

This study represents the first functional assignment of the connector domain of a Mononegavirales L protein. Furthermore, this study localizes P polymerase cofactor activity to specific amino acids. The functional necessity of this interaction, combined with the uniqueness of L and P proteins to the order Mononegavirales, makes disruption of the P-connector site a potential target for developing antivirals against other negative-strand RNA viruses. Furthermore, the connector domain as an acceptor site for the P protein represents a new understanding of Mononegavirales L protein biology.
KEYWORDS: RNA-dependent RNA polymerase, mass spectrometry, nonsegmented negative-strand RNA viruses, nuclear magnetic resonance, replication, transcription, vesicular stomatitis virus
ABSTRACT
Vesicular stomatitis virus (VSV) is an archetypical member of Mononegavirales, viruses with a genome of negative-sense single-stranded RNA (−ssRNA). Like other viruses of this order, VSV encodes a unique polymerase, a complex of viral L (large, the enzymatic component) protein and P (phosphoprotein, a cofactor component). The L protein has a modular layout consisting of a ring-shaped core trailed by three accessory domains and requires an N-terminal segment of P (P N-terminal disordered [PNTD]) to perform polymerase activity. To date, a binding site for P on L had not been described. In this report, we show that the connector domain of the L protein, which previously had no assigned function, binds a component of PNTD. We further show that this interaction is a positive regulator of viral RNA synthesis, and that the interfaces mediating it are conserved in other members of Mononegavirales. Finally, we show that the connector-P interaction fits well into the existing structural information of VSV L.
IMPORTANCE This study represents the first functional assignment of the connector domain of a Mononegavirales L protein. Furthermore, this study localizes P polymerase cofactor activity to specific amino acids. The functional necessity of this interaction, combined with the uniqueness of L and P proteins to the order Mononegavirales, makes disruption of the P-connector site a potential target for developing antivirals against other negative-strand RNA viruses. Furthermore, the connector domain as an acceptor site for the P protein represents a new understanding of Mononegavirales L protein biology.
INTRODUCTION
The order Mononegavirales includes numerous important human pathogens, such as Ebola, measles, and human parainfluenza viruses. These viruses harbor a single genomic segment of RNA with negative polarity that encodes a functionally conserved set of proteins that are fundamental to viral replication. Among these proteins are the large (L) protein and phosphoprotein (P) which form the viral polymerase complex. Genomic replication and transcription of mononegaviruses are completely reliant on L-P (L-VP35 in Filoviridae) complex formation; however, limited information is available regarding the essential protein-protein interactions required for viral RNA synthesis.
The highly conserved (1, 2) L protein supplies enzymatic activities for viral RNA synthesis, including RNA-dependent RNA polymerase (RdRP) (3–6), GDP polyribonucleotidyltransferase (PRNTase) (7), guanidine-N-7 and ribose 2′-O-methyltransferases (8, 9), and polyadenylate synthetase (10). The latter three enzymatic functions are associated with 5′-capping and mRNA maturation. The enzymatic activities of L proteins localize to 6 conserved regions (CRs) in the L proteins of Mononegavirales (2). CRI to CRIII comprise the RdRP domain, CRIV and CRV the capping PRNTase domain, and CRVI the methyltransferase domain. In vesicular stomatitis virus (VSV), the only mononegavirus for which a structure of the full-length L protein is available, an ordered domain, known as the connector domain, was observed to lie between CRV and CRVI (11). The connector domain, a helical bundle, was determined to be necessary for complete L function in VSV (12). Multiple studies have suggested that the VSV L connector domain could accommodate the P protein (11, 12), but this has never been validated. More recent reports have described L-P complex structures in respiratory syncytial virus (RSV) and human metapneumovirus (HMPV); however, in these structures, the putative connector, methyltransferase, and C-terminal domains were not visible (13, 14). In contrast, it was suggested that these structures represented an elongating L-P complex, with the VSV L-P study describing a preinitiation complex.
Phosphoproteins and other self-oligomerizing cofactors are also a conserved feature throughout Mononegavirales. In VSV P, several distinct functional regions are present. The ordered N terminus of P (residues 1 to 35) plays a chaperone role by binding nucleocapsid (N) protein prior to encapsidation of viral RNA (denoted by N0) and subsequent capsid formation (15, 16). An ordered central region (residues 107 to 177) mediates self-oligomerization (17, 18), while the ordered C terminus (residues 195 to 265) binds the nucleocapsid RNA template (19 to 21). An N-terminal disordered region (PNTD, residues 35 to 106), enables processivity in the L protein through a mechanism associated with the conformational compaction of L (22). Interestingly, the carboxy terminus of PNTD (P L-stimulatory domain [PLSD], residues 81 to 106) enables more limited processivity on an initiation site-like template (22) and was shown to be significant in the enzymatically distinct formation of the first phosphodiester bond in VSV RNA synthesis (23). Functional analogs of PNTD have been found across Mononegavirales; for instance, phosphoprotein and phosphoprotein-like proteins act as polymerase cofactors in Newcastle disease virus (24), Marburg virus (25), Sendai virus (26), and human metapneumovirus (27). These P and P-like proteins also harbor both ordered and disordered regions (28–30). A structural overview of VSV L and P proteins is provided in Fig. 1.
FIG 1.

Structures of the VSV L and P proteins. (A and B) VSV L protein (PDB ID 5A22) (A) and VSV P (a composite of PDB ID 3HHW, 2FQM, and 3PMK) (B) are shown in cartoon format with domains labeled and colored individually. Domain boundaries by residue number are provided below each cartoon. conn, connector. IDRc, C-terminal intrinsically disordered region.
Because of the extensive structural and functional characterization of VSV L and P, we elected to use them as a model system for characterizing L-P interactions in Mononegavirales. In the cryo-electron microscopy (cryo-EM) single-particle reconstruction of the VSV L-PNTD complex (11), the P protein was not resolved. However, volumes of unassigned density were suggested to potentially correspond to PNTD. These volumes were found proximal to the connector domain among other locations (11).
In this paper, we report that P protein residues 76 to 106 interact with the connector domain of VSV L protein and provide a docking model for the structure of this interaction. We validate the functional significance of P-connector interactions for RNA synthesis using a minigenome reporter assay. A bioinformatic analysis indicates that the connector-P interface shows conservation among vesiculo- and spriviviruses and that the residues of the connector interface specifically show conservation across Mononegavirales. This discovery represents the functional assignment for a connector domain in a mononegavirus L protein and implicates this domain as a potential target for antivirals.
RESULTS
The L connector domain interacts directly with PNTD.
In a previous structural study of the L-PNTD complex by cryo-EM, the structures of L protein domains were assigned (11); however, the structure of PNTD was not resolved. Additional volumes of unassigned density were observed near the PRNTase, connector, methyltransferase (MTase), and C-terminal domains in reconstructed density maps. Due to intense but unassignable atomic density near the connector domain, we elected to evaluate the connector domain as a binding partner for PNTD. We produced a construct comprising VSV L residues 1334 to 1595. Hydrogen-deuterium exchange mass spectrometry (HDX-MS) showed the construct to be largely structured with a more disordered N terminus, and circular dichroism (CD) indicated that the construct was largely helical (data not shown). These observations were consistent with existing structural information for this domain (11). Interestingly, major flexibility was not observed for the C terminus of the construct.
To experimentally determine if the PNTD directly interacts with the connector domain of L, we used nuclear magnetic resonance (NMR) chemical shift perturbation analysis (CSP), a method that has been used extensively to determine protein-ligand interaction sites with amino acid residue resolution (31). High-quality triple-resonance spectra for PNTD allowed backbone resonance assignments to be obtained for 62 of the 66 (94%) nonproline residues (Fig. 2A). Analysis of 13Cα chemical shifts (32) indicates that PNTD is intrinsically disordered, consistent with previous structural information on PNTD (16, 29).
FIG 2.

VSV P residues 76 to 106 interact with the VSV L connector domain. (A) HSQC spectra of PNTD, residues 35 to 106, showing amide shift migration and intensity loss upon the addition of the connector domain (apo spectra in black, connector added in green). (B) The changes to position (top) and intensity (bottom) of amide peaks upon connector addition are presented. On the upper graph, a red line is present at our quantitative cutoff (see Materials and Methods) at 0.071 ppm, and residues with significant amide peak migrations are indicated in red asterisks. In the intensity plot, a region of greater than 80% signal depletion is noted in residues 97 to 106 (red bar). Due to difficulty in tracking severely weakened peaks, migrations are not provided for residues 97 to 106. (C) Fluorescence anisotropy using a peptide identical in sequence to VSV P 80 to 106 with an N-terminal FITC label was performed with various concentrations of the connector domain. mPU, millipolarization units.
Upon addition of the connector domain of L, perturbations were observed for multiple residues compared to the reference 1H-15N HSQC spectra of PNTD alone. Normalized weighted average 1H and 15N shift differences were plotted as a function of residue position (Fig. 2B). Multiple significant movements of amide cross-peaks occurred. Furthermore, we observed a significant loss of peak intensity (>90%) for residues 97 to 106 (Fig. 2B). This loss in intensity prevented the measurement of 1H and 15N shift differences for residues 97 to 106; thus, these measurements are not included. These results strongly suggest that the connector domain of L directly interacts with amino acids 76 to 106 of PNTD, with the directly implicated residues comprising V76, I88, Q89, D97, E98, D99, V100, D101, V102, V103, F104, T105, and S106.
Having implicated the connector domain as a phosphoprotein binding site, we sought to test our hypothesis that the functionally assigned PLSD (22, 23), comprising residues 81 to 106, would be sufficient for binding to the connector domain. We approached this hypothesis using fluorescence anisotropy. Experiments were carried out using this peptide and increasing concentrations of the connector domain. Addition of connector resulted in an increase in polarization, consistent with our previously observed interaction (Fig. 2C). This demonstrated that PLSD was sufficient for connector domain interaction.
Connector residues involved in phosphoprotein interaction are located near the connector-CTD interface.
Having localized the P-connector interface on the P protein, we proceeded to identify the reciprocal interface on the connector domain. Assigning heteronuclear single quantum coherence (HSQC) spectra for the connector domain was infeasible, though peak migrations and intensity changes upon PLSD addition were visible (data not shown). We instead used HDX-MS, in which differences in the uptake of solvent-supplied deuterons can be used to locate binding sites (33). Following an initial time course which showed that changes dissipated quickly, consistent with a weak interaction (data not shown), we carried out a 1- to 5-min time course and found that the addition of PNTD lowered the exchange rate of the connector domain at residues 1414 to 1436 and 1529 to 1547. These residues cluster in space at the surface of the connector domain, are localized near the interface of the connector domain and C-terminal domain (CTD) in the intact L protein, and are largely solvent accessible in the cryo-EM structure of L (Fig. 3). The implicated region harbors a number of solvent-accessible basic residues (R1419, R1420, H1424, K1426, R1427, K1534, K1540, K1542, and R1546) and hydrophobic/aromatic residues (Y1433, L1529, I1531, Y1533, F1536, and L1537), consistent with the acidic and hydrophobic character of P76–106.
FIG 3.

Hydrogen-deuterium exchange of the connector-P interaction. (A) HDX-MS time course experiments were conducted in the 1- to 5-min range using the connector domain as an analyte and PNTD as a variable. A volcano plot, which quantifies the significance of a change in deuteron uptake against the magnitude of the change for each time point, is shown. The box in the upper left corner contains time points which showed a significant decrease in deuteron uptake of the PNTD sample relative to the apo sample at a confidence of P value of ≤0.01. (B) Representative peptide uptake plots are shown from the HDX-MS time course. Peptides indicated in the volcano plot are displayed (i, ii), as is a representative peptide not indicated as significant (iii). Error bars display 95% confidence intervals for deuteron uptake. (C) i, the connector domain (yellow) is displayed with the significant regions highlighted (1414 to 1436, pink; 1529 to 1547, cyan); ii, the connector is shown in the context of L with other domains shaded in gray. The images of L are based on PDB ID 5A22.
Docking studies provide a model of the connector domain-P complex.
Following our structural analysis, we sought to use computational modeling to determine what the structural characteristics of the connector-PLSD interaction might be. A peptide model for VSV P 97 to 104 (the most strongly interacting and conserved fragment) was generated using I-TASSER (34). We noted that the cryo-EM reconstruction of VSV L contained unassigned density in contact with the surface of the connector implicated by HDX-MS. The modeled P fragment was placed within this density before carrying out docking against the surface using FlexPepDock (35, 36). The top 10 models remained remarkably well converged in the unassigned density (Fig. 4A to C). The highly conserved VxF motif of P (described below) was positioned proximal to the connector aromatic residues Y1433, Y1533, and F1536. Though our construct length could not include the entire PLSD (residues 81 to 106) due to wider conformational freedom and secondary/tertiary structure imposed on longer peptides in I-TASSER and performance issues with flexible docking of longer constructs, we expect that the acidic residues in the remainder of the acidic stretch will interact with other surface-exposed basic residues of L 1414 to 1436 and 1529 to 1547 (Fig. 4D to F).
FIG 4.
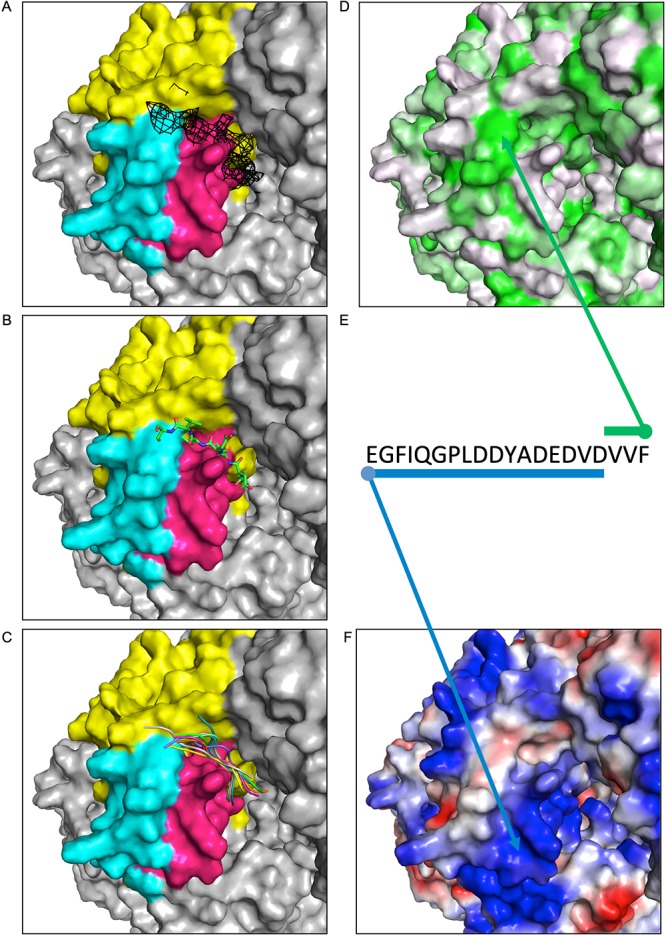
Docking studies using VSV P and L. A fixed view of the connector domain is provided in each window. (A) The connector domain (yellow) is displayed with residues implicated in P binding by HDX highlighted in pink (1414 to 1436) or cyan (1529 to 1547). Unassigned density from the VSV L reconstruction (11) is shown in black mesh at a contour level of 2σ. (B) Starting model of VSV P 97 to 104 is shown positioned proximal to the density. (C) The top 10 docked models are displayed as cartoons, each in a different color. (D) The surface of L is colored by hydrophobicity using the PyMOL color h script with a normalized consensus hydrophobicity scale (white to green, hydrophilic to hydrophobic). (E) The sequence of the VSV P acidic stretch and VxΩ motif is shown, with arrows and bars indicating suspected interactions of P with hydrophobic (green) or basic (blue) patches of the L protein. (F) The surface of L is displayed by charge using the vacuum electrostatics feature of PyMOL (blue to red, basic to acidic).
The connector-PNTD interface is conserved in and functionally consequential.
Having identified a novel interaction between the VSV P and L proteins, we next sought to examine the extent to which the binding interfaces of L and P were conserved within the Vesiculovirus and closely related Sprivivirus genera. We aligned phospho- and large protein sequences for members of the Vesiculovirus and Sprivivirus genera using ClustalX2 (37). An alignment of the Vesiculovirus and Sprivivirus phosphoproteins against residues 76 to 106 of VSV P showed that they share two major conserved features. Consensus sequences are displayed using extended single-letter codes (Ω, aromatic; Φ, hydrophobic; ζ, hydrophilic; +, basic; −, acidic). On the carboxy terminus, a VxΩ motif was present, which displayed a consensus of VxF for Vesiculovirus and VxW for Sprivivirus. VxΩ was part of a more extended but weakly conserved ΦxVxΩ motif, with only 8 viruses displaying a conventional hydrophobic side chain (2 aromatics and 6 aliphatics) and the other residues in the Φ position comprising proline (twice), tyrosine (twice), and glutamic acid (once). The other conserved feature was an acidic stretch encompassing the roughly 15 amino acids leading into the (Φx)VxΩ motif. The total acidic side-chain content of this region varied from as high as 67% observed in Cocal and Maraba viruses to as low as 27% in Isfahan virus. Considerable variability was observed for the spacing of the acidic residues; contiguous lengths range from 1 to 4 and interacid distances from 1 to 7 residues (Fig. 5A).
FIG 5.

Conservation of L and P proteins. (A to C) P and L proteins from members of the Vesiculovirus and Sprivivirus genera were aligned in Clustal (37). (A) P proteins were aligned against VSV P 76 to 106. The most strongly interacting fragment, P 97 to 106, is highlighted with a red bar. (B and C) L proteins were aligned against VSV L 1414 to 1436 (B) and 1529 to 1547 (C), the two fragments implicated by HDX-MS. (D to F) L proteins from members of Mononegavirales were aligned in Clustal and subjected to secondary structure prediction. (D and E) L proteins were aligned against VSV L 1414 to 1436 (D) and 1529 to 1547 (E). (F) Secondary structure prediction was carried out for each putative connector sequence using PSIPRED (38). The predicted helical content (Helpred) is listed for each protein, except for VSV L, in which the authentic helical content is provided from the existing structure (PDB ID 5A22). VSIV, Indiana vesicular stomatitis virus; VSNV, New Jersey vesicular stomatitis virus; VSAV, Alagoas vesicular stomatitis virus; CARV, Carajas virus; CHPV, Chandipura virus; COCV, Cocal virus; ISFV, Isfahan virus; MARAV, Maraba virus; PIRYV, Piry virus; JURV, Jurona virus; PERIV, Perinet virus; YUBOV, Yug Bogdanovac virus; PFRV, pike fry virus; SVCV, spring viremia of carp virus; hRSV, human respiratory syncytial virus; hMPV, human metapneumovirus; ZEBV, Ebola virus; MARV, Marburg virus; PIV3, parainfluenza virus type 3; SeV, Sendai virus; HENV, Hendra virus; NIPV, Nipah virus; SV5, simian virus 5; MuV, mumps virus; MeV, measles virus; NDV, Newcastle disease virus. Absolute (*), strong (:), and weak (.) conservation is denoted above the alignments.
Alignment of the viral L proteins from Vesiculovirus and Sprivivirus showed more dramatic conservation of the interacting regions. The first stretch implicated by HDX-MS, residues 1414 to 1436, displayed a consensus sequence of (C/V)Q(V/A)(I/L)HRRxxxx(L/I)++(P/T)A(N/D)(A/V)(V/I)YGG, in which ++ represents a partially conserved dibasic motif. The second implicated region of residues 1529 to 1547 showed a consensus of ΦxΦLY+xxL(S/T)x+−+xxΦRζ (Fig. 5B and C). Of note, a large number of the conserved basic residues mapped to the surface of the connector domain. In the VSV L connector domain, these two sequences comprise separate helix-turn-helix motifs which contact by their turn-proximal ends, with an aforementioned high level of steric accessibility.
Given the presence of both catalytic large proteins and charged, disordered polymerase cofactors in Mononegavirales, we used protein sequence data to examine whether or not the connector-P interaction was likely to be present in other mononegaviruses. Secondary structure prediction was carried out on the PSIPRED server (38) using representative L proteins from across Mononegavirales. The known secondary structure of VSV L, a member of the Rhabdoviridae, was also supplied. Potential connector regions were defined as the sequences which aligned against VSV L residues 1334 to 1595 in ClustalX2 (37) (Fig. 5D and E). All viruses displayed a large amount of helical propensity (Fig. 5F), from 37 to 61%, indicating that structured, helical connector domains might be a general feature of Mononegavirales and not just of VSV and its close relatives within Rhabdoviridae.
The two regions implicated in P-binding on the VSV connector surface, VSV L residues 1414 to 1436 and 1529 to 1547, were examined in the sequence alignment of the proteins. We noted the partial conservation of four basic residues or pairs thereof in the order, two of which corresponded to surface-exposed basics in VSV L, 1419-RR-1420 and R1546. Hydrophobic residues showed variable conservation across the L proteins, and an aromatic residue (Y1533 in VSV) also showed conservation. Though basic amino acid residues were present, absolute conservation was difficult to define due to shifts in their primary position. However, this is interesting given the striking number of acidic residues in the sequence alignments with viral phosphoproteins presented above. We hypothesize that these basic residues accommodate charged cofactors in other mononegaviruses, but testing this hypothesis will require functional testing of mutants in viruses other than VSV.
We next sought to validate the functional significance of the implicated amino acids in L and P. We approached this question with a minigenome reporter system with both wild-type and mutagenized P. Multiple amino acid residues within the P segment 76 to 106 were found to decrease reporter production when substituted with alanine or deleted (Fig. 6A). Large decreases of 65% and 73% were observed for V102A and F104A, respectively. Intermediate decreases of 40% and 34% were observed for D94A and delV103, and last, the I88A, D97A, E98A, and T105A substitutions showed relatively small (but still statistically significant, P < 0.05) decreases in the 10 to 20% range. Our collective results indicate that conserved residues in PNTD confirmed to interact with the connector domain in NMR and HDX experiments have a functional role in viral RNA synthesis. Examination of mutant P expression using Western blotting (Fig. 6C) showed that mutant proteins are expressed at a level similar to that of the wild type.
FIG 6.

Minigenome reporter assay using mutagenized VSV L and P. (A and B) Using a minigenome reporter system, the effects of alanine substitutions or residue deletion in VSV L (A) and P (B) on reporter activity were quantified and compared against the wild-type (PosCon) sample. Error bars represent standard deviations (SD) for n = 3, and asterisks denote a significant difference (P < 0.05) of the sample relative to WT in a two-tailed t test. (C) Western blots against wild-type and mutagenized P are provided for each sample, along with a control blot against glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Similarly, we examined the functional consequences of mutating residues in the connector domain (Fig. 6B). In the aromatic pocket suspected of accommodating the VxΩ motif, the Y1533A and F1536E substitutions greatly suppressed reporter activity (98% and 99%, respectively), while the Y1433A and F1536A substitutions had only a minor effect (14% and 19% decrease, respectively). Multiple basic residue substitutions, H1428A, R1546A, K1540A, and K1542A, had significant effects. K1540A showed ∼60% suppression in contrast to the near-total suppression of the others. Mutants intended to disrupt the general structure of the binding interface (A1429F, A1429E, and G1434W) showed a decrease in RNA synthesis, with the G1434W mutation having a 21% decrease, while A1429 mutations nearly eliminated reporter activity. Interestingly, disruption of a conserved basic residue with the R1420A substitution had no effect on RNA synthesis. The levels of L in the minigenome reporter system are below the level of detection with our antibodies. Despite this limitation, the number of functionally consequential positions in this region supports its significance in viral RNA synthesis.
DISCUSSION
RNA synthesis of nonsegmented negative-strand RNA viruses relies on a complex series of protein-protein interactions. Due to conformational variability, difficulty of expression, and resistance to many conventional structural approaches, studying the interaction between the L and P proteins has historically been difficult. Despite this, L-P interactions merit extensive study due to the complete reliance of mononegaviral replication cycles on the interactions of these proteins. The L protein is a modular protein of five domains, with most having defined functions. However, the connector domain(s) of L proteins from mononegaviruses had no assigned function to date but had been suggested to be a binding site for P (11, 12). In this study, we have shown that the connector domain from VSV L is a binding site for the C terminus of the cofactor PNTD which, while known to bind L, did not have a known binding site. This interaction is mediated by conserved amino acid residues in both L and P and is a positive regulator of viral RNA synthesis.
Different approaches to L-P studies in the past have yielded disparate results across multiple viruses. In rabies virus, the C-terminal domain of L was implicated to bind P (39–41), while in the paramyxoviruses simian virus 5 (42) and Sendai virus (43, 44), the N terminus of L was implicated. In the filoviruses Marburg virus (45) and Ebola virus (46), the N terminus of L was similarly implicated in L-VP35 interactions. In recent Lcore-P structures in RSV and HMPV (13, 14), P bound the core of the polymerase, though the accessory domains were not visible. In the context of resolving disparate implications across the field, it bears keeping in mind that our study implicates only a partial binding site of the full-length PNTD. Given that the conformational effects of PNTD on L are explainable solely with L-P interactions (22), another binding site or sites for PNTD must be present on L. Consistent with this idea, multiple regions of unassigned density were present in the L-PNTD structure beyond the one in which we modeled residues 97 to 104, including at the C-terminal domain and capping-connector interface (11).
Though we have shown it to be a positive regulator of RNA synthesis, the mechanism by which the connector-P interaction affects the polymerase is not clear. As we have only accounted for the C terminus of the cofactor in connector binding, we hypothesize that full-length PNTD binds the L protein at more than one position, anchoring the connector to another position in the polymerase. This most likely occurs in or around the accessory domains, which undergo conformational compaction upon the addition of PNTD (22). Studies similar to those conducted in this report but using different L domains, or alternatively, a full-length L-P structure in which P can be resolved, are needed to test this hypothesis.
The VSV L structure (11) is believed to correspond to a preinitiation conformation, given that the priming-capping loop, a feature which mediates first phosphodiester bond synthesis (23), is blocking the putative product exit channel. Interestingly, recent structures of RSV and HMPV L-P complexes (13, 14) show a putative postinitiation state. This structure does not account for the accessory connector, methyltransferase, and C-terminal domains, but it does show P bound to the core of the polymerase. It is interesting to note that these two structures do not implicitly suggest differentiated modes of L-P binding in RSV/HMPV and VSV; in other words, it is possible that an L-P interaction involving the connector domain takes place before initiation in both viruses and an L-P interaction involving the polymerase core takes place after.
The L and P proteins of Mononegavirales represent absolute requirements for viral replication cycles that do not have host analogs and are therefore attractive targets for antivirals. Furthermore, the conservation of residues in the VSV connector-P interaction across the order indicates that a connector-P interaction may be a widespread feature. Future work to address both this idea and the aforementioned potential for multiple L-P states illuminated by the structures of RSV and HMPV L-P complexes will require additional L-P structures in other branches of the order, as well as the structures of polymerase-template complexes at different stages of RNA synthesis.
MATERIALS AND METHODS
Plasmids.
Using primers purchased from Integrated DNA Technologies (Skokie, IL) with the sequences 5′-ACGGTCTCAAGGTGGTTCGTGGGGACAAGAGATAAAACAG and 5′-CCCAAGGGGGATAGCTCATCTCGAGTTAGTCTTTATTATTATCCTTAGCAA, a region of VSV L comprising amino acids 1334 to 1595 was amplified from a cDNA clone of the vesicular stomatitis Indiana virus (VSIV) L gene. The PCR product was digested using BsaI and XhoI restriction enzymes and ligated into the SUMOpro gene fusion technology kanamycin (SpGFTk) vector (LifeSensors, Malvern, PA). Amino acids 35 to 106 of VSV P were similarly amplified from a cDNA clone of the VSV P gene using primers 5′-ACGGTCTCAAGGTTCCAATTATGAGTTGTTCCAAGAG and 5′-CACCGCTCGAGTTACGAAGTGAATACAACATCCACGTC and likewise cloned into SpGFTk. The plasmids were identified as Sp-L1334-1595 and Sp-P35-106, respectively. Amplification of plasmid DNA was carried out in Escherichia coli DH5α following a heat shock transformation with the plasmid using a QIAprep spin miniprep kit (Qiagen, Hilden, Germany).
Genes encoding the VSV N, P, or L protein were cloned into pT7CFE1-CHis. Plasmids used in the minigenome reporter system consisted of a T7-controlled expression vector for VSV N, P, or L protein or a minigenome reporter consisting of a NanoLuc luciferase gene (Promega) with a 3′ VSV leader sequence and 5′ VSV trailer sequence. All plasmids were validated using Sanger sequencing at the University of Alabama at Birmingham (UAB) Heflin Center for Genomic Science.
Synthetic peptides.
Peptides comprising VSV P residues 76 to 106, 80 to 106, or 95 to 106 with the sequence N-VPDPEAEQVEGFIQGPLDDYADEDVDVVFTS, N-EAEQVEGFIQGPLDDYADEDVDVVFTS, or N-YADEDVDVVFTS, respectively, were synthesized by GenScript (Piscataway, NJ) both with and without an N-terminal fluorescein isothiocyanate (FITC) label.
Protein expression and purification.
E. coli BL21-CodonPlus(DE3) was transformed with Sp-L1334-1595 or Sp-P35-106 using heat shock transformation. A single transformant colony was used to prepare a starter culture, grown overnight at 37°C with shaking at 250 rpm in either LB broth (Fisher, Fair Lawn, NJ) or M9 minimal medium made with 15N-ammonium chloride and 13C-glucose (99% isotopic content; Cambridge Isotope Laboratories, Andover, MA) when isotopic labeling was required. The growth medium contained kanamycin at 50 μg/ml and chloramphenicol at 35 μg/ml. Overnight starter cultures were pelleted and the bacteria resuspended in fresh medium with antibiotic after growth. Resuspended starter cultures were diluted 1:100 into expression cultures which were grown to an optical density at 600 nm (OD600) of 0.4 to 0.6 (LB) or 0.7 (M9). Large cultures were cooled to 18°C and induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). After induction, the cultures were incubated overnight at 18°C with shaking at 250 rpm.
Following expression, bacteria were pelleted at 6,000 × g for 15 min. Bacterial pellets were resuspended in 30 ml of binding buffer (500 mM sodium chloride, 10% glycerol [vol/vol], 5 mM imidazole, 20 mM Tris [pH 7.9]) per liter of culture. Resuspended pellets were lysed using sonication, and sonicates were pelleted at 4°C and 27,000 × g for 30 min. Supernatants were purified using nickel affinity chromatography. Removal of the His-SUMO tag was carried out overnight at 4°C with shaking by adding a 2:1,000 weight ratio of SUMO protease to the sample. For P 35 to 106, protease treatment was followed by buffer exchange; the protein was exchanged to MQA buffer (150 mM sodium chloride, 20 mM Tris, 0.02% sodium azide [wt/vol] [pH 7.5]). Following buffer exchange, an additional round of nickel affinity chromatography was used to remove the SUMO tag from P 35 to 106.
Additional purification of L 1334 to 1595 was carried out using heparin affinity chromatography on a HiTrap heparin 5-ml column (GE, Pittsburgh, PA). The digested sample was exchanged to HA buffer [150 mM sodium chloride, 10% glycerol (vol/vol), 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 0.02% sodium azide (wt/vol) (pH 7.0)] and loaded onto the column. A buffer of the same composition but with 1 M sodium chloride, labeled HB buffer, was used to form a salt gradient, with the protein eluting at a 50% combination of the buffers. The final purification step was done using a HiLoad 16/600 Superdex 200-pg column (GE) in HB buffer.
Following the second round of nickel affinity chromatography, P 35 to 106 was further purified using anion exchange on a Mono Q 1-ml column (GE). In addition to MQA buffer, a salt gradient was formed using MQB buffer (1 M sodium chloride, 20 mM Tris, 0.02% sodium azide [wt/vol] [pH 7.5]), and the protein was eluted at ∼65% MQA buffer and 35% MQB buffer. The final step of purification was size exclusion chromatography on a HiLoad 16/600 Superdex 75-pg column (GE) using HB buffer.
NMR spectroscopy.
All NMR experiments were carried out at 25°C on Bruker Avance III spectrometers with TCI CryoProbes operating at 1H frequencies of 600 MHz or 850 MHz. Data analysis and visualization were carried out using the NMRPipe (47) and NMR View (48) softwares. Proteins were exchanged into NMR buffer (200 mM sodium chloride, 10% glycerol [vol/vol], 20 mM HEPES, 1 mM TCEP [pH 6.8]) using a HiPrep 26/10 desalting column (GE). Backbone 1H, 13C, and 15N resonances were assigned using standard triple-resonance assignment experiments including HNCACB and CBCA(CO)NH (49). Interactions between the VSV P protein and L protein were assessed using 1H-15N HSQC-based chemical shift perturbation experiments. Changes to chemical shift position were described by the equation below.
A cutoff value for determining statistically significant changes in chemical shift was calculated using a previously described statistical method (50).
Circular dichroism.
A sample of VSV L 1334 to 1595 at 1.1 mg/ml was prepared in CD buffer (75 mM NaCl, 10% glycerol, 20 mM HEPES, 0.02% NaN3 [pH 7.0]). The sample was diluted 1:1 with water and placed into a 0.1-mm-pathlength cuvette, which was read in a Jasco J-815 circular dichroism spectrometer from 190 to 260 nm. The reading was performed with an increment step of 1 nm and a collection time of 32 s per increment.
Fluorescence anisotropy.
Before use in fluorescence anisotropy, protein samples were exchanged to MS buffer (200 mM sodium chloride, 20 mM HEPES, 1 mM TCEP, 0.02% sodium azide [pH 7.0]) using a HiPrep 26/10 desalting column (GE) and concentrated using centrifugal filtration. Samples of VSV L 1334 to 1595 and P 80 to 106 with an N-terminal FITC label were prepared with a labeled peptide concentration of 5 nM and L 1334 to 1595 at concentrations of 0, 50, 100, 150, 200, or 300 nM. Samples of 100 μl were loaded into 96-well plates and measured for fluorescence anisotropy using a BioTek Synergy 2 plate reader. All measurements were taken in 3 technical replicates.
HDX-MS.
Mass spectrometry (MS) experiments were carried out on a G2-Si Synapt (Waters Corp.) and a Leap HD/X-PAL (Trajan) fluidics system. All samples contained 30 pmol L 1334 to 1595, with or without 60 pmol P 35 to 106. Experiments were in MS buffer (200 mM sodium chloride, 20 mM HEPES, 1 mM TCEP, 0.02% sodium azide [pH 7.0]) or an equivalent made with deuterium oxide. Exchange was carried out at 20°C for 0, 1, 2, 3, 4, or 5 min. Quenching was performed at 4°C with a 1:1 dilution of 100 mM monobasic potassium phosphate (pH 2.5). The quenched exchanged protein was digested by passage over an Enzymate pepsin column at 0.1 ml/minute (pore size, 300 Å; particle size, 5 μm; column size, 2.1 mm by 30 mm; Waters Corp.). The digestion duration was 2 min, at a temperature of 15°C. Peptide separation was carried out over an Acclaim PepMap C18 trap column (Thermo Fisher) and an Acquity BEH C18 reverse-phase column (pore size, 130 Å; particle size, 1.7 μm; column size, 1 mm by 50 mm; Waters Corp.), with all liquid chromatography (LC) carried out using a Shimadzu SPD-20 series pump system. LC was performed with two solutions, SA (water with 0.1% [vol/vol] formic acid) and SB (acetonitrile with 0.1% [vol/vol] formic acid). Loading onto the pepsin column was carried out with SA solution. Loading was carried out at 0.1 ml/min and resolving at 0.07 to 0.1 ml/min.
Peptides were selected in PLGS (Waters) with a quality score of 6 or greater and a charge state of +1 to +4. Levels of deuterium exchange were measured using HDExaminer software (Sierra Analytics, Modesto, CA). Significant uptake changes were indicated by a confidence threshold of a P value of ≤0.01. A full-exchange time point was used to measure system-intrinsic back exchange for VSV L 1334 to 1595, with an inferred average back exchange of 19%. A summary of the collection statistics for our deuterium exchange experiments is provided in Table 1.
TABLE 1.
Hydrogen-deuterium exchange collection statistics
| Criterion | Connector only | Connector + PNTD |
|---|---|---|
| HDX reaction details | 200 mM NaCl, 20 mM HEPES, 1 mM TCEP, 0.02% NaN3, pHread 7.00, 20°C | |
| HDX time course (min) | 0, 1, 2, 3, 4, 5 | |
| HDX control samples | Fully labeled control (9-day exchange) | |
| Back exchange (%) (mean/SD) | 19/12 | |
| No. of peptides | 50 | |
| Sequence coverage (%) | 90.5 | |
| Avg peptide length/redundancy (amino acid residues) | 14.5, 2.8 | |
| No. of technical replicates | 3 | |
| Repeatability (%D) (avg SD) | 1.2 | 1.3 |
| Significance criterion for HDX (no. of D) | 0.961 (99% confidence) | |
Minigenome luciferase assay.
Transfections were carried out in a BSRT7 cell line. Cells were grown to 90% confluence in 24-well plates and subsequently transfected using TranslT-LT1 with plasmids encoding VSV N, P, and L proteins, as well as a minigenome reporter expressing NanoLuc (Promega). Plasmids for viral proteins featured an internal ribosome entry site (IRES) to enable expression. A pGL4.53(luc2/PGK) plasmid expressing firefly luciferase was also transfected. Following incubation at 37°C and 5% CO2 for 24 h, the cells were harvested of protein, and a luciferase assay was performed using a GloMax instrument (Promega). Luminescence values from NanoLuc were normalized to the firefly luciferase values on a well-by-well basis. Each transfection was carried out and measured in triplicate. The expression of VSV P was determined using Western blotting, essentially as previously described (7).
Modeling of protein-peptide interactions.
A starting model for P was generated for VSV P in I-TASSER (34) using the sequence of VSV Indiana P 97 to 106, N-DEDVDVVFTS. The L model was supplied from PDB 5A22 (11) with heteroatoms removed manually, and the analytes were positioned in spatial proximity using Coot (51). Docking was carried out on the FlexPepDock server (35, 36) under default parameters for both low- and high-resolution model generation.
Protein sequence alignments.
All sequence alignments were performed using ClustalX2 (37). Sequences for viral phosphoproteins were retrieved from the UniProt database.
Sequences for viral phosphoproteins were retrieved from the UniProt database under the following accession codes: P04880 (Indiana vesicular stomatitis virus), I7CGJ1 (New Jersey vesicular stomatitis virus), B3FRL2 (Alagoas vesicular stomatitis virus), A0A0D3R1C6 (Carajas virus), E3T3B5 (Chandipura virus), B3FRK7 (Cocal virus), Q5K2K6 (Isfahan virus), F8SPF1 (Maraba virus), Q01769 (Piry virus), I1SV83 (Jurona virus), I1SV88 (Perinet virus), K4FFQ0 (Yug Bogdanovac virus), C3VM12 (pike fry virus), and A8VJA2 (spring viremia of carp virus).
Sequences for large proteins from Vesiculovirus and Sprivivirus spp. were retrieved from the UniProt database under the following accession codes: P03523 (Indiana vesicular stomatitis virus), P13615 (New Jersey vesicular stomatitis virus), B3FRL5 (Alagoas vesicular stomatitis virus), A0A0D3R1M8 (Carajas virus), P13179 (Chandipura virus), B3FRL0 (Cocal virus), Q5K2K3 (Isfahan virus), F8SPF5 (Maraba virus), A0A1I9L1X2 (Piry virus), I1SV85 (Jurona virus), I1SV91 (Perinet virus), C3VM15 (pike fry virus), Q91DR9 (spring viremia of carp virus), and K7DVB8 (Yug Bogdanovac virus).
Sequences for large proteins from other members of Mononegavirales were retrieved from UniProt under the following accession codes: O89344 (Hendra virus), Q997F0 (Nipah virus), P11205 (Newcastle disease virus), Q88434 (human parainfluenza virus 5), P06447 (Sendai virus), P12576 (measles virus), P30929 (mumps virus), P35262 (Marburg virus), and Q05318 (Ebola virus).
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grants AI116738 to T.J.G., AI1346931 to C.M.P., and AI146172 to T.O.). The Bruker 850 MHz and 600 MHz instruments used herein were funded by the National Cancer Institute (grant 1P30 CA-13148) and the National Center for Research Resources (grant 1s10 RR022994-01A1). Antibodies were generously provided by Amiya Banerjee.
REFERENCES
- 1.Whelan SPJ, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. [DOI] [PubMed] [Google Scholar]
- 2.Poch O, Blumberg BM, Bougueleret L, Tordo N. 1990. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J Gen Virol 71:1153–1162. doi: 10.1099/0022-1317-71-5-1153. [DOI] [PubMed] [Google Scholar]
- 3.Emerson SU, Wagner RR. 1972. Dissociation and reconstitution of the transcriptase and template activities of vesicular stomatitis B and T virions. J Virol 10:297–309. doi: 10.1128/JVI.10.2.297-309.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emerson SU, Wagner RR. 1973. L protein requirement for in vitro RNA synthesis by vesicular stomatitis virus. J Virol 12:1325–1335. doi: 10.1128/JVI.12.6.1325-1335.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emerson SU, Yu Y. 1975. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J Virol 15:1348–1356. doi: 10.1128/JVI.15.6.1348-1356.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naito S, Ishihama A. 1976. Function and structure of RNA polymerase from vesicular stomatitis virus. J Biol Chem 251:4307–4314. [PubMed] [Google Scholar]
- 7.Ogino T, Banerjee AK. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol Cell 25:85–97. doi: 10.1016/j.molcel.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Rhodes DP, Moyer SA, Banerjee AK. 1974. In vitro synthesis of methylated messenger RNA by the virion-associated RNA polymerase of vesicular stomatitis virus. Cell 3:327–333. doi: 10.1016/0092-8674(74)90046-4. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Wang JT, Whelan S. 2006. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc Natl Acad Sci U S A 103:8493–8498. doi: 10.1073/pnas.0509821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banerjee AK, Rhodes DP. 1973. In vitro synthesis of RNA that contains polyadenylate by virion-associated RNA polymerase of vesicular stomatitis virus. Proc Natl Acad Sci U S A 70:3566–3570. doi: 10.1073/pnas.70.12.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, Whelan S. 2015. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162:314–327. doi: 10.1016/j.cell.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruedas JB, Perrault J. 2014. Putative domain-domain interactions in the vesicular stomatitis virus L polymerase protein appendage region. J Virol 88:14458–14466. doi: 10.1128/JVI.02267-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilman MSA, Liu C, Fung A, Behera I, Jordan P, Rigaux P, Ysebaert N, Tcherniuk S, Sourimant J, Eléouët J-F, Sutto-Ortiz P, Decroly E, Roymans D, Jin Z, McLellan JS. 2019. Structure of the respiratory syncytial virus polymerase complex. Cell 179:193–204. doi: 10.1016/j.cell.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan J, Qian X, Lattmann S, Sahili AE, Yeo T, Jia H, Cressey T, Ludeke B, Noton S, Kalocsay M, Fearns R, Lescar J. 2020. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 577:275–279. doi: 10.1038/s41586-019-1759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masters PS, Banerjee AK. 1988. Complex formation with vesicular stomatitis virus phosphoprotein NS prevents binding of nucleocapsid protein N to nonspecific RNA. J Virol 62:2658–2664. doi: 10.1128/JVI.62.8.2658-2664.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yabukarski F, Leyrat C, Martinez N, Communie G, Ivanov I, Ribeiro EA, Buisson M, Gerard FC, Bourhis J-M, Jensen MR, Bernadó P, Blackledge M, Jamin M. 2016. Ensemble structure of the highly flexible complex formed between vesicular stomatitis virus unassembled nucleoprotein and its phosphoprotein chaperone. J Mol Biol 428:2671–2694. doi: 10.1016/j.jmb.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Ding H, Green TJ, Lu S, Luo M. 2006. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J Virol 80:2808–2814. doi: 10.1128/JVI.80.6.2808-2814.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerard FCA, de Almeida Ribeiro E Jr, Albertini AAV, Gutsche I, Zaccai G, Ruigrok RWH, Jamin M. 2007. Unphosphorylated Rhabdoviridae phosphoproteins form elongated dimers in solution. Biochemistry 46:10328–10338. doi: 10.1021/bi7007799. [DOI] [PubMed] [Google Scholar]
- 19.Emerson SU, Schubert M. 1987. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc Natl Acad Sci U S A 84:5655–5659. doi: 10.1073/pnas.84.16.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green TJ, Macpherson S, Qiu S, Lebowitz J, Wertz GW, Luo M. 2000. Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J Virol 74:9515–9524. doi: 10.1128/jvi.74.20.9515-9524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc Natl Acad Sci U S A 106:11713–11718. doi: 10.1073/pnas.0903228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahmeh AA, Morin B, Schenk AD, Liang B, Heinrich BS, Brusic V, Walz T, Whelan S. 2012. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc Natl Acad Sci U S A 109:14628–14633. doi: 10.1073/pnas.1209147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogino M, Gupta N, Green TJ, Ogino T. 2019. A dual-functional priming-capping loop of rhabdoviral RNA polymerases directs terminal de novo initiation and capping intermediate formation. Nucleic Acids Res 47:299–309. doi: 10.1093/nar/gky1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamaguchi M, Yoshida T, Nishikawa K, Naruse H, Nagai Y. 1983. Transcriptive complex of Newcastle disease virus I. Both L and P proteins are required to constitute an active complex. Virology 128:105–117. doi: 10.1016/0042-6822(83)90322-7. [DOI] [PubMed] [Google Scholar]
- 25.Mühlberger E, Lötferin B, Klenk H-D, Becker S. 1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J Virol 72:8756–8764. doi: 10.1128/JVI.72.11.8756-8764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamb RA, Mahy BWJ, Choppin PW. 1976. The synthesis of Sendai virus polypeptides in infected cells. Virology 69:116–131. doi: 10.1016/0042-6822(76)90199-9. [DOI] [PubMed] [Google Scholar]
- 27.Yu Q, Hardy RW, Wertz GW. 1995. Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J Virol 69:2412–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlin D, Ferron F, Canard B, Longhi S. 2003. Structural disorder and modular organization in Paramyxovirinae N and P J Gen Virol 84:3239–3252. doi: 10.1099/vir.0.19451-0. [DOI] [PubMed] [Google Scholar]
- 29.Leyrat C, Schneider R, Ribeiro EA Jr, Yabukarski F, Yao M, Gérard FCA, Jensen MR, Ruigrok RWH, Blackledge M, Jamin M. 2012. Ensemble structure of the modular and flexible full-length vesicular stomatitis virus phosphoprotein. J Mol Biol 423:182–197. doi: 10.1016/j.jmb.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Leung DW, Borek DM, Luthra P, Binning JM, Anantpadma M, Liu G, Harvey IB, Su Z, Endlich-Frazier A, Pan J, Shabman RS, Chiu W, Davey R, Otwinowski Z, Basler CF, Amarasinghe GK, Louis S. 2015. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep 11:376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster MP, Wuttke DS, Clemens KR, Jahnke W, Radhakrishnan I, Tennant L, Reymond M, Chung J, Wright PE. 1998. Chemical shift as a probe of molecular interfaces: NMR studies of DNA binding by the three amino-terminal zinc finger domains from transcription factor IIIA. J Biomol NMR 12:51–71. doi: 10.1023/a:1008290631575. [DOI] [PubMed] [Google Scholar]
- 32.Wishart DS, Sykes BD. 1994. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR 4:171–180. doi: 10.1007/bf00175245. [DOI] [PubMed] [Google Scholar]
- 33.Lam TT, Lanman JK, Emmett MR, Hendrickson CL, Marshall AG, Prevelige PE. 2002. Mapping of protein:protein contact surfaces by hydrogen/deuterium exchange, followed by on-line high-performance liquid chromatography-electrospray ionization Fourier-transform ion-cyclotron-resonance mass analysis. J Chromatogr A 982:85–95. doi: 10.1016/s0021-9673(02)01357-2. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raveh B, London N, Schueler-Furman O. 2010. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins 78:2029–2040. doi: 10.1002/prot.22716. [DOI] [PubMed] [Google Scholar]
- 36.London N, Raveh B, Cohen E, Fathi G, Schueler-Furman O. 2011. Rosetta FlexPepDock Web server—high resolution modeling of peptide-protein interactions. Nucleic Acids Res 39:W249–W253. doi: 10.1093/nar/gkr431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 38.Buchan DWA, Jones DT. 2019. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res 47:W402–W407. doi: 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chenik M, Schnell M, Conzelmann KK, Blondel D. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930. doi: 10.1128/JVI.72.3.1925-1930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castel G, Chtéoui M, Caignard G, Préhaud C, Méhouas S, Réal E, Jallet C, Jacob Y, Ruigrok RWH, Tordo N. 2009. Peptides that mimic the amino-terminal end of the rabies virus phosphoprotein have antiviral activity. J Virol 83:10808–10820. doi: 10.1128/JVI.00977-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa K, Kobayashi Y, Ito N, Suzuki Y, Okada K, Makino M, Goto H, Takahashi T, Sugiyama M. 2017. Molecular function analysis of rabies virus RNA polymerase L protein by using an L gene-deficient virus. J Virol 91:e00826-17. doi: 10.1128/JVI.00826-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parks GD. 1994. Mapping of a region of the paramyxovirus L protein required for the formation of a stable complex with the viral phosphoprotein P. J Virol 68:4862–4872. doi: 10.1128/JVI.68.8.4862-4872.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandrika R, Horikami SM, Smallwood S, Moyer SA. 1995. Mutations in conserved domain I of the Sendai virus L polymerase protein uncouple transcription and replication. Virology 213:352–363. doi: 10.1006/viro.1995.0008. [DOI] [PubMed] [Google Scholar]
- 44.Holmes DE, Moyer SA. 2002. The phosphoprotein (P) binding site resides in the N terminus of the L polymerase subunit of Sendai virus. J Virol 76:3078–3083. doi: 10.1128/jvi.76.6.3078-3083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker S, Rinne C, Hofsäss U, Klenk HD, Mühlberger E. 1998. Interactions of Marburg virus nucleocapsid proteins. Virology 249:406–417. doi: 10.1006/viro.1998.9328. [DOI] [PubMed] [Google Scholar]
- 46.Trunschke M, Conrad D, Enterlein S, Olejnik J, Brauburger K, Mühlberger E. 2013. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology 441:135–145. doi: 10.1016/j.virol.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. doi: 10.1007/bf00197809. [DOI] [PubMed] [Google Scholar]
- 48.Johnson BA, Blevins RA. 1994. NMR View: a computer program for the visualization and analysis of NMR data. J Biomol NMR 4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 49.Muhandiram DR, Kay LE. 1994. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J Magn Reson B 103:203–216. doi: 10.1006/jmrb.1994.1032. [DOI] [Google Scholar]
- 50.Schumann FH, Riepl H, Maurer T, Gronwald W, Neidig K-P, Kalbitzer HR. 2007. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J Biomol NMR 39:275–289. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]