

Abstract
Proteolytic fragmentation of polyglutamine-expanded ataxin-3 is a concomitant and modifier of the molecular pathogenesis of Machado–Joseph disease (MJD), the most common autosomal dominant cerebellar ataxia. Calpains, a group of calcium-dependent cysteine proteases, are important mediators of ataxin-3 cleavage and implicated in multiple neurodegenerative conditions. Pharmacologic and genetic approaches lowering calpain activity showed beneficial effects on molecular and behavioural disease characteristics in MJD model organisms. However, specifically targeting one of the calpain isoforms by genetic means has not yet been evaluated as a potential therapeutic strategy. In our study, we tested whether calpains are overactivated in the MJD context and if reduction or ablation of calpain-1 expression ameliorates the disease-associated phenotype in MJD cells and mice. In all analysed MJD models, we detected an elevated calpain activity at baseline. Lowering or removal of calpain-1 in cells or mice counteracted calpain system overactivation and led to reduced cleavage of ataxin-3 without affecting its aggregation. Moreover, calpain-1 knockout in YAC84Q mice alleviated excessive fragmentation of important synaptic proteins. Despite worsening some motor characteristics, YAC84Q mice showed a rescue of body weight loss and extended survival upon calpain-1 knockout. Together, our findings emphasize the general potential of calpains as a therapeutic target in MJD and other neurodegenerative diseases.
Introduction
Calpains have been characterized as modulators of proteotoxicity and degenerative processes in several neurological disorders, including the autosomal dominantly inherited Machado–Joseph disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3) (1,2). These calcium-dependent cysteine proteases, which perform vital regulatory functions in healthy cells, have been shown to be overactivated in neurodegenerative conditions like Alzheimer’s disease, Huntington’s disease or ischemic stroke. Calpains can excessively cleave disease-causing proteins and fragment neuronal substrates both in the pre- and post-synapse, thus inducing a detrimental neurotoxic cascade that ultimately leads to neuronal death (3–5).
In our previous studies, we reported on the calpain-mediated cleavage of the MJD protein ataxin-3 (6,7). By knocking out the gene for the endogenous calpain inhibitor calpastatin (CAST), we induced an aggravation of the neurological phenotype in MJD mice concomitant with increased neuronal cell death in the cerebellum and elevated aggregation of polyglutamine (polyQ)-expanded ataxin-3 in the brain (7). Moreover, we showed that both calpain-1 and calpain-2 cleave wild-type (WT) and polyQ-expanded ataxin-3 at specific cleavage sites, thus giving rise to breakdown products of different toxicities and aggregation propensities. More importantly, the resulting fragments were also detected in post-mortem brain tissue of MJD patients (6), underlining the functional relevance of the calpain system in the molecular pathogenesis of MJD. In addition, our investigations highlighted three distinct strategies for lowering calpain-mediated cleavage of ataxin-3 leading to the amelioration of the MJD disease phenotype. The first strategy is based on mutating identified cleavage sites within ataxin-3 to render the protein resistant to cleavage. This approach was successfully tested in our cell-based assays by performing triple tryptophan substitutions on the identified calpain recognition motifs (6). Alternatively, albeit with reportedly negative repercussions on the protein structure and function, antisense oligonucleotide-mediated exon-skipping strategies were used to remove a central 88-amino acid large region of ataxin-3 that harbours several cleavage sites for both calpains and caspases (8). The second approach involved the suppression of calpain activity either by pharmacological means or by the overexpression of CAST. The application of calpain-inhibiting compounds, such as BDA-410 or calpeptin, as well as the overexpression of CAST by injecting respective adeno-associated viral vectors, yielded beneficial effects on the molecular, histological and behavioural phenotypes of MJD mice or zebrafish (9–11). Finally, the third strategy aims at the direct knockout of one of the calpain isoforms, which was guided by our previously findings that calpain-1 efficiently cleaves ataxin-3 in vitro (6,7). Therefore, we sought to investigate the effect of calpain-1 lowering or removal in MJD cell lines and animal models.
Early investigations on the inactivation of the calpain-1-encoding gene in Capn1 knockout (Capn1−/−) mice demonstrated impaired platelet functions without any negative impact on viability and fertility (12). A later study based on the same mouse model showed neuroprotective effects of the calpain-1 loss in traumatic brain injury, suggesting that targeted inhibition of this protease may provide a therapeutic approach against neurodegenerative diseases in general (13). Interestingly, later examinations of the same Capn1−/− mice revealed a mild form of ataxia, starting at 3 months of age, seemingly caused by abnormal cerebellar development (14).
Notwithstanding the ambiguous outcomes of previous studies, we evaluated the impact of knocking out calpain-1 on polyQ-expanded ataxin-3 and, consequently, on MJD. First, we investigated the molecular pathogenesis of the disease to ascertain whether calpains are overactivated in MJD consistent with their known activation in other neurodegenerative disorders. Next, we tested our calpain-1 lowering hypothesis in vitro by performing gene knockdowns in cells overexpressing WT and polyQ-expanded human ataxin-3 and analysing their effects on calpain system activation and ataxin-3 cleavage. These molecular markers, extended by ataxin-3 aggregation and known neuronal substrates of calpains, were examined in MJD mice endowed with a genetic ablation of Capn1. Finally, we evaluated the phenotype of the genetically modified mice by performing body weight measurements, gait and activity analyses and assessment of motor skills and survival to tease out the specific role of calpain-1 in MJD.
Results
The calpain system is overactivated in cell models of MJD
A potentially indirect indication for a dysregulated calpain system in MJD was previously shown by detecting reduced CAST levels in mice and post-mortem brain of patients (10). To further explore this molecular condition, we analysed primary fibroblasts of healthy control subjects and MJD patients, and HEK 293T ATXN3−/− cells transiently transfected with V5-tagged WT and polyQ-expanded ataxin-3. Western blot analysis of cell lysates showed a significantly increased fragmentation of α-spectrin, a known substrate of calpains and indicator of calpain activation (15), in both MJD fibroblasts and ataxin-3 70Q-overexpressing cells compared to their respective control cells (Fig. 1A–D). In the latter model, the enhanced α-spectrin cleavage was furthermore accompanied by an elevated auto-proteolysis of calpain-1 (Fig. 1C and D). The observation of an increased α-spectrin cleavage in patient-derived cells and an MJD cell model suggests that calpain overactivation is triggered by the expression of polyQ-expanded ataxin-3 representing a molecular hallmark of the disease.
Figure 1.
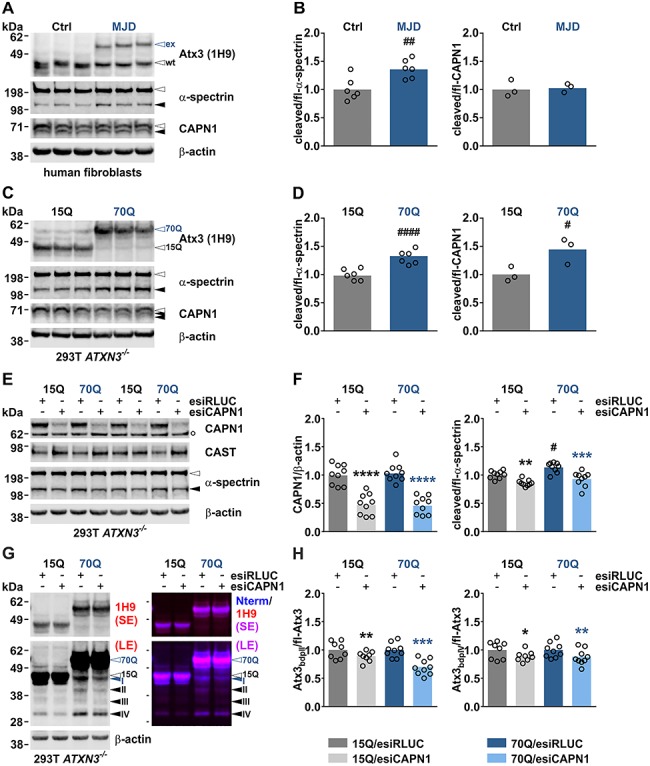
Calpain system overactivation and ataxin-3 cleavage in MJD cell models is lowered by calpain-1 knockdown. (A) Western blot of cleaved α-spectrin and processed calpain-1 in fibroblasts derived from healthy control (Ctrl) subjects and MJD patients. Genotypes were confirmed by detecting ataxin-3. Actin served as a loading control. (B) Densitometric quantification of α-spectrin cleavage (n = 6) and calpain processing (n = 3). #:Ctrl versus MJD; ##P ≤ 0.01; Student’s t-test. (C) Western blot of cleaved α-spectrin and processed calpain-1 in HEK 293T ATXN3−/− cells (293T ATXN3−/−) transfected with V5-tagged ataxin-3 15Q or 70Q. Genotypes were confirmed by detection of ataxin-3. Actin served as a loading control. (D) Densitometric quantification of α-spectrin cleavage (n = 6) and calpain-1 processing (n = 3). #:15Q versus 70Q; #P ≤ 0.05, ####P ≤ 0.0001; Student’s t-test. (E) Western blot of calpain-1, CAST and cleaved α-spectrin in HEK 293T ATXN3−/− cells double transfected with V5-tagged ataxin-3 15Q or 70Q and esiRNA directed against human calpain-1 (esiCAPN1) or control esiRNA against Renilla luciferase (esiRLUC). Actin served as a loading control. (F) Densitometric quantification of calpain-1 levels (n = 9) and α-spectrin cleavage (n = 9). *(black): 15Q/esiRLUC versus 15Q/esiCAPN1, *(blue): 70Q/esiRLUC versus 70Q/esiCAPN1, #:15Q/esiRLUC versus 70Q/esiRLUC; #P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001; one-way ANOVA with Tukey post-test. (G) Western blot analysis of ataxin-3 cleavage in ATXN3−/− cells double transfected with V5-tagged ataxin-3 15Q or 70Q and esiRNA directed against human calpain-1. Ataxin-3 breakdown products (bdp) were detected using antibodies specific against the centre (1H9) or the N-terminus (Nterm) of the protein. Full-length ataxin-3 15Q or 70Q is indicated by black- or blue-lined white arrowheads, respectively. Black arrowheads indicate N-terminal fragments, the blue one a polyQ-containing ataxin-3 breakdown product. Actin served as a loading control. (H) Densitometric quantification of two prominent ataxin-3 fragments bdp II and bdp IV (for both 15Q: n = 8; 70Q: n = 9). Values were normalized to respective controls ataxin-3 15Q/esiRLUC or ataxin-3 70Q/esiRLUC. *(black): 15Q/esiRLUC versus 15Q/esiCAPN1, *(blue): 70Q/esiRLUC versus 70Q/esiCAPN1; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; Student’s t-test. White arrowheads indicate full-length proteins, blue and black arrowheads protein fragments. White bullet (°) indicates an unspecific band. Bdp = breakdown product, ex = polyQ-expanded ataxin-3, fl = full-length, LE = long exposure, SE = short exposure, wt = wild-type ataxin-3. Columns represent means.
Calpain-1 knockdown lowers calpain activation and ataxin-3 cleavage in HEK 293T cells
WT and polyQ-expanded ataxin-3 were identified as substrates of calpains, and proteolytic fragments containing the expanded polyQ stretch may contribute to the molecular pathogenesis of MJD (2,16). In our previous studies, we proved that the best-characterized and ubiquitously expressed calpain isoforms, calpain-1 and calpain-2, are both capable of cleaving ataxin-3 in vitro, and proteolysis by either of the isoforms leads to comparable ataxin-3 fragmentation patterns (6,7). Now, we sought to investigate the effect of the calpain isoform-specific ablation on overall calpain system activity and on ataxin-3 cleavage by firstly knocking down calpain-1 in HEK 293T ATXN3−/− cells, overexpressing WT and polyQ-expanded ataxin-3. For this purpose, cells were double transfected with V5-tagged ataxin-3 15Q or 70Q and esiRNA directed against human calpain-1 for 48 h, and the repercussion of the knockdown in thereof generated lysates was analysed by western blotting. Immunodetection showed reduction of calpain-1 protein levels by more than 50%, which subsequently led to a robust reduction in α-spectrin cleavage, but without clearly altering CAST levels (Fig. 1E and F, Supplementary Material, Fig. S1A). Analysis of ataxin-3 cleavage using antibodies directed against the centre (1H9) or the N-terminus (Nterm) of the protein detected up to four specific fragments (Fig. 1G, Supplementary Material, Fig. S1B). Based on size comparisons between the cleaved forms of ataxin-3 15Q and 70Q, three of these fragments represent most-likely N-terminal breakdown products (black arrowheads II—IV), whereas one may constitute a polyQ-containing fragment (blue arrowhead I). Quantification of two prominent fragments (II and IV) showed a significant decrease upon calpain-1 knockdown (Fig. 1H). Overall, knockdown of the calpain-1 isoform was sufficient to reduce total calpain system activation and the generation of specific WT and polyQ-expanded ataxin-3 fragments in a human cell line.
Calpain-1 knockout in MJD mice attenuates calpain system overactivation
After successfully proving that calpain-1 knockdown lowers ataxin-3 fragmentation in cells, we aimed for reproducing this finding in vivo. For this, we crossbred a well-established YAC transgenic mouse model for MJD (SCA3-YAC-84Q, hereinafter referred to as YAC84Q) with previously characterized mice featuring a genetically inactivated Capn1 gene (12,17). To confirm knockout of calpain-1 in single (Capn1−/−) and double-mutant animals (YAC84Q/Capn1−/−), we performed western blot analysis of whole brain lysates generated from mice at 3 months of age. Detection of calpain-1 showed the complete absence of the specific full-length protein band in knockout-positive animals (Fig. 2A). As a response to the genetic depletion of calpain-1, levels of CAST increased, and both calpain-specific cleavage of α-spectrin and conversion of p35, another neuronal substrate of calpains (18), to p25 were reduced (Fig. 2A and B). This observation points to a substantial contribution of the calpain-1 enzyme to the baseline fragmentation of analysed substrates. Moreover, as shown before in our calpain-1 knockdown experiments in HEK 293T cells, expression of human polyQ-expanded ataxin-3 triggered significantly increased calpain activation in YAC84Q animals compared to WT mice. This was indicated by elevated levels of cleaved α-spectrin and p25 and lowered levels of CAST at baseline (Fig. 2A and B). As Capn1−/− animals exhibit a global loss of calpain-1 expression in all tissues, we investigated the impact of the calpain-1 knockout in a peripheral tissue. For this, we analysed the calpain activation in lysates of femoral muscle tissue. Western blot analysis confirmed the knockout of calpain-1 on protein level. The lack of calpain-1 expression led to a distinct elevation of CAST levels, whereas alterations of α-spectrin cleavage were not clearly detectable (Supplementary Material, Fig. S2A).
Figure 2.
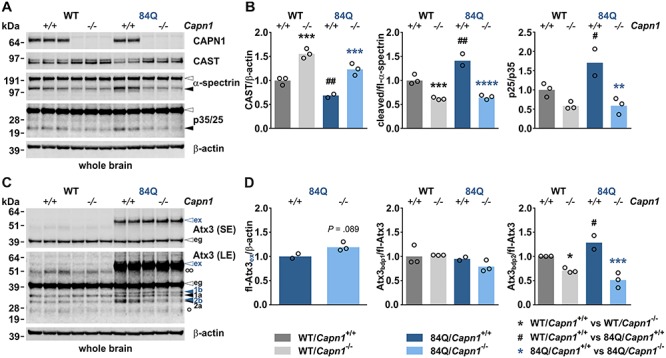
Calpain-1 knockout in MJD mice lowers calpain system overactivation and cleavage of ataxin-3. (A) Western blot of calpain-1, CAST and substrate proteins α-spectrin and p35 in whole brain of 3-month-old mice. Actin served as a loading control. (B) Densitometric quantification of CAST levels, α-spectrin cleavage and p35-to-p25 conversion (WT/Capn1+/+: n = 3; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 2; 84Q/Capn1−/−: n = 3). *(black): WT/Capn1+/+ versus WT/Capn1−/−, *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−, #:WT/Capn1+/+ versus 84Q/Capn1+/+; #P ≤ 0.05, **/##P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001; one-way ANOVA with Fisher’s LSD post-test. (C) Western blot analysis of ataxin-3 levels and cleavage in whole brain of 3-month-old mice. Ataxin-3 fragmentation was detected using the centre-binding antibody 1H9. Ataxin-3 breakdown products (bdp) are indicated with black and blue arrowheads (labelled with 1a, 1b, 2a and 2b). Blue arrowheads indicate ataxin-3 fragments specifically formed in YAC84Q animals. Actin served as a loading control. (D) Densitometric quantification of full-length polyQ-expanded ataxin-3 and of two prominent ataxin-3 fragments bdp 1 (a and b) and bdp 2 (a and b) (WT/Capn1+/+: n = 3; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 2; 84Q/Capn1−/−: n = 3). *(black): WT/Capn1+/+ versus WT/Capn1−/−, *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−, #:WT/Capn1+/+ versus 84Q/Capn1+/+; *,#P ≤ 0.05, ***P ≤ 0.001; Student’s t-test (for full-length polyQ-expanded ataxin-3) and one-way ANOVA with Fisher’s LSD post-test (for all other comparisons). White arrowheads indicate full-length proteins, black arrowheads protein fragments. White bullets indicate heavy (°°) and light (°) IgG chains. eg = endogenous ataxin-3, ex = polyQ-expanded ataxin-3, fl = full-length, LE = long exposure, SE = short exposure. Columns represent means.
Calpain-1 knockout in MJD mice lowers cleavage of ataxin-3
After confirming that knockout of calpain-1 resulted in reduced global calpain activation in vivo, we further analysed whole brain lysates of 3-month-old mice by western blotting to survey full-length levels and fragments of ataxin-3. Detection with the ataxin-3-specific antibody 1H9 revealed a distinct fragmentation pattern for ataxin-3 in mouse brain (Fig. 2C, black arrowheads 1a and 2a). Compared to WT animals, a variation of this pattern could be observed in YAC84Q mice, which featured additional cleavage bands (Fig. 2C, blue arrowheads 1b and 2b). These marginally bigger fragments appeared to be specific N-terminal breakdown products of the polyQ-expanded human ataxin-3, which cannot be explained by an expectedly much stronger polyQ-dependent size shift and may point to variations between calpain recognition sites within the murine and human ataxin-3. Importantly, the 1H9 immunostaining showed a significant reduction of fragments 2a and 2b in Capn1−/− animals and double-mutant YAC84Q/Capn1−/− mice, which was increased in YAC84Q animals compared to WT (Fig. 2C and D). Fragments 1a and 1b showed only a trend towards a decrease in double-mutant animals. This observation confirms our earlier findings that ataxin-3 fragments are not only generated in a calpain-1-dependent manner, as we were able to show that calpain-2 can also cleave WT and polyQ-expanded ataxin-3. In addition, we investigated the effects of the calpain-1 knockout on the ataxin-3 fragmentation in femoral muscle tissue samples. Here, the main cleavage pattern was comparable to the one found in mouse brain (Supplementary Material, Fig. S2B). However, due to a higher variability and low sample number, our results remained inconclusive.
Moreover, we analysed polyQ-expanded human ataxin-3 levels in brain of double-mutant animals using both western blotting and a specific and highly sensitive TR-FRET approach. Our measurements showed an increase of full-length polyQ-expanded ataxin-3 in YAC84Q/Capn1−/− mice compared to YAC84Q animals at 3 months of age (Fig. 2C and D and 3A), which might be a direct effect of the lowered calpain-dependent fragmentation. On the other side, TR-FRET analysis of polyQ-expanded ataxin-3 levels in 15-month-old animals did not show the effects observed in young animals (Fig. 3D).
Figure 3.
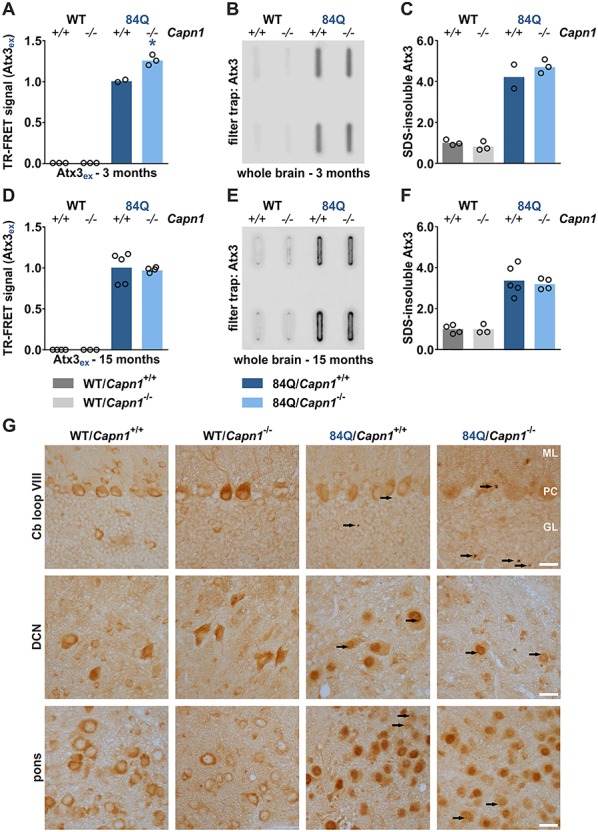
Increased soluble levels of polyQ-expanded ataxin-3 but no alterations in ataxin-3 aggregates upon calpain-1 knockout. (A, D) TR-FRET analysis of polyQ-expanded ataxin-3 levels in whole brain of 3-month (WT/Capn1+/+: n = 3; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 2; 84Q/Capn1−/−: n = 3) and 15-month-old mice (WT/Capn1+/+: n = 4; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 5; 84Q/Capn1−/−: n = 4). *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−; *P ≤ 0.05; Student’s t-test. (B, C) Filter trap analysis and densitometric quantification of SDS-insoluble polyQ-expanded ataxin-3 in whole brain of 3-month-old mice (WT/Capn1+/+: n = 3; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 2; 84Q/Capn1−/−: n = 3). (E, F) Filter trap analysis and densitometric quantification of SDS-insoluble polyQ-expanded ataxin-3 in whole brain of 15-month-old mice (WT/Capn1+/+: n = 4; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 5; 84Q/Capn1−/−: n = 4). (G) Immunohistochemical analysis of cerebellum (Cb) loop VIII, deep cerebellar nuclei (DCN) and pons using the ataxin-3 antibody 1H9. Aggregates are indicated by black arrows. ML = molecular layer, PC = Purkinje cells, GL = granular layer. Scale bar = 20 μm. ex = polyQ-expanded ataxin-3. Columns represent means.
Ataxin-3 aggregation levels are not altered in double-mutant mice
Proteolytic fragmentation of polyQ proteins has been suggested as the source of not only toxic but also aggregation-prone breakdown products and thus as a contributing factor to the formation of protein aggregates (2). To investigate the effects of the calpain-1 knockout on ataxin-3 aggregation in vivo, we performed filter trap assays of brain homogenates from 3- and 15-month-old mice. Rather surprisingly, detection with the ataxin-3-specific antibody 1H9 showed no major differences in the aggregate levels between both genotypes neither at 3 months nor at 15 months of age (Fig. 3B, C, E and F). Additional immunohistochemical stainings of brain sections from animals at 15 months of age detected comparably low amounts of microscopically distinguishable ataxin-3-positive neuronal inclusions in cerebellum (Cb loop VIII), deep cerebellar nuclei (DCN) and pons, without any indication of a changed aggregate number in double-mutant mice (Fig. 3G). Interestingly, the histological analysis revealed a more intensive nuclear staining for ataxin-3 in neurons of both YAC84Q and YAC84Q/Capn1−/− mice, best seen in DCN and pons, whereas in WT and Capn1−/− mice ataxin-3 was mainly present in the cytoplasm (Fig. 3G). This observation corroborates baseline differences in the primary subcellular localization of (murine) WT and (human) polyQ-expanded ataxin-3, which has been described earlier for this model (19).
Calpain-1 knockout counteracts excessive cleavage of synaptic proteins in MJD animals
As calpains are known to cleave and regulate multiple neuronal proteins and thereby modulate, e.g. synaptic function (20–22), we analysed a selection of known substrate proteins at the pre- and post-synapse in whole brain lysates of 3-month-old animals. Western blot analyses of two presynaptic proteins synapsin-1 and synapsin-2a, the synaptosomal-associated protein 25 (SNAP-25) and the postsynaptic density protein 95 (PSD-95) demonstrated distinct alterations in full-length levels and/or fragmentation. Synapsin-1 and -2a full-length levels were reduced in YAC84Q mice and partly rescued by the calpain-1 knockout in double-mutant animals. These alterations were accompanied by elevated synapsin fragment levels in YAC84Q mice at baseline, which were markedly lowered by the loss of calpain-1, not only in YAC84Q/Capn1−/− but also in Capn1−/− mice (Fig. 4A and B). This observation suggests a calpain cleavage-dependent origin of the detected breakdown products, dovetailing with our previous findings in Huntington’s disease mice (23). Similar to synapsin-1 and 2a, SNAP-25 fragmentation was increased in YAC84Q mice and strongly diminished upon calpain-1 knockout (Fig. 4C and D). We did not detect fragmentation of PSD-95; however, despite not showing differences between WT and YAC84Q mice at baseline, lack of calpain-1 led to higher full-length levels in respective animals (Fig. 4C and D). These results show that YAC84Q mice feature dysregulations of different synaptic proteins at an early age, and these changes could be counteracted by the genetic inactivation of calpain-1.
Figure 4.
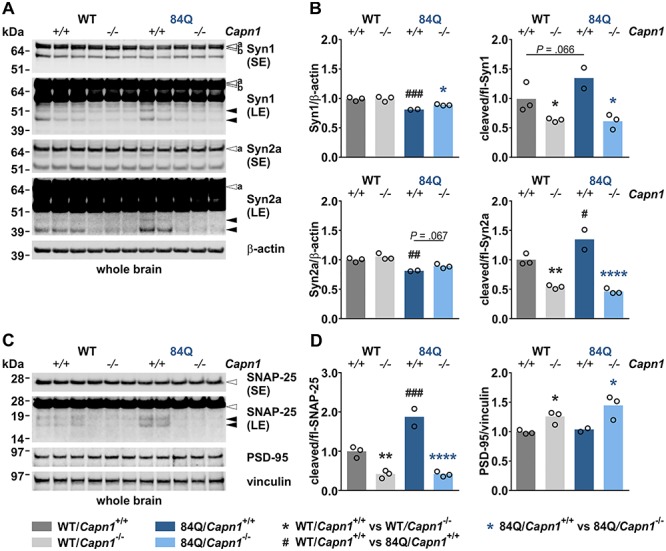
Calpain-1 knockout counteracts excessive cleavage of synaptic proteins in MJD animals. (A) Western blot of synapsin-1 and synapsin-2a in whole brain of 3-month-old mice. Actin served as a loading control. (B) Densitometric quantification of synapsin-1 and -2a full-length levels and fragmentation (WT/Capn1+/+: n = 3; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 2; 84Q/Capn1−/−: n = 3). *(black): WT/Capn1+/+ versus WT/Capn1−/−, *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−, #:WT/Capn1+/+ versus 84Q/Capn1+/+; */#P ≤ 0.05, **/##P ≤ 0.01, ###P ≤ 0.001, ****P ≤ 0.0001; one-way ANOVA with Fisher’s LSD post-test. (C) Western blot of SNAP-25 and PSD-95 in whole brain of 3-month-old mice. Vinculin served as a loading control. (D) Densitometric quantification of SNAP-25 cleavage and PSD-95 levels (WT/Capn1+/+: n = 3; WT/Capn1−/−: n = 3; 84Q/Capn1+/+: n = 2; 84Q/Capn1−/−: n = 3). *(black): WT/Capn1+/+ versus WT/Capn1−/−, *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−, #:WT/Capn1+/+ versus 84Q/Capn1+/+; *P ≤ 0.05, **P ≤ 0.01, ###P ≤ 0.001, ****P ≤ 0.0001; one-way ANOVA with Fisher’s LSD post-test. White arrowheads indicate full-length proteins, black arrowheads protein fragments, ‘a’ and ‘b’ mark respective synapsin isoforms. fl = full-length, LE = long exposure, SE = short exposure. Columns represent means.
Calpain-1 knockout mice feature a gait phenotype and an early reduced ambulatory activity
Primarily, Capn1−/− mice were characterized as viable and fertile animals with no obvious behavioural phenotype but an impairment in platelet function (12). In a later study, Wang and colleagues demonstrated a mild form of ataxia due to a slightly abnormal cerebellar development (14). To further examine a potential neurological phenotype of Capn1−/− mice, we performed comprehensive behaviour analyses every 3 months starting at 3 months of age. Body weight assessment both for males and females revealed no significant weight differences between Capn1−/− mice and WT controls over a period of 56 weeks (Supplementary Material, Fig. S6A). Likewise, no discrepancies in the food or water intake were detected (Fig. 5D, Supplementary Material, Fig. S6B). Interestingly, gait analyses using the CatWalk system showed a reduced footprint area in hind limbs, which was most prominent in females, but without alterations in average speed and only a trend towards an increase in total number of steps per run in females (Fig. 5E, Supplementary Material, Figs S3 and S6C). Reduced footprint area in hind limbs was accompanied by tendency in increased base of support in females (Supplementary Material, Fig. S4). Moreover, Capn1−/− mice did not show deficits in rotarod performance test (Fig. 5F). However, at 3 months of age, impaired learning was shown during rotarod training sessions together with decreased ambulatory activity as assessed by LabMaster analysis (Supplementary Material, Fig. S5). Taken together, the assessed characteristics of Capn1−/− mice point towards a slight gait, learning and activity phenotype, already beginning at an early age.
Figure 5.
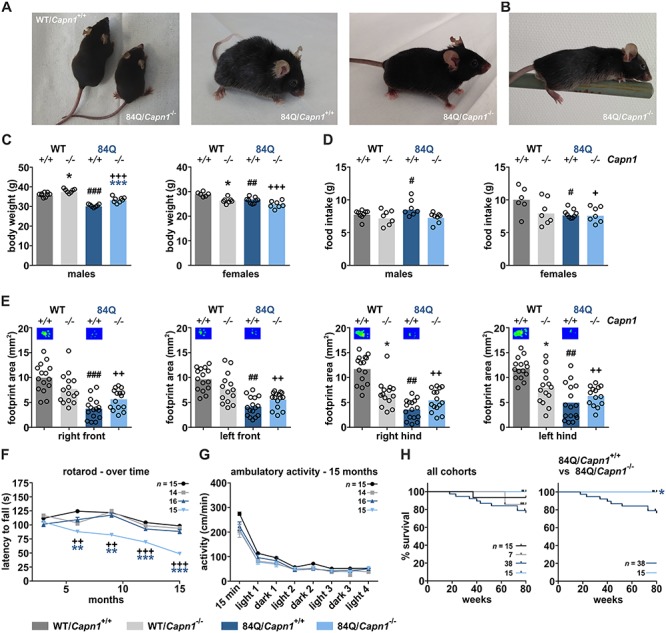
Calpain-1 knockout worsens motor deficits but rescues body weight loss and improves survival of MJD mice. (A) Representative pictures of mice at 9 months of age. (B) Picture of a YAC84Q/Capn1−/− mouse performing a pen test. (C) Sex-dependent body weight analysis at 56 weeks of age (WT/Capn1+/+: n = 9 ♂/6 ♀; WT/Capn1−/−: n = 7 ♂/7 ♀; 84Q/Capn1+/+: n = 7 ♂/9 ♀; 84Q/Capn1−/−: n = 8 ♂/7 ♀). *(black): WT/Capn1+/+ versus WT/Capn1−/−, *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−, #(black): WT/Capn1+/+ versus 84Q/Capn1+/+, +: WT/Capn1+/+ versus 84Q/Capn1−/−; *P ≤ 0.05, ##P ≤ 0.01, ***/###/+++P ≤ 0.001; ANOVA with Fisher’s LSD post-test. (D) Sex-dependent home cage-based LabMaster analysis of food intake at 15 months of age (WT/Capn1+/+: n = 9 ♂/6 ♀; WT/Capn1−/−: n = 7 ♂/7 ♀; 84Q/Capn1+/+: n = 7 ♂/9 ♀; 84Q/Capn1−/−: n = 8 ♂/7 ♀). #(black): WT/Capn1+/+ versus 84Q/Capn1+/+, +: WT/Capn1+/+ versus 84Q/Capn1−/−; #/+P ≤ 0.05; ANOVA with Fisher’s LSD post-test. (E) Footprint area determination of all four paws using CatWalk gait analysis at 15 months of age (WT/Capn1+/+: n = 15; WT/Capn1−/−: n = 14; 84Q/Capn1+/+: n = 16; 84Q/Capn1−/−: n = 15). *(black): WT/Capn1+/+ versus WT/Capn1−/−, #(black): WT/Capn1+/+ versus 84Q/Capn1+/+, +: WT/Capn1+/+ versus 84Q/Capn1−/−; *P ≤ 0.05, ##/++P ≤ 0.01, ###P ≤ 0.001; two-way ANOVA with Bonferroni post-test. (F) Analysis of motor deficits using the rotarod performance test as assessed every 3 months between 3 and 15 months of age. Number of animals is indicated in the panel. *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−, +: WT/Capn1+/+ versus 84Q/Capn1−/−; **/++P ≤ 0.01, ***/+++P ≤ 0.001; two-way ANOVA with Bonferroni post-test. (G) Ambulatory activity assessment during light and dark phases using the home cage-based LabMaster system at 15 months of age. Number of animals is indicated in the panel. (H) Survival analysis of all cohorts or YAC84Q and YAC84Q/Capn1−/− mice until an age of 80 weeks using the Kaplan–Meier estimator. Number of animals is indicated in the panels. *(blue): 84Q/Capn1+/+ versus 84Q/Capn1−/−; *P ≤ 0.05; log-rank test. Columns represent means and data points show means ± SEM.
Double-mutant mice show impaired motor performance, but rescued body weight and improved survival
After investigating repercussions of the calpain-1 knockout in MJD mice on several molecular markers, we analysed its manifestation on physical characteristics and the behavioural level. Like the other cohorts, double-mutant mice were viable and fertile and showed normal weight at birth, unaffected posture, groomed hair coat and no body tremor (Fig. 5A). General health assessment each second week revealed marked body weight loss of male YAC84Q mice starting at 36 weeks of age, which was counteracted by the calpain-1 knockout in double-mutant animals (Fig. 5C, Supplementary Material, Fig. S6A). Body weight in females was varying to a lesser extent, without observing a decline but generally lower values not only for double-mutant but also for single-mutant and YAC84Q animals compared to WT controls (Fig. 5C, Supplementary Material, Fig. S6A). In contrast to female mice, where the lower body weight could be correlated to a lower food intake, in male mice both body weight loss in YAC84Q animals and rescue in YAC84Q/Capn1−/− mice could not be linked to differences in feeding behaviour as assessed by the home cage-based LabMaster system at 15 months of age (Fig. 5D). Gait analysis using the CatWalk system did not show significant differences at 6 months and only slight effects in double-mutant mice at 12 months (data not shown). At 15 months, however, an irregular footprint pattern of all four limbs in YAC84Q mice was detected, which showed no obvious improvement in double-mutant animals (Fig. 5E). These characteristics were comparable for both male and female animals (Supplementary Material, Fig. S3A, C and D). Additional parameters like speed, number of steps and base of support were unchanged or nearly unchanged in all analysed genotypes and sexes (Supplementary Material, Fig. S3B, S4 and S6C). However, YAC84Q and double-mutant mice appeared to show wide-based hind limbs during sitting and walking in the normal home cage and difficulties in holding fast onto a pen in the respective test (Fig. 5A and B). Interestingly, rotarod performance test from 3 to 15 months of age demonstrated significant and progressive coordination deficits present in double-mutant mice but not in YAC84Q or single-mutant animals (Fig. 5F). This effect could be observed likewise in both sexes (Supplementary Material, Fig. S6D). Activity measurements for 70 h in the home cage-based LabMaster system, by contrast, revealed no abnormalities between all genotypes at 15 months of age (Fig. 5G). Separation by sex showed only a few isolated discrepancies in males and females without indication of a clear trend (Supplementary Material, Fig. S6E). Finally, we assessed the impact of a calpain-1 knockout on mouse overall survival until 80 weeks of age. Notably, the YAC84Q cohort showed a reduced lifespan, with mice beginning to die at 20 weeks of age. In contrast to this, double-mutant animals showed a normal lifespan without any losses within the investigated timeframe (Fig. 5H). In summary, behavioural analysis of double-mutant mice in comparison with YAC84Q animals showed, on the one hand, a progressive worsening of the motor phenotype as assessed by rotarod performance test, but on the other, rescue of body weight loss in males, and an improved overall survival.
Discussion
In this study, we employed esiRNA-mediated knockdown and genetic inactivation of calpain-1 in human cells and in the YAC84Q mouse model of MJD, respectively, as a potential therapeutic strategy for suppressing calpain activation and calpain-mediated cleavage of polyQ-expanded ataxin-3. Ataxin-3 fragmentation has been implicated as the source for toxic fragments seeding formation of aggregates, and its reduction might alleviate the disease progression (2). Our previous investigations have shown that calpain-1 and calpain-2 can cleave both WT and polyQ-expanded ataxin-3. Moreover, the induced overactivation of the calpain system by knocking out endogenous inhibitor CAST led to a worsening MJD phenotype in vivo (6,7). Other studies in mouse and zebrafish models of MJD demonstrated that diminishing calpain activity by CAST overexpression or by pharmacological inhibition lowered ataxin-3 cleavage and fragmentation and prevented cerebellar degeneration and motor behavioural deficits (9–11). Thus, genetic ablation of one of the calpain isoforms associated with polyQ-expanded ataxin-3 cleavage may represent an option to ameliorate disease-related molecular and phenotypic abnormalities.
Calpain overactivation is present in cell and animal models of MJD
To determine if the calpain activity is modulated at baseline in MJD, we first evaluated calpain substrates in polyQ-expanded ataxin-3-transfected cells, MJD patient-derived fibroblasts and brains of the YAC84Q mouse model by western blotting. Evidenced by the increased cleavage of multiple substrates, including α-spectrin and p35, we found an elevated activation of the calpain system in the tested model systems. Our results are consistent with previous findings in neurodegenerative disorders such as Alzheimer’s disease, amyotrophic lateral sclerosis or Huntington’s disease, where calpains have been shown to be overactivated in cell and animal models, and in patient tissue samples (24–30). Autoproteolysis of calpain-1, a concomitant of calpain activation without any direct relevance of the enzyme’s activation state (31), was raised in polyQ-expanded ataxin-3-transfected cells and YAC84Q brains, but not in patient fibroblasts. This discrepancy may indicate a divergent global calpain activation, potentially based on a different calpain isoform. Our observation of relatively low CAST levels in YAC84Q mice comply with evidence from another transgenic MJD mouse model and post-mortem patient brain tissue analyses demonstrating reduced levels of the endogenous calpain inhibitor (10). The increased calpain-mediated p35-to-p25 conversion, which we observed in YAC84Q mice, is known to result in p25-mediated hyperactivation of cyclin-dependent kinase 5 (CDK5) associated with neurodegenerative effects in Alzheimer’s disease (18,32). Our results further emphasize the general validity of calpain overactivation as a unifying pathological mechanism for therapeutic targeting in neurodegeneration.
Calpain-1 ablation as an efficient countermeasure for preventing ataxin-3 cleavage
Since calpain-1 efficiently cleaves polyQ-expanded ataxin-3, we tested if its depletion would lead to the suppression of ataxin-3 fragmentation. We performed both calpain-1 knockdown in cells expressing polyQ-expanded ataxin-3 and crossbred Capn1−/− mice with hemizygous YAC84Q animals. Western blot analyses showed that calpain-1 knockdown in cells and knockout in mice led to a robust lowering of calpain system activation as assessed by α-spectrin and p35 fragmentation. Moreover, CAST levels were significantly increased in mice featuring a calpain-1 knockout, which points to CAST’s suggested mode of action as a suicidal substrate type inhibitor of calpains (33). Both approaches reduced both WT and polyQ-expanded ataxin-3 cleavage and, consequently, increased full-length levels of polyQ-expanded ataxin-3 in 3-month-old mice. Similar results have been reported by the CAST overexpression and administration of pharmacological calpain inhibitors in different MJD cell and animal models (6,9–11,34). Our western blot analyses showed that counteracting calpain-1-mediated cleavage reduced but not totally abolished the formation of several most-likely N-terminal fragments in both MJD cells and mice. The absence of complete abrogation can be attributed to the circumstance that not only calpain-1, but also additional calpains and further proteases have been associated with cleavage of the ataxin-3 protein (2,6). As observed in our previous work (6), C-terminal ataxin-3 fragments and effects on them were hardly detectable, thwarting direct evaluation on breakdown products comprising the polyQ stretch-containing portion of the disease protein. However, our readout on N-terminal fragments implies consequential repercussions on their C-terminal counterparts. Furthermore, lowering the generation of N-terminal fragment species of ataxin-3 may have beneficial effects on the disease phenotype as C-terminally truncated forms of ataxin-3 lacking the polyQ stretch were also demonstrated as potentially relevant for MJD’s molecular pathology (35,36). As C-terminal fragments of polyQ-expanded ataxin-3 have been shown to feature an increased aggregation propensity and may act as a trigger for neuronal inclusion formation, we analysed the effects of calpain-1 knockout on ataxin-3 aggregate load in mouse brain using filter trap assays and immunohistochemistry. In contrast to previous studies that employed non-isoform-specific pharmacological or CAST-dependent inhibition of calpains (10,11,34,37), we did not observe the reduction of ataxin-3 aggregation in response to lower ataxin-3 cleavage in YAC84Q/Capn1−/− mice. The lack of a discernible effect of calpain-1 knockout on ataxin-3 aggregation at both 3 and 15 months of age might be a consequence of multiple factors: the absence of observed changes in the aggregate levels at earlier time points may be due to the comparatively low load of ataxin-3 inclusions despite both hemizygous and homozygous YAC84Q mice showing robust intranuclear accumulation of polyQ-expanded ataxin-3 as early as 6–8 weeks of age (38). This circumstance, however, does not explain the negative results observed at 15 months, which also include the lacking effects on full-length ataxin-3 levels as measured by TR-FRET, and is noteworthy considering the often-described role of calpains as negative regulators of autophagy and protection against polyQ toxicity via the calpain inhibition-dependent induction of autophagy (39–41). In fact, our immunohistochemical analysis demonstrated a generally low occurrence of ataxin-3-positive inclusions in the mouse brain even at the later age of 15 months. A further conceivable—but not yet investigated—effect-levelling factor in older animals might be an aging-dependent attenuation of the calpain-system overactivation, which has been earlier reported in a Huntington’s disease mouse model (42).
Impact of the calpain-1 knockout on synaptic markers
Calpains are known to function as important modulators of synaptic function. They regulate stability and activity of various substrates within the pre- and post-synapse, including multiple enzymes, receptors and scaffolding proteins, and potentially contribute to pathological conditions (20–22). In neurodegenerative disorders such as Huntington’s disease, compromised synaptic connectivity and transmission are thought to play a crucial early role in disease pathology (43). In Alzheimer’s disease, dementia with Lewy bodies and Parkinson’s disease, proteomic studies have revealed impairment of synaptic proteins, thus pointing to their predictive and therapeutic potential (44). However, in MJD and respective model organisms thereof, the precise role of synaptic dysfunction remains poorly understood. Therefore, we evaluated a set of synaptic proteins, including known calpain substrates, such as presynaptic synapsins (45), the pre- and post-synaptic protein SNAP-25 (46,47) and postsynaptic scaffolding protein PSD-95 (48). We detected reduced levels of synapsin-1 and -2a in YAC84Q animals, concomitant with increased levels of respective protein fragments. The elevated fragmentation was lowered, and the full-length protein levels were partly rescued by the ablation of calpain-1 in the double-mutant animals. These findings correspond to one of our earlier studies on Huntington’s disease, where we detected increased calpain-dependent synapsin cleavage in a respective mouse model (23). Moreover, we observed comparable effects on SNAP-25, where the increased protein fragmentation at baseline was abrogated by the calpain-1 knockout. Changes in SNAP-25 have earlier been shown in post-mortem dentate nucleus of MJD patients (49) Lastly, the at baseline unaltered levels of PSD-95 were raised and the elevated cleavage of α-spectrin, an essential neuronal and synaptic scaffolding protein (50,51), was attenuated in YAC84Q/Capn1−/− mice. The loss of synaptic proteins and their increased cleavage may suggest a pathologically increased turnover of key regulators of synapses triggering detrimental effects on neuronal functions in MJD and contributing to the progression of the pathogenesis. However, this phenotype could also represent a compensatory effect against a polyQ-expanded ataxin-3-dependent neurotoxicity. Despite some evidence on synaptic dysfunctions in MJD (52–54), the lack of comprehensive experimental data demands further investigations in additional MJD models and patient tissue.
Repercussions of calpain-1 gene inactivation on the MJD mouse phenotype
To examine functional consequences of the calpain-1 knockout and the associated reduction in cleavage of ataxin-3 and other synaptic substrates, we assessed several physical and behavioural characteristics in our cohorts. Despite a normal appearance, unchanged body weight and no difference in the food intake, the Capn1−/− mice showed reduced ambulatory activity, most prominent in females, and an impaired learning phenotype in the rotarod training at 3 months of age. These changes were, however, not detected at 15 months of age. Initial characterizations of this mouse model described them as fertile and viable, without obvious (neurological) abnormalities, but an impairment in platelet function in adult mice (12). Consistent with our observations in the young—but not old—animals, a recent study reported an impaired learning phenotype in the rotarod performance test (14). Moreover, our Capn1−/− mice demonstrated a reduced hind limb footprint pattern and thus indicating an irregular use of hind limbs in gait analyses at 15 months of age, which confirms the previously described mild ataxic phenotype (14). Correspondingly, it was shown that knockout or knockdown of the orthologues of calpain-1 is linked to locomotor deficits in Drosophila melanogaster, Caenorhabditis elegans and zebrafish, accompanied by axonal abnormalities and demise of GABAergic motor neurons (55). Importantly, mutations in the CAPN1 gene lead to an ataxic phenotype in Parson Russell Terriers and autosomal recessive hereditary spastic paraplegia or spastic ataxia in humans (14,55–58).
Guided by the interesting neurological phenotypes in Capn1−/− mice, we further analysed the ramifications of the calpain-1 knockout on the MJD mice phenotype. Our YAC84Q/Capn1−/− mice revealed a pronounced motor phenotype as assessed by rotarod performance test, starting at 6 months of age and most prominent in 15-month-old animals. Interestingly, just like Capn1−/− mice, YAC84Q animals did not show any overt motor deficits in this assay throughout the experimentation time, suggesting that only the combination of both genetic modifications potentiated into a detectable phenotype. Despite the deficiencies in ambulatory activity in Capn1−/− mice at 3 months of age, none of the cohorts displayed significant changes in the respective assessments at 15 months. In contrast to the initial characterization (17), lack of motor and ambulatory activity impairments in hemizygous YAC84Q mice has been reported, showing that a homozygosity for the transgene is required for producing a robust phenotype (38). Gait analyses revealed abnormally small footprint areas in both YAC84Q mice and double-mutant animals at 15 months of age, not only for front limbs but also for hind limbs. Further hallmarks of male and female YAC84Q mice are an early-onset body weight deficit and a weight loss setting in at 30 weeks of age, which has been reproduced in later studies (17,38,52). In our cohorts, we observed only small body weight differences at baseline. Notwithstanding this phenotype, a weight loss in male YAC84Q mice set in at 36 weeks of age, which was clearly rescued by the lack of calpain-1 in double-mutant animals. Finally, our MJD model had a decreased survival rate with mice starting to die already with 20 weeks of age. Remarkably, the YAC84Q/Capn1−/− did not show any losses throughout the total assessment time of 80 weeks, indicating an improved survival due to the lack of calpain-1 in this animal model. It is imaginable that this amelioration of both diseases hallmarks body weight loss and reduced survival in our mice may represent beneficial ramifications of the calpain-1 knockout on the yet insufficiently investigated peripheral pathologies in MJD.
Is targeting calpains a promising therapeutic approach?
Because of extensive literature supporting a functional role of calpains in neurodegenerative processes, there is much interest in this class of cysteine proteases as promising therapeutic targets. The complexity of the calpain system, the compensatory feature of respective calpain isoforms and the current lack of highly specific pharmacological inhibitors continue to evade therapeutic evaluation in vivo. In this study, we assessed the outcomes of a genetic removal of calpain-1 in the context of cell and animal models of MJD, a disorder whose causative protein ataxin-3 is a known calpain substrate. Here, we provide new evidence that the calpain system is overactivated at baseline in the investigated models. Furthermore, ablation of calpain-1 did not only reduce disease protein fragmentation in vitro and in vivo but also suppressed the excessive cleavage of synaptic proteins in MJD mice, which may represent an additional aspect of the molecular pathology. The repercussions on the disease phenotype in mice were complex, featuring on the one hand a progressive worsening of the motor phenotype but on the other a rescue of body weight loss and improved survival. Recent findings show that mutations in the calpain-1 gene lead to forms of hereditary spastic paraplegia or spastic ataxia (14,55), calling for special caution when therapeutically targeting this protease. The proposed antagonism of a neuroprotective calpain-1 and the neurodegenerative calpain-2 implies that the latter isoform might represent a more promising candidate for developing treatments for neurological diseases (21). However, as proposed earlier, the overactivation of calpain-1—as probably the case in MJD or Alzheimer’s disease—may still be neurotoxic and could require, at least, partial inhibition (59).
In summary, here we report on the overactivation of the calpain system as a distinct pathological hallmark in cell and animal models of MJD. Moreover, we describe the corrective and balancing combination of a mouse model deficient in calpain-1 function with MJD mice that feature a calpain overactivation, which resulted—to a certain degree—in an alleviated molecular, physical and behavioural MJD phenotype. Overall, targeting the calpain system remains a promising approach in treating MJD and other neurodegenerative disorders sharing this unifying pathological mechanism.
Materials and Methods
Ethical statement on human and animal research
Informed consent was acquired for all patients from whom skin biopsies were obtained. Skin biopsies were approved by the ethics committee of the University of Tübingen (NEUROMICS No. 598/2011BO1).
All mouse experiments were approved by the local ethics committee at the Regierungspräsidium Tübingen (HG9/12) and performed in accordance with the German Animal Welfare Act and the guidelines of the Federation of European Laboratory Animal Science Associations, based on European Union legislation (Directive 2010/63/EU). The study was conducted in accordance with the ARRIVE guidelines (60).
Expression constructs and esiRNAs
For ataxin-3 overexpression, pcDNA3.1 vectors carrying cDNA for V5-tagged full-length human ataxin-3 (cDNA for isoform 2; UniProt identifier: P54252–2) with 15 or 70 glutamines were employed. Knockdown of calpain-1 was achieved using endoribonuclease-prepared siRNA (esiRNA) directed against human calpain-1 (MISSION® esiRNA EHU032581) or control esiRNA against Renilla luciferase (MISSION® esiRNA EHURLUC) (both Sigma-Aldrich).
Cell culture and transfection
Cell culture experiments were performed using HEK 293T cells (ATCC: CRL-11268) featuring a knockout of the ataxin-3 gene (293T ATXN3−/−) to circumvent difficulties in interpreting ataxin-3 fragmentation patterns when overexpressing the disease protein. Generation of this cell line was described elsewhere (61). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), high glucose, GlutaMAX™ supplemented with 10% fetal calf serum (FCS), 1% non-essential amino acids (MEM NEAA) and 1% antibiotic–antimycotic (all Gibco®, Thermo Fisher Scientific) at 37°C in 5% CO2. Transient transfections were carried out for 48 h using the Attractene reagent (Qiagen) following the manufacturer’s protocol. Human fibroblasts were obtained from skin biopsies of three MJD patients and three healthy control subjects as described earlier (6). Relevant clinical data of the subjects are respectively listed in Supplementary Material, Table S1. Fibroblasts were maintained in DMEM, high glucose, supplemented with 10% FCS and at 37°C in 5% CO2.
Animals
Homozygous Capn1−/− and hemizygous YAC transgenic MJD (YAC84Q) mice bred on a C57BL/6 background were maintained and genotyped as previously described (12,17). YAC84Q mice were obtained from Jackson Laboratory (B6;CBA-Tg(ATXN3*)84.2Cce/IbezJ, Stocl No. 012705) and Capn1−/− mice were kindly provided by Athar Chishti. Both models were crossbred to obtain four groups of mice: WT, Capn1−/−, hemizygous YAC84Q and YAC84Q/Capn1−/−. All animals were housed in a specific pathogen-free environment under standard conditions with 12 h light–dark cycle, food and water ad libitum. For assessment of the behavioural phenotype, animals were kept in groups of 3–5 mice per cage without enrichment up to an age of 18 months and analysed using the subsequently described methodology.
For protein analysis, animals were sacrificed by CO2 inhalation. Whole brains were immediately dissected on ice, snap-frozen in liquid nitrogen and stored at −80°C for further analysis.
For immunohistochemical analysis, animals were transcardially perfused first with phosphate-buffered saline (PBS; 4.3 mm Na2HPO4 and 1.4 mm KH2PO4 pH 7.5, 137 mm NaCl, 2.7 mM KCl) and then with 4% cold paraformaldehyde diluted in PBS. Subsequently, brains were removed, post-fixed overnight and embedded in paraffin.
Protein extraction
Cell lysates were prepared as follows. Transfected HEK 293T were rinsed off and resuspended in cell culture media by gentle pipetting. Cell suspensions were transferred into reaction tubes and pelleted at 500×g for 5 min. Then, pellets were washed with cold Dulbecco’s PBS (DPBS; Gibco®, Thermo Fisher Scientific) and centrifuged again at 300×g for 5 min. After aspiration of DPBS, RIPA buffer (50 mm Tris pH 7.5, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate and 1% Triton X-100, containing cOmplete® protease inhibitor cocktail (Roche)) was added. Cells were homogenized by ultrasonication for 3 s and then incubated at 10 min on ice. For lysate preparation, cell homogenates were centrifuged for 10 min at 4°C and 16 100×g and supernatants were transferred to fresh pre-cooled tubes and mixed with glycerol to a final concentration of 10%. Deviating from this, human fibroblasts were washed once with DPBS after aspirating cell culture media and scraped off in RIPA buffer before lysis.
Homogenates and lysates of mouse brain and femoral muscle tissue were obtained by homogenizing samples in RIPA buffer using an ULTRA-TURRAX® disperser (VWR). Homogenates were incubated for 25 min on ice and stored at −80°C for further analysis. For lysate preparation, homogenates were centrifuged for 30 min at 4°C and 13 200×g. Supernatants were transferred into pre-cooled reaction tubes and mixed with glycerol to a final concentration of 10%.
Until used, all protein samples were stored at −80°C. Protein concentrations of RIPA homogenates and lysates were measured spectrophotometrically using Bradford reagent (Bio-Rad Laboratories).
Western blot analysis
Western blotting was performed according to standard procedures. Briefly, 30 μg of protein for cell culture, mouse brain and muscle lysates were mixed with 4× LDS sample buffer (1 M Tris pH 8.5, 2 mm EDTA, 8% LDS, 40% glycerol, 0.075% CBB G, 0.025% phenol red) in a ratio 3:1 and supplemented with 0.1 M dithiothreitol. After heat denaturing for 10 min at 70°C, protein samples were electrophoretically separated using homemade 10% Bis-Tris gels or 4–12% Bolt Bis-Tris gradient gels (Thermo Fisher Scientific) with the respective electrophoresis buffers, MES buffer (50 mm MES, 50 mm Tris pH 7.3, 0.1% SDS, 1 mm EDTA) or MOPS buffer (50 mm MOPS, 50 mm Tris pH 7.7, 0.1% SDS, 1 mm EDTA). SeeBlue Plus2 and Novex Sharp pre-stained protein standards (both Thermo Fisher Scientific) were used for molecular weight estimation. Proteins were transferred on Amersham Protran Premium 0.2 μm nitrocellulose membranes (GE Healthcare) using Bicine/Bis-Tris transfer buffer (25 mm Bicine, 25 mm Bis-Tris pH 7.2, 1 mm EDTA, 15% methanol) and a TE22 Transfer Tank (Hoefer) at 80 V and a maximal amperage of 250 mA for 1.5 h.
After transfer, membranes were blocked for 1 h with 5% skim milk powder (Merck) in TBS (Tris-buffered saline) at room temperature and probed overnight at 4°C with following primary antibodies diluted in TBS-T (TBS with 0.1% Tween 20): mouse anti-β-actin (1:10000; clone AC-15, A5441, Merck), mouse anti-ataxin-3 (1:2000; clone 1H9, MAB5360, Merck), rabbit anti-ataxin-3 (1:500; ab96316, Abcam), rabbit anti-calpain-1 (1:1000; ab39170, Abcam), rabbit anti-calpastatin (1:1000; #4146, Cell Signaling), rabbit anti-citrate synthase (1:1000; ab96600, Abcam), rabbit anti-p35 (1:500; clone C64B10, #2680, Cell Signaling), goat anti-PSD-95 (1:1000; ab12093, Abcam), mouse anti-α-spectrin (1:1000; clone AA6, MAB1622, Merck), mouse anti-SNAP-25 (1:1000; clone SMI 81, SMI 81R-100, Convance), rabbit anti-synapsin-1a/b (1:500; sc-20 780, Santa Cruz) and rabbit anti-synapsin-2a (1:500; sc-25 538, Santa Cruz). Afterwards, western blots were washed with TBS-T and incubated at room temperature for 1 h with the respective HRP-conjugated secondary antibodies goat anti-mouse (1:5000; 115–035-003, Jackson ImmunoResearch) and goat anti-rabbit (1:10000; ab97051, Abcam) or by secondary IRDye antibodies goat anti-mouse 680LT, goat anti-mouse 800CW and goat anti-rabbit 800CW (all 1:10000; LI-COR Biosciences). After final washing with TBS-T, chemiluminescence and fluorescence signals were detected using the LI-COR ODYSSEY FC and quantified with Image Studio 4.0 software (both LI-COR Biosciences).
TR-FRET analysis
Ataxin-3 measurements via time-resolved Förster resonance energy transfer (TR-FRET) were performed using a combination of an anti-ataxin-3 antibody (clone 1H9, MAB5360, Merck) labelled with Tb (donor) fluorophore and an anti-polyQ antibody (clone MW1, Developmental Studies Hybridoma Bank) labelled with d2 (acceptor) fluorophore (labelling by Cisbio) as previously described (62). Briefly, mouse brain RIPA homogenates were diluted in DPBS containing cOmplete protease inhibitor cocktail (Roche) to a final concentration of 1 μg/μl. Next, 5 μl of diluted sample were incubated with 1 μl of the TR-FRET antibody mix (0.3 ng 1H9-Tb + 10 ng MW1-d2 in 50 mm NaH2PO4, 400 mm NaF, 0.1% BSA, 0.05% Tween-20) in a low-volume white ProxiPlate 384 TC Plus plate (PerkinElmer) at 4°C for 22 h. Signals were detected at 620 and 665 nm using an EnVision Multimode Plate Reader with a TRF-laser unit (PerkinElmer).
Filter trap assay
Detection of SDS-insoluble ataxin-3 species was performed as previously described (6). Briefly, 12.5 μg total protein of mouse brain homogenates were diluted in DPBS containing 2% SDS and 50 mm dithiothreitol. Afterwards, samples were heat denatured for 5 min at 95°C and filtered through a nitrocellulose membrane (Amersham Protran 0.45 μm, GE Healthcare) using a Minifold II Slot Blot System (Schleicher & Schuell). Afterwards, membranes were detected using the primary antibody mouse anti-ataxin-3 (1:2000; clone 1H9, MAB5360, Merck) and secondary antibody goat anti-mouse 800CW (1:5000; LI-COR Biosciences). Fluorescence signals were detected using the LI-COR ODYSSEY FC and quantified with Image Studio 4.0 software (both LI-COR Biosciences).
Immunohistochemistry and microscopy
To analyse the distribution and aggregation of ataxin-3, 7 μm sections of paraffin-embedded brain tissue were immunostained according to standard procedures. Briefly, rehydrated sections were microwaved for 3× 5 min in 0.1 M citric acid and 0.1 M sodium citrate. After washing 3× in PBS, the endogenous peroxidase was inhibited by 4% H2O2 for 20 min. Sections were blocked in 5% normal goat serum for 1 h at room temperature after three additional washing steps. Afterwards, sections were incubated with anti-ataxin-3 antibody (1:2000; clone 1H9, MAB5360, Merck) overnight at 4°C, followed by incubation with a biotin-coupled anti-mouse secondary antibody (1:200; Vector laboratories). Staining was visualized by an avidin–biotin enhancer complex coupled with peroxidase (ABC Elite; Vector Laboratories) and 3,3′-diaminobenzidine (DAB; Sigma Aldrich). Light microscopic images of three mice per genotype were acquired using Axioplan 2 imaging microscope equipped with an AxioCam HR colour digital camera at a 630× magnification using the AxioVision 4.3 imaging software (all Zeiss).
Behavioural analysis
As basic health screen, the body weight of a standard group of 43 WT, 15 Capn1−/−, 38 YAC84Q and 21 YAC84Q/Capn1−/− animals was measured each second week starting at the age of 4 weeks. To assess the neurological phenotype, a battery of behavioural tests, including CatWalk gait analysis (Noldus), LabMaster (TSE) and rotarod (TSE), was done including both sexes in a standard group of 15 WT, 14 Capn1−/−, 16 YAC84Q and 15 YAC84Q/Capn1−/− mice. All tests were performed during the dark phase and by investigators blinded to the genotypes as follows. Lifespan was assessed by recording the age of spontaneous death or when mice reached the euthanization criteria strictly following the recommendations presented in the Guide for Care and Use of Laboratory Animals of the University of Tübingen.
Rotarod performance tests
To assess coordination deficits, mice were placed on an accelerating rod (rotarod, TSE) at the same time of a day for four consecutive days. Three trainings sessions each with two trials (with acceleration from 4 to 16 rpm in 2 min) were followed by one test session with two trials. Each test trial started with rotation at 4 rpm and accelerated to a maximum of 40 rpm within 5 min. Elapsed time until the mouse fell off the rod was recorded. The trials at the same day were performed with a waiting time of 1 h between sessions. To analyse coordination deficits longitudinally, mice were placed at the rotarod every 3 months starting at 3 months of age and ending with 15 months of age.
LabMaster analysis
General movement activity and consumption of food and water were assessed in an automated home cage-based system (LabMaster System, TSE). For this, mice were maintained under a 12 h light–dark cycle and drinking, food consumption and activity were continuously measured for 70 h. For the latter, total distance travelled in the cage was assessed and analysed over time. Food and water intake were measured by automated instruments, simultaneously. LabMaster analyses were performed starting at 3 months (baseline) and at 9 and 15 months of age.
CatWalk gait analysis
To measure abnormalities in gait, gait analyses were performed at 6, 12 and 15 months using the CatWalk XT system with the CatWalk XT 8.1 software (both Noldus). Mice were placed in a walkway and allowed to freely ambulate along an illuminated glass plate within a confined corridor of 20 cm × 8 cm. Footprints were recorded with a high-speed camera. Run criteria were set up with a maximum of 50s walk time and speed variation less than 45%. The print area of front and hind limbs, number of steps and average speed were calculated from five evaluable runs at each time point for each mouse. Footprint area is defined as the total floor area in pixels contacted by the paw during stance phase.
Primary and secondary outcome parameters
Molecular markers like fragmentation pattern and aggregation propensity were chosen as the primary outcome parameter. Additionally, behavioural changes linked to the molecular pathology served as the secondary outcome parameter.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 6.00 for Windows (GraphPad Software Inc.). If not explicitly stated otherwise, data are shown as arithmetic means ± SEM. Numbers of animals/samples are indicated within the respective figure or figure legend. Statistical significance of data sets was determined using Student’s t-test, one-way- or two-way ANOVA with the respective post-test. Statistical comparison of survival curves was performed using the log-rank (Mantel-Cox) test. P values ≤0.05 were considered as statistically significant. More detailed information is given in the respective figure legend.
Supplementary Material
Acknowledgements
We want to thank Yvonne Schelling for her technical assistance.
Funding
This work was supported by the German Research Foundation (DFG, research grant number HU1770/3-1). JJW was funded by the Baden-Wuerttemberg Foundation (research grant number P-BWS-SPII/3-08). AHC received financial support from the National Institutes of Health (NIH, research grant number HL089517) for the generation of the calpain-1 null mouse model.
Conflict of Interest statement: The authors declare that they have no competing interests.
Author Contributions
J.J.W., O.R. and J.H.-S. conceptualized the experimental design of the study. J.J.W., E.H., Y.M. and J.H.-S conducted the experiments and analysed the data. S.H. provided the human fibroblast samples. N.L.P.C. analysed the LabMaster and CatWalk data. A.H.C. provided the Capn1−/− knockout mouse model and protocols for breeding and genotyping. J.J.W. and J.H.-S. generated the figures and wrote the manuscript. All authors read and approved the final manuscript.
References
- 1. Ono Y., Saido T.C. and Sorimachi H. (2016) Calpain research for drug discovery: challenges and potential. Nat. Rev. Drug Discov., 15, 854–876. [DOI] [PubMed] [Google Scholar]
- 2. Matos C.A., Almeida L.P. de and Nobrega C. (2017) Proteolytic cleavage of Polyglutamine disease-causing proteins: revisiting the toxic fragment hypothesis. Curr. Pharm. Des., 23, 753–775. [DOI] [PubMed] [Google Scholar]
- 3. Curcio M., Salazar I.L., Mele M., Canzoniero L.M.T. and Duarte C.B. (2016) Calpains and neuronal damage in the ischemic brain: the Swiss knife in synaptic injury. Prog. Neurobiol., 143, 1–35. [DOI] [PubMed] [Google Scholar]
- 4. Mahaman Y.A.R., Huang F., Kessete Afewerky H., Maibouge T.M.S., Ghose B. and Wang X. (2019) Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med. Res. Rev., 39, 608–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bano D., Zanetti F., Mende Y. and Nicotera P. (2011) Neurodegenerative processes in Huntington’s disease. Cell Death Dis., 2, e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber J.J., Golla M., Guaitoli G., Wanichawan P., Hayer S.N., Hauser S., Krahl A.-C., Nagel M., Samer S., Aronica E. et al. (2017) A combinatorial approach to identify calpain cleavage sites in the Machado-Joseph disease protein ataxin-3. Brain, 140. [DOI] [PubMed] [Google Scholar]
- 7. Hübener J., Weber J.J., Richter C., Honold L., Weiss A., Murad F., Breuer P., Wüllner U., Bellstedt P., Paquet-Durand F. et al. (2013) Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum. Mol. Genet., 22, 508–518. [DOI] [PubMed] [Google Scholar]
- 8. Toonen L.J.A., Schmidt I., Luijsterburg M.S., Attikum H. and Roon-Mom W.M.C. (2016) Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3. Sci. Rep., 6, 35200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watchon M., Yuan K.C., Mackovski N., Svahn A.J., Cole N.J., Goldsbury C., Rinkwitz S., Becker T.S., Nicholson G.A. and Laird A.S. (2017) Calpain inhibition is protective in Machado-Joseph disease Zebrafish due to induction of autophagy. J. Neurosci., 37, 7782–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simões A.T., Goncalves N., Koeppen A., Deglon N., Kugler S., Duarte C.B. and Pereira de Almeida L. (2012) Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain, 135, 2428–2439. [DOI] [PubMed] [Google Scholar]
- 11. Simões A.T., Gonçalves N., Nobre R.J., Duarte C.B. and Pereira de Almeida L. (2014) Calpain inhibition reduces ataxin-3 cleavage alleviating neuropathology and motor impairments in mouse models of Machado-Joseph disease. Hum. Mol. Genet., 23, 4932–4944. [DOI] [PubMed] [Google Scholar]
- 12. Azam M., Andrabi S.S., Sahr K.E., Kamath L., Kuliopulos A. and Chishti A.H. (2001) Disruption of the mouse mu-calpain gene reveals an essential role in platelet function. Mol. Cell. Biol., 21, 2213–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamada K.H., Kozlowski D.A., Seidl S.E., Lance S., Wieschhaus A.J., Sundivakkam P., Tiruppathi C., Chishti I., Herman I.M., Kuchay S.M. et al. (2012) Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress. J. Biol. Chem., 287, 13182–13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y., Hersheson J., Lopez D., Hammer M., Liu Y., Lee K.-H., Pinto V., Seinfeld J., Wiethoff S., Sun J. et al. (2016) Defects in the CAPN1 gene result in alterations in cerebellar development and cerebellar Ataxia in mice and humans. Cell Rep., 16, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Czogalla A. and Sikorski A.F. (2005) Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell. Mol. Life Sci., 62, 1913–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weber J.J., Sowa A.S., Binder T. and Hübener J. (2014) From pathways to targets: understanding the mechanisms behind polyglutamine disease. Biomed Res. Int., 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cemal C.K., Carroll C.J., Lawrence L., Lowrie M.B., Ruddle P., Al-Mahdawi S., King R.H.M., Pook M.a., Huxley C. and Chamberlain S. (2002) YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum. Mol. Genet., 11, 1075–1094. [DOI] [PubMed] [Google Scholar]
- 18. Lee M.S., Kwon Y.T., Li M., Peng J., Friedlander R.M. and Tsai L.H. (2000) Neurotoxicity induces cleavage of p 35 to p 25 by calpain. Nature, 405, 360–364. [DOI] [PubMed] [Google Scholar]
- 19. Chen X., Tang T.-S., Tu H., Nelson O., Pook M., Hammer R., Nukina N. and Bezprozvanny I. (2008) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci., 28, 12713–12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu J., Liu M.C. and Wang K.K.W. (2008) Calpain in the CNS: from synaptic function to neurotoxicity. Sci. Signal., 1, re1. [DOI] [PubMed] [Google Scholar]
- 21. Baudry M. and Bi X. (2016) Calpain-1 and Calpain-2: the yin and Yang of synaptic plasticity and Neurodegeneration. Trends Neurosci., 39, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Briz V. and Baudry M. (2017) Calpains: master regulators of synaptic plasticity. Neurosci., 23, 221–231. [DOI] [PubMed] [Google Scholar]
- 23. Weber J.J., Kloock S.J., Nagel M., Ortiz-Rios M.M., Hofmann J., Riess O. and Nguyen H.P. (2018) Calpastatin ablation aggravates the molecular phenotype in cell and animal models of Huntington disease. Neuropharm., 133, 94–106. [DOI] [PubMed] [Google Scholar]
- 24. Rao M.V., Campbell J., Palaniappan A., Kumar A. and Nixon R.A. (2016) Calpastatin inhibits motor neuron death and increases survival of hSOD1 G93A mice. J. Neurochem., 137, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber J.J., Ortiz Rios M.M., Riess O., Clemens L.E. and Nguyen H.P. (2016) The calpain-suppressing effects of olesoxime in Huntington’s disease. Rare Dis., 4, e1153778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clemens L.E., Weber J.J., Wlodkowski T.T., Yu-Taeger L., Michaud M., Calaminus C., Eckert S.H., Gaca J., Weiss A., Magg J.C.D. et al. (2015) Olesoxime suppresses calpain activation and mutant huntingtin fragmentation in the BACHD rat. Brain, 138. [DOI] [PubMed] [Google Scholar]
- 27. Saito K., Elce J.S., Hamos J.E. and Nixon R.A. (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc. Natl. Acad. Sci., 90, 2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kurbatskaya K., Phillips E.C., Croft C.L., Dentoni G., Hughes M.M., Wade M.A., Al-Sarraj S., Troakes C., O’Neill M.J., Perez-Nievas B.G. et al. (2016) Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol. Commun., 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gafni J. and Ellerby L.M. (2002) Calpain activation in Huntington’s disease. J. Neurosci., 22, 4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamashita T., Hideyama T., Hachiga K., Teramoto S., Takano J., Iwata N., Saido T.C. and Kwak S. (2012) A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun., 3, 1307. [DOI] [PubMed] [Google Scholar]
- 31. Campbell R.L. and Davies P.L. (2012) Structure–function relationships in calpains. Biochem. J., 447, 335–351. [DOI] [PubMed] [Google Scholar]
- 32. Kimura T., Ishiguro K. and Hisanaga S. (2014) Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci., 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blomgren K., Hallin U., Andersson A.L., Puka-Sundvall M., Bahr B.a., McRae A., Saido T.C., Kawashima S. and Hagberg H. (1999) Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. J. Biol. Chem., 274, 14046–14052. [DOI] [PubMed] [Google Scholar]
- 34. Haacke A., Hartl F.U. and Breuer P. (2007) Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J. Biol. Chem., 282, 18851–18856. [DOI] [PubMed] [Google Scholar]
- 35. Hübener J., Vauti F., Funke C., Wolburg H., Ye Y., Schmidt T., Wolburg-Buchholz K., Schmitt I., Gardyan A., Drießen S. et al. (2011) N-terminal ataxin-3 causes neurological symptoms with inclusions, endoplasmic reticulum stress and ribosomal dislocation. Brain, 134, 1925–1942. [DOI] [PubMed] [Google Scholar]
- 36. Harmuth T., Prell-Schicker C., Weber J.J., Gellerich F., Funke C., Drießen S., Magg J.C.D., Krebiehl G., Wolburg H., Hayer S.N. et al. (2018) Mitochondrial morphology, function and homeostasis are impaired by expression of an N-terminal calpain cleavage fragment of ataxin-3. Front. Mol. Neurosci., 11, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koch P., Breuer P., Peitz M., Jungverdorben J., Kesavan J., Poppe D., Doerr J., Ladewig J., Mertens J., Tüting T. et al. (2011) Excitation-induced ataxin-3 aggregation in neurons from patients with Machado–Joseph disease. Nature, 480, 543–546. [DOI] [PubMed] [Google Scholar]
- 38. Carmo Costa M., Luna-Cancalon K., Fischer S., Ashraf N.S., Ouyang M., Dharia R.M., Martin-Fishman L., Yang Y., Shakkottai V.G., Davidson B.L. et al. (2013) Toward RNAi therapy for the polyglutamine disease Machado–Joseph disease. Mol. Ther., 21, 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weber J.J., Pereira Sena P., Singer E. and Nguyen H.P. (2019) Killing two angry birds with one stone: autophagy activation by inhibiting calpains in neurodegenerative diseases and beyond. Biomed Res. Int., 2019, 4741252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams A., Sarkar S., Cuddon P., Ttofi E.K., Saiki S., Siddiqi F.H., Jahreiss L., Fleming A., Pask D., Goldsmith P. et al. (2008) Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol., 4, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Menzies F.M., Garcia-Arencibia M., Imarisio S., O’Sullivan N.C., Ricketts T., Kent B.a., Rao M.V., Lam W., Green-Thompson Z.W., Nixon R.a. et al. (2015) Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ., 22, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dau A., Gladding C.M., Sepers M.D. and Raymond L.a. (2014) Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol. Dis., 62, 533–542. [DOI] [PubMed] [Google Scholar]
- 43. Milnerwood A.J. and Raymond L.A. (2010) Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci., 33, 513–523. [DOI] [PubMed] [Google Scholar]
- 44. Bereczki E., Branca R.M., Francis P.T., Pereira J.B., Baek J.-H., Hortobágyi T., Winblad B., Ballard C., Lehtiö J. and Aarsland D. (2018) Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain, 141, 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cesca F., Baldelli P., Valtorta F. and Benfenati F. (2010) The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol., 91, 313–348. [DOI] [PubMed] [Google Scholar]
- 46. Antonucci F., Corradini I., Fossati G., Tomasoni R., Menna E. and Matteoli M. (2016) SNAP-25, a known presynaptic protein with emerging postsynaptic functions. Front. Synaptic Neurosci., 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ando K., Kudo Y. and Takahashi M. (2005) Negative regulation of neurotransmitter release by calpain: a possible involvement of specific SNAP-25 cleavage. J. Neurochem., 94, 651–658. [DOI] [PubMed] [Google Scholar]
- 48. Lu X., Rong Y. and Baudry M. (2000) Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci. Lett., 286, 149–153. [DOI] [PubMed] [Google Scholar]
- 49. Koeppen A.H., Dickson A.C., Lamarche J.B. and Robitaille Y. (1999) Synapses in the hereditary ataxias. J. Neuropathol. Exp. Neurol., 58, 748–764. [DOI] [PubMed] [Google Scholar]
- 50. Das A. and Dubreuil R.R. (2009) Spectrin: organization and function in neurons. Encycl. Neurosci. doi: 10.1016/B978-008045046-9.00731-2. [DOI] [Google Scholar]
- 51. Pielage J., Fetter R.D. and Davis G.W. (2005) Presynaptic Spectrin is essential for synapse stabilization. Curr. Biol., 15, 918–928. [DOI] [PubMed] [Google Scholar]
- 52. Toonen L.J.A., Overzier M., Evers M.M., Leon L.G., Zeeuw S.A.J., Mei H., Kielbasa S.M., Goeman J.J., Hettne K.M., Magnusson O.T. et al. (2018) Transcriptional profiling and biomarker identification reveal tissue specific effects of expanded ataxin-3 in a spinocerebellar ataxia type 3 mouse model. Mol. Neurodegener., 13, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chou A.-H., Yeh T.-H., Ouyang P., Chen Y.-L., Chen S.-Y. and Wang H.-L. (2008) Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis., 31, 89–101. [DOI] [PubMed] [Google Scholar]
- 54. Chou A.-H., Chen S.-Y., Yeh T.-H., Weng Y.-H. and Wang H.-L. (2011) HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol. Dis., 41, 481–488. [DOI] [PubMed] [Google Scholar]
- 55. Gan-Or Z., Bouslam N., Birouk N., Lissouba A., Chambers D.B., Vérièpe J., Androschuck A., Laurent S.B., Rochefort D., Spiegelman D. et al. (2016) Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am. J. Hum. Genet., 98, 1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Forman O.P., De Risio L., Mellersh C.S., Guttman M. and Lander E. (2013) Missense mutation in CAPN1 is associated with Spinocerebellar Ataxia in the parson Russell terrier dog breed. PLoS One, 8, e64627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen Y., Cen Z., Zheng X., Xie F., Chen S. and Luo W. (2019) A novel homozygous CAPN1 pathogenic variant in a Chinese patient with pure hereditary spastic paraplegia. J. Clin. Neurol., 15, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kocoglu C., Gundogdu A., Kocaman G., Kahraman-Koytak P., Uluc K., Kiziltan G., Caglayan A.O., Bilguvar K., Vural A. and Basak A.N. (2018) Homozygous CAPN1 mutations causing a spastic-ataxia phenotype in 2 families. Neurol. Genet., 4, e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gan-Or Z. and Rouleau G.A. (2016) Calpain 1 in neurodegeneration: a therapeutic target? Lancet. Neurol., 15, 1118. [DOI] [PubMed] [Google Scholar]
- 60. Kilkenny C., Browne W.J., Cuthill I.C., Emerson M. and Altman D.G. (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol., 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weishäupl D., Schneider J., Peixoto Pinheiro B., Ruess C., Dold S.M., von Zweydorf F., Gloeckner C.J., Schmidt J., Riess O. and Schmidt T. (2019) Physiological and pathophysiological characteristics of ataxin-3 isoforms. J. Biol. Chem., 294, 644–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nguyen H.P., Hübener J., Weber J.J., Grueninger S., Riess O. and Weiss A. (2013) Cerebellar soluble mutant ataxin-3 level decreases during disease progression in Spinocerebellar Ataxia type 3 mice. PLoS One, 8, e62043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.