, and is occasionally associated with constitutional symptoms of fever, sweats, and weight loss. Physical exam findings are similar to AEP, with wheeze and or crackles being present in most cases. Radiological findings typically show peripheral infiltrates. Bronchoalveolar lavage fluid (BALF) shows an eosinophil predominance. While patients may respond initially to corticosteroids, many relapse and may require long-term corticosteroid treatment. Peripheral eosinophilia of >1000/mm3 is common, but not universal.32
Eosinophilic Hepatitis
Eosinophilia of the liver may occur in primary liver diseases, such as primary biliary cirrhosis or autoimmune hepatitis, in the setting of hepatobiliary involvement by helminth infections, including clonorchiasis and schistosomiasis, or secondary to a wide variety of prescription and nonprescription drugs. Primary eosinophilic hepatitis with or without peripheral eosinophilia is, however, relatively rare. Early studies using immunohistochemistry implicated eosinophils in the pathogenesis of progression to chronic hepatitis by demonstrating major basic protein and activated, degranulated eosinophils in close proximity to affected hepatocytes.33 Hepatic cholangiopathy, fibrosis and liver failure have been reported, although most patients in the literature have been responsive to corticosteroids or hydroxyurea.
Eosinophilic Cystitis
Eosinophilic cystitis is a rare disorder, primarily seen in children, that presents with dysuria, gross hematuria, or suprapubic pain. Urinalysis may reveal microscopic hematuria or pyuria. Cystoscopy often reveals bladder wall erythema, edema, or nodules, and the diagnosis is made histologically from biopsies of the bladder.34 Infrequently, necrosis or fibrosis can be present with delayed diagnosis. It is important to exclude bladder cancer, since eosinophilic cystitis may be present in the setting of bladder cancer. Treatment with antihistamines, nonsteroidal anti-inflammatory medications, fulguration and steroids were reported to be successful in one series.35
Overlap Syndromes with Hypereosinophilic Syndrome
A number of systemic disorders have overlapping clinical presentations with HES. Of these, the two most common are SM and CSS. SM is a myeloproliferative disorder, most commonly associated with a D816V mutation in KIT. Patients typically present with symptoms related to mast cell tissue infiltration and mediator release, including anaphylaxis, diarrhea, flushing, and urticaria. Diagnosis is based on the presence of major and minor criteria according to the WHO classification.2 D816V-positive SM with eosinophilia (SM-eo) is clinically distinct from FIP1L1-PDGFRA-positive CEL and can be distinguished using molecular and clinical findings.18
CSS is a distinct multisystem disorder that is characterized by the presence of eosinophilic vasculitis in the setting of asthma, pulmonary infiltrates, sinusitis with polyps, and marked peripheral eosinophilia. ANCA may be present and appears to be associated with a more severe course, often with renal involvement. Definitive diagnosis can be made by tissue biopsy demonstrating granulomata and necrotizing eosinophilic vasculitis. Significant overlap in clinical presentation with idiopathic HES can cause diagnostic confusion, particularly since corticosteroids must often be initiated to prevent serious end-organ damage before appropriate biopsies can be obtained.36
Other Disorders Associated with Eosinophilia
Marked eosinophilia can be seen in association with immunodysregulation of varied etiologies, including primary immunodeficiencies, secondary immunodeficiencies [e.g., human immunodeficiency virus (HIV) infection], and autoimmune disorders (Table 13.9.1 ). In general, the peripheral eosinophilia seen in these disorders is not associated with characteristic end-organ manifestations. Exceptions include Omenn syndrome and Dock8 deficiency, which may present with eosinophilic tissue infiltration involving the skin or other organs.37., 38. In some disorders, including autoimmune lymphoproliferative syndrome (ALPS) and HIV, peripheral eosinophilia reflects more profound immunodysregulation and in some cases may portend a worse prognosis.39., 40.
TABLE 13.9.1.
Selected Disorders Associated with Immune Deficiency or Dysregulation and Eosinophilia
| Disorders Associated with Eosinophilia∗ |
| Autoimmune disorders |
| ALPS |
| Connective tissue disorders (e.g., dermatomyositis and rheumatoid arthritis) |
| Human immunodeficiency virus infection |
| Inflammatory bowel disease |
| Neoplasms (e.g., adenocarcinoma, lymphocytic leukemia, and lymphoma) |
| Primary immunodeficiencies |
| IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome) |
| DIDS (Dock8 immunodeficiency syndrome) |
| Omenn syndrome |
| Hyper-IgE syndrome (Job syndrome) |
| Atypical DiGeorge syndrome |
| Sarcoidosis |
This is only a partial list
Novel Therapies for Hypereosinophilic Syndrome
Improved understanding of the pathogenesis of eosinophilic disorders, including eosinophilic asthma, has led to the development of a number of novel agents, including agents targeting IL-5 and its receptor, although none are currently FDA approved. These are discussed in Chapter 15. Additional agents, including alemtuzumab, a monoclonal antibody that targets CD52 on aberrant T cells, and tyrosine kinase inhibitors with activity against imatinib-resistant FIP1L1/PDGFRA-positive mutations, have also been used with success in small numbers of patients with L-HES and CEL, respectively.24
Conclusion
The complexity of diagnosing and treating HES arises from the difficulty in excluding secondary forms of eosinophilia and knowing when to treat and with what medications. An understanding of underlying pathogenesis has improved genotype–phenotype classification of some eosinophilic disorders and reinforced the perception of HES as a heterogeneous group of rare disorders with differing pathogenesis, prognosis, and treatment options. Advancing knowledge of molecular pathogenesis and new genetic discoveries will provide the foundation for development of novel targeted therapies, as well as a promising outlook for the care of patients with HES.
References
- 1.Chusid M.J., Dale D.C., West B.C., Wolff S.M. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975;54:1–27. [PubMed] [Google Scholar]
- 2.Swerdlow S.H., International Agency for Research on Cancer, World Health Organization . International Agency for Research on Cancer; Lyon, France: 2008. WHO classification of tumours of haematopoietic and lymphoid tissues. [Google Scholar]
- 3.Klion A.D., Bochner B.S., Gleich G.J., Nutman T.B., Rothenberg M.E., Simon H.U. Approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J Allergy Clin Immunol. 2006;117:1292–1302. doi: 10.1016/j.jaci.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 4.Simon H.U., Rothenberg M.E., Bochner B.S., Weller P.F., Wardlaw A.J., Wechsler M.E. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol. 2010;126:45–49. doi: 10.1016/j.jaci.2010.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crane M.M., Chang C.M., Kobayashi M.G., Weller P.F. Incidence of myeloproliferative hypereosinophilic syndrome in the United States and an estimate of all hypereosinophilic syndrome incidence. J Allergy Clin Immunol. 2010;126:179–181. doi: 10.1016/j.jaci.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogbogu P.U., Bochner B.S., Butterfield J.H., Gleich G.J., Huss-Marp J., Kahn J.E. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol. 2009;124 doi: 10.1016/j.jaci.2009.09.022. 1319–1325, e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravoet M., Sibille C., Gu C., Libin M., Haibe-Kains B., Sotiriou C. Molecular profiling of CD3-CD4+ T cells from patients with the lymphocytic variant of hypereosinophilic syndrome reveals targeting of growth control pathways. Blood. 2009;114:2969–2983. doi: 10.1182/blood-2008-08-175091. [DOI] [PubMed] [Google Scholar]
- 8.Raghavachar A., Fleischer S., Frickhofen N., Heimpel H., Fleischer B. T lymphocyte control of human eosinophilic granulopoiesis. Clonal analysis in an idiopathic hypereosinophilic syndrome. J Immunol. 1987;139:3753–3758. [PubMed] [Google Scholar]
- 9.Roufosse F., Cogan E., Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am. 2007;27:389–413. doi: 10.1016/j.iac.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Simon H.U., Plotz S.G., Dummer R., Blaser K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med. 1999;341:1112–1120. doi: 10.1056/NEJM199910073411503. [DOI] [PubMed] [Google Scholar]
- 11.Edelman J., Meyerson H.J. Diminished CD3 expression is useful for detecting and enumerating Sezary cells. Am J Clin Pathol. 2000;114:467–477. doi: 10.1093/ajcp/114.3.467. [DOI] [PubMed] [Google Scholar]
- 12.Brugnoni D., Airo P., Rossi G., Bettinardi A., Simon H.U., Garza L. A case of hypereosinophilic syndrome is associated with the expansion of a CD3-CD4+ T-cell population able to secrete large amounts of interleukin-5. Blood. 1996;87:1416–1422. [PubMed] [Google Scholar]
- 13.Schandene L., Roufosse F., de Lavareille A., Stordeur P., Efira A., Kennes B. Interferon alpha prevents spontaneous apoptosis of clonal Th2 cells associated with chronic hypereosinophilia. Blood. 2000;96:4285–4292. [PubMed] [Google Scholar]
- 14.Roufosse F., de Lavareille A., Schandene L., Cogan E., Georgelas A., Wagner L. Mepolizumab as a corticosteroid-sparing agent in lymphocytic variant hypereosinophilic syndrome. J Allergy Clin Immunol. 2010;126 doi: 10.1016/j.jaci.2010.06.049. 828–835, e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cools J., DeAngelo D.J., Gotlib J., Stover E.H., Legare R.D., Cortes J. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 16.Buitenhuis M., Verhagen L.P., Cools J., Coffer P.J. Molecular mechanisms underlying FIP1L1-PDGFRA-mediated myeloproliferation. Cancer Res. 2007;67:3759–3766. doi: 10.1158/0008-5472.CAN-06-4183. [DOI] [PubMed] [Google Scholar]
- 17.Robyn J., Lemery S., McCoy J.P., Kubofcik J., Kim Y.J., Pack S. Multilineage involvement of the fusion gene in patients with FIP1L1/PDGFRA-positive hypereosinophilic syndrome. Br J Haematol. 2006;132:286–292. doi: 10.1111/j.1365-2141.2005.05863.x. [DOI] [PubMed] [Google Scholar]
- 18.Maric I., Robyn J., Metcalfe D.D., Fay M.P., Carter M., Wilson T. KIT D816V-associated systemic mastocytosis with eosinophilia and FIP1L1/PDGFRA-associated chronic eosinophilic leukemia are distinct entities. J Allergy Clin Immunol. 2007;120:680–687. doi: 10.1016/j.jaci.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 19.Klion A.D., Robyn J., Akin C., Noel P., Brown M., Law M. Molecular remission and reversal of myelofibrosis in response to imatinib mesylate treatment in patients with the myeloproliferative variant of hypereosinophilic syndrome. Blood. 2004;103:473–478. doi: 10.1182/blood-2003-08-2798. [DOI] [PubMed] [Google Scholar]
- 20.Sakamoto K., Erdreich-Epstein A., deClerck Y., Coates T. Prolonged clinical response to vincristine treatment in two patients with idiopathic hypereosinophilic syndrome. Am J Pediatr Hematol Oncol. 1992;14:348–351. [PubMed] [Google Scholar]
- 21.Ueno N.T., Anagnostopoulos A., Rondon G., Champlin R.E., Mikhailova N., Pankratova O.S. Successful non-myeloablative allogeneic transplantation for treatment of idiopathic hypereosinophilic syndrome. Br J Haematol. 2002;119:131–134. doi: 10.1046/j.1365-2141.2002.03771.x. [DOI] [PubMed] [Google Scholar]
- 22.Butterfield J.H. Treatment of hypereosinophilic syndromes with prednisone, hydroxyurea, and interferon. Immunol Allergy Clin North Am. 2007;27:493–518. doi: 10.1016/j.iac.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Jabbour E., Verstovsek S., Giles F., Gandhi V., Cortes J., O’Brien S. 2-Chlorodeoxyadenosine and cytarabine combination therapy for idiopathic hypereosinophilic syndrome. Cancer. 2005;104:541–546. doi: 10.1002/cncr.21186. [DOI] [PubMed] [Google Scholar]
- 24.Verstovsek S., Tefferi A., Kantarjian H., Manshouri T., Luthra R., Pardanani A. Alemtuzumab therapy for hypereosinophilic syndrome and chronic eosinophilic leukemia. Clin Cancer Res. 2009;15:368–373. doi: 10.1158/1078-0432.CCR-08-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitini V., Teti D., Arrigo C., Righi M. Alemtuzumab therapy for refractory idiopathic hypereosinophilic syndrome with abnormal T cells: a case report. Br J Haematol. 2004;127:477. doi: 10.1111/j.1365-2141.2004.05206.x. [DOI] [PubMed] [Google Scholar]
- 26.Orson F.M. Intravenous immunoglobulin therapy suppresses manifestations of the angioedema with hypereosinophilia syndrome. Am J Med Sci. 2003;326:94–97. doi: 10.1097/00000441-200308000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Donald C.E., Kahn M.J. Successful treatment of hypereosinophilic syndrome with cyclosporine. Am J Med Sci. 2009;337:65–66. doi: 10.1097/MAJ.0b013e318169569a. [DOI] [PubMed] [Google Scholar]
- 28.Lee J.H., Lee J.W., Jang C.S., Kwon E.S., Min H.Y., Jeong S. Successful cyclophosphamide therapy in recurrent eosinophilic colitis associated with hypereosinophilic syndrome. Yonsei Med J. 2002;43:267–270. doi: 10.3349/ymj.2002.43.2.267. [DOI] [PubMed] [Google Scholar]
- 29.Klion A.D., Law M.A., Riemenschneider W., McMaster M.L., Brown M.R., Horne M. Familial eosinophilia: a benign disorder? Blood. 2004;103:4050–4055. doi: 10.1182/blood-2003-11-3850. [DOI] [PubMed] [Google Scholar]
- 30.Rioux J.D., Stone V.A., Daly M.J., Cargill M., Green T., Nguyen H. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086–1094. doi: 10.1086/302053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Philit F., Etienne-Mastroianni B., Parrot A., Guerin C., Robert D., Cordier J.F. Idiopathic acute eosinophilic pneumonia: a study of 22 patients. Am J Respir Crit Care Med. 2002;166:1235–1239. doi: 10.1164/rccm.2112056. [DOI] [PubMed] [Google Scholar]
- 32.Marchand E., Reynaud-Gaubert M., Lauque D., Durieu J., Tonnel A.B., Cordier J.F. Idiopathic chronic eosinophilic pneumonia. A clinical and follow-up study of 62 cases. The Groupe d’Etudes et de Recherche sur les Maladies ‘Orphelines’ Pulmonaires (GERM‘O’P) Medicine (Baltimore) 1998;77:299–312. doi: 10.1097/00005792-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 33.Foong A., Scholes J.V., Gleich G.J., Kephart G.M., Holt P.R. Eosinophil-induced chronic active hepatitis in the idiopathic hypereosinophilic syndrome. Hepatology. 1991;13:1090–1094. [PubMed] [Google Scholar]
- 34.Itano N.M., Malek R.S. Eosinophilic cystitis in adults. J Urol. 2001;165:805–807. [PubMed] [Google Scholar]
- 35.Kilic S., Erguvan R., Ipek D., Gokce H., Gunes A., Aydin N.E. Eosinophilic cystitis. A rare inflammatory pathology mimicking bladder neoplasms. Urol Int. 2003;71:285–289. doi: 10.1159/000072680. [DOI] [PubMed] [Google Scholar]
- 36.Wechsler M.E. Pulmonary eosinophilic syndromes. Immunol Allergy Clin North Am. 2007;27:477–492. doi: 10.1016/j.iac.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q., Davis J.C., Dove C.G., Su H.C. Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome. Dis Markers. 2010;29:131–139. doi: 10.3233/DMA-2010-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villa A., Notarangelo L.D., Roifman C.M. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122:1082–1086. doi: 10.1016/j.jaci.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 39.Kim Y.J., Dale J.K., Noel P., Brown M.R., Nutman T.B., Straus S.E. Eosinophilia is associated with a higher mortality rate among patients with autoimmune lymphoproliferative syndrome. Am J Hematol. 2007;82:615–624. doi: 10.1002/ajh.20851. [DOI] [PubMed] [Google Scholar]
- 40.Rosenthal D., LeBoit P.E., Klumpp L., Berger T.G. Human immunodeficiency virus-associated eosinophilic folliculitis. A unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206–209. [PubMed] [Google Scholar]
Chapter 13.10. Eosinophils and Cancer
Ramin Lotfi, Neal Spada, Michael Thomas Lotze
Common findings within tumors are areas of microthrombosis and micronecrosis. Necrosis and most forms of stress are typically associated with subsequent release of damage-associated molecular pattern molecules (DAMPs), including high mobility group B1 (HMGB1), S100 proteins, DNA, and heat shock proteins. When evaluating a tumor, it is important to assess not only the tumor cells themselves but also their microenvironment, consisting of1 the quantity and quality of tumor-surrounding stromal and inflammatory cells,2 their state of activation, and3 released factors, including DAMPs. Eosinophilia is frequently associated with several tumor types and eosinophils are also found at increased numbers within the blood and tumor following tumor immunotherapy with interleukin-2 (IL-2), IL-4, granulocyte-macrophage colony-stimulating factor, or antibody to cytotoxic T-lymphocyte protein 4. Candidates for promoting eosinophil chemotaxis into tumor tissue are the eosinophil chemotactic factors eotaxin and C-C motif chemokine 24/eotaxin 2, as well as DAMPs which are released following necrosis or during periods of hypoxic, nutrient, or oxidant stress. By acting as danger signals, DAMPs not only induce angiogenesis but also activate eosinophils that are capable of oxidizing and thus inactivating DAMPs. Tumor-associated eosinophils have at least two dominant nonoverlapping activities:1 destructive effector functions potentially limiting tumor growth as well as causing recruitment and activation of other leukocytes; and2 immunoregulatory and remodeling activities that may suppress immune responses.
Inflammation and Necrosis are Major Components of the Epithelial Tumor Microenvironment
Advanced epithelial tumors typically undergo necrosis with subsequent release of damage-associated molecular pattern molecules (DAMPs).1 Tumor cells are dependent on the host-created microenvironment, including endothelial cells, inflammatory cells, and stromal cells. This makes it difficult to cultivate more than a minority of tumor cells in vitro but, as is increasingly being understood, provides unique opportunities for cancer therapy. Thus, when evaluating a tumor, it is important to assess three elements within the microenvironment:
-
1.
Factors released by tumor cells and their surrounding cells, consisting of epithelial and endothelial cells, fibroblasts, specialized local mesenchymal cells, and infiltrating leukocytes;
-
2.
The quantity and quality of tumor-associated cells, specifically leukocytes;
-
3.
Their state of activation.
The mammalian immune system reciprocally interacts within dynamic networks of tissue-associated nonimmune cells, enabling metabolic homeostasis, orderly scheduled cellular maturation and replacement, the timely eradication of effete cells, the repair of damage, and protection against pathogens. The simultaneous tolerance to self-antigens and reciprocal reactivity to new or occult antigens often occurs in settings of tissue damage and wound healing. When tissue homeostasis is perturbed, mast cells, granulocytes, and macrophages are recruited and rapidly release mediators such as cytokines, chemokines, matrix remodeling proteases and reactive oxygen species (ROS), and bioactive mediators such as histamine. These in turn induce mobilization and infiltration of additional leukocytes into damaged tissue (causing inflammation). Subsequently, the process of wound healing begins, characterized by phagocytosis of cellular debris and apoptotic cells, immune suppression, reepithelialization, and synthesis of extracellular matrix (ECM). Thus, inflammation is resolved, thereby restoring tissue homeostasis. Tumor cells, paradoxically growing in the setting of substantial necrosis and (chronic) inflammation, harness the collaborative capabilities (see below) of immune cells and local nonmutated but injured tissues to promote cell survival and proliferation, partly by releasing factors such as transforming growth factor β (TGF-β) and interleukin-10 (IL-10), leading to restoration of barrier function in epithelial tissues. The host therefore enables tumor cells to escape from eradication and to release tissue-healing factors, thereby providing neovascularization and subsequent nutritional supply to tumor cells. Wound healing and tumor stroma formation share many important properties. Nevertheless, wound healing is itself a self-limiting process, whereas tumors addicted to death 1 release DAMPs, thereby sustaining tissue proliferation, angiogenesis and continuous leukocyte recruitment.
Prolonged (chronic) inflammation is often associated with carcinogenesis. ROS generated largely intracellularly or at the cell membrane by NADPH oxidases can also promote mutagenic changes in cells when aerobic denaturation of extracellular DAMPs is ineffective.2 Tumor necrosis factor α (TNF-α)3 and matrix metalloproteinases promote recruitment of inflammatory cells and tissue remodeling, but also facilitate tumor metastasis. A state of metabolic symbiosis4., 5., 6., 7., 8. between the tumor and the surrounding stroma, or within central hypoxic tumor cells and those at the growing rim of the tumor, allows regional variations in oxidative phosphorylation and autophagy that depend on nutrient supply and availability of molecular oxygen.
Biology of Eosinophils within Normal and Damaged Tissues
Compartments with abundant resident populations of eosinophils include tissues with substantial cellular turnover and regenerative capacity, such as the bone marrow, primary and secondary lymphoid tissues (e.g., lymph nodes, spleen, and thymus),9 the uterus,10 and almost the entire gastrointestinal tract (with the exception of the esophagus, except under abnormal states).9., 11. This link with cell turnover and tissue repair may also explain the presence of eosinophils at sites of wound repair12 and the commonality of an eosinophil infiltrate among solid tumors.13 Eosinophil localization to the lamina propria of the gastrointestinal tract is critically regulated by eotaxin/C-C motif chemokine 11 (CCL11), a chemokine constitutively expressed throughout the gastrointestinal tract.14 Nevertheless, eotaxin expression within the gastrointestinal tract (e.g., the esophagus) is by itself insufficient to induce eosinophil accumulation, because the esophagus is normally devoid of these granulocytes. This suggests the potential involvement of other eosinophil chemoattractants and activating factors that contribute to tissue-specific accumulation and degranulation. In particular, the correlation of eosinophil recruitment/activation with tissue damage and cell death associated with these inflammatory responses suggests that DAMPs may represent previously overlooked signaling molecules that elicit eosinophil agonist activities (Fig. 13.10.1 ). Consistently, high mobility group box 1 (HMGB1; a prototypic DAMP molecule) serves as a chemoattractant and survival factor for eosinophilic granulocytes.15
FIGURE 13.10.1.

Eosinophilic and neutrophilic granulocytes are recruited to neoplastic tissue by DAMPs.
DAMPs released by (necrotic) neoplastic cells recruit eosinophils and neutrophils. They in turn produce and release reactive oxygen species (ROS), inducing oxidation and degradation of tumor-related DAMPs. In addition, they initiate an acute inflammatory response to recruit, activate, and polarize B cells, cytotoxic lymphocytes, dendritic cells (DCs), and monocytes. Chronic exposure to active DAMPs promotes angiogenesis and immunosuppression by recruiting and activating endothelial cells, fibroblasts, and leukocytes such as T-regulatory (Treg) cells, and myeloid-derived suppressor (MDS) cells, which are important for promoting wound healing. ATP, adenosine 5′-triphosphate; HMGB1, high mobility group box 1; HSP, heat shock protein; S100, protein S100 Th2, T-helper type 2.
Eosinophilic granulocytes are found within necrotic tissues and the pseudocapsule surrounding tumors.16 These immune cells contain, and can release, several cationic proteins that, in addition to their toxic tissue damaging character, are also potentially important for tissue remodeling and clearing cellular debris.17 Eosinophils are thought to be, in part, responsible for the cell death and tissue damage commonly observed in disease states that are associated with increased eosinophil numbers and tissue-specific eosinophil recruitment.18
Since the mid-1980s, eosinophils have been known to mediate their effects via at least three independent mechanisms in addition to the release of cytotoxic granule proteins, which enable them to modulate the intensity of inflammation, as well as to elicit cell death leading to the loss of tissue integrity:
-
1.
Eosinophils may act as potent regulators of local inflammatory responses.19
-
2.
Recruited eosinophils are a source of reactive oxygenated species20 and established small molecule lipid mediators of inflammation. In particular, eosinophils generate cysteinyl leukotrienes (i.e., LTB4, LTC4, LTD4, and LTE4,21., 22. 5-HETE,23 PGE2,24 and platelet-activating factor (PAF).25 The capability of cysteinyl leukotrienes to mediate primary inflammatory responses such as edema,26 the recruitment of other proinflammatory leukocytes,27 and the induction of tissue histopathology28 uniquely positions these molecules as mediators of inflammation.
-
3.
Eosinophils are a prodigious source of cytokines associated with tissue repair and remodeling.
A growing body of literature suggests that both immunoregulation and tissue repair/remodeling may represent important nonoverlapping eosinophil effector functions.17 A quantitative assessment of eosinophil recruitment/accumulation in solid tumors showed that the tissue eosinophilia is apparently mediated by one or more factors released directly from necrotic tissues within the tumor. Studies linking eosinophil recruitment and activations with cell death and necrosis abound.15., 16., 29. Thus, DAMPs released from damaged/dying epithelial cells may represent a previously underappreciated signaling event capable of mediating both eosinophil recruitment and the execution of effector functions leading to (and/or promoting) tissue repair and remodeling. In addition to their capacity to synthesize and release a variety of immunoregulatory molecules,30 some studies have suggested that eosinophils may function as antigen presenting cells (APCs)31 or enhance DC maturation.19 Eosinophils may also affect local T cell responses by modulating the balance of T-helper type 1 (Th1) and Th2 immune responses [e.g., through eosinophil-derived indoleamine 2,3-dioxygenase (IDO) production of kynurenine32]. Importantly, IDO appears to be essential for the induction of tolerance by tissue recruited T cells. Thus, similar to other eosinophil-mediated immunosupressive activities (e.g., the potential induction of T-regulatory cells through TGF-β production),33 eosinophil-derived IDO may also play a crucial role in immunosuppression and potentially facilitate tissue repair and, as a by-product, tumor growth.
Eosinophils and the Immunotherapy of Patients with Cancer
The eosinophil plays a somewhat passive role in the tumor. Eosinophils are frequently observed following immunotherapy with IL-2,34 IL-4,35., 36. GM-CSF,37 repeated vaccination,38 and antibodies to CTLA-4, but the significance of their appearance remains largely unknown. In particular, the antitumor effects of successful cytokine therapy of cancer with IL-2 has been associated with the identification of degranulating eosinophils within the tumor,34 suggesting that eosinophil effector functions (e.g., direct or antibody-dependent tumor cell lysis or the immunoregulatory capacity of eosinophils modulating the local tumor microenvironment) may play a role in the anticancer activities mediated by systemic IL-2 administration.34., 39. However, despite the promise of these potential eosinophil-mediated antitumor activities, the presence of eosinophils is not an important prognostic indicator in high-dose IL-2-treated patients.
Mouse studies suggesting a link between eosinophils and the therapeutic value of antitumor responses associated with IL-4 administration40 have led to clinical trials to evaluate these responses in cancer patients. In a phase I clinical trial of IL-4 administered to cancer patients, Sosman and colleagues35 showed that IL-4 therapy induced systemic eosinophil degranulation associated with increased serum and urine major basic protein (MBP) levels. Moreover, the increase in serum MBP was IL-4 dose dependent. Unfortunately, the link of antitumor activities with eosinophil numbers in these patients is only correlative and similar to observations made in patients following IL-2 administration. No definitive conclusions can be made as to whether and how eosinophils modulate tumor growth.
Efforts to demonstrate experimentally a role for eosinophils in tumor immunity have also been fraught with complicating factors yielding qualified interpretations. Most notably, considerable excitement was generated by data from the elegant studies of Tepper and colleagues36., 40., 41. that demonstrated in athymic nude mice that malignant cell lines transfected for constitutive expression of interleukin 4 (IL-4) elicited a tumor associated macrophage and eosinophil infiltrate that led to the attenuation of tumor growth. This provoked a series of tumor xenograft studies that attempted to define the cellular and molecular mechanisms of the apparent IL-4-mediated antitumor effect. Although these studies have shown that spontaneous tumors showed evidence of tumor regression, associated with tumor-infiltrating eosinophils, none of these studies has resolved the role(s) of eosinophils in tumor rejection reactions. Recently, colon tumor eradication by eosinophils in murine models suggested a more conventional cytolytic response.42 In this study, coculture of eosinophils with colorectal tumor cells led to the release of eosinophil cationic protein and eosinophil-derived neurotoxin, as well as TNF-α secretion. Interestingly, this may be related to the ability of eosinophils to both lyse and promote clearance of stressed or damaged cells.43 Eosinophils accumulate at unique sites in response to cell turnover, thus regulating tissue homeostasis, and are regulators of Local Immunity and/or Remodeling/Repair according to the LIAR hypothesis, suggesting a more nuanced and interesting role for these cells in damaged tissues and tumors.44
Within several tumor types, including gastrointestinal tumors, tumor-associated tissue eosinophilia (TATE) is associated with a significantly better prognosis.30 The converse is true in other tumor types, such as differentiated oral squamous cell carcinoma30 or Hodgkin lymphoma,45., 46. where eosinophils may be involved in promoting cancer cell growth. The mechanism by which eosinophils, in particular, are recruited into tumor tissues is largely unknown. We could characterize DAMPs, including the nuclear protein HMGB1, as candidate factors eliciting eosinophil chemotaxis into tumor tissue.47 Thus, eosinophil activities are likely to have multiple roles, dictated by specific circumstances, which were originally adapted to maintain tissue homeostasis. Eosinophils are not only able to destroy tissue but are also attracted and activated by stressed and damaged cells, as we and others have demonstrated.16., 47. It is likely that stressed cells attract and activate eosinophils by expressing molecules such as NKG2D ligand 4 (LETAL), major histocompatibility complex class I chain related A (MICA), and MICB,11 as well as other NKG2D ligands or UL16-binding proteins (ULBPs). These stress-associated molecules all serve as ligands for NKG2D, first described on natural killer (NK) cells45 and subsequently on eotaxin-activated eosinophils48 and T cells.49 Thus, tumor-associated eosinophils appear to have at least two dominant nonoverlapping activities:
-
1.
Destructive effector functions that may limit tumor growth and induce recruitment and activation of other leukocytes;
-
2.
Immunoregulatory and remodeling activities that suppress immune response and release cytokines, thus promoting wound healing.
Consistent with the hypothesis that DAMPs initiate innate immune cell activation when encountering microbes or parasites,50 eosinophils are often first responders to tissue damage and likely mediate some aspects of tissue remodeling and repair. The presence of DAMPs such as HMGB1 in the necrotic areas of tumors may, in part, elicit both eosinophil tissue recruitment and localized execution of effector functions such as degranulation. The available data, however, suggest that while all threatened epithelial cells, including cancer cells, release DAMPs, not all eosinophil tissue infiltration is associated with tumor eradication.51., 53. This conclusion again suggests that the relationship of eosinophils with the modulation of tumor onset/growth is complex and that expression of DAMPs, the emergent role of autophagy, and redox chemistry54., 55., 56., 57., 58., 59., 60., 61., 62., 63., 64., 65., 66., 67., 68. is likely to be only one of several inflammatory mechanisms capable of eliciting eosinophil effector functions.
Interestingly, of all tested biologic activities, eosinophils respond most sensitively to the presence of DAMPs, including HMGB1, with generation of peroxide leading to oxidative degradation and thus inactivation of DAMPs.15 These DAMPs with or without oxidation, play differential roles in the recruitment of mesenchymal stem cells as well as T regulatory cells which may confound and limit the antitumor response.69., 70., 71., 72.
Conclusion
In summary, while the role of eosinophils in tumor onset and growth is unresolved, recent studies suggest that eosinophils are a common and robust tumor infiltrate and that much interesting biology remains to be explored. Specifically, do eosinophil activities limit tumor growth through destructive effector functions or do eosinophil-derived immunoregulation and tissue repair/remodeling promote tumor growth and metastasis? How does the critical role of redox49 inform eosinophil function in relation to DAMPs? The resolution of these questions could inform the initiation of eosinophil-based modalities and, in turn, novel therapeutic approaches to treat cancer patients.
References
- 1.Zeh H.J., III, Lotze M.T. Addicted to death: invasive cancer and the immune response to unscheduled cell death. J Immunother. 2005;28(1):1–9. doi: 10.1097/00002371-200501000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Cerutti P.A. Prooxidant states and tumor promotion. Science (New York, NY. 1985;227(4685):375–381. doi: 10.1126/science.2981433. [DOI] [PubMed] [Google Scholar]
- 3.Moore R.J., Owens D.M., Stamp G., Arnott C., Burke F., East N. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nature medicine. 1999;5(7):828–831. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 4.Sonveaux P., Vegran F., Schroeder T., Wergin M.C., Verrax J., Rabbani Z.N. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008 Dec;118(12):3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza G.L. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008 Dec;118(12):3835–3837. doi: 10.1172/JCI37373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009 Sep;92(3):329–333. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 7.Le A., Cooper C.R., Gouw A.M., Dinavahi R., Maitra A., Deck L.M. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi: 10.1073/pnas.0914433107. Feb 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Outschoorn U.E., Whitaker-Menezes D., Pavlides S., Chiavarina B., Bonuccelli G., Casey T. The autophagic tumor stroma model of cancer or ‘battery-operated tumor growth’: A simple solution to the autophagy paradox. Cell Cycle. 2010 Nov;9(21):4297–4306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothenberg M.E., Mishra A., Brandt E.B., Hogan S.P. Gastrointestinal eosinophils. Immunological Reviews. 2001;179:139–155. doi: 10.1034/j.1600-065x.2001.790114.x. [DOI] [PubMed] [Google Scholar]
- 10.Jones R.L., Hannan N.J., Kaitu’u T.J., Zhang J., Salamonsen L.A. Identification of chemokines important for leukocyte recruitment to the human endometrium at the times of embryo implantation and menstruation. J Clin Endocrinol Metab. 2004 Dec;89(12):6155–6167. doi: 10.1210/jc.2004-0507. [DOI] [PubMed] [Google Scholar]
- 11.Matthews A.N., Friend D.S., Zimmermann N., Sarafi M.N., Luster A.D., Pearlman E. Eotaxin is required for the baseline level of tissue eosinophils. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6273–6278. doi: 10.1073/pnas.95.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J., Torio A., Donoff R.B., Gallagher G.T., Egan R., Weller P.F. Depletion of eosinophil infiltration by anti-IL-5 monoclonal antibody (TRFK-5) accelerates open skin wound epithelial closure. Am J Pathol. 1997 Sep;151(3):813–819. [PMC free article] [PubMed] [Google Scholar]
- 13.Samoszuk M. Eosinophils and human cancer. Histol Histopathol. 1997;12(3):807–812. [PubMed] [Google Scholar]
- 14.Rothenberg M.E. Eotaxin. An essential mediator of eosinophil trafficking into mucosal tissues. Am J Respir Cell Mol Biol. 1999;21(3):291–295. doi: 10.1165/ajrcmb.21.3.f160. [DOI] [PubMed] [Google Scholar]
- 15.Lotfi R., Herzog G.I., DeMarco R.A., Beer-Stolz D., Lee J.J., Rubartelli A. Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J Immunol. 2009 Oct 15;183(8):5023–5031. doi: 10.4049/jimmunol.0900504. [DOI] [PubMed] [Google Scholar]
- 16.Cormier S.A., Taranova A.G., Bedient C., Nguyen T., Protheroe C., Pero R. Pivotal advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. Journal of Leukocyte Biology. 2006;79(6):1131–1139. doi: 10.1189/jlb.0106027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J.J., Lee N.A. Eosinophil degranulation: an evolutionary vestige or a universally destructive effector function? Clin Exp Allergy. 2005;35(8):986–994. doi: 10.1111/j.1365-2222.2005.02302.x. [DOI] [PubMed] [Google Scholar]
- 18.Afshar K., Vucinic V., Sharma O.P. Eosinophil cell: pray tell us what you do! Current Opinion in Pulmonary Medicine. 2007;13(5):414–421. doi: 10.1097/MCP.0b013e328224b90b. [DOI] [PubMed] [Google Scholar]
- 19.Lotfi R., Lotze M.T. Eosinophils induce DC maturation, regulating immunity. Journal of Leukocyte Biology. 2008;83(3):456–460. doi: 10.1189/jlb.0607366. [DOI] [PubMed] [Google Scholar]
- 20.Nagata M., Sedgwick J.B., Bates M.E., Kita H., Busse W.W. Eosinophil adhesion to vascular cell adhesion molecule-1 activates superoxide anion generation. J Immunol. 1995 Aug 15;155(4):2194–2202. [PubMed] [Google Scholar]
- 21.Bandeira-Melo C., Woods L.J., Phoofolo M., Weller P.F. Intracrine cysteinyl leukotriene receptor-mediated signaling of eosinophil vesicular transport-mediated interleukin-4 secretion. J Exp Med. 2002 Sep 16;196(6):841–850. doi: 10.1084/jem.20020516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson W.R., Harley J.B., Fauci A.S. Arachidonic acid metabolism in normal and hypereosinophilic syndrome human eosinophils: generation of leukotrienes B4, C4, D4 and 15-lipoxygenase products. Immunology. 1984 Apr;51(4):679–686. [PMC free article] [PubMed] [Google Scholar]
- 23.Turk J., Maas R.L., Brash A.R., Roberts L.J., Oates J.A. Arachidonic acid 15-lipoxygenase products from human eosinophils. J Biol Chem. 1982 Jun 25;257(12):7068–7076. [PubMed] [Google Scholar]
- 24.Bruijnzeel P., Kok P., Kijne G., Verhagen J. Leukotriene synthesis by isolated granulocytes from intrinsic and extrinsic asthmatics and age-matched controls. Agents Actions Suppl. 1989;28:191–194. [PubMed] [Google Scholar]
- 25.Lee T., Lenihan D.J., Malone B., Roddy L.L., Wasserman S.I. Increased biosynthesis of platelet-activating factor in activated human eosinophils. J Biol Chem. 1984 May 10;259(9):5526–5530. [PubMed] [Google Scholar]
- 26.Hay D.W., Torphy T.J., Undem B.J. Cysteinyl leukotrienes in asthma: old mediators up to new tricks. Trends Pharmacol Sci. 1995 Sep;16(9):304–309. doi: 10.1016/s0165-6147(00)89059-8. [DOI] [PubMed] [Google Scholar]
- 27.Ford-Hutchinson A.W., Bray M.A., Doig M.V., Shipley M.E., Smith M.J., Leukotriene B. a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980 Jul 17;286(5770):264–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 28.Busse W.W. Leukotrienes and inflammation. Am J Respir Crit Care Med. 1998 Jun;157(6 Pt 2):S210–S213. [PubMed] [Google Scholar]
- 29.Stenfeldt A.L., Wenneras C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology. 2004;112(4):605–614. doi: 10.1111/j.1365-2567.2004.01906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lotfi R., Lee J.J., Lotze M.T. Eosinophilic granulocytes and damage-associated molecular pattern molecules (DAMPs): role in the inflammatory response within tumors. J Immunother. 2007 Jan;30(1):16–28. doi: 10.1097/01.cji.0000211324.53396.f6. [DOI] [PubMed] [Google Scholar]
- 31.Akuthota P., Wang H., Weller P.F. Eosinophils as antigen-presenting cells in allergic upper airway disease. Curr Opin Allergy Clin Immunol. 2010 Feb;10(1):14–19. doi: 10.1097/ACI.0b013e328334f693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odemuyiwa S.O., Ghahary A., Li Y., Puttagunta L., Lee J.E., Musat-Marcu S. Cutting edge: human eosinophils regulate T cell subset selection through indoleamine 2,3-dioxygenase. J Immunol. 2004;173(10):5909–5913. doi: 10.4049/jimmunol.173.10.5909. [DOI] [PubMed] [Google Scholar]
- 33.Schramm C., Huber S., Protschka M., Czochra P., Burg J., Schmitt E. TGFbeta regulates the CD4+CD25+ T-cell pool and the expression of Foxp3 in vivo. Int Immunol. 2004 Sep;16(9):1241–1249. doi: 10.1093/intimm/dxh126. [DOI] [PubMed] [Google Scholar]
- 34.Huland E., Huland H. Tumor-associated eosinophilia in interleukin-2-treated patients: evidence of toxic eosinophil degranulation on bladder cancer cells. J Cancer Res Clin Oncol. 1992;118(6):463–467. doi: 10.1007/BF01629431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sosman J.A., Bartemes K., Offord K.P., Kita H., Fisher S.G., Kefer C. Evidence for eosinophil activation in cancer patients receiving recombinant interleukin-4: effects of interleukin-4 alone and following interleukin-2 administration. Clin Cancer Res. 1995;1(8):805–812. [PubMed] [Google Scholar]
- 36.Tepper R.I., Coffman R.L., Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257(5069):548–551. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 37.Bristol J.A., Zhu M., Ji H., Mina M., Xie Y., Clarke L. In vitro and in vivo activities of an oncolytic adenoviral vector designed to express GM-CSF. Mol Ther. 2003;7(6):755–764. doi: 10.1016/s1525-0016(03)00103-5. [DOI] [PubMed] [Google Scholar]
- 38.Schaefer J.T., Patterson J.W., Deacon D.H., Smolkin M.E., Petroni G.R., Jackson E.M. Dynamic changes in cellular infiltrates with repeated cutaneous vaccination: a histologic and immunophenotypic analysis. J Transl Med. 2010 Aug;20(8):79. doi: 10.1186/1479-5876-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivoltini L., Viggiano V., Spinazze S., Santoro A., Colombo M.P., Takatsu K. In vitro anti-tumor activity of eosinophils from cancer patients treated with subcutaneous administration of interleukin 2. Role of interleukin 5. International Journal of Cancer. 1993;54(1):8–15. doi: 10.1002/ijc.2910540103. [DOI] [PubMed] [Google Scholar]
- 40.Tepper R.I., Pattengale P.K., Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 1989 May 5;57(3):503–512. doi: 10.1016/0092-8674(89)90925-2. [DOI] [PubMed] [Google Scholar]
- 41.Tepper R.I. The eosinophil-mediated antitumor activity of interleukin-4. The Journal of Allergy and Clinical Immunology. 1994;94(6 Pt 2):1225–1231. doi: 10.1016/0091-6749(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 42.Legrand F., Driss V., Delbeke M., Loiseau S., Hermann E., Dombrowicz D. Human Eosinophils Exert TNF-{alpha} and Granzyme A-Mediated Tumoricidal Activity toward Colon Carcinoma Cells. J Immunol. 2010 Nov 10;185(12):7443–7451. doi: 10.4049/jimmunol.1000446. [DOI] [PubMed] [Google Scholar]
- 43.Kim H.J., Alonzo E.S., Dorothee G., Pollard J.W., Sant’Angelo D.B. Selective depletion of eosinophils or neutrophils in mice impacts the efficiency of apoptotic cell clearance in the thymus. PLoS One. 2010 Jul 6;5(7) doi: 10.1371/journal.pone.0011439. e11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee J.J., Jacobsen E.A., McGarry M.P., Schleimer R.P., Lee N.A. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy. 2010 Apr;40(4):563–575. doi: 10.1111/j.1365-2222.2010.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costello R.T., Fauriat C., Sivori S., Marcenaro E., Olive D. NK cells: innate immunity against hematological malignancies? Trends in Immunology. 2004;25(6):328–333. doi: 10.1016/j.it.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Enblad G., Molin D., Glimelius I., Fischer M., Nilsson G. The potential role of innate immunity in the pathogenesis of Hodgkin’s lymphoma. Hematology/oncology Clinics of North America. 2007;21(5):805–823. doi: 10.1016/j.hoc.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 47.Lotfi R., Herzog G.I., DeMarco R.A., Beer-Stolz D., Lee J.J., Rubartelli A. Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J Immunol. 2009 Oct 15;183(8):5023–5031. doi: 10.4049/jimmunol.0900504. [DOI] [PubMed] [Google Scholar]
- 48.Kataoka S., Konishi Y., Nishio Y., Fujikawa-Adachi K., Tominaga A. Antitumor activity of eosinophils activated by IL-5 and eotaxin against hepatocellular carcinoma. DNA Cell Biol. 2004;23(9):549–560. doi: 10.1089/dna.2004.23.549. [DOI] [PubMed] [Google Scholar]
- 49.Burgess S.J., Marusina A.I., Pathmanathan I., Borrego F., Coligan J.E. IL-21 Down-Regulates NKG2D/DAP10 Expression on Human NK and CD8+ T Cells. J Immunol. 2006;176(3):1490–1497. doi: 10.4049/jimmunol.176.3.1490. [DOI] [PubMed] [Google Scholar]
- 50.Matzinger P. An innate sense of danger. Ann N Y Acad Sci. 2002 Jun;961:341–342. doi: 10.1111/j.1749-6632.2002.tb03118.x. [DOI] [PubMed] [Google Scholar]
- 51.Coussens L.M., Pollard J.W. Leukocytes in mammary development and cancer. Cold Spring Harb Perspect Biol. 2011 Mar 1;3(3) doi: 10.1101/cshperspect.a003285. pii:a003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curran C.S., Evans M.D., Bertics P.J. GM-CSF production by glioblastoma cells has a functional role in eosinophil survival, activation, and growth factor production for enhanced tumor cell proliferation. J Immunol. 2011 Aug 1;187(3):1254–1263. doi: 10.4049/jimmunol.1001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudson S.A., Herrmann H., Du J., Cox P., Haddad E, Butler B. Developmental, malignancy-related, and cross-species analysis of eosinophil, mast cell, and basophil Siglec-8 expression. J Clin Immunol. 2011 Dec;31(6):1045–1053. doi: 10.1007/s10875-011-9589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang R., Loux T., Tang D., Schapiro N.E., Vernon P., Livesey K.M. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A. 2012 May 1;109(18):7031–7036. doi: 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchser W.J., Laskow T.C., Pavlik P.J., Lin H.M., Lotze M.T. Cell-mediated Autophagy Promotes Cancer Cell Survival. Cancer Res. 2012 Apr 17 doi: 10.1158/0008-5472.CAN-11-3396. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weiner L.M., Lotze M.T. Tumor-cell death, autophagy, and immunity. N Engl J Med. 2012 Mar 22;366(12):1156–1158. doi: 10.1056/NEJMcibr1114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Livesey K.M., Kang R., Vernon P., Buchser W., Loughran P., Watkins S.C. p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer Res. 2012 Apr 15;72(8):1996–2005. doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang X., de Vera M.E., Buchser W.J., Romo d, V., Loughran P., Beer-Stolz D. Inhibiting Autophagy During Interleukin 2 Immunotherapy Promotes Long Term Tumor Regression. Cancer Res. 2012 Apr 3 doi: 10.1158/0008-5472.CAN-12-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang R., Livesey K.M., Zeh H.J., III, Lotze M.T., Tang D. Metabolic regulation by HMGB1-mediated autophagy and mitophagy. Autophagy. 2011 Oct;7(10):1256–1258. doi: 10.4161/auto.7.10.16753. [DOI] [PubMed] [Google Scholar]
- 60.Tang D., Kang R., Livesey K.M., Kroemer G., Billiar T.R., van H.B. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011 Jun 8;13(6):701–711. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang R., Livesey K.M., Zeh H.J., III, Lotze M.T., Tang D. HMGB1 as an autophagy sensor in oxidative stress. Autophagy. 2011 Aug;7(8):904–906. doi: 10.4161/auto.7.8.15704. [DOI] [PubMed] [Google Scholar]
- 62.Tang D., Kang R., Livesey K.M., Zeh H.J., III, Lotze M.T. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid Redox Signal. 2011 Oct 15;15(8):2185–2195. doi: 10.1089/ars.2010.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amaravadi R.K., Lippincott-Schwartz J., Yin X.M., Weiss W.A., Takebe N., Timmer W. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011 Feb 15;17(4):654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang R., Tang D., Lotze M.T., Zeh H.J., III RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy. 2011 Apr;7(4):442–444. doi: 10.4161/auto.7.4.14681. [DOI] [PubMed] [Google Scholar]
- 65.Kang R., Zeh H.J., Lotze M.T., Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011 Apr;18(4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang D., Lotze M.T., Kang R., Zeh H.J. Apoptosis promotes early tumorigenesis. Oncogene. 2011 Apr 21;30(16):1851–1854. doi: 10.1038/onc.2010.573. [DOI] [PubMed] [Google Scholar]
- 67.Kang R., Tang D., Livesey K.M., Schapiro N.E., Lotze M.T., Zeh H.J., III The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid Redox Signal. 2011 Oct 15;15(8):2175–2184. doi: 10.1089/ars.2010.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang D., Kang R., Zeh H.J., III, Lotze M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011 Apr 1;14(7):1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bergmann C., Wild C.A., Narwan M., Lotfi R., Lang S., Brandau S. Human tumor-induced and naturally occurring Treg cells differentially affect NK cells activated by either IL-2 or target cells. Eur J Immunol. 2011 Dec;41(12):3564–3573. doi: 10.1002/eji.201141532. [DOI] [PubMed] [Google Scholar]
- 70.Lotfi R., Eisenbacher J., Solgi G., Fuchs K., Yildiz T., Nienhaus C. Human mesenchymal stem cells respond to native but not oxidized damage associated molecular pattern molecules from necrotic (tumor) material. Eur J Immunol. 2011 Jul;41(7):2021–2028. doi: 10.1002/eji.201041324. [DOI] [PubMed] [Google Scholar]
- 71.Wild C.A., Brandau S., Lotfi R., Mattheis S., Gu X., Lang S. HMGB1 is overexpressed in tumor cells and promotes activity of regulatory T cells in patients with head and neck cancer. Oral Oncol. 2012 May;48(5):409–416. doi: 10.1016/j.oraloncology.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 72.Wild C.A., Bergmann C., Fritz G., Schuler P., Hoffmann T.K., Lotfi R. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012 Apr 3 doi: 10.1093/intimm/dxs051. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Chapter 13.11. Eosinophils and Chronic Rhinosinusitis
Robert P. Schleimer, Atsushi Kato, Robert Kern
Chronic rhinosinusitis (CRS) affects nearly 10% of the population in Europe and America and has been recognized as a profoundly eosinophilic disease since shortly after the eosinophil was first recognized. Due to access to sinonasal tissue from CRS patients undergoing surgery to correct this condition, studies of sinus tissue or nasal polyp tissues have been very helpful in the elucidation and in vivo validation of pathways of eosinophil biology. Studies in CRS have shown a role for eosinophil progenitors and local expansion of eosinophils; elucidated the endothelial adhesion molecules, chemokines, and cytokines involved in the recruitment and activation of eosinophils in the airways; and have been important in establishing the role of eosinophils and their derived factors in the mediation of pathogenic responses. Recent studies suggest that targeting the eosinophil may have therapeutic utility in the treatment of CRS, and the eosinophil thus continues to stand as a central cell in models of the pathogenesis of this disease. Further studies on the role of eosinophils in CRS will clearly be of value in documenting the precise mechanisms of the processes that bring them to tissue and by which they mediate pathology.
Introduction
Eosinophils have been associated with numerous diseases affecting multiple organ systems and, as this volume attests, their activities have been implicated as a cause of tissue pathology in a wide range of these conditions. Eosinophils are strongly implicated in the pathogenesis of allergic diseases, including allergic rhinitis, asthma, atopic dermatitis, food allergy, and others. Although chronic rhinosinusitis (CRS) is not strictly an allergic disease, it is often associated with allergy and the appearance of intense eosinophilia in tissues from the upper airways and sinuses of patients suffering from this condition has been known for the better part of a century. The purpose of this subchapter is to review the evidence showing an association between the eosinophil and CRS; the mechanisms of recruitment of eosinophils into sinonasal tissue in CRS patients; the roles that eosinophils play in the pathogenesis of this disease; and the potential for treatment modalities based on targeting the eosinophil.
CRS is a disease of the upper airways and paranasal sinuses that affects 5–10% of the general population. It is typically characterized by one or more of the following symptoms: copious secretion of mucus, facial pressure and headache, fatigue, loss of smell, and nasal obstruction. The physical exam may be unremarkable, although in some cases nasal endoscopy may reveal the presence of purulent drainage and/or nasal polyps. Historically, CRS has been subdivided according to many of its pathophysiological features, including appearance of fungal mucins, comorbidity with asthma, formation of polyps, hyperplasia of connective tissue, and sensitivity to aspirin. More recently, partly for the sake of simplifying the study of this condition, it has generally been divided into CRS with nasal polyps (CRSwNP) and CRS without nasal polyps (CRSsNP), a terminology that we will use here. The vast majority of CRS cases are idiopathic with an unpredictable clinical course; however, a minority have a more characteristic clinical picture. Specifically, CRS with fungal mucins together with fungal atopy is often viewed as a distinct condition known as AFS (allergic fungal sinusitis). Patients with aspirin allergy, asthma, and nasal polyps have a syndrome known as Samter’s triad, characterized by a severe form of recurrent polyposis. Lastly, CRS occurs in nearly all patients with cystic fibrosis, commonly with nasal polyposis, and this is frequently categorized as a discrete entity as well. In the United States, there are in excess of 300,000 surgeries performed annually to relieve the suffering of patients with CRS, and the discarded tissue from these procedures has provided access to tissue for those interested in studying the pathogenesis of the disease. The formation of nasal polyps occurs in roughly 15–20% of patients with CRS and the inflammatory process within the polyp is particular intense and may differ from inflammation that occurs in CRSsNP. The level of eosinophilia is greater in CRSwNP and this form of the disease has been the most extensively studied. Consequently, we primarily focus the discussion in this review on the role of the eosinophil in CRS with nasal polyps.
Association of Eosinophils with Sinonasal Inflammation in Chronic Rhinosinusitis
The appearance of eosinophils in pathological conditions in the airways was noticed shortly after Paul Ehrlich developed the acidic dyes that continue to be used to detect these cells today.1 CRS is one of the most intensely eosinophilic diseases, rivaling hypereosinophilic syndromes, eosinophilic esophagitis and eosinophilic gastroenteritis, in terms of tissue density of eosinophils (Fig. 13.11.1 ).2., 3. Bachert and his colleagues have shown that average eosinophil numbers are elevated in both CRSsNP and CRSwNP, but they are higher in CRSwNP and highest within nasal polyps themselves.4 In our own studies, eosinophilia is the highest in patients with Samter’s triad (see below—unpublished observations). Although some controversy exists as to whether there are relative differences in the eosinophilia of nasal polyps in populations of African, Caucasian, and Chinese descent, Bachert has reported low levels of eosinophilia in Chinese patients with CRS.5., 6., 7. Such studies are often complicated by the fact that the degree of eosinophilia in sinonasal tissue of patients with CRS can be dramatically altered by therapies that patients receive prior to surgery, especially oral glucocorticoids, which can diminish the eosinophil number by an order of magnitude (see below). It has been suggested that some intranasal Chinese herbal remedies may also alter eosinophil numbers.6 Along with elevated levels of eosinophils, nasal polyps also demonstrate elevated levels of B cells, dendritic cells, neutrophils, macrophages, mast cells, and T cells, and thus represent a tissue undergoing a robust immune and inflammatory response. Although mast cells are not the subject of this volume, it must be acknowledged that they are likely to play an important role in the pathogenesis of CRS, especially in the tissue swelling and formation of nasal polyps that occur.8., 9. Detection of eosinophils and eosinophilia in CRS has employed assays for eosinophil granule proteins, such as eosinophil cationic protein (ECP), as well as specific antibodies against eosinophil granule proteins, notably EG2, which is considered to specifically stain activated eosinophils.10., 11. Studies using these approaches have confirmed the presence of eosinophilia in CRS and provided evidence for activation of the tissue-resident eosinophils.12., 13., 14. Persson and colleagues have emphasized the importance of cytolytic eosinophil degranulation and release of clusters of free granules. Evidence has been provided to demonstrate that eosinophils in sinonasal tissue of CRS patients undergo both piecemeal degranulation and cytolytic degranulation.15., 16. Recently, a method has been developed to purify eosinophils from human tissues, including nasal polyps, that should allow further studies into the activation state of eosinophils and gene expression in CRS.17
FIGURE 13.11.1.
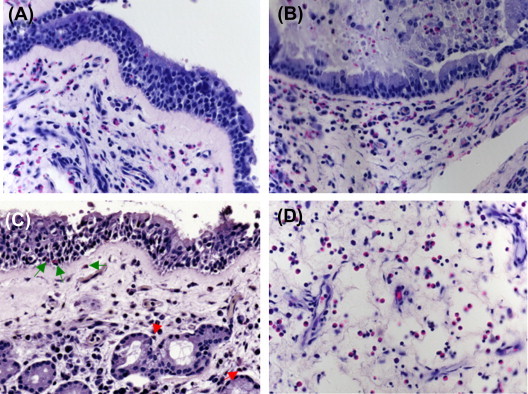
A–D, Hematoxylin and eosin stain of different views of typical nasal polyps from a patient with chronic rhinosinusitis. C, Green arrows denote clearly stained, classic eosinophils and red arrows refer to eosinophils that, while not as clearly visible as the eosinophils denoted by green arrows, nevertheless do have the pink granules surrounding their nuclei that are characteristic of eosinophilia. (magnification ×400)
(Courtesy of Roderick Carter.)
CRS is often comorbid with asthma, and one of the most severe forms of the disease, nasal polyps with asthma and aspirin sensitivity (referred to often as Samter’s triad), manifests both severe polyposis and severe, often glucocorticoid-resistant, asthma.18 CRS with nasal polyps is also comorbid with Churg-Strauss syndrome, a systemic vasculitic disorder in which eosinophils feature prominently.19 Several groups have compared the level of eosinophils in sinonasal tissue with asthma severity, levels of eosinophils in bronchial lavage, or sputum samples from the lower airways. In one study, levels of eosinophils in lavage from the middle meatus correlated with FEV1 (forced expiratory volume in one second) in asthmatics with CRS.20 In another study, the presence of eosinophils in sinonasal tissue was higher in CRS patients undergoing sinus surgery who had comorbid asthma than in those who did not have asthma.21 Mehta et al. found that the extent of CRS disease as measured by sinus computed tomography (CT) scores correlated with eosinophils in blood and sputum, suggesting that systemic elevation and activation of eosinophils may be a feature of CRS.22 Patients with CRSwNP were found to have increased asthma prevalence, as well as increased exhaled nitric oxide.23
The etiology of CRS has yet to be definitively ascribed to infection with any single pathogen or class of pathogens. It is clear that patients with CRS experience frequent acute, presumably infectious, exacerbations of disease, and CRS patients are generally treated frequently with antibiotics, suggesting that treating physicians suspect the presence of bacteria. Along these lines, it has been suggested that patients with CRS may have a defect in the innate immune barrier, making them more susceptible to infection or colonization with microorganisms in general.24., 25. Another line of investigation implicates fungi, or at least allergy to fungi, in CRS, based on the presence of activation of lymphocytes to express cytokines in response to fungal extracts.26., 27. While atopy to aeroallergens is frequently seen in patients with CRS, nearly half of CRS patients are nonatopic according to standard tests, and based on this present state of knowledge, CRS should not be viewed as a classical allergic disease.28 Yet another line of investigation suggests that Staphylococcus aureus and the toxins it produces are important inciting factors in CRS.29., 30., 31., 32. According to the superantigen hypothesis of CRS, staphylococcus-derived superantigens drive a T-helper type 2 (Th2)-skewed inflammatory response that is responsible for the eosinophilia observed in CRS (the mechanism of eosinophilia in CRS is discussed below). Superantigens activate large numbers of T cells by cross-linking class II MHC on antigen-presenting cells with specific Vβ regions on the T-cell receptor, leading to profound release of cytokines, in some cases skewed toward Th2.33 Reports on the extent of staphylococcal colonization in CRS have been variable but most have demonstrated normal or supranormal frequencies. Bachert et al. and Desrosiers et al. have reported that 50–90% of CRS patients with nasal polyps have colonization with staphylococcus.34., 35. Staphylococcal superantigens can also act as allergens, and several reports have detected staphylococcal superantigens in the airways of CRS patients and/or demonstrated the presence of functional immunoglobulin E (IgE) antibodies directed against the staphylococcal superantigens.36., 37., 38., 39. A recent study by Bachert et al. has implicated the presence of interleukin-5 (IL-5) and IgE directed to staphylococcal enterotoxins in CRS comorbid with asthma.40 A longitudinal study demonstrated that high numbers of eosinophils in the nasal mucosa or in mucus collected from CRS patients are strong risk factors for recurrence of disease and the need for subsequent surgery in a 5-year follow-up study.41 In summary, eosinophil numbers are increased in sinonasal tissue of patients with CRS and increased the most in those with nasal polyps, those with asthma, and especially those with all three conditions. The correlation of eosinophils with disease severity implicates them as either a biomarker of the pathogenic process or a mediating cell responsible for disease.
Mechanisms for Recruitment and Activation of Eosinophils in Chronic Rhinosinusitis
The molecular mechanisms by which eosinophils are recruited to tissues have been reviewed and are the topic of another subchapter in this volume. We restrict our comments therefore to the available evidence on recruitment of eosinophils in CRS. In general, several important processes occur. One is the priming and survival-promoting effects of cytokines on eosinophils, especially including granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-5, both of which also play a role in the generation of eosinophils. Another is the local expression of eosinophil-attracting chemokines by epithelium and other tissue cells. The expression of adhesion molecules by endothelium, especially vascular cell adhesion protein 1 (VCAM-1), is equally important in the rolling, arrest and transmigration of these cells into the affected tissue. An extensive literature that shows the elevation of these factors in sinonasal tissue of patients with CRS will be summarized next.
The primary receptor driving chemokine-mediated recruitment of eosinophils is C-C chemokine receptor 3 (CCR3), which has a number of ligands, notably RANTES (or C-C motif chemokine 5; CCL5), eotaxin (CCL11), eotaxin-2 (CCL24), eotaxin-3 (CCL26), and MCP-4 (CCL13), with less activity for MCP-1, MCP-2, and MCP-3.42., 43. Mice lacking CCR3 have severe, but not totally diminished, eosinophil infiltration in allergic inflammation, and CCR3 antagonists are under investigation in clinical trials. Beck et al. found elevated levels of RANTES mRNA and tissue staining in nasal polyp tissue but not in normal turbinate tissue.44 RANTES expression is primarily found in epithelial cells, an observation leading to speculation that localization of eosinophils to the epithelium and lamina propria may be related to epithelial chemokine expression.45., 46., 47. Similar findings have subsequently been made with other CCR3 ligands, including eotaxins 1–3 and MCP-4.48., 49., 50., 51. A careful study by Jahnsen et al. found that levels of mRNA for eotaxin-1, eotaxin-2, and MCP-4 are elevated in nasal polyp tissue and that levels of eotaxin-2 and MCP-4 in turbinates from CRSwNP patients are higher than in normal turbinates.49 Taken together, it is clear that both surrounding tissue (i.e., turbinate) and affected tissue (nasal polyps, which commonly emerge from the ethmoid sinuses and other deeper tissues) in patients with CRS have elevated levels of CCR3 ligands that are likely to play a role in eosinophil recruitment to both of these regions in CRS. Regulation of chemokine expression by epithelium is complex, but both NF-κB and signal transducer and activator of transcription 6 (STAT6) play important roles in the response, and Th2 cytokines including IL-4 and IL-13 are important inducers.52., 53. Other stimuli may also be important in activating CCR3 ligand expression in CRS, and chitin has recently been shown to induce eotaxin-3 in human sinonasal epithelial cells in vitro.54
Eosinophils not only respond to CCR3 agonists but also release eotaxin, eotaxin-2, eotaxin-3, and RANTES.51., 55. The release of CCR3 agonists from eosinophils may be involved in the local eosinophil accumulation in nasal polyps. Recently, our group has found that MPIF-1 (CCL23) is elevated in eosinophilic nasal polyps and that EG2+ eosinophils are major MPIF-1-producing cells in nasal polyps.55a MPIF-1 is a ligand for CCR1 and is known to recruit monocytes, macrophages, and dendritic cells.56., 57. These findings indicate that activation of eosinophils may further enhance local inflammation via secondary recruitment of additional cells in nasal polyps.
Exposure of eosinophils to GM-CSF or IL-5 leads to several notable phenotypic changes that are likely to be relevant to their accumulation in CRS, including increased expression and function of adhesion molecules, increased transendothelial migration, and increased survival. The earliest studies on CRS were by Denburg and colleagues, and showed elevated levels of GM-CSF in eosinophils in nasal polyp tissue,58., 59. and that GM-CSF is prominently expressed by epithelial cells, in agreement with earlier in vitro studies showing that epithelial cells are a rich source of this cytokine.60 Several studies have shown that conditioned medium from nasal polyps stimulated with antigen or from cultured nasal polyp epithelial cells can prolong eosinophil survival or activate eosinophils in vitro and that the activity can be significantly blocked by anti-GM-CSF antibodies.61., 62., 63., 64. Additional studies have shown that IL-5 is also an important eosinophil priming and survival factor in nasal polyp tissue.13., 65., 66., 67. This finding is supported by success in recent clinical trials by Bachert et al. using anti-IL-5 antibodies (see below). The relative importance of IL-5 and GM-CSF as priming cytokines in sinonasal tissue is unknown. Liu and Busse found that lung-migrating eosinophils have reduced expression of the IL-5 receptor in asthmatics and postulated that the main effects of IL-5 (and therefore probably anti-IL-5) may be in the bone marrow and circulation.68 Whether the same is true in the upper airways and sinuses in patients with CRS requires further investigation.
In vitro, VCAM-1 is known to be an important and relatively selective endothelial adhesion molecule that mediates eosinophil rolling, firm adhesion, and transendothelial migration. Several groups have demonstrated increased endothelial expression of VCAM-1 in nasal polyps and shown that VCAM-1 levels correlate with the presence of eosinophils, leading to the hypothesis that recruitment of eosinophils in CRS is partially mediated by VCAM-1.46., 69., 70., 71., 72., 73. Using the Stamper–Woodruff assay, Symon, Wardlaw, and collaborators have demonstrated that eosinophils bind to nasal polyp tissue in vitro via interactions with P-selectin and on this basis suggested a role for P-selectin as well.74 In general, the T cell cytokine milieu that most drives eosinophilic inflammation is a Th2 pattern, including the VCAM-1 activators IL-4 and IL-13 as well as the eosinophil priming cytokine IL-5. Although most CRS is probably Th2 driven, it has been suggested that in some cases of CRS, including nonallergic CRS, other T-cell cytokines may be involved.75., 76. At present, there is controversy about whether Th17 are elevated in CRS. Saitoh et al. have reported increased IL-17 in CRS, while Peters et al. (our group) and Bachert and Geveart have not found evidence for a role of IL-17 in CRS.77., 78., 79. As far as eosinophil recruitment and activation in CRS is concerned, there are reports of a possible role for complement activation, IL-33, and activation of protease-activated receptors (PAR), eicosanoids, and stem cell factor. In some cases, these pathways may activate local resident or infiltrating cells to express eosinophil priming and recruitment factors, including chemokines and cytokines such as GM-CSF.80., 81., 82., 83., 84., 85.
Potential Roles of Eosinophils in the Pathogenesis of Chronic Rhinosinusitis
The discussion above firmly establishes that eosinophils are present in high numbers in the sinonasal tissue of patients with CRS, especially the polypoid form, and that these tissue eosinophils are activated. Together, these two lines of investigation provide circumstantial evidence that eosinophils may play a role in alterations to nasal physiology, tissue structure, and clinical phenotype. This section discusses some features of CRS that may be mediated by activated eosinophils. In most cases, these discussions should be viewed as hypothetical or speculative, as there is no validated animal model of CRS and in most cases it is not possible to definitively assess mechanisms in human subjects (other than some recent studies with antibodies against IL-5 or IgE, discussed in the next section). In our view, it is clear that CRSsNP and CRSwNP are distinct disease entities that are primarily related by their frequent resistance to treatment, need for surgery (probably greater in CRSwNP), and eosinophilia (probably greater in CRSwNP). However, they are distinct in many of the clinical and inflammatory processes that we and others have been investigating at the molecular level.86., 87., 88., 89. As to the roles of eosinophils in pathogenesis, by far the most data are available for polypoid CRS, and we will restrict our comments here to CRSwNP, even though they may in many cases apply to CRSsNP. Although eosinophils are elevated in CRS and express the activation marker EG2 (see above), EG2 has not proven to be a reliable marker of degranulation; eosinophils in nasal polyps are undoubtedly activated, as studies of eosinophil morphology by Erjefält and Persson have demonstrated extensive piecemeal degranulation and cytolytic degranulation in eosinophils in nasal polyp tissue.16., 90.
The most obvious and prominent feature of CRSwNP is the formation of the polyp itself, although details of the mechanism of this metamorphosis of sinonasal tissue are not well understood. Perhaps the best studies have been those by Bachert et al., who studied early-phase and established nasal polyps.91 They found formation of a subepithelial eosinophilic cap at the upper surface of the tissue outgrowth and implicated fibronectin and deposition of albumin and extracellular matrix proteins in the early and late phase, respectively. The presence of IL-5 and eotaxin-2 correlated with the process, thus leading them to propose an important role for eosinophils.91 Based on the deposition of albumin, we can presume that vascular leakage is occurring in CRS, although the stimuli for the leak are not well understood. Studies by Steinke, Borish, and others have shown that the 5-lipoxygenase (5-LO) pathway is activated in CRS, especially in aspirin-sensitive disease, and that eosinophils express 5-LO and LTC4 synthase within the polyp tissue.92., 93. Increased eicosanoid metabolism has been reported in CRS and correlates with ECP and IL-5.82 Although eosinophil cyclooxygenase and lipoxygenase metabolites might be important in vascular leakage driving polyp formation, in general the clinical experience with inhibitors of both of these pathways has been disappointing and they are unlikely to be the primary mediators driving vascular leak in a forming polyp. The relative importance of mast cell and eosinophil mediators in polyp formation is unknown. Di Lorenzo et al. measured ECP, histamine, and tryptase in nasal lavage and found only tryptase and ECP to correlate with symptom scores.94 Since mast cells are highly elevated in nasal polyps and are capable of releasing numerous mediators of vascular leakage, their potential role in polyp formation must be seriously considered.
Considerable evidence has accumulated to demonstrate elevations of both IgE and IgA in nasal polyp tissue, and it is not unreasonable to speculate that these antibodies play a role in activating mast cells and eosinophils, respectively, in CRSwNP. With respect to eosinophils, activation by IgE, if it occurs at all, most likely occurs indirectly as a result of the action of mediators released from basophils, mast cells, and other cells that clearly express a functional IgE receptor (e.g., inflammatory macrophage-like cells). Kita and colleagues have shown that IgG and IgA, but not IgE, can prolong eosinophil survival, and induce cytokine expression and effector function.95., 96. However, convincing data demonstrate that eosinophils are activated to degranulate by cross-linking receptors for IgA.97., 98. More uncertain is the nature of the antigen systems that might drive IgA-mediated degranulation in nasal polyp eosinophils. As discussed above, only half of patients with CRSwNP are atopic, and it is difficult to implicate typical aeroallergens as anything more than exacerbating factors. Recent studies by Tan and colleagues have demonstrated the presence of autoantibodies of both IgG and IgA isotypes in patients with recalcitrant CRS requiring revision surgery.99 Suh et al. found correlations between both total IgG and total IgE with the number of EG2+ cells, but not IgA or secretory IgA.100 Clearly, further work is required to establish the importance of immunoglobulins in eosinophil activation in CRS.
In both asthma and CRS there is evidence for epithelial damage mediated by eosinophils. Gleich and colleagues demonstrated that the presence of eosinophils and extracellular levels of the toxic eosinophil granule protein major basic protein (MBP) correspond to regions of epithelial injury in patients with CRS.101 This occurs in asthma as well, and there is ample evidence for a similar pathological process occurring in the two diseases. Chanez, Bousquet, and colleagues and Gaga et al. demonstrated a strong correlation between eosinophils in the nose and lung in asthmatics.101., 102., 103. Bresciani et al. found that 70% of asthmatics have CRS, as determined using clinical and CT scores, and observed a correlation of clinical scores to blood eosinophils in those with mild and moderate asthma.104 Eosinophil-derived granule mediators are well established to be toxic to epithelium, to activate mast cells and basophils, and to drive inflammation. An ultrastructural investigation of epithelial damage in asthmatic and nonasthmatic nasal polyps revealed reduced length of desmosomes in allergic CRSwNP and in asthmatics.105 Several studies suggest that the epithelium in both asthma and CRS presents a poor barrier.24., 25., 106. Whether damage from eosinophils occurs as a result of the cationic granular proteins, the respiratory burst and peroxidase activation, protein nitration, or another mechanism is worthy of further investigation.107., 108. Recent studies indicate reduced SPINK5 in epithelial tissue from CRS patients.109 SPINK5 is a protease inhibitor that can regulate barrier function in the skin via preventing activation of PAR2 and subsequent induction of thymic stromal lymphopoietin (TSLP).110 Briot et al. found that high levels of TSLP in the skin drive a highly eosinophil and mast cell rich inflammation even in the absence of T cells.110 Kamekura et al. presented evidence that TSLP itself can induce claudins and occludins and enhance tight junction function in nasal epithelial cells in vitro, suggesting that its influences may be complex. TSLP is elevated in CRS, and whether TSLP regulates barrier function in CRS and/or contributes to eosinophilia needs further clarification.111., 112., 113.
Studies in the lower airways indicate that eosinophilic inflammation is often associated with fibrotic changes, including the laying down of extracellular matrix proteins or repair proteins (e.g., lumican, procollagen, and tenascin—see Chapter 12).114., 115. Some nasal polyps are characterized by the deposition of collagen and other matrix proteins, and it is possible that eosinophils may play a role in this process. Studies by Ohno et al. have demonstrated that transforming growth factors α and β1 (TGF-α and TGF-β1), as well as platelet-derived growth factor receptor (PDGF), are expressed by eosinophils in nasal polyps and suggest that eosinophil-derived growth factors may alter the structure of the affected nasal mucosa.116., 117. Eosinophil production of TGF in nasal polyps was confirmed by Elovic et al.118 Ultrastructural studies in nasal polyposis using anti-IL-5 antibodies, such as the ones used by Flood-Page, Kay et al. in asthma, will be required to assess the role of eosinophils in remodeling of the sinonasal tissue in CRS.114
Eosinophils as a Therapeutic Target for Management of Chronic Rhinosinusitis
At present, there are no specific approved therapies for the treatment of CRS. In general, a significant number of patients that present with the diagnosis have failed treatment with antibiotics and/or intranasal glucocorticoids. Since nasal polyps commonly grow out of the sinuses, it is a challenge for intranasal glucocorticoid sprays to penetrate to the area of relevant inflammation. Treatment with oral steroids is often effective, especially in CRSwNP, and is frequently used to treat CRSwNP, although most patients and physicians prefer to avoid chronic treatment with oral glucocorticoids.
Glucocorticoids continue to be the most effective anti-inflammatory drugs available for a wide variety of autoimmune and allergic chronic inflammatory illnesses. Their mechanisms are pleiotropic and represent actions exerted upon numerous cell types known to participate in CRS, including epithelial, Th1, and Th2 cells. Other important cells, such as endothelial cells, mast cells, neutrophils, and Th17 cells, are relatively unresponsive to glucocorticoid treatment and are unlikely to be important targets in CRS.119 Among the steroid responsive cells are eosinophils, and the actions of glucocorticoids on eosinophils have been reviewed.120., 121. In 2007, Patiar and Reece performed a Cochrane review of the literature and identified only one randomized-controlled study comparing oral steroids with no intervention or placebo.122., 123. As found in earlier uncontrolled trials, oral treatment with prednisone or a similar systemic steroid shrank polyps, reduced symptoms, and improved olfaction in this study. Alobid et al. also found that continued treatment with intranasal steroid maintained the benefits of the 2-week treatment with oral steroids for nearly 1 year.123 Subsequently, Hissaria et al. performed a controlled trial testing a short course of prednisolone in CRSwNP, and found improved symptom scores and a reduction in polyp size, as determined both subjectively and objectively by MRI.124 Recently, Van Zele, Bachert, and collaborators performed a double-blind placebo-controlled multicenter trial of oral methylprednisolone in CRSwNP.125 In addition to shrinking polyps, improving olfaction and reducing symptoms, the oral steroids improved congestion, nasal peak flow rates, postnasal drip, and rhinorrhea. Importantly, in this study methylprednisolone reduced ECP, IgE, and IL-5 in nasal secretions and decreased blood eosinophils, ECP, and soluble IL-5Ra in the serum.125
In vitro studies have identified numerous effects of glucocorticoids on eosinophils (for a summary, see119). Glucocorticoids diminish eosinophil survival in vitro by promoting apoptosis and this effect is blocked by survival-promoting cytokines such as GM-CSF or IL-5. Glucocorticoids also diminish production of these specific cytokines by many cell types, including epithelial cells and T lymphocytes. Steroids have a similar suppressive effect in vitro on the expression of cytokines that activate endothelial adhesion molecule expression [e.g., IL-1, IL-4, IL-13, and tumor necrosis factor (TNF)] and of chemokines known to cause eosinophil migration (e.g., CCR3 agonists). Thus, theoretically, glucocorticoids should diminish eosinophil recruitment to the sinuses and nasal cavity and hasten the disappearance of the eosinophils once they arrive in the sinonasal cavity. Numerous in vivo studies have addressed these theoretical effects of glucocorticoids by administering intranasal or oral glucocorticoids to patients with CRS prior to surgery or biopsy.
A general feature of these studies is that glucocorticoids uniformly reduce the number of eosinophils found in nasal polyps. Kanai et al. also found treatment with budesonide to reduce the proportion of activated (EG2+) eosinophils, along with reducing levels of total eosinophils, T cells, the ICAM-1 adhesion molecule, and HLA-DR, a marker of adaptive immune activation.126 Similar findings were made by Hamilos and coworkers, who additionally found reduced P-selectin (but not IL-1β, TNF, or VCAM-1) in polyps from patients treated with intranasal fluticasone propionate.127 They also found reduced numbers of cells expressing Th2 cytokines (IL-4 and IL-13). Delbrouck et al. reported that budesonide decreases levels of both ICAM-1 and VCAM-1, and to a lesser extent P-selectin.128 As mentioned above, Denburg, Dolovich, and collaborators identified GM-CSF as an important eosinophil-priming and survival-promoting cytokine in the nose of patients with allergic rhinitis and CRS. Nonaka et al. from this group found that budesonide reduces survival of peripheral blood eosinophils in vitro but not eosinophils extracted from nasal polyps, suggesting that the polyp eosinophils are primed and rendered glucocorticoid resistant as a result of the GM-CSF exposure within the polyp.129 It is unclear whether GM-CSF or IL-5 is the most important eosinophil survival cytokine in CRSwNP, and this is not a question that can be easily addressed, since both of these cytokines have effects on eosinophils outside of the sinonasal cavity. Nonetheless, Bolard et al. found a correlation between levels of IL-5 and eosinophils in the nasal secretions of patients with nasal polyps and that intranasal steroids reduce both of these parameters.130 Whether glucocorticoid treatment directly induces apoptosis in tissue eosinophils or indirectly induces eosinophil death as a result of suppressing survival-promoting cytokines is still an open question. As to the expression of chemokines that attract eosinophils, Jahnsen et al. found that nasal polyp tissue expresses highly elevated levels of eotaxin, eotaxin-2, MCP-4 and RANTES, and that after treatment with oral glucocorticoid these factors are all profoundly reduced along with a reduction in eosinophil numbers in the tissue.49 Importantly, in association with the reduction of eosinophils and of the cytokines that induce their accumulation, treatment of CRS with intranasal glucocorticoids also leads to a reduction in damage to the epithelium that is thought to be mediated by eosinophils and is accompanied by the restoration of epithelium integrity.131
Perhaps the most valuable data implicating eosinophils in the pathogenesis of CRSwNP is from the group of Gevaert, Bachert, and collaborators.132., 133., 134. These researchers noted increased levels of IL-5 and soluble IL-5RA in patients with nasal polyposis and found that treatment of CRSwNP patients with an antibody against IL-5 led to a reduction in nasal polyp size, particularly in patients with elevated IL-5. The same patients had a corresponding reduction in the level of eosinophilia, as measured by total eosinophils in the blood and levels of ECP in both the serum and in nasal secretions.132., 133., 134.
Prospects for new Therapies Targeting Eosinophilic Inflammation in Chronic Rhinosinusitis
Studies using anti-IL-5 antibodies to reduce eosinophilic inflammation are still ongoing and this approach continues to show promise for the treatment of CRS. As kinase inhibitors that block the development of eosinophils are developed, it is possible that some will emerge that have some specificity for the eosinophilic lineage of cells and will thus have utility in a variety of diseases including CRS. Treatment of mice with antibodies against Fas caused widespread apoptosis of eosinophils but lacked specificity and thus killed many other important cell types.135 Treatments based on Siglec-8 activation may induce apoptosis more selectively in eosinophils, and in the related allergic cells mast cells and basophils, and thereby provide some benefit without undue safety concerns136 (see also Chapter 15.4). In addition, recent studies from our group demonstrate that B cells and immunoglobulins, especially IgA which is a potent eosinophil activator, are highly elevated in CRS.137., 138. Therapies targeting B cells, such as antibodies against B cell activating factor belonging to the TNF family (BAFF) or B cells (e.g., antiCD20), may prove to diminish the activation of eosinophils in CRS if IgA and secretory IgA are important in activating eosinophils in CRS.
References
- 1.Mahmoud A.A.F., Austen K.F. Grune and Stratton Inc.; New York: 1980. The Eosinophil in Health and Disease. 3–354. [Google Scholar]
- 2.Taillens J.P. Nasal polyposis; value of blood eosinophils in surgical indications and in sinusal surgical therapy. Rev Laryngol Otol Rhinol (Bord) 1952;73:20–59. [PubMed] [Google Scholar]
- 3.Taillens J.P. Polyposis of nasal sinuses; pathogenetic study with therapeutic conclusions. Pract Otorhinolaryngol (Basel) 1953;15:211–242. [PubMed] [Google Scholar]
- 4.Van Zele T., Claeys S., Gevaert P., Van Maele G., Holtappels G., Van Cauwenberge P. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61:1280–1289. doi: 10.1111/j.1398-9995.2006.01225.x. [DOI] [PubMed] [Google Scholar]
- 5.Zhang N., Holtappels G., Claeys C., Huang G., van Cauwenberge P., Bachert C. Pattern of inflammation and impact of Staphylococcus aureus enterotoxins in nasal polyps from southern China. Am J Rhinol. 2006;20:445–450. doi: 10.2500/ajr.2006.20.2887. [DOI] [PubMed] [Google Scholar]
- 6.Lacroix J.S., Zheng C.G., Goytom S.H., Landis B., Szalay-Quinodoz I., Malis D.D. Histological comparison of nasal polyposis in black African, Chinese and Caucasian patients. Rhinology. 2002;40:118–121. [PubMed] [Google Scholar]
- 7.Zhang N., Liu S., Lin P., Li X., van Bruaene N., Zhang J. Remodeling and inflammation in Chinese versus white patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125:507. doi: 10.1016/j.jaci.2009.10.015. author reply 507–508. [DOI] [PubMed] [Google Scholar]
- 8.Drake-Lee A.B., McLaughlan P., Baker T.H. Histamine, asthma, and nasal polyps. Lancet. 1982;2:213. doi: 10.1016/s0140-6736(82)91055-8. [DOI] [PubMed] [Google Scholar]
- 9.Pawliczak R., Kowalski M.L., Danilewicz M., Wagrowska-Danilewicz M., Lewandowski A. Distribution of mast cells and eosinophils in nasal polyps from atopic and nonatopic subjects: a morphometric study. Am J Rhinol. 1997;11:257–262. doi: 10.2500/105065897781446711. [DOI] [PubMed] [Google Scholar]
- 10.Stoop A.E., van der Heijden H., Biewenga J., van der Baan S. Eosinophils in nasal polyps and nasal mucosa: An immunohistochemical study. J Allergy Clin Immunol. 1993;91:616–620. doi: 10.1016/0091-6749(93)90267-j. [DOI] [PubMed] [Google Scholar]
- 11.Tai P.C., Spry C.J., Peterson C., Venge P., Olsson I. Monoclonal antibodies distinguish between storage and secreted forms of eosinophil cationic protein. Nature. 1984;309:182–184. doi: 10.1038/309182a0. [DOI] [PubMed] [Google Scholar]
- 12.Appenroth E., Gunkel A.R., Muller H., Volklein C., Schrott-Fischer A. Activated and non-activated eosinophils in patients with chronic rhinosinusitis. Acta Otolaryngol. 1998;118:240–242. doi: 10.1080/00016489850154964. [DOI] [PubMed] [Google Scholar]
- 13.Bachert C., Wagenmann M., Hauser U., Rudack C. IL-5 synthesis is upregulated in human nasal polyp tissue. J Allergy Clin Immunol. 1997;99:837–842. doi: 10.1016/s0091-6749(97)80019-x. [DOI] [PubMed] [Google Scholar]
- 14.Sun D.I., Joo Y.H., Auo H.J., Kang J.M. Clinical significance of eosinophilic cationic protein levels in nasal secretions of patients with nasal polyposis. Eur Arch Otorhinolaryngol. 2009;266:981–986. doi: 10.1007/s00405-008-0872-9. [DOI] [PubMed] [Google Scholar]
- 15.Armengot M., Garin L., Carda C. Eosinophil degranulation patterns in nasal polyposis: an ultrastructural study. Am J Rhinol Allergy. 2009;23:466–470. doi: 10.2500/ajra.2009.23.3357. [DOI] [PubMed] [Google Scholar]
- 16.Erjefält J.S., Andersson M., Greiff L., Korsgren M., Gizycki M., Jeffery P.K. Cytolysis and piecemeal degranulation as distinct modes of activation of airway mucosal eosinophils. J Allergy Clin Immunol. 1998;102:286–294. doi: 10.1016/s0091-6749(98)70098-3. [DOI] [PubMed] [Google Scholar]
- 17.Ben Efraim A.H., Munitz A., Sherman Y., Mazer B.D., Levi-Schaffer F., Eliashar R. Efficient purification of eosinophils from human tissues: a comparative study. J Immunol Methods. 2009;343:91–96. doi: 10.1016/j.jim.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Samter M. Nasal polyps: their relationship to allergy, particularly to bronchial asthma. Med Clin North Am. 1958;42:175–179. doi: 10.1016/s0025-7125(16)34334-6. [DOI] [PubMed] [Google Scholar]
- 19.Bacciu A., Buzio C., Giordano D., Pasanisi E., Vincenti V., Mercante G. Nasal polyposis in Churg-Strauss syndrome. Laryngoscope. 2008;118:325–329. doi: 10.1097/MLG.0b013e318159889d. [DOI] [PubMed] [Google Scholar]
- 20.Ragab A., Clement P., Vincken W. Correlation between the cytology of the nasal middle meatus and BAL in chronic rhinosinusitis. Rhinology. 2005;43:11–17. [PubMed] [Google Scholar]
- 21.Dhong H.J., Kim H.Y., Cho D.Y. Histopathologic characteristics of chronic sinusitis with bronchial asthma. Acta Otolaryngol. 2005;125:169–176. doi: 10.1080/00016480410015767. [DOI] [PubMed] [Google Scholar]
- 22.Mehta V., Campeau N.G., Kita H., Hagan J.B. Blood and sputum eosinophil levels in asthma and their relationship to sinus computed tomographic findings. Mayo Clin Proc. 2008;83:671–678. doi: 10.4065/83.6.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guida G., Rolla G., Badiu I., Marsico P., Pizzimenti S., Bommarito L. Determinants of exhaled nitric oxide in chronic rhinosinusitis. Chest. 2010;137:658–664. doi: 10.1378/chest.09-0667. [DOI] [PubMed] [Google Scholar]
- 24.Kern R., Conley D., Walsh W., Chandra R., Kato A., Tripathi-Peters A. Perspectives on the etiology of chronic rhinosinusitis: An immune barrier hypothesis. Am J Rhinol. 2008;22(6):549–559. doi: 10.2500/ajr.2008.22.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tieu D.D., Kern R.C., Schleimer R.P. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124:37–42. doi: 10.1016/j.jaci.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponikau J.U., Sherris D.A., Kephart G.M., Adolphson C., Kita H. The role of ubiquitous airborne fungi in chronic rhinosinusitis. Clin Rev Allergy Immunol. 2006;30:187–194. doi: 10.1385/CRIAI:30:3:187. [DOI] [PubMed] [Google Scholar]
- 27.Shin S.H., Ponikau J.U., Sherris D.A., Congdon D., Frigas E., Homburger H.A. Chronic rhinosinusitis: an enhanced immune response to ubiquitous airborne fungi. J Allergy Clin Immunol. 2004;114:1369–1375. doi: 10.1016/j.jaci.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Pearlman A., Chandra R, Chang D., Conley D., Tripathi-Peters A., Grammer L. Relationships between severity of chronic rhinosinusitis and nasal polyposis, asthma, and atopy. American Journal of Rhinology & Allergy. 2009;23(2):145–148. doi: 10.2500/ajra.2009.23.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bachert C., Gevaert P., van Cauwenberge P. Staphylococcus aureus superantigens and airway disease. Curr Allergy Asthma Rep. 2002;2:252–258. doi: 10.1007/s11882-002-0027-9. [DOI] [PubMed] [Google Scholar]
- 30.Bachert C., van Zele T., Gevaert P., De Schrijver L., Van Cauwenberge P. Superantigens and nasal polyps. Curr Allergy Asthma Rep. 2003;3:523–531. doi: 10.1007/s11882-003-0065-y. [DOI] [PubMed] [Google Scholar]
- 31.Bernstein J.M., Ballow M., Schlievert P.M., Rich G., Allen C., Dryja D. A superantigen hypothesis for the pathogenesis of chronic hyperplastic sinusitis with massive nasal polyposis. Am J Rhinol. 2003;17:321–326. [PubMed] [Google Scholar]
- 32.Bernstein J.M., Kansal R. Superantigen hypothesis for the early development of chronic hyperplastic sinusitis with massive nasal polyposis. Curr Opin Otolaryngol Head Neck Surg. 2005;13:39–44. doi: 10.1097/00020840-200502000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Herman A., Kappler J.W., Marrack P., Pullen A.M. Superantigens: mechanism of T-cell stimulation and role in immune responses. Annu Rev Immunol. 1991;9:745–772. doi: 10.1146/annurev.iy.09.040191.003525. [DOI] [PubMed] [Google Scholar]
- 34.Van Zele T., Gevaert P., Watelet J.B., Claeys G., Holtappels G., Claeys C. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol. 2004;114:981–983. doi: 10.1016/j.jaci.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Stephenson M.F., Mfuna L., Dowd S.E., Wolcott R.D., Barbeau J., Poisson M. Molecular characterization of the polymicrobial flora in chronic rhinosinusitis. J Otolaryngol Head Neck Surg. 2010;39:182–187. [PubMed] [Google Scholar]
- 36.Perez-Novo C.A., Kowalski M.L., Kuna P., Ptasinska A., Holtappels G., van Cauwenberge P. Aspirin sensitivity and IgE antibodies to Staphylococcus aureus enterotoxins in nasal polyposis: studies on the relationship. Int Arch Allergy Immunol. 2004;133:255–260. doi: 10.1159/000076832. [DOI] [PubMed] [Google Scholar]
- 37.Rohde G., Gevaert P., Holtappels G., Borg I., Wiethege A., Arinir U. Increased IgE-antibodies to Staphylococcus aureus enterotoxins in patients with COPD. Respir Med. 2004;98:858–864. doi: 10.1016/j.rmed.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Seiberling K.A., Conley D.B., Tripathi A., Grammer L.C., Shuh L., Haines G.K., 3rd Superantigens and chronic rhinosinusitis: detection of staphylococcal exotoxins in nasal polyps. Laryngoscope. 2005;115:1580–1585. doi: 10.1097/01.mlg.0000168111.11802.9c. [DOI] [PubMed] [Google Scholar]
- 39.Suh Y.J., Yoon S.H., Sampson A.P., Kim H.J., Kim S.H., Nahm D.H. Specific immunoglobulin E for staphylococcal enterotoxins in nasal polyps from patients with aspirin-intolerant asthma. Clin Exp Allergy. 2004;34:1270–1275. doi: 10.1111/j.1365-2222.2004.02051.x. [DOI] [PubMed] [Google Scholar]
- 40.Bachert C., Zhang N., Holtappels G., De Lobel L., van Cauwenberge P., Liu S. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J Allergy Clin Immunol. 2010;126:962–968. doi: 10.1016/j.jaci.2010.07.007. e966. [DOI] [PubMed] [Google Scholar]
- 41.Matsuwaki Y., Ookushi T., Asaka D., Mori E., Nakajima T., Yoshida T. Chronic rhinosinusitis: risk factors for the recurrence of chronic rhinosinusitis based on 5-year follow-up after endoscopic sinus surgery. Int Arch Allergy Immunol. 2008;146(Suppl 1):77–81. doi: 10.1159/000126066. [DOI] [PubMed] [Google Scholar]
- 42.Combadiere C., Ahuja S., Tiffany H.L., Murphy P.M. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1α, MIP-1β, and RANTES. J Leuk Biol. 1996;60:147–152. doi: 10.1002/jlb.60.1.147. [DOI] [PubMed] [Google Scholar]
- 43.Daugherty B.L., Siciliano S.J., DeMartino J.A., Malkowitz L., Sirotina A., Springer M.S. Cloning, expression, and characterization of the human eosinophil eotaxin receptor. J Exp Med. 1996;183:2349–2354. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beck L.A., Stellato C., Beall L.D., Schall T.J., Leopold D., Bickel C.A. Detection of the chemokine RANTES and endothelial adhesion molecules in nasal polyps. J Allergy Clin Immunol. 1996;98:766–780. doi: 10.1016/s0091-6749(96)70126-4. [DOI] [PubMed] [Google Scholar]
- 45.Allen J.S., Eisma R., LaFreniere D., Leonard G., Kreutzer D. Characterization of the eosinophil chemokine RANTES in nasal polyps. Ann Otol Rhinol Laryngol. 1998;107:416–420. doi: 10.1177/000348949810700510. [DOI] [PubMed] [Google Scholar]
- 46.Beck L.A., Stellato C., Beall L.D., Schall T.J., Leopold D., Bickel C.A. Detection of the chemokine RANTES and endothelial adhesion molecules in nasal polyps. J Allergy Clin Immunol. 1996;98:766–780. doi: 10.1016/s0091-6749(96)70126-4. [DOI] [PubMed] [Google Scholar]
- 47.Meyer J.E., Bartels J., Gorogh T., Sticherling M., Rudack C., Ross D.A. The role of RANTES in nasal polyposis. Am J Rhinol. 2005;19:15–20. [PubMed] [Google Scholar]
- 48.Bartels J., Maune S., Meyer J.E., Kulke R., Schluter C., Rowert J. Increased eotaxin-mRNA expression in non-atopic and atopic nasal polyps: comparison to RANTES and MCP-3 expression. Rhinology. 1997;35:171–174. [PubMed] [Google Scholar]
- 49.Jahnsen F.L., Haye R., Gran E., Brandtzaeg P., Johansen F.-E. Glucocorticosteroids inhibit mRNA expression for eotaxin, eotaxin-2, and MCP-4 in airway inflammation with eosinophilia. J Immunol. 1999;163:1545–1551. [PubMed] [Google Scholar]
- 50.Molinaro R.J., Bernstein J.M., Koury S.T. Localization and quantitation of eotaxin mRNA in human nasal polyps. Immunol Invest. 2003;32:143–154. doi: 10.1081/imm-120022975. [DOI] [PubMed] [Google Scholar]
- 51.Yao T., Kojima Y., Koyanagi A., Yokoi H., Saito T., Kawano K. Eotaxin-1, -2, and -3 immunoreactivity and protein concentration in the nasal polyps of eosinophilic chronic rhinosinusitis patients. Laryngoscope. 2009;119:1053–1059. doi: 10.1002/lary.20191. [DOI] [PubMed] [Google Scholar]
- 52.Matsukura S., Stellato C., Plitt J., Bickel C., Miura K., Georas S. Activation of eotaxin gene transcription by NF-kB and STAT6 in human airway epithelial cells. J Immunol. 1999;163:6876–6883. [PubMed] [Google Scholar]
- 53.Kuperman D.A., Schleimer R.P. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8:384–392. doi: 10.2174/156652408785161032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lalaker A., Nkrumah L., Lee W.K., Ramanathan M., Lane A.P. Chitin stimulates expression of acidic mammalian chitinase and eotaxin-3 by human sinonasal epithelial cells in vitro. Am J Rhinol Allergy. 2009;23:8–14. doi: 10.2500/ajra.2009.23.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ying S., Meng Q., Taborda-Barata L., Corrigan C.J., Barkans J., Assoufi B. Human eosinophils express messenger RNA encoding RANTES and store and release biologically active RANTES protein. Eur J Immunol. 1966;26:70–76. doi: 10.1002/eji.1830260111. [DOI] [PubMed] [Google Scholar]
- 55.Poposki J.A., Uzzaman A., Nagarkar D.A., Chustz R.T., Pefers A.T., Suh L.A. Increased expression of the chemokine CCk23 in eosinophilic chronic rhinosinusitiswith nasal polyps. J Allergy Clin Immunol. 2011;128:73–81. doi: 10.1016/j.jaci.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nardelli B., Tiffany H.L., Bong G.W., Yourey P.A., Morahan D.K., Li Y. Characterization of the signal transduction pathway activated in human monocytes and dendritic cells by MPIF-1, a specific ligand for CC chemokine receptor 1. J Immunol. 1999;162:435–444. [PubMed] [Google Scholar]
- 57.Patel V.P., Kreider B.L., Li Y., Li H., Leung K., Salcedo T. Molecular and functional characterization of two novel human C-C chemokines as inhibitors of two distinct classes of myeloid progenitors. J Exp Med. 1997;185:1163–1172. doi: 10.1084/jem.185.7.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gauldie J., Cox G., Jordana M., Ohno I., Kirpalani H. Growth and colony-stimulating factors mediate eosinophil fibroblast interactions in chronic airway inflammation. Ann N Y Acad Sci. 1994;725:83–90. doi: 10.1111/j.1749-6632.1994.tb39792.x. [DOI] [PubMed] [Google Scholar]
- 59.Ohnishi M., Ruhno J., Bienenstock J., Milner R., Dolovich J., Denburg J.A. Human nasal polyp epithelial basophil/mast cell and eosinophil colony-stimulating activity. Am Rev Respir Dis. 1988;138:560–564. doi: 10.1164/ajrccm/138.3.560. [DOI] [PubMed] [Google Scholar]
- 60.Churchill L., Friedman B., Schleimer R.P., Proud D. Production of granulocyte-macrophage colony-stimulating factor by cultured human tracheal epithelial cells. Immunology. 1992;75:189–195. [Google Scholar]
- 61.Hamilos D.L., Leung D.Y., Wood R., Meyers A., Stephens J.K., Barkans J. Chronic hyperplastic sinusitis: association of tissue eosinophilia with mRNA expression of granulocyte-macrophage colony-stimulating factor and interleukin-3. J Allergy Clin Immunol. 1993;92:39–48. doi: 10.1016/0091-6749(93)90035-e. [DOI] [PubMed] [Google Scholar]
- 62.Park H.S., Jung K.S., Shute J., Roberts K., Holgate S.T., Djukanovic R. Allergen-induced release of GM-CSF and IL-8 in vitro by nasal polyp tissue from atopic subjects prolongs eosinophil survival. Eur Respir J. 1997;10:1476–1482. doi: 10.1183/09031936.97.10071476. [DOI] [PubMed] [Google Scholar]
- 63.Shin S.H., Lee S.H., Jeong H.S., Kita H. The effect of nasal polyp epithelial cells on eosinophil activation. Laryngoscope. 2003;113:1374–1377. doi: 10.1097/00005537-200308000-00020. [DOI] [PubMed] [Google Scholar]
- 64.Xaubet A., Mullol J., López E., Ferrer J.R., Rozman M., Carrión T. Comparison of the role of nasal polyp and normal nasal mucosal epithelial cells on in vitro eosinohpil survival. medication by GM-CSF and inhibition by dexamethasone. Clin Exp Allergy. 1994;24:307–317. doi: 10.1111/j.1365-2222.1994.tb00240.x. [DOI] [PubMed] [Google Scholar]
- 65.Hamilos D.L., Leung D.Y., Huston D.P., Kamil A., Wood R., Hamid Q. GM-CSF, IL-5 and RANTES immunoreactivity and mRNA expression in chronic hyperplastic sinusitis with nasal polyposis (NP) Clin Exp Allergy. 1998;28:1145–1152. doi: 10.1046/j.1365-2222.1998.00380.x. [DOI] [PubMed] [Google Scholar]
- 66.Lamblin C., Bolard F., Gosset P., Tsicopoulos A., Perez T., Darras J. Bronchial interleukin-5 and eotaxin expression in nasal polyposis. Relationship with (a)symptomatic bronchial hyperresponsiveness. Am J Respir Crit Care Med. 2001;163:1226–1232. doi: 10.1164/ajrccm.163.5.2004197. [DOI] [PubMed] [Google Scholar]
- 67.Simon H.-U., Yousefi S., Schranz C., Schapowal A., Bachert C., Blaser K. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J Immunol. 1997;158:3902–3908. [PubMed] [Google Scholar]
- 68.Liu L.Y., Sedgwick J.B., Bates M.E., Vrtis R.F., Gern J.E., Kita H. Decreased expression of membrane IL-5 receptor alpha on human eosinophils: II. IL-5 down-modulates its receptor via a proteinase-mediated process. J Immunol. 2002;169:6459–6466. doi: 10.4049/jimmunol.169.11.6459. [DOI] [PubMed] [Google Scholar]
- 69.Beck L.A., Schall T.J., Beall L.D., Leopold D., Bickel C., Baroody F. Detection of the chemokine RANTES and activation of vascular endothelium in nasal polyps. J Allergy Clin Immunol. 1994;93:A234. doi: 10.1016/s0091-6749(96)70126-4. [DOI] [PubMed] [Google Scholar]
- 70.Corsi M.M., Pagani D., Dogliotti G., Perona F., Sambataro G., Pignataro L. Protein biochip array of adhesion molecule expression in peripheral blood of patients with nasal polyposis. Int J Biol Markers. 2008;23:115–120. doi: 10.1177/172460080802300208. [DOI] [PubMed] [Google Scholar]
- 71.Eweiss A., Dogheim Y., Hassab M., Tayel H., Hammad Z. VCAM-1 and eosinophilia in diffuse sino-nasal polyps. Eur Arch Otorhinolaryngol. 2009;266:377–383. doi: 10.1007/s00405-008-0762-1. [DOI] [PubMed] [Google Scholar]
- 72.Hamilos D.L., Leung D.Y., Wood R., Bean D.K., Song Y.L., Schotman E. Eosinophil infiltration in nonallergic chronic hyperplastic sinusitis with nasal polyposis (CHS/NP) is associated with endothelial VCAM-1 upregulation and expression of TNF-alpha. Am J Respir Cell Mol Biol. 1996;15:443–450. doi: 10.1165/ajrcmb.15.4.8879177. [DOI] [PubMed] [Google Scholar]
- 73.Jahnsen F.L., Haraldsen G., Aanesen J.P., Haye R., Brandtzaeg P. Eosinophil infiltration is related to increased expression of vascular cell adhesion molecule-1 in nasal polyps. Am J Respir Cell Mol Biol. 1995;12:624–632. doi: 10.1165/ajrcmb.12.6.7539273. [DOI] [PubMed] [Google Scholar]
- 74.Symon F.A., Walsh G.M., Watson S.R., Wardlaw A.J. Eosinophil adhesion to nasal polyp endothelium is P-selectin-dependent. J Exp Med. 1994;180:371–376. doi: 10.1084/jem.180.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bachert C., Gevaert P., Holtappels G., van Cauwenberge P. Mediators in nasal polyposis. Curr Allergy Asthma Rep. 2002;2:481–487. doi: 10.1007/s11882-002-0088-9. [DOI] [PubMed] [Google Scholar]
- 76.Hamilos D.L., Leung D.Y., Wood R., Cunningham L., Bean D.K., Yasruel Z. Evidence for distinct cytokine expression in allergic versus nonallergic chronic sinusitis. J Allergy Clin Immunol. 1995;96:537–544. doi: 10.1016/s0091-6749(95)70298-9. [DOI] [PubMed] [Google Scholar]
- 77.Van Bruaene N., Perez-Novo C.A., Basinski T.M., Van Zele T., Holtappels G., De Ruyck N. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol. 2008;121:1435–1441. doi: 10.1016/j.jaci.2008.02.018. 1441 e1431–1433. [DOI] [PubMed] [Google Scholar]
- 78.Peters A.T., Kato A., Zhang N., Conley D.B., Suh L., Tancowny B. Evidence for altered activity of the IL-6 pathway in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2010;125:397–403. doi: 10.1016/j.jaci.2009.10.072. e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saitoh T., Kusunoki T., Yao T., Kawano K., Kojima Y., Miyahara K. Role of interleukin-17A in the eosinophil accumulation and mucosal remodeling in chronic rhinosinusitis with nasal polyps associated with asthma. Int Arch Allergy Immunol. 2010;151:8–16. doi: 10.1159/000232566. [DOI] [PubMed] [Google Scholar]
- 80.Buysschaert I.D., Grulois V., Eloy P., Jorissen M., Rombaux P., Bertrand B. Genetic evidence for a role of IL33 in nasal polyposis. Allergy. 2010;65:616–622. doi: 10.1111/j.1398-9995.2009.02227.x. [DOI] [PubMed] [Google Scholar]
- 81.Kowalski M.L., Lewandowska-Polak A., Wozniak J., Ptasinska A., Jankowski A., Wagrowska-Danilewicz M. Association of stem cell factor expression in nasal polyp epithelial cells with aspirin sensitivity and asthma. Allergy. 2005;60:631–637. doi: 10.1111/j.1398-9995.2005.00753.x. [DOI] [PubMed] [Google Scholar]
- 82.Perez-Novo C.A., Claeys C., Van Cauwenberge P., Bachert C. Expression of eicosanoid receptors subtypes and eosinophilic inflammation: implication on chronic rhinosinusitis. Respir Res. 2006;7:75. doi: 10.1186/1465-9921-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shin S.H., Lee Y.H., Jeon C.H. Protease-dependent activation of nasal polyp epithelial cells by airborne fungi leads to migration of eosinophils and neutrophils. Acta Otolaryngol. 2006;126:1286–1294. doi: 10.1080/00016480500395179. [DOI] [PubMed] [Google Scholar]
- 84.Van Zele T., Coppieters F., Gevaert P., Holtappels G., Van Cauwenberge P., Bachert C. Local complement activation in nasal polyposis. Laryngoscope. 2009;119:1753–1758. doi: 10.1002/lary.20484. [DOI] [PubMed] [Google Scholar]
- 85.Vandermeer J., Sha Q., Lane A.P., Schleimer R.P. Innate immunity of the sinonasal cavity: expression of messenger RNA for complement cascade components and toll-like receptors. Arch Otolaryngol Head Neck Surg. 2004;130:1374–1380. doi: 10.1001/archotol.130.12.1374. [DOI] [PubMed] [Google Scholar]
- 86.Jankowski R., Bouchoua F., Coffinet L., Vignaud J.M. Clinical factors influencing the eosinophil infiltration of nasal polyps. Rhinology. 2002;40:173–178. [PubMed] [Google Scholar]
- 87.Polzehl D., Moeller P., Riechelmann H., Perner S. Distinct features of chronic rhinosinusitis with and without nasal polyps. Allergy. 2006;61:1275–1279. doi: 10.1111/j.1398-9995.2006.01132.x. [DOI] [PubMed] [Google Scholar]
- 88.Van Cauwenberge P., Van Hoecke H., Bachert C. Pathogenesis of chronic rhinosinusitis. Curr Allergy Asthma Rep. 2006;6:487–494. doi: 10.1007/s11882-006-0026-3. [DOI] [PubMed] [Google Scholar]
- 89.Schleimer R.P., Kato A., Peters A., Conley D., Kim J., Liu M.C. Epithelium, inflammation, and immunity in the upper airways of humans: studies in chronic rhinosinusitis. Proc Am Thorac Soc. 2009;6:288–294. doi: 10.1513/pats.200808-088RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Erjefalt J.S., Greiff L., Andersson M., Adelroth E., Jeffery P.K., Persson C.G. Degranulation patterns of eosinophil granulocytes as determinants of eosinophil driven disease. Thorax. 2001;56:341–344. doi: 10.1136/thorax.56.5.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bachert C., Gevaert P., Holtappels G., Cuvelier C., van Cauwenberge P. Nasal polyposis: from cytokines to growth. Am J Rhinol. 2000;14:279–290. doi: 10.2500/105065800781329573. [DOI] [PubMed] [Google Scholar]
- 92.Adamjee J., Suh Y.J., Park H.S., Choi J.H., Penrose J.F., Lam B.K. Expression of 5-lipoxygenase and cyclooxygenase pathway enzymes in nasal polyps of patients with aspirin-intolerant asthma. J Pathol. 2006;209:392–399. doi: 10.1002/path.1979. [DOI] [PubMed] [Google Scholar]
- 93.Steinke J.W., Bradley D., Arango P., Crouse C.D., Frierson H., Kountakis S.E. Cysteinyl leukotriene expression in chronic hyperplastic sinusitis-nasal polyposis: importance to eosinophilia and asthma. J Allergy Clin Immunol. 2003;111:342–349. doi: 10.1067/mai.2003.67. [DOI] [PubMed] [Google Scholar]
- 94.Di Lorenzo G., Drago A., Esposito Pellitteri M., Candore G., Colombo A., Gervasi F. Measurement of inflammatory mediators of mast cells and eosinophils in native nasal lavage fluid in nasal polyposis. Int Arch Allergy Immunol. 2001;125:164–175. doi: 10.1159/000053811. [DOI] [PubMed] [Google Scholar]
- 95.Bartemes K.R., Cooper K.M., Drain K.L., Kita H. Secretory IgA induces antigen-independent eosinophil survival and cytokine production without inducing effector functions. J Allergy Clin Immunol. 2005;116:827–835. doi: 10.1016/j.jaci.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 96.Muraki M., Gleich G.J., Kita H. Antigen-Specific IgG and IgA, but Not IgE, Activate the Effector Functions of Eosinophils in the Presence of Antigen. Int Arch Allergy Immunol. 2011;154:119–127. doi: 10.1159/000320226. [DOI] [PubMed] [Google Scholar]
- 97.Abu-Ghazaleh R.I., Fujisawa T., Mestecky J., Kyle R.A., Gleich G.J. IgA-induced eosinophil degranulation. J Immunol. 1989;142:2393–2400. [PubMed] [Google Scholar]
- 98.Pleass R.J., Lang M.L., Kerr M.A., Woof J.M. IgA is a more potent inducer of NADPH oxidase activation and degranulation in blood eosinophils than IgE. Mol Immunol. 2007;44:1401–1408. doi: 10.1016/j.molimm.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 99.Tan B.K., Li Q., Suh L., Kato A., Conley D.B., Chandra R. Evidence for intranasal antinuclear autoantibodies with patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128:1198–1206. doi: 10.1016/j.jaci.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Suh K.S., Park H.S., Nahm D.H., Kim Y.K., Lee Y.M., Park K. Role of IgG, IgA, and IgE antibodies in nasal polyp tissue: their relationships with eosinophilic infiltration and degranulation. J Korean Med Sci. 2002;17:375–380. doi: 10.3346/jkms.2002.17.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harlin S.L., Ansel D.G., Lane S.R., Myers J., Kephart G.M., Gleich G.J. A clinical and pathologic study of chronic sinusitis: the role of the eosinophil. J Allergy Clin Immunol. 1988;81:867–875. doi: 10.1016/0091-6749(88)90944-x. [DOI] [PubMed] [Google Scholar]
- 102.Chanez P., Vignola A.M., Vic P., Guddo F., Bonsignore G., Godard P. Comparison between nasal and bronchial inflammation in asthmatic and control subjects. Am J Respir Crit Care Med. 1999;159:588–595. doi: 10.1164/ajrccm.159.2.9801022. [DOI] [PubMed] [Google Scholar]
- 103.Gaga M., Lambrou P., Papageorgiou N., Koulouris N.G., Kosmas E., Fragakis S. Eosinophils are a feature of upper and lower airway pathology in non-atopic asthma, irrespective of the presence of rhinitis. Clin Exp Allergy. 2000;30:663–669. doi: 10.1046/j.1365-2222.2000.00804.x. [DOI] [PubMed] [Google Scholar]
- 104.Bresciani M., Paradis L., Des Roches A., Vernhet H., Vachier I., Godard P. Rhinosinusitis in severe asthma. J Allergy Clin Immunol. 2001;107:73–80. doi: 10.1067/mai.2001.111593. [DOI] [PubMed] [Google Scholar]
- 105.Shahana S., Jaunmuktane Z., Asplund M.S., Roomans G.M. Ultrastructural investigation of epithelial damage in asthmatic and non-asthmatic nasal polyps. Respir Med. 2006;100:2018–2028. doi: 10.1016/j.rmed.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 106.Holgate S.T. Epithelium dysfunction in asthma. J Allergy Clin Immunol. 2007;120:1233–1244. doi: 10.1016/j.jaci.2007.10.025. quiz 1245–1236. [DOI] [PubMed] [Google Scholar]
- 107.Gleich G.J. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol. 2000;105:651–663. doi: 10.1067/mai.2000.105712. [DOI] [PubMed] [Google Scholar]
- 108.Bernardes J.F., Shan J., Tewfik M., Hamid Q., Frenkiel S., Eidelman D.H. Protein nitration in chronic sinusitis and nasal polyposis: role of eosinophils. Otolaryngol Head Neck Surg. 2004;131:696–703. doi: 10.1016/j.otohns.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 109.Richer S.L., Truong-Tran A.Q., Conley D.B., Carter R., Vermylen D., Grammer L.C. Epithelial genes in chronic rhinosinusitis with and without nasal polyps. Am J Rhinol. 2008;22:228–234. doi: 10.2500/ajr.2008.22.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Briot A., Deraison C., Lacroix M., Bonnart C., Robin A., Besson C. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206:1135–1147. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Allakhverdi Z., Comeau M.R., Jessup H.K., Yoon B.R., Brewer A., Chartier S. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kato A., Favoreto S., Jr., Avila P.C., Schleimer R.P. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu T., Li T.L., Zhao F., Xie C., Liu A.M., Chen X. Role of Thymic Stromal Lymphopoietin in the Pathogenesis of Nasal Polyposis. Am J Med Sci. 2011;341(1):40–47. doi: 10.1097/MAJ.0b013e3181f20489. [DOI] [PubMed] [Google Scholar]
- 114.Flood-Page P., Menzies-Gow A., Phipps S., Ying S., Wangoo A., Ludwig M.S. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laitinen L.A., Laitinen A. Structural and cellular changes in asthma. Eur Respir Rev. 1994;4:348–351. [Google Scholar]
- 116.Ohno I., Nitta Y., Yamauchi K., Hoshi H., Honma M., Woolley K. Eosinophils as a potential source of platelet-derived growth factor B-chain (PDGF-B) in nasal polyposis and bronchial asthma. Am J Respir Cell Mol Biol. 1995;13:639–647. doi: 10.1165/ajrcmb.13.6.7576701. [DOI] [PubMed] [Google Scholar]
- 117.Ohno I., Nitta Y., Yamauchi K., Hoshi H., Honma M., Woolley K. Transforming growth factor β1 (TGFβ1) gene expression by eosinophils in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1996;15:404–409. doi: 10.1165/ajrcmb.15.3.8810646. [DOI] [PubMed] [Google Scholar]
- 118.Elovic A., Wong D.T., Weller P.F., Matossian K., Galli S.J. Expression of transforming growth factors-alpha and beta 1 messenger RNA and product by eosinophils in nasal polyps. J Allergy Clin Immunol. 1994;93:864–869. doi: 10.1016/0091-6749(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 119.Schleimer R.P. Mosby Elsevier; New York: 2009. Pharmacology of Glucocorticoids in Allergic Disease. In: Middleton's Allergy, 7th ed. [Google Scholar]
- 120.Gleich G.J., Hunt L.W., Bochner B.S.a., Schleimer R.P. Marcel Dekker, Inc.; New York, NY: 1997. Glucocorticoid effects on human eosinophils. pp.279–308. [Google Scholar]
- 121.Schleimer R.P., Bochner B.S. The effects of glucocorticoids on human eosinophils. J Allergy Clin Immunol. 1994;94:1202–1213. doi: 10.1016/0091-6749(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 122.Patiar S., Reece P. Oral steroids for nasal polyps. Cochrane Database Syst Rev:CD005232. 2007 doi: 10.1002/14651858.CD005232.pub2. [DOI] [PubMed] [Google Scholar]
- 123.Alobid I., Benitez P., Pujols L., Maldonado M., Bernal-Sprekelsen M., Morello A. Severe nasal polyposis and its impact on quality of life. The effect of a short course of oral steroids followed by long-term intranasal steroid treatment. Rhinology. 2006;44:8–13. [PubMed] [Google Scholar]
- 124.Hissaria P., Smith W., Wormald P., Taylor J. Short Course of Systemic Corticosteroids in Sinonasal Polyposis: A double-blind, randomized, placebo-controlled trial with evaluation of outcome measures. J Allergy Clin Immunol. 2006;118:118–128. doi: 10.1016/j.jaci.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 125.Van Zele T., Gevaert P., Holtappels G., Beule A., Wormald P.J., Mayr S. Oral steroids and doxycycline: two different approaches to treat nasal polyps. J Allergy Clin Immunol. 2010;125 doi: 10.1016/j.jaci.2010.02.020. 1069–1076, e1064. [DOI] [PubMed] [Google Scholar]
- 126.Kanai N., Denburg J., Jordana M., Dolovich J. Nasal polyp inflammation. Effect of topical nasal steroid. Am J Respir Crit Care Med. 1994;150:1094–1100. doi: 10.1164/ajrccm.150.4.7921442. [DOI] [PubMed] [Google Scholar]
- 127.Hamilos D.L., Thawley S.E., Kramper M.A., Kamil A., Hamid Q.A. Effect of intranasal fluticasone on cellular infiltration, endothelial adhesion molecule expression, and proinflammatory cytokine mRNA in nasal polyp disease. J Allergy Clin Immunol. 1999;103:79–87. doi: 10.1016/s0091-6749(99)70529-4. [DOI] [PubMed] [Google Scholar]
- 128.Delbrouck C., Kaltner H., Danguy A., Nifant’ev N.E., Bovin N.V., Vandenhoven G. Glucocorticoid-induced differential expression of the sialylated and nonsialylated Lewis(a) epitopes and respective binding sites in human nasal polyps maintained under ex vivo tissue culture conditions. Ann Otol Rhinol Laryngol. 2002;111:1097–1107. doi: 10.1177/000348940211101207. [DOI] [PubMed] [Google Scholar]
- 129.Nonaka R., Nonaka M., Takanashi S., Jordana M., Dolovich J. Eosinophil activation in the tissue: synthetic steroid, budesonide, effectively inhibits the survival of eosinophils isolated from peripheral blood but not nasal polyp tissues. J Clin Lab Immunol. 1999;51:39–53. [PubMed] [Google Scholar]
- 130.Bolard F., Gosset P., Lamblin C., Bergoin C., Tonnel A.B., Wallaert B. Cell and cytokine profiles in nasal secretions from patients with nasal polyposis: effects of topical steroids and surgical treatment. Allergy. 2001;56:333–338. doi: 10.1034/j.1398-9995.2001.00835.x. [DOI] [PubMed] [Google Scholar]
- 131.Mastruzzo C., Greco L.R., Nakano K., Nakano A., Palermo F., Pistorio M.P. Impact of intranasal budesonide on immune inflammatory responses and epithelial remodeling in chronic upper airway inflammation. J Allergy Clin Immunol. 2003;112:37–44. doi: 10.1067/mai.2003.1586. [DOI] [PubMed] [Google Scholar]
- 132.Gevaert P., Bachert C., Holtappels G., Novo C.P., Van der Heyden J., Fransen L. Enhanced soluble interleukin-5 receptor alpha expression in nasal polyposis. Allergy. 2003;58:371–379. doi: 10.1034/j.1398-9995.2003.00110.x. [DOI] [PubMed] [Google Scholar]
- 133.Gevaert P., Hellman C., Lundblad L., Lundahl J., Holtappels G., van Cauwenberge P. Differential expression of the interleukin 5 receptor alpha isoforms in blood and tissue eosinophils of nasal polyp patients. Allergy. 2009;64:725–732. doi: 10.1111/j.1398-9995.2008.01885.x. [DOI] [PubMed] [Google Scholar]
- 134.Gevaert P., Lang-Loidolt D., Lackner A., Stammberger H., Staudinger H., Van Zele T. Nasal IL-5 levels determine the response to anti-IL-5 treatment in patients with nasal polyps. J Allergy Clin Immunol. 2006;118:1133–1141. doi: 10.1016/j.jaci.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 135.Tsuyuki S., Bertrand C., Erard F., Trifilieff A., Tsuyuki J., Wesp M. Activation of the fas receptor on lung eosinophils leads to apoptosis and the resolution of eosinophilic inflammation of the airways. J Clin Invest. 1995;96:2924–2931. doi: 10.1172/JCI118364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kikly K.K., Bochner B.S., Freeman S.D., Tan K.B., Gallagher K.T., D’Alessio K. Identification of SAF-2, a novel Siglec expressed on eosinophils, mast cells, and basophils. J Allergy Clin Immunol. 2000;105:1093–1100. doi: 10.1067/mai.2000.107127. [DOI] [PubMed] [Google Scholar]
- 137.Kato A., Peters A., Suh L., Carter R., Harris K.E., Chandra R. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121:1385–1392. doi: 10.1016/j.jaci.2008.03.002. 1392 e1381–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kato A., Xiao H., Chustz R.T., Liu M.C., Schleimer R.P. Local release of B cell-activating factor of the TNF family after segmental allergen challenge of allergic subjects. J Allergy Clin Immunol. 2009;123:369–375. doi: 10.1016/j.jaci.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chapter 13.12. Eosinophils and Vascular Healing
Mark C. Lavigne, Michael J. Eppihimer
The deployment of both bare metal and drug-eluting stents (BMS and DES) has revolutionized the treatment options for revascularization of occluded atherosclerotic coronary arteries. However, stent implantation can promote vessel damage and engage a series of exaggerated vascular healing events that may promote thrombosis and/or reocclusion or restenosis within targeted vessels. In some patients, additional hypersensitivity immune reactions to stent components may elevate the risk of developing thrombosis and restenosis compared to nonallergic patients. Eosinophils appear to contribute to hypersensitivity-driven adverse vascular reactions to stent residency, because they have been observed in the vicinity of stent struts in the context of thrombotic and restenotic events following BMS or DES implantation. Furthermore, eosinophil granule protein actions toward thrombosis and restenosis regulators suggest direct, deleterious roles of eosinophils in stent-associated thrombosis and restenosis. However, before any eosinophil-targeted changes in stent composition or post-stenting medicinal strategies are made, more investigations are warranted to provide incontrovertible evidence that eosinophils actively make significant contributions to stent-associated thrombosis and restenosis.
The ability of organisms to repair themselves is an indispensable requirement for their survival. Most members of the animal kingdom inevitably encounter external or internal environmental pressures that can threaten their existence. Examples of these are, respectively, injury inflicted by a traumatic event, such as a stab wound, and parasites that inappropriately reside in various tissues. A complex series of reactions to these survival challenges has evolved among vertebrates to correct tissue damage traumatically imposed by a foreign object (i.e., a knife) or to completely remove an injurious agent (i.e., an infection). These shared mechanisms of self-preservation are collectively referred to as healing. In many individuals, healing events in the coronary vasculature occur and can be characterized by both types of reactions, including correction of vessel damage inflicted by circulating substances (i.e., coronary artery disease) and subsequent introduction of a foreign agent (i.e., a stent), as well as to mount an immune response (i.e., hypersensitivity) aimed at eliminating such a foreign body.
The most common type of healing occurrence in blood vessels is, paradoxically, also responsible for the most common type of coronary artery disease. Indeed, atherosclerosis is likely the result of a response to injury that occurs predominantly in medium-sized muscular vessels, including the left anterior descending, left circumflex, and right coronary arteries.1 The injury is thought to be primarily inflicted on endothelial cells (ECs), which, from their position as a lining of the vessel lumen, normally provide a nonthrombogenic, nonadhesive surface despite their direct contact with flowing blood. Agents suspected of damaging ECs include elevated blood cholesterol levels and oxidized low-density lipoprotein. The outcome of vessel healing is often manifest as an eccentrically oriented lesion consisting of, in its most mature form, lipid-laden macrophages and smooth muscle cells (or so-called foam cells), and lymphocytes enclosed by a fibrous cap comprised of collagen, elastin, and proteoglycans.1 Consequently, these lesions, or plaques, in their stable form can compromise critical oxygen supply to the energetic myocardium by significantly impeding blood flow and, in their unstable or vulnerable forms, can cause myocardial infarction and death by providing sites for platelet adherence and activation of clotting factors to promote formation of a fully occlusive thrombus. Although lifestyle changes and medications, including cholesterol synthesis inhibitors (or statins), effectively reduce the risk factors for developing atherosclerosis,2 its place as a predominant cause of morbidity and mortality in industrialized nations has not wavered. Indeed, its recurrent detection is afforded by sophisticated angiographic evidence through catheterization of vessels suspected of harboring lesions. Once detected, a patient’s physician has several choices for revascularizing occluded coronary vessels.
Since the late 1970s, a variety of minimally invasive, catheter-based procedures have evolved to displace occlusive atherosclerotic plaques in coronary vessels. Andreas Gruentzig and colleagues performed the first such procedure in 1977 by using a catheter to guide a balloon to the site of atherosclerosis.3 Once positioned, the balloon was expanded to crush the plaque against the vessel wall. This procedure, referred to as percutaneous transluminal coronary angioplasty (PTCA), or simply angioplasty, was at least acutely successful for providing a less-invasive treatment, compared to traditionally used coronary artery bypass grafting (CABG), to restore adequate blood flow to the myocardium. Approximately 6 months following the procedure, however, approximately 35% of patients experienced a renarrowing (restenosis) of the blood vessel at the original atheromatous site.4 Since then, two major modifications to PTCA have been made in an effort to reduce the incidence of restenosis, including balloon-mediated deployment of bare metal stents (BMS) and drug and polymer addition to such stents to form a device collectively referred to as a drug-eluting stent (DES). The former amendment to PTCA originated with the recognition that restenosis was at least partially attributable to elastic recoil of the vessel as an immediate reaction to balloon-induced vessel expansion, while the addition of drug came with an appreciation for the significant contributions of vascular smooth muscle cell (VSMC) proliferation5 and migration6 to restenosis following BMS insertion into coronary arteries. The cell proliferative component is similar to that which promotes benign tumor formation and, therefore, was as a good candidate for being disrupted by antiproliferative agents such as those on first-generation DES that emerged from Johnson and Johnson in 2003 (Cypher DES; Cordis, Miami Lakes, FL)7 and Boston Scientific Corporation in 2004 (Taxus Express2, Maple Grove, MN),8 and which included sirolimus and paclitaxel, respectively.
Vascular responses to stent implantation in coronary arteries occur in sequence to culminate in what is collectively referred to as healing. In the traditional sense, vascular wound healing is orchestrated by platelet- and clotting factor-mediated hemostasis, cytokine-/chemokine- and leukocyte-mediated inflammation, and VSMC- and fibroblast-mediated tissue remodeling.9 Although healing naturally connotes a process that is favorable, it can be characterized by an exaggeration of the processes outlined above, leading to unfavorable consequences, such as reocclusion, or restenosis of the vessel and/or thrombosis. Restenosis can occur in conjunction with stent use.10 Moreover, thrombosis rates associated with DES implantation are higher than those related to BMS deployment after 1 year,11 possibly owing to insufficient EC coverage of the stent itself and of the local vascular area in which a DES resides.12 The fate of the vessel wall localized to the area of stent implantation is determined, for the most part, by a balance between the interactions of cells and the soluble factors that they secrete. These events, in turn, may be dictated by the circumstances of DES deployment itself, such as the extent of mechanical injury imposed by stent insertion.13 Additionally, DES composition may play a significant role in determining the prognosis of DES use as a therapy to alleviate occlusive coronary vascular disease.
Along with their differential abilities to prevent restenosis and association with thrombosis, DES and BMS platforms may be distinguished by inflammatory cell infiltrates, based on the extent of eosinophil presence that follows deployment of each. Clinically used variations of DES currently feature controlled drug release from polymers that coat the metal stent skeleton. Twenty-eight days following their deployment, overlapping Taxus and Cypher DES each had more eosinophils associated with them compared to their respective overlapping BMS controls in rabbit iliac artery.14 These DES are composed of different drugs and nonerodible polymers, but the same metals (316L stainless steel), suggesting that drugs and/or polymers may ultimately attract eosinophils to stent insertion sites. John and colleagues found that a critical amount of polymer may selectively incite eosinophil recruitment in rabbit iliac artery. Cypher was associated with significantly more luminal eosinophils than a polymer-free sirolimus-eluting stent, a polymer-free sirolimus–estradiol-eluting stent, or a BMS, but only when each stent platform was overlapped upon itself (i.e., one stent on top of another of the same kind) in the vessel.15 Eosinophil recruitment following DES implantation in rabbits appears not to be specific to this species, since eosinophil accumulation was observed around Cypher stent struts, followed by less infiltration near both Taxus and BM stents, in porcine coronary arteries.16 Table 13.12.1 provides a summary of studies citing the occurrence of eosinophil infiltration in response to a variety of catheter-based revascularization interventions, including purely balloon-mediated, or plain old balloon angioplasty (POBA), and BMS or DES implantation. Such reactions to foreign-body stent substances suggest an eosinophil-mediated hypersensitivity,17 which has been implicated in thrombosis and restenosis following stent insertion into blood vessels, as described below.
TABLE 13.12.1.
Eosinophil Recruitment Following Catheter-Based Revascularization of Coronary Vessels
| Species | Interventions | Extent of Eosinophil Inflammation | Reference Number |
|---|---|---|---|
| Rabbit | BMS and DES | Taxus > Cypher DES > BMS |
14 |
| Rabbit | BMS, DES (no polymer), DES (polymer) | DES (polymer) > DES (no polymer) and BMS | 15 |
| Pig | BMS and DES | Cypher > Taxus DES > BMS |
16 |
| Pig | POBA and BMS | BMS > POBA | 22 |
| Pig | POBA and BMS | BMS > POBA | 23 |
| Human | POBA and BMS | BMS > POBA | 24 |
BMS, bare metal stent; DES, drug-eluting stent; POBA, plain old balloon angioplasty.
Several instances of thrombosis occurring after BMS or DES implantation and their correlation with histological evidence of eosinophil infiltration have been recorded. Zavolloni and colleagues18 reported that inflammatory infiltrate observed in thrombectomy material retrieved from right coronary artery previously implanted (10 years) with a BMS for myocardial infarction contained a prevalence of eosinophils. Data such as these contrast with previous reports that associated stent thrombosis and eosinophil recruitment particularly with DES rather than BMS. Virmani and colleagues provided histological evidence of thrombus formation in left circumflex coronary artery of a 58-year-old male who had received overlapping Cypher stents in that vessel 18 months earlier.19 Aneurysm formation and inflammatory prominence consisting of eosinophils, giant cells, lymphocytes, macrophages, and plasma cells within the vessel area localized to stent placements were evident. The authors referred to these phenomena as a ‘hypersensitivity vasculitis,’ which has since been described by other investigators following DES implantation in conjunction with repeated thromboses associated with implantation of Cypher stents into the left circumflex20 and also in autopsy specimens.21 The latter study is especially informative, given that it compared eosinophil presence among thrombotic events according to classification, and in the context of acute myocardial infarction or not, and time-frame, including early (≤30 days) after BMS or DES [Cypher, Taxus, and Endeavor (Medtronic, Inc. Minneapolis, MN); drug is zotarolimus] deployment, late (31–365 days) after BMS deployment, and very late (>1 year) after DES deployment. Here, eosinophil accumulation and the fraction of leukocytes accounted for by eosinophils around Cypher stents was substantially greater than that measured in association with Taxus or Endeavor stents. Furthermore, eosinophils were most evident in thrombi that occurred very late following DES (predominantly of Cypher) deployment. Interestingly, vessel remodeling was exclusively related to very late stent thrombosis that occurred after DES, with its extent varying according to the number of eosinophils found in thrombi. The authors speculated that vessel remodeling likely caused stent malapposition and subsequent thrombosis. Taken together, these studies indicate eosinophilic reactions to BMS or DES deployment and suggest a role for eosinophils in stent thrombosis. At least two questions may be drawn from these conclusions:
-
1.
What factors related to coronary stents and/or their deployment attract eosinophils to stented vessel segments?
-
2.
How might eosinophils contribute to stent-related thrombosis?
Eosinophilic inflammation occurs following both POBA and stent insertion. However, eosinophilic recruitment is more robust following stent placement,22., 23., 24. suggesting that balloon-mediated vessel expansion, which is common to both POBA and stent placement, is not completely responsible for enhanced eosinophilic recruitment associated with stent placement. Indeed, stent components and prophylactic dual antiplatelet (medicinal) therapies (DAPT) that are prescribed for use after stent implantation, including the platelet adenosine diphosphate (ADP) receptor antagonist, clopidogrel (Plavix; Bristol-Myers Squibb, New York, NY, and Sanofi–Aventis, Paris, France) and acetylsalicylic acid (aspirin), an inhibitor of prostaglandin G/H synthase 1 (inhibits thromboxane production), can elicit hypersensitivity reactions.25 The well-recognized role of eosinophils in allergic reactions17 and their suggested role in a localized hypersensitivity reaction to stent placement19 are in agreement with positive correlations between allergic sensitivity to metal stent components, including nickel20., 26. and molybdenum,26 and eosinophil recruitment to stented vessel segments.20., 26. However, restenosis incidence may27 or may not28 positively correlate with allergic reactions to nickel and molybdenum. In addition, drug-eluting stent-related hypersensitivity reactions appear not to guarantee subsequent thrombosis, as revealed by a study29 showing that a minority (approximately 1.5%) of total DES-specific hypersensitivity reactions (based on 262 hypersensitivity cases reported) were accompanied by thrombosis. These hypersensitive, thrombotic cases, however, were characterized by eosinophilic inflammation and incomplete stent coverage, or so-called delayed healing, 100% of the time. Overall, the majority of DES-specific hypersensitivity cases were associated with Cypher implantation (approximately 80%) and likely attributable to the poly n-butyl-methacrylate and polyethylene-vinyl acetate Cypher copolymer allergens,25 but not to sirolimus, since the latter can reduce eosinophil infiltration.30 Consistent with this, Finn and colleagues reported that 5/105 cases of late stent thrombosis were associated with hypersensitivity reactions, with four occurring after Cypher and one after Taxus implantation.12 In these episodes, too, eosinophilic inflammation was always present. Taken together, these studies suggest that the risk of hypersensitivity to stent components is likely to be small among patients that have or will receive DES, with the onset of thrombosis occurring in such hypersensitivity cases also to be small. However, histological evidence shows that the incidence of such rare cases is consistently associated with eosinophil accumulation at stent sites and seems to be especially common with Cypher implantation.12., 29. At least one study found the combination of hypersensitivity and delayed healing (i.e., endothelialization) of stent struts to be a risk factor for developing late-stent thrombosis.31 Identification of patients prone to each of these circumstances is not performed and may, in fact, be unpredictable considering that hypersensitivity reactions to stents appear not to be unequivocally associated with thrombosis29 and, further, the incidence of eosinophil involvement in such reactions, which correlates with hypersensitivity-related thrombosis,12., 29. is also unclear. Thus, without objective, basic study- and clinical trial-based reasoning for differential postprocedural prescription to DES recipients, it is essential that all patients that receive DES comply with using DAPT medicines for their full recommended terms.
Currently, the exact duration of DAPT therapy following stent deployment is questionable, but would appear to be optimized by an awareness of when stent struts are completely healed (or endothelialized) such that the risk of platelet adhesion and activation is dramatically diminished. Unfortunately, the time required for adequate, antithrombotic endothelialization of stent struts and the local vascular wall likely varies among patients, because endothelialization rates among DES patients afflicted with complicating clinical backgrounds, such as diabetes,32 may vary, especially compared to DES patients without complicating ancillary medical conditions. The concern, then, pertaining to eosinophil involvement in thrombus formation associated with stent deployment is that DAPT may be suspended before stent struts, whether covered by polymer or not, are masked enough by healing processes so as not to be a potentially chronic stimulus for eosinophil recruitment and activation. Indeed, the nonspecific nature of antiproliferative drugs currently included on clinically used DES inhibit not only VSMC proliferation and migration, two major contributors to restenosis,5., 6. but also attenuate the same properties of ECs.33., 34. Therefore, the risk of eosinophil-related thrombosis following DES deployment may be assessed by (1) whether the stent is BMS or DES and (2) evidence of allergies to stent components that may predispose the patient to a hypersensitivity reaction that involves eosinophils. Fortunately, research efforts have provided suggestive evidence concerning the mechanism of eosinophil recruitment to stented vessel segments and the mechanism through which eosinophils contribute to thrombus formation. These pieces of information may be used to direct future therapies intended to mitigate the purported role of eosinophils in promoting thrombosis following stent deployment.
Descriptions of two hypersensitivity reactions (I and IV) have been suggested to provide hypothetical explanations for eosinophilic involvement in inflammatory responses to stent insertion. As suggested by Virmani and coworkers, eosinophil recruitment to segments stented with Cypher and their association with thrombosis19 in this context may be due to a type IV hypersensitivity reaction, in which T-helper lymphocyte liberation of T-helper type 2 (Th2) cytokines and interleukins 4 and 13 attract eosinophils.35 Another hypothesis25 includes a two-phase cascade of cell- and soluble factor-mediated reactions being initially orchestrated by IgE-activated mast cells and the mediators that these cells elaborate (approximately 1–24 h post-stent deployment) to promote secondary infiltration of basophils, eosinophils, macrophages, neutrophils, and T lymphocytes (approximately 12–24 h post-stent deployment) that can remain chronically situated around stent struts. Thus, occurrence of the first phase of hypersensitivity would coincide with onset of acute stent thrombosis, while the latter time frame would fit the onset of both late and very late stent thromboses. Evidence of a role for eosinophils in thrombogenesis includes observations made in hypereosinophilic patients.36 Furthermore, endothelial cells are likely targets for the highly basic-charged major basic and eosinophil cationic proteins (MBP and ECP) of eosinophils, considering that ECs express a negatively charged glycocalyx on their luminal surface. Once bound to ECs, these proteins may inflict damage or even kill these cells, as suggested by their cytotoxic capabilities.17 Alternatively, direct activation of platelets by MBP and/or eosinophil peroxidase (EPO),37 or disruption of thrombomodulin function by MBP,38 may explain the contribution made by eosinophils to stent thrombosis. Eosinophil presence in the context of stent-related thrombosis cases that may be secondarily related to tissue remodeling and consequent stent malapposition21 raises the possibility that eosinophils directly or indirectly possess tissue-remodeling properties. Both may be true, since eosinophils express matrix metalloproteinase-9 (MMP-9),39 which is a collagenase capable of degrading type IV collagen, a major component of subendothelial layer basement membranes. Furthermore, eosinophils secrete interleukin-8,17 a chemokine that has been shown to induce release of MMP-2 (a collagenase) and MMP-9 from ECs.40 Eosinophils also express vascular endothelial growth factor (VEGF)41 and heparanase.42 These factors are likely to partially mediate the ability of eosinophils to promote angiogenesis,43 by inducing EC growth (VEGF) and degradation of perlecan (heparanase), a heparan sulfate proteoglycan component of basement membranes. The relevance of this is that angiogenesis can occur in the context of the granulation stage of vascular healing following stent insertion. The cumulative occurrence of any thrombi that may form as a result of angiogenic events may culminate as a thrombus of significant size, with the ability to dramatically or completely block blood flow in the main stented vessel. Table 13.12.2 summarizes investigations that have linked stent-related thrombosis with eosinophilic inflammation. Kawano and coworkers reported that a patient who received a BMS to relieve total occlusion of the left coronary artery experienced repeated episodes of restenosis after stent implantation.26 Examination of the restenotic lesion revealed granulation tissue with eosinophil infiltration. The patient displayed positive reactions to allergic patch tests for nickel and molybdenum, both of which are components of the 316L stainless steel BMS that the patient received. Thus, through their direct association with granulation tissue formation, eosinophils may contribute to both thrombotic and restenotic mechanisms that pertain to stent deployment. Details concerning the former are outlined above, while the latter may be due to activation of platelets by eosinophilic proteins37 and subsequent platelet degranulation to release promitogenic factors, such as platelet-derived growth factor and fibroblast growth factor, capable of stimulating VSMC migration and proliferation.
TABLE 13.12.2.
Association of Eosinophils with Stent Thrombosis
| Species | Intervention(s) | Observations | Reference Number |
|---|---|---|---|
| Human | BMS | VLST, eosinophil infiltrate | 18 |
| Human | Overlapping Cypher | LST, eosinophils, T lymphocytes, aneurysm | 19 |
| Human | Overlapping Cypher | Repeated thrombosis, eosinophils | 20 |
| Human | Cypher and Taxus | VLST, eosinophils, vessel remodeling, stent malapposition Cypher > Taxus |
21 |
| Human | Cypher and Taxus | Occurrence of eosinophil inflammation in DES-specific, HS-associated thrombosis = 100% | 29 |
| Human | Cypher and Taxus | Occurrence of eosinophil inflammation in DES-related, HS-associated thrombosis = 100% | 12 |
BMS, bare metal stent; DES, drug-eluting stent; HS, hypersensitivity; LST, late stent thrombosis (1 month–1 year postprocedure); VLST, very late stent thrombosis (>1 year postprocedure).
Restenosis of revascularization attempts by BMS or DES implantation are associated with eosinophil infiltration. This was documented nearly 20 years ago, when the transition from POBA to POBA with BMS deployment was being tested for clinical use. Karas and colleagues compared histological patterns of restenosis between POBA and insertion of a BM tantalum stent into swine coronary arteries.22 Inflammation accompanied by greater VSMC proliferation was observed in association with in-stent restenosis. The inflammatory infiltrate consisted of eosinophils, macrophage-like histiocytes, and T lymphocytes surrounding stent struts. Consistent with this, a more recent report found VSMC proliferation to be primarily responsible for restenosis following stenting, but not following POBA, in swine.23 Macrophages were selectively found in stented lesions and were accompanied by neutrophils and eosinophils. In another investigation, T lymphocytes were found in restenotic lesions of both POBA and BMS; however, significantly more VSMCs and eosinophils were associated with in-stent restenosis.24 These studies suggest a positive correlation between inflammation characterized by eosinophil presence and VSMC proliferation and, similar to observations made relating eosinophils to stent thrombosis, a hypersensitivity reaction to a stent component that ultimately attracts eosinophils to the stented vessel segment. Such a relationship between the VSMC proliferative component of restenosis and eosinophil infiltration is significant, given the importance of VSMC growth in restenosis5 and the fact that DES are loaded specifically with antiproliferative compounds primarily intended to block VSMC mitogenesis. Interestingly, other studies have shown the selective association of eosinophils, among other circulating cells including inflammatory and bone marrow-derived progenitor cells, with in-stent restenosis. Gabbasov and colleagues found that the number of osteonectin-positive progenitor cells, but not granulocytes, in blood were higher in patients afflicted with ischemic heart disease (IHD; n = 38) than in healthy individuals (n = 17).44 In contrast, only elevations in eosinophils were detected in IHD patients that subsequently received Cypher DES and experienced restenosis (n = 15). Blood eosinophil levels were not increased in patients that did not undergo restenosis (n = 23). Together, histological and blood analyses have established a link between eosinophil presence and restenosis that is particularly associated with stent deployment, compared to POBA. These study observations suggest that eosinophils are equipped to contribute to the mechanism of restenosis. However, the question remains: are eosinophilic contents biomarkers of restenosis and thrombosis or do they actively contribute to these processes?
Niccoli and colleagues45 showed that serum ECP levels prior to Cypher or Taxus implantation predicted whether patients would experience a major adverse cardiac event (MACE), including cardiac death, recurrent myocardial infarction, or target lesion revascularization (TLR), which was defined as being necessary if >50% stenosis occurred within 5 mm upstream or downstream of the stent. The majority (60%) of MACE onset occurred 180 days after stent insertion, while 27% of such cases happened more than 1 year following deployment. Clopidogrel was prescribed for 9 months and aspirin for a lifetime after stent insertion. Some patients had allergies, none of which were confirmed to stent components, such as metals or polymers. Furthermore, ECP levels were nearly equivalent between allergic and nonallergic patients that did not experience MACE. What factor(s) could preelevate ECP levels in individuals who had not yet been exposed to potential allergens contained in DES? Elevated ECP levels may be explained by prior observations indicating that eosinophil count positively correlated with IHD development46 and that eotaxin/C-C motif chemokine 11 (CCL11) levels may play a role in atherosclerosis.47 Taken together, these studies suggest that ECP may play a causative role in MACE, particularly TLR, which represented the majority of MACE cases in this study, since it was elevated before stent implantation. Related to this issue, experimental evidence implicates eosinophilic contents as having the potential to promote prorestenotic events during the healing process post-stent deployment. For example, as discussed earlier, activation of platelets by both MBP and EPO37 may liberate growth factors from platelets that are capable of inducing migration and proliferation of VSMCs. By secreting eotaxin and transforming growth factor β (TBF- β),17 eosinophils may directly stimulate VSMC migration48 and extracellular matrix production by fibroblasts and VSMCs,49 respectively. Each of these manifestations of eosinophilic secretory products would contribute to restenotic lesion development. Masu and colleagues described that a heat-labile, unidentified constituent of <10 kDa in eosinophilic lysates can promote proliferation of airway smooth muscle cells (SMCs),50 suggesting the ability of eosinophils to also stimulate vascular SMC growth. Table 13.12.3 provides a summary of studies that have reported an association between eosinophilic inflammation and restenosis. Further studies involving genetic deficiency or mRNA silencing may delineate which specific eosinophil component is responsible for this and other prorestenotic activities.
TABLE 13.12.3.
Association of Eosinophils with Restenosis
| Species | Intervention(s) | Observations | Reference Number |
|---|---|---|---|
| Pig | POBA and BMS | Eosinophils associated with BMS only; also histiocytes and macrophages present | 22 |
| Pig | POBA and BMS | Eosinophils associated with BMS only; neutrophils also present; VSMC hyperplasia | 23 |
| Human | POBA and BMS | Eosinophils, VSMCs, T lymphocytes: BMS > POBA |
24 |
| Human | BMS | Eosinophils, VSMC and matrix accumulation | 26 |
BMS, bare metal stent; POBA, plain old balloon angioplasty; VSMC, vascular smooth muscle cell.
Conclusion
In summary, healing in coronary vessels can be a deleterious phenomenon in two instances, including forming atherosclerotic lesions following EC injury and forming thrombi and restenotic lesions following stent implantation. Ironically, treatments to alleviate atherosclerotic burden have evolved to include use of a minimally invasive therapeutic mode, namely deployment of BMS and DES, that potentially incites yet further, exaggerated, healing responses that can manifest as clinical concerns. This subchapter explains that healing events associated with stent deployment may be two-fold, including standard hemostatic, inflammatory, and tissue-remodeling phenomena that are likely common to all occurrences of stent implantation and, in a minority of individuals, superimposition of such standard healing responses by hypersensitivity reactions to the stent itself. To date, data suggest that both BM and polymer components of DES are candidates to stimulate involvement of factors and cells that mediate such hypersensitivity reactions, including eosinophils. Basic science studies have revealed the possibility that eosinophils contribute to thrombosis and restenosis associated with stent implantation, by virtue of the potentially prothrombotic effects of their granule proteins, such as ECP, EPO, and MBP, on platelets and thrombomodulin, and on SMC growth. As the incidence of hypersensitivity reactions as sequelae to stent implantation becomes more evident, routine prophylactic measures may be warranted, in addition to postprocedural DAPT prescription, in candidate stent recipients, to include prescreening individuals for allergies to stent components. Of course, the integrity of such tests is encumbered by the caveat that positive allergic patch tests may not predict the occurrence of stent-related thrombotic or restenotic events.28 Alternatively, the use of next-generation bioabsorbable stents, which have relatively limited residency times in vessels compared to nonbioabsorbable stents such as Cypher and Taxus, would theoretically eliminate the stimulus for eosinophilic responses relatively quickly and serve as a reasonable and useful way to reduce complications due to hypersensitivity in vulnerable patients. Clearly, observations made of eosinophils in the vicinity of stent struts in association with thrombosis and restenosis are highly suggestive of a role for these cells in such adverse events. Further work, perhaps involving eosinophil-deficient animals or cultured eosinophils deficient in granule proteins, is needed to more precisely define the role of eosinophils and the extent of medical attention deemed necessary to mitigate their presumed involvement in stent-associated thrombosis and in restenosis.
References
- 1.Ross R. Rous-Whipple Award Lecture Atherosclerosis: A defense mechanism gone awry. Am J Pathol. 1993;143:987–1002. [PMC free article] [PubMed] [Google Scholar]
- 2.Mantell G. Lipid lowering drugs in atherosclerosis—the HMG-CoA reductase inhibitors. Clin Exp Hypertens. 1989;11:927–941. doi: 10.3109/10641968909035383. [DOI] [PubMed] [Google Scholar]
- 3.Gruentzig A.R., Senning A., Siegenthaler W.E. Nonoperative dilatation of coronary-artery stenosis: percutaneous transluminal coronary angioplasty. N Engl J Med. 1979;301:61–68. doi: 10.1056/NEJM197907123010201. [DOI] [PubMed] [Google Scholar]
- 4.Holmes D.R., Jr., Vlietstra R.E., Smith H.C., Vetrovec G.W., Kent K.M., Cowley M.J. Restenosis after percutaneous transluminal coronary angioplasty (PTCA): a report from the PTCA Registry of the National Heart, Lung, and Blood Institute. Am J Cardiol. 1984;53:C77–C81. doi: 10.1016/0002-9149(84)90752-5. [DOI] [PubMed] [Google Scholar]
- 5.Komatsu R., Ueda M., Naruko T., Kojima A., Becker A.E. Neointimal tissue response at sites of coronary stenting in humans. Macroscopic, histological and immunohistochemical analysis. Circulation. 1998;98:224–233. doi: 10.1161/01.cir.98.3.224. [DOI] [PubMed] [Google Scholar]
- 6.Casscells W. Migration of smooth muscle and endothelial cells: critical events in restenosis. Circulation. 1992;86:723–729. doi: 10.1161/01.cir.86.3.723. [DOI] [PubMed] [Google Scholar]
- 7.Regar E., Serruys P.W., Bode C., Holubarsch C., Guermonprez J.L., Wijns W., RAVEL Study Group Angiographic findings of the multicenter Randomized Study with the Sirolimus-Eluting Bx Velocity Balloon-Expandable Stent (RAVEL): sirolimus-eluting stents inhibit restenosis irrespective of the vessel size. Circulation. 2002;106:1949–1956. doi: 10.1161/01.cir.0000034045.36219.12. [DOI] [PubMed] [Google Scholar]
- 8.Colombo A., Drzewiecki J., Banning A., Grube E., Hauptmann K., Silber S., TAXUS II Study Group Randomized study to assess the effectiveness of slow- and moderate release polymer-based paclitaxel-eluting stents for coronary artery lesions. Circulation. 2003;108:788–794. doi: 10.1161/01.CIR.0000086926.62288.A6. [DOI] [PubMed] [Google Scholar]
- 9.Scott N.A. Restenosis following implantation of bare metal coronary stents: pathophysiology and pathways involved in the vascular response to injury. Adv Drug Del Rev. 2006;58:358–376. doi: 10.1016/j.addr.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Auer J., Leitner A., Berent R., Lamm G., Lassnig E., Krennmair G. Long-term outcomes following coronary drug-eluting- and bare-metal-stent implantation. Atherosclerosis. 2010;210:503–509. doi: 10.1016/j.atherosclerosis.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Stone G.W., Moses J.W., Ellis S.G., Schofer J., Dawkins K.D., Morice M.C. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med. 2007;356:998–1008. doi: 10.1056/NEJMoa067193. [DOI] [PubMed] [Google Scholar]
- 12.Finn A.V., Nakazawa G., Joner M., Kolodgie F.D., Mont E.K., Gold H.K. Vascular responses to drug eluting stents. Importance of delayed healing. Arterioscler Thromb Vasc Biol. 2007;27:1500–1510. doi: 10.1161/ATVBAHA.107.144220. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz R.S., Huber K.C., Murphy J.G., Edwards W.D., Camrud A.R., Vlietstra R.E. Restenosis and the proportional neointimal response to coronary artery injury: results in a porcine model. J Am Coll Cardiol. 1992;19:267–274. doi: 10.1016/0735-1097(92)90476-4. [DOI] [PubMed] [Google Scholar]
- 14.Finn A.V., Kolodgie F.D., Harnek J., Guerrero L.J., Acampado E., Tefera K. Differential response of delayed healing and persistent inflammation at sites of overlapping sirolimus- or paclitaxel-eluting stents. Circulation. 2005;112:270–278. doi: 10.1161/CIRCULATIONAHA.104.508937. [DOI] [PubMed] [Google Scholar]
- 15.John M.C., Wessely R., Kastrati A., Schomig A., Joner M., Uchihashi M. Differential healing response in polymer- and nonpolymer-based sirolimus-eluting stents. J Am Coll Cardiol Cardiovascular Interventions. 2008;1:535–544. doi: 10.1016/j.jcin.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Wilson G.J., Nakazawa G., Schwartz R.S., Huibregtse B., Poff B., Herbst T.J. Comparison of inflammatory response after implantation of sirolimus- and paclitaxel-eluting stents in porcine coronary arteries. Circulation. 2009;120:141–149. doi: 10.1161/CIRCULATIONAHA.107.730010. [DOI] [PubMed] [Google Scholar]
- 17.Hogan S.P., Rosenberg H.F., Moqbel R., Phipps S., Foster P.S., Lacy P. Eosinophils: Biological properties and role in health and disease. Clin Exp Allergy. 2008;38:709–750. doi: 10.1111/j.1365-2222.2008.02958.x. [DOI] [PubMed] [Google Scholar]
- 18.Zavolloni D., Bossi P., Rossi M.L., Gasparini G.L., Lisignoli V., Presbitero P. Inflammatory substrate with eosinophils may be present in bare-metal stent thrombosis. J Cardiovasc Med. 2009;10:942–943. doi: 10.2459/JCM.0b013e32832f3f8e. [DOI] [PubMed] [Google Scholar]
- 19.Virmani R., Guagliumi G., Farb A., Musumeci G., Grieco N., Motta T. Localized hypersensitivity and late coronary thrombosis secondary to a sirolimus-eluting stent. Should we be cautious? Circulation. 2004;109:701–705. doi: 10.1161/01.CIR.0000116202.41966.D4. [DOI] [PubMed] [Google Scholar]
- 20.Yokouchi Y., Oharaseki T., Ihara I., Naoe S., Sugawara S., Takahashi K. Repeated stent thrombosis after DES implantation and localized hypersensitivity to a stent implanted in the distal portion of a coronary aneurysm thought to be sequela of Kawasaki disease: autopsy report. Pathol International. 2009;60:112–118. doi: 10.1111/j.1440-1827.2009.02484.x. [DOI] [PubMed] [Google Scholar]
- 21.Cook S., Ladich E., Nakazawa G., Eshtehardi P., Neidhart M., Vogel R. Correlation of intravascular ultrasound findings with histopathological analysis of thrombus aspirates in patients with very late drug-eluting stent thrombosis. Circulation. 2009;120:391–399. doi: 10.1161/CIRCULATIONAHA.109.854398. [DOI] [PubMed] [Google Scholar]
- 22.Karas S.P., Gravanis M.B., Santoian E.C., Robinson K.A., Anderberg K.A., King S.B., III Coronary intimal proliferation after balloon injury and stenting in swine: and animal model of restenosis. J Am Coll Cardiol. 1992;20:467–474. doi: 10.1016/0735-1097(92)90119-8. [DOI] [PubMed] [Google Scholar]
- 23.Nakatani M., Takeyama Y., Shibata M., Yorozuya M., Suzuki H., Koba S. Mechanisms of restenosis after coronary intervention. Difference between plain old balloon angioplasty and stenting. Cardiovasc Pathol. 2003;12:40–48. doi: 10.1016/s1054-8807(02)00135-7. [DOI] [PubMed] [Google Scholar]
- 24.Rittersma S.Z.H., Meuwissen M., van der Loos C.M., Koch K.T., de Winter R.J., Piek J.J. Eosinophilic infiltration in restenotic tissue following coronary stent implantation. Atherosclerosis. 2006;184:157–162. doi: 10.1016/j.atherosclerosis.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 25.Chen J.P., Hou D., Pendyala L., Goudevenos J.A., Kounis N.G. Drug-eluting stent thrombosis. The Kounis hypersensitivity-associated acute coronary syndrome revisited. J Am Coll Cardiol: Cardiovascular Interventions. 2009;2:583–593. doi: 10.1016/j.jcin.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 26.Kawano H., Koide Y., Baba T., Nakamizo R., Toda G., Takenaka M. Granulation tissue with eosinophil infiltration in the restenotic lesion after coronary stent implantation—a case report. Circ J. 2004;68:722–723. doi: 10.1253/circj.68.722. [DOI] [PubMed] [Google Scholar]
- 27.Koster R., Vieluf D., Kiehn M., Sommerauer M., Kahler J., Baldus S. Nickel and molybdenum contact allergies in patients with coronary in-stent restenosis. Lancet. 2000;356:1895–1897. doi: 10.1016/S0140-6736(00)03262-1. [DOI] [PubMed] [Google Scholar]
- 28.Hillen U., Haude M., Erbel R., Goos M. Evaluation of metal allergies in patients with coronary stents. Contact Dermatitis. 2002;47:353–356. doi: 10.1034/j.1600-0536.2002.470607.x. [DOI] [PubMed] [Google Scholar]
- 29.Nebeker J.R., Virmani R., Bennett C.L., Hoffman J.M., Samore M.H., Alvarez J. Hypersensitivity cases associated with drug-eluting coronary stents. A review of available cases from the Research on Adverse Drug Events and Reports (RADAR) project. J Am Coll Cardiol. 2006;47:175–181. doi: 10.1016/j.jacc.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 30.Francischi J.N., Conroy D., Maghni K., Sirois P. Rapamycin inhibits airway leukocyte infiltration and hyperreactivity in guinea pigs. Agents Actions. 1993;39:C139–C141. doi: 10.1007/BF01972746. [DOI] [PubMed] [Google Scholar]
- 31.Joner M., Finn A.V., Farb A., Mont E.K., Kolodgie F.D., Ladich E. Pathology of drug-eluting stents in humans. Delayed healing and late thrombotic risk. J Am Coll Cardiol. 2006;48:193–202. doi: 10.1016/j.jacc.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 32.Cagliero E., Roth T., Taylor A.W., Lorenzi M. The effects of high glucose on human endothelial cell growth and gene expression are not mediated by transforming growth factor-beta. Lab Invest. 1995;73:667–673. [PubMed] [Google Scholar]
- 33.Parry T.J., Brosius R., Thyagarajan R., Carter D., Argentieri D., Falotico R. Drug-eluting stents: sirolimus and paclitaxel differentially affect cultured cells and injured arteries. Euro J Pharmacol. 2005;524:19–29. doi: 10.1016/j.ejphar.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 34.Wessely R., Blaich B., Belaiba R.S., Merl S., Gorlach A., Kastrati A. Comparative characterization of cellular and molecular anti-restenotic profiles of paclitaxel and sirolimus. Thromb Haemost. 2007;97:1003–1012. [PubMed] [Google Scholar]
- 35.Grunig G., Warnock M., Wakil A.E., Venkayya R., Brombacher F., Rennick D.M. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogbogu P., Rosing D.R., Horne M.K., III Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin North Am. 2007;27:457–475. doi: 10.1016/j.iac.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rohrbach M.S., Wheatley C.L., Slifman N.R., Gleich G.J. Activation of platelets by eosinophil granule proteins. J Exp Med. 1990;172:1271–1274. doi: 10.1084/jem.172.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukai H.Y., Ninomiya H., Ohtani K., Nagasawa T., Abe T. Major basic protein binding to thrombomodulin potentially contributes to the thrombosis in patients with eosinophilia. Br J Haematol. 1995;90:892–899. doi: 10.1111/j.1365-2141.1995.tb05211.x. [DOI] [PubMed] [Google Scholar]
- 39.Ohno I., Ohtani H., Nitta Y., Suzuki J., Hoshi H., Honma M. Eosinophils as a source of matrix metalloproteinase-9 in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1997;16:212–219. doi: 10.1165/ajrcmb.16.3.9070604. [DOI] [PubMed] [Google Scholar]
- 40.Li A., Dubey S., Varney M.L., Dave B.J., Singh R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 41.Horiuchi T., Weller P.F. Expression of vascular endothelial growth factor by human eosinophils: upregulation by granulocyte macrophage colony-stimulating factor and interleukin-5. Am J Respir Cell Mol Biol. 1997;17:70–77. doi: 10.1165/ajrcmb.17.1.2796. [DOI] [PubMed] [Google Scholar]
- 42.Temkin V., Aingorn H., Puxeddu I., Goldshmidt O., Zcharia E., Gleich G.J. Eosinophil major basic protein: first identified natural heparanase-inhibiting protein. J Allergy Clin Immunol. 2004;113:703–709. doi: 10.1016/j.jaci.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 43.Puxeddu I., Alian A., Piliponsky A.M., Ribatti D., Panet A., Levi-Schaffer F. Human peripheral blood eosinophils induce angiogenesis. Int J Biochem Cell Biol. 2005;37:628–636. doi: 10.1016/j.biocel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Gabbasov Z.A., Kozlov S.G., Saburova O.S., Titiov V.N., Liakishev A.A. Stromal progenitor cells and blood leukocytes after implantation of drug-eluting stents. Kardiologiia. 2010;50:36–41. [PubMed] [Google Scholar]
- 45.Niccoli G., Schiavino D., Belloni F., Ferrante G., La Torre G., Conte M. Pre-intervention eosinophil cationic protein serum levels predict clinical outcomes following implantation of drug-eluting stents. Eur Heart J. 2009;30:1340–1347. doi: 10.1093/eurheartj/ehp120. [DOI] [PubMed] [Google Scholar]
- 46.Sweetnam P.M., Thomas H.F., Yarnell J.W., Baker I.A., Elwood P.C. Total and differential leukocyte counts as predictors of ischemic heart disease: the Caerphilly and Speedwell studies. Am J Epidemiol. 1997;145:416–421. doi: 10.1093/oxfordjournals.aje.a009123. [DOI] [PubMed] [Google Scholar]
- 47.Emanuele E., Falcone C., D’Angelo A., Minoretti P., Buzzi M.P., Bertona M. Association of plasma eotaxin levels with the presence and extent of angiographic coronary artery disease. Atherosclerosis. 2006;186:140–145. doi: 10.1016/j.atherosclerosis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 48.Kodali R.B., Kim W.J., Galaria I.I., Miller C., Schecter A.D., Lira S.A. CCL11 (Eotaxin) induces CCR3-dependent smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2004;24:1211–1216. doi: 10.1161/01.ATV.0000131654.90788.f5. [DOI] [PubMed] [Google Scholar]
- 49.Leask A., Abraham D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 50.Masu K., Ohno I., Suzuki K., Okada S., Hattori T., Shirato K. Proliferative effects of eosinophil lysates on cultured human airway smooth muscle cells. Clin Exp Allergy. 2002;32:595–601. doi: 10.1046/j.0954-7894.2002.01243.x. [DOI] [PubMed] [Google Scholar]
Chapter 13.13. Eosinophils and Allograft Rejection
Florence Roufosse, Annick Massart, Fleur Samantha Benghiat, Philippe Lemaitre, Alain Le Moine
Three decades ago, eosinophils were described as having a role in transplant rejection or graft-versus-host disease. Although the concept of an experimental alloreactive T-helper type 2-mediated effector pathway involving interleukin-4 (IL-4), IL-5, and eosinophils is widely accepted, the precise role of eosinophils in these clinical settings is still unclear. Indeed, eosinophils are potentially important mediators in processes of tissue damage, but also in tissue repair and remodeling, as well as immune regulation. Besides, eosinophilia could be interpreted as an outcome predictor. Herein, relevant clinical and experimental observations of eosinophil infiltration in solid organ and hematopoietic stem cell transplantation are discussed in a friend or foe perspective.
Brief Overview of Eosinophil Biology
Eosinophils are a minor leukocyte subset in healthy subjects, representing less than 5% of circulating white blood cells, and present in discrete locations, specifically the bone marrow, digestive tract, mammary glands, thymus, and uterus.1., 2. Eosinophils belong to the myeloid lineage, and their differentiation and proliferation in the marrow is controlled successively by specific transcription factors [including GATA binding protein 1 (GATA1) and transcription factor PU.1] and growth factors [granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), and IL-5]. Among the latter, only IL-5 is specific for the eosinophil lineage in humans, as eosinophil precursors express the ligand-binding IL-5RA on their surface. Increased production of IL-5 in vivo by CD4+ T cells3 or transformed cells (e.g., carcinomas4 or Reed Sternberg cells5) results in increased eosinophilopoiesis and peripheral eosinophilia in blood and/or tissues. Various factors contribute to preferential eosinophil transendothelial migration and homing in tissues,1 including adhesion molecules (vascular cell adhesion protein 1; VCAM-1), cytokines (IL-5), chemokines [specifically, eotaxins 1, 2 and 3, as well as RANTES (C-C motif chemokine 5) and monocyte chemoattractant protein (MCP)], and arachidonic acid metabolites [leukotriene B4 (LTB4), cysteinyl-leukotrienes, and prostaglandin D2 (PGD2)], in addition to the more general signals generated under conditions of cell stress and death.
Eosinophils were long considered as exclusively effector cells, able to induce significant tissue damage and dysfunction by releasing preformed, highly cytotoxic mediators, including the granule proteins, major basic protein and eosinophil cationic protein, producing reactive oxygen species, and generating arachidonic acid metabolites such as platelet activating factor (PAF) and LTC4 (reviewed in 1). These effector functions were considered potentially beneficial in the setting of parasitic infections, and harmful in the setting of allergic responses to environmental antigens. Recent studies have shattered this paradigm. It is now well established that eosinophils are active participants in ongoing immune responses through the production of cytokines and chemokines, and through previously unrecognized functions of their granule proteins [e.g., the ability of eosinophil-derived neurotoxin, a natural Toll-like receptor 2 (TLR2) ligand, to induce dendritic cell maturation and to promote antigen-specific T-helper type 2 (Th2)-biased immune responses6], and that they possess many characteristics of antigen-presenting cells, enabling them to elicit antigen-specific CD4+ T cell responses. Indeed, eosinophils express major histocompatibility molecule (MHC) class II and co-stimulatory molecules (CD40, CD80, and CD86), and were shown to be able to process antigen (ovalbumin; OVA) and present it to naive CD4+ T cells in lymph nodes, in a murine model of allergic pulmonary inflammation.7 Further upstream in allergic pulmonary inflammation, eosinophils have been shown to be required for the recruitment of antigen-specific effector CD4+ T cells to the lungs and the development of typical histopathological changes after allergen challenge in mice.8 Their ability to induce production of the Th2 chemokines thymus and activation-regulated chemokine (TARC) and macrophage-derived chemokine (MDC) in the lung is critical in this process.
In parallel with these proimmune functions, eosinophils also have the potential to modulate local inflammatory responses, for example dampening Th1-dominated inflammation through release of IL-4 and IL-6.9 Production of galectin-10 (also known as Charcot–Leyden crystal protein), IL-10, indoleamine 2,3-dioxygenase, and transforming growth factor β (TGF-β) may confer eosinophils with a regulatory role on effector T-cell responses.10., 11., 12. Eosinophils also contribute to processes of remodeling and repair. Although eosinophils produce several potentially relevant factors, including fibroblast growth factor 2 (FGF-2), matrix metalloproteinase-9 (MMP-9), and vascular endothelial growth factor (VEGF), mechanistic studies establishing a causal role in remodeling are lacking; in contrast, several studies strongly suggest that eosinophil-derived TGF-β contributes to airway remodeling in allergic asthma.13., 14.
A series of observations on eosinophil behavior in health and disease unaccounted for by our current understanding of eosinophil biology, together with the paucity of experimental data supporting a causal relationship between eosinophil cytotoxicity and tissue damage and/or disease, have generated the Local Immunity and/or Remodeling/Repair (LIAR) hypothesis, which has recently been presented for scientific scrutiny.15 The authors propose a central role for eosinophils in modulating LIAR, with recruitment of these cells to sites of cell death and turnover where stem cell activities are operative, in order to maintain tissue homeostasis. The accumulation and functions of eosinophils are dependent on various factors in the local microenvironment, including the presence of other specific immune effector cells, and of soluble growth factors liable to sustain eosinophil survival and activation. At physiological sites of high cell turnover, such as the endometrial lining and the gut, it is assumed that eosinophils dampen immune responses that could be triggered by such active metabolic activity. Similarly, eosinophils may inhibit the immune response elicited by tissue-infiltrating helminth parasites, favoring cohabitation between host and pathogen. In contrast, the local production of eosinophil growth-promoting and activating cytokines by other immune cells in allergic inflammation may favor positive feedback loops that sustain and amplify the immune response.7., 16., 17.
Finally, in addition to the increasing complexity of eosinophil contributions to adaptive immunity, a role for eosinophils in innate immunity was recently suggested by a study showing that eosinophils express variable levels of CD3 and γ/δ T-cell receptors, which are involved in antimycobacterial and antitumor immune responses.18 The inflammation that develops in solid organ transplants in the setting of an alloimmune response (i.e., transplant rejection) may contain, and in some instances be dominated by, eosinophilic infiltrates.19 Whether eosinophils are directly involved in the damage to foreign tissue and thus actively contribute to rejection through release of their cytotoxic mediators, or are engaged in LIAR activities, is currently unknown. The local release of small molecule mediators, such as damage-associated molecular pattern molecules, by dying cells within the transplant may contribute to very early eosinophil recruitment.15 Eosinophils could theoretically contribute to initiation of the allogeneic response by cross-presentation of foreign MHC antigens; they may also favor local recruitment of allospecific effector T cells, as is seen in allergic inflammation.
Eosinophils in Experimental Allograft Rejection
In mouse models of acute graft rejection in the setting of deficient CD8+ T-cell effector functions, namely MHC class II-incompatible skin grafts20 and fully histoincompatible cardiac transplants in CD8-deficient recipients,21 marked eosinophilic infiltrates emerge that are dependent on IL-5 produced by antidonor CD4 T cells. However, IL-5 neutralization or silencing, and associated eosinophil depletion, fails to prevent rejection in these stringent alloreactive models, indicating at most a partial contribution of eosinophils to rejection.19 This is not surprising, since allograft rejections are mediated by multiple redundant pathways.22 Nevertheless, turning off T-cell cytotoxicity revealed a role for IL-5 and eosinophils in a model of acute and chronic rejection of MHC class II-incompatible skin grafts.19., 20., 23. In the chronic rejection model, the Th1 component of alloreactivity and T-cell cytotoxicity were dampened by repeated injections of anti-CD3 antibody. Similarly, treatment with anti-CD154 and a depleting CD8 monoclonal antibody Ab resulted in eosinophilic infiltration in a model of transplant arteriosclerosis.24., 25. In other experiments, the adoptive transfer of alloreactive non-cytotoxic Th2 clones into T cell-deficient mice induced the rejection of skin or cardiac allografts characterized by a dense eosinophil infiltrate.26., 27. Finally, IL-4 or IL-5 neutralization, as well as eosinophil depletion through repeated injections of anti-CCR3 (C-C chemokine receptor type 3) antibody, prevented the rejection of weakly immunogenic skin grafts bearing a single minor antigen disparity in MHC class I-deficient recipients.28
Eosinophils in Solid Organ Transplantation
Eosinophils and Kidney Transplantation
The role of eosinophils in renal transplantation has aroused little attention. Although publications described the presence of eosinophils during renal allograft rejection in the early 1980s,29 the first systematic reviews on the subject only appeared in the mid-1980s.30., 31. This may be due to technical reasons. Indeed, conventional staining techniques, like hematoxylin–eosin (H&E), underestimate the presence of tissue eosinophils and do not usually detect their degranulation.32 Motivated by a case report of marked hypereosinophilia and eosinophilic infiltration in a rejected renal allograft, in 1986 Weir and coworkers decided to investigate retrospectively a cohort of 132 renal transplant recipients (124 biopsies) with 187 episodes of acute rejection.30 They concluded that increased eosinophils in the blood or renal biopsy represented an adverse prognostic factor for renal outcome. This was followed by a prospective study by Kormendi and coworkers analyzing cellular infiltrates using fine-needle aspiration in a cohort of 83 renal allograft recipients during the first month posttransplantation.31 Tissue eosinophilia exceeding 4% was considered a useful cutoff with a predictive accuracy for serious or irreversible rejections of 71% (sensitivity: 78%; specificity: 91%, with a prevalence of acute rejections of 32.5%). In contrast, blood eosinophil counts were found to be less reliable. In another study, Ten and coworkers showed eosinophils in the kidney interstitium and in tubular casts.33 Of note, eosinophil degranulation was evaluated by the extracellular localization of the eosinophil granule major basic protein (MBP) as revealed by immunofluorescence. In this small cohort of 16 patients, eosinophils and extracellular MBP were more frequently observed in acute rejection (94% and 87%, respectively) than in cyclosporine toxicity (6 patients; 17% and 17%), whereas both features were absent in controls (normal kidney donors). Similarly, urinary levels of MBP were also elevated in acute rejections and acute interstitial nephritis while they remained normal in cyclosporine nephrotoxicity. In another study, eosinophils and extracellular eosinophil cationic protein (ECP) were prominent features of acute vascular rejection rather than interstitial rejection, and eosinophil density increased in areas bordering necrotic tissue and in arteries with necrotic lesions.34 A correlation between eosinophil infiltrates and rejection severity was also observed by Meleg and coworkers, who reviewed 29 allograft nephrectomies.35 They concluded that a significant interstitial graft eosinophil infiltrate called SIGE was statistically associated with vascular rejections but not iatrogenic interstitial nephritis. This was already reported by Hongwei and coworkers, who also found a correlation between the density of the eosinophil infiltrate and the rate of graft loss by rejection.36 All together, these observations reinforce the possibility of a nonincidental, causative association between eosinophils and acute allograft rejection (Table 13.13.1 ).
TABLE 13.13.1.
Summary of Observations
| Type of Transplantation | Supposed Role∗ | Marker of Severity | Predictive of Good or Bad Outcome | References |
|---|---|---|---|---|
| Kidney | Effector | Yes | Bad | 30., 31., 32., 33., 34., 35., 35. |
| Lung | Effector | Yes | Bad | 37., 40. |
| Liver | Effector | Yes | Bad | 41., 42., 43., 44., 45., 46., 47., 48., 49., 50. |
| HSC | ? | Yes No |
Bad Good |
55., 56., 59., 72., 73., 74. 57., 58., 61., 63. |
Based on each author’s report, although the role of eosinophils might be more complex (see text).
Eosinophils are also linked to chronic allograft rejection characterized by interstitial fibrosis, obliterative arteriopathy, and tubular atrophy. Indeed, Nolan and coworkers reported the presence of eosinophils in 93% of renal allografts undergoing chronic rejection.32 They were located in the intimal and adventitial space of the thickened arteries, as well as in the interstitium. Interestingly, in vitro experiments revealed that eosinophil by-products (see above) enhanced fibroblast and vascular smooth muscle cell proliferation in murine and human experiments, perhaps reflecting a pathogenic mechanism involved in obliterative arteriopathy. Although these data strongly suggest a pathogenic role for eosinophils and by-products in kidney allografts, their presence may be related to other functions, as evoked in the LIAR hypothesis.
Eosinophils and Lung Transplantation
Although eosinophils are described in chronic and acute lung allograft rejection, their role in these processes remains unclear.37., 38., 39. Acute lung rejection classically occurs during the first year after transplantation and its diagnosis is essentially based on transbronchial biopsies (TBB). The current international guidelines, published in 2010 by the International Heart and Lung Transplantation Society (ISHLT),40 establish acute lung rejection as the presence of, firstly, perivascular and interstitial mononuclear cell infiltrates (grade A) and, second, small-airway inflammation, namely lymphocytic bronchiolitis (grade B). Grading is scaled depending on the composition, extension, and intensity of the infiltrate. Eosinophils are not a feature of grade A1 (minimal rejection), but are found in grade A2 (mild vascular rejection) and, importantly, are considered to be a common finding in severe vascular rejection (grade A3). Regarding airway inflammation, eosinophils are considered to be occasional in low-grade (B1R) and common in high-grade (B2R) inflammation.
Chronic lung allograft rejection, synonymous with bronchiolitis obliterans syndrome (BOS), occurs in up to 50% of recipients. BOS is characterized by a persistent decrease in expiratory flow and is potentially life threatening, requiring retransplantation. Although eosinophils are not taken into consideration in the ISHLT working formulation for this condition, a recent prospective cohort study reported that recurrent tissue eosinophilia (with higher concentrations of IL-6 and IL-8) is significantly associated with an increased risk of developing BOS.37
Increased eosinophilia in lung transplant recipients should be interpreted with caution for two reasons.39 Firstly, there are numerous nonrejection-related causes of graft eosinophilia. Among these, infectious diseases are dominant, including fungi (e.g., aspergillus and coccidioidomycosis), bacteria (e.g., tuberculosis), helminths (e.g., Toxocara canis and Ascaris lumbricoides), and even viruses (e.g., cocksackies). High-dose steroid treatment could be detrimental under these conditions. Drug reactions are also a common cause of pulmonary eosinophilia (e.g., antibiotics, diuretics, or methotrexate). The second reason is that blood eosinophilia does not always reflect bronchoalveolar lavage fluid or lung tissue eosinophilia. Lung biopsies are therefore crucial for assessing the role of eosinophils after lung transplantation.
Eosinophils and Liver Transplantation
There are many similarities regarding eosinophilia in liver transplantation compared with other transplanted organs already discussed. IL-5 and eosinophils were rapidly identified during liver allograft rejection.41., 42., 43., 44., 45. There was a consensus for considering eosinophils and IL-5 as mediators of a nonclassical pathway of rejection (i.e., non-Th1-mediated rejection).46., 47. Indeed, liver allografts with evidence of rejection showed concomitant intragraft IL-5 mRNA and activated eosinophils releasing MBP.46 In pediatric recipients, elevated biliary and serum IL-5 correlate with rejection.47 Along the same lines, another study reported that blood eosinophilia and serum ECP are early indicators of acute liver allograft rejection and precede alterations of conventional liver function tests by several days.48 However, the use of increased serum ECP as a rejection marker is limited by its association with infections.48 These pioneering findings were confirmed and refined by more recent studies that identified graft and blood eosinophilia as an independent highly specific marker of acute liver allograft rejection.41., 42., 45., 49., 50. In addition, an elevated blood eosinophil count may predict the severity of rejection, just as in the case of lung and kidney transplantation.49 However, the use of this potential marker of rejection is limited in patients with hepatitis C infection, and those treated with corticosteroids, as both these circumstances decrease eosinophil levels.49
Eosinophils and Hematopoietic Stem Cell Transplantation
Inflammation plays a pivotal role in the complex pathogenesis of acute and chronic graft-versus-host disease (GVHD). Conventionally, acute GVHD (aGVHD) is described as a Th1 disease associated with the release of interferon γ (IFN-γ), IL-2, IL-12, and TNF-α.51., 52., 53. The gastrointestinal tract, liver, and skin are the most common targets of GVHD, and diarrhea, jaundice, and skin rash are its most common manifestations. Chronic GVHD has features resembling autoimmune and other immunological disorders, such as bronchiolitis obliterans (see above), chronic immunodeficiency, Sjögren syndrome, and systemic sclerosis. Although the pathophysiology has not been fully elucidated, chronic GVHD appears to be mediated by the overproduction of Th2-type cytokines, namely IL-4 and IL-5.53., 54.
The role of eosinophils in acute and chronic GVHD is a matter for speculation. Thirty years ago, Shulman and coworkers showed for the first time that eosinophilia after allogeneic hematopoietic stem cell transplantation (HSCT) was often present at the time of diagnosis of chronic GVHD.55 Afterwards, it was shown that eosinophilia could precede, sometimes by several months, the onset of chronic GVHD symptoms, and seemed to have a strong predictive value for the subsequent development of this condition.56., 57. Should this be confirmed by additional prospective trials, this observation may have significant clinical impact. More recently, eosinophilia was also observed among patients who developed aGVHD and, similar to chronic GVHD, was seen before the beginning of symptoms in some cases.58., 59. The pathophysiology behind eosinophilia in the setting of GVHD remains unclear. Th2 cytokine production may be involved, as suggested by the finding that serum IL-5 concentrations are elevated in patients with symptoms of aGVHD.60., 61. However, no correlation with blood eosinophilia has been observed.62
Studies focusing on the prognostic importance of eosinophilia have produced conflicting data. In chronic GVHD, some retrospective data suggest a better outcome for HSCT recipients with eosinophilia, while others found no correlation, suggesting that eosinophilia may just be a bystander of cGVHD rather than a prognostic biomarker.56., 61., 63. Among young patients with malignant diseases treated by HSCT, those with eosinophilia showed increased event-free survival and a lower relapse rate than those without eosinophilia, suggesting that eosinophils could be involved in the graft versus leukemia effect.61 In aGVHD, observational studies showed that patients with eosinophilia after allogeneic HSCT have a milder disease than patients without eosinophilia.58
One could hypothesize that improved prognosis of aGVHD when eosinophils are present is related to the fact that this reflects Th2, rather than Th1 (classically considered as the effectors of aGVHD cell activation). In agreement, murine studies have shown that several cytokines that are known to favor Th2 polarization of donor T cells, such as GM-CSF, IL-4, or rapamycin, can reduce aGVHD.64., 65., 66., 67.
In humans, it has been shown that an increased number of IL-4- and IL-10-producing cells are associated with reduced severity or absence of aGVHD,68., 69. also suggesting a possible protective role for Th2 cytokines in aGVHD. Nevertheless, reports indicate that Th2 subsets may actually cause aGVHD by targeting other organs than those targeted by Th1 subsets.70., 71. This argues against a rigid paradigm according to which aGVHD is a Th1 process and chronic GVHD a Th2 process. In support of this hypothesis, prospective data have shown that bone marrow eosinophilia after HSCT may be a predictive marker of severe aGVHD.72 Similarly, the presence of eosinophils in duodenal biopsy specimens taken during acute flares correlates with intestinal GVHD severity.73 There is also evidence that eosinophils show signs of activation in both blood and target organs of patients during aGVHD flares.74., 75. The bad reputation of eosinophils in GVHD generated by these observations has led some investigators to test the efficacy of montelukast (an orally active leukotriene antagonist that inhibits eosinophils) as a supplement to standard therapy for patients with chronic GVHD, with promising preliminary results.76 For the time being, the role of eosinophils in target organ damage still needs to be assessed in the setting of well-conducted experimental studies.77
Conclusion
In conclusion, compelling evidence links activated eosinophils with allograft rejection and graft-versus-host disease. In contrast to mast cells, they are not (yet) linked to transplantation tolerance.78 Their presence seems to be correlated with the gravity of tissue damage, which may reflect effector functions contributing to rejection, or eosinophil accumulation in response to tissue damage, in agreement with the LIAR hypothesis. To date, the roles played by IL-5 and eosinophils in the redundant pathways of allograft rejection and the potential graft healing processes remain cryptic. The recent availability of genetically engineered mice lacking eosinophils (PHIL and ΔdblGATA) provides a unique opportunity to clarify their contribution to these processes.79., 80.
References
- 1.Blanchard C., Rothenberg M.E. Biology of the eosinophil. Adv Immunol. 2009;101:81–121. doi: 10.1016/S0065-2776(08)01003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothenberg M.E., Hogan S.P. The eosinophil. Annu Rev Immunol. 2006;24:147–174. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 3.Romagnani S. Human TH1 and TH2 subsets: doubt no more. Immunol Today. 1991;12:256–257. doi: 10.1016/0167-5699(91)90120-I. [DOI] [PubMed] [Google Scholar]
- 4.Pandit R., Scholnik A., Wulfekuhler L., Dimitrov N. Non-small-cell lung cancer associated with excessive eosinophilia and secretion of interleukin-5 as a paraneoplastic syndrome. Am J Hematol. 2007;82:234–237. doi: 10.1002/ajh.20789. [DOI] [PubMed] [Google Scholar]
- 5.Samoszuk M., Nansen L. Detection of interleukin-5 messenger RNA in Reed-Sternberg cells of Hodgkin’s disease with eosinophilia. Blood. 1990;75:13–16. [PubMed] [Google Scholar]
- 6.Yang D., Chen Q., Su S.B., Zhang P., Kurosaka K., Caspi R.R. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J Exp Med. 2008;205:79–90. doi: 10.1084/jem.20062027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H.B., Ghiran I., Matthaei K., Weller P.F. Airway eosinophils: allergic inflammation recruited professional antigen-presenting cells. J Immunol. 2007;179:7585–7592. doi: 10.4049/jimmunol.179.11.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobsen E.A., Ochkur S.I., Pero R.S., Taranova A.G., Protheroe C.A., Colbert D.C. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J Exp Med. 2008;205:699–710. doi: 10.1084/jem.20071840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spencer L.A., Szela C.T., Perez S.A., Kirchhoffer C.L., Neves J.S., Radke A.L. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J Leukoc Biol. 2009;85:117–123. doi: 10.1189/jlb.0108058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobsen E.A., Taranova A.G., Lee N.A., Lee J.J. Eosinophils: singularly destructive effector cells or purveyors of immunoregulation? J Allergy Clin Immunol. 2007;119:1313–1320. doi: 10.1016/j.jaci.2007.03.043. [DOI] [PubMed] [Google Scholar]
- 11.Odemuyiwa S.O., Ghahary A., Li Y., Puttagunta L., Lee J.E., Musat-Marcu S. Cutting edge: human eosinophils regulate T cell subset selection through indoleamine 2,3-dioxygenase. J Immunol. 2004;173:5909–5913. doi: 10.4049/jimmunol.173.10.5909. [DOI] [PubMed] [Google Scholar]
- 12.Kubach J., Lutter P., Bopp T., Stoll S., Becker C., Huter E. Human CD4+CD25+ regulatory T cells: proteome analysis identifies galectin-10 as a novel marker essential for their anergy and suppressive function. Blood. 2007;110:1550–1558. doi: 10.1182/blood-2007-01-069229. [DOI] [PubMed] [Google Scholar]
- 13.Kay A.B., Phipps S., Robinson D.S. A role for eosinophils in airway remodelling in asthma. Trends Immunol. 2004;25:477–482. doi: 10.1016/j.it.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Phipps S., Flood-Page P., Menzies-Gow A., Ong Y.E., Kay A.B. Intravenous anti-IL-5 monoclonal antibody reduces eosinophils and tenascin deposition in allergen-challenged human atopic skin. J Invest Dermatol. 2004;122:1406–1412. doi: 10.1111/j.0022-202X.2004.22619.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee J.J., Jacobsen E.A., McGarry M.P., Schleimer R.P., Lee N.A. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy. 2010;40:563–575. doi: 10.1111/j.1365-2222.2010.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacKenzie J.R., Mattes J., Dent L.A., Foster P.S. Eosinophils promote allergic disease of the lung by regulating CD4(+) Th2 lymphocyte function. J Immunol. 2001;167:3146–3155. doi: 10.4049/jimmunol.167.6.3146. [DOI] [PubMed] [Google Scholar]
- 17.Sabin E.A., Kopf M.A., Pearce E.J. Schistosoma mansoni egg-induced early IL-4 production is dependent upon IL-5 and eosinophils. J Exp Med. 1996;184:1871–1878. doi: 10.1084/jem.184.5.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Legrand F., Driss V., Woerly G., Loiseau S., Hermann E., Fournie J.J. A functional gammadeltaTCR/CD3 complex distinct from gammadeltaT cells is expressed by human eosinophils. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005926. e5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldman M., Le Moine A., Braun M., Flamand V., Abramowicz D. A role for eosinophils in transplant rejection. Trends Immunol. 2001;22:247–251. doi: 10.1016/s1471-4906(01)01893-2. [DOI] [PubMed] [Google Scholar]
- 20.Le Moine A., Surquin M., Demoor F.X., Noel J.C., Nahori M.A., Pretolani M. IL-5 mediates eosinophilic rejection of MHC class II-disparate skin allografts in mice. J Immunol. 1999;163:3778–3784. [PubMed] [Google Scholar]
- 21.Braun M.Y., Desalle F., Le Moine A., Pretolani M., Matthys P., Kiss R. IL-5 and eosinophils mediate the rejection of fully histoincompatible vascularized cardiac allografts: regulatory role of alloreactive CD8(+) T lymphocytes and IFN-gamma. Eur J Immunol. 2000;30:1290–1296. doi: 10.1002/(SICI)1521-4141(200005)30:5<1290::AID-IMMU1290>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 22.Le Moine A., Goldman M., Abramowicz D. Multiple pathways to allograft rejection. Transplantation. 2002;73:1373–1381. doi: 10.1097/00007890-200205150-00001. [DOI] [PubMed] [Google Scholar]
- 23.Le Moine A., Flamand V., Demoor F.X., Noel J.C., Surquin M., Kiss R. Critical roles for IL-4, IL-5, and eosinophils in chronic skin allograft rejection. J Clin Invest. 1999;103:1659–1667. doi: 10.1172/JCI5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ensminger S.M., Spriewald B.M., Witzke O., Morrison K., van Maurik A., Morris P.J. Intragraft interleukin-4 mRNA expression after short-term CD154 blockade may trigger delayed development of transplant arteriosclerosis in the absence of CD8+ T cells. Transplantation. 2000;70:955–963. doi: 10.1097/00007890-200009270-00013. [DOI] [PubMed] [Google Scholar]
- 25.Ensminger S.M., Spriewald B.M., Sorensen H.V., Witzke O., Flashman E.G., Bushell A. Critical role for IL-4 in the development of transplant arteriosclerosis in the absence of CD40-CD154 costimulation. J Immunol. 2001;167:532–541. doi: 10.4049/jimmunol.167.1.532. [DOI] [PubMed] [Google Scholar]
- 26.Matesic D., Valujskikh A., Pearlman E., Higgins A.W., Gilliam A.C., Heeger P.S. Type 2 immune deviation has differential effects on alloreactive CD4+ and CD8+ T cells. J Immunol. 1998;161:5236–5244. [PubMed] [Google Scholar]
- 27.Honjo K., Xu X.Y., Bucy R.P. Heterogeneity of T cell clones specific for a single indirect alloantigenic epitope (I-Ab/H-2Kd54–68) that mediate transplant rejection. Transplantation. 2000;70:1516–1524. doi: 10.1097/00007890-200011270-00020. [DOI] [PubMed] [Google Scholar]
- 28.Surquin M., Le Moine A., Flamand V., Rombaut K., Demoor F.X., Salmon I. IL-4 deficiency prevents eosinophilic rejection and uncovers a role for neutrophils in the rejection of MHC class II disparate skin grafts. Transplantation. 2005;80:1485–1492. doi: 10.1097/01.tp.0000176486.01697.3f. [DOI] [PubMed] [Google Scholar]
- 29.Shalev O., Rubinger D., Barlatzky Y., Kopolovic J., Drukker A. Eosinophilia associated with acute allograft kidney rejection. Nephron. 1982;31:182–183. doi: 10.1159/000182641. [DOI] [PubMed] [Google Scholar]
- 30.Weir M.R., Hall-Craggs M., Shen S.Y., Posner J.N., Alongi S.V., Dagher F.J. The prognostic value of the eosinophil in acute renal allograft rejection. Transplantation. 1986;41:709–712. doi: 10.1097/00007890-198606000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Kormendi F., Amend W.J., Jr. The importance of eosinophil cells in kidney allograft rejection. Transplantation. 1988;45:537–539. doi: 10.1097/00007890-198803000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Nolan C.R., Saenz K.P., Thomas C.A., III, Murphy K.D. Role of the eosinophil in chronic vascular rejection of renal allografts. Am J Kidney Dis. 1995;26:634–642. doi: 10.1016/0272-6386(95)90601-0. [DOI] [PubMed] [Google Scholar]
- 33.Ten R.M., Gleich G.J., Holley K.E., Perkins J.D., Torres V.E. Eosinophil granule major basic protein in acute renal allograft rejection. Transplantation. 1989;47:959–963. doi: 10.1097/00007890-198906000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Hallgren R., Bohman S.O., Fredens K. Activated eosinophil infiltration and deposits of eosinophil cationic protein in renal allograft rejection. Nephron. 1991;59:266–270. doi: 10.1159/000186563. [DOI] [PubMed] [Google Scholar]
- 35.Meleg-Smith S., Gauthier P.M. Abundance of interstitial eosinophils in renal allografts is associated with vascular rejection. Transplantation. 2005;79:444–450. doi: 10.1097/01.tp.0000147318.48620.44. [DOI] [PubMed] [Google Scholar]
- 36.Hongwei W., Nanra R.S., Stein A., Avis L., Price A., Hibberd A.D. Eosinophils in acute renal allograft rejection. Transpl Immunol. 1994;2:41–46. doi: 10.1016/0966-3274(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 37.Scholma J., Slebos D.J., Boezen H.M., van den Berg J.W., van der B.W., de Boer W.J. Eosinophilic granulocytes and interleukin-6 level in bronchoalveolar lavage fluid are associated with the development of obliterative bronchiolitis after lung transplantation. Am J Respir Crit Care Med. 2000;162:2221–2225. doi: 10.1164/ajrccm.162.6.9911104. [DOI] [PubMed] [Google Scholar]
- 38.Mogayzel P.J., Jr., Yang S.C., Wise B.V., Colombani P.M. Eosinophilic infiltrates in a pulmonary allograft: a case and review of the literature. J Heart Lung Transplant. 2001;20:692–695. doi: 10.1016/s1053-2498(00)00218-7. [DOI] [PubMed] [Google Scholar]
- 39.Yousem S.A. Graft eosinophilia in lung transplantation. Hum Pathol. 1992;23:1172–1177. doi: 10.1016/0046-8177(92)90036-3. [DOI] [PubMed] [Google Scholar]
- 40.Stewart S., Fishbein M.C., Snell G.I., Berry G.J., Boehler A., Burke M.M. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26:1229–1242. doi: 10.1016/j.healun.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 41.Datta G.S., Hudson M., Burroughs A.K., Morris R., Rolles K., Amlot P. Grading of cellular rejection after orthotopic liver transplantation. Hepatology. 1995;21:46–57. [PubMed] [Google Scholar]
- 42.de Groen P.C., Kephart G.M., Gleich G.J., Ludwig J. The eosinophil as an effector cell of the immune response during hepatic allograft rejection. Hepatology. 1994;20:654–662. doi: 10.1016/0270-9139(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 43.Foster P.F., Sankary H.N., Williams J.W., Bhattacharyya A., Coleman J., Ashmann M. Morphometric inflammatory cell analysis of human liver allograft biopsies. Transplantation. 1991;51:873–876. doi: 10.1097/00007890-199104000-00026. [DOI] [PubMed] [Google Scholar]
- 44.Foster P.F., Bhattacharyya A., Sankary H.N., Coleman J., Ashmann M., Williams J.W. Eosinophil cationic protein’s role in human hepatic allograft rejection. Hepatology. 1991;13:1117–1125. [PubMed] [Google Scholar]
- 45.Foster P.F., Sankary H.N., Hart M., Ashmann M., Williams J.W. Blood and graft eosinophilia as predictors of rejection in human liver transplantation. Transplantation. 1989;47:72–74. doi: 10.1097/00007890-198901000-00016. [DOI] [PubMed] [Google Scholar]
- 46.Martinez O.M., Ascher N.L., Ferrell L., Villanueva J., Lake J., Roberts J.P. Evidence for a nonclassical pathway of graft rejection involving interleukin 5 and eosinophils. Transplantation. 1993;55:909–918. doi: 10.1097/00007890-199304000-00041. [DOI] [PubMed] [Google Scholar]
- 47.Lang T., Krams S.M., Berquist W., Cox K.L., Esquivel C.O., Martinez O.M. Elevated biliary interleukin 5 as an indicator of liver allograft rejection. Transpl Immunol. 1995;3:291–298. doi: 10.1016/0966-3274(95)80014-x. [DOI] [PubMed] [Google Scholar]
- 48.Hughes V.F., Trull A.K., Joshi O., Alexander G.J. Monitoring eosinophil activation and liver function after liver transplantation. Transplantation. 1998;65:1334–1339. doi: 10.1097/00007890-199805270-00009. [DOI] [PubMed] [Google Scholar]
- 49.Barnes E.J., Abdel-Rehim M.M., Goulis Y., Abou R.M., Davies S., Dhillon A. Applications and limitations of blood eosinophilia for the diagnosis of acute cellular rejection in liver transplantation. Am J Transplant. 2003;3:432–438. doi: 10.1034/j.1600-6143.2003.00083.x. [DOI] [PubMed] [Google Scholar]
- 50.Dollinger M.M., Plevris J.N., Bouchier I.A., Harrison D.J., Hayes P.C. Peripheral eosinophil count both before and after liver transplantation predicts acute cellular rejection. Liver Transpl Surg. 1997;3:112–117. doi: 10.1002/lt.500030203. [DOI] [PubMed] [Google Scholar]
- 51.Paczesny S., Hanauer D., Sun Y., Reddy P. New perspectives on the biology of acute GVHD. Bone Marrow Transplant. 2010;45:1–11. doi: 10.1038/bmt.2009.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferrara J.L., Levy R., Chao N.J. Pathophysiologic mechanisms of acute graft-vs.-host disease. Biol Blood Marrow Transplant. 1999;5:347–356. doi: 10.1016/s1083-8791(99)70011-x. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka J., Imamura M., Kasai M., Hashino S., Kobayashi S., Noto S. Th2 cytokines (IL-4, IL-10 and IL-13) and IL-12 mRNA expression by concanavalin A-stimulated peripheral blood mononuclear cells during chronic graft-versus-host disease. Eur J Haematol. 1996;57:111–113. doi: 10.1111/j.1600-0609.1996.tb00501.x. [DOI] [PubMed] [Google Scholar]
- 54.Martin P.J. Biology of chronic graft-versus-host disease: implications for a future therapeutic approach. Keio J Med. 2008;57:177–183. doi: 10.2302/kjm.57.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shulman H.M., Sullivan K.M., Weiden P.L., McDonald G.B., Striker G.E., Sale G.E. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69:204–217. doi: 10.1016/0002-9343(80)90380-0. [DOI] [PubMed] [Google Scholar]
- 56.Kalaycioglu M.E., Bolwell B.J. Eosinophilia after allogeneic bone marrow transplantation using the busulfan and cyclophosphamide preparative regimen. Bone Marrow Transplant. 1994;14:113–115. [PubMed] [Google Scholar]
- 57.Kim D.H., Popradi G., Xu W., Gupta V., Kuruvilla J., Wright J. Peripheral blood eosinophilia has a favorable prognostic impact on transplant outcomes after allogeneic peripheral blood stem cell transplantation. Biol Blood Marrow Transplant. 2009;15:471–482. doi: 10.1016/j.bbmt.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 58.Imahashi N., Miyamura K., Seto A., Watanabe K., Yanagisawa M., Nishiwaki S. Eosinophilia predicts better overall survival after acute graft-versus-host-disease. Bone Marrow Transplant. 2010;45:371–377. doi: 10.1038/bmt.2009.135. [DOI] [PubMed] [Google Scholar]
- 59.McNeel D., Rubio M.T., Damaj G., Emile J.F., Belanger C., Varet B. Hypereosinophilia as a presenting sign of acute graft-versus-host disease after allogeneic bone marrow transplantation. Transplantation. 2002;74:1797–1800. doi: 10.1097/00007890-200212270-00028. [DOI] [PubMed] [Google Scholar]
- 60.Fujii N., Hiraki A., Aoe K., Murakami T., Ikeda K., Masuda K. Serum cytokine concentrations and acute graft-versus-host disease after allogeneic peripheral blood stem cell transplantation: concurrent measurement of ten cytokines and their respective ratios using cytometric bead array. Int J Mol Med. 2006;17:881–885. [PubMed] [Google Scholar]
- 61.Sato T., Kobayashi R., Nakajima M., Iguchi A., Ariga T. Significance of eosinophilia after stem cell transplantation as a possible prognostic marker for favorable outcome. Bone Marrow Transplant. 2005;36:985–991. doi: 10.1038/sj.bmt.1705168. [DOI] [PubMed] [Google Scholar]
- 62.Imoto S., Oomoto Y., Murata K., Das H., Murayama T., Kajimoto K. Kinetics of serum cytokines after allogeneic bone marrow transplantation: interleukin-5 as a potential marker of acute graft-versus-host disease. Int J Hematol. 2000;72:92–97. [PubMed] [Google Scholar]
- 63.Aisa Y., Mori T., Nakazato T., Shimizu T., Yamazaki R., Ikeda Y. Blood eosinophilia as a marker of favorable outcome after allogeneic stem cell transplantation. Transpl Int. 2007;20:761–770. doi: 10.1111/j.1432-2277.2007.00509.x. [DOI] [PubMed] [Google Scholar]
- 64.Fowler D.H., Kurasawa K., Smith R., Eckhaus M.A., Gress R.E. Donor CD4-enriched cells of Th2 cytokine phenotype regulate graft-versus-host disease without impairing allogeneic engraftment in sublethally irradiated mice. Blood. 1994;84:3540–3549. [PubMed] [Google Scholar]
- 65.Pan L., Delmonte J., Jr., Jalonen C.K., Ferrara J.L. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood. 1995;86:4422–4429. [PubMed] [Google Scholar]
- 66.Foley J.E., Jung U., Miera A., Borenstein T., Mariotti J., Eckhaus M. Ex vivo rapamycin generates donor Th2 cells that potently inhibit graft-versus-host disease and graft-versus-tumor effects via an IL-4-dependent mechanism. J Immunol. 2005;175:5732–5743. doi: 10.4049/jimmunol.175.9.5732. [DOI] [PubMed] [Google Scholar]
- 67.Tawara I., Maeda Y., Sun Y., Lowler K.P., Liu C., Toubai T. Combined Th2 cytokine deficiency in donor T cells aggravates experimental acute graft-vs-host disease. Exp Hematol. 2008;36:988–996. doi: 10.1016/j.exphem.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weston L.E., Geczy A.F., Briscoe H. Production of IL-10 by alloreactive sibling donor cells and its influence on the development of acute GVHD. Bone Marrow Transplant. 2006;37:207–212. doi: 10.1038/sj.bmt.1705218. [DOI] [PubMed] [Google Scholar]
- 69.Takabayashi M., Kanamori H., Takasaki H., Yamaji S., Koharazawa H., Taguchi J. A possible association between the presence of interleukin-4-secreting cells and a reduction in the risk of acute graft-versus-host disease. Exp Hematol. 2005;33:251–257. doi: 10.1016/j.exphem.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 70.Nikolic B., Lee S., Bronson R.T., Grusby M.J., Sykes M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest. 2000;105:1289–1298. doi: 10.1172/JCI7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yi T., Chen Y., Wang L., Du G., Huang D., Zhao D. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. 2009;114:3101–3112. doi: 10.1182/blood-2009-05-219402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Basara N., Kiehl M.G., Fauser A.A. Eosinophilia indicates the evolution to acute graft-versus-host disease. Blood. 2002;100:3055. doi: 10.1182/blood-2002-04-1118. [DOI] [PubMed] [Google Scholar]
- 73.Daneshpouy M., Socie G., Lemann M., Rivet J., Gluckman E., Janin A. Activated eosinophils in upper gastrointestinal tract of patients with graft-versus-host disease. Blood. 2002;99:3033–3040. doi: 10.1182/blood.v99.8.3033. [DOI] [PubMed] [Google Scholar]
- 74.Rumi C., Rutella S., Bonini S., Bonini S., Lambiase A., Sica S. Immunophenotypic profile of peripheral blood eosinophils in acute graft-vs.-host disease. Exp Hematol. 1998;26:170–178. [PubMed] [Google Scholar]
- 75.Daneshpouy M., Facon T., Jouet J.P., Janin A. Acute flare-up of conjunctival graft-versus-host disease with eosinophil infiltration in a patient with chronic graft-versus-host disease. Leuk Lymphoma. 2002;43:445–446. doi: 10.1080/10428190290006323. [DOI] [PubMed] [Google Scholar]
- 76.Or R., Gesundheit B., Resnick I., Bitan M., Avraham A., Avgil M. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation. 2007;83:577–581. doi: 10.1097/01.tp.0000255575.03795.df. [DOI] [PubMed] [Google Scholar]
- 77.Janin A., Ertault-Daneshpouy M., Socie G. Eosinophils and severe forms of graft-versus-host disease. Blood. 2003;101:2073. doi: 10.1182/blood-2002-12-3650. [DOI] [PubMed] [Google Scholar]
- 78.Lu L.F., Lind E.F., Gondek D.C., Bennett K.A., Gleeson M.W., Pino-Lagos K. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 79.Lee J.J., Dimina D., Macias M.P., Ochkur S.I., McGarry M.P., O’Neill K.R. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;17:1773–1776. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 80.Yu C., Cantor A.B., Yang H., Browne C., Wells R.A., Fujiwara Y. Deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J Exp Med. 2002;195:1387–1395. doi: 10.1084/jem.20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chapter 13.14. Eosinophils and Calpain-3 Mutation: A Genetic Cause Implicated in Idiopathic Eosinophilic Myositis
Martin Krahn, Marc Bartoli, Nicolas Levy
Eosinophilic myositis is a rare histopathological entity characterized by infiltration of skeletal muscle tissue by eosinophils, possibly in association with peripheral blood and/or bone marrow hypereosinophilia. Eosinophilic myositis may be associated with parasite infections, systemic disorders, or toxic causes. The exclusion of these different etiologies determines the diagnosis of idiopathic eosinophilic myositis. Calpain-3 gene mutations have been identified as the first genetic change involved in eosinophilic myositis, whereas eosinophilic infiltration is not known to be a typical feature of limb-girdle muscular dystrophy type 2A, also caused by CAPN3 mutations. Different reports now raise the question of transient eosinophilic infiltration being a possible feature in a wider range of muscular dystrophies.
Eosinophilic Myositis
Eosinophilic myositis is a rare histopathological entity characterized by infiltration of skeletal muscle tissue by eosinophils, possibly in association with peripheral blood and/or bone marrow hypereosinophilia. These characteristics distinguish eosinophilic myositis from other idiopathic inflammatory myopathies, such as polymyositis and dermatomyositis. The diagnosis of eosinophilic myositis is based on the histological examination of muscle biopsy sections. Infiltration of skeletal muscle tissue by eosinophils is an unusual event, observed especially during parasite infections (including Taenia solium, Trichinella spiralis, and sarcocystosis)1 or more rarely bacterial infections (borreliosis). Several immune disorders, such as sarcoidosis or rheumatoid arthritis, may also be accompanied by eosinophilic infiltration of the skeletal muscle tissue. These forms must be identified as they may benefit from specific therapeutic management. Additionally, some toxic causes have been implicated in the formation of eosinophilic infiltrates in muscle tissue. Those include, in particular, the ingestion of certain plant oils (which caused Spanish toxic oil syndrome in 1981)2 and the eosinophilia-myalgia syndrome, caused by the ingestion of L-tryptophan and presenting with a histological aspect resembling eosinophilic fasciitis, but associated with multisystemic manifestations.3., 4.
Idiopathic Eosinophilic Myositis
The exclusion of the aforementioned different etiologies determines the diagnosis of idiopathic eosinophilic myositis. Depending on the localization of the eosinophilic infiltrate, idiopathic eosinophilic myositis can be classified into three subgroups (reviewed in5., 6.): focal eosinophilic myositis, eosinophilic polymyositis, and eosinophilic perimyositis.
Focal Eosinophilic Myositis
Focal eosinophilic myositis includes eosinophilic infiltration of muscle tissue with invasion of muscle fibers, and is associated with necrotic fibers.5 Clinically, myopathy preferentially affects the lower limbs, without involvement of skin or fascia. Eosinophilia is usually observed, and is associated with elevated serum creatine phosphokinase (CPK) levels. Spontaneous or corticosteroid-induced recovery may be observed, but with frequent relapses.
Eosinophilic Polymyositis
At the histological level, eosinophilic polymyositis combines diffuse eosinophilic infiltration of the muscle tissue and at perivenular locations, associated with necrotic fibers.5., 7. Unlike focal eosinophilic myositis, infiltration is instead located at the perimysium, without muscle fiber invasion. Clinical presentation associates myositis with severe systemic symptoms, including possible cardiac and skin involvement. Myopathy is preferentially proximal and high serum CPK levels are observed, reflecting extensive muscle damage. Corticosteroid treatment can allow recovery, but with possible relapses if not continued long term.
Eosinophilic Perimyositis
In eosinophilic perimyositis, infiltrates predominate at the superficial fascia and perimysium.5., 8. There is usually no damage to muscle fibers and, in particular, no necrosis. Clinically, a prodromal phase (abdominal pain, arthralgia, and fever) precedes muscle damage, which involves preferential impairment of lower limbs with localized induration. Eosinophilia is rare, and serum CPK levels are usually normal. The evolution can be spontaneously favorable.
Shulman's Syndrome
Shulman’s syndrome,9 or eosinophilic fasciitis, is a distinct entity characterized by eosinophilic infiltration of the deep fascia, without systemic manifestations, and about half the cases are responsive to corticosteroid treatment.10
A Genetic Cause for a Subset of Idiopathic Eosinophilic Myositis
At the current state of knowledge, eosinophilic myopathies constitute a heterogeneous group of rare diseases without a causal factor being identified in most cases.
In 2006, our group reported an unexpected clinical observation: a 4-year-old boy was diagnosed with idiopathic eosinophilic myositis and muscle biopsy analysis revealed a calpain-3 protein defect.11 Calpain-3 is a muscle-specific protein, belonging to the family of calpains, nonlysosomal calcium-dependent cysteine proteases.12 The most well studied of the calpains are the ubiquitous heterodimeric calpains (μ-calpain and m-calpain). The human calpain-3 gene is located on chromosome 15q15.1–q21.1. The predominant product of this gene is encoded by 24 exons corresponding to a 3316 bp mRNA expressed mainly in adult skeletal muscles.13
The translation of the main calpain-3 gene (CAPN3) product leads to the formation of a 94 kDa protein comprising 821 amino acids and consisting of a short N-terminal region (domain I), a papain-type proteolytic domain (domains IIa and IIb), a C2-like domain (domain III), and a calcium-binding domain composed of five EF-hand motifs (domain IV). In addition, calpain-3 possesses three unique sequences not found in other calpains: the NS (N-terminal sequence), and the IS1 and IS2 (inserted sequences 1 and 2) sequences.14 IS1 is a polypeptide of about 50 amino acids encoded by an alternative exon 6. It is composed of an α-helix flanked by loops that close the catalytic cleft, thus blocking access to substrates and inhibitors.15 It also contains autolytic sites involved in the initiation of calpain-3 proteolysis by opening the catalytic cleft.16 Immunolocalization studies carried out in human and mouse demonstrated that calpain-3 located in the N2A, M-line, and Z-line regions of the sarcomere. In addition to these localizations, calpain-3 has also been found at costameres and near the triad of the T-tubule.17 Calpain-3 functions to promote proteolysis of several substrates located in the costameres, sarcolemma, and sarcomere and seems to be most important in fully differentiated fibers (for review see14). In adult fibers, calpain-3 participates in sarcomere adaptation by cleaving cytoskeletal proteins during muscular adjustment, in accordance with the distribution of known substrates.17
Mutations in the CAPN3 gene cause the most prevalent form of autosomal recessive limb-girdle muscular dystrophy (LGMD), type 2A (LGMD2A),18 also referred to as calpainopathy. Based on the calpain-3 protein defect identified in the boy with idiopathic eosinophilic myositis mentioned above, the CAPN3 gene was analyzed, revealing mutations and thus a genetic cause associated with eosinophilic infiltration in this case. Nonspecific inflammatory features may be associated with a variety of muscle dystrophies, possibly leading to misdiagnosis of polymyositis in some cases (e.g., congenital muscle dystrophy, fascioscapulohumeral muscular dystrophy, and dysferlinopathies). However, eosinophilic infiltration had not been previously characterized as a component of inflammatory features in muscular dystrophies.
We subsequently identified five additional children diagnosed with idiopathic eosinophilic myositis (exemplified in one patient in Fig. 13.14.1) and identified CAPN3 disease-causing mutations in all cases, either in a homozygous or compound heterozygous state. Following our publication of these pediatric cases, two adult cases of idiopathic eosinophilic myositis and CAPN3 mutations were reported by Amato.19 In 2009, Oflazer and colleagues reported another pediatric case of idiopathic eosinophilic myositis associated with CAPN3 mutations,20 in which a positive effect of immunosuppressive therapy was observed. Since our initial report, we have characterized five additional unrelated patients with idiopathic eosinophilic myositis and CAPN3 mutations (one adult and four children, unpublished data). Importantly, except for the initially identified boy presenting with a calpain-3 defect, inclusion criteria for CAPN3 mutation screening of the other cases were based only on the particular histopathological presentation, without any identified etiological factors.
FIGURE 13.14.1.
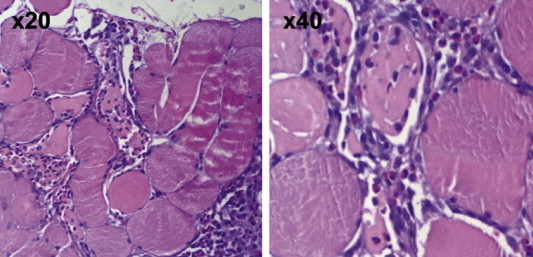
Eosinophilic myositis caused by CAPN3 mutations in a pediatric patient.
Depicted are histological findings (hematoxylin and eosin staining) on muscle biopsy samples evidencing mild myopathic changes with focal inflammatory lesions including abundant eosinophilic infiltration, involving necrotic fibers.
(Reproduced with permission from Krahn et al., 2006; Annals of Neurology; Wiley.)
Noteworthy, CAPN3 mutations identified in patients with idiopathic eosinophilic myositis do not appear to constitute any particular mutational spectrum as compared to typical LGMD2A. Our findings demonstrate that at least a subset of idiopathic eosinophilic myositis has a genetic origin, caused by mutations in the CAPN3 gene and with an autosomal recessive mode of inheritance. On the other hand, as eosinophilic infiltration is not known to be a typical feature of LGMD2A, it is possible that eosinophilia may be a transient feature in the natural course of this disease.
The explanation for eosinophilic infiltration correlating with defective calpain-3 needs to be further evaluated. T lymphocytes may be a key component in this process21 as:
-
1.
Together with macrophages, they are the main components of inflammatory lesions in the vicinity of damaged muscle fibers.
-
2.
They play a central role in the chemoattraction of eosinophils by secreting interleukin-5, which induces local eosinophil accumulation22 and they express calpain-3.23
Regarding the latter, no relevant function for calpain-3 in T lymphocytes has been identified to date. The presence of eosinophils has been previously reported to be a component of muscular dystrophy in mdx mice, promoted by perforin-dependent cytotoxicity of effector T cells.24 In addition, eosinophils play a specific role in muscle fiber degradation, due to degradation of myofibrillar and membrane-associated proteins by eosinophil cationic protein.25
The identification of CAPN3 mutations as the first genetic cause involved in eosinophilic myositis indicates that mutations in other genes may also be causal, or act as modifiers, of this pathophysiology. In this regard, a case of dystrophinopathy in which a muscle biopsy shows the appearance of eosinophilic myositis has been described,26 and Baumeister and coworkers recently reported a case of idiopathic eosinophilic myositis caused by a homozygous mutation in the γ-sarcoglycan gene,27 which is implicated in another form of LGMD. Interestingly, these reports suggest that early, but transient, eosinophilic infiltration may be a feature of a wider range of muscular dystrophies. The role of eosinophils in the natural history of diverse muscular dystrophies should therefore be further investigated, and it is possible that eosinophils may represent a novel therapeutic target.
References
- 1.Bocanegra T.S., Vasey F.B. Musculoskeletal syndromes in parasitic diseases. Rheum Dis Clin North Am. 1993 May;19(2):505–513. [PubMed] [Google Scholar]
- 2.Kilbourne E.M., Rigau-Perez J.G., Heath C.W., Jr., Zack M.M., Falk H., Martin-Marcos M. Clinical epidemiology of toxic-oil syndrome. Manifestations of a New Illness. N Engl J Med. 1983 Dec 8;309(23):1408–1414. doi: 10.1056/NEJM198312083092302. [DOI] [PubMed] [Google Scholar]
- 3.Hertzman P.A., Blevins W.L., Mayer J., Greenfield B., Ting M., Gleich G.J. Association of the eosinophilia-myalgia syndrome with the ingestion of tryptophan. N Engl J Med. 1990 Mar 29;322(13):869–873. doi: 10.1056/NEJM199003293221301. [DOI] [PubMed] [Google Scholar]
- 4.Hollander D., Adelman L.S. Eosinophilia-myalgia syndrome associated with ingestion of L-tryptophan: muscle biopsy findings in 4 patients. Neurology. 1991 Feb;41 doi: 10.1212/wnl.41.2_part_1.319. (2(Pt 1)):319–21. [DOI] [PubMed] [Google Scholar]
- 5.Hall F.C., Krausz T., Walport M.J. Idiopathic eosinophilic myositis. QJM. 1995 Aug;88(8):581–586. [PubMed] [Google Scholar]
- 6.Pickering M.C., Walport M.J. Eosinophilic myopathic syndromes. Curr Opin Rheumatol. 1998 Nov;10(6):504–510. doi: 10.1097/00002281-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Layzer R.B., Shearn M.A., Satya-Murti S. Eosinophilic polymyositis. Ann Neurol. 1977 Jan;1(1):65–71. doi: 10.1002/ana.410010106. [DOI] [PubMed] [Google Scholar]
- 8.Serratrice G., Pellissier J.F., Cros D., Gastaut J.L., Brindisi G. Relapsing eosinophilic perimyositis. J Rheumatol. 1980 Mar-Apr;7(2):199–205. [PubMed] [Google Scholar]
- 9.Shulman L.E. Diffuse fasciitis with eosinophilia: a new syndrome? Trans Assoc Am Physicians. 1975;88:70–86. [PubMed] [Google Scholar]
- 10.Barnes L., Rodnan G.P., Medsger T.A., Short D. Eosinophilic fasciitis. A pathologic study of twenty cases. Am J Pathol. 1979 Aug;96(2):493–518. [PMC free article] [PubMed] [Google Scholar]
- 11.Krahn M., Lopez de Munain A., Streichenberger N., Bernard R., Pécheux C., Testard H. CAPN3 mutations in patients with idiopathic eosinophilic myositis. Ann Neurol. 2006 Jun;59(6):905–911. doi: 10.1002/ana.20833. [DOI] [PubMed] [Google Scholar]
- 12.Goll D.E., Thompson V.F., Li H., Wei W., Cong J. The calpain system. Physiol Rev. 2003 Jul;83(3):731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 13.Sorimachi H., Suzuki K. Sequence comparison among muscle-specific calpain, p94, and calpain subunits. Biochim Biophys Acta. 1992 Nov 10;1160(1):55–62. doi: 10.1016/0167-4838(92)90037-e. [DOI] [PubMed] [Google Scholar]
- 14.Duguez S., Bartoli M., Richard I. Calpain 3: a key regulator of the sarcomere? FEBS J. 2006 Aug;273(15):3427–3436. doi: 10.1111/j.1742-4658.2006.05351.x. [DOI] [PubMed] [Google Scholar]
- 15.Diaz B.G., Moldoveanu T., Kuiper M.J., Campbell R.L., Davies P.L. Insertion sequence 1 of muscle-specific calpain, p94, acts as an internal propeptide. J Biol Chem. 2004 Jun 25;279(26):27656–27666. doi: 10.1074/jbc.M313290200. [DOI] [PubMed] [Google Scholar]
- 16.Ono Y., Torii F., Ojima K., Doi N., Yoshioka K., Kawabata Y. Suppressed disassembly of autolyzing p94/CAPN3 by N2A connectin/titin in a genetic reporter system. J Biol Chem. 2006 Jul 7;281(27):18519–18531. doi: 10.1074/jbc.M601029200. [DOI] [PubMed] [Google Scholar]
- 17.Taveau M., Bourg N., Sillon G., Roudaut C., Bartoli M., Richard I. Calpain 3 is activated through autolysis within the active site and lyses sarcomeric and sarcolemmal components. Mol Cell Biol. 2003 Dec;23(24):9127–9135. doi: 10.1128/MCB.23.24.9127-9135.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richard I., Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., Bourg N. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995 Apr 7;81(1):27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 19.Amato A.A. Adults with eosinophilic myositis and calpain-3 mutations. Neurology. 2008 Feb 26;70(9):730–731. doi: 10.1212/01.wnl.0000287138.89373.6b. [DOI] [PubMed] [Google Scholar]
- 20.Oflazer P.S., Gundesli H., Zorludemir S., Sabuncu T., Dincer P. Eosinophilic myositis in calpainopathy: could immunosuppression of the eosinophilic myositis alter the early natural course of the dystrophic disease? Neuromuscul Disord. 2009 Apr;19(4):261–263. doi: 10.1016/j.nmd.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Brown R.H., Jr., Amato A. Calpainopathy and eosinophilic myositis. Ann Neurol. 2006 Jun;59(6):875–877. doi: 10.1002/ana.20900. [DOI] [PubMed] [Google Scholar]
- 22.Murata K., Sugie K., Takamure M., Fujimoto T., Ueno S. Eosinophilic major basic protein and interleukin-5 in eosinophilic myositis. Eur J Neurol. 2003 Jan;10(1):35–38. doi: 10.1046/j.1468-1331.2003.00481.x. [DOI] [PubMed] [Google Scholar]
- 23.De Tullio R., Stifanese R., Salamino F., Pontremoli S., Melloni E. Characterization of a new p94-like calpain form in human lymphocytes. Biochem J. 2003 Nov 1;375(Pt 3):689–696. doi: 10.1042/BJ20030706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai B., Spencer M.J., Nakamura G., Tseng-Ong L., Tidball J.G. Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. Am J Pathol. 2000 May;156(5):1789–1796. doi: 10.1016/S0002-9440(10)65050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugihara R., Kumamoto T., Ito T., Ueyama H., Toyoshima I., Tsuda T. Human muscle protein degradation in vitro by eosinophil cationic protein (ECP) Muscle Nerve. 2001 Dec;24(12):1627–1634. doi: 10.1002/mus.1198. [DOI] [PubMed] [Google Scholar]
- 26.Weinstock A., Green C., Cohen B.H., Prayson R.A. Becker muscular dystrophy presenting as eosinophilic inflammatory myopathy in an infant. J Child Neurol. 1997 Feb;12(2):146–147. doi: 10.1177/088307389701200214. [DOI] [PubMed] [Google Scholar]
- 27.Baumeister S.K., Todorovic S., Milic-Rasic V., Dekomien G., Lochmuller H., Walter M.C. Eosinophilic myositis as presenting symptom in γ-sarcoglycanopathy. Neuromuscul Disord. 2009 Feb;19(2):167–171. doi: 10.1016/j.nmd.2008.11.010. [DOI] [PubMed] [Google Scholar]