

Abstract
Poor aqueous solubility and dissolution of drug candidates drive key decisions on lead series optimization during drug discovery, on formulation optimization, and clinical studies planning during drug development. The interpretation of the in vivo relevance of early pharmaceutical profiling is often confounded by the multiple factors affecting oral systemic exposure. There is growing evidence that in vitro drug solubility may underestimate the true in vivo solubility and lead to drug misclassification. Based on 10 poorly water‐soluble tyrosine kinase inhibitors, this paper demonstrates the use of physiologically‐based pharmacokinetic (PK) analysis in combination with early clinical PK data to identify drugs whose absorption is truly limited by solubility in vivo and, therefore, expected to exhibit food effect. Our study supports a totality of evidence approach using early clinical data to guide decisions on conducting drug interaction studies with food and acid‐reducing agents.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The solubility of a drug candidate significantly impacts decisions in drug development. The Biopharmaceutics Classification System classification based solely on in vitro solubility can be conservative with respect to impact of solubility on a compound's absorption.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Deconvolution of the mechanisms underlying the gut bioavailability is hindered by parameter nonidentifiability. This research uses a combined in vitro, in vivo, and in silico analysis to understand the in vivo relevance of in vitro–measured solubility for a better prediction of food and proton pump inhibitor effects on oral drug exposure.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ A more precise classification based on a sound mechanistic understanding and totality of evidence is proposed for efficient drug development.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Identification of compounds that have sufficient in vivo solubility despite low in vitro solubility can support decisions on the need and timing of studies and help save valuable resources.
In drug discovery, solubility of drug candidates in aqueous media is one of the pivotal physicochemical properties to optimize a chemical series. A candidate drug is commonly required to have solubility above 10 µM to facilitate preclinical testing.1 Low, highly variable oral bioavailability and less‐than‐dose‐proportional exposure are among the consequences of poor drug solubility (Table S1). Therefore, pharmaceutical companies strive to increase the solubility of a drug candidate during lead optimization.1, 2 An analysis of the early clinical data to understand the in vivo relevance of in vitro solubility could be valuable in assessing the need for resource‐intensive formulation development and/or clinical trials.
Based on the Biopharmaceutics Classification System (BCS),3 the solubility class threshold is determined using the highest strength that can be completely dissolved in 250 mL of an aqueous medium (pH 1–6.8 at 37°C). More recently, the Developability Classification System (DCS),4 recognized the need to differentiate between dissolution rate (IIa) and solubility (IIb) limitation. However, a precise classification of drug candidates may not be feasible at the end of lead optimization because the active dose range at this phase can only be estimated.5
Evidence suggests that oral bioavailability may not always depend on the solubility of poorly soluble drugs. First, several BCS II/IV drugs, such as naproxen, phenytoin, and diazepam, have an absolute bioavailability (F) > 90%.6, 7 Second, although poorly water‐soluble, lipophilic compounds are generally expected to show a better solubilization in gastrointestinal fluids in the presence of food and, thus, a better oral absorption in the fed state,9 poorly water‐soluble anticancer drugs, such as imatinib and trametinib, show only a modest food effect, if any.10 Last, enabling formulations meant to improve the kinetic solubility of poorly water‐soluble active ingredients do not always enhance oral bioavailability,11 indicating that poor oral bioavailability may be caused by other factors.12, 13
Physiologically‐based pharmacokinetic (PBPK) models are powerful tools that describe drug pharmacokinetic (PK) through the integration of PK mechanisms, compound data, and physiology.14 The objective of the current study is to conduct PBPK analysis of 10 poorly soluble anticancer drugs, as described by Peters,15 along with an analysis of gut bioavailability (fraction of the administered oral dose reaching the portal vein) and dose‐exposure proportionality, to evaluate the in vivo relevance of in vitro solubility data in determining their oral absorption. Such an evaluation could the pave the way for better predicting the impact of food and acid‐reducing agents on the exposure of poorly water‐soluble drugs.
Results
Biopharmaceutical properties
Table 1 shows the fasted state simulated intestinal fluid (FaSSIF) and simulated gastric fluid (SGF) solubility of the model drugs, permeability, and reported biopharmaceutical properties. The FaSSIF solubility of these compounds is generally < 1 mg/mL, except for imatinib. The measured solubility is consistent with literature, except for pazopanib whose measured solubility in SGF at pH 1.6 is higher than the value reported at pH 1.1.16
Table 1.
Summary of biopharmaceutical properties and metabolizing CYP enzymes
| Parameters | Imatinib mesylate | M1 | Crizotinib | Trametinib | Dabrafenib mesylate | Gefitinib | Erlotinib HCl | Lapatinib ditosylate | Vemurafenib | Pazopanib HCl |
|---|---|---|---|---|---|---|---|---|---|---|
| Measured solubility in FaSSIF pH 6.5 (mg/mL) | ≥ 5 | 0.0728 | 0.7430 | N.A. | 0.0037 | 0.0887 | 0.0124 | 0.0350 | 0.0054 | 0.0012 |
| Measured solubility in SGF pH 1.6 (mg/mL) | N.A. | 0.0213 | N.A. | N.A. | 0.0117 | N.A. | 0.1665 | 0.0055 | 0.0003 | 1.0779 |
| Measured Caco‐2 Papp passive (10−6 cm/second) | 25.90 | 14.70 | 15.35 | 15.44 | 12.45 | 10.41 | 40.43 | 0.21 | 0.11 | 28.46 |
| cPeff [10−4 cm/second] | 2.61 | 6.19 | 2.22 | 1.07 | 2.38 | 5.23 | 3.58 | 1.84 | 2.54 | 0.85 |
| pKa values (basic) | 8.07, 1.5235 | 9.5, 2.8 | 9.4, 5.620 | N.A. | 2.2, 1.536 | 7.2, 5.437 | 5.4238 | 4.6, 6.739 | N.A. | 6.4, 2.118 |
| pKa values (acidic) | N.A. | N.A. | N.A. | N.A. | 6.636 | N.A. | N.A. | N.A. | 7.9, 11.140 | 10.218 |
| Clinical dose in food effect study (mg)a | 400 | Clinical dose is yet to be established | 250 | 2 | 150 | 250 | 150 | 1,500 | 960 | 800 |
| BCS classification | II10 | Likely IV | IV20 | II24 | II36 | II41 | II38 | IV42 | IV40 | II18 |
| Dose number | 0.38 | > 1 | 1.35 | 11.43 | 193.55 | 11.27 | 53.10 | 277.78 | 711.11 | 2,909.09 |
| SLAD (mg)a | 6,527.96 | > dose in food effect study | 985.72 | 0.45b | 4.41 | 227.43 | 24.18 | 23.75 | 8.20 | 0.55 |
| DCS classification | Ila | Likely IV | IV | Ilb | Ilb | Ila | Ilb | IV | IV | Ilb |
| Major metabolizing CYP enzymes | CYP3A443 |
CYP3A4 CYP2C8 |
CYP3A4 CYP3A520 |
— |
CYP2C8 CYP3A436 |
CYP3A4 CYP2D644 |
CYP3A438 |
CYP3A4 CYP3A542 |
CYP3A440 | CYP3A418 |
Dose number: dose number < 1 indicates that the dose is soluble in 250 mL FaSSIF, solubility criterion for BCS classification (Eq. 2).
BCS, Biopharmaceutical Classification System; Caco‐2 Papp passive, apparent passive permeability determined in Caco‐2 assay; cPeff, calculated effective intestinal permeability; DCS, Developability Classification System; FaSSIF, fasted state intestinal fluid; N.A., not applicable; pKa, acid dissociation constant; SGF, simulated gastric fluid; SLAD, solubility limited absorbable dose (amount of drug soluble in 500 mL FaSSIF, solubility criterion for DCS IIa/IIb classification (Eq. 3)).
Dose given as free base. bCalculated using literature solubility from the US Food and Drug Administration Clinical Pharmacology and Biopharmaceutics Review.24
PBPK absorption models
PBPK modeling for all model drugs is exemplified by pazopanib (Figure 1). The PBPK simulation of oral PK profiles using clearance and distribution parameters derived from i.v. simulation, coupled with FaSSIF solubility and Caco‐2 permeability, may not capture the observed exposure (Figure 1 b). To capture the observed exposure, a 20‐fold increase in the input solubility (Figure 1 c) and reduced colonic absorption were needed (Figure 1 d). PBPK simulation using a hypothetically high solubility results in maximum plasma concentration (Cmax) and area under the plasma concentration‐time curve (AUC) that are fourfold higher than the observed (Table 2 and Figure 1 e). This indicates that pazopanib exposure is limited by solubility. Poor sensitivity to a hypothetically high permeability shows that its exposure is not limited by permeability (Figure 1 f).
Figure 1.
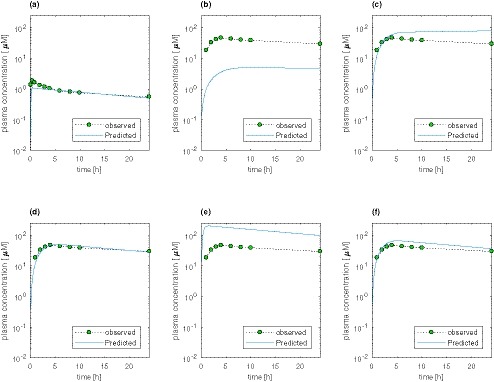
Physiologically‐based pharmacokinetic (PBPK) simulations of pharmacokinetic profiles of pazopanib. (a) PBPK simulation of i.v. infusion of 5 mg pazopanib over 5 minutes to obtain the intrinsic clearance and multiplicative factor, for simultaneously scaling all tissue distribution coefficients. (b) Pazopanib 800 mg oral administration simulated with in vitro fasted state simulated intestinal fluid (FaSSIF) solubility and Caco‐2 permeability. (c) Pazopanib 800 mg oral administration simulated with Caco‐2 permeability and an input solubility that is 20‐fold higher than FaSSIF solubility. (d) Pazopanib 800 mg oral administration simulated with Caco‐2 permeability, an input solubility that is 20‐fold higher than FaSSIF solubility and reduced colonic absorption. (e) Pharmacokinetic profile using hypothetical Biopharmaceutics Classification System class I‐like solubility in the pazopanib PBPK model with good fit in (d). (f) Pharmacokinetic profile using hypothetically high permeability of 10*10−4 cm/second in the pazopanib PBPK model with good fit in d. [Colour figure can be viewed at https://www.wileyonlinelibrary.com]
Table 2.
Observed vs. predicted pharmacokinetic parameters at doses used in their clinical food effect studies and simulation using hypothetical BCS class I‐like solubility
| Observed PK parameters | PBPK simulated PK parameters using Caco‐2 permeability | PBPK simulated PK using calculated P eff | AUC and Cmax ratios (hypothetical BCS class I‐like solubility to best‐fit solubility) from PBPK simulations | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Caco‐2 permeability | Calculated P eff | ||||||||||||||
| AUC0–24 (hour*µM) | Cmax (µM) | Tmax (hour) | AUC0–24 (hour*µM) | Cmax (µM) | Tmax (hour) | Solubility increase (mg/mL) | AUC0–24 (hour*µM) | Cmax (µM) | Tmax (hour) | Solubility increase (mg/mL) | AUC0–24 ratio | Cmax ratio | AUC0–24 ratio | Cmax ratio | |
| Imatinib | 28.91 | 3.51 | 1.51 | 36.4 | 2.78 | 1.6 | None | 36 | 2.65 | 2.24 | None | 1.0 | 1.0 | 1.0 | 1.0 |
| M1 | 1.073 | 0.0621 | 8 | 1.75 | 0.312 | 0.96 | None | 1.76 | 0.352 | 0.8 | None | 1.0 | 1.0 | 1.0 | 1.0 |
| Crizotinib | 3.635 | 0.311 | 5.05 | 3.36 | 0.261 | 3.6 | None | 3.34 | 0.257 | 4 | None | 1.0 | 1.0 | 1.0 | 1.0 |
| Trametinib | 0.1162 | 0.0136 | 1.5 | 0.0912 | 0.0052 | 2.16 | 0.0008 → 0.0069 | 0.0896 | 0.0054 | 1.76 | 0.0008 → 0.0416 | 1.1 | 1.7 | 1.0 | 1.0 |
| Dabrafenib | 25.38 | 3.99 | 2 | 18.6 | 3.89 | 2.48 | 0.0037 → 0.0616 | 18.4 | 3.52 | 2.72 | 0.0037 → 0.0616 | 1.0 | 1.3 | 1.0 | 1.3 |
| Gefitinib | 2.549 | 0.161 | 3.72 | 2.24 | 0.142 | 3.82 | None | 2.27 | 0.145 | 3.42 | None | 1.0 | 1.0 | 1.0 | 1.0 |
| Erlotinib | 21.3 | 1.94 | 4.13 | 53.9 | 4.03 | 3.67 | None | 35.6 | 1.98 | 4.71 | None | 1.1 | 2.1 | 1.6 | 3.4 |
| Lapatinib | 18.07 | 1.46 | 3.96 | 15.3 | 1.18 | 2.88 | 0.0350 → 6.6044 | 22.7 | 1.4 | 3.92 | 0.0350 → 0.0943 | 1.0 | 1.0 | 4.6 | 11.0 |
| Vemurafenib | 94.71 | 6.41 | 4.08 | 109 | 5.91 | 4.7 | 0.0054 → 0.6124 | 133 | 7.3 | 6.14 | None | 2.6 | 2.6 | 54.1 | 53.8 |
| Pazopanib | 843.6 | 47.3 | 4 | 889 | 50.7 | 5.44 | 0.0012 → 0.0237 | 978 | 55.7 | 5.12 | 0.0012 → 0.2370 | 3.9 | 4.0 | 2.7 | 2.7 |
AUC0–24, area under the plasma concentration time curve from 0 to 24 hours; BCS, Biopharmaceutical Classification System; Cmax, maximum plasma concentration; PBPK, physiologically‐based pharmacokinetic; P eff, effective intestinal permeability; PK, pharmacokinetic; Tmax, time to reach the maximum plasma concentration.
The intrinsic microsomal clearance (CLint) and a multiplicative factor (K p) to scale the tissue distribution coefficients, derived from i.v. PBPK simulations are reported in Table S2. In the absence of i.v. PK for vemurafenib and lapatinib, the volume of distribution for these two compounds were estimated (Table S3). Oral PBPK simulations of all model drugs under fasting conditions at doses used in their clinical food effect studies show that the simulated PK metrics are adequately captured (Table 2, Figure S1) except for erlotinib and M1, a development compound, whose exposures were overpredicted due to an intestinal loss of drug. Gastric emptying rate was decreased to capture the observed profiles of crizotinib, gefitinib, and imatinib. Colonic absorption of vemurafenib and pazopanib had to be decreased. Similar to the pazopanib case, the FaSSIF solubilities of dabrafenib, lapatinib, trametinib, and vemurafenib were increased to accurately predict their plasma exposure. The elimination slope of the predicted dabrafenib oral PK profile derived from the i.v. PK is steeper than the observed, suggesting prolonged absorption.17
Measured absolute bioavailability and calculated gut bioavailability
The gut bioavailability of vemurafenib and lapatinib cannot be determined in the absence of i.v. PK. All other compounds, including M1, have a relatively high gut bioavailability (> 0.75) at the dose tested, except for pazopanib and erlotinib (Table S4). Gut bioavailability > 1 can result from the use of mean values.
Effect of solubility and permeability on PK metrics based on PBPK model simulations
AUC and Cmax ratios (hypothetical BCS class I‐like solubility to best‐fit solubility) from PBPK simulations are summarized in Table 2. Crizotinib, dabrafenib, gefitinib, imatinib, M1, and trametinib are insensitive to an increase in the input solubility. In contrast, the oral exposures of pazopanib and vemurafenib were sensitive to solubility with either measures of permeability (Caco‐2 or calculated effective intestinal permeability (P eff)). For erlotinib, Cmax was increased using the hypothetical BCS class I‐like solubility but not the AUC, indicating that its rate of absorption depends on dissolution.
The use of Caco‐2 and calculated P eff led to similar results for all drugs except for the poorly permeable compounds vemurafenib and lapatinib, for which calculated P eff in PBPK models led to a significant increase in exposure.
Clinical PK analysis on dose‐exposure proportionality and food effect
Based on the clinical PK data presented in Table 3, systemic exposure increases in a dose‐proportional or supra‐dose‐proportional manner for all compounds except for pazopanib > 800 mg18, 19 and M1. Even though the single‐dose PK of crizotinib was reported to be less‐than‐dose‐proportional,20 the proportionality based on the data presented seems linear in our assessment.
Table 3.
Evaluation of dose‐exposure proportionality for model compounds from the FDA's Clinical Pharmacology and Biopharmaceutics Reviews and food effect
| Dose range tested (mg) | Dosing schedule | Increase in exposure | Food effect study dose (mg) | AUC fed/fasted ratio | Cmax fed/fasted ratio | |
|---|---|---|---|---|---|---|
| Imatinib | 25–1,000 | N.A. | Dose proportional43 | 400 | 0.9245 | 0.8945 |
| M1 | 30–1,400 | Steady‐state | Less than proportional (food effect dose is within the linear region) | 30 | 1.17 | 1.29 |
| Crizotinib |
50–300 50–200 200–300 |
Single dose Steady state Steady state |
Less than proportional More than proportional More than proportional20 |
250 | 0.8646 | 0.8646 |
| Trametinib | 0.125–10 | Single Dose |
More than proportional (Cmax proportional)24 |
2 | 0.89747 | 0.30147 |
| Dabrafenib | 12–300 |
Single dose Steady state |
Dose proportional Less than proportional36 |
150 | 0.7023 | 0.4923 |
| Gefitinib |
50–500 50–400 50–700 |
Single dose (HV) Steady state (pat.) Steady state (pat.) |
Dose proportional Dose proportional More than proportional41 |
250 | 1.3721 | 1.3221 |
| Erlotinib | 100–1,000 | N.A. | Dose proportional38 | 150 | 1.97/0.9322 | 1.57/1.1522 |
| Lapatinib | Approx. 600–1,800 | Steady state | Dose proportional42 | 1,500 | 4.2548 | 3.0348 |
| Vemurafenib | 240–960 |
Single dose Steady state |
Dose proportional Dose proportional40 |
960 | 4.749 | 2.549 |
| Pazopanib | 50–2,000 |
Single dose Steady state |
Less than proportional Less than proportional18 |
800 | 2.3450 | 2.0850 |
AUC, area under the plasma concentration time curve; Cmax, maximum plasma concentration; FDA, US Food and Drug Administration; HV, healthy volunteers; N.A., not applicable; pat., patients.
A strong positive food effect on systemic exposure was only observed for lapatinib, vemurafenib, and pazopanib (Table 3). High intersubject variability prevents the detection of significant food effect on gefitinib exposure.21 The exposure of 150 mg erlotinib22 doubled under fed conditions in the first period but slightly decreased in the second period compared with fasted conditions. This anomaly is not further explained by the investigators.
Discussion
In vitro solubility measurement within the biopharmaceutical framework (BCS and DCS)
Although the primary regulatory purpose of the BCS is guiding in vivo bioequivalence study waivers, its scientific rationale is used in other areas, including food effect predictions.9 As per the BCS specifications, all 10 model anticancer drugs have been classified as poorly soluble based on the drug substance.10 Of note, the dose number for imatinib is less than unity in our calculation (Table 1), which would qualify it as a BCS I drug. Under the DCS framework, imatinib, M1, crizotinib, and gefitinib have a solubility‐limited absorbable dose (SLAD) higher than the clinical dose used in their food effect studies. Therefore, oral exposure of these drugs is not expected to be solubility‐limited.
Gut bioavailability
Gut bioavailability ≥ 0.75 indicates that systemic exposure of the most model compounds may not depend on the drug's solubility at the tested dose levels. Only erlotinib (0.61) and pazopanib (0.21) have a relatively low gut bioavailability pointing to an intestinal drug loss attributable to efflux, gut metabolism, and/or solubility limitation.
In silico PBPK modeling probing the role of solubility in oral drug absorption
Hypothesis testing with PBPK modeling15 has been used to evaluate the role of in vitro solubility in oral absorption of poorly water‐soluble drugs. During model development, a 2 to 200‐fold increase over the measured in vitro solubility was necessary to simulate the observed exposure of 5 extremely poorly water‐soluble (solubility < 0.01 mg/mL) drugs (Table 2). The in vitro solubility in a defined solvent could underestimate the true in vivo solubility, which may be influenced by supersaturation (especially for weak bases), and formulation effects. For example, hydroxypropyl methylcellulose presented in the shell of dabrafenib capsules shows an inhibitory effect on precipitation of supersaturated dabrafenib solution,23 or the presence of sodium dodecyl sulfate in trametinib24 tablets may increase the apparent drug solubility.
The PBPK simulations of vemurafenib, lapatinib, and pazopanib show a significantly higher‐than‐observed exposure when a hypothetically high BCS class I‐like solubility is used as input (Table 2). This suggests that the absorption of these compounds is limited by solubility. BCS class IV drugs, vemurafenib and lapatinib, exhibit significantly higher‐than‐observed exposure with a hypothetically high solubility as well as permeability, indicating a solubility‐limited and/or permeability‐limited exposure. On the contrary, the Cmax and AUC ratios with hypothetically high solubility are close to one for imatinib, trametinib, crizotinib, dabrafenib, gefitinib, and M1 (Table 2), indicating that the exposures of these drugs are not solubility‐limited.
The need to reduce the gastric emptying rate for crizotinib, imatinib, and gefitinib for better fit to the observed profile is possibly due to drug‐induced delayed gastric emptying. The use of the generic PBPK model to identify delayed gastric emptying was already validated in the rat.25
PBPK simulations of erlotinib show an intestinal drug loss that is likely due to gut metabolism or efflux. Erlotinib, a CYP3A substrate, is known to be metabolized in the gut.26 With only 1% of the dose as parent drug in the feces after oral administration,26 the possibility of transporter‐mediated intestinal loss may be ruled out. The in vitro FaSSIF solubility was sufficient to explain the observed erlotinib PK profile but using a hypothetical BCS class I‐like solubility enables a higher Cmax, whereas AUC remains unchanged (Figure S3 c), indicating a slow in vivo dissolution but nonsolubility limited absorption. Other model drugs are substrates of CYP3A4 as well (Table 1), but their exposures are probably not limited by gut metabolism, as evidenced by the good gut bioavailability (> 0.75) for most of these compounds and by the PBPK analysis.
Both the calculation of gut bioavailability as well PBPK analysis relies on i.v. PK data at doses leading to concentrations comparable to the oral administration. When such i.v. PK data are not available, PBPK analysis can still be carried out if elimination parameters can be estimated from well‐characterized, single‐dose oral PK profiles with < 20% AUC extrapolation.
Analysis of dose‐exposure relationship
A less‐than‐dose‐proportional exposure observed in dose‐escalating PK studies can help identify solubility‐limited drug absorption. However, identifying dose‐exposure trends is confounded by high intersubject variability in clinical studies, insufficient number of subjects per dose group, or insufficient number of doses in the dose range of interest (Figure S2).
Identification of solubility‐limited exposure and food effect predictions
Table 4 distinguishes drugs with solubility‐limited absorption (shown in red) from those that are not (shown in green). On the right, the food effect for these drugs are also color‐coded to distinguish drugs that show a positive food effect (red) from those that do not (green).
Table 4.
Heatmap summarizing the properties in the BCS and DCS frameworks along with properties/methods used in this analysis for the identification of solubility‐limited exposure
| Compound | Dose number (BCS) | SLAD (DCS) | Gut bioavailability | PBPK modeling | Dose proportionality in single‐dose PK studies | Food effect |
|---|---|---|---|---|---|---|
| Imatinib | II | I | IIa | |||
| M1 | Likely IV | Likely IV | ||||
| Crizotinib | IV | IV | ||||
| Gefitinib | II | IIa | ||||
| Trametinib | II | IIb | ||||
| Dabrafenib | II | IIb | ||||
| Erlotinib | II | IIb | ||||
| Lapatinib | IV | IV | i.v. PK data required for the calculation is not available | solubility and/or permeability | ||
| Vemurafenib | IV | IV | solubility and/or permeability | |||
| Pazopanib | II | IIb |
Every property/method is color‐coded for the drugs selected in this study to distinguish solubility‐limited drugs (red) from those that are not (green). On the far right, the food effects for these drugs are also color‐coded red or green to distinguish drugs that show a positive food effect from those that do not. Green – No solubility limitation: Dose number < 1 (BCS I/III), SLAD > clinical dose, gut bioavailability > 0.75, PBPK model not sensitive to increase in solubility, proportional, or more than dose proportional increase of AUC and Cmax in the food effect dose range, absence of positive food effect (AUC and Cmax fed/fasted ratio ≤ 1). Red – solubility‐limited: Dose number > 1 (BCS II/IV), SLAD < clinical dose, gut bioavailability < 0.75, better exposure in PBPK model using increased input solubility (solubility and/or permeability limitation of vemurafenib and lapatinib absorption is nonidentifiable), less than dose proportional increase of AUC and Cmax, ≥ 2‐fold positive food effect. Yellow – Anomalies: Food effect (~ 30%21 increase in AUC and Cmax) of gefitinib is confounded by a high intersubject variability, the food effect of erlotinib is inconsistent between the two periods of the 150 mg single‐dose food effect study.22
AUC, area under the plasma concentration time curve; BCS, Biopharmaceutical Classification System; Cmax, maximum plasma concentration; DCS, Developability classification system; i.v., intravenous; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetic; SLAD, solubility limited absorbable dose (amount of drug soluble in 500 mL fasted state simulated intestinal fluid, solubility criterion for DCS IIa/IIb classification (Eq. 3)).
Dose number, dose number < 1 indicates that the dose is soluble in 250 mL FaSSIF, solubility criterion for BCS classification (Eq. 2); gut bioavailability, the fraction of the administered dose reaching the portal vein, deconvoluted from the absolute bioavailability using the systemic clearance.
Although the BCS classification identifies all the 10 study compounds to have solubility‐limited absorption, the DCS is less conservative and able to predict lack of food effect in crizotinib, gefitinib, M1, and imatinib. However, this a very small number to draw any comparative conclusions.
When phase I clinical data are available, calculation of gut bioavailability and PBPK analysis become possible. Apart from erlotinib, gut bioavailability and PBPK analysis show similar trends of solubility‐limited absorption for the model drugs. Although a gut bioavailability < 1 can identify an intestinal drug loss, it cannot distinguish between mechanisms contributing to intestinal loss (e.g., solubility‐limitation, transporter‐mediated efflux, and gut metabolism). Hypothesis testing with PBPK analysis can distinguish among intestinal loss mechanisms, gut metabolism, and poor aqueous solubility, as exemplified by erlotinib (Figure S3). Another advantage of the PBPK approach over gut bioavailability method is that it can be applied even when i.v. PK data are not available, provided the oral PK profile is sufficiently well characterized. When PK data from the entire dose range of interest becomes available, it is possible to distinguish solubility limitation (less‐than‐dose‐proportional exposure) from gut metabolism and efflux (supra dose‐proportional exposure), and to confirm the PBPK analysis. The drugs identified by PBPK to have solubility‐limited absorption are also those that show a positive food effect. The extent of food effect correlates with the AUC and Cmax ratios (hypothetical BCS class I‐like solubility to best fit) from PBPK simulations. It should be noted, however, that the “in vivo solubility” derived from the PBPK modeling represents a minimum solubility. True in vivo solubility at a given dose may be even higher.
As pointed out earlier, establishing a dose‐exposure relationship is often challenged by high variability and insufficient dose groups, commonly encountered in oncology drug development. This is true for BCS IV vemurafenib and lapatinib, where the dose linearity seems to contradict the outcome from PBPK analysis. For both compounds, the dose range in the dose‐escalation studies does not cover that tested in food effect studies well, especially in the upper end. In the case of M1, less‐than‐dose‐proportional exposure indicates solubility‐limitation at doses much higher than the food effect study, which is why no food effect was expected.
Our study demonstrates that if the oral absorption of a drug candidate can be accurately identified as solubility‐limited, it is straightforward to predict the solubilization‐driven food effect.
Awareness of the impact and limitation of measured solubility on predicting the oral drug absorption of a drug candidate in the early development phase is critical to saving valuable resources in drug development. Not all poorly water‐soluble compounds defined under the biopharmaceutical framework have solubility‐limited oral absorption. Our analysis demonstrates that deconvolution of the key mechanisms driving intestinal loss with PBPK analysis reliably identifies those poorly water‐soluble drugs whose oral absorption is truly solubility‐limited and are, therefore, likely to show a positive food effect. Deconvolution of mechanisms contributing to intestinal loss can further complement existing deconvolution methods for an improved in vitro–in vivo correlation.
Our study supports a totality of evidence approach using early clinical data to guide decisions on conducting drug interaction studies with food and acid‐reducing agents (Figure 2). Further validation with a larger dataset could enhance confidence in the approach.
Figure 2.
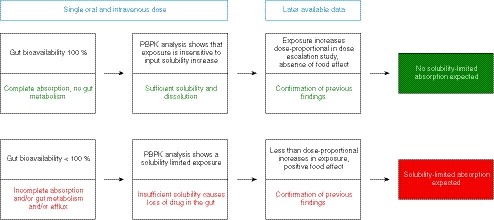
Schematic illustration of the proposed methods to assess solubility limited absorption behavior through a mechanistic analysis of human clinical pharmacokinetic data. When pharmacokinetic data from one i.v. and one oral dose are available, the gut bioavailability can be easily calculated to assess whether there is a loss of drug in the gut. Using a physiologically‐based pharmacokinetic (PBPK) model, it can be analyzed, if this loss of drug is related to insufficient solubilization. When pharmacokinetic data from ascending oral doses or food effect study become available, the outcomes of the PBPK modeling approach can be confirmed by the manner exposure behaves after increasing the dose or food intake.
Materials and Methods
Materials
Dabrafenib mesylate and vemurafenib were purchased from Chem Shuttle (Hayward, CA), lapatinib ditosylate monohydrate and pazopanib hydrochloride (HCl) from Ark Pharm (Arlington Heights and Libertyville), erlotinib HCl from Activate Scientific (Prien‐Chiemsee, Germany), imatinib mesylate from abcr GmbH (Karlsruhe, Germany), and trametinib *DMSO solvate from Asta Tech (Bristol, PA). Crizotinib and gefitinib were obtained from internal batches (Merck KGaA, Darmstadt, Germany). Simulated intestinal fluid powder (version 1) was purchased from http://Biorelevant.com (London, UK), and the water was purified by a Milli‐Q water purification system (Merck KGaA). All other chemicals, solvents at the liquid chromatography grades, and high‐performance liquid chromatography (HPLC) columns were obtained from Merck KGaA.
Model drug selection
Based on the clinical summary of oral anticancer drugs reported by Willemsen et al.,10 crizotinib, dabrafenib mesylate, erlotinib HCl, gefitinib, imatinib mesylate, pazopanib HCl, and trametinib were selected as model drugs due to their poor aqueous solubility (BCS II or IV) based on the US Food and Drug Administration's (FDA's) reviews, availability of i.v. PK data in humans, and the absence of special handling requirements for safety reasons. M1, a development compound, which meets the above selection criteria, was also included in the analysis. Although the i.v. PK data are not available for vemurafenib and lapatinib ditosylate, they were included in the analysis due to the strong food effect observed in the clinical studies. The analysis was performed at clinically relevant doses.
Human in vivo PK data
The clinical PK data used in the current study are summarized in Table S4. The i.v. PK and absolute bioavailability studies were used for the calculation of the gut bioavailability. PBPK simulations were performed at doses used in food effect studies using the observed profiles in the fasted state. The dose‐exposure proportionality information was obtained from single ascending dose studies.
In the absence of appropriate human in vivo PK data for erlotinib HCl and imatinib mesylate, closest substitutes were used. For erlotinib, the plasma clearance in healthy volunteers is not given.27 Instead, the renal clearance in patients after 100 mg i.v.28 was used to estimate the worst‐case (i.e., lowest) gut bioavailability. For imatinib, because the plasma‐concentration time profiles of the food studies are not available, the oral PK profiles in the absolute bioavailability study were used.29
Thermodynamic solubility measurement
The thermodynamic solubility of the model compounds in their marketed salt forms (in case of vemurafenib amorphous co‐precipitate from ground market product), was measured in FaSSIF pH 6.5 (containing 3 mM sodium taurocholate and 0.75 mM phospholipids) and SGF pH 1.6. An excess amount of drug substance was weighed into an Erlenmeyer flask with 10 mL of the media. The flask was incubated in a shaking water bath (250 movements per minute) at 37°C for 24 hours. Samples were taken after 24 hours and centrifuged at 20,817 g and 37°C for 5 minutes. The supernatant was diluted with an organic solvent and analyzed by HPLC. If possible, a largely universally applicable HPLC method using a gradient of eluent A (water + 0.1% formic acid) and eluent B (acetonitrile + 0.1% formic acid) and Chromolith Performance RP18e 100 × 3 mm column with a low flow of 0.85 mL/minute was used. Otherwise, an alternative method was applied using a Chromolith High Resolution RP18e 100 × 4,6 mm column, a flow rate of 3 mL/minute, and a gradient of eluent A (1,900 mL water + 100 mL acetonitrile + 2 mL trifluoroacetic acid) and eluent B (1,900 mL acetonitrile + 100 mL water + 2 mL trifluoroacetic acid).
Effective human intestinal permeability from apparent permeability in Caco‐2 cells
Permeability across a TC7 Caco‐2 monolayer on a microporous polycarbonate membrane filter was measured bidirectionally (i.e., apical (A) → basolateral (B) and B → A) in a 24‐well plate and up to 5 compounds per well. The apparent passive permeability (geometric mean of Papp A → B and B → A) was measured in the presence of cyclosporin A that inhibits P‐glycoprotein. Hank's balanced salt solution (pH 7.4) was used as the reservoir for apical and basolateral matrices. The compounds were added as dimethyl sulfoxide stock solutions resulting in a final drug concentration of 1 µM. The apical and basolateral drug concentrations after the reaction were quantified via liquid‐chromatography mass spectrometry. A factor of 25 was used to scale the measured Caco‐2 permeability to the effective human intestinal permeability.
Calculated effective human intestinal permeability
The effective human intestinal permeability was calculated, using log P, the polar surface area and the number of hydrogen bond donors, according to Eq. 1.30 The input values used for the calculation are shown in Table S5.
| (1) |
BCS/DCS classification
The BCS classification presented in literature10 is summarized in Table 1. Additionally, the dose number (D o) was calculated to confirm whether the highest clinical dose (M 0) can be completely dissolved in 250 mL intestinal fluid according to Amidon et al. 3 with Eq. 2 using the thermodynamic solubility in FaSSIF (Cs) and volume (V 0) of 250 mL.
| (2) |
The compounds were further classified based on the DCS classification by Butler et al.,4 calculating the SLAD using Eq. 3 with the thermodynamic solubility in FaSSIF and volume (V) of 500 mL, where M p is equal to A n (Eq. 4)3 for high permeability compounds (calculated P eff > 1 × 10‐4 cm/s) and equal to 1 for others. A n was calculated from Eq. 4 using a tube radius R of 1 cm3 and residence time (t res) of 3.32 hours.4
| (3) |
| (4) |
PBPK modeling
A generic whole‐body PBPK model built in MATLAB software (version R2017a; The MathWorks, Natick, MA) has been used for a line‐shape analysis of the observed i.v. and oral PK profiles at doses used in their clinical food effect studies, as shown in Figure 3. The in vitro microsomal clearance generally tends to underpredict the human clearance.31 The clearance along with the distribution parameters were estimated by optimizing CLint and K p factors to best fit the i.v. PK profiles. These parameters are uncorrelated and have unique influences on the line shape. Hence, confidence in the estimated parameters is high.13, 25 This principle of obtaining CLint and K p is similar to estimating CLint using retrograde calculation and K p factor in Simcyp (https://www.certara.com/) in a top‐down analysis.
Figure 3.
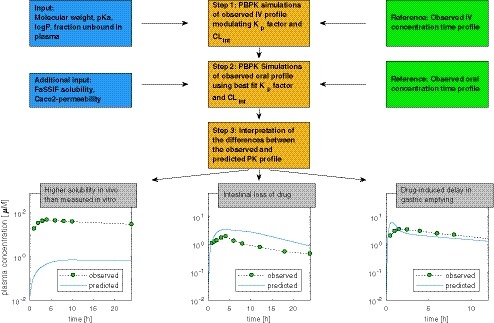
Schematic illustration of the physiologically‐based pharmacokinetic (PBPK) modeling approach. Clint, intrinsic microsomal clearance; K p factor, multiplicative factor, to scale the tissue distribution coefficients; logP, decadic logarithm of the partition coefficient; PK, pharmacokinetic; pKa, acid dissociation constant.
Because the i.v. PK data of vemurafenib and lapatinib ditosylate were not available, the clearance was estimated from the elimination rate constant (k e) (Eq. 5) based on the slope of the elimination phase in the oral PK profiles (i.e., the linear part of the log PK profiles at time points well beyond the absorption phase). This is based on the assumption that the volume of distribution per kilogram body weight, when corrected for plasma protein binding, is the same across different species,32 as shown in Table S3.
| (5) |
Clearance and volume of distribution along with FaSSIF solubility and effective permeability were used to simulate oral PK profiles. The input parameters are shown in Table S2. Effective permeability was derived from apparent permeability in Caco‐2 cells or calculated from structural properties. Under the assumption of linear PK, the difference between the observed and simulated AUC can generate an understanding of the mechanisms underlying exposure (Figure 3). A simulated profile that cannot match the much steeper upswing of the observed profile and is characterized by a poor sensitivity to permeability, identifies an in vivo solubility that is greater than the measured solubility used as input.
The solubility value that best captures the observed profile represents the least in vivo solubility. The actual in vivo solubility could be higher. For BCS class IV drugs and those with borderline permeability, a higher permeability could have also achieved a similar good fit to the observed exposure.
PBPK simulations repeated with a hypothetically high solubility (dose/250 mL, analog BCS class I criteria, but at least 1 mg/mL) can identify any solubility‐limited drug exposure. If the AUC or Cmax ratio of exposure simulated by PBPK model using hypothetical BCS class I‐like solubility to exposure derived from good fit is 1 or close to 1, exposure is not solubility‐limited. A significantly higher ratio identifies solubility‐limited exposure.
For drugs that induce gastric emptying delay, it may be necessary to reduce the gastric emptying rate in order to match the observed profile (Figure 3). When the observed profile is characterized by a significantly lower AUC compared with the simulated profile, gut metabolism or efflux could be responsible for the loss of the compound in the intestine (Figure 3). These two intestinal loss mechanisms cannot be distinguished.
Estimation of gut bioavailability
Oral bioavailability is a product of the fraction absorbed into the enterocytes (F a), the fraction escaping intestinal metabolism (F g), and the fraction escaping hepatic metabolism (F h) (Eq. 6).34 The product of F a × F g, the gut bioavailability, represents the fraction of the administered dose reaching the portal vein.
When PK data from i.v. and single‐dose oral studies are available, the gut bioavailability can be calculated according to Eq. 7, using a liver blood flow (Q) of 90 L/hour,33 assuming that the systemic clearance (Cl) is driven only by hepatic metabolism.
| (6) |
Rearranging Eq. 6 leads to Eq. 7:
| (7) |
Analysis of dose‐exposure PK linearity and food effect
The dose‐exposure linearity of the model drugs based on dose‐escalating studies obtained from the FDA's Clinical Pharmacology and Biopharmaceutics Reviews and the reported food effect are summarized in Table 3.
Funding
This work was supported by Merck Healthcare KGaA, Darmstadt, Germany.
Conflict of Interest
C.F., K.W., M.S., H.B., H.D., and S.‐A.P. are employees of Merck Healthcare KGaA, Darmstadt, Germany. D.S. is employed by Merck KGaA.
Author Contributions
C.F., D.S., S.A.P., and H.D. wrote the manuscript. S.‐A.P. designed the research. C.F. performed the research. All authors analyzed the data.
Supporting information
Figure S1. Simulated and observed intravenous and oral (Caco‐2 permeability) profiles.
Figure S2. Dose proportionality of AUC and Cmax.
Figure S3. Hypothesis testing with physiologically based pharmacokinetic (PBPK) modeling to identify that the loss of erlotinib in the gut is mediated by gut metabolism and not due to solubility‐limitation.
Table S1. Potential issues arising from poor drug solubility and their consequences.
Table S2. Summary of input parameters for the physiologically based pharmacokinetic simulations.
Table S3. Preclinical data to estimate the human clearance and volume of distribution of lapatinib and vemurafenib.
Table S4. Clinical pharmacokinetic data analyzed to assess solubility‐limited absorption.
Table S5. Input parameters for calculation of the effective permeability of the 10 model compounds.
Acknowledgments
The authors would like to thank Anita Nair for her support and the scientific discussions. The authors acknowledge the leaderships of the Merck Healthcare KGaA departments, Chemical Pharmaceutical Development, as well as the former Quantitative Pharmacology and Drug Disposition for providing the framework of this research. The authors would like to thank Annabel Mehl for her support in the laboratory and during the literature research for pharmacokinetic data. The authors also thank the colleagues from Analytical Development for the technical support and the colleagues from DMPK for providing the Caco‐2 permeability data.
References
- 1. Di, L. , Fish, P.V. & Mano, T. Bridging solubility between drug discovery and development. Drug Discov. Today 17, 486–495 (2012). [DOI] [PubMed] [Google Scholar]
- 2. Stegemann, S. , Leveiller, F. , Franchi, D. , De Jong, H. & Lindén, H. When poor solubility becomes an issue: from early stage to proof of concept. Eur. J. Pharm. Sci. 31, 249–261 (2007). [DOI] [PubMed] [Google Scholar]
- 3. Amidon, G.L. , Lennernas, H. , Shah, V.P. & Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12, 413–420 (1995). [DOI] [PubMed] [Google Scholar]
- 4. Butler, J.M. & Dressman, J.B. The Developability Classification System: application of biopharmaceutics concepts to formulation development. J. Pharm. Sci. 99, 4940–4954 (2010). [DOI] [PubMed] [Google Scholar]
- 5. Morgan, P. et al. Impact of a five‐dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discov. 17, 167 (2018). [DOI] [PubMed] [Google Scholar]
- 6. Yazdanian, M. , Briggs, K. , Jankovsky, C. & Hawi, A. The “high solubility” definition of the current FDA guidance on biopharmaceutical classification system may be too strict for acidic drugs. Pharm. Res. 21, 293–299 (2004). [DOI] [PubMed] [Google Scholar]
- 7. Faassen, F. & Vromans, H. Biowaivers for oral immediate‐release products. Clin. Pharmacokinet. 43, 1117–1126 (2004). [DOI] [PubMed] [Google Scholar]
- 8. Brunton, L.L. , Chabner, B.A. & Knollmann, B.C . Goodman and Gilman's The Pharmacological Basis of Therapeutics 12th edn (McGraw‐Hill Education/Medical, New York, NY, 2011). [Google Scholar]
- 9. Lentz, K.A. Current methods for predicting human food effect. AAPS J. 10, 282–288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Willemsen, A.E. , Lubberman, F.J. , Tol, J , Gerritsen, W.R. , van Herpen, C.M. & van Erp, N.P. Effect of food and acid‐reducing agents on the absorption of oral targeted therapies in solid tumors. Drug Discov. Today 21, 962–976 (2016). [DOI] [PubMed] [Google Scholar]
- 11. Kuentz, M. , Nick, S. , Parrott, N. & Röthlisberger, D. A strategy for preclinical formulation development using GastroPlus™ as pharmacokinetic simulation tool and a statistical screening design applied to a dog study. Eur. J. Pharm. Sci. 27, 91–99 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Thummel, K.E. et al. Oral first‐pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A‐mediated metabolism. Clin. Pharmacol. Ther. 59, 491–502 (1996). [DOI] [PubMed] [Google Scholar]
- 13. Peters, S.A. Identification of intestinal loss of a drug through physiologically based pharmacokinetic simulation of plasma concentration‐time profiles. Clin. Pharmacokinet. 47, 245–259 (2008). [DOI] [PubMed] [Google Scholar]
- 14. Jones, H. et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 97, 247–262 (2015). [DOI] [PubMed] [Google Scholar]
- 15. Peters, S.A. Evaluation of a generic physiologically based pharmacokinetic model for lineshape analysis. Clin. Pharmacokinet. 47, 261–275 (2008). [DOI] [PubMed] [Google Scholar]
- 16. Budha, N.R. et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: Is pH‐dependent solubility the Achilles heel of targeted therapy? Clin. Pharmacol. Ther. 92, 203–213 (2012). [DOI] [PubMed] [Google Scholar]
- 17. GlaxoSmithKline (GSK) . Clinical Study Report 113479 ‐ Determination of the Absolute Bioavailability of GSK2118436 Following a Single Oral Dose Co‐Administered with an Intravenous Radiolabelled Microtracer of GSK2118436 in Subjects with BRAF Mutant Solid Tumors <https://www.gsk-clinicalstudyregister.com/study/113479?search=study%26search_terms=113479#csr> (2012). Accessed August 31, 2017.
- 18. US Food and Drug Administration . Pazopanib (votrient) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000TOC.cfm> (2008). Accessed August 12, 2016.
- 19. Hurwitz, H.I. et al. Phase I trial of pazopanib in patients with advanced cancer. Clin. Cancer Res. 15, 4220–4227 (2009). [DOI] [PubMed] [Google Scholar]
- 20. US Food and Drug Administration . Crizotinib (Xalkori) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000TOC.cfm> (2011). Accessed July 6, 2016.
- 21. Swaisland, H.C. et al. Single‐dose clinical pharmacokinetic studies of gefitinib. Clin. Pharmacokinet. 44, 1165–1177 (2005). [DOI] [PubMed] [Google Scholar]
- 22. Ling, J. , Fettner, S. , Lum, B.L. , Riek, M. & Rakhit, A. Effect of food on the pharmacokinetics of erlotinib, an orally active epidermal growth factor receptor tyrosine‐kinase inhibitor, in healthy individuals. Anticancer Drug. 19, 209–216 (2008). [DOI] [PubMed] [Google Scholar]
- 23. Ouellet, D. et al. Effects of particle size, food, and capsule shell composition on the oral bioavailability of dabrafenib, a BRAF inhibitor, in patients with BRAF mutation‐positive tumors. J. Pharm. Sci. 102, 3100–3109 (2013). [DOI] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Trametinib (Mekinist) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000TOC.cfm> (2013). Accessed January 11, 2017.
- 25. Peters, S.A. & Hultin, L. Early identification of drug‐induced impairment of gastric emptying through physiologically based pharmacokinetic (PBPK) simulation of plasma concentration‐time profiles in rat. J. Pharmacokinet. Pharmacodyn. 35, 1–30 (2008). [DOI] [PubMed] [Google Scholar]
- 26. Ling, J. et al Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 34, 420–426 (2006). [DOI] [PubMed] [Google Scholar]
- 27. Ranson, M. et al. A phase I dose‐escalation and bioavailability study of oral and intravenous formulations of erlotinib (Tarceva®, OSI‐774) in patients with advanced solid tumors of epithelial origin. Cancer Chemother. Pharmacol. 66, 53–58 (2010). [DOI] [PubMed] [Google Scholar]
- 28. Frohna, P. et al. Evaluation of the absolute oral bioavailability and bioequivalence of erlotinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in a randomized, crossover study in healthy subjects. J. Clin. Pharmacol. 46, 282–290 (2006). [DOI] [PubMed] [Google Scholar]
- 29. Peng, B. et al. Absolute bioavailability of Imatinib (Glivec®) orally versus intravenous infusion. J. Clin. Pharmacol. 44, 158–162 (2004). [DOI] [PubMed] [Google Scholar]
- 30. Winiwarter, S. , Bonham, N.M. , Ax, F. , Hallberg, A. , Lennernäs, H. & Karlén, A. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J. Med. Chem. 41, 4939–4949 (1998). [DOI] [PubMed] [Google Scholar]
- 31. Brown, H.S. , Griffin, M. & Houston, J.B. Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab. Dispos. 35, 293–301 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Berry, L.M. , Li, C. & Zhao, Z. Species differences in distribution and prediction of human Vss from preclinical data. Drug Metab. Dispos. 39, 2103–2116 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Davies, B. & Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 10, 1093–1095 (1993). [DOI] [PubMed] [Google Scholar]
- 34. Rowland, M. & Tozer, T.N. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications 4th edn (Lippincott Williams & Wilkins, Philadelphia, PA, 2010). [Google Scholar]
- 35. Peng, B. , Lloyd, P. & Schran, H. Clinical pharmacokinetics of Imatinib. Clin. Pharmacokinet. 44, 879‐894 (2005). [DOI] [PubMed] [Google Scholar]
- 36. US Food and Drug Administration . Dabrafenib (Tafinlar) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/202806Orig1s000TOC.cfm> (2013). Accessed February 20, 2017.
- 37. Swaisland, H. et al. Pharmacokinetics and tolerability of the orally active selective epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in healthy volunteers. Clin. Pharmacokinet. 40, 297–306 (2001). [DOI] [PubMed] [Google Scholar]
- 38. US Food and Drug Administration . Erlotinib (Tarceva) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21-743_Tarceva.cfm> (2004). Accessed January 25, 2017.
- 39. Koch, K.M. et al. Effects of esomeprazole on the pharmacokinetics of lapatinib in breast cancer patients. Clin. Pharmacol. Drug Develop. 2, 336–341 (2013). [DOI] [PubMed] [Google Scholar]
- 40. US Food and Drug Administration . Vemurafenib (zelboraf) clinical pharmacology and biopharmaceutics review <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202429Orig1s000ClinPharmR.pdf> (2011). Accessed January 27, 2017.
- 41. US Food and Drug Administration . Gefitinib (Iressa) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021399_iressa.cfm> (2002). Accessed May 17, 2017.
- 42. US Food and Drug Administration . Lapatinib (Tykerb) clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022059s000TOC.cfm> (2007). Accessed July 7, 2018.
- 43. US Food and Drug Administration . Imatinib (Gleevec) clinical pharmacology and biopharmaceutics review <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/021588s000_GleevecTOC.cfm> (2003). Accessed January 25, 2017.
- 44. Scheffler, M. , Di Gion, P. , Doroshyenko, O. , Wolf, J. & Fuhr, U. Clinical pharmacokinetics of tyrosine kinase inhibitors. Clin. Pharmacokinet. 50, 371–403 (2011). [DOI] [PubMed] [Google Scholar]
- 45. European Medicines Agency . European Public Assessment Report (EPAR) Glivec ‐ summary of product characteristics (Annex 1) <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000406/WC500022207.pdf> (2009). Accessed August 31, 2017.
- 46. Xu, H. et al. Evaluation of crizotinib absolute bioavailability, the bioequivalence of three oral formulations, and the effect of food on crizotinib pharmacokinetics in healthy subjects. J. Clin. Pharmacol. 55, 104–113 (2015). [DOI] [PubMed] [Google Scholar]
- 47. Cox, D.S. et al. Evaluation of the effects of food on the single‐dose pharmacokinetics of trametinib, a first‐in‐class MEK inhibitor, in patients with cancer. J. Clin. Pharmacol. 53, 946–954 (2013). [DOI] [PubMed] [Google Scholar]
- 48. Koch, K.M. et al. Effects of food on the relative bioavailability of lapatinib in cancer patients. J. Clin. Oncol. 27, 1191–1196 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ribas, A. et al. The effects of a high‐fat meal on single‐dose vemurafenib pharmacokinetics. J. Clin. Pharmacol. 54, 368–374 (2014). [DOI] [PubMed] [Google Scholar]
- 50. Heath, E.I. et al. A phase I study of the pharmacokinetic and safety profiles of oral pazopanib with a high‐fat or low‐fat meal in patients with advanced solid tumors. Clin. Pharmacol. Ther. 88, 818–823 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Simulated and observed intravenous and oral (Caco‐2 permeability) profiles.
Figure S2. Dose proportionality of AUC and Cmax.
Figure S3. Hypothesis testing with physiologically based pharmacokinetic (PBPK) modeling to identify that the loss of erlotinib in the gut is mediated by gut metabolism and not due to solubility‐limitation.
Table S1. Potential issues arising from poor drug solubility and their consequences.
Table S2. Summary of input parameters for the physiologically based pharmacokinetic simulations.
Table S3. Preclinical data to estimate the human clearance and volume of distribution of lapatinib and vemurafenib.
Table S4. Clinical pharmacokinetic data analyzed to assess solubility‐limited absorption.
Table S5. Input parameters for calculation of the effective permeability of the 10 model compounds.