

Abstract
As the field of artificial intelligence and machine learning (AI/ML) for drug discovery is rapidly advancing, we address the question “What is the impact of recent AI/ML trends in the area of Clinical Pharmacology?” We address difficulties and AI/ML developments for target identification, their use in generative chemistry for small molecule drug discovery, and the potential role of AI/ML in clinical trial outcome evaluation. We briefly discuss current trends in the use of AI/ML in health care and the impact of AI/ML context of the daily practice of clinical pharmacologists.
The field of artificial intelligence and machine learning (AI/ML) has witnessed many changes recently, particularly with respect to deep learning (DL) methods (Figure 1; Table 1), enabled both by hardware improvements and the availability of very large training datasets (“big data”). As biomedical data are becoming increasingly more available in ML‐ready digital formats, due to technological advances, public policy efforts, and community engagement, it is now possible to deploy AI/ML techniques to support healthcare research and services. This includes, for example, risk‐based guidance with DL‐models used for predicting avoidable hospital readmissions, clinical trial participation selection with optimized patient selection and recruiting techniques, often paired with more effective patient monitoring during clinical trials, or medical devices accessing individual patient data and informing medical decisions.
Figure 1.
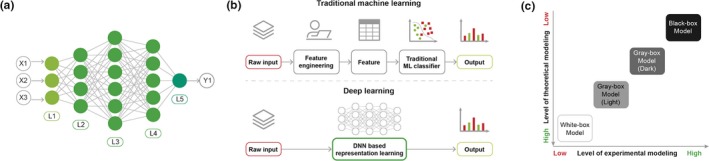
(a) A deep neural network (DNN) is a collection of neurons organized in a sequence of multiple layers. There are three types of layers. The input layer (L1), which contains the features extracted from the input data. Second, there are the hidden layers (L2, L3, and L4). Each of them is a set of nodes acting as computational units. The neurons implement a nonlinear mapping from the input to the output. This mapping is learned from the data by adapting the weights of each neuron. The output layer (L5) is similar to the hidden layer but produces the final output. The number of nodes in the output layer depends on the type of task to be solved. (b) Traditional machine learning (ML) relies on feature engineering, which transforms raw data into features that better represent the predictive task. DNNs discover the mapping from representation to output and learn the most informative features from data. This ability to automatically extract high‐dimensional abstract information from a data without the need to hand‐design features and the flexibility and adaptability of the model architecture are two advantages of DNN in the context of molecular design. (c) Depending on the balance between the levels of experimental and theoretical modeling, the outputs of ML methods can be difficult for humans to interpret (Table 1). For standard ML, the features are interpretable and the role of the algorithm is to map the representation to output. An interpretation for a decision made can be retrieved by scrutinizing the inference process. For deep learning methods, although the input domain of the DNN is also interpretable, the learned internal representations and the flow of information through the network are harder to analyze and modules must be implemented to interpret the output.
Table 1.
A summary of common terms in machine learning
| Machine learning (ML): ML refers to algorithms that learn from and make predictions on data by building a model from sample inputs. ML is used for computing tasks where designing and programming explicit algorithms with good performance is difficult or infeasible. Today, most common traditional ML methods are k‐nearest neighbors (kNNs), logistic regression (LR), support vector machines (SVMs), gradient boosting machines (GBMs), and random forest (RF). The performance of ML methods can vary depending on the type of task (regression or classification), types, and amount of data to handle. |
| Deep learning (DL): DL refers to a class of ML techniques that exploit many layers of nonlinear computational units to model complex relationships among data. These architectures, composed of multiple layers, are commonly called deep neural networks (DNNs), or sometimes stacked neural networks. The difference between the single‐hidden‐layer artificial neural networks (ANNs) and DNNs is the depth; that is, the number of layers of nodes through which data are processed. Usually, more than three layers (including input and output) qualify as “deep” learning. Thus, “deep” is a technical term that means more than one hidden layer. DNNs use a cascade of many layers of nonlinear processing units for feature extraction. Each successive layer uses the output from the previous layer as input. Higher level features are derived from lower level features to form a hierarchical representation. This hierarchy of features is called a deep architecture. These methods are capable of learning multiple levels of representations that correspond to different levels of abstraction. These levels form a hierarchy of concepts. |
| Generative Adversarial Networks (GANs): GANs are structured, probabilistic models for generating data. Being an unsupervised technique, GANs can be used to generate data similar to the dataset that the GAN was trained on. A GAN consists of two DNNs called Discriminator and Generator. The discriminator estimates the probability that a given sample is coming from the real dataset. It works as a critic and is optimized to distinguish the fake samples from the real ones. The generator outputs synthetic samples using a noise variable as input following a distribution. It is trained to capture the real data distribution so that it can generate samples with distribution, which are as real as possible. The generator should improve its output until the discriminator is unable to distinguish the generated output from the real ones. The two models compete against each other during the training process. The goal of the generator is to try to trick the discriminator while the discriminator attempts to not be cheated. This process happening between the two models motivates them to improve their functionalities in order to obtain generated samples indistinguishable from the real data. |
| Reinforcement learning (RL): RL refers to goal‐oriented algorithms, which learn how to attain a complex objective or maximize along a particular dimension over many steps. RL algorithms operate in a delayed return environment, where it is not straightforward to figure out which action leads to which outcome over many time steps. Thus, RL aims at correlating immediate actions with the delayed returns they produce. The reinforcement takes place in the sense that RL algorithms are penalized when making the wrong decisions, and they get rewarded when making the right one. RL algorithms are expected to increase performance in more ambiguous, real‐life environments. |
| Interpretability: Understanding the output of a model in terms of its inputs and intrinsic properties. Interpretability is the idea that humans should understand, at some level, the decisions being made by algorithms. Interpretability is useful to understand whether a model is working properly and how the model could be modified to be improved. |
| Knowledge graphs: A knowledge graph is a database that stores information in a graphical format and can be used to generate a graphical representation of the relationships between any of its data points. The advantage over the older, relational style database is that the relationships between any data points can be calculated far more quickly and with less compute power overheads, regardless of whether the data points fit neatly together into a table. |
This begs the question, what is the potential impact of AI/ML developments in the area of clinical pharmacology? In its broadest terms, clinical pharmacology can be defined1 as “all aspects of the study and use of drugs in humans,” and covers a wide range of disciplines, from molecular pharmacology and the way drugs act at the molecular level, to epidemiology and related disciplines at the population level. Here, we briefly discuss specific AI/ML trends in target identification and small molecule drug discovery and their potential impact for clinical pharmacologists in the next decade.
AI/ML in Target Discovery
To date, the choice of novel biomolecular drug targets in pharmaceutical research and development (i.e., target selection or target prioritization) remains an uncertain process. Although the majority of therapeutics are associated with, and act via protein targets, novel mechanistic target classes and procedures could be successfully exploited to cure disease (Figure 2). In a broad definition, “drug targets” are material entities with a quantifiable mass, typically macromolecules that physically interact with the therapeutic agent. Whereas drug targets are characteristically native to the biological system on which the drug acts (“native” can also imply a disease state such as mutated or fused genes/proteins), a variety of drug targets are also not native (e.g., microbial infections or parasitic infestations). Furthermore, the physical interaction between therapeutic agents and the intended targets causes detectable effects in living systems, although the clinical outcomes may be due to downstream effects.
Figure 2.
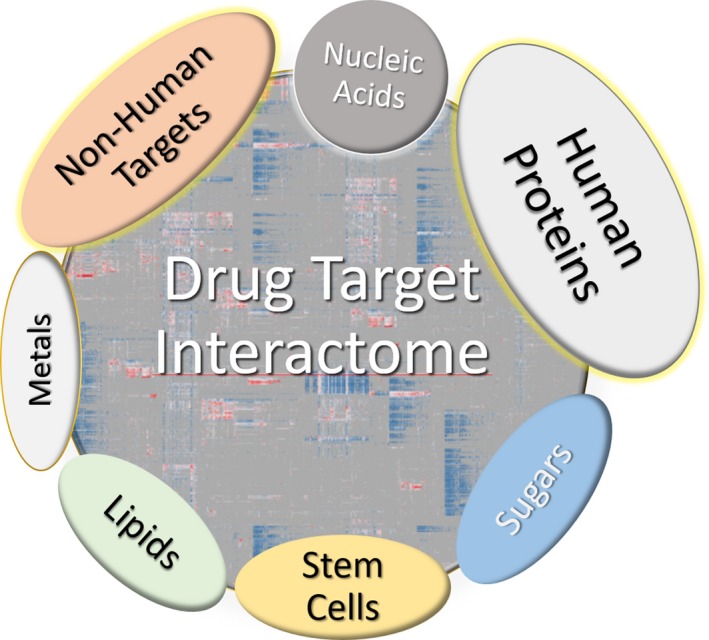
Therapeutic target categories. Although sugars and, to a great extent, lipids are not currently targeted by approved drugs, they are the subject to drug discovery research.
Against this backdrop, it is important to accurately map the interactions between approved drugs and their efficacy targets (i.e., the targets through which medicines exert their therapeutic effect).2 Currently, 11% of the human proteome is annotated with small molecule probes, whereas one in three proteins remains understudied (i.e., their function and role in human biology and disease is not understood).3 The fact that in‐depth biological insights are limited to a small number of proteins4 is (for the most part) due to a causality dilemma: The study of specific genes and proteins requires specific biomolecular tools (e.g., antibodies, chemical probes, etc.). For understudied genes, the availability of specific tools is unlikely. The lack of such tools perpetuates the lack of interest in exploring the dark corners of the genome.
With target prioritization in mind, there is an increasing trend to apply network (topological) methods, such as knowledge graphs (Table 1), to impute novel protein‐phenotype or protein‐function associations by seeking different network paths that connect proteins/genes to specific properties.3 One such methodology, MetaPath,5 can be used to prioritize novel drug targets. These AI/ML techniques are well suited for big data (e.g., ‐omics datasets, phenotypic and expression data, disease associations, etc.). Such methods can identify combinations of genetic variants or abnormalities that cause disease, including cases where causal genes are known or unknown.6 Indeed, progress in data integration combined with novel AI/ML algorithms and disease causality modeling7 will likely shift the paradigm and establish unbiased ways for target selection and prioritization.
AI/ML for Clinical Trial Outcome Prediction and Design
The failure rate of clinical trials contributes to the inefficiency of the drug development cycle: Clinical trials can take up to 7–10 years, with costs of $1.46 billion of $2.56 billion in capitalized costs for bringing a new drug to market. Each failed trial impairs not only the investment into the trial itself but also preclinical development costs, rendering the loss per failed clinical trial at $800 million to $1.4 billion. Inefficient patient selection and recruiting, combined with the inability to effectively monitor patients during trials, are two of the main causes for high failure rates. AI/ML technologies have begun to be deployed within key steps of clinical trial design from study preparation to execution, leading to trial success rate improvement.8
For instance, IBM Watson has developed a system for Clinical Trial Matching, which uses the large quantity of structured and unstructured patient electronic medical record data and the abundance of available trials to create detailed profiles of clinical findings for the patients to compare to trial eligibility criteria. As the system incorporates all the complex protocol criteria to consider, it eliminates the need to manually sort through and analyze complex enrollment criteria and enables clinicians to optimize their search for clinical trials for an eligible patient or for finding patients eligible for a given trial. Those two tasks are otherwise challenging and time‐consuming. The improvement in screening efficiency and more effective patient recruitment help increase clinical trial enrollment targets. This system can also be useful to help manage and track patients through the recruitment process and share progress across networks in near real‐time.
Clinical trials outcome prediction via AI/ML models could further reduce the cost of clinical trials by improving success rate. The purpose of such models is to predict the likelihood of success by analyzing compound responses, side effects, etc. For instance, a DL‐based model was used to predict the outcomes of phase I/II clinical trials.9 This pipeline predicted the drug‐induced pathway activation and related side effects. Side effect probabilities and pathway activation scores were used to train a model to predict clinical trial outcomes. Separate models were used to predict bioavailability and target‐related properties, such as tissue selectivity, and chemical features combined with target‐based features to predict clinical drug toxicity.10 We anticipate that improved AI/ML models could be used for clinical trials outcome prediction.
Other projects, such as the Virtual Physiological Human, aim to develop in silico approaches to support in silico clinical trials. 11 The development of such AI/ML frameworks can be used to synthesize the physiological and pathological information about a patient at different scales of space and time, with the ultimate aim of producing patient‐specific and subpopulation‐specific predictions for diagnosis, prognosis, posology, and treatment planning.
Generative Chemistry: AI/ML for Small Molecule Drug Discovery
One of the first applications of AI/ML in the context of early drug discovery was the quantification of “druglikeness,” which attempts to mimic the intuition of medicinal chemists in estimating the likelihood that novel chemical structures are more likely to become “drugs.” Although “drug (in the regulatory sense) is not an intrinsic property of chemicals,”12 the process of estimating druglikeness using AI/ML remains a practical chemical space navigation tool, as the number of possible drug‐like chemicals can exceed 1023. Regardless of algorithms, such methods do not predict “drug” as an attribute, but rather score a multiparametric similarity, interpreted as “these molecules are similar to other molecules that chemists consider to be of pharmaceutical interest.” The lack of true negative data has hindered a more rigorous approach in this direction, because > 40% of “non‐drugs” do contain druglike fragments,13 due to the fact that chemical vendors adapt their own AI/ML druglikeness models to boost sales to pharmaceutical companies. Which raises the question, can AI/ML suggest what compounds to make next? Considering the encouraging results obtained with Generative Adversarial Networks (GANs)14 combined with reinforcement learning (RL)15 (Table 1) and other architectures, such as variational auto‐encoder, AI/ML has gained credibility as an alternative to standard drug discovery tools.
Indeed, GAN/RL methods have successfully been applied to design generate molecules in silico, given a desired set of chemical properties. From the first DL‐based molecular generator using an adversarial auto‐encoder to generate molecular fingerprints,16 significant progress was made in several areas: First, molecular representation. Beginning with the Wisswesser line notation, several ways to encode molecular structures are available, such as chemical fingerprints,17 Simplified Molecular Input Line System (SMILES),18 and molecular graphs.19 Thus, GAN methods using fingerprints,16 SMILES,20 chemical graphs, and 3D wave‐transforms21 have been proposed, with different strengths concerning the amount and type of structural information and features kept or lost during encoding. Second, molecular property profiles: GAN architectures need to generate not just valid chemical structures, but also molecules matching certain bioactivity, novelty, diversity profiles, and, preferably, other features. These constraints led to the emergence of generative models, which eventually were combined with RL (Figure 3). Various architectures have been designed so far (Figure 4). These methods ensure that the generated molecules have suitable chemical properties using filters, reward functions, and evaluation metrics run in parallel with model architectures. Despite the increasing number of models, architectures, data types, and learning methods, it is difficult to assess model performance without a “gold standard” benchmark and evaluation metrics. Molecular Sets (MOSES) is one such platform22 that offers ways to evaluate and compare generative model performance.
Figure 3.
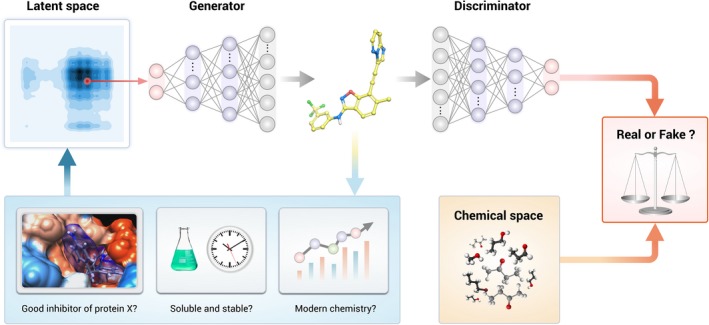
A Generative Adversarial Network–reinforcement learning (RL) model is made of three main components. (1) The generator captures the real‐data and generates synthetic samples as real as possible. (2) The discriminator estimates the probability that a sample is coming from the real dataset. The generator should improve its output until the discriminator is unable to distinguish the generated from the real ones and the discriminator is optimized to distinguish the synthetic samples from the real ones. (3) The RL module rewards the discriminator based on how accurate it is in distinguishing the synthetic samples from the real ones. The RL optimization procedure drives the hidden space, a multimodal distribution of compressed molecular structures used for the generation of novel compounds, toward the desired objective of generating novel molecules with specific properties.
Figure 4.
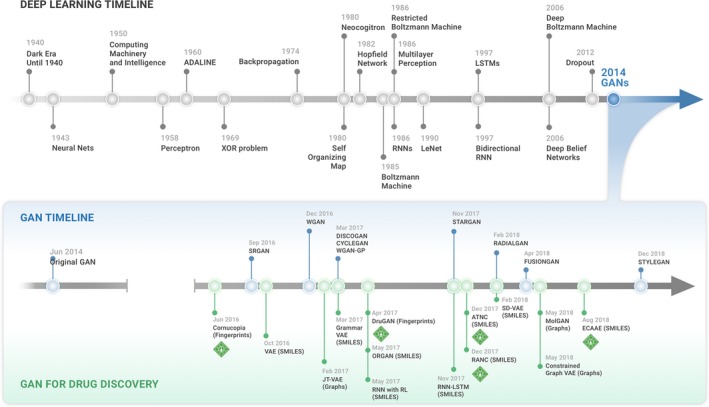
Timeline summarizing the key advances in designing molecular generator models. The creation of the Generative Adversarial Network (GAN) architecture was a turning point as this enabled to build architectures with unprecedented generative capabilities. SMILES, Simplified Molecular Input Line System; VAE, variational auto‐encoder.
An important milestone for the use of generative chemistry in drug discovery was recently accomplished, demonstrating that generated molecules can be synthesized, are active in vitro, metabolically stable, and show in vivo activity in disease‐relevant models. The first example of an in vitro active molecule generated using the conditional adversarial autoencoder GAN was the JAK3 inhibitor.23 Another generative model, Generative Tensorial Reinforcement Learning (GENTRL) produced in vivo active DDR1 and DDR2 inhibitors.24 Here, DDR1 and DDR2 inhibitors with different property and selectivity profiles were assayed in vitro, followed by in vivo mouse experiments that validate the pharmacokinetics of DDR1 inhibitor. To date, the design/make/test/evaluate feedback loop in drug discovery can afford a limited number of cycles because each step is resource and time‐consuming. Given these promising in vitro and in vivo results derived from generative reinforcement learning technologies, AI/ML methods are poised to become an integral part of the drug design cycle. However, the time it takes to validate the molecules produced by generative and other ML systems by far exceeds the time it takes to build and train the models. In addition, while multiple groups worldwide are working on the automated synthesis and in vitro validation machinery, synthesis and experimental validation will remain the main gating factor for the transformation of drug discovery using AI/ML over the next few years.
AI/ML in Clinical Pharmacology: Challenges and Outlook
Currently, the key challenges in drug discovery are related to chemical structures (e.g., toxicity, side effects, or even intellectual property), choosing the right drug target or the appropriate dosage for a specific patient subpopulation. Despite major advances in clinical pharmacology, the silo‐ing of late preclinical and clinical data remains a critical issue blocking the deployment of AI/ML technologies. The pharmaceutical industry does not typically share pharmacokinetic and pharmacodynamic measurements for most of the candidate drugs or their combinations, unless the drugs are approved for human use. Indeed, unlike other areas of research and development, only a small fraction of the drug discovery data overall is available for training AI/ML models. This is particularly with respect to true negative data. The problem pervades not only for, for example, discriminating “drugs” from “nondrugs,” but also with respect to AI/ML models for novel target‐disease associations, for understanding why clinical trials were discontinued, why drugs were withdrawn, or even accessing complete datasets from successful clinical trials. Nonetheless, there is no doubt that clinical development is currently moving through major transformation and that the usage of AI/ML is accelerating.
On a daily basis, “a clinical pharmacologist is a medically qualified practitioner who teaches, does research, frames policy, and gives information and advice about the actions and proper uses of medicines in humans, and implements that knowledge in clinical practice.”1 We can safely state that recent AI/ML developments are likely to impact clinical pharmacologists at all levels in the next decade:
In teaching, the use of data‐intensive methodologies for example, literature searching and processing, as well as interactions with on‐line predictive AI/ML models, are likely to increase. All‐encompassing resources, such as ADMETLab, http://admet.scbdd.com/#, or specific resources, such as software for drug‐drug interactions25 are likely to benefit and co‐evolve with increased usage of AI/ML methods.
In research, the use of AI/ML methods is accelerating pace, as detailed above.
In framing policy, AI/ML methods are anticipated to influence healthcare in most countries.
For the proper use of medicines, most “digital natives” (i.e., patients and scientists who started to use computers/tablets/smartphones from an early age) are seeking healthcare and drug information via social media and web‐based platforms. Predictive analytics (e.g., anticipating query terms and personalized searches) for drug‐related information are already available. Advanced AI/ML systems, tailored to an individual’s medical and genomic history are around the corner.
One of the major challenges of the pharmaceutical industry is the many areas of drug discovery and development can take years, cost hundreds of millions of dollars, and are often disconnected from the planning perspective and managed by different people. The time it takes to identify a disease target and formulate a biological hypothesis may take decades and cost billions. The typical time from identifying a hit molecule for a given target to a marketed product usually takes 12.5 years. In addition, although the amount of clinical pharmacology data is rapidly increasing, it is likely to be substantially less abundant than the amount of experimental chemistry and in vitro high‐throughput screening data. There are very few studies with measuring a large number of parameters in vitro, in mice and in humans with published freely available data. The majority of these studies was conducted by the pharmaceutical companies that rightfully treat these data as a competitive advantage making clinical pharmacology more difficult for the advances in AI/ML to disrupt.
The next decade may witness seamless integration between human (“natural”) intelligence and AI systems. Intelligent personal assistants (e.g., Siri, Alexa, etc.) already offer support with respect to navigation, entertainment, and shopping. Medical advice, specifically regarding the use of medicines, is quite likely on the “to do” list for most developers competing in this sector. It remains to be seen which one (if any) will be endorsed by professional associations, such as American Society for Clinical Pharmacology and Therapeutics (ASCPT; https://www.ascpt.org/) on the basis of accuracy and precision. Although most AI/ML developments are expected to benefit patients and scientists alike, we do anticipate that these trends will not replace clinical pharmacologists in the next decade, and clinical pharmacology in general will be disrupted slower than the other fields of drug discovery and development. There is, however, a strong incentive for clinical pharmacologists to adapt, to monitor such trends, and to become cognizant on the usage and perils of AI/ML systems, particularly for those that influence their daily practice. There needs to be a community and regulatory effort for open data sharing to make larger number of preclinical and clinical pharmacology data available for the ML community to use in order to accelerate the AI/ML progress in the area. Note: The GENTRL‐generated molecule is similar to ponatinib. Following the publication of the GENTRL results, guidelines for assessing AI‐generated molecules have been proposed by Walters and Murcko.26
Funding
Part of this work was supported by the National Institutes of Health (NIH) grants P30 CA118100, U24 CA224370, U01 CA239108, and U24 TR002278 (T.I.O.).
Conflict of Interest
A.Z. and Q.V. are affiliated with Insilico Medicine Inc., a company developing an AI‐based end‐to‐end integrated pipeline for drug discovery and development and engaged in aging and cancer research. T.I.O. is a former full‐time employee of AstraZeneca (1996–2002) and has received honoraria, or consulted for, Abbott, AstraZeneca, Chiron, Genentech, Infinity Pharmaceuticals, Merz Pharmaceuticals, Merck Darmstadt, Mitsubishi Tanabe, Novartis, Ono Pharmaceuticals, Pfizer, Roche, Sanofi, and Wyeth. His spouse was a full‐time employee of AstraZeneca (2002–2014) and is a full‐time employee of Genentech Inc.
References
- 1. Aronson, J.K. A manifesto for clinical pharmacology from principles to practice. Br. J. Clin. Pharmacol. 70, 3–13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Santos, R. et al A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oprea, T.I. et al Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 17, 317–332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Edwards, A.M. et al Too many roads not taken. Nature 470, 163–165 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Sun, Y. , Barber, R. , Gupta, M. , Aggarwal, C.C. & Han, J. Co‐author relationship prediction in heterogeneous bibliographic networks. International Conference on Advances in Social Networks Analysis and Mining, Kaohsiung, Taiwan, July 25, 2011–July 27, 2011.
- 6. Papadimitriou, S. et al Predicting disease‐causing variant combinations. Proc. Natl. Acad. Sci. USA 116, 11878–11887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bang, S. , Kim, J.‐H. & Shin, H. Causality modeling for directed disease network. Bioinformatics 32, i437–i444 (2016). [DOI] [PubMed] [Google Scholar]
- 8. Harrer, S. , Shah, P. , Antony, B. & Hu, J. Artificial intelligence for clinical trial design. Trends Pharmacol. Sci. 40, 577–591 (2019). [DOI] [PubMed] [Google Scholar]
- 9. Artemov, A.V. , Putin, E. , Vanhaelen, Q. , Aliper, A. , Ozerov, I.V. , & Zhavoronkov, A. Integrated deep learned transcriptomic and structure‐based predictor of clinical trials outcomes. bioRxiv (2016). 10.1101/095653 [DOI] [Google Scholar]
- 10. Gayvert, K.M. , Madhukar, N.S. & Elemento, O. A data‐driven approach to predicting successes and failures of clinical trials. Cell Chem. Biol. 23, 1294–1301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Viceconti, M. , Henney, A. & Morley‐Fletcher, E. In silico clinical trials: how computer simulation will transform the biomedical industry. Int. J. Clin. Trials 3, 37 (2016). [Google Scholar]
- 12. Ursu, O. , Rayan, A. , Goldblum, A. & Oprea, T.I. Understanding drug‐likeness. WIREs Comput. Mol. Sci. 1, 760–781 (2011). [Google Scholar]
- 13. Ursu, O. & Oprea, T.I. Model‐free drug‐likeness from fragments. J. Chem. Inf. Model. 50, 1387–1394 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Goodfellow, J. et al Generative adversarial nets. Adv. Neural Inf. Process. Syst. 27, 2672–2680 (2014). [Google Scholar]
- 15. Kulkarni, P. Reverse Hypothesis Machine Learning. (Springer, Cham, Switzerland, 2017). [Google Scholar]
- 16. Kadurin, A. et al The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8, 10883–10890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durant, J.L. , Leland, B.A. , Henry, D.R. & Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280 (2002). [DOI] [PubMed] [Google Scholar]
- 18. Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988). [Google Scholar]
- 19. Akutsu, T. & Nagamochi, H. Comparison and enumeration of chemical graphs. Comput. Struct. Biotechnol. J. 5, e201302004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Putin, E. et al Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 15, 4386–4397 (2018). [DOI] [PubMed] [Google Scholar]
- 21. Kuzminykh, D. et al 3D Molecular representations based on the wave transform for convolutional neural networks. Mol. Pharm. 15, 4378–4385 (2018). [DOI] [PubMed] [Google Scholar]
- 22. Polykovskiy, D. et alMolecular sets (MOSES): a benchmarking platform for molecular generation models. arXiv [cs.LG] (2018) <http://arxiv.org/abs/1811.12823>. [DOI] [PMC free article] [PubMed]
- 23. Polykovskiy, D. et al Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm. 15, 4398–4405 (2018). [DOI] [PubMed] [Google Scholar]
- 24. Zhavoronkov, A. et al Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019). [DOI] [PubMed] [Google Scholar]
- 25. Kheshti, R. , Aalipour, M. & Namazi, S. A comparison of five common drug‐drug interaction software programs regarding accuracy and comprehensiveness. Am. J. Pharmacogenomics 5, 257–263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walters, W.P. & Murcko, M. Assessing the impact of generative AI on medicinal chemistry. Nat. Biotechnol. 38, 143–145 (2020). [DOI] [PubMed] [Google Scholar]