

Recurring ocular infections caused by HSV-1 can cause corneal scarring and blindness. A major function of the HSV-1 latency-associated transcript (LAT) is to establish high levels of latency and reactivation, thus contributing to the development of eye disease. Here, we show that the host CD80 T cell costimulatory molecule functions similarly to LAT and can restore the ability of LAT to establish latency, reactivation, and immune exhaustion as well as induce the expression of caspase 3, caspase 8, caspase 9, and Bcl2. Our results suggest that, in contrast to several other previously tested genes, CD80-expressing virus can completely compensate for all known and tested LAT functions.
KEYWORDS: corneal scarring, T cells, primary infection, exhaustion, apoptosis, latency, ocular infection, reactivation
ABSTRACT
High rates of wild-type (WT) herpes simplex virus 1 (HSV-1) latency reactivation depend on the anti-apoptotic activities of latency-associated transcript (LAT). Replacing LAT with the baculovirus inhibitor of apoptosis protein (cpIAP) or cellular FLIP (FLICE-like inhibitory protein) gene restored the WT latency reactivation phenotype to that of a LAT-minus [LAT(−)] virus, while similar recombinant viruses expressing interleukin-4 (IL-4) or interferon gamma (IFN-γ) did not. However, HSV-1 recombinant virus expressing cpIAP did not restore all LAT functions. Recently, we reported that a similar recombinant virus expressing CD80 in place of LAT had higher latency reactivation than a LAT-null virus. The present study was designed to determine if this CD80-expressing recombinant virus can restore all LAT functions as observed with WT virus. Our results suggest that overexpression of CD80 fully rescues LAT function in latency reactivation, apoptosis, and immune exhaustion, suggesting that LAT and CD80 have multiple overlapping functions.
IMPORTANCE Recurring ocular infections caused by HSV-1 can cause corneal scarring and blindness. A major function of the HSV-1 latency-associated transcript (LAT) is to establish high levels of latency and reactivation, thus contributing to the development of eye disease. Here, we show that the host CD80 T cell costimulatory molecule functions similarly to LAT and can restore the ability of LAT to establish latency, reactivation, and immune exhaustion as well as induce the expression of caspase 3, caspase 8, caspase 9, and Bcl2. Our results suggest that, in contrast to several other previously tested genes, CD80-expressing virus can completely compensate for all known and tested LAT functions.
INTRODUCTION
Eye disease associated with ocular herpes simplex virus 1 (HSV-1) infection is a major cause of herpes keratitis that can lead to blindness (1–3). The incidence of eye disease associated with HSV-1 is globally on the rise (4), and over 90% of herpes-associated eye disease is associated with HSV-1 rather than HSV-2 infection (2, 3, 5–7).
Although the cornea is considered an immune-privileged site, HSV-1 ocular infections lead to the recruitment of inflammatory infiltrates into the cornea (8). This results in the activation of CD4+ and CD8+ T cells, which are important for controlling primary virus infection (9). T cell activation occurs following the binding of CD28 on the T cell surface to CD80 (B7-1) or CD86 (B7-2) on antigen-presenting cells (APCs) (10). This binding induces T cell proliferation, differentiation, and cytokine secretion (11, 12). However, the activation of T cells is responsible for initiating eye disease (9, 13–17). After initial infection, HSV-1 travels to the trigeminal ganglia (TG), where it establishes lifelong latency. HSV-1 can periodically reactivate from the TG, and repeated reactivation leads to corneal scarring (18, 19). The HSV-1 latency-associated transcript (LAT) is necessary for high levels of latency and reactivation. LAT is the only HSV-1 transcript abundantly expressed during latency (20, 21). It is well established that LAT controls apoptosis (22–29), which likely occurs via the type I interferon (IFN) pathway (30). In addition, LAT is responsible for CD8+ T cell immune exhaustion, which is also essential for latency establishment (31, 32). The anti-apoptotic activity of LAT is crucial for latency reactivation because the replacement of LAT with other antiapoptotic genes, such as the bovine herpesvirus 1 (BHV-1) latency-related (LR) gene (23), the open reading frame (ORF) of the baculovirus inhibitor of apoptosis protein gene (cpIAP) (27), or cellular FLICE-like inhibitory protein (cFLIP) (29, 33), can restore wild-type (WT) reactivation to a LAT-null mutant of HSV-1 strain McKrae. However, the mechanism by which LAT controls apoptosis is likely unique, because the replacement of LAT with cpIAP does not rescue the expression of caspase 8 or Bcl2 (30). Furthermore, infection of mice with a recombinant virus expressing cpIAP in place of LAT did not restore LAT-dependent immune exhaustion (30).
We recently showed that viral expression of CD80, a host T cell costimulatory molecule, restores latency and reactivation in a LAT-null virus (34). We also demonstrated that CD80, but not CD86, is downregulated upon HSV-1 ocular infection in mice (35). This downregulation was dependent on ICP22 binding to the CD80 promoter (35, 36). Suppression of CD80 delayed viral clearance but also dampened the inflammatory response, thus reducing damage to the eye (35, 36). Our findings are exciting as they establish a novel pathway used by HSV-1 to escape immune clearance. Our results also suggested that CD80 has overlapping functions with LAT.
The goal of the present study was to establish whether CD80 can compensate for other known LAT functions. We used a recombinant virus expressing CD80 under the control of the LAT promoter in place of LAT to compare levels of virus replication, latency, reactivation, immune exhaustion, and apoptosis markers in C57BL/6 mice ocularly infected with HSV-CD80 {LAT-minus [LAT(−)]} or WT McKrae {LAT-plus [LAT(+)]}. C57BL/6 mice were used in this study because they are refractory to HSV-1 infection. Our results showed that despite displaying similar replication in mouse tears, animals infected with HSV-CD80 virus had significantly more corneal scarring (CS) than did those infected with WT McKrae. We also found that HSV-CD80 (LAT-minus) recombinant virus displayed levels of latency and reactivation similar to those of WT McKrae virus. Similar to LAT, CD80 was also expressed in the TG of HSV-CD80 latently infected mice. Additionally, CD80 restored T cell exhaustion in the absence of LAT. Together, these results demonstrate that CD80 and LAT share a remarkable amount of functional overlap.
RESULTS
Replication of HSV-CD80 recombinant virus in mouse tears is similar to that of WT McKrae virus.
Viral replication during ocular infection was measured by determining PFU in the tear films of ocularly infected mice. C57BL/6 mice were infected ocularly with 2 × 105 PFU/eye of HSV-CD80 and McKrae viruses as described in Materials and Methods. Tear films were collected daily for 7 days, and the amount of virus in tear films was determined using standard plaque assays on rabbit skin (RS) cells. Virus titers in the HSV-CD80-infected mouse group were similar to those in the WT McKrae-infected group (P > 0.05 at all time points) (Fig. 1). Virus titers peaked on day 5 postinfection (p.i.) and declined thereafter. Thus, the replacement of LAT with CD80 did not affect viral replication. Similar results were obtained in BALB/c mice (34).
FIG 1.

Virus replication in the eyes of infected mice. Mice were ocularly infected with 2 × 105 PFU/eye of HSV-CD80 [LAT(−)] or McKrae [LAT(+)] virus without corneal scarification. Tear films were collected on days 1 to 7 p.i., and virus titers were determined by standard plaque assays. Each point represents the mean titers for 30 eyes from two separate experiments.
Overexpression of CD80 does not affect viral load during acute infection.
To investigate the effects of CD80 overexpression on viral load, mice were infected ocularly as described above. TG and cornea were harvested from infected mice on day 4 p.i., and the gB copy number was determined by quantitative reverse transcriptase PCR (qRT-PCR). The HSV-1 gB copy number in the TG and cornea of mice infected with HSV-CD80 was similar to that seen in WT McKrae-infected mice {P > 0.05 for HSV-CD80 [LAT(−)] versus McKrae [LAT(+)]} (Fig. 2A).
FIG 2.
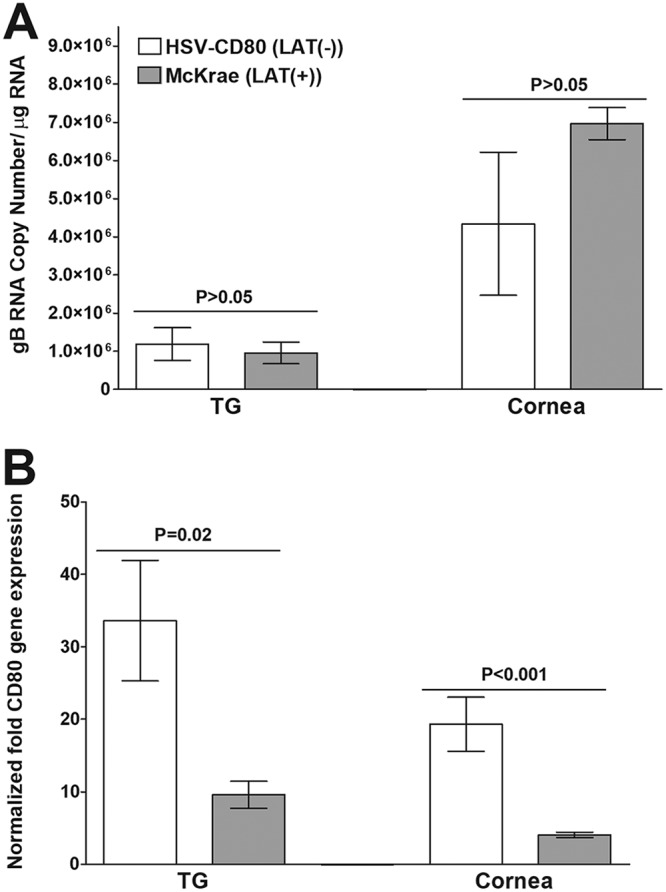
Effect of CD80 overexpression on viral gB copy number and CD80 expression on day 4 p.i. Mice were ocularly infected with 2 × 105 PFU/eye of HSV-CD80 [LAT(−)] and McKrae [LAT(+)] viruses as described in the Fig. 1 legend. (A) gB RNA copy number. On day 4 p.i., corneas and TG were harvested from infected mice. qPCR was performed on individual mouse corneas and TG. TG and corneas from one mouse were pooled. In each experiment, the estimated relative gB copy number was calculated using standard curves generated using pAC-gB1 (75). Each point represents the mean ± the standard error of the mean (SEM) from 20 samples. (B) Relative CD80 gene expression in mice infected ocularly with HSV-CD80 [LAT(−)] and WT McKrae [LAT(+)] viruses. On day 4 p.i., corneas and TG were harvested from infected mice. TG and corneas from one mouse were pooled, and qRT-PCR was performed to estimate CD80 gene expression. GAPDH expression was used to normalize the relative expression of CD80 in TG and cornea. Each point represents the mean ± SEM from 20 samples.
We have shown previously that CD80 is expressed in the corneas and TG of BALB/c mice ocularly infected with HSV-CD80 virus (34). To verify that CD80 is also expressed in the corneas and TG of C57BL/6 mice ocularly infected with HSV-CD80 virus, qRT-PCR was performed on isolated RNA as described above (Fig. 2A), and CD80 expression was measured. As expected, the level of CD80 expression was significantly higher in the TG and cornea of HSV-CD80-infected mice than in WT McKrae-infected mice (P = 0.02 in TG and P < 0.001 in cornea) (Fig. 2B). Together, these results suggest that viral replication is not affected by overexpression of CD80 and verify that CD80 is expressed from the LAT promoter during acute ocular infection in C57BL/6 mice.
Exacerbation of corneal scarring in mice ocularly infected with HSV-CD80 recombinant virus.
We recently reported exacerbated eye disease in BALB/c mice infected with HSV-CD80 compared to mice infected with the parental, LAT(−) virus (35). In contrast to BALB/c mice, C57BL/6 mice are refractory to eye disease following ocular infection with HSV-1 strain McKrae or McKrae-derived recombinant viruses (37, 38). To determine whether HSV-CD80 infection exacerbates eye disease in C57BL/6 mice, mice were ocularly infected with 2 × 105 PFU per eye of HSV-CD80 or WT McKrae. The mortality rate of infected C57BL/6 mice was similar, and more than 95% of infected mice survived ocular infection (data not shown). Interestingly, HSV-CD80-infected mice developed significantly more CS than McKrae-infected mice recorded at 28 days p.i. (P < 0.05 by Student’s t test) (Fig. 3). Thus, similar to BALB/c mice (35), overexpression of CD80 in C57BL/6 mice, which are less susceptible to eye disease, also exacerbated eye disease compared with WT virus. These results confirm our overall finding that CD80 plays a pathogenic role in the eye (34–36). Thus, in contrast to LAT, CD80 overexpression exacerbates eye disease in ocularly infected mice.
FIG 3.
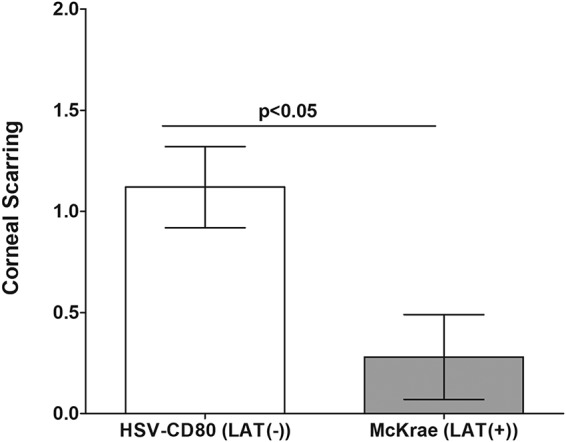
Effect of CD80 gene overexpression on corneal scarring (CS) on day 28 p.i. Mice were infected ocularly with HSV-CD80 [LAT(−)] and McKrae [LAT(+)] viruses as described in the Fig. 1 legend. CS in surviving mice was examined on day 28 p.i. as described in Materials and Methods. The CS score represents the average ± SEM from 30 eyes for each virus.
CD80 is expressed by the LAT promoter in TG of HSV-CD80-infected mice.
The above-described studies demonstrated that CD80 is expressed by HSV-CD80 virus during primary infection in cornea and TG of infected mice (Fig. 2B). To verify that CD80 is expressed in TG of latently infected mice, mice were infected ocularly as described above, and TG from infected mice were harvested on day 28 p.i. Levels of CD80 gene expression were determined by qRT-PCR. There was significantly more CD80 expression in HSV-CD80-infected mice than in WT McKrae-infected mice (P < 0.05) (Fig. 4A). Thus, similar to LAT, CD80 driven by the LAT promoter is expressed during latency.
FIG 4.

Effect of CD80 overexpression on CD80 expression and viral gB copy number on day 28 p.i. Mice were ocularly infected with 2 × 105 PFU/eye of HSV-CD80 [LAT(−)] and McKrae [LAT(+)] viruses as described in the Fig. 1 legend. (A) CD80 fold gene expression in mice infected ocularly with HSV-CD80 and WT McKrae virus. On day 28 p.i., TG were harvested from infected mice. TG from individual mice were extracted, and qRT-PCR was performed to measure CD80 gene expression. GAPDH expression was used to normalize relative CD80 expression in TG. Each bar represents the mean CD80 gene expression level ± SEM from 20 TG. (B) gB DNA copy number. On day 28 p.i., TG were harvested from infected mice. qPCR was performed on each individual mouse TG. In each experiment, the estimated gB relative copy number was calculated using standard curves generated using pAC-gB1 (75). Each bar represents the mean ± SEM from 20 TG.
In the absence of LAT, CD80 completely restores latency levels to those of LAT-plus virus.
LAT-null viruses do not establish latency as efficiently as WT viruses (18, 19, 31). We previously showed that BALB/c mice infected with HSV-CD80 had higher levels of latency than mice infected with the parental, LAT(−) virus (34). To determine if CD80 could rescue LAT function to establish latency, mice were ocularly infected as described above, and the gB copy number was determined in TG on day 28 p.i. by quantitative PCR (qPCR). There was no significant difference in gB copy numbers in mice infected with HSV-CD80 or WT McKrae {P > 0.05 for HSV-CD80 [LAT(−)] versus McKrae [LAT(+)]} (Fig. 4B), suggesting that CD80 overexpression could completely restore latency to the levels of WT McKrae virus in the absence of LAT.
CD80 rescues reactivation in the absence of LAT to WT McKrae levels.
It is well established that the presence of LAT is important for effective latency and reactivation in mice (18, 19, 31) and that latency levels correlate with the time of reactivation (39). To determine whether CD80 can rescue LAT function with regard to reactivation, TG were obtained from mice that had been ocularly infected with HSV-CD80 [LAT(−)] or McKrae [LAT(+)] virus as described above. Virus reactivation was analyzed by explanting individual TG from infected mice on day 28 p.i. as described in Materials and Methods. The reactivation time for HSV-CD80 [LAT(−)] virus was similar to that of WT McKrae [LAT(+)]-infected mice (4.9 ± 0.3 days for McKrae versus 4.1 ± 0.3 days for HSV-CD80; P > 0.05) (Fig. 5). These results suggest that CD80 can rescue the reactivation function of LAT.
FIG 5.
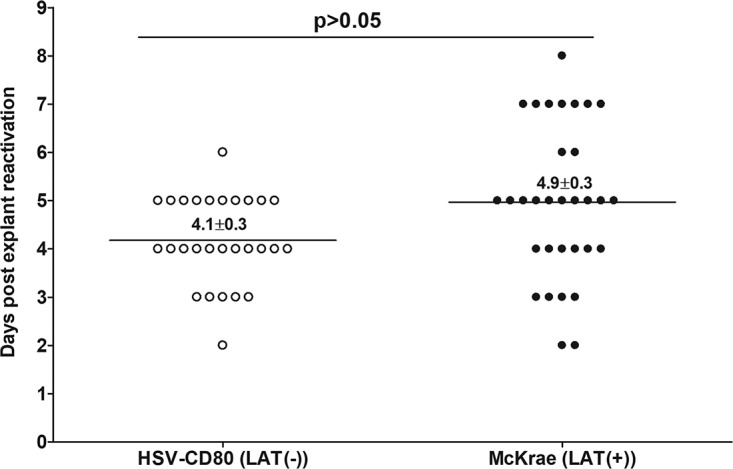
Effect of CD80 gene expression on the kinetics of induced reactivation in explanted TG from latently infected mice. TG from latently infected HSV-CD80 [LAT(−)] and McKrae [LAT(+)] mice were individually isolated on day 28 p.i. Each individual TG was incubated in 1.5 ml of tissue culture medium at 37°C. Medium aliquots were removed from each culture daily for up to 15 days and plated on indicator RS cells to assess the appearance of reactivated virus. The results are plotted as the number of TG that reactivated daily. Numbers indicate the average time that the TG from each group first showed cytopathic effect (CPE) ± SEM. Reactivation is based on 27 to 30 TG for each virus from two separate experiments.
CD80 overexpression rescues the T cell exhaustion function of LAT.
We have previously shown that HSV-1 infection results in LAT-dependent CD8+ T cell exhaustion (31, 40). To determine whether CD80 can replace LAT function in T cell exhaustion, the expression of the exhaustion markers CD4, CD8, Tim-3, PD-1 (programmed death 1), IFN-γ, tumor necrosis factor alpha (TNF-α), interleukin-2 (IL-2), and IL-21 was determined using qRT-PCR in TG of mice at day 28 after ocular infection, as described above. The expression levels of CD8, PD-1, TNF-α, and IL-2 genes were similar in the two infected groups {P > 0.05 for HSV-CD80 [LAT(−)] versus McKrae [LAT(+)]} (Fig. 6). In contrast, the expression levels of CD4, Tim-3, IFN-γ, and IL-21 transcripts in HSV-CD80-infected mice were higher than those in WT McKrae-infected mice {P < 0.05 for HSV-CD80 [LAT(−)] versus McKrae [LAT(+)]} (Fig. 6). These results indicate that while CD80 likely has functions beyond those of LAT, CD80 can restore T cell exhaustion in the absence of LAT.
FIG 6.

Effect of CD80 expression on T cell exhaustion in the TG of latently infected mice. TG from latently infected HSV-CD80 [LAT(−)] and McKrae [LAT(+)] mice were individually isolated on day 28 p.i., and qRT-PCR was performed using total RNA as described in Materials and Methods. CD4, CD8, PD-1, Tim-3, IL-2, IL-21, TNF-α, and IFN-γ expression in naive mice was used as a baseline to estimate the relative expression of each transcript in TG of latently infected mice. GAPDH expression was used to normalize the relative expression of each transcript. Each point represents the mean ± SEM from 10 TG.
Similar to LAT, CD80 also activates apoptosis pathway components.
It is well accepted that LAT has anti-apoptotic function in vitro and in vivo (22–29). We have recently shown that the expression of apoptosis pathway components such as caspase 3, caspase 8, caspase 9, and Bcl2 are LAT dependent (30). To determine whether CD80 can restore the expression of these transcripts in the absence of LAT, levels of caspase 3, caspase 8, caspase 9, and Bcl2 were determined in latently infected mouse TG by qRT-PCR. The expression of these genes did not differ significantly between HSV-CD80- and WT McKrae-infected mice {P > 0.05 for HSV-CD80 [LAT(−)] versus McKrae [LAT(+)]} (Fig. 7), suggesting that CD80 and LAT both function to induce the expression of these transcripts. Thus, our results suggest that CD80 and LAT control apoptosis via a similar mechanism.
FIG 7.

Apoptotic effect of CD80 on the presence or absence of LAT in the TG of latently infected mice. TG from latently infected HSV-CD80 [LAT(−)] and McKrae [LAT(+)] mice were individually isolated on day 28 p.i., and qRT-PCR was performed using total RNA as described in Materials and Methods. Caspase 3 (Casp 3), caspase 8, caspase 9, and Bcl2 fold gene expression was assessed for each group of mice. GAPDH expression was used to normalize the relative expression of each transcript. Each point represents the mean ± SEM from 10 TG.
DISCUSSION
Infectious agents have developed mechanisms to escape host immune responses. Among the 80 known HSV-1 genes (41), ICP-0 (42, 43), ICP47 (44, 45), gE/gI (46, 47), and gC (48, 49) have been shown to interfere with the host immune response. Recently, we described a novel HSV-1 mechanism of immune evasion via ICP22-mediated suppression of CD80 expression in corneas of infected mice (35). The CD80 molecule is expressed by B cells, macrophages, dendritic cells, and T cells (50–52) and binds to CD28 (53), CTLA4 (11), and PD-L1 (54, 55). CD80 is upregulated later during the immune response (50, 51) and is critical in prolonging primary immune responses or costimulating secondary immune responses (56, 57).
In this study, we have shown that the host T cell costimulatory molecule CD80 and HSV-1 LAT have significant functional overlap, and the expression of CD80 can restore all tested functions of LAT. LAT is the only known HSV-1 gene expressed at high levels during both acute and latent infections (20, 58–60). While viruses lacking LAT replicate to levels similar to those of WT viruses during acute infection, they do not establish latency and do not reactivate as efficiently as WT viruses (18, 19, 61). LAT controls the latency reactivation cycle at least in part by inhibiting apoptosis and is responsible for establishing a state of immune exhaustion (31, 32). We demonstrated that the replication of HSV-CD80 recombinant virus in the eyes of C57BL/6 mice was indistinguishable from that of WT virus (Fig. 1). We also showed that CD80 can be expressed from the viral LAT promoter during acute and latent infection, because levels of CD80 expression were higher in the cornea and TG of mice infected with HSV-CD80 than in those infected with WT McKrae virus (Fig. 2B and Fig. 4A). Furthermore, infection with HSV-CD80 exacerbated eye disease, demonstrating the fact that CD80 also has functions distinct from those of LAT (Fig. 3). The exacerbated eye disease is of special interest because C57BL/6 mice are refractory to infections with WT McKrae. These results agree with our previous reports in BALB/c mice (34–36, 62). Furthermore, while CD80 can mimic all tested functions of LAT, CD80 has additional functions that are responsible for exacerbated eye disease.
It is well established that LAT plays an important role in T cell exhaustion (31, 32). Previously, we have shown a LAT-dependent elevation of the immune exhaustion markers CD4, CD8-α, CD8-β, PD-1, and Tim-3 as well as their associated cytokines IL-2, IL-21, IFN-γ, and TNF-α (31, 32, 40). In this study, we showed that in the absence of LAT, the expression of CD80 restores the expression of these markers to the levels of or higher than those of WT McKrae, suggesting that CD80 can also restore the function of LAT responsible for exhaustion. In addition, these results are similar to previous studies from our group and others that established a strong relationship between higher latency reactivation, increased CD8+ T cell expression, and T cell exhaustion (31, 32, 40).
Our finding that CD80 overexpression upregulates the expression of the anti-apoptotic Bcl-2 gene (Fig. 7) agrees with a report showing that CD80 engagement of CD28 can promote T cell survival by upregulating the expression of another member of the antiapoptotic Bcl-2 protein family, Bcl-XL (63). A proinflammatory environment has been shown to promote the expression of CD80 on the surface of neurons (64); however, it is not known if CD80 has anti-apoptotic activity in neurons. Our results are also consistent with our previous report showing that caspase 3, caspase 8, caspase 9, and Bcl-2 are all upregulated in TG of mice latently infected with HSV-1 in a LAT-dependent manner (30). In this study, we showed that the expression levels of these members of the apoptosis pathway are similar in mice infected with WT McKrae and HSV-CD80, suggesting that CD80 and LAT function in a similar manner to control apoptosis.
CD80 is the only tested gene that can restore all known LAT functions. While it is not clear if CD80 and LAT function via identical mechanisms because infection with HSV-CD80 resulted in higher expression levels of PD-1, IFN-γ, and IL-21 than infection with WT McKrae, the expression of CD80 results in the rescue of all known functions of LAT. The HSV-CD80 recombinant virus used in this study was generated by inserting the complete ORF of the CD80 gene at the 5′ end of both copies of the LAT gene (one in each viral long repeat) and was under the control of the LAT promoter (55). The LAT promoter was chosen to overexpress CD80 because it is the only viral promoter that sustains high levels of transcription during latent as well as primary infection (65–67). Several recombinant viruses have been generated using the same approach as the one that we used in this study; however, none have restored all known functions of LAT. For example, while the replacement of LAT with cpIAP restored latency and reactivation (27), this virus failed to restore the immune exhaustion activity of LAT (30). Because cpIAP is a baculovirus anti-apoptotic gene, the absence of T cell exhaustion in the presence of cpIAP despite latency levels similar to those of WT McKrae virus could be due to the fact that this is a nonmammalian gene. In another study to evaluate the anti-apoptotic function of LAT, two copies of the bovine herpesvirus 1 (BHV-1) LR gene, which is known to have anti-apoptotic activity, were inserted into two normal copies of the HSV-1 LAT gene to create the chimeric virus CJLAT (23). Despite the fact that both the BHV-1 LR gene and the HSV-1 LAT gene have anti-apoptotic activity, these genes were not functionally identical, as latently CJLAT-infected mice showed more mortality and CS than did animals infected with WT McKrae virus (23).
Other HSV-1 genes have also failed to replace LAT. Previously, we found that the replacement of LAT with HSV-1 glycoprotein K (gK) also produced significant eye disease in C57BL/6 mice (9, 65, 68, 69). CD80 is unique among tested host genes expressed by recombinant HSV-1, as we have shown that replacing LAT with the host cytokine IFN-γ or IL-4 did not restore latency, reactivation, or immune exhaustion (70). Thus, our results demonstrated that unlike any other tested gene, CD80 expression completely compensated for all known functions of LAT in LAT-minus viruses.
MATERIALS AND METHODS
Viruses, cells, and mice.
Triple-plaque-purified WT McKrae [LAT(+)] and HSV-CD80 [LAT(−)] viruses were used in this study. HSV-CD80 recombinant virus was generated by homologous recombination using WT virulent HSV-1 strain McKrae dLAT2903 DNA and the plasmid pLAT-CD80 as we described previously (55). Rabbit skin (RS) cells (used to prepare virus stocks, culture mouse tear films, and determine growth kinetics) were grown in Eagle’s minimal essential medium (MEM) supplemented with 5% fetal calf serum (FCS). Mice used in this study were female 6-week-old inbred C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME). All animal procedures adhered to the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and vision research (16) and according to institutional animal care and use guidelines. The animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (protocol number 5030).
Ocular infection.
Mice were infected ocularly with 2 × 105 PFU of HSV-1 strain McKrae per eye in 2 μl of tissue culture medium as an eye drop without corneal scarification, as we described previously (37, 38).
Titration of virus in tears.
Tear films were collected from 30 eyes on days 1 to 7 p.i. for each group as described previously (71). Each swab was placed in 1 ml of tissue culture medium, and the amount of virus in the medium was determined using a standard plaque assay on RS cells.
Evaluation of corneal scarring.
The severity of corneal scarring (CS) in surviving mice on day 28 p.i. was scored in a masked fashion on a scale of 0 to 4 (0, no disease; 1, 25% involvement; 2, 50% involvement; 3, 75% involvement; 4, 100% involvement) as described previously (40).
In vitro explant reactivation assay.
Mice were sacrificed on day 28 p.i., and individual TG were extracted and cultured in tissue culture medium. Aliquots of medium were removed from each culture daily for up to 12 days and plated on indicator RS cells to assay for the appearance of reactivated virus as previously described (72).
DNA extraction and PCR analysis for HSV-1 gB DNA.
DNA was isolated from homogenized individual TG on day 28 p.i. as we described previously (65, 73). PCR analyses were performed using gB-specific primers (forward primer 5′-AACGCGACGCACATCAAG-3′, reverse primer 5′-CTGGTACGCGATCAGAAAGC-3′, and probe 5′-FAM [6-carboxyfluorescein]-CAGCCGCAGTACTACC-3′). The resulting amplicon for this primer set is 72 bp long. Relative copy numbers of the gB gene were calculated using standard curves generated from the plasmid pAc-gB1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize the transcripts in all experiments.
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
Individual TG from ocularly infected mice were isolated at 28 days p.i. Total RNA was extracted and processed as we described previously (65, 73). Primer-probe sets consisted of two unlabeled PCR primers and the FAM dye-labeled TaqMan MGB probe formulated into a single mixture. The following assays were used in this study: (i) CD8 (α chain) (ABI assay identification [ID] Mm01182108_m1; amplicon length of 67 bp), (ii) PD-1 (programmed death 1) (also known as CD279) (ABI assay ID Mm00435532_m1; amplicon size of 65 bp), (iii) CD80 (ABI assay ID Mm00711660_m1; amplicon length of 117 bp), (iv) GAPDH (ABI assay ID Mm999999.15_G1; amplicon length of 107 bp), (v) IFN-γ (ABI assay ID Mm00801778_m1; amplicon size of 101 bp), (vi) CD4 (ABI assay ID Mm00442754_m1; amplicon length of 78 bp), (vii) IL-2 (ABI assay ID Mm00434256_m1; amplicon length of 82 bp), (viii) TNF-α (ABI assay ID Mm00443258_m1; amplicon size of 81 bp), (ix) IL-21 (ABI assay ID Mm00517640_m1; amplicon length of 67 bp), (x) Tim-3 (ABI assay ID Mm00454540_m1; amplicon length of 98 bp), (xi) caspase 9 (ABI assay ID Mm00516563_m1; amplicon length of 68 bp), (xii) caspase 8 (ABI assay ID Mm00802247_m1; amplicon length of 96 bp), (xiii) caspase 3 (ABI assay ID Mm01195085_m1; amplicon length of 70 bp), and (xiv) Bcl2 (ABI assay ID Mm00477631_m1; amplicon length of 85 bp). Quantitative real-time RT-PCR (qRT-PCR) was performed using the QuantStudio 5 system (Applied Biosystems, Foster City, CA) in 384-well plates as we described previously (38, 55, 74). GAPDH RNA was used to normalize transcripts in all experiments.
Statistical analysis.
Student’s t test was used to calculate P values, except where otherwise indicated. Protective parameters were analyzed as we described previously (38, 74). Results were considered statistically significant if the P value was <0.05.
ACKNOWLEDGMENTS
This work was supported by NIH grants 1RO1EY026944 and 1RO1EY029160.
REFERENCES
- 1.Dawson CR. 1984. Ocular herpes simplex virus infections. Clin Dermatol 2:56–66. doi: 10.1016/0738-081x(84)90066-x. [DOI] [PubMed] [Google Scholar]
- 2.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Doyle Stulting R, Asbell PA, for the Herpetic Eye Disease Study Group. 1994. Herpetic eye disease study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 101:1871–1882. doi: 10.1016/s0161-6420(13)31155-5. [DOI] [PubMed] [Google Scholar]
- 3.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br J Ophthalmol 80:969–972. doi: 10.1136/bjo.80.11.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liesegang TJ. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Hill TJ. 1987. Ocular pathogenicity of herpes simplex virus. Curr Eye Res 6:1–7. doi: 10.3109/02713688709020060. [DOI] [PubMed] [Google Scholar]
- 8.Mo JS, Streilein JW. 2001. Immune privilege persists in eyes with extreme inflammation induced by intravitreal LPS. Eur J Immunol 31:3806–3815. doi:. [DOI] [PubMed] [Google Scholar]
- 9.Jaggi U, Wang S, Tormanen K, Matundan H, Ljubimov AV, Ghiasi H. 2018. Role of herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) pathogenic CD8(+) T cells in exacerbation of eye disease. Front Immunol 9:2895. doi: 10.3389/fimmu.2018.02895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenfield EA, Nguyen KA, Kuchroo VK. 1998. CD28/B7 costimulation: a review. Crit Rev Immunol 18:389–418. doi: 10.1615/critrevimmunol.v18.i5.10. [DOI] [PubMed] [Google Scholar]
- 11.Sharpe AH, Freeman GJ. 2002. The B7-CD28 superfamily. Nat Rev Immunol 2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 12.Bretscher PA. 1999. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A 96:185–190. doi: 10.1073/pnas.96.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanke T, Graham FL, Rosenthal KL, Johnson DC. 1991. Identification of an immunodominant cytotoxic T-lymphocyte recognition site in glycoprotein B of herpes simplex virus by using recombinant adenovirus vectors and synthetic peptides. J Virol 65:1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin S, Zhu XX, Silverstein SJ, Courtney RJ, Yao F, Jenkins FJ, Rouse BT. 1990. Murine cytotoxic T lymphocytes specific for herpes simplex virus type 1 recognize the immediate early protein ICP4 but not ICP0. J Gen Virol 71:2391–2399. doi: 10.1099/0022-1317-71-10-2391. [DOI] [PubMed] [Google Scholar]
- 15.Ghiasi H, Cai S, Perng GC, Nesburn AB, Wechsler SL. 2000. Both CD4+ and CD8+ T cells are involved in protection against HSV-1 induced corneal scarring. Br J Ophthalmol 84:408–412. doi: 10.1136/bjo.84.4.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osorio Y, Cai S, Hofman FM, Brown DJ, Ghiasi H. 2004. Involvement of CD8+ T cells in exacerbation of corneal scarring in mice. Curr Eye Res 29:145–151. doi: 10.1080/02713680490504632. [DOI] [PubMed] [Google Scholar]
- 17.Mercadal CM, Bouley DM, DeStephano D, Rouse BT. 1993. Herpetic stromal keratitis in the reconstituted scid mouse model. J Virol 67:3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol 61:3820–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 22.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 23.Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. 2002. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol 76:1224–1235. doi: 10.1128/jvi.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, Jones C. 2002. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol 8(Suppl 2):103–111. doi: 10.1080/13550280290101085. [DOI] [PubMed] [Google Scholar]
- 25.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 79:9019–9025. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenter D, Hsiang C, Brown DJ, Jin L, Osorio N, BenMohamed L, Jones C, Wechsler SL. 2007. Stable cell lines expressing high levels of the herpes simplex virus type 1 LAT are refractory to caspase 3 activation and DNA laddering following cold shock induced apoptosis. Virology 369:12–18. doi: 10.1016/j.virol.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin L, Perng GC, Carpenter D, Mott KR, Osorio N, Naito J, Brick DJ, Jones C, Wechsler SL. 2007. Reactivation phenotype in rabbits of a herpes simplex virus type 1 mutant containing an unrelated antiapoptosis gene in place of latency-associated transcript. J Neurovirol 13:78–84. doi: 10.1080/13550280601164333. [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Chentoufi AA, Hsiang C, Carpenter D, Osorio N, BenMohamed L, Fraser NW, Jones C, Wechsler SL. 2011. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol 85:2325–2332. doi: 10.1128/JVI.01791-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, Jones C, Wechsler SL. 2008. Cellular FLIP can substitute for the herpes simplex virus type 1 latency-associated transcript gene to support a wild-type virus reactivation phenotype in mice. J Neurovirol 14:389–400. doi: 10.1080/13550280802216510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tormanen K, Allen S, Mott KR, Ghiasi H. 2019. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J Virol 93:e00103-19. doi: 10.1128/JVI.00103-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. 2011. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol 85:9127–9138. doi: 10.1128/JVI.00587-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. 2005. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol 79:12286–12295. doi: 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tormanen K, Wang S, Ghiasi H. 16 October 2019. CD80 plays a critical role in increased inflammatory responses in HSV-1 infected mouse corneas. J Virol doi: 10.1128/JVI.01511-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matundan H, Ghiasi H. 2019. Herpes simplex virus 1 ICP22 suppresses CD80 expression by murine dendritic cells. J Virol 93:e01803-18. doi: 10.1128/JVI.01803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matundan H, Jaggi U, Wang S, Ghiasi H. 2019. Loss of ICP22 in HSV-1 elicits immune infiltration and maintains stromal keratitis despite reduced primary and latent virus infectivity. Invest Ophthalmol Vis Sci 60:3398–3406. doi: 10.1167/iovs.19-27701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. doi: 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, Mott KR, Cilluffo M, Kilpatrick CL, Murakami S, Ljubimov AV, Kousoulas KG, Awasthi S, Luscher B, Ghiasi H. 2018. The absence of DHHC3 affects primary and latent herpes simplex virus 1 infection. J Virol 92:e01599-17. doi: 10.1128/JVI.01599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matundan HH, Mott KR, Allen SJ, Wang S, Bresee CJ, Ghiasi YN, Town T, Wechsler SL, Ghiasi H. 2016. Interrelationship of primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol 90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69:1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- 42.Heilingloh CS, Muhl-Zurbes P, Steinkasserer A, Kummer M. 2014. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J Gen Virol 95:1366–1375. doi: 10.1099/vir.0.062810-0. [DOI] [PubMed] [Google Scholar]
- 43.van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, Kurt-Jones EA. 2010. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J Virol 84:10802–10811. doi: 10.1128/JVI.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature 375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 45.Goldsmith K, Chen W, Johnson DC, Hendricks RL. 1998. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med 187:341–348. doi: 10.1084/jem.187.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bell S, Cranage M, Borysiewicz L, Minson T. 1990. Induction of immunoglobulin G Fc receptors by recombinant vaccinia viruses expressing glycoproteins E and I of herpes simplex virus type 1. J Virol 64:2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson DC, Frame MC, Ligas MW, Cross AM, Stow ND. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J Virol 62:1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenberg RJ, Ponce de Leon M, Friedman HM, Fries LF, Frank MM, Hastings JC, Cohen GH. 1987. Complement component C3b binds directly to purified glycoprotein C of herpes simplex virus types 1 and 2. Microb Pathog 3:423–435. doi: 10.1016/0882-4010(87)90012-x. [DOI] [PubMed] [Google Scholar]
- 49.Friedman HM, Wang L, Fishman NO, Lambris JD, Eisenberg RJ, Cohen GH, Lubinski J. 1996. Immune evasion properties of herpes simplex virus type 1 glycoprotein gC. J Virol 70:4253–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lenschow DJ, Su GH, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, Miller J, Bluestone JA. 1993. Expression and functional significance of an additional ligand for CTLA-4. Proc Natl Acad Sci U S A 90:11054–11058. doi: 10.1073/pnas.90.23.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. 1994. Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J Exp Med 180:631–640. doi: 10.1084/jem.180.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larsen CP, Ritchie SC, Hendrix R, Linsley PS, Hathcock KS, Hodes RJ, Lowry RP, Pearson TC. 1994. Regulation of immunostimulatory function and costimulatory molecule (B7-1 and B7-2) expression on murine dendritic cells. J Immunol 152:5208–5219. [PubMed] [Google Scholar]
- 53.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. 1994. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1:793–801. doi: 10.1016/S1074-7613(94)80021-9 (Erratum, 2:203, 1995). [DOI] [PubMed] [Google Scholar]
- 54.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. 2007. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mott KR, Allen SJ, Zandian M, Akbari O, Hamrah P, Maazi H, Wechsler SL, Sharpe AH, Freeman GJ, Ghiasi H. 2014. Inclusion of CD80 in HSV targets the recombinant virus to PD-L1 on DCs and allows productive infection and robust immune responses. PLoS One 9:e87617. doi: 10.1371/journal.pone.0087617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freeman GJ, Gray GS, Gimmi CD, Lombard DB, Zhou LJ, White M, Fingeroth JD, Gribben JG, Nadler LM. 1991. Structure, expression, and T cell costimulatory activity of the murine homologue of the human B lymphocyte activation antigen B7. J Exp Med 174:625–631. doi: 10.1084/jem.174.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr, Lombard LA, Gray GS, Nadler LM. 1993. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 262:909–911. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 58.Wagner EK, Guzowski JF, Singh J. 1995. Transcription of the herpes simplex virus genome during productive and latent infection. Prog Nucleic Acid Res Mol Biol 51:123–165. doi: 10.1016/s0079-6603(08)60878-8. [DOI] [PubMed] [Google Scholar]
- 59.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and non-coding RNA: a critical retrospective. J Neuroimmunol 308:65–101. doi: 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 60.Sawtell NM. 1997. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol 71:5423–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicoll MP, Proenca JT, Connor V, Efstathiou S. 2012. Influence of herpes simplex virus 1 latency-associated transcripts on the establishment and maintenance of latency in the ROSA26R reporter mouse model. J Virol 86:8848–8858. doi: 10.1128/JVI.00652-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mott KR, Allen SJ, Zandian M, Ghiasi H. 2014. Coregulatory interactions among CD8α dendritic cells, the latency-associated transcript, and programmed death 1 contribute to higher levels of herpes simplex virus 1 latency. J Virol 88:6599–6610. doi: 10.1128/JVI.00590-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. 1995. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. 2006. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med 12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 65.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zwaagstra J, Ghiasi H, Nesburn AB, Wechsler SL. 1989. In vitro promoter activity associated with the latency-associated transcript gene of herpes simplex virus type 1. J Gen Virol 70:2163–2169. doi: 10.1099/0022-1317-70-8-2163. [DOI] [PubMed] [Google Scholar]
- 67.Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsler SL. 1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol 64:5019–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matundan HH, Mott KR, Akhtar AA, Breunig JJ, Ghiasi H. 2015. Mutations within the pathogenic region of herpes simplex virus 1 gK signal sequences alter cell surface expression and neurovirulence. J Virol 89:2530–2542. doi: 10.1128/JVI.03506-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghiasi H, Cai S, Slanina S, Nesburn AB, Wechsler SL. 1997. Nonneutralizing antibody against the glycoprotein K of herpes simplex virus type-1 exacerbates herpes simplex virus type-1-induced corneal scarring in various virus-mouse strain combinations. Invest Ophthalmol Vis Sci 38:1213–1221. [PubMed] [Google Scholar]
- 70.Lee DH, Ghiasi H. 2018. An M2 rather than a TH2 response contributes to better protection against latency reactivation following ocular infection of naive mice with a recombinant herpes simplex virus 1 expressing murine interleukin-4. J Virol 92:e00051-18. doi: 10.1128/JVI.00051-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 72.Mott KR, Ghiasi H. 2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin Vaccine Immunol 15:1859–1867. doi: 10.1128/CVI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi H. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol Vis 13:1802–1812. [PubMed] [Google Scholar]
- 74.Wang S, Mott KR, Wawrowsky K, Kousoulas KG, Luscher B, Ghiasi H. 2017. Binding of herpes simplex virus 1 UL20 to GODZ (DHHC3) affects its palmitoylation and is essential for infectivity and proper targeting and localization of UL20 and glycoprotein K. J Virol 91:e00945-17. doi: 10.1128/JVI.00945-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. doi: 10.1016/0168-1702(92)90087-p. [DOI] [PubMed] [Google Scholar]