

Abstract
Chromatin immunoprecipitation coupled to next generation sequencing (ChIP-seq) has served as the central method for the study of histone modifications for the last decade. In ChIP-seq, antibodies are used to selectively capture nucleosomes bearing a modification of interest and the associated DNA can then be mapped to the genome to determine the distribution of the mark. However, this approach suffers from a number of serious drawbacks: 1.) ChIP interpretation necessitates the assumption of perfect antibody specificity, despite growing evidence that this is often a fallacy. 2.) The common methods for evaluating antibody specificity in other formats have little or no bearing on specificity within a ChIP experiment. 3.) Uncalibrated ChIP is reported as relative enrichment, which is biologically meaningless outside the experimental reference frame defined by a discrete IP, thereby preventing facile comparison across experimental conditions or modifications. 4.) The act of differential library amplification and loading on next generation sequencers, as well as computational normalization, can further compromise quantitative relationships that may exist between samples. Consequently, the ChIP experimenter is presented with a series of potential pitfalls and is blind to nearly all of them. Here, we provide a detailed protocol for Internally Calibrated ChIP (ICeChIP), a method we have recently developed to resolve these serious problems by spiking-in defined nucleosomal standards within a ChIP procedure. This protocol is optimized for specificity and quantitative power, allowing for the measurement of both antibody specificity and an absolute measurement of histone modification density (HMD) at genomic loci on a biologically meaningful scale that enables unambiguous comparisons. We also provide guidance on optimal conditions for next-generation sequencing and instructions for analysis of ICeChIP-seq data. This protocol takes between 17–18 hours to complete, excluding time for sequencing or bioinformatic analysis. The ICeChIP procedure permits accurate measurement of histone post-translational modifications genome-wide in mammalian cells but has also been successfully applied to Drosophila melanogaster and Caenorhabditis elegans, indicating suitability for eukaryotic cells more broadly.
Introduction
Histone post-translational modifications (PTMs) have long been recognized as critical regulators of genomic structure and function1–3. One of the most important methods for studying histone PTMs has been chromatin immunoprecipitation (ChIP), wherein antibodies are used to capture histones with a particular modification along with any DNA bound to the captured nucleosomes4. The DNA can then be mapped to the genome to determine the genomic distribution of the captured DNA and, by proxy, the targeted modification. Over the course of decades, ChIP has been a critical component of studies providing insight into the biological roles of histone modifications5–43. However, ChIP, as traditionally practiced, has several critical drawbacks that render its use in continuing to drive chromatin biology problematic, particularly for quantitative analyses or comparisons.
Chief amongst these drawbacks is the output of the experiment itself. ChIP results are often reported as either a fold-change over input or mock IgG pull-down38,43,44. However, this metric is not presented in biologically meaningful units; it is merely a measure, in that one experiment, of the relative amount of histone modification at a given locus. Consequently, different experiments cannot be readily compared to each other; two different ChIPs targeting different PTMs are scaled differently (due to epitope abundance, antibody affinity and specificity), so it is impossible to measure which modification is dominant at a given locus or whether they may coexist43,45. Similarly, traditional ChIP analysis and normalization are insensitive to global changes in histone PTM abundance, so even if the actual amount of histone PTM is changing dramatically, traditional ChIP will not detect a difference at loci where the relative abundances are constant38,43,45.
Another pitfall with traditional ChIP lies with the question of antibody specificity. Traditional ChIP-seq signal is a conflation of on-target capture by the antibody, off-target capture of similar epitopes, and non-specific background, and yet, the experimenter is blind to the relative proportions of each of these components. Worse still, ChIP analyses are predicated on the assumption that antibodies used are perfectly specific towards their targets, despite increasing evidence that many commercially available antibodies are not specific towards their putative targets43,45–51. Previous studies in other experimental formats have found that histone PTM antibodies frequently bind modifications other than the intended target46–51, and our recent work has shown that many commonly used antibodies demonstrate poor specificity within ChIP contexts as well43,45. Indeed, there is no effective basis besides ICeChIP to evaluate antibody quality within a ChIP experiment.
To address these problems, we have developed internally calibrated ChIP (ICeChIP)43,45, schematized in Fig. 1. In ICeChIP, a defined set of semisynthetic nucleosome standards, each bearing a PTM of interest and several uniquely-identifiable DNA sequences (or “barcodes”), are spiked into a ChIP experiment and subjected to immunoprecipitation with the genomic chromatin43,45. The pulldown efficiency of the nucleosome standard bearing the target modification can be measured in order to determine the expected pulldown efficiency of a locus that is 100% modified with the target modification43,45. This is computed as the amount of DNA recovered from the standard in the IP sample relative to the amount of such DNA in the Input sample, and we refer to this as the on-target enrichment (e.g. Fig 1a, ladder tags). This target pulldown efficiency can then be used to calibrate the genomic pulldown efficiencies, yielding the histone modification density (HMD), or the proportion of nucleosomes at an arbitrary locus bearing the modification of interest43,45. This is computed as the enrichment at a locus divided by the enrichment of the on-target standard. Crucially, unlike the fold-change output of traditional ChIP, HMD is an absolute measure of histone modification levels, meaning that different ICeChIP experiments can be quantitatively compared to each other to determine which modification is dominant at any given locus. Ideally, the ladder is spiked in at a quantity such that the number of reads from each locus falls between the extremes in number of reads from the different ladder members (Fig. 1b).
Figure 1 |. The ICeChIP-seq workflow.
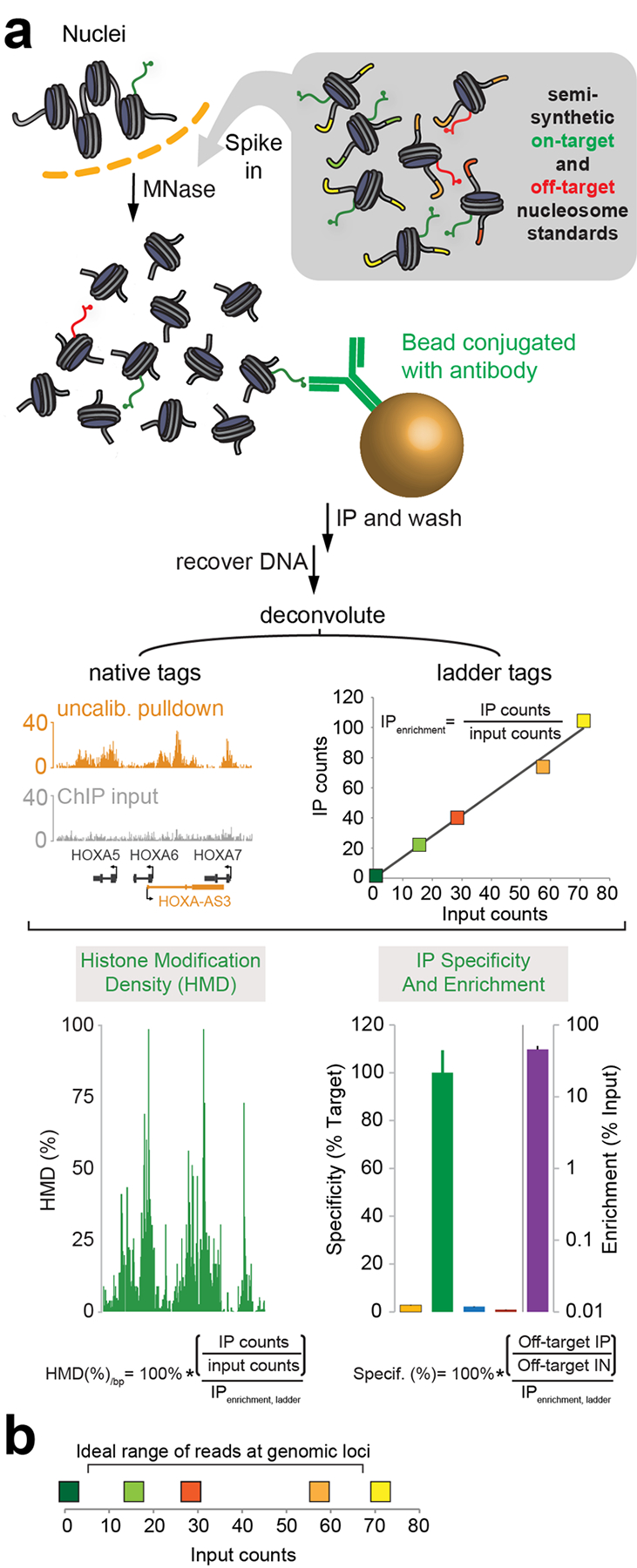
(a) Purified nuclei are spiked with barcoded nucleosome standards representing on- and off-target modifications before chromatin is fragmented by MNase digestion. Fragmented chromatin is then incubated with antibody conjugated to magnetic beads to purify targeted nucleosomes. DNA is then recovered, sequenced, and mapped to the genome or to nucleosome standard barcodes. Histone modification density can be computed as the ratio of locus enrichment to on-target enrichment, and antibody specificity can be computed as the ratio of off-target to on-target enrichment. Adapted from Grzybowski et al.43 and Shah et al.45 Error bars represent standard deviation, as described in the original publications. (b) Example scatterplot showing reads from genomic loci falling between the reads from the highest- and lowest-abundance ladder members for optimal quantification. Ideally, the range of the reads from the barcodes on the target modification standard should encompass the range of input counts at the genomic loci. Different colors each refer to different ladder members comprising the nucleosome ladder.
Second, ICeChIP also provides information on antibody specificity43,45, which has often been highlighted as a contributor to the “reproducibility crisis” in biological sciences45,52–54. When different standards with different modifications are spiked into a ChIP experiment, then the pulldown efficiency of those modifications can also be measured and compared to the pulldown efficiency of the target in order to determine the specificity of the antibody in situ43,45. In its most straightforward form, this would consist of dividing the enrichment of each off-target standard by the enrichment of the on-target standard. A highly specific antibody will pull-down the target modification ~100 times more efficiently than the off-target modifications (i.e. the off-target enrichment will be ~1% of the on-target enrichment; e.g. Fig. 1a, lower right panel); a poorly-specific antibody will have off-target pulldown efficiencies that are a considerable fraction of the on-target efficiencies, and occasionally, may even pull down an off-target modification more than the on-target43,45. Whether an antibody is serviceable will depend on both the specificity factor, and the genomic abundance and location of the off-target species captured. For example, an H3K4me3 antibody that captures 30% of H3K4me2 is unsuitable, as the latter is several-fold more abundant and resides in similar genomic loci. Traditionally, the researcher is blind to this pitfall; with ICeChIP, they are able to detect the inadequacies of their antibodies – or gain confidence that their results are accurate.
Expertise Needed to Implement the Protocol
The ICeChIP-qPCR protocol can be implemented by a competent bench scientist familiar with immunoprecipitation and quantitative PCR. The ICeChIP-seq protocol can be implemented by a competent bench scientist through to the NGS library preparation. For the sequencing, use of a specialized core facility is recommended. The analysis of ICeChIP-seq data can be accomplished by any data scientist, bioinformatician, or individual reasonably familiar with a UNIX command line environment, self-taught or otherwise.
Limitations
ICeChIP is currently not compatible with ChIP targeting more loosely bound factors, such as transcription factors and unstable nucleosomes, due both to the native protocol and the lack of plausible usable internal standards. As such, at the moment, ICeChIP can only target histone modifications and stable variants.
Additionally, although this native procedure is highly effective for modifications on the histone tail, it is problematic for modifications on the highly-structured globular domain of the nucleosome43, where the local environment and structure of a PTM may differ greatly from linear, unstructured peptides against which most commercially-available antibodies are raised. As such, a denaturative version of this protocol may be needed to effectively query internal histone modifications, and this remains an active focus of our research.
This protocol is also not compatible with crosslinking for the reasons noted above – namely, that crosslinked ChIP yields more off-target artefacts and, when combined with sonication, causes more epitope damage. Additionally, crosslinking of standards would not occur in the same molecular context as the native chromatin, rendering quantitative comparisons between the two difficult. This limitation, however, does mean that chromatin must be prepared from native sources, and previously crosslinked samples (e.g. FFPE) samples cannot be used in our ICeChIP procedure.
This protocol also uses a very large number of cells due to the extensive nuclei purification, whereas many other ChIP procedures use far fewer cells. Though the high degree of nuclei purity yields more consistent and specific ChIP experiments, the cell number may present a barrier. Though 108 cells or more is feasible for research using cell lines or in primary cell studies with high cell numbers, other studies involving cells that cannot be harvested in such great numbers may be more difficult to conduct using this protocol. In principle, this can be mitigated by skipping sucrose cushion purification and conducting more aggressive MNase digestion, but this too is a factor that must be optimized by the end user in such situations. It should be possible to apply nucleosome calibrants to single-cell, or very small cell number ChIP methods43, but this protocol does not cover the input preparation for such low-cell applications.
For maximum specificity, the chromatin concentration relative to the amount of antibody should be optimized, and we have provided guidelines for approximate chromatin concentrations in Box 1. In the worst case, this may require a good deal of fine-tuning and the concomitant labor. In practice, we have found that the conditions provided by the guidelines in Box 1 often provide adequate specificity and recovery45.
Box 1: Determining the Amount of Chromatin for ICeChIP.
The chromatin concentration is important for optimal ICeChIP specificity. This is the parameter that will likely require the most optimization from the end-user, as the amounts of chromatin to be used tend to be highly idiosyncratic across different targets, antibodies, and cell types or treatments. In general, however, there are some guidelines that can be offered. These guidelines assume that the antibody to be used is of reasonable efficiency (>10% recovery) and high specificity (as a guideline, at least 10-fold greater recovery of the target over off-target modifications). We highly recommend optimizing the amount of chromatin used for the ICeChIP experiment, but if this is ultimately not feasible for reasons of cost or time, then the recommendations below may be used so long as the antibody displays high specificity. This is highly suboptimal because the specificity will likely not be as high as possible, but if optimization is not possible, then the guidelines below may help.
For highly punctate and sparse modifications, such as H3K4me3, more chromatin will be needed than for more broadly-distributed marks. As such, we would recommend using approximately 6 μg of chromatin per 6 μg of antibody. For very broadly-distributed marks, such as H3K27me3 or H4K20me2, we would recommend using much less chromatin – on the order of 1.5 μg of chromatin per 6 μg of antibody. For marks with intermediate distributions, such as H3K4me1 or H3K36me3, we would recommend using intermediate amounts of chromatin – approximately 3 μg of chromatin per 6 μg of antibody.
Again, however, these are only guidelines. We strongly recommend optimization by the end user given their cell types, modifications, and most importantly, antibodies.
For ICeChIP-seq experiments, the sequencing depth needed for maximum quantitative power tends to be very high, particularly for the input samples, as discussed in Box 2. This is to ensure that the ratiometric comparisons between the IP and the input can remain quantitatively useful as compared to the statistical noise inherent in read quantification and comparison. As per-read sequencing costs continue to decrease with sub-Moore’s law scaling, this requirement becomes less expensive.
Box 2: Computing Sequencing Reads Needed and Planning Multiplexing.
To decide on a multiplexing scheme, the most important consideration is the number of reads to be devoted to each sample. In general, the input is going to be the most complex sample (i.e. the sample with the greatest diversity of DNA fragments and genomic loci represented), so to obtain sufficient read depth for quantification, the input sample will need the most reads. As such, for mammalian genomes, we generally aim for 350–450 million paired-end sequencing reads or more for the input. In principle, this could be reduced, but at significant cost of quantitative power—the uncertainty scales inversely proportional to the square root of the input depth.
Deciding upon the sequencing depth for the IPs is complicated by the fact that different IPs have different complexities. In general, we use the following guidelines for mammalian genomes. If we are confident that the mark of interest is highly punctate, sparse and peak-like (e.g. H3K4me3), then we aim for approximately 50 million paired-end sequencing reads. If we know that the mark of interest is very broadly distributed and has high complexity (e.g. H3K27me3), then we aim for almost as many reads as used for input -- approximately 250–350 million paired-end reads. If we believe the mark to have complexity between the two extremes (e.g. H3K4me1, H3K4me2) or if we are unsure of the complexity of the mark, we aim for 100–150 million paired-end reads, and if that is insufficient, we re-sequence to obtain more reads.
With these numbers in mind, and with an understanding of the number of reads expected from the sequencing machine you will use, decide upon the multiplexing scheme you wish to use. For example, if the sequencer will yield approximately 450 million reads per lane, and if you have to sequence H3K27me3, H3K4me1, and H3K4me3 IPs, then one lane can be devoted to input (450 million paired-end reads), and another lane can be devoted to a mixture of H3K27me3, H3K4me1, and H3K4me3 IPs, with the libraries mixed as 5/9 H3K27me3, 1/3 H3K4me1, and 1/9 H3K4me3 (250 million, 150 million, and 50 million paired-end reads, respectively). As such, the first lane has to only use one index, and the second lane has to use three indices for multiplexing. Care should be taken to maximize Hamming Distance associated with multiplexed adapters (Supplementary Table 3).
For non-mammalian organisms, these numbers can be scaled roughly linearly with genome size.
The specificity evaluation of the antibodies will be limited by the diversity of standards used in the ICeChIP experiment. For example, if an antibody changes its binding characteristics based on the presence or absence of flanking combinatorial modifications (e.g. H3K4me3K9acK27ac vs. H3K4me3 alone), then this effect will not be detected without standards representing both of those modification patterns. However, we expect this limitation to diminish as more standards with different modification states become available and as antibody development proceeds apace.
Finally, these studies are critically dependent on the antibody reagents used. Unfortunately, our previous studies have shown that many of the validation metrics that antibody companies and large-scale consortia alike use are not effective predictors of antibody performance in ChIP experiments45, so in some respects, unless a previous study or group has previously validated an appropriate antibody with ICeChIP or its commercial equivalent, finding an antibody that is sufficiently specific for a ChIP experiment may require a certain degree of trial and error on the part of the end user.
Other Spike-In Methods
Other methods making use of exogenous spike-ins for ChIP have been described38,44. In particular, a previous study employed chromatin from Drosophila melanogaster as an exogenous spike-in to normalize ChIP-seq datasets targeting both tail and internal histone modifications38. This method (named ChIP-Rx) is similar to ICeChIP, but rather than calibrating with defined semisynthetic nucleosomes, nuclei or cells from a different organism than being studied are spiked into the ChIP experiment at the beginning of the workflow. In downstream analyses, the reads from this exogenous chromatin are used for normalization of the target ChIP enrichment much as our ICeChIP nucleosome standards are employed.
The primary advantage of the ChIP-Rx38 method relative to ICeChIP is that it is not inherently incompatible with fixed cell samples because both the target and exogenous cells can be crosslinked identically, especially if they are combined prior to crosslinking and sonication. This is in contrast with ICeChIP where, as previously mentioned, cells cannot be crosslinked. Additionally, at present, ICeChIP is only applicable for histone modifications and stable variants. However, given enough epitope similarity between transcription factors in the target and exogenous cells, the exogenous cell spike-in method could be applied to normalize ChIP-seq datasets targeting transcription factors or other targets not presently compatible with ICeChIP. To this end, another study described a “spike adjustment procedure (SAP)” based on a similar principle: the use of exogenous chromatin as a spike-in to normalize ChIP-seq towards non-histone targets44. By doing so, they enable themselves to detect global changes in PolII occupancy through normalized ChIP-seq44.
However, the normalized read density obtained from exogenous cell spike-in methods such as ChIP-Rx38, SAP44, or similar procedures55–57, will not be an absolute measurement, but rather, a relative measurement, unlike the HMD obtained from ICeChIP. As such, normalized ChIP-seq datasets cannot be compared between different modifications or antibodies employed. Additionally, out of these methods, only ICeChIP will provide meaningful information about the specificity of the antibody in situ, which is of critical importance. The ChIP-Rx also tends to show higher variability than does ICeChIP; the original work describing the use of D. melanogaster cell spike-ins showed massive quantitative differences in normalized ChIP-seq density across replicates38. The normalization only allows comparison, furthermore, between datasets that used the same set of cells as exogenous spike-ins; once this population of cells is depleted and a new lot of cells must be grown, then any comparisons between datasets normalized with different sets of cells will be more challenging. Additionally, the current exogenous cell spike-in methods are presently designed for NGS analysis rather than qPCR38,44,55–57, whereas ICeChIP is compatible with both NGS and qPCR43,45.
For targets or samples with which ICeChIP is not presently compatible, spiking-in exogenous cells will offer more normalization and comparability than no normalization whatsoever, and in that sense, is valuable. On balance, however, ICeChIP represents a more powerful tool to quantify histone modifications or stable variants for which nucleosome standards currently exist.
ICeChIP Experimental Design
This procedure owes a debt of gratitude to the seminal native ChIP protocol published more than a decade ago in this journal, geared towards qPCR readout4. In this work and our prior publications, we have expanded our ICeChIP method to be compatible both with qPCR readouts as well as Illumina next-generation sequencing readouts, as illustrated by the bioinformatic workflow in Fig. 2. While we have since optimized MNase digests for DNA quantification by deep sequencing (with higher conversion to mononucleosomes and greater genomic DNA recovery to minimize potential euchromatin bias, Fig. 3a–b), this protocol is highly similar to that of Brand et al.,4 but with internal standards spiked-in, and the attendant exigencies of sequencing library preparation and calibrated analysis.
Figure 2 |. ICeChIP-seq Bioinformatics Analysis Pipeline.
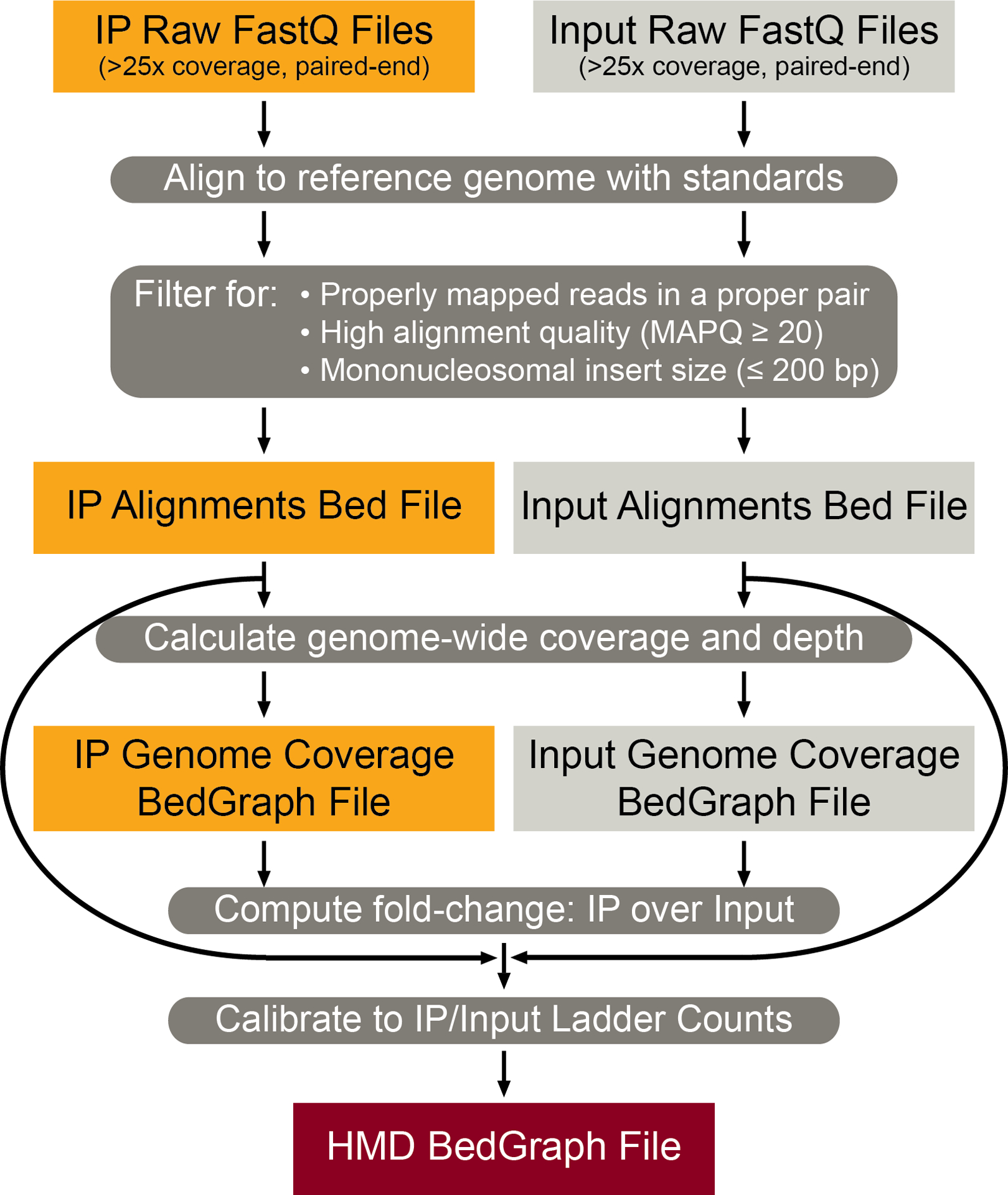
Workflow for analysis of ICeChIP-seq data. IP and Input FastQ paired-end reads are mapped and filtered for quality and mononucleosomal size into their respective alignment Bed files. From these Bed files, genome-wide coverage BedGraph files are generated and merged to compute the fold change of the IP over Input at genomic loci as a BedGraph file. This ratiometric BedGraph file is then calibrated to the nucleosome standard enrichment to generate the final HMD BedGraph file.
Figure 3 |. Representative results of the ICeChIP procedure.
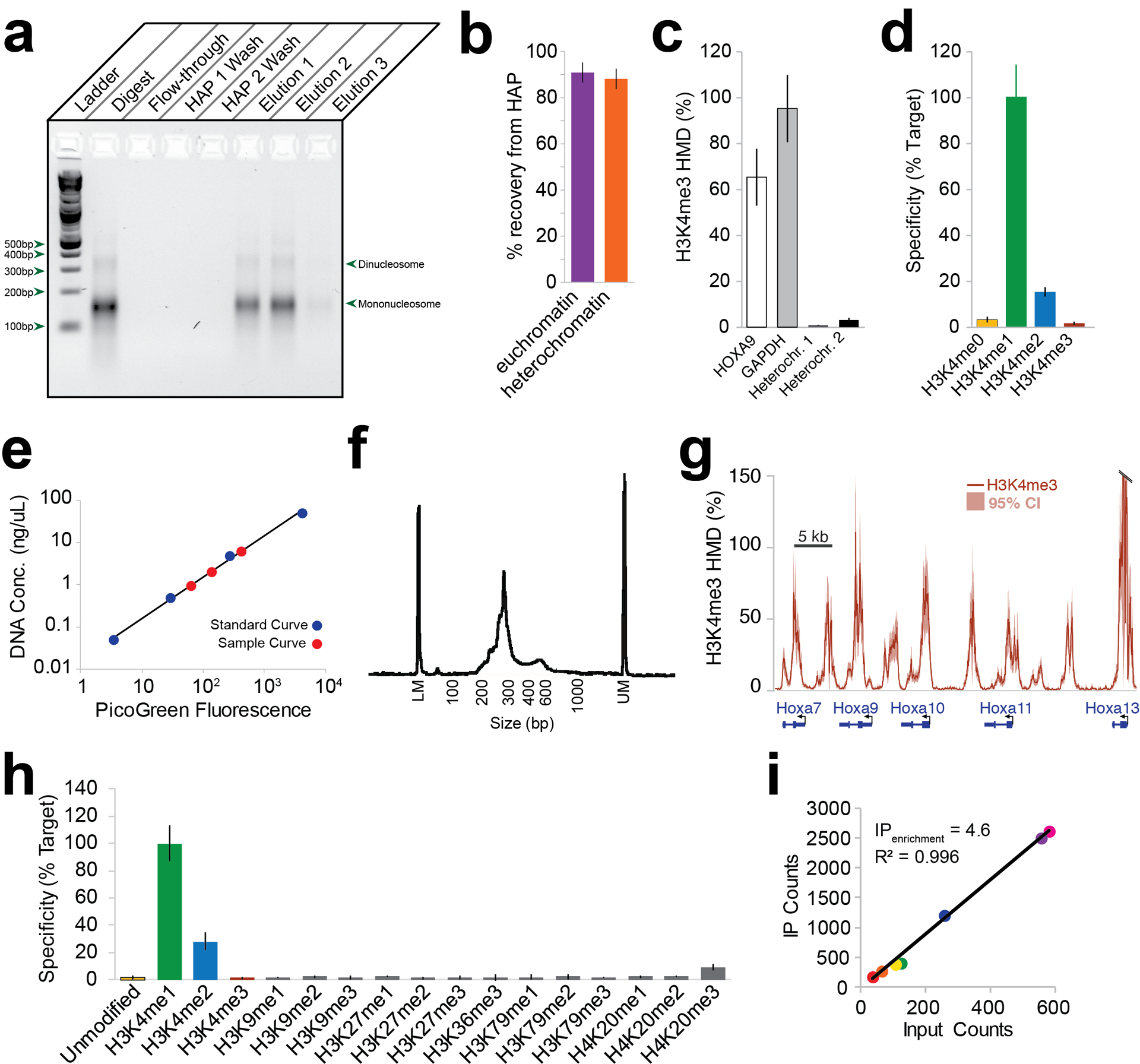
(a) Agarose gel showing anticipated HAP chromatography purification of native mononucleosomes4. Lanes, from left to right: DNA ladder, pooled digested elutions (Step 35), flow-through of binding resin (Step 31), pooled HAP Buffer 1 washes (Step 32), pooled HAP Buffer 2 washes (Step 33), HAP Elution #1 (Step 34), HAP Elution #2 (Step 34), and HAP Elution #3 (Step 34). The gel shows a largely mononucleosome-sized distribution of DNA in the elutions (pooled and individual) and no detectable DNA in the flow-through of resin or the washes. (b) Recovery of chromatin from HAP chromatography measured with euchromatin- and heterochromatin-specific primers. The euchromatic and heterochromatic nucleosomes are recovered in highly similar proportions. (c) H3K4me3 histone modification density measurements at four loci by qPCR. Error bars represent standard deviation of three qPCR measurements from one ICeChIP experiment. The samples all show HMDs between 0–100%, and there is greater H3K4me3 HMD at gene promoters (HOXA9, GAPDH) than at heterochromatic regions. (d) Specificity of anti-H3K4me1 antibody measured by qPCR. Black error bars represent standard deviation of replicates. Colored error bar shows qPCR standard deviation as percent of target. The binding of the antibody to off-target species is small compared to binding of antibody to the target, indicating that this is a high-specificity antibody. (e) Example of PicoGreen DNA quantification with both calibration curve (blue) and sample quantification curve (red). The calibration curve can be used to measure the DNA concentrations of the samples and spans a greater range of concentrations than do the samples. (f) Bioanalyzer capillary electrophoresis trace of appropriately-sized NGS IP library. This trace shows a peak largely between 200–350 bp, corresponding to a mononucleosome-sized insert (after adapter ligation, accounting for the greater size of the fragment). (g) H3K4me3 HMD genomic browser view from E14 mESCs. Shaded bars represent 95% confidence interval about HMD; solid line represents HMD measurement. The genome-browser view shows biologically meaningful HMDs between 0–100%, with peaks at genic regions, as expected for this modification. (h) Full specificity panel in NGS of H3K4me1 ICeChIP. Error bars represent standard deviation of enrichment of ladder members. The antibody shows high specificity (i.e. low binding to off-target modifications relative to the target), as anticipated from the ICeChIP-qPCR specificity testing in Fig. 3d. (i) Example of IP vs. Input count scatterplot from ICeChIP-seq, showing linearity of IP for H3K4me1 ladder. The scatterplot shows high linear correlation between the number of reads in the IP and the number of reads in the input for this given modification, which is expected. Panels (b), (c), and (g) adapted from Grzybowski et al.43 using AM39159 in E14 mESCs; panels (d) and (h) adapted from Shah et al.45 using EPG A-4031–050, Lot 606359. Panel (i) based on reanalysis of sequencing data accompanying Shah et al.45 using EPG A-4031–050, Lot 606359.
There are a number of key elements to an ICeChIP experiment that should be considered and, as necessary, optimized by the end-user.
Input Preparation
The first of these is digestion of the chromatin to a mononucleosomal pool. Our previous studies have shown that a small proportion of di- and oligonucleosomes are pulled down in ChIP disproportionately compared to mononucleosomal substrates, likely due to avidity biases43. As such, the digestion conditions should be optimized to ensure that the MNase digestion results in an overwhelmingly mononucleosomal population (>95%, Fig. 3a). Though we offer some guidance in MNase conditions that we have found useful, the digestion should nonetheless be confirmed and optimized by titrating either MNase units or the length of the digest by the end-user to yield the most accurate results.
It is important that this procedure should be conducted natively as opposed to on crosslinked and sonicated chromatin. We have found that if digestion is conducted completely, chromatin recovery is roughly equal between heterochromatin and euchromatin (Fig. 3b)43. Even if this was not the case, ICeChIP uses ratiometric comparison between the IP and Input for each locus, which would compensate for any differences in recovery between heterochromatin and euchromatin. Conversely, crosslinking has been previously noted to reduce the specificity of immunoprecipitations, and sonication can damage epitopes, which reduces ChIP efficiency58–61. In our experience, we have also found sonication to yield highly variable ICeChIP pulldowns, and the size distribution of fragments is often not fully mononucleosomal, which will lead to oligonucleosome avidity biases and distortions.
Scale of Experiment
Also important is the amount of chromatin and antibody used for the ChIP experiment. Overloading chromatin on a pulldown should be avoided, as higher amounts of chromatin can increase the amount of nonspecific precipitation onto the beads and will thereby reduce the specificity of the pulldown45. At the same time, the beads should not be substantially underloaded, because if the on-target modifications are not able to saturate the antibody surfaces, then off-target species may be able to bind more easily, again reducing specificity45. Though these effects are mild45, for optimal results, the amount of chromatin added should be optimized by the end user, because each antibody will behave differently and have different pulldown efficiencies. We offer guidance to this end in Box 1.
The scale of the experiment is particularly important for NGS applications. For ideal ICeChIP-seq library preparation, we have found that using 10 ng of DNA is ideal for mammalian or similarly-sized genomes, although if absolutely necessary, as little as 0.5ng of DNA can be used43,45. To achieve this, the approximate enrichment of an antibody should be known; some antibodies we have tested have target enrichments as low as 0.05% recovery, whereas some antibodies have enrichments as high as 95% recovery45. The optimal amount of DNA may be proportionally different for other genome sizes or samples of reduced complexity. If this amount of DNA cannot be obtained feasibly, the use of molecular barcodes in sequencing library preparation allows for the removal of PCR duplicates without introducing bias in NGS data analyses62, but sequencing depth at each locus may suffer in these cases. To maximize the power of ICeChIP-seq, we generally use very high-depth sequencing, as discussed in Box 2.
Standard Selection and Antibody Specificity Testing
The nucleosome standards used for the ICeChIP experiment should be ordered from commercial sources (see Reagents). The modifications represented by these nucleosome standards should be carefully considered, as the selection and design of these standards is important for obtaining the most reliable results. The specificity information that can be gleaned from an ICeChIP experiment is only as complete as the set of nucleosome standards spiked into the experiment, and to that end, under ideal circumstances, all plausibly cross-reacting standards should be spiked into the ICeChIP experiment. For example, under ideal circumstances, an anti-H3K4me3 ICeChIP experiment would make use of all the available lysine methylation nucleosome standards for antibody specificity testing. Based on these guidelines, we recommend ordering nucleosome standards representing all plausibly cross-reacting modifications.
However, testing all these standards for every experiment may prove to be both costly and laborious, particularly for a qPCR-dependent analysis method. For this reason, we highly recommend broadly testing an antibody using as many standards as feasible to test with qPCR, and then for subsequent ICeChIP-qPCR experiments, only the most important and similar cross-reacting marks can be tested. For example, in an anti-H3K4me3 ICeChIP, this may consist of all the standards bearing the H3K4 methylation states as well as select H3 trimethylation nucleosome standards. For ICeChIP-seq experiments, we recommend using all available standards, both to provide additional validation of the antibody and because with next-generation sequencing (NGS), each individual standard does not need to be manually probed by qPCR, so analysis of additional off-target standards is more straightforward and significantly less laborious.
Additionally, the standards that can be commercially ordered may have different numbers of uniquely-identifying DNA “barcode” sequences assigned to each modification. For example, some sets of commercial standards may have only two DNA barcodes per nucleosome standards at roughly equal concentrations (e.g. two DNA sequences bound to H3K4me3, another two DNA sequences bound to H3K9me3, and so on). For applications such as ICeChIP-qPCR, where it is likely that only a few sequences will be manually probed, or for applications in which antibody specificity is the primary evaluation metric, this is likely to be fine, and as such, these two-barcode nucleosome standards can be ordered and used.
However, for applications in ICeChIP-seq, where the actual quantification of the HMD is important, we recommend ordering a formulation of nucleosomes with at least 4–5 DNA barcodes per modification at different concentrations. This way, the linearity of the IP can be more easily assessed by a scatterplot after ICeChIP-seq (Fig. 1a) and, if the number of genomic reads falls within the range spanned by the different DNA sequences on the target modification (Fig. 1b), then this will offer increased confidence that the HMD measurements are appropriately quantitative. Ideally, for ICeChIP-seq, all of the nucleosome standards will carry a “ladder’ of 4–5 DNA barcodes, but at the very least, the nucleosome standards for the target modifications should be marked with 4–5 DNA barcodes; the other standards, if necessary, can rely on more limited numbers of DNA barcodes (e.g. 2 equimolar sequences), as the purpose of these standards is to evaluate specificity rather than to directly calibrate the pulldown.
In general, because of the ratiometric nature of ICeChIP calibration, the method is robust to the amount of each standard added45. Care should be taken to avoid adding too little of each standard, because if the standards are not being sampled with high enough numbers, then the result may be significantly increased noise in downstream analyses. This problem can be avoided by adding approximately 25 femtomoles of each nucleosome ladder per 100ug of crude chromatin. However, if more calibrant is added (inadvertently or otherwise), this should not pose a problem beyond consuming marginally more sequencing reads than are strictly necessary45. Though we spike in approximately the same amount of each calibrant to the chromatin, in evaluating the specificity of the antibody, it is important to consider the relative abundance differences of the on- and off-target modifications, as this may greatly affect the relative capture from genomic loci. For example, if H3K4me2 represents 5% of all genomic nucleosomes and H3K4me3 represents 1% of all genomic nucleosomes, then an anti-H3K4me3 antibody that pulls down 20% H3K4me2 relative to H3K4me3 will capture equivalent amounts of DNA from genomic nucleosomes bearing H3K4me2 and H3K4me345,63.
It is important that nucleosome standards be added prior to MNase digestion. As noted above, the exact amount of each standard that is added is not of critical important, subject to the caveats previously listed. However, the calibration method assumes that any DNA belonging to a standard in the input sample after hydroxyapatite (HAP) purification is bound to a nucleosome and is, therefore, available for immunoprecipitation by the antibody. If there is DNA in the sample that is not bound to the nucleosomes, due to handling or manufacturing, and this free DNA is not digested prior to the HAP purification, then it will appear in the input but not in the IP because free DNA will not be targeted by the IP. As such, it would appear that there was more of the standard to begin with than there actually was, which will cause a deflated enrichment computation and an inflated HMD computation. This problem can be avoided by adding standards prior to the MNase digestion step; in this case, the free DNA will be digested and will not appear in the input DNA, preserving the quantitative calibration by the standards.
qPCR Primer and Probe Design
Finally, primers and probes for qPCR should be designed to span a single nucleosome that is well-positioned; if MNase-seq datasets exist for the cell type of study, then those datasets should be used to guide primer and hydrolysis probe placement. In our experience, we have found 5’ FAM, 3’ BHQ1 hydrolysis probes to work well, and we have found success when using the Integrated DNA Technologies (IDT) PrimerQuest tool to design primer and probe sets. We recommend using hydrolysis probes, but SYBR-based qPCR should also work, albeit with higher background signal. Several primer and probe sets that we have previously validated43 for use in qPCR for HEK293 cells and E14 mESCs are listed in Supplementary Table 1. These can serve as examples or positive controls, but different cell types and treatment conditions may cause nucleosomes to be shifted, so validation or de novo MNase-seq may be needed for unusual cell types or treatment conditions.
Controls Needed
The major advantage of ICeChIP is that the internal standards allow for in situ controls of the antibody specificity and IP efficiency. As such, as noted above, we recommend conducting all ICeChIP experiments with standards both towards the targets as well as towards targets that may present a significant specificity challenge towards the antibody in question. Additionally, we recommend that all standards available be used for ICeChIP-seq experiments.
Particularly if troubleshooting, it may also be useful to use other controls that have been previously shown to work. For example, if MNase digestion proves to be difficult, or if the antibody shows very low efficiency or specificity, conducting the ICeChIP protocol with HEK293 cells (listed under “Reagents”) will serve as a positive control, as we have found that these cells work well for ICeChIP. Additionally, if datasets have been previously published with ICeChIP showing that a particular modification is or is not present at a given locus in a particular cell type, then conducting ICeChIP in that cell line and probing that locus may also serve as a useful positive or negative control.
Materials
Biological Materials
HEK293 Cells (ATCC, cat. number CRL-1573)
CAUTION: The cell lines used in your research should be regularly checked to ensure they are authentic and not infected with mycoplasma.
Reagents
Trizma hydrochloride (Sigma, cat. number T3253)
Sodium chloride (Sigma, cat. number S7653)
Potassium chloride (Sigma, cat. number P9333)
Sucrose (Sigma, cat. number S7903)
Hydrochloric acid (Sigma, cat. number H1758)
CAUTION: Corrosive to skin and acutely toxic. Wear gloves when handling and thoroughly wash any skin exposed to this chemical.
Sodium hydroxide (Sigma, cat. number 72068)
CAUTION: Corrosive to skin. Wear gloves when handling and thoroughly wash any skin exposed to this chemical.
Magnesium chloride hexahydrate (Sigma, cat. number 2670)
Calcium chloride dihydrate (Sigma, cat. number C8106)
Dithiothreitol (DTT) (Gold Biotechnology, cat. number DTT)
Phenylmethylsulfonyl fluoride (PMSF) (Gold Biotechnology, cat. number P-470)
CAUTION: Corrosive to skin and acutely toxic. Wear gloves when handling and thoroughly wash any skin exposed to this chemical.
cOmplete Protease Inhibitor Cocktail (Pierce, cat. number 78425)
Bovine Serum Albumin (BSA) (New England BioLabs, cat. number B9000).
Nonidet P-40 (NP-40) Substitute (Sigma, cat. number 74385)
CAUTION: Corrosive to skin. Wear gloves when handling and thoroughly wash any skin exposed to this chemical.
Ethylenediaminetetraacetic acid (EDTA) (Sigma, cat. number EDS)
Ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA) (Sigma, cat. number E3889)
Sodium phosphate monobasic (Sigma, cat. number S8282)
Potassium phosphate monobasic (Sigma, cat. number P5655)
Sodium dodecyl sulfate (Sigma, cat. number L3771)
CAUTION: Corrosive to skin and respiratory irritant. Wear gloves when handling solution. Wear both gloves and face mask when handling powder to prevent inhalation. Thoroughly wash any skin exposed to this chemical.
Glycerol (Sigma, cat. number G5516)
Tween 20 (Sigma, cat. number P9416)
Sucrose (Sigma, cat. number S0389)
Boric acid (Sigma, cat. number B7901)
Lithium chloride (Sigma, cat. number L4408)
Sodium deoxycholate monohydrate (Sigma, cat. number D5670)
TaqMan Gene Expression Master Mix (Applied Biosystems, cat. number 4369016)
Ethanol (Sigma, cat. number E7023)
CAUTION: Flammable. Keep away from open flames.
Sera-Mag Magnetic Speed-Beads (Fischer Scientific, cat. number 09-981-123)
Polyethylene glycol 8000 (Sigma, cat. number 89510)
Proteinase K (Invitrogen, cat. number 25530049)
Agarose (Invitrogen, cat. number 16500100)
Invitrogen 1 kb Plus DNA Ladder (Invitrogen, cat. number 10787018)
SYBR Gold (Invitrogen, cat. number S11494)
CAUTION: DNA intercalating dye. Wear gloves when handling and thoroughly wash any skin exposed to this chemical.
Micrococcal Nuclease (Worthington, cat. number LS004797)
CRITICAL: Different enzyme manufacturers use different unit definitions and quality control metrics for MNase, so substitutions of this reagent may require tuning of a wider parameter space to optimize.
Dynabeads Protein G (Invitrogen, cat. number 10003D)
CHT Ceramic Hydroxyapatite Type I, 20 μm particle size (Bio-Rad, cat. number 1582000)
Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher, cat. number P7589)
NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, cat. number E7645)
NEBNext Multiplex Oligos for Illumina (New England Biolabs, cat. number E7335/E7500)
DNA ladder (Invitrogen, cat. number 15628-050)
Primers and hydrolysis probes (IDT custom order; see Experimental Design, and Supplementary Table 1 for further information)
Semisynthetic nucleosome standards (Epicypher custom order; see Experimental Design)
Anti-histone PTM antibody (variable)
Equipment
UltraFree-MC Centrifugal Filter, 0.45 μm pore size (Millipore, cat. number UFC30HV25)
Siliconized low retention microcentrifuge tubes (Fisher, cat. number 02-681-321)
PCR tubes (Denville, cat. number C18064)
Conical centrifuge tubes, 15 mL (Fisher, cat. number 14-959-53A)
NBS 384 well microplates (Corning, cat. number 3575)
384 Well PCR Plate (Thermo Fisher, cat. number AB-1384)
Sorvall Legend XTR Centrifuge (or equivalent)
Thermo Fisher TX-750 4 + 750 mL Swinging Bucket Rotor (or equivalent)
Sorvall Legend Micro 17 R Centrifuge (or equivalent)
Branson 2800 Ultrasonic Cleaner (water bath sonicator) (or equivalent)
NanoDrop ND-1000 Spectrophotometer (or equivalent)
Bio-Rad C1000 Touch Thermal Cycler (or equivalent)
Bio-Rad CFX384 Real-Time System (or equivalent)
Eppendorf Thermomixer (or equivalent)
ATR Rotamix (or equivalent)
Labnet Spectrafuge Mini centrifuge (or equivalent)
DynaMag-96 Side Magnet (or equivalent)
Scientific Industries Vortex-Genie 2 (or equivalent)
Tecan Infinite F200 Pro Plate Reader (or equivalent)
Reagent Setup
CRITICAL: Do not store and reuse supplemented solutions.
PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM NaH2PO4, 1.8 mM KH2PO4. Adjust pH to 7.5 with HCl and NaOH solutions, autoclave and store at 25°C for up to six months.
PMSF stock: 200 mM PMSF in ethanol, store at −20°C for years. Bring to room temperature (20–25°C) and mix well by vortexing prior to use.
DTT stock: 1 M DTT solution in water, aliquot and store at −20°C for years.
1x TBE: 89 mM Tris-HCl, 89 mM boric acid, 2 mM EDTA. Adjust pH to 8.3 with HCl and NaOH solutions, store at 25°C for up to six months.
TE Buffer: 10 mM Tris-HCl, 1 mM EDTA. Adjust pH to 8.0 with HCl and NaOH solutions, filter sterilize, and store at 25°C for up to six months.
0.1x TE Buffer: 1 mM Tris-HCl, 100 μM EDTA. Dilute 1xTE 10-fold in sterile water, and store at 25°C for up to six months.
Buffer N: 15 mM Tris-HCl, 15 mM NaCl, 60 mM KCl, 8.5% (w/v) sucrose, 5 mM MgCl2, 1 mM CaCl2. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize, store at −20°C for years or 4°C for up to six months. At time of use, supplement with 1 mM DTT, 200 μM PMSF, 1x Protease Inhibitor Cocktail, 50 ug/mL BSA.
2x Lysis Buffer: 15 mM Tris base, 15 mM NaCl, 60 mM KCl, 8.5% (w/v) sucrose, 5 mM MgCl2, 1 mM CaCl2. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize, store at −20°C for years or 4°C for up to six months. At time of use, supplement an aliquot of this buffer with1 mM DTT, 200 uM PMSF, 1x Protease Inhibitor Cocktail, 50 μg/mL BSA, 0.6% (v/v) NP-40 substitute.
Sucrose Cushion: 15 mM Tris-HCl, 15 mM NaCl, 60 mM KCl, 30% (w/v) sucrose, 5 mM MgCl2, 1 mM CaCl2. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize, store at −20°C for years or 4°C for up to six months. At time of use, supplement with 1 mM DTT, 200 μM PMSF, 1x Protease Inhibitor Cocktail, 50 μg/mL BSA.
MNase Solution: Dilute to 15 Worthington units per μL. Aliquot and store at −80°C for years. CRITICAL: Treat as single-use aliquots and discard after use. MNase conditions tend to be robust for a given suspended batch if stored this way.
10x MNase Stop Buffer: 100 mM EDTA, 100 mM EGTA. Filter sterilize and store at room temperature for years.
HAP Buffer 1: 5 mM NaH2PO4, 600 mM NaCl, 1 mM EDTA. Adjust pH to 7.2 with HCl and NaOH solutions, filter sterilize, and store at room temperature for years. At time of use, supplement with 200 μM PMSF.
HAP Buffer 2: 5 mM NaH2PO4, 100 mM NaCl, 1 mM EDTA. Adjust pH to 7.2 with HCl and NaOH solutions, filter sterilize, and store at room temperature for years. At time of use, supplement with 200 μM PMSF.
HAP Elution Buffer: 500 mM NaH2PO4, 100 mM NaCl, 1 mM EDTA. Adjust pH to 7.2 with HCl and NaOH solutions, filter sterilize, and store at room temperature for years. At time of use, supplement with 200 μM PMSF.
ChIP Buffer 1: 25 mM Tris-HCl, 5 mM MgCl2, 100 mM KCl, 10% (v/v) glycerol, 0.1% (v/v) NP-40 Substitute. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize. At time of use, supplement with 200 μM PMSF and 50 μg/mL BSA.
ChIP Buffer 2: 25 mM Tris-HCl, 5 mM MgCl2, 300 mM KCl, 10% (v/v) glycerol, 0.1% (v/v) NP-40 Substitute. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize. At time of use, supplement with 200 μM PMSF.
ChIP Buffer 3: 10 mM Tris-HCl, 250 mM LiCl, 1 mM EDTA, 0.5% (w/v) sodium deoxycholate, 0.5% (v/v) NP-40 Substitute. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize. At time of use, supplement with 200 μM PMSF.
ChIP Elution Buffer: 50 mM Tris-HCl, 1 mM EDTA, 1% (w/v) SDS. Adjust pH to 7.5 with HCl and NaOH solutions, filter sterilize. Store at 4°C for up to six months.
SeraPure64: 0.1% Sera-Mag Magnetic Speed-Beads (washed twice with water on magnetic rack), 18% PEG-8000 (w/v), 1 M NaCl, 10 mM Tris-HCl, 1 mM EDTA, 0.05% Tween 20. Store at 4°C for up to six months.
Software
Bowtie265 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
Samtools66 (http://samtools.sourceforge.net/)
BEDTools67 (http://bedtools.readthedocs.io/en/latest/)
UCSC Tools (http://hgdownload.soe.ucsc.edu/admin/exe/)
icechip (http://github.com/shah-rohan/icechip)
computeHMDandError (http://github.com/shah-rohan/icechip)
Procedure
Nuclei Preparation | Timing: 2h
-
1
Culture cells to a quantity of 108 cells in log phase growth, adding fresh media 3–6 hours prior to collection. ? Troubleshooting
-
2
Collect cells as per passaging instructions (e.g. trypsinization and quenching for adherent cells), pellet by table-top centrifugation (500g, 5 minutes, 4°C), and discard supernatant.
-
3
Resuspend pellet in 5 mL of ice-cold PBS, pellet by spinning (500g, 5 minutes, 4°C), and remove supernatant. Repeat this wash.
Pause Point: Cell pellets can be flash-frozen and stored at −80°C at this point for several years. When ready to proceed, thaw cells in hand and proceed with resuspension, pipetting well until there are no visible clumps.
-
4
Resuspend pellet in 5 mL of ice-cold Buffer N, pellet by spinning (500g, 5 minutes, 4°C), and remove supernatant. Repeat this wash.
-
5
Resuspend cells to a single-cell suspension in roughly two packed cell volumes (PCV) of ice-cold Buffer N. For 108 cells at the beginning of the protocol, this is approximately 2 mL. Measure volume of cell suspension.
Critical Step: Cells must be in a single-cell suspension prior to addition of lysis buffer. Typically, this means pipetting up and down for at least twice the number of strokes required to obtain a visually homogenous solution. If uncertain, examine the suspension by hemocytometry to confirm absence of clumps.
-
6
Add 1 volume of ice-cold 2x Lysis Buffer to the cell suspension and mix by pipetting up and down at least 10 times. Incubate on ice for 10 minutes.
-
7
Pellet nuclei by table-top centrifugation (500g, 5 minutes, 4°C) and discard supernatant. ? Troubleshooting
Critical Step: The nuclei pellet should be whiter in color than the cell pellet was prior to lysis. If color remains slightly yellow, examine the crude nuclei under a light microscope and confirm lysis by trypan staining68.
-
8
To a fresh 15 mL conical tube, add 7.5 mL of sucrose cushion. Resuspend nuclei in 2.5 mL of Buffer N and gently layer the suspension on top of the sucrose cushion by pipette. If the boundary between the crude nuclear suspension and the sucrose is really sharply defined, submerge a pipette tip just a few millimeters into the cushion and slowly stir to ensure a “fuzzy” interface. Too sharp of a phase boundary can cause excessive hang-up of nuclei and defeat the purpose of the cushion. Pellet nuclei by spinning (500g, 12 minutes, 4°C, swinging-bucket rotor). Remove supernatant by first completely removing the top ~5 mL, then aspirating the remaining supernatant. ? Troubleshooting
Critical Step: Ensure that the cellular debris at the top of the supernatant is completely removed before removing the remainder of the supernatant. If you fail to completely remove the cellular debris, it will interfere with chromatin concentration measurements.
-
9
Resuspend nuclei in 6 pellet volumes of Buffer N and place on ice.
-
10
Gently mix nuclei suspension and immediately aliquot 2 μL into each of three separate tubes. Add 18 μL of 2 M NaCl to all three aliquots to lyse nuclei. Also prepare a spectrophotometric blank with 2 μL of Buffer N and 18 μL of 2M NaCl.
-
11
Mix by vigorous vortexing and sonicate for 10–15 minutes to solubilize chromatin in a room temperature water bath and default sonication settings.
Critical Step: Sonication settings will vary by sonicator. If using a sonicator other than the Branson 2800 Ultrasonic Cleaner, then the time and settings may vary and need to be empirically optimized. Sonication is complete when the solution is nonviscous and can be pipetted.
-
12
Measure total nucleic acid concentration by Nanodrop, using the aliquot of Buffer N with 2 M NaCl as blank solution. This can be approximated as C ng/μL = A260, 10 mm path length * 50 ng/μL (the concentration provided by the Nanodrop). ? Troubleshooting
-
13
Average the three measurements of chromatin concentration and multiply by 10 (accounting for the 10-fold dilution of the aliquots) to obtain the chromatin concentration in the nuclei suspension. Dilute the suspension to an approximate concentration of 1 μg/μL of nucleic acid with Buffer N. ? Troubleshooting
-
14
If possible, aliquot nuclei into 100 μL aliquots into siliconized, low-adhesion Eppendorf microcentrifuge tubes. From 108 mammalian cells, this nuclei preparation procedure yields approximately 7–8 aliquots of nuclei suspensions.
Pause Point: Nuclei can be flash-frozen and stored at −80°C at this point for several years. However, this may cause increased clumping of the nuclei, which may decrease MNase efficiency. If the nuclei are flash-frozen and stored at −80°C at this stage, extra care should be taken to adequately resuspend them at the time of use by thawing in hand and gently pipetting up and down to resuspend until there are no visible clumps. To verify adequate resuspension, examine the nuclei under a light microscope to confirm that the nuclei are evenly suspended.
Antibody Conjugation to Beads | Timing: 2h
CRITICAL: The following steps (15–21) should be undertaken for each different antibody to be deployed.
-
15
Aliquot 25 μL of gently resuspended Protein G magnetic beads (30 mg/mL). Collect beads with rare earth magnetic rack and remove supernatant. ? Troubleshooting
Critical Step: If you will be conducting more than one IP, then ideally, these steps should be followed in parallel for each IP. However, for convenience, if two or more IPs use the same antibody, then it is possible to increase the volume of beads and amount of antibody in the conjugation steps (Steps 15–20), then split the beads as 25 μL per IP in Step 21.
-
16
Resuspend beads in 200 μL of ChIP Buffer 1 by pipetting, place on magnetic rack once again, and remove supernatant. Repeat this wash once more.
-
17
Dilute 6 μg of antibody to a final volume of 100 μL with ChIP Buffer 1 and add to beads. Resuspend beads by pipetting.
-
18
Incubate on a rotator at 4°C for at least one hour.
-
19
Collect beads with magnetic rack and remove supernatant.
-
20
Resuspend beads in 200 μL of ChIP Buffer 1 and, on magnetic rack, remove supernatant. Repeat this wash once more.
-
21
Resuspend beads in 25 μL of ChIP Buffer 1 and place on ice for up to 6 hours while digesting chromatin and conducting hydroxyapatite (HAP) purification of nucleosomes (Steps 22–37).
MNase Digestion and HAP Purification | Timing: 1.5h
-
22
If nuclei (from Step 14) were frozen, thaw and mix well by pipetting.
Critical Step: If you will need more than ~30 μg of HAP-purified chromatin for your IPs, then process multiple crude nuclear aliquots representing 100 μg of total nucleic acid from Step 14 in parallel. HAP chromatography in this format is most consistent with aliquots processed in parallel rather than as one large batch of nuclei. If you will be processing crude nuclear aliquots representing less than 100 μg of total nucleic acid, scale down this section linearly.
-
23
Add the manufacturer recommended quantity of each nucleosome standard to nuclei containing 100 μg of chromatin (measured in Step 13) and mix by pipetting.
Critical Step: Standards must be added prior to MNase digestion. If you add standards after MNase digestion, then any free DNA left in the standards (from reconstitution, handling or thawing) will be present in input but not in the IP. As such, the enrichment of the nucleosome standards will be deflated, and the HMD measurements of genomic loci will be inflated accordingly. We have found 25 femtomoles of each nucleosome standard to be broadly adequate for 100 μg of crude chromatin.
-
24
Prewarm nuclei for 2 minutes at 37°C while shaking at 900 rpm in a thermomixer.
-
25
While still at 37°C, add 1.5 μL of MNase Enzyme Solution. Incubate for 12 minutes at 37°C while shaking at 900 rpm. After incubation is complete, immediately place on ice.
Critical Step: Ensure nuclei are not clumped at this stage; if they are, then digestion may not go to completion. Use multiple pipette strokes to gently break clumps. However, insoluble pellets may occur in freeze-thawed nuclei. If there is an insoluble pellet at this stage, it is particularly crucial to ensure the digestion progressed to completion by gel visualization (Steps 56–57).
-
26
Stop digestion by adding 0.1 volumes of MNase Stop Buffer while gently vortexing. For example, if the volume of the sample after adding standards is 110 μL, add 11 μL of MNase Stop Buffer.
Critical Step: MNase Stop Buffer should be added on vortex to prevent locally high concentrations of EDTA and EGTA salts from causing nucleosomes to fall apart.
-
27
Lyse nuclei by adding 0.12 volumes of 5 M NaCl while gently vortexing. For example, if the volume of the sample after adding standards and stopping digestion is 121 μL, add 14.5 μL of 5 M NaCl.
Critical Step: Nuclei lysis should be done on the vortex to prevent a locally high concentration of NaCl from causing nucleosomes to fall apart. The solution should demonstrably and materially clarify.
-
28
Pellet insoluble fraction of lysed nuclei by centrifugation (18000g, 1 minute, 4°C). Transfer soluble fraction, which includes chromatin, to a fresh tube and place on ice. Discard insoluble pellet.
-
29
In a separate microcentrifuge tube, rehydrate 66 mg of hydroxyapatite (HAP) resin with 200 μL of HAP Buffer 1 and mix by pipetting.
-
30
Add soluble chromatin fraction from Step 28 to rehydrated HAP slurry from Step 29. Rotate end-over-end at 10–15 rpm for 10 minutes at 4°C.
-
31
Transfer HAP slurry into 0.45 μm UltraFree-MC centrifugal filter unit and spin (1000g, 30 seconds, 4°C). Save 5μL of flow-through on ice for purification and visualization (Steps 47–57) and discard remainder.
-
32
Add 200 uL of HAP Buffer 1 to HAP resin and spin (1000g, 30 seconds, 4°C),. Repeat this step three times for a total of four washes with HAP Buffer 1. Pool flow-throughs, save 5 μL on ice for purification and visualization (Steps 47–57), and discard remainder.
-
33
Add 200 uL of HAP Buffer 2 to HAP resin and spin (1000g, 30 seconds, 4°C). Repeat this step three times for a total of four washes with HAP Buffer 2. Pool flow-throughs, save 5 μL on ice for purification and visualization (Steps 47–57), and discard remainder.
-
34
Transfer filter unit to a fresh microcentrifuge tube. Add 100 μL of HAP Elution Buffer to HAP resin and spin (1000g, 30 seconds, 4°C). Save elution. Repeat this step two times for a total of three elutions.
-
35
Pool elutions and mix well by pipetting or gentle vortexing. Set aside 5 μL of elution on ice for purification and visualization (Steps 47–57), which should show a largely mononucleosomal pool.
-
36
Measure fragmented and purified chromatin concentration in triplicate by Nanodrop, with HAP Elution Buffer used as a blank. The chromatin concentration can be approximated as 50*Abs260nm.
-
37
Adjust concentration of chromatin to 20 μg/mL with ChIP Buffer 1.
Critical Step: If chromatin concentration is less than 40 μg/mL, then add one volume of ChIP Buffer 1 to stabilize nucleosomes. However, native nucleosomes are inherently unstable below 5 ng/uL and will likely fall apart. If this is the case, your HMD values will likely be deflated because the semisynthetic nucleosomes reconstituted with the 601 DNA-binding sequence are more stable than native genomic nucleosomes. If your chromatin concentration is below 5 ng/uL, then we recommend repeating the procedure with scaled down the HAP chromatography so that you can elute in a smaller volume.
Immunoprecipitation | Timing: 1h
-
38
Save 15 μL of the chromatin and set it aside on ice to be used as Input (Step 47).
-
39
Add antibody conjugated beads (prepared in Steps 15–21) for each IP to an appropriate amount of chromatin, taken from the chromatin not set aside in Step 38.
Critical Step: Determining the amount of chromatin to be used is critical, as discussed in Box 1. At this stage, samples can be moved to PCR strips for convenience, but care should be taken to avoid cross-contamination between samples.
-
40
Incubate samples with gentle end-over-end rotation for 10 minutes at 4°C. Collect beads with a magnetic rack and remove supernatant.
Critical Step: Do not over-incubate the antibody with the sample – long incubations tend to reduce specificity. For most decent antibodies, the kinetics of binding will saturate the antibody in minutes, whereas nonspecific precipitation on the surface of the beads will occur more slowly, accumulating over time. Therefore, antibody binding should be limited in time.
-
41
Resuspend beads in 200 μL of ChIP Buffer 2, transfer the slurry to fresh siliconized tubes, and rotate for 10 minutes at 4°C. Collect beads with a magnetic rack and remove supernatant.
-
42
Repeat Step 41 for a second ChIP Buffer 2 wash.
-
43
Resuspend beads in 200 μL of ChIP Buffer 3, transfer the slurry to fresh siliconized tubes, and rotate for 10 minutes at 4°C. Collect beads with a magnetic rack and remove supernatant.
-
44
Resuspend beads in 200 μL of ChIP Buffer 1 and transfer to new siliconized tubes. Collect beads with a magnetic rack and remove supernatant.
-
45
Resuspend beads in 200 μL of TE Buffer. Collect beads with a magnetic rack and remove supernatant.
-
46
Resuspend beads in 50 μL of ChIP Elution Buffer in PCR strips and incubate in a thermocycler at 55°C with the hot lid at ≥65°C for 5–10 minutes.
-
47
Collect beads with a magnetic rack and transfer supernatant to new siliconized tubes. Add 35 μL of ChIP Elution Buffer to the Input from Step 38, bringing it to a total of 50 μL, and transfer to new siliconized tube. Add 45 μL of ChIP Elution Buffer to the 5 μL of samples saved for visualization (from Steps 31, 32, 33 and 35), bringing each to a total of 50 μL, and transfer to new siliconized tubes.
Pause Point: All samples can be stored at −20°C at this point for up to 1 month.
DNA Purification | Timing: 3h
-
48
Add 2 μL of 5 M NaCl, 1 μL of 500 mM EDTA, and 1 μL of 20 mg/mL Proteinase K to each sample.
-
49
Incubate in a thermocycler at 55°C with the hot lid at ≥65°C for 2 hours to conduct Proteinase K digestion.
-
50
After 1.5 hours of Proteinase K digestion, place SeraPure beads on a rotator at room temperature to equilibrate temperature and resuspend beads.
-
51
After 2 hours of Proteinase K digestion, add 150 μL of SeraPure beads to each sample and mix well by pipetting. Incubate at room temperature for 15 minutes.
Critical Step: SeraPure is highly viscous, so pipetting and mixing should be done carefully to ensure accurate volume delivery.
-
52
Collect beads on magnetic rack for at least 5 minutes, then remove and discard supernatant.
-
53
Without removing tubes/beads from the magnetic rack, add 200 μL of 70% ethanol to the beads. Without resuspending the beads, remove all the ethanol. Let dry for 1 minute.
-
54
Repeat Step 53 twice.
-
55
Remove beads from magnetic rack and resuspend in 50 μL of 0.1x TE Buffer. Collect beads with magnetic rack and transfer supernatant, containing purified DNA, to new siliconized tubes.
Pause Point: Samples can be stored at −20°C at this point for up to 1 month or at −80°C for several years.
Visualization of DNA | Timing: 2h
-
56
Run 100bp marker ladder and half of the purified DNA from samples saved for visualization on a 2% agarose in 1x TBE gel at a constant rate of 5–7 V/cm for 1 hour.
-
57
Stain gel in 1x SYBR Gold for 1 hour and visualize with an ultraviolet transilluminator to ensure the DNA primarily is present in the elutions and is mononucleosome-sized (150–200 bp in size). ? Troubleshooting
Pause Point: Samples can be stored at −20°C at this point for up to 1 month or at −80°C for several years.
DNA Quantification with qPCR | Timing: 4h
-
58
Make up qPCR 20x Primer-Probe Mix for each qPCR primer and probe combination, both for genomic loci and nucleosome standards, as 18 μM of each primer and 5 μM probe.
-
59
For each qPCR 20x Primer-Probe Mix, make Reaction-Specific Master Mix with 1.1*3*(number of samples)*5 μL of 2x TaqMan Gene Expression Master Mix and 1.1*3*(number of samples)*0.5 μL of qPCR 20x Primer-Probe Mix.
-
60
Pipette 5.5 μL of each Reaction-Specific Master Mix into a qPCR plate to set up reactions in triplicate for each of the samples, including input.
-
61
Add 4.5 μL of 1:10 dilutions of each sample (input and each IP from Step 55) to the appropriate wells of the qPCR plate containing Reaction-Specific Master Mix. Mix well by pipetting up and down at least 10 times.
The qPCR setup described in Steps 59–61 is summarized in the table below.
| Component | Amount (μL) | Final Concentration |
|---|---|---|
| 20x qPCR Primer-Probe Mix | 0.5 | 900 nM each primer 250 nM hydrolysis probe |
| TaqMan Gene Expression Master Mix | 5 | |
| Sample DNA | 4.5 | |
| Total | 10 (one reaction) |
-
62
Cover the qPCR plate with transparent cover sheet and spin briefly to collect liquid at bottom.
-
63
Conduct qPCR with the following reaction conditions:
| Cycle Number | Denature | Anneal/Extend | Plate Read |
|---|---|---|---|
| 1 | 95°C, 10 min | No | |
| 2–40 | 95°C, 15 s | 60°C, 1 min | Yes |
-
64
Compute enrichment of each standard and genomic locus using the equation: Enrichment = 2IN-IP*(dilution)*100%, where IN and IP represent Cq values of the IP and the Input and the dilution represents the fold dilution of the Input chromatin (15 μL saved from Step 38) over the IP chromatin (however much was added to the IP). This enrichment value represents the amount of DNA found in the IP divided by the amount of DNA found in the Input for the probed species – in essence, the pulldown efficiency for nucleosomes at each locus or for each nucleosome standard. As such, enrichment values should be computed for each probed genomic locus (EnrichmentLocus) and each relevant barcoded standard, particularly amplicons for the on-target nucleosome (EnrichementOn-Target), and other related off-target nucleosomes (EnrichmentOff-Target). ? Troubleshooting
-
65
Compute HMD as EnrichmentLocus/EnrichmentOn-Target*100%.
-
66
Compute Off-Target Binding as EnrichmentOff-target/EnrichmentOn-Target*100%. We find it useful to display this as a bar graph to evaluate specificity (Fig. 3c). ? Troubleshooting
Pause Point: Samples can be stored at −20°C at this point for up to 1 month or at −80°C for several years.
DNA Quantification with PicoGreen prior to Illumina Library preparation | Timing: 1h
-
67
Make up 300 + 50*(number of inputs and IPs) μL of 1x TE buffer from nuclease-free 10x TE buffer provided with Quant-iT PicoGreen dsDNA Assay Kit using nuclease-free water.
-
68
Make up 150 + 25*(number of inputs and IPs) μL of 1x PicoGreen with 1x TE buffer.
-
69
Dilute 1 μL of stock 100 ng/μL Lambda dsDNA (provided with PicoGreen Kit) with 49 μL of 1x TE buffer to make 50 μL of 2 ng/uL Lambda dsDNA solution.
-
70
Pipette 27 uL of 1x TE buffer into three separate tubes. Serially dilute 3 uL of the 2 ng/uL Lambda dsDNA solution into each of these tubes sequentially. The end result is four solutions, with respective Lambda dsDNA concentrations of 2 ng/uL, 0.2 ng/μL, 0.02 ng/μL, and 0.002 ng/μL.
-
71
Using a Corning plate with wells appropriate for a fluorescent plate reader, transfer 25 μL of the Lambda dsDNA serial dilutions into separate wells of the plate reader. Transfer 25 μL of 1x TE buffer to another well of the plate reader for background measurement.
-
72
Transfer 24 μL of 1x TE buffer into as many wells as the number of inputs and IPs.
-
73
Transfer 1 μL of each input or IP DNA (Step 55) into a well containing TE buffer.
-
74
Add 25 μL of 1x PicoGreen to each well containing Lambda dsDNA, input, or IP. Mix by pipetting up and down 10 times in the well.
-
75
Wrap the plate in aluminum foil and spin briefly to collect liquid at the bottom of the well. Incubate the plate at room temperature for approximately 2–3 minutes.
-
76
Read fluorescence in triplicate with a Tecan Infinite F200 Pro Plate Reader plate reader using the following settings: excitation at 485 nm, emission at 535 nm, optimal gain, 10 flashes, 20 μs integration time, 0 ms lag time, 10 ms settle time.
-
77
For each sample, average the fluorescence measurements of the three reads, then subtract the average fluorescence reading from the blank background sample.
-
78
Using the average fluorescence readings minus the blank fluorescence reading for the serially diluted Lambda dsDNA samples, compute a linear regression slope between the fluorescence readings and the amount of DNA added to each well (i.e. 50 ng, 5 ng, 0.5 ng, and 0.05 ng) in EXCEL or R. Use the regression slope to compute the amount of DNA in each well of the input and IP samples (scaled by the 25-fold dilution factor), which is also the concentration of input and IP samples in ng/μL.
-
79
Multiply the concentration of each sample by the volume of the sample to obtain the amount of DNA for each input or IP sample. ? Troubleshooting
Pause Point: Samples can be stored at −20°C at this point for up to 1 month or at −80°C for several years.
Next-Generation Sequencing Library Adaptor Ligation | Timing: 2.5h
-
80
For samples containing at least 10 ng of DNA, carry forward 10 ng of DNA and dilute with water to a final volume of 50 μL in PCR tubes or strips. Store any remaining DNA at −80°C for several years. For samples containing less than 10 ng of DNA, proceed with all of the material diluted with MilliQ water to a final volume of 50 μL.
-
81
To each 50 μL sample, add 7 μL of NEBNext Ultra II End Prep Reaction Buffer and 3 μL of NEBNext Ultra II End Prep Enzyme Mix. Mix well by pipetting up and down 10 times.
-
82
Incubate samples in a thermocycler at 20°C for 30 minutes, at 65°C for 30 minutes, then holding at 4°C, with the heated lid set to ≥75°C throughout.
-
83
Remove the SeraPure beads from refrigerator so they will be at room temperature when needed and rotate end-over-end.
-
84
Dilute NEBNext Adaptors for Illumina to a final volume of 2.5 μL per sample. For samples with 5–10 ng of DNA, dilute NEBNext Adaptors for Illumina 10-fold from the stock solution. For samples with less than 5 ng of DNA, dilute NEBNext Adaptors for Illumina 25-fold from the stock solution.
Critical Step: Ensure that the samples receive the proper dilution of NEBNext Adaptors for Illumina, as described in this step (Step 84) and manufacturer recommendations. If the samples receive over-diluted adaptors, then the library preparation will have lower yield. If the samples receive under-diluted adaptors, then adaptor dimers can form, which are troublesome to remove from the sample and may consume many sequencing reads.
-
85
To each sample, add 30 μL of NEBNext Ultra II Ligation Master Mix, 1 μL of NEBNext Ligation Enhancer, and 2.5 μL of diluted NEBNext Adaptors for Illumina. Mix well by pipetting up and down 10 times.
Critical Step: NEBNext Ultra II Ligation Master Mix is highly viscous, so pipetting and mixing should be done carefully. If making pre-mixes of the reagents to add to each sample, do not add NEBNext Adaptors for Illumina to the pre-mix.
-
86
Incubate samples in a thermocycler at 20°C for 15 minutes with the heated lid off.
Critical Step: Do not incubate samples for longer than 15 minutes. If samples are incubated for too long, then adaptor dimers can form, which are troublesome to remove from the sample and may take up many sequencing reads.
-
87
Add 3 μL of USER Enzyme to each sample and mix well by pipetting up and down 10 times. Incubate samples in a thermocycler at 37°C for 15 minutes with the heated lid set to ≥47°C.
-
88
Add 82 μL of SeraPure Beads to each sample. Mix well by pipetting up and down at least 10 times and incubate at room temperature for 15 minutes.
Critical Step: SeraPure is highly viscous, so pipetting and mixing should be done carefully.
-
89
Collect beads on magnetic rack for at least 5 minutes. Remove and discard supernatant.
-
90
Without removing beads from the magnetic rack, add 200 μL of 70% ethanol to the beads. Without resuspending the beads, remove all the ethanol. Let dry for 1 minute.
Critical Step: Disrupting SeraPure resin pellet will dissociate the bound DNA, markedly reducing yield.
-
91
Repeat Step 90 twice.
-
92
Remove beads from magnetic rack and resuspend in 17 μL of 0.1x TE Buffer. Collect beads with magnetic rack and transfer supernatant, containing purified DNA, to new siliconized tubes.
Next-Generation Sequencing Library Amplification | Timing: 1.5h
-
93
Decide upon multiplexing scheme for DNA library generation, as described in Box 2.
-
94
For each lane requiring multiplexing, use Supplementary Table 1 to find sets of indices that have high Hamming distances to minimize improper demultiplexing after sequencing.
-
95
To each sample, add the following:
| Component | Amount (μL) | Final Concentration |
|---|---|---|
| NEB Index Primer | 5 | 960 nM |
| NEB Universal PCR Primer | 5 | 960 nM |
| NEBNext Ultra II Q5 Master Mix | 25 | |
| Sample DNA | 17 | |
| Total | 52 (one reaction) |
-
96For libraries initially prepared from 10 ng of DNA, conduct PCR with the following reaction conditions:
Cycle Number Denature Anneal/Extend 1 98°C, 30 s 2–8 98°C, 10 s 65°C, 75 s For libraries initially receiving 5 ng of DNA, use 8 cycles of amplification. For libraries initially receiving 1 ng of DNA, use 9–10 cycles of amplification. For libraries initially receiving less than 1 ng of DNA, use 10–11 cycles of amplification.
-
97
Repeat Steps 88–92, using 60 μL beads, and resuspending in 25 μL of 0.1x TE Buffer.
-
98
Quantify library concentrations using Quant-iT PicoGreen dsDNA Assay Kit as per Steps 67–79. ? Troubleshooting
-
99
Mix libraries as per the multiplexing scheme (Step 93 and Box 2) to the final concentration required by the sequencing facility and submit for paired-end sequencing with reads of at least 42 bp in length.
Critical Step: It is absolutely crucial that the sequencing be paired-end. Without paired-end sequencing, it is impossible to know the length of the inserts, meaning that the samples cannot be filtered to exclude non-mononucleosomal fragments, inevitably distorting the data and reducing the accuracy of quantification.
Pause Point: At this point, any remaining libraries can be stored at −80°C for several years, and it is necessary to wait for sequencing to complete prior to data analysis. It is highly recommended that prior to sequencing, the sequencing facility should evaluate the size distribution of the library fragments by capillary electrophoresis so as to ensure that the fragments correspond to a primarily nucleosomal population of DNA (Fig. 3f).
Next-Generation Sequencing Data Analysis | Timing: Variable
Critical: For those unfamiliar with navigation and file manipulation in a UNIX command line environment, we recommend familiarization with basic UNIX commands (e.g. see: http://mally.stanford.edu/~sr/computing/basic-unix.html) The following steps all assume that the files being discussed are all in the same directory. If this is not true, then it is recommended that you copy (with the cp command) or move (with the mv command) the files into a single directory. Alternatively, files can be referenced with the full path if moving the files into the same directory is not desirable or possible. Additionally, we assume that the PATH variables pointing to the locations of various tools listed in SOFTWARE are appropriately set in the shell. Alternatively, the full path to these tools can be prepended to the commands listed below. This workflow is outlined in Fig. 2.
-
100If there is more than one fastq file per sample, then concatenate together the fastq files for each read so there is only one fastq file per read per sample using the cat command. For example, if the files for Read 1 are given as read1_A.fq, read1_B.fq, read1_C.fq, and the files for Read 2 as read2_A.fq, read2_B.fq, read2_C.fq, then these should be concatenated as:
cat read1_A.fq read1_B.fq read1_C.fq > read1.fq cat read2_A.fq read2_B.fq read2_C.fq > read2.fq ? Troubleshooting
-
101
Create a concatenated genome fasta file containing the reference genome of interest (e.g. hg38, mm10, dm6) and the barcodes used for the standards (Supplementary Table 2). For example, if both these fastq files are in the same folder:
cat hg38.fq barcodes.fq > hg38_w_barcodes.fq ? Troubleshooting
-
102In a text editor or EXCEL, build a calibration table as a tab-delimited file such that the first column contains each barcode name and the second column contains the corresponding histone mark. For example, in a text editor, this can be typed out as:
[DNA sequence 1] [tab] [corresponding nucleosome A] [DNA sequence 2] [tab] [corresponding nucleosome A] [DNA sequence 3] [tab] [corresponding nucleosome B] …
An example calibration table that was specific for our previous work has been included to serve as a template (Supplementary Table 2), but this specific table will not work in your application without customization unless you used the same barcodes and nucleosomes that we used. ? Troubleshooting
-
103From the concatenated genome-barcodes fasta file, build a chromosome length index file. For example:
samtools faidx hg38_w_barcodes.fq && awk ‘{print $1”\t”$2}’ \ hg38_w_barcodes.fq.fai > hg38_w_barcodes.len ? Troubleshooting -
104From the concatenated genome-barcodes fasta file, build Bowtie2 indices by using the command:
bowtie2-build hg38_w_barcodes.fq hg38_w_barcodes
The expected output is a set of files with the extension “.bt2” placed into the directory you are in. ? Troubleshooting
-
105Run the provided icechip script (graphically depicted in Fig. 2) to automatically align the fastq files, filter for length and quality, and generate genome coverage maps and calibration files, all using the command:
./icechip -p <number of cores> -x hg38_w_barcodes −1 <fastq for PE read 1> \ −2 <fastq for PE read 2> -g hg38_w_barcodes.len \ -c <calibration table> -o <output file base name>
The expected output is a genome coverage bedgraph file, a calibration file with the extension “.cal” for computation of HMD and specificity, a genome coverage bigwig file for genome browser visualization, and a log file that gives statistics on the number of reads processed and their fates. ? Troubleshooting
-
106After conducting Step 105 for both the IP and the associated input, run the computeHMDandError script to compute HMD and 95% confidence intervals by using the command:
./computeHMDandError -m <name of modification> \ −1 <IP genome coverage bedgraph> −2 <Input genome coverage bedgraph> \ -g <length file>
The expected output is an HMD bedgraph file as computed per Fig. 1a, a 95% Confidence Interval magnitude bedgraph file, and bigwig files for visualization on a genome browser. ? Troubleshooting
-
107
Using the calibrant read numbers in the cal file output for the IP and the Input (Step 105), compute the specificity of the IP in the sequencing experiment as per Fig. 1a. Plot the input vs. IP read counts for the target nucleosome standard as a scatterplot to assess for linearity of ICeChIP. ? Troubleshooting
Timing
Steps 1–14, nuclei preparation: 2 hours.
Steps 15–21, antibody conjugation to beads: 2 hours.
Steps 22–37, MNase digestion and HAP purification: 1.5 hours (should be done in parallel with antibody conjugation to beads).
Steps 38–47, immunoprecipitation: 1 hour.
Steps 48–55, DNA purification: 3 hours.
Steps 56–57, DNA visualization: 2 hours.
Steps 58–66, DNA quantification with qPCR: 4 hours.
Steps 67–79, DNA quantification with PicoGreen: 1 hour.
Steps 80–92, Next-Generation Sequencing Library Adaptor Ligation: 2.5 hours.
Steps 93–99, Next-Generation Sequencing Library Amplification: 1.5 hours.
Steps 100–107, Next-Generation Sequencing Data Analysis: Variable, based on computational resources available and sequencing depths.
There are pause points after Steps 14, 47, 55, 57, 66, and 79. Step 99 represents a necessary pause point for submission of samples for NGS.
Steps 67–79 are not necessary for ICeChIP-qPCR. Steps 80–107 are not applicable to ICeChIP-qPCR.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Anticipated Results
Steps 1–14, nuclei preparation, are anticipated to yield ~500–1000 μg of chromatin from 108 mammalian cells. This corresponds to ~5–10 nuclei aliquots of 100 μg of chromatin each. Steps 15–21 do not yield directly measurable results but should result in the conjugation of the antibody to the magnetic beads for use in ChIP.
Steps 22–37, MNase digestion and HAP purification, should yield ~30 μg of purified chromatin. Digestion to mononucleosomes is critical to avoid oligonucleosome avidity biases in the pulldown and corresponds to an approximate resolution of ~150 bp for the ChIP experiment. As such, before proceeding with expensive or time-consuming procedures such as NGS library preparation or NGS itself, the samples saved in this stage for visualization should be observed on an agarose gel as described in Steps 56–57. The resultant gel should ideally look similar to that displayed in Fig. 3a. Ideally, using heterochromatin-specific or euchromatin-specific sequencing primers, it should also be confirmed that there is no bias in the recovery of heterochromatin and euchromatin in the input (Fig. 3b). However, this is not critical; if there is bias in HAP recovery, the ratiometric comparison with the IP will compensate for the bias in the HMD computation.
Steps 38–47, immunoprecipitation, yield no directly observable results, but should result in nucleosomes with the modification of interest being pulled down.
Steps 48–55, DNA quantification with qPCR, should yield target enrichment as a percent of input, HMD at genomic loci (Fig. 3c), and off-target binding as percent of target binding, a measure of the specificity of the antibody (Fig. 3d). These values will all be highly variable, depending on the loci chosen for analysis, the modification of interest, and the quality of the antibody used.
The remaining steps of the protocol are all devoted to preparation for and analysis of next-generation sequencing. Steps 58–66, DNA quantification with PicoGreen, will provide the DNA yields of the input and the IPs. Approximately 250–300 ng of DNA should be expected from the input, but the amount of DNA expected from each IP will vary dramatically based on the genomic abundance of the modification of interest, the specificity of the antibody, and the efficiency of the pulldown. An example of a PicoGreen standard curve with sample measurements is shown in Fig. 3e.
Steps 67–99 are dedicated to generating the DNA libraries for sequencing and will produce, by the end, multiplexed libraries that can be submitted to a sequencing facility for paired-end Illumina sequencing at the appropriate read depth. The library preparation itself should yield approximately 250–750 ng of DNA library, depending on the amount of DNA initially put into the library preparation. Additionally, the majority of the library sample should have a fragment size of 200–350 base pairs, as per Fig. 3f, which corresponds to a mononucleosomal insert size with adaptors added. This can be assessed by capillary electrophoresis, which should be done prior to sequencing.
Steps 100–107, next-generation sequencing analysis, will yield genome-wide histone modification density and confidence interval files for each IP. Examples of these are displayed as genome browser views in Fig. 3g. They will also yield antibody specificity measurements for all targets spiked into the experiment, as exemplified in Fig. 3h, and a scatterplot showing linearity of target IP, as exemplified in Fig. 3i.
Table 1.
Troubleshooting
| Step | Problem | Possible Reasons | Solutions |
|---|---|---|---|
| 1 | It is not feasible to obtain 108 cells. | The cells are not expandable or are difficult to expand up to 108 cells. | 108 cells are recommended for convenience to ensure sufficient recovery of nuclei. However, it is possible to use fewer cells – we have successfully conducted nuclei preparations with ~10 million cells using smaller volumes of Buffer N and Sucrose Cushions. However, if the cell numbers are less than that, then it may be necessary to skip the sucrose cushion purification of nuclei, which may marginally reduce efficiency of digestion and IP specificity. |
| 7 | The pellet is not whiter in color. | The cells may not have lysed. | To confirm lysis, it is advisable to observe the cells under a microscope with trypan exclusion staining. If the cells are confirmed to not have fully lysed, it is possible your cell line is somewhat resistant to 0.3% NP-40, in which case the amount of NP-40 in the Lysis Buffer can be increased. |
| 8 | The pellet will not resuspend in Buffer N. | Some of the nuclei may have lysed, releasing the chromatin. | If possible, it is highly recommended to start the procedure over with new cells and to use less NP-40 in the Lysis Buffer and carefully adhere to the listed amount of time that cells and crude nuclei are exposed to detergent containing buffer. If starting over is not possible due to a lack of cells, then in principle, you can go directly to chromatin digestion and HAP chromatography. This is challenging because optimizing MNase digestion conditions without knowing the amount of chromatin in the pellet is difficult, but if the cells are irreplaceable or difficult to reobtain, then it is possible. A small sample can be titrated with MNase empirically, if there is enough sample for this to occur. |
| 8 | There is no nuclei pellet after the purification by Sucrose Cushion. | The cells do not have intact nuclei (e.g. granulocytes), the nuclei do not pellet through the Sucrose Cushion at 500g, too few cells were used at the beginning of the protocol, or too many cells were lost in the nuclei purification process. | If your cells do not have intact nuclei (e.g. granulocytes), then nuclei purification by Sucrose Cushion is not possible. In this case, you should proceed immediately from cell lysis to quantification and MNase digestion without a Sucrose Cushion purification. If your cells do have intact nuclei, then it is possible that your nuclei do not pellet through the Sucrose Cushion at 500g. In such circumstances, you can try a second spin up to 1300g to pellet nuclei. A white film atop the sucrose cushion is diagnostic of the flux of nuclei crossing the boundary being too high, such that nuclei encounter one another and stick. In this case, remove the whole buffer layer and the film above the cushion with as little cushion as possible, dilute 2x further in Buffer N and reapply to a new cushion, also recovering any nuclei that did pellet. If a pellet still does not appear, then it is likely that you started with too few cells, or you have lost too many cells to yield a sizeable pellet. In this case, it is recommended that you start over and either use more cells or skip Sucrose Cushion purification and instead move straight to chromatin quantification (Steps 9–11). |
| 12 | During UV-vis quantitation, the chromatin remains too viscous to pipette even after 10–15 minutes of sonication. | The chromatin has not completely solubilized and remains too intact to pipette easily. | Sonicate for longer, in increments of 5 minutes. Depending on the age and quality of the water bath sonicator, sonicating enough to permit sufficient pipetting can take quite some time. You can also dilute all your aliquots with additional 2 M NaCl and sonicate longer. If using a water bath sonicator other than the Branson 2800 Ultrasonic Cleaner, then optimization of sonication conditions and time may be needed. |
| 13 | The chromatin concentrations are vastly different across the three replicates. | Some nuclei may have settled as you were taking aliquots, so some aliquots may not be representative of the sample as a whole. Alternatively, the chromatin in some samples is still not sufficiently solubilized. |
Try to repeat the process of UV-vis sample preparation (Steps 10–13), resuspending and mixing nuclei well (but gently) while taking aliquots. Alternatively, it is possible that your chromatin in some samples is still too viscous, which is preventing it from being easily pipetted. In this case, continue sonicating for longer (see above). |
| 13 | The amount of chromatin is not detectable by Nanodrop. | The chromatin concentration of the aliquot may be below the sensitivity of the Nanodrop instrument (~5ng/μL), particularly if working with a very small number of nuclei (~1 million or fewer). | You can prepare aliquots again by diluting with less 2 M NaCl (or using a smaller volume of 5 M NaCl to maintain the same level of salt), you can pellet and resuspend nuclei in a smaller volume of Buffer N, or you can use a more sensitive DNA quantification tool (e.g. Quant-iT PicoGreen dsDNA Assay Kit). If none of these are possible for you, then you can “guesstimate” the concentration of chromatin. From our experience, there are about 1μg of crude mammalian chromatin per 60k-100k nuclei (assuming 3–3.5 Gbp per genome). Needless to say, this method is not the most accurate and should be reserved only for situations in which no other alternative presents itself. |
| 15 | The antibody does not bind Protein G well, per antibody characterization by the manufacturer or based on the antibody isoform. | Some isotypes of antibodies do not bind Protein G well. | Try using Protein A magnetic beads instead. |
| 57 | Digested chromatin is not a mononucleosomal population. | The MNase may not have the activity that is anticipated, the nuclei may be clumped, or excess debris in the nuclei solutions is preventing accurate quantification or complete digestion. | If there is a >5% population of di- or oligonucleosomes, repeat the protocol but titrating several up to several fold increases in of MNase used or employ a freshly thawed aliquot of MNase from stock solution. Conversely, if there is a large population of DNA fragments smaller than ~150 bp corresponding to mononucleosome fragments, the digestion has progressed too far (often characteristic ~115 bp or ~80 bp). If the DNA fragment population displays both over and underdigestion, then the nuclei are not sufficiently uniform-- mix nuclei well to ensure that the nuclei are not forming clumps during MNase digestion. If even mononucleosomal digestion remains a problem, begin nuclei preparation with fresh cells, but use two sucrose cushions sequentially to improve their homogeneity. |
| 57 | Nucleosomes are appearing in HAP wash flow-throughs. | The pH may be incorrect, or there may be too much phosphate in the wash buffers. | Ensure that the buffers have a pH of 7.2 and ensure that the phosphate concentrations are reasonably accurate. This may require preparing fresh HAP Buffers. |
| 64 | High Cq values (>32 cycles) are observed in both the IP and the Input. | Insufficient primer and probe were added to the qPCR reaction. Alternatively, the primer and probe may not span a well-positioned nucleosome. | Remake your qPCR 20x Primer-Probe Master Mix to rule out the possibility that there was insufficient primer and probe. If you still observe high Cq values, it is very likely that your primer and probe set are inappropriate. Try creating and using a different primer and probe set. Primers and probes should ideally line up with a well-positioned nucleosome, which would appear as regions of buildup in MNase-seq datasets, if available for your cell type. |
| 64 | Enrichment of the target standards for the IP is low. | The antibody may not have saturated the Protein G magnetic beads, or it may be an inherently low-enrichment antibody in these conditions. | Ensure that the amount of antibody used was enough to saturate the beads, per the recommendation of the manufacturer of the beads; if this was not the case, then increase the amount of antibody used. For the beads we recommend, this is approximately 6 μg of antibody per 25 μL of beads. Additionally, if the IP showed high specificity (see Step 64), then it may be possible to increase enrichment by reducing the number of washes (e.g. one wash with ChIP Buffer 2 rather than two washes, no washes with ChIP Buffer 3 rather than one wash). As a practical matter, increasing the target enrichment percentage of an IP is difficult and often not worth the trouble. If possible, obtain a new antibody or scale up the reaction with more antibody and more chromatin instead. If, as measured by sensitive DNA quantification methods (e.g. Quant-iT PicoGreen dsDNA Assay), you are still obtaining more than 10 ng of DNA in the IP, then you are certainly recovering enough DNA even for next-generation sequencing library preparation, and the enrichment is not worth worrying about. |
| 66 | Specificity of the antibody is low/the antibody binds substantial off-target modifications. | The antibody is of poor quality. | Try adding washes with ChIP Buffer 2 and 3, but understand that this will come at the expense of enrichment. If at all possible, get a different antibody. |
| 79 | There is little to no DNA found in my sample, as measured by PicoGreen. | Nucleases may have contaminated the samples. Alternatively, the antibody used has low enrichment for the target. | Redoing the experiment with fresh, nuclease-free buffers may work if the sample was contaminated with nucleases. However, it is much more likely that your antibody has low enrichment for target. If there is at least 5 ng of DNA, then your sequencing will probably be fine. If there are 1–5 ng of DNA, then your sequencing may be slightly overamplified but may be workable. With less than 1 ng of DNA, it is likely that the samples will be dramatically overamplified. If you have low DNA yield, it is recommended that you either obtain a better antibody or scale up the reaction with more antibody and more chromatin. |
| 98 | There is not enough DNA found in my library, as measured by PicoGreen. | The library preparation has likely failed. | If all of your library preparations have failed, then it is likely that a step was done incorrectly or that your kit has spoiled. In this case, the preparation can be redone if enough sample DNA remains, or the entire experiment can be redone. If only one library preparation has failed, then if enough DNA remains, it can be redone, but if the initial DNA concentration was so low that not enough remains, that is likely a major reason the library preparation has failed. |
| 100–107 | The file was not found (or file could not be opened). | The path pointing to the file may be incorrect, or you may not have the necessary permissions to open that file. | Ensure that the path pointing to the file is correct. For example, if the file is not in the directory that you are currently working in, then you need to list the full path of the file in question. Also ensure that you have the necessary permissions to open that file. |
| 100–107 | A command has returned an error: [command]: command not found |
The program being run may not be properly added to the PATH. | Ensure that the program being run is appropriately added to the PATH. To do this, first find the path of the program installation (different for each program, but generally listed in the online documentation for each). Then run the command: nano ~/.bashrc Scroll to the end of the file using the “PgDn” key. Enter the following at the end of the file (without the brackets): export PATH=$PATH:[full path to program folder] Then press Ctrl+x to close the file, press “Y” and press enter to save the file. Finally, run the command: source ~/.bashrc |
| 106 | The specificity or calibration is not what was expected or a divide-by-zero error was reported. | You may be using an incorrect calibration table. | Ensure that you are using a custom calibration table for your set of barcodes and corresponding nucleosomal marks rather than the calibration table we have provided strictly as a formatting example. |
| 107 | The terminal window cleared and displayed unusual characters. | Your calibration table may not be a properly formatted plain text tab-delimited file. | Please ensure that the calibration table being used is a custom calibration table for your specific application that is provided in a plain text tab-delimited file rather than an Excel file. |
Supplementary Material
Supplementary Table 2 | Example of Calibration Table Formatting. Tab-delimited file showing example of how calibration table can be formatted for ICeChIP-seq.
Supplementary Table 3 | Hamming Distances for NEBNext Illumina Adapters. Excel file listing the Hamming distances between the distinct NEBNext Illumina adapters, for use in designing multiplexing schemes.
Supplementary Material
Supplementary Table 1 | Primer and Probe Sequences. List of primer and probe sequences validated for use in ICeChIP43.
Acknowledgements
This work was funded by the National Institutes of Health under award number R01-GM115945 to A.J.R.
Footnotes
Data Accessibility and Accession
The sequencing data in this work has been previously published by Grzybowski et al.43 and Shah et al.45. Sequencing data can be found at GEO under accession numbers GSE60378 and GSE103543.
Code Availability
All code used in this work is listed under Analytical Tools. Scripts provided here are under GNU General Public License. The most updated versions of the tools and scripts described in this work can be found at http://github.com/shah-rohan/icechip.
Competing Interests
The authors declare competing interests. A.J.R. and A.T.G. hold partial intellectual property rights to ICeChIP as inventors. R.N.S., A.T.G., and A.J.R. have previously served in a compensated consulting role to Epicypher, Inc., the commercial developer and supplier of ICeChIP barcoded nucleosomes (i.e. SNAP-ChIP™ and CAP-ChIP™, both under license from the University of Chicago [Patent #US20160341743]).
References
- 1.Luger K, Mäder AW, Richmond RK, Sargent DF & Richmond TJ Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 18(1997). [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Kouzarides T Chromatin Modifications and Their Function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Brand M, Rampalli S, Chaturvedi C-P & Dilworth FJ Analysis of epigenetic modifications of chromatin at specific gene loci by native chromatin immunoprecipitation of nucleosomes isolated using hydroxyapatite chromatography. Nat. Protoc 3, 398–409 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S & Carroll JS FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 17, 2715–2723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rada-Iglesias A et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heintzman ND et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet 39, 311–318 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Heintzman ND et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calo E & Wysocka J Modification of Enhancer Chromatin: What, How, and Why? Mol. Cell 49, 825–837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C et al. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci 113, 11871–11876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng J et al. A Role for H3K4 Monomethylation in Gene Repression and Partitioning of Chromatin Readers. Mol. Cell 53, 979–992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Li X & Hu H H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 103, 222–228 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Fang R et al. Human LSD2/KDM1b/AOF1 Regulates Gene Transcription by Modulating Intragenic H3K4me2 Methylation. Mol. Cell 39, 222–233 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pekowska A et al. H3K4 tri‐methylation provides an epigenetic signature of active enhancers. EMBO J. 30, 4198–4210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popova EY, Pinzon-Guzman C, Salzberg AC, Zhang SS-M & Barnstable CJ LSD1-Mediated Demethylation of H3K4me2 Is Required for the Transition from Late Progenitor to Differentiated Mouse Rod Photoreceptor. Mol. Neurobiol 53, 4563–4581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Parvin J & Huang K Redistribution of H3K4me2 on neural tissue specific genes during mouse brain development. BMC Genomics 13, S5(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrero MJ et al. Macrohistone Variants Preserve Cell Identity by Preventing the Gain of H3K4me2 during Reprogramming to Pluripotency. Cell Rep. 3, 1005–1011 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Bergmann JH et al. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP‐A assembly on a synthetic human kinetochore. EMBO J. 30, 328–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siklenka K et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 350, aab2006(2015). [DOI] [PubMed] [Google Scholar]
- 20.Santos-Rosa H et al. Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Schneider J et al. Molecular Regulation of Histone H3 Trimethylation by COMPASS and the Regulation of Gene Expression. Mol. Cell 19, 849–856 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Sims RJ & Reinberg D Histone H3 Lys 4 methylation: caught in a bind? Genes Dev. 20, 2779–2786 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Sims III RJ et al. Recognition of Trimethylated Histone H3 Lysine 4 Facilitates the Recruitment of Transcription Postinitiation Factors and Pre-mRNA Splicing. Mol. Cell 28, 665–676 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruthenburg AJ, Allis CD & Wysocka J Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Mol. Cell 25, 15–30 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Davie JR, Xu W & Delcuve GP Histone H3K4 trimethylation: dynamic interplay with pre-mRNA splicing. Biochem. Cell Biol 94, 1–11 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Shimazaki N & Lieber MR Histone methylation and V(D)J recombination. Int. J. Hematol 100, 230–237 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Vallianatos CN & Iwase S Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 7, 503–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen E, Shulha H, Weng Z & Akbarian S Regulation of histone H3K4 methylation in brain development and disease. Phil Trans R Soc B 369, 20130514(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deb M et al. Chromatin dynamics: H3K4 methylation and H3 variant replacement during development and in cancer. Cell. Mol. Life Sci 71, 3439–3463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilmour DS & Lis JT Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc. Natl. Acad. Sci. U. S. A 81, 4275–4279 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solomon MJ & Varshavsky A Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc. Natl. Acad. Sci. U. S. A 82, 6470–6474 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solomon MJ, Larsen PL & Varshavsky A Mapping proteinDNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell 53, 937–947 (1988). [DOI] [PubMed] [Google Scholar]
- 33.Chen H, Lin RJ, Xie W, Wilpitz D & Evans RM Regulation of Hormone-Induced Histone Hyperacetylation and Gene Activation via Acetylation of an Acetylase. Cell 98, 675–686 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Barski A et al. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Gifford CA et al. Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell 153, 1149–1163 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guenther MG et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 22, 3403–3408 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guenther MG, Levine SS, Boyer LA, Jaenisch R & Young RA A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell 130, 77–88 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orlando DA et al. Quantitative ChIP-Seq Normalization Reveals Global Modulation of the Epigenome. Cell Rep. 9, 1163–1170 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Xie W et al. Epigenomic Analysis of Multilineage Differentiation of Human Embryonic Stem Cells. Cell 153, 1134–1148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikkelsen TS et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landt SG et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grzybowski AT, Chen Z & Ruthenburg AJ Calibrating ChIP-Seq with Nucleosomal Internal Standards to Measure Histone Modification Density Genome Wide. Mol. Cell 58, 886–899 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonhoure N et al. Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res. 24, 1157–1168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah RN et al. Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies. Mol. Cell 72, 162–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bock I et al. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 6, 256–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egelhofer TA et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol 18, 91–93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuchs SM, Krajewski K, Baker RW, Miller VL & Strahl BD Influence of Combinatorial Histone Modifications on Antibody and Effector Protein Recognition. Curr. Biol 21, 53–58 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishikori S et al. Broad Ranges of Affinity and Specificity of Anti-Histone Antibodies Revealed by a Quantitative Peptide Immunoprecipitation Assay. J. Mol. Biol 424, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hattori T et al. Recombinant antibodies to histone post-translational modifications. Nat. Methods 10, 992–995 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothbart SB et al. An Interactive Database for the Assessment of Histone Antibody Specificity. Mol. Cell 59, 502–511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker M Reproducibility crisis: Blame it on the antibodies. Nat. News 521, 274(2015). [DOI] [PubMed] [Google Scholar]
- 53.Baker M 1,500 scientists lift the lid on reproducibility. Nat. News 533, 452(2016). [DOI] [PubMed] [Google Scholar]
- 54.Harris R Rigor Mortis: How Sloppy Science Creates Worthless Cures, Crushes Hopes, and Wastes Billions. (Basic Books, 2017). [Google Scholar]
- 55.Guertin MJ, Cullen AE, Markowetz F & Holding AN Parallel factor ChIP provides essential internal control for quantitative differential ChIP-seq. Nucleic Acids Res. 46, e75–e75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Egan B et al. An Alternative Approach to ChIP-Seq Normalization Enables Detection of Genome-Wide Changes in Histone H3 Lysine 27 Trimethylation upon EZH2 Inhibition. PLOS ONE 11, e0166438(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu C et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kasinathan S, Orsi GA, Zentner GE, Ahmad K & Henikoff S High-resolution mapping of transcription factor binding sites on native chromatin. Nat. Methods 11, 203–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teytelman L et al. Impact of Chromatin Structures on DNA Processing for Genomic Analyses. PLOS ONE 4, e6700(2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teytelman L, Thurtle DM, Rine J & Oudenaarden A van. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci 110, 18602–18607 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan X & Struhl K Where Does Mediator Bind In Vivo? PLOS ONE 4, e5029(2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng Q, Vijaya Satya R, Lewis M, Randad P & Wang Y Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genomics 16, 589(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.LeRoy G et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 6, 20(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rohland N & Reich D Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. (2012). doi: 10.1101/gr.128124.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Strober W Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol 21, A.3B.1–A.3B.2 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 2 | Example of Calibration Table Formatting. Tab-delimited file showing example of how calibration table can be formatted for ICeChIP-seq.
Supplementary Table 3 | Hamming Distances for NEBNext Illumina Adapters. Excel file listing the Hamming distances between the distinct NEBNext Illumina adapters, for use in designing multiplexing schemes.
Supplementary Material
Supplementary Table 1 | Primer and Probe Sequences. List of primer and probe sequences validated for use in ICeChIP43.