

Abstract
Activation of the transcription factor estrogen receptor α (ERα) and the subsequent regulation of estrogen-responsive genes play a crucial role in the development and progression of the majority of breast cancers. One gene target of ERα, growth regulation by estrogen in breast cancer 1 (GREB1), is associated with proliferation and regulation of ERα activity in estrogen-responsive breast cancer cells. The GREB1 gene encodes three distinct isoforms: GREB1a, GREB1b and GREB1c, whose molecular functions are largely unknown. Here, we investigate the role of these isoforms in regulation of ERα activity and proliferation. Interaction between GREB1 and ERα was mapped to the amino terminus shared by all GREB1 variants. Analysis of isoform-specific regulation of ERα activity suggests none of the GREB1 isoforms possess potent co-regulator activity. Exogenous expression of GREB1a resulted in elevated expression of some ER-target genes, independent of ERα activity. Despite this slight specificity of GREB1a for gene regulation, exogenous expression of either GREB1a or GREB1b resulted in decreased proliferation in both ER-positive and ER-negative breast carcinoma cell lines, demonstrating an ER-independent function of GREB1. Interestingly, we show an increase in the expression of GREB1b and GREB1c mRNA in malignant breast tissue compared to normal patient samples, suggesting a selective preference for these isoforms during malignant transformation. Together, these data suggest GREB1a has an isoform-specific function as a transcriptional regulator while all isoforms share an ER-independent activity that regulates proliferation.
Keywords: breast cancer, estrogen receptor, proliferation, gene expression
Introduction
The majority (~70%) of breast cancers rely on estrogen receptor (ER) activity for cell growth and survival (Ali & Coombes 2002, Dixon 2014, Tryfonidis et al. 2016). Once activated by estrogens, ER translocates to the nucleus where it dimerizes on estrogen response elements (EREs) to regulate the transcription of target genes (Heldring et al. 2007). Two ER isoforms exist (α and β), each encoded by distinct genes (Heldring et al. 2007). In estrogen-dependent breast cancer, the activation of ERα ultimately leads to proliferation (Dixon 2014). ER-positive breast cancers are treated with endocrine therapies that disrupt the activity of ERα (Heldring et al. 2007). Unfortunately, patients develop resistance to endocrine therapies through re-activation of ERα, allowing for estrogen-responsive gene expression and progression of the disease (Ali & Coombes 2002, Heldring et al. 2007, Dixon 2014, Clarke et al. 2015, Tryfonidis et al. 2016). Therefore, a better understanding of how gene targets of ERα allow for increased proliferation of breast cancer cells is needed in order to develop new and innovative therapies.
One primary gene target of ERα, growth regulation by estrogen in breast cancer 1 (GREB1), is of particular interest in the modulation of proliferation in ER-positive breast cancer. Initially identified as a gene upregulated by estrogen and repressed by an ERα antagonist in ER-positive breast cancer cells, GREB1 expression is controlled by three distal EREs (Ghosh et al. 2000, Deschenes et al. 2007, Sun et al. 2007). The expression of GREB1 is highly correlated to ER-positivity in breast cancer cell lines and patient tumor samples, suggesting a functional role for this protein in ER-positive breast cancer (Ghosh et al. 2000, Rae et al. 2005, Hnatyszyn et al. 2010, Mohammed et al. 2013). Loss of GREB1 expression in the estrogen-dependent MCF7 breast cancer cell line reduces proliferation and anchorage-independent growth, suggesting GREB1 is essential for hormone-dependent proliferation in ER-positive breast cancer cells (Rae et al. 2005, Mohammed et al. 2013).
A previous study suggested that GREB1 may act as a co-factor for ERα, interacting preferentially with agonist-bound ERα (Mohammed et al. 2013). Chromatin immunoprecipitation experiments demonstrated that over 95% GREB1 chromatin-binding sites overlapped with ERα chromatin-binding sites (Mohammed et al. 2013). These data suggest that GREB1 has the potential to regulate the activity of ERα through modulation of co-factor binding.
Despite evidence suggesting an important functional role of GREB1 in breast cancer, no study has differentiated between the three distinct protein isoforms encoded by the GREB1 gene: GREB1a, GREB1b and GREB1c (Ghosh et al. 2000). Each transcript is estrogen dependent and contains a unique 5′ untranslated region, which splices to a conserved exon that encodes the translational start site (Ghosh et al. 2000). Thus, the amino terminus of the three protein isoforms is identical. The transcripts for GREB1b and GREB1c differ from GREB1a due to alternative splicing after exon 10 and exon 9, respectively (Ghosh et al. 2000). In each instance, the alternative exon encodes a stop codon resulting in truncated versions of the full-length isoform, GREB1a (Supplementary Fig. 1, see section on supplementary data given at the end of this article). Despite the alternative splicing events, GREB1b and GREB1c contain only 8 and 23 unique amino acids, respectively (Supplementary Fig. 1). None of the GREB1 isoforms have any homology to other proteins or known functional domains that may suggest their molecular function and the GREB1b and GREB1c isoforms have never been investigated.
Here, we set out to better characterize the contribution of the three different GREB1 isoforms to the modulation of ERα activity and proliferation in breast cancer cell lines. To this end, we mapped the binding of GREB1 to ERα protein. Despite the interaction of all GREB1 isoforms with ERα protein, none of the GREB1 isoforms potently regulate ERα transcriptional activity. Further, we show that both GREB1a and GREB1b have the ability to regulate proliferation of breast cancer cell lines independent of ERα expression. These data suggest that GREB1 has additional molecular functions beyond acting as a transcriptional co-regulator of ERα.
Materials and methods
Cell lines and reagents
HEK-293AD, HEK-293T, MCF7, T47D, MDA-MB-231 and MDA-MB-468 cells were validated using Short Tandem Repeat analysis by the Genomics Core in the Research Technology Support Facility (Michigan State University, East Lansing, MI, USA). Cell lines were maintained in DMEM supplemented with phenol red (Gibco by Life Technologies), 5% (vol/vol) fetal bovine serum (FBS; Sigma), 1% (vol/vol) penicillin-streptomycin (Corning) and 2 mM L-glutamine (HyClone, GE Healthcare). For hormone-free conditions, cells were cultured in phenol-red-free DMEM (Gibco by Life Technologies) supplemented with 5% (vol/vol) charcoal-dextran-treated FBS (CDT, Sigma), 1% (vol/vol) penicillin-streptomycin and 2 mM l-glutamine. Cells were treated with either vehicle control (ethanol) or 10 nM estradiol (E2; Sigma) for the indicated time.
Plasmids
pcDNA-ERα, H2B-GFP, 3XERE-Luciferase and PS2-Luciferase have been previously described (Zacharewski et al. 1994, Norris et al. 1997, Hall & Korach 2002, Burd et al. 2005). GREB1a, GREB1b and GREB1c cDNA were amplified from MCF7 reverse-transcribed RNA and cloned into pJET 2.1 vector (Thermo). GREB1a, GREB1b and GREB1c inserts were removed from pJet2.1 vectors by restriction digestion and inserted into pcDNA 3.1 vector (Thermo) with a 3XFLAG coding sequence in front of the multiple cloning site. pcDNA 3XFLAG-GREB1 (1–500), (492–992), (984–1477) and (1469–1949) were generated by PCR amplification of the specific fragments from pcDNA 3XFLAG-GREB1a and inserted into pcDNA 3XFLAG via Gibson cloning (NEB, Iswich, MA, USA). GIPZ Lentiviral non-specific shRNA (# RHS4346) and GREB1-targeted shRNA plasmids (V2LHS_139192 and V3LHS_372339) were purchased from Open Biosystems. CMV-Renilla luciferase reporter construct was purchased from Promega.
Immunoblot analysis and antibodies
Cells were lysed in Buffer E (10 mM Tris-HCl, pH 8.0, 60 mM NaCl, 1 mM EDTA, 0.3% IGEPAL) with added protease inhibitors (Sigma, P8340). Lysates were incubated with Laemmli buffer at 37°C for 30 min as incubation of cell lysates at higher than 55°C causes GREB1a to aggregate (Supplementary Fig. 2). Lysates were subjected to SDS-PAGE and immunoblots visualized using Licor Odyssey as previously described (Patterson et al. 2015). Immunoblots were probed with the following antibodies: GREB1 (Abcam; ab72999), FLAG (Sigma; F1804), ERα (GeneTex, Irvine, CA, USA; GTX62423), ERα (Santa Cruz; SC-8005) and β-actin (Cell Signaling; 3700).
Adenovirus
GREB1a and GREB1b were moved from 3XFLAG plasmids to a pshuttle-IRES GFP 3XFLAG plasmid (Agilent). Shuttle vectors were recombined with pAdeasy using BJ5183-AD1 bacteria (Agilent). Adenovirus was produced and amplified in HEK-293AD cells (Agilent) then purified by CsCl gradient. Ad5-CMV-eGFP adenovirus (Baylor College of Medicine Vector Development Labs, Houston, Texas) was used as a control.
Immunoprecipitation
Endogenous GREB1 was immunoprecipitated from MCF7 cells. Individual isoforms or fragments were immunoprecipitated from HEK-293AD cells transfected with H2BGFP, ERα and indicated GREB1 plasmids using PEI transfection reagent (Polysciences, Inc., Warrington, PA, USA). Cells were lysed in Buffer E and immunoprecipitation was performed with anti-FLAG M2 Magnetic Beads (Sigma, M8823) or GREB1 antibody with PureProteome Protein A magnetic beads (Abcam; ab72999 and EMD Millipore). Beads were washed in 250 mM NaCl Buffer E, incubated with Laemmli buffer at 37°C for 30 min, and immunoblot analysis performed as described.
Gene expression analysis
Total RNA was harvested using RiboZol (VWR, Radnor, PA, USA) and reverse transcribed using the RevertAid RT Kit (Thermo). Relative gene expression was analyzed via real-time PCR using SYBR Green (Bio-Rad) and the indicated primers (Supplementary Table 1). Data are relative to RPL13a, normalized to control and are depicted as mean ± s.e. Data represent a minimum of three biological replicates, and statistical significance was determined using either a one-way ANOVA with post hoc Tukey’s HSD test (knockdown experiment) or two-way ANOVA with a post hoc Tukey’s HSD test (estrogen-induced gene expression experiments).
GREB1 isoform expression
Tissue samples were provided by the ‘Total Cancer Care Protocol: A Lifetime Partnership with Patients of the James/OSUCCC’, IRB protocol 2013H0197 (The James Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio). RNA was harvested using RiboZol (Amresco) and was converted to cDNA using the RevertAid RT Kit (Thermo). Absolute mRNA copy number was determined from a standard curve generated using TaqMan probes for GREB1a, GREB1b and GREB1c (Supplementary Table 2). The Kruskal–Wallis multiple comparison method was used to determine statistical significance.
Cancer RNA-seq Nexus (Li et al. 2016) was used to determine mRNA levels of GREB1a, GREB1b and GREB1c from a publically available data set of transcript expression from breast cancer patients (Varley et al. 2014).
Reporter assay
HEK-293AD cells were cultured in hormone-depleted media and transfected with CMV-Renilla, PS2-Luciferase or 3XERE-Luciferase, pcDNA-ERα and pcDNA 3XFLAG (EV), pcDNA 3XFLAG-GREB1a or pcDNA 3XFLAG-GREB1b. The following day, cells were treated with 10 nM estradiol (E2) or vehicle control (ethanol) for 48 h. Lysates were harvested and analyzed using the Dual-Luciferase kit (Promega). Data represent three biological replicates and statistical significance was determined using a two-way ANOVA with a post hoc Tukey’s HSD test.
MTT assays
Cells transduced with GFP, GREB1a or GREB1b adenovirus were treated with 5 mg/mL MTT solution (Sigma) at 37°C for 3 h. Formazan was solubilized in 4 mM HCl, 0.1% IGEPAL in isopropanol and absorbance was measured at 570 nm. Data are depicted as mean absorbance normalized to day 0 ± s.d. for each condition from three biological replicates. Statistical significance for MTT assays was determined using either a one-way ANOVA with post hoc Tukey’s HSD test (shRNA experiments), two-way ANOVA with a post hoc Tukey’s HSD test (exogenous expression in presence and absence of hormone) or a two-tailed Student’s t-test (exogenous expression in cell line panels).
EdU incorporation assay
MCF7 cells were transduced with GFP, GREB1a, or GREB1b adenovirus. Proliferation was measured after 6 h of EdU labeling using the Click-iT EdU Imaging Kit (Invitrogen) following the manufacturers protocol. Images of EdU-stained cells were blindly quantified and statistical significance was determined using a one-way ANOVA with a post hoc Tukey’s HSD test.
Cell viability assay
MCF7 cells were transduced with GFP, GREB1a or GREB1b adenovirus. After 72 h, the cells were harvested and stained with a LIVE/Dead Fixable Dead Cell Stain Kit (Invitrogen). Cells were analyzed on a LSR II flow cytometer (BD Biosciences). Unstained MCF7 cells and stained, heat-treated MCF7 cells served as controls. Statistical significance was determined using one-way ANOVA with a post hoc Tukey’s HSD test.
Results
The amino terminus of GREB1 isoforms interact with ERα
To confirm previous studies indicating that GREB1 interacts with ERα(Mohammed et al. 2013), we performed immunoprecipitations from MCF7 breast carcinoma whole-cell lysate. As previously reported (Mohammed et al. 2013), immunoblot analysis revealed an interaction between GREB1a and ERα (Fig. 1A). To map this interaction, plasmids expressing roughly 500 amino acid fragments of GREB1a tagged with 3XFLAG (Fig. 1B) were transfected with ERα plasmid into HEK-293AD cells and immunoprecipitated from whole-cell lysate with anti-FLAG magnetic beads. Immunoblot analysis showed Fragment #1 (60 kDa) immunoprecipitated with ERα, while no interaction was observed with Fragment #2 (56 kDa), which contains the LxxLL motif known to bind nuclear receptors, Fragment #3 (62 kDa), nor Fragment #4 (56 kDa) (Fig. 1C). These data suggest that the amino terminus of GREB1a interacts with ERα.
Figure 1.
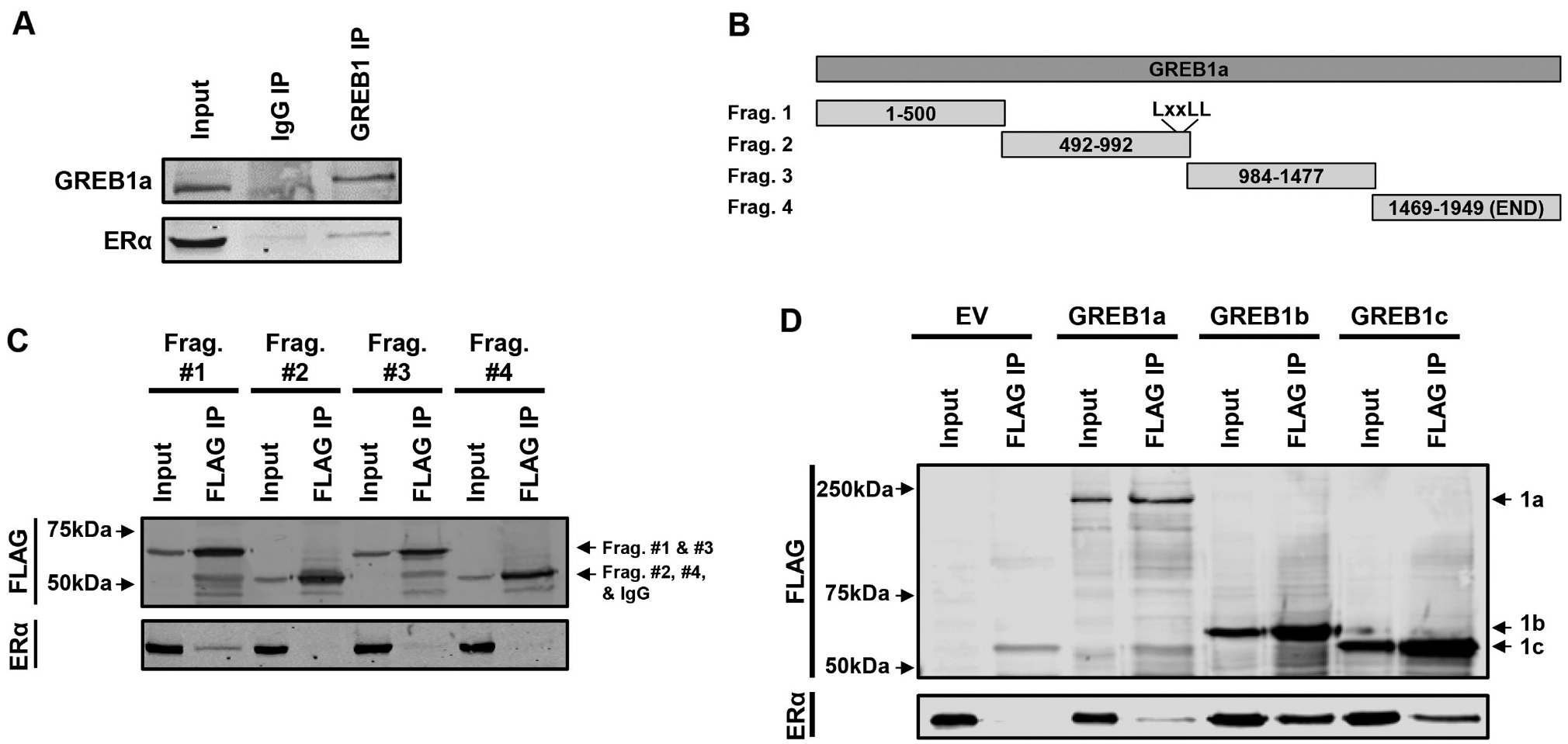
The amino terminus of GREB1 isoforms interact with ERα. (A) Endogenous GREB1 was immunoprecipitated from MCF7 whole-cell lysate and purified complexes were subjected to immunoblot analysis with GREB1 and ERα antibodies. (B) 3xFLAG-tagged fragments of GREB1a were generated according to the schematic. (C) Plasmids expressing the individual fragments were co-transfected with ERα into HEK-293AD cells and immunoprecipitated from whole-cell lysate using anti-FLAG magnetic beads. Immunoblot analysis of input and FLAG IP lysates was performed with FLAG and ERα antibodies. (D) 3xFLAG-tagged GREB1a, GREB1b, GREB1c or empty vector (EV) was co-transfected with ERα into HEK-293AD cells. Immunoprecipitation with anti-FLAG magnetic beads was followed by immunoblot analysis of input and FLAG IP lysates with FLAG and ERα antibodies.
The interaction between ERα and the amino terminus of GREB1 was particularly interesting as this region is shared by all three isoforms of GREB1 (Supplementary Fig. 1). To determine if all three isoforms of GREB1 are able to bind to ERα each isoform or empty vector was co-transfected with ERα plasmid into HEK-293AD cells. Using FLAG antibody, each isoform was immunoprecipitated from whole-cell lysate and subjected to immunoblot analysis. While ERα was not pulled-down with the empty vector control (EV), 3XFLAG-GREB1a (228 kDa), 3XFLAG-GREB1b (56 kDa) and 3XFLAG-GREB1c (49 kDa) were all able to pull down ERα (Fig. 1D). These data indicate that all three protein isoforms of GREB1 are able to interact with ERα.
GREB1 isoforms are not robust regulators of ERα activity
Previous studies have suggested that GREB1 expression is an essential co-regulator of ERα activity (Mohammed et al. 2013). Our binding data (Fig. 1) suggest the potential for all three GREB1 isoforms to regulate the transcriptional activity of ERα. To determine the contribution of the GREB1 isoforms to ERα activity, we utilized shRNA-mediated knockdown of GREB1 with constructs targeting either all isoforms (shRNA #1) or only GREB1a (shRNA #2; Fig. 2A). The homology between GREB1b and GREB1c makes targeting these specific isoforms difficult. Knockdown of GREB1a in MCF7 cells was confirmed via immunoblot analysis. Compared to non-specific shRNA control, expression of GREB1a protein was reduced by 88% with shRNA #1 and by 79% with shRNA #2 when normalized to the loading control, beta actin (Fig. 2B). Knockdown of GREB1b and GREB1c protein was unable to be confirmed as endogenous levels of these isoforms are undetectable via immunoblot.
Figure 2.

GREB1 isoforms are not robust regulators of ERα activity. MCF7 cells were transduced with a non-specific shRNA (shNS) or one of the two shRNAs targeting GREB1. (A) Diagram showing the relative targeting location of the shRNAs used to knockdown all isoforms of GREB1 (shRNA #1) or GREB1a specifically (shRNA #2). (B) Immunoblot depicting the relative expression of GREB1a in MCF7 cells treated with shRNA. Densitometry analysis of GREB1 normalized to beta actin, relative to shNS. (C and D) Real-time PCR was used to assess the relative expression of control genes (C) and known ERα target genes (D). Results are displayed as expression relative to RPL13a and presented as mean ± s.e.; n = 5; *P ≤ 0.05.
Expression of control genes and knockdown of GREB1 isoforms was assessed via real-time PCR. There was no change in the expression of either ERα or HPRT (control) when either shRNA #1 or shRNA #2 was used compared to control shRNA (Fig. 2C).When using the shRNA targeted to all isoforms of GREB1 (shRNA #1), there was a significant reduction in the expression of GREB1a and a non-significant decrease in GREB1b and GREB1c (Fig. 2C). The shRNA specifically targeted to GREB1a (shRNA #2) resulted in a significant decrease in the expression of only GREB1a (Fig. 2C). The effect of GREB1 knockdown on ERα activity was assessed via real-time PCR at gene targets previously identified to be regulated by GREB1 (Mohammed et al. 2013). Knockdown of GREB1 using shRNA #1 resulted in a slight, but non-significant, decrease in the expression of C-MYC, CASP7, CAV1 and EGR3 (Fig. 2D). Knockdown of GREB1a using shRNA#2 resulted in a slight, but significant, decrease in the expression of CAV1 (Fig. 2D). However, some ER-target genes (PS2, PR and SDF1 (i.e. CXCL12)) were unaffected by knockdown with either shRNA (Fig. 2D). Further, knockdown of GREB1 had no impact on ERα activity at these same genes when cells were hormone-starved and stimulated with estrogen (Supplementary Fig. 3). These data suggest that while GREB1a is predominantly responsible for the modest co-activator function at some genes (CAV1), it does not significantly regulate most ER-target genes.
Elevated expression of GREB1a and GREB1b differentially regulate ERα target genes
In order to better assess the co-regulatory potential of specific GREB1 isoforms we expressed exogenous GREB1a or GREB1b in MCF7 cells. As the amino acid sequence of GREB1b is extremely similar to that of GREB1c and is more abundant (Supplementary Fig. 4), we focused only on the elevated expression of GREB1b. MCF7 cells were transduced with adenovirus expressing GREB1a, GREB1b or GFP control. Relative expression of the GREB1 isoforms compared to GFP control was determined using immunoblot analysis 24-h post infection (Fig. 3A). Transduced cells were treated with 10 nM estradiol (+E2) or vehicle control (ethanol, −E2) for 2 h to measure early estrogen response genes. Transcript expression of estrogen-responsive genes previously shown to be regulated by GREB1 (EGR3, PR, PS2 and SDF1) (Mohammed et al. 2013) was measured using real-time PCR. Treatment of GFP-transduced MCF7 cells with estradiol resulted in the expected increase in expression of estrogen-responsive genes, EGR3, PR, PS2 and SDF1 (Fig. 3B). As this analysis is performed after only 2 h of estradiol stimulation, accumulation of some target genes was not robust. When GREB1a was expressed at elevated levels in the absence of estradiol, there was an increase in the basal expression levels of two estrogen-responsive genes, PS2 and SDF1, compared to GFP-expressing and GREB1b-expressing cells (Fig. 3B, −E2). Following estradiol treatment, cells that expressed GREB1a at elevated levels had significantly higher induction of the same estrogen-responsive genes, PS2 and SDF1, compared to GFP-expressing and GREB1b-expressing cells (Fig. 3B). Exogenous GREB1b expression had no effect on the expression of estrogen-responsive genes in the absence or presence of estradiol in comparison to GFP-expressing cells (Fig. 3B). These data suggest that GREB1a and GREB1b differentially regulate the expression of some estrogen-responsive genes, with only GREB1a potentiating their expression.
Figure 3.

GREB1a and GREB1b differentially regulate ERα target genes. (A) MCF7 cells were transduced with GFP, GREB1a or GREB1b adenovirus. Immunoblot depicting overexpression of GREB1a or GREB1b in comparison to control, GFP treated MCF7 cells. (B) MCF7 cells were transduced with GFP, GREB1a, or GREB1b adenovirus then starved of hormone for 72 h and treated with ethanol (−E2) or 10 nM estradiol (+E2) for 2 h. Real-time PCR was used to measure relative levels of known ERα target genes. Results are displayed as expression relative to RPL13a mean + s.e.; n = 3. (C) The indicated reporters were transfected with 3xFLAG-tagged GREB1a, GREB1b or empty vector (EV), and ERα, into HEK 293 cells and luciferase activity was measured after 48 h with either ethanol (−E2) or 10 nM estradiol (+E2). Results are displayed as firefly luciferase activity relative to Renilla luciferase activity + s.e.; n = 3. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
As exogenous GREB1a expression increased the expression of some ER-target genes in the absence of ligand, we investigated whether GREB1 was able to interact with ERα under these conditions. We found that GREB1a preferentially interacts with ERα in the absence of ligand and that this interaction occurs primarily in the cytoplasm (Supplementary Fig. 5), suggesting that GREB1a may potentiate ligand-independent ER activity. Thus, luciferase assays were used to test receptor activity in a cell line with no endogenous expression of ERα or GREB1. Luciferase reporter assays using PS2 and 3X-ERE promoters were performed in HEK-293AD cells with exogenous expression of EV, GREB1a or GREB1b. Expression of neither GREB1a nor GREB1b significantly impacted the activity of ERα on either the PS2- or 3XERE-luciferase reporters in the presence or absence of hormone compared to EV control (Fig. 3C). These data confirm that neither GREB1a nor GREB1b are potent co-activators of ERα at target genes and indicate that GREB1a can potentiate transcription of some ER-target genes independent of the receptor.
GREB1 expression is not sufficient to drive hormone-independent growth
GREB1 has long been implicated in the regulation of proliferation. While knockdown experiments have demonstrated the requirement of GREB1 for estrogen-dependent proliferation (Supplementary Fig. 6) (Rae et al. 2005, Mohammed et al. 2013), here we tested if GREB1a or GREB1b expression was sufficient to drive proliferation under hormone-deprived conditions. As GREB1b encompasses the majority of GREB1c, only ectopic expression of GREB1a and GREB1b was investigated. MCF7 cells were transduced with adenovirus expressing GREB1a, GREB1b or GFP and immunoblot analysis confirmed elevated expression in transduced cells (Fig. 4A). As expected, GREB1a protein expression decreased in cells cultured in CDT media (Fig. 4A). CDT media inhibited the proliferation uninfected and GFP-transduced MCF7 cells compared to MCF7 cells grown in FBS media, demonstrating the hormone-dependence of these ER-positive breast cancer cells (Fig. 4B). Elevated expression of either GREB1a or GREB1b was unable to promote hormone-independent proliferation (Fig. 4B). These data suggest that while GREB1 has been shown to be necessary for proliferation (Supplementary Fig. 6) (Rae et al. 2005, Mohammed et al. 2013), exogenous expression of either GREB1a or GREB1b is insufficient to drive hormone-independent proliferation.
Figure 4.

GREB1 expression is not sufficient to drive hormone-independent growth. MCF7 cells were grown in full-serum media (FBS) or hormone-depleted media (CDT) and transduced with GFP, GREB1a or GREB1b adenovirus. (A) Immunoblot depicts GREB1 expression under each condition. (B) Cell proliferation under the above conditions was measured via MTT assay. Data are plotted as mean absorbance normalized to MTT Day 0 ± s.d.; n = 3. ****P<0.0001.
GREB1a and GREB1b modulate proliferation of breast cancer cells independent of ERα status
Although elevated expression of GREB1 isoforms did not induce hormone-independent proliferation, these isoforms could still contribute a growth advantage in the presence of estrogen. Thus, we tested the ability of exogenous GREB1a and GREB1b to enhance proliferation of ER-positive breast cancer cell lines (MCF7, T47D and ZR751) and ER-negative breast cancer cell lines (MDA-MB-231 and MDA-MB-468), which do not express high levels of endogenous GREB1 (Supplementary Fig. 4). Immunoblot analysis with GREB1 antibody confirmed the elevated expression of GREB1a or GREB1b in all cell lines following transduction with adenovirus (Fig. 5A and Supplementary Fig. 7A). Proliferation was measured over the course of 5 days in all cell lines using MTT assays. Interestingly, elevated expression of GREB1a and GREB1b significantly impaired the proliferation of all three ER-positive cell lines and both ER-negative cell lines (Fig. 5B and Supplementary Fig. 7B).
Figure 5.
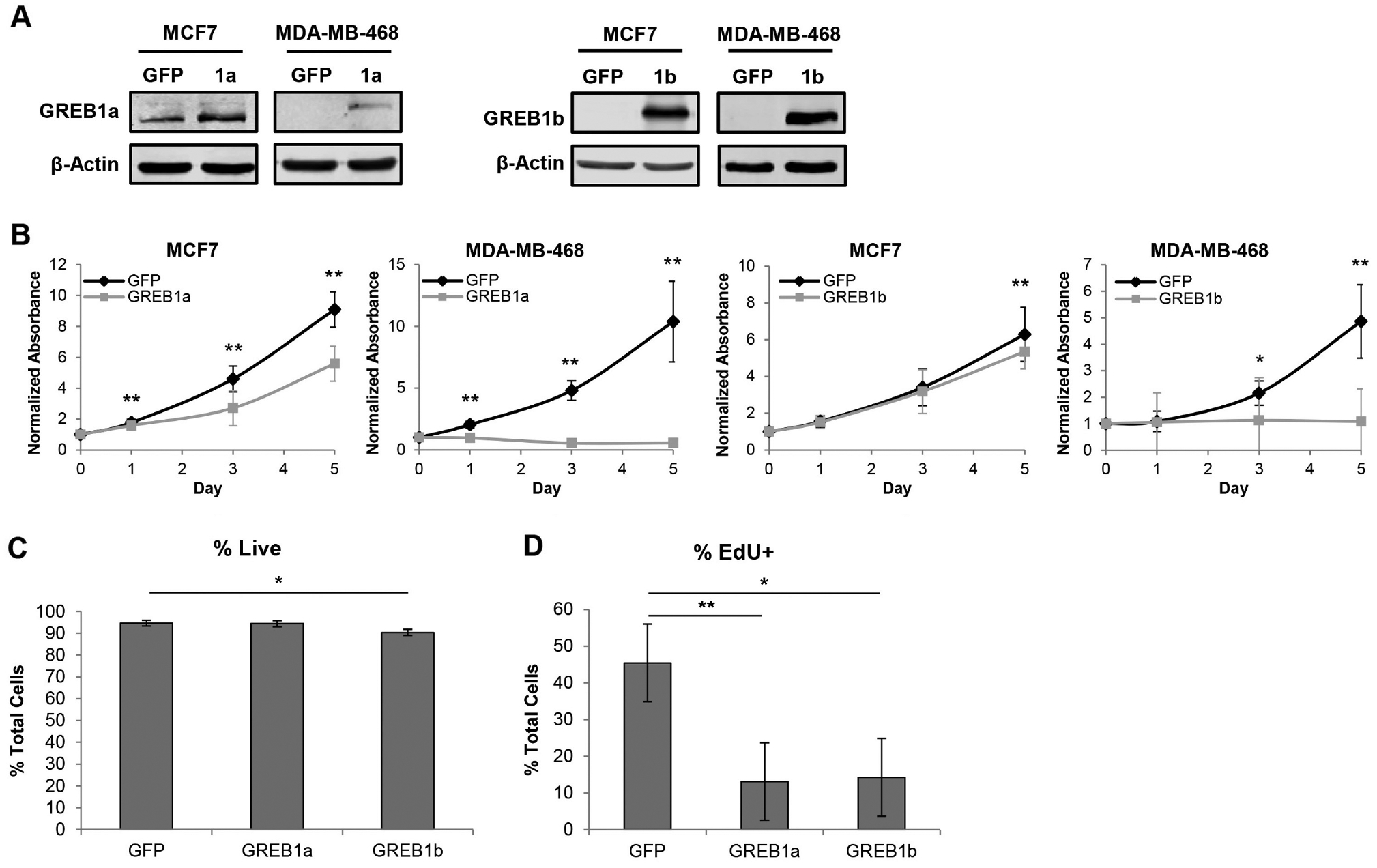
Elevated GREB1a and GREB1b expression reduces proliferation of breast cancer cells independent of ERα status. ER-positive (MCF7) and ER-negative (MDA-MB-468) cells were transduced with adenovirus expressing GFP, GREB1a, or GREB1b. (A) Immunoblot showing elevated expression of GREB1a and GREB1b in transduced cells compared to GFP-treated MCF7 cells. (B) Proliferation of MCF7 and MDA-MB-468 cells after transduction with GREB1a, GREB1b or GFP adenovirus was measured by MTT assay. Data are plotted as mean absorbance normalized to MTT Day 0 ± s.d.; n = 3 for each cell line. (C) MCF7 cells were transduced with GREB1a, GREB1b or GFP adenovirus. After 72 h, cells were stained with LIVE/DEAD Fixable Dead Cell Stain and analyzed by flow cytometry. Data are depicted as live cells as a mean percent total cells ± s.e.; n = 3. (D) MCF7 cells were transduced with GREB1a, GREB1b or GFP adenovirus. After 72 h, cells were treated with EdU and fixed after 6 h. Data are displayed as mean percent of EdU-positive cells ± s.e.; n = 3. *P ≤ 0.05, **P ≤ 0.01.
In order to determine if exogenous GREB1 expression affected cell viability, MCF7 cells were stained with LIVE/DEAD viability dye and analyzed by flow cytometry 72 h post transduction with GFP, GREB1a or GREB1b adenovirus. No toxicity was associated with exogenous GREB1a expression and only a slight, but significant, decrease in cell viability was associated with exogenous GREB1b expression (Fig. 5C).
To confirm that GREB1 was driving changes in proliferation rather than metabolism, as measured by the MTT assay, proliferation of MCF7 cells was measured by EdU incorporation following transduction with GFP, GREB1a or GREB1b adenovirus. There was a significant decrease in the incorporation of EdU into cells transduced with GREB1a and GREB1b in comparison to GFP-transduced cells (Fig. 5D). Together, these data indicate that elevated expression of GREB1 inhibits proliferation of breast cancer cell lines independent of ERα status. This activity is conserved between both GREB1a and GREB1b isoforms.
GREB1b and GREB1c expression is increased in primary and metastatic patient tumor samples
Based on the finding that the GREB1 isoforms have different effects on gene expression (Fig. 3), we examined isoform expression in human breast cancer cell lines and breast cancer patient samples using absolute quantification of mRNA copy number. As expected, and in line with immunoblot data (Fig. 5 and Supplementary Fig. 7), GREB1a was the predominant form in ER-positive cell lines (Supplementary Fig. 4) and patient samples (Supplementary Fig. 8). High levels of GREB1 expression were limited to ER-positive breast cancer cell lines T47D, MCF7 and ZR751 in which GREB1a represented at least 90% of all GREB1 transcripts (Supplementary Fig. 4D). Similarly, ER-positive cancer samples had higher levels of GREB1 expression than ER-negative breast cancer samples indicative of the increased ER expression in cancer (Supplementary Fig. 8A, B and C). Expression of all GREB1 isoforms increased from normal to primary disease (Supplementary Fig. SA, B and C). Remarkably, the proportion of GREB1b and GREB1c mRNA increased significantly from normal to primary and metastatic tissue in matched patient samples, although GREB1a was still the most highly expressed isoform (Fig. 6). We calculated the proportion of GREB1 isoform expression using a larger, publically available data set of transcript expression in normal and malignant breast tissue (Varley et al. 2014, Li et al. 2016). These data showed a similar trend in which the proportion of GREB1b and GREB1c mRNA was significantly higher in tumor samples compared to normal breast tissue. Together, these data suggest that increased expression of all GREB1 isoforms is associated with both ER positivity and primary or metastatic disease state.
Figure 6.
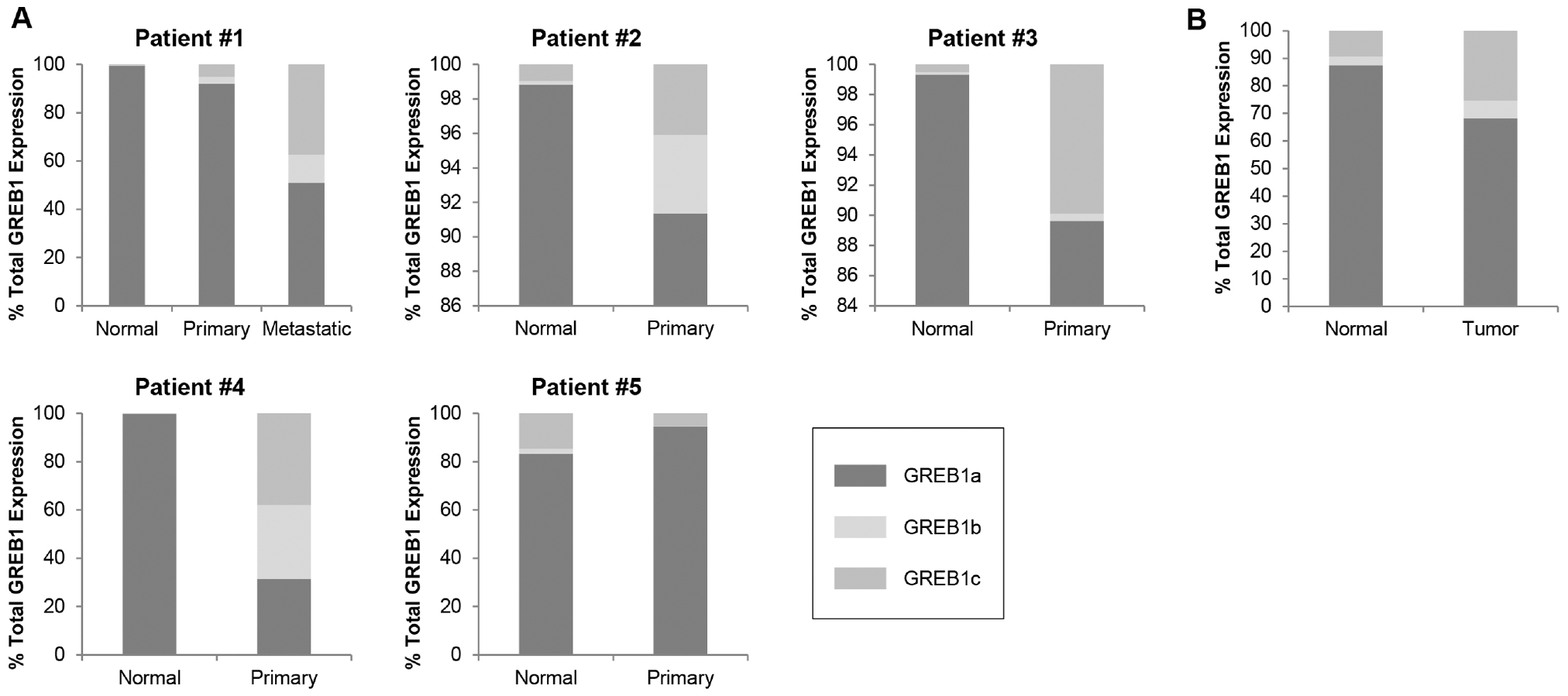
Expression of GREB1b and GREB1c is higher in malignant tissue. (A) Absolute mRNA copy number was determined from a standard curve generated using Taqman probes for GREB1a, GREB1b and GREB1c in patient-matched normal and tumor tissue samples. The percentage of total GREB1 expression of each isoform was compared between normal and primary or metastatic tissue from each patient using the Kruskal–Wallis multiple comparison method. The percent of GREB1b and GREB1c expression is significantly higher in primary tumor tissue in four of the five matched patient samples (P ≤ 0.05). (B) GREB1a, GREB1b and GREB1c transcript expression was generated from a publically available dataset of normal and tumor tissue from breast cancer patients (Varley et al. 2014, Li et al. 2016). The percentage of total GREB1 expression of each isoform was compared between normal and tumor samples using differential expression analysis (Li et al. 2016). Expression of GREB1b and GREB1c is significantly higher in tumor tissue than in normal tissue (P ≤ 0.005).
Discussion
Many efforts have been made to identify ERα-regulated genes that control proliferation and GREB1 was identified as one such gene (Ghosh et al. 2000). Despite routinely being used as a molecular marker for ERα activity, and nearly two decades of research, the molecular functions of this protein remain unclear beyond its reported ERα co-regulatory activity. Further, published studies have failed to differentiate between the three known protein isoforms encoded by the GREB1 gene, making it unclear which isoform may be involved in regulation of ERα activity and proliferation of breast cancer. Here, we demonstrate that all three isoforms have the ability to interact with ERα (Fig. 1), although none are potent regulators of ERα activity (Figs 2 and 3). We further show that only GREB1a has the ability to enhance expression of ERα target genes (Fig. 3). Our data demonstrate the surprising ability of exogenously expressed GREB1a and GREB1b to repress proliferation of breast cancer cell lines, independent of ERα expression (Fig. 5 and Supplementary Fig. 7). In an effort to determine the significance of GREB1 isoform expression, we quantified the transcript abundance of GREB1a, -b and -c. All three isoforms were detected in a panel of breast cancer cell lines (Supplementary Fig. 4) and patient normal and tumor samples (Fig. 6 and Supplementary Fig. 8). While GREB1a is universally the most expressed transcript, breast cancer progression is associated with increased expression of the GREB1b and GREB1c isoforms (Fig. 6). These findings underscore the notion that not all isoforms of GREB1 have conserved functional roles in breast cancer.
GREB1 isoforms are not potent regulators of ERα activity
Previous studies have suggested that GREB1 may interact with ERα to regulate the receptor’s activity through modulation of co-factor binding (Mohammed et al. 2013). We demonstrate that ER interacts with the amino terminus of all three isoforms of GREB1 (Fig. 1) independent of the LxxLL motif within GREB1a (Fig. 1C). However, the ability of these isoforms to enhance ERα activity was disparate. Elevated expression of only GREB1a was able to enhance the expression of ERα target genes (Fig. 3B). Notably, this pronounced effect on the expression of known ERα target genes was evident both in the presence and absence of hormone (Fig. 3B). Previous reports suggest that GREB1 interaction with ERα is ligand inducible (Mohammed et al. 2013); however, our data show that GREB1 preferentially interacts with ERα in the absence of ligand and in the cytoplasm (Supplementary Fig. 5). The increase in basal expression of some ER-target genes seems to account for most of the transcriptional changes at these targets and appears to be receptor-independent. In contrast to GREB1a, elevated expression of GREB1b had no effect on ERα target gene expression (Fig. 3B). While GREB1c was not tested, the protein isoform differs from GREB1b by the absence of exon 10 in the coding region and would not be expected to have additional functions than those seen in GREB1b.
Surprisingly, the overall effect of GREB1 on ERα target gene expression was modest at best. In fact, knockdown of all GREB1 isoforms resulted in only a 3–30% decrease in the expression of target genes in the presence of endogenous hormone (Fig. 2D) and knockdown of GREB1 isoforms had no effect on estrogen-induced expression of these target genes (Supplementary Fig. 3B). Previous studies using microarray analysis following knockdown of GREB1 with siRNA had demonstrated a 26–91% change in these same genes (Mohammed et al. 2013). However, the previous microarray study also showed a 43% decrease in ERα gene expression upon knockdown of GREB1 (6-h time point) (Mohammed et al. 2013), while our data did not demonstrate a similar decrease in receptor levels (Fig. 2C). Thus, the impact of GREB1 knockdown on the expression of estrogen-responsive genes in the previous publication may be a result of reduced ERα expression that may be dependent upon the methodology.
GREB1 regulates proliferation through ERα independent activities
Previous studies have established GREB1 as a critical regulator of proliferation in breast cancer (Rae et al. 2005, Mohammed et al. 2013), prostate cancer (Rae et al. 2006) and ovarian cancer (Laviolette et al. 2014). However, it was unclear which of the individual isoforms of GREB1 are responsible for this proliferative phenotype.
Specifically, one study found that knockdown of GREB1 in the presence or absence of hormone reduces proliferation of MCF7 breast cancer cell lines to that of estrogen-depleted conditions of control cells suggesting the necessity of GREB1 for hormone-induced proliferation (Rae et al. 2005). Thus, we hypothesized that elevated expression of GREB1 may circumvent the requirement of hormone for the proliferation of MCF7 breast cancer cells, presumably through activation of estrogen-responsive genes. We show that exogenous GREB1a expression can increase target gene expression even under estrogen-depleted conditions (Fig. 3B). However, we found that elevated expression of either of the predominant GREB1 isoforms, GREB1a or GREB1b, did not induce hormone-independent growth of MCF7 breast cancer cells (Fig. 4). These data suggest that the modest role of GREB1 in the modulation of ER-target genes (Fig. 3) is not sufficient to drive proliferation of breast cancer cells in hormone-free conditions.
Surprisingly, elevated expression of GREB1a or GREB1b significantly reduced the proliferation of all ER-positive breast cancer cell lines even in hormone-containing media (Fig. 5 and Supplementary Fig. 7), suggesting that the role of GREB1 in regulating proliferation is not solely growth promoting. The proliferation of ER-negative breast cancer cell lines was also severely inhibited by exogenous expression of GREB1a and GREB1b (Fig. 5 and Supplementary Fig. 7), suggesting GREB1 is able to modulate proliferation independent of ERα expression and activity. These data demonstrate that GREB1 has alternative functions beyond being a nuclear receptor co-regulator. This growth inhibitory role of GREB1 may explain why higher GREB1 expression in prostate cancer patients and hypomethylation of the GREB1 promoter in ovarian cancer patients correlates with better outcome (Bauerschlag et al. 2011). We postulate that there is a homeostatic level of GREB1 that is growth promoting while reduced or elevated expression results in growth inhibition in breast cancer cells.
Interestingly, previous analysis in tamoxifen-resistant MCF7 cells suggested that therapy resistance can be attributed to a loss of GREB1 co-activator function (Mohammed et al. 2013). While exogenous restoration of GREB1 caused a decrease in proliferation in the presence of tamoxifen, our data suggest this decrease may not be caused by restoration of tamoxifen sensitivity but rather an overexpression of GREB1. As we demonstrate herein, exogenous GREB1 expression causes a decrease in proliferation of most breast cancer cell lines regardless of ERα status and activity (Fig. 5 and Supplementary Fig. 7). Thus, this previously published data supports the notion that an optimal level of GREB1 expression is necessary for proliferation of breast cancer cells.
While there remains a need for further research to be done in order to determine the discrete molecular function of the GREB1 isoforms, the data herein suggest a function of GREB1 isoforms outside of the canonical hormone receptor transcriptional network.
Supplementary Material
Acknowledgements
The authors thank C Wormsbaecher, X Guan, R Hennessey, A Holderbaum, K Lapak and B Murphy for critical reading of the manuscript. They also thank A Shapiro and K Wernke for help in generating preliminary data for this work.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Footnotes
Supplementary data
This is linked to the online version of the paper at https://doi.org/10.1530/ERC-17-0496.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Ali S & Coombes RC 2002. Endocrine-responsive breast cancer and strategies for combating resistance. Nature Reviews Cancer 2 101–112. ( 10.1038/nrc721) [DOI] [PubMed] [Google Scholar]
- Bauerschlag DO, Ammerpohl O, Brautigam K, Schem C, Lin Q, Weigel MT, Hilpert F, Arnold N, Maass N, Meinhold-Heerlein I, et al. 2011. Progression-free survival in ovarian cancer is reflected in epigenetic DNA methylation profiles. Oncology 80 12–20. ( 10.1159/000327746) [DOI] [PubMed] [Google Scholar]
- Burd CJ, Petre CE, Moghadam H, Wilson EM & Knudsen KE 2005. Cyclin D1 binding to the androgen receptor (AR) NH2-terminal domain inhibits activation function 2 association and reveals dual roles for AR corepression. Molecular Endocrinology 19 607–620. ( 10.1210/me.2004-0266) [DOI] [PubMed] [Google Scholar]
- Clarke R, Tyson JJ & Dixon JM 2015. Endocrine resistance in breast cancer – an overview and update. Molecular and Cellular Endocrinology 418 220–234. ( 10.1016/j.mce.2015.09.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes J, Bourdeau V, White JH & Mader S 2007. Regulation of GREB1 transcription by estrogen receptor alpha through a multipartite enhancer spread over 20 kb of upstream flanking sequences. Journal of Biological Chemistry 282 17335–17339. ( 10.1074/jbc.C700030200) [DOI] [PubMed] [Google Scholar]
- Dixon JM 2014. Endocrine resistance in breast cancer. New Journal of Science 2014 1–27. ( 10.1155/2014/390618) [DOI] [Google Scholar]
- Ghosh MG, Thompson DA & Weigel RJ 2000. PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Research 60 6367–6375. [PubMed] [Google Scholar]
- Hall JM & Korach KS 2002. Analysis of the molecular mechanisms of human estrogen receptors alpha and beta reveals differential specificity in target promoter regulation by xenoestrogens. Journal of Biological Chemistry 277 44455–44461. ( 10.1074/jbc.M200849200) [DOI] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, et al. 2007. Estrogen receptors: how do they signal and what are their targets. Physiological Reviews 87 905–931. ( 10.1152/physrev.00026.2006) [DOI] [PubMed] [Google Scholar]
- Hnatyszyn HJ, Liu M, Hilger A, Herbert L, Gomez-Fernandez CR, Jorda M, Thomas D, Rae JM, El-Ashry D & Lippman ME 2010. Correlation of GREB1 mRNA with protein expression in breast cancer: validation of a novel GREB1 monoclonal antibody. Breast Cancer Research and Treatment 122 371–380. ( 10.1007/s10549-009-0584-x) [DOI] [PubMed] [Google Scholar]
- Laviolette LA, Hodgkinson KM, Minhas N, Perez-Iratxeta C & Vanderhyden BC 2014. 17beta-Estradiol upregulates GREB1 and accelerates ovarian tumor progression in vivo. International Journal of Cancer 135 1072–1084. ( 10.1002/ijc.28741) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JR, Sun CH, Li W, Chao RF, Huang CC, Zhou XJ & Liu CC 2016. Cancer RNA-Seq Nexus: a database of phenotype-specific transcriptome profiling in cancer cells. Nucleic Acids Research 44 D944–D951. ( 10.1093/nar/gkv1282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, D’Santos C, Serandour AA, Ali HR, Brown GD, Atkins A, Rueda OM, Holmes KA, Theodorou V, Robinson JL, et al. 2013. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Reports 3 342–349. ( 10.1016/j.celrep.2013.01.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Fan D, Kerner SA & McDonnell DP 1997. Identification of a third autonomous activation domain within the human estrogen receptor. Molecular Endocrinology 11 747–754. ( 10.1210/mend.11.6.0008) [DOI] [PubMed] [Google Scholar]
- Patterson AR, Mo X, Shapiro A, Wernke KE, Archer TK & Burd CJ 2015. Sustained reprogramming of the estrogen response after chronic exposure to endocrine disruptors. Molecular Endocrinology 29 384–395. ( 10.1210/me.2014-1237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae JM, Johnson MD, Scheys JO, Cordero KE, Larios JM & Lippman ME 2005. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Research and Treatment 92 141–149. ( 10.1007/s10549-005-1483-4) [DOI] [PubMed] [Google Scholar]
- Rae JM, Johnson MD, Cordero KE, Scheys JO, Larios JM, Gottardis MM, Pienta KJ & Lippman ME 2006. GREB1 is a novel androgen-regulated gene required for prostate cancer growth. Prostate 66 886–894. ( 10.1002/pros.20403) [DOI] [PubMed] [Google Scholar]
- Sun J, Nawaz Z & Slingerland JM 2007. Long-range activation of GREB1 by estrogen receptor via three distal consensus estrogen-responsive elements in breast cancer cells. Molecular Endocrinology 21 2651–2662. ( 10.1210/me.2007-0082) [DOI] [PubMed] [Google Scholar]
- Tryfonidis K, Zardavas D, Katzenellenbogen BS & Piccart M 2016. Endocrine treatment in breast cancer: cure, resistance and beyond. Cancer Treatment Reviews 50 68–81. ( 10.1016/j.ctrv.2016.08.008) [DOI] [PubMed] [Google Scholar]
- Varley KE, Gertz J, Roberts BS, Davis NS, Bowling KM, Kirby MK, Nesmith AS, Oliver PG, Grizzle WE, Forero A, et al. 2014. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Research and Treatment 146 287–297. ( 10.1007/s10549-014-3019-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharewski TR, Bondy KL, McDonell P & Wu ZF 1994. Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on 17 beta-estradiol-induced pS2 expression. Cancer Research 54 2707–2713. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.