

HSV-1 is a common cause of recurrent viral infections in humans. The virus can cause a range of mucosal pathologies. Efficient viral egress from infected cells is an important step for HSV-1 transmission and virus-associated pathologies. Host mechanisms that contribute to HSV-1 egress from infected cells are poorly understood. Syndecan-1 is a common heparan sulfate proteoglycan expressed by many natural target cells. Despite its known connection with heparanase, a recently identified mediator of HSV-1 release, syndecan-1 has not been previously investigated in HSV-1 release. In this study, we demonstrate that the shedding of syndecan-1 by MMP-3 and MMP-7 supports viral egress. We show that the mechanism behind the activation of these MMPs is mediated by heparanase, which is upregulated upon HSV-1 infection. Our study elucidates a new connection between HSV-1 egress, heparanase, and matrix metallopeptidases; identifies new molecular markers of infection; and provides potential new targets for therapeutic interventions.
KEYWORDS: heparan sulfate, heparanase, syndecan, herpes simplex virus, metalloenzymes
ABSTRACT
Herpes simplex virus 1 (HSV-1) can infect virtually all cell types in vitro. An important reason lies in its ability to exploit heparan sulfate (HS) for attachment to cells. HS is a ubiquitous glycosaminoglycan located on the cell surface and tethered to proteoglycans such as syndecan-1. Previously, we have shown that heparanase (HPSE) facilitates the release of viral particles by cleaving HS. Here, we demonstrate that HPSE is a master regulator where, in addition to directly enabling viral release via HS removal, it also facilitates cleavage of HS-containing ectodomains of syndecan-1, thereby further enhancing HSV-1 egress from infected cells. Syndecan-1 cleavage is mediated by upregulation of matrix metalloproteases (MMPs) that accompanies higher HPSE expression in infected cells. By overexpressing HPSE, we have identified MMP-3 and MMP-7 as important sheddases of syndecan-1 shedding in corneal epithelial cells, which are natural targets of HSV-1 infection. MMP-3 and MMP-7 were also naturally upregulated during HSV-1 infection. Altogether, this paper shows a new connection between HSV-1 release and syndecan-1 shedding, a phenomenon that is regulated by HPSE and executed by the MMPs. Our results also identify new molecular markers for HSV-1 infection and new targets for future interventions.
IMPORTANCE HSV-1 is a common cause of recurrent viral infections in humans. The virus can cause a range of mucosal pathologies. Efficient viral egress from infected cells is an important step for HSV-1 transmission and virus-associated pathologies. Host mechanisms that contribute to HSV-1 egress from infected cells are poorly understood. Syndecan-1 is a common heparan sulfate proteoglycan expressed by many natural target cells. Despite its known connection with heparanase, a recently identified mediator of HSV-1 release, syndecan-1 has not been previously investigated in HSV-1 release. In this study, we demonstrate that the shedding of syndecan-1 by MMP-3 and MMP-7 supports viral egress. We show that the mechanism behind the activation of these MMPs is mediated by heparanase, which is upregulated upon HSV-1 infection. Our study elucidates a new connection between HSV-1 egress, heparanase, and matrix metallopeptidases; identifies new molecular markers of infection; and provides potential new targets for therapeutic interventions.
INTRODUCTION
Herpesviruses are a family of enveloped, double-stranded DNA viruses (1). More than 90% of adults worldwide are infected with a herpesvirus, and roughly 73% of Americans are infected with herpes simplex virus 1 (HSV-1) or HSV-2 alone (1, 2). HSV-1 has been reported to infect the mouth, gums, genitals, skin, brain, and eyes (3–5). Ocular HSV-1 infections can lead to a host of diseases, including blepharitis, epithelial keratitis, conjunctivitis, and anterior uveitis (6–8). Indeed, HSV-1-induced keratitis represents one of the leading causes of infectious blindness in the United States (6, 9, 10).
HSV-1 entry into human cells begins with the attachment of its glycoproteins gB and gC to heparan sulfate proteoglycans (HSPGs) located on the cell surface, although proteoglycan-independent entry pathways do exist (11–14). HSPGs are composed of multiple chains of heparan sulfate (HS), a glycosaminoglycan found on the surface of most mammalian cell types, attached to a core protein by trisaccharide linkages (13, 15, 16). The most common family of HSPGs are the syndecans, made up of syndecan-1, -2, -3, and -4 (13, 17, 18). Syndecan-1 and syndecan-2 production have been shown to be upregulated by HSV-1 upon infection, and reduction of these syndecans impedes HSV-1 entry into HeLa cells (13). In addition, the increase in syndecan levels results in more HS moieties being present on the cell surface (13). A single HS chain has the ability to bind multiple viruses, and a syndecan-1 protein has three HS attachment sites, giving it the potential to trap many HSV-1 virions in its structure (19, 20). In addition, syndecan-1 is commonly found on natural target cells for HSV-1 infection (21–23). Thus, syndecans are heavily implicated in mediating successful HSV-1 infection (19, 24).
Heparanase (HPSE) is an endo-β,d-glycosidase that is the only known mammalian enzyme capable of cleaving HS (25–27). HPSE targets heparan sulfate at limited sites and does not cleave the entire HS chain from syndecan-1 proteoglycans (28, 29). HPSE has previously been shown to be upregulated upon HSV-1 infection via NF-κB signaling and is required for the release of HSV-1 from host cells (30). It is believed that HPSE upregulation by the virus shifts the host cell into a “detachment mode” and cleaves HS chains on the cell surface, which allows for an efficient release of newly generated virions (30, 31). In addition, given the connection between HPSE and syndecan shedding (30), the egress of the newly made viruses could be due in part to the shedding of the entire syndecan HSPG from the extracellular surface. Loss of HSPGs may enhance the rate of viral release from the cells and facilitate pathogenesis to neighboring cells. HPSE has been reported to increase syndecan-1 shedding in myeloma cells via the phosphorylation of extracellular signal-regulated kinase-1 (ERK) and the downstream activation of the sheddase matrix metalloproteinase 9 (MMP-9) (32, 33). Under various physiological or pathological conditions, the HS-expressing ectodomains of syndecans are also shed by additional sheddases that include MMP-3 and MMP-7 (34–36). However, to our knowledge, a role of MMPs in syndecan shedding during HSV-1 infection has not yet been elucidated. Likewise, the synchrony between HPSE and syndecan shedding to facilitate HSV-1 egress is not yet established. The results from this study target these gaps in the literature.
Here, we demonstrate that HPSE overexpression results in the increased cleavage of syndecan-1 from the cell surface. The transfection of HPSE or infection with HSV-1 upregulates specific MMPs, which facilitate the shedding of syndecan-1. Our evidence suggests that HPSE plays a dual role in the viral life cycle by directly cleaving HS chains from the cell surface to promote entry and mediating the shedding of syndecan-1 to promote egress. We also implicate MMP-3 and MMP-7 as the sheddases that regulate syndecan shedding in response to HSV-1 infection. These MMPs may provide new targets for antiviral therapies.
RESULTS
HSV-1 infection of human corneal epithelial cells increases shedding of syndecan-1.
Given that HSV-1 infection has been reported to decrease HS expression on the cell surface (30), we sought to identify the effects of HSV-1 on syndecan levels on immortalized human corneal epithelial (HCE) cells. Using flow cytometry, we observed the loss of syndecan-1 on HCE cells infected with HSV-1 at both 24 and 36 hours postinfection (hpi) relative to uninfected controls (Fig. 1A). Quantification of the syndecan loss at each time point revealed a roughly 25% decrease of cell surface syndecan-1 at 24 hpi, which progressed to 50% by 36 hpi (Fig. 1B). We next wanted to verify that the loss of syndecan-1 seen in immortal HCE cells was not specific to the cell line used, so we extracted primary human corneal epithelial cells from human donor corneas and infected them with HSV-1. Similar to our initial observation, we found that syndecan-1 levels decreased in primary HCE cells with infection, again with roughly a 50% loss in the levels of syndecan-1 on the cell surface (Fig. 1C and D). We also performed immunofluorescence microscopy on HCE cells that were either uninfected or infected with HSV-1 for 24 h. After staining the cells for syndecan-1, we observed a loss of syndecan-1 from the cell surface in the infected cells (Fig. 1E).
FIG 1.

HSV-1 infection of human corneal epithelial cells increases syndecan-1 shedding. (A) Syndecan-1 expression is decreased on the HCE cell surface at different time points postinfection, measured using flow cytometry. Cells were infected with KOS-WT at an MOI of 0.1 and were stained for syndecan-1 at 24 and 36 hpi. (B) Fluorescence intensity measurements based on flow cytometry results. Integrated mean fluorescence intensity of the whole cell population was measured, and fold change was normalized to uninfected mock samples for each time point. (C, left) Syndecan-1 expression on the cell surface is decreased in primary human corneal epithelial cells. Cells were infected with 17-GFP at MOI 0.1 and were stained for syndecan-1 at 48 hpi. Mean fluorescence intensity (MFI) values for the uninfected and infected cells were 122,353.86 and 66,945.33, respectively. (C, right) Dot plot of the flow cytometry results. Q1 and Q2 represent FITC-positive populations (infected). Q2 and Q3 represent syndecan-1-positive populations (right). (D) Fluorescence intensity measurements based on flow cytometry results. Integrated mean fluorescence intensity of the whole cell population was measured using flow cytometry at 36 hpi. Uninfected and 36-hpi samples are compared. (E, left) Immunofluorescence microscopy images show decreased expression of syndecan-1 on the cell surface of infected cells. HCE cells were grown on imaging dishes and infected with KOS-WT at an MOI of 0.1 for 24 h. The cells were stained for syndecan-1 (red) and 4′,6-diamidino-2-phenylindole (DAPI) (blue). Merged images of syndecan-1, DAPI, and brightfield microscopy are also shown. (E, right) Images were quantified for syndecan-1 intensity. A hundred cells were picked at random for each sample, and the intensity of syndecan-1 was measured for each cell using ImageJ.
HSV-1 infection causes the shedding of syndecan-1 from the cell surface.
Although the above experiments demonstrated that there was a loss of syndecan-1 from HCE cells after HSV-1 infection, they did not conclusively show that the syndecan-1 was shed and not simply endocytosed from the cell surface. In order to understand the fate of the syndecan-1 on the cell surface, we performed a slot blot assay on the supernatant of HCE cells 24 and 36 h postinfection. We found that the supernatant of infected HCE cells showed more unbound syndecan-1 protein than uninfected cells, suggesting that the syndecan-1 was intact and merely cleaved from the surface of infected cells (Fig. 2A). Densitometry quantification of the slot blot assay results revealed that the syndecan-1 in the supernatant increased 1.5-fold 24 hpi and about 2.5-fold 36 hpi (Fig. 2B). As a result, we concluded that syndecan-1 was being shed from cells upon infection.
FIG 2.
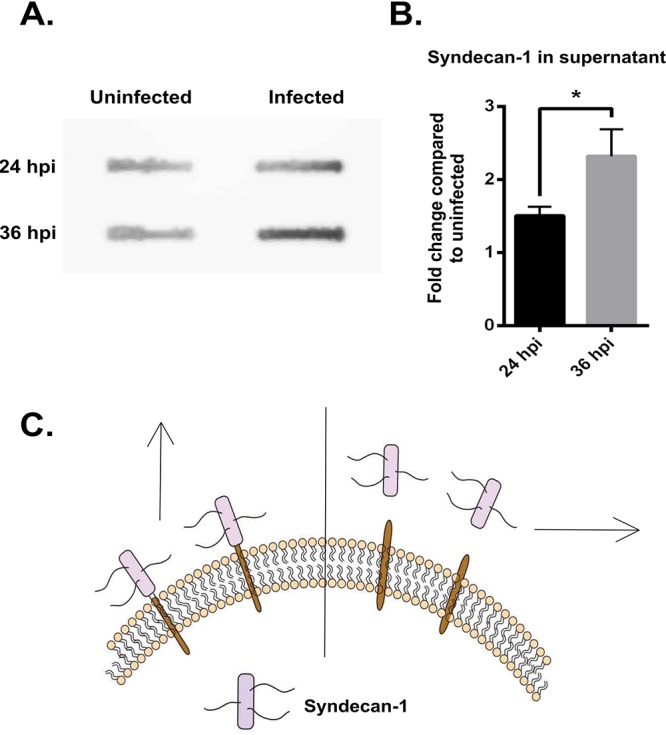
HSV-1 infection causes the shedding of syndecan-1 from the cell surface. (A) Slot blot assay shows increased syndecan-1 shedding in HCE cells. HCE cells were infected with KOS-WT at an MOI of 0.1 for 24 and 36 h. The supernatant was used to measure the levels of syndecan-1 that was shed from the cell surface. (B) Densitometry quantification of syndecan-1 shed in supernatant based on slot blot assay. The fold change was normalized to uninfected mock samples at each time point. (C) Image representing shedding of syndecan-1. The left side of the perpendicular line shows syndecan-1 bound to the cell surface, which was measured by flow cytometry. The right side of the perpendicular line shows syndecan-1 shed in the supernatant, which was measured by slot blot assay and is described above in panel A.
The loss of syndecan-1 from the cell surface during HSV-1 infection is not due to an increase in the synthesis of syndecan-1.
The increase in syndecan-1 shedding could have been caused by an upregulation of its synthesis in infected cells. It may have been the case that the upregulation of syndecan-1, in conjunction with a normal rate of shedding, was responsible for the increase in shed syndecan previously observed (Fig. 2). HSV-1 infection has previously been shown to upregulate syndecan-1 production in HeLa cells (13). In order to test if more syndecan-1 protein was being produced by infected cells, we performed both quantitative PCR (qPCR) and Western blotting on infected HCE cells. Neither the syndecan-1 transcripts nor protein were increased in infected cells (Fig. 3A and B). Hence, we concluded that the infection of human corneal epithelial cells by HSV-1 results in the shedding of syndecan-1 to the extracellular matrix, not the upregulation of syndecan-1. The lack of changes in syndecan transcripts in HCE cells compared to HeLa cells suggests that the effects of HSV-1 infection are highly sensitive to the cell type used.
FIG 3.

Syndecan-1 loss during HSV-1 infection is not due to transcriptional changes in the syndecan protein. (A) mRNA expression shows no significant change in syndecan-1 transcripts at either 24 or 36 hpi. HCE cells were infected with KOS-WT at an MOI of 0.1, and mRNA was extracted from cells at the indicated time points. Results are shown as fold change of infected over uninfected cells at each time point, normalized to GAPDH. (B, left) Western blotting (WB) shows no significant change in protein expression of syndecan-1 after 24 and 36 hpi. HCE cells were infected with KOS-WT at an MOI of 0.1. (B, right) Densitometry quantification of syndecan-1 normalized to GAPDH. The fold change of infected over uninfected cells is shown for each time point.
Upregulation of HPSE decreases cell surface syndecan-1 in human corneal epithelial cells.
Syndecan-1 has been reported to shed by numerous mechanisms (37, 38). Because HPSE is activated during corneal HSV-1 infection, we thought that it may play a role in mediating the effects of HSV-1 on syndecan-1 shedding (30). To test whether HPSE is responsible for syndecan-1 shedding, HCE cells were transfected with a plasmid containing the human HPSE-green fluorescent protein (GFP) gene. Flow cytometry was performed on the HPSE-transfected cells, and we found that the cells which overexpressed HPSE had less syndecan-1 present on their cell surfaces than cells transfected with an empty vector (Fig. 4A). Quantification of the flow results demonstrated that the HPSE-GFP cells had less than half of the syndecan-1 on the cell surface compared to the control cells (Fig. 4A). This ratio of cell surface syndecan-1 in HPSE-transfected to empty vector (EV)-transfected cells is remarkably similar to the ones seen in immortalized and primary HCE cells at 36 hpi compared to the uninfected group (Fig. 1B and C). Immunofluorescence microscopy using the HPSE-GFP cells also revealed a loss of syndecan-1 (Fig. 4B).
FIG 4.

HPSE upregulation decreases cell surface syndecan-1 in human corneal epithelial cells. (A) HCE cells were overexpressed with enhanced green fluorescent protein (EGFP)-HPSE, and the successfully transfected cells were isolated and stained with syndecan-1 antibody on the cell surface. Flow cytometry analysis shows that HPSE-overexpressed cells show decreased syndecan-1 when compared to cells transfected with only EGFP plasmid. MFI values for the EGFP- and EGFP-HPSE-transfected cells were 366,772.09 and 271,691.79, respectively. (B) Immunofluorescence microscopy shows decreased syndecan-1 expression on the surface of EGFP-HPSE-overexpressed cells when compared to control vector EGFP-overexpressed cells. Green indicates either EGFP HPSE or EGFP control vector, blue indicates 4′,6-diamidino-2-phenylindole (DAPI), and red indicates syndecan-1. Merged images are also shown.
MMP-3 and MMP-7 are upregulated upon HPSE overexpression or HSV-1 infection.
MMPs are sheddases that are capable of cleaving syndecans from the cell surface. Multiple studies have reported that MMP-1, -2, -3, -7, and -9 are involved with upregulation of HPSE in various cell lines (32, 39–41). Additionally, decreasing the levels of MMP-9 has been shown to ameliorate herpes simplex keratitis in vitro and in vivo (42, 43). However, these MMPs are specific to certain cell types. To investigate which of the MMPs are upregulated in HCE cells, we transfected the HPSE plasmid into HCE cells and measured the number of MMP transcripts produced 48 h posttransfection using qPCR. Out of the 5 MMPs selected, only MMP-3 and MMP-7 significantly increased upon HPSE overexpression (Fig. 5A). Because HSV-1 upregulates HPSE expression in HCE cells, we wanted to test whether infecting cells would similarly increase their expression of these MMPs. Upon infection with HSV-1, we observed a similar increase in MMP-3 and MMP-7 in HCE cells (Fig. 5B). We then wanted to investigate whether the rise in MMP transcripts led to an increase in active, functional enzymes on the cell surface. Both infected and uninfected cells were stained with either MMP-3 or MMP-7 antibodies. Using flow cytometry and immunofluorescence microscopy, we observed significant increases in both MMP-3 and MMP-7 on the infected cell surfaces (Fig. 5C to F). Quantification of MMP staining from the microscopy images demonstrated a highly significant (P < 0.0001) increase of both MMP-3 and MMP-7 on the surface of the HCE cells at 24 hpi (Fig. 5G and H). These results suggest a chain of events in which HSV-1 upregulates HPSE, which proceeds to increase MMP-3 and MMP-7 transcription, and these two MMPs migrate to the cell surface to facilitate the shedding of syndecan-1.
FIG 5.

MMP-3 and MMP-7 are upregulated upon HPSE overexpression or HSV-1 infection. (A) HPSE overexpression increases production of MMP-3 and MMP-7 transcripts. Cells were transfected with HPSE vector, and mRNA was collected 48 h posttransfection. Quantitative PCR was performed with the primers MMP-1, MMP-2, MMP-3, MMP-7, and MMP-9. (B) When cells were infected with HSV-1 for 24 h, the increase in MMP profile was similar to that of HPSE-overexpressed cells with significant increases in MMP-3 and MMP-7. (C) Flow cytometry analysis shows progressively increased expression of MMP-3 on the surface of HCE cells when infected with HSV-1. The gray histogram shows the isotype antibody; black indicates uninfected, and red shows infected cell populations. Cells were analyzed at 24 hpi. (D) Flow cytometry analysis shows progressively increased expression of MMP-7 on the surface of HCE cells when infected with HSV-1. The gray histogram shows the isotype antibody; black and red indicate uninfected and infected cell populations, respectively. Cells were analyzed at 24 hpi. (E) Immunofluorescence imaging shows a similar increase in the expression of MMP-3 on the surface of infected cells. Red represents surface MMP-3, and blue shows 4′,6-diamidino-2-phenylindole (DAPI). Images were taken at 0 and 36 hpi. (F) Immunofluorescence imaging shows a similar increase in the expression of MMP-7 on the surface of infected cells. Red represents surface MMP-7, and blue shows DAPI. Images were taken at 0 and 36 hpi. (G) Quantification of the immunofluorescence results demonstrates a significant increase in surface MMP3 expression at 36 hpi. (H) Quantification of the immunofluorescence results demonstrates a significant increase in surface MMP7 expression at 36 hpi.
DISCUSSION
HSV-1 infection can cause many mucosal and systemic diseases, including infection of the eye (44). Depending on the location of viral replication, the latter can manifest itself as corneal ulcers, keratitis, conjunctivitis, iridocyclitis, and acute retinal necrosis (45, 46). One major process underlying the spread of HSV-1 throughout the eye is the transmission of viral progeny from infected cells to nearby healthy cells (9, 47). It is known that HSV-1 uses ubiquitously expressed HS moieties on heparan sulfate proteoglycans as attachment receptors for infection of epithelial cells in the cornea (19). Additionally, it has been shown that heparanase, which is upregulated by HSV-1 during infection, aids in viral egress by clipping heparan sulfate chains on the cell surface (30). Both the loss of HS moieties and the upregulation of HPSE are seen in the later stages of infection at both 24 and 36 hpi (30). For more efficient transmission of viral progeny to the surroundings, we hypothesized that the entire syndecan-1 proteoglycan is shed during HSV-1 infection. The loss of HS from the cell surface may be due in part to the cleavage of the whole syndecan-1 proteoglycan, not just individual HS chains. Additionally, a previous study has reported that the upregulation of enzymatically active HPSE was sufficient to enhance the rate of syndecan-1 shedding in myeloma cells (38). We thought that a parallel mechanism may be at work in HCE cells during HSV-1 infection.
We have discovered that when infected with HSV-1, HCE cells upregulate HPSE, which stimulates the expression of the enzymes MMP-3 and MMP-7. These MMPs have been shown to cleave syndecan-1 ectodomains, located either 6- or 15-amino-acid residues from the transmembrane domain (34). The mechanism by which HPSE upregulates MMP-3 and MMP-7 in HCE cells could be mediated by the ERK signaling pathway. In multiple myeloma cells, the upregulation of HPSE stimulated ERK signaling and subsequently the expression of MMPs (32, 48). In the model proposed by Purushothaman et al., enzymatically active HPSE stimulates the phosphorylation of ERK, which proceeds to activate MMP-9 (32). MMP-9 then cleaves syndecan-1 proteoglycans from the surface of myeloma cells (32). A similar mechanism could be present in HCE cells, with the activation of MMP-3 and MMP-7 facilitating the cleavage of syndecan-1. Activation of ERK signaling can be initiated by the binding of growth factors to specific receptors on the cell (49). Since HS can bind and interact with a variety of growth factors, its cleavage by heparanase may release ligands for their attachment to an appropriate receptor and begin an ERK signaling cascade (48, 50–52).
As we demonstrate in this study, the upregulation of HPSE is sufficient to stimulate MMP-3 and MMP-7 activity, which increases the rate of syndecan-1 shedding. We propose that the stimulation of this signaling pathway occurs as the result of the clipping of heparan sulfate chains by heparanase. As heparanase cleaves HS chains on syndecan-1, cryptic epitopes within the heparan sulfate chains or the ectodomain of the core protein are exposed and may interact with new ligands to facilitate signaling (53, 54). Not only does HSV-1 deplete HS on the surface of an infected cell, but the downstream signaling resulting from the cleavage of HS also leads to the snipping of the syndecan-1 protein. The loss of HS via HPSE overexpression has been reported to decrease viral binding at the cell surface, reduce the number of new virions that can attach to the exterior of the cell, and increase viral egress (30). Previous work suggests that HSV-1 infection can be divided into an “attachment phase” during entry when HS is abundant and a “detachment phase” during egress when HS levels are diminished (30). The findings of this study expand the current model and demonstrate that the entire HSPG, not just individual HS chains, is initially abundant and subsequently lost during the attachment and detachment phases, respectively. Because one syndecan-1 protein can bind to three HS chains, it may be more efficient for the virus to promote shedding of entire proteoglycans instead of only the HS moieties on them (19, 20).
Several studies have described how matrix metalloproteinases regulate the inflammatory response and that MMPs are upregulated in nearly every case of inflamed human tissue (40, 55–57). The HSV-1-induced upregulation of MMP-3 and MMP-7 may be responsible for the inflammation observed in ocular tissues. Consistent with these findings, overexpression of HPSE has been shown to stimulate the inflammatory response in vivo, manifesting as swollen ipsilateral lymph nodes and delayed wound healing (31). It is known that the γ34.5 gene, transcribed in the later stages of lytic HSV-1 infection, is required for the upregulation of HPSE (31). Taken together, the findings suggest that once HSV-1 infects a cell and produces the γ34.5 gene product, HPSE becomes upregulated and proceeds to increase MMP-3 and MMP-7 expression. These two MMPs facilitate syndecan shedding and likely play a major role in creating inflammatory symptoms during an ocular HSV-1 infection. Aside from observing the phenotype of an upregulation of HPSE via infection or transfection, we also tested the effects of a broad-spectrum MMP inhibitor, Batimastat, on HCE cells during infection. Plaque assay results for viral release revealed a 2-fold decrease in plaques in cells incubated with Batimastat compared to a dimethyl sulfoxide (DMSO) control (data not shown).
In addition to facilitating viral egress, syndecan shedding can result in several angiogenic factors to be released, resulting in the formation of new blood vessels (58, 59). Syndecans are involved in the formation of chemokine gradients, and they serve as coreceptors for growth factors such as fibroblast growth factor 2 (FGF-2) (48). In addition, matrix metalloproteinases have been implicated in angiogenesis by liberating vascular endothelial growth factor (VEGF) and insulin-like growth factor-1 (IGF-1) from the extracellular matrix (60, 61). HPSE-mediated stimulation of MMPs could be a powerful driver of angiogenesis (62). Corneal neovascularization is a major complication of herpes simplex keratitis, and syndecan-1 shedding could play a key role in this pathophysiology (44). Inhibition of VEGF has been shown to reduce the severity of corneal neovascularization, and similar results may arise from preventing MMP-3 and MMP-7 from degrading syndecan-1 in corneal cells (63).
Future studies should investigate the mechanism by which HPSE upregulates these two MMPs and whether the inhibition of syndecan-1 shedding reduces the severity of corneal disease in murine models. In conclusion, we propose that the shedding of syndecan-1 promotes viral egress by reducing the amount of viral progeny trapped near the cell surface by heparan sulfate moieties. This is produced by the initial activation of HPSE by HSV-1 and its upregulation of MMP-3 and MMP-7, which translocate to the cellular membranes and cleave the ectodomain of syndecan-1. Upregulation of MMP-3 and MMP-7 may highlight new molecular markers for the infection and future therapeutic targeting of matrix metalloproteinases and other members of this HPSE-mediated pathway may prove to be an effective new method of controlling HSV-1 infections.
MATERIALS AND METHODS
Cell and viruses.
A human corneal epithelial cell line (RCB1834 HCE-T) was obtained from Kozaburo Hayashi (National Eye Institute, Bethesda, MD) and cultured in minimal essential medium (MEM) (Life Technologies, Carlsbad, CA) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All infections were performed with HSV-1 KOS-wild type (WT) or 17-GFP at a multiplicity of infection (MOI) of 0.1 on HCE cells unless mentioned otherwise.
Antibodies, plasmids, enzymes, and drugs.
HPSE antibody HP130 (Advanced Targeting Systems, San Diego, CA) was used for Western blot analysis (1:200), imaging (1:100), and flow cytometry studies (1:100). The syndecan-1 antibody (catalog no. sc-6532; Santa Cruz Biotechnology) was used for Western blotting, a supernatant slot blot, and immunofluorescence microscopy. MMP-3 (clone 50647; Novus Biologicals) and MMP-7 (catalog no. AF907; R&D Systems) antibodies were used for immunofluorescence microscopy. Human HPSE expression plasmid pIRES2 EGFP-HPSE1 and the control empty vector pIRES2 EGFP plasmid (64) were provided by Dr. Ralph Sanderson (University of Alabama at Birmingham, Birmingham, AL). Transfection efficiencies ranged from 50–60%. The MMP inhibitor Batimastat (BB-94; Selleckchem) was used for a plaque assay.
Western blot analysis.
Proteins from samples in this study were collected using a radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s protocol. After gel electrophoresis, membranes were blocked in 5% milk/Tris-buffered saline with Tween 20 (TBS-T) for 1 h followed by incubation with the primary antibody overnight. After washes and incubation with respective horseradish peroxidase-conjugated secondary antibodies (anti-mouse, 1:10,000; anti-rabbit, 1:20,000) for 1 h, protein bands were visualized using the SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific, Waltham, MA) with an ImageQuant LAS 4000 biomolecular imager (GE Healthcare Life Sciences, Pittsburgh, PA). The densities of the bands were quantified using the ImageQuant TL image analysis software (GE Healthcare Life Sciences) (65).
Supernatant slot blot.
Cell culture supernatants of infected or mock-treated HCE cells were vacuum filtered through Immobilon-Ny+ nylon membrane (Millipore Corporation, Billerica, MA) using the Bio-Dot SF apparatus (Bio-Rad, Hercules, CA). Membranes were blotted overnight with an antibody specific for syndecan-1 (catalog no. sc-6532; Santa Cruz Biotechnology) followed by species-specific secondary antibody and chemiluminescence detection using the ImageQuant LAS 4000 biomolecular imager, as described above (30).
PCR.
RNA was extracted from cells using TRIzol (Life Technologies) according to the manufacturer’s protocol. RNA was then transcribed to DNA using the High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Real-time quantitative PCR was performed using Fast SYBR green master mix (Applied Biosystems) using QuantStudio 7 Flex (Applied Biosystems) (31). The following primers were purchased from Integrated DNA Technologies: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-TGCACCACCAACTGCTTA-3′, and reverse, 5′-GGATGCAGGGATGATGTTC-3′), MMP-1 (forward, 5′-GGCTGTTTTGTACTGCCTGCT-3′, and reverse, 5′-AGGAGACACAGGCTCTAGGGAA-3′), MMP-2 (forward, 5′-AGGACTACGACCGCGACAAG-3′, and reverse, 5′-GTTCCCACCAACAGTGGACAT-3′), MMP-3 (forward, 5′-AACCTGTCCCTCCAGAACCT-3′, and reverse, 5′-CAGCATCAAAGGACAAAGCA-3′), MMP-7 (forward, 5′-GTATGGGACATTCCTCTGATCC-3′, and reverse, 5′-CCAATGAATGAATGAATG GATG-3′), and MMP-9 (forward, 5′-GGCGCTCATGTACCCTATGT-3′, and reverse, 5′-GCCATTCACGTCGTCCTTAT-3′).
Flow cytometry.
Syndecan-1, MMP3, and MMP-7 cell surface expression was detected after HSV-1 KOS-WT infection. Monolayers of HCE cells were infected at 0.1 MOI and harvested at 24 h and 36 h postinfection. For the detection of protein on the cell surface, HCE cells were harvested and incubated with 3 μg of primary antibody per sample. The samples were then diluted in PBS with 1% bovine serum albumin (BSA) for 1 h at 4°C and incubated with fluorescein isothiocyanate (FITC)-conjugated or allophycocyanin (APC)-conjugated anti-mouse IgG diluted to 1:100 for 30 min at 4°C. Cells stained with their respective secondary antibodies were used as background controls. Entire cell populations were used for the mean fluorescence intensity calculations (65).
Immunofluorescence microscopy.
HCE cells were cultured in glass-bottom dishes (MatTek Corporation, Ashland, MA). Cells were fixed in 10% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 10 min for intracellular labeling. This was followed by incubation with the primary antibody for 2 h at 4°C. When a secondary antibody was needed, cells were incubated with FITC-conjugated secondary antibody at a dilution of 1:150 for 1 h and were examined under the Zeiss 710 confocal microscope (Germany) using a × 63 oil immersion objective lens. The pinhole was set to 1 Airy unit. For cell surface staining, cells were incubated with their respective primary and secondary antibodies before fixation with paraformaldehyde for imaging. The fluorescence intensity of the images was calculated using ImageJ software (30).
Human corneas.
Human donor corneoscleral buttons were obtained from the Illinois Eye Bank, Chicago, IL. The corneas had unknown seropositivity for HSV and came from anonymous donors. The corneal stem cells were extracted and cultured in a keratinocyte serum-free medium (SFM). After 3 days, the medium was changed to MEM, which allowed for the differentiation of stem cells into corneal epithelial cells. Infection of these cells was done at an MOI of 0.1 with HSV-1 KOS-WT for 36 h (66).
Statistics.
The data were analyzed using GraphPad Prism version 6.01 for Windows 10 (GraphPad Software, La Jolla, CA). Error bars of all figures represent the standard error of the mean (SEM) of three independent experiments (n = 3), unless otherwise specified. Asterisks denote a significant difference as determined by Student's t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (P values are for Fig. 1B and D, Fig. 2B, and Fig. 5A, B, and G, and H).
ACKNOWLEDGMENTS
This work was supported by RO1 grants from the NIH (EY029426 and EY024710) to D.S. and a core grant (P30 EY001792).
We acknowledge Ruth Zhelka for help with using the departmental imaging facilities.
REFERENCES
- 1.Whitley RJ. 1996. Herpesviruses In Baron S. (ed), Medical microbiology, 4th ed. University of Texas Medical Branch at Galveston, Galveston, TX. [PubMed] [Google Scholar]
- 2.Xu F, Schillinger JA, Sternberg MR, Johnson RE, Lee FK, Nahmias AJ, Markowitz LE. 2002. Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988–1994. J Infect Dis 185:1019–1024. doi: 10.1086/340041. [DOI] [PubMed] [Google Scholar]
- 3.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. doi: 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 5.Whitley R, Kimberlin DW, Prober CG. 2007. Pathogenesis and disease In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K and (eds), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge. [PubMed] [Google Scholar]
- 6.Farooq AV, Shah A, Shukla D. 2010. The role of herpesviruses in ocular infections. Virus Adapt Treat 2:115–123. doi: 10.2147/VAAT.S9500. [DOI] [Google Scholar]
- 7.Wald A, Corey L. 2007. Persistence in the population: epidemiology, transmission In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K and (eds), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge. [PubMed] [Google Scholar]
- 8.Jaggi U, Wang S, Tormanen K, Matundan H, Ljubimov AV, Ghiasi H. 2018. Role of herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) pathogenic CD8+ T cells in exacerbation of eye disease. Front Immunol 9:2895. doi: 10.3389/fimmu.2018.02895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farooq AV, Valyi-Nagy T, Shukla D. 2010. Mediators and mechanisms of herpes simplex virus entry into ocular cells. Curr Eye Res 35:445–450. doi: 10.3109/02713681003734841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas J, Rouse BT. 1997. Immunopathogenesis of herpetic ocular disease. Immunol Res 16:375–386. doi: 10.1007/BF02786400. [DOI] [PubMed] [Google Scholar]
- 11.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/jvi.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trybala E, Liljeqvist J, Svennerholm B, Bergström T. 2000. Herpes simplex virus types 1 and 2 differ in their interaction with heparan sulfate. J Virol 74:9106–9114. doi: 10.1128/jvi.74.19.9106-9114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacsa S, Karasneh G, Dosa S, Liu J, Valyi-Nagy T, Shukla D. 2011. Syndecan-1 and syndecan-2 play key roles in herpes simplex virus type-1 infection. J Gen Virol 92:733–743. doi: 10.1099/vir.0.027052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banfield BW, Leduc Y, Esford L, Schubert K, Tufaro F. 1995. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J Virol 69:3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Donnell CD, Shukla D. 2008. The importance of heparan sulfate in herpesvirus infection. Virol Sin 23:383–393. doi: 10.1007/s12250-008-2992-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esko JD, Lindahl U. 2001. Molecular diversity of heparan sulfate. J Clin Invest 108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tumova S, Woods A, Couchman JR. 2000. Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol 32:269–288. doi: 10.1016/S1357-2725(99)00116-8. [DOI] [PubMed] [Google Scholar]
- 18.Muto T, Miyoshi K, Munesue S, Nakada H, Okayama M, Matsuo T, Noma T. 2007. Differential expression of syndecan isoforms during mouse incisor amelogenesis. J Med Invest 54:331–339. doi: 10.2152/jmi.54.331. [DOI] [PubMed] [Google Scholar]
- 19.Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest 108:503–510. doi: 10.1172/JCI200113799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langford JK, Stanley MJ, Cao D, Sanderson RD. 1998. Multiple heparan sulfate chains are required for optimal syndecan-1 function. J Biol Chem 273:29965–29971. doi: 10.1074/jbc.273.45.29965. [DOI] [PubMed] [Google Scholar]
- 21.Szatmári T, Ötvös R, Hjerpe A, Dobra K. 2015. Syndecan-1 in cancer: implications for cell signaling, differentiation, and prognostication. Dis Markers 2015:796052. doi: 10.1155/2015/796052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karasneh GA, Shukla D. 2011. Herpes simplex virus infects most cell types in vitro: clues to its success. Virol J 8:481. doi: 10.1186/1743-422X-8-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rautemaa R, Helander T, Meri S. 2002. Herpes simplex virus 1 infected neuronal and skin cells differ in their susceptibility to complement attack. Immunology 106:404–411. doi: 10.1046/j.1365-2567.2002.01421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiwari V, Liu J, Valyi-Nagy T, Shukla D. 2011. Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J Biol Chem 286:25406–25415. doi: 10.1074/jbc.M110.201103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivara S, Milazzo FM, Giannini G. 2016. Heparanase: a rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med Chem 8:647–680. doi: 10.4155/fmc-2016-0012. [DOI] [PubMed] [Google Scholar]
- 26.Bame KJ. 2001. Heparanases: endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology 11:91R–98R. doi: 10.1093/glycob/11.6.91R. [DOI] [PubMed] [Google Scholar]
- 27.Vlodavsky I, Singh P, Boyango I, Gutter-Kapon L, Elkin M, Sanderson RD, Ilan N. 2016. Heparanase: from basic research to therapeutic applications in cancer and inflammation. Drug Resist Updat 29:54–75. doi: 10.1016/j.drup.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanderson RD, Elkin M, Rapraeger AC, Ilan N, Vlodavsky I. 2017. Heparanase regulation of cancer, autophagy and inflammation: new mechanisms and targets for therapy. FEBS J 284:42–55. doi: 10.1111/febs.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fux L, Ilan N, Sanderson RD, Vlodavsky I. 2009. Heparanase: busy at the cell surface. Trends Biochem Sci 34:511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, Shukla D. 2015. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat Commun 6:6895. doi: 10.1038/ncomms7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agelidis AM, Hadigal SR, Jaishankar D, Shukla D. 2017. Viral activation of heparanase drives pathogenesis of herpes simplex virus-1. Cell Rep 20:439–450. doi: 10.1016/j.celrep.2017.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purushothaman A, Chen L, Yang Y, Sanderson RD. 2008. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem 283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ilan N, Elkin M, Vlodavsky I. 2006. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol 38:2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Manon-Jensen T, Multhaupt HAB, Couchman JR. 2013. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J 280:2320–2331. doi: 10.1111/febs.12174. [DOI] [PubMed] [Google Scholar]
- 35.Manon-Jensen T, Itoh Y, Couchman JR. 2010. Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J 277:3876–3889. doi: 10.1111/j.1742-4658.2010.07798.x. [DOI] [PubMed] [Google Scholar]
- 36.Chen P, Abacherli LE, Nadler ST, Wang Y, Li Q, Parks WC. 2009. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting a2ß1 integrin activation. PLoS One 4:e6565. doi: 10.1371/journal.pone.0006565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Macleod V, Miao H, Theus A, Zhan F, Shaughnessy JD, Sawyer J, Li J, Zcharia E, Vlodavsky I, Sanderson RD. 2007. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem 282:13326–13333. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 39.Hirata A, Katayama K, Tsuji T, Natsume N, Sugahara T, Koga Y, Takano K, Otsuki Y, Nakamura H. 2013. Heparanase localization during palatogenesis in mice. Biomed Res Int 2013:760236. doi: 10.1155/2013/760236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi W, Liu J, Li M, Gao H, Wang T. 2010. Expression of MMP, HPSE, and FAP in stroma promoted corneal neovascularization induced by different etiological factors. Curr Eye Res 35:967–977. doi: 10.3109/02713683.2010.502294. [DOI] [PubMed] [Google Scholar]
- 41.Zcharia E, Jia J, Zhang X, Baraz L, Lindahl U, Peretz T, Vlodavsky I, Li J-P. 2009. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS One 4:e5181. doi: 10.1371/journal.pone.0005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heiligenhaus A, Li HF, Yang Y, Wasmuth S, Steuhl KP, Bauer D. 2005. Transplantation of amniotic membrane in murine herpes stromal keratitis modulates matrix metalloproteinases in the cornea. Invest Ophthalmol Vis Sci 46:4079–4085. doi: 10.1167/iovs.05-0192. [DOI] [PubMed] [Google Scholar]
- 43.Rajasagi NK, Bhela S, Varanasi SK, Rouse BT. 2017. Frontline Science: aspirin-triggered resolvin D1 controls herpes simplex virus-induced corneal immunopathology. J Leukoc Biol 102:1159–1171. doi: 10.1189/jlb.3HI1216-511RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koganti R, Yadavalli T, Shukla D. 2019. Current and emerging therapies for ocular herpes simplex virus type-1 infections. Microorganisms 7:429. doi: 10.3390/microorganisms7100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowe AM, St Leger AJ, Jeon S, Dhaliwal DK, Knickelbein JE, Hendricks RL. 2013. Herpes keratitis. Prog Retin Eye Res 32:88–101. doi: 10.1016/j.preteyeres.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaye S, Choudhary A. 2006. Herpes simplex keratitis. Prog Retin Eye Res 25:355–380. doi: 10.1016/j.preteyeres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Akhtar J, Shukla D. 2009. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J 276:7228–7236. doi: 10.1111/j.1742-4658.2009.07402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zong F, Fthenou E, Wolmer N, Hollósi P, Kovalszky I, Szilák L, Mogler C, Nilsonne G, Tzanakakis G, Dobra K. 2009. Syndecan-1 and FGF-2, but not FGF receptor-1, share a common transport route and co-localize with heparanase in the nuclei of mesenchymal tumor cells. PLoS One 4:e7346. doi: 10.1371/journal.pone.0007346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blume-Jensen P, Hunter T. 2001. Oncogenic kinase signalling. Nature 411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 50.Rapraeger AC, Krufka A, Olwin BB. 1991. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 51.Forsten-Williams K, Chu C, Fannon M, Buczek-Thomas J, Nugent M. 2008. Control of growth factor networks by heparan sulfate proteoglycans. Ann Biomed Eng 36:2134–2148. doi: 10.1007/s10439-008-9575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Billings PC, Pacifici M. 2015. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: mechanisms and mysteries. Connect Tissue Res 56:272–280. doi: 10.3109/03008207.2015.1045066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramani VC, Pruett PS, Thompson CA, DeLucas LD, Sanderson RD. 2012. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem 287:9952–9961. doi: 10.1074/jbc.M111.330803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma P, Beck SL, Raab RW, McKown RL, Coffman GL, Utani A, Chirico WJ, Rapraeger AC, Laurie GW. 2006. Heparanase deglycanation of syndecan-1 is required for binding of the epithelial-restricted prosecretory mitogen lacritin. J Cell Biol 174:1097–1106. doi: 10.1083/jcb.200511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manicone AM, McGuire JK. 2008. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol 19:34–41. doi: 10.1016/j.semcdb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nissinen L, Kähäri V. 2014. Matrix metalloproteinases in inflammation. Biochim Biophys Acta 1840:2571–2580. doi: 10.1016/j.bbagen.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 57.Chen Q, Jin M, Yang F, Zhu J, Xiao Q, Zhang L. 2013. Matrix metalloproteinases: inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm 2013:1. doi: 10.1155/2013/928315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamorte S, Ferrero S, Aschero S, Monitillo L, Bussolati B, Omedè P, Ladetto M, Camussi G. 2012. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia 26:1081–1090. doi: 10.1038/leu.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Rossi G, Whiteford JR. 2014. Syndecans in angiogenesis and endothelial cell biology. Biochem Soc Trans 42:1643–1646. doi: 10.1042/BST20140232. [DOI] [PubMed] [Google Scholar]
- 60.Kreuger J, Phillipson M. 2016. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat Rev Drug Discov 15:125–142. doi: 10.1038/nrd.2015.2. [DOI] [PubMed] [Google Scholar]
- 61.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. 1999. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. 2001. Heparanase as mediator of angiogenesis: mode of action. FASEB J 15:1661–1663. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- 63.Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. 2001. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol 75:9828–9835. doi: 10.1128/JVI.75.20.9828-9835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelly T, Miao H, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. 2003. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res 63:8749–8756. [PubMed] [Google Scholar]
- 65.Hopkins J, Yadavalli T, Agelidis AM, Shukla D. 2018. Host Enzymes heparanase and cathepsin L promote herpes simplex virus 2 release from cells. J Virol 92:e01179-18. doi: 10.1128/JVI.01179-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Djalilian A, Milani B, Sagha H, Hematti P. 2013. Bone marrow derived mesenchymal stem cells for reconstructing the limbal niche. Invest Ophthalmol Vis Sci 54:557–557. [Google Scholar]