

The phosphoprotein (P) is an essential and conserved component of all nonsegmented negative-sense RNA viruses, including some major human pathogens (e.g., rabies virus, measles virus, respiratory syncytial virus [RSV], Ebola virus, and Nipah virus). P is a modular protein with intrinsically disordered regions and folded domains that plays specific and similar roles in the replication of the different viruses and, in some cases, hijacks cell components to the advantage of the virus and is involved in immune evasion. All P proteins are multimeric, but the role of this multimerization is still unclear. Here, we demonstrate that the dimerization domain of VSV P is dispensable for the expression of virally encoded proteins and for virus growth in cell culture. This provides new insights into and raises questions about the functioning of the RNA-synthesizing machinery of the nonsegmented negative-sense RNA viruses.
KEYWORDS: phosphoprotein, SAXS, protein domains, two-hybrid screening, vesicular stomatitis virus, viral replication
ABSTRACT
The phosphoprotein (P) of the nonsegmented negative-sense RNA viruses is a multimeric modular protein that is essential for RNA transcription and replication. Despite great variability in length and sequence, the architecture of this protein is conserved among the different viral families, with a long N-terminal intrinsically disordered region comprising a nucleoprotein chaperone module, a central multimerization domain (PMD), connected by a disordered linker to a C-terminal nucleocapsid-binding domain. The P protein of vesicular stomatitis virus (VSV) forms dimers, and here we investigate the importance of its dimerization domain, PMD, for viral gene expression and virus growth. A truncated P protein lacking the central dimerization domain (PΔMD) loses its ability to form dimers both in vitro and in a yeast two-hybrid system but conserves its ability to bind N. In a minireplicon system, the truncated monomeric protein performs almost as well as the full-length dimeric protein, while a recombinant virus harboring the same truncation in the P protein has been rescued and follows replication kinetics similar to those seen with the wild-type virus, showing that the dimerization domain of P is dispensable for viral gene expression and virus replication in cell culture. Because RNA viruses have high mutation rates, it is unlikely that a structured domain such as a VSV dimerization domain would persist in the absence of a function(s), but our work indicates that it is not required for the functioning of the RNA polymerase machinery or for the assembly of new viruses.
IMPORTANCE The phosphoprotein (P) is an essential and conserved component of all nonsegmented negative-sense RNA viruses, including some major human pathogens (e.g., rabies virus, measles virus, respiratory syncytial virus [RSV], Ebola virus, and Nipah virus). P is a modular protein with intrinsically disordered regions and folded domains that plays specific and similar roles in the replication of the different viruses and, in some cases, hijacks cell components to the advantage of the virus and is involved in immune evasion. All P proteins are multimeric, but the role of this multimerization is still unclear. Here, we demonstrate that the dimerization domain of VSV P is dispensable for the expression of virally encoded proteins and for virus growth in cell culture. This provides new insights into and raises questions about the functioning of the RNA-synthesizing machinery of the nonsegmented negative-sense RNA viruses.
INTRODUCTION
A recent literature survey revealed the existence of 41 different species of nonsegmented negative-sense RNA viruses capable of infecting humans (1). Among these viruses, some naturally infect humans, with measles virus and mumps virus causing diseases in children and respiratory syncytial virus, metapneumovirus, and different types of parainfluenza viruses causing respiratory diseases (pneumonia, bronchiolitis, croup). Others, such as rabies virus (RABV), Ebola virus, Marburg virus, Nipah virus, and Hendra virus, are zoonotic viruses that naturally infect animals but accidentally cross the species barrier and infect humans.
All these viruses share similar genome organizations, similar forms of packaging of their genomic RNA into a helical homopolymer of nucleoproteins (N) named nucleocapsids (NC), and similar mechanisms for transcription and replication of their genomes and for assembly of their NC. Today, many nonsegmented negative-sense RNA viruses are classified in eight different families within the order Mononegavirales (MNV), the most prominent ones being the Rhabdoviridae, Paramyxoviridae, Pneumoviridae, and Filoviridae (2).
Vesicular stomatitis virus (VSV) is the archetype of the family Rhabdoviridae, which also includes the rabies virus. VSV has served as a model system to decipher many aspects of the replication mechanisms of the MNVs (3). Its genome comprises five transcription units that are common to all MNVs and are always arranged in the same order along the genomic RNA strand from the 3′ end to the 5′ end, successively encoding the nucleoprotein (N), the phosphoprotein (P), the matrix (M), the surface glycoprotein (G), and the RNA-dependent RNA polymerase (L). Three of these proteins, N, P, and L, are essential for RNA transcription and replication, and together with the genomic RNA, they constitute the infectious core of the viral particle. Processive RNA synthesis by the viral polymerase requires both a ribonucleoprotein template, where the RNA is entirely coated by a homopolymer of N proteins (N-RNA), and the presence of P (4, 5). After the release of the N-RNA/P/L infectious core in the cytoplasm of a host cell, the polymerase first transcribes the genome into messenger RNAs, which are then translated by the cellular translation machinery. In a second stage of the infectious process, the polymerase synthesizes full-length positive copies of the genome, which then serve for the generation of negative-sense genome progeny. During this replication phase, the process of synthesis of genomic or antigenomic RNAs is concomitant with their encapsidation by unassembled nucleoprotein (N0), yielding genomic or antigenomic NC that are competent as templates for new rounds of RNA synthesis by the viral polymerase. This process requires the production of unassembled RNA-free nucleoprotein (N0), which is chaperoned and delivered by P in the form of a soluble N0-P complex (6, 7).
The P protein is thus a multifunctional protein that acts as a cofactor of the polymerase and as a chaperone of N0 (6, 8) and that, for many viruses, also plays a role in innate immune evasion (9). In the process of viral RNA synthesis, the P protein acts as a hub protein, linking both L and N0 to the ribonucleoprotein template (10–12), and the usual view is that multimerization of P is essential for this function, in particular for keeping L attached to the ribonucleoprotein complex when it moves along the template RNA and synthesizes cRNA and when it scans intergenic regions during transcription (13). Recently revealed structures of the L/P polymerase complex of different MNVs, including VSV and RABV but also respiratory syncytial virus and human metapneumovirus, have started to unveil how P interacts with L and manipulates the conformation of L (12, 14–16).
All MNV P proteins are modular proteins with a conserved architecture, despite showing high variability in both sequence and length (17, 18). They have a long, intrinsically disordered N-terminal region (PNTR) that includes an N0 chaperone module (PCM) of about 30 residues at the N-terminal end (7, 10, 19) and a site of interaction with the L polymerase (12, 20–22) (Fig. 1A and B). The C-terminal region (PCTR) is composed of a folded multimeric domain (PMD) (23), a flexible linker, and a folded C-terminal domain (PCTD) that binds to NC (24, 25) (Fig. 1A and B). In Rhabdoviridae, PMD forms dimers both in isolation (23, 26) and in the full-length protein (27, 28), whereas it forms tetramers in Paramyxoviridae and Pneumoviridae (29–31) and forms trimers or tetramers in Filoviridae (32, 33). However, a comparison of the crystal structures of VSV and RABV PMD revealed different folds and even different topologies of the polypeptide chain, showing that the only common feature of these domains is their dimeric state and suggesting the possibility of convergent evolution (23, 26).
FIG 1.

Modular organization and molecular mass of VSV P constructs. (A) Structural model of P. One chain is colored from blue (N-terminal end) to red (C-terminal end), while the other is shown in gray. The know crystal structures are shown as surface structures, and the corresponding Protein Data Bank (PDB) codes are indicated. (B) Schematic architectures of VSV P and of the variants used. PNTR, N-terminal region of P; PCTR, C-terminal region of P; PCM, chaperon module; PMD, multimerization domain; PCTD, C-terminal NC-binding domain. Boxes and lines show structured domains and intrinsically disordered regions, respectively. (C) Multiple-sequence alignment. Multiple-sequence alignments of full-length P sequences were performed with Clustal Omega (67) and rendered using the Espript 3 webserver (68). The excerpt shows the region that includes PMD, whose structure is known (23). The green stars indicate the conserved hydrophobic residues. (D) Structure mapping of conserved residues in PMD. One chain is shown in surface representation and the other in cartoon representation. In both chains, residues are colored according to their conservation, using the alignment shown in panel C. (E) SEC-MALLS-RI analysis. Size exclusion chromatography (SEC) was performed on a Superdex 75 column (GE Healthcare) equilibrated in 20 mM Tris–HCl buffer at pH 7.5 containing 150 mM NaCl. Separations were performed at 20°C with a flow rate of 0.5 ml·min−1. The lines show elution profiles monitored by excess refractive index (left ordinate axis). The crosses show the molecular masses (right ordinate axis) derived from multiangle laser light scattering (MALLS) and refractometry measurements. The theoretical molecular masses calculated for VSV PFL and PΔMD are 30.9 kDa and 23.1 kDa, respectively. The inset shows the Coomassie blue-stained SDS-PAGE gel; the right lane (M) shows the molecular mass markers (kDa).
In a previous study using a reverse genetics approach (34), the level of expression of a reporter gene in a minireplicon assay measured with a RABV P protein deleted from residues 61 to 175, which included PMD, was above 90% of the level measured with the wild-type (w.t.) P protein, showing PMD to be dispensable for the transcriptional activity of the polymerase (34). However, more recently, a binding site for the focal adhesion kinase was found in RABV PMD, unraveling a role for PMD residues in hijacking this cellular component (35). Additionally, transcription and replication of RABV take place within viral factories (36), called Negri bodies (NBs), which are membraneless cytoplasmic inclusions formed by a mechanism of liquid-liquid phase separation, and the presence of PMD was found to be required for the formation of Negri body-like structures in cells coexpressing N and P proteins (37). For VSV, no additional function has been currently assigned to PMD, and the issue arising is thus whether VSV PMD has a role in the viral cycle.
Here, we set out to determine whether the dimerization domain (PMD) of VSV P plays a role in viral replication. We purified a recombinant P protein lacking PMD and used different biophysical methods to characterize its oligomeric state, size, and structural properties. We implemented a yeast two-hybrid experiment to determine the capacity of the truncated form of P to interact with itself, with full-length P, and with N. Finally, we used two reverse genetics approaches, namely, a minireplicon assay and the production and characterization of a recombinant virus, to evaluate the effect of the deletion of PMD on the activity of the polymerase machinery and on the fitness of the mutated virus, respectively.
RESULTS
The dimerization domain of P is conserved among vesiculoviruses.
Multiple-sequence alignment (Fig. 1C) revealed that the dimerization domain (PMD) identified by limited proteolysis (38), whose structure was solved by X-ray crystallography (23), is conserved throughout the genus Vesiculovirus. Conserved residues are mainly hydrophobic residues involved in the dimerization interface (Fig. 1C and D). In accordance with the absence of other known functions, surface residues are less conserved (Fig. 1D).
Recombinant PΔMD is monomeric and highly flexible in solution.
The truncated form of VSV P protein lacking residues 108 to 177 (Fig. 1B), named PΔMD here, was expressed in Escherichia coli BL21 RIL cells in fusion with a C-terminal 6×His tag and purified by affinity chromatography on a Ni2+-nitrilotriacetic acid (Ni2+-NTA) column and by size exclusion chromatography (SEC) on a Superdex S200 column. Analysis by SEC on a Superdex S75 column combined with multiangle laser light scattering (MALLS) and refractometry revealed that the protein eluted in a single peak at 9.3 ml (Fig. 1E). The molecular mass of 23.8 ± 0.5 kDa calculated from the light-scattering intensity was in perfect agreement with the theoretical mass of 23.1 kDa calculated from the amino acid sequence (Fig. 1E). In contrast, full-length protein (PFL) eluted at 8.0 ml. The molecular mass calculated from the light-scattering intensity of 62 ± 2 kDa was in agreement with the theoretical mass of 61.8 kDa calculated for a dimer (Fig. 1E). For both proteins, the molecular mass calculated from light-scattering intensity was constant throughout the chromatographic peak (Fig. 1E), and the molecular weight/molecular number (Mw/Mn) values were 1.00 ± 0.03 and 1.00 ± 0.03, respectively, indicating monodispersal compounds. Both proteins migrated in SDS-PAGE like proteins of higher molecular mass, as previously reported for VSV P (25, 39) and for another fragment of P (residues 1 to 177) that included PNTR and PMD (39) (inset in Fig. 1E). Such aberrant mobility on SDS-PAGE is known for proteins containing regions rich in positive or negative charges (40). VSV PNTR has an isoelectric point of 3.8 and contains 31 negatively charged residues (18 Glu and 13 Asp) for only 8 positively charged residues (3 Arg, 4 Lys, and 1 His) with several densely charged patches such as 58DDSDTESEPEIED70, offering a plausible explanation for the aberrant mobility of P fragments comprising this region.
A PΔMD sample (500 μl) at a concentration of 5.8 mg·ml−1 was analyzed by SEC combined with online detection by small-angle X-ray scattering (SAXS) (Table 1). The protein eluted as a single peak (Fig. 2A), and SAXS curves were recorded at regular intervals along the elution profile for scattering vectors (Q) ranging from 0.05 to 5.00 nm−1. The curve shape and the radius of gyration (Rg) of the particle were constant throughout the elution peak, confirming the absence of concentration dependence and the presence of a single conformational ensemble in solution (Fig. 2A). Panel B of Fig. 2 shows the SAXS curve obtained by averaging data throughout the chromatographic peak. The molecular mass of 27 ± 1 kDa determined from the volume of correlation is in agreement with the theoretical mass (41). The scattered intensity decreased monotonously with increasing Q, as is typically the case for intrinsically disordered proteins (Fig. 2B) (42). The Guinier plot at a low Q value is linear, yielding an average Rg value of 4.1 ± 0.1 nm in the range Q·Rg ≤ 1.3 (Fig. 2C), significantly lower that the Rg of 5.6 ± 0.1 nm measured previously for dimeric PFL (27, 28). This value is, however, higher than that calculated for a globular protein of the same molecular mass and is of the same order as that calculated for an intrinsically disordered protein (4.0 nm) (43), in agreement with the presence of the more extended conformations expected for a protein with a long disordered region. The molecular mass scales with the volume of the particle, and thus the observed increase of Rg results in a 2.5-fold increase in volume, which accounts for the difference in molecular mass between monomeric PΔMD and dimeric PFL. The pair distribution function generated by Fourier transformation of the experimental curve exhibits an asymmetrical shape, with a large Dmax value of 17.5 nm (Fig. 2D), which is similar to the value of 17.5 nm reported for PFL (28). On the other hand, the far-UV circular dichroism (CD) spectrum of PFL (Fig. 3A) exhibited minima at 208 nm and 222 nm, indicative of α-helical content of more than 40%, in agreement with the available structural data (23, 24, 28). The spectra of PΔMD showed a minimum at 208 nm and a prominent shoulder near 222 nm, which indicate that the protein conserved some α-helical structure but has proportionally higher content in disordered regions than PFL (Fig. 3A). The difference spectrum (Fig. 3B) showed that the deleted region had a high level of α-helical structure content, as expected from the crystal structure of PMD (23). The CD spectrum of PΔMD likely reflects the presence of folded PCTD, which is an autonomous folding unit with an α-helix-rich fold (24). If transient helices have been detected in PNTR, in particular at the N terminus in the N0-binding region, this region of the protein is mainly unfolded both in isolation and in the context of full-length P (7, 28) and thus contributes marginally to the helical structure detected by CD spectroscopy.
TABLE 1.
SAXS data collection and scattering-derived parameters for PΔMD
| Parametera | Result |
|---|---|
| Data collection parameters | |
| Instrument | ESRF—BM29 |
| Beam size at sample (μM) | 700 by 700 |
| Wavelength (Å) | 0.9919 |
| q range (Å−1) | 0.25–50 |
| Detector | Pilatus 1M |
| Detector distance (m) | 2.867 |
| Exposure (s per image) | 2 |
| Column | S200 increase 10/300 GL |
| Flow rate (ml·min−1) | 0.5 |
| Injected sample concn (mg·ml−1) | 5.8 |
| Injected volume (μl) | 500 |
| Temperature (K) | 293 |
| Structural parameters | |
| Rg (nm) from P(r) | 4.3 |
| Rg (nm) from Guinier | 4.1 |
| Dmax (nm) | 17.5 |
| Porod volume estimate (nm−3) | 33.5 |
| Mol mass MR (kDa) from vol of correlation | 27.6 |
| Calculated MR (kDa) from sequence | 23.1 |
| Software employed | |
| Primary data reduction | Primus |
| Data processing | Primus |
| Ab initio analysis | EOM and flexible-meccano |
| Validation and averaging | Primus |
| 3D graphics representation | PyMOL |
| EOM χexp | 0.445 |
3D, three-dimensional; EOM, ensemble optimization method.
FIG 2.

Solution structure characterization of VSV PΔMD. (A) SEC-SAXS analysis of PΔMD. The SAXS data were recorded at the BioSAXS ESRF BM29 beamline. The black line and dots show the elution profile monitored by X-ray-scattering intensity (left axis). The red circles show the Rg values determined by using the Guinier approximation for individual curves recorded at different times (right axis). (B) Averaged experimental SAXS data. The upper portion of the panel shows the scattering intensity I(Q) as a function of the scattering vector, Q (nm−1), obtained by averaging the data of frames 940 to 949 in panel A. The red line shows the fit obtained for a subensemble of 10 conformers with the ensemble optimization method (EOM). The lower portion of the panel shows the plot of residuals between experimental data and the fitted curve. The vertical red bars show the experimental error ranges. (C) Guinier plot. The upper panel shows the Guinier plot at small Q values (Q·Rg < 1.3). The red line shows the linear regression. The lower panel shows the plot of residuals between experimental data and linear regression. The red bars show the experimental errors. (D) Pair distance distribution function. The pair distance distribution function P(r) was calculated with the program GNOM for the averaged curve shown in panel B. (E) Ensemble representation of VSV PΔMD. A representative ensemble of the 10 conformers selected by GAJOE (Genetic Algorithm Judging Optimization of Ensembles; 45) is shown in two different orientations. The different conformers were aligned by superimposing their PCTD data (shown as gray-colored surface representation). The flexible N-terminal tails of the individual conformers are shown in different colors. (F) Rg distributions. The black curve shows the Rg distribution calculated for the initial pool of 10,000 conformers. The red curve shows the Rg distribution for the selected subensemble of 10 conformers that collectively fits the experimental SAXS data. Typically, the selection process performed with GAJOE involves 50 successive cycles, and the distribution shown in red corresponds to the distribution of Rg values for the models selected in the best ensemble of each of these successive selections. (G) Dmax distributions. The black curve shows the Dmax distribution calculated for the initial pool of 10,000 conformers, and the red curve shows the Dmax distribution for the selected ensemble of 10 conformers that fits the experimental SAXS data. (H) Electrostatic surface potential of PΔMD. The surface potential was calculated with the APBS program, and the calculated data are color coded on the surface from red (negatively charged residues, −3 kcal/mol) to blue (positively charged residues, +3 kcal/mol).
FIG 3.
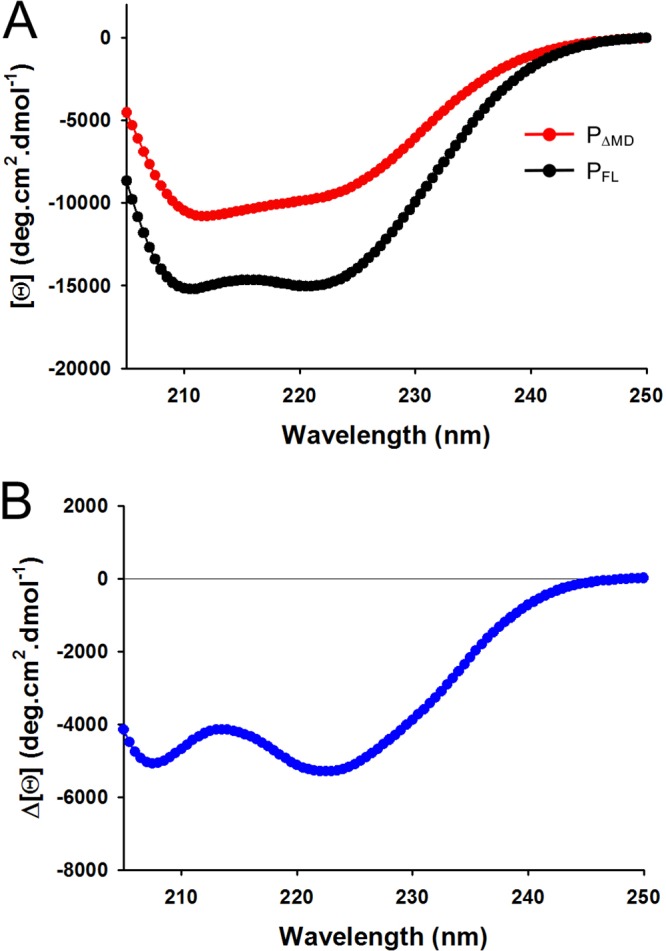
Far-UV circular dichroism spectra. (A) Far-UV CD spectra of VSV PFL and PΔMD. The spectra were recorded at 25°C in 20 mM Tris-HCl (pH 7.5) in a cuvette of 1 mm. The figure shows the molar ellipticity per residue [Θ] as a function of the wavelength (nm). The red line and dots show the spectrum of PΔMD, and the black line and dots show the spectrum of PFL. The presence of minima of similar degrees of molar ellipticity at 210 and 222 nm indicates high α-helix content. (B) Difference spectrum. The curve shows the difference spectrum obtained by subtracting the red spectrum from the black spectrum shown in panel A. Again, the presence of minima of similar degrees of molar ellipticity at 210 and 222 nm indicates that, in agreement with the crystal structure, PMD is rich in α-helix.
Our previous structural characterizations of full-length VSV P (17, 27, 28), in accordance with the SEC-MALLS, SEC-SAXS, and far-UV CD results reported here, suggest a model for PΔMD where the folded PCTD is connected to a long N-terminal disordered, flexible tail. To test this hypothesis, we set out to reproduce the experimental SAXS curve with an ensemble of conformers. Our approach consisted of (i) generating a large ensemble of 10,000 physically possible conformers with the program flexible-meccano (44) by conserving the structure of PCTD and allowing the N-terminal tail to take various conformations and (ii) selecting subensembles with the program GAJOE (Genetic Algorithm Judging Optimization of Ensembles) (45), which collectively reproduced the scattering profile. Subensembles of 10 conformers were found to reproduce the experimental curve with a global χ2 value of 0.45 (Fig. 2B). Independent rounds of selection yielded different subensembles with similar qualities of fit. Panel E of Fig. 2 shows a representative subensemble of 10 conformers that reproduces the experimental SAXS curve and demonstrates the large conformational diversity of the protein. The Rg and Dmax distributions for the initial ensemble had maximum values at 3.5 nm (average Rg value of 3.9 nm) and 11.3 nm (average Dmax value of 12.3 nm), respectively (Fig. 2F and G). The corresponding distributions for the selected ensemble contained higher numbers of extended molecules and lower numbers of compact molecules than the initial ensemble (see arrows in Fig. 2F and G) such as we had seen before for full-length P (28). One possible explanation for this bias toward extended conformations is the high content in negatively charged residues in PNTR, as seen in the electrostatic surface potential calculated at pH 7.0 (Fig. 2H) and the strong electrostatic repulsions operating between these residues.
PΔMD is monomeric and binds to nucleoprotein N in cellula.
To investigate the role of PMD in interactions with itself and with N in a more relevant environment, yeast two-hybrid (Y2H) experiments were performed on agar plates (Fig. 4A). Previous data showed that full-length P had the ability to interact with both P and N (22, 46, 47), and those interactions were confirmed here. Two deletion mutations were used to investigate the importance of PMD in these interactions. First, a fragment encompassing residues 1 to 184 of P (P1–184) (Fig. 1B), which included PNTR and PMD but lacked PCTD, also conserved the ability to interact with both P and N (Fig. 4A). Second, the deletion mutant PΔMD still interacted with N but interacted neither with itself nor with P1–184 nor with PFL, confirming that, even in a cellular environment, the central region of P (amino acids [aa] 108 to 177) is required for forming dimers and that no other region of P appeared to rescue this defect (Fig. 4A).
FIG 4.

Functional assays. (A) Interaction of VSV PΔMD with P and N by yeast two-hybrid assay. L40 yeast cells were cotransformed with the indicated combination of plasmids and were streaked onto plates containing minimal medium lacking tryptophan and leucine for selecting double transformants. The interactions among PFL, P1–184, and PΔMD fused to the DNA-binding domain (BD) of LexA (rows in the figure) and PFL, PΔMD and N fused to the GAL4 activation domain (AD) (columns in the figure) were assessed by induction of the lacZ reporter gene and the appearance of blue colonies in the presence of X-Gal. (B) Minireplicon assay. BSR cells infected with vaccinia-vTF7-3 were cotransfected with four plasmids, coding respectively for PFL or PΔMD, N, L, and a minireplicon construct that served as a template for the VSV polymerase and resulted in the expression of eGFP as described in Materials and Methods. Quantification of fluorescence intensity and of fractions of GFP-positive cells was performed by flow cytometry. Background fluorescence values were obtained in the absence of L protein and were subtracted from all conditions. Error bars represent standard errors of the means of results from two independent experiments.
The self-interaction of P through the dimerization domain (Fig. 1E) and the interactions between N and P are strong interactions, as evidenced by coelution on SEC columns (25, 48, 49). The Y2H assay clearly demonstrated that PΔMD had lost its ability to form such strong self-interactions even if we cannot exclude the possibility that it forms weak interactions (equilibrium dissociation constant [KD] values below the micromolar level) that would not be detected in our assay. Alternatively, weak self-association could be explored by tandem affinity purification (TAP) or by bimolecular fluorescence complementation (BiFC) in vivo or by nuclear magnetic resonance (NMR) spectroscopy in vitro. These results also clearly confirmed that dimerization of P is not required for the interaction with N (10, 22, 25, 48, 49).
The dimerization domain of P is dispensable for viral gene expression.
To determine whether PMD is essential for the function of P in the viral RNA polymerase machine, we used a minireplicon system relying on plasmids under the control of the T7 promoter. BSR cells infected with vaccinia virus V-T7-3, expressing the T7 RNA polymerase, were transfected with (i) a negative-sense replicon construct encoding the enhanced green fluorescent protein (eGFP) flanked by VSV leader and trailer sequences under the control of VSV transcription signals and (ii) three plasmids that express the VSV N, P, and L proteins, allowing the reconstitution of the viral RNA polymerase complex. The negative-sense minigenome RNA is encapsidated by N proteins and is thus transcribed and replicated by the reconstituted viral RNA polymerase complex (L/P). Expression of the eGFP reporter protein occurs only in the presence of a functional VSV RNA synthesis complex.
Deletion of PMD resulted in no significant difference in either the fluorescence intensity (fluorescence intensity of PΔMD = 94% of PFL level) or the fraction of cells expressing eGFP (Fig. 4B), suggesting that the presence of PMD (and therefore the dimerization of P) is dispensable for the functioning of the viral RNA polymerase machine.
The dimerization domain of P is dispensable for virus growth.
To further assess the role of the dimerization domain in viral replication, we used the reverse genetics system to generate a recombinant virus with a P gene with the central dimerization domain truncated. For this, the gene encoding PΔMD was subcloned into full-length VSV cDNA, and a recombinant virus was recovered and passaged in cell culture (50).
We infected cultured BSR cells at a multiplicity of infection (MOI) of 3 either with the wild-type (w.t.) virus or with the mutated virus and monitored over time both expression of full-length P and PΔMD with a polyclonal anti-P antibody using tubulin as a Western blot loading control (Fig. 5A and B) and the production of new infectious viral particles at 5, 6, and 8 h p.i. (Fig. 5C). Given the MOI used in these experiments and the replication rate of VSV, the infection cycle was completed in less than 8 h, resulting in cell lysis. Our results thus report on overall viral fitness with respect to replication and on the capacity of the RNA polymerase machinery to both transcribe and replicate the viral genome without distinguishing between these two activities. For both w.t. and mutant viruses, expression of P was detected from 3 h p.i. The kinetics of the intracellular appearance of both P proteins and the amounts of P proteins relative to the tubulin reference were quite similar during the viral cycle (Fig. 5A and B). For both proteins, a smaller form appeared 3 h p.i. as a well-defined band on the SDS-PAGE, suggesting that it corresponded to a form of discrete length (Fig. 5A and B). Given the offset observed for this additional band between the different protein samples, it might have resulted from N-terminal proteolysis and might correspond to a fragment that included PMD and PCTD in the PFL sample and to a fragment that included PCTD in the PΔMD sample.
FIG 5.

Fitness of recombinant w.t. and mutant viruses in cell culture. (A and B) Time course analysis of BSR cells infected with VSV PFL (A) or VSV PΔMD (B), respectively. BSR cells were infected at an MOI of 3, and cell extracts were analyzed by Western blotting at different times postinfection. Expression of VSV P (green) was monitored by using a specific polyclonal anti-P antibody. Tubulin (red) was detected using a mouse monoclonal anti-α-tubulin antibody. The area density of P and tubulin bands was measured, and the P/tubulin ratios are presented at the bottom of the Western blot. The molecular ladder Precision Plus Protein dual color (Biolabs) was used, and the positions of the bands for two reference proteins are provided on the right side of panel B. (C) Growth kinetics of VSV PFL and VSV PΔMD. BSR cells were infected as described for panel A, and supernatants were harvested at the indicated times postinfection. The virus titers were quantified by plaque assays and represent averages of results from at least three independent experiments.
The comparisons of the viral titers over time clearly showed that the truncation of PMD had no effect on virus fitness (Fig. 5C); the w.t. and mutated viruses replicated at the same rate and yielded almost identical titers at each time point.
DISCUSSION
The structural organization and dynamics of VSV P have been characterized previously (7, 17, 27, 28, 48). Full-length proteins as well as the isolated fragment encompassing residues 107 to 177, PMD, were shown to form dimers (23, 27, 51). We have demonstrated that PMD is required for the dimerization of full-length P and that the removal of this domain generates a soluble, stable, monomeric protein, PΔMD.
PΔMD conserves the ability to bind to unassembled N0 and/or to NC, which was expected, since this protein construct contains both an N-terminal N0-binding region (48) and a C-terminal NC-binding region (24), and since both binding interactions were shown to occur independently of other P parts. More surprisingly, the RNA transcription/replication machine reconstituted with PΔMD was able to express the reporter gene from the VSV minireplicon, and a recombinant virus harboring the gene coding for PΔMD (instead of that coding for PFL) grew at the same rate and reached the same viral titer as the w.t. virus in cultured cells. Previous studies focusing on the role of the dimerization of VSV P concluded that dimerization is essential for transcription and replication (47, 52). However, those works were performed at a time when the structural organization of the protein had not yet been properly established and the localization and structure of the folded dimerization (aa 107 to 177) and NC binding (aa 195 to 265) domains were not yet available (23, 24). On the basis of a limited number of deletion mutants, the dimerization region had been localized between residues 161 and 210 (47). On the basis of our current structural and functional knowledge, we can argue that the deletion of region 161 to 210 (PΔ161–210) had two effects: (i) it abrogated dimerization and (ii) it reduced transcriptional activity significantly, suggesting that the two elements were correlated (47). The described effects are not correlated but rather result from the concomitant truncations of independent functional regions (7, 17, 23, 24, 28). The removal of the C-terminal beta-strand of PMD (aa 161 to 177) likely results in the misfolding of PMD (Fig. 6A) and thus interferes with dimerization, whereas the removal of the N-terminal part of PCTD (aa 195 to 210) likely results in the misfolding of PCTD (Fig. 6A), preventing proper interaction with NC and explaining the effect of this deletion on transcriptional activity. In contrast, the deletion of residues 138 to 161 (PΔ138–160), which removed most of the central helix of PMD (Fig. 6B), had no major effect on transcription (47), in agreement with the observations reported here. In another study, deletions between residues 130 and 201 had moderate effects on replication activity (40% to 80% of w.t. activity), whereas deletions involving residues downstream of residue 201 (aa 201 to 221) that likely affected the structural integrity of PCTD completely abrogated replication activity (52). In this study, we did not attempt to directly compare the transcription and replication activities of PΔMD and PFL, but if there was a reduction in activity associated with the truncation of PMD, it had no impact either on viral gene expression or on viral growth. It is plausible that transcription and replication of the viral genome are not rate limiting in the viral multiplication process and therefore that a slight reduction in the polymerase activity has no effect on viral fitness under these conditions.
FIG 6.

Effects of deletion mutants on the structural integrity of PMD and PCTD. (A) Model of PΔ161–210. The deleted region (aa 161–210) is shown in orange in a model of full-length P and in cartoon representations of PMD and PCTD. (B) Model of PΔ138–160. The deleted region (aa 138 to 160) is shown in yellow in a model of full-length P and in a cartoon representation of PMD.
Widely accepted functions of P are the establishment of a connection between L and the template NC and the maintenance of this connection when L moves along NC during transcription and replication of the genome, thus acting as a processivity factor. Recent data obtained with measles virus suggest that this connection plays an especially important role when L scans intergenic regions during transcription (53). Also widely accepted is the concept that multimerization of P is essential for these functions, as follows. (i) The association of dimeric PFL with the NC can be assumed to occur in two successive binding events. The first binding event, which corresponds to the association of the first P protomer (PCTD) with one site on the NC, is bimolecular, whereas the second binding event, corresponding to the association of the second P protomer (PCTD) with a second site on the NC, is unimolecular. It follows that the dimerization of P probably increases the binding affinity of P for the NC by providing a second intramolecular binding interaction that is structurally equivalent to the first one. It also provides two points of attachment to the template lattice for each P protein and thus probably increases the residence time of P at the surface of the NC, representing an overall expectation for a processivity factor. (ii) The polymerase L/P complex could thus move along the NC by alternative association/dissociation of the two P protomers, always keeping at least one P protomer attached to the template while the polymerase moves forward or pauses at the intergenic region. The results reported here show that multimerization of P is dispensable for viral gene expression and is thus not required for the functional operation of the RNA transcription/replication machine. Additionally, because truncation of P also has no effect on viral growth in mammalian cells, we can also infer that dimeric P is not required in NC packaging, in particle assembly, or in cell infection. However, it is possible that multimerization of P confers an advantage to the virus under conditions other than those used in our studies. Under more-stringent conditions or in a competition situation, multimerization may provide an advantage. The MNV RNA polymerase is error prone, with an error rate of the order of 10−6, close to the error catastrophe rate (54). According to experiments carried out by Haruna and Spiegelman with different viral RNA polymerases, a competition situation should provide an advantage to shorter genomes, which replicate faster than longer ones, and should lead to the streamlining of the genetic information to reduce the generation time (55). Unnecessary features such as a useless dimerization domain should be readily eliminated in such situations.
Numerous studies have shown that all MNV P are multimeric. For RABV, it has been shown that PMD is required for the assembly of viral factories in infected cells (37). For Paramyxoviridae, it has been found that PMD is important for binding to the viral polymerase and for RNA synthesis (56, 57). For VSV, no function other than dimerization of P has so far been found to be associated with PMD. Sequence alignments revealed that the dimerization domain is conserved among the vesiculoviruses. The conservation of PMD thus suggests that this domain provides a selective advantage to the virus and is not eliminated. It is possible that because VSV replicates more quickly than other viruses, the proper assembly of viral factories is less important.
MATERIALS AND METHODS
Plasmid constructions.
The cDNAs encoding full-length P (Indiana serotype), P with a deletion of the 81 C-terminal residues (P1–184), and P with a deletion of the dimerization domain from residue 108 to residue 177 (PΔMD) were fused to the sequence encoding the DNA binding domain (BD) of Lex A and cloned into pLex (Clontech) as described previously (58). The plasmid pGAD-P was obtained by fusing the full-length P gene with the GAL4 activation domain (AD) of pGAD (Clontech). The plasmid pGAD-N was described previously (58).
Plasmids for expressing full-length VSV P (Indiana serotype) in bacteria were previously described (27). For inserting the gene coding for PΔMD into the expression plasmid pET22b(+), a blunt end cloning approach was performed using the following primers: 5′-GTTGTATTTACTAGTGACTATAAGGTCACTCCAGTGATGAACACACAT-3′ and 5′-CACTGGAGTGACCTTATAGTCACTAGTAAATACAACATCCACTTCCTCATCTGCATAGTC-3′. For each construct, a His6 tag and a linker of two amino acids (Glu-Leu) were introduced at the C-terminal extremity. All plasmids were verified by standard dideoxy sequencing.
Two-hybrid system.
Yeast L40 cells were cotransformed with the plasmids encoding wild-type P or N fused to the AD of GAL4 and the P mutants fused to the DB of Lex A. The interaction was assayed by the appearance of blue colonies in the presence of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). An X-Gal mixture containing 0.5% agar, 0.1% SDS, 6% dimethylformamide, and 0.04% X-Gal (5-bromo-4-chloro-3-indolyl-βΔ-galactosidase) was overlaid on freshly transformed cells grown on Trp-Leu dishes, and blue clones were detected after 60 min to 18 h at 30°C.
Production and purification of VSV PFL and VSV PΔMD.
The plasmids for expressing full-length PFL or PΔMD (deleted from residues 108 to 177) were transformed into E. coli BL21(DE3)-RIL cells. Cells were grown in LB medium containing 100 μg·ml−1 ampicillin and 35 μg·ml−1 kanamycin at 37°C until the optical density at 600 nm (OD600) reached 0.6 to 0.8 absorbance units (A.U.). At that point, IPTG (isopropyl β-d-1-thiogalactopyranoside) was added at a final concentration of 0.5 mM and the temperature was shifted to 18°C. After overnight incubation, cells were harvested by centrifugation and suspended in a mixture containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.2 mM TCEP [Tris(2-carboxyethyl)phosphine hydrochloride] (buffer A) supplemented with antiproteases (Complete protease inhibitor cocktail tablets; Roche), and the cells were disrupted by sonication. The extract was centrifuged at 20,000 × g for 45 min at 4°C. The supernatant was loaded onto a Ni2+ resin column that had been preequilibrated with buffer A. The resin was washed with 3 bed volumes of buffer A and then with 3 bed volumes of a mixture containing 20 mM Tris-HCl (pH 7.5), 1 M NaCl, and 10 mM imidazole, and the protein was eluted using buffer A supplemented with 250 mM imidazole. The recombinant protein was concentrated using a Centricon filtration unit (Amicon) (10,000-molecular-weight cutoff [MWCO] for PFL) and a Vivaspin ultrafiltration unit (Sartorius) (5,000 MWCO for PΔMD) and loaded onto a size exclusion chromatography column (Superdex 200 HR 16/600; GE Healthcare) equilibrated with buffer A. Separations were performed at a flow rate of 1 ml·min−1. The protein solution was subjected to a final concentration step. The purity of the protein samples was assessed by SDS-PAGE. Protein concentrations were measured by spectrophotometry using extinction coefficients calculated from the amino acid sequence (59).
Circular dichroism spectroscopy.
A J-810 CD spectropolarimeter (Jasco) equipped with a temperature controller (Peltier system) was used to record the far-UV CD spectra at 20°C, with the data accumulated at least 10 times. VSV PFL or VSV PΔMD was diluted to final concentrations ranging from 10 μM to 40 μM in 20 mM Tris-HCl (pH 7.5) using a cuvette with a path length of 1 mm.
SEC-MALLS-RI.
Size-exclusion chromatography (SEC) was performed with a Superdex 75 HR 10/30 GL column (S75; GE Healthcare) equilibrated with buffer A. Separations were performed at 20°C with a flow rate of 0.5 ml·min−1. Typically, 50 μl of a protein solution at a concentration of 5 to 10 mg·ml−1 was injected. Online multiangle laser light-scattering refractive index (MALLS-RI) detection was performed with a DAWN-EOS II detector (Wyatt Technology Corp., Santa Barbara, CA) using a laser with emissions at 690 nm. Protein concentrations were measured using an Optilab T-rEX detector (Wyatt Technology) and a refractive-index increment, dn/dc, of 0.185 ml g−1. Data were analyzed and weight-averaged molar masses (Mw) were calculated using ASTRA software (Wyatt Technology Corp., Santa Barbara, CA) (60).
SAXS.
(i) Data recording and processing. SAXS data were collected at the BioSAXS beamline BM29 (ESRF, Grenoble, Switzerland) using an in-line analytical SEC S200 column (10/300 GL; GE Healthcare). SAXS measurements were performed every second with a Pilatus 1M detector at a distance of 2.867 m, allowing measurements for momentum transfer (Q) ranging from 0.03 to 4.94 nm, with a wavelength of 0.9919 Å [Q = (4 π sinθ)/λ, where θ = scattering angle and λ = wavelength]. The scattering measured for the buffer before and after each sample measurement was used for background subtraction. Data were analyzed using Primus (ATSAS) (61) and the ScÅtter (Bioisis) program suite (62). The volume of correlation was used to determine the molecular mass (41). The radius of gyration (Rg) was calculated using both the Guinier approximation and the pair distribution function. SAXS data sets were scaled and averaged to produce a single I(Q) curve.
(ii) Model generation and selection. An initial ensemble of all atom conformers suitable for fitting the SAXS data was built by the use of flexible-meccano (44). The known structure was used for PCTD, and residues in the N-terminal disordered tail were stochastically assigned amino acid-specific backbone dihedral angles in agreement with random-coil statistics. Side chains in this disordered region(s) were constructed using the program SCCOMP (63). Scattering curves were calculated with CRYSOL (64), and selections were performed with the program GAJOE (45).
The VSV minireplicon assay.
Plasmids pBS-N, pBS-P, and pBS-L of the VSV (Indiana serotype) were kindly provided by John K. Rose (Yale University). Plasmid pBS-PΔMD was obtained by insertion of the PΔMD gene into the pBS vector at EcoRI and XhoI sites. The previously described (65) VSV replicon plasmid encoding enhanced green fluorescent protein (eGFP) was from Sean Whelan (Harvard Medical School, Boston, MA). The VSV gene expression replicon assay was performed as previously described (65). Briefly, BSR cells were infected with a recombinant vaccinia virus (vTF7-3) producing T7 RNA polymerase. Plasmids pBS-L and pBS-N and wild-type pBS-P or mutant pBS-P (pBS-PΔMD) was transfected into cells 1 h postinfection (p.i.) by using Lipofectamine 2000 (Life Technologies, Grand Island, NY) along with a negative-sense replicon construct encoding eGFP. Expression of eGFP was detected 48 h posttransfection by measuring fluorescence at 488 nm using a BD Accuri C6 flow cytometer.
Rescue of recombinant viruses.
The w.t. or mutant (PΔMD) gene of the VSV Indiana serotype was inserted into original full-length genomic plasmid pVSV-FL(+) (66) using two unique sites of pVSV-FL(+), namely, BstZ17I in the 3′-terminal region of the N gene and Afe I in the 5′-terminal region of the gene, after removal of the corresponding VSV Indiana genes. Recombinant VSV was recovered as described previously (50). As in the replicon assay, BSR cells were infected with vT7F-3. Plasmids pBS-L, pBS-N, pBS-P, and pVSV-FL(+) and w.t. or pVSV-FL(PΔMD) were transfected into cells 1 h postinfection by using Lipofectamine 2000 (Life Technologies, Grand Island, NY) in the presence of 10 μg/ml of AraC (Sigma). Working stocks of recombinant virus were prepared as follows. Monolayers of BSR cells were infected at an MOI of 0.1. After 18 to 24 h, supernatants were collected and cellular debris was removed by low-speed centrifugation; titers were determined by the use of a standard plaque assay performed on BSR cells, and the P gene was sequenced.
Western blotting.
Infected cells were washed in phosphate-buffered saline (PBS) and lysed in cold lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% NP-40, supplemented with a cocktail of protease inhibitors [Roche]) for 30 min on ice. Lysates were clarified by centrifugation at 15,000 × g for 20 min at 4°C. Proteins were separated with 12% SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% skimmed milk–Tris-buffered saline (TBS) and incubated with rabbit polyclonal anti-P antibody (kindly provided by Gail Wertz, Charlottesville, VA, USA) or mouse monoclonal anti-α-tubulin antibody (DM1A from Sigma), followed by incubation with the appropriate Fluor 800- or Fluor 680-conjugated secondary antibody (Cell Signaling). The membrane was scanned using an Odyssey infrared imaging system (Li-Cor, Lincoln, NE) at a wavelength of 700 or 800 nm.
ACKNOWLEDGMENTS
This work was supported by a grant from the French Agence Nationale de la Recherche to D.B. and M.J. (ANR BSV8-2012—NNViPol) and by a grant from the Fond de la Recherche Médicale (FRM; Equipe 2017 DEQ20170336754) and used the platforms of the Grenoble Instruct-ERIC Center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INBS-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). F.C.A.G. was supported by a postdoctoral fellowship from the ANR program (ANR BSV8-2012—NNViPol).
REFERENCES
- 1.Woolhouse MEJ, Brierley L. 20 February 2018, posting date Epidemiological characteristics of human-infective RNA viruses. Sci Data doi: 10.1038/sdata.2018.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maes P, Amarasinghe GK, Ayllón MA, Basler CF, Bavari S, Blasdell K, Briese T, Brown P, Bukreyev A, Balkema-Buschmann A, Buchholz U, Chandran K, Crozier I, de Swart R, Dietzgen R, Dolnik O, Domier L, Drexler J, Dürrwald R, Dundon W, Duprex W, Dye J, Easton A, Fooks A, Formenty P, Fouchier R, Freitas-Astúa J, Ghedin E, Griffiths A, Hewson R, Horie M, Hurwitz J, Hyndman T, Jiāng D, Kobinger G, Kondō H, Kurath G, Kuzmin I, Lamb R, Lee B, Leroy E, Lǐ J, Marzano S, Mühlberger E, Netesov S, Nowotny N, Palacios G, Pályi B, Pawęska J, Payne S, et al. 2019. Taxonomy of the order Mononegavirales: second update 2018. Arch Virol 164:1233–1244. doi: 10.1007/s00705-018-04126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyles DS, Kuzmin IV, Rupprecht CE. 2013. Rhabdoviridae, p 885–922. In Knipe DM, Howley PM, Cohen JL, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Arnheiter H, Davis NL, Wertz G, Schubert M, Lazzarini RA. 1985. Role of the nucleocapsid protein in regulating vesicular stomatitis virus RNA synthesis. Cell 41:259–267. doi: 10.1016/0092-8674(85)90079-0. [DOI] [PubMed] [Google Scholar]
- 5.Morin B, Rahmeh AA, Whelan SP. 2012. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J 31:1320–1329. doi: 10.1038/emboj.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peluso RW, Moyer SA. 1988. Viral proteins required for the in vitro replication of vesicular stomatitis virus defective interfering particle genome RNA. Virology 162:369–376. doi: 10.1016/0042-6822(88)90477-1. [DOI] [PubMed] [Google Scholar]
- 7.Leyrat C, Jensen MR, Ribeiro EA, Gérard FCA, Ruigrok RWH, Blackledge M, Jamin M. 2011. The N0-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient α-helices. Protein Sci 20:542–556. doi: 10.1002/pro.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emerson SU, Yu Y. 1975. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J Virol 15:1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chatterjee S, Basler CF, Amarasinghe GK, Leung DW. 2016. Molecular mechanisms of innate immune inhibition by non-segmented negative-sense RNA viruses. J Mol Biol 428:3467–3482. doi: 10.1016/j.jmb.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen MZ, Ogino T, Banerjee AK. 2007. Interaction of vesicular stomatitis virus P and N proteins: identification of two overlapping domains at the N terminus of P that are involved in N0-P complex formation and encapsidation of viral genome RNA. J Virol 81:13478–13485. doi: 10.1128/JVI.01244-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mellon MG, Emerson SU. 1978. Rebinding of transcriptase components (L and NS proteins) to the nucleocapsid template of vesicular stomatitis virus. J Virol 27:560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenni S, Bloyet L, Diaz-Avalos R, Liang B, Whelan SPJ, Grigorieff N, Harrison SC. 2019. Structure of the vesicular stomatitis virus L protein in complex with its phosphoprotein cofactor. bioRxiv 10.1101/792960. [DOI] [PMC free article] [PubMed]
- 13.Kolakofsky D, Le Mercier P, Iseni F, Garcin D. 2004. Viral RNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 318:463–473. doi: 10.1016/j.virol.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 14.Gilman MSA, Liu C, Fung A, Behera I, Jordan P, Rigaux P, Ysebaert N, Tcherniuk S, Sourimant J, Eléouët J-F, Sutto-Ortiz P, Decroly E, Roymans D, Jin Z, McLellan JS. 2019. Structure of the respiratory syncytial virus polymerase complex. Cell 179:193–204.e14. doi: 10.1016/j.cell.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan J, Qian X, Lattmann S, El Sahili A, Yeo TH, Jia H, Cressey T, Ludeke B, Noton S, Kalocsay M, Fearns R, Lescar J. 7 November 2019, posting date Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature doi: 10.1038/s41586-019-1759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horwitz JA, Jenni S, Harrison SC, Whelan S. 2019. Structure of a rabies virus polymerase complex from electron cryo-microscopy. bioRxiv 10.1101/794073. [DOI] [PMC free article] [PubMed]
- 17.Gerard FCA, Ribeiro EDA, Leyrat C, Ivanov I, Blondel D, Longhi S, Ruigrok RWH, Jamin M. 2009. Modular organization of rabies virus phosphoprotein. J Mol Biol 388:978–996. doi: 10.1016/j.jmb.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 18.Karlin D, Ferron F, Canard B, Longhi S. 2003. Structural disorder and modular organization in paramyxovirinae N and P. J Gen Virol 84:3239–3252. doi: 10.1099/vir.0.19451-0. [DOI] [PubMed] [Google Scholar]
- 19.Mavrakis M, Iseni F, Mazza C, Schoehn G, Ebel C, Gentzel M, Franz T, Ruigrok RW. 2003. Isolation and characterisation of the rabies virus N degrees-P complex produced in insect cells. Virology 305:406–414. doi: 10.1006/viro.2002.1748. [DOI] [PubMed] [Google Scholar]
- 20.Rahmeh AA, Morin B, Schenk AD, Liang B, Heinrich BS, Brusic V, Walz T, Whelan S. 2012. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc Natl Acad Sci U S A 109:14628–14633. doi: 10.1073/pnas.1209147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, Whelan S. 2015. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162:314–327. doi: 10.1016/j.cell.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takacs AM, Das T, Banerjee AK. 1993. Mapping of interacting domains between the nucleocapsid protein and the phosphoprotein of vesicular stomatitis virus by using a two-hybrid system. Proc Natl Acad Sci U S A 90:10375–10379. doi: 10.1073/pnas.90.21.10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding H, Green TJ, Lu S, Luo M. 2006. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J Virol 80:2808–2814. doi: 10.1128/JVI.80.6.2808-2814.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ribeiro EA, Favier A, Gerard FCA, Leyrat C, Brutscher B, Blondel D, Ruigrok RWH, Blackledge M, Jamin M. 2008. Solution structure of the C-terminal nucleoprotein-RNA binding domain of the vesicular stomatitis virus phosphoprotein. J Mol Biol 382:525–538. doi: 10.1016/j.jmb.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 25.Green TJ, Macpherson S, Qiu S, Lebowitz J, Wertz GW, Luo M. 2000. Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J Virol 74:9515–9524. doi: 10.1128/jvi.74.20.9515-9524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanov I, Crépin T, Jamin M, Ruigrok R. 2010. Structure of the dimerization domain of the rabies virus phosphoprotein. J Virol 84:3707–3710. doi: 10.1128/JVI.02557-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerard FCA, Ribeiro EDA, Albertini AAV, Gutsche I, Zaccai G, Ruigrok RWH, Jamin M. 2007. Unphosphorylated Rhabdoviridae phosphoproteins form elongated dimers in solution. Biochemistry 46:10328–10338. doi: 10.1021/bi7007799. [DOI] [PubMed] [Google Scholar]
- 28.Leyrat C, Schneider R, Ribeiro EA, Yabukarski F, Yao M, Gérard FCA, Jensen MR, Ruigrok RWH, Blackledge M, Jamin M. 2012. Ensemble structure of the modular and flexible full-length vesicular stomatitis virus phosphoprotein. J Mol Biol 423:182–197. doi: 10.1016/j.jmb.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Communie G, Crépin T, Maurin D, Jensen MR, Blackledge M, Ruigrok R. 2013. Structure of the tetramerization domain of measles virus phosphoprotein. J Virol 87:7166–7169. doi: 10.1128/JVI.00487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. 2014. Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure 22:136–148. doi: 10.1016/j.str.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox R, Green TJ, Purushotham S, Deivanayagam C, Bedwell GJ, Prevelige PE, Luo M. 2013. Structural and functional characterization of the mumps virus phosphoprotein. J Virol 87:7558–7568. doi: 10.1128/JVI.00653-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruhn JF, Kirchdoerfer RN, Urata SM, Li S, Tickle IJ, Bricogne G, Saphire EO. 2016. Crystal structure of the Marburg virus VP35 oligomerization domain. J Virol 91:e01085-16. doi: 10.1128/JVI.01085-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zinzula L, István N, Massimiliano O, Elisabeth W-S, Andreas B, Wolfgang B. 2 January 2019, posting date Structures of Ebola and Reston virus VP35 oligomerization domains and comparative biophysical characterization in all Ebolavirus species. Structure doi: 10.1016/j.str.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 34.Jacob Y, Real E, Tordo N. 2001. Functional interaction map of lyssavirus phosphoprotein: identification of the minimal transcription domains. J Virol 75:9613–9622. doi: 10.1128/JVI.75.20.9613-9622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fouquet B, Nikolic J, Larrous F, Bourhy H, Wirblich C, Lagaudrière-Gesbert C, Blondel D. 2015. Focal adhesion kinase is involved in rabies virus infection through its interaction with viral phosphoprotein P. J Virol 89:1640–1651. doi: 10.1128/JVI.02602-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nikolic J, Le Bars R, Lama Z, Scrima N, Lagaudrière-Gesbert C, Gaudin Y, Blondel D. 2017. Negri bodies are viral factories with properties of liquid organelles. Nat Commun 8:58. doi: 10.1038/s41467-017-00102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding H, Qiu S, Li S, Symersky J, Green TJ, Luo M. 2003. Expression, purification, crystallization of fragments from the C-terminal region of DFF45/ICAD. Acta Crystallogr D Biol Crystallogr 59:1323–1326. doi: 10.1107/s0907444903010692. [DOI] [PubMed] [Google Scholar]
- 39.Yabukarski F, Leyrat C, Martinez N, Communie G, Ivanov I, Ribeiro EAJ, Buisson M, Gérard FC, Bourhis J-M, Jensen MR, Bernadó P, Blackledge M, Jamin M. 21 April 2016, posting date Ensemble structure of the highly flexible complex formed between vesicular stomatitis virus unassembled nucleoprotein and its phosphoprotein chaperone. J Mol Biol doi: 10.1016/j.jmb.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Armstrong D, Roman A. 1993. The anomalous electrophoretic behavior of the human papillomavirus type 16 E7 protein is due to the high content of acidic amino acid residues. Biochem Biophys Res Commun 192:1380–1387. doi: 10.1006/bbrc.1993.1569. [DOI] [PubMed] [Google Scholar]
- 41.Rambo RP, Tainer JA. 2013. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kikhney AG, Svergun DI. 29 August 2015, posting date A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett doi: 10.1016/j.febslet.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 43.Bernadó P, Blackledge M. 2009. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys J 97:2839–2845. doi: 10.1016/j.bpj.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozenne V, Bauer F, Salmon L, Huang JR, Jensen MR, Segard S, Bernado P, Charavay C, Blackledge M. 2012. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 28:1463–1470. doi: 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- 45.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. 2007. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 46.Emerson SU, Schubert M. 1987. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc Natl Acad Sci U S A 84:5655–5659. doi: 10.1073/pnas.84.16.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen M, Ogino T, Banerjee AK. 2006. Mapping and functional role of the self-association domain of vesicular stomatitis virus phosphoprotein. J Virol 80:9511–9518. doi: 10.1128/JVI.01035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. 2011. Structure of the vesicular stomatitis virus N-P complex. PLoS Pathog 7:e1002248. doi: 10.1371/journal.ppat.1002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor P. Proc Natl Acad Sci U S A 106:11721–11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schnell MJ, Buonocore L, Whitt MA, Rose JK. 1996. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol 70:2318–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding H, Green TJ, Luo M. 2004. Crystallization papers crystallization and preliminary X-ray analysis of a proteinase-K-resistant domain within the phosphoprotein of vesicular stomatitis virus (Indiana). Acta Crystallogr D Biol Crystallogr 60(Pt 11):2087–2090. doi: 10.1107/S0907444904024102. [DOI] [PubMed] [Google Scholar]
- 52.Das SC, Pattnaik AK. 2005. Role of the hypervariable hinge region of phosphoprotein P of vesicular stomatitis virus in viral RNA synthesis and assembly of infectious virus particles. J Virol 79:8101–8112. doi: 10.1128/JVI.79.13.8101-8112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox RM, Krumm SA, Thakkar VD, Sohn M, Plemper RK. 2017. The structurally disordered paramyxovirus nucleocapsid protein tail domain is a regulator of the mRNA transcription gradient. Sci Adv 3:e1602350. doi: 10.1126/sciadv.1602350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biebricher CK, Eigen M. 2005. The error threshold. Virus Res 107:117–127. doi: 10.1016/j.virusres.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 55.Haruna I, Spiegelman S. 1965. Specific template requirements of RNA replicases. Proc Natl Acad Sci U S A 54:579–587. doi: 10.1073/pnas.54.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowman MC, Smallwood S, Moyer SA. 1999. Dissection of individual functions of the Sendai virus phosphoprotein in transcription. J Virol 73:6474–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruhn JF, Hotard AL, Spiropoulou CF, Lo MK, Saphire EO. 2019. A conserved basic patch and central kink in the Nipah virus phosphoprotein multimerization domain are essential for polymerase function. Structure 27:660–669. doi: 10.1016/j.str.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raux H, Flamand A, Blondel D. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol 74:10212–10216. doi: 10.1128/jvi.74.21.10212-10216.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edelhoch H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 60.Wyatt PJ. 1998. Submicrometer particle sizing by multiangle light scattering following fractionation. J Colloid Interface Sci 197:9–20. doi: 10.1006/jcis.1997.5215. [DOI] [PubMed] [Google Scholar]
- 61.Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI. 2017. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 50:1212–1225. doi: 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rambo R. 2010. Bioisis. Elixir bio.tools. http://www.bioisis.net/.
- 63.Eyal E, Najmanovich R, McConkey BJ, Edelman M, Sobolev V. 2004. Importance of solvent accessibility and contact surfaces in modeling side-chain conformations in proteins. J Comput Chem 25:712–724. doi: 10.1002/jcc.10420. [DOI] [PubMed] [Google Scholar]
- 64.Svergun D, Barberato C, Koch MH. 1995. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28:768–773. doi: 10.1107/S0021889895007047. [DOI] [Google Scholar]
- 65.Heinrich BS, Morin B, Rahmeh AA, Whelan S. 2012. Structural properties of the C terminus of vesicular stomatitis virus N protein dictate N-RNA complex assembly, encapsidation, and RNA synthesis. J Virol 86:8720–8729. doi: 10.1128/JVI.00990-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci U S A 92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539–544. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42(Web Server issue):W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]