

Anatomical anomalies
Agenesis of liver
This condition is incompatible with life. It has been reported in stillborn fetuses, usually in association with other severe anomalies.1
Absence (agenesis) of a lobe of the liver
Absence of the left lobe has been described,2 in one case associated with floating gallbladder.3 Radin et al. collected 19 cases of absence of the right lobe from the literature.4 The anomaly was associated with biliary tract disease in 12 patients, portal hypertension in seven patients, other congenital anomalies in four patients, but it was an incidental finding in five patients. Inoue et al.5 reported a case of hypogenesis of the right hepatic lobe associated with portal hypertension and reviewed 31 other cases of agenesis or hypogenesis in the Japanese literature. Injury to bile ducts occurred at cholecystectomy in a case of right lobe agenesis.6
Hypoplasia of the right lobe
This rare anomaly has been associated with suprahepatic7 or retrohepatic8 gallbladder, the latter complicated by hepatolithiasis and liver abscess.
Anomalies of position
In situs inversus totalis or abdominalis, the liver is found in the left hypochondrium, with the falciform ligament coursing from the left anterior margin towards the umbilicus. Hepatolithiasis has been reported in a patient with situs inversus.9 Adenocarcinoma of the distal common bile duct was reported in a 68-year-old woman with total situs inversus.10 A liver in a left or transverse position is one of the anomalies which may be associated with biliary atresia.11
Varying amounts of hepatic tissue may be displaced into congenital diaphragmatic hernias or omphalocoeles. Hepatic tissue was present in three of the 19 omphalocoeles studied by Soper and Green.12 The liver tissue may be the seat of non-parasitic cysts13 and may be detached from the rest of the liver.14 Several cases of hepatic herniation through defects in the diaphragm have been reported.15, 16, 17 A unique case of a supradiaphragmatic right liver lobe and gallbladder has been reported.18 Partial eventration of the right hemidiaphragm is a congenital lesion caused by aplasia or hypoplasia of part of the musculature of the diaphragm with resultant bulging of the affected portion from intra-abdominal pressure.19 The underlying portion of the liver prolapses into the diaphragmatic pouch where it may become strangulated.
Accessory lobes
Riedel lobe is a tongue-like caudal projection from the right lobe of the liver, which may be palpated in the right upper quadrant. In the 31 cases of Reitemeier et al.20 all the patients except one were women, their ages ranging from 31 to 77 years. Supernumerary lobes are relatively frequent findings, particularly on the inferior surface of the liver. They are connected to the liver by hepatic tissue or by a mesentery containing branches of the portal vein, hepatic vein and hepatic artery, and a bile duct.21 Intrathoracic accessory lobes, with their vascular supply perforating the diaphragm, have been reported.22 Accessory lobes may rarely require surgical intervention because of their large size, torsion of a pedicle, or the presence of other associated defects.23 Pedunculated hepatocellular carcinoma may arise in accessory lobes.
Ectopic hepatic tissue
Ectopic hepatic tissue may be found in the suspensory ligaments of the liver, lung, wall of the gallbladder, splenic capsule, retroperitoneal space, adrenal gland and greater omentum.24, 25, 26, 27, 28, 29, 30 A unique case of ectopic liver in the placenta was reported by Willis.31 In most instances, the hepatic tissue in these ectopic sites is microscopically normal, but when the liver is abnormal (fatty change, chronic hepatitis, cirrhosis) the ectopic liver tissue reflects the same changes.32, 33 Infantile haemangioendothelioma arising in an ectopic intrathoracic liver has been reported.25 Twenty-three cases of hepatocellular carcinoma, mostly from Japan, have arisen in ectopic livers.33, 34
Heterotopias of the liver
Most of the so-called adrenal heterotopias are actually examples of adrenal-hepatic fusion. Dolan and Janovski made a distinction between ‘adhesion’ and ‘fusion’ on the basis of presence or absence of a capsule interposed between the two organs.35 In both types, there was a marked diminution or complete absence of medullary tissue. The fusion is unilateral and is not associated with clinical evidence of adrenal impairment. It was found in 9.9% of unselected autopsy cases in one study.36 The incidence was much higher in older age groups suggesting that the condition may be an ageing phenomenon. There is a single description of hepatolienal fusion.37
Pancreatic heterotopia in the liver is rare (Fig. 3.1 ). In the case reported by Ballinger,38 an islet cell carcinoma arose in the aberrant pancreatic tissue. Retention cyst has been recorded in an obstructed duct within the heterotopic pancreatic tissue.39 Foci of exocrine pancreas in the liver of a 41-year-old patient with cirrhosis were attributed to a metaplastic process.39 Pancreatic acini were found intermingled with peribiliary glandular acini in 4% of autopsy livers, probably representing an intrinsic component of these glands.40 Splenic heterotopia presenting as a mass lesion was reported by Lacerda et al.41 With one exception,42 the rare cases recently reported are examples of splenosis or subcapsular splenic implants following trauma or surgical splenectomy43 (see Chapter 16). Thyroid heterotopia has been occasionally encountered (Fig. 3.2 ) and has been reported in a fetus with trisomy 18.44
Figure 3.1.
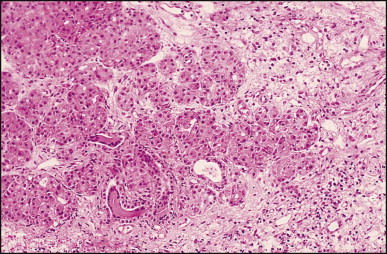
Pancreatic heterotopia. Pancreatic acini and several small ducts are present but there are no islets of Langerhans. (H&E)
Figure 3.2.
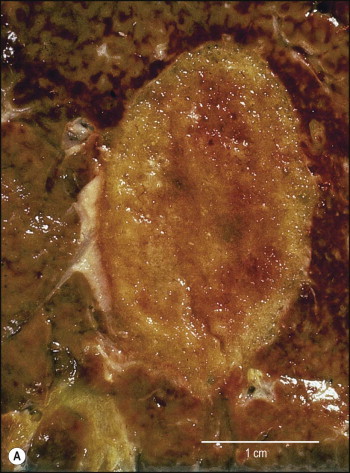
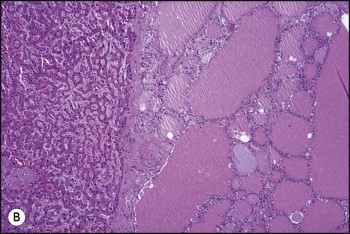
Thyroid heterotopia. Section of nodule in liver (A) that is composed microscopically of thyroid acini (B). (H&E)
Vascular anomalies
Hepatic artery
Aberrant hepatic arteries occur in a significant proportion of individuals.45 Anomalous origin of the hepatic artery has been described in association with biliary atresia.46 Congenital duplication of the gallbladder has been reported in association with an anomalous right hepatic artery.47 The existence of an accessory right hepatic artery (arising from the left and passing behind the portal vein bifurcation) must be recognized and appropriately managed during split liver transplantation, in order to ensure a complete vascular supply to both grafts.48 Rupture of an aberrant hepatic artery can occur rarely.48 Congenital hepatoportal arteriovenous fistulas have been reported in infants.49, 50, 51 Surgical correction or embolization using a variety of materials has been followed by reversal of the haemodynamic complications.51, 52
Portal vein
Anatomical variations of the portal vein are almost as common as those of the hepatic artery, and their recognition is important to radiologists and transplant surgeons.53 Preduodenal portal vein is the result of a variation in the normal developmental pattern of the embryonic precursors of the portal vein, i.e. the right and left vitelline veins and their three anastomotic channels. It may lead to duodenal obstruction54 and this anomaly was the apparent cause of gastric outlet obstruction in an adult.55 A vascular complex consisting of absent inferior vena cava, anomalous origin of the hepatic artery and preduodenal portal vein was reported in three children with biliary atresia.56
Obstructing valves within the lumen of the splenic vein, the portal vein, or both, may be a rare cause of portal hypertension in children.57
A case of reduplication of the portal vein was reported by Hsia and Gellis.57 One branch ran anterior to the pancreas and was obliterated by an old organized thrombus.
Atresia or hypoplasia of the portal vein has been recognized by a number of investigators.58, 59 It may involve the entire length of the vessel or be limited to the point of entrance into the liver, or it may occur just proximal to the division into two branches. Microscopic study of the atretic segments has generally shown no evidence of inflammation. When associated with biliary atresia, it may occur whether or not a Kasai portoenterostomy has been performed.
Congenital absence of the portal vein is rare and is diagnosed mainly in children, although a few cases in adults are reported.60, 61, 62, 63 Cardiac and inferior vena caval anomalies and polysplenia may occur simultaneously. The patient of Marois et al. had an associated hepatoblastoma.64 Focal nodular hyperplasia,65 hyperplastic nodules66 and nodular regenerative hyperplasia67 have all been reported in association with congenital absence of the portal vein. One patient presented with hepatopulmonary syndrome.68 Children can also present with pulmonary hypertension and congenital absence of the portal vein. Liver transplantation is technically possible though more difficult.69
Congenital shunts (portocaval, portohepatic and between the left portal vein and internal mammary veins) have been reported.70, 71, 72, 73, 74, 75, 76 Congenital portosystemic venous shunts (PSVS) may lead to hepatic encephalopathy. They can be intra- or extrahepatic. Persistence of the ductus venosus can lead to hypergalactosaemia without an enzyme deficiency on newborn screening.73 Patients may present with hepatic impairment associated with a severe steatosis, which is reversed by surgical closure.75
Uchino et al.77 reviewed their experience with 51 cases of portosystemic shunts; 67% were intrahepatic. Twelve patients had hepatic encephalopathy, the frequency of which increased in individuals after 60 years of age. A total of 75% of the newborns had hypergalactosaemia. Children with hepatic encephalopathy had shunt ratios over 60%; no encephalopathy occurred with a shunt ratio <30%. Haemangiomas were present in 10% of patients; 10% of patients (all over the age of 20) had a portal vein aneurysm; 20% of patients had a patent ductus venosus and <10% had absence of the portal vein. The extrahepatic PSVS never closed spontaneously. The liver histopathology was usually characterized by steatosis or mild fibrosis. Five patients had hepatic atrophy, and three patients had focal nodular hyperplasia.
A congenital aneurysmal malformation of the extrahepatic portal vein was described by Thompson et al.78 Another aneurysm of the left branch of the portal vein was diagnosed in utero by ultrasonography.79 An intrahepatic portal vein aneurysm communicating with the hepatic veins was associated with intrahepatic haemangiomas and an intracranial arteriovenous malformation.80
Cavernous transformation of the portal vein (‘portal cavernoma’) is a condition in which the vein is replaced by a spongy trabeculated venous lake with extension into the gastroduodenal ligament.81, 82, 83 It is a major cause of portal hypertension and may account for as many as 30% of all children with bleeding oesophageal varices.84 A thrombotic diathesis due to inherited abnormalities of anticoagulant proteins is very rarely a factor in the pathogenesis.85 Haemorrhage is common but some children present with asymptomatic splenomegaly.83 An early report of marked growth retardation in children with extrahepatic portal vein obstruction86 has not been confirmed in subsequent studies.87 Obstructive jaundice is an uncommon complication.88, 89 It was detected in 8 of 121 children presenting over a 14-year period with cavernous transformation and regressed after surgical decompression.90 A study in 25 adults with portal cavernoma found that 25% had clinically significant features of biliary obstruction and nearly all patients had cholangiographic evidence of bile duct damage,91 cases now referred to as portal biliopathy.92, 93
Pancytopenia of varying degrees occurs in the majority of cases.94 Colour Doppler ultrasonography, computed tomography, or contrast enhanced magnetic resonance imaging95, 96 obviate the need for splenoportography or angiography. Percutaneous or open liver biopsy specimens are either normal or show minimal fibrosis. Klemperer81 reported multiple ‘adenomas’, but the description of the nodules and their designation as ‘regenerative formations’ suggest that the condition was nodular regenerative hyperplasia.
Two theories have been proposed for the pathogenesis of cavernous transformation, namely a sequel to portal vein thrombosis due to omphalitis, umbilical vein catheterization or intra-abdominal sepsis, or an angiomatous malformation of the portal vein;97 however, the latter is not supported by hard data and in most instances thrombotic occlusion with subsequent recanalization of the vein appears the likely cause. Experimental evidence in support of occlusion of the portal vein and the subsequent opening up of adjacent collateral vessels has been reported by Williams and Johnston.98 In a prospective ultrasonographic evaluation of neonates undergoing umbilical catheterization, portal vein thrombosis was detected in 56% of the patients of whom 20% went on to partial or complete resolution.99 Although not all cases of cavernous transformation have a history of umbilical catheterization, such a procedure, especially when catheter use is prolonged, seems to account for a significant number of cases. Conversely, congenital abnormalities, in particular atrial septal defects, malformations of the biliary tract, and anomalous inferior vena cava, have been observed in 12 of 30 cases studied by Odièvre et al.100 Several cases of cavernous transformation have also been found associated with congenital hepatic fibrosis.101, 102 It would appear that in some cases an anatomical anomaly of the portal vein may underpin the thrombotic process. The rapidly changing haemodynamics occurring during birth in regard to the portal circulation may play an additional role.
Hepatic veins
Membranous obstruction of the hepatic portion of the inferior vena cava, which may be associated with occlusion of hepatic veins, was thought to have a congenital aetiology,103 but this has been disputed.104, 105 Kage et al. have demonstrated histological evidence of thrombus formation and occlusion of hepatic vein orifices in eight cases, suggesting an acquired rather than a congenital lesion.105 Okuda et al. suggested that membranous obstruction of the vena cava be termed ‘obliterative hepatocavopathy’ to distinguish it from the classic Budd–Chiari syndrome. The condition is frequently reported from Japan and South Africa.105, 106, 107, 108 Patients present with the Budd–Chiari syndrome and are prone to develop hepatocellular carcinoma.106 Membranous obstruction of the vena cava occurs mainly in adults but a series of nine cases in children was reported from Namibia.109 Rare instances of congenital Budd–Chiari syndrome, attributed to maternal drug use110 or ingestion of herbal tea111 or idiopathic,112, 113 have been reported. Veno-occlusive disease in children with cellular and humoral immune deficiency has also been described.114 In the cases of Mellis and Bale, consanguinity and early age of onset suggested an inherited cause for the veno-occlusive disease.115
Hereditary haemorrhagic telangiectasia (Osler–Rendu–Weber disease)
Hereditary haemorrhagic telangiectasia (HHT) is an autosomal dominant disorder characterized by an aberrant vascular development.116, 117 The resulting vascular lesions range from smaller mucocutaneous telangiectases to large visceral arteriovenous malformations. The estimated frequency ranges from 1 to 20 per 100 000.117 Mutations in the genes encoding endoglin (ENG, chromosome 9q34),118 and activin receptor type-like kinase 1 (ACVRL1 or ALK-1, chromosome 12q13),119, 120 both of which mediate signalling by transforming growth factor-β ligands in vascular endothelial cells, are associated with HHT1 and HHT2 subtypes, respectively. Mutations in the gene MADH4 encoding SMAD4, an important member of the TGFβ pathway, are seen in patients with the combined syndrome of juvenile polyposis and HHT and should be tested for in HHT patients found negative for ENG and ALK-1 mutations as they are bound to have undiagnosed juvenile polyposis.121 A fourth putative HHT gene has been localized to chromosome 5.122 The disease is characterized by telangiectases (skin, mucous membranes), arteriovenous fistulas in the liver (30% of cases), lungs and central nervous system, and aneurysms. Hepatic involvement is characterized clinically by pain in the right upper quadrant of the abdomen, and a large, sometimes pulsatile, liver. High-output cardiac failure may result from arteriovenous shunting in the liver.123, 124 Portal hypertension and encephalopathy may occur.125 Intrahepatic lithiasis is a rare complication.126
The hepatic lesions can be demonstrated by angiography127 and colour Doppler ultrasonography128 and are readily visualized during laparoscopy.129 Arterial embolization has been used successfully in treatment of the disease.123, 124, 130 Liver transplantation131, 132 performed in a few patients has been shown to reverse the hyperdynamic circulatory state, but disease recurrence has been observed in one patient.133
Macroscopically, the liver is nodular, fibrotic or rarely cirrhotic. Spider-like arrangements of minute blood vessels may be noted on the surface. Microscopically, three fibrovascular patterns were reported by Daly and Schiller.134 One pattern comprises a honeycomb meshwork of dilated sinusoidal channels lined by endothelial cells set either directly upon hepatocyte cords or amid a loose fibrous stroma (Fig. 3.3A ); the distribution of these foci is haphazard. A second pattern consists of tortuous thick-walled veins flanked by numerous wide-calibre arteries that course randomly through the parenchyma amid variable amounts of fibrous tissue. A third pattern is evident in the enlarged portal areas in which numerous dilated vessels (veins, arteries and lymphatics) show prominently against a background of fibrous tissue (Fig. 3.3B). Regenerative nodules (nodular transformation) were described in the cases reported by Zelman135 and Wanless and Gryfe.136 A high prevalence of focal nodular hyperplasia has been detected by Doppler ultrasonography.137 Blewitt et al.138 described marked disruption of the hepatic architecture with hepatocyte necrosis and ischaemic bile duct injury in a patient with HHT and intra-abdominal sepsis. One woman treated with ethinyl oestradiol, multiple blood transfusions and iron dextran developed hepatocellular carcinoma.139 Mouse models have been reported for the two main genomic mutations with histological changes recapitulating those observed in man.140, 141, 142
Figure 3.3.
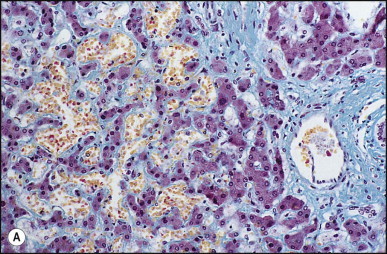
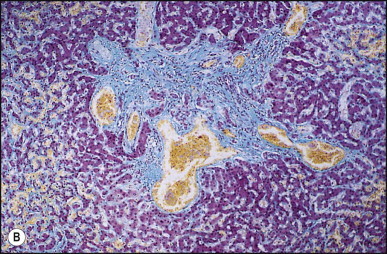
Hereditary haemorrhagic telangiectasia. (A) Dilated anastomosing sinusoidal channels, lined by endothelial cells with some subendothelial fibrous tissue. (B) The same case as (A) showing dilated vessels within a portal tract; foci of dilated sinusoidal vessels are also present (left and upper right). (Masson trichrome)
Ataxia telangiectasia
This autosomal recessive disorder is characterized by cerebellar ataxia, oculocutaneous telangiectases, IgA and IgE deficiency, recurrent infections, and an increased incidence of cancer. Homozygotes have an incidence of cancer that is 100 times higher than that of unaffected age-matched subjects.143 There is also an increased incidence (particularly breast cancer) in heterozygotes.143 Mutations in ATM (ataxia telangiectasia mutated), a protein kinase shown to be a crucial nexus for the cellular response to DNA double-stranded breaks, have been identified as the underlying cause of the disease.144 Several cases of hepatocellular carcinoma145 and two cases of veno-occlusive disease146 have been reported in patients with this disease. Other hepatic involvement includes hepatitis with periportal fibrosis, telangiectases and cirrhosis.146
von Hippel–Lindau disease
This autosomal dominant multisystemic cancer syndrome, due to a mutation of the VHL tumour suppressor gene on chromosome 3, rarely involves the liver.147 Multiple cavernous haemangiomas were reported by Zeitlin,148 and multiple ‘haemangioblastomas’ are described.149, 150 Occasionally, pancreatic cysts may lead to jaundice by obstruction of the common bile duct.151
Focal nodular hyperplasia
Wanless et al. have shown that focal nodular hyperplasia is a hyperplastic response of the hepatic parenchyma to a pre-existing arterial, spider-like malformation.152 Multiple focal nodular hyperplasia may be associated with haemangioma of the liver, meningioma, astrocytoma, telangiectases of the brain, berry aneurysm, dysplastic systemic arteries and portal vein atresia. Wanless et al. proposed that this was a new syndrome resulting from underlying systemic abnormality of blood vessel development.153 Components of the syndrome (berry aneurysms, cerebral telangiectases) have subsequently been reported in several patients who only had a solitary focal nodular hyperplasia.154, 155
Bile duct anomalies
Congenital abnormalities of the biliary tract are best demonstrated and studied by radiographic methods. In a series of 3845 operative cholangiograms, Puente and Bannura demonstrated anatomical variations (defined as those having no pathological significance) in 24% and congenital abnormalities (defined as pathologically significant deviations from the normal pattern) in 18.4%.156 The latter included left-sided cystic duct (9.5%), aberrant hepatic ducts (4.6%) and accessory hepatic ducts (1.7%).
Agenesis of the common bile duct
In this very rare anomaly, the common hepatic duct empties directly into the gallbladder, while the latter drains by a long cystic duct into the second part of the duodenum.157
Agenesis of the common hepatic duct
Several patients with this anomaly have been described.158, 159, 160 They presented with obstructive jaundice in very early infancy and at operation were found to have no proximal biliary tree. There were no ductal structures in the hepatic hilum. Palliative surgery appears ineffective and liver transplantation is preferred.
Anomalous insertion of the right hepatic duct
Nomura et al. reported a case with this rare anomaly.161 Cholangiography prior to laparoscopic cholecystectomy demonstrated insertion of the right hepatic duct into the cystic duct.
Anomalous (‘accessory’) bile ducts
These are found in about 15% of individuals when the bile ducts are dissected.162 Anomalous hepatic ducts which pass beyond the porta hepatis almost invariably arise from the right lobe and frequently from a dorsal segment. The mode of termination is variable; final entry may be into the gallbladder, cystic duct, right or common hepatic duct, junction of the cystic and common hepatic ducts or even the common bile duct.163 A rare case of an anomalous right posterior segmental hepatic duct associated with a stricture and hepatolithiases was reported by Cullingford et al.164 Abnormal sectorial distribution of the right hepatic duct was found in 36% of 139 patients evaluated for potential live liver donation and a classification proposed to recognize risk areas when performing right graft live donor liver transplantation and tumour resections involving the right lobe of the liver.165
Cholecystohepatic ducts are rare and occur when there is persistence of the fetal connections between the gallbladder and liver parenchyma, with failure of recanalization of the right and left hepatic ducts.166 Two cases were reported in association with oesophageal atresia.167 Failure to recognize the presence of cholecystohepatic ducts at cholecystectomy may lead to a persistent biliary fistula, bile peritonitis, stricture or death.168 Cholangiography is mandatory whenever there is any doubt about the anatomy of the biliary tree in order to avoid the increased morbidity and mortality of reoperation.168 Anomalous bile ducts may occur in association with biliary cystadenoma of the liver.169 Two examples of a long accessory hepatic duct have been found among 23 cases of congenital dilatation of the common bile duct.170 Atresia of the common hepatic duct with an accessory duct has been reported.158 Accessory hepatic ducts may occasionally contain calculi.171
Duplication of the bile ducts
These lesions were reviewed by Swartley and Weeder.172 One duct may empty into the pylorus or both may drain into the duodenum. In one case a choledochal cyst was associated.172 Patients are usually free of symptoms until obstruction by stone or infection occurs. A case of congenital duplication involved the cystic duct and the common hepatic duct, which were both lined by gastric mucosa.173
Congenital bronchobiliary and tracheobiliary fistulas
Several cases of bronchobiliary and tracheobiliary fistula have been reported.174 One patient had a bronchobiliary fistula, as well as a tracheo-oesophageal fistula and oesophageal atresia.175 Bronchobiliary fistula has been reported with biliary atresia.176, 177 The bronchobiliary fistula typically arises from the proximal part of the right main stem bronchus, a short distance below the carina, and generally joins the biliary system at the level of the left hepatic duct. The proximal part of the fistula resembles a bronchus while the distal part is lined by columnar and/or squamous epithelium. Patients present in early infancy with aspiration pneumonia and atelectasis.
Ciliated hepatic foregut cyst
Some 60 cases of this rare cyst have been reviewed.178 The cyst is generally found incidentally at laparotomy or by imaging studies. It occurs more frequently in men and is most commonly in the medial segment of the left hepatic lobe. In one case, the cyst appeared closely associated with the left hepatic vein.179 In most instances the cyst is unilocular (Fig. 3.4 ). The mean cyst diameter is approximately 3 cm. One large cyst was associated with an elevated serum CA 19–9 level.180 There have been a few isolated reports of squamous cell carcinoma arising in a ciliated foregut cyst.181, 182
Figure 3.4.
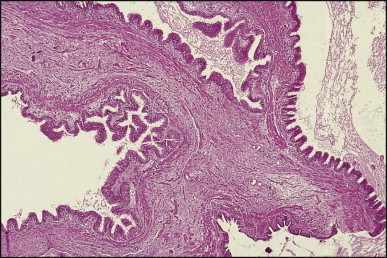
Ciliated foregut cyst. This multiloculated cyst is very much less common than the unilocular type. (H&E)
Histologically, the cyst wall consists of four layers: pseudostratified ciliated columnar epithelium with mucous cells, subepithelial connective tissue, bundles of smooth muscle, and an outermost fibrous capsule (Figure 3.4, Figure 3.5 ).178, 183, 184, 185 Endocrine cells are present in the cyst epithelium.185 In all Chatelain and colleagues' seven cases, immunoreactivity of some cells for CD10 suggested the presence of Clara cells.185 The cysts are believed to arise from the embryonic foregut and to differentiate toward bronchial structures in the liver.
Figure 3.5.
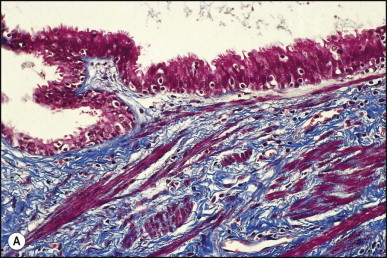
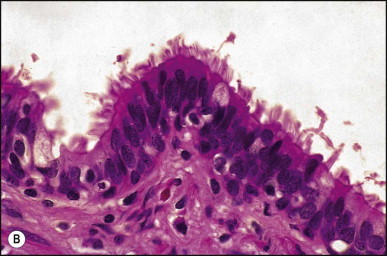
(A) Ciliated foregut cyst. The lining of pseudostratified epithelium is supported by fibrous tissue with scattered bundles of smooth muscle. (Masson trichrome) (B) Cyst lining consists of pseudostratified columnar epithelium. Cilia are clearly seen at high magnification. (H&E)
Spontaneous bile duct perforation
In the first few months of life, spontaneous bile peritonitis may occur from leakage of bile at the junction of the cystic and common bile ducts. In patients without other bile duct structural lesions, the aetiology is unknown, although congenital weakness of the bile duct walls, mucous plugs and gallstones have been suggested.186, 187, 188 The clinical presentation consists of jaundice, ascites, failure to thrive, and signs of peritoneal irritation. The diagnosis can be suspected by finding bile on abdominal paracentesis and confirmed by hepatobiliary scanning with a technetium-99m labelled iminodiacetic acid derivative.188 Treatment is surgical and the prognosis excellent. Pathological descriptions of the liver are usually consistent with obstruction, with ductular reaction and oedematous portal tracts.
Biliary atresia
This is one of the most important causes of severe neonatal liver disease and the major indication for liver transplantation in young children. Initially the extrahepatic biliary tree is affected, evident as an obstructive picture both clinically and histopathologically. This is the defining lesion of this disorder. Biliary cirrhosis develops early in life. Those children who survive infancy because of having a successful Kasai portoenterostomy continue to have intrahepatic bile duct damage which leads eventually to profound loss of small intrahepatic bile ducts and recurrent cholestasis due to bile duct paucity. Accordingly, this hepatobiliary disorder is now called simply biliary atresia, without specifying an extrahepatic (or intrahepatic) location. The term ‘biliary atresia’ (BA) is used in this section interchangeably with the former term ‘extrahepatic biliary atresia’ (EHBA). The term ‘intrahepatic biliary atresia’ to denote intrahepatic bile duct paucity has been obsolete for years and should be abandoned.
Classification and aetiopathogenesis
Approximately 30% of infants presenting with conjugated hyperbilirubinaemia in the neonatal period have biliary atresia (BA), the overall incidence being approximately 1 in 6000 to 1 in 19 000 live births.189, 190, 191, 192 There is no clear-cut racial predilection although some ethnicities appear to have a higher incidence: African-Americans193 and Polynesians189 compared with Caucasian infants. BA is more common in girls than boys.46, 191 Seasonal variation in the occurrence of this disease has been suggested in North American studies,193, 194 although this pattern has not been confirmed in Europe189 or Japan.195 Unquestionably, there are multiple disease mechanisms which can produce BA since it rather than BA may occur as an isolated lesion or in association with various types of congenital structural abnormalities or specific chromosomal abnormalities. It can be conceptually useful to classify BA in two general patterns: ‘early’ or embryonal/fetal accounting for 15–35% of cases and ‘late’ or generally perinatal (acquired) in 65–85% of cases. This aetiopathogenic heterogeneity of BA was delineated by a study of 237 children by Silveira et al.11 Forty-seven of the children (20%) had associated congenital anomalies (28 cardiovascular, 22 digestive and 19 splenic). The splenic malformations included 13 with polysplenia syndrome and two with asplenia. Karyotypic abnormalities were found in two of eight children studied. Silveira et al. divided BA into four distinct subgroups, three involving a congenital form that could arise through a malformation, a disruption or a chromosomal abnormality, while the fourth is attributable to agents active in the perinatal period (the acquired form).11 Most infants have this ‘late’ acquired pattern: their apparently normal biliary system has been subjected to a fibrosing inflammatory process toward the end of gestation or shortly after birth. Discordance for BA in HLA identical twins supports a postnatal event being of primary importance in the pathogenesis of late-pattern BA.196 By contrast, approximately 10–30% of infants with BA have extrahepatic congenital abnormalities such as polysplenia, left atrial isomerism, double-sided left lung, pre-duodenal portal vein, intestinal malrotation, and/or congenital heart defects.46, 197, 198 These congenital defects are sometimes grouped as a ‘laterality complex’. Abnormalities of the spleen are not invariably present in infants who have other typical features of the laterality complex, and thus splenic disorder does not define this association.199 A subset of infants with BA, comprising 6–10% of patients overall, have damage to the biliary tree which produces cystic dilatation200, 201, 202; most of these patients seem to have a choledochal cyst, but exceptionally the cystic change is confined to intrahepatic ducts. This type of BA, now termed cystic BA (formerly called ‘correctable’ BA), may be identified on sonography of the fetus in approximately 40% of cases.202 It may represent yet another disease mechanism for BA, distinct from those represented by early- and late-pattern BA, although cystic BA has been identified in patients with early-pattern (fetal/embryonal) BA. Some infants with BA have specific chromosomal abnormalities such as trisomy 17–18,203 Turner syndrome,204 or cat-eye syndrome.205 A lethal autosomal recessive syndrome with intra-uterine growth retardation, intra- and extrahepatic BA, and oesophageal and duodenal atresia was reported in one family.206 BA has been reported with the Kabuki make-up syndrome,207 in one patient with Zimmermann–Laband syndrome and brachydactyly,208 and in Martinas–Frias syndrome.209 Other genetic disorders may be associated with BA as familial occurrence has been reported,210, 211 and in one case a woman who had had BA gave birth to a daughter with BA.212
Viral infection in combination with a genetic predisposition to a robust or disordered inflammatory response may play a role in the development of late-pattern (perinatal/acquired) BA. Chance occurrence of a viral infection during a limited period of susceptibility would explain the rarity of BA; however, no consensus exists as to which viruses are pre-eminent in such an aetiopathogenesis and some recent observations suggest that viral infection is a secondary phenomenon.213 Cytomegalovirus (CMV) infection is found in a high proportion of children with BA. Tarr et al.214 found evidence for viral infection in five of 23 patients with BA. The diagnosis was based on histopathological evidence of CMV infection, serology (IgM antibodies) or culture. The detection of CMV infection by the polymerase chain reaction (PCR) is higher in neonatal hepatitis than in BA.215 In a set of identical twins both infected with CMV, one twin had BA and the other had neonatal hepatitis.216 Infants with BA and concurrent CMV infection may have a worse prognosis.217 Reovirus 3 was suggested as a cause of BA and neonatal hepatitis on the basis of clinical and experimental studies218, 219, 220, 221 but that association was questioned by other investigators.222, 223 More compelling evidence for an aetiological association of BA and reoviruses was provided by Tyler et al.224 These investigators detected reovirus RNA from hepatobiliary tissues of 55% of patients with BA and 78% of patients with choledochal cysts. A possible relationship between group C rotavirus and BA was suggested by Riepenhoff-Talty et al.225 Subsequently, however, no evidence of group A, B or C rotaviruses was detected by PCR in BA.226 Human papilloma virus has been detected in neonatal hepatitis and BA by nested PCR for DNA,227 but the role, if any, of this virus needs to be clarified.
Recent progress has focused on immune mechanisms in the pathogenesis of BA, especially late-pattern (perinatal/acquired) BA.228, 229 The envisioned disease mechanism is that during the perinatal period a viral infection occurs and it targets the biliary epithelium and provokes an aberrant autoimmune injury to the bile ducts which persists long after the viral infection is gone. This proposed mechanism entails an active, ongoing immune response which can be documented empirically, and it accounts for the conflicting reports of viral infection in BA as well as the absence of detectable ongoing viral infection in liver or biliary tissue from BA patients. The hepatic inflammatory infiltrate in BA was found remarkable for evidence of lymphocyte activation.230 Studies in liver tissue from infants with BA showed a Th1-type of cytokine expression pattern with CD4+ and CD8+ lymphocytes, CD68+ macrophages in portal tracts and increased IL-2, IL-12, interferon-γ and tumour necrosis factor-α.231 Determinants on the activated T cells are typical of an oligoclonal expansion, consistent with being evoked by a specific antigen.232 Upregulation of osteopontin expression in intrahepatic biliary epithelium correlated with portal fibrosis and ductular reaction.233 At the same time, genomic studies of liver from infants with BA have shown upregulation of genes involved in regulating lymphocyte differentiation, mainly of those with Th1-commitment.234 Upregulation of the expression of interferon-γ and osteopontin was notable. Another study, which included somewhat older patients, also found upregulation of genes involved in morphogenesis, cell signalling and regulation of gene transcription.235 Further studies suggested that the pattern of regulatory gene expression in late-pattern (perinatal/acquired) BA is not equivalent to that in the early-pattern; these data also failed to show a pattern relating to laterality genes in early-pattern (embryonic/fetal) BA.236 Both forms of BA appear to induce a strong immunological response. HLA studies in BA might support a disease mechanism involving autoimmunity, but results to date are contradictory. One early study showed that infants with late-pattern (perinatal/acquired) BA have a high prevalence of the HLA-B12 determinant compared both to normal controls and to infants with BA plus congenital anomalies;237 haplotypes A9-B5 and A28-B35 were more common in infants with late-pattern (perinatal/acquired) BA. A subsequent study failed to confirm any characteristic HLA pattern in BA.238 Yet additional studies have shown an association with HLA-DR2239 and with HLA-B8 and HLA-DR3.240 Further insight into the possible mechanism of late-pattern (perinatal/acquired) BA has come from recent work in the Rhesus rotavirus (RRV) murine model of BA. It can be simulated in Balb/c-mice which have been infected with rotavirus.241, 242 This model shares many features with the human disease. Interferon-γ plays an important role in bile duct damage: knockout mice not expressing interferon-γ failed to get severe duct damage after infection with RRV despite a brief hepatitis whereas wild type animals did; administration of recombinant interferon-γ abrogated the protective effect of not being able to produce interferon-γ.243 Certain chemokines may also contribute to biliary damage;244 IL-12 seems to play a lesser role;245 tumour necrosis factor-α appears not to be involved.246 In this mouse model, primed neonatal CD8 T-cells appear capable of initiating damage to bile ducts.247 When T cells from RRV-disease mice were transferred into naive syngeneic SCID mice, the recipients developed bile duct-specific inflammation without previous RRV infection.248 Some autoantibodies have been detected in this model (directed to α-enolase or vimentin).249 Thus the combination of observations in infants with BA and in this mouse model strongly suggests that a complex pattern of immune reactivity appears to be important in late-pattern (perinatal/acquired) BA. In the early-pattern (embryonic/fetal) form, other genes may play a more direct role.
Other theories have been proposed for the pathogenesis of BA. Landing250 first proposed that neonatal hepatitis, biliary atresia, infantile choledochal cyst and possibly some instances of ‘intrahepatic biliary atresia’ (meaning congenital duct paucity syndromes) are all manifestations of a single basic disease process that he named ‘infantile obstructive cholangiopathy’. He considered biliary atresia (and choledochal cyst) to be the result of an inflammatory rather than a maldevelopmental process and postulated that the most probable cause was a viral infection. Since the aetiopathogenesis of an important congenital bile duct paucity syndrome, namely Alagille syndrome, has since been elucidated as genetic, this theory clearly requires modification. Nevertheless, as previously discussed, the role of viral infection at least in the pathogenesis of late-pattern (perinatal/acquired) BA is an important focus of current research. Certain chromosomal abnormalities may give rise to complex syndromes including altered immune reactivity and thus predispose to hepatobiliary disease mediated by viral infection. Microchimerism has recently been proposed as part of the mechanism of hepatobiliary damage in BA;251 it might initiate an immune response in the absence of viral infection. The various manifestations of infantile obstructive cholangiopathy may depend on the timing of the insult. Specifically, rats given the drug 1,4-phenylenediiso-thiocyanate during fetal life developed stenotic or atretic bile ducts due to thickening and fibrosis, whereas those given the drug after birth had dilatation of the ducts with inflammation.252 It is difficult to generalize from this rat model to human infants. BA with features of the ductal plate malformation might reflect a different disease mechanism.253 Recent observations, however, have argued against the presence of ductal plate malformation in BA as unique to patients with early-pattern (embryonic/fetal) BA.254
Whereas the pathogenesis of the late-pattern (perinatal/acquired) BA probably involves immunogenetic susceptibility and exposure to an instigating factor, such as viral infection, during a limited period of susceptibility, the aetiopathogenesis of the early-pattern (embryonal/fetal) BA appears to be much more diverse. Extrahepatic congenital abnormalities such as polysplenia, congenital heart defects, and disturbed rotation of the intestines suggest an extensive and early developmental abnormality. Some infants with BA and these extrahepatic findings show features of the ductal plate lesion on liver biopsy.253, 255, 256 This abnormal configuration of small bile ducts is attributed to disorganization in the fetal development of the biliary tree: failure of remodelling of the ductal plate leads to residual embryonic bile duct structures in this rather striking configuration. Finding the ductal plate lesion in extrahepatic biliary atresia is consistent with a destructive hepatobiliary process beginning early in gestation. Abnormal cilia have been reported in children with the polysplenia syndrome and BA.257, 258 While the association with abnormal cilia is unclear, ciliary function appears to be important in left/right asymmetry.259 There is a pathogenic role for multiple defects in the laterality sequence.260 Early studies focused on the inversion (inv) gene, one of three genes that control left/right asymmetry in the mouse.261 Beginning in early embryonic development the liver is a predominant site for this gene expression. A transgenic mouse with recessive deletion of inv develops situs inversus and jaundice; the early fetal lesion is a complete obstruction with cystic change of the biliary tree.262 Few of the various genes which have been found mutated in human laterality disorders (ZIC3, CFC1, LEFTYA, ACVR2B, NODAL) have been investigated in BA; however, mutations in CFC1 263 and ZIC3 264 have been found in infants with BA and major laterality defects. Three children were described with ultrastructural abnormalities of the canalicular microvilli and no expression of villin; phenotypically they had BA without laterality complex or ductal plate malformation.265
The extrahepatic biliary tree in BA may be totally atretic or the atresia may involve only proximal or distal segments. The intrahepatic bile ducts are gradually destroyed with progression of the disease. Most infants with BA have conjugated hyperbilirubinaemia from an early age, but clinical jaundice is not always apparent or appreciated. Indeed in many infants jaundice is initially physiological and merges with the jaundice of advancing liver disease. Infants typically have dark urine and pale stools, but the stools may retain enough colour to be falsely reassuring. The infants look well and generally gain weight adequately. At clinical presentation they have hepatomegaly and usually some degree of splenomegaly, unless polysplenia is present. The infant who presents with congenital heart disease and conjugated hyperbilirubinaemia requires intensive evaluation since the leading hepatic diagnoses will be BA or Alagille syndrome. Untreated BA rapidly progresses to hepatic fibrosis and cirrhosis with all the complications of portal hypertension, in addition to malnutrition and fat-soluble vitamin deficiency. The median age of death is 12 months if BA is not diagnosed and treated.266
Clinically, the differential diagnosis is the broad spectrum of disorders constituting the neonatal hepatitis syndrome267 (see Table 3.1 ). Congenital infection should be excluded, although CMV may be found along with BA. Systemic bacterial infection should be ruled out, including a silent urinary tract infection. Inherited metabolic diseases require specific attention, especially α1-antitrypsin deficiency, which can be associated with severe cholestasis and acholic stools and very rarely has been associated with BA. Cystic fibrosis can generate a duct lesion indistinguishable clinically from BA. These two conditions as well as galactosaemia may produce a histological picture closely resembling that of BA. Structural abnormalities of the extrahepatic biliary tree cause the clinical presentation like BA: choledocholithiasis, idiopathic perforation of the biliary tract,186, 187, 268 true choledochal cyst, and extrahepatic biliary hypoplasia or ‘hair-like’ bile duct syndrome. Some infants with Alagille syndrome show ductular reaction, rather than duct paucity, on liver biopsy taken early in the course of the disease.
Table 3.1.
Classification of infantile conjugated bilirubinaemia disorders (neonatal hepatitis syndrome) with major histological and diagnostic investigations
| Categories | Specific diseases/causes | Comments | Liver histology | Diagnostic investigations |
|---|---|---|---|---|
| Infection |
Toxoplasmosis (congenital) | NSH (calcifications) | Maternal infection/IgM specific Abs | |
| PCR on amniotic fluid | ||||
| Rubella (congenital) | NSH | IgM specific Abs | ||
| Cytomegalovirus (congenital) | NSH ‘owl-eye’ nuclear inclusions | Urine for viral culture, IgM Abs, PCR | ||
| Herpes simplex (congenital) | Acute-pattern neonatal liver failure | Necrotizing hepatitis/viral inclusions (IHC) | Liver biopsy | |
| Viral culture (scrapings from skin vesicles) | ||||
| Syphilis (congenital) | Diffusely fibrosing hepatitis | Standard test, VDRL, fluorescent treponema Abs | ||
| Human herpesvirus-6 | Acute-pattern neonatal liver failure | Necrotizing hepatitis/viral inclusions (rare) | Serology, PCR | |
| Herpes zoster | NSH | Serology, PCR | ||
| Hepatitis B (mainly vertical) | Acute-pattern neonatal liver failure | Severe hepatitis | Mother's serum eAg positive (or negative due to precore mutant) | |
| Hepatitis C (mainly vertical) | Rarely cause of NHS | Screening of infants born to HCV +ve mothers by RT PCR | ||
| Human immunodeficiency virus (vertical) | Rarely cause of NHS | NSH (opportunistic infections, in particular CMV) | Anti-HIV, CD4 count | |
| Parvovirus 19 infection | Chronic-pattern neonatal liver failure | NSH, marked haemopoiesis, siderosis, perisinusoidal fibrosis, few GC | Severe anaemia | |
| IgM Abs, PCR | ||||
| Syncytial giant cell hepatitis (?paramyxovirus) | NSH, prominent syncytial GC (EM paramyxovirus-like inclusions) | Liver ultrastructure (no supportive serology) | ||
| Enteric viral sepsis (echoviruses, Coxsackie viruses, adenoviruses) | Acute-pattern neonatal liver failure | NSH GC/cholestasis | Appropriate serology, viral culture or direct fluorescent assay | |
| Bacterial infection (extrahepatic or sepsis) | Acute-pattern neonatal liver failure | Nonspecific hepatitis/cholestasis (ductular bile casts) | Blood, urine or CNS culture | |
| Listeriosis | NSH, focal necrosis granulomas | Listeria isolation from blood, CSF or liver | ||
| Tuberculosis |
Caseating granulomas (acid-fast bacilli) |
High index of suspicion | ||
| Mantoux test | ||||
| Structural | Biliary atresia | Biliary features: loose portal fibroplasia, ductular reaction and cholestasis including ductular bile plugs/GC (15%); DPM-like (20%) | Acholic stools/liver histology | |
| No excretion on hepatobiliary scan | ||||
| ERCP | ||||
| Laparotomy | ||||
| Choledochal cyst | Differentiate from biliary atresia | Biliary features | Ultrasound, cholangiography | |
| Caroli disease/syndrome | Biliary features/DPM | Ultrasound, cholangiography | ||
| Choledocholithiasis | Differentiate from biliary atresia | Biliary features | Ultrasound, cholangiography | |
| Neonatal sclerosing cholangitis | Biliary features/periductal fibrosis inconstant | Cholangiography | ||
| Extrahepatic biliary hypoplasia (‘hair-like bile duct syndrome’) | Differentiate from biliary atresia | Biliary features | Cholangiography | |
| Spontaneous perforation of common bile duct | Differentiate from biliary atresia | Biliary features | Imaging | |
| Bile stained ascites (paracentesis) | ||||
| Non-syndromic duct paucity (idiopathic) | Paucity of intrahepatic bile ducts | Liver biopsy | ||
| Cholestasis | ||||
| Alagille syndrome |
Differentiate from biliary atresia |
Paucity of intrahepatic bile ducts Cholestasis |
Extrahepatic syndromic features | |
| Identifiable bile ducts ± mild ductular reaction occasionally seen in early biopsy |
High serum cholesterol | |||
| Liver biopsy | ||||
| JAG1 mutations (20p) or Notch2 (1p13) | ||||
| Metabolic genetic (Chapter 4) | α1-antitrypsin deficiency | Differentiate from biliary atresia | Variable. Biliary features mimicking BA, duct paucity or NSH (DPAS not diagnostic before 8–12 weeks of age); GC rare; periportal steatosis | Serum α1-antitrypsin concentration α1-antitrypsin phenotype (PI type) |
| Cystic fibrosis | Differentiate from biliary atresia | Steatosis/cholestasis/biliary features (focal fibrosis)/cholangiolar eosinophilic casts | Sweat chloride/extrahepatic complications | |
| Gene mutation (7q31.2 – CFTR protein) | ||||
| Galactosaemia | Acute- or chronic-pattern neonatal liver failure | Steatosis/biliary features/fibrosis Severe parenchymal damage and loss Later cirrhosis (now rare) |
Galactose-1–6-phosphate uridyl transferase assay Erythrocyte galactose-1-phosphate level |
|
| Tyrosinaemia, type 1 | Acute- or chronic-pattern neonatal liver failure | Severe parenchymal injury and loss, steatosis, GC, regenerative nodules cell dysplasia; fibrosis, cirrhosis | Elevated serum tyrosine, phenylalanine, methionine/elevated succinylacetone in urine/FAA activity in fibroblasts or lymphocytes | |
| Gene mutation (15q23–25) | ||||
| Hereditary fructosaemia | Chronic-pattern neonatal liver failure | Steatosis, biliary rosettes, GC, fibrosis, later cirrhosis | Liver biopsy (enzyme analysis) | |
| Gene mutation (9q22) | ||||
| Glycogen storage disease, type IV | Early perinatal variant, very rare | Eosinophilic (‘ground glass’) cytoplasmic inclusions, PAS positive, diastase resistant (amylopectin-like) | Liver biopsy | |
| Brancher enzyme in liver, white blood cells or cultured fibroblasts | ||||
| Niemann–Pick, type A | Hepatocyte and macrophage storage (lipidic, microvesicular/foamy) | Sphingomyelinase assay (peripheral blood cells or liver) | ||
| Niemann–Pick, type C | Chronic-pattern neonatal liver failure | Hepatocytic/macrophage storage as type A, but few cells; biliary features | Storage cells in bone marrow aspirate | |
| Cultured fibroblasts/cholesterol esterification studies | ||||
| Wolman disease | Cholesterol ester stored mainly in macrophages (cholesterol crystals)/neutral lipids in hepatocytes | Liver biopsy | ||
| Lysosomal acid lipase activity | ||||
| Gaucher disease | Macrophage storage (foamy/striated cytoplasm)/variable perisinusoidal fibrosis | Liver biopsy | ||
| Acid β-glucosidase activity (white blood cells or cultured fibroblasts) | ||||
| FIC-1 deficiency (PFIC type 1) | Bland cholestasis, mild disease | Low GGT cholestatic syndrome | ||
| EM: granular bile (‘Byler bile’) | Liver biopsy (EM) | |||
| Genomic mutation (18q21–22) | ||||
| Bile salt export pump (BSEP) deficiency (PFIC type 2) | Severe parenchymal injury, ballooned (cholate-static) hepatocytes/GC/absent canalicular BSEP (IHC staining) | Low GGT cholestatic syndrome | ||
| Liver biopsy | ||||
| Gene mutation (2q24) | ||||
| Multidrug resistant 3 (MDR3) deficiency (PFIC type 3) | Biliary features; progressive fibrosis; absent canalicular MDR3 (IHC staining) | High GGT cholestatic syndrome | ||
| Histology IHC | ||||
| Gene mutation (7q21) | ||||
| North American Indian familial cholestasis | Bland cholestasis | Gene mutation (16q22/cirhin) | ||
| Later progressive fibrosis | ||||
| Aagenaes syndrome | Very rare | Cholestasis | Cholestatic syndrome | |
| Lymphoedema (lower limb) | ||||
| Primary disorders of bile acid synthesis: | Resemble PFIC type 2, but canalicular | Low GGT cholestatic syndrome | ||
| BSEP present (IHC) | Urine and plasma bile acids | |||
| 3β-hydroxy-Δ5-C27-steroid dehydrogenase/isomerase deficiency | ||||
| Δ4−3-oxosteroid 5β-reductase deficiency | ||||
| Arthrogryposis, renal dysfunction, and cholestasis (ARC) | Variable cholestasis | Ichthyosis, recurrent infection, failure to thrive, | ||
| Fanconi-like renal tubular dysfunction; arthrogryposis may not be evident | ||||
| Gene mutation (VPS33B on 15q26) | ||||
| Peroxisomal disorders (e.g. Zellweger syndrome) | Variable | Dysmorphic features | ||
| Cholestasis/fibrosis/haemosiderosis | Very long chain fatty acid study/red cell plasmalogens | |||
| Absence of peroxisomes on EM | ||||
| X-linked adrenoleukodystrophy | Chronic-pattern neonatal liver failure | Absent or reduced peroxisomes on EM | Dysmorphic features (less striking than Zellweger) | |
| Perinatal haemochromatosis | Chronic-pattern neonatal liver failure | Severe parenchymal injury and loss; GC – haemosiderin pigment in hepatocytes | High serum ferritin, TIBC | |
| Iron accumulation in heart and/or pancreas on computerized tomography or magnetic resonance imaging | ||||
| Liver biopsy/lip biopsy for extrahepatic iron storage (accessory salivary glands) | ||||
| Mitochondrial DNA depletion syndrome | Chronic-pattern neonatal liver failure | Microvesicular steatosis, oxyphilic cells, siderosis, cirrhosis | Lactic acidosis hypoglycaemia | |
| Abnormally low ratio of mtDNA/nDNA in tissue | ||||
| Mitochondrial anomalies (EM) | ||||
| Citrullinaemia, type II | Mainly Asian descent | Cholestasis/steatosis/siderosis | Gene mutation (SLC25A13) | |
| Adenosine deaminase deficiency | Very rare | |||
| Panhypopituitarism (septo-optic dysplasia) | NS hepatitis/no distinctive features | Low cortisol, TSH and T4 | ||
| Hypothyroidism | NSH/cholestasis | High TSH titre, low T4, free T4, T3 | ||
| Genetic (gross chromosomal abnormalities) |
Trisomy 18 | Associated with biliary atresia | Biliary features | Karyotype |
| Cat-eye syndrome | Associated with biliary atresia | Biliary features | Karyotype | |
| Trisomy 21 | Chronic-pattern neonatal liver failure (rare); associated biliary atresia (occasional) | Fibrosing hepatitis with leukaemoid cell infiltration | Karyotype | |
| Kabuki syndromea, b | Rarely associated with biliary atresia | Biliary features | Congenital anomalies/mental retardation | |
| Neoplasia (Chapter 15) |
Neonatal leukaemia | Acute-pattern neonatal liver failure | Leukaemic infiltration | Peripheral blood, bone marrow aspirate |
| Neuroblastoma | Acute-pattern neonatal liver failure | Small cell tumour, rosettes | Imaging | |
| IHS, EM | Urinary vanilmandelic and homovanillic acid | |||
| Langerhans cell histiocytosis | Biliary features similar to sclerosing cholangitis/Langerhans cells (CD1a) inconstant | Involvement of other systems (skin, bone, lung) | ||
| Haemophagocytic lymphohistiocytosis |
Chronic-pattern neonatal liver failure |
Haemophagocytic activity |
High level of macrophage derived cytokines in serum | |
| Elevated ferritin; elevated triglycerides | ||||
| Toxic |
Total parenteral nutrition-associated cholestasis | Differentiate from biliary atresia |
Biliary features, cholestasis |
|
| Drug-induced (via breast-milk or other) | ||||
| Vascular |
Budd–Chiari syndrome | Rare | Features of venous outflow block | |
| Severe congestive heart failure | Perivenular cell dropout/congestion | |||
| Perinatal/neonatal asphyxia | Differentiate from biliary atresia | Ischaemic necrosis | ||
| Immune |
Inspissated bile syndrome associated with ABO incompatibility | Differentiate from biliary atresia | Biliary features | Coombs test |
| Neonatal lupus erythematosus | NSH | Maternal history/congenital heart block/skin rash (discoid lupus) | ||
| Anti-Ro – anti-La in liver tissue (IHC) | ||||
| Anti-Ro and anti-La antibodies | ||||
| Neonatal hepatitis with autoimmune haemolytic anaemia | Acute or chronic pattern liver disease | Prominent syncytial GC, necrosis, inflammation, fibrosis | Coombs positive haemolytic anaemia | |
| Idiopathic | Idiopathic neonatal hepatitis (sero-negative) | Differentiate from biliary atresia | GC, parenchymal loss with stromal collapse of variable severity | Liver biopsy |
| ‘Le foie vide’ (infantile hepatic non-regenerative disorder) | Chronic-pattern neonatal liver failure | Total parenchymal cell dropout, scant ductular reaction | Liver biopsy | |
| Hardikar syndromec | Differentiate from biliary atresia, Alagille syndrome | Biliary features | Associated with cleft palate, pigmentary retinitis, hydronephrosis | |
Note: Acute-pattern neonatal liver failure denotes metabolic instability, coagulopathy, and extremely elevated serum aminotransferases in a neonate; chronic-pattern neonatal liver failure denotes metabolic instability, coagulopathy, hypoalbuminaemia, near-normal serum aminotransferases in a neonate; conditions which require specific consideration vis-à-vis biliary atresia are denoted ‘Differentiate from biliary atresia’.
NSH, non-specific hepatitis; GC, multinucleated giant cells; IHC, immunohistochemistry; Abs, antibodies; EM, electron microscopy; DPM, ductal plate malformation; PFIC, progressive familial intrahepatic cholestasis; FAA, fumarylacetoacetase; GGT, γ-glutamyl-transpeptidase; TIBC, total iron binding capacity; mtDNA, mitochondrial DNA; nDNA nuclear DNA; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone.
Selicorni A, Colombo C, Bonato S, et al. Biliary atresia and Kabuki syndrome: another case with long-term follow-up. (Letter) Am J Med Genet 2001; 100:251.
van Haelst MM, Brooks AS, Hoogeboom J, et al. Unexpected life-threatening complications in Kabuki syndrome. Am J Med Genet 2000; 94:170–173.
Nydegger A, Van Dyck M, Fisher RA, et al. Hardikar syndrome: long term outcome of a rare genetic disorder. Am J Med Genet A. 2008;146A:2468–72.
Preoperative diagnosis relies on demonstrating the presence or absence of bile secretion in the intestine. Hepatic sonography may reveal a dilated extrahepatic biliary tree, consistent with distal, ‘correctable’ atresia, but it is unusual to find dilated intrahepatic bile ducts. Hepatobiliary scanning, using a technetium-99m labelled iminodiacetic acid derivative such as DISIDA or PIPIDA, fails to demonstrate passage of the radiolabelled substance into the intestinal tract over a 24-hour period. Although hepatobiliary scanning has high sensitivity, scanning may appear normal if performed very early in the disease process in late-pattern BA.269, 270 Hepatobiliary scanning is informative if it shows that tracer, and thus bile, reaches the intestine; it is objective, recorded, and can be quantified. A negative or non-draining scan does not mean that the disorder is necessarily BA because non-draining hepatobiliary scans may be found with severe idiopathic neonatal hepatitis, small duct paucity syndromes, such as Alagille syndrome, with severe α1-antitrypsin deficiency or with TPN-associated cholestasis. The role of endoscopic retrograde cholangiopancreatography (ERCP) remains controversial: ERCP is technically feasible in infants and may be useful in selected cases.271, 272, 273 Percutaneous liver biopsy is essential and has high diagnostic specificity in the range of 60–95%, depending on the timing of the biopsy, adequacy of the specimen and expertise of the pathologist.274, 275 In our experience, the majority of non-diagnostic biopsy specimens are taken within the first few weeks of life. In fact incidental liver biopsy specimens taken at laparotomy for duodenal stricture within the first week of life in three infants with BA showed only trivial liver abnormalities.276 This may imply that although bile duct destruction is likely to have started in utero, the actual liver damage may not occur until the placenta discontinue the clearance of biliary products, in particular bile salts.
Pathological features at surgical intervention
Portoenterostomy (the Kasai procedure) was introduced in 1959 and remains the only potentially corrective procedure, other than liver transplantation.277 In this operation, the atretic biliary tree is resected, and bile drainage is re-established via a broad anastomosis of the end of an intestinal Roux-en-Y loop to the bare edge of the transected porta hepatis. The efficacy of the recently introduced laparoscopic version of the Kasai portoenterostomy currently remains uncertain. Extensive retrospective studies have shown that the prognosis for a good long-term result from the conventional Kasai procedure depends primarily on operation before 60 days of age and the absence of cholangitis,191, 278, 279, 280, 281, 282, 283 but there is potential for reasonable medium-term survival in about one-third of infants coming to primary corrective surgery 100 days or older. Most centres continue to favour the Kasai procedure as the first therapeutic option, rather than subjecting patients to immediate liver transplantation simply on the basis of age.284 The lack of significant fibrosis at the time of operation may play a role in a good long-term outcome.285 Computerized quantification of fibrosis on liver biopsy obtained at Kasai portoenterostomy may discriminate between negligible fibrosis and sufficient fibrosis to portend a bad prognosis.286 One group suggested that histological features on the initial liver biopsy specimen can predict the success of portoenterostomy.287 Another group reported that the extent of ductular reaction, based on keratin 7 (K7) immunostaining, found in the liver biopsy specimen obtained at the time of Kasai procedure was predictive of native liver survival over the following year.288 Histological studies of the extrahepatic bile ducts removed at surgery have been performed by several groups of investigators.289, 290, 291, 292, 293 In a study of 98 cases, Gautier and Eliot classified the biliary remnants into three types: in the first the duct is completely atretic, with few or no inflammatory cells in the surrounding connective tissue (Fig. 3.6A ); in the second type it is present as a cleft-like lumen lined by occasional cuboidal or low columnar epithelium which is variably necrotic, hyperplastic, or focally absent (Fig. 3.6B,C); the altered ducts are sometimes very numerous, usually having lumina of <50 µm, periluminal neutrophilic infiltration is characteristic and cellular debris, and less often bile, may be found in the lumen. Epithelial necrosis is evident in ducts with a diameter exceeding 300 µm. The third type consists of altered bile duct incompletely lined by columnar epithelium, in addition to numerous smaller epithelial structures (Fig. 3.6D). These histological types were evaluated by Gautier et al.291 at three levels – porta hepatis, junction of the cystic and common hepatic ducts and an intermediate level; completely atretic duct becomes increasingly more frequent from the porta hepatis to the junction of the hepatic duct and cystic ducts. This classification, which may help pathologists to describe the changes observed in the biliary remnants removed at portoenterostomy, is of limited clinical significance because in individual cases serial sectioning often shows atretic ducts alternating with variably destroyed ducts in a random fashion. In addition the numerous smaller structures intermingled with variably altered ducts are likely to represent anastomosing channels recruited from peribiliary glands, of which effectiveness in bypassing the atretic duct is uncertain; anastomoses between ramified peribiliary glands are well demonstrated in normal adult livers using injection techniques (see Chapter 10). Correlations between the size and number of residual ducts and establishment of bile flow after surgery have yielded conflicting results. Two groups of investigators believed that bile flow is most likely to occur when the diameter of the residual ducts exceeds 150 µm.278, 289, 294 However, another study of the extrahepatic biliary remnants of 204 cases of BA showed that the patterns of bile duct obliteration are not indicative of prognosis.293
Figure 3.6.
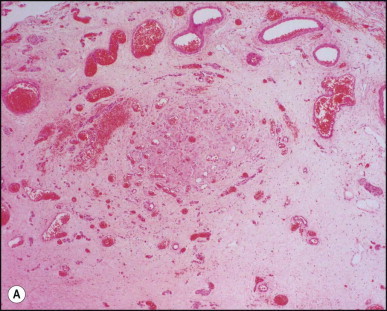
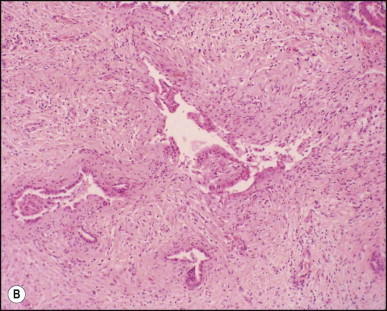
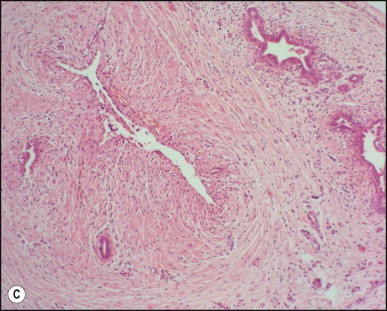
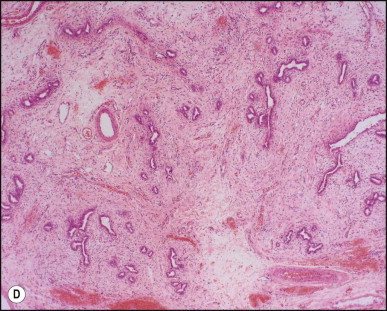
Biliary atresia. Transverse sections of biliary remnants removed at portoenterostomy. (A) Atretic common hepatic duct showing luminal occlusion by vascular fibrous tissue with very few inflammatory cells. (B) Distorted bile duct inconsistently lined by desquamated columnar epithelium and surrounded by fibroplasia with a light inflammatory cell infiltrate. (C) Cleft-like lumen devoid of epithelial lining side by side with duct structures which may represent adjacent segments of the same ducts or hyperplastic peribiliary glands. (D) Hilar region close to the surgical resection line showing numerous ducts or glandular structures set in a loose, mildly inflamed fibrous tissue. (H&E)
Although the Kasai procedure is essentially a palliative operation, many children enjoy prolonged good health afterwards and approximately 20–35% of patients who undergo portoenterostomy will survive into adulthood without liver transplantation. Approximately 30–35% of patients drain bile but develop complications of cirrhosis and require liver transplantation before age 10.295 For the remaining patients, bile flow is inadequate following portoenterostomy and cirrhosis rapidly develops. Survival in BA with a functioning Kasai portoenterostomy but without orthotopic liver transplantation is 10–20% by the age of 20 years.296 In a recent survey of 271 patients, 23% were alive with their native liver 20 years after surgery, all but two having signs of cirrhosis; after age 20 years, two patients died of liver failure and 14 underwent, or were waiting for, a liver transplant. Women who are long-term survivors with BA and have not yet had a liver transplant may have normal pregnancies, but in general such pregnancies must be treated as high-risk because of complications from portal hypertension or hepatic decompensation.297 Liver transplantation has become the treatment of choice for infants and children in whom the Kasai portoenterostomy has failed. The safety and results of liver transplantation with the use of livers from living related donors and cadaveric donors are excellent. One-year survival is >90%, with better results obtained under elective conditions and in children who are over 10 kg in weight.296, 298 Recent data indicate that the overall survival at 10 years for all surgical treatment is of the order of 75–80%;191, 299 general health 10 years after paediatric liver transplantation is good, although renal impairment and cardiovascular disease may develop.300
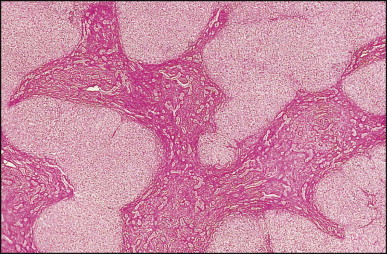
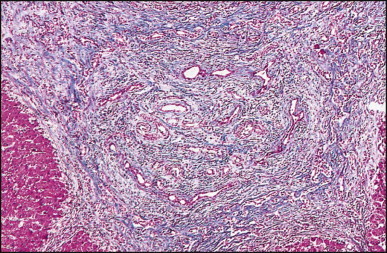
The macroscopic appearance of the liver in BA varies according to the stage of the disease. At first it is enlarged and dark green in colour, becoming finely nodular as cirrhosis develops (Fig. 3.7 ). In untreated cases, the cirrhosis may take between 1 and 6 months from birth to develop. Dilated bile ducts filled with inspissated bile may be seen in sections of large portal areas (Fig. 3.8 ). The cystically dilated bile ducts may resemble Caroli disease.301 They only occur after the age of 3 months and are not amenable to surgical drainage procedures. There may be portal lymphadenopathy. The median maximum node dimension in six cases studied by Hübscher and Harrison was 14 mm.302 These lymph nodes are brown in colour and full of pigment-laden macrophages. Livers removed at transplantation after an apparently successful Kasai procedure (loss of jaundice), but with subsequent development of cirrhosis and portal hypertension, are often coarsely nodular with areas of macronodular hypertrophy and broad intervening or peripherally located scars resembling the gross appearance of focal nodular hyperplasia (Fig. 3.9 ).
Figure 3.7.

Liver removed at transplantation 6 months after a failed Kasai procedure. Cirrhosis is characterized by small and dark green parenchymal nodules.
Figure 3.8.
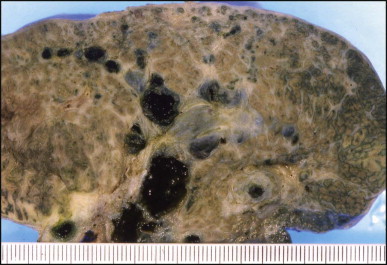
Bisected liver from patient with biliary atresia. Multiple dilated bile ducts are filled with black bilirubin casts. Some of the ducts have thick fibrous walls. The hepatic parenchyma is cirrhotic with a periseptal distribution of the cholestasis (right of the field).
Figure 3.9.
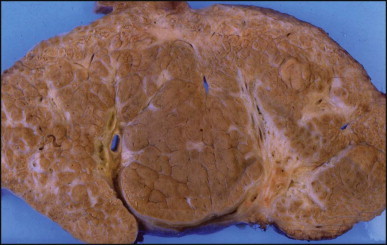
Liver removed at transplantation 8 years after ‘successful’ Kasai procedure. Bisected specimen showing macronodular areas of re-expanded parenchyma (centre of the field) with more fibrotic micronodular areas particularly at the periphery. Note the large fibrotic and seemingly stretched portal areas which contain well-identifiable bile duct branches (yellow and light green).
Pathology of intrahepatic changes
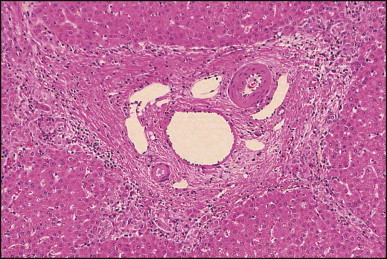
The histological features of BA include cholestasis, portal tract expansion by oedematous fibroplasia and periportal ductular reaction with the presence of bile plugs in dilated lumens of cholangioles that are distorted and often form an irregularly anastomosing network at the portal periphery (Figure 3.10, Figure 3.11 ). Arterial branches are unusually prominent and portal vein branches appear attenuated. Giant cell transformation of hepatocytes is seen in some cases (Fig. 3.12 ), and may occasionally be prominent. Loose fibrosis is progressive and periportal/perilobular in location, with linkage of portal areas and eventual development of a secondary biliary cirrhosis (Fig. 3.13 ). A study of the extracellular and cellular components of the connective tissue matrix in BA by de Freitas et al. suggested that activation of a connective tissue cellular clone by the reactive ductules may be responsible for the portal fibroplasia.303 Ho et al. reported an arteriopathy (hyperplasia and hypertrophy) of the common hepatic artery and its peripheral branches supplying the entire biliary tree in 11 cases of biliary atresia.304 Thickening of the medial layer of small hepatic arteries may be present.305 Subsequent studies have indicated that the transcription factor HNF6 plays an important role in intrahepatic bile duct and arterial development.306, 307 HNF6 and HNF1ß appear necessary for ductal plate formation.306 Large perihilar bile ducts may show ulceration with loss of epithelial lining, bile impregnation of the wall and bile sludge formation in the lumen. In addition to the severe cholestasis, the cirrhotic stage of BA is characterized by marked pseudo-xanthomatous transformation, the presence of bile lakes, Mallory–Denk bodies and variable accumulation of copper and copper-associated protein in liver cells (Figure 3.14, Figure 3.15 ). In one study, copper concentrations were increased in over two-thirds of liver samples obtained during portoenterostomy and decreased in some patients after successful biliary drainage.308 However, copper deposition in liver is elevated in the first 2 months of life and periportal deposition of copper on liver biopsy specimens does not discriminate extra from intrahepatic causes of cholestasis in early infancy. Acute and chronic inflammation is noted in portal/periportal areas in BA in both pre-cirrhotic and cirrhotic stages, and bile duct degeneration and inflammation may be evident (Fig. 3.16 ). The mononuclear infiltrate in portal and lobular areas of livers with end-stage BA is similar to normal adult liver and very different from that associated with autoimmune hepatitis or chronic hepatitis B infection. The growth of large perihilar regenerative nodules, probably as a consequence of functioning intrahepatic ducts in this region, may be important for maintaining biliary drainage after Kasai procedure.309 Bile lakes occur after the age of 3 months, by which time irreversible hepatic damage has occurred.310
Figure 3.10.
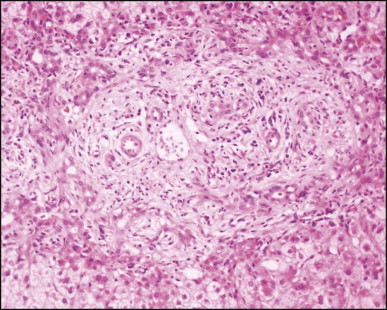
Biliary atresia. Liver biopsy performed at 3 weeks of age. Characteristic portal tract expansion by loose fibrous tissue containing irregularly anastomosing bile ductules at the periphery, some being dilated with inspissated bile in their lumen. (H&E)
Figure 3.11.
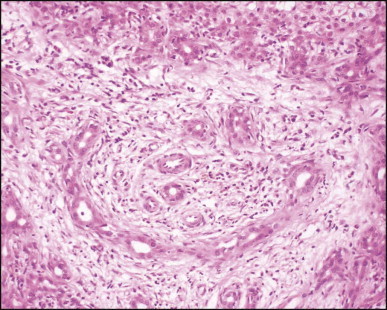
Biliary atresia. Cholangiolar reaction forming crescent shape profile reminiscent of the embryonal ductal plate. In our experience, this pattern may be observed irrespective of the patient having associated extrahepatic malformation. (H&E)
Figure 3.12.
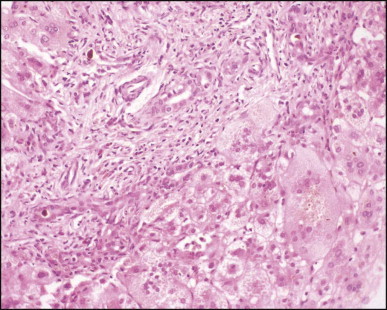
Biliary atresia. Portal features of distal obstructive cholangiopathy are associated with giant cell transformation in the parenchyma. (H&E)
Figure 3.13.
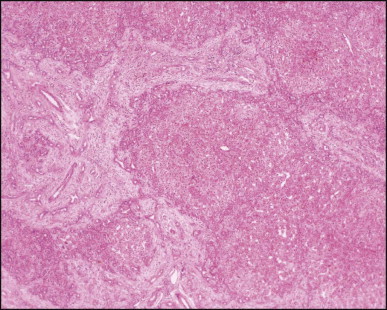
Biliary atresia, cirrhotic transformation. Porto-portal bridging septa made of oedematous fibrous tissue show a marginal ductular reaction and delineate small and irregularly shaped parenchymal nodules. (H&E)
Figure 3.14.
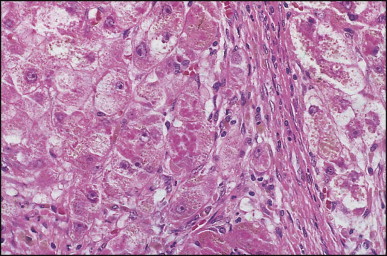
Biliary atresia. The same case as illustrated in Fig. 3.13. Mallory–Denk bodies in ballooned periseptal hepatocytes. (H&E)
Figure 3.15.
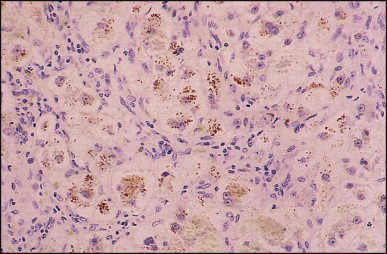
Biliary atresia. Same case as illustrated in Figure 3.13, Figure 3.14. Marked copper accumulation in liver cells (red granules). (Rhodanine)
Figure 3.16.
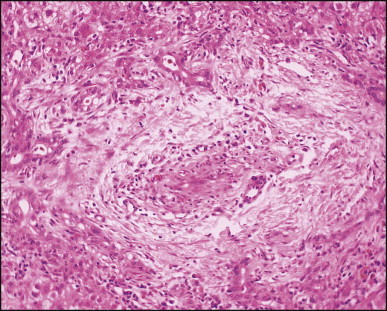
Biliary atresia. Inflamed and arteriole-rich, lemon-shaped fibrous tissue marks the site of a small, largely destroyed, intrahepatic bile duct. (H&E)
Interlobular bile ducts become few in number as early as the 4th or 5th month after birth (Fig. 3.17 ) and advanced duct loss accompanies progressive fibrosis by the age of 8 or 9 months.250 Activated hepatic stellate cells are responsible for increased collagen production.311, 312 In addition to paucity of ducts, Raweily et al.256 identified concentric tubular ductal structures in 21.6% of cases of BA; these bore some resemblance to those seen in ductal plate malformations (Fig. 3.18 ). Similar observations had been made earlier by Desmet and Callea,313 who hypothesized that this subgroup has more severe and rapidly progressive liver damage. It is of interest that children in this subgroup reveal a histopathological picture resembling that of congenital hepatic fibrosis 4 or 5 years after portoenterostomy313, 314 (Figure 3.18, Figure 3.19 ). The interlobular bile ducts continue to disappear and cirrhosis can develop despite satisfactory bile drainage after portoenterostomy. The bile duct loss has been attributed to persistent interference with bile flow,315 recurrent cholangitis or continuation of the immune process causing the atresia. Detailed histopathological study of sequential liver specimens taken at the time of Kasai operation, relaparotomy and/or transplantation has provided evidence that the bile duct loss is due to an unpredictable and uneven obliteration of bile ducts in the porta hepatis during wound healing and scarring after portoenterostomy.315
Figure 3.17.
Biliary atresia. A portal area is bereft of bile ducts. There is periportal cholangiolar reaction with an associated neutrophilic response. (H&E)
Figure 3.18.
Biliary atresia. Liver resection 1 year after portoenterostomy. Pattern of fibrosis and reactive small bile ducts and ductules are reminiscent of congenital hepatic fibrosis. There were only minimal chronic cholestatic features in this liver. PAS after diastase digestion.
Figure 3.19.
Biliary atresia. Same liver as illustrated in Fig. 3.18. Note absence of an interlobular bile duct and a pattern of ductular reaction reminiscent of a ductal plate malformation. (Masson trichrome)
Ultrastructural degenerative changes affecting the intrahepatic bile ducts and ductules in BA have been described in detail by Ito et al.316 who found that the degree of obstruction of the lumen of these ducts appears to be an important determinant of prognosis following corrective surgery.
Malignant epithelial tumours of hepatobiliary origin rarely complicate biliary cirrhosis associated with biliary atresia, but both hepatocellular carcinoma317, 318 and cholangiocarcinoma319 have been reported. Focal nodular hyperplasia after portoenterostomy has been reported in a few children with BA.320, 321 Macroregenerative nodules may also develop.322
Neonatal hepatitis
Neonatal hepatitis is a term that was coined for presumed viral infections of the liver in early infancy. It has become evident that these disorders are by no means exclusively viral, or even infectious, in aetiology. Neonatal hepatitis represents a clinical pattern of neonatal liver disease, hence the designation ‘neonatal hepatitis syndrome’. Other diseases such as galactosaemia, hereditary fructose intolerance, cystic fibrosis and the conditions discussed under biliary atresia and paucity of the intrahepatic bile ducts may also present with pathological changes in the liver resembling an infectious process. Giant cell transformation, a frequent histological component of neonatal hepatitis, has been seen in all cholestatic conditions in infancy including pure haemolytic anaemias and endotoxic injury.323 Although clinical jaundice is not present in every case of neonatal hepatitis syndrome, conjugated hyperbilirubinaemia is invariably present. Therefore the entire spectrum of these diseases might best be called ‘infantile conjugated bilirubinaemia disorders’, a term which avoids the inherent disadvantages of each of the component terms of ‘neonatal hepatitis syndrome’. A classification of infantile conjugated bilirubinaemia disorders (neonatal hepatitis syndrome) is shown in Table 3.1. In the past 20 years the proportion of cases with no known aetiology has fallen substantially from 50–60%324 to approximately 30% or less. Many of the disorders dissected out of the idiopathic category are inherited metabolic diseases, discussed in detail in Chapter 4.
Nomenclature for neonatal liver disease is problematic. The simplest term ‘neonatal jaundice’ may be confused with physiological jaundice in the newborn. The term ‘neonatal cholestasis’ is not precise because in the first 3–4 months of life, every infant has some degree of cholestasis physiologically. This physiological cholestasis occurs because uptake of bile acids and other organic anions by hepatocytes is immature and thus inefficient, leading to high concentrations of bile acids in blood. In addition, hepatocellular pathways for bile acid conjugation and biliary secretion are also immature, in part because bile canalicular transporters are also regulated developmentally. The circulating bile acid pool is contracted, and ileal uptake of bile acids is under-developed. The term ‘neonatal hepatitis’ is obviously imprecise because hepatic inflammation is not a feature of every condition. The term ‘neonatal hepatitis syndrome’ emphasizes the uniformity of the clinical presentation and similarity of pathological findings as well as the broad spectrum of causative disease processes. The terminology ‘infantile conjugated bilirubinaemia disorders’ (ICBRD) not only identifies the definitional finding but conveys inclusion of a broad spectrum of infectious, metabolic, structural and other aetiologies.
Numerous infections, usually congenital, are implicated in neonatal hepatitis syndrome, including CMV, rubella virus, hepatitis B virus, herpes simplex virus, herpes zoster virus, Coxsackie and Echo viruses, paramyxovirus,325 toxoplasma and Treponema pallidum. An unusually high incidence of CMV infection (49%) was reported in a series of 45 cases from Taiwan.326 Herpes simplex virus, enteroviruses, adenovirus and hepatitis B virus may cause neonatal liver failure. In addition to genetic metabolic disorders, endocrine disorders may cause neonatal hepatitis syndrome. An association with hypopituitarism was reported in two infants by Herman et al.,327 and later Sheehan et al.328 Immunological disorders may cause neonatal hepatitis syndrome: neonatal lupus erythematosus is most common. Rarely a Coombs-positive haemolytic anaemia defines a severe form of giant cell hepatitis which rapidly progresses to cirrhosis or death.329 Early and sustained immunosuppressive therapy may control the disease in some patients.330 The liver lesion has been shown to recur in the allograft of the few cases transplanted.331, 332 Infants with birth asphyxia may develop severe neonatal hepatitis syndrome.333, 334 Conjugated hyperbilirubinaemia typically occurs at 1 week of age, lasts 3–4 months, and the hepatomegaly and liver tests return to normal by 1 year of age. Various hepatobiliary structural abnormalities, of which the most important is biliary atresia, are associated with the neonatal hepatitis syndrome. Furthermore certain chromosomal defects predispose to the neonatal hepatitis syndrome.
In approximately 30% of infants with conjugated hyperbilirubinaemia no aetiology is found. The prognosis for so-called idiopathic neonatal hepatitis is generally good; mortality runs at 13–25%.326, 335 In the study of Dick and Mowat,324 two of 29 patients with idiopathic neonatal hepatitis died, and only a further two had signs of persisting liver disease. Overall, predictors of poor prognosis include persisting severe jaundice, acholic stools, prominent hepatomegaly, severe inflammation on liver biopsy, and familial occurrence. Numerous inherited disorders causing the neonatal hepatitis syndrome have recently been described in terms of gene defect, such as the bile canalicular transporter disorders and bile acid synthesis disorders, and these had previously been classified as idiopathic neonatal hepatitis. Undoubtedly other such disorders remain to be delineated. A hereditary form with giant cell transformation and lymphoedema resulting from abnormal deep lymphatics has been reported but the gene abnormality has not yet been determined.336, 337 A further identified aetiology is adenosine deaminase deficiency, with recovery after enzyme replacement.338 Citrullinaemia type 2 due to deficiency of citrin can cause neonatal hepatitis syndrome (then also known as NICCD, neonatal intrahepatic cholestasis caused by citrin deficiency);339, 340, 341, 342 it may occur in ethnicities other than East Asian. ‘Le foie vide’ describes a severe neonatal liver disorder characterized by failure of hepatocellular regeneration.343
Histopathological features
Liver biopsy specimens are characterized by varying degrees of cholestasis (with or without pseudoglandular structures), giant cell transformation, ballooning, apoptotic bodies, extramedullary haemopoiesis, lobular and portal inflammation, and progressive fibrosis in some cases. Unusually severe inflammation and hepatocellular damage may be found in α1-antitrypsin deficiency, hereditary tyrosinaemia type 1, Niemann–Pick disease type C, syncytial giant cell hepatitis, citrullinaemia type 2, primary disorders of bile acid synthesis (mainly Δ4 −3-oxosteroid 5β-reductase deficiency), bile salt export pump (BSEP) deficiency (progressive familial intrahepatic cholestasis type 2) and idiopathic neonatal hepatitis. Associated macrovesicular steatosis favours a metabolic disorder, e.g. tyrosinaemia. Confluent hepatocyte necrosis or loss with bridging collapse is seen in the rare patients presenting with acute-pattern neonatal liver failure or having a subacute clinical course associated with perinatal haemochromatosis, non-Wilsonian copper toxicosis, or other metabolic disorders such as tyrosinaemia (Chapter 4), and occasionally viral hepatitis, in particular infants born to mothers carrying the precore mutant of hepatitis B, and seronegative (idiopathic) neonatal hepatitis.
The pathological aspects of giant cell transformation, a frequent and often dominant finding in neonatal hepatitis, have been reviewed extensively.344, 345, 346 The change is seen throughout the parenchyma but it is often more marked in the perivenular areas. The giant cells contain four or more nuclei, sometimes as many as 40 per cell, have ill-defined outlines and may be detached from other cells in the hepatic plate (Figure 3.20, Figure 3.21 ). The cytoplasm of some giant cells may contain remnants of cell membranes. It is partially rarefied and often contains bile and/or haemosiderin. The cells may have more glycogen than normal hepatocytes and a greater activity of a variety of enzymes, such as glucose 6-phosphatase, acid phosphatase and succinic dehydrogenase. Death of the giant cells is associated with a neutrophilic inflammatory response (Fig. 3.22 ). In severe forms, extensive bridging cell loss may divide the parenchyma into micronodules which are highlighted by a reticulin stain (Fig. 3.23A,B ). The number of giant cells decreases as patients grow older, and are rare after the age of 1 year. Formation of giant cells is considered to be a characteristic change resulting from mitotic inhibition of the young, growing liver tissue by a number of agents such as viruses, drugs, or hereditary abnormalities,347 or from dissolution of cell membranes, as suggested by Craig and Landing,348 who first described this entity. Negative nuclear staining for cell proliferation markers and the demonstration of canalicular remnants using carcinoembryonic antigen (CEA) immunostaining support a fusion of rosette-forming hepatocytes as the likely mechanism of giant cell formation.346
Figure 3.20.
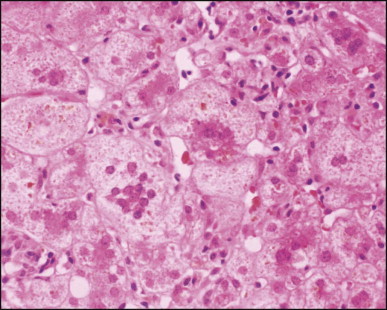
Neonatal hepatitis, idiopathic, with giant cell transformation. (H&E)
Figure 3.21.
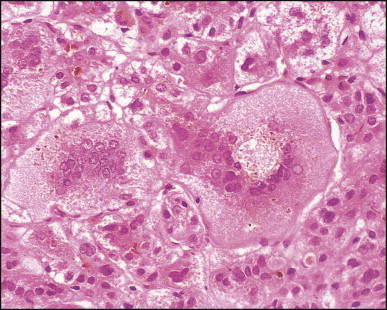
The same case as illustrated in Figure 3.20. Detail of multinucleated giant hepatocyte. Periphery of cell (left) suggests fusion with several smaller cells. (H&E)
Figure 3.22.
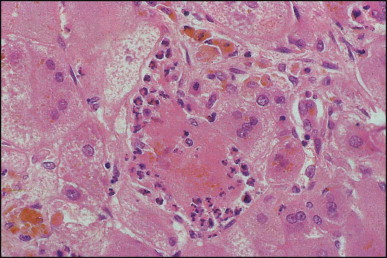
The same case as illustrated in Figure 3.20, Figure 3.21. Neutrophilic satellitosis of degenerated giant cell. (H&E)
Figure 3.23.
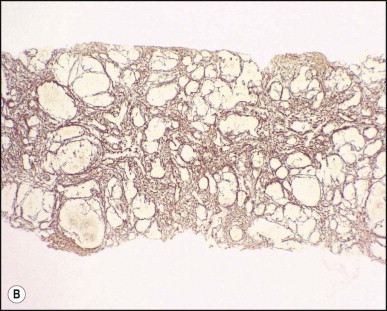
Severe neonatal hepatitis of undetermined cause. Extensive bridging parenchymal loss with concomitant reticulin collapse divides the parenchyma into minutes nodules with prominent giant cell transformation. (A) H&E; (B) Gordon-Sweet reticulin.
Paucity of the intrahepatic bile ducts
Paucity of the intrahepatic bile ducts has been reported in many conditions, either congenital or acquired, affecting all age groups, especially infants and children. The relevant finding is a reduction in the number of interlobular bile ducts, that is, in the small bile ducts with in portal tracts. The normal ratio of small bile ducts per portal tract in full-term infants, children and adults is 0.9–1.8 ducts per tract. In duct paucity syndromes this ratio is <0.5 given an adequate number of portal tracts (at least 10) examined on biopsy. Premature infants have a reduced number of small bile ducts per portal tract and if they have duct paucity with cholestasis, it may be physiological. In addition, it must be noted that early biopsy specimens in a few cases of clinically undisputed paucity of the intrahepatic bile ducts have shown not only identifiable ducts, but significant ductular reaction. Liver disorders with paucity of the intrahepatic bile ducts are generally divided into two groups: ‘syndromic’, which refers to Alagille syndrome (AGS or ALGS), and ‘non-syndromic’, which refers to all the rest of these diseases. The non-syndromic duct paucity conditions include numerous diseases where portal small duct paucity is associated with another identifiable disease. These include infection (congenital rubella or cytomegalovirus infection), immune abnormality (graft-versus-host disease, liver allograft chronic rejection), and hepatotoxicity (due to carbamazepine or amoxicillin-clavulinic acid),349 the latter two groups being generally referred to as ductopenia, formerly vanishing bile duct syndromes (see Chapters 10 and 15). Various inherited metabolic diseases such as Zellweger syndrome, α1-antitrypsin deficiency, and inborn errors of bile acid metabolism (discussed in Chapter 4) may display paucity of the intrahepatic ducts. Chromosomal defects, such as 45XO Turner syndrome,350 trisomy 17–18, trisomy 21,351 and prune belly syndrome,352 may have duct paucity. More importantly, the term non-syndromic duct paucity may be used to refer to isolated, idiopathic paucity of interlobular bile ducts in infancy and childhood; this condition may be the same as idiopathic adulthood ductopenia. Finally, paucity of the intrahepatic ducts or ductopenia is frequently found as a late feature of certain chronic diseases such as biliary atresia, sclerosing cholangitis, Langerhans cell histiocytosis, and primary biliary cirrhosis (Chapter 10).
Patients in the syndromic group have Alagille syndrome (ALGS), also known as arteriohepatic dysplasia,353 Comprehensive descriptions have been reported by Alagille and colleagues354, 355 and others.356, 357, 358 Two distinct genetic mechanisms are known to be responsible for Alagille syndrome. The vast majority of cases (ALGS1) are due to mutations in JAGGED1 (JAG1, encoding the ligand for the Notch 1 receptor) on chromosome 20p12359, 360 and may be associated with a macroscopic deletion of the short arm of chromosome 20 in some patients or microdeletions of 20p in others. The pattern of genetic transmission is autosomal dominant due to haploinsufficiency or dominant negative effect;360 gene penetrance is high but expression is extremely variable.361 The reported incidence of arteriohepatic dysplasia as 1 : 70 000 live births was an underestimate, and it is actually 1 : 30 000.358 Approximately 50–70% of patients have new mutations, rather than genetic transmission within the family. Crosnier et al.362 found mutations of the JAG1 gene in 69 of 109 patients (63%) with Alagille syndrome, and transmission analysis showed a high frequency of sporadic cases (70%). Numerous mutations have now been defined.363
The common clinical findings in Alagille syndrome include cholestatic liver disease due to paucity of the intrahepatic bile ducts (94%); congenital heart disease, usually peripheral pulmonary stenosis although complex congenital heart disease may occur (92%); a typical facies (91%); posterior embryotoxon in the eye (80–93%); and butterfly-shaped vertebral arch deficits (40–67%).357 The facies of Alagille syndrome consists of an inverted triangle shape, slight hypertelorism, deep-set eyes, broad and rather prominent forehead, and beak-like nose. Although the specificity of the facies has been questioned,364 it is accepted as a typical finding,365 better appreciated in the actual clinical setting than in photographs. The facies is sometimes not evident in the first months of life, and in adults the facies is somewhat different from that described in children (longer face, rather coarse features, prominent forehead, and comparatively small nose). Most patients have a systolic murmur related to stenosis of the pulmonary arterial system. More severe conditions include: tetralogy of Fallot, pulmonary valve stenosis, aortic stenosis, ventricular septal defect, atrial septal defect, anomalous pulmonary venous return, and complex problems involving a single right ventricle with pulmonary valve atresia.366, 367 Up to 15% of patients may have life-threatening cardiac complications.355 In addition to posterior embryotoxon or Axenfeld anomaly, abnormal retinal pigmentation may be found.368 Strabismus, ectopic pupil, and hypotrophic optic discs have also been reported. Optic disc drusen, which are calcified deposits in the extracellular space of the optic nerve head, occur commonly in Alagille syndrome. These can be found by ocular ultrasound examination and it may facilitate the diagnosis.369 Other skeletal abnormalities include short distal phalanges and clinodactyly.370, 371, 372
Other systems apart from those in the cardinal criteria for the syndrome may be affected. Renal abnormalities, such as tubulo-interstitial nephropathy, membranous nephropathy, single kidney and renovascular hypertension, may be prominent.373, 374, 375 The most frequent finding is mesangiolipidosis.376 Nephrolithiasis may occur, and various types of renal cystic disease have been reported.377 Renal tubular acidosis may develop.358 Two children with Alagille syndrome and nephroblastoma were reported.378 Systemic vascular disease appears to be more prevalent than originally appreciated. Several cases of moya-moya disease in association with Alagille syndrome have been reported.379, 380, 381 The propensity to intracranial bleeding found in the first few years of life in Alagille syndrome may be due to abnormal intracranial vessels.382, 383, 384 Abnormalities in the large intra-abdominal vessels have been found, and these abnormalities may complicate liver transplantation.381, 385 Skin changes relate mainly to formation of xanthomas, which regress after successful liver transplantation.386
Abnormalities of the biliary tract include hypoplasia of the extrahepatic bile ducts,387, 388, 389, 390, 391 hypoplasia of the gallbladder371 and cholelithiasis.387 Endoscopic retrograde cholangiopancreatography has demonstrated narrowing of the intrahepatic ducts, reduced arborization and focal areas of dilatation, as well as narrowing of the extrahepatic ducts.387, 388, 389. Portal venous disease may also be present, specifically hypoplasia of the portal vein.392
Many patients develop growth retardation prior to adolescence,393, 394 especially if they have persisting clinical jaundice. Short stature is found in some affected children whose nutrition is normal. Although some display some degree of mental retardation, in general children with Alagille syndrome show a broad range of cognitive and intellectual ability. Nearly all patients have pruritus, although it may be mild. They have elevated serum bile acids, the levels of cholic acid being greater than those of chenodeoxycholic acid. Variable hyperlipidaemia (sometimes severe, with xanthomas) is frequent. Treatments for the pruritus include cholestyramine, rifampicin or surgical diversion of bile flow.358, 395
In general, the prognosis is good for children whose jaundice resolves; however, approximately 25% of patients succumb in childhood to severe cardiac disease or progressive liver disease. The outcome in 92 patients in the series of Emerick et al.357 was as follows: the 20-year predicted life expectancy was 75% for all patients, 80% for those not requiring liver transplantation, and 60% for those who required transplantation. Liver transplantation has been reserved for patients with chronic liver failure, intolerable pruritus unresponsive to medical treatment, and severe growth failure. Transplantation for hepatic decompensation was necessary in 19 of 92 cases (21%) with 79% alive 1 year post-transplantation; the mortality was 17%. In the Bicêtre series of 163 patients followed over a 40-year period, 102 of 132 patients presenting with cholestatic jaundice in infancy remained jaundiced at the study endpoint, and one-third required liver transplantation; cirrhosis was found in some children with Alagille syndrome but no neonatal hepatitis syndrome and none of them underwent liver transplantation (two succumbing to end-stage liver disease before the advent of liver transplantation).393 Overall survival in this series was 62% at 20 years and seemed unaffected by availability of liver transplantation.393 The mortality is higher among patients who have more severe cardiac disease or intracranial bleeding, or who had previously undergone portoenterostomy.396, 397 A recent retrospective study suggested that affected pre-school-aged children with total serum bilirubin >111 µmol/L, serum conjugated bilirubin >77 µmol/L and serum cholesterol >13.5 mmol/L were likely to have a severe outcome, irrespective of actual JAG1 mutation.398 Liver transplantation is an option for those in end-stage liver failure, or with severe chronic cholestasis or intractable pruritus; however, severity of cardiac and renal involvement, as well as intra-abdominal vascular abnormalities, must be evaluated as contributing to increased surgical risk.399 Catch-up growth after transplantation is variable.400, 401 Long-term complications of Alagille syndrome not requiring liver transplantation have been reported including renal failure and intracranial bleeding or stroke.357, 391, 393 Hepatocellular carcinoma has been reported in children and adults with Alagille syndrome.402, 403, 404, 405, 406 Although rare, hepatic malignancy may occur in infants with Alagille syndrome.
Histopathological aspects of Alagille syndrome are described in various reports and reviews.354, 355, 389, 407, 408, 409, 410, 411, 412, 413 The major finding is absence of bile ducts from portal areas (Figure 3.24, Figure 3.25 ). The ratio of interlobular bile ducts to the number of portal areas is between 0.0 and 0.4 compared with 0.9 and 1.8 in normal children. A reduced number of portal areas has been noted.407 The loss of bile ducts is progressive from early infancy to childhood.357, 389, 412, 413, 414 Cholangiodestructive lesions have been observed in infants between 3 and 6 months of age.389, 413 The degree of cholestasis is variable in intensity and is especially prominent in the first 12 months of life. Immunohistochemical staining for endopectidase (CD10) is typically absent from the canalicular surface in children with Alagille syndrome.415 Giant cell transformation may be seen in early infancy.389, 412, 413 There is usually patchy pseudoxanthomatous change and accumulation of stainable copper and copper-associated protein in periportal hepatocytes.389, 412, 413 Copper accumulation has been demonstrated by quantitative methods in both syndromic and non-syndromic types of paucity of the intrahepatic bile ducts.408 Periportal fibrosis is mild, and it may remain unchanged long-term,354 possibly due to the absence of ductular reaction, known to play a role in periportal fibrogenesis. Nonetheless, although progression to cirrhosis is rare, occasional patients do develop extensive fibrosis or cirrhosis (Fig. 3.26 ). Portal inflammation and periportal ductular reaction, when present, are seen mainly in early infancy and can suggest the presence of distal duct obstruction.389, 413, 416
Figure 3.24.
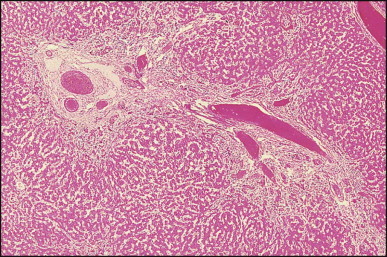
Arteriohepatic dysplasia. Two fused portal areas lack bile ducts. (H&E)
Figure 3.25.
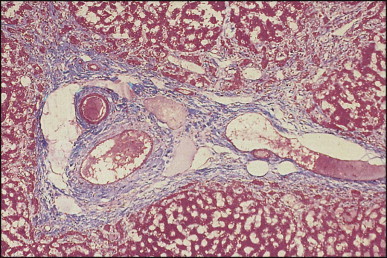
The same case as illustrated in Figure 3.24. Portal area contains vessels but no ducts. There is patchy periportal fibrosis but no cholangiolar proliferation. (Masson trichrome)
Figure 3.26.
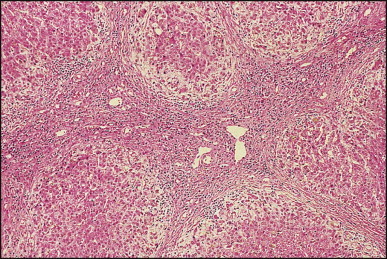
Arteriohepatic dysplasia. Micronodular cirrhosis. Note ductopenia, moderate septal inflammation and cholate stasis. (H&E)
Ultrastructural studies of the intrahepatic bile ducts in Alagille syndrome have been reported.410, 412, 413, 417 Bile canalicular changes are controversial. In one study of 12 biopsies from 10 patients, distinctive ultrastructural changes were noted.410 Bile pigment retention was found in the cytoplasm of liver cells, especially in lysosomes and in vesicles of the cis-Golgi, but only rarely in bile canaliculi or the immediate pericanalicular region. It was suggested that the basic defect in Alagille syndrome involves the bile secretory apparatus.410 The aetiopathogenesis of the cholangiodestructive lesions described histopathologically and ultrastructurally remains to be elucidated, but the possibility of ‘disuse atrophy’ has been raised by two groups of investigators.410, 413 Recent studies suggest defective branching of intrahepatic bile ducts in the postnatal period.418
Alagille syndrome is the first childhood disorder identified with a mutation in a ligand for a Notch protein. JAG1 encodes a ligand of Notch 1, one of a family of transmembrane proteins with epidermal growth factor (EGF)-like motifs. Notch proteins are highly conserved and have a role in determining cell fate during differentiation, especially in tissues where epithelial-mesenchymal interactions are important.419 Jagged/Notch interactions are known to be critical for determination of cell fates in early development. Notch 4 expression during embryogenesis is seen in endothelial cells of vessels forming the dorsal aorta, intersegmental vessels, cephalic vessels and the heart. The expression of Notch 1 and its ligand includes many of the organs potentially abnormal in Alagille syndrome.420 JAG1 plays an important role in embryogenesis of the heart, kidneys and blood vessels; in embryonic and fetal liver it is expressed in portal vascular tissue. Recent studies in mice indicate that Notch signalling regulates the remodelling of the ductal plate and bile duct morphogenesis and that Jagged 1 plays an important role in this complex process.421 Jagged 1 on ductal plate and Notch 3 on portal tract mesenchyme and hepatic arterial endothelium interact for ductal plate remodelling and development of intrahepatic bile ducts.422, 423 Mice with defects in murine Jagged 1 and Notch 2 expression have abnormalities similar to human Alagille syndrome; zebrafish with knockdowns of jagged ± notch genes have biliary, pancreatic, cardiac, renal, and craniofacial developmental abnormalities; these studies suggest that Notch may promote biliary epithelial cell evolution from a bipotential precursor cells.424 In humans, mutations in JAG1 result in truncated, and inactive proteins; since residual gene expression cannot compensate, there is haploinsufficiency.363 Dose of Notch ligands is critical and this may contribute to the clinical diversity of Alagille syndrome. No clear relationship between genotype and phenotype has been found, but the Delta/Serrate/Lag-2 (DSL) domain in the JAG1 protein may influence the severity of liver disease.425, 426, 427
A second genetic basis for Alagille syndrome has been reported. Children with mutations in the gene encoding Notch 2 (found on chromosome 1p13) have a clinical disorder like conventional Alagille syndrome, except for a somewhat more severe renal disease.428 Notch 2 appears to have an important role in bile duct development.429, 430 This second version of Alagille syndrome is designated ALGS2, but it accounts for a very small proportion of cases.
In contrast to patients with Alagille syndrome, those with non-syndromic bile duct paucity do not have a constellation of extrahepatic disorders. Children with non-syndromic bile duct paucity are supposed to have a less favourable outlook than children with Alagille syndrome. They present with persistent cholestasis and severe pruritus. Growth retardation is common. No associated aetiological agent, defined genetic factors or congenital anomalies have been found in this group, except for one study of 10 patients with a high rate of consanguinity.431 A chronic cholestatic disease with duct paucity, called idiopathic adulthood ductopenia, has been described in adults.432, 433, 434 Most of these patients are young adults, although patients over 60 have been reported.435 Non-syndromatic paucity of bile ducts in infancy and idiopathic adulthood ductopenia may be related diseases. The outlook for younger patients with idiopathic adulthood ductopenia is poor; approximately 50% succumb to progressive liver disease or require transplantation. Hepatocellular carcinoma has been reported in a woman with intrahepatic biliary hypoplasia, apparently distinct from Alagille syndrome.436
A histopathological study of 17 children with non-syndromic paucity of bile ducts was reported by Kahn et al.437 Before 90 days of age there was paucity of ducts and periportal fibrosis as well as nonspecific parenchymal changes (cholestasis, giant cell transformation, perisinusoidal fibrosis and haematopoiesis). After 90 days the duct paucity and fibrosis persisted but cholestasis was mild or no longer apparent. Kahn et al.437 suggested that the paucity in non-syndromic cases may result from a primary ductal insult with destruction and disappearance of the ducts. The differential diagnosis of paucity of the intrahepatic bile ducts in children and adults has been reviewed by West and Chatila.438
Congenital dilatations of the bile ducts
Congenital dilatations of the bile ducts are classified into five types, both extrahepatic and intrahepatic:439
-
•Type I – a dilatation of the common bile duct which may present three anatomical variations:
-
aLarge saccular
-
bSmall localized
-
cDiffuse fusiform.
-
a
-
•
Type II – diverticulum of the common bile duct or the gallbladder
-
•
Type III – choledochocoele
-
•
Type IV – multiple intrahepatic and extrahepatic dilatations (Caroli disease)
-
•
Type V – fusiform intrahepatic and extrahepatic dilatations.
Types I and IV account for the majority of reported cases although types IV and V may prevail in the Far East, where the disease occurs more frequently. Type IV cysts are more frequent in adults than in children.440 Although this classification remains in common use, its value has been questioned by Visser et al.,441 who consider the distinction between type I and IV arbitrary; they suggest that the term ‘choledochal cyst’ should be reserved for congenital dilatation of the extrahepatic and intrahepatic bile ducts, other forms being referred to by their name, for example choledochocoele and bile duct diverticulum. Caroli disease, assimilated to type IV in Hadad classification, is not clearly related to ‘choledochal cyst’, given its common association with both congenital hepatic fibrosis and fibrocystic lesions in the kidney and its distinct morphological features (see below).
Choledochal cyst
The classic clinical triad of pain, a mass in the right upper quadrant and jaundice, occurs in less than a third of patients with a choledochal cyst.442, 443 In children, jaundice is the most common presentation, while in adults the signs and symptoms are those of ascending cholangitis.444 In the early years of life cholestasis is usually associated with cystic dilatation of the common bile duct and accounts for 2% of infants presenting with cholestasis. Up to 60% of choledochal cysts are diagnosed before age 10 years, but diagnosis can be made at any age and some cases may present for the first time at as late as the 8th decade of life.445 Several cases have been diagnosed antenatally.446, 447 Some 80% of the patients are female. Differences in presentation between children and adults with choledochal cysts have been emphasized in two large series.448, 449, 450 The preoperative diagnosis can be made in the majority of patients by cholangiographic studies, ultrasonography and isotope scanning.451 Dynamic magnetic resonance cholangiopancreatography (MRCP), including secretin stimulation, contributes to the understanding of the pathophysiology.452
Complications include perforation, liver abscesses, stone formation, secondary biliary cirrhosis, pancreatitis, amyloidosis and carcinoma of the biliary tree.442, 453, 454, 455, 456, 457 Regression of biliary cirrhosis following drainage of a choledochal cyst has been reported.458 One case presented with anaemia secondary to bleeding from erosions of the duodenal mucosa between the ampullary sphincter and the sphincters of the common bile duct and pancreatic duct.459 Biliary tract anomalies reported in association with a choledochal cyst include double common bile duct, double gallbladder, absent gallbladder, annular pancreas, biliary atresia or stenosis,460 stenoses of the intrahepatic bile ducts,461 and most commonly anomalies of the pancreaticobiliary junction.462, 463, 464 In a series of 104 choledochal cysts from Japan, 25% were found to have co-existing biliary anomalies.465 Differences between (1) isolated choledochal cysts and (2) choledochal cysts associated with biliary atresia have been noted by ultrasonography. In the former the cysts are larger, intrahepatic ducts are dilated and the gallbladder is not atretic as compared to those with choledochal cysts and biliary atresia.466 In general, the apparent choledochal cyst associated with biliary atresia is actually proximal duct dilatation associated with focal atresia of the distal common bile duct, so-called ‘correctable’ or cystic atresia.
Maljunction of the pancreaticobiliary ductal system (common channel) remains the most plausible aetiopathogenic mechanism for choledochal cysts, and this is supported by experimental studies.467 The lesion is defined as a junction of the pancreatic and bile ducts located outside the duodenal wall, usually forming a markedly long common channel. The anomaly allows pancreatobiliary regurgitation since hydrostatic pressure within the pancreatic duct is usually higher than that in the common bile duct. As a result, high pancreatic enzyme levels are found in the bile. Common channels may occur without bile duct dilatation and lead to primarily gallbladder rather than biliary complications including malignancy. In this instance prophylactic cholecystectomy might be sufficient whereas biliary complications and the risk of cholangiocarcinoma underpin the need for radical surgical resection in those associated with choledochal cysts.464
Reovirus 3 RNA sequences have been recovered from resected choledochal cysts,224 but the implication of reovirus infection in the aetiology of choledochal cyst is unclear. A single report of choledochal cysts in association with familial adenomatous polyposis raises the possibility of a genetic basis for the cysts.468
Treatment is by complete cyst resection, cholecystectomy and Roux-en-Y hepaticojejunostomy as a preventive measure against the subsequent development of carcinoma.469, 470 Laparoscopic repair has been successful in two recent series.471, 472 Dilemmas may arise when the cysts involve the intrahepatic or intrapancreatic segments, requiring more extensive surgery, given the small risk of malignancy developing in cystic remnant at the anastomotic site or in the dilated intrahepatic bile duct of type IV or V cysts.443, 473 In a report of 48 Japanese patients treated by total or subtotal excision, no malignant change occurred after a mean follow-up of 9.1 years.474 Choledochal cysts vary greatly in size with some of the larger ones containing 5–10 litres of bile (Figure 3.27, Figure 3.28 ). Histopathologically, the wall is usually thickened by inflammation and fibrosis, and is bile stained. Smooth muscle fibres may be identified in the lower portion of the cyst but not in the narrow (intrapancreatic) portion.475 There is generally no epithelial lining but islets of cylindrical or columnar epithelium may be preserved (Fig. 3.29 ). Intestinal metaplasia with mucous gland proliferation, as well as the presence of goblet and Paneth cells and neuroendocrine differentiation, have been reported.476, 477 According to Komi et al.477 the intestinal metaplasia increases with age, so that it can be demonstrated in almost all cysts from patients over 15 years of age. Kusunoki et al. noted the absence of ganglion cells in the narrow portion of a choledochal cyst, and suggested that the cyst could be the result of postganglionic neural dysfunction.478
Figure 3.27.
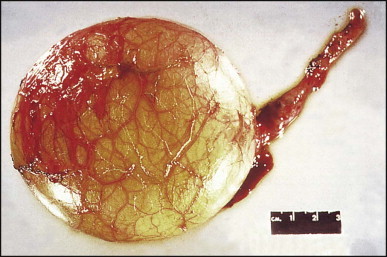
Choledochal cyst measuring about 10 cm in diameter. Note numerous vessels over the surface.
Figure 3.28.
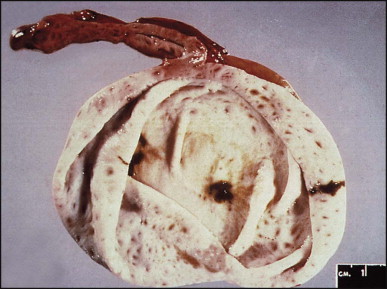
Same cyst as illustrated in Fig. 3.27. Opened, collapsed cyst has a smooth inner lining. It did not contain bile.
Figure 3.29.
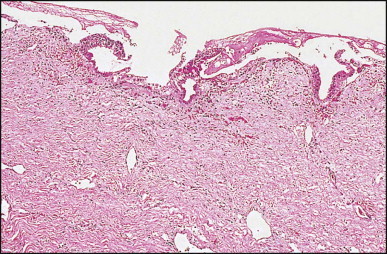
Segment of wall of choledochal cyst shows inflammation and focal epithelial ulceration. (H&E)
The majority of tumours arising in congenital cystic dilatations of the bile ducts are adenocarcinomas, but some anaplastic and several squamous carcinomas have been reported479, 480, 481 and one report mentioned sarcomatous changes.482 The overall incidence of carcinoma arising in all cystic dilatations of the bile ducts is about 3%.480 The risk is age-related, increasing from 0.7% in the first decade to 6.8% in the second decade to 14.3% in later decades.481 The complication is thus usually seen in adults; only three patients reviewed by Iwai et al. were under 18 years of age.483 An 11-year-old boy is the youngest so far reported.484 Reveille et al.485 found that stasis of bile in the choledochal cyst contributes to bacterial overgrowth and the generation of unconjugated secondary bile acids, a possible cause of biliary metaplasia and carcinoma. Interestingly, bile from congenital choledochal cyst patients is shown to promote the proliferation of human cholangiocarcinoma QBC939 cells.486 Certain bile acid fractions together with reflux of pancreatic enzymes may play a primary role as pancreaticobiliary maljunction is associated with an increased risk of biliary tree malignancy irrespective of the presence of cysts.487
Hereditary fibropolycystic disease of the liver (ductal plate malformation)
The term fibropolycystic diseases of the liver – not to be confused with cystic fibrosis – is used to describe a heterogeneous group of genetic disorders in which segmental dilatations of the intrahepatic bile ducts and associated fibrosis can be interpreted as sequelae of persistence and/or aberrant remodelling of the embryonal ductal plate (Chapter 1). They represent a spectrum of microscopic and/or macroscopic cystic lesions often associated with fibrocystic anomalies in the kidneys. The severity of the renal lesions may overshadow the liver disease, as in the early presentation of autosomal recessive polycystic kidney disease. Conversely, portal hypertension with a preserved liver function may later in life dominate the picture, as exemplified by congenital hepatic fibrosis. Cholangitis may develop, especially when the cysts communicate with the biliary system. These abnormalities are classified as ductal plate malformation,488, 489 a term which refers to the histological changes of circumferentially disposed and variably ectatic bile ducts and ductules, often directly abutting the hepatocytic plates, which resemble an exuberant embryonal ductal plate. The main disorders, in particular autosomal recessive polycystic kidney disease (ARPKD), the closely associated congenital hepatic fibrosis and Caroli disease, and autosomal dominant polycystic kidney disease (ADPKD), are discussed in detail, whereas the rarer associated syndromes, which have been comprehensively reviewed by Knisely,490 are briefly mentioned. Over the past decade genes and encoded proteins for several of these disorders have been identified and these are summarized in Table 3.2 . The recognition that the ‘cystoproteins’, the mutation of which causes polycystic disease, have been localized in the primary cilia or basal bodies of tubular epithelial cells has led to a renewed interest in these forgotten structures. The clinicopathological discussion of specific disorders is therefore preceded by a short account on cilia and cystogenesis; more details and comprehensive references can be found in recent reviews.491, 492
Table 3.2.
Genes and encoded proteins in polycystic diseases of kidney and liver
| Disorder | Gene | Product | Localized in cilium |
|---|---|---|---|
| ARPKD | PKHD | Fibrocystin/polyductin | Yes |
| ADPKD |
PKD1 | Polycystin 1 | Yes |
| PKD2 | Polycystin 2 | Yes | |
| PCLD | PRKCSH | β-subunit of glucosidase II | No |
| SEC63 | Component of translocon in endoplasmic reticulum | No | |
ARPKD, autosomal recessive polycystic kidney disease; ADPKD, Autosomal dominant polycystic kidney disease; PCLD, polycystic liver disease without kidney abnormalities.
Primary cilia and cystogenesis
Most studies on mechanisms of cyst development in hereditary polycystic disease have been conducted in the kidney, but there are strong suggestions that a number of pathways are common to the conversion of tubes into cysts in general, and especially in the kidney tubules and the hepatic bile ducts. Primary (or solitary) cilia arise from centrioles and form a finger-like extension of the cytoplasm covered with the cell membrane. In their axis primary cilia contain a system of nine pairs of longitudinal microtubules arranged in a circle (the axoneme); in contrast, motile cilia (such as those in ciliated respiratory epithelium) contain a set of two centrally-located microtubules within the circle of the nine pairs of longitudinal microtubules (‘9+2 pattern’). The pattern typical of, and defining, primary ciliary is the ‘9+0 pattern’.493 The microtubules secure the circulation of non-membrane bound macromolecules – intraflagellar transport – which is essential for the assembly and maintenance of cilia and also acts as a sensory process, in that the particles and associated peptides are changed in the cilium and carry a message back to the cell body. The solitary cilium is at present considered to represent a sensory antenna, functioning through the polycystin complex as a mechanotransducer. Loss of function of the complex results in perturbation of normal intracellular Ca2+ concentration which underlies a multitude of pathological reactions.494 Severe alterations in structure and function of tubular or ductal cells will follow involving cell–cell contact, actin cytoskeleton organization, cell-extracellular matrix interactions, cell proliferation and apoptosis.495
An enticing finding of recent years has been the demonstration that most proteins mutated in experimental polycystic disease have been localized in the primary cilia or basal bodies of tubular epithelial cells.496 They include those responsible for human disease, in particular polycystin-1 and polycystin-2,497 fibrocystin/polyductin498 but not the cystoproteins of isolated PCLD: hepatocystin and SEC63.499 Mutational analysis best studied in ADPKD suggests that both germline and somatic mutations may be involved in a two-hit model, either as a prerequisite initiating event or as somatic later event needed for cyst expansion and progression.496 Although the ciliary model is most attractive, it may represent only one of several parallel pathways that control essential cellular functions such as proliferation, apoptosis, adhesion, and differentiation.500
Cyst epithelial lining cells show profound pathological alterations. An increased proliferation has been observed in ADPKD, comprising formation of papillary projections and overexpression of Ki-67 and PCNA (proliferating cell nuclear antigen), a loss of their ‘planar or tissue polarity’ (the ability to sense their position and orientation relative to the overall orientation of the epithelial sheet) and a switch from an absorptive into a secretory cell type, resulting in fluid secretion that is responsible (together with epithelial proliferation) for further cyst expansion.501 Although studies of cyst development have been mainly performed in kidney, primary cilia have been explored in cholangiocytes502, 503 and evidence produced that – at least to some extent – cystogenesis in bile ducts and ductules may be similar to that in the kidney. In ADPKD, PKD1 and PKD2 are co-localized in bile duct epithelial cells504 and the two-hit model of germ-line plus somatic mutations of PKD1 in focal liver cyst development appears applicable, as in the kidney cysts.505 Biliary cyst, like kidney cyst, expansion occurs by secretion (as well as proliferation) of the cyst lining epithelial cells. Autocrine and paracrine factors, including IL-8, epithelial neutrophil attractant 78, IL-6, and vascular endothelial growth factor, secreted into the cystic lumen modulate the rate of errant hepatic cyst growth.506 In ARPKD, findings suggestive of similarity with renal cystogenesis include: the localization of PKHD1 in the cilia of cholangiocytes in the PCK rat,502 which is a model for Caroli syndrome,507 the finding that hepatocyte nuclear factor1β (HNF1β) localized in bile duct epithelium regulates expression of PKHD1,508 together with other ‘cystoproteins’ that co-localize into the cilium.509
Differences in cystogenetic pathways between liver and kidney in ARPKD are indicated in animal models of the disease.491 The mouse cpk mutation is the most extensively characterized murine model, closely resembling human ARPKD, with the exception that the B6-cpk/cpk homozygotes do not express the lesion of ductal plate malformation (DPM). However, homozygous mutants from outcrosses to some other strains, express the DPM. Genetic analysis supports a loss-of-function model (two-hit model) for biliary cysts developing in an age-dependent fashion. There is no correlation between the severity of the DPM and the renal cystic disease. Expression of the biliary lesion is modulated by genetic background, and the specific biliary phenotype (DPM-predominant or cyst-predominant) is determined by whether loss of function of the cpk gene occurs as a germline or a somatic event.510
In the mouse model generated by targeted mutation of Pkdh1, the animals develop severe malformation of their intrahepatic bile ducts, but develop normal kidneys.511 The cholangiocytes maintain a proliferative state secreting TGFβ1 that continuously stimulates mesenchymal cells to synthesize and put down extracellular matrix, resulting in fibrosis. The findings suggest that fibrocystin/polyductin, already expressed in the embryonic ductal plate stage512 acts as a matrix sensor and signal receptor during intrahepatic bile duct development, and that its mutation results in a congenital hepatic fibrosis (CHF)-like picture.513 Its role in liver and kidney appears to be functionally divergent, because protein domains essential for bile duct development do not affect nephrogenesis in this model.511
Autosomal recessive polycystic kidney disease (ARPKD)
Infantile presentation
This disease is inherited in an autosomal recessive manner. The prevalence is estimated to be between 1: 10 000 and 1: 60 000 live births.514 The gene for this disorder, PKHD1 (polycystic kidney and hepatic disease 1), was mapped to chromosome 6p 21.2–p12,515, 516 both the severe and mild form of the disease mapped to the same locus.517, 518 The gene encodes a 4074-amino-acid protein, called fibrocystin519 or polyductin520; this large transmembrane polypeptide, also known to localize to the cilia498 may be a receptor that acts in the differentiation of renal collecting-duct and bile duct. An animal model is widely used.510 In humans there is an equal sex incidence. Depending on the age of presentation and the degree of renal involvement, ARPKD has been divided into four types by Blyth and Ockenden521 – perinatal, neonatal, infantile and juvenile. These authors proposed that four different mutant alleles are responsible, and that there may be a fifth group in which the onset of symptoms is later than juvenile. ARPKD has been reviewed by a number of authors.253, 522, 523
The perinatal type is the most severe form. Fifty per cent of patients are diagnosed perinatally and die shortly after birth.517 In the series of Blyth and Ockenden521, no infant survived beyond 6 weeks of age. The majority of patients were admitted with signs of respiratory distress and had marked abdominal distension due to huge symmetrical renal masses. Liver function test abnormalities were uncommon. Surviving patients with the neonatal type of the disease develop gradually increasing renal insufficiency and systemic hypertension. Pyelonephritis is common. Portal fibrosis and cystic dilatation of bile ducts are severe, and cholangitis is a frequent complication. In the infantile group the clinical picture is either of chronic renal failure or of increasing portal hypertension. Portal fibrosis is moderate. The juvenile group of Blyth and Ockenden521 typically includes children (1–5 years old) who present with portal hypertension. Liver histopathological changes are marked. It is likely that this group represents cases of congenital hepatic fibrosis, as suggested by Landing et al.524 Gang and Herrin525 have found that ARPKD has a spectrum of phenotype expression with prognostic implications, but suggest that not all cases fit into the sharply defined subgroups of Blyth and Ockenden. In their study of 11 patients, four had 90% or more renal cystic change; these patients did not survive beyond 20 days of birth. In contrast, five of the seven less severely affected patients with a 20–75% range of cystic changes in the kidneys were all alive at 6–21 years of age.
The liver in ARPKD does not appear abnormal macroscopically, although it may be enlarged and firm. Histologically, the changes range from a persistent, circular ductal plate (Fig. 3.30 ) to a striking increase in the number of biliary channels which arise in portal areas and extend irregularly and deeply into the parenchyma (Fig. 3.31A ). They appear to branch or ‘anastomose’ and often show polypoid projections (Fig. 3.31B). Normal interlobular ducts with corresponding arteries are not seen. According to Witzleben,522 the biliary channels are in continuity with the rest of the biliary system, similar to the cystic spaces of Caroli disease (‘communicating’ cystic disease), in contrast to the non-communicating ADPKD. The supporting connective tissue is very scanty and, in the intralobular extensions, the basement membrane of the epithelium appears to be in direct contact with the liver cell plates. The epithelial lining consists of a single layer of low columnar to cuboidal cells. Cyst formation is uncommon. The dilated channels may contain a small quantity of a pink or orange-coloured material or, rarely, pus. Reconstruction studies by Adams et al.526 have shown irregularly dilated ducts running longitudinally at the periphery of the portal tract and anastomosing so extensively that they formed a single annular channel; no main interlobular duct could be identified in that portal tract or in several others from the same liver. A stereological study of 10 cases by Jörgensen527 showed ductal structures that could be divided into two groups: one consisted of irregular tubular structures shaped like circular cylinders, and the other of elliptical cylinders (the ‘ductal plates’). These were dilated but cysts were rare. In patients who survive for months or years there is marked fibrosis in the liver as well as kidney lesions which appear to have been progressive lesions.
Figure 3.30.
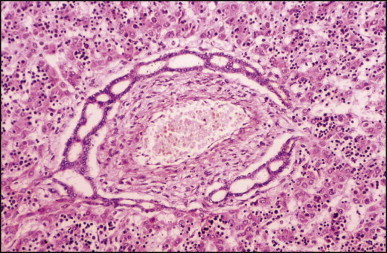
Autosomal recessive polycystic kidney disease: unusually prominent ductal plate in the liver of a stillborn fetus with huge polycystic kidneys. (H&E)
Figure 3.31.
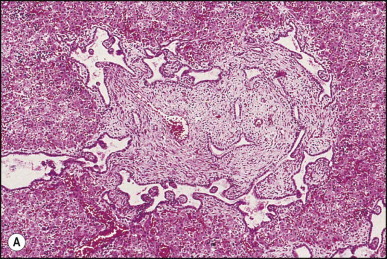
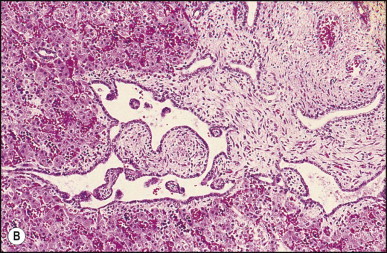
Autosomal recessive polycystic kidney disease. (A) The bile ducts form an interrupted ring at the periphery of what should eventually become a portal area. There is no interlobular bile duct. Note branch of portal vein, as well as smaller vessels. (H&E) (B) The same case, with higher magnification of part of the ductal plate showing irregularity in outline of the ducts with polypoid projections into a dilated lumen. The lining epithelium is low cuboidal. (H&E)
Juvenile and adult presentation – congenital hepatic fibrosis (CHF)
Congenital hepatic fibrosis is considered a variant of ARPKD affecting predominantly children and adolescents, some cases being identical to the ‘juvenile’ form. The inheritance pattern is not a simple autosomal recessive one. PKHD1 missense mutation has been identified in cases of congenital hepatic fibrosis with minimal kidney involvement.528 It may be associated with dilatation of the intra- or extrahepatic bile ducts,529, 530 so-called Caroli syndrome (see below), the intrahepatic cysts being detectable by ultrasonography or magnetic resonance cholangiography.531 Infants present with abdominal distension from enlarged organs, respiratory distress and systemic hypertension. Older patients come to medical attention because of hepatosplenomegaly or bleeding from oesophageal varices532, but asymptomatic cases have been reported.533 Cholangitis as a manifestation of CHF has been emphasized by Fauvert and Benhamou,533 who recognized four clinical forms – portal hypertensive, cholangitic, mixed portal hypertensive-cholangitic and latent forms. The pure cholangitic form is rare. In the mixed form patients suffer from recurrent bouts of cholangitis, with or without jaundice, in addition to the manifestations of portal hypertension.
In one series of 42 children with ARPKD from 20 sibships, 12 patients presented in the perinatal period, nine in the neonatal period, 13 in the infantile period, and eight in the juvenile period; the presentation and course of the disease was disparate in over 50% of patients.534 The organ predominantly affected may vary within the same family. Routine liver enzymes are usually normal, although alkaline phosphatase levels may be increased.
When present, the usual renal disease in CHF is medullary tubular ectasia, a fusiform or cystic dilation of tubules (particularly the collecting ducts). Occasionally, patients have cystic kidneys typical of adult-type polycystic disease (ADPKD), an autosomal dominant disease, and others may have nephronophthisis.535, 536, 537, 538
The prognosis in patients surviving beyond the neonatal period is generally good and depends on the extent of hepatic and renal disease. Death related to renal failure or uncontrollable variceal bleed is now exceptional.539 The combination of a patent portal vein and well-preserved liver function makes patients with CHF ideal candidates for standard portosystemic shunt surgery. Follow-up examination of 16 patients who underwent portosystemic shunt surgery540 revealed no impairment of liver function or hepatic encephalopathy. Development of cholangiocarcinoma may have been a chance occurrence in an adult case not associated with Caroli disease.541, 542
Pathological descriptions of CHF have been published by many authors.522, 533, 543 Grossly, the liver is enlarged, has a firm to hard consistency, and shows a fine reticular pattern of fibrosis; no cysts are visible to the naked eye. Although the entire liver is usually involved, occasional lobar cases of CHF are described.540
Microscopically, there is diffuse periportal fibrosis, the bands of fibrous tissue varying in thickness. Irregularly shaped islands of hepatic tissue, some incorporating several lobules, may be seen (Fig. 3.32 ). When the bands of fibrous tissue are thick, hepatic venules may be encroached upon and become incorporated within the fibrous tissue; thus, portal hypertension in this condition may not always be presinusoidal. The fibrous bands may encircle single or groups of lobules; occasionally, a small islet of hepatic tissue becomes separated from an acinus and encircled by fibrous tissue. Numerous uniform and generally small bile ducts are scattered in the fibrous tissue (Fig. 3.33 ). An interrupted circular arrangement of the ducts (ductal plate malformation) is often recognizable (Fig. 3.34 ). The ducts are lined by cuboidal to low columnar epithelium and may contain bile or traces of mucin. They may be slightly dilated and irregular in outline. Cholestasis is not a feature of the uncomplicated case of CHF. Reduction in the number of portal vein branches is often apparent, the likely cause of the portal hypertension. There is generally little inflammation in CHF except in cases associated with cholangitis, when numerous neutrophils infiltrate ducts, ductules and surrounding connective tissue (Fig. 3.35 ); rupture of the ducts can result in micro-abscess formation. Histopathologically, the latter cases may be difficult to differentiate from extrahepatic biliary obstruction with ascending infection, particularly since there may be an associated tissue cholestasis. The correct diagnosis must be based on the history, clinical findings and the results of imaging studies. Cholestasis is particularly prominent in those cases associated with microscopic dilatation of the intrahepatic bile ducts, too subtle to be seen on imaging (‘microscopical Caroli’). Recurrent cholangitis may lead to progressive fibrosis with functional impairment similar to cirrhosis.
Figure 3.32.
Congenital hepatic fibrosis. Jigsaw pattern of hepatic parenchyma and fibrous tissue. (PAS)
Figure 3.33.
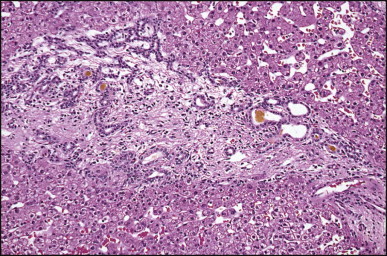
Congenital hepatic fibrosis. Fibrous tissue contains small bile ducts, a few of which are dilated and contain bile. Note absence of inflammation. The adjacent hepatic parenchyma shows no evidence of cholestasis and the patient is jaundice-free. (H&E)
Figure 3.34.
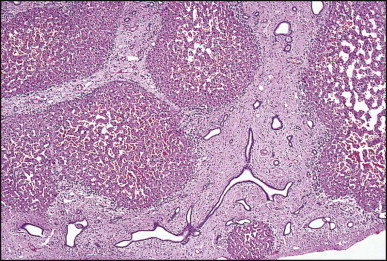
Congenital hepatic fibrosis. The same case as illustrated in Figure 3.33. Appearance of ducts suggests part of a ductal plate malformation. (H&E)
Figure 3.35.
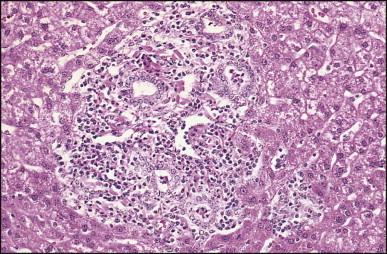
Congenital hepatic fibrosis, ‘cholangitic type’. The ducts are involved by an acute cholangitis. (H&E)
A number of malformation syndromes, characterized by hepatic morphological changes which resemble those of CHF (ductal plate malformation), can be differentiated by the associated findings. They include:
-
•
Medullary cystic kidney disease 1 (MCKD1),544 which has an autosomal dominant inheritance, has been mapped to chromosome 1q21
-
•
Medullary cystic kidney disease 2 – via uromodulin (UMOD 2),545 also autosomal dominant, mapped to chromosome 16p12
-
•
Nephronophthisis – congenital hepatic fibrosis,546 a recessive disorder, whose gene NPHP1 has been mapped to chromosome 16p12
-
•
Meckel syndrome 547 with encephalocoele, polydactyly and cystic kidneys
-
•
Renal-hepatic-pancreatic dysplasia 548 with polycystic kidney disease, ductal plate malformation in liver and pancreatic dysplasia, incompatible with postnatal survival, candidate genetic locus on chromosome 3
-
•
Asplenia with cystic liver, kidney and pancreas, which represents Ivemark syndrome with variants,549 possibly in the same spectrum as renal-hepatic-pancreatic dysplasia
-
•
Ellis–van Creveld syndrome or chondroectodermal dysplasia 550 with polydactyly, short limbs, short ribs, postaxial polydactyly, and dysplastic nails and feet, which has been mapped to chromosome 4p16
-
•
Sensenbrenner syndrome (cranio-ectodermal dysplasia)551 with dwarfism and progressive tubulo-interstitial nephritis
-
•
Asphyxiating thoracic dystrophy (Jeune syndrome) 552 with skeletal dysplasia, pulmonary hypoplasia and retinal lesions
-
•
Congenital disorder of glycosylation, type Ib (phosphomannose isomerase deficiency),553 sometimes with protein-losing enteropathy (see Chapter 4)
-
•
Joubert syndrome with cerebellar vermis hypoplasia/aplasia, colobomata, and psychomotor retardation;554 recently defined by the acronym COACH syndrome (Cerebellar vermis hypoplasia, Oligophrenia, Ataxia, Coloboma, and Hepatic fibrosis) shown to carry mutation in a Meckel syndrome gene (MKS3)555, 556
-
•
Vaginal atresia syndrome 524
-
•
Tuberous sclerosis.524
Caroli disease
This disease generally involves the entire liver, but it may be segmental or lobar.557, 558, 559 The inheritance is autosomal recessive. By 1982, some 99 cases had been reported; they were evaluated together with 10 personally studied cases by Mercadier et al.560 Since then, many other case reports, as well as small series, have been reported.561, 562 Clinically, patients suffer from bouts of recurrent fever and pain. Jaundice occurs only when sludge or stones block the common bile duct. Liver enzymes are generally normal except during episodes of biliary obstruction. The diagnosis is established by a variety of imaging modalities including ERCP, ultrasonography, computed tomography and MRCP.563, 564, 565, 566
The complications of Caroli disease resemble those of choledochal cyst and include recurrent cholangitis, abscess formation, septicaemia, intrahepatic lithiasis and amyloidosis. Spontaneous rupture of a bile duct was reported by Chalasani et al.567 Adenocarcinomas, including some arising in cases with a lobar distribution, have also been reported;542, 568 their incidence is 7%.569 Hepatocellular carcinoma occurs rarely.570 Medical treatment consists of symptomatic, or prophylactic, treatment of cholangitis and promotion of bile flow with ursodiol. Surgical treatment includes internal or external drainage procedures.560 Transhepatic decompression has been advocated.571 Segmental or lobar forms of Caroli disease can be treated by partial hepatectomy.560, 572 Extracorporeal shock wave lithotripsy has been utilized for disintegration of intrahepatic stones.573 Liver or combined liver and kidney transplantation has been successfully performed in patients with mono- and multilobar disease with portal hypertension, respectively 96 and 8 having received liver or combined liver and kidney grafts from 1987 to 2006 in the USA;574 the authors propose an algorithm for evaluation and treatment of Caroli disease.
Macroscopically, the intrahepatic cystic dilatations are round or lanceolate, 1.0–4.5 cm in diameter, and may be separated by stretches of essentially normal duct (Fig. 3.36 ).557 Transluminal fibrovascular bridges are reminiscent of the periportal location of the cysts (ductal plate) and explain in part the central dot sign observed on computed tomography.565 Inspissated bile or soft and friable bilirubin calculi may be found in the lumen. Microscopically, the dilated ducts usually show severe chronic inflammation, with or without superimposed acute inflammation, and varying degrees of fibrosis (Fig. 3.37 ). The epithelium may appear normal (cuboidal to tall columnar), partly or completely ulcerated, or focally hyperplastic; all of these changes can be found in different ducts in the same liver (Fig. 3.38 ). Mucous glands (sometimes in abundance) may be present in the fibrotic and inflamed wall. Areas of severe epithelial dysplasia are seen rarely.575 The lumen contains admixtures of inspissated mucin and bile, calcareous material, or frank pus during bouts of acute cholangitis (Fig. 3.39 ). Caroli disease is frequently associated with congenital hepatic fibrosis (in which case it is termed Caroli syndrome), rarely with infantile polycystic disease (ARPKD)576 and even adult polycystic disease (ADPKD).577
Figure 3.36.
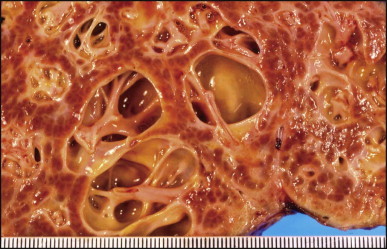
Caroli disease. Section of liver shows cystically dilated bile ducts. The cystic cavities are traversed by fibrous cords, known to contain the portal vessels. The lining of the ducts is bile stained.
Figure 3.37.
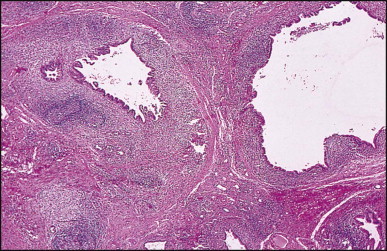
Caroli disease. Dilated bile ducts have thickened walls due to marked chronic inflammation. Note lymphoid follicles in the wall of one duct (left). (H&E)
Figure 3.38.
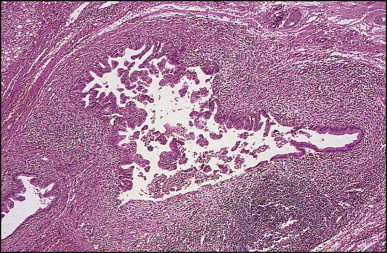
Caroli disease. The thickened bile duct wall reveals marked chronic inflammation. The lining epithelium is hyperplastic, except for a small segment of the duct (right). There is an area of ulceration (bottom). (H&E)
Figure 3.39.
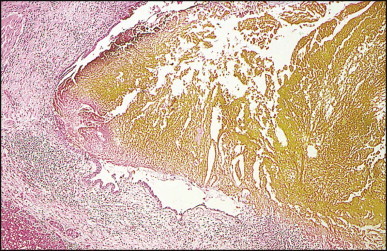
Caroli disease. Markedly dilated duct contains golden-yellow inspissated bile. It shows marked inflammation, as well as ulceration (bottom). (H&E)
According to Desmet,488 the pathogenesis of Caroli disease seems to involve total or partial arrest of remodelling of the ductal plate of the larger intrahepatic bile ducts. In Caroli syndrome, the hereditary factor causing the arrest of remodelling seems to exert its influence not only during the early period of bile duct embryogenesis, but also later on during development of the more peripheral biliary ramifications (the interlobular bile ducts).
There is a single case described of Marfan syndrome with diffuse ectasia of the biliary tree.578 The authors suggested that the defect of connective tissue in that disease could have led to weakness of the wall of the bile ducts with resultant ectasia.
Autosomal dominant polycystic kidney disease (ADPKD)
Mutations mainly in two different genes PKD1 and PKD2 can lead to ADPKD, one of the most common genetic disorders worldwide. The PKD1 gene lies on the short arm of chromosome 16 (16p 13.3), immediately adjacent to the TSC2, a gene responsible for approximately 50% of tuberous sclerosis.496, 579 The incidence of ADPKD due to mutations in PKD1 is 1 in 1000 live birth, comprising 90% of cases. The PKD1 gene encodes a protein called polycystin.580 It is present in plasma membranes of renal tubular cells, bile ductules, pancreatic ducts,581 hepatocytes and cells of large bile ducts. In the kidney, localization at the contact points (only between neighbouring cells) indicates that its main function is cell to cell interaction, such that loss of function contributes to cyst formation.582 The PKD2 gene is located on chromosome 4 (4q 21–23) and encodes a 100 kD protein, polycystin 2 with sequence homology to α subunits of voltage-activated calcium channels.583 As mentioned earlier, localization of polycystin-1 and polycystin-2 to primary cilia in cultured renal epithelial cells513 and demonstration that the proteins function as a ciliary flow-sensitive mechanosensors584 implicate defects in ciliary structure and function as an important mechanism of cyst formation.509 There is at least one unmapped focus, which accounts for 5% of the disease population.585 Although PKD2 is clinically milder than PKD1, it does have a deleterious impact on overall life expectancy and cannot be regarded as a benign disorder.586 The mean age to death or end-stage renal disease is 53 years in PKD1-associated disease, 69 years in PKD2-associated disease and 78 years in controls. PKD2 patients are less likely to have systemic hypertension, urinary tract infections and haematuria.586 Cerebral aneurysms are found in 10–15% of patients with PKD1 and represent a significant risk of subarachnoidal haemorrhage causing death and disability. Familial clustering of bleeding from intracranial aneurysms is recorded.587 In an earlier mutational analysis of 58 ADPKD families with vascular complications 51 were PKD1 (88%) and seven PKD2 (12%). The authors found that the position of the mutation in PKD1 is predictive for development of intracranial aneurysms (59 mutations are more commonly associated with vascular disease) and is therefore of prognostic importance.588
ADPKD is a multisystem disease with cysts and connective-tissue abnormalities involving multiple organs.589 Associated conditions include colonic diverticula (70%), cardiac valve complications (25%), ovarian cysts (40%), inguinal hernia (15%) and intracranial aneurysms (10%), suggesting a diffusely abnormal matrix.590 Mitral valve prolapse was found in 12% of affected children.591
ADPKD is the cause of end-stage renal disease in 8–10% of adults; the number of cysts is age-related.592 The renal disease can be present at birth but hepatic manifestations are rare before 16 years of age.593 The average age at first admission for liver-related problems was 52.8 years with an average duration of symptoms of 3 years.594 The symptoms included a gradually enlarging abdominal mass, upper abdominal pain or discomfort, and rare episodes of severe pain with or without nausea, vomiting, and occasionally fever. The most frequent physical finding is hepatomegaly, which can be massive.595 Liver tests are often normal. Jaundice is unusual,596, 597 and portal hypertension is rare.598 At times this can be related to hepatic outflow obstruction.599, 600 There is an increased risk of gallstones in patients with hepatic cysts.601 Rarely is treatment needed by excision602 or a combination of excision and fenestration.603 Liver or combined liver and renal transplantation has been performed successfully in patients with ADPKD.604
The incidence of liver involvement and its complications have been reviewed in a large series including 132 patients on, and 120 patients not on, haemodialysis.604 Liver cysts were found by non-invasive imaging procedures in 85 of 124 patients on dialysis; sex distribution was equal. In contrast, the non-dialyzed population demonstrated a 75% incidence of liver cysts in females and 44% incidence in males, the peak incidence occurring 10 years earlier in females. The cysts were larger and greater in number in the non-dialyzed females, and there was a correlation with the number of pregnancies. Nineteen autopsies were reported in which five deaths were liver-related. Risk factors for the development of hepatic cysts in ADPKD were also examined in 39 patients and 189 unaffected family members by Gabow.605 The hepatic expression of the disease was found to be modulated by age, female gender, pregnancy, and severity of the renal lesion and functional impairment. Oestrogen treatment of postmenopausal women with ADPKD is associated with selective liver enlargement and abdominal symptoms.606
The leading complication in ADPKD is infection of the liver cysts, with cholangiocarcinoma the second most common complication. A study examining hepatic cyst infection suggested that the incidence increases from 1% to 3% during end-stage renal failure.607 Enterobacteriaceae were cultured from the infected cysts in 9 of 12 patients. In the case of Ikei et al., Pseudomonas aeruginosa was cultured from the infected cysts.608 Positron emission tomography scan will probably make the diagnosis of cyst infections easier and more accurate. This may allow early treatment with appropriately selected antibiotics, often including a fluoroquinolone; the timely drainage of large infected cysts remains an option.609
Hepatic cysts are rarely detected before puberty and increase with age (ultimately to 75%) in individuals over 70.610 However, cysts have been found in early childhood and even in the first year of life.611, 612 Prior to availability of a genetic probe, the criteria for identification of this disorder in children included a positive paternal history, cysts in any portion of the renal tubule or Bowman space, macroscopic cysts in the liver and cerebral aneurysms. Molecular diagnosis is useful in individuals in whom the diagnosis of ADPKD is uncertain due to lack of family history or equivocal imaging results and in younger at-risk individuals who are being evaluated as living-related kidney donors.613
Grossly, the liver in ADPKD is enlarged and diffusely cystic, the cysts varying from <1 mm to 12 cm or more in diameter (Fig. 3.40 ). One liver reported by Kwok and Lewin weighed 7.7 kg.595 Occasionally only one lobe, usually the left, is affected. Diffuse dilatation of the intra- and extrahepatic bile ducts has been reported in some cases.614 The cysts contain a clear, colourless or yellow fluid. Analysis of cyst fluid in one case disclosed similarities to the ‘bile salt-independent’ fraction of human bile, suggesting that such cysts are lined by a functioning secretory bile duct epithelium.615
Figure 3.40.
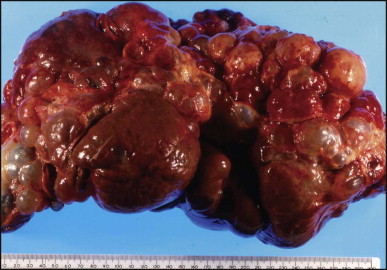
Autosomal dominant polycystic kidney disease. Numerous cysts of varied size are studded throughout the liver. They contained clear fluid.
Microscopically, the cysts are lined by columnar or cuboidal epithelium, but the larger cysts have a flat epithelium (Fig. 3.41 ). Collapsed cysts resemble corpora atretica of the ovary (Fig. 3.42 ). The supporting connective tissue is scanty except in relation to von Meyenburg complexes (Fig. 3.41), a frequently associated lesion, where it may be dense and hyalinized. A small number of inflammatory cells, usually lymphocytes, may infiltrate the supporting stroma. Infected cysts contain pus and may rupture (Fig. 3.43 ). Calcification of the wall of hepatic (and renal) cysts in ADPKD has been reported.616
Figure 3.41.
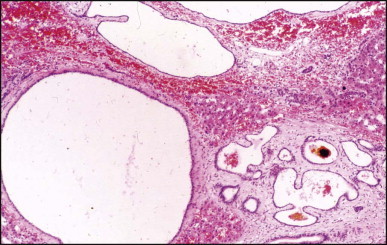
Autosomal dominant polycystic kidney disease. Section of liver shows multiple cysts of varied size that are lined by a flattened epithelium adjacent to a von Meyenburg complex. (H&E)
Figure 3.42.
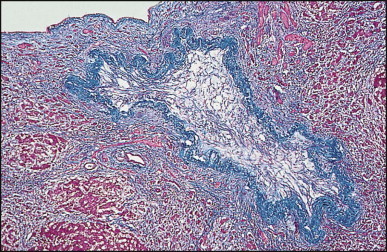
Autosomal dominant polycystic kidney disease. Collapsed cyst has a corrugated wall and the lumen is filled in with a loose connective tissue. (Masson trichrome)
Figure 3.43.
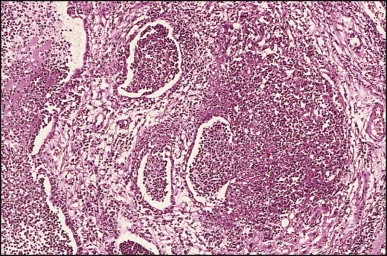
Autosomal dominant polycystic kidney disease. Infected cysts are filled with pus. The cyst to the right has ruptured with formation of a small cholangitic abscess. (H&E)
Von Meyenburg complexes are considered part of the spectrum of adult polycystic disease, and Melnick was of the opinion that polycystic disease of the liver develops progressively over the years by gradual cystic dilatation of these complexes.617 That view is supported by a histomorphometric and clinicopathological study of 28 cases of ADPKD reported by Ramos et al.618 Kida et al. have suggested that cystic dilatation of peribiliary glands also may lead to formation of the cysts in ADPKD.619 Von Meyenburg complexes are small (<0.5 cm in diameter), greyish white or green, and are usually scattered in both lobes.620 They have an abnormal vascular pattern in angiographic studies and are occasionally associated with cavernous haemangiomas. Microscopically, the lesions are discrete, round to irregular in shape and typically periportal in location. The constituent ducts or ductules are embedded in a collagenous stroma, are round or irregular in shape, and have a slightly dilated lumen (Fig. 3.44 ). They are lined by low columnar or cuboidal epithelium and contain pink amorphous material that may be bile-stained, or actual bile (Fig. 3.45 ). Cholangiocarcinomas have been reported in association with von Meyenburg complexes,621, 622 as well as with multiple hepatic cysts considered part of the spectrum of ADPKD.623, 624 Congenital hepatic fibrosis has been found in some cases.625 Intraductal papillary-mucinous neoplasm of the pancreas has been reported.626
Figure 3.44.
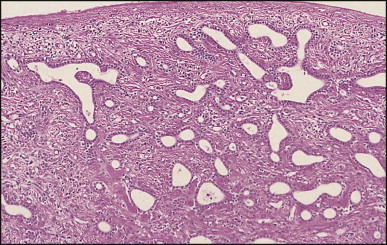
Von Meyenburg complex. Small bile ducts are embedded in a fibrous stroma. Note irregular shape of two of the ducts, each of which contains a polypoid projection. (H&E)
Figure 3.45.
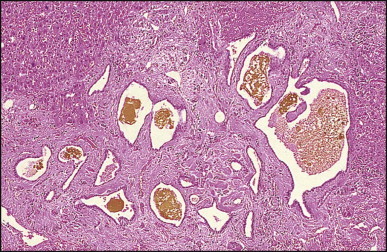
Von Meyenburg complex. The ducts are variably dilated and contain altered bile. (H&E)
Polycystic liver disease without kidney abnormalities (PCLD)
Isolated polycystic liver disease (PCLD), not linked to PKD1 or PKD2, has been reported.627, 628, 629 Germline mutations in protein kinase C substrate 80K-H gene (PRKCSH), a known gene encoding for a previously described human protein ‘noncatalytic beta subunit of glucosidase II’(GIIβ), have been associated with ADPKD. This protein now renamed hepatocystin, seems to segregate in families with PCLD.630, 631 The protein is widely distributed in tissues and predicted to be an endoplasmic reticulum luminal protein that recycles from the Golgi. The mutations found in PCLD are all predicted to cause premature chain termination, rendering loss-of-function changes most likely. The two-hit hypothesis for cyst formation in ADPKD appears applicable to PCLD. Glucosidase II plays a major role in regulation of proper folding and maturation of glycoproteins. Polycystin-1, polycystin-2, and fibrocystin/polyductin are all glycoproteins. Mutations could compromise the processing of the N-linked oligosaccharide chains of the newly synthesized glycoproteins.631 Improper association and trafficking of the polycystins due to defective glycosylation by mutant GIIβ may link PCLD to the ADPKD pathway. This would be consistent with the marked similarity in clinical liver disease in both conditions.630 The second gene involved in PCLD, SEC63, encodes a protein that is part of the multicomponent translocon of the endoplasmic reticulum (Sec63p) which is required in both the post-translational and the co-translational (or signal recognition particle-dependent) targeting pathway and may explain a functional link between the two genes known to be associated with PCLD. Respective lack of expression of hepatocystin and Sep63p in PCLD cyst lining appears to correlate with mutational results.632 Immunohistochemical analysis of cyst lining suggests that the disease involves overexpression of growth factor receptors and loss of adhesion, but proliferation or deregulated apoptosis do not seem to be implicated in PLCD. Differential findings for PRKCSH- and SEC63-PCLD suggest a divergent mechanism for cystogenesis in the two groups.633
Solitary (non-parasitic) bile duct cyst
Solitary bile duct cyst is defined as a unilocular cyst lined by a single layer of columnar or low cuboidal epithelium resting on a basement membrane and a layer of fibrous tissue. The cysts occur at all ages though the majority present in the 4th to the 6th decades. They are rare in the paediatric age group; in the Boston Children's Hospital series 31 solitary non-parasitic cysts (26 unilocular and 5 multilocular) were diagnosed in 63 years.634 The female to male ratio is 4 : 1.635 Cysts smaller than 8–10 cm rarely cause symptoms. When present, symptoms include fullness or an upper abdominal mass, nausea and occasional vomiting. Rapid enlargement has been reported in infancy.636 Jaundice occurs infrequently.637 An acute abdominal crisis may result from torsion, strangulation, haemorrhage into the cyst or rupture.638 Diagnosis is usually established by ultrasonography, computed tomography or other imaging modalities. Solitary bile duct cysts involve the right lobe twice as often as the left. Rarely, they can arise in the falciform ligament.639 They are usually round and rarely are pedunculated; the lining is typically smooth (Fig. 3.46 ). The larger ones may contain one to several litres of fluid which is usually clear, but may be mucoid, purulent (if the cyst is infected), haemorrhagic or rarely bile-stained.
Figure 3.46.
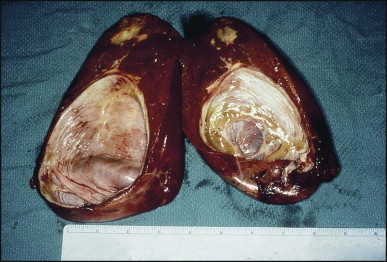
Solitary bile duct cyst. The sectioned cyst has a smooth lining.
Microscopically, the cyst lining usually consists of a single layer of columnar, cuboidal or flat epithelium (Fig. 3.47 ). The epithelium rests on a basement membrane that in turn is supported by a layer of fibrous tissue. Adenocarcinomas may arise in the cyst.640 Other malignancies reported to arise in solitary cysts include squamous cell carcinoma,641 carcinosarcoma642 and carcinoid tumour.643
Figure 3.47.
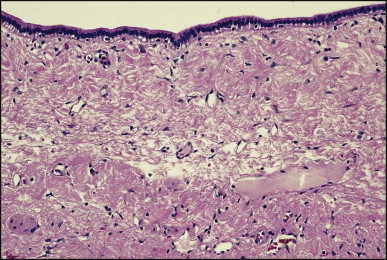
Solitary bile duct cyst. The cyst is lined by a single layer of columnar cells. (H&E)
The pathogenesis of solitary bile duct cyst is unknown. A congenital origin is supported by the occurrence of the cysts in fetuses and newborns,636 by a case presenting as a congenital diaphragmatic hernia,644 and the association of another case with the Peutz–Jeghers syndrome.645
In the past, the treatment of choice of solitary cysts was excision,646 but this has been supplanted by aspiration and sclerotherapy,647, 648 or laparoscopic fenestration.649, 650
Reye syndrome
The syndrome of fatty liver and encephalopathy, first described in 1963 by Reye et al.,651 based on post-mortem findings in 17 children from Australia, was soon recognized to have a worldwide distribution.652 The observation of an association between aspirin and Reye syndrome,653 subsequent advice from the Surgeon General of the USA that the use of salicylates be avoided in children suffering from influenza or varicella, and confirmatory prospective studies654, 655 have been followed by a steady decline in the incidence of ‘classic’ or ‘idiopathic’ Reye syndrome worldwide.656, 657 At the present time, all patients presenting with a ‘Reye-like syndrome’ are presumed to have an inherited metabolic disorder unless there is clear evidence of the classical combination of a flu-like illness and salicylate therapy. In addition, metabolic disorders were subsequently diagnosed in many patients who had survived an acute disease attributed to Reye syndrome.658 Consequently, the following account may be in part historical as it is largely based on data collected at a time when our knowledge of metabolic disorders and their diagnostic tools were limited. Nevertheless, it seems likely that Reye syndrome as such exists and may reflect a pharmacogenetic or immunogenetic susceptibility in those who do not have a demonstrable metabolic disorder. Which drugs or environmental toxins and which viruses may contribute to development of classic Reye syndrome remain a conundrum.
With few exceptions,659, 660 idiopathic Reye syndrome occurs almost exclusively in children, has no sex predilection, and is usually preceded by a resolving viral illness, particularly influenza B or varicella. Initial symptoms include vomiting (usually repetitive), lethargy and changes in mental status. Hepatic dysfunction can be overlooked since the liver is minimally enlarged, and there is no jaundice or splenomegaly. Subsequent symptoms and signs are predominantly neurological and terminate in coma. Seizures sometimes occur, particularly in younger children with hypoglycaemia. Most patients have received multiple medications, including aspirin, for the symptoms of the viral illness. Initial laboratory screening tests disclose elevated serum aminotransferase levels, hyperammonaemia and coagulopathy. Although the liver disease is benign and transient, the encephalopathy (secondary to a hypoxic/metabolic insult resulting in cerebral oedema) can be life-threatening and can result in permanent neurological disability.652 Early diagnosis based on clinical findings and liver biopsy, along with meticulous supportive care, results in decreased mortality and morbidity.
Grossly, liver biopsy or autopsy specimens from patients with Reye syndrome are yellow. The major histopathological finding is diffuse microvesicular steatosis.661 In the typical case the hepatocytes are swollen and packed with multiple small vacuoles (Fig. 3.48 ); neutral lipid can be demonstrated in frozen sections stained with Oil Red O or Sudan Black B (Fig. 3.49A ). The lipid droplets are consistently smaller in the perivenular zone than in other areas.661 Vacuolization may not be evident in biopsy material obtained less than 24 h after the onset of encephalopathy, even though appropriately stained frozen sections will reveal an abundance of fat. Nuclei of the liver cells are enlarged and centrally located. Mitotic activity of variable degree may be seen. Hepatocellular necrosis is generally absent or is mild and spotty. Periportal necrosis is occasionally found,662 preceded by ballooning degeneration of periportal hepatocytes. Variation in the severity of glycogen depletion of liver cells in early biopsies correlates with other histological measures of severity, and with the occurrence of hypoglycaemia, severity of encephalopathy at the time of admission, and the mortality rate. Portal inflammation is either minimal or absent. Cholestasis is rarely observed; in some instances it may be related to an associated pancreatitis.663 Reduction of succinic dehydrogenase and cytochrome oxidase activities (and other mitochondrial enzymes) has been demonstrated by enzymatic stains and is most helpful in confirming the diagnosis664 (Fig. 3.49B).
Figure 3.48.
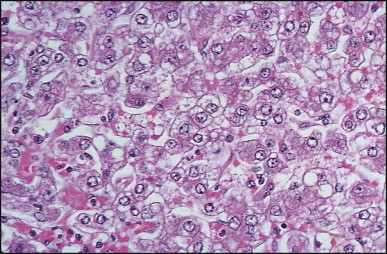
Reye syndrome. Liver cells show microvesicular steatosis. (H&E)
Figure 3.49.
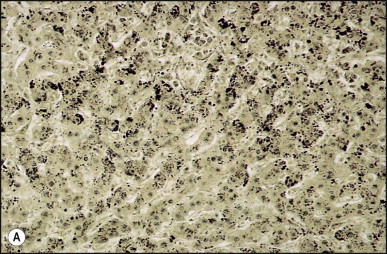
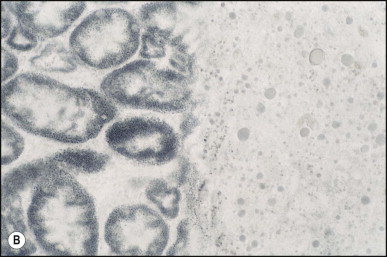
Reye syndrome. (A) Microvesicular steatosis is shown in this frozen section stained with Sudan black B. (B) Succinyl dehydrogenase is absent in the liver (right of the field), contrasting with the finely granular staining in the tubules of a control rat kidney (left of the field), in which the liver biopsy specimen has been embedded.
At the ultrastructural level, the most dramatic changes are seen in hepatocellular mitochondria,664, 665 which are enlarged and misshapen. The degree of enlargement correlates to some extent with the stage of encephalopathy.665 The severity of the disease may also be correlated with lucency of the mitochondrial matrix and loss of matrical dense bodies (Fig. 3.50 ). Other mitochondrial changes in initial hepatic biopsy specimens from patients who die or have severe neurological sequelae include a decrease in size and number of cristae (Fig. 3.50). There may be an increase in the amount of smooth endoplasmic reticulum with varying degrees of dilatation, and 50% of hepatic biopsy specimens reveal flocculent peroxisomes.666
Figure 3.50.
Reye syndrome. Electron micrograph showing swollen mitochondria with loss of matrix density and both fragmentation and reduction in the number of cristae. Note absence of dense granules in the mitochondria (×20 000).
(Courtesy of Dr Cynthia C Daugherty, Children's Hospital Medical Center, Cincinnati, Ohio, USA)
Microvesicular steatosis has been reported in several inherited metabolic disorders (Chapter 4) and in drug- and toxin-induced liver injury, such as with tetracycline, nucleoside analogues, aflatoxin, pyrrolizidine alkaloids, margosa oil, hypoglycin A, pentanoic acid and valproic acid (Chapter 13). Aspirin toxicity may mimic Reye syndrome.666 At the light microscopic level, the liver findings in children with fatal salicylate intoxication resemble those of Reye syndrome,667 but the ultrastructural changes appear to be different.665
Animal models of Reye syndrome with encephalomyocarditis virus668 or influenza B669 infection, and the spontaneous ‘Reye-like syndrome’ in BALB/cByJ mice (associated in the majority with coronavirus infection) have been described.670 In another murine model, treatment with 4-pentenoic acid produced a Reye-like syndrome.671
Kawasaki disease
First described as acute febrile mucocutaneous lymph node syndrome in 1967,672 the clinical features, diagnostic criteria and management of Kawasaki disease have now been reviewed.673 The principal diagnostic criteria of this acute, self-limited, multisystem vasculitis of childhood are fever; bilateral non-exudative conjunctivitis; erythema of lips and oropharyngeal cavity; erythematous rash and desquamation of the fingertips and toes; and cervical lymphadenopathy.674 Activated T lymphocytes play a key role in the disease mechanism, and a genetic susceptibility locus has recently been identified.675 Gastrointestinal signs and symptoms include abdominal tenderness, vomiting, diarrhoea, bloody stools and mild jaundice. Hepatobiliary complications comprise hepatosplenomegaly, mild ascites and hydrops of the gallbladder. Jaundice is a rare presentation.676 The laboratory data include abnormal electrocardiograms, neutrophil leucocytosis with a shift to the left, thrombocytosis and an increased erythrocyte sedimentation rate. Mortality in boys during the first 2 months of the illness is twice the average rate for the disease generally.677 Coronary aneurysms and myocarditis occur in 10–20% of patients, with a higher incidence in boys.678 Thus it is a major cause of acquired heart disease in children. Early therapy with high-dose intravenous γ-globulin and perhaps aspirin has proved beneficial, the former seemingly reducing the frequency of coronary artery abnormalities.679, 680 Male sex, abnormal liver enzymes, low albumin and platelets, and the percentage leukocytes/lymphocytes have been found as risk factors for treatment non-response.681 Patients less than 1 year old or more than 9 years old at diagnosis may have poorer outcomes.682 Atypical cases not fulfilling the conventional diagnostic criteria have a much higher mortality rate, perhaps because of the lack of therapy.683
The hepatobiliary complications of Kawasaki disease have been underestimated. The most common problem results from a hydrops of the gallbladder, which is detectable by ultrasonography with an incidence ranging from 2.5 to 13.7%.684 Prolonged pain has been attributed to poor emptying of the gallbladder.685 The patients have right upper quadrant pain sometimes associated with vomiting. A majority have hepatomegaly and, at times, a palpable gallbladder. Resolution usually occurs in 4 weeks. Perforation is a rare complication. Cholangitis with bile duct injury and ductular reaction has been observed on liver biopsy in three cases.686 Pathological findings at laparotomy include inflammation of the gallbladder, with or without evidence of vasculitis, and occasionally cystic duct obstruction due to inflammatory oedema or lymph node compression.
In addition to vasculitis, inflammation has been noted in autopsy cases in the oral cavity (including ductal structures), small bowel, and the pancreas (including the ducts);687 acute cholangitis or bile duct damage has also been observed.687, 688, 689 Mild bile ductular reaction has been noted but, to date, ductopenia has not been a feature. Cholangitis may be associated with sinusoidal neutrophilic infiltrates (Figure 3.51, Figure 3.52 ), and proliferation and swelling of Kupffer cells are commonly seen.
Figure 3.51.
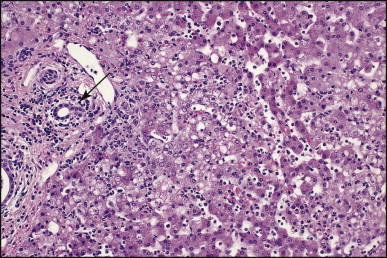
Kawasaki disease. Portal inflammation (left) with an acute cholangitis (arrow). Note presence of neutrophils in sinusoids and periportal steatosis. (H&E)
Figure 3.52.
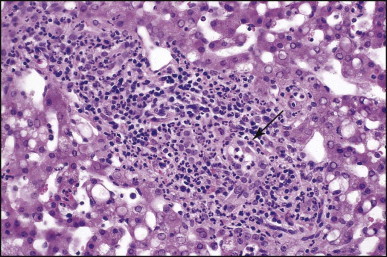
Kawasaki disease. Marked portal inflammation, predominantly neutrophilic, and acute cholangitis (arrow). (H&E)
Similarities between this condition and leptospirosis have been raised; although the Weil–Felix reaction may be positive, direct tests for leptospirosis are negative.690 The close resemblance of fatal cases to infantile periarteritis nodosa has also been emphasized in several studies.687, 691 Affected vessels include the larger coronary arteries, splenic, renal, pulmonary, pancreatic, spermatic, periadrenal, hepatic and mesenteric vessels. Microscopically, the vessels show periarterial inflammation, necrosis and destruction of the media, and intimal inflammation. Lesions of varying stages can be observed in different vessels in the same patient and in different areas of the same type of vessel (e.g. coronary arteries), suggesting that the inflammatory process continues over a period of time during the course of the disease. Aneurysmal dilatation of coronary and other vessels (brachial, iliac, renal and pulmonary) may occur687, 692. A case of a 4-year-old boy with a hepatic artery aneurysm that caused obstructive jaundice was reported by Marks et al.693
Other than leptospirosis, Propionobacterium acnes,694 toxin-secreting Staphylococcus aureus,695 HIV,696 Epstein–Barr virus (EBV),697 parvovirus B19,698 Yersinia infection,699 and an unusual response in a susceptible individual to an infectious agent such as the measles virus700 have all been proposed as causes of Kawasaki disease but are not supported by hard data.
Langerhans cell histiocytosis
This rare disorder (previously termed histiocytosis X) is characterized by infiltration of various tissues and organs by Langerhans cell histiocytes. Langerhans cell histiocytosis (LCH) primarily affects bone, but lung, skin and lymph node involvement is not uncommon. Hepatic involvement is also well recognized, especially in children, with sclerosing cholangitis occurring in 10–15% of those with multisystemic involvement,701, 702 whereas LCH confined to the liver appears very unusual.703, 704 Kaplan et al. reported nine cases of hepatobiliary LCH and found another 85 acceptable cases in the literature.705 Ages ranged from 7 days to 62 years of age, with a median of 18 months, and a 2 : 1 female preponderance. Hepatosplenomegaly, jaundice, liver dysfunction and ascites were the most common clinical presentations.
Gross findings vary with the stage of disease and type of hepatic involvement. Large aggregates of Langerhans cells, eosinophils and other inflammatory cells may form tumour-like masses. Infiltration of large ducts may lead to grossly visible cystic dilatation and rupture. Other cases show irregularly distributed biliary fibrosis, while a biliary cirrhosis is already present in a few cases.705
The diagnostic feature of all cases was the presence of Langerhans cells.706 They typically have an abundant pink cytoplasm, lobulated, coffee-bean shaped or contorted nuclei with a fine chromatin pattern and no discernible nucleoli (Figure 3.53, Figure 3.54 ). In the vast majority of cases the Langerhans cells are accompanied by varying numbers of eosinophils (Fig. 3.55 ), lymphocytes, neutrophils, plasma cells, non-Langerhans histiocytes, and occasional multinucleated giant cells. Immunostains were useful in confirming the nature of the Langerhans cells using antibodies to S-100 protein, CD1a (Fig. 3.56 )707, 708 and CD31.709 CD1a is important as activated Kupffer cells may acquire S100 positivity.710 The lectin receptor CD207/langerin is useful, but it seems to mark dermal cells unrelated to Langerhans cells.711 Typical Birbeck granules may be found ultrastructurally in Langerhans cells.
Figure 3.53.
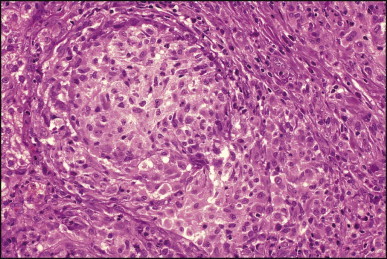
Langerhans cell histiocytosis. Granulomatoid aggregate of Langerhans cell. (H&E)
Figure 3.54.
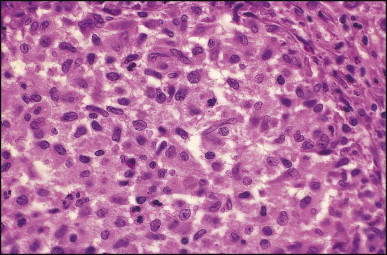
Langerhans cell histiocytosis. Higher magnification of Langerhans cell. (H&E)
Figure 3.55.
Langerhans cell histiocytosis. Infiltrate contains many eosinophils. (H&E)
Figure 3.56.
Langerhans cell histiocytosis. Langerhans cells are strongly immunoreactive to anti-CD1a.
Many with disseminated disease have aggregates of Langerhans cells that range from granulomatous foci to large nodules in the liver (Figure 3.57, Figure 3.58 ) with eosinophils present in abundance in these lesions.705, 712 The majority of cases show some degree of active bile duct infiltration, injury and destruction by Langerhans cells (Figure 3.59, Figure 3.60 ); this is the most characteristic feature of hepatic involvement. Small and medium-size bile ducts are often infiltrated by the Langerhans cells with displacement of the epithelial cells. Some ducts may be entirely replaced by masses of Langerhans cells within the pre-existing basement membrane. Injury to large bile ducts by the Langerhans cells may produce cystic dilatation and rupture with a xanthogranulomatous inflammatory response.
Figure 3.57.
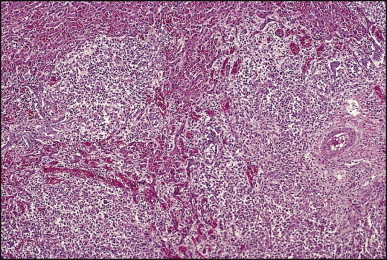
Langerhans cell histiocytosis. The section is from a case that had a grossly visible tumour. (H&E)
Figure 3.58.
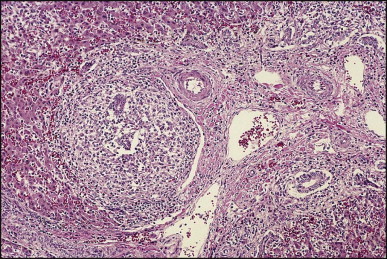
Langerhans cell histiocytosis. The same case depicted in Figure 3.56 showing heavy infiltration of portal area and bile duct necrosis (left). (H&E)
Figure 3.59.
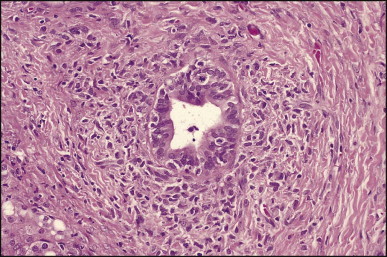
Langerhans cell histiocytosis. Bile duct showing degenerative changes is surrounded by Langerhans cells. (H&E)
Figure 3.60.
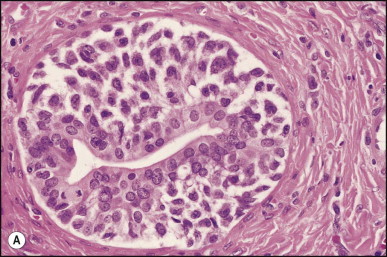
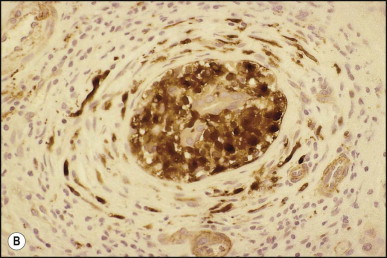
Langerhans cell histiocytosis. (A) High magnification of degenerating bile duct surrounded by a cuff of Langerhans cells. (H&E) (B) Bile duct is heavily infiltrated by Langerhans cells that are strongly immunoreactive to anti-CD1a.
Concentric periductal fibrosis is a prominent feature in the majority of cases. When the ductal infiltration by Langerhans cells is pronounced, there is often marked surrounding fibrosis with demonstrable Langerhans cells in the fibrous tissue. Ducts at a distance from the Langerhans cell lesions may also show epithelial injury and periductal fibrosis, consistent with a secondary sclerosing cholangitis.
Changes due to the bile duct lesions include periportal ductular reaction, ductopenia (Fig. 3.61 ), chronic cholestatic features with periportal cholate stasis (‘pseudoxanthomatous change’) and deposition of copper and copper-binding protein. Some degree of periportal or bridging fibrosis is common and progress to a biliary cirrhosis (Fig. 3.62 ). It must be noted that needle biopsy specimens may show changes due to a distal cholangiopathy only, without demonstration of Langerhans cells, despite direct LCH infiltration of the major bile ducts.713
Figure 3.61.
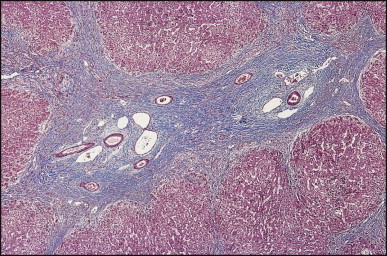
Langerhans cell histiocytosis. Two fused portal areas are bereft of bile ducts. (Masson trichrome)
Figure 3.62.
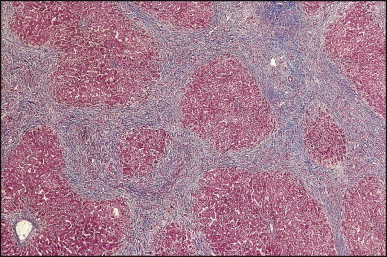
Langerhans cell histiocytosis. End-stage micronodular cirrhosis. (Masson trichrome)
The differential diagnosis of LCH in childhood includes other forms of sclerosing cholangitis: primary sclerosing cholangitis, with or without chronic inflammatory bowel disease, as well as sclerosing cholangitis associated with immunodeficiency (see Chapter 10). In view of the morphological overlap with LCH it is recommended that immunostains for S-100 and CD1a be performed in all cases clinically and/or morphologically diagnosed as sclerosing cholangitis in children; however, extrahepatic manifestations of LCH will generally help reaching the correct diagnosis.
The aetiology and pathogenesis of LCH remain undetermined. Evidence suggests an immune dysregulation with uncontrolled clonal proliferation of dendritic cells exhibiting Langerhans cell characteristics.714 A low susceptibility to apoptosis715 and high level of diverse cytokines production by Langerhans cells,716 with sustained stimulation of T cells, lead to the unique pathological picture, which combines features of a neoplasm and chronic inflammation.717 The demonstrated clonality of LCH does not prove the neoplastic nature of the lesion.718, 719 Viral proteins and DNA sequences of human cytomegalovirus,720 Epstein–Barr virus721 and human herpes virus 6722 have been detected by immunohistochemistry, in situ hybridization and PCR in LCH tissue, but whether these represent causal agents or opportunistic infections secondary to immune dysregulation remains uncertain.
Although patients with LCH have a good overall prognosis,723 approximately 20% of patients with multisystem involvement will show a progressive disease course despite treatment.724, 725 In particular, patients with hepatic involvement have a worse prognosis with survival at 3 years of 96.7% for those without, but only 51.8% for those with liver involvement.726 In patients with cirrhosis and end-stage liver failure secondary to LCH sclerosing cholangitis, orthotopic liver transplantation has been proven a successful therapeutic option713, 727, 728 with 67% of patients living long-term after surgery (median follow-up 5.8 years, range 2.1–7.5 years). A significant number of LCH patients have developed post-transplant lymphoproliferative disorders, which seems related to a higher incidence of refractory rejection.729 Disease recurrence in the liver allograft occurs in some 30% of patients and seems easily managed.729, 730
Juvenile xanthogranuloma, a histiocytic disorder, primarily but not exclusively seen throughout the first two decades of life and principally as a solitary cutaneous lesion, can manifest with multiple small systemic lesions – xanthoma disseminatum – in which case the liver can occasionally be involved. The infiltrating cells are factor XIIIa, fascin and CD68 positive, but S100 and CD1a negative.731 Touton cells are rarely seen in extracutaneous sites. There appears to be an overlap with LCH since skin lesions with characteristics of juvenile xanthogranuloma may be found in children with LCH.702
Erdheim–Chester disease is a rare non-Langerhans type of histiocytosis with xanthogranulomas. It appears to be neoplastic.732 Liver involvement is uncommon; biliary tract obstruction has been reported.733
Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease)
This disorder was first reported by Rosai and Dorfman734 and subsequently reviewed by Foucar et al.735 It usually affects patients between the ages of 10 and 20 years. In addition to massive enlargement of cervical and other lymph nodes, there is fever, leucocytosis, an elevated sedimentation rate and hyperglobulinaemia. The course is protracted, lasting from 3 to 9 months, but the prognosis is usually excellent. The disease can involve extranodal sites, including soft tissues, the oral cavity, lower respiratory tract, genitourinary system, and the liver. Involvement of the last three organs is associated with a poor prognosis.735 Liver involvement is uncommon.736 Lauwers et al. studied 11 patients in whom the disease involved the intestinal tract (five cases), liver (five cases) and pancreas (one case).737 Most patients also had evidence of disease in other extranodal sites, as well as in one or more lymph node groups.
In the review of Foucar et al., hepatomegaly was noted in 27 of 157 cases.735 Four patients had histopathological evidence of hepatic involvement. In most instances gross evidence of disease is lacking, except for one case in which a 1.5 cm, well-circumscribed white nodule was found in the right lobe of the liver.735 The hallmark of the disease is the proliferation of histiocytes which have an abundant cytoplasm and normal-appearing nuclei (Figs 3.63A,B ). The cells are seen in portal areas and in hepatic sinusoids. They display avid leucophagocytosis, but can also phagocytose red cells. According to Eisen et al.,738 the cells express: S-100 protein (Fig. 3.63C); pan-macrophage antigens such as EBM11, HAM56 and Leu-M3; antigens functionally associated with phagocytosis (Fc receptor for IgG and complement receptor); antigens functionally associated with lysosomal activity (lysozyme, α1-antitrypsin and α1-antichymotrypsin); antigens associated with early inflammation (Mac-387, 27E10); antigens commonly found on monocytes, but not tissue macrophages (OKM5, Leu-M1); and activation antigens (Ki-1 and receptors for transferrin and interleukin-2). They are negative for CD1a. The cells thus appear to be true, functionally activated macrophages derived recently from circulating monocytes.738
Figure 3.63.
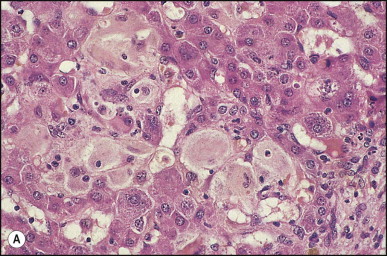
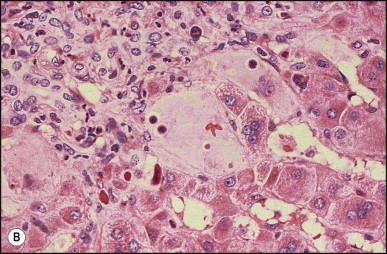
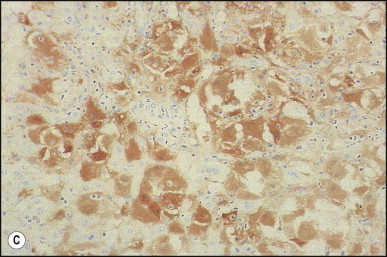
Rosai–Dorfman disease. (A) Sinusoids are filled with large histiocytes showing leucophagocytosis. (H&E) (B) The same case as illustrated in (A). Histiocytes display both erythro- and leucophagocytosis. (H&E) (C) The same case as illustrated in (A), (B). Histiocytes are immunoreactive for S-100 protein.
Haemophagocytic syndromes
Haemophagocytic lymphohistiocytosis (familial haemophagocytic reticulosis)
Familial haemophagocytic lymphohistiocytosis (FLH), a fatal inherited form of haemophagocytic lymphohistiocytosis (HLH) syndrome, is a defect in cell-mediated cytotoxicity characterized by the overwhelming activation of T lymphocytes and macrophages. FLH is a heterogeneous autosomal recessive disorder; one causative gene (PFR1, localized to chromosome 10q21–22) encoding for perforin, a cytotoxic effector protein, is identified in a subset of patients.739, 740 Genes implicated are all important for the exocytosis of cytotoxic granules which contain perforin and granzymes and induce apoptosis upon entering the target cell and therefore are important for the downregulation of the immune response.741 FLH can usually be distinguished from other infantile causes of histiocytosis by the absence of skin lesions and the high incidence of central nervous system involvement. The disease is characterized by fever, anorexia, irritability and pallor in infancy and early childhood. Jaundice and hepatosplenomegaly may develop later. FLH may present as liver failure in infancy; since FLH often shows very high serum ferritin levels, distinction from so-called neonatal haemochromatosis can be difficult.742, 743 Tissue specimens are critical in distinguishing these two diseases as liver transplantation is contraindicated in FHL, whereas it might be life-saving in neonatal haemochromatosis.743 Markedly abnormal coagulation studies reflect low fibrinogen levels and thrombocytopenia, but other coagulation factors are normal. Neurological symptoms may develop from histiocytic involvement of the brain. Although clinical manifestations may remit temporarily, the disease is eventually fatal. The diagnosis is based on identifying erythrophagocytosis by histiocytes in bone marrow biopsy material, or occasionally on liver biopsy. Perforin expression by peripheral lymphocytes, assessment of 2B4 lymphocyte receptor, and natural killer (NK) cell activity have been proposed as rapid tests to assist differentiating FLH from HLH subtypes, therefore highlighting patients with a poor prognosis who may benefit from bone marrow transplantation.739
In an autopsy series haemophagocytosis was most commonly observed in the spleen, lymph nodes and bone marrow.744 Hepatic involvement is characterized by hypertrophy of Kupffer cells and portal macrophages which manifest striking erythrophagocytosis (Fig. 3.64 ), but this may be inconspicuous or even absent.745 In one study of 19 patients, hepatic histology showed portal and sinusoidal infiltrates of CD3, CD8, granzyme B+ lymphocytes admixed with CD68, CD1a- histiocytes showing haemophagocytosis in all specimens.746 Portal areas may show lymphohistiocytic infiltrates with a predominance of T lymphocytes, in a pattern reminiscent of chronic hepatitis (Fig. 3.64). In some cases there is striking enlargement of endothelial cell nuclei and perivenous erythropoiesis resembling graft rejection.745 Other changes include mild hepatocellular damage in the vicinity of the lymphohistiocytic infiltrates, variable cholestasis and epithelioid granulomas.745
Figure 3.64.
Haemophagocytic lymphohistiocytosis. (A) Portal area (left) infiltrated by lymphocytes. (H&E) There is mild periportal steatosis. (B) Higher magnification showing erythrophagocytosis by a Kupffer cell (arrow). (H&E)
Infection-associated (reactive) haemophagocytic syndrome
In this condition there is proliferation of morphologically non-malignant histiocytes showing phagocytosis of haemopoietic cells, with associated fever and pancytopenia. There is a wide range of clinical severity from the incidental observation during infection or at autopsy that macrophages throughout the body are large and activated to the full-blown haemophagocytic syndrome (HLH) that includes fever, hepatosplenomegaly, coagulopathy, and various cytopenias.702 Epstein–Barr virus infection (virus-associated haemophagocytic syndrome) remains the most frequent association, but since the first report in 1979, many other viruses747, 748 and other infectious agents have been incrimiated.749, 750 There are a few reports of simultaneous development of HLH and systemic lupus erythematous.751 Patients with rheumatic disorders, especially juvenile chronic arthritis, appear particularly at risk, the condition being often referred to as macrophage activation syndrome in this setting.752, 753 Haemophagocytic features are often seen in livers of patients with AIDS (Fig. 3.65 ). The syndrome may also develop in diseases other than infections, such as lymphoma and disseminated carcinoma.754, 755 Reactive (secondary) HLH and macrophage activation syndrome manifest as a massive cytokine and chemokine release, mostly TNFα, interferon (IFN)γ, soluble interleukin-2 (IL-2) receptor, IL-6, and other cytokines whose levels in the blood may have a prognostic value.756 This cytokine profile distinguishes HLH from the primary familial conditions in which an underlying genetic defect is identified. In contrast, natural killer cell function is preserved in the reactive form but defective in the inherited syndromes.702
Figure 3.65.
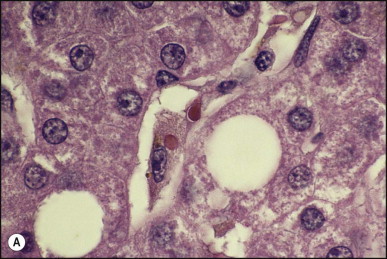
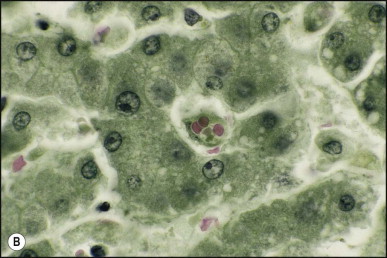
Reactive haemophagocytosis in a patient with AIDS. (A) Kupffer cell has phagocytosed two erythrocytes. (H&E) (B) Kupffer cell contains three erythrocytes. (Biebrich scarlet)
Histologically, there is infiltration of the portal tracts with normal-appearing histiocytes, lymphocytes and plasma cells, together with marked hypertrophy of Kupffer cells displaying prominent haemophagocytosis. The Kupffer cells are only weakly stained with PAS, a feature helpful in distinguishing this change from a response to necro-inflammatory conditions, in which the Kupffer cells are often strongly PAS positive.757 Additionally, Kupffer cells frequently show siderosis, possibly related to the erythrophagocytosis itself or to blood transfusions.757 There is a lack of correlation between the degree of peripheral cytopenia and the degree of hepatic haemophagocytosis.757 The hepatic manifestations in 30 patients with haemophagocytic syndrome were reviewed.758 Immunohistochemical findings on liver tissues have provided direct evidence for the involvement of activated CD8+ lymphocytes through the production of IFNγ, and of macrophages through the haemophagocytosis and production of both interleukin 6 and TNFα, irrespective of the associated condition.759
Down syndrome
Severe liver disease can occur in Down syndrome or trisomy 21 mosaicism.760, 761 Stillbirth, hydrops fetalis, or liver failure at or within a few weeks of birth are recognized presentations.762, 763, 764, 765, 766 Morphologically, there is diffuse fibrosis surrounding reactive bile ductules and residual hepatocytes.760 Extensive hepatic necrosis has also been observed. In most livers a large number of megakaryocytes or megakaryoblasts, staining positively for factor VIII-related antigen and binding to Ulex europaeus 1, as well as other haematopoietic elements, are present in the sinusoids.760, 762 Parenchymal iron deposition of variable degree was noted in most of the cases studied by Ruchelli et al.760 Arai et al.765 have suggested that megakaryocyte-derived TGFβ1 is one of the candidates in the activation of hepatic stellate cells leading to the hepatic fibrosis. High levels of N-terminal peptide of procollagen III, type IV collagen, and hyaluronic acid were found in the serum of a lethal case at 5 days of life.767
The liver disease generally accompanies a haematological disorder unique to Down syndrome called transient abnormal myelopoiesis, transient myeloproliferative disorder or transient leukaemia.768, 769 The condition consists of a clonal proliferation of blast cells exhibiting megakaryocytic features and historically associated with a high rate of spontaneous remission. Its true incidence remains to be determined. A number of cases may occur as an incidental finding of abnormal cell counts and blast cells in the peripheral blood in an otherwise well child, but in approximately 20% of cases the disease is manifest as hydrops fetalis and liver or multi-organ failure resulting in death. Of those children who enter a spontaneous remission, 13–33% have been found to develop acute megakaryoblastic leukemia, usually within the first 3 years of life, which if left untreated is fatal. In trisomy 21 fetal haemopoietic progenitors acquire N-terminal truncating mutations in the key megakaryocyte-erythroid transcription factor GATA1. Mutations are found in blast cells of both transient myeloproliferative disorder and megakaryoblastic leukaemia,770 suggesting that additional as yet unidentified (epi)genetic mutations are required for progression to AMKL.771 A recent multicentre survey suggest a good response of leukaemic cases to early therapy with standard-dose intensive chemotherapy including cytarabine and anthracyclines.772
Neonatal lupus erythematosus
Hepatic involvement may occur as part of the spectrum of neonatal lupus erythematosus (NLE), which is due to passage of maternal anti-Ro and anti-La antibodies across the placenta. Fetal tissues expressing Ro and La antigenic determinants may be damaged. The heart, skin and liver are most likely to be involved, rarely with thrombocytopenia and leukopenia.773, 774 Congenital heart block is the most important cardiac manifestation, and some infants develop a rash which resembles discoid lupus erythematosus in the newborn period or some weeks later. In approximately 10% hepatic disease occurs, usually typical neonatal hepatitis syndrome.775, 776, 777 Occasionally, liver involvement is severe enough to suggest extrahepatic biliary tract obstruction, with acholic stools and non-draining hepatobiliary scan.778, 779 Increased iron accumulation in the liver was found on liver biopsy in the first infant recognized as having NLE with liver involvement,775 and subsequently an infant was reported with severe liver disease showing features of perinatal haemochromatosis.780 Transient elevation of serum aminotransferases only or unexplained isolated conjugated hyperbilirubinaemia in the perinatal period and later presentation at 2–3 months of age with transient elevations of serum aminotransferases are recognized.781 In most infants, the liver disease resolves completely between 6 and 12 months of age, as the maternal antibodies are degraded. Mild fibrosis was found in one child with NLE on repeat liver biopsy.
The diagnosis of NLE is difficult in the absence of congenital heart block or typical skin rash. Some infants have only transient jaundice and myocarditis with abnormal electrocardiogram. The diagnosis should be suspected if the mother is known to have systemic lupus erythematosus or Sjögren syndrome. Frequently, however, the mother is asymptomatic with respect to rheumatological disease. Routine methods may fail to detect anti-Ro and anti-La in the infant; these studies should be performed at as young an age as possible. Very high titres of ANA in the infant may be due to NLE. Deposits of associated antibodies (anti-Ro and/or anti-La) may be found in affected liver tissue by immunofluorescence.782 Most infants with unexplained neonatal hepatitis syndrome do not have NLE.783 One infant was reported with neonatal hepatitis syndrome and transplacental transfer of anti-mitochondrial antibodies.784
References
- 1.Grosfeld JL, Clatworthy HW. Hepatic agenesis. In: Bergsma D, editor. Birth defects atlas and compendium. Alan R Liss; New York: 1973. p. 517. [Google Scholar]
- 2.Belton RL, Van Zandt TF. Congenital absence of the left lobe of the liver: a radiologic diagnosis. Radiology. 1983;147:184. doi: 10.1148/radiology.147.1.6828725. [DOI] [PubMed] [Google Scholar]
- 3.Maeda N, Horie Y, Shiota G. Hypoplasia of the left hepatic lobe associated with floating gallbladder. A case report. Hepatogastroenterology. 1998;45:1100–1103. [PubMed] [Google Scholar]
- 4.Radin DR, Colletti PM, Ralls PW. Agenesis of the right lobe of the liver. Radiology. 1987;164:639–642. doi: 10.1148/radiology.164.3.3303118. [DOI] [PubMed] [Google Scholar]
- 5.Inoue T, Matsuzaki Y, Okauchi Y. Hypogenesis of right hepatic lobe accompanied by portal hypertension: case report and review of 31 Japanese cases. J Gastroenterol. 1997;32:836–842. doi: 10.1007/BF02936965. [DOI] [PubMed] [Google Scholar]
- 6.Fields RC, Heiken JP, Strasberg SM. Biliary injury after laparoscopic cholecystectomy in a patient with right liver agenesis: case report and review of the literature. J Gastrointest Surg. 2008;12:1577–1581. doi: 10.1007/s11605-008-0576-x. [DOI] [PubMed] [Google Scholar]
- 7.Faintuch J, Machado MCC, Raia AA. Suprahepatic gallbladder with hypoplasia of the right lobe of the liver. Arch Surg. 1980;115:658–659. doi: 10.1001/archsurg.1980.01380050080019. [DOI] [PubMed] [Google Scholar]
- 8.Hsu KL, Cheng YF, Ko SF. Hypoplastic right hepatic lobe with retrohepatic gallbladder complicated by hepatolithiasis and liver abscess: a case report. Hepatogastroenterology. 1997;44:803–807. [PubMed] [Google Scholar]
- 9.Sato M, Watanabe Y, Iseki S. Hepatolithiasis with situs inversus: first case report. Surgery. 1996;119:534–537. doi: 10.1016/s0039-6060(96)80274-1. [DOI] [PubMed] [Google Scholar]
- 10.Organ BC, Skandalakis LJ, Gray SW, Skandalakis JE. Cancer of the bile duct with situs inversus. Arch Surg. 1991;126:1150–1153. doi: 10.1001/archsurg.1991.01410330112017. [DOI] [PubMed] [Google Scholar]
- 11.Silveira TR, Salzano FM, Howard ER, Mowat AP. Congenital structural abnormalities in biliary atresia: evidence for etiopathogenic heterogeneity and therapeutic implications. Acta Paediatr Scand. 1991;80:1192–1199. doi: 10.1111/j.1651-2227.1991.tb11808.x. [DOI] [PubMed] [Google Scholar]
- 12.Soper RT, Green EW. Omphalocele. Surg Gynecol Obstet. 1961;113:501–508. [Google Scholar]
- 13.Desser PL, Smith S. Nonparasitic liver cysts in children. J Pediatr. 1956;49:297–305. doi: 10.1016/s0022-3476(56)80186-8. [DOI] [PubMed] [Google Scholar]
- 14.Fock G. Ectopic liver in omphalocele. Acta Paediatr. 1963;52:288–292. doi: 10.1111/j.1651-2227.1963.tb03781.x. [DOI] [PubMed] [Google Scholar]
- 15.Feist JH, Lasser EC. Identification of uncommon liver lobulations. JAMA. 1959;169:1859–1862. doi: 10.1001/jama.1959.03000330031005. [DOI] [PubMed] [Google Scholar]
- 16.Korobkin MT, Miller SW, deLorimier AA. Hepatic herniation through the Morgagni foramen. Am J Dis Child. 1973;126:217–219. doi: 10.1001/archpedi.1973.02110190191017. [DOI] [PubMed] [Google Scholar]
- 17.Rendina EA, Venuta F, Pescarmona EO. Intrathoracic lobe of the liver: case report and review of the literature. Eur J Cardiothorac Surg. 1989;3:75–78. doi: 10.1016/1010-7940(89)90015-8. [DOI] [PubMed] [Google Scholar]
- 18.Organ CH, Hayes DF. Supradiaphragmatic right liver lobe and gallbladder. Arch Surg. 1980;115:989–990. doi: 10.1001/archsurg.1980.01380080079017. [DOI] [PubMed] [Google Scholar]
- 19.Vogel A, Small A. Partial eventration of the right diaphragm (congenital diaphragmatic herniation of the liver) Ann Intern Med. 1955;43:63–82. doi: 10.7326/0003-4819-43-1-61. [DOI] [PubMed] [Google Scholar]
- 20.Reitemeier RJ, Butt HR, Baggenstoss AH. Riedel's lobe of the liver. Gastroenterology. 1958;34:1090–1095. [PubMed] [Google Scholar]
- 21.Cullen TSL. Accessory lobes of the liver. Arch Surg. 1925;11:718–764. [Google Scholar]
- 22.Naganuma H, Ishida H, Niizawa M. Intrathoracic accessory lobes of the liver. J Clin Ultrasound. 1993;21:143–146. doi: 10.1002/jcu.1870210213. [DOI] [PubMed] [Google Scholar]
- 23.Peter H, Strohm WD. Torquierter akzessorischer Leberlappen als Ursache eines akuten Abdomens. Leber Magen Darm. 1980;4:203–206. [PubMed] [Google Scholar]
- 24.Mendoza A, Voland J, Wolf P, Benirschke K. Supradiaphragmatic liver in the lung. Arch Pathol Lab Med. 1986;110:1085–1086. [PubMed] [Google Scholar]
- 25.Shah KD, Beck AR, Jhaveri MK. Infantile hemangioendothelioma of heterotopic intrathoracic liver associated with diaphragmatic hernia. Hum Pathol. 1987;18:754–756. doi: 10.1016/s0046-8177(87)80250-2. [DOI] [PubMed] [Google Scholar]
- 26.Buck FS, Koss MN. Heterotopic liver in an adrenal gland. Pediatr Pathol. 1988;8:535–540. doi: 10.3109/15513818809022309. [DOI] [PubMed] [Google Scholar]
- 27.Tejada E, Danielson C. Ectopic or heterotopic liver (choristoma) associated with the gallbladder. Arch Pathol Lab Med. 1989;113:950–952. [PubMed] [Google Scholar]
- 28.Shapiro JL, Metlay LA. Heterotopic supradiaphragmatic liver formation in association with congenital cardiac anomalies. Arch Pathol Lab Med. 1991;115:238–240. [PubMed] [Google Scholar]
- 29.Boyle L, Gallivan MVE, Chan B, Lack EE. Heterotopia of gastric mucosa and liver involving the gallbladder. Arch Pathol Lab Med. 1992;116:138–142. [PubMed] [Google Scholar]
- 30.Ikoma A, Tanaka K, Hamada N. Left-sided gallbladder with accessory liver accompanied by intrahepatic cholangiocarcinoma. J Jpn Surg Soc. 1992;93:434–436. [PubMed] [Google Scholar]
- 31.Willis RA. Some unusual developmental heterotopias. Br Med J. 1968;2:267–272. doi: 10.1136/bmj.3.5613.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lieberman MK. Cirrhosis in ectopic liver tissue. Arch Pathol. 1966;82:443–446. [PubMed] [Google Scholar]
- 33.Arakawa M, Kimura Y, Sakata K. Propensity of ectopic liver to hepatocarcinogenesis: case reports and a review of the literature. Hepatology. 1999;29:57–61. doi: 10.1002/hep.510290144. [DOI] [PubMed] [Google Scholar]
- 34.Le Bail B, Carles J, Saric J. Ectopic liver and hepatocarcinogenesis. Hepatology. 1999;30:585–586. doi: 10.1002/hep.510300227. [DOI] [PubMed] [Google Scholar]
- 35.Dolan MF, Janovski NA. Adreno-hepatic union (adrenal dystopia) Arch Pathol. 1960;86:22–24. [PubMed] [Google Scholar]
- 36.Honma K. Adreno-hepatic fusion: an autopsy study. Zentralb Pathol. 1991;137:117–122. [PubMed] [Google Scholar]
- 37.Cotelingam JD, Saito R. Hepatolienal fusion: case report of an unusual lesion. Hum Pathol. 1978;9:234–236. doi: 10.1016/s0046-8177(78)80116-6. [DOI] [PubMed] [Google Scholar]
- 38.Ballinger J. Hypoglycemia from metastasizing insular carcinoma of aberrant pancreatic tissue in liver. Arch Pathol. 1941;32:277–285. [Google Scholar]
- 39.Schaefer B, Meyer G, Arnholdt H, Hohlbach G. Heterotope. Pankreaspseudocyste in der Leber. Chirurg. 1989;60:556–558. [PubMed] [Google Scholar]
- 40.Terada T, Nakanuma Y, Kakita A. Pathologic observations of intrahepatic peribiliary glands in 1000 consecutive autopsy livers. Heterotopic pancreas in the liver. Gastroenterology. 1990;98:1333–1337. doi: 10.1016/s0016-5085(12)90353-4. [DOI] [PubMed] [Google Scholar]
- 41.Lacerda MA, Ludwig J, Ward EM. Intrahepatic spleen presenting as a mass lesion. Am J Gastroenterol. 1993;88:2116–2117. [PubMed] [Google Scholar]
- 42.Chun JM, Hwang YJ, Kim JY. Intrahepatic splenic tissue without medical history of splenic injury or splenectomy. Hepatogastroenterology. 2007;54:944–945. [PubMed] [Google Scholar]
- 43.Nakajima T, Fujiwara A, Yamaguchi M. Intrahepatic splenosis with severe iron deposition presenting with atypical magnetic resonance images. Intern Med. 2008;47:743–746. doi: 10.2169/internalmedicine.47.0689. [DOI] [PubMed] [Google Scholar]
- 44.Sekine S, Nagata M, Hamada H, Watanabe T. Heterotopic thyroid tissue at the porta hepatis in a fetus with trisomy 18. Virchows Arch. 2000;436:498–501. doi: 10.1007/s004280050479. [DOI] [PubMed] [Google Scholar]
- 45.Michels N. The hepatic, cystic and retroduodenal arteries and their relation to the biliary ducts. Ann Surg. 1951;133:503–524. doi: 10.1097/00000658-195104000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carmi R, Magee CA, Neill CA, Karrer FM. Extrahepatic biliary atresia and associated anomalies: etiologic heterogeneity suggested by distinctive patterns of associations. Am J Med Genet. 1993;45:683–693. doi: 10.1002/ajmg.1320450606. [DOI] [PubMed] [Google Scholar]
- 47.Udelsman R, Sugarbaker PH. Congenital duplication of the gallbladder associated with an anomalous right hepatic artery. Am J Surg. 1985;149:812–815. doi: 10.1016/s0002-9610(85)80193-8. [DOI] [PubMed] [Google Scholar]
- 48.Rela M, McCall JL, Karani J, Heaton ND. Accessory right hepatic artery arising from the left. Implications for split liver transplantation. Transplantation. 1998;66:792–794. doi: 10.1097/00007890-199809270-00014. [DOI] [PubMed] [Google Scholar]
- 49.Helikson MA, Shapiro DL, Seashore JH. Hepatoportal arteriovenous fistula and portal hypertension in an infant. Pediatrics. 1977;60:921–924. [PubMed] [Google Scholar]
- 50.Vauthey J-N, Tomczak RJ, Helmberger T. The arterioportal fistula syndrome: clinicopathologic features, diagnosis and therapy. Gastroenterology. 1997;113:1390–1401. doi: 10.1053/gast.1997.v113.pm9322535. [DOI] [PubMed] [Google Scholar]
- 51.Lamireau T, Chateil JF, Portier F. Successful embolization of congenital intrahepatic arterioportal fistula in two infants. J Pediatr Gastroenterol Nutr. 1999;29:211–214. doi: 10.1097/00005176-199908000-00021. [DOI] [PubMed] [Google Scholar]
- 52.Bilbao J, Longo JM, Aquerreta J. Platinum wire embolization of an intrahepatic arterioportal fistula. Am J Gastroenterol. 1990;85:859–860. [PubMed] [Google Scholar]
- 53.Covey AM, Brody LA, Getrajdman GI. Incidence, patterns, and clinical relevance of variant portal vein anatomy. AJR Am J Roentgenol. 2004;183:1055–1064. doi: 10.2214/ajr.183.4.1831055. [DOI] [PubMed] [Google Scholar]
- 54.Boles ET, Jr, Smith B. Preduodenal portal vein. Pediatrics. 1978;28:805–809. [Google Scholar]
- 55.John AK, Gur U, Aluwihare A, Cade D. Pre duodenal portal vein as a cause of duodenal obstruction in an adult. ANZ J Surg. 2004;74:1032–1033. doi: 10.1111/j.1445-1433.2004.03226.x. [DOI] [PubMed] [Google Scholar]
- 56.Lilly JR, Starzl TE. Liver transplantation in children with biliary atresia and vascular anomalies. J Pediatr Surg. 1974;9:707–714. doi: 10.1016/0022-3468(74)90109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsia DYY, Gellis SS. Portal hypertension in infants and children. Am J Dis Child. 1955;90:290–298. doi: 10.1001/archpedi.1955.04030010292011. [DOI] [PubMed] [Google Scholar]
- 58.Bell JW. Portal-vein hypoplasia with inferior mesenteric hypertension. N Engl J Med. 1970;283:1149–1150. doi: 10.1056/NEJM197011192832108. [DOI] [PubMed] [Google Scholar]
- 59.Raffensperger JG, Shkolnik AA, Boggs JD, Swenson O. Portal hypertension in children. Arch Surg. 1972;105:249–254. doi: 10.1001/archsurg.1972.04180080101017. [DOI] [PubMed] [Google Scholar]
- 60.Matsuoka Y, Ohtomo K, Okubo T. Congenital absence of the portal vein. Gastrointest Radiol. 1992;17:31–33. doi: 10.1007/BF01888504. [DOI] [PubMed] [Google Scholar]
- 61.Morgan G, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portasystemic vascular anomalies. J Pediatr Surg. 1994;29:1239–1241. doi: 10.1016/0022-3468(94)90812-5. [DOI] [PubMed] [Google Scholar]
- 62.Appel H, Loddenkemper C, Schirmacher P. Congenital absence of the portal vein with splenomegaly and hypersplenism in a young woman. Digestion. 2003;67:105–110. doi: 10.1159/000070398. [DOI] [PubMed] [Google Scholar]
- 63.De Gaetano AM, Gui B, Macis G. Congenital absence of the portal vein associated with focal nodular hyperplasia in the liver in an adult woman: imaging and review of the literature. Abdom Imaging. 2004;29:455–459. doi: 10.1007/s00261-003-0131-x. [DOI] [PubMed] [Google Scholar]
- 64.Marois D, van Heerden JA, Carpenter HA, Sheedy PF. Congenital absence of the portal vein. Mayo Clin Proc. 1979;54:55–59. [PubMed] [Google Scholar]
- 65.Guariso G, Fiorio S, Altavilla G. Congenital absence of the portal vein associated with focal nodular hyperplasia of the liver and cystic dysplasia of the kidney. Eur J Pediatr. 1998;157:287–290. doi: 10.1007/s004310050812. [DOI] [PubMed] [Google Scholar]
- 66.Tanaka Y, Takayanagi M, Shiratori Y. Congenital absence of portal vein with multiple hyperplastic nodular lesions in the liver. J Gastroenterol. 2003;38:288–294. doi: 10.1007/s005350300050. [DOI] [PubMed] [Google Scholar]
- 67.Tsuji K, Naoki K, Tachiyama Y. A case of congenital absence of the portal vein. Hepatol Res. 2005;31:43–47. doi: 10.1016/j.hepres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 68.Cheung KM, Lee CY, Wong CT, Chan AK. Congenital absence of portal vein presenting as hepatopulmonary syndrome. J Paediatr Child Health. 2005;41:72–75. doi: 10.1111/j.1440-1754.2005.00542.x. [DOI] [PubMed] [Google Scholar]
- 69.Andreani P, Srinivasan P, Ball CS. Congenital absence of the portal vein in liver transplantation for biliary atresia. Int J Surg Investig. 2000;2:81–84. [PubMed] [Google Scholar]
- 70.Raskin NK, Price JB, Fishman RA. Portal-systemic encephalopathy due to congenital intrahepatic shunts. N Engl J Med. 1964;270:225–229. doi: 10.1056/NEJM196401302700503. [DOI] [PubMed] [Google Scholar]
- 71.Shawker TH, Chang R, Garra B. An unusual anomalous intrahepatic connection between the left portal vein and internal mammary veins. J Clin Ultrasound. 1988;16:425–435. doi: 10.1002/jcu.1870160612. [DOI] [PubMed] [Google Scholar]
- 72.Lewis AM, Aquino NM. Congenital portohepatic vein fistula that resolved spontaneously in a neonate. AJR Am J Roentgenol. 1992;159:837–838. doi: 10.2214/ajr.159.4.1529852. [DOI] [PubMed] [Google Scholar]
- 73.Gitzelmann R, Arbenz OU, Willi UV. Hypergalactosemia and portosystemic encephalopathy due to persistence of the ductus venosus Aranti. Eur J Pediatr. 1992;151:564–568. doi: 10.1007/BF01957721. [DOI] [PubMed] [Google Scholar]
- 74.Fiane AE, Gjestvang FT, Smevik B. Hepatoportal arteriovenous fistula and bleeding oesophageal varices in a child. Eur J Surg. 1993;159:185–186. [PubMed] [Google Scholar]
- 75.Uchino T, Endo F, Ikeda S. Three brothers with progressive hepatic dysfunction and severe hepatic steatosis due to a patent ductus venosus. Gastroenterology. 1996;110:1964–1968. doi: 10.1053/gast.1996.v110.pm8964424. [DOI] [PubMed] [Google Scholar]
- 76.Ozbek SS, Killi MR, Pourbagher MA. Portal venous system aneurysms: Report of five cases. J Ultrasound Med. 1999;18:417–422. doi: 10.7863/jum.1999.18.6.417. [DOI] [PubMed] [Google Scholar]
- 77.Uchino T, Matsuda I, Endo F. The long-term prognosis of congenital portosystemic venous shunt. J Pediatr. 1999;135:254–256. doi: 10.1016/s0022-3476(99)70031-4. [DOI] [PubMed] [Google Scholar]
- 78.Thompson PB, Oldham KT, Bedi DG. Aneurysmal malformation of the extrahepatic portal vein. Am J Gastroenterol. 1986;81:695–697. [PubMed] [Google Scholar]
- 79.Gallagher DM, Leiman S, Hux CH. In utero diagnosis of a portal vein aneurysm. J Clin Ultrasound. 1993;21:147–151. doi: 10.1002/jcu.1870210214. [DOI] [PubMed] [Google Scholar]
- 80.Ito Y, Tarao K, Tamai S. Portal vein aneurysm in the liver associated with multiple vascular malformations. J Gastroenterol. 1994;29:776–781. doi: 10.1007/BF02349287. [DOI] [PubMed] [Google Scholar]
- 81.Klemperer P. Cavernous transformation of the portal vein. Arch Pathol. 1928;6:353–377. [Google Scholar]
- 82.Myers NA, Robinson MJ. Extrahepatic portal hypertension in children. J Pediatr Surg. 1973;8:467–473. doi: 10.1016/0022-3468(73)90208-x. [DOI] [PubMed] [Google Scholar]
- 83.Berdon WE, Baker DH, Casarella W. Liver disease in children: portal hypertension, hepatic masses. Semin Roentgenol. 1975;10:207–214. doi: 10.1016/0037-198x(75)90063-2. [DOI] [PubMed] [Google Scholar]
- 84.Maksoud JG, Goncalves ME, Porta G. The endoscopic and surgical management of portal hypertension in children: analysis of 123 cases. J Pediatr Surg. 1991;26:177–181. doi: 10.1016/0022-3468(91)90904-8. [DOI] [PubMed] [Google Scholar]
- 85.Pinto RB, Silveira TR, Bandinelli E, Roehsig L. Portal vein thrombosis in children and adolescents: the low prevalence of hereditary thrombophilic disorders. J Pediatr Surg. 2004;39:1356–1361. doi: 10.1016/j.jpedsurg.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 86.Sarin SK, Bansal A, Sasan S, Nigem A. Portal-vein obstruction in children leads to growth retardation. Hepatology. 1992;15:229–233. doi: 10.1002/hep.1840150210. [DOI] [PubMed] [Google Scholar]
- 87.Bellomo-Brandao MA, Morcillo AM, Hessel G. Growth assessment in children with extra-hepatic portal vein obstruction and portal hypertension. Arq Gastroenterol. 2003;40:247–250. doi: 10.1590/s0004-28032003000400009. [DOI] [PubMed] [Google Scholar]
- 88.Perlemuter G, Béjanin H, Fritsch J. Biliary obstruction caused by portal cavernoma: study of 8 cases. J Hepatol. 1996;25:58–63. doi: 10.1016/s0168-8278(96)80328-x. [DOI] [PubMed] [Google Scholar]
- 89.Chandra R, Kapoor D, Tharakan A. Portal biolopathy. J Gastroenterol Hepatol. 2001;16:1086–1092. doi: 10.1046/j.1440-1746.2001.02562.x. [DOI] [PubMed] [Google Scholar]
- 90.Gauthier-Villars M, Franchi S, Gauthier F. Cholestasis in children with portal vein obstruction. J Pediatr. 2005;146:568–573. doi: 10.1016/j.jpeds.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 91.Condat B, Vilgrain V, Asselah T. Portal cavernoma-associated chaloangiopathy: a clinical and MR cholangiography couped with MR portography imaging study. Hepatology. 2003;37:1302–1308. doi: 10.1053/jhep.2003.50232. [DOI] [PubMed] [Google Scholar]
- 92.Rosenthal M, White G, Stephen M. Vascular biliopathy as a cause of common bile duct obstruction successfully treated by mesocaval shunt and endoscopic retrograde cholangiopancreatography biliary stent placement. Vascular. 2008;16:356–358. doi: 10.2310/6670.2008.00066. [DOI] [PubMed] [Google Scholar]
- 93.Ozkavukcu E, Erden A, Erden I. Imaging features of portal biliopathy: frequency of involvement patterns with emphasis on MRCP. Eur J Radiol. 2009;71:129–134. doi: 10.1016/j.ejrad.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 94.Voorhees AB, Jr, Harris RC, Britton RC. Portal hypertension in children: 98 cases. Surgery. 1965;58:540–549. [PubMed] [Google Scholar]
- 95.Ros PR, Viamonte M, Soila K. Demonstration of cavernomatous transformation of the portal vein by magnetic resonance imaging. Gastrointest Radiol. 1986;11:90–92. doi: 10.1007/BF02035040. [DOI] [PubMed] [Google Scholar]
- 96.Bradbury MS, Kavanagh PV, Chen MY. Noninvasive assessment of portomesenteric venous thrombosis: current concepts and imaging strategies. J Comput Assist Tomogr. 2002;26:392–404. doi: 10.1097/00004728-200205000-00014. [DOI] [PubMed] [Google Scholar]
- 97.Marks C. Developmental basis of the portal venous system. Am J Surg. 1969;117:671–681. doi: 10.1016/0002-9610(69)90404-8. [DOI] [PubMed] [Google Scholar]
- 98.Williams AO, Johnston GV. Cavernous transformation of the portal vein in rhesus monkeys. J Pathol Bacteriol. 1965;90:613–618. doi: 10.1002/path.1700900230. [DOI] [PubMed] [Google Scholar]
- 99.Kim JH, Lee YS, Kim SH. Does umbilical vein catheterization lead to portal venous thrombosis? Prospective US evaluation in 100 neonates. Radiology. 2001;219:645–650. doi: 10.1148/radiology.219.3.r01jn17645. [DOI] [PubMed] [Google Scholar]
- 100.Odièvre M, Pige G, Alagille D. Congenital abnormalities associated with extrahepatic portal hypertension. Arch Dis Child. 1977;52:383–385. doi: 10.1136/adc.52.5.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bayraktar Y, Balkanci F, Kayhan B. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology. 1997;44:1588–1594. [PubMed] [Google Scholar]
- 102.El-Matary W, Roberts EA, Kim P. Management of portal hypertensive biliopathy by sequential portal-systemic shunt and hepaticojejunostomy. Eur J Pediatr. 2008;167:1339–1342. doi: 10.1007/s00431-008-0675-4. [DOI] [PubMed] [Google Scholar]
- 103.Yamamoto S, Yokoyama Y, Takeshige K, Iwatsuki S. Budd-Chiari syndrome with obstruction of the inferior vena cava. Gastroenterology. 1968;54:1070–1084. [PubMed] [Google Scholar]
- 104.Terabayashi H, Okuda K, Nomura F. Transformation of inferior vena caval thrombosis to membranous obstruction in a patient with the lupus anticoagulant. Gastroenterology. 1986;91:219–224. doi: 10.1016/0016-5085(86)90462-2. [DOI] [PubMed] [Google Scholar]
- 105.Kage M, Arakawa M, Kojiro M, Okuda K. Histopathology of membranous obstruction of the inferior vena cava in the Budd-Chiari syndrome. Gastroenterology. 1992;102:2081–2090. doi: 10.1016/0016-5085(92)90336-w. [DOI] [PubMed] [Google Scholar]
- 106.Okuda K. Membranous obstruction of the inferior vena cava: etiology and relation to hepatocellular carcinoma. Gastroenterology. 1982;82:376–379. [PubMed] [Google Scholar]
- 107.Okuda K, Kage M, Shrestha SM. Proposal for a new nomenclature for Budd-Chiari syndrome: hepatic vein thrombosis versus thrombosis of the inferior vena cava at its hepatic portion. Hepatology. 1998;28:1191–1198. doi: 10.1002/hep.510280505. [DOI] [PubMed] [Google Scholar]
- 108.Simson IW. Membranous obstruction of the inferior vena cava and hepatocellular carcinoma in South Africa. Gastroenterology. 1982;82:171–178. [PubMed] [Google Scholar]
- 109.Hoffman HD, Stockland B, von der Heyden U. Membranous obstruction of the inferior vena cava with Budd-Chiari syndrome in children: a report of nine cases. J Pediatr Gastroenterol Nutr. 1987;6:878–884. doi: 10.1097/00005176-198711000-00010. [DOI] [PubMed] [Google Scholar]
- 110.Jaffe R, Yunis EJ. Congenital Budd-Chiari syndrome. Pediatr Pathol. 1983;1:187–192. doi: 10.3109/15513818309040656. [DOI] [PubMed] [Google Scholar]
- 111.Roulet M, Laurini R, Rivier L, Calame A. Hepatic veno-occlusive disease in newborn infant of a woman drinking herbal tea. J Pediatr. 1988;112:433–436. doi: 10.1016/s0022-3476(88)80330-5. [DOI] [PubMed] [Google Scholar]
- 112.Gentil-Kocher S, Bernard O, Brunelle F. Budd-Chiari syndrome in children: report of 22 cases. J Pediatr. 1988;113:30–38. doi: 10.1016/s0022-3476(88)80524-9. [DOI] [PubMed] [Google Scholar]
- 113.Sergi C, Beedgen B, Linderkamp O, Hofmann WJ. Fatal course of veno-occlusive disease of the liver (endophlebitis hepatica obliterans) in a preterm infant. Pathol Res Pract. 1999;195:847–851. doi: 10.1016/S0344-0338(99)80108-3. [DOI] [PubMed] [Google Scholar]
- 114.Etzioni A, Benderly A, Rosenthal E. Defective humoral and cellular immune functions associated with veno-occlusive disease of the liver. J Pediatr. 1987;110:549–554. doi: 10.1016/s0022-3476(87)80546-2. [DOI] [PubMed] [Google Scholar]
- 115.Mellis C, Bale PM. Familial hepatic veno-occlusive disease with probable immune deficiency. J Pediatr. 1976;88:236–242. doi: 10.1016/s0022-3476(76)80988-2. [DOI] [PubMed] [Google Scholar]
- 116.Haitjema T, Westermann CJJ, Overtoom TTC. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease). New insights in pathogenesis, complications, and treatment. Arch Intern Med. 1996;156:714–719. [PubMed] [Google Scholar]
- 117.Sharma VK, Howden CW. Gastrointestinal and hepatic manifestations of hereditary hemorrhagic telangiectasia. Dig Dis Sci. 1998;16:169–174. doi: 10.1159/000016861. [DOI] [PubMed] [Google Scholar]
- 118.McAllister KA, Grogg KM, Johnson DW. Endoglin, a TGFβ binding protein of endothelial cells, in the gene for hereditary hemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 119.Vincent P, Plauchu H, Hazan J. A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet. 1995;4:945–949. doi: 10.1093/hmg/4.5.945. [DOI] [PubMed] [Google Scholar]
- 120.Harrison RE, Flanagan JA, Sankelo M. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40:865–871. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gallione CJ, Repetto GM, Legius E. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 122.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–100. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Caselitz M, Wagner S, Chavan A. Clinical outcome of transfemoral embolisation in patients with arteriovenous malformations of the liver in hereditary haemorrhagic telangiectasia (Weber-Rendu-Osler disease) Gut. 1998;42:123–126. doi: 10.1136/gut.42.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Trotter JF, Suhocki PV. Hereditary hemorrhagic telangiectasia causing high output failure: treatment with transcatheter embolization. Am J Gastroenterol. 1998;93:1569–1571. doi: 10.1111/j.1572-0241.1998.00486.x. [DOI] [PubMed] [Google Scholar]
- 125.Fagel WJ, Perlberger R, Kauffmann RH. Portosystemic encephalopathy in hereditary hemorrhagic telangiectasia. Am J Med. 1988;85:858–860. doi: 10.1016/s0002-9343(88)80036-6. [DOI] [PubMed] [Google Scholar]
- 126.Mendoza A, Oliff S, Elias E. Hereditary haemorrhagic telangiectasia and secondary biliary cirrhosis. Eur J Gastroenterol Hepatol. 1995;7:999–1002. doi: 10.1097/00042737-199510000-00017. [DOI] [PubMed] [Google Scholar]
- 127.Hashimoto M, Tate E, Nishii T. Angiography of hepatic vascular malformations associated with hereditary hemorrhagic telangiectasia. Cardiovasc Intervent Radiol. 2003;26:177–180. doi: 10.1007/s00270-002-1507-y. [DOI] [PubMed] [Google Scholar]
- 128.Ralls PW, Johnson MB, Radin R. Hereditary hemorrhagic telangiectasia: findings in the liver with color Doppler sonography. Am J Roentgenol. 1992;159:59–61. doi: 10.2214/ajr.159.1.1609722. [DOI] [PubMed] [Google Scholar]
- 129.Solis-Herruzo JA, Garcia-Cabezudo J, Santalla-Pecina F. Laparoscopic findings in hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu disease) Endoscopy. 1984;16:137–139. doi: 10.1055/s-2007-1018558. [DOI] [PubMed] [Google Scholar]
- 130.Chavan A, Caselitz M, Gratz KF. Hepatic artery embolization for treatment of patients with hereditary hemorrhagic telangiectasia and symptomatic hepatic vascular malformations. Eur Radiol. 2004;14:2079–2085. doi: 10.1007/s00330-004-2455-5. [DOI] [PubMed] [Google Scholar]
- 131.Boillot O, Bianco F, Viale J-P. Liver transplantation resolves the hyperdynamic circulation in hereditary hemorrhagic telangiectasia with hepatic involvement. Gastroenterology. 1999;116:187–192. doi: 10.1016/s0016-5085(99)70243-x. [DOI] [PubMed] [Google Scholar]
- 132.Azoulay D, Precetti S, Emile JF. Liver transplantation for intrahepatic Rendu–Osler–Weber's disease: the Paul Brousse hospital experience. Gastroenterol Clin Biol. 2002;26:828–834. [PubMed] [Google Scholar]
- 133.Cura MA, Postoak D, Speeg KV, Vasan R. Transjugular intrahepatic portosystemic shunt for variceal hemorrhage due to recurrent of hereditary hemorrhagic telangiectasia in a liver transplant. J Vasc Interv Radiol. 2010;21:135–139. doi: 10.1016/j.jvir.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 134.Daly JJ, Schiller AL. The liver in hemorrhagic telangiectasia (Osler-Weber-Rendu disease) Am J Med. 1976;60:723–726. doi: 10.1016/0002-9343(76)90510-6. [DOI] [PubMed] [Google Scholar]
- 135.Zelman S. Liver fibrosis in hereditary hemorrhagic telangiectasia. Fibrosis of diffuse insular character. Arch Pathol. 1962;74:66–72. [PubMed] [Google Scholar]
- 136.Wanless IR, Gryfe A. Nodular transformation of the liver in hereditary hemorrhagic telangiectasia. Arch Pathol Lab Med. 1986;110:331–335. [PubMed] [Google Scholar]
- 137.Buscarini E, Danesino C, Plauchu H. High prevalence of hepatic focal nodular hyperplasia in subjects with hereditary hemorrhagic telangiectasia. Ultrasound Med Biol. 2004;30:1089–1097. doi: 10.1016/j.ultrasmedbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 138.Blewitt R, Brown CM, Wyatt JI. The pathology of acute hepatic disintegration in hereditary haemorrhagic telangiectasia. Histopathology. 2003;42:265–269. doi: 10.1046/j.1365-2559.2003.01579.x. [DOI] [PubMed] [Google Scholar]
- 139.Sussman EB, Sternberg SS. Hereditary hemorrhagic telangiectasia, a case with hepatocellular carcinoma and acquired hepatocerebral degeneration. Arch Pathol. 1975;99:95–100. [PubMed] [Google Scholar]
- 140.Bourdeau A, Faughan M, McDonald ML. Potential role of modifier genes influencing TGFβ1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am J Pathol. 2001;180:2011–2020. doi: 10.1016/s0002-9440(10)64673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Torsney E, Charlton R, Diamond AG. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation. 2003;107:1653–1657. doi: 10.1161/01.CIR.0000058170.92267.00. [DOI] [PubMed] [Google Scholar]
- 142.Srinivasan S, Hanes MA, Dickens T. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet. 2003;12:473–482. doi: 10.1093/hmg/ddg050. [DOI] [PubMed] [Google Scholar]
- 143.Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med. 1991;325:1831–1836. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- 144.McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weinstein S, Scottolini AG, Loo SYT. Ataxia telangiectasia with hepatocellular carcinoma in a 15-year-old girl and studies of her kindred. Arch Pathol Lab Med. 1985;109:1000–1004. [PubMed] [Google Scholar]
- 146.Srisirirojanakorn N, Finegold MJ, Gopalakrishna GS, Klish WJ. Hepatic veno-occlusive disease in ataxia telangiectasia. J Pediatr. 1999;134:786–788. doi: 10.1016/s0022-3476(99)70301-x. [DOI] [PubMed] [Google Scholar]
- 147.Joerger M, Koeberle D, Neumann HP, Gillessen S. Von Hippel–Lindau disease – a rare disease important to recognize. Onkologie. 2005;28:159–163. doi: 10.1159/000083860. [DOI] [PubMed] [Google Scholar]
- 148.Zeitlin H. Hemangioblastomas of the meninges and their relationship to Lindau's disease. J Neuropathol Exp Neurol. 1942;1:14–23. [Google Scholar]
- 149.Rojiani AM, Owen DA, Berry K. Hepatic hemangioblastoma: an unusual presentation in a patient with von Hippel-Lindau disease. Am J Surg Pathol. 1991;15:81–86. [PubMed] [Google Scholar]
- 150.McGrath FP, Gibney RG, Morris DC. Case report: Multiple hepatic and pulmonary hemangioblastomas – a new manifestation of von Hippel-Lindau disease. Clin Radiol. 1992;42:37–39. doi: 10.1016/s0009-9260(05)81467-9. [DOI] [PubMed] [Google Scholar]
- 151.Deboever G, Dewulf P, Maerteus J. Common bile duct obstruction due to pancreatic involvement in the von Hippel-Lindau syndrome. Am J Gastroenterol. 1992;87:1866–1868. [PubMed] [Google Scholar]
- 152.Wanless IR, Mawdsley C, Adams R. On the pathogenesis of focal nodular hyperplasia of the liver. Hepatology. 1985;5:1194–1200. doi: 10.1002/hep.1840050622. [DOI] [PubMed] [Google Scholar]
- 153.Wanless IR, Albrecht S, Bilbao J. Multiple focal nodular hyperplasia of the liver associated with vascular malformations of various organs and neoplasia of the brain: a new syndrome. Mod Pathol. 1989;3:456–462. [PubMed] [Google Scholar]
- 154.Goldin RD, Rose DSC. Focal nodular hyperplasia of the liver associated with intracranial vascular malformations. Gut. 1990;31:554–555. doi: 10.1136/gut.31.5.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Forns X, Castella A, Bruix J. Hiperplasia nodular focal associada a malformaciones vasculares cerebrales. Gastroenterol Hepatol. 1992;15:405–407. [Google Scholar]
- 156.Puente SG, Bannura GC. Radiological anatomy of the biliary tract: variations and congenital abnormalities. World J Surg. 1983;7:271–276. doi: 10.1007/BF01656159. [DOI] [PubMed] [Google Scholar]
- 157.Markle GB. Agenesis of the common bile duct. Arch Surg. 1981;116:350–352. doi: 10.1001/archsurg.1981.01380150068018. [DOI] [PubMed] [Google Scholar]
- 158.Nygren EJ, Barnes WA. Atresia of the common hepatic duct with shunt via an accessory duct: report of a case. Arch Surg. 1954;68:337–343. doi: 10.1001/archsurg.1954.01260050339009. [DOI] [PubMed] [Google Scholar]
- 159.Stellin GP, Karner FM, Toyama WM, Lilly JR. Biliary agenesis (abstr) Hepatology. 1986;6:1218. [Google Scholar]
- 160.Schwartz MZ, Hall RJ, Reubner B. Agenesis of the extrahepatic bile ducts: report of five cases. J Pediatr Surg. 1990;25:805–807. doi: 10.1016/s0022-3468(05)80026-0. [DOI] [PubMed] [Google Scholar]
- 161.Nomura T, Shirai Y, Sasagawa M. Anomalous insertion of the right hepatic duct into the cyst duct: report of a case before laparoscopic cholecystectomy. Surg Laparosc Endosc. 1999;9:211–212. [PubMed] [Google Scholar]
- 162.Dowdy GS, Waldron GW, Brown WG. Surgical anatomy of the pancreaticobiliary system: observations. Arch Surg. 1962;84:229–246. doi: 10.1001/archsurg.1962.01300200077006. [DOI] [PubMed] [Google Scholar]
- 163.Hand BH. Anatomy and function of the extrahepatic biliary system. Clin Gastroenterol. 1973;2:3–29. [PubMed] [Google Scholar]
- 164.Cullingford G, Davidson B, Dooley J, Habit N. Hepatolithiasis with anomalous biliary anatomy and a vascular compression. HPB Surg. 1991;3:129–137. doi: 10.1155/1991/23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Radtke A, Sgourakis G, Sotiropoulos GC. A new systematic classification of peripheral anatomy of the right hepatic duct: experience from adult live liver donor transplantation. Transplant Proc. 2008;40:3158–3160. doi: 10.1016/j.transproceed.2008.08.049. [DOI] [PubMed] [Google Scholar]
- 166.Jackson JB, Kelly TB. Cholecystohepatic ducts: case report. Ann Surg. 1964;159:581–584. doi: 10.1097/00000658-196404000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Redkar R, Davenport M, Myers N, Howard ER. Association of oesophageal atresia and cholecystohepatic duct. Pediatr Surg Int. 1999;15:21–23. doi: 10.1007/s003830050503. [DOI] [PubMed] [Google Scholar]
- 168.Stokes TL, Old L. Cholecystohepatic duct. Ann Surg. 1978;135:703–705. doi: 10.1016/0002-9610(78)90140-x. [DOI] [PubMed] [Google Scholar]
- 169.Ishak KG, Willis GW, Cummins SD, Bullock AA. Biliary cystadenoma and cystadenocarcinoma. Report of 14 cases and review of the literature. Cancer. 1977;39:322–338. doi: 10.1002/1097-0142(197701)39:1<322::aid-cncr2820390149>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 170.Ng JWT, Wong MK, Kong CK. Long accessory hepatic duct with congenital dilation of the common bile duct. Am J Gastroenterol. 1993;88:619–621. [PubMed] [Google Scholar]
- 171.Walters W. Surgical lesions of the biliary tract. Arch Surg. 1960;81:1–13. [Google Scholar]
- 172.Swartley WB, Weeder SD. Choledochus cyst with a double common bile duct. Ann Surg. 1935;101:912–920. doi: 10.1097/00000658-193503000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee CMJ. Duplication of the cystic and common hepatic ducts, lined with gastric mucosa: a rare congenital anomaly. N Engl J Med. 1957;256:927–931. doi: 10.1056/NEJM195705162562003. [DOI] [PubMed] [Google Scholar]
- 174.Hourigan JS, Carr MG, Burton EM, Ledbetter JC. Congenital bronchobiliary fistula: MRI appearance. Pediatr Radiol. 2004;34:348–350. doi: 10.1007/s00247-003-1091-6. [DOI] [PubMed] [Google Scholar]
- 175.Kalayoglu M, Olcay I. Congenital bronchobiliary fistula associated with esophageal atresia and tracheoesophageal fistula. J Pediatr Surg. 1976;11:463–464. doi: 10.1016/s0022-3468(76)80205-9. [DOI] [PubMed] [Google Scholar]
- 176.Chan YT, Ng WD, Mak WP. Congenital bronchobiliary fistula associated with biliary atresia. Br J Surg. 1984;71:240–241. doi: 10.1002/bjs.1800710329. [DOI] [PubMed] [Google Scholar]
- 177.Gunlemez A, Tugay M, Elemen L. Surgical experience in a baby with congenital broncho-biliary fistula. Ann Thorac Surg. 2009;87:318–320. doi: 10.1016/j.athoracsur.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 178.Jakowski JD, Lucas JG, Seth S, Frankel WL. Ciliated hepatic foregut cyst: a rare but increasingly reported liver cyst. Ann Diagn Pathol. 2004;8:342–346. doi: 10.1053/j.anndiagpath.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 179.Momin TA, Milner R, Sarmiento JM. Ciliated hepatic foregut cyst of the left hepatic vein. J Gastrointest Surg. 2004;8:601–603. doi: 10.1016/j.gassur.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 180.Wu MLC, Abecassis MM, Rao MS. Ciliated hepatic foregut cyst mimicking neoplasm. Am J Gastroenterol. 1998;93:2212–2214. doi: 10.1111/j.1572-0241.1998.00616.x. [DOI] [PubMed] [Google Scholar]
- 181.Vick DJ, Goodman ZD, Ishak KG. Squamous cell carcinoma arising in a ciliated hepatic foregut cyst. Arch Pathol Lab Med. 1999;23:1115–1117. doi: 10.5858/1999-123-1115-SCCAIA. [DOI] [PubMed] [Google Scholar]
- 182.Furlanetto A, Dei Tos AP. Squamous cell carcinoma arising in a ciliated hepatic foregut cyst. Virchows Arch. 2002;441:296–298. doi: 10.1007/s00428-002-0668-z. [DOI] [PubMed] [Google Scholar]
- 183.Vick DJ, Goodman ZD, Devers MT. Ciliated hepatic foregut cyst: a study of six cases and review of the literature. Am J Surg Pathol. 1999;23:671–677. doi: 10.1097/00000478-199906000-00006. [DOI] [PubMed] [Google Scholar]
- 184.Terada T, Nakanuma Y, Kono N. Ciliated hepatic foregut cyst. A mucus histological, immunohistochemical, and ultrastructural study in three cases in comparison with normal bronchi and intrahepatic bile ducts. Am J Surg Pathol. 1990;14:356–463. [PubMed] [Google Scholar]
- 185.Chatelain D, Chailley-Heu B, Terris B. The ciliated hepatic foregut cyst, an unusual bronchiolar foregut malformation: a histological histochemical, and immunohistochemical study of 7 cases. Hum Pathol. 2000;31:241–246. doi: 10.1016/s0046-8177(00)80227-0. [DOI] [PubMed] [Google Scholar]
- 186.Haller JO, Condon VR, Berdon WE. Spontaneous perforation of the common bile duct in children. Radiology. 1989;172:621–624. doi: 10.1148/radiology.172.3.2672089. [DOI] [PubMed] [Google Scholar]
- 187.Davenport M, Heaton N, Howard E. Spontaneous perforation of the bile duct in infants. Br J Surg. 1991;78:1068–1070. doi: 10.1002/bjs.1800780912. [DOI] [PubMed] [Google Scholar]
- 188.Evans K, Marsden N, Desai A. Spontaneous perforation of the bile duct in infancy and childhood: a systematic review. J Pediatr Gastroenterol Nutr. 2010;50:677–681. doi: 10.1097/MPG.0b013e3181d5eed3. [DOI] [PubMed] [Google Scholar]
- 189.Chardot C, Carton M, Spire-Bendelac N. Epidemiology of biliary atresia in France: a national study 1986–1996. J Hepatol. 1999;31:1006–1013. doi: 10.1016/s0168-8278(99)80312-2. [DOI] [PubMed] [Google Scholar]
- 190.McKiernan PJ, Baker AJ, Kelly DA. The frequency and outcome of biliary atresia in the UK and Ireland. Lancet. 2000;355:25–29. doi: 10.1016/S0140-6736(99)03492-3. [DOI] [PubMed] [Google Scholar]
- 191.Schreiber RA, Barker CC, Roberts EA. Biliary atresia: the Canadian experience. J Pediatr. 2007;151:659–665. doi: 10.1016/j.jpeds.2007.05.051. 65 e1. [DOI] [PubMed] [Google Scholar]
- 192.Hsiao CH, Chang MH, Chen HL. Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology. 2008;47:1233–1240. doi: 10.1002/hep.22182. [DOI] [PubMed] [Google Scholar]
- 193.Yoon PW, Bresee JS, Olney RS. Epidemiology of biliary atresia: a population-based study. Pediatrics. 1997;99:376–382. doi: 10.1542/peds.99.3.376. [DOI] [PubMed] [Google Scholar]
- 194.Strickland AD, Shannon K. Studies in the etiology of extrahepatic biliary atresia: time-space clustering. J Pediatr. 1982;100:749–753. doi: 10.1016/s0022-3476(82)80576-3. [DOI] [PubMed] [Google Scholar]
- 195.Wada H, Muraji T, Yokoi A. Insignificant seasonal and geographical variation in incidence of biliary atresia in Japan: a regional survey of over 20 years. J Pediatr Surg. 2007;42:2090–2092. doi: 10.1016/j.jpedsurg.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 196.Hyams JS, Glaser JH, Leichtner AM, Morecki R. Discordance for biliary atresia in two sets of monozygotic twins. J Pediatr. 1985;107:420–422. doi: 10.1016/s0022-3476(85)80524-2. [DOI] [PubMed] [Google Scholar]
- 197.Chandra RS. Biliary atresia and other structural anomalies in the congenital polysplenic syndrome. J Pediatr. 1974;85:649–655. doi: 10.1016/s0022-3476(74)80508-1. [DOI] [PubMed] [Google Scholar]
- 198.Davenport M, Mowat AP, Howard ER. Biliary atresia splenic malformation syndrome: an etiology and prognostic subgroup. Surgery. 1993;113:662–668. [PubMed] [Google Scholar]
- 199.Guttman OR, Roberts EA, Schreiber RA. Biliary atresia and associated structural anomalies: clinical and outcome findings Canada-wide (abstr) Can J Gastroenterol. 2008;22(Suppl. A):125A. [Google Scholar]
- 200.De Matos V, Erlichman J, Russo PA, Haber BA. Does ‘cystic’ biliary atresia represent a distinct clinical and etiological subgroup? A series of three cases. Pediatr Dev Pathol. 2005;8:725–731. doi: 10.1007/s10024-005-0018-7. [DOI] [PubMed] [Google Scholar]
- 201.Muise AM, Turner D, Wine E. Biliary atresia with choledochal cyst: implications for classification. Clin Gastroenterol Hepatol. 2006;4:1411–1414. doi: 10.1016/j.cgh.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 202.Caponcelli E, Knisely AS, Davenport M. Cystic biliary atresia: an etiologic and prognostic subgroup. J Pediatr Surg. 2008;43:1619–1624. doi: 10.1016/j.jpedsurg.2007.12.058. [DOI] [PubMed] [Google Scholar]
- 203.Alpert LI, Strauss L, Hirschorn K. Neonatal hepatitis and biliary atresia associated with trisomy 17–18. N Engl J Med. 1969;280:16–20. doi: 10.1056/NEJM196901022800104. [DOI] [PubMed] [Google Scholar]
- 204.Molland EA, Purcell M. Biliary atresia and the Dandy-Walker anomaly in a neonate with 45,X Turner's syndrome. J Pathol. 1975;115:227–230. doi: 10.1002/path.1711150407. [DOI] [PubMed] [Google Scholar]
- 205.Allotey J, Lacaille F, Lees MM. Congenital bile duct anomalies (biliary atresia) and chromosome 22 aneuploidy. J Pediatr Surg. 2008;43:1736–1740. doi: 10.1016/j.jpedsurg.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 206.Annéren G, Mearling S, Lilja H. Lethal autosomal recessive syndrome with intrauterine growth retardation, intra- and extra- hepatic biliary atresia and esophageal and duodenal atresia. Am J Med Genet. 1998;78:306–309. doi: 10.1002/(sici)1096-8628(19980707)78:3<306::aid-ajmg22>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 207.Selicorni A, Colombo C, Bonato S. Biliary atresia and Kabuki syndrome: another case with long-term follow-up. Am J Med Genet. 2001;100:251. doi: 10.1002/ajmg.1253. [DOI] [PubMed] [Google Scholar]
- 208.Balasubramanian M, Parker MJ. Zimmermann–Laband syndrome in a child previously described with brachydactyly, extrahepatic biliary atresia, patent ductus arteriosus and seizures. Clin Dysmorphol. 2010;19:48–50. doi: 10.1097/MCD.0b013e328333c239. [DOI] [PubMed] [Google Scholar]
- 209.Galan-Gomez E, Sanchez EB, Arias-Castro S, Cardesa-Garcia JJ. Intrauterine growth retardation, duodenal and extrahepatic biliary atresia, hypoplastic pancreas and other intestinal anomalies: further evidence of the Martinez–Frias syndrome. Eur J Med Genet. 2007;50:144–148. doi: 10.1016/j.ejmg.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 210.Lachaux A, Descos B, Plauchu H. Familial extrahepatic biliary atresia. J Pediatr Gastroenterol Nutrit. 1988;7:280–283. doi: 10.1097/00005176-198803000-00020. [DOI] [PubMed] [Google Scholar]
- 211.Smith BM, Laberge J-M, Schreiber R. Familial biliary atresia in three siblings including twins. J Pediatr Surg. 1991;26:1331–1333. doi: 10.1016/0022-3468(91)90613-x. [DOI] [PubMed] [Google Scholar]
- 212.Kobayashi K, Kubota M, Okuyama N. Mother-to-daughter occurrence of biliary atresia: a case report. J Pediatr Surg. 2008;43:1566–1568. doi: 10.1016/j.jpedsurg.2008.03.051. [DOI] [PubMed] [Google Scholar]
- 213.Rauschenfels S, Krassmann M, Al-Masri AN. Incidence of hepatotropic viruses in biliary atresia. Eur J Pediatr. 2009;168:469–476. doi: 10.1007/s00431-008-0774-2. [DOI] [PubMed] [Google Scholar]
- 214.Tarr PI, Haas JE, Christie DL. Biliary atresia, cytomegalovirus, and age at referral. Pediatrics. 1996;97:828–831. [PubMed] [Google Scholar]
- 215.Chang MH, Huang HH, Huang ES. Polymerase chain reaction to detect human cytomegalovirus in livers of infants with neonatal hepatitis. Gastroenterology. 1992;103:1022–1025. doi: 10.1016/0016-5085(92)90038-z. [DOI] [PubMed] [Google Scholar]
- 216.Hart MH, Kaufman SS, Vanderhoof JA. Neonatal hepatitis and extrahepatic biliary atresia associated with cytomegalovirus infection in twins. Am J Dis Child. 1991;145:302–305. doi: 10.1001/archpedi.1991.02160030070024. [DOI] [PubMed] [Google Scholar]
- 217.Shen C, Zheng S, Wang W, Xiao XM. Relationship between prognosis of biliary atresia and infection of cytomegalovirus. World J Pediatr. 2008;4:123–126. doi: 10.1007/s12519-008-0024-8. [DOI] [PubMed] [Google Scholar]
- 218.Morecki R, Glaser JH, Cho S. Biliary atresia and reovirus type 3 infection. N Eng J Med. 1982;307:481–484. doi: 10.1056/NEJM198208193070806. [DOI] [PubMed] [Google Scholar]
- 219.Rosenberg DP, Morecki R, Lollini LO. Extrahepatic biliary atresia in a Rhesus monkey (Macaca mulatta) Hepatology. 1983;3:577–580. doi: 10.1002/hep.1840030417. [DOI] [PubMed] [Google Scholar]
- 220.Glaser JH, Balistreri WF, Morecki R. Role of reovirus type 3 in persistent infantile cholestasis. J Pediatr. 1984;105:912–915. doi: 10.1016/s0022-3476(84)80076-1. [DOI] [PubMed] [Google Scholar]
- 221.Morecki R, Glaser JH, Johnson AB, Kress Y. Detection of reovirus type 3 in the porta hepatis of an infant with extrahepatic biliary atresia: ultrastructural and immunocytochemical study. Hepatology. 1984;4:1137–1142. doi: 10.1002/hep.1840040608. [DOI] [PubMed] [Google Scholar]
- 222.Brown WR, Sokol RJ, Levin MJ. Lack of correlation between infection with reovirus 3 and extrahepatic biliary atresia or neonatal hepatitis. J Pediatr. 1988;113:670–676. doi: 10.1016/s0022-3476(88)80376-7. [DOI] [PubMed] [Google Scholar]
- 223.Steele MI, Marshall CM, Lloyd RE, Randolph VE. Reovirus 3 not detected by reverse transcriptase-mediated polymerase chain reaction analysis of preserved tissue from infants with cholestatic liver disease. Hepatology. 1995;21:697–702. [PubMed] [Google Scholar]
- 224.Tyler KL, Sokol KJ, Oberhans SM. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology. 1998;27:1475–1482. doi: 10.1002/hep.510270603. [DOI] [PubMed] [Google Scholar]
- 225.Riepenhoff-Talty M, Gouvea V, Evans MJ. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis. 1996;174:8–15. doi: 10.1093/infdis/174.1.8. [DOI] [PubMed] [Google Scholar]
- 226.Bobo L, Ojeh C, Chiu D. Lack of evidence for rotavirus by polymerase chain reaction/enzyme immunoassay of hepatobiliary samples from children with biliary atresia. Pediatr Res. 1997;41:229–234. doi: 10.1203/00006450-199702000-00013. [DOI] [PubMed] [Google Scholar]
- 227.Drut R, Drut RM, Gomez MA. Presence of human papilloma virus in EBA. J Pediatr Gastroenterol Nutr. 1998;27:530–535. doi: 10.1097/00005176-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 228.Schreiber RA, Kleinman RE. Genetics, immunology, and biliary atresia: an opening or a diversion? J Pediatr Gastroenterol Nutr. 1993;16:111–113. doi: 10.1097/00005176-199302000-00001. [DOI] [PubMed] [Google Scholar]
- 229.Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233–242. doi: 10.1055/s-2007-985068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Davenport M, Gonde C, Redkar R. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg. 2001;36:1017–1025. doi: 10.1053/jpsu.2001.24730. [DOI] [PubMed] [Google Scholar]
- 231.Mack CL, Tucker RM, Sokol RJ. Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation. Pediatr Res. 2004;56:79–87. doi: 10.1203/01.PDR.0000130480.51066.FB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mack CL, Falta MT, Sullivan AK. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133:278–287. doi: 10.1053/j.gastro.2007.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Whitington PF, Malladi P, Melin-Aldana H. Expression of osteopontin correlates with portal biliary proliferation and fibrosis in biliary atresia. Pediatr Res. 2005;57:837–844. doi: 10.1203/01.PDR.0000161414.99181.61. [DOI] [PubMed] [Google Scholar]
- 234.Bezerra JA, Tiao G, Ryckman FC. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360:1653–1659. doi: 10.1016/S0140-6736(02)11603-5. [DOI] [PubMed] [Google Scholar]
- 235.Chen L, Goryachev A, Sun J. Altered expression of genes involved in hepatic morphogenesis and fibrogenesis are identified by cDNA microarray analysis in biliary atresia. Hepatology. 2003;38:567–576. doi: 10.1053/jhep.2003.50363. [DOI] [PubMed] [Google Scholar]
- 236.Zhang DY, Sabla G, Shivakumar P. Coordinate expression of regulatory genes differentiates embryonic and perinatal forms of biliary atresia. Hepatology. 2004;39:954–962. doi: 10.1002/hep.20135. [DOI] [PubMed] [Google Scholar]
- 237.Silveira TR, Salzano FM, Donaldson PT. Association between HLA and extrahepatic biliary atresia. J Pedaitr Gastroenterol Nutr. 1993;16:114–117. doi: 10.1097/00005176-199302000-00002. [DOI] [PubMed] [Google Scholar]
- 238.Donaldson PT, Clare M, Constantini PK. HLA and cytokine gene polymorphisms in biliary atresia. Liver. 2002;22:213–219. doi: 10.1046/j.0106-9543.2002.01647.x. [DOI] [PubMed] [Google Scholar]
- 239.Yuasa T, Tsuji H, Kimura S. Human leukocyte antigens in Japanese patients with biliary atresia: retrospective analysis of patients who underwent living donor liver transplantation. Hum Immunol. 2005;66:295–300. doi: 10.1016/j.humimm.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 240.A-Kader HH, El-Ayyouti M, Hawas S. HLA in Egyptian children with biliary atresia. J Pediatr. 2002;141:432–433. doi: 10.1067/mpd.2002.127506. [DOI] [PubMed] [Google Scholar]
- 241.Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. J Hepatol. 1998;28:603–607. doi: 10.1016/s0168-8278(98)80283-3. [DOI] [PubMed] [Google Scholar]
- 242.Petersen C, Kuske M, Bruns E. Progress in developing animal models for biliary atresia. Eur J Pediatr Surg. 1998;8:137–141. doi: 10.1055/s-2008-1071140. [DOI] [PubMed] [Google Scholar]
- 243.Shivakumar P, Campbell KM, Sabla GE. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114:322–329. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mohanty SK, Ivantes CA, Mourya R. Macrophages are targeted by rotavirus in experimental biliary atresia and induce neutrophil chemotaxis by Mip2/Cxcl2. Pediatr Res. 2010;67:345–351. doi: 10.1203/PDR.0b013e3181d22a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mohanty SK, Shivakumar P, Sabla G, Bezerra JA. Loss of interleukin-12 modifies the pro-inflammatory response but does not prevent duct obstruction in experimental biliary atresia. BMC Gastroenterol. 2006;6:14. doi: 10.1186/1471-230X-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Tucker RM, Hendrickson RJ, Mukaida N. Progressive biliary destruction is independent of a functional tumor necrosis factor-alpha pathway in a rhesus rotavirus-induced murine model of biliary atresia. Viral Immunol. 2007;20:34–43. doi: 10.1089/vim.2006.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Shivakumar P, Sabla G, Mohanty S. Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133:268–277. doi: 10.1053/j.gastro.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mack CL, Tucker RM, Lu BR. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology. 2006;44:1231–1239. doi: 10.1002/hep.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lu BR, Brindley SM, Tucker RM. α-enolase autoantibodies cross-reactive to viral proteins in a mouse model of biliary atresia. Gastroenterology. 2010;139:1753–1761. doi: 10.1053/j.gastro.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Landing BH. Considerations of the pathogenesis of neonatal hepatitis, biliary atresia and choledochal cyst–the concept of infantile obstructive cholangiopathy. Prog Pediatr Surg. 1974;6:113–139. [PubMed] [Google Scholar]
- 251.Muraji T, Hosaka N, Irie N. Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes of maternal cells in the liver. Pediatrics. 2008;121:517–521. doi: 10.1542/peds.2007-0568. [DOI] [PubMed] [Google Scholar]
- 252.Ogawa T, Suruga K, Kojima Y. Experimental study of the pathogenesis of infantile obstructive cholangiography and its clinical evaluation. J Pediatr Surg. 1983;18:131–135. doi: 10.1016/s0022-3468(83)80533-8. [DOI] [PubMed] [Google Scholar]
- 253.Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme ‘ductal plate malformation’. Hepatology. 1992;16:1069–1083. doi: 10.1002/hep.1840160434. [DOI] [PubMed] [Google Scholar]
- 254.Pacheco MC, Campbell KM, Bove KE. Ductal plate malformation-like arrays in early explants after a Kasai procedure are independent of splenic malformation complex (heterotaxy) Pediatr Dev Pathol. 2009;12:355–360. doi: 10.2350/09-01-0598-OA.1. [DOI] [PubMed] [Google Scholar]
- 255.Desmet VJ. Cholangiopathies: past, present and future. Semin Liver Dis. 1987;7:67–76. doi: 10.1055/s-2008-1040566. [DOI] [PubMed] [Google Scholar]
- 256.Raweily EA, Gibson AAM, Burt AD. Abnormalities of intrahepatic bile ducts in extrahepatic biliary atresia. Histopathology. 1990;17:521–527. doi: 10.1111/j.1365-2559.1990.tb00791.x. [DOI] [PubMed] [Google Scholar]
- 257.Teichberg S, Markowitz J, Silverberg M. Abnormal cilia in a child with the polysplenia syndrome and extrahepatic biliary atresia. J Pediatr. 1982;100:399–401. doi: 10.1016/s0022-3476(82)80438-1. [DOI] [PubMed] [Google Scholar]
- 258.Gershoni-Baruch R, Gottfried E, Pery M. Immotile cilia syndrome including polysplenia, situs inversus, and extrahepatic biliary atresia. Am J Med Genet. 1989;33:390–393. doi: 10.1002/ajmg.1320330320. [DOI] [PubMed] [Google Scholar]
- 259.Chen J, Knowles IFU, Herbert JL, Hackett BP. Mutations of the mouse hepatocyte nuclear factor 1 fork head homologue 4 gene results in absence of cilia and random left/right asymmetry. J Clin Invest. 1998;102:1077–1082. doi: 10.1172/JCI4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Peeters H, Devriendt K. Human laterality disorders. Eur J Med Genet. 2006;49:349–362. doi: 10.1016/j.ejmg.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 261.Morgan D, Turnpenny L, Goodslub J. Inversion, a novel gene in the vertebrate left-right axis pathway is partially deleted in the inv mouse. Nat Genet. 1998;20:149–156. doi: 10.1038/2450. [DOI] [PubMed] [Google Scholar]
- 262.Mazziotti MV, Willis LK, Heuckeroth RO. Anomalous development of the hepatobiliary system in the Inv mouse. Hepatology. 1999;30:372–378. doi: 10.1002/hep.510300223. [DOI] [PubMed] [Google Scholar]
- 263.Davit-Spraul A, Baussan C, Hermeziu B. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2008;46:111–112. doi: 10.1097/01.mpg.0000304465.60788.f4. [DOI] [PubMed] [Google Scholar]
- 264.Ware SM, Peng J, Zhu L. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74:93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Phillips MJ, Azuma T, Meredith SL. Abnormalities in villin gene expression and canalicular microvillus structure in progressive cholestatic liver disease of childhood. Lancet. 2003;362:1112–1119. doi: 10.1016/S0140-6736(03)14467-4. [DOI] [PubMed] [Google Scholar]
- 266.Adelman S. Prognosis of uncorrected biliary atresia: an update. J Pediatr Surg. 1978;13:389–391. doi: 10.1016/s0022-3468(78)80462-x. [DOI] [PubMed] [Google Scholar]
- 267.Roberts EA. Neonatal hepatitis syndrome. Semin Neonatol. 2003;8:357–374. doi: 10.1016/S1084-2756(03)00093-9. [DOI] [PubMed] [Google Scholar]
- 268.Howard ER, Johnston DI, Mowat AP. Spontaneous perforation of the common bile duct in infants. Arch Dis Child. 1976;51:883–886. doi: 10.1136/adc.51.11.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lloyd DA, Mickel RE. Spontaneous perforation of the extrahepatic bile ducts in neonates and infants. Br J Surg. 1980;67:621–623. doi: 10.1002/bjs.1800670905. [DOI] [PubMed] [Google Scholar]
- 270.Gilmour SM, Hershkop M, Reifen R. Outcome of hepatobiliary scanning in neonatal hepatitis syndrome. J Nucl Med. 1997;38:1279–1282. [PubMed] [Google Scholar]
- 271.Clarke B, O'Donovan AN, Coates G. Delayed excretion of radionuclide in scanning with diisopropyl iminodiacetic acid does not exclude the possibility of primary biliary atresia: case report. Can Assoc Radiol J. 1997;48:42–43. [PubMed] [Google Scholar]
- 272.Petersen C, Meier PN, Schneider A. Endoscopic retrograde cholangiopancreaticography prior to explorative laparotomy avoids unnecessary surgery in patients suspected for biliary atresia. J Hepatol. 2009;51:1055–1060. doi: 10.1016/j.jhep.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 273.Shanmugam NP, Harrison PM, Devlin J. Selective use of endoscopic retrograde cholangiopancreatography in the diagnosis of biliary atresia in infants younger than 100 days. J Pediatr Gastroenterol Nutr. 2009;49:435–441. doi: 10.1097/MPG.0b013e3181a8711f. [DOI] [PubMed] [Google Scholar]
- 274.Keil R, Snajdauf J, Rygl M. Diagnostic efficacy of ERCP in cholestatic infants and neonates–a retrospective study on a large series. Endoscopy. 2010;42:121–126. doi: 10.1055/s-0029-1215372. [DOI] [PubMed] [Google Scholar]
- 275.Manolaki AG, Larcher VF, Mowat AP. The prelaparotomy diagnosis of extrahepatic biliary atresia. Arch Dis Child. 1983;58:591–594. doi: 10.1136/adc.58.8.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Rastogi A, Krishnani N, Yachha SK. Histopathological features and accuracy for diagnosing biliary atresia by prelaparotomy liver biopsy in developing countries. J Gastroenterol Hepatol. 2009;24:97–102. doi: 10.1111/j.1440-1746.2008.05737.x. [DOI] [PubMed] [Google Scholar]
- 277.Kasai M, Kimura S, Asakura Y. Surgical treatment of biliary atresia. J Pediatr Surg. 1968;3:665–675. [Google Scholar]
- 278.Kasai M, Watanabe I, Ohi R. Follow-up studies of long-term survivors after hepatic portoenterostomy for ‘non-correctable’ biliary atresia. J Pediatr Surg. 1975;10:173–182. doi: 10.1016/0022-3468(75)90275-4. [DOI] [PubMed] [Google Scholar]
- 279.Mieli-Vergani G, Howard ER, Portmann B, Mowat AP. Late referral for biliary atresia –missed opportunities for effective surgery. Lancet. 1989:421–423. doi: 10.1016/s0140-6736(89)90012-3. [DOI] [PubMed] [Google Scholar]
- 280.Ohi R, Nio M, Chiba T. Long-term follow-up after surgery for patients with biliary atresia. J Pediatr Surg. 1990;25:442–445. doi: 10.1016/0022-3468(90)90390-u. [DOI] [PubMed] [Google Scholar]
- 281.Karrer FM, Price MR, Bensard DD. Long-term results with the Kasai operation for biliary atresia. Arch Surg. 1996;131:493–496. doi: 10.1001/archsurg.1996.01430170039006. [DOI] [PubMed] [Google Scholar]
- 282.Valayer J. Conventional treatment of biliary atresia: long-term results. J Pediatr Surg. 1996;31:1546–1551. doi: 10.1016/s0022-3468(96)90174-8. [DOI] [PubMed] [Google Scholar]
- 283.Serinet MO, Wildhaber BE, Broue P. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009;123:1280–1286. doi: 10.1542/peds.2008-1949. [DOI] [PubMed] [Google Scholar]
- 284.Davenport M, Puricelli V, Farrant P. The outcome of the older (> or =100 days) infant with biliary atresia. J Pediatr Surg. 2004;39:575–581. doi: 10.1016/j.jpedsurg.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 285.Schweizer P, Lunzmann K. Extrahepatic bile duct atresia: how efficient is hepato-porto-enterostomy? Eur J Pediatr Surg. 1998;8:150–154. doi: 10.1055/s-2008-1071143. [DOI] [PubMed] [Google Scholar]
- 286.Pape L, Olsson K, Petersen C. Prognostic value of computerized quantification of liver fibrosis in children with biliary atresia. Liver Transpl. 2009;15:876–882. doi: 10.1002/lt.21711. [DOI] [PubMed] [Google Scholar]
- 287.Azarow KS, Phillips MJ, Sandler AD. Biliary atresia: should all patients undergo portoenterostomy? J Pediatr Surg. 1997;32:168–170. doi: 10.1016/s0022-3468(97)90173-1. [DOI] [PubMed] [Google Scholar]
- 288.Santos JL, Kieling CO, Meurer L. The extent of biliary proliferation in liver biopsies from patients with biliary atresia at portoenterostomy is associated with the postoperative prognosis. J Pediatr Surg. 2009;44:695–701. doi: 10.1016/j.jpedsurg.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 289.Chandra RS, Altman RP. Ductal remnants in extrahepatic biliary atresia: a histopathological study with clinical correlation. J Pediatr. 1978;93:196–200. doi: 10.1016/s0022-3476(78)80495-8. [DOI] [PubMed] [Google Scholar]
- 290.Witzleben CL, Buck BE, Schnaufer L, Brzosko WJ. Studies on the pathogenesis of biliary atresia. Lab Invest. 1978;38:525–532. [PubMed] [Google Scholar]
- 291.Gautier M, Jehan P, Odievre M. Histologic study of biliary fibrous remnants in 48 cases of extrahepatic biliary atresia: correlation with postoperative bile flow restoration. J Pediatr. 1976;89:704–709. doi: 10.1016/s0022-3476(76)80787-1. [DOI] [PubMed] [Google Scholar]
- 292.Ohi R, Shikes RH, Stellin GP, Lilly JR. In biliary atresia duct histology correlates with bile flow. J Pediatr Surg. 1984;19:467–470. doi: 10.1016/s0022-3468(84)80277-8. [DOI] [PubMed] [Google Scholar]
- 293.Tan CE, Davenport M, Driver M, Howard ER. Does the morphology of the extrahepatic biliary remnants in biliary atresia influence survival? A review of 205 cases. J Pediatr Surg. 1994;29:1459–1464. doi: 10.1016/0022-3468(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 294.Altman RP, Lilly JR, Greenfeld J. A multivariable risk factor analysis of the portoenterostomy (Kasai) procedure for biliary atresia: twenty-five years of experience from two centers. Ann Surg. 1997;226:348–353. doi: 10.1097/00000658-199709000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Matsuo S, Suita S, Kubota M, Shono K. Long-term results and clinical problems after portoenterostomy in patients with biliary atresia. Eur J Pediatr Surg. 1998;8:142–145. doi: 10.1055/s-2008-1071141. [DOI] [PubMed] [Google Scholar]
- 296.Ryckman FC, Alonso MH, Bucavalas JC, Balistreri WF. Biliary atresia – surgical management and treatment options as they relate to outcome. Liver Transpl Surg. 1998;4(Suppl. 1):524–533. [PubMed] [Google Scholar]
- 297.Sasaki H, Nio M, Hayashi Y. Problems during and after pregnancy in female patients with biliary atresia. J Pediatr Surg. 2007;42:1329–1332. doi: 10.1016/j.jpedsurg.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 298.Otte JB, de Ville de Goyet J, Reding R. Sequential treatment of biliary atresia with Kasai portoenterostomy and liver transplantation: a review. Hepatology. 1994;20:41S–48S. doi: 10.1016/0270-9139(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 299.McKiernan PJ, Baker AJ, Lloyd C. British paediatric surveillance unit study of biliary atresia: outcome at 13 years. J Pediatr Gastroenterol Nutr. 2009;48:78–81. doi: 10.1097/MPG.0b013e31817d80de. [DOI] [PubMed] [Google Scholar]
- 300.Avitzur Y, De Luca E, Cantos M. Health status ten years after pediatric liver transplantation – looking beyond the graft. Transplantation. 2004;78:566–573. doi: 10.1097/01.tp.0000131663.87106.1a. [DOI] [PubMed] [Google Scholar]
- 301.Fain JS, Lewin KJ. Intrahepatic biliary cysts in congenital biliary atresia. Arch Pathol Lab Med. 1989;113:1383–1386. [PubMed] [Google Scholar]
- 302.Hübscher SG, Harrison RF. Portal lymphadenopathy associated with lipofuscin in chronic cholestatic liver disease. J Clin Pathol. 1989;42:1160–1165. doi: 10.1136/jcp.42.11.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.de Freitas LA, Chevallier M, Louis D, Grimaud JA. Human extrahepatic biliary atresia: portal connective tissue activation related to ductular proliferation. Liver. 1986;6:253–261. doi: 10.1111/j.1600-0676.1986.tb00288.x. [DOI] [PubMed] [Google Scholar]
- 304.Ho CW, Shioda K, Shirasaki K. The pathogenesis of biliary atresia: a morphological study of the hepatobiliary system and the hepatic artery. J Pediatr Gastroenterol Nutr. 1993;16:53–60. doi: 10.1097/00005176-199301000-00011. [DOI] [PubMed] [Google Scholar]
- 305.dos Santos JL, da Silveira TR, da Silva VD. Medial thickening of hepatic artery branches in biliary atresia. A morphometric study. J Pediatr Surg. 2005;40:637–642. doi: 10.1016/j.jpedsurg.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 306.Clotman F, Lannoy VJ, Reber M. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–1828. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 307.Clotman F, Libbrecht L, Gresh L. Hepatic artery malformations associated with a primary defect in intrahepatic bile duct development. J Hepatol. 2003;39:686–692. doi: 10.1016/s0168-8278(03)00409-4. [DOI] [PubMed] [Google Scholar]
- 308.Ohi R, Lilly JR. Copper kinetics in infantile hepatobiliary disease. J Pediatr Surg. 1980;15:509–512. doi: 10.1016/s0022-3468(80)80763-9. [DOI] [PubMed] [Google Scholar]
- 309.Hussein A, Wyatt J, Guthrie A, Stringer MD. Kasai portoenterostomy – new insights from hepatic morphology. J Pediatr Surg. 2005;40:322–326. doi: 10.1016/j.jpedsurg.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 310.Fonkelsrud EW, Arima E. Bile lakes in congenital biliary atresia. Surgery. 1975;77:384–390. [PubMed] [Google Scholar]
- 311.Ramm GA, Nair VG, Bridle KR. Contribution of hepatic parenchymal and nonparenchymal cells to hepatic fibrogenesis in biliary atresia. Am J Pathol. 1998;153:527–535. doi: 10.1016/S0002-9440(10)65595-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Lamireau T, Le Bail B, Broussarie L. Expression of collagens type I and IV, osteonectin and transforming growth factor beta-1 (TGFβ1) in biliary atresia and paucity of intrahepatic bile ducts during infancy. J Hepatol. 1999;31:248–255. doi: 10.1016/s0168-8278(99)80221-9. [DOI] [PubMed] [Google Scholar]
- 313.Desmet VJ, Callea F. Cholestatic syndromes of infancy and childhood. In: Zakim D, Boyer TD, editors. W B Saunders; Philadelphia: 1990. pp. 1355–1395. [Google Scholar]
- 314.Callea F, Facchetti F, Lucini L. Liver morphology in anicteric patients at long-term follow-up after Kasai operation: a study of 16 cases. In: Ohi R, editor. Professional Postgraduate Services; Tokyo: 1987. [Google Scholar]
- 315.Nietgen GW, Vacanti JP, Perez-Atayde AR. Intrahepatic bile duct loss in biliary atresia despite portoenterostomy: a consequence of ongoing obstruction? Gastroenterology. 1992;102:2126–2133. doi: 10.1016/0016-5085(92)90342-v. [DOI] [PubMed] [Google Scholar]
- 316.Ito T, Horisawa M, Ando H. Intrahepatic bile ducts in biliary atresia – a possible factor determining prognosis. J Pediatr Surg. 1983;18:124–130. doi: 10.1016/s0022-3468(83)80532-6. [DOI] [PubMed] [Google Scholar]
- 317.Brunati A, Feruzi Z, Sokal E. Early occurrence of hepatocellular carcinoma in biliary atresia treated by liver transplantation. Pediatr Transplant. 2007;11:117–119. doi: 10.1111/j.1399-3046.2006.00623.x. [DOI] [PubMed] [Google Scholar]
- 318.Hol L, van den Bos IC, Hussain SM. Hepatocellular carcinoma complicating biliary atresia after Kasai portoenterostomy. Eur J Gastroenterol Hepatol. 2008;20:227–231. doi: 10.1097/MEG.0b013e3282cfb716. [DOI] [PubMed] [Google Scholar]
- 319.Kulkarni PB, Beatty E., Jr Cholangiocarcinoma associated with biliary cirrhosis due to congenital biliary atresia. Am J Dis Child. 1977;131:442–444. doi: 10.1001/archpedi.1977.02120170068013. [DOI] [PubMed] [Google Scholar]
- 320.Ohtomo K, Itai Y, Hasizume K. CT and MR appearance of focal nodular hyperplasia of the liver in children with biliary atresia. Clin Radiol. 1991;43:88–90. doi: 10.1016/s0009-9260(05)81584-3. [DOI] [PubMed] [Google Scholar]
- 321.Okugawa Y, Uchida K, Inoue M. Focal nodular hyperplasia in biliary atresia patient after Kasai hepatic portoenterostomy. Pediatr Surg Int. 2008;24:609–612. doi: 10.1007/s00383-007-2090-8. [DOI] [PubMed] [Google Scholar]
- 322.Liang JL, Cheng YF, Concejero AM. Macro-regenerative nodules in biliary atresia: CT/MRI findings and their pathological relations. World J Gastroenterol. 2008;14:4529–4534. doi: 10.3748/wjg.14.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Campbell LV, Jr, Gilbert EF. Experimental giant cell transformation in the liver induced by E. coli endotoxin. Am J Pathol. 1967;51:855–864. [PMC free article] [PubMed] [Google Scholar]
- 324.Dick MC, Mowat AP. Hepatitis syndrome in infancy – an epidemiological survey with 10 year follow-up. Arch Dis Child. 1985;60:512–516. doi: 10.1136/adc.60.6.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Hicks J, Barrish J, Zhu SH. Neonatal syncytial giant cell hepatitis with paramyxoviral-like inclusions. Ultrastruct Pathol. 2001;25:65–71. doi: 10.1080/019131201300004708. [DOI] [PubMed] [Google Scholar]
- 326.Chang MH, Hsu HC, Lee CY. Neonatal hepatitis: a follow-up study. J Pediatr Gastroenterol Nutr. 1987;6:203–207. doi: 10.1097/00005176-198703000-00006. [DOI] [PubMed] [Google Scholar]
- 327.Herman SP, Baggenstoss AH, Cloutier MD. Liver dysfunction and histologic abnormalities in neonatal hypopituitarism. J Pediatr. 1975;87:892–895. doi: 10.1016/s0022-3476(75)80900-0. [DOI] [PubMed] [Google Scholar]
- 328.Sheehan AG, Martin SR, Stephure D, Scott RB. Neonatal cholestasis, hypoglycemia and congenital hypopituitarism. J Pediatr Gastroenterol Nutr. 1992;14:426–430. doi: 10.1097/00005176-199205000-00009. [DOI] [PubMed] [Google Scholar]
- 329.Bernard O, Hadchouel M, Scotto J. Severe giant cell hepatitis with autoimmune hemolytic anemia in early childhood. J Pediatr. 1981;99:704–711. doi: 10.1016/s0022-3476(81)80388-5. [DOI] [PubMed] [Google Scholar]
- 330.Brichard B, Sokal E, Gosseye S. Coombs-positive giant cell hepatitis of infancy: effect of steroids and azathioprine therapy. Eur J Pediatr. 1991;150:314–317. doi: 10.1007/BF01955929. [DOI] [PubMed] [Google Scholar]
- 331.Pappo O, Yunis E, Jordan JA. Recurrent and de novo giant cell hepatitis after orthotopic liver transplantation. Am J Surg Pathol. 1994;18:804–813. doi: 10.1097/00000478-199408000-00007. [DOI] [PubMed] [Google Scholar]
- 332.Vilca Menedez H, Rela M, Baker A. Liver transplant for giant cell hepatitis with autoimmune haemolytic anaemia. Arch Dis Child. 1997;7:249–251. doi: 10.1136/adc.77.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Vajro P, Amelio A, Stagni A. Cholestasis in newborn infants with perinatal asphyxia. Acta Paediatr. 1997;86:895–898. doi: 10.1111/j.1651-2227.1997.tb08619.x. [DOI] [PubMed] [Google Scholar]
- 334.Jacquemin E, Lykavieris P, Chaoui N. Transient neonatal cholestasis: origin and outcome. J Pediatr. 1998;133:563–567. doi: 10.1016/s0022-3476(98)70070-8. [DOI] [PubMed] [Google Scholar]
- 335.Suita S, Arima T, Ishii K. Fate of infants with neonatal hepatitis: pediatric surgeons’ dilemma. J Pediatr Surg. 1992;27:696–699. doi: 10.1016/s0022-3468(05)80093-4. [DOI] [PubMed] [Google Scholar]
- 336.Aagenaes O. Hereditary cholestasis with lymphoedema (Aagenaes syndrome, cholestasis-lymphoedema syndrome). New cases and follow-up from infancy to adult age. Scand J Gastroenterol. 1998;33:335–345. doi: 10.1080/00365529850170955. [DOI] [PubMed] [Google Scholar]
- 337.Fruhwirth M, Janecke AR, Muller T. Evidence for genetic heterogeneity in lymphedema-cholestasis syndrome. J Pediatr. 2003;142:441–447. doi: 10.1067/mpd.2003.148. [DOI] [PubMed] [Google Scholar]
- 338.Bollinger ME, Arredondo-Vega F, Santisteban I. Brief report: hepatic dysfunction as a complication of adenosine deaminase deficiency. N Engl J Med. 1996;334:1367–1371. doi: 10.1056/NEJM199605233342104. [DOI] [PubMed] [Google Scholar]
- 339.Tazawa Y, Kobayashi K, Ohura T. Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. J Pediatr. 2001;138:735–740. doi: 10.1067/mpd.2001.113264. [DOI] [PubMed] [Google Scholar]
- 340.Ben-Shalom E, Kobayashi K, Shaag A. Infantile citrullinemia caused by citrin deficiency with increased dibasic amino acids. Mol Genet Metab. 2002;77:202–208. doi: 10.1016/s1096-7192(02)00167-1. [DOI] [PubMed] [Google Scholar]
- 341.Tazawa Y, Kobayashi K, Abukawa D. Clinical heterogeneity of neonatal intrahepatic cholestasis caused by citrin deficiency: case reports from 16 patients. Mol Genet Metab. 2004;83:213–219. doi: 10.1016/j.ymgme.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 342.Ohura T, Kobayashi K, Tazawa Y. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) J Inherit Metab Dis. 2007;30:139–144. doi: 10.1007/s10545-007-0506-1. [DOI] [PubMed] [Google Scholar]
- 343.Gilmour SM, Hughes-Benzie R, Silver MM, Roberts EA. Le foie vide: a unique case of neonatal liver failure. J Pediatr Gastroenterol Nutr. 1996;23:618–623. doi: 10.1097/00005176-199612000-00019. [DOI] [PubMed] [Google Scholar]
- 344.Montgomery CK, Ruebner BH. Neonatal hepatocellular giant cell transformation: a review. Perspect Pediatr Pathol. 1976;3:85–101. [PubMed] [Google Scholar]
- 345.Ruebner B, Thaler MM. Giant cell transformation in infantile liver disease. In: Javitt NB, editor. Neonatal hepatitis and biliary atresia. US Government Printing Press; Washington DC: 1979. pp. 299–311. [Google Scholar]
- 346.Koukoulis G, Mieli-Vergani G, Portmann B. Infantile liver giant cells: immunohistological study of their proliferative state and possible mechanisms of formation. Pediatr Dev Pathol. 1999;2:353–359. doi: 10.1007/s100249900134. [DOI] [PubMed] [Google Scholar]
- 347.Oledzka-Slotwinska H, Desmet V. Morphologic study on neonatal liver ‘giant’ cell transformation. Exp Mol Biol. 1969;10:162–175. doi: 10.1016/0014-4800(69)90037-9. [DOI] [PubMed] [Google Scholar]
- 348.Craig JM, Landing BH. Form of hepatitis in neonatal period simulating biliary atresia. Arch Pathol. 1952;54:321–323. [PubMed] [Google Scholar]
- 349.Richardet JP, Mallat A, Zafrani ES. Prolonged cholestasis with ductopenia after administration of amoxicillin/clavulanic acid. Dig Dis Sci. 1999;44:1997–2000. doi: 10.1023/a:1026610015899. [DOI] [PubMed] [Google Scholar]
- 350.Gardner LJ. Intrahepatic bile stasis in 45/X Turner's syndrome. N Engl J Med. 1974;290:406. doi: 10.1056/nejm197402142900723. [DOI] [PubMed] [Google Scholar]
- 351.Puri P, Guiney EJ. Intrahepatic biliary atresia in Down's syndrome. J Pediatr Surg. 1975;10:423–424. doi: 10.1016/0022-3468(75)90107-4. [DOI] [PubMed] [Google Scholar]
- 352.Aanpreung P, Beckwith B, Galansky SH. Association of paucity of interlobular bile ducts with prune belly syndrome. J Pediatr Gastroenterol Nutr. 1993;16:81–86. doi: 10.1097/00005176-199301000-00017. [DOI] [PubMed] [Google Scholar]
- 353.Watson GH, Miller V. Arteriohepatic dysplasia. Familial pulmonary arterial stenosis with neonatal liver disease. Arch Dis Child. 1973;48:459–466. doi: 10.1136/adc.48.6.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, et al. J Pediatr. 1975;86:63–71. doi: 10.1016/s0022-3476(75)80706-2. [DOI] [PubMed] [Google Scholar]
- 355.Alagille D, Estrada A, Hadchouel M. Syndromatic paucity of interlobular bile ducts Alagille syndrome or arteriohepatic dysplasia: review of 80 cases. J Pediatr. 1987;110:195–200. doi: 10.1016/s0022-3476(87)80153-1. [DOI] [PubMed] [Google Scholar]
- 356.Deprettere A, Portmann B, Mowat AP. Syndromic paucity of the intrahepatic bile ducts: diagnostic difficulty; severe morbidity throughout early childhood. J Pediatr Gastroenterol Nutr. 1987;6:865–871. doi: 10.1097/00005176-198711000-00008. [DOI] [PubMed] [Google Scholar]
- 357.Emerick KM, Rand EB, Goldmuntz E. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–829. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- 358.Kamath BM, Loomes KM, Piccoli DA. Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50:580–586. doi: 10.1097/MPG.0b013e3181d98ea8. [DOI] [PubMed] [Google Scholar]
- 359.Oda T, Elkahloun AG, Pike BL. Mutations in the human Jagged 1 gene are responsible for the Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 360.Boyer J, Crosnier C, Driancourt C. Expression of mutant JAGGED1 alleles in patients with Alagille syndrome. Hum Genet. 2005;116:445–453. doi: 10.1007/s00439-005-1262-7. [DOI] [PubMed] [Google Scholar]
- 361.LaBrecque DR, Mitros FA, Nathan RJ. Four generations of arteriohepatic dysplasia. Hepatology. 1982;2:467–474. doi: 10.1002/hep.1840020413. [DOI] [PubMed] [Google Scholar]
- 362.Crosnier C, Driancourt C, Raynaud N. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology. 1999;116:1141–1148. doi: 10.1016/s0016-5085(99)70017-x. [DOI] [PubMed] [Google Scholar]
- 363.Spinner NB, Colliton RP, Crosnier C. Jagged1 mutations in Alagille syndrome. Hum Mutat. 2001;17:18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 364.Sokol RJ, Heubi JE, Ballistreri WF. Intrahepatic ‘cholestasis facies’: is it specific for Alagille syndrome? J Pediatr. 1983;103:205–208. doi: 10.1016/s0022-3476(83)80345-x. [DOI] [PubMed] [Google Scholar]
- 365.Kamath BM, Loomes KM, Oakey RJ. Facial features in Alagille syndrome: specific or cholestasis facies? Am J Med Genet. 2002;112:163–170. doi: 10.1002/ajmg.10579. [DOI] [PubMed] [Google Scholar]
- 366.Silberbach M, Lashley D, Reller MD. Arteriohepatic dysplasia and cardiovascular malformations. Am Heart J. 1991;127:695–699. doi: 10.1016/0002-8703(94)90684-x. [DOI] [PubMed] [Google Scholar]
- 367.McElhinney DB, Krantz ID, Bason L. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567–2574. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- 368.Hingorani M, Nischal KK, Davies A. Ocular abnormalities in Alagille syndrome. Ophthalmology. 1999;106:330–337. doi: 10.1016/S0161-6420(99)90072-6. [DOI] [PubMed] [Google Scholar]
- 369.Nischal KK, Hingorani M, Bentley CR. Ocular ultrasound in Alagille syndrome. A new sign. Ophthalmology. 1997;104:79–85. doi: 10.1016/s0161-6420(97)30358-3. [DOI] [PubMed] [Google Scholar]
- 370.Rosenfield NS, Kelley MJ, Jensen PS. Arteriohepatic dysplasia: radiologic features of a new syndrome. Am J Roentgenol. 1980;135:1217–1223. doi: 10.2214/ajr.135.6.1217. [DOI] [PubMed] [Google Scholar]
- 371.Levin SE, Zarvos P, Milner S, Schmaman A. Arteriohepatic dysplasia: association of liver disease with pulmonary arterial stenosis as well as facial and skeletal abnormalities. Pediatrics. 1980;66:876–883. [PubMed] [Google Scholar]
- 372.Sanderson E, Newman V, Haigh SF. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002;32:114–119. doi: 10.1007/s00247-001-0599-x. [DOI] [PubMed] [Google Scholar]
- 373.Harendza S, Hubner CA, Glaser C. Renal failure and hypertension in Alagille syndrome with a novel JAG1 mutation. J Nephrol. 2005;18:312–317. [PubMed] [Google Scholar]
- 374.Shrivastava R, Williams A, Mikhail A. An unusual cause of hypertension and renal failure: a case series of a family with Alagille syndrome. Nephrol Dial Transplant. 2010;25:1501–1506. doi: 10.1093/ndt/gfp692. [DOI] [PubMed] [Google Scholar]
- 375.Davis J, Griffiths R, Larkin K. Glomerular basement membrane lipidosis in Alagille syndrome. Pediatr Nephrol. 2010;25:1181–1184. doi: 10.1007/s00467-009-1426-0. [DOI] [PubMed] [Google Scholar]
- 376.Russo PA, Ellis E, Hashida Y. Renal histopathology in Algille's syndrome. Pediatr Pathol. 1987;7:557–568. doi: 10.3109/15513818709161419. [DOI] [PubMed] [Google Scholar]
- 377.Martin SR, Garel L, Alvarez F. Alagille's syndrome associated with cystic renal disease. Arch Dis Child. 1996;74:232–235. doi: 10.1136/adc.74.3.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Bourdeaut F, Guiochon-Mantel A, Fabre M. Alagille syndrome and nephroblastoma: unusual coincidence of two rare disorders. Pediatr Blood Cancer. 2008;50:908–911. doi: 10.1002/pbc.21255. [DOI] [PubMed] [Google Scholar]
- 379.Woolfenden AR, Albers GW, Steinberg GK. Moyamoya syndrome in children with Alagille syndrome: additional evidence of a vasculopathy. Pediatrics. 1999;103:505–508. doi: 10.1542/peds.103.2.505. [DOI] [PubMed] [Google Scholar]
- 380.Connor SE, Hewes D, Ball C, Jarosz JM. Alagille syndrome associated with angiographic moyamoya. Childs Nerv Syst. 2002;18:186–190. doi: 10.1007/s00381-001-0518-3. [DOI] [PubMed] [Google Scholar]
- 381.Kamath BM, Spinner NB, Emerick KM. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109:1354–1358. doi: 10.1161/01.CIR.0000121361.01862.A4. [DOI] [PubMed] [Google Scholar]
- 382.Berard E, Triolo V. Intracranial hemorrhages in Alagille syndrome. J Pediatr. 2000;136:708–710. [PubMed] [Google Scholar]
- 383.Lykavieris P, Crosnier C, Trichet C. Bleeding tendency in children with Alagille syndrome. Pediatrics. 2003;111:167–170. doi: 10.1542/peds.111.1.167. [DOI] [PubMed] [Google Scholar]
- 384.Emerick KM, Krantz ID, Kamath BM. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;41:99–107. doi: 10.1097/01.mpg.0000162776.67758.2f. [DOI] [PubMed] [Google Scholar]
- 385.Kayhan A, Ilkhchoui Y, Venu N. Multiple abdominal vascular anomalies in a patient with Alagille syndrome. J Vasc Interv Radiol. 2010;21:937–940. doi: 10.1016/j.jvir.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 386.Garcia MA, Ramonet M, Ciocca M. Alagille syndrome: cutaneous manifestations in 38 children. Pediatr Dermatol. 2005;22:11–14. doi: 10.1111/j.1525-1470.2005.22102.x. [DOI] [PubMed] [Google Scholar]
- 387.Gorelick FS, Dobbins JW, Burrell M, Riely CA. Biliary tract abnormalities in patients with arteriohepatic dysplasia. Dig Dis Sci. 1982;27:815–820. doi: 10.1007/BF01391375. [DOI] [PubMed] [Google Scholar]
- 388.Markowitz J, Daum F, Kahn FI. Arteriohepatic dysplasia. I. Pitfalls in diagnosis and management. Hepatology. 1983;3:74–76. doi: 10.1002/hep.1840030112. [DOI] [PubMed] [Google Scholar]
- 389.Kahn EI, Daum F, Markowitz J. Arteriohepatic dysplasia. II. Hepatobiliary morphology. Hepatology. 1983;3:77–84. doi: 10.1002/hep.1840030113. [DOI] [PubMed] [Google Scholar]
- 390.Morelli A, Pelli MA, Vedovelli A. Endoscopic retrograde cholangiopancreatography study in Alagille's syndrome: first report. Am J Gastroenterol. 1983;78:241–244. [PubMed] [Google Scholar]
- 391.Schwarzenberg SJ, Grothe RM, Sharp HL. Long-term complications of arteriohepatic dysplasia. Am J Med. 1992;93:171–176. doi: 10.1016/0002-9343(92)90047-f. [DOI] [PubMed] [Google Scholar]
- 392.Cruz M, Corretger JM, Arquer A. [Intrahepatic portal venous hypoplasia in Alagille's syndrome] Arch Fr Pediatr. 1985;42:107–109. [PubMed] [Google Scholar]
- 393.Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001;49:431–435. doi: 10.1136/gut.49.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Arvay JL, Zemel BS, Gallagher PR. Body composition of children aged 1 to 12 years with biliary atresia or Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;40:146–150. doi: 10.1097/00005176-200502000-00012. [DOI] [PubMed] [Google Scholar]
- 395.Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35:1501–1506. doi: 10.1053/jhep.2002.33332. [DOI] [PubMed] [Google Scholar]
- 396.Tzakis AG, Reyes J, Tepetes K. Liver transplantation for Alagille's syndrome. Arch Surg. 1993;128:337–339. doi: 10.1001/archsurg.1993.01420150093017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Kaye AJ, Rand EB, Munoz PS. Effect of Kasai procedure on hepatic outcome in Alagille Syndrome. J Pediatr Gastroenterol Nutr. 2010;51:319–321. doi: 10.1097/MPG.0b013e3181df5fd8. [DOI] [PubMed] [Google Scholar]
- 398.Kamath BM, Munoz PS, Bab N. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50:526–530. doi: 10.1097/MPG.0b013e3181cea48d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Kamath BM, Schwarz KB, Hadzic N. Alagille syndrome and liver transplantation. J Pediatr Gastroenterol Nutr. 2010;50:11–15. doi: 10.1097/MPG.0b013e3181c1601f. [DOI] [PubMed] [Google Scholar]
- 400.Cardona J, Houssin D, Gauthier F. Liver transplantation in children with Alagille syndrome – a study of twelve cases. Transplantation. 1995;60:339–342. doi: 10.1097/00007890-199508270-00007. [DOI] [PubMed] [Google Scholar]
- 401.Quiros-Tejeira RE, Ament ME, Heyman MB. Does liver transplantation affect growth pattern in Alagille syndrome? Liver Transpl. 2000;6:582–587. doi: 10.1053/jlts.2000.9739. [DOI] [PubMed] [Google Scholar]
- 402.Adams PC. Hepatocellular carcinoma associated with arteriohepatic dysplasia. Dig Dis Sci. 1986;31:438–442. doi: 10.1007/BF01311683. [DOI] [PubMed] [Google Scholar]
- 403.Rabinovitz M, Imperial JC, Schade RR, Van Thiel DH. Hepatocellular carcinoma in Alagille's syndrome: a family study. J Pediatr Gastroenterol Nutr. 1989;8:26–30. doi: 10.1097/00005176-198901000-00006. [DOI] [PubMed] [Google Scholar]
- 404.Bhadri VA, Stormon MO, Arbuckle S. Hepatocellular carcinoma in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;41:676–678. doi: 10.1097/01.mpg.0000179759.60048.c4. [DOI] [PubMed] [Google Scholar]
- 405.Kim B, Park SH, Yang HR. Hepatocellular carcinoma occurring in Alagille syndrome. Pathol Res Pract. 2005;201:55–60. doi: 10.1016/j.prp.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 406.Tsai S, Gurakar A, Anders R. Management of large hepatocellular carcinoma in adult patients with Alagille syndrome: a case report and review of literature. Dig Dis Sci. 2010 doi: 10.1007/s10620-009-1123-7. [DOI] [PubMed] [Google Scholar]
- 407.Hadchouel M, Hugon RN, Gautier M. Reduced ratio of portal tracts to paucity of intrahepatic bile ducts. Arch Pathol Lab Med. 1978;102:402–403. [PubMed] [Google Scholar]
- 408.Perrault J. Copper overload in paucity of interlobular bile ducts syndrome. Gastroenterology. 1980;75:875–878. [Google Scholar]
- 409.Witzleben CL. Bile duct paucity (‘intrahepatic atresia’) Perspect Pediatr Pathol. 1982;7:185–201. [PubMed] [Google Scholar]
- 410.Valencia-Mayoral P, Weber J, Cutz E. Possible defect in the bile secretory apparatus in arteriohepatic dysplasia (Alagille's syndrome): a review with observations on the ultrastructure of liver. Hepatology. 1984;4:691–698. doi: 10.1002/hep.1840040422. [DOI] [PubMed] [Google Scholar]
- 411.Hashida Y, Yunis EJ. Syndromatic paucity of inter-lobular bile ducts: hepatic histopathology of the early and endstage liver. Pediatr Pathol. 1988;8:1–15. doi: 10.3109/15513818809022275. [DOI] [PubMed] [Google Scholar]
- 412.Berman MD, Ishak KG, Schaefer EJ. Syndromatic hepatic ductular hypoplasia (arteriohepatic dysplasia): a clinical and hepatic histologic study of three patients. Dig Dis Sci. 1981;26:485–497. doi: 10.1007/BF01308096. [DOI] [PubMed] [Google Scholar]
- 413.Dahms B, Petrelli M, Wyllie R. Arteriohepatic dysplasia in infancy and childhood: a longitudinal study of six patients. Hepatology. 1982;2:350–358. doi: 10.1002/hep.1840020311. [DOI] [PubMed] [Google Scholar]
- 414.Kahn E. Paucity of interlobular bile ducts. Arteriohepatic dysplasia and nonsyndromic duct paucity. Perspect Pediatr Pathol. 1991;14:168–215. [PubMed] [Google Scholar]
- 415.Byrne JA, Meara NJ, Rayner AC. Lack of hepatocellular CD10 along bile canaliculi is physiologic in early childhood and persistent in Alagille syndrome. Lab Invest. 2007;87:1138–1148. doi: 10.1038/labinvest.3700677. [DOI] [PubMed] [Google Scholar]
- 416.Deutsch GH, Sokol RJ, Stathos TH, Knisely AS. Proliferation to paucity: evolution of bile duct abnormalities in a case of Alagille syndrome. Pediatr Dev Pathol. 2001;4:559–563. doi: 10.1007/s10024001-0102-6. [DOI] [PubMed] [Google Scholar]
- 417.Witzleben CL, Finegold M, Piccoli DA, Treem WR. Bile canalicular morphometry in arteriohepatic dysplasia. Hepatology. 1987;7:1262–1266. doi: 10.1002/hep.1840070614. [DOI] [PubMed] [Google Scholar]
- 418.Libbrecht L, Spinner NB, Moore EC. Peripheral bile duct paucity and cholestasis in the liver of a patient with Alagille syndrome: further evidence supporting a lack of postnatal bile duct branching and elongation. Am J Surg Pathol. 2005;29:820–826. doi: 10.1097/01.pas.0000161325.36348.25. [DOI] [PubMed] [Google Scholar]
- 419.Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- 420.Crosnier C, Attie-Bitach T, Encha-Razavi F. JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology. 2000;32:574–581. doi: 10.1053/jhep.2000.16600. [DOI] [PubMed] [Google Scholar]
- 421.Lozier J, McCright B, Gridley T. Notch signaling regulates bile duct morphogenesis in mice. PLoS One. 2008;3:e1851. doi: 10.1371/journal.pone.0001851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Flynn DM, Nijjar S, Hübscher SG. The role of Notch receptor expression in bile duct development and disease. J Pathol. 2004;204:55–64. doi: 10.1002/path.1615. [DOI] [PubMed] [Google Scholar]
- 423.Kodama Y, Hijikata M, Kageyama R. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology. 2004;127:1775–1786. doi: 10.1053/j.gastro.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 424.Lorent K, Yeo SY, Oda T. Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development. 2004;131:5753–5766. doi: 10.1242/dev.01411. [DOI] [PubMed] [Google Scholar]
- 425.Crosnier C, Driancourt C, Raynaud N. Fifteen novel mutations in the JAGGED1 gene of patients with Alagille syndrome. Hum Mutat. 2001;17:72–73. doi: 10.1002/1098-1004(2001)17:1<72::AID-HUMU11>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 426.Yuan ZR, Okaniwa M, Nagata I. The DSL domain in mutant JAG1 ligand is essential for the severity of the liver defect in Alagille syndrome. Clin Genet. 2001;59:330–337. doi: 10.1034/j.1399-0004.2001.590506.x. [DOI] [PubMed] [Google Scholar]
- 427.Colliton RP, Bason L, Lu FM. Mutation analysis of Jagged1 (JAG1) in Alagille syndrome patients. Hum Mutat. 2001;17:151–152. doi: 10.1002/1098-1004(200102)17:2<151::AID-HUMU8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 428.McDaniell R, Warthen DM, Sanchez-Lara PA. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Tchorz JS, Kinter J, Muller M. Notch2 signaling promotes biliary epithelial cell fate specification and tubulogenesis during bile duct development in mice. Hepatology. 2009;50:871–879. doi: 10.1002/hep.23048. [DOI] [PubMed] [Google Scholar]
- 430.Sparks EE, Huppert KA, Brown MA. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology. 2010;51:1391–1400. doi: 10.1002/hep.23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Kocak N, Gurukun F, Yuce A. Non-syndrome paucity of the interlobular bile ducts: clinical and laboratory findings in 10 cases. J Pediatr Gastroenterol Nutr. 1997;24:44–48. doi: 10.1097/00005176-199701000-00011. [DOI] [PubMed] [Google Scholar]
- 432.Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia: a cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol. 1988;7:193–199. doi: 10.1016/s0168-8278(88)80482-3. [DOI] [PubMed] [Google Scholar]
- 433.Zafrani ES, Metreau JM, Douvin C. Idiopathic biliary ductopenia in adults: a report of five cases. Gastroenterology. 1990;99:1823–1828. doi: 10.1016/0016-5085(90)90494-l. [DOI] [PubMed] [Google Scholar]
- 434.Bruguera M, Llach J, Rodes J. Nonsyndromic paucity of intrahepatic bile ducts in infancy and idiopathic ductopenia in adulthood: the same syndrome? Hepatology. 1992;15:830–834. doi: 10.1002/hep.1840150514. [DOI] [PubMed] [Google Scholar]
- 435.Muller C, Ulrich W, Penner E. Manifestation late in life of idiopathic adulthood ductopenia. Liver. 1995;15:213–218. doi: 10.1111/j.1600-0676.1995.tb00673.x. [DOI] [PubMed] [Google Scholar]
- 436.Bach N, Kahn H, Thung S. Hepatocellular carcinoma in a long-term survivor of intrahepatic biliary hypoplasia. Am J Gastroenterol. 1991;86:1527–1530. [PubMed] [Google Scholar]
- 437.Kahn E, Daum F, Markowitz J. Nonsyndromatic paucity of interlobular bile ducts: light and electron microscopic evaluation of sequential liver biopsies in early childhood. Hepatology. 1986;6:890–901. doi: 10.1002/hep.1840060514. [DOI] [PubMed] [Google Scholar]
- 438.West AB, Chatila R. Differential diagnosis of bile duct injury and ductopenia. Semin Diagn Pathol. 1998;15:270–284. [PubMed] [Google Scholar]
- 439.Hadad AR, Westbrook KC, Campbell GS. Congenital dilatation of the bile ducts. Am J Surg. 1976;132:799–804. doi: 10.1016/0002-9610(76)90462-1. [DOI] [PubMed] [Google Scholar]
- 440.Soreide K, Korner H, Havnen J, Soreide JA. Bile duct cysts in adults. Br J Surg. 2004;91:1538–1548. doi: 10.1002/bjs.4815. [DOI] [PubMed] [Google Scholar]
- 441.Visser BC, Suh I, Way LW, Kang SM. Congenital choledochal cysts in adults. Arch Surg. 2004;139:855–860. doi: 10.1001/archsurg.139.8.855. [DOI] [PubMed] [Google Scholar]
- 442.Sherman P, Kolster E, Davies C. Choledochal cysts: heterogeneity of clinical presentation. J Pediatr Gastroenterol Nutr. 1986;5:867–872. [PubMed] [Google Scholar]
- 443.Metcalfe MS, Wemyss-Holden SA, Maddern GJ. Management dilemmas with choledochal cysts. Arch Surg. 2003;138:333–339. doi: 10.1001/archsurg.138.3.333. [DOI] [PubMed] [Google Scholar]
- 444.Shukri N, Hasegawa T, Wasa M. Characteristics of infantile cases of congenital dilatation of the bile duct. J Pediatr Surg. 1998;33:1794–1797. doi: 10.1016/s0022-3468(98)90287-1. [DOI] [PubMed] [Google Scholar]
- 445.Sela-Herman S, Scharschmidt BF. Choledochal cyst: a disease for all ages. Lancet. 1996;347:779. doi: 10.1016/s0140-6736(96)90864-8. [DOI] [PubMed] [Google Scholar]
- 446.Bancroft JD, Bucuvalas JC, Ryckman FC. Antenatal diagnosis of choledochal cyst. J Pediatr Gastroenterol Nutr. 1994;18:142–145. doi: 10.1097/00005176-199402000-00004. [DOI] [PubMed] [Google Scholar]
- 447.Hamada V, Tanano A, Sato K. Rapid enlargement of a choledochal cyst: antenatal diagnosis and delayed primary excision. Pediatr Surg Int. 1998;5–6:419–421. doi: 10.1007/s003830050354. [DOI] [PubMed] [Google Scholar]
- 448.Lipsett PA, Pitt HA, Colombani PM. Choledochal cyst disease. A changing pattern of presentation. Ann Surg. 1994;220:644–652. doi: 10.1097/00000658-199411000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Chaudhary A, Dhar P, Sachdev A. Choledochal cysts – differences in children and adults. Br J Surg. 1996;83:186–188. [PubMed] [Google Scholar]
- 450.Tsai MS, Lin WH, Hsu WM. Clinicopathological feature and surgical outcome of choledochal cyst in different age groups: the implication of surgical timing. J Gastrointest Surg. 2008;12:2191–2195. doi: 10.1007/s11605-008-0593-9. [DOI] [PubMed] [Google Scholar]
- 451.Takaya J, Muneyuki M, Tokuhara D. Congenital dilatation of the bile duct: changes in diagnostic tools over the past 19 years. Pediatr Int. 2003;45:383–387. doi: 10.1046/j.1442-200x.2003.01736.x. [DOI] [PubMed] [Google Scholar]
- 452.Matos C, Nicaise N, Deviere J. Choledochal cysts: comparison of findings at MR cholangiopancreatography and endoscopic retrograde cholangiopancreatography in eight patients. Radiology. 1998;209:443–448. doi: 10.1148/radiology.209.2.9807571. [DOI] [PubMed] [Google Scholar]
- 453.Robertson JFR, Raine PAM. Choledochal cyst: a 33-year review. Br J Surg. 1988;75:799–801. doi: 10.1002/bjs.1800750826. [DOI] [PubMed] [Google Scholar]
- 454.Swisher SG, Cates JA, Hunt KK. Pancreatitis associated with adult choledochal cysts. Pancreas. 1994;9:633–637. [PubMed] [Google Scholar]
- 455.Tomomasa T, Tabata K, Myashite M. Acute pancreatitis in Japanese and Western children: etiologic comparisons. J Pediatr Gastroenterol Nutr. 1994;19:109–110. doi: 10.1097/00005176-199407000-00018. [DOI] [PubMed] [Google Scholar]
- 456.Karnak I, Tanyei FC, Buyukpamukcu N, Hicsonmez A. Spontaneous rupture of choledochal cyst: an unusual cause of acute abdomen in children. J Pediatr Surg. 1997;32:736–738. [PubMed] [Google Scholar]
- 457.Bismuth H, Kriscut J. Choledochal cystic malignancies. Ann Oncol. 1999;10(Suppl. 4):94–98. [PubMed] [Google Scholar]
- 458.Yeong ML, Nicholson GI, Lee SP. Regression of biliary cirrhosis following choledochal cyst drainage. Gastroenterology. 1982;82:332–335. [PubMed] [Google Scholar]
- 459.Krepel HP, Siersema PD, Tilanus HW. Choledochocele presenting with anaemia. Eur J Gastroenterol Hepatol. 1997;9:641–643. doi: 10.1097/00042737-199706000-00022. [DOI] [PubMed] [Google Scholar]
- 460.Barlow B, Tabor E, Blanc WA. Choledochal cyst: a review of 19 cases. J Pediatr. 1976;89:934–940. doi: 10.1016/s0022-3476(76)80599-9. [DOI] [PubMed] [Google Scholar]
- 461.Ando H, Takahiro I, Kaneko K. Congenital stenosis of the intrahepatic bile duct associated with choledochal cysts. J Am Coll Surg. 1995;181:426–430. [PubMed] [Google Scholar]
- 462.Okada A, Nakamura T, Okumura K. Surgical treatment of congenital dilatation of bile duct (choledochal cyst) with technical considerations. Surgery. 1987;101:239–443. [PubMed] [Google Scholar]
- 463.Young WT, Thomas GV, Blethyn AJ, Lawrie BW. Choledochal cyst and congenital anomalies of the pancreatico-biliary junction: the clinical findings, radiology and outcome in nine cases. Br J Radiol. 1992;65:33–38. doi: 10.1259/0007-1285-65-769-33. [DOI] [PubMed] [Google Scholar]
- 464.Kamisawa T, Takuma K, Anjiki H. Pancreaticobiliary maljunction. Clin Gastroenterol Hepatol. 2009;7:S84–S88. doi: 10.1016/j.cgh.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 465.Todani T, Watanabe Y, Toki A. Co-existing biliary anomalies and anatomical variants in choledochal cyst. Br J Surg. 1998;85:760–763. doi: 10.1046/j.1365-2168.1998.00697.x. [DOI] [PubMed] [Google Scholar]
- 466.Kim WS, Kim ID, Yeon KM. Choledochal cyst with or without biliary atresia in neonates and young infants: US differentiation. Radiology. 1998;209:465–469. doi: 10.1148/radiology.209.2.9807575. [DOI] [PubMed] [Google Scholar]
- 467.Yamashiro Y, Mijano T, Suruga K. Experimental study of the pathogenesis of choledochal cyst and pancreatitis, with special reference to the role of bile acids and pancreatic enzymes in the anomalous choledocho-pancreatico ductal junction. J Pediatr Gastroenterol Nutr. 1984;3:721–727. doi: 10.1097/00005176-198411000-00015. [DOI] [PubMed] [Google Scholar]
- 468.Behrns KE, Shaheen NJ, Grimm IS. Type 1 choledochal cyst in association with familial adenomatous polyposis. Am J Gastroenterol. 1998;93:1377–1379. doi: 10.1111/j.1572-0241.1998.425_f.x. [DOI] [PubMed] [Google Scholar]
- 469.Miyano T, Yamataka A, Koto Y. Hepaticoenterostomy after excision of choledochal cyst in children: a 30-year experience with 180 cases. J Pediatr Surg. 1996;31:1417–1421. doi: 10.1016/s0022-3468(96)90843-x. [DOI] [PubMed] [Google Scholar]
- 470.de Vries JS, de Vries S, Aronson DC. Choledochal cysts: age of presentation, symptoms, and late complications related to Todani's classification. J Pediatr Surg. 2002;37:1568–1573. doi: 10.1053/jpsu.2002.36186. [DOI] [PubMed] [Google Scholar]
- 471.Hong L, Wu Y, Yan Z. Laparoscopic surgery for choledochal cyst in children: a case review of 31 patients. Eur J Pediatr Surg. 2008;18:67–71. doi: 10.1055/s-2008-1038486. [DOI] [PubMed] [Google Scholar]
- 472.Thanh L, Hien P, Dung LA, Son TN. Laparoscopic repair for choledochal cyst: lessons learned from 190 cases. J Pediatr Surg. 2010;45:540–544. doi: 10.1016/j.jpedsurg.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 473.Goto N, Yasuda I, Uematsu T. Intrahepatic cholangiocarcinoma arising 10 years after the excision of congenital extrahepatic biliary dilation. J Gastroenterol. 2001;36:856–862. doi: 10.1007/s005350170010. [DOI] [PubMed] [Google Scholar]
- 474.Ishibashi T, Kasahara Y, Yasuda Y. Malignant change in the biliary tract after excision of choledochal cyst. Br J Surg. 1997;84:1687–1691. [PubMed] [Google Scholar]
- 475.Ando H, Ito T, Sugito T. Histological study of the choledochal cyst wall. Jpn J Gastroenterol. 1987;84:1797–1801. [PubMed] [Google Scholar]
- 476.Kozuka S, Kurashima M, Tsubone M. Significance of intestinal metaplasia for evolution of cancer in the biliary tract. Cancer. 1984;54:2277–2285. doi: 10.1002/1097-0142(19841115)54:10<2277::aid-cncr2820541037>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 477.Komi N, Tamura T, Tsuge S. Relation of patient age to premalignant changes in choledochal cyst epithelium: histochemical and immunohistochemical studies. J Pediatr Surg. 1986;21:430–433. doi: 10.1016/s0022-3468(86)80514-0. [DOI] [PubMed] [Google Scholar]
- 478.Kusunoki M, Yamamura T, Takahashi T. Choledochal cyst: its possible autonomic involvement in the bile duct. Arch Surg. 1987;122:997–1000. doi: 10.1001/archsurg.1987.01400210035004. [DOI] [PubMed] [Google Scholar]
- 479.Gallagher PJ, Mills RR, Mitchinson MJ. Congenital dilatation of the intrahepatic bile ducts with cholangiocarcinoma. J Clin Pathol. 1972;25:804–808. doi: 10.1136/jcp.25.9.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Kagawa Y, Kashihara S, Kuramoto S, Maetani S. Carcinoma arising in a congenitally dilated biliary tract. Report of a case and review of the literature. Gastroenterology. 1978;74:1286–1294. [PubMed] [Google Scholar]
- 481.Voyles CR, Smadja C, Shands C, Blumgart LH. Carcinoma in choledochal cysts: age-related incidence. Arch Surg. 1983;118:986–988. doi: 10.1001/archsurg.1983.01390080088022. [DOI] [PubMed] [Google Scholar]
- 482.Nonomura A, Mizukami Y, Matsubara F. A case of choledochal cyst associated with adenocarcinoma exhibiting sarcomatous features. J Gastroenterol. 1994;29:669–675. doi: 10.1007/BF02365455. [DOI] [PubMed] [Google Scholar]
- 483.Iwai N, Deguchi E, Yanagihara J. Cancer arising in a choledochal cyst in a 12-year-old girl. J Pediatr Surg. 1990;25:1261–1263. doi: 10.1016/0022-3468(90)90525-e. [DOI] [PubMed] [Google Scholar]
- 484.Tanaka S, Kubota M, Yagi M. An 11-year-old male patient demonstrating cholangiocarcinoma associated with congenital biliary dilatation. J Pediatr Surg. 2006;41:e15–e19. doi: 10.1016/j.jpedsurg.2005.10.066. [DOI] [PubMed] [Google Scholar]
- 485.Reveille RM, Van Stiegmann G, Everson GT. Increased secondary bile acids in a choledochal cyst. Possible role in biliary metaplasia and carcinoma. Gastroenterology. 1990;99:525–527. doi: 10.1016/0016-5085(90)91036-6. [DOI] [PubMed] [Google Scholar]
- 486.Wu GS, Zou SQ, Luo XW. Proliferative activity of bile from congenital choledochal cyst patients. World J Gastroenterol. 2003;9:184–187. doi: 10.3748/wjg.v9.i1.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Matsumoto Y, Fujii H, Itakura J. Recent advances in pancreaticobiliary maljunction. J Hepatobiliary Pancreat Surg. 2002;9:45–54. doi: 10.1007/s005340200004. [DOI] [PubMed] [Google Scholar]
- 488.Desmet VJ. What is congenital hepatic fibrosis? Hepatology. 1992;20:465–477. doi: 10.1111/j.1365-2559.1992.tb01031.x. [DOI] [PubMed] [Google Scholar]
- 489.Desmet VJ. Ludwig symposium on biliary disorders – part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 490.Knisely AS. Biliary tract malformations. Am J Med. 2003;122:343–350. doi: 10.1002/ajmg.a.20479. [DOI] [PubMed] [Google Scholar]
- 491.Desmet VJ, Roskams TAD. The cholangiopathies. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver Disease in Children. 3rd edn. Williams and Wilkins; Philadelphia: 2007. pp. 35–70. [Google Scholar]
- 492.Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:118–130. doi: 10.1053/j.ackd.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Ibanez-Tallon I, Heintz N, Omran H. To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet. 2003;12:R27–R35. doi: 10.1093/hmg/ddg061. [DOI] [PubMed] [Google Scholar]
- 494.Delmas P, Padilla F, Osorio N. Polycystins, calcium signaling, and human diseases. Biochem Biophys Res Commun. 2004;322:1374–1383. doi: 10.1016/j.bbrc.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 495.Mai W, Chen D, Ding T. Inhibition of Pkhd1 impairs tubulomorphogenesis of cultured IMCD cells. Mol Biol Cell. 2005;16:4398–4409. doi: 10.1091/mbc.E04-11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 497.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, et al. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 498.Wang S, Luo Y, Wilson PD. The autosomal recessive polycystic kidney disease protein is localized to primary cilia, with concentration in the basal body area. J Am Soc Nephrol. 2004;15:592–602. doi: 10.1097/01.asn.0000113793.12558.1d. [DOI] [PubMed] [Google Scholar]
- 499.Davila S, Furu L, Gharavi AG. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 500.Ong AC, Wheatley DN. Polycystic kidney disease – the ciliary connection. Lancet. 2003;361:774–776. doi: 10.1016/S0140-6736(03)12662-1. [DOI] [PubMed] [Google Scholar]
- 501.Sutters M, Germino GG. Autosomal dominant polycystic kidney disease: molecular genetics and pathophysiology. J Lab Clin Med. 2003;141:91–101. doi: 10.1067/mlc.2003.13. [DOI] [PubMed] [Google Scholar]
- 502.Masyuk TV, Huang BQ, Ward CJ. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 503.Masyuk TV, Huang BQ, Masyuk AI. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–1730. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Ong AC, Ward CJ, Butler RJ. Coordinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue. Am J Pathol. 1999;154:1721–1729. doi: 10.1016/S0002-9440(10)65428-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Watnick TJ, Torres VE, Gandolph MA. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell. 1998;2:247–251. doi: 10.1016/s1097-2765(00)80135-5. [DOI] [PubMed] [Google Scholar]
- 506.Nichols MT, Gidey E, Matzakos T. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 507.Sanzen T, Harada K, Yasoshima M. Polycystic kidney rat is a novel animal model of Caroli's disease associated with congenital hepatic fibrosis. Am J Pathol. 2001;158:1605–1612. doi: 10.1016/S0002-9440(10)64116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Hiesberger T, Shao X, Gourley E. Role of the hepatocyte nuclear factor-1beta (HNF-1beta) C-terminal domain in Pkhd1 (ARPKD) gene transcription and renal cystogenesis. J Biol Chem. 2005;280:10578–10586. doi: 10.1074/jbc.M414121200. [DOI] [PubMed] [Google Scholar]
- 509.Gresh L, Fischer E, Reimann A. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Guay-Woodford LM, Green WJ, Lindsey JR, Beier DR. Germline and somatic loss of function of the mouse cpk gene causes biliary ductal pathology that is genetically modulated. Hum Mol Genet. 2000;9:769–778. doi: 10.1093/hmg/9.5.769. [DOI] [PubMed] [Google Scholar]
- 511.Moser M, Matthiesen S, Kirfel J. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2005;41:1113–1121. doi: 10.1002/hep.20655. [DOI] [PubMed] [Google Scholar]
- 512.Nagasawa Y, Matthiesen S, Onuchic LF. Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J Am Soc Nephrol. 2002;13:2246–2258. doi: 10.1097/01.asn.0000030392.19694.9d. [DOI] [PubMed] [Google Scholar]
- 513.Yoder BK, Mulroy S, Eustace H. Molecular pathogenesis of autosomal dominant polycystic kidney disease. Expert Rev Mol Med. 2006;8:1–22. doi: 10.1017/S1462399406010362. [DOI] [PubMed] [Google Scholar]
- 514.McDonald RA, Avner EA. Inherited polycystic kidney disease in childhood. Semin Nephrol. 1991;11:632–642. [PubMed] [Google Scholar]
- 515.Zerres K, Mucher G, Bachner L. Mapping the gene for autosomal recessive polycystic kidney disease (ARPKD) to chromosome 6p21cm. Nat Genet. 1994;7:429–432. doi: 10.1038/ng0794-429. [DOI] [PubMed] [Google Scholar]
- 516.Mucher G, Wirth B, Zerres K. Refining the map and defining flanking markers of the gene for autosomal recessive polycystic kidney on chromosome 6p 21.2-p12. Am J Hum Genet. 1994;55:1281–1284. [PMC free article] [PubMed] [Google Scholar]
- 517.Guay-Woodford LK, Meucha G, Hopkins SD. The severe perinatal form-of autosomal recessive polycystic kidney disease. Am J Hum Genet. 1995;56:1101–1107. [PMC free article] [PubMed] [Google Scholar]
- 518.Park JH, Dixit MP, Onuchic LF. A 1-Mb BAC/PAC-based physical map of the autosomal recessive polycystic kidney disease gene (PKHD1) region on chromosome 6. Genomics. 1999;57:249–255. doi: 10.1006/geno.1999.5777. [DOI] [PubMed] [Google Scholar]
- 519.Ward CJ, Hogan MC, Rossetti S. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 520.Onuchic LF, Furu L, Nagasawa Y. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Blyth H, Ockenden BG. Polycystic disease of kidneys and liver presenting in childhood. J Med Genet. 1971;81:257–284. doi: 10.1136/jmg.8.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Witzleben CL. Cystic diseases of the liver. In: Zakim D, Boyer TD, editors. Hepatology A textbook of liver disease. WB Saunders; Philadelphia: 1990. pp. 1395–1411. [Google Scholar]
- 523.Lonergan GJ, Rice RR, Suarez ES. Autosomal recessive polycystic kidney disease: radiologic-pathologic correlation. Radiographics. 2000;20:837–855. doi: 10.1148/radiographics.20.3.g00ma20837. [DOI] [PubMed] [Google Scholar]
- 524.Landing BH, Walls TR, Claireaux AE. Morphometric analysis of liver lesions in cystic diseases of childhood. Hum Pathol. 1980;11:549–560. [PubMed] [Google Scholar]
- 525.Gang DL, Herrin JT. Infantile polycystic disease of the liver and kidneys. Clin Nephrol. 1986;25:28–36. [PubMed] [Google Scholar]
- 526.Adams CM, Danks DM, Campbell PE. Comments upon the classification of infantile polycystic diseases of the liver and kidney, based upon three dimensional reconstruction of the liver. J Med Genet. 1974;11:234–243. doi: 10.1136/jmg.11.3.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Jörgensen MJ. The ductal plate malformation. APMIS (Suppl) 1977;257:1–88. [PubMed] [Google Scholar]
- 528.Adeva M, El-Youssef M, Rossetti S. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD) Medicine (Baltimore) 2006;85:1–21. doi: 10.1097/01.md.0000200165.90373.9a. [DOI] [PubMed] [Google Scholar]
- 529.Nakanuma Y, Terada T, Ohta G. Caroli's disease in congenital hepatic fibrosis and infantile polycystic disease. Liver. 1982;2:346–354. doi: 10.1111/j.1600-0676.1982.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 530.Summerfield JA, Nagafuchi Y, Sherlock S. Hepatobiliary fibropolycystic diseases. A clinical and histological review of 51 patients. J Hepatol. 1986;2:141–156. doi: 10.1016/s0168-8278(86)80073-3. [DOI] [PubMed] [Google Scholar]
- 531.Jung G, Benz-Bohm G, Kugel H. MR cholangiography in children with autosomal recessive polycystic kidney disease. Pediatr Radiol. 1999;29:463–466. doi: 10.1007/s002470050618. [DOI] [PubMed] [Google Scholar]
- 532.Averback P. Congenital hepatic fibrosis: asymptomatic adults without renal anomaly. Arch Pathol Lab Med. 1977;101:260–261. [PubMed] [Google Scholar]
- 533.Fauvert R, Benhamou JP. Congenital hepatic fibrosis. In: Schaffner F, Sherlock S, Leevy CM, editors. The liver and its diseases. Intercontinental Medical Book; New York: 1974. pp. 283–288. [Google Scholar]
- 534.Deget F, Rudnik-Schoneborn S, Zerres K. Course of autosomal recessive polycystic kidney disease (ARPKD) in siblings: a clinical comparison of 20 sibships. Clin Genet. 1995;47:248–253. doi: 10.1111/j.1399-0004.1995.tb04305.x. [DOI] [PubMed] [Google Scholar]
- 535.Kerr DNS, Harrison CV, Sherlock S, Walker RM. Congenital hepatic fibrosis. Q J Med. 1960;30:91–117. [PubMed] [Google Scholar]
- 536.Sommerschild HC, Langmark F, Maurseth K. Congenital hepatic fibrosis: report of two new cases and review of the literature. Surgery. 1973;73:53–58. [PubMed] [Google Scholar]
- 537.Boichis H, Passwell J, David R, Miller H. Congenital hepatic fibrosis and nephronophthisis. Q J Med. 1973;42:221–233. [PubMed] [Google Scholar]
- 538.Cobben J, Breuning M, Schoots C. Congenital hepatic fibrosis in autosomal-dominant polycystic kidney disease. Kidney Int. 1990;38:880–885. doi: 10.1038/ki.1990.286. [DOI] [PubMed] [Google Scholar]
- 539.Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:302–306. doi: 10.1007/s004670050281. [DOI] [PubMed] [Google Scholar]
- 540.Alvarez F, Bernard O, Brunelle F. Congenital hepatic fibrosis in children. J Pediatr. 1981;99:370–375. doi: 10.1016/s0022-3476(81)80320-4. [DOI] [PubMed] [Google Scholar]
- 541.Scott J, Shousha S, Thomas HC, Sherlock S. Bile duct carcinoma: a late complication of congenital hepatic fibrosis. Case report and review of literature. Am J Gastroenterol. 1980;73:113–119. [PubMed] [Google Scholar]
- 542.Chen KTK. Adenocarcinoma of the liver: association with congenital hepatic fibrosis and Caroli's disease. Arch Pathol Lab Med. 1981;105:294–295. [PubMed] [Google Scholar]
- 543.De Vos BF, Cuvelier C. Congenital hepatic fibrosis. J Hepatol. 1988;6:222–228. doi: 10.1016/s0168-8278(88)80036-9. [DOI] [PubMed] [Google Scholar]
- 544.Christodoulou K, Tsingis M, Stavrou C. Chromosome 1 localization of a gene for autosomal dominant medullary cystic kidney disease. Hum Mol Genet. 1998;7:905–911. doi: 10.1093/hmg/7.5.905. [DOI] [PubMed] [Google Scholar]
- 545.Hart TC, Gorry MC, Hart PS. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39:882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Donaldson JC, Dise RS, Ritchie MD, Hanks SK. Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J Biol Chem. 2002;277:29028–29035. doi: 10.1074/jbc.M111697200. [DOI] [PubMed] [Google Scholar]
- 547.Sergi C, Adam S, Kahl P, Otto HF. Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr Dev Pathol. 2000;3:568–583. doi: 10.1007/s100240010104. [DOI] [PubMed] [Google Scholar]
- 548.Fiskerstrand T, Houge G, Sund S. Identification of a gene for renal-hepatic-pancreatic dysplasia by microarray-based homozygosity mapping. J Mol Diagn. 2010;12:125–131. doi: 10.2353/jmoldx.2010.090033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Torra R, Alos L, Ramos J, Estivill X. Renal-hepatic-pancreatic dysplasia: an autosomal recessive malformation. J Med Genet. 1996;33:409–412. doi: 10.1136/jmg.33.5.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Böhm N, Fukuda M, Staudt R, Helwig H. Chondroectodermal dysplasia (Ellis-van Creveld syndrome) with dysplasia of the renal medulla and bile ducts. Histopathology. 1978;2:267–281. doi: 10.1111/j.1365-2559.1978.tb01719.x. [DOI] [PubMed] [Google Scholar]
- 551.Zaffanello M, Diomedi-Camassei F, Melzi ML. Sensenbrenner syndrome: a new member of the hepatorenal fibrocystic family. Am J Med Genet A. 2006;140:2336–2340. doi: 10.1002/ajmg.a.31464. [DOI] [PubMed] [Google Scholar]
- 552.Labrune P, Fabre M, Trioche P. Jeune syndrome and liver disease: report of three cases treated with ursodeoxycholic acid. Am J Med Genet. 1999;87:324–328. doi: 10.1002/(sici)1096-8628(19991203)87:4<324::aid-ajmg8>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 553.Freeze HH. New diagnosis and treatment of congenital hepatic fibrosis. J Pediatr Gastroenterol Nutr. 1999;29:104–106. doi: 10.1097/00005176-199907000-00027. [DOI] [PubMed] [Google Scholar]
- 554.Lewis SM, Roberts EA, Marcon MA. Joubert syndrome with congenital hepatic fibrosis: an entity in the spectrum of oculo-encephalo-hepato-renal disorders. Am J Med Genet. 1994;52:419–426. doi: 10.1002/ajmg.1320520406. [DOI] [PubMed] [Google Scholar]
- 555.Brancati F, Iannicelli M, Travaglini L. MKS3/TMEM67 mutations are a major cause of COACH syndrome, a Joubert syndrome related disorder with liver involvement. Hum Mutat. 2009;30:E432–E442. doi: 10.1002/humu.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Doherty D, Parisi MA, Finn LS. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis) J Med Genet. 2010;47:8–21. doi: 10.1136/jmg.2009.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Caroli J. Diseases of the intrahepatic biliary tree. Clin Gastroenterol. 1973;2:147–161. [PubMed] [Google Scholar]
- 558.Ramond MJ, Huguet C, Danan G. Partial hepatectomy in the treatment of Caroli's disease. Report of a case and review of the literature. Dig Dis Sci. 1984;29:367–370. doi: 10.1007/BF01318526. [DOI] [PubMed] [Google Scholar]
- 559.Sandle GI, Lodge JPA. A 32-year-old man with recurrent cholangitis. Lancet. 1997;350:408. doi: 10.1016/s0140-6736(97)06172-2. [DOI] [PubMed] [Google Scholar]
- 560.Mercadier M, Chigot JP, Clot JP. Caroli's disease. World J Surg. 1984;8:22–29. doi: 10.1007/BF01658359. [DOI] [PubMed] [Google Scholar]
- 561.Nagusue N. Successful treatment of Caroli's disease by hepatic resection. Ann Surg. 1984;200:718–723. doi: 10.1097/00000658-198412000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Tandon RK, Grewal H, Amand AC, Vashisht S. Caroli's syndrome: a heterogeneous entity. Am J Gastroenterol. 1990;85:170–173. [PubMed] [Google Scholar]
- 563.Marchal GJ, Desmet VJ, Proesmans WC. Caroli's disease: high-frequency US and pathologic findings. Radiology. 1986;158:507–511. doi: 10.1148/radiology.158.2.3510448. [DOI] [PubMed] [Google Scholar]
- 564.Hopper KD. The role of computed tomography in the evaluation of Caroli's disease. Clin Imaging. 1989;13:68–73. doi: 10.1016/0899-7071(89)90129-0. [DOI] [PubMed] [Google Scholar]
- 565.Choi BI, Mo-Yeon K, Kim SH, Han MC. Caroli disease: central dot sign in CT. Radiology. 1990;174:161–163. doi: 10.1148/radiology.174.1.2294544. [DOI] [PubMed] [Google Scholar]
- 566.Asselah T, Ernst O, Sergent G. Caroli's disease: a magnetic resonance cholangiopancreatography diagnosis. Am J Gastroenterol. 1998;93:109–110. doi: 10.1111/j.1572-0241.1998.109_c.x. [DOI] [PubMed] [Google Scholar]
- 567.Chalasani N, Nguyen C, Gitlin N. Spontaneous rupture of a bile duct and its endoscopic management in a patient with Caroli's syndrome. Am J Gastroenterol. 1997;92:1062–1063. [PubMed] [Google Scholar]
- 568.Chevillotte G, Sastre B, Sahel J. Maladie de Caroli localisée et associée à un adénocarcinome papillaire mucosécrétant: interêt de la résection hépatique. Presse Med. 1984;13:1137–1139. [PubMed] [Google Scholar]
- 569.Taylor ACF, Plamer K. Caroli's disease. Eur J Gastroenterol Hepatol. 1998;10:105–108. doi: 10.1097/00042737-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 570.Kchir N, Haouet S, Boubaker S. [Caroli's disease associated with hepatocarcinoma. A case report and review of the literature] Arch Anat Cytol Pathol. 1990;38:95–99. [PubMed] [Google Scholar]
- 571.Witlin LT, Gadacz TR, Zuidema GD, Kridelbaugh WW. Transhepatic decompression of the biliary tree in Caroli's disease. Surgery. 1982;91:205–209. [PubMed] [Google Scholar]
- 572.Giovanardi RO. Monolobar Caroli's disease in an adult. Case report. Hepatogastroenterology. 2003;50:2185–2187. [PubMed] [Google Scholar]
- 573.Lointier PH, Kauffmann P, Francannet P. Management of intrahepatic calculi in Caroli's disease by extracorporeal shock wave lithotripsy. Br J Surg. 1980;77:987–988. doi: 10.1002/bjs.1800770909. [DOI] [PubMed] [Google Scholar]
- 574.Millwala F, Segev DL, Thuluvath PJ. Caroli's disease and outcomes after liver transplantation. Liver Transpl. 2008;14:11–17. doi: 10.1002/lt.21366. [DOI] [PubMed] [Google Scholar]
- 575.Fozard JBJ, Wyatt JI, Hall RI. Epithelial dysplasia in Caroli's disease. Gut. 1989;30:1150–1153. doi: 10.1136/gut.30.8.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Hussman KL, Friedwald JP, Gollub MJ, Melamed J. Caroli's disease associated with infantile polycystic kidney disease: prenatal sonographic appearance. J Ultrasound. 1991;10:235–237. doi: 10.7863/jum.1991.10.4.235. [DOI] [PubMed] [Google Scholar]
- 577.Jordon D, Harpaz N, Thung SN. Caroli's disease and adult polycystic kidney disease: a rarely recognized association. Liver. 1989;9:30–35. doi: 10.1111/j.1600-0676.1989.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 578.Merza AP, Raiser MW. Biliary tract manifestations of the Marfan syndrome. Am J Gastroenterol. 1987;82:779–782. [PubMed] [Google Scholar]
- 579.Consortium E. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell. 1994;77:881–894. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 580.Hughes J, Ward CJ, Peral B. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 581.Geng L, Segal Y, Peissel B. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest. 1996;98:2674–2682. doi: 10.1172/JCI119090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Peters DJM, van DeWall A, Spruit L. Cellular localization and tissue distribution of polycystin-1. J Pathol. 1999;188:439–446. doi: 10.1002/(SICI)1096-9896(199908)188:4<439::AID-PATH367>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 583.Mochizuki T, Wu G, Hayashi T. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 584.Nauli SM, Alenghat FJ, Luo Y. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 585.Daoust MC, Reynolds DM, Bichet DG, Somlo S. Evidence for a third genetic locus for autosomal dominant polycystic kidney disease. Genomics. 1995;25:733–736. doi: 10.1016/0888-7543(95)80020-m. [DOI] [PubMed] [Google Scholar]
- 586.Hateboer N, v Dijk MA, Bogdanova M. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet. 1999;353:103–107. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 587.Ring T, Spiegelhalter D. Risk of intracranial aneurysm bleeding in autosomal-dominant polycystic kidney disease. Kidney Int. 2007;72:1400–1402. doi: 10.1038/sj.ki.5002488. [DOI] [PubMed] [Google Scholar]
- 588.Rossetti S, Chauveau D, Kubly V. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361:2196–21201. doi: 10.1016/S0140-6736(03)13773-7. [DOI] [PubMed] [Google Scholar]
- 589.Gabow PA. Autosomal dominant polycystic kidney disease. N Eng J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 590.Perron RD. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Kidney Int. 1997;51:2022–2036. [Google Scholar]
- 591.Ivy D, Shaffer E, Johnson A. Cardiovascular abnormalities in children with austosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;5:2032–2036. doi: 10.1681/ASN.V5122032. [DOI] [PubMed] [Google Scholar]
- 592.Thomsen HS, Thaysen JH. Frequency of hepatic cysts in adult polycystic kidney disease. Acta Med Scand. 1998;224:381–384. doi: 10.1111/j.0954-6820.1988.tb19598.x. [DOI] [PubMed] [Google Scholar]
- 593.Fick GM, Johnson AM, Strain JD. Characteristics of very early onset autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3:1863–1876. doi: 10.1681/ASN.V3121863. [DOI] [PubMed] [Google Scholar]
- 594.Henson SW, Jr, Gray HK, Dockerty MB. Benign tumours of the liver III. Solitary cysts. Surg Gynecol Obstet. 1956;103:607–612. [PubMed] [Google Scholar]
- 595.Kwok MK, Lewin KJ. Massive hepatomegaly in adult polycystic liver disease. Am J Surg Pathol. 1988;12:321–324. doi: 10.1097/00000478-198804000-00010. [DOI] [PubMed] [Google Scholar]
- 596.Garber S, Mathieson J, Cooperberg PL. Percutaneous sclerosis of hepatic cysts to treat obstructive jaundice in a patient with polycystic liver disease. AJR Am J Roentgenol. 1993;161:77–78. doi: 10.2214/ajr.161.1.8517326. [DOI] [PubMed] [Google Scholar]
- 597.Dumot JA, Fields MS, Meyer RA. Alcohol sclerosis for polycystic liver disease and obstructive jaundice: use of a nasobiliary catheter. Am J Gastroenterol. 1994;89:1555–1557. [PubMed] [Google Scholar]
- 598.van Erpecum KJ, Janssens AR, Terpstra JL. TR. Highly symptomatic adult polycystic disease of the liver. A report of fifteen cases. J Hepatol. 1987;5:109–117. doi: 10.1016/s0168-8278(87)80068-5. [DOI] [PubMed] [Google Scholar]
- 599.Torres VE, Rastogi S, King BF. Hepatic venous outflow obstruction in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1994;5:1186–1192. doi: 10.1681/ASN.V551186. [DOI] [PubMed] [Google Scholar]
- 600.Uddin W, Ramage JK, Portmann B. Hepatic venous outflow obstruction in patients with polycystic liver disease: pathogenesis and treatment. Gut. 1995;36:142–145. doi: 10.1136/gut.36.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Harris RA, Gray DW, Britton BJ. Hepatic cystic disease in an adult polycystic kidney disease transplant population. Aust N Z J Surg. 1996;66:166–168. doi: 10.1111/j.1445-2197.1996.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 602.Vons C, Chauveau D, Martinod E. Résection hépatique chez les malades atteints de polykystose hépatique [Liver resection in patients with polycystic liver disease] Gastroenterol Clin Biol. 1998;22:50–54. [PubMed] [Google Scholar]
- 603.Que F, Nagorney DM, Gross JB, Jr, Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108:487–894. doi: 10.1016/0016-5085(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 604.Everson GT, Taylor MR. Management of polycystic liver disease. Curr Gastroenterol Rep. 2005;7:19–25. doi: 10.1007/s11894-005-0061-6. [DOI] [PubMed] [Google Scholar]
- 605.Gabow PA. Autosomal dominant polycystic kidney disease: more than a renal disease. Am J Kidney Dis. 1990;16:403–413. doi: 10.1016/s0272-6386(12)80051-5. [DOI] [PubMed] [Google Scholar]
- 606.Sherstha R, McKinley C, Russ P. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282–1286. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- 607.Telenti A, Torres VE, Gross JB., Jr Hepatic cyst infection in autosomal dominant polycystic kidney disease. Mayo Clin Proc. 1990;65:933–942. doi: 10.1016/s0025-6196(12)65154-4. [DOI] [PubMed] [Google Scholar]
- 608.Ikei S, Yamaguchi Y, Mori K. Infection of hepatic cysts with Pseudomonas aeroginosa in polycystic liver disease. Dig Surg. 1990;7:117–121. [Google Scholar]
- 609.Sallee M, Rafat C, Zahar JR. Cyst infections in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4:1183–1189. doi: 10.2215/CJN.01870309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Kaehny WD, Everson GT. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Semin Nephrol. 1991;11:661–670. [PubMed] [Google Scholar]
- 611.Cole BR, Conley SB, Stapleton FB. Polycystic kidney disease in the first year of life. J Pediatr. 1987;111:693–699. doi: 10.1016/s0022-3476(87)80244-5. [DOI] [PubMed] [Google Scholar]
- 612.Milutinovic J, Schabel SI, Ainsworth SK. Autosomal dominant polycystic kidney disease with liver and pancreatic involvement in early childhood. Am J Kidney Dis. 1989;13:340–344. doi: 10.1016/s0272-6386(89)80043-5. [DOI] [PubMed] [Google Scholar]
- 613.Zhao X, Paterson AD, Zahirieh A. Molecular diagnostics in autosomal dominant polycystic kidney disease: utility and limitations. Clin J Am Soc Nephrol. 2008;3:146–152. doi: 10.2215/CJN.03430807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Terada T, Nakanuma Y. Congenital biliary dilatation in autosomal dominant adult polycystic disease of the liver and kidneys. Arch Pathol Lab Med. 1988;112:1113–1116. [PubMed] [Google Scholar]
- 615.Patterson M, Gonzalez-Vitale JC, Fagan CJ. Polycystic liver disease: a study of cyst fluid constituents. Hepatology. 1982;2:475–478. doi: 10.1002/hep.1840020414. [DOI] [PubMed] [Google Scholar]
- 616.Coffin B, Hadengue A, Gegos F, Benhamou J-P. Calcified hepatic and renal cysts in adult dominant polycystic kidney disease. Dig Dis Sci. 1990;35:1172–1175. doi: 10.1007/BF01537592. [DOI] [PubMed] [Google Scholar]
- 617.Melnick PJ. Polycystic liver. Arch Pathol. 1955;59:162–172. [PubMed] [Google Scholar]
- 618.Ramos A, Torres VE, Holley KE. The liver in autosomal dominant polycystic kidney disease. Arch Pathol Lab Med. 1990;114:180–184. [PubMed] [Google Scholar]
- 619.Kida T, Nakanuma Y, Terada T. Cystic dilatation of peribiliary glands in livers with adult polycystic disease and livers with solitary nonparasitic cysts: an autopsy study. Hepatology. 1992;16:334–340. doi: 10.1002/hep.1840160209. [DOI] [PubMed] [Google Scholar]
- 620.Redston MS, Wanless IR. The hepatic von Meyenburg complex: prevalence and association with hepatic and renal cysts among 2843 autopsies [corrected] Mod Pathol. 1996;9:233–237. [PubMed] [Google Scholar]
- 621.Honda N, Cobb C, Lechago J. Bile duct carcinoma associated with multiple von Meyenburg complexes in the liver. Hum Pathol. 1986;17:1287–1290. doi: 10.1016/s0046-8177(86)80575-5. [DOI] [PubMed] [Google Scholar]
- 622.Bruns CD, Kuhms JG, Wieman J. Cholangiocarcinoma in association with multiple biliary microhamartomas. Arch Pathol Lab Med. 1990;114:1287–1289. [PubMed] [Google Scholar]
- 623.Rossi RL, Silverman ML, Braasch JW. Carcinomas arising in cystic conditions of the bile ducts. Ann Surg. 1987;205:377–384. doi: 10.1097/00000658-198704000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Theise N, Miller F, Worman HJ. Biliary cystadenocarcinoma arising in a liver with fibropolycystic disease. Arch Pathol Lab Med. 1993;117:163–165. [PubMed] [Google Scholar]
- 625.Coffen T, Breuning M, Shoots C. Congenital hepatic fibrosis in ADPKD. Kidney Int. 1990;38:880–885. doi: 10.1038/ki.1990.286. [DOI] [PubMed] [Google Scholar]
- 626.Sato Y, Mukai M, Sasaki M. Intraductal papillary-mucinous neoplasm of the pancreas associated with polycystic liver and kidney disease. Pathol Int. 2009;59:201–204. doi: 10.1111/j.1440-1827.2009.02352.x. [DOI] [PubMed] [Google Scholar]
- 627.Pirson Y, Lannoy N, Peters D. Isolated polycystic liver disease as a distinct genetic disease, unlinked to polycystic kidney disease 1 and polycystic kidney disease 2. Hepatology. 1996;23:249–252. doi: 10.1002/hep.510230208. [DOI] [PubMed] [Google Scholar]
- 628.Iglesias DM, Palmitano JA, Arrizurieta E. Isolated polycystic liver disease not linked to polycystic kidney disease 1 and 2. Dig Dis Sci. 1999;44:385–388. doi: 10.1023/a:1026623005401. [DOI] [PubMed] [Google Scholar]
- 629.Qian Q, Li A, King BF. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–171. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 630.Li A, Davila S, Furu L. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Drenth JP, Martina JA, Te Morsche RH. Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology. 2004;126:1819–1827. doi: 10.1053/j.gastro.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 632.Waanders E, te Morsche RH, de Man RA. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum Mutat. 2006;27:830. doi: 10.1002/humu.9441. [DOI] [PubMed] [Google Scholar]
- 633.Waanders E, Van Krieken JH, Lameris AL, Drenth JP. Disrupted cell adhesion but not proliferation mediates cyst formation in polycystic liver disease. Mod Pathol. 2008;21:1293–1302. doi: 10.1038/modpathol.2008.115. [DOI] [PubMed] [Google Scholar]
- 634.Donovan MJ, Kozakewich H, Perez-Atayde A. Solitary non parasitic cysts of the liver. The Boston Children's Hospital experience. Pediatr Pathol Lab Med. 1995;15:419–428. doi: 10.3109/15513819509026977. [DOI] [PubMed] [Google Scholar]
- 635.Geist DC. Solitary nonparasitic cyst of the liver; review of the literature and report of two patients. Arch Surg. 1955;71:867–880. doi: 10.1001/archsurg.1955.01270180073010. [DOI] [PubMed] [Google Scholar]
- 636.Byrne WJ, Fonkalsrud EW. Congenital solitary nonparasitic cyst of the liver: a rare cause of rapidly enlarging abdominal mass in infancy. J Pediatr Surg. 1982;17:316–317. doi: 10.1016/s0022-3468(82)80022-5. [DOI] [PubMed] [Google Scholar]
- 637.Miyamoto M, Oka M, Izumiya T. Nonparasitic solitary giant hepatic cyst causing obstructive jaundice was successfully treated with monoethanolamine oleate. Intern Med. 2006;45:621–625. doi: 10.2169/internalmedicine.45.1408. [DOI] [PubMed] [Google Scholar]
- 638.Akriviadis EA, Steindel H, Ralls P, Redeker AG. Spontaneous rupture of nonparasitic cyst of the liver. Gastroenterology. 1989;97:213–215. doi: 10.1016/0016-5085(89)91438-8. [DOI] [PubMed] [Google Scholar]
- 639.Brock JS, Pachter HL, Schreiber J, Hofstetter SR. Surgical diseases of the falciform ligament. Am J Gastroenterol. 1992;67:757–758. [PubMed] [Google Scholar]
- 640.Lin CC, Lin SC, Ko WC. Adenocarcinoma and infection in a solitary hepatic cyst: a case report. World J Gastroenterol. 2005;11:1881–1883. doi: 10.3748/wjg.v11.i12.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Pliskin A, Cualing H, Stenger RJ. Primary squamous cell carcinoma originating in congenital cysts of the liver. Arch Pathol Lab Med. 1992;166:105–107. [PubMed] [Google Scholar]
- 642.Terada T, Notsumata K, Nakanuma Y. Biliary carcinosarcoma arising in nonparasitic simple cyst of the liver. Virchows Arch. 1994;424:331–335. doi: 10.1007/BF00194620. [DOI] [PubMed] [Google Scholar]
- 643.Ueyama T, Ding J, Hashimoto H. Carcinoid tumor arising in the wall of a congenital bile duct cyst. Arch Pathol Lab Med. 1992;116:291–293. [PubMed] [Google Scholar]
- 644.Chu DY, Olson AL, Mishalany HG. Congenital liver cyst presenting as congenital diaphragmatic hernia. J Pediatr Surg. 1986;21:897–899. doi: 10.1016/s0022-3468(86)80019-7. [DOI] [PubMed] [Google Scholar]
- 645.Thrasher S, Adelman S, Chang C-H. Hepatic cyst associated with Peutz-Jeghers syndrome. Arch Pathol Lab Med. 1990;114:1278–1280. [PubMed] [Google Scholar]
- 646.Deziel DJ, Rossi RL, Munson JL. Management of bile duct cysts in adults. Arch Surg. 1986;121:410–415. doi: 10.1001/archsurg.1986.01400040046006. [DOI] [PubMed] [Google Scholar]
- 647.Furuta T, Yoshida Y, Saku M. Treatment of symptomatic non-parasitic liver cyst-surgical treatment versus alcohol injection therapy. HPB Surg. 1990;2:269–279. doi: 10.1155/1990/60495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Pozniczek M, Wysocki A, Bobrzynski A. Sclerosant therapy as first-line treatment for solitary liver cysts. Dig Surg. 2004;21:452–454. doi: 10.1159/000083473. [DOI] [PubMed] [Google Scholar]
- 649.Fabiani P, Mazza D, Toouli J. Laparoscopic fenestration of symptomatic non-parasitic cysts of the liver. Br J Surg. 1997;84:321–322. [PubMed] [Google Scholar]
- 650.Fiamingo P, Tedeschi U, Veroux M. Laparoscopic treatment of simple hepatic cysts and polycystic liver disease. Surg Endosc. 2003;17:623–626. doi: 10.1007/s00464-002-9088-z. [DOI] [PubMed] [Google Scholar]
- 651.Reye RDK, Morgan G, Baral J. Encephalopathy and fatty degeneration of the viscera, a disease entity in childhood. Lancet. 1963;ii:749–752. doi: 10.1016/s0140-6736(63)90554-3. [DOI] [PubMed] [Google Scholar]
- 652.Crocker JF. Reye's syndrome. Semin Liver Dis. 1982;2:340–352. doi: 10.1055/s-2008-1040720. [DOI] [PubMed] [Google Scholar]
- 653.Starko KM, Ray CG, Dominguez LB. Reye's syndrome and salicylate use. Pediatrics. 1980;66:859–864. [PubMed] [Google Scholar]
- 654.Hurwitz ES, Barrett MJ, Bregman D. Public Health Service study on Reye's syndrome and medications. Report of the pilot phase. N Engl J Med. 1985;313:849–857. doi: 10.1056/NEJM198510033131403. [DOI] [PubMed] [Google Scholar]
- 655.Hurwitz ES, Barrett MJ, Bregman D. Public Health Service study of Reye's syndrome and medications. Report of the main study. JAMA. 1987;257:1905–1911. [PubMed] [Google Scholar]
- 656.Belay ED, Bresee JS, Holman RC. Reye's syndrome in the United States from 1981 through 1997. N Engl J Med. 1999;340:1377–1382. doi: 10.1056/NEJM199905063401801. [DOI] [PubMed] [Google Scholar]
- 657.Hall SM, Lynn R. Reye's syndrome. N Engl J Med. 1999;341:845–846. doi: 10.1056/NEJM199909093411112. [DOI] [PubMed] [Google Scholar]
- 658.Orlowski JP. Whatever happened to Reye's syndrome? Did it ever really exist? Crit Care Med. 1999;27:1582–1587. doi: 10.1097/00003246-199908000-00032. [DOI] [PubMed] [Google Scholar]
- 659.Varma RR, Riedel DR, Komorowski RA. Reye's syndrome in nonpediatric age groups. JAMA. 1979;242:1373–1375. [PubMed] [Google Scholar]
- 660.Stillman A, Gitter H, Shillington D. Reye's syndrome in the adult: case report and review of the literature. Am J Gastroenterol. 1983;78:365–368. [PubMed] [Google Scholar]
- 661.Bove KE, McAdams AJ, Partin JC. The hepatic lesion in Reye's syndrome. Gastroenterology. 1975;69:685–697. [PubMed] [Google Scholar]
- 662.Bentz MS, Cohen C. Periportal hepatic necrosis in Reye's syndrome. One case in a review of eight patients. Am J Gastroenterol. 1980;73:49–53. [PubMed] [Google Scholar]
- 663.Ellis GH, Mirkin LD, Mills MC. Pancreatitis and Reye's syndrome. Am J Dis Child. 1979;133:1014–1016. doi: 10.1001/archpedi.1979.02130100038007. [DOI] [PubMed] [Google Scholar]
- 664.Phillips MJ, Poucell S, Patterson J, Valencia P. Raven Press; New York: 1987. The liver. An atlas and text of ultrastructural pathology. [Google Scholar]
- 665.Partin JC, Schubert WK, Partin JS. Mitochondrial ultrastructure in Reye's syndrome (encephalopathy and fatty degeneration of the viscera) N Engl J Med. 1971;285:1339–1343. doi: 10.1056/NEJM197112092852402. [DOI] [PubMed] [Google Scholar]
- 666.Bradel EJ, Reiner CB. The fine structure of hepatocytes in Reye's syndrome. In: D PJ, editor. Reye's syndrome. Grune & Stratton; New York: 1975. pp. 147–158. [Google Scholar]
- 667.Starko KM, Mullick FG. Hepatic and cerebral pathology findings in children with fatal salicylate intoxication: further evidence for a causal relation between salicylate and Reye's syndrome. Lancet. 1983;1:326–329. doi: 10.1016/s0140-6736(83)91629-x. [DOI] [PubMed] [Google Scholar]
- 668.Hug G, Bosken J, Bove K. Reye's syndrome simulacra in liver of mice after treatment with chemical agents and encephalomyocarditis virus. Lab Invest. 1981;45:89–109. [PubMed] [Google Scholar]
- 669.Davis LE, Cole LL, Lockwood SJ, Kornfeld M. Experimental influenza B virus toxicity in mice. Lab Invest. 1983;48:140–147. [PubMed] [Google Scholar]
- 670.Brownstein DG, Johnson EA, Smith AL. Spontaneous Reye's-like syndrome in BALB/cByJ mice. Lab Invest. 1984;51:386–395. [PubMed] [Google Scholar]
- 671.Sakaida N, Senzaki H, Shikata N, Morii S. Microvesicular fatty liver in rats with resembling Reye's syndrome induced by 4-pentenoic acid. Acta Pathol Jpn. 1990;40:635–642. doi: 10.1111/j.1440-1827.1990.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 672.Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi. 1967;16:178–222. [PubMed] [Google Scholar]
- 673.Royle J, Burgner D, Curtis N. The diagnosis and management of Kawasaki disease. J Paediatr Child Health. 2005;41:87–93. doi: 10.1111/j.1440-1754.2005.00555.x. [DOI] [PubMed] [Google Scholar]
- 674.Melish ME, Hicks RV. Kawasaki syndrome: clinical features. Pathophysiology, etiology and therapy. J Rheumatol. 1990;24(Suppl.):2–10. [PubMed] [Google Scholar]
- 675.Onouchi Y, Gunji T, Burns JC. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. 2008;40:35–42. doi: 10.1038/ng.2007.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Valentini P, Ausili E, Schiavino A. Acute cholestasis: atypical onset of Kawasaki disease. Dig Liver Dis. 2008;40:582–584. doi: 10.1016/j.dld.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 677.Nakamura Y, Yanagawa H, Kawasaki T. Mortality among children with Kawasaki disease in Japan. N Engl J Med. 1992;326:1246–1249. doi: 10.1056/NEJM199205073261903. [DOI] [PubMed] [Google Scholar]
- 678.Nakamura Y, Fujita Y, Nagai M. Cardiac sequelae of Kawasaki disease in Japan: statistical analysis. Pediatrics. 1991;88:1144–1147. [PubMed] [Google Scholar]
- 679.Newburger JW, Takahashi M, Burns JC. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315:341–347. doi: 10.1056/NEJM198608073150601. [DOI] [PubMed] [Google Scholar]
- 680.Onouchi Z, Hamaoka K, Sakata K. Long-term changes in coronary artery aneurysms in patients with Kawasaki disease: comparison of therapeutic regimens. Circ J. 2005;69:265–272. doi: 10.1253/circj.69.265. [DOI] [PubMed] [Google Scholar]
- 681.Do YS, Kim KW, Chun JK. Predicting factors for refractory kawasaki disease. Korean Circ J. 2010;40:239–242. doi: 10.4070/kcj.2010.40.5.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Manlhiot C, Yeung RS, Clarizia NA. Kawasaki Disease at the extremes of the age spectrum. Pediatrics. 2009;124:e410–e415. doi: 10.1542/peds.2009-0099. [DOI] [PubMed] [Google Scholar]
- 683.Levy M, Koren G. Atypical Kawasaki disease; analysis of clinical presentation and diagnostic clues. Pediatr Infect Dis J. 1990;9:122–126. [PubMed] [Google Scholar]
- 684.Suddleson EA, Reid B, Woolley MM, Takahashi M. Hydrops of the gallbladder associated with Kawasaki syndrome. J Pediatr Surg. 1987;22:956–959. doi: 10.1016/s0022-3468(87)80600-0. [DOI] [PubMed] [Google Scholar]
- 685.Bishop WP, Kao SC. Prolonged postprandial abdominal pain following Kawasaki syndrome with acute gallbladder hydrops: association with impaired gallbladder emptying. J Pediatr Gastroenterol Nutr. 1991;13:307–311. doi: 10.1097/00005176-199110000-00013. [DOI] [PubMed] [Google Scholar]
- 686.Bader-Meunier B, Hadchouel M, Fabre M. Intrahepatic bile duct damage in children with Kawasaki disease. J Pediatr. 1992;120:750–752. doi: 10.1016/s0022-3476(05)80239-2. [DOI] [PubMed] [Google Scholar]
- 687.Amano S, Hazama F, Kubagawa H. General pathology of Kawasaki disease. On the morphological alterations corresponding to the clinical manifestations. Acta Pathol Jpn. 1980;30:681–694. [PubMed] [Google Scholar]
- 688.Edwards KM, Glick AD, Greene HL. Intrahepatic cholangitis associated with mucocutaneous lymph node syndrome. J Pediatr Gastroenterol Nutr. 1985;4:140–142. doi: 10.1097/00005176-198502000-00025. [DOI] [PubMed] [Google Scholar]
- 689.Gear JH, Meyers KE, Steele M. Kawasaki disease manifesting with acute cholangitis. A case report. S Afr Med J. 1992;81:31–33. [PubMed] [Google Scholar]
- 690.Bergeson PS, Serlin SP, Corman LI. Mucocutaneous lymph-node syndrome with positive Weil-Felix reaction but negative Leptospira studies. Lancet. 1978;1:720–721. doi: 10.1016/s0140-6736(78)90836-x. [DOI] [PubMed] [Google Scholar]
- 691.Ahlström H, Lundstrom NR, Mortensson W. Infantile periarteritis nodosa or mucocutaneous lymph node syndrome. A report on four cases and diagnostic considerations. Acta Paediatr Scand. 1977;66:193–198. doi: 10.1111/j.1651-2227.1977.tb07832.x. [DOI] [PubMed] [Google Scholar]
- 692.Kato H, Koike S, Yamamoto M. Coronary aneurysms in infants and young children with acute febrile mucocutaneous lymph node syndrome. J Pediatr. 1975;86:892–898. doi: 10.1016/s0022-3476(75)80220-4. [DOI] [PubMed] [Google Scholar]
- 693.Marks WH, Coran AG, Wesley JR. Hepatic artery aneurysm associated with the mucocutaneous lymph node syndrome. Surgery. 1985;98:598–601. [PubMed] [Google Scholar]
- 694.Kato H, Fujimoto T, Inoue O. Variant strain of Propionobacterum acnes: a clue to the aetiology of Kawasaki disease. Lancet. 1983;ii:1383–1388. doi: 10.1016/s0140-6736(83)90921-2. [DOI] [PubMed] [Google Scholar]
- 695.Leung DY, Meissner HC, Fulton DR. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. 1993;342:1385–1388. doi: 10.1016/0140-6736(93)92752-f. [DOI] [PubMed] [Google Scholar]
- 696.Viraben A, Dupre A. Kawasaki disease associated with HIV infection. Lancet. 1987;1:1430–1431. doi: 10.1016/s0140-6736(87)90616-7. [DOI] [PubMed] [Google Scholar]
- 697.Kikuta H, Taguchi Y, Tomizawa K. Epstein-Barr virus genome-positive T-lymphocytes in a boy with chronic active EBV infection associated with Kawasaki-like disease. Nature. 1988;333:455–457. doi: 10.1038/333455a0. [DOI] [PubMed] [Google Scholar]
- 698.Nigro G, Zerbini M, Krzysztofiak A. Active or recent parvovirus B19 infection in children with Kawasaki disease. Lancet. 1994;343:1260–1261. doi: 10.1016/s0140-6736(94)92154-7. [DOI] [PubMed] [Google Scholar]
- 699.Chou CT, Chang JS, Ooi SE. Serum anti-Yersinia antibody in Chinese patients with Kawasaki disease. Arch Med Res. 2005;36:14–18. doi: 10.1016/j.arcmed.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 700.Whitby D, Hoad JG, Tizard EJ. Isolation of measles virus from child with Kawasaki disease. Lancet. 1991;338:1215. doi: 10.1016/0140-6736(91)92085-g. [DOI] [PubMed] [Google Scholar]
- 701.Pagnoux C, Hayem G, Roux F. [Sclerosing cholangitis as a complication of Langerhans’cell histiocytosis] Rev Med Interne. 2003;24:324–327. doi: 10.1016/s0248-8663(03)00063-8. [DOI] [PubMed] [Google Scholar]
- 702.Jaffe R. Liver involvement in the histiocytic disorders of childhood. Pediatr Dev Pathol. 2004;7:214–225. doi: 10.1007/s10024-003-9876-z. [DOI] [PubMed] [Google Scholar]
- 703.Finn LS, Jaffe R. Langerhans’ cell granuloma confined to the bile duct. Pediatr Pathol Lab Med. 1997;17:461–468. [PubMed] [Google Scholar]
- 704.Buza N, Lagarde DC, Dash S, Haque S. Langerhans cell histiocytosis: report of a single organ involvement in a child. J Cell Mol Med. 2004;8:397–401. doi: 10.1111/j.1582-4934.2004.tb00329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Kaplan KJ, Goodman ZD, Ishak KG. Liver involvement in Langerhans’ cell histiocytosis. A study of nine cases. Mod Pathol. 1999;12:370–378. [PubMed] [Google Scholar]
- 706.Favara BE, Jaffe R, Egeler RM. Macrophage activation and hemophagocytic syndrome in Langerhans cell histiocytosis: report of 30 cases. Pediatr Dev Pathol. 2002;5:130–140. doi: 10.1007/s10024001-0159-2. [DOI] [PubMed] [Google Scholar]
- 707.Krenacs L, Tiszaluicz L, Krenacs T, Boumsell L. Immunohistochemical detection of CD1a antigen in formalin-fixed and paraffin-embedded tissue sections with monoclonal antibody 010. J Pathol. 1993;171:99–104. doi: 10.1002/path.1711710206. [DOI] [PubMed] [Google Scholar]
- 708.Emile JF, Wechsler J, Brousse N. Langerhans’ cell histiocytosis. Definitive diagnosis with the use of monoclonal antibody O10 on routinely paraffin-embedded samples. Am J Surg Pathol. 1995;19:636–641. doi: 10.1097/00000478-199506000-00003. [DOI] [PubMed] [Google Scholar]
- 709.Slone SP, Fleming DR, Buchino JJ. Sinus histiocytosis with massive lymphadenopathy and Langerhans cell histiocytosis express the cellular adhesion molecule CD31. Arch Pathol Lab Med. 2003;127:341–344. doi: 10.5858/2003-127-0341-SHWMLA. [DOI] [PubMed] [Google Scholar]
- 710.Niki T, Oka T, Shiga J. Increased S-100 protein-immunoreactivity of Kupffer cells is associated with lymphohematological malignancy. Pathol Int. 1995;45:742–747. doi: 10.1111/j.1440-1827.1995.tb03391.x. [DOI] [PubMed] [Google Scholar]
- 711.Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev. 2010;234:120–141. doi: 10.1111/j.0105-2896.2009.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Cavazza A, Pasquinelli G, Carlinfante G. Nodular Langerhans’ cell histiocytosis of the liver in an adult with colonic adenocarcinoma. Histopathology. 1999;34:273–275. [PubMed] [Google Scholar]
- 713.Braier J, Ciocca M, Latella A. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell histiocytosis. Med Pediatr Oncol. 2002;38:178–182. doi: 10.1002/mpo.1306. [DOI] [PubMed] [Google Scholar]
- 714.Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: current concepts and treatments. Cancer Treat Rev. 2010;36:354–359. doi: 10.1016/j.ctrv.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 715.Marchal J, Kambouchner M, Tazi A. Expression of apoptosis-regulatory proteins in lesions of pulmonary Langerhans cell histiocytosis. Histopathology. 2004;45:20–28. doi: 10.1111/j.1365-2559.2004.01875.x. [DOI] [PubMed] [Google Scholar]
- 716.Annels NE, Da Costa CE, Prins FA. Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med. 2003;197:1385–1390. doi: 10.1084/jem.20030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Laman JD, Leenen PJ, Annels NE. Langerhans-cell histiocytosis ‘insight into DC biology’. Trends Immunol. 2003;24:190–196. doi: 10.1016/s1471-4906(03)00063-2. [DOI] [PubMed] [Google Scholar]
- 718.Willman CL, Busque L, Griffith BB. Langerhans’-cell histiocytosis (histiocytosis X): a clonal proliferative disease. N Engl J Med. 1994;331:154–160. doi: 10.1056/NEJM199407213310303. [DOI] [PubMed] [Google Scholar]
- 719.Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet. 1994;343:767–768. doi: 10.1016/s0140-6736(94)91842-2. [DOI] [PubMed] [Google Scholar]
- 720.Kawakubo Y, Kishimoto H, Sato Y. Human cytomegalovirus infection in foci of Langerhans cell histiocytosis. Virchows Arch. 1999;434:109–115. doi: 10.1007/s004280050313. [DOI] [PubMed] [Google Scholar]
- 721.Shimakage M, Sasagawa T, Kimura M. Expression of Epstein-Barr virus in Langerhans’ cell histiocytosis. Hum Pathol. 2004;35:862–868. doi: 10.1016/j.humpath.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 722.Glotzbecker MP, Carpentieri DF, Dormans JP. Langerhans cell histiocytosis: a primary viral infection of bone? Human herpes virus 6 latent protein detected in lymphocytes from tissue of children. J Pediatr Orthop. 2004;24:123–129. [PubMed] [Google Scholar]
- 723.Lieberman PH, Jones CR, Steinman RM. Langerhans cell (eosinophilic) granulomatosis: a clinicopathologic study encompassing 50 years. Am J Surg Pathol. 1996;20:519–522. doi: 10.1097/00000478-199605000-00001. [DOI] [PubMed] [Google Scholar]
- 724.Howarth DM, Gilchrist GS, Mullan BP. Langerhans cell histiocytosis. Diagnosis, natural history, management and outcome. Cancer. 1999;85:2278–2290. doi: 10.1002/(sici)1097-0142(19990515)85:10<2278::aid-cncr25>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 725.Jubran RF, Marachelian A, Dorey F, Malogolowkin M. Predictors of outcome in children with Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005;45:37–42. doi: 10.1002/pbc.20364. [DOI] [PubMed] [Google Scholar]
- 726.A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch Dis Child. 1996;75:17–24. doi: 10.1136/adc.75.1.17. Anonymous. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Zandi P, Panis Y, Debray D. Pediatric liver transplantation for Langerhans’ cell histiocytosis. Hepatology. 1995;21:129–133. [PubMed] [Google Scholar]
- 728.Griffiths W, Davies S, Gibbs P. Liver transplantation in an adult with sclerosing cholangitis due to Langerhans cell histiocytosis. J Hepatol. 2006;44:829–831. doi: 10.1016/j.jhep.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 729.Newell KA, Alonso EM, Kelly SM. Association between liver transplantation for Langerhans cell histiocytosis, rejection, and development of posttransplant lymphoproliferative disease in children. J Pediatr. 1997;131:98–104. doi: 10.1016/s0022-3476(97)70131-8. [DOI] [PubMed] [Google Scholar]
- 730.Hadzic N, Pritchard J, Webb D. Recurrence of Langerhans cell histiocytosis in the graft after pediatric liver transplantation. Transplantation. 2000;70:815–819. doi: 10.1097/00007890-200009150-00019. [DOI] [PubMed] [Google Scholar]
- 731.Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol. 2003;27:579–593. doi: 10.1097/00000478-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 732.Chetritt J, Paradis V, Dargere D. Chester-Erdheim disease: a neoplastic disorder. Hum Pathol. 1999;30:1093–1096. doi: 10.1016/s0046-8177(99)90228-9. [DOI] [PubMed] [Google Scholar]
- 733.Gundling F, Nerlich A, Heitland WU, Schepp W. Biliary manifestation of Erdheim-Chester disease mimicking Klatskin's carcinoma. Am J Gastroenterol. 2007;102:452–454. doi: 10.1111/j.1572-0241.2006.00893.x. [DOI] [PubMed] [Google Scholar]
- 734.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63–70. [PubMed] [Google Scholar]
- 735.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19–73. [PubMed] [Google Scholar]
- 736.Maheshwari A, Seth A, Choudhury M. Rosai-Dorfman disease: a case with lymphadenopathy and liver involvement. J Pediatr Hematol Oncol. 2009;31:200–202. doi: 10.1097/MPH.0b013e31818e5369. [DOI] [PubMed] [Google Scholar]
- 737.Lauwers GY, Perez-Atayde A, Dorfman RF, Rosai J. The digestive system manifestations of Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy): review of 11 cases. Hum Pathol. 2000;31:380–385. doi: 10.1016/s0046-8177(00)80254-3. [DOI] [PubMed] [Google Scholar]
- 738.Eisen R, Buckley PJ, Rosai J. Immunophenotypic characterization of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) Semin Diagn Pathol. 1990;7:74–82. [PubMed] [Google Scholar]
- 739.Arico M, Allen M, Brusa S. Haemophagocytic lymphohistiocytosis: proposal of a diagnostic algorithm based on perforin expression. Br J Haematol. 2002;119:180–188. doi: 10.1046/j.1365-2141.2002.03773.x. [DOI] [PubMed] [Google Scholar]
- 740.Feldmann J, Le Deist F, Ouachee-Chardin M. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2002;117:965–972. doi: 10.1046/j.1365-2141.2002.03534.x. [DOI] [PubMed] [Google Scholar]
- 741.Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6:137–154. doi: 10.1586/eci.09.58. [DOI] [PubMed] [Google Scholar]
- 742.Parizhskaya M, Reyes J, Jaffe R. Hemophagocytic syndrome presenting as acute hepatic failure in two infants: clinical overlap with neonatal hemochromatosis. Pediatr Dev Pathol. 1999;2:360–366. doi: 10.1007/s100249900135. [DOI] [PubMed] [Google Scholar]
- 743.Natsheh SE, Roberts EA, Ngan B. Liver failure with marked hyperferritinemia: ‘ironing out’ the diagnosis. Can J Gastroenterol. 2001;15:537–540. doi: 10.1155/2001/651470. [DOI] [PubMed] [Google Scholar]
- 744.Ost A, Nilsson-Ardnor S, Henter J-I. Autopsy findings in 27 children with haemophagocytic lymphohistiocytosis. Histopathology. 1998;32:310–316. doi: 10.1046/j.1365-2559.1998.00377.x. [DOI] [PubMed] [Google Scholar]
- 745.Favara BE. Histopathology of the liver in histiocytosis syndromes. Pediatr Pathol Lab Med. 1996;16:413–433. doi: 10.1080/15513819609168681. [DOI] [PubMed] [Google Scholar]
- 746.Chen JH, Fleming MD, Pinkus GS. Pathology of the liver in familial hemophagocytic lymphohistiocytosis. Am J Surg Pathol. 2010;34:852–867. doi: 10.1097/PAS.0b013e3181dbbb17. [DOI] [PubMed] [Google Scholar]
- 747.Imashuku S, Ueda I, Teramura T. Occurrence of haemophagocytic lymphohistiocytosis at less than 1 year of age: analysis of 96 patients. Eur J Pediatr. 2005;164:315–319. doi: 10.1007/s00431-005-1636-9. [DOI] [PubMed] [Google Scholar]
- 748.Maakaroun NR, Moanna A, Jacob JT, Albrecht H. Viral infections associated with haemophagocytic syndrome. Rev Med Virol. 2010;20:93–105. doi: 10.1002/rmv.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Woda BA, Sullivan JL. Reactive histiocytic disorders. Am J Clin Pathol. 1993;99:459–463. doi: 10.1093/ajcp/99.4.459. [DOI] [PubMed] [Google Scholar]
- 750.Yoshiyama M, Kounami S, Nakayama K. Clinical assessment of Mycoplasma pneumoniae-associated hemophagocytic lymphohistiocytosis. Pediatr Int. 2008;50:432–435. doi: 10.1111/j.1442-200X.2008.02701.x. [DOI] [PubMed] [Google Scholar]
- 751.Taki H, Shinoda K, Hounoki H. Presenting manifestations of hemophagocytic syndrome in a male patient with systemic lupus erythematosus. Rheumatol Int. 2010;30:387–388. doi: 10.1007/s00296-009-0932-5. [DOI] [PubMed] [Google Scholar]
- 752.Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85:421–426. doi: 10.1136/adc.85.5.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Cortis E, Insalaco A. Macrophage activation syndrome in juvenile idiopathic arthritis. Acta Paediatr Suppl. 2006;95:38–41. doi: 10.1111/j.1651-2227.2006.tb02414.x. [DOI] [PubMed] [Google Scholar]
- 754.Chan JKC, Ng CS, Law CK. Reactive hemophagocytic syndrome. A study of 7 fatal cases. Pathology. 1987;19:43–50. doi: 10.3109/00313028709065134. [DOI] [PubMed] [Google Scholar]
- 755.Celkan T, Berrak S, Kazanci E. Malignancy-associated hemophagocytic lymphohistiocytosis in pediatric cases: a multicenter study from Turkey. Turk J Pediatr. 2009;51:207–213. [PubMed] [Google Scholar]
- 756.Fujiwara F, Hibi S, Imashuku S. Hypercytokinemia in hemophagocytic syndrome. Am J Pediatr Hematol Oncol. 1993;15:92–98. doi: 10.1097/00043426-199302000-00012. [DOI] [PubMed] [Google Scholar]
- 757.Tsui WMS, Wong KF, Tse CCH. Liver changes in reactive haemophagocytic syndrome. Liver. 1992;12:363–367. doi: 10.1111/j.1600-0676.1992.tb00590.x. [DOI] [PubMed] [Google Scholar]
- 758.de Kerguenec C, Hillaire S, Molinie V. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96:854–857. doi: 10.1111/j.1572-0241.2001.03632.x. [DOI] [PubMed] [Google Scholar]
- 759.Billiau AD, Roskams T, Van Damme-Lombaerts R. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood. 2005;105:1648–1651. doi: 10.1182/blood-2004-08-2997. [DOI] [PubMed] [Google Scholar]
- 760.Ruchelli ED, Uri A, Dimmick JE. Severe perinatal liver disease and Down syndrome: an apparent relationship. Hum Pathol. 1991;22:1274–1280. doi: 10.1016/0046-8177(91)90111-2. [DOI] [PubMed] [Google Scholar]
- 761.Schwab M, Niemeyer C, Schwarzer U. Down syndrome, transient myeloproliferative disorder, and infantile liver fibrosis. Med Pediatr Oncol. 1998;31:159–165. doi: 10.1002/(sici)1096-911x(199809)31:3<159::aid-mpo6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 762.Becroft DMO, Zwi J. Perinatal visceral fibrosis accompanying the megakaryoblastic leukemoid reaction of Down syndrome. Pediatr Pathol. 1990;10:397–406. doi: 10.3109/15513819009067127. [DOI] [PubMed] [Google Scholar]
- 763.Tsuda T, Komiyama A, Aonuma K. Transient abnormal myelopoiesis and diffuse hepatic necrosis in Down's syndrome with bleeding diathesis. Acta Paediatr Scand. 1990;79:241–244. doi: 10.1111/j.1651-2227.1990.tb11449.x. [DOI] [PubMed] [Google Scholar]
- 764.Gilson JP, Bendon RW. Megakaryocytosis of the liver in a trisomy 21 stillbirth. Arch Pathol Lab Med. 1993;117:738–739. [PubMed] [Google Scholar]
- 765.Arai H, Ishida A, Nakajima W. Immunohistochemical study on transforming growth factor-beta 1 expression in liver fibrosis of Down's syndrome. Hum Pathol. 1999;30:474–476. doi: 10.1016/s0046-8177(99)90125-9. [DOI] [PubMed] [Google Scholar]
- 766.Anuk D, Tarcan A, Alioglu B. Hydrops fetalis in a neonate with Down syndrome, transient myeloproliferative disorder and hepatic fibrosis. Fetal Pediatr Pathol. 2007;26:223–228. doi: 10.1080/15513810701818379. [DOI] [PubMed] [Google Scholar]
- 767.Shiozawa Y, Fujita H, Fujimura J. A fetal case of transient abnormal myelopoiesis with severe liver failure in Down syndrome: prognostic value of serum markers. Pediatr Hematol Oncol. 2004;21:273–278. doi: 10.1080/08880010490277088. [DOI] [PubMed] [Google Scholar]
- 768.Dormann S, Kruger M, Hentschel R. Life-threatening complications of transient abnormal myelopoiesis in neonates with Down syndrome. Eur J Pediatr. 2004;163:374–377. doi: 10.1007/s00431-004-1452-7. [DOI] [PubMed] [Google Scholar]
- 769.Massey GV. Transient leukemia in newborns with Down syndrome. Pediatr Blood Cancer. 2005;44:29–32. doi: 10.1002/pbc.20141. [DOI] [PubMed] [Google Scholar]
- 770.Greene ME, Mundschau G, Wechsler J. Mutations in GATA1 in both transient myeloproliferative disorder and acute megakaryoblastic leukemia of Down syndrome. Blood Cells Mol Dis. 2003;31:351–356. doi: 10.1016/j.bcmd.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 771.Vyas P, Roberts I. Down myeloid disorders: a paradigm for childhood preleukaemia and leukaemia and insights into normal megakaryopoiesis. Early Hum Dev. 2006;82 doi: 10.1016/j.earlhumdev.2006.09.016. 767-763. [DOI] [PubMed] [Google Scholar]
- 772.Tandonnet J, Clavel J, Baruchel A. Myeloid leukaemia in children with Down syndrome: report of the registry-based French experience between 1990 and 2003. Pediatr Blood Cancer. 2010;54:927–933. doi: 10.1002/pbc.22515. [DOI] [PubMed] [Google Scholar]
- 773.Silverman ED, Laxer RM. Neonatal lupus erythematosus. Rheum Dis Clin North Am. 1997;23:599–618. doi: 10.1016/s0889-857x(05)70349-5. [DOI] [PubMed] [Google Scholar]
- 774.Lee LA. The clinical spectrum of neonatal lupus. Arch Dermatol Res. 2009;301:107–110. doi: 10.1007/s00403-008-0896-4. [DOI] [PubMed] [Google Scholar]
- 775.Laxer RM, Roberts EA, Gross KR. Liver disease in neonatal lupus erythematosus. J Pediatr. 1990;116:238–242. doi: 10.1016/s0022-3476(05)82880-x. [DOI] [PubMed] [Google Scholar]
- 776.Evans N, Gaskin K. Liver disease in association with neonatal lupus erythematosus. J Paediatr Child Health. 1993;29:478–480. doi: 10.1111/j.1440-1754.1993.tb03026.x. [DOI] [PubMed] [Google Scholar]
- 777.Lin SC, Shyur SD, Huang LH. Neonatal lupus erythematosus with cholestatic hepatitis. J Microbiol Immunol Infect. 2004;37:131–134. [PubMed] [Google Scholar]
- 778.Rosh JR, Silverman ED, Groisman G. Intrahepatic cholestasis in neonatal lupus erythematosus. J Pediatr Gastroenterol Nutr. 1993;17:310–312. doi: 10.1097/00005176-199310000-00014. [DOI] [PubMed] [Google Scholar]
- 779.Kanitkar M, Rohini KP, Puri B, Nair MN. Neonatal lupus mimicking extra hepatic biliary atresia. Indian Pediatr. 2004;41:1252–1254. [PubMed] [Google Scholar]
- 780.Schoenlebe J, Buyon JP, Zitelli BJ. Neonatal hemochromatosis associated with maternal autoantibodies against Ro/SS-A and La/SS-B ribonucleoproteins. Am J Dis Child. 1993;147:1072–1075. doi: 10.1001/archpedi.1993.02160340058014. [DOI] [PubMed] [Google Scholar]
- 781.Lee LA, Sokol RJ, Buyon JP. Hepatobiliary disease in neonatal lupus: prevalence and clinical characteristics in cases enrolled in a national registry. Pediatrics. 2002;109:E11. doi: 10.1542/peds.109.1.e11. [DOI] [PubMed] [Google Scholar]
- 782.Selander B, Cedergren S, Domanski H. A case of severe neonatal lupus erythematosus without cardiac or cutaneous involvement. Acta Paediatr. 1998;87:105–107. doi: 10.1080/08035259850157994. [DOI] [PubMed] [Google Scholar]
- 783.Burch JM, Sokol RJ, Narkewicz MR. Autoantibodies in mothers of children with neonatal liver disease. J Pediatr Gastroenterol Nutr. 2003;37:262–267. doi: 10.1097/00005176-200309000-00012. [DOI] [PubMed] [Google Scholar]
- 784.Hannam S, Bogdanos DP, Davies ET. Neonatal liver disease associated with placental transfer of anti-mitochondrial antibodies. Autoimmunity. 2002;35:545–550. doi: 10.1080/0891693021000054057. [DOI] [PubMed] [Google Scholar]