

A factor other than the virus receptor expressed by target cells has been found to accelerate the loss of an enterovirus (CVB3/28) infectious titer, with little effect on nearly identical mutant strains. The destabilizing factor in fetal bovine serum, identified as albumin, does not interfere with the catalytic activity of soluble receptor at saturating receptor concentrations and amplifies the catalytic activity of the soluble receptor at a concentration that otherwise produces about one-third the saturated receptor-catalyzed rate of virus decay. This finding evidences the possibility that other virus-“priming” ligands may also be nonessential activating cofactors that serve to accelerate receptor-catalyzed viral eclipse.
KEYWORDS: coxsackievirus, enterovirus, kinetics, receptor, uncoating
ABSTRACT
Three strains of coxsackievirus B3 (CVB3) differ by single mutations in capsid protein VP1 or VP3 and also differ in stability at 37°C in tissue culture medium. Among these strains, the CVB3/28 parent strain has been found to be uniquely sensitive to a component in fetal bovine serum (FBS) identified as serum albumin. In cell culture medium, serum increased the rate of CVB3/28 conversion to noninfectious particles at least 2-fold. The effect showed a saturable dose response. Rates of conversion to noninfectious virus with high concentrations of soluble coxsackievirus and adenovirus receptor (sCAR) were similar with and without FBS, but FBS amplified the catalytic effect of 100 nM sCAR nearly 3-fold. Such effects in other systems are due to nonessential activating cofactors.
IMPORTANCE A factor other than the virus receptor expressed by target cells has been found to accelerate the loss of an enterovirus (CVB3/28) infectious titer, with little effect on nearly identical mutant strains. The destabilizing factor in fetal bovine serum, identified as albumin, does not interfere with the catalytic activity of soluble receptor at saturating receptor concentrations and amplifies the catalytic activity of the soluble receptor at a concentration that otherwise produces about one-third the saturated receptor-catalyzed rate of virus decay. This finding evidences the possibility that other virus-“priming” ligands may also be nonessential activating cofactors that serve to accelerate receptor-catalyzed viral eclipse.
INTRODUCTION
Enteroviruses are comprised of icosahedral capsids built from 60 copies each of proteins VP1 to VP4 and an internal single-strand positive-sense RNA genome. VP4 has a myristylated amino terminus, and most of these viruses contain a second lipid moiety called pocket factor inside a hydrophobic pocket within VP1 (1–4). Pocket factor and drugs that occupy the pocket stabilize the virus and are believed to impede capsid conformational dynamics referred to as “breathing” (5–12).
While there is probably an ensemble of intermediate states, two temperature-dependent breathing conformations based on differences in antigenicity and capsid permeability to small molecules have been observed (6, 13–16). The impermeable closed conformation predominates at ambient temperatures and below; VP4 is fully internal to the capsid, and the amino-terminal region of VP1 is buried beneath the capsid surface. At physiological temperatures, the transient open conformation, in which the capsid is permeable to small molecules and in which the amino-terminal regions of both VP1 and VP4 are exposed and available for binding by antibodies and to proteolysis, has been observed (13–16). At temperatures that enable breathing, the viruses transition with first-order kinetics to altered particles (A-particles) that are indistinguishable from A-particles produced by incubation with the receptor (9, 17). The A-particle is expanded and porous, has exposed VP1 amino termini, is missing VP4, and still contains the viral genome (1, 3, 17, 18). A-particles typically bind the cell surface receptor poorly if at all and are consequently highly attenuated since they are not infectious by the receptor-mediated mechanism (1, 3, 19). The open and closed conformations exist in a temperature-dependent equilibrium; transition to the A-particle is irreversible (3). Human rhinovirus and poliovirus have been found to bind to their respective cell surface receptors with greater affinity when in the open conformation than when in the closed conformation, establishing conditions of an allosteric system (20–22).
The relationship between measured virus stability and the ability of the virus receptor to catalyze the conformational transition from infectious virus to A-particles has not received much attention, with the exception of work describing the stabilizing effects of pocket-binding drugs (12, 23, 24). We have been interested in (i) the influence of the stability of coxsackievirus B3 (CVB3, Picornaviridae, Enterovirus, Human enterovirus B, serotype 3) on virus interaction with the cell surface receptor that mediates infection and (ii) the combined influences of virus stability, environment, and the coxsackievirus and adenovirus receptor (CAR) on virion conformational transitions that precede genome release (17, 25–27). In this study, we report the destabilizing effect of bovine serum albumin on CVB3/28 and its effect on receptor-catalyzed conversion of the virus to A-particles.
RESULTS
Albumin and fatty acids (present in serum) and divalent cations (present in Dulbecco’s modified Eagle’s medium [DMEM] and serum) have been shown to stabilize enteroviruses (5, 11, 28), so we conducted experiments to determine the effect of fetal bovine serum (FBS) on the stability of CVB3/28 in DMEM. The experiments revealed that CVB3/28 was more stable in DMEM (apparent first-order rate constant [kapp] = 0.04 h−1) than in DMEM-10 (DMEM with 8.9% FBS; kapp = 0.11 h−1), showing that some component in FBS destabilized the virus (Table 1; Fig. 1). The destabilizing effect was dose-dependent and saturable (Fig. 2 and 3). Equation 10 (see Materials and Methods) for kapp was fit to the data in Fig. 3 using a k of 2 h−1 (the average of 1.8 and 2.1 h−1 from the two best solutions in reference 25) and approximating k′ as equal to k, where k and k′ are the first-order rate constants in equation 4 (see Materials and Methods). The fit parameters were calculated as follows: K1 = 0.02 (standard error [SE] = 0.002), K2 = 2.29 × 105 M−1 (SE = 0.68 × 105 M−1), and K3 = 0.067 (SE = 0.003). K3 is equivalent to the conformational equilibrium constant (Keq) from the report of Carson (25), which had an average value of 0.054 from the two best models. The value for K1 is the same as if solved directly from kapp when the bovine serum albumin (BSA [B]) concentration is 0.
TABLE 1.
CVB3 decay experiments
| Strain and medium (Figure) | FBS (%) | BSA (μM) | No. of decay expts | kapp (h−1) | SE (h−1) |
|---|---|---|---|---|---|
| CVB3/28 in DMEM and DMEM-10 (Fig. 1) | |||||
| DMEM | 0 | 0 | 2 | 0.041 | 0.003 |
| DMEM | 8.9 | 24 | 2 | 0.111 | 0.006 |
| Varied concns of FBS in DMEM (Fig. 2) | |||||
| DMEM (same as shown in Fig. 1A) | 0 | 0 | 2 | 0.041 | 0.003 |
| DMEM | 0.1 | 0.27 | 2 | 0.047 | 0.003 |
| DMEM | 0.5 | 1.4 | 2 | 0.051 | 0.004 |
| DMEM | 1 | 2.7 | 2 | 0.076 | 0.005 |
| DMEM | 5 | 14 | 2 | 0.113 | 0.003 |
| DMEM | 8.9 | 24 | 1 | 0.113 | 0.008 |
| DMEM | 10 | 27 | 1 | 0.11 | 0.006 |
| DMEM | 20 | 54 | 2 | 0.118 | 0.004 |
| CVB3/28 L1092V and CVB3/28 A3180T (Fig. 4) | |||||
| CVB3/28 L1092V | |||||
| DMEM | 0 | 0 | 2 | 0.03 | 0.004 |
| DMEM | 10 | 27 | 2 | 0.03 | 0.004 |
| CVB3/28 L3180T | |||||
| DMEM | 0 | 0 | 2 | 0.038 | 0.003 |
| DMEM | 10 | 27 | 2 | 0.057 | 0.004 |
| PBE 94 pool A BSA and Sigma BSA in PBS (Fig. 8) | |||||
| PBS, PBE 94 pool A | 0 | 4 | 2 | 0.198 | 0.013 |
| PBS, Sigma BSA | 0 | 4 | 2 | 0.27 | 0.019 |
| PBS, FBS, BSA (from Affi-Gel blue) (Fig. 10) | |||||
| PBS | 0 | 0 | 10 | 0.038 | 0.002 |
| PBS | 10 | 27 | 6 | 0.276 | 0.009 |
| PBS | 0 | 3.8 | 3 | 0.212 | 0.007 |
| Varied concns of FBS in PBSa (Fig. 11) | |||||
| PBS | 0 | 0 | 1 | 0.043 | 0.005 |
| PBS | 0.1 | 0.27 | 1 | 0.043 | 0.007 |
| PBS | 1 | 2.7 | 1 | 0.294 | 0.03 |
| PBS | 5 | 14 | 1 | 0.35 | 0.028 |
| PBS | 10 | 27 | 1 | 0.289 | 0.031 |
| PBS | 20 | 54 | 1 | 0.252 | 0.018 |
| TBS-10, Ca-Mg, EDTA (Fig. 13) | |||||
| TBS | 10 | 27 | 1 | 0.125 | 0.014 |
| TBS + 1.8 mM Ca + 0.8 mM Mg | 10 | 27 | 1 | 0.085 | 0.013 |
| DMEM + 5 mM EDTA | 10 | 27 | 1 | 0.127 | 0.016 |
| FBS effect is reversibleb (Fig. 14) | |||||
| PBS | 0 | 0 | 2 | 0.038 | 0.004 |
| PBS | 20 | 54 | 2 | 0.038 | 0.004 |
| PBS + 1 mM EDTA | 20 | 54 | 1 | 0.045 | 0.005 |
| FBS potentiates sCARc (Fig. 15) | |||||
| DMEM, 100 nM sCAR | 0 | 2 | 0.558 | 0.097 | |
| DMEM, 250 nM sCAR | 0 | 2 | 1.41 | 0.094 | |
| DMEM, 500 nM sCAR | 0 | 2 | 1.543 | 0.107 | |
| DMEM, 1,000 nM sCAR | 0 | 2 | 1.512 | 0.13 | |
| DMEM, 100 nM sCAR | 9.69/10 | 26/27 | 2 | 1.526 | 0.09 |
| DMEM, 250 nM sCAR | 9.62/10 | 26/27 | 2 | 1.536 | 0.101 |
| DMEM, 500 nM sCAR | 9.51/10 | 26/27 | 2 | 1.743 | 0.12 |
| DMEM, 1,000 nM sCAR | 9.28/10 | 25/27 | 2 | 1.699 | 0.129 |
Data are fit for two virus populations; see the details in Fig. 11.
Two hours at 37°C and then centrifuged through sucrose and assay stability.
FBS was diluted by added sCAR in the first of two experiments.
FIG 1.

CVB3/28 decay in DMEM and DMEM-10 (8.9% FBS) shows that FBS has a destabilizing effect on the virus. (A) CVB3/28 decay in DMEM without FBS (kapp = 0.041 h−1, SE = 0.003 h−1); (B) CVB3/28 decay in DMEM-10 (kapp = 0.111 h−1, SE = 0.006 h−1).
FIG 2.

CVB3/28 decay over time in DMEM with varied amounts of FBS. (A) FBS = 0, kapp = 0.041 h−1, SE = 0.003 h−1 (the data in panel A are the same as shown in Fig. 1A); (B) FBS = 0.1%, kapp = 0.047 h−1, SE = 0.003 h−1; (C) FBS = 0.5%, kapp = 0.051 h−1, SE = 0.004 h−1; (D) FBS = 1%, kapp = 0.076 h−1, SE = 0.005 h−1; (E) FBS = 5%, kapp = 0.113 h−1, SE = 0.003 h−1; (F) FBS = 8.9%, kapp = 0.113 h−1, SE = 0.008 h−1; (G) FBS = 10%, kapp = 0.110 h−1, SE = 0.006 h−1; (H) FBS = 20%, kapp = 0.118 h−1, SE = 0.004 h−1.
FIG 3.

kapp versus FBS at 0.1% to 20% in DMEM showed that the destabilizing effect is saturable. From 5% to 20% FBS, kapp values were statistically equivalent. Each point is derived from decay time courses shown in Fig. 2. The line was fit using the model equations 10 and 13 (derived in Materials and Methods).
To characterize the destabilizing component(s), FBS was fractionated using several protein separation methods in sequence, and the fractions were assessed for virus-destabilizing activity. All of the destabilizing fractions following gel filtration contained proteins consistent with BSA. The destabilizing fractions did not contain bands consistent with IgM, and the destabilizing activity was not removed with protein A/G agarose. FBS at 10% did not diminish the infectious titer of CVB3/28 L1092V, which differs from that of CVB3/28 by a single Leu-to-Val mutation at position 92 of VP1 (Table 1; Fig. 4 and 5) (27). It is therefore unlikely that the destabilizing component is an antibody. The destabilizing pool after gel filtration was applied to a column of Affi-Gel blue using conditions that are highly selective for removing albumin from serum. The bound protein, eluted with sodium chloride, accelerated CVB3/28 decay, whereas proteins not bound to the Affi-Gel blue were no more destabilizing than the phosphate-buffered saline (PBS) control. SDS-PAGE analysis of nonreduced proteins that eluted from the column (Fig. 6A, lanes 3 and 4) revealed proteins near 50 and 100 kDa, appropriate for the nonreduced BSA monomer and dimer. When the samples were reduced (Fig. 6B), the principal stained band in both eluate pools was near 68 kDa, consistent with reduced BSA. A Western blot of the combined eluate pools (Fig. 6C) confirmed that the proteins in the fractions that destabilized CVB3/28 were in fact BSA and BSA oligomers. The 38 μM BSA preparation contained 24 μM fatty acids. In an additional attempt to separate the CVB3/28-destabilizing activity from BSA, the Affi-Gel blue eluate was subjected to anion-exchange chromatography. One predominant protein peak, followed by a very small peak, was eluted from PBE 94 (GE Healthcare Life Sciences) (Fig. 7). CVB3/28-destabilizing activity corresponded to the amount of BSA in the pools. A time course of CVB3/28 decay in the presence of 4 μM BSA from pool A gave a kapp of 0.20 h−1 (Table 1; Fig. 8). These experiments showed that the principal component of FBS responsible for destabilizing CVB3/28 is BSA. As confirmation that BSA destabilizes CVB3/28, monomeric BSA isolated from commercial essentially fatty acid-free BSA (Fig. 9) was also found to accelerate CVB3/28 decay with first-order kinetics (kapp = 0.27 h−1) (Table 1; Fig. 8).
FIG 4.

Decay rates for CVB3/28 L1092V (A, B) and CVB3/28 A3180T (C, D) in DMEM (A, C) and DMEM with 10% FBS (B, D) showed that the FBS had no measurable effect on CVB3/28 L1092V (for both conditions, kapp = 0.030 h−1, SE = 0.004 h−1). (C, D) The kapp for CVB3/28 A3180T in DMEM (0.038 h−1, SE = 0.003 h−1) (C) was not statistically different from the kapp measured for CVB3/28 L1092V, but the rate measured in 10% FBS (0.057 h−1, SE = 0.004 h−1) (D) was significantly higher than the rate in DMEM alone.
FIG 5.
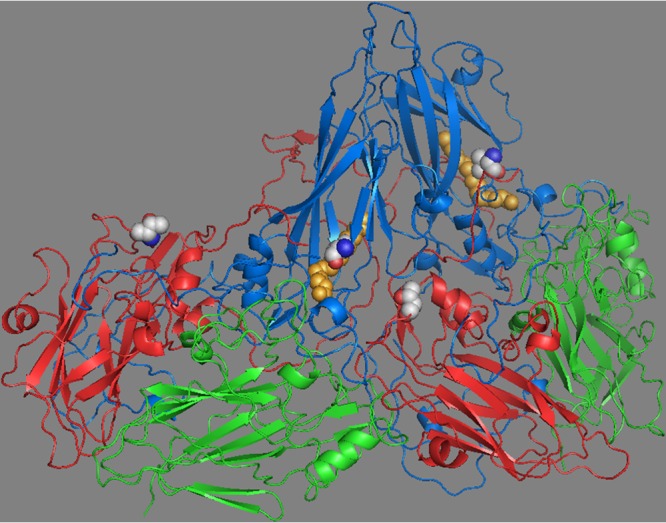
Two protomers of the CVB3 capsid showing VP1 (blue), VP2 (green), and VP3 (red). VP4 lies on the inner capsid surface and is not shown. Pocket factor is shown (orange spheres) within the hydrophobic pocket of VP1. The mutation (Leu to Val) in CVB3/28 L1092V is shown as spheres (side chain carbons are light gray) in VP1 adjacent to the pocket factor. The side chain of leucine at VP1 residue 92 extends into the pocket and clashes with the pocket factor, and there is evidence that side chains extending into the pocket limit the length of pocket factor alkyl chains (49). Valine at residue 92 does not conflict with the longer pocket factor. The mutation (Ala to Thr) in CVB3/28 A3180T is shown as spheres in VP3 at a protomer-protomer interface with VP1 (near Pro1146, which is adjacent to the CAR contact residue Pro1145) (17). Both mutations were identified in a population derived from CVB3/28 after selection for survival at 37°C (27). The structure is from 1COV and rendered in PyMOL (50).
FIG 6.
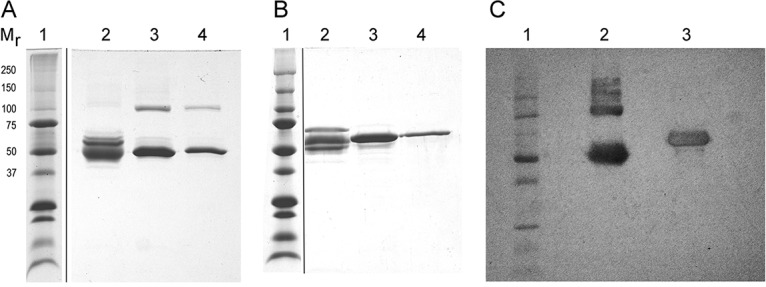
SDS-PAGE of Affi-Gel blue pools that were not reduced (A), reduced (B), and (C) Western blot probed with anti-BSA. Mr indicates the molecular masses (in kilodaltons) of the marker proteins in lane 1 of all three panels. (A and B) Lanes 2 contain proteins not bound to the Affi-Gel blue, and lanes 3 and 4 contain proteins in the two pools (one broad protein peak) that eluted from the column. (C) The blot shows the combined eluate pools analyzed that were not reduced (lane 2) and reduced (lane 3).
FIG 7.

BSA purified from FBS through Affi-Gel blue was chromatographed on PBE 94. Destabilizing activity corresponded to the amount of albumin in pools A to C. t, time.
FIG 8.

(A) CVB3/28 decay in PBS with 4 μM BSA from PBE 94 pool A (kapp = 0.198 h−1, SE = 0.013 h−1); (B) BSA monomer isolated from essentially fatty acid-free BSA (Sigma A-0281) (kapp = 0.27 h−1, SE = 0.019 h−1).
FIG 9.
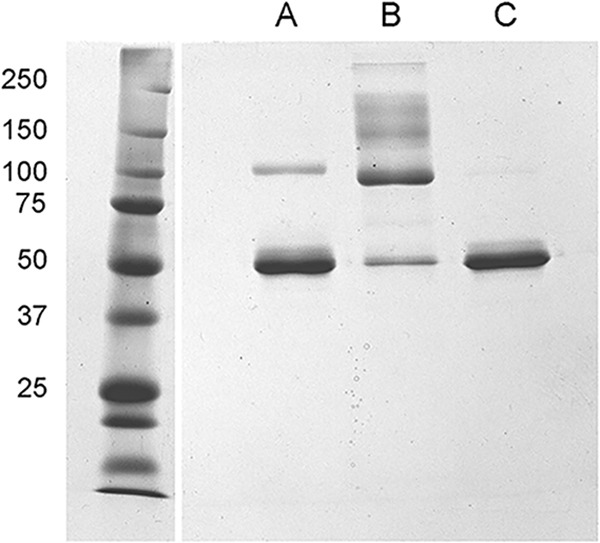
Coomassie blue-stained gel of nonreduced bovine serum albumin (2.6 μg/lane) (A) purified from FBS following chromatography on PBE 94; (B) high-molecular-weight fraction obtained by gel filtration of commercial essentially fatty acid-free bovine serum albumin (Sigma A-0281); (C) monomeric BSA fraction obtained by gel filtration chromatography of the Sigma BSA. Protein size markers are in the left lane, with their molecular masses indicated to the left in kilodaltons. The band in lane A near 100 kDa corresponds to the BSA dimer as shown in Fig. 6.
The finding that albumin destabilizes CVB3/28 requires reconsideration of mechanisms by which MOPS (morpholinepropanesulfonic acid) might stabilize CVB3/28 (26). The original model involved MOPS binding to virus, possibly in the VP1 pocket. Now, it is possible that MOPS binds albumin, rather than virus, and interferes with its ability to destabilize CVB3/28 (29). The MOPS models were written by adding MOPS (M) and the virus closed conformation (V) in equilibrium with the virus-MOPS complex (VM), or MOPS and BSA (B) in equilibrium with MOPS bound to BSA (MB) to equation 4, to give equation 16 or 18 (see Materials and Methods), respectively, and analyzed by testing the resulting solutions for kapp against the data from the work of Carson et al. (26). In the former model, equation 17 applies (see Materials and Methods). K1, K2, and K3 were solved in the fit of equation 10 to the data shown in Fig. 3. This model, in which B is approximately total B (BT) again fit the data from 2017, as expected since equations 15 and 17 in Materials and Methods are equivalent when B is saturating and k times U is much less than k′ times UB (where U is the open virus conformation). From the regression analysis of equation 17 applied to the 2017 data, r was 0.8769, the reference hydrogen ion concentration (H0+) as described in reference 26 was 14.7 times 10−9 M (SE = 7 × 10−9 M), K was 45.6 × 10−9 (SE = 21.6 × 10−9), and K4 was 30.3 M−1 (SE ≪ 1 M−1). In the latter model, equation 25 in Materials and Methods applies. This model also fit the data as well as the model presented in the original work, with r being 0.8800, (H0+) being 13.2 × 10−9 M (SE = 7.7 × 10−9 M), K being 52.6 times 10−9 M (SE = 24.4 × 10−9 M), and K4 being 63.3 M−1 (SE ≪ 1 M−1). The information currently available does not preclude either the model where MOPS stabilizes CVB3/28 by binding the virus or the model where MOPS binds albumin and interferes with its ability to destabilize CVB3/28.
In PBS, the purified BSA had greater destabilizing activity at 3.8 μM (equivalent to 1.4% FBS; kapp = 0.212 h−1) than was measured for 5% FBS (14 μM BSA; kapp = 0.11 h−1) or 20% FBS (54 μM BSA; kapp = 0.12 h−1) in DMEM (Table 1; Fig. 3 and 10). Direct experimental comparison showed that 10% FBS had a greater effect on CVB3/28 stability in PBS (kapp = 0.28 h−1) than has typically been observed with FCS in DMEM (kapp = 0.11 to 0.12 h−1) (Table 1; Fig. 1 and 10) (17). A limited dose-response analysis (Fig. 11; Table 1) showed that the rate of CVB3/28 decay increased sharply between 0.1% FBS and 1% FBS and was saturable (Fig. 12), achieving a maximum kapp more than two times greater than was observed in DMEM with FBS (Fig. 3; Table 1).
FIG 10.

CVB3/28 decay in PBS alone (kapp = 0.038 h−1, SE = 0.002 h−1) (A), with 10% FBS (kapp = 0.276 h−1, SE = 0.009 h−1) (B), or with 3.8 μM BSA purified from FBS (kapp = 0.212 h−1, SE = 0.007 h−1) (C).
FIG 11.

CVB3/28 decay curves for varied FBS concentrations in PBS. These curves are similar to those previously reported for preparations of CVB3/28 under selection for more-stable variants (27). Viruses with stabilizing mutations exist as minor populations in stocks of CVB3/28 (27). The lines in panels A (no FBS) and B (FBS = 0.1%) were fit by linear regression to all of the data points. (C to F) FBS at 1%, 5%, 10%, and 20%, respectively. The dotted lines were obtained by linear regression using only points where ln(V/V0) is −7 or greater. The solid lines were fit by using nonlinear regression and the equation ln(V/V0) = ln[A · exp(–k1 · t) + (1 – A) · exp(–k2 · t)] + Int, where A is the proportion of CVB3/28 in the population, k1 is the kapp for CVB3/28, k2 is the kapp for the more stable virus population, Int is the y intercept, and t is elapsed time in hours. k1 values are presented in Table 1. The average value for A was 0.999 ± 0.0009. The plot in panel G presents kapp values calculated from the two-population model (circles) and the kapp from linear regression fitted to the truncated data sets (+). The average k2 for panels C through F was 0.034 h−1 (standard deviation [SD] = 0.024 h−1).
FIG 12.

The destabilizing effect of FBS in PBS increases sharply between 0.1% and 1% FBS and shows saturation. The line is an empirical fit.
Elevated concentrations of magnesium have been shown to stabilize poliovirus (30), so the calcium and magnesium in DMEM but absent in PBS might stabilize CVB3/28 and moderate the FBS effect. This was tested by measuring the decay of CVB3/28 in Tris-buffered saline (TBS) (0.05 M Tris, 0.1 M NaCl, pH 7.2), in TBS with calcium (1.8 mM) and magnesium (0.8 mM), and in DMEM containing 5 mM EDTA, all with 10% FBS (Fig. 13). The kapp values for virus decay in TBS with 10% FBS (0.13 h−1), TBS with calcium-magnesium and 10% FBS (0.09 h−1), and DMEM with 10% FBS and EDTA (0.13 h−1) were all similar to the kapp observed for CVB3/28 decay in DMEM-10 (Table 1). The addition of calcium and magnesium provided some stabilization, but the effect was not statistically different from that of either the sample without added metals or the sample with EDTA added to the DMEM and FCS. PBS alone is not destabilizing (Table 1; Fig. 10). There is an as-yet-unexplained synergy between FBS and phosphate that enhances CVB3/28 destabilization by FBS.
FIG 13.

(A to C) CVB3/28 decay in TBS with 10% FBS (A), in TBS with 10% FBS, 1.8 mM CaCl2, and 0.8 mM MgCl2 (B), and in DMEM with 10% FBS and 5 mM EDTA (C). The kapp values were 0.125 h−1 (SE = 0.014 h−1) (A), 0.085 h−1 (SE = 0.013 h−1) (B), and 0.127 h−1 (SE = 0.016 h−1) (C) and were not statistically different.
Mutant strains CVB3/28 L1092V and CVB3/28 A3180T differ from CVB3/28 by single amino acids in VP1 and VP3, respectively, and are significantly more stable than CVB3/28 in DMEM-10 (Fig. 1 and 4) (27). Moreover, the 17-h half-life reported for CVB3/28 L1092V in DMEM-10 is equivalent to a kapp of 0.04 h−1, the same as determined here for CVB3/28 decay in DMEM without FBS (Table 1). Evaluation of both mutant strains in DMEM alone and in DMEM with 10% FBS found that FBS had a small but statistically significant effect on CVB3/28 A3180T (0.04 h−1 versus a kapp of 0.06 h−1, respectively), but there was no measurable effect on CVB3/28 L1092V (kapp = 0.03 h−1 in DMEM with and without 10% FBS) (Table 1; Fig. 4). Notably, in the absence of FBS, the kapp values for CVB3/28 and both mutant strains are comparable (Table 1), indicating that the apparent lesser stabilities of the CVB3/28 parent strain and the A3180T strain are condition-dependent and, in this instance, the result of a destabilizing interaction of CVB3/28 with the BSA in FBS. Capsids of both CVB3/28 and the A3180T strain contain leucine at position 92 of VP1 (27).
It is possible that the FBS effect on CVB3/28 is irreversible if the active component forms a tight destabilized complex with the virus or if it destabilizes virus by sequestering pocket factor. To test these hypotheses, CVB3/28 was placed in PBS, PBS with 20% FBS, and PBS with 1 mM EDTA. After 2 h at 37°C, the samples were layered over 30% sucrose (lacking magnesium for the EDTA-containing sample), and the virus was reisolated by ultracentrifugation. The reisolated CVB3/28 strain was assessed for posttreatment stability in PBS without FBS to determine whether the treatments had altered the inherent stability (Fig. 14). The first-order rate constants were not significantly different among the treatments (kapp values equal 0.04 to 0.05 h−1) and no different than had been determined previously for untreated CVB3/28 in DMEM or PBS without FBS (Table 1). These results showed that the FBS effect is reversible and that destabilized virus does not accumulate.
FIG 14.

CVB3/28 was suspended in PBS (A), PBS with 20% FBS (B), or PBS with 1 mM EDTA (C) and placed at 37°C for 2 h. The samples were layered on top of 30% sucrose for reisolation of virus by ultracentrifugation (the sucrose for the sample with EDTA lacked magnesium). Pelleted virus was suspended in PBS and placed at 37°C to assess the rate of decay. CVB3/28 preincubated in PBS had a kapp of 0.038 h−1 (SE = 0.004 h−1) (A), CVB3/28 preincubated in PBS with 20% FBS had a kapp of 0.038 h−1 (SE = 0.004 h−1) (B), and CVB3/28 preincubated in PBS with 1 mM EDTA had a kapp of 0.045 h–1 (SE = 0.005 h−1) (C) after separation from FBS. The first-order rate constants were not significantly different.
The possibility that FBS destabilization of CVB3/28 influences the sCAR-catalyzed conversion to A-particles was tested by measuring CVB3/28 decay in DMEM and DMEM with 10% FBS at four concentrations of sCAR, focused on concentrations that define k and just below those concentrations (Fig. 15) (25). Although the differences at each concentration of sCAR were not statistically significant, kapp in DMEM with sCAR at 250 to 1,000 nM was consistently lower than kapp in DMEM with FBS at the same concentrations of sCAR. FBS augments sCAR activity, but as the rates appear to converge with increasing sCAR concentration (implies that k is approximately k′), FBS apparently does not significantly affect the catalytic activity of saturating amounts of sCAR (Fig. 16). Once the concentration of sCAR was decreased to 100 nM, however, kapp for CVB3/28 decay in DMEM was nearly 3-fold lower than that for CVB3/28 in DMEM with FBS (Table 1; Fig. 16). Thus, there is significant synergy between the FBS and sCAR when sCAR is below saturating concentrations.
FIG 15.

CVB3/28 decay in DMEM (A to D) and DMEM with 9.28% to 10% FBS (E to H) with sCAR at 100 nM (A, E), 250 nM (B, F), 500 nM (C, G), and 1,000 nM (D, H). Values for kapp and standard errors were 0.558 and 0.097 h−1 (A), 1.41 and 0.094 h−1 (B), 1.543 and 0.107 h−1 (C), 1.512 and 0.13 h−1 (D), 1.526 and 0.09 h−1 (E), 1.536 and 0.101 h−1 (F), 1.743 and 0.12 h−1 (G), and 1.699 and 0.129 h−1 (H), respectively. The linearity assumed in panels D and H is an approximation in this data set.
FIG 16.

kapp values at various concentrations of sCAR (monomer calculated using Kd from the work of Patzke et al. [46]) in DMEM (open circles) and DMEM with FBS (9.3% to 10%; solid circles). Data obtained with DMEM-10 and published in reference 17 are shown for comparison (gray circles). The kapp values with and without FBS at 95 nM sCAR monomer (100 nM total sCAR) are significantly different. Differences with and without FBS at 224, 413, and 728 nM sCAR monomer (250, 500, and 1,000 nM total sCAR) are not statistically significant at a P of 0.05.
DISCUSSION
Of several proteins that have been reported to bind CVB strains, only the cell surface receptor, CAR, has been shown to catalyze virus conversion to A-particles as well as support virus infection of host cells (31). However, sialylated glycans, which are not required for the infection of neurons or astrocytes, are able to support a conformational intermediate of EV-D68, and bovine serum albumin can render some enteroviruses (e.g., echovirus 1) more permeable to small molecules. These intermediate conformations were less stable than those of the untreated viruses and were described as “primed” for transition to A-particles (32–34).
Results herein show that FBS contains a CVB3/28-destabilizing activity that, like that of the receptor, accelerates the loss of infectivity with first-order kinetics but with lesser efficiency. Relative to the rate in DMEM without serum, the rate at which CVB3/28 loses infectious activity in DMEM containing 5% or more FBS was increased by more than 2.5-fold. In comparison, in the absence of FBS, the rate at which infectious titer was lost increased from about 0.04 h−1 (sCAR = 0) to about 1.5 h−1, or 37-fold, when total sCAR was 250 nM or greater. The destabilizing effect was attenuated in CVB3/28 A3180T and not measurable in CVB3/28 L1092V, so the effect appears to be varied and restricted to a subset of CVB3 strains.
Enterovirus breathing is believed to require equilibrium between occupied and empty pockets; i.e., pocket factor must be displaced for the capsid conformation to open (6, 7). Albumin is the principal fatty acid-binding protein in serum, and defatted albumin has been shown to promote an echovirus conversion to A-particles and to increase permeability to small molecules (33, 35). The measured fatty acid content of the BSA purified from FBS was substantially less than saturating, so the BSA in these experiments has ample reserve capacity to bind fatty acids (36). It is possible that albumin might destabilize CVB3/28 by binding exposed pocket factor and sequestering it away from the capsid, as has been proposed for echovirus 1 (33). When CVB3/28 was incubated with FBS at 37°C and then reisolated by centrifugation through sucrose, the stability of the recovered virus was the same as that of virus that had not been exposed to FBS. Thus, the effect of FBS on CVB3/28 stability is readily reversible, and destabilized capsids lacking pocket factor did not accumulate during incubation with BSA. Notably, similar enteroviruses do not spontaneously convert to A-particles when the pocket is vacant (32, 37). It is therefore most likely that CVB3/28 is destabilized by a reversible association between capsids and albumin, for which destabilization of EV-D68 by glycans containing sialic acid and antibody destabilization of EV-71 and rhinovirus B14 provide precedents (32, 38, 39).
The discovery that serum destabilizes CVB3/28 necessitates refinement of our working model for receptor-catalyzed conversion to A-particles to account for the destabilized virus (25, 26). The experiments that supported that model were all done in DMEM-10 (8.9% FBS) using CVB3/28 (17, 26). There is almost certainly an ensemble of virus conformations between the closed state and the open state that precedes the A-particle. We propose that albumin interacts with one of these intermediate states of CVB3/28 and, as with the interaction with receptor, shifts the conformational equilibrium to a state nearer the threshold for conversion to A-particles. This interpretation is consistent with the increased permeability (time spent in an open conformation) of echovirus 1 in the presence of albumin, the intermediate breathing structure for EV-D68 in the presence of sialic acid-containing glycan, and the antibody-accelerated conversion of EV-71 and rhinovirus B14 to A-particles (32, 33, 38, 39).
The revision of the allosteric model (25) includes an albumin-supported intermediate open conformation, O, with increased affinity for sCAR, stabilized by association with albumin, B, to form complex OB. The O conformation is expected to show increased affinity for sCAR relative to the fully closed conformation, V (an expectation informed by the BSA-sCAR synergy and by the temperature-related affinities for poliovirus binding of its receptor [21]). From derivations presented in Materials and Methods, the revised model for equilibria and conversion to A-particles is represented as
(where U is the open conformation and A is the A-particle [see equation 4]) and kapp (the measured rate at which infectious virus is lost) is solved as
(see equation 10). This equation, using the formulas BT − VT ∼ BT and k ∼ k′, was used to fit the data shown in Fig. 3.
The albumin (FBS) effect is dose dependent and saturable. Since albumin was saturating in the previous work, the published model is simply reconsidered as (when B is saturating, V is ∼0 and VT is ∼OB plus UB). Consequently, Keq and k in the previous model are equivalent to K3 and k′ in equations 4 and 10. The premise of the working model for receptor-catalyzed conversion of CVB3 to A-particles still holds, but now there are intermediate conformations that can be stabilized by ligands other than the receptor; i.e., there can be strain-specific nonessential activating cofactors (positive heterotropic allosteric ligands), the effects of which have simply been described elsewhere as priming the virus for genome release (32, 33).
There is now evidence from multiple systems that molecules other than the cell surface receptors can bind enteroviruses and decrease their stability, accompanied by a shift of conformational equilibrium toward the open conformation. This investigation shows that albumin not only destabilizes, or primes, CVB3/28 but acts kinetically as a nonessential activating cofactor for sCAR-catalyzed conversion to A-particles.
MATERIALS AND METHODS
Cultured cells were maintained in DMEM-10 medium prepared by combining 1 liter of Dulbecco’s modified Eagle’s medium (DMEM) containing glucose at 4.5 g/liter with 100 ml heat-inactivated fetal bovine serum (FBS; final actual concentration, 8.9%, by volume), 10 ml penicillin-streptomycin (10,000 U/ml and 10 mg/ml, respectively), 5 ml 200 mM glutamine, and 7.5 ml gentamicin (10 mg/ml), all from GIBCO/ThermoFisher. Culture conditions were 37°C and 6% CO2–94% air in a humidified incubator.
CVB3/28 and mutant strains CVB3/28 L1092V (CVB3/28 with leucine in protein VP1 at residue 92 replaced with valine) and CVB3/28 A3180T (CVB3/28 with alanine in protein VP3 at residue 180 replaced with threonine) were propagated in lab strain HeLa cells from infectious cDNA clones (passage 1) and used at passage 2 (27, 40, 41). Viruses were partially purified from freeze-thawed cells by low-speed centrifugation to remove cell debris, followed by centrifugation in a Beckman SW28 rotor for 30 min at 25,000 rpm or in a Beckman SW41 rotor for 15 min at 35,000 rpm. Supernatants were extracted with chloroform (1:1, vol/vol), and the upper phase was collected after low-speed centrifugation to separate the aqueous and organic phases. Virus was pelleted by centrifugal sedimentation (overnight, Beckman SW28 rotor, 25,000 rpm, 4 to 8°C) through 30% (wt/vol) sucrose dissolved in 1 M NaCl, 1 mM MgCl2, 0.025 M Tris, pH 7.6, with approximately 200 μl glycerol beneath the sucrose (42). Virus was suspended in 0.1 M NaCl, aliquoted, and stored at –76°C. Virus stocks ranged from 8 × 108 to 2 × 109 50% tissue culture infectious doses (TCID50)/ml.
Concentrations of infectious virus were determined as numbers of TCID50 per milliliter using RDt3 cells (43, 44). RDt3 cells, i.e., RD cells (CCL-136) that express a C-terminal truncated coxsackievirus and adenovirus receptor (CAR), were seeded into 96-well plates at 4,000 cells/well in 100 μl DMEM-10. The following day, virus was diluted serially into chilled DMEM-10 prior to inoculation of the RDt3 cells. Cytopathic effect (CPE) was ascertained 72 h postinoculation, and results are reported as the geometric means of results from three assays.
Soluble CAR (sCAR) was produced from NS-1 cells (a mouse plasmacytoma line; ATCC TIB-18) transfected with pEF-ECAR and purified by immunoaffinity chromatography using monoclonal antibody RmcB (produced from hybridoma ATCC CRL-2379) coupled to Affi-Gel 10 (Bio-Rad) (42, 45). Purified sCAR protein was stored in 0.1 M sodium bicarbonate at –20°C. sCAR concentration was determined with the bicinchoninic acid (BCA) protein assay (Pierce ThermoFisher) and a peptide molecular weight of 25 kDa (the protein runs larger upon SDS-PAGE due to glycosylation). Monomeric sCAR concentrations were calculated from the total sCAR concentration using the dissociation constant (Kd) from the work of Patzke et al. (46).
Virus decay experiments were set up by combining DMEM or buffer with FBS to give the desired concentration of FBS (e.g., for 10% FBS, 720 μl of DMEM, and 80 μl of FBS) in a sterile screw-cap 1.5-ml tube and placed into the 37°C CO2 (6%) incubator with the caps loosened. CVB3 (20 μl) was added the next day, and a time zero sample (75 μl) was collected into a cold, sterile 1.5-ml tube and placed into the freezer at –76°C. The experimental samples were returned to the incubator pending resampling at timed intervals. For time courses in DMEM over 12 h in duration, two additional tubes without virus were used to control for evaporation; these tubes were weighed at the times that virus-containing tubes were sampled, and the TCID50 per milliliter results were corrected for evaporation. A single lot of FBS was used for all of these experiments, but the results with DMEM-10 were remarkably similar to results accumulated since 2011 using multiple lots of FBS (0.097 h−1 [42], 0.10 h−1 [17], and 0.099 h−1 [27]). Observed decay was first order, so plots of ln(V/V0) versus time, where V0 is total infectious virus at time zero and V is the concentration of infectious virus at subsequent time points, are linear, with the slope equal to negative kapp, the measured apparent first-order rate constant. Data for individual decay curves were adjusted to provide a regression line with an intercept equal to zero, which corrects for errors in V0 values without affecting the slope of the fit line. Regression and statistical analyses were done using PSI-Plot or Pro-Stat (Poly Software International). Simple Bonferroni corrections were made when appropriate; a P of ≤0.05 was considered statistically significant. Unless otherwise noted, decay experiments were done independently at least twice, and the combined data were used for a single regression to determine the apparent first-order rate constant, kapp, and the standard error of regression (SE).
The CVB3/28-destabilizing component in FBS was isolated by passing 1 ml FBS over a column (1.5 by 45 cm) of Sephacryl S300 (GE Healthcare Life Sciences) in PBS (0.05 M phosphate, 0.1 M NaCl, pH 7.2), with collection of 1-ml fractions. Protein was detected using the BCA assay, and destabilizing activity was detected by incubating CVB3/28 with each fraction at 37°C for 24 h and comparing losses of infectious virus. Proteins in the fractions were visualized by Coomassie blue staining after SDS-PAGE (47). Peak virus-destabilizing fractions were pooled and applied to a column (1 by 11 cm) of Affi-Gel blue (Bio-Rad) using the protocol recommended for highly specific removal of serum albumin. The column was washed with 10 ml PBS and then with a gradient from 20 ml PBS to 20 ml PBS plus 1.5 M NaCl, and 1-ml fractions were collected. Proteins not bound to the column were pooled, and protein-containing fractions eluted by the gradient were combined separately and concentrated to 2.5 mg/ml with a Centricon 10 (10-kDa-cutoff centrifugal concentrator; Amicon). To further test whether the destabilizing activity could be separated from albumin, the Affi-Gel blue eluate was diluted to 0.5× PBS and applied to a column (0.75 by 8 cm) of PBE 94 (GE Healthcare Life Sciences). The column was washed with 0.5× PBS and eluted with sequential gradients (9 ml for each component) from 0.5× PBS to PBS plus 0.2 M NaCl, from PBS with 0.2 M NaCl to PBS with 0.5 M NaCl, and from PBS with 0.5 M NaCl to PBS with 1 M NaCl. Fractions were assessed for protein content by the BCA assay and for virus-destabilizing activity.
Commercial essentially fatty acid-free BSA was obtained from Sigma (A-0281). SDS-PAGE revealed proteins larger than monomeric albumin and not consistent with dimeric BSA (Fig. 9). Monomeric albumin was isolated from 25 mg (25 mg/ml) using gel filtration on Sephacryl S-200 (GE Healthcare Life Sciences) in PBS (column, 1.5 by 72 cm).
Bovine serum albumin concentrations in the FBS were measured by radial immunodiffusion using rabbit anti-BSA serum (Sigma B-1520), 50 μl in 2% (wt/vol) SeaPlaque GTG agarose (FMC BioProducts) in 20 ml TBS (0.05 M Tris, 0.1 M NaCl, pH 7.6), with commercial, purified BSA as a standard (Pierce, ThermoFisher) (48). Protein concentrations were determined by the BCA assay. Fatty acid concentrations were determined using the EnzyChrom free fatty acid assay kit (BioAssay Systems). The samples, 80 μl in 1.5-ml tubes, were combined with 200 μl methanol and 100 μl chloroform and vortexed for 1 min. An additional 100 μl of water and 100 μl of chloroform were added, and the samples were centrifuged briefly to separate the phases. The upper, aqueous layer was removed and discarded; the lower, organic phase was collected and evaporated in a SpeedVac. The dried residue was dissolved in 40 μl 5% (wt/vol) Triton X-100 in water with vigorous mixing. Palmitic acid was used as the fatty acid standard. The FBS lot used in these experiments contained 271 μM BSA and 72 μM fatty acids.
Derivations of the equations to fit previous and revised models were as follows (25, 26). The current working model for CVB3 breathing and conversion to A-particles in the absence of a receptor is
| (1) |
V is the closed conformation, U is the open conformation, and A is the noninfectious A-particle. k is rate limiting, and the equilibrium is rapid (25).
| (2) |
There is probably an ensemble of conformations between V and U such that
| (3) |
The model for CVB3/28 with FBS (or albumin) is as follows. When BSA (B) is present and the virus is CVB3/28,
| (4) |
O is an intermediate conformation that interacts with B.
| (5) |
| (6) |
| (7) |
| (8) |
Placing values from equation 8 into equation 7,
| (9) |
solving for U/VT and UB/VT, and placing these values into equation 6, we get
| (10) |
An exact solution for B can be written from
| (11) |
After using substitutions from equation 8 and rearranging,
| (12) |
Rearranging equation 12 results in the quadratic equation in B
| (13) |
This quadratic is not easily solved because VT is not constant (dVT/dt = –dA/dt). However, experimentally, BT is present in millimolar concentrations and VT is present in picomolar concentrations, so a close approximation is available using BT – VT ∼ BT. B for use in equation 10 is then the appropriate root of equation 13.
The model(s) when MOPS is present is as follows. The previous model for MOPS stabilization of CVB3/28 was based on MOPS binding to the virus, possibly in the VP1 pocket (26).
| (14) |
with solution for kapp
| (15) |
Considering the FBS (albumin) effect on CVB3/28, equation 4 needs to be rewritten to include MOPS, represented as M.
| (16) |
Now, K4 equals VM/(V · M), while K1 through K3 remain as in equation 8. (Note that K4 is equivalent to 1/Kd in equation 15.) Solving equation 16 for kapp and adding the pH correction term from reference 26 results in
| (17) |
The current findings also present the possibility that MOPS associates with BSA, rather than virus, and interferes with the BSA destabilization of CVB3/28.
Equation 4 is modified to
| (18) |
K1, K2, and K3 are the same as in equation 8.
| (19) |
| (20) |
VT remains as in equation 7.
Equation 10 still provides the correct expression for kapp, but because the model predicts that increasing M will deplete B, the simple approximation B ∼ BT is not appropriate. The interaction between B and M necessitates a new solution for B. From the definitions for K1 to K4 (equations 8 and 19) and VT, MT, and BT (equations 7, 19, and 20),
| (21) |
| (22) |
Placing equations 21 and 22 into equation 20 gives
| (23) |
from which
| (24) |
Equation 24 lacks a simple solution for B because dVT/dt equals −dA/dt, but, experimentally, MT > 0 is in millimolar units, BT > 0 is in micromolar units, and VT is in picomolar units, so a solution is approximated by ignoring VT in the regression. Using the second root of equation 24 for B, and values for K1, K2, and K3, from analysis of data in Fig. 3, equation 10 with the pH correction term (K + )/(K + H+),
| (25) |
ACKNOWLEDGMENTS
This work was partially supported by grant R03 AI30174 from the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
We thank Doug Stickle for many helpful conversations and suggested approaches. Susan Hafenstein suggested that serum might alter CVB stability.
REFERENCES
- 1.Fry E, Stuart D. 2010. Virion structure, p 59–72. In Ehrenfeld E, Domingo E, Roos R (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 2.Bergelson J. 2010. Receptors, p 73–86. In Ehrenfeld E, Domingo E, Roos R (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 3.Levy H, Bostina M, Filman D, Hogle J. 2010. Cell entry: a biochemical and structural perspective, p 87–160. In Ehrenfeld E, Domingo E, Roos R (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 4.Hogle JM. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ismail-Cassim N, Chezzi C, Newman JF. 1990. Inhibition of the uncoating of bovine enterovirus by short chain fatty acids. J Gen Virol 71:2283–2289. doi: 10.1099/0022-1317-71-10-2283. [DOI] [PubMed] [Google Scholar]
- 6.Lewis JK, Bothner B, Smith TJ, Siuzdak G. 1998. Antiviral agent blocks breathing of the common cold virus. Proc Natl Acad Sci U S A 95:6774–6778. doi: 10.1073/pnas.95.12.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smyth M, Tate J, Hoey E, Lyons C, Martin S, Stuart D. 1995. Implications for viral uncoating from the structure of bovine enterovirus. Nat Struct Biol 2:224–231. doi: 10.1038/nsb0395-224. [DOI] [PubMed] [Google Scholar]
- 8.Reisdorph N, Thomas JJ, Katpally U, Chase E, Harris K, Siuzdak G, Smith TJ. 2003. Human rhinovirus capsid dynamics is controlled by canyon flexibility. Virology 314:34–44. doi: 10.1016/s0042-6822(03)00452-5. [DOI] [PubMed] [Google Scholar]
- 9.Tsang SK, Danthi P, Chow M, Hogle JM. 2000. Stabilization of poliovirus by capsid-binding antiviral drugs is due to entropic effects. J Mol Biol 296:335–340. doi: 10.1006/jmbi.1999.3483. [DOI] [PubMed] [Google Scholar]
- 10.Oliveira MA, Zhao R, Lee WM, Kremer MJ, Minor I, Rueckert RR, Diana GD, Pevear DC, Dutko FJ, McKinlay MA. 1993. The structure of human rhinovirus 16. Structure 1:51–68. doi: 10.1016/0969-2126(93)90008-5. [DOI] [PubMed] [Google Scholar]
- 11.Dorval BL, Chow M, Klibanov M. 1989. Stabilization of poliovirus against heat inactivation. Biochem Biophys Res Commun 159:1177–1183. doi: 10.1016/0006-291X(89)92234-1. [DOI] [PubMed] [Google Scholar]
- 12.Plevka P, Perera R, Yap ML, Cardosa J, Kuhn RJ, Rossmann MG. 2013. Structure of human enterovirus 71 in complex with a capsid-binding inhibitor. Proc Natl Acad Sci U S A 110:5463–5467. doi: 10.1073/pnas.1222379110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roivainen M, Piirainen L, Rysa T, Narvanen A, Hovi T. 1993. An immunodominant N-terminal region of VP1 protein of poliovirion that is buried in crystal structure can be exposed in solution. Virology 195:762–765. doi: 10.1006/viro.1993.1427. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Yafal AG, Lee YM, Hogle J, Chow M. 1994. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J Virol 68:3965–3970. doi: 10.1128/JVI.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broo K, Wei J, Marshall D, Brown F, Smith TJ, Johnson JE, Schneemann A, Siuzdak G. 2001. Viral capsid mobility: a dynamic conduit for inactivation. Proc Natl Acad Sci U S A 98:2274–2277. doi: 10.1073/pnas.051598298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, Lee LY, Roivainen M, Filman DJ, Hogle JM, Belnap DM. 2012. Structure of the Fab-labeled “breathing” state of native poliovirus. J Virol 86:5959–5962. doi: 10.1128/JVI.05990-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Organtini LJ, Makhov AM, Conway JF, Hafenstein S, Carson SD. 2014. Kinetic and structural analysis of coxsackievirus B3 receptor interactions and formation of the A-particle. J Virol 88:5755–5765. doi: 10.1128/JVI.00299-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren J, Wang X, Hu Z, Gao Q, Sun Y, Li X, Porta C, Walter TS, Gilbert RJ, Zhao Y, Axford D, Williams M, McAuley K, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2013. Picornavirus uncoating intermediate captured in atomic detail. Nat Commun 4:1929. doi: 10.1038/ncomms2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curry S, Chow M, Hogle JM. 1996. The poliovirus 135S particle is infectious. J Virol 70:7125–7131. doi: 10.1128/JVI.70.10.7125-7131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casasnovas JM, Springer TA. 1995. Kinetics and thermodynamics of virus binding to receptor. Studies with rhinovirus, intercellular adhesion molecule-1 (ICAM-1), and surface plasmon resonance. J Biol Chem 270:13216–13224. doi: 10.1074/jbc.270.22.13216. [DOI] [PubMed] [Google Scholar]
- 21.McDermott BM Jr, Rux AH, Eisenberg RJ, Cohen GH, Racaniello VR. 2000. Two distinct binding affinities of poliovirus for its cellular receptor. J Biol Chem 275:23089–23096. doi: 10.1074/jbc.M002146200. [DOI] [PubMed] [Google Scholar]
- 22.Xing L, Tjarnlund K, Lindqvist B, Kaplan GG, Feigelstock D, Cheng RH, Casasnovas JM. 2000. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J 19:1207–1216. doi: 10.1093/emboj/19.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsang SK, McDermott BM, Racaniello VR, Hogle JM. 2001. Kinetic analysis of the effect of poliovirus receptor on viral uncoating: the receptor as a catalyst. J Virol 75:4984–4989. doi: 10.1128/JVI.75.11.4984-4989.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelnabi R, Geraets JA, Ma Y, Mirabelli C, Flatt JW, Domanska A, Delang L, Jochmans D, Kumar TA, Jayaprakash V, Sinha BN, Leyssen P, Butcher SJ, Neyts J. 2019. A novel druggable interprotomer pocket in the capsid of rhino- and enteroviruses. PLoS Biol 17:e3000281. doi: 10.1371/journal.pbio.3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carson SD. 2014. Kinetic models for receptor-catalyzed conversion of coxsackievirus B3 to A-particles. J Virol 88:11568–11575. doi: 10.1128/JVI.01790-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carson SD, Hafenstein S, Lee H. 2017. MOPS and coxsackievirus B3 stability. Virology 501:183–187. doi: 10.1016/j.virol.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carson SD, Tracy S, Kaczmarek ZG, Alhazmi A, Chapman NM. 2016. Three capsid amino acids notably influence coxsackie B3 virus stability. J Gen Virol 97:60–68. doi: 10.1099/jgv.0.000319. [DOI] [PubMed] [Google Scholar]
- 28.McGeady ML, Siak JS, Crowell RL. 1979. Survival of coxsackievirus B3 under diverse environmental conditions. Appl Environ Microbiol 37:972–977. doi: 10.1128/AEM.37.5.972-977.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta BS, Taha M, Lee M-J. 2015. Buffers more than buffering agent: introducing a new class of stabilizers for the protein BSA. Phys Chem Chem Phys 17:1114–1133. doi: 10.1039/c4cp04663c. [DOI] [PubMed] [Google Scholar]
- 30.Wallis C, Melnick JL. 1961. Stabilization of poliovirus by cations. Tex Rep Biol Med 19:683–700. [PubMed] [Google Scholar]
- 31.Freimuth P, Philipson L, Carson SD. 2008. The coxsackievirus and adenovirus receptor. Curr Top Microbiol Immunol 323:67–87. doi: 10.1007/978-3-540-75546-3_4. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Sheng J, Baggen J, Meng G, Xiao C, Thibaut HJ, van Kuppeveld FJ, Rossmann MG. 2015. Sialic acid-dependent cell entry of human enterovirus D68. Nat Commun 6:8865. doi: 10.1038/ncomms9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruokolainen V, Domanska A, Laajala M, Pelliccia M, Butcher SJ, Marjomaki V. 2019. Extracellular albumin and endosomal ions prime enterovirus particles for uncoating that can be prevented by fatty acid saturation. J Virol 93:e00599-19. doi: 10.1128/JVI.00599-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenfeld AB, Warren AL, Racaniello VR. 2019. Neurotropism of enterovirus D68 isolates is independent of sialic acid and is not a recently acquired phenotype. mBio 10:e02370-19. doi: 10.1128/mBio.02370-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ward T, Powell RM, Chaudhry Y, Meredith J, Almond JW, Kraus W, Nelsen-Salz B, Eggers HJ, Evans DJ. 2000. Fatty acid-depleted albumin induces the formation of echovirus A particles. J Virol 74:3410–3412. doi: 10.1128/jvi.74.7.3410-3412.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Vusse GJ. 2009. Albumin as fatty acid transporter. Drug Metab Pharmacokinet 24:300–307. doi: 10.2133/dmpk.24.300. [DOI] [PubMed] [Google Scholar]
- 37.Strauss M, Filman DJ, Belnap DM, Cheng N, Noel RT, Hogle JM. 2015. Nectin-like interactions between poliovirus and its receptor trigger conformational changes associated with cell entry. J Virol 89:4143–4157. doi: 10.1128/JVI.03101-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plevka P, Lim PY, Perera R, Cardosa J, Suksatu A, Kuhn RJ, Rossmann MG. 2014. Neutralizing antibodies can initiate genome release from human enterovirus 71. Proc Natl Acad Sci U S A 111:2134–2139. doi: 10.1073/pnas.1320624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong Y, Liu Y, Jiang W, Smith TJ, Xu Z, Rossmann MG. 2017. Antibody-induced uncoating of human rhinovirus B14. Proc Natl Acad Sci U S A 114:8017–8022. doi: 10.1073/pnas.1707369114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tu Z, Chapman NM, Hufnagel G, Tracy S, Romero JR, Barry WH, Zhao L, Currey K, Shapiro B. 1995. The cardiovirulent phenotype of coxsackievirus B3 is determined at a single site in the 5′ nontranslated region. J Virol 69:4607–4618. doi: 10.1128/JVI.69.8.4607-4618.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carson SD, Pirruccello S. 2013. HeLa cell heterogeneity and coxsackievirus B3 cytopathic effect: implications for inter-laboratory reproducibility of results. J Med Virol 85:677–683. doi: 10.1002/jmv.23528. [DOI] [PubMed] [Google Scholar]
- 42.Carson SD, Chapman NM, Hafenstein S, Tracy S. 2011. Variations of coxsackievirus B3 capsid primary structure, ligands, and stability are selected for in a coxsackievirus and adenovirus receptor-limited environment. J Virol 85:3306–3314. doi: 10.1128/JVI.01827-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reed LJ, Muench H. 1938. A simple method for estimating fifty percent endpoints. Am J Hyg 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 44.Cunningham KA, Chapman NM, Carson SD. 2003. Caspase-3 activation and ERK phosphorylation during CVB3 infection of cells: influence of the coxsackievirus and adenovirus receptor and engineered variants. Virus Res 92:179–186. doi: 10.1016/s0168-1702(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 45.Hsu KH, Lonberg-Holm K, Alstein B, Crowell RL. 1988. A monoclonal antibody specific for the cellular receptor for the group B coxsackieviruses. J Virol 62:1647–1652. doi: 10.1128/JVI.62.5.1647-1652.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patzke C, Max KE, Behlke J, Schreiber J, Schmidt H, Dorner AA, Kroger S, Henning M, Otto A, Heinemann U, Rathjen FG. 2010. The coxsackievirus-adenovirus receptor reveals complex homophilic and heterophilic interactions on neural cells. J Neurosci 30:2897–2910. doi: 10.1523/JNEUROSCI.5725-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Mancini G, Carbonara AO, Heremans JF. 1965. Immunochemical quantification of antigens by single radial immunodiffusion. Immunochemistry 2:235–254. doi: 10.1016/0019-2791(65)90004-2. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Sheng J, Fokine A, Meng G, Shin WH, Long F, Kuhn RJ, Kihara D, Rossmann MG. 2015. Structure and inhibition of EV-D68, a virus that causes respiratory illness in children. Science 347:71–74. doi: 10.1126/science.1261962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muckelbauer JK, Kremer M, Minor I, Tong L, Zlotnick A, Johnson JE, Rossmann MG. 1995. Structure determination of coxsackievirus B3 to 3.5 A resolution. Acta Crystallogr D Biol Crystallogr 51:871–877. doi: 10.1107/S0907444995002253. [DOI] [PubMed] [Google Scholar]