

Here, we report the discovery, via deep sequencing, of numerous noncoding RNAs (ncRNAs) derived from both EBOV and MARV during infection of both bat and human cell lines. In addition to identifying several novel ncRNAs from both viruses, we identified two EBOV ncRNAs in our sequencing data that were near-matches to computationally predicted viral miRNAs reported in the literature. Using molecular and immunological techniques, we assessed the potential of EBOV ncRNAs to function as viral miRNAs. Importantly, we found little evidence supporting this hypothesis. Our work is significant because it represents the first rigorous assessment of the potential for EBOV to encode viral miRNAs and provides evidence contrary to the existing paradigm regarding the biological role of computationally predicted EBOV ncRNAs. Moreover, our work highlights further avenues of research regarding the nature and function of EBOV ncRNAs.
KEYWORDS: filoviruses, Ebola virus, Marburg virus, microRNA, noncoding RNA, bats, RNA interference, deep sequencing
ABSTRACT
The question as to whether RNA viruses produce bona fide microRNAs (miRNAs) during infection has been the focus of intense research and debate. Recently, several groups using computational prediction methods have independently reported possible miRNA candidates produced by Ebola virus (EBOV). Additionally, efforts to detect these predicted RNA products in samples from infected animals and humans have produced positive results. However, these studies and their conclusions are predicated on the assumption that these RNA products are actually processed through, and function within, the miRNA pathway. In the present study, we performed the first rigorous assessment of the ability of filoviruses to produce miRNA products during infection of both human and bat cells. Using next-generation sequencing, we detected several candidate miRNAs from both EBOV and the closely related Marburg virus (MARV). Focusing our validation efforts on EBOV, we found evidence contrary to the idea that these small RNA products function as miRNAs. The results of our study are important because they highlight the potential pitfalls of relying on computational methods alone for virus miRNA discovery.
IMPORTANCE Here, we report the discovery, via deep sequencing, of numerous noncoding RNAs (ncRNAs) derived from both EBOV and MARV during infection of both bat and human cell lines. In addition to identifying several novel ncRNAs from both viruses, we identified two EBOV ncRNAs in our sequencing data that were near-matches to computationally predicted viral miRNAs reported in the literature. Using molecular and immunological techniques, we assessed the potential of EBOV ncRNAs to function as viral miRNAs. Importantly, we found little evidence supporting this hypothesis. Our work is significant because it represents the first rigorous assessment of the potential for EBOV to encode viral miRNAs and provides evidence contrary to the existing paradigm regarding the biological role of computationally predicted EBOV ncRNAs. Moreover, our work highlights further avenues of research regarding the nature and function of EBOV ncRNAs.
INTRODUCTION
MicroRNAs (miRNAs) are important regulators of posttranscriptional gene regulation in plants and animals. Endogenous miRNAs are transcribed in the nucleus from discrete genomic loci, undergo early processing by the nuclear Microprocessor machinery (Drosha/DGCR8), and are exported into the cytoplasm, where they are processed into mature miRNA duplexes by the cytoplasmic RNase III enzyme Dicer. These duplexes are then loaded into the RNA-induced silencing complex (RISC), where thermodynamic instability favors a single strand, known as the guide strand, for retention in the RISC, while the complementary strand, known as the passenger strand, is discarded from the RISC and is rapidly degraded (1). The RISC is led to cognate mRNA transcripts via sequence complementarity between the target and the guide strand, resulting in either translational repression or degradation of the transcript (reviewed in reference 2). A noncanonical, Dicer-independent pathway responsible for the biogenesis of at least one miRNA (hsa-mir-451) has also been reported (3). In the past two decades, the importance of small RNA (sRNA) pathways, particularly the miRNA pathway, in disease, as well as in the maintenance of cellular homeostasis, has become clear. miRNAs and their dysregulation have been implicated in a multitude of diseases, from cancer (4) to neurodegenerative disease (5). More recently, the role of miRNAs in a number of diseases caused by viral agents and their importance in pathogenesis has been considered. Perhaps unsurprisingly, given the importance of miRNAs in cell homeostasis, numerous viruses have devised methods to exploit or perturb the host miRNA pathway for their own benefit (6–9). For example, hepatitis C virus sequesters hsa-miR-122 to two binding sites at the 5′ end of the viral genome, which aids in genomic stability, enhances translation, and protects the viral RNA from degradation by host factors (10–13), while certain DNA viruses (i.e., herpesviruses) and retroviruses (i.e., bovine leukemia virus) produce their own miRNAs to facilitate infection (14, 15). RNA viruses have historically been thought to be incapable of generating their own miRNAs (16, 17). Nonetheless, several reports describe miRNAs produced by positive-strand viruses (18–21), although in the case of flaviviruses, the validity of these findings has been debated (22, 23). Additionally, small viral noncoding RNAs (vncRNAs) produced during infection by influenza A virus (IAV) have been reported (24–26), and a miRNA-like small RNA from IAV has been functionally validated (27). Thus, despite some ongoing controversy, evidence of the ability of RNA viruses to produce miRNA-like molecules is growing.
The filoviruses are nonsegmented negative-stranded RNA viruses in the order Mononegavirales and include the ebolaviruses Ebola virus (EBOV), Sudan virus (SUDV), Bundibugyo virus (BDBV), Tai Forest virus (TAFV), and Reston virus (RESTV) and the marburgviruses Marburg virus (MARV) and Ravn virus (RAVV). All of these viruses except RESTV are capable of causing severe and often fatal febrile disease in humans and all but EBOV lack approved vaccines or therapeutics. In contrast, these viruses cause only subclinical infection in bats, which are likely a natural reservoir of EBOV (28–31) and are a proven natural reservoir of MARV (32). Within the order Mononegavirales, which also includes highly pathogenic members of the families Rhabdoviridae and Paramyxoviridae, no biologically verified miRNA-producing viruses have been reported. However, several recent reports using in silico analyses have suggested that EBOV is able to encode pre-miRNAs and mature miRNAs (33–36). Moreover, several reports have described the detection of these RNAs in reverse transcription-quantitative PCR (qRT-PCR) and/or sequencing data from biological samples from outbreaks and nonhuman primate (NHP) or rodent infection models (37, 38), as well as in experimentally infected human retinal pigment epithelial cells (39), although importantly, none of these studies address the biogenic origin of these RNAs and their association with the RISC. In addition, EBOV possesses no fewer than three proteins (VP30, VP35, and VP40) that are reported to function as viral suppressors of RNA interference (RNAi), or VSRs, although mechanistic validation of RNAi suppressor activity has been published only for VP30 and VP35 (40–42). Known VSRs are most commonly employed by viruses infecting plants or arthropods, which face a robust RNAi-mediated immune response to infection in those hosts (43, 44). In mammals, the role played by RNAi in antiviral immunity is considerably less well understood and has been a subject of controversy (45–52). It has long been assumed that mammals (and presumably other vertebrates) have largely dispensed with an RNAi-based innate immune response in favor of a potent interferon (IFN)-mediated response (53). Unlike insects, which possess two Dicer enzymes that function largely independently in the miRNA and small interfering RNA (siRNA) pathways, mammals possess only a single Dicer enzyme (54). Biochemical evidence suggests that mammalian Dicer processes long double-stranded RNAs (dsRNAs) (such as those formed during RNA virus replication) into siRNAs in a very inefficient manner, in contrast to the action of invertebrate Dicer-2 (55–57). Additionally, several older studies failed to find strong evidence of a mammalian antiviral RNAi response (45, 58–60). Despite this, more recent studies have described a functional siRNA pathway in mammalian cells following virus infection (61–64).
Accordingly, we sought to determine whether mammalian cells from disparate host species (bats and humans) mount an antiviral RNAi response following infection with filoviruses. Using next-generation sequencing (NGS) technology, we profiled the virus-derived small RNA (vsRNA) populations in one human and two bat cell lines. We identified several high-abundance vncRNA sequences from both EBOV- and MARV-infected cells. We focused our efforts on EBOV vncRNAs in order to determine if they function as virus-derived miRNAs. Our analysis demonstrates that EBOV vncRNAs are processed independently of Dicer involvement and appear to lack interaction with the Argonaute (AGO) family of proteins, the catalytic component of the RISC. Moreover, we found no evidence that EBOV vncRNAs are involved in the suppression of host cell mRNA transcripts, and they do not inhibit virus replication by acting as antiviral siRNAs. Taken together, our data suggest that contrary to the conclusions of previous in silico studies, EBOV small vncRNAs likely do not represent products of host miRNA/siRNA pathway biogenesis and do not play a role in host transcript silencing or the suppression of viral replication. Given that two of the EBOV vncRNAs identified in our sequencing data closely match in silico-predicted RNAs presumed to function as viral miRNAs, our data emphasize the requirement for rigorous biological validation of computationally predicted viral miRNA candidates.
RESULTS
Virus-derived sRNA profiles of filovirus-infected bat and human cell lines.
We profiled the EBOV- and MARV-derived vsRNA populations in a human hepatocarcinoma-derived cell line (HepG2) and two bat cell lines, RO6E/J (derived from Rousettus aegyptiacus fetal tissue) (65) and EpoNi/22.1 (derived from Epomops buettikoferi adult kidney) (66). Both bat cell lines have been shown previously to be susceptible to EBOV and MARV infections, although the growth kinetics of these viruses differ both from each other and from that of Vero E6 cells (66, 67). Cells were infected for 12 or 24 h with either recombinant wild-type EBOV (wt rEBOV), wt rMARV, or mutants exhibiting a point mutation in VP35 that disables the interferon antagonism and dsRNA-binding properties of the protein (68–71); derivatives of these viruses expressing enhanced green fluorescent protein (eGFP) (72, 73) were used for the visualization of infections. Total RNA was harvested at either 12 or 24 h postinfection (hpi). Cell lines differed in their susceptibilities to filovirus infections, as measured by plaque assay titration of cell culture supernatants (Fig. 1A and E), qRT-PCR of intracellular viral genomes and mRNAs (Fig. 1B, C, D, and F), and visualization of the eGFP signal by fluorescence microscopy (data not shown). Both wt EBOV and wt MARV tended to outgrow VP35 mutant viruses at one or both time points, with the exception of wt MARV at 24 hpi in EpoNi/22.1 cells, which had a significantly lower titer than the VP35 mutant virus. The ratio of EBOV VP40 mRNA to vRNA, a measure of transcriptional activity, also differed significantly between human and bat cell lines and between time points in the two bat cell lines (Fig. 1D). After construction of cDNA libraries and deep sequencing, the adapter-trimmed libraries were aligned against the reference genome for each virus. Bioinformatics analyses of sequencing data are summarized in Fig. 2. At 12 hpi, vsRNAs from all cell lines infected with either EBOV (Fig. 3A to C) or MARV (Fig. 3D to F) were predominantly 22 nucleotides (nt) long. At 24 hpi, vsRNAs were more evenly distributed by length in EBOV-infected cells; however, they were almost entirely composed of 22-nt reads in MARV-infected cells. In all libraries from EBOV-infected cells, the percentage of 19- to 32-nt vsRNAs relative to the entire population of 19- to 32-nt sequences in each library increased significantly from 12 to 24 hpi (Fig. 4A), although only EpoNi/22.1 and RO6E/J cells showed increases across time points following infection with MARV (Fig. 4B). Normalization of the abundance of vsRNA to that of viral genomic RNA (vRNA) (Fig. 4C) showed that the relative proportion of vsRNAs remained static from 12 to 24 h in EBOV-infected HepG2 and EpoNi/22.1 cells; however, the relative proportion increased dramatically in RO6E/J cells. It should be noted that in wt rEBOV–eGFP-infected RO6E/J cells, vsRNAs were barely detectable at 12 hpi (<100 reads per biological replicate). vsRNAs derived from MARV tended to decrease in relative abundance in all cell lines, although the decrease was significant only for wt rMARV–eGFP in HepG2 cells, rMARV VP35 R301A–eGFP in EpoNi/22.1 cells, and both viruses in RO6E/J cells. Across all cell lines, vsRNAs were derived predominantly from the positive (antigenomic) strand of the viral RNA for all viruses, with the exception of wt rEBOV–eGFP-infected RO6E/J cells at 12 hpi (Fig. 4D and E). This lack of strong strand bias was likely due to the extremely limited number of reads aligning to the virus at this time point.
FIG 1.

Viral titers and RNA abundances from human and bat cell lines following infection with EBOV or MARV. (A and E) Supernatants from EBOV (A)- or MARV (E)-infected cells used to make sRNA sequencing libraries were assayed by plaque titration. (B, C, and F) Total RNAs from cell monolayers were assayed by qRT-PCR for the presence of vRNA/cRNA and mRNA for EBOV (B and C) or vRNA/cRNA only for MARV (F). RNA quantities are expressed as copy numbers per nanogram of total RNA. (D) For EBOV, the ratio of mRNA to vRNA was calculated for each virus and time point. For all panels, virus–time point combinations were assigned numbers 1 to 4, and cell lines were assigned letters A to C. Within a cell line, comparisons were made between the 12- and 24-h time points for each virus or between viruses within a time point. Comparisons between cell lines were made for each virus at a given time point. The data plotted represent the means of three biological replicates ± standard deviations. Numbers or letters above bars indicate significant differences from other time points (assigned the corresponding number) or cell lines (assigned the corresponding letter) as measured by two-way analysis of variance (α = 0.05), followed by Tukey’s multiple-comparison posttest. For a comprehensive list of comparisons and P values, refer to Data Set S2 in the supplemental material.
FIG 2.
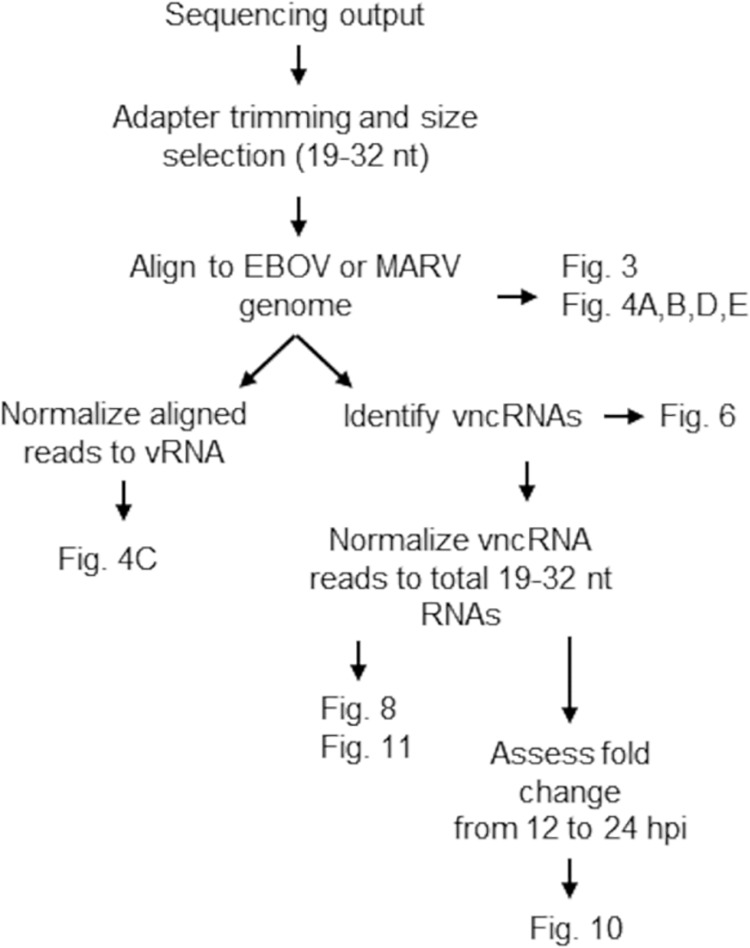
Schematic of bioinformatics analyses of sequencing data.
FIG 3.

Length distribution profiles of EBOV and MARV vsRNAs in human and bat cells. Reads aligning to the EBOV (A to C) or MARV (D to F) genomes were plotted by length as a percentage of the total number of reads aligned to the viral genomes. The data plotted are the means for three biological replicate sequencing libraries at each time point ± standard deviations. (A and D) HepG2 cells; (B and E) EpoNi/22.1 cells; (C and F) RO6E/J cells.
FIG 4.

EBOV and MARV vsRNAs exhibit differential temporal abundance relative to viral genomic RNA, and are predominately derived from positive-strand RNA. vsRNA reads for each virus and cell line are expressed as a percentage of the total 19- to 32-nt RNAs sequenced from each library (A and B) or normalized to vRNA abundance (C). The normalization procedure is described under “Bioinformatics” in Materials and Methods. (A and B) For each virus, time points were assigned numbers, and cell lines were assigned letters. Within a cell line, comparisons were made between the 12- and 24-h time points for each virus or between viruses within a time point. Comparisons between cell lines were made for each virus at a given time point. Numbers or letters above bars indicate significant differences between either time points (assigned the corresponding number) or cell lines (assigned the corresponding letter) as measured by two-way analysis of variance (α = 0.05), followed by Tukey’s multiple-comparison posttest. For a comprehensive list of comparisons and P values, please refer to Data Set S2 in the supplemental material. (C) The data from panels A and B were normalized to viral genomic RNA (see “Bioinformatics” in Materials and Methods), and the ratio of vsRNA reads sequenced at 24 hpi to those at 12 hpi was plotted. Statistical comparisons were made between the 12- and 24-h time points for each virus within each cell line, and significance was determined by the unpaired t test with Welch’s correction. (P values are indicated by letters above bars: A, P = 0.0231; B, P = 0.0161; C, P = 0.0147; D, P = 0.0274; E, P = 0.0221; F, P = 0.0058). (D and E) The percentage of vsRNA reads aligning to either the positive or the negative strand of each virus per time point is plotted for EBOV (D) and MARV (E). For all panels, the means of three biological replicates ± standard deviations are plotted.
The lack of a more symmetric distribution of reads mapping to both the positive and negative strand, particularly of 19- to 23-nt reads, led us to hypothesize that the majority of vsRNA reads were being produced from mRNA rather than dsRNA viral replicative intermediates. In all cell lines, a substantial proportion of EBOV- and MARV-derived vsRNA reads mapped to the 5′ untranslated regions (UTRs) of several viral genes (Fig. 5) (74–77). In EBOV-infected cells, these reads were aligned primarily to the transcription start sites (TSS) of VP40, eGFP or glycoprotein (GP), VP30, VP24, and L (Fig. 6A; Fig. 7A and B). Since the first 59 nt of eGFP and GP 5′ UTRs are identical in the recombinant EBOV we used, it was not possible to differentiate the proportion of TSS-derived reads from each, because the analysis software evenly split the reads between the two sites. Reads aligning to the TSS of nucleoprotein (NP) were only rarely observed, and reads from the TSS of VP35 were almost completely absent among all replicates and time points (Fig. 6C to E). For MARV, TSS-derived reads were aligned primarily to the NP, VP24, and GP genes (Fig. 6B; Fig. 7C and D); VP40- and VP35-derived reads were rarely sequenced; and there was a complete absence of reads aligning to VP30 and L TSS (Fig. 6F to H). The eGFP TSS-derived read from the recombinant MARV we used is unique in that it extends into the open reading frame (ORF) of eGFP, so it was possible to determine that a minority population of reads derived from this TSS was present in most libraries. Using previously predicted secondary structures for EBOV and MARV mRNA 5′ ends, we found very few reads corresponding to what would be predicted to be the 3p arm of the precursor stem-loop from either EBOV or MARV in all cell lines. These data indicate that these RNA species either may have been size selected out of the analysis or may be subject to rapid degradation.
FIG 5.
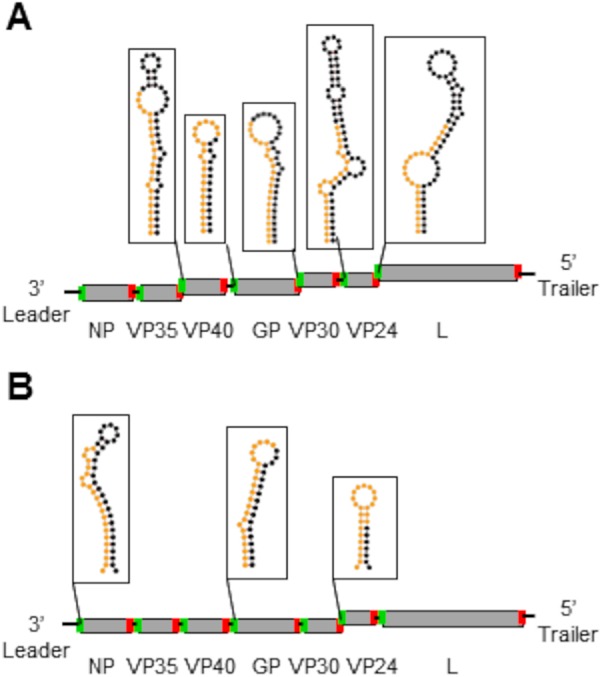
Filovirus vsRNAs are derived primarily from structured elements in the 5′ UTRs of viral mRNAs. Shown are schematic representations of EBOV (A) and MARV (B) genome organization, with predicted secondary structures for the 5′ ends of genes from which the highest proportions of sequenced vsRNAs were derived. The nucleotides making up the 22-nt vncRNA derived from each gene are color-coded orange, and the rest of the stem-loop structure is color-coded black. The structures depicted for EBOV and MARV were computed in references 73 to 75 and were drawn here using Forna (76). Transcription start and stop signals for each gene are depicted in green and red, respectively. Black connections between genes reflect intergenic regions; overlaps between start and stop signals are staggered. For both viruses, the eGFP transgene is omitted from the schematic.
FIG 6.

Filovirus vncRNAs make up a substantial portion of the total vsRNA population. (A) Proportion of combined EBOV VP40, eGFP/GP, VP30, VP24, and L vncRNAs relative to the total vsRNA population in all cell lines. (B) Proportion of combined MARV NP, GP, and VP24 vncRNAs relative to the total vsRNA population in all cell lines. For each virus, time points were assigned numbers, and cell lines were assigned letters. Within a cell line, comparisons were made between the 12- and 24-h time points for each virus or between viruses within a time point. Comparisons between cell lines were made for each virus at a given time point. Numbers or letters above bars indicate significant differences between either time points (assigned the corresponding number) or cell lines (assigned the corresponding letter) as measured by two-way analysis of variance (α = 0.05), followed by Tukey’s multiple-comparison posttest. For a comprehensive list of comparisons and P values, refer to Data Set S2 in the supplemental material. (C to E) Histograms plotting individual proportions of EBOV vncRNAs relative to the total vsRNA population in HepG2 (C), EpoNi/22.1 (D), and RO6E/J (E) cells. (F to H) Histograms plotting individual proportions of MARV vncRNAs relative to the total vsRNA population in HepG2 (F), EpoNi/22.1 (G), and RO6E/J (H) cells. For all panels, the means for three biological replicate sequencing libraries for each group ± standard deviations are plotted.
FIG 7.

vsRNA read frequency plots for rEBOV- and rMARV-infected cells at 12 and 24 hpi. The mean numbers of reads aligning to the virus genome from three biological replicate libraries for each cell line, virus, and time point are plotted. Dashed lines indicate transcription start sites, except for the last line, which represents the border between the 3′ UTR of L and the 5′ trailer. (A) wt rEBOV–eGFP; (B) rEBOV VP35 R312A–eGFP; (C) wt MARV–eGFP; (D) MARV VP35 R301A–eGFP. Note that the y axis denotes the number of reads aligning at each position; a single read may have multiple alignments.
The relative abundances of vncRNAs in EpoNi/22.1 and RO6E/J cells were compared with those in HepG2 cells by calculating reads per million (RPM) for each vncRNA and normalizing the RPM to the vRNA abundance for each library relative to that in HepG2 cells (Fig. 8). In addition, we determined the absolute abundance of the EBOV GP vncRNA from each library using qRT-PCR (Fig. 9). For rEBOV-infected EpoNi/22.1 cells, the abundance of each vncRNA was statistically equal to that in HepG2 cells, with the exception of the GP vncRNA derived from wt rEBOV–eGFP (Fig. 8A to D; Fig. 9A and B). Strikingly, at 24 hpi, for both wt and VP35 mutant rEBOV, RO6E/J cells displayed a substantial increase in the relative abundance of vncRNAs over that in HepG2 cells (Fig. 8A and B; Fig. 9B). A more moderate fold increase in the relative proportion of vncRNAs was observed in rMARV-infected RO6E/J cells at 24 hpi (Fig. 8E to G). We next calculated the fold change in vRNA-normalized RPM values from each virus from 12 to 24 hpi (Fig. 9C and D; Fig. 10). For rEBOV-infected HepG2 and EpoNi/22.1 cells, individual vncRNAs tended to decrease substantially in abundance between the two time points (Fig. 9C and D; Fig. 10A and B). Conversely, the relative abundance of vncRNAs increased by 24 hpi in RO6E/J cells infected with rEBOV VP35 R312A–eGFP, a finding congruent with our observation that EBOV vsRNAs overall increase over time in this cell line, although this analysis was not conducted using RPM normalization for RO6E/J cells infected with wt rEBOV–eGFP, due to the low number of reads from the 12-hpi time point. However, when the vRNA-normalized absolute abundance of the GP vncRNA from wt rEBOV–eGFP-infected cells at 12 hpi was compared to that at 24 hpi, a difference was seen, although it failed to reach significance due to the presence of an outlier identified by Grubbs's test for outliers (Fig. 9D [outlier not removed from figure data]). In contrast, rMARV-derived vncRNAs tended to decrease in relative abundance between 12 and 24 hpi in all cell lines, although these changes were not significant in EpoNi/22.1 cells infected with wt rMARV–eGFP or in HepG2 cells infected with rMARV VP35 R301A–eGFP (Fig. 10C and D). Last, we assessed the degree of agreement between deep-sequencing read normalization using the RPM method and absolute quantitation of the GP vncRNA using qRT-PCR, and we found a significant degree of correlation (Fig. 9E). Taken together, these data suggest that the efficiency of vsRNA production is, at least in part, cell line dependent and that disabling of the dsRNA-binding activity of EBOV VP35 or MARV VP35 does not dramatically affect the abundance of vsRNAs over time. Additionally, due to the impaired replicative ability of EBOV in RO6E/J cells relative to that in HepG2 and EpoNi/22.1 cells, this analysis implicates enhanced vsRNA production as a potential restriction factor for rEBOV infection. When grouped by size, EBOV vncRNAs were found to be primarily 22 or 25 nt long, depending on the gene from which they were derived, across all cell lines (Fig. 11). MARV vncRNAs were almost entirely 22 nt long, with the exception of NP, which included a substantial proportion of larger species (Fig. 11).
FIG 8.

Filovirus vncRNAs exhibit cell line-dependent differential abundances. For each virus and time point, the abundance of each vncRNA relative to vRNA abundance from the EpoNi/22.1 and RO6E/J libraries was compared to the abundance in HepG2 cells. The normalization procedure is described under “Bioinformatics” in Materials and Methods. All statistical comparisons were made against HepG2 libraries by using two-way analysis of variance followed by Dunnett’s multiple-comparison posttest, except in the case of panel A, where Sidak’s multiple-comparison posttest was used. Letters above bars indicate significance; P values are given in the legend for each panel. (A) wt rEBOV–eGFP at 12 hpi. A, P = 0.0067. Note that RO6E/J cells were omitted from this analysis. (B) wt rEBOV–eGFP at 24 hpi. A, P ≤ 0.0001; B, P = 0.0172. (C) rEBOV VP35 R312A–eGFP at 12 hpi. A, P = 0.0448. (D) rEBOV VP35 R312A–eGFP at 24 hpi. A, P ≤ 0.0001; B, P ≤ 0.0001; C, P = 0.0012; D, P ≤ 0.0001. (E) rMARV wt–eGFP at 12 hpi. A, P ≤ 0.0001. (F) rMARV wt–eGFP at 24 hpi. A, P = 0.0245; B, P ≤ 0.0001; C, P = 0.0102; D, P ≤ 0.0001. (G) rMARV VP35 R301A–eGFP at 12 hpi. A, P = 0.0026. (H) rMARV VP35 R301A–eGFP at 24 hpi. A, P = 0.0472; B, P = 0.0341; C, P ≤ 0.0001; D, P ≤ 0.0001; E, P ≤ 0.0001. For all panels, the means for three biological replicate sequencing libraries for each group ± standard deviations are plotted.
FIG 9.

Absolute quantitation of EBOV GP vncRNA by qRT-PCR. Total RNA from the same samples used to generate the wt rEBOV–eGFP and rEBOV VP35 R312A–eGFP small-RNA sequencing libraries was used for absolute quantitation of the EBOV GP vncRNA using qRT-PCR. (A) EBOV GP vncRNA copy number per nanogram of total RNA. For statistical comparison, time points were assigned numbers and cell lines were assigned letters. Within a cell line, comparisons were made between the 12- and 24-h time points for each virus or between viruses within a time point. Comparisons between cell lines were made for each virus at a given time point. Numbers or letters above bars indicate significant differences between either time points (assigned the corresponding number) or cell lines (assigned the corresponding letter) as measured by two-way analysis of variance (α = 0.05), followed by Tukey’s multiple-comparison posttest. For a comprehensive list of comparisons and P values, refer to Data Set S2 in the supplemental material. (B) For each virus and time point, the abundance of the GP vncRNA relative to the vRNA abundance from EpoNi/22.1 and RO6E/J cells was compared to the abundance in HepG2 cells (which was set to 1). The normalization procedure is described under “Bioinformatics” in Materials and Methods. All statistical comparisons were made against HepG2 cells by using one-way analysis of variance followed by Dunnett’s multiple-comparison posttest. Specific P values, indicated by the letters above the bars, are as follows: A, P = 0.0230; B, P = 0.0039; C, P = 0.0133; D, P = 0.0052. (C and D) For either wt rEBOV–eGFP (C) or rEBOV VP35 R312A–eGFP (D), the proportion of GP vncRNA relative to vRNA abundance at 24 hpi was compared with the abundance at 12 hpi (set to 1). The normalization procedure is described under “Bioinformatics” in Materials and Methods. Statistical significance was computed using independent unpaired t tests (α = 0.05) with the Holm-Sidak correction for multiple comparisons. For panels A to D, the data plotted represent the means for three biological replicates ± standard deviations. (E) The degree of agreement between the methods of RPM normalization and absolute quantitation of the GP vncRNA was assessed by plotting the mean RPM of three biological replicates from each sample group against the mean GP vncRNA copy number (per nanogram of total RNA) from each sample group. The coefficient of correlation was calculated by the Pearson method (P = 0.0054), and a best-fit line was drawn using linear regression.
FIG 10.

Filovirus vncRNA abundance changes over time. For each virus, the proportion of each vncRNA relative to vRNA abundance at 24 hpi was compared to the abundance at 12 hpi (set to 1). The normalization procedure is described under “Bioinformatics” in Materials and Methods. All statistical comparisons were made using two-way analysis of variance followed by Sidak’s multiple-comparison posttest. Letters above bars indicate significance as given in the legend to each panel. (A) wt rEBOV–eGFP. A, P ≤ 0.0001; B, P ≤ 0.0001; C, P ≤ 0.0001; D, P ≤ 0.0001; E, P ≤ 0.0001; F, P = 0.0044; G, P = 0.0134; H, P = 0.0022; I, P = 0.0017; J, P = 0.0098. Note that RO6E/J cells were omitted from this analysis. (B) rEBOV VP35 R312A–eGFP. A, P = 0.0175; B, P = 0.0205; C, P = 0.0322; D, P = 0.0065; E, P = 0.0037; F, P=-0.0079; G, P ≤ 0.0001. (C) wt rMARV–eGFP. A, P = 0.0007; B, P ≤ 0.0001; C, P ≤ 0.0001; D, P = 0.0091; E, P = 0.0056; F, P = 0.0182. (D) rMARV VP35 R312A–eGFP. A, P = 0.0002; B, P ≤ 0.0001; C, P ≤ 0.0001; D, P = 0.0054; E, P = 0.0007; F, P = 0.0020. For all panels, the means for three biological replicate sequencing libraries for each group ± standard deviations are plotted.
FIG 11.

Length distribution profiles of EBOV and MARV vncRNAs. vncRNA sequences ranging from 19 to 32 nt, including reads with SNPs, were plotted as percentages of the total number of reads derived from each TSS. The mean for three biological replicate sequencing libraries for each group ± standard deviation is plotted.
We further observed a remarkable abundance of a G→U single nucleotide polymorphism (SNP) at position 1 (the 5′ end) in all EBOV and MARV vncRNA sequences (Table 1). In EBOV-infected HepG2 and EpoNi/22.1 cells, the frequency of this SNP ranged from 15 to 20% of all other derivations of the same sequence (including the “wt” sequence and other, more minor SNPs), particularly in the 22-nt size class (data not shown). In RO6E/J cells, the abundance of the 1U SNP was far lower, on par with the abundance of 1U variants in MARV vncRNAs from all cell lines. Another peculiar finding observed in libraries from all cell lines, and for both wt and VP35 mutant viruses, was that the vast majority of 26-nt reads corresponding to the EBOV eGFP or GP vncRNA contained a SNP at the 3′ end (G→U). In some cases, the proportion of this read to the wt sequence exceeded 100:1 (data not shown). We analyzed the individual base quality scores across randomly selected libraries for each of the vncRNA sequences using BamView, v.1.2.11, and found the maximum expected error rate at these positions to be 0.1 to 1%, far below the frequency observed.
TABLE 1.
TSS-derived ncRNA sequences and their features
| Virus | Sequence (positive sense)a | Gene | hsa-miR homolog | No. of targets predicted in miRDBb |
|---|---|---|---|---|
| EBOV | GATGAAGATTAAGAAAAACCTA | VP40 | hsa-miR-3142 | 197 for hsa-miR-3142, 673 for VP40 |
| GATGAAGATTAAGCCGACAGTG | GP | None | 632 | |
| GATGAAGATTAATGCGGAGGTC | VP24 | hsa-miR-3679-3p | 517 for hsa-miR-3679-3p, 632 for VP24 | |
| GATGAAGATTAAGAAAAAGGTA | VP30 | None | 673 | |
| GAGGAAGATTAAGAAAAACTGC | L | None | 1,150 | |
| MARV | GAAGAATATTAACATTGACATT | NP | None | 766 |
| GAAGAACATTAATTGCTGGATG | GP | hsa-miR-424-5p | 77 for hsa-miR-424-5p, 917 for GP | |
| GAAGAACATTAAGAAAAAGGAT | VP24 | None | 981 |
For each ncRNA, the transcription start signal is italicized, and the putative seed sequence for the ncRNA is underlined. The boldface first base denotes the position of the G → U SNP observed in a proportion of reads for each TSS ncRNA.
Targets were predicted using the custom prediction tool in the MicroRNA Target Prediction Database (miRDB).
We next asked whether EBOV vncRNAs were present in the tissues of animals following infection with EBOV. Total RNA was extracted from archived liver tissue from a rhesus macaque vaccinated against EBOV that survived lethal challenge, and from a control animal that succumbed to infection at 8 days postchallenge (78). These RNAs were subjected to miRNA-specific qRT-PCR. The EBOV GP vncRNA was detectable in the liver from the control macaque; however, no detectable GP vncRNA was present in tissue from the vaccinated animal (Fig. 12A). Melt curve analysis revealed a single amplification product that was nearly identical for RNA extracted from the liver of the control animal and RNA extracted from EpoNi/22.1 cells at 24 h after infection with wt rEBOV–eGFP (Fig. 12B). The control animal exhibited high circulating viremia upon euthanasia, while the vaccinated animal had no detectable viremia when euthanized 28 days postchallenge. This demonstrates that EBOV vncRNAs are produced in vivo during the course of infection.
FIG 12.

miRNA-specific qRT-PCR detection and quantitation of EBOV GP vncRNA in nonhuman primate liver tissue. Total RNA was extracted from archived liver tissues from EBOV-vaccinated and unvaccinated rhesus macaques and was subjected to miRNA-specific qRT-PCR. qRT-PCRs for unknowns were performed in triplicate; qRT-PCRs for serially diluted standards were performed in duplicate. Total RNA extracted from EpoNi/22.1 cells at 24 h after infection with wt rEBOV–eGFP was used as a positive control. A synthetic ssRNA oligonucleotide homologous to GP vncRNA was spiked into total human brain RNA at a concentration of 0.1 ng/μl (total human brain RNA concentration, 10 ng/μl) and was used as a standard for absolute quantitation (undiluted; 10−7). (A) Amplification curves for a wild-type human parainfluenza virus type 3 (HPIV3)-vaccinated control rhesus macaque (moribund animal euthanized 8 days postchallenge; extrapolated GP vncRNA copy number, 3.06 × 103 copies/ng total RNA), an HPIV3/EboGP-vaccinated rhesus macaque (surviving animal euthanized 28 days postchallenge, at study endpoint; extrapolated GP vncRNA copy number, 0 copies/ng total RNA), wt rEBOV–eGFP-infected EpoNi/22.1 cells at 24 hpi (extrapolated GP vncRNA copy number, 2.78 × 104 copies/ng total RNA), and the EBOV GP vncRNA standard (ssRNA GP Oligo) (10−3 dilution; 8.4 × 106 copies/ng total RNA). (B) Melt curve analysis of GP vncRNA qRT-PCR amplicons. For clarity, the melt curve for only a single technical replicate is shown for each sample and is representative of all technical replicates for that sample. The average amplicon Tm values of three technical replicates for each sample (two for the standard) were as follows: for the HPIV3-vaccinated control rhesus macaque, 70.45°C; for the HPIV3/EboGP-vaccinated rhesus macaque, 65.19°C; for EpoNi/22.1 total RNA at 24 h after infection with wt rEBOV–eGFP, 70.50°C; for the EBOV GP vncRNA standard (10−3 dilution; ∼8.4 × 105 copies/ng total RNA), 70.54°C. Rn, ratio of the fluorescence of the reporter dye (SYBR Green) to the fluorescence of the passive ROX dye. ΔRn, normalized Rn value obtained by subtracting the baseline fluorescence of the reporter dye from Rn. -R', negative derivative reporter, obtained as the first negative derivative of the the normalized Rn value. All values are relative fluorescent units reported and plotted by the Applied Biosystems StepOne software.
EBOV vncRNAs are produced independently of the host miRNA machinery.
We next asked whether the biogenesis of the EBOV vncRNAs that we identified was dependent on processing by miRNA pathway-associated endoribonucleases (i.e., Dicer and Drosha). We infected a Dicer knockout cell line (NoDice 4-25) and the Dicer-competent parental line (293T-P) (79) with wt rEBOV–eGFP and probed for the presence of EBOV vncRNAs by miRNA-specific qRT-PCR. At 20 hpi, the production of the VP40 and GP vncRNAs was approximately 4- and 10-fold higher, respectively, in 293T-P cells than in NoDice 4-25 cells (Fig. 13A). The L vncRNA was omitted from this analysis because it could not be reliably detected in all biological and technical replicates in NoDice 4-25 cells. When the viral load was measured by qRT-PCR detection of viral genomic RNA (vRNA) and mRNA, which were approximately 6- and 10-fold higher, respectively, in 293T-P cells than in NoDice 4-25 cells (Fig. 13B), the apparent difference in the relative abundance of viral vncRNAs between cell lines was negligible. In contrast, in NoDice 4-25 cells, production of the endogenous mature miRNAs hsa-miR103a-3p and hsa-let-7a-5p was impaired by approximately 2,000- and 200-fold, respectively, relative to that in the parental cell line. However, the former may be impacted by factors other than the lack of Dicer (79). There was no difference in the relative abundance of the endogenous U6 snRNA, which is not dependent on Dicer for its biogenesis, between cell lines.
FIG 13.

EBOV vncRNAs are produced in a Dicer-independent manner. 293T-P or Dicer-null NoDice 4-25 cells were infected with wt rEBOV at an MOI of 2 PFU/cell and were lysed in TRIzol at 20 hpi. (A) miRNA-specific qRT-PCR analysis of the VP40 and GP vncRNAs as well as the endogenous cellular miRNAs hsa-miR-103a-3p and hsa-let-7a-5p. The Dicer-independent U6 snRNA was assayed as a negative control. The relative fold change was determined using the 2–ΔΔCT method of approximation, with an exogenous spike-in RNA used as the reference. Asterisks indicate significance as determined by two-way analysis of variance (α = 0.05) followed by Sidak’s multiple-comparison posttest (*, P < 0.05; ***, P < 0.001). (B) Tag-based strand-specific qRT-PCR was performed on the same samples to determine the viral RNA copy number. RNA equivalents for vRNA and mRNA were determined using a standard curve obtained by serial dilutions of strand-specific standards. Data are expressed as the fold change from the level for NoDice 4-25 cells. Asterisks indicate a significant difference (P < 0.05) from NoDice 4-25 cells as measured by two-way analysis of variance (α = 0.05) followed by Sidak’s multiple-comparison test (***, P = 0.0005; ****, P < 0.0001). (C) Tag-based strand-specific qRT-PCR was performed on total RNA from Drosha, Ago2, RNase L, DIS3, CPSF3L, and scrambled siRNA-transfected cells, as well as mock-transfected cells, to determine viral RNA copy numbers. Data were first normalized to those for mock-transfected cells and are expressed as the fold change from the value for the scrambled siRNA control. Asterisks indicate significance (P < 0.05) as measured by one-way analysis of variance (α = 0.05; calculated separately for mRNA and vRNA) followed by Dunnett’s multiple-comparison test, with all comparisons made to the scrambled control (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (D) miRNA-specific qRT-PCR of EBOV GP vncRNA. The relative fold change (as measured by the 2–ΔΔCT method of approximation) was normalized first to the value for mock-transfected cells for all samples and then to either mock-normalized viral mRNA fold change values or mock-normalized vRNA fold change values, as indicated. The fold change values of the endogenous hsa-miR-103a-3p miRNA were not normalized to viral titers, since we assumed that its levels were not directly affected by viral RNA levels. Asterisks indicate significance (P < 0.05) as determined by one-way analysis of variance (α = 0.05; calculated separately for mRNA-normalized GP vncRNA, vRNA-normalized GP vncRNA, and hsa-miR-103a-3p) followed by Dunnett’s multiple-comparison test, with all comparisons made to the scrambled control (*, P < 0.05; **, P < 0.01). For panels A to D, the means of three biological replicates ± standard deviations are plotted. (E and F) The degree of correlation between the GP vncRNA and either viral mRNA (E) or vRNA (F) was plotted by taking the mean fold change of the GP vncRNA for each siRNA transfection normalized to the scrambled siRNA and plotting it against the corresponding values of the viral RNA. Deming regression was performed to fit a line through the data points, and the coefficient of correlation (r) was calculated using the Pearson method.
We next used siRNA-mediated knockdown of a panel of host proteins with RNase activity, including RNase L, DIS3, CPSF3L, Drosha, and AGO, to determine their effects, if any, on the production of EBOV vncRNAs. The degree of knockdown was verified by Western blotting or, in the case of AGO2 and Drosha (which we were not able to reliably detect by Western blotting), qRT-PCR detection of miRNA products and mRNA transcripts (Fig. 13D and Fig. 14C, respectively). Cells were infected with wt rEBOV at 48 h posttransfection (hpt) and were harvested at 24 hpi (72 hpt). For all nucleases in the panel except CPSF3L, gene-specific knockdown resulted in levels of EBOV vRNA and mRNA significantly higher than those for the scrambled control (Fig. 13C). For each siRNA-treated biological replicate, including the scrambled siRNA, the fold abundance of the GP vncRNA relative that in to mock-transfected cells was normalized separately from the relative fold abundance of EBOV vRNA and mRNA so as to generate a vncRNA/vRNA or vncRNA/mRNA ratio. For cells in which GP vncRNA was normalized to viral mRNA, only CPSF3L siRNA-transfected cells displayed a GP vncRNA/mRNA ratio significantly lower than that for the scrambled control (Fig. 13D). For cells in which GP vncRNA was normalized to vRNA, knockdown of RNase L showed a significant increase in the relative abundance of the GP vncRNA, likely explained by the importance of RNase L as an antiviral effector. While the fold abundance of the GP vncRNA remained decreased in CPSF3L siRNA-treated cells, the decrease was not significant when GP vncRNA was normalized to vRNA. The relative abundance of hsa-miR103a-3p, which is not dependent on CPSF3L for processing, export, or maturation, remained unchanged. When the fold change for each gene-specific siRNA knockdown was standardized relative to the scrambled control, the ratios of fold change in vncRNA to fold change in mRNA or vRNA were effectively 1:1 (Fig. 13E and F), supporting our hypothesis that viral mRNA is the biogenic substrate for EBOV vncRNAs. In contrast, no such association was observed between the fold change in the abundances of hsa-miR-103a-3p and that of either viral mRNA or vRNA (data not shown). Taken together, these data suggest that EBOV vncRNAs are produced in a Dicer-independent manner but still involve interaction with host nucleases.
FIG 14.

Validation of siRNA knockdown by Western blotting and qRT-PCR. (A and B) (Top) Western blots and band pixel density analysis of RNase L, DIS3, and CPSF3L in siRNA-transfected cells at 48 h (A) and 72 h (B) posttransfection. For both panels, siRNA knockdowns were performed in triplicate (numbers above the blot images). GS, gene-specific siRNA; Scr, scrambled (nonspecific) siRNA. (Bottom) Quantitative data on the levels of proteins in siRNA-transfected cells, derived from Western blotting and normalized to those for mock-transfected cells. nd, not determined. (C) qRT-PCR of Drosha and Ago2 mRNA transcripts. For all panels, bars represent the means for three biological replicates and error bars represent standard deviations.
EBOV vncRNAs are not associated with Argonaute/RISC and do not have an effect on virus replication.
A critical requirement of miRNA functionality is the ability to be loaded and selected for in the RISC. To determine whether EBOV vncRNAs were associated with host Argonaute proteins, we performed RNA-binding protein immunoprecipitation (RIP) on wt rEBOV–eGFP-infected 293T cells at 20 hpi, using antibodies individually targeting human AGO1 to AGO4, as well a pan-AGO antibody. Following the isolation of Ago-associated RNAs, we probed for the presence of four of the EBOV vncRNAs (VP40, GP, VP24, L) using miRNA-specific qRT-PCR. Of six independent experimental repetitions, EBOV GP vncRNA was enriched in two experiments, and EBOV L vncRNA was enriched in one (data not shown). Subsequent experiments failed to enrich for any of the four EBOV vncRNAs profiled (Fig. 15A [only data for EBOV GP vncRNA are shown]). Endogenous miRNAs (hsa-let-7a-5p and/or hsa-miR-103a-3p) were used as positive controls and were highly enriched for AGO2 and pan-AGO precipitations but were not associated with AGO1, AGO3, or AGO4.
FIG 15.

EBOV vncRNAs are not associated with the RISC and do not positively or negatively affect virus replication. (A) qRT-PCR analysis of EBOV vncRNAs following RNA-binding protein immunoprecipitation of AGO1 to -4 with individual AGO antibodies and a pan-AGO antibody. Combined data from two independent experiments are shown; the data for pan-AGO RIP are representative of five separate experiments. SAM-68 was used as an irrelevant RNA-binding protein control. Of the four EBOV vncRNAs profiled, only data for the EBOV GP vncRNA are shown. (B) EBOV titers in supernatants of 293T-P cells at 48 hpi. Prior to infection, cells were transfected with the indicated total concentrations of siRNAs homologous to the VP40 and L vncRNAs. siRNAs were transfected individually or combined (combined concentrations, 12.5 nM each or 25 nM each). Scrambled control siRNA-transfected or mock-transfected cells were used as controls for comparison. Significance was tested by two-way analysis of variance (α = 0.05) followed by Dunnett’s multiple-comparison posttest, and separate independent comparisons were made to the mock-transfected and scrambled siRNA-transfected controls. The means for three biological replicates ± standard deviations are plotted.
To further investigate the potential for EBOV vncRNAs to act as either proviral or antiviral elements, we identified two potential binding sites, one each in the 5′ UTRs of GP and VP30 mRNAs, for the VP40-, GP-, VP30-, and VP24-derived vncRNAs and a single binding site in the ORF of GP and the 5′ UTR of L mRNAs for the L-derived vncRNA; we then transfected 293T-P cells with synthetic siRNA duplexes with the guide strands designed to be homologous to the VP40 and L vncRNAs, followed by infection with wt rEBOV at 4 hpt. At 48 hpi, we observed no significant difference between the virus titers in the supernatants of cells treated with VP40, L, or combined siRNAs and those in the scrambled negative-control siRNA- or mock-transfected cells at either 25 nM or 50 nM concentrations (Fig. 15B). Taken together, these experiments indicate that EBOV vncRNAs are not associated with the host miRNA machinery and do not positively or negatively affect virus replication. Moreover, we demonstrate that EBOV effectively resists suppression by siRNAs complementary to putative binding sites in several mRNAs.
EBOV vncRNAs lack the ability to effectively suppress a reporter transcript with multiple miRNA-binding elements.
We next asked whether EBOV vncRNAs could suppress the expression of a luciferase reporter with miRNA-binding elements (MBEs) in the 3′ UTR of the reporter. We designed three separate synthetic 3′ UTRs containing EBOV GP vncRNA MBEs and cloned them into the pmirGLO dual-luciferase vector (Promega). MBEs were designed to be either perfectly complementary to the EBOV GP vncRNA, partially complementary (containing mismatched nucleotides at positions 11 to 13 of the MBE [“bulged”]), or complementary to only the first 10 nt of the EBOV GP vncRNA. Following transfection with each of the plasmids, 769-P (human renal adenocarcinoma) and EpoNi/22.1 cells were infected with wt rEBOV at a multiplicity of infection (MOI) of 2 PFU/cell. Cell lysates were collected at 24 hpi and were assessed for firefly luciferase (FLuc) expression, with normalization to Renilla luciferase (RLuc). FLuc expression was significantly lower in rEBOV-infected 769-P cells that had been transfected with either the perfect or the 10-nt MBE construct than in empty-vector-transfected cells; however, cells transfected with the perfect MBE construct and subsequently mock infected showed a similar reduction in the FLuc signal (Fig. 16A). FLuc expression was significantly higher in rEBOV-infected EpoNi/22.1 cells transfected with the perfect MBE construct than in empty-vector-transfected cells (Fig. 16B). As a control experiment, we transfected a synthetic siRNA homologous to the EBOV GP vncRNA 4 h after transfection with each of the reporter plasmids. In both 769-P and EpoNi/22.1 cells, transfection of the GP vncRNA reduced the expression of the FLuc reporter relative to that in cells transfected with a scrambled siRNA, except in EpoNi/22.1 cells transfected with the 10-nt MBE construct (Fig. 16C and D). These data suggest that EBOV vncRNAs do not demonstrate silencing activity in either bat or human cells.
FIG 16.

Dual-luciferase reporter assay for EBOV vncRNA function. 769-P (A and C) or EpoNi/22.1 (B and D) cells were transfected with the indicated pmirGLO dual-luciferase reporter constructs and were either infected with wt rEBOV (A and B) or subsequently transfected with a synthetic siRNA homologous to the EBOV GP vncRNA at a final concentration of 50 nM (C and D). For each assay, FLuc values were normalized to the corresponding RLuc values to generate a ratio; the data are expressed as the mean FLuc/RLuc ratio ± standard deviation proportional to that for the empty-vector control construct. All assays were performed in biological quadruplicate (n = 4). One-way analysis of variance followed by a Dunnett multiple-comparison posttest was used to assess statistical significance, and all comparisons were made to the empty-vector-transfected group. Multiplicity-adjusted P values are indicated by letters above the bars. (A) A, P < 0.0001; B, P < 0.0001; C, P < 0.0001; (B) A, P = 0.0065; (C) A, P < 0.0001; B, P = 0.0145; C, P = 0.0064; (D) A, P < 0.0001; B, P = 0.0017; C, P = 0.0018. Data are representative of two independent experiments.
DISCUSSION
Whether or not RNA viruses produce functional miRNA-like molecules has been a matter of controversy for several years, and the question remains contentious (80). While it has been demonstrated that recombinant RNA viruses can be engineered to express exogenous, functional miRNAs (81–84), more-recent reports of viral miRNAs produced during the course of natural infection by flaviviruses have been met with intense criticism (17, 23). Despite the skepticism about the existence of RNA virus-derived miRNAs, several recent in silico studies have predicted numerous potential EBOV-derived pre-miRNA and mature miRNA products (33–36).
In the present study, we utilized deep sequencing to profile the small-RNA populations of bat and human cells following infection with the filoviruses EBOV and MARV, along with interferon antagonism-impaired mutants of each virus. We found that the vast majority of virus-derived reads from all viruses in all cell lines were derived from the TSS of several viral genes, namely, VP40, GP, VP30, VP24, and L in EBOV and NP, GP, and VP24 in MARV and that several of these vncRNAs have human miRNA homologs (Fig. 6A and B; Table 1). In EBOV-infected cells, these RNA products were primarily 22 or 25 nt long, depending on the gene, while in MARV-infected cells, they were almost entirely composed of 22-nt reads, congruent with the typical size of cellular miRNAs. The range of sizes for each vncRNA product was highly consistent between replicates and cell lines, strongly indicating that filovirus vncRNAs are not simply products of stochastic processes. In addition, a strong positive-strand bias was observed for all virus-derived reads, suggesting that mRNAs, not dsRNA replicative intermediates, are the substrate utilized to produce these products. Consequently, we also interpreted this unipolar strand bias as evidence against an siRNA-mediated response to filovirus infection. The effectiveness of a mammalian antiviral siRNA response is a matter of considerable debate; however, several groups have described a functional pathway. Most recently, Qiu et al. reported that the apparent lack of a noticeable siRNA-mediated immune response in mammalian cells reported in older studies was due largely to potent VSR activity masking the siRNA signature (64). Although this must be considered a possible explanation for our observations given that EBOV possesses three proteins with reported VSR activity (40–42), it should be noted that in all of the studies demonstrating a mammalian siRNA response to virus infection, the viruses utilized have dsRNA replicative intermediates that are unencapsidated and thus likely to be accessible to host RNAi machinery, i.e., Dicer. With regard to filoviruses, the positive-sense replicative strand (cRNA) is encapsidated during replication (85, 86), which likely shields it from nucleases. In contrast, viral mRNAs are not encapsidated and are therefore more vulnerable to nucleolytic digestion.
An unexpectedly high proportion of vncRNA sequences from EBOV and, to a lesser degree, MARV exhibited a G→U SNP at the first position of the vncRNA. The mechanism by which these SNPs are produced and their biological relevance, if any, is unclear. Given the high degree of sequence conservation of filovirus transcriptional control elements and their critical importance to the efficient transcription of viral mRNAs, it is unlikely that the SNP is incorporated into the genomic template. A more plausible explanation is that a proportion of mRNAs are transcribed with a misincorporated base at position 1, possibly through a process such as template misalignment (87). We attempted to distinguish between the wt and 1U SNP variants by qRT-PCR; however, primers designed for the SNP variant detected the wt vncRNA, and vice versa, at roughly equal efficiencies, as determined by qRT-PCR of human brain total RNA spiked with synthetic miRNA mimic oligonucleotides homologous to the wt GP-and 1U SNP GP vncRNAs. It was determined by Qiagen/Exiqon technical support that this limitation in the discrimination of targets was not likely to be overcome by repositioning of the locked nucleic acid in the SNP-specific primers. The overwhelming proportion, relative to the wt, of a G-to-U SNP at position 26 of only the 26-nt variant of the EBOV eGFP/GP vncRNA was also surprising. While it is tempting to conclude that this was the result of PCR-biased overrepresentation, this SNP was present in all EBOV libraries from all three cell lines and was always the majority sequence, casting doubt on this conclusion. Instead, its presence may be better explained as a result of misincorporation during transcription.
A defining characteristic of miRNAs and siRNAs is their incorporation into the RISC, in the absence of which their silencing effect is not known to occur. A recent report by Duy et al. validated the presence of several of the previously computationally predicted EBOV “miRNAs” from nonhuman primate, mouse, and human serum samples (38). However, neither in that study nor in any of the preceding studies was the direct association with the RISC investigated, although a single report did imply that production of two of the predicted mature miRNAs is dependent on Dicer-mediated digestion of the predicted pre-miRNA (33). With regard to this, we investigated the involvement of the host miRNA machinery in the production of the EBOV vncRNAs discovered in our study. Using a stable Dicer knockout cell line, we were able to determine that the production of mature EBOV vncRNAs occurs in a Dicer-independent manner (Fig. 13A and B). Dicer-independent, AGO2-dependent processing of at least one endogenous pre-miRNA is known to occur (3, 88), and this has similarly been reported for a miRNA-like small RNA produced by IAV subtype H5N1 (27). However, in our study, siRNA-mediated knockdown of AGO2 had no deleterious effect on the abundance of EBOV vncRNAs relative to that with the scrambled siRNA control. This was also true of siRNA-mediated knockdowns of RNase L and DIS3; however, knockdown of Drosha and CPSF3L resulted in moderate suppression of the GP vncRNA, although this result was significant only for CPSF3L (Fig. 13D). RNase L has been reported to be involved in the noncanonical processing of viral RNAs into siRNA-like molecules (89). DIS3, the catalytic component of the exosome complex, a 3′–5′ exoRNase, was selected because we reasoned that 3′–5′ nucleolytic digestion of filovirus mRNAs may result in the production of pre-miRNA-like molecules if the exosome complex stalls upon reaching the stem-loop structure in the 5′ UTR. While DIS3 is primarily nuclear, several reports indicate its presence, albeit in smaller amounts, in the cytoplasm (90). Both Drosha and CPSF3L are reported to process viral RNAs into small RNA molecules, albeit through mechanisms distinct from one another. In the case of Drosha, translocation from the nucleus occurs upon infection by a diverse group of RNA viruses, including vesicular stomatitis virus (VSV), a nonsegmented negative-strand virus (nsNSV) (91). Additionally, an exclusively cytoplasmic isoform of Drosha has been described (92). The cleavage specificity of Drosha, which uses a “molecular ruler” to count approximately 11 bp up the dsRNA stem-loop from the basal dsRNA–single-stranded RNA (ssRNA) junction and approximately 22 bp down from the apical junction of the terminal loop (2, 93, 94), would not appear to produce pre-miRNAs consistent with the vncRNAs that we sequenced, assuming that the predicted secondary structures for the 5′ ends of EBOV and MARV mRNAs are at least mostly accurate. Intriguingly, CPSF3L, a component of the Integrator complex, has been reported to produce snRNAs and miRNAs via cleavage of stem-loop structures in transcribed precursor molecules produced by Herpesvirus saimiri (95, 96). Although in this example processing occurs in the nucleus and is dependent on Dicer for miRNA maturation, CPSF3L is also found diffusely throughout the cytoplasm (97), and it is possible that it similarly recognizes the 5′ UTR stem-loop structures in filovirus mRNAs as suitable substrates. While our findings do not conclusively implicate any specific nuclease as being responsible for the production of filovirus vncRNAs, they do exclude Dicer dependency for vncRNA biogenesis. Furthermore, the nearly perfectly linear relationship that we observed between fold changes in viral mRNA and vncRNA across the knockdown panel adds strength to the conclusion that viral mRNA is the biogenic substrate for filovirus vncRNAs.
We next investigated the association of EBOV vncRNAs with host Argonaute proteins, the catalytic component of the RISC. Of the four EBOV vncRNAs we profiled by qRT-PCR of Ago immunoprecipitations, we were able to detect modest enrichment of GP vncRNAs in two independent experiments and of L vncRNAs in one. However, in four subsequent independent repetitions of this experiment, no enrichment of any of the four EBOV vncRNAs profiled by qRT-PCR was observed, despite our observation of robust enrichment for endogenous host miRNAs. The reasons for the disparity between the initial experiments and subsequent attempts are not clear. All immunoprecipitation experiments utilized the same pan-AGO antibody, and infections and immunoprecipitation protocols were performed identically across all experiments. However, given that only the GP vncRNA was detected in more than one experiment, and two vncRNAs were not detected at all, we must conclude that the association of EBOV vncRNAs with the RISC is most likely to be transient and nonspecific in nature, and that therefore, they are not expected to be functional as silencing RNAs in any biologically meaningful context. This is of particular importance because while none of the computationally predicted mature miRNA sequences in previous studies were exact matches for the vncRNAs identified in our sequencing data, two were strikingly similar. EBV-miR-T2-5p, predicted by Teng et al. (34), was shifted by a single nucleotide downstream from the sequenced VP40 vncRNA, while ZEBOV-miR-1-5p, predicted by Liu et al. (35), was shifted by 7 bases relative to the sequenced VP24 vncRNA. Considering that the authors of these and subsequent studies have assumed that these predicted RNAs actually function as miRNAs (and thus associate with the host miRNA machinery), our finding of a lack of association of highly similar sequences produced during experimental infection with EBOV casts doubt on those conclusions. Additionally, we failed to find an appreciable abundance of any of the other predicted EBOV “miRNAs” from previous studies, and most were completely absent from our sequencing data sets. As mentioned above, Duy et al. were able to detect most of the predicted EBOV small RNAs in biological samples from infected humans, mice, and NHPs (38), and we too were able to detect EBOV GP vncRNA in the liver of an EBOV-infected rhesus macaque (attempts to detect additional EBOV vncRNAs that we identified were not made), indicating that these RNAs are produced during the course of natural infection. Our results therefore provide a useful cautionary note with regard to the use of computational modeling for the prediction of viral miRNAs. A common inference of sRNA deep-sequencing data has been that highly abundant mature miRNA sequences are likely to be associated with the RISC, because the lack of a 5′ cap or poly(A) tail would make them susceptible to nucleolytic degradation. However, it has been reported that only a relatively small proportion of the total cellular miRNA pool is actually associated with the RISC (98, 99). Moreover, a subsequent publication provided evidence that the association of mature miRNA with the RISC was predicated on several factors, including the abundance of highly complementary mRNA targets (100). However, despite these potential alternative explanations for a lack of association with AGO, and given the clear independence of their biogenesis from Dicer, we conclude that there is insufficient evidence that EBOV vncRNAs represent true miRNAs.
In addition to a lack of association with canonical host miRNA machinery, the EBOV vncRNAs identified in our studies failed to show any measurable effect on either virus replication or the silencing of a reporter transcript. In HepG2 and EpoNi/22.1 cells, the estimated abundances of the GP vncRNAs ranged from several hundred to more than a thousand copies per cell, respectively, as assessed by qRT-PCR, assuming a total RNA content of 10 pg/cell (Fig. 9A). Despite this, infection of both human and bat cells with EBOV failed to suppress the expression of a luciferase reporter with EBOV GP vncRNA MBEs in the 3′ UTR of the reporter mRNA. This stands in contrast to our results with an siRNA homologous to the GP vncRNA. Effective silencing of mRNAs, especially in instances of partial complementarity to their mRNA target, is in large part dependent on stoichiometry; i.e., high levels of a given miRNA are required in order to exert an effect on levels of an imperfectly complementary target (101). However, even a reporter with MBEs perfectly complementary to the EBOV GP vncRNA was not significantly silenced during infection with EBOV. In RO6E/J cells, it appears that the enhanced production of vncRNAs relative to vRNA or mRNA may in fact function as a restriction factor, given the impaired ability of EBOV to replicate in these cells relative to that in EpoNi/22.1 and HepG2 cells. Importantly, Rousettus aegyptiacus bats have been described as refractory to EBOV infection (102, 103). This is perhaps not surprising, since enhanced digestion of mRNA substrates to produce vncRNAs would likely impose a replicative burden on the virus. This striking difference in relative abundance was not observed in MARV-infected cells; thus, their enhanced production in a species of bat known to be refractory to EBOV infection may in fact act as a restriction factor to productive infection.
Given their apparent lack of function as viral miRNAs (summarized in Table 2), what might the origin and biological function of EBOV vncRNAs be? As with other nsNSVs, 5′ capping of filovirus mRNAs is coupled with transcription. For the prototypical nsNSV VSV, nascent mRNAs are capped after the addition of the 31st nucleotide (104). Most small-RNA sequencing methods, including the method we used, require 5′-monophosphate and 3′-hydroxl moieties for adapter ligation to occur; therefore, if the same were true for EBOV as for VSV, one could hypothesize that the vncRNAs identified in our sequencing data may represent precapped mRNAs. However, the consistency in proportional abundance between biological replicates, and even between wild-type and mutant viruses in our sequencing and qRT-PCR studies, implies that these are not simply randomly captured immature mRNAs. Proper addition of the 5′ cap in part serves as a quality control step; in the absence of capping, elongation of the mRNA halts, resulting in an aborted transcript (105). However, premature termination of transcription does not occur immediately, and aborted VSV mRNA transcripts range in length from 40 to 500 nt (105–107), considerably larger than the RNAs sequenced in our study. Premature termination of transcripts in respiratory syncytial virus (RSV) similarly starts after the addition of nt 40 (108, 109). Assuming that EBOV mRNAs exhibit a minimal capping length and a gradient of premature transcription termination similar to those of other nsNSVs, the RNAs sequenced in our study likely do not represent products of abortive transcription. Indeed, the relative lack of abundance of 40- to 50-nt vncRNAs from either EBOV or MARV (data not shown) implies that if such products exist, they were size selected out during sequencing library preparation, and our analysis clearly shows a predominance of 22- to 25-nt RNAs for most of the TSS-derived vncRNAs that we identified, with relative abundance declining with increased length.
TABLE 2.
Summary of EBOV vncRNA findings
| Characteristic | Presence or absencea
in: |
|
|---|---|---|
| Genuine miRNAs | EBOV ncRNAs | |
| Biogenesis | ||
| Drosha processing | ✓ | X |
| Dicer processing | ✓ | X |
| Function | ||
| RISC loading | ✓ | X |
| Transcript silencing | ✓ | X |
| Translational repression | ✓ | X |
| Biochemistry | ||
| 5′-Phosphate | ✓ | ✓ |
| 3′-OH | ✓ | ✓ |
| 22-nt length | ✓ | Variable |
✓, presence; X, absence.
Another consideration is that EBOV TSS-derived ncRNAs function similarly to the short leader RNAs (leRNAs) produced by other NSVs (24, 110, 111). IAV is reported to produce small RNA products from the 5′ ends of cRNAs. However, instead of functioning to posttranscriptionally regulate host or viral gene expression, these small RNAs regulate the switch from transcription of viral mRNAs to replication of the IAV genome by acting as enhancers of IAV polymerase activity (24, 25). Additionally, these viral small RNAs are not reported to associate with the RISC. Although EBOV and MARV are thought to regulate the switch between transcription and replication through the phosphorylation state of VP30 (112, 113), the exact mechanism of this process is not fully understood, and this does not preclude reliance on additional means of regulation. Therefore, it is possible that EBOV utilizes TSS-derived ncRNAs for a similar purpose.
Last, we must consider the possibility that EBOV vncRNAs are by-products of the secondary structure in the 5′ UTRs of viral mRNAs without conferring any selective advantage of their own, i.e., that they are spandrels (114). Although only the stem-loop in the 5′ UTR of EBOV NP is currently known to be critical for VP30-dependent transcriptional initiation (86, 115), the stable 5′ UTR stem-loop structures predicted for all mammal-hosted filovirus mRNAs (which are formed in part by the TSS of each mRNA) suggest that their maintenance is under some selective constraint. According to this hypothesis, the production of vncRNAs through a process such as nucleolytic digestion would be little more than a cost to filoviruses for maintaining the topology of the 5′ UTR. Any biological activity would therefore be incidental to their primary function, likely as an element of transcriptional regulation. If these vncRNA products confer no selective advantage, it would make sense for the virus to develop strategies to hinder this process; i.e., if the fitness cost of modifying the secondary structure in the 5′ UTR is insurmountable, the evolution of mechanisms to suppress the degradation of vncRNAs may be favored. In support of this notion, while we observed that the absolute amount of TSS-derived vncRNAs increased from 12 to 24 hpi, the proportion of vncRNAs relative to the viral burden decreased. Conversely, the stem-loops themselves may have undergone selection to make them less amenable to nucleolytic digestion while still maintaining their primary function.
It should be noted that because our validation was focused only on EBOV vncRNAs, we cannot exclude the possibility that MARV vncRNAs are associated with the miRNA pathway. The marburgviruses are phylogenetically basal to the ebolaviruses and differ from them in several key aspects of their biology, such as the lack of an editing site in GP. It is therefore possible that MARV retains a mechanism of RNAi that was lost in EBOV. Therefore, prudence dictates that the biological origin and function, if any, of the MARV TSS-derived vncRNAs identified in our studies be assessed independently of the conclusions drawn for EBOV.
Here, we present, to our knowledge, the first rigorous biological assessment of the capacity for EBOV to produce viral miRNA-like molecules. Importantly, despite predictions of such molecules in the literature, we found little evidence to suggest that EBOV vncRNAs, whether previously predicted or from our own sequencing data, function as miRNAs or are processed through the canonical miRNA pathway. Importantly, in addition to describing several novel vncRNAs produced by both EBOV and MARV, this work validated the existence of two previously predicted EBOV miRNA-like molecules in the context of live virus infection, while challenging assumptions that they play a role in host mRNA silencing. Taken together, these studies suggest that EBOV vncRNAs likely serve biological functions unrelated to small-RNA-mediated gene silencing or, conversely, exist as nonfunctional spandrels. Avenues of further investigation should include research into the biological activity of filovirus vncRNAs and their potential as biomarkers of infection.
MATERIALS AND METHODS
Cells.
RO6E/J cells were a kind gift from Ingo Jordan (CureVac AG, Germany) and were maintained in Dulbecco’s modified Eagle medium (DMEM)–F-12 plus GlutaMAX (Thermo Fisher) complete medium (10% heat-inactivated [56°C, 30 min] fetal bovine serum [FBS] and 50 mg/liter [final concentration] gentamicin sulfate [Cellgro]) at 37°C under 5% CO2. EpoNi/22.1 cells were a kind gift from Christian Drosten (The Charité—Universitätsmedizin Berlin) and were maintained identically to RO6E/J cells. HepG2 cells were obtained from the ATCC (HB-8065) and were maintained in DMEM low-glucose (1 g/liter) complete medium (Thermo Fisher) at 37°C under 5% CO2. HEK 293T/17 cells (referred to below as “293T cells”) were obtained from the ATCC (CRL-11268) and were maintained in DMEM high-glucose (4.5 g/liter) complete medium (Thermo Fisher) at 37°C under 5% CO2. 769-P cells were acquired from the ATCC (CRL-1933) and were maintained in RPMI 1640 complete medium (Thermo Fisher) at 37°C under 5% CO2. Dicer knockout NoDice 4-25 cells and the parental cell line (referred to as “293T-P cells” to distinguish them from the 293T cells obtained from the ATCC) were a kind gift from Bryan Cullen (Duke University School of Medicine) and were maintained identically to the 293T cells obtained from the ATCC. BHK-21 C13 (CCL-10) and Vero E6 (CRL-1586) cells were obtained from the ATCC and were maintained identically to HepG2 cells.
Viruses.
All experiments involving infectious EBOV and MARV were conducted in the Galveston National Laboratory biosafety level 4 (BSL-4) facility by trained staff with the appropriate U.S. government permissions and registrations for work with the EBOV and MARV full-length cDNA clones (FLC) and viruses. Recombinant wt EBOV (strain Mayinga) and the VP35 R312A mutant virus expressing eGFP were rescued and propagated as described previously (116) using EBOV FLC provided by John Towner and Stuart Nichol (CDC) and the EBOV NP, VP35, L, VP30, and T7 polymerase plasmids provided by Yoshihiro Kawaoka (University of Wisconsin) and Heinz Feldmann (NIH). The wt EBOV FLC lacking the eGFP transgene was prepared by restriction digestion of the eGFP-flanking BsiWI sites followed by religation of the linearized vector. The MARV reverse genetics system used in these studies (73) was a kind gift from Jonathan Towner and Stuart Nichol (CDC). Plasmids containing the MARV FLC expressing eGFP (strain Uganda 371Bat2007, isolate 811277), rodent cell codon-optimized NP, VP30, VP35, and L genes, and T7 polymerase were transfected into BHK-21 C13 cells using the TransIT LT1 transfection reagent (Mirus Bio). Rescued viruses (passage 0) were further amplified by 2 to 3 passages in Vero E6 cells to generate working stocks for infection experiments. The VP35 R301A virus was generated by subcloning the VP35-containing AfeI-KasI fragment of the FLC into pCAGGS MCS. Site-directed mutagenesis was performed using the QuikChange kit (Agilent), after which the mutated fragment was reinserted into the FLC and rescued as described above. The titers of recovered viruses were determined in Vero E6 cell monolayers. Plaques were visualized 11 to 12 days postinfection by crystal violet staining.
Experimental infections.
To generate samples for small-RNA sequencing, 90% confluent monolayers of RO6E/J, EpoNi/22.1, or HepG2 cells were inoculated with EBOV or MARV at an MOI of 2 PFU/cell or were mock infected (complete medium) in biological triplicate, allowed to adsorb for 1 h, washed twice with cold phosphate-buffered saline (PBS), and then replenished with fresh complete medium. At 12 hpi and 24 hpi, cell supernatants were collected and frozen at −80°C for subsequent titering, and cell monolayers were lysed in 1.0 ml of TRIzol reagent for removal from the BSL-4 facility and RNA isolation. Titers of supernatants were determined as for virus stocks. For qRT-PCR comparison of EBOV miRNA abundances in Dicer-competent and Dicer knockout cells, cells were inoculated as described above with wt EBOV–eGFP at an MOI of 2 PFU/cell or were mock infected, and cell monolayers were collected in TRIzol at 20 hpi. Procedures for other experiments involving live filoviruses are described separately.
Virus inactivation and RNA extraction with TRIzol.
After removal of the supernatant, monolayers of filovirus-infected or mock-infected cells were treated with 1 ml TRIzol (Ambion), incubated for 10 min at room temperature, and removed from the BSL-4 facility. A 200-μl volume of chloroform per 1 ml of TRIzol used was added to each sample, and the samples were incubated at room-temperature for 5 min, after which they were centrifuged at 12,000 × g for 20 min. The aqueous phase was removed to fresh nuclease-free 1.5-ml tubes, to which 15 μg of linear acrylamide (Ambion) and 1.0 ml of 2-propanol per ml of TRIzol used was added. Samples were incubated at −20°C overnight to precipitate RNA. Following incubation, the samples were centrifuged at maximum speed (16,900 × g) at 4°C for 30 min and were then washed three times with 80% ethanol (each wash was followed by centrifugation at maximum speed for 30 min at 4°C). Precipitated RNA pellets were allowed to air dry for ∼5 min before being resuspended in nuclease-free water. Resuspended total RNA was quantified and was assessed for purity by a NanoDrop system (Thermo Fisher).
Small-RNA library preparation.
Total RNA integrity was assayed using an RNA 6000 Nano chip on the Agilent 2100 bioanalyzer. All RNAs used to make libraries had an RNA integrity number (RIN) of ≥8. Small-RNA libraries were made using the TruSeq Small RNA Sample Prep kit (Illumina) according to the manufacturer’s suggested protocol. Briefly, 1 μg of total RNA was 3′ and 5′ adapter ligated, reverse transcribed using SuperScript II (Invitrogen), and then PCR amplified, during which procedure unique index sequences were added to the libraries. PCR-amplified libraries were electrophoresed on 2.5%-to-3% agarose gels, and bands corresponding to ∼18- to 35-bp inserts were excised. The size-selected libraries were gel purified using the Monarch gel extraction kit (New England Biolabs). Purified libraries were validated for sequencing by qRT-PCR, Qubit, and Bioanalyzer and were then single-end sequenced for 50 cycles on an Illumina HiSeq 2500 system on rapid run mode. Detailed information regarding total reads passing the filter, read quality information, and virus alignment counts can be found in Data Set S1 in the supplemental material.
Bioinformatics.
For small-RNA analysis, FASTQ files containing the raw sequence reads were trimmed of the 3′ adapter sequence using the FASTX Toolkit, size selected to include only reads between 19 and 32 bp, and aligned to the appropriate infectious clone reference genome using Bowtie, v.0.12.8 (117) in the -k –best mode, allowing for a single mismatch and limiting reported alignments to a single instance. SAM output files from Bowtie were fed into SAMTools (118) to generate SAM, BAM, and mpileup files. To determine the fold change in vsRNA from 12 hpi to 24 hpi, vsRNAs for each library were normalized by dividing the total number of 19- to 32-nt vsRNA reads by the total number of 19- to 32-nt reads to obtain a vsRNA frequency. The following equation was then used to generate a vRNA-normalized vsRNA frequency:
where sf is the vsRNA frequency of a single biological replicate at either 12 or 24 hpi, s1 to s3 are the vsRNA frequencies for each of three biological replicates at 12 hpi, vf is the vRNA abundance for a single replicate at 12 or 24 hpi, v1 to v3 are the vRNA abundances for each of three biological replicates at 12 hpi (this equation calculates the vRNA-adjusted vsRNA fold change), and r1 to r3 are the individual vsRNA fold changes for each of three biological replicates at 12 hpi. Normalization for individual vncRNA abundance in EpoNi/22.1 and RO6E/J cells versus HepG2 cells was performed using the following equation:
where ni is the number of 19- to 32-nt reads for a given vncRNA for a given library, oi is the total number of 19- to 32-nt reads processed for a given library (reads per million [RPM] was calculated by multiplying the resulting value by 106), vi is the vRNA copy number for a given library (per nanogram of total RNA), v1 to v3 are the vRNA copy numbers (per nanogram of total RNA) for each HepG2 biological replicate (this equation results in the vRNA-adjusted RPM), and r1 to r3 are the vRNA-adjusted RPM for each of three HepG2 biological replicates. To determine the fold change from 12 hpi to 24 hpi for individual vncRNAs, we used the following equation:
where RPMf is the RPM for a given vncRNA at 12 or 24 hpi from an individual biological replicate, RPM1 to RPM3 are the RPM for each biological replicate at 12 hpi, vf is the vRNA copy number (per nanogram of total RNA) for an individual biological replicate at 12 or 24 hpi, v1 to v3 are the vRNA copy numbers (per nanogram of total RNA) for each of three biological replicates at 12 hpi (this equation calculates the vRNA-adjusted fold change for a given vncRNA), and r1 to r3 are the individual vRNA-adjusted fold change values for each of three biological replicates at 12 hpi. Additional analysis and statistics were performed using viRome, a package for R (119), Microsoft Excel, and GraphPad Prism 6. Statistical analyses for all experiments were performed using GraphPad Prism 6. Unless otherwise noted, P values for all statistical analyses are two tailed.
miRNA-specific qRT-PCR.
Primer sequences and/or unique identification (ID) numbers for all qRT-PCR experiments are provided in Data Set S3 in the supplemental material. SYBR green qRT-PCR was performed using the miRCURY LNA RT and miRNA PCR System kits (Qiagen) according to the manufacturer’s suggested protocol. Briefly, 10 ng input total RNA (unless otherwise specified) was first poly(A) tailed and then reverse transcribed with a universal miRNA RT primer, which adds a tag sequence to the RNAs. A kit-provided synthetic miRNA spike-in (UniSp6) was used in all reactions as a control for monitoring the efficiency of reverse transcription. The resulting miRNA cDNA libraries were diluted 1:60 in nuclease-free water, and 3 μl of diluted cDNA (50 pg) was used as the input for triplicate qRT-PCRs using EBOV miRNA-specific primers or primers to endogenous (hsa-miR-103a-3p) and exogenous (UniSp6) miRNAs. Controls without reverse transcriptase (–RT) and no-template controls (NTC) were also included for all primer sets. qRT-PCR was performed on StepOnePlus or QuantStudio 6 Flex real-time PCR systems (Thermo Fisher) using cycling settings specified in the miRCURY LNA miRNA PCR protocol, and data were collected in the associated StepOne or QuantStudio software (Thermo Fisher). The UniSp6 synthetic miRNA was used as the reference miRNA, since its “expression” was consistent between all biological and qRT-PCR technical replicates and experimental conditions, and spike-ins have been shown to be reliable references for qRT-PCR normalization (120, 121). The relative fold change between samples was calculated in Microsoft Excel using the 2–ΔΔCT method of approximation. For the purposes of determining fold change, targets without threshold cycle (CT) values (i.e., undetectable) were arbitrarily assigned a CT value of 40 (the last cycle). Thus, fold change approximations represent a minimum fold change comparison. For absolute quantitation, a synthetic ssRNA oligonucleotide exhibiting 5′-monophosphate and 3′-hydroxyl moieties and homologous to the 22-nt form of the EBOV GP vncRNA (Integrated DNA Technologies [IDT]) was spiked into human brain total RNA (TaKaRa Bio) at a concentration of 0.1 ng/10 ng total RNA (corresponding to ∼8.35 × 109 copies of the synthetic GP vncRNA per 10 ng total RNA). Ten nanograms of spiked human brain total RNA was reverse transcribed as described above, diluted 1:60 in nuclease-free water, and then further serially diluted in 1:10 increments. Three microliters of each serial dilution was then used as a standard for absolute quantitation.
Detection of viral vRNA/cRNA/mRNA by TaqMan (hydrolysis probe) qRT-PCR.
Tag-based strand-specific qRT-PCR assays were designed for EBOV as described previously (122). After RNA extraction, first-strand cDNA synthesis was performed using the SuperScript IV system (Thermo Fisher Scientific) and followed a modified version of the manufacturer’s protocol. Twenty nanograms of RNA was used as the input. Primer annealing was performed in the absence of deoxynucleoside triphosphates (dNTPs), which were added to the reverse transcription master mix. Reverse transcription was performed at 50°C. Following reverse transcription, qPCRs were set up with the TaqMan Universal PCR master mix (Thermo Fisher Scientific) using 4 ng of cDNA. Custom primer-probe sets (labeled with 6-carboxyfluorescein [FAM]) were obtained from Thermo Fisher Scientific. AT-rich flaps were added to the primers to improve efficiency, as described previously (123). Absolute quantitation was performed based on DNA oligonucleotide standards obtained from Integrated DNA Technologies.
RIP assay.
Near-confluent (∼90%) T225 flasks of 293T cells were either infected with wt rEBOV–eGFP at an MOI of 2 or mock infected. At 20 hpi, cells were washed twice with cold PBS, lysed in Magna RIP lysis buffer with protease and RNase inhibitors added (EMD Millipore), and subjected to one freeze-thaw cycle at −80°C. A single T225 flask inoculated with wt rEBOV–eGFP was used for all target protein and control immunoprecipitations (IPs) in a given independent experiment. Prior to immunoprecipitation, 10-μl volumes of the wt rEBOV–eGFP-infected and mock-infected lysates were collected for use as pre-IP input controls for qPCR. Argonaute (AGO) proteins were immunoprecipitated from lysates corresponding to ∼1.0 × 107 to 2.0 × 107 cells by use of 5 μg of each antibody (Data Set S3 in the supplemental material) and the EZ-Magna RIP RNA-binding protein immunoprecipitation kit (EMD Millipore) according to the manufacturer’s suggested protocol. IgG isotype control immunoprecipitation was performed using 5 μg of the kit-supplied normal mouse IgG. Immunoprecipitations were performed overnight at 4°C with gentle agitation. Following proteinase K digestion, 1 ml of TRIzol was added to the RNA or protein eluates and pre-IP lysates for residual virus inactivation and removal from the BSL-4 facility. The concentrations of purified RNAs from immunoprecipitations were too low to allow quantification using the Qubit HS RNA assay kit (Thermo Fisher), so equal volumes of each immunoprecipitate (6.5 μl) were used as the input for the miRCURY LNA RT kit. Ten nanograms of total RNA from the pre-IP input was used for reverse transcription.
siRNA-mediated knockdown.
A list of siRNAs utilized, their manufacturers, and their final concentrations used in our experiments can be found in Data Set S3 in the supplemental material. For siRNA transfection, we used the TransIT-siQuest reagent (Mirus Bio). To determine transfection efficiency, the siGLO fluorescent reporter (Dharmacon) was used in tandem with all transfections. All siRNA transfections were carried out in triplicate.
Western blotting.
Antibodies, manufacturers, and dilutions are provided in Data Set S3 in the supplemental material. One hundred micrograms of radioimmunoprecipitation assay (RIPA) lysate was run on 4-to-12% Bis-Tris PAGE gels (Invitrogen) and was dry blotted onto nitrocellulose membranes using the iBlot 2 system (Invitrogen). Membranes were blocked in 5% nonfat dry milk dissolved in 1× Tris-buffered saline (TBS) with 0.1% (final concentration) Tween 20 (TBS-T) for 1 h. Membranes were incubated overnight at 4°C with the primary antibody diluted to the indicated concentration (provided in Data Set S3, “Antibodies” tab) in either 5% bovine serum albumin (BSA) dissolved in 1× TBS-T or blocking buffer, depending on the manufacturer’s suggested protocol. Membranes were then washed three times in TBS-T, after which the secondary antibody was diluted in blocking buffer and was allowed to incubate with the membranes for 1 h at room temperature. Following a wash with TBS-T, membranes were developed using the Amersham ECL Western blotting reagent (GE Healthcare Life Sciences) and were imaged on an Odyssey Fc imaging system (Li-Cor Biotechnology) using 10-min exposures in the chemiluminescent and 700-nm channels. Band pixel density analysis was performed using Li-Cor Image Studio Lite, v.5.2.
Plaque immunostaining assays.
The antibodies used for plaque immunostaining can be found in Data Set S3 in the supplemental material. Confluent monolayers of Vero E6 cells were inoculated with serially diluted samples and were allowed to adsorb for 1 h at 37°C, after which a 0.6% methylcellulose–2% fetal bovine serum (FBS) MEM overlay was added. After 5 days of incubation, monolayers were fixed with a 10% formalin solution and were incubated overnight in the BSL-4 facility prior to being removed. Fixed monolayers were blocked with 5% milk in TBS-T for 1 h, washed, incubated with a primary antibody for 1 h, washed, incubated with a secondary antibody for 1 h, washed, and developed using a 4CN peroxidase substrate reagent (SeraCare).
Luciferase reporter assays.
We utilized the pmirGLO dual-luciferase reporter assay system (Promega), which expresses both firefly luciferase (FLuc; the “reporter”) and mammalian codon-optimized Renilla luciferase (hRLuc) as an internal control for normalization. Artificial 3′ UTRs were designed by generating a 456-nt scrambled sequence lacking homology to any known sequence in GenBank and embedding two miRNA-binding elements (MBEs) within the sequence, for a total insert length of 486 nt. MBEs were designed either to be perfectly complementary to the EBOV GP vncRNA, to contain mismatched base pairs within the center of the sequence (outside the seed [“bulged”]), or to be complementary to only the first 10 nt of the EBOV GP vncRNA (all sequences are provided in Data Set S3 in the supplemental material). Synthetic dsDNA fragments (“gBlocks”) were synthesized by IDT. Artificial 3′ UTRs were cloned immediately downstream of the FLuc reporter. For live-virus experiments, 769-P and EpoNi/22.1 cells were transfected with each of the reporter plasmids using the TransIT LT-1 reagent (Mirus), incubated for 4 h, and then infected with wt rEBOV at an MOI of 2 PFU/cell. At 24 hpi, cells were lysed in passive lysis buffer, subjected to a −80°C freeze-thaw cycle, and assayed on a luminometer using the Dual-Luciferase reporter assay system (Promega), and statistical comparisons were made to the empty-vector plasmid (no 3′ UTR). A control experiment was performed by transfecting a synthetic siRNA homologous to the EBOV GP vncRNA (Dharmacon) at a final concentration of 50 nM 4 h after transfection of the reporter constructs. siRNA transfection was performed using the Trans-IT siQuest reagent (Mirus). Cells were lysed 24 h after siRNA transfection and were processed identically to cells infected with live virus. Statistical comparisons for the siRNA experiment were made to cells transfected with both the pmirGLO construct containing the perfectly complementary MBE and a scrambled siRNA. For both experiments, a 10-s integration step was used for both FLuc and hRLuc readings, and FLuc luminescence values were normalized to corresponding hRLuc values. Both experiments were performed independently twice, with each treatment being performed in quadruplicate.
Data availability.
Adapter-trimmed, virus genome-aligned sequences have been deposited under NCBI SRA BioProject number PRJNA508298.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the U.S. Defense Threat Reduction Agency (DTRA), grant HDTRA1-14-1-0013, “Comparative immunology of Rousettus aegyptiacus reservoir with filoviruses,” to C.F.B. and A.B.
Adult Epomops buettikoferi kidney-derived cells EpoNi/22.1 cells were kindly provided by Christian Drosten (The Charité–Universitätsmedizin Berlin). The Egyptian rousette cell line R06E/J was generated and kindly provided by Ingo Jordan (ProBioGen AG, Berlin, Germany). The 293T-P and NoDice 4-25 cells were kindly provided by Bryan Cullen (Duke University School of Medicine). We thank Gregory Ebel and Tyler Eike at Colorado State University (Fort Collins, CO) for providing access to bioinformatics tools, as well as the laboratories of Sanjeev Sahni and Grant Hughes at The University of Texas Medical Branch (UTMB) for the use of StepOnePlus real-time PCR thermocyclers (Thermo Fisher Scientific). We also thank the staff and faculty of UTMB Next-Generation Sequencing Core for performing the sequencing for this project and Ivan Kuzmin for generating the rMARV VP35 R301A-eGFP infectious clone. Last, we thank the members of the Bukreyev laboratory for helpful discussions regarding the content and preparation of the manuscript and Shelton Bradrick (UTMB) for critical readings of the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. 2003. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 2.Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 3.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, Wolfe SA, Giraldez AJ. 2010. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng Y, Croce CM. 2016. The role of microRNAs in human cancer. Signal Transduct Target Ther 1:15004. doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Femminella GD, Ferrara N, Rengo G. 2015. The emerging role of microRNAs in Alzheimer’s disease. Front Physiol 6:40. doi: 10.3389/fphys.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 7.Backes S, Shapiro JS, Sabin LR, Pham AM, Reyes I, Moss B, Cherry S, tenOever BR. 2012. Degradation of host microRNAs by poxvirus poly(A) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell Host Microbe 12:200–210. doi: 10.1016/j.chom.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck AH, Perot J, Chisholm MA, Kumar DS, Tuddenham L, Cognat V, Marcinowski L, Dolken L, Pfeffer S. 2010. Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA 16:307–315. doi: 10.1261/rna.1819210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazalla D, Yario T, Steitz JA. 2010. Down-regulation of a host microRNA by a herpesvirus saimiri noncoding RNA. Science 328:1563–1566. doi: 10.1126/science.1187197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. 2012. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci U S A 109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M. 2008. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machlin ES, Sarnow P, Sagan SM. 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci U S A 108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amador-Cañizares Y, Bernier A, Wilson JA, Sagan SM. 2018. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res 46:5139–5158. doi: 10.1093/nar/gky273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grundhoff A, Sullivan CS. 2011. Virus-encoded microRNAs. Virology 411:325–343. doi: 10.1016/j.virol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kincaid RP, Sullivan CS. 2012. Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog 8:e1003018. doi: 10.1371/journal.ppat.1003018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asgari S. 2014. Role of microRNAs in arbovirus/vector interactions. Viruses 6:3514–3534. doi: 10.3390/v6093514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cullen BR. 2010. Five questions about viruses and microRNAs. PLoS Pathog 6:e1000787. doi: 10.1371/journal.ppat.1000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hussain M, Asgari S. 2014. MicroRNA-like viral small RNA from dengue virus 2 autoregulates its replication in mosquito cells. Proc Natl Acad Sci U S A 111:2746–2751. doi: 10.1073/pnas.1320123111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussain M, Torres S, Schnettler E, Funk A, Grundhoff A, Pijlman GP, Khromykh AA, Asgari S. 2011. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res 40:2210–2223. doi: 10.1093/nar/gkr848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weng KF, Hung CT, Hsieh PT, Li ML, Chen GW, Kung YA, Huang PN, Kuo RL, Chen LL, Lin JY, Wang RY, Chen SJ, Tang P, Horng JT, Huang HI, Wang JR, Ojcius DM, Brewer G, Shih SR. 2014. A cytoplasmic RNA virus generates functional viral small RNAs and regulates viral IRES activity in mammalian cells. Nucleic Acids Res 42:12789–12805. doi: 10.1093/nar/gku952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales L, Oliveros JC, Fernandez-Delgado R, tenOever BR, Enjuanes L, Sola I. 2017. SARS-CoV-encoded small RNAs contribute to infection-associated lung pathology. Cell Host Microbe 21:344–355. doi: 10.1016/j.chom.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bogerd HP, Skalsky RL, Kennedy EM, Furuse Y, Whisnant AW, Flores O, Schultz KL, Putnam N, Barrows NJ, Sherry B, Scholle F, Garcia-Blanco MA, Griffin DE, Cullen BR. 2014. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J Virol 88:8065–8076. doi: 10.1128/JVI.00985-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skalsky RL, Olson KE, Blair CD, Garcia-Blanco MA, Cullen BR. 2014. A “microRNA-like” small RNA expressed by dengue virus? Proc Natl Acad Sci U S A 111:E2359. doi: 10.1073/pnas.1406854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, Garcia-Sastre A, tenOever BR. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci U S A 107:11525–11530. doi: 10.1073/pnas.1001984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez JT, Zlatev I, Aggarwal S, Subramanian S, Sachidanandam R, Kim B, Manoharan M, tenOever BR. 2012. A small-RNA enhancer of viral polymerase activity. J Virol 86:13475–13485. doi: 10.1128/JVI.02295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umbach JL, Yen H-L, Poon LLM, Cullen BR. 2010. Influenza A virus expresses high levels of an unusual class of small viral leader RNAs in infected cells. mBio 1:e00204-10. doi: 10.1128/mBio.00204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Fu Z, Liang H, Wang Y, Qi X, Ding M, Sun X, Zhou Z, Huang Y, Gu H, Li L, Chen X, Li D, Zhao Q, Liu F, Wang H, Wang J, Zen K, Zhang CY. 2018. H5N1 influenza virus-specific miRNA-like small RNA increases cytokine production and mouse mortality via targeting poly(rC)-binding protein 2. Cell Res 28:157–171. doi: 10.1038/cr.2018.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leendertz SA, Gogarten JF, Dux A, Calvignac-Spencer S, Leendertz FH. 2016. Assessing the evidence supporting fruit bats as the primary reservoirs for Ebola viruses. Ecohealth 13:18–25. doi: 10.1007/s10393-015-1053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leroy EM, Epelboin A, Mondonge V, Pourrut X, Gonzalez JP, Muyembe-Tamfum JJ, Formenty P. 2009. Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis 9:723–728. doi: 10.1089/vbz.2008.0167. [DOI] [PubMed] [Google Scholar]
- 30.Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Delicat A, Paweska JT, Gonzalez JP, Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 31.Olival KJ, Hayman DT. 2014. Filoviruses in bats: current knowledge and future directions. Viruses 6:1759–1788. doi: 10.3390/v6041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Towner JS, Amman BR, Sealy TK, Carroll SA, Comer JA, Kemp A, Swanepoel R, Paddock CD, Balinandi S, Khristova ML, Formenty PB, Albarino CG, Miller DM, Reed ZD, Kayiwa JT, Mills JN, Cannon DL, Greer PW, Byaruhanga E, Farnon EC, Atimnedi P, Okware S, Katongole-Mbidde E, Downing R, Tappero JW, Zaki SR, Ksiazek TG, Nichol ST, Rollin PE. 2009. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog 5:e1000536. doi: 10.1371/journal.ppat.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang H, Zhou Z, Zhang S, Zen K, Chen X, Zhang C. 2014. Identification of Ebola virus microRNAs and their putative pathological function. Sci China Life Sci 57:973–981. doi: 10.1007/s11427-014-4759-2. [DOI] [PubMed] [Google Scholar]
- 34.Teng Y, Wang Y, Zhang X, Liu W, Fan H, Yao H, Lin B, Zhu P, Yuan W, Tong Y, Cao W. 2015. Systematic genome-wide screening and prediction of microRNAs in EBOV during the 2014 Ebolavirus outbreak. Sci Rep 5:9912. doi: 10.1038/srep09912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Sun J, Zhang H, Wang M, Gao GF, Li X. 2016. Ebola virus encodes a miR-155 analog to regulate importin-α5 expression. Cell Mol Life Sci 73:3733–3744. doi: 10.1007/s00018-016-2215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu P-C, Chiou B-H, Huang C-M. 2018. On revealing the gene targets of Ebola virus microRNAs involved in the human skin microbiome. PeerJ 6:e4138. doi: 10.7717/peerj.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Liang H, Chen X, Ke Y, Zhou Z, Yang M, Zen K, Yang R, Liu C, Zhang CY. 2016. An Ebola virus-encoded microRNA-like fragment serves as a biomarker for early diagnosis of Ebola virus disease. Cell Res 26:380–383. doi: 10.1038/cr.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duy J, Honko AN, Altamura LA, Bixler SL, Wollen-Roberts S, Wauquier N, O’Hearn A, Mucker EM, Johnson JC, Shamblin JD, Zelko J, Botto MA, Bangura J, Coomber M, Pitt ML, Gonzalez J-P, Schoepp RJ, Goff AJ, Minogue TD. 2018. Virus-encoded miRNAs in Ebola virus disease. Sci Rep 8:6480. doi: 10.1038/s41598-018-23916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliver GF, Orang AV, Appukuttan B, Marri S, Michael MZ, Marsh GA, Smith JR. 2019. Expression of microRNA in human retinal pigment epithelial cells following infection with Zaire ebolavirus. BMC Res Notes 12:639. doi: 10.1186/s13104-019-4671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haasnoot J, de Vries W, Geutjes EJ, Prins M, de Haan P, Berkhout B. 2007. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog 3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fabozzi G, Nabel CS, Dolan MA, Sullivan NJ. 2011. Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J Virol 85:2512–2523. doi: 10.1128/JVI.01160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Y, Cherukuri NC, Jackel JN, Wu Z, Crary M, Buckley KJ, Bisaro DM, Parris DS. 2012. Characterization of the RNA silencing suppression activity of the Ebola virus VP35 protein in plants and mammalian cells. J Virol 86:3038–3049. doi: 10.1128/JVI.05741-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Neal ST, Samuel GH, Adelman ZN, Myles KM. 2014. Mosquito-borne viruses and suppressors of invertebrate antiviral RNA silencing. Viruses 6:4314–4331. doi: 10.3390/v6114314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao JH, Hua CL, Fang YY, Guo HS. 2016. The dual edge of RNA silencing suppressors in the virus-host interactions. Curr Opin Virol 17:39–44. doi: 10.1016/j.coviro.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Backes S, Langlois RA, Schmid S, Varble A, Shim JV, Sachs D, tenOever BR. 2014. The mammalian response to virus infection is independent of small RNA silencing. Cell Rep 8:114–125. doi: 10.1016/j.celrep.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding SW, Voinnet O. 2014. Antiviral RNA silencing in mammals: no news is not good news. Cell Rep 9:795–797. doi: 10.1016/j.celrep.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cullen BR, Cherry S, tenOever BR. 2013. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell Host Microbe 14:374–378. doi: 10.1016/j.chom.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 48.Cullen BR. 2014. Viruses and RNA interference: issues and controversies. J Virol 88:12934–12936. doi: 10.1128/JVI.01179-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.tenOever BR. 2014. Response to Voinnet et al. Cell Rep 9:798–799. doi: 10.1016/j.celrep.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.tenOever BR. 2017. Questioning antiviral RNAi in mammals. Nat Microbiol 2:17052. doi: 10.1038/nmicrobiol.2017.52. [DOI] [PubMed] [Google Scholar]
- 51.Jeffrey KL, Li Y, Ding SW. 2017. Reply to ‘Questioning antiviral RNAi in mammals’. Nat Microbiol 2:17053. doi: 10.1038/nmicrobiol.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cullen BR. 2017. RNA interference in mammals: the virus strikes back. Immunity 46:970–972. doi: 10.1016/j.immuni.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Umbach JL, Cullen BR. 2009. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev 23:1151–1164. doi: 10.1101/gad.1793309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tijsterman M, Plasterk RH. 2004. Dicers at RISC; the mechanism of RNAi. Cell 117:1–3. doi: 10.1016/s0092-8674(04)00293-4. [DOI] [PubMed] [Google Scholar]
- 55.Ma E, MacRae IJ, Kirsch JF, Doudna JA. 2008. Autoinhibition of human Dicer by its internal helicase domain. J Mol Biol 380:237–243. doi: 10.1016/j.jmb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flemr M, Malik R, Franke V, Nejepinska J, Sedlacek R, Vlahovicek K, Svoboda P. 2013. A retrotransposon-driven Dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 155:807–816. doi: 10.1016/j.cell.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Taylor DW, Ma E, Shigematsu H, Cianfrocco MA, Noland CL, Nagayama K, Nogales E, Doudna JA, Wang HW. 2013. Substrate-specific structural rearrangements of human Dicer. Nat Struct Mol Biol 20:662–670. doi: 10.1038/nsmb.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 59.tenOever BR. 2013. RNA viruses and the host microRNA machinery. Nat Rev Microbiol 11:169–180. doi: 10.1038/nrmicro2971. [DOI] [PubMed] [Google Scholar]
- 60.Parameswaran P, Sklan E, Wilkins C, Burgon T, Samuel MA, Lu R, Ansel KM, Heissmeyer V, Einav S, Jackson W, Doukas T, Paranjape S, Polacek C, dos Santos FB, Jalili R, Babrzadeh F, Gharizadeh B, Grimm D, Kay M, Koike S, Sarnow P, Ronaghi M, Ding SW, Harris E, Chow M, Diamond MS, Kirkegaard K, Glenn JS, Fire AZ. 2010. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog 6:e1000764. doi: 10.1371/journal.ppat.1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Lu J, Han Y, Fan X, Ding SW. 2013. RNA interference functions as an antiviral immunity mechanism in mammals. Science 342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, Ding SW, Voinnet O. 2013. Antiviral RNA interference in mammalian cells. Science 342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Basavappa M, Lu J, Dong S, Cronkite DA, Prior JT, Reinecker HC, Hertzog P, Han Y, Li WX, Cheloufi S, Karginov FV, Ding SW, Jeffrey KL. 2016. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat Microbiol 2:16250. doi: 10.1038/nmicrobiol.2016.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qiu Y, Xu Y, Zhang Y, Zhou H, Deng Y-Q, Li X-F, Miao M, Zhang Q, Zhong B, Hu Y, Zhang F-C, Wu L, Qin C-F, Zhou X. 2017. Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 46:992–1004.e5. doi: 10.1016/j.immuni.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 65.Jordan I, Horn D, Oehmke S, Leendertz FH, Sandig V. 2009. Cell lines from the Egyptian fruit bat are permissive for modified vaccinia Ankara. Virus Res 145:54–62. doi: 10.1016/j.virusres.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuhl A, Hoffmann M, Muller MA, Munster VJ, Gnirss K, Kiene M, Tsegaye TS, Behrens G, Herrler G, Feldmann H, Drosten C, Pohlmann S. 2011. Comparative analysis of Ebola virus glycoprotein interactions with human and bat cells. J Infect Dis 204(Suppl 3):S840–S849. doi: 10.1093/infdis/jir306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krähling V, Dolnik O, Kolesnikova L, Schmidt-Chanasit J, Jordan I, Sandig V, Günther S, Becker S. 2010. Establishment of fruit bat cells (Rousettus aegyptiacus) as a model system for the investigation of filoviral infection. PLoS Negl Trop Dis 4:e802. doi: 10.1371/journal.pntd.0000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bale S, Julien JP, Bornholdt ZA, Kimberlin CR, Halfmann P, Zandonatti MA, Kunert J, Kroon GJ, Kawaoka Y, MacRae IJ, Wilson IA, Saphire EO. 2012. Marburg virus VP35 can both fully coat the backbone and cap the ends of dsRNA for interferon antagonism. PLoS Pathog 8:e1002916. doi: 10.1371/journal.ppat.1002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cardenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartman AL, Towner JS, Nichol ST. 2004. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 328:177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 71.Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, Otwinowski Z, Liu G, Huh J, Basler CF, Amarasinghe GK. 2012. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A 109:20661–20666. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Towner JS, Paragas J, Dover JE, Gupta M, Goldsmith CS, Huggins JW, Nichol ST. 2005. Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology 332:20–27. doi: 10.1016/j.virol.2004.10.048. [DOI] [PubMed] [Google Scholar]
- 73.Albarino CG, Uebelhoer LS, Vincent JP, Khristova ML, Chakrabarti AK, McElroy A, Nichol ST, Towner JS. 2013. Development of a reverse genetics system to generate recombinant Marburg virus derived from a bat isolate. Virology 446:230–237. doi: 10.1016/j.virol.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanchez A, Kiley MP, Holloway BP, Auperin DD. 1993. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res 29:215–240. doi: 10.1016/0168-1702(93)90063-s. [DOI] [PubMed] [Google Scholar]
- 75.Muhlberger E, Trommer S, Funke C, Volchkov V, Klenk HD, Becker S. 1996. Termini of all mRNA species of Marburg virus: sequence and secondary structure. Virology 223:376–380. doi: 10.1006/viro.1996.0490. [DOI] [PubMed] [Google Scholar]
- 76.Bukreyev AA, Belanov EF, Blinov VM, Netesov SV. 1995. Complete nucleotide sequences of Marburg virus genes 5 and 6 encoding VP30 and VP24 proteins. Biochem Mol Biol Int 35:605–613. [PubMed] [Google Scholar]
- 77.Kerpedjiev P, Hammer S, Hofacker IL. 2015. Forna (force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics 31:3377–3379. doi: 10.1093/bioinformatics/btv372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meyer M, Garron T, Lubaki NM, Mire CE, Fenton KA, Klages C, Olinger GG, Geisbert TW, Collins PL, Bukreyev A. 2015. Aerosolized Ebola vaccine protects primates and elicits lung-resident T cell responses. J Clin Invest 125:3241–3255. doi: 10.1172/JCI81532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bogerd HP, Whisnant AW, Kennedy EM, Flores O, Cullen BR. 2014. Derivation and characterization of Dicer- and microRNA-deficient human cells. RNA 20:923–937. doi: 10.1261/rna.044545.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aguado LC, tenOever B. 2018. RNA virus building blocks—miRNAs not included. PLoS Pathog 14:e1006963. doi: 10.1371/journal.ppat.1006963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rouha H, Thurner C, Mandl CW. 2010. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res 38:8328–8337. doi: 10.1093/nar/gkq681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Langlois RA, Shapiro JS, Pham AM, tenOever BR. 2012. In vivo delivery of cytoplasmic RNA virus-derived miRNAs. Mol Ther 20:367–375. doi: 10.1038/mt.2011.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shapiro JS, Varble A, Pham AM, Tenoever BR. 2010. Noncanonical cytoplasmic processing of viral microRNAs. RNA 16:2068–2074. doi: 10.1261/rna.2303610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Varble A, Chua MA, Perez JT, Manicassamy B, Garcia-Sastre A, tenOever BR. 2010. Engineered RNA viral synthesis of microRNAs. Proc Natl Acad Sci U S A 107:11519–11524. doi: 10.1073/pnas.1003115107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muhlberger E, Lotfering B, Klenk HD, Becker S. 1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J Virol 72:8756–8764. doi: 10.1128/JVI.72.11.8756-8764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. doi: 10.1128/JVI.73.3.2333-2342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pomerantz RT, Temiakov D, Anikin M, Vassylyev DG, McAllister WT. 2006. A mechanism of nucleotide misincorporation during transcription due to template-strand misalignment. Mol Cell 24:245–255. doi: 10.1016/j.molcel.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. 2010. A Dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Girardi E, Chane-Woon-Ming B, Messmer M, Kaukinen P, Pfeffer S. 2013. Identification of RNase L-dependent, 3′-end-modified, viral small RNAs in Sindbis virus-infected mammalian cells. mBio 4:e00698-13. doi: 10.1128/mBio.00698-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lykke-Andersen S, Tomecki R, Jensen TH, Dziembowski A. 2011. The eukaryotic RNA exosome: same scaffold but variable catalytic subunits. RNA Biol 8:61–66. doi: 10.4161/rna.8.1.14237. [DOI] [PubMed] [Google Scholar]
- 91.Shapiro JS, Schmid S, Aguado LC, Sabin LR, Yasunaga A, Shim JV, Sachs D, Cherry S, tenOever BR. 2014. Drosha as an interferon-independent antiviral factor. Proc Natl Acad Sci U S A 111:7108–7113. doi: 10.1073/pnas.1319635111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dai L, Chen K, Youngren B, Kulina J, Yang A, Guo Z, Li J, Yu P, Gu S. 2016. Cytoplasmic Drosha activity generated by alternative splicing. Nucleic Acids Res 44:10454–10466. doi: 10.1093/nar/gkw668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Han J, Lee Y, Yeom K-H, Nam J-W, Heo I, Rhee J-K, Sohn SY, Cho Y, Zhang B-T, Kim VN. 2006. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 94.Zeng Y, Yi R, Cullen BR. 2005. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J 24:138–148. doi: 10.1038/sj.emboj.7600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cazalla D, Xie M, Steitz JA. 2011. A primate herpesvirus uses the Integrator complex to generate viral microRNAs. Mol Cell 43:982–992. doi: 10.1016/j.molcel.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie M, Zhang W, Shu MD, Xu A, Lenis DA, DiMaio D, Steitz JA. 2015. The host Integrator complex acts in transcription-independent maturation of herpesvirus microRNA 3′ ends. Genes Dev 29:1552–1564. doi: 10.1101/gad.266973.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jodoin JN, Sitaram P, Albrecht TR, May SB, Shboul M, Lee E, Reversade B, Wagner EJ, Lee LA. 2013. Nuclear-localized Asunder regulates cytoplasmic dynein localization via its role in the Integrator complex. Mol Biol Cell 24:2954–2965. doi: 10.1091/mbc.E13-05-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Janas MM, Wang B, Harris AS, Aguiar M, Shaffer JM, Subrahmanyam YV, Behlke MA, Wucherpfennig KW, Gygi SP, Gagnon E, Novina CD. 2012. Alternative RISC assembly: binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA 18:2041–2055. doi: 10.1261/rna.035675.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J, Fritzsche A, Aeschimann F, Pfanzagl V, Basselet P, Weiler J, Hintersteiner M, Morrissey DV, Meisner-Kober NC. 2013. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J 32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flores O, Kennedy EM, Skalsky RL, Cullen BR. 2014. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic Acids Res 42:4629–4639. doi: 10.1093/nar/gkt1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. 2012. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones ME, Schuh AJ, Amman BR, Sealy TK, Zaki SR, Nichol ST, Towner JS. 2015. Experimental inoculation of Egyptian rousette bats (Rousettus aegyptiacus) with viruses of the Ebolavirus and Marburgvirus genera. Viruses 7:3420–3442. doi: 10.3390/v7072779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paweska JT, Storm N, Grobbelaar AA, Markotter W, Kemp A, Jansen van Vuren P. 2016. Experimental inoculation of Egyptian fruit bats (Rousettus aegyptiacus) with Ebola virus. Viruses 8:E29. doi: 10.3390/v8020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tekes G, Rahmeh AA, Whelan SP. 2011. A freeze frame view of vesicular stomatitis virus transcription defines a minimal length of RNA for 5′ processing. PLoS Pathog 7:e1002073. doi: 10.1371/journal.ppat.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stillman EA, Whitt MA. 1999. Transcript initiation and 5′-end modifications are separable events during vesicular stomatitis virus transcription. J Virol 73:7199–7209. doi: 10.1128/JVI.73.9.7199-7209.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li J, Rahmeh A, Brusic V, Whelan SP. 2009. Opposing effects of inhibiting cap addition and cap methylation on polyadenylation during vesicular stomatitis virus mRNA synthesis. J Virol 83:1930–1940. doi: 10.1128/JVI.02162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ogino T. 2014. Capping of vesicular stomatitis virus pre-mRNA is required for accurate selection of transcription stop–start sites and virus propagation. Nucleic Acids Res 42:12112–12125. doi: 10.1093/nar/gku901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Braun MR, Deflubé LR, Noton SL, Mawhorter ME, Tremaglio CZ, Fearns R. 2017. RNA elongation by respiratory syncytial virus polymerase is calibrated by conserved region V. PLoS Pathog 13:e1006803. doi: 10.1371/journal.ppat.1006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liuzzi M, Mason SW, Cartier M, Lawetz C, McCollum RS, Dansereau N, Bolger G, Lapeyre N, Gaudette Y, Lagace L, Massariol MJ, Do F, Whitehead P, Lamarre L, Scouten E, Bordeleau J, Landry S, Rancourt J, Fazal G, Simoneau B. 2005. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA-dependent RNA polymerase. J Virol 79:13105–13115. doi: 10.1128/JVI.79.20.13105-13115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kurilla MG, Cabradilla CD, Holloway BP, Keene JD. 1984. Nucleotide sequence and host La protein interactions of rabies virus leader RNA. J Virol 50:773–778. doi: 10.1128/JVI.50.3.773-778.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leppert M, Rittenhouse L, Perrault J, Summers DF, Kolakofsky D. 1979. Plus and minus strand leader RNAs in negative strand virus-infected cells. Cell 18:735–747. doi: 10.1016/0092-8674(79)90127-2. [DOI] [PubMed] [Google Scholar]
- 112.Modrof J, Muhlberger E, Klenk HD, Becker S. 2002. Phosphorylation of VP30 impairs Ebola virus transcription. J Biol Chem 277:33099–33104. doi: 10.1074/jbc.M203775200. [DOI] [PubMed] [Google Scholar]
- 113.Tigabu B, Ramanathan P, Ivanov A, Lin X, Ilinykh PA, Parry CS, Freiberg AN, Nekhai S, Bukreyev A. 2018. Phosphorylated VP30 of Marburg virus is a repressor of transcription. J Virol 92:e00426-18. doi: 10.1128/JVI.00426-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gould SJ, Lewontin RC. 1979. The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc R Soc Lond B Biol Sci 205:581–598. [DOI] [PubMed] [Google Scholar]
- 115.Weik M, Modrof J, Klenk HD, Becker S, Muhlberger E. 2002. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J Virol 76:8532–8539. doi: 10.1128/jvi.76.17.8532-8539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lubaki NM, Ilinykh P, Pietzsch C, Tigabu B, Freiberg AN, Koup RA, Bukreyev A. 2013. The lack of maturation of Ebola virus-infected dendritic cells results from the cooperative effect of at least two viral domains. J Virol 87:7471–7485. doi: 10.1128/JVI.03316-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Watson M, Schnettler E, Kohl A. 2013. viRome: an R package for the visualization and analysis of viral small RNA sequence datasets. Bioinformatics 29:1902–1903. doi: 10.1093/bioinformatics/btt297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Johnston S, Gallaher Z, Czaja K. 2012. Exogenous reference gene normalization for real-time reverse transcription-polymerase chain reaction analysis under dynamic endogenous transcription. Neural Regen Res 7:1064–1072. doi: 10.3969/j.issn.1673-5374.2012.14.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roberts TC, Coenen-Stass AML, Wood M. 2014. Assessment of RT-qPCR normalization strategies for accurate quantification of extracellular microRNAs in murine serum. PLoS One 9:e89237. doi: 10.1371/journal.pone.0089237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J Virol Methods 173:1–6. doi: 10.1016/j.jviromet.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Afonina I, Ankoudinova I, Mills A, Lokhov S, Huynh P, Mahoney W. 2007. Primers with 5′ flaps improve real-time PCR. Biotechniques 43:770–774. doi: 10.2144/000112631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Adapter-trimmed, virus genome-aligned sequences have been deposited under NCBI SRA BioProject number PRJNA508298.