

Summary
Coronaviruses (CoV) are enveloped positive‐strand RNA viruses that induce different membrane rearrangements in infected cells in order to efficiently replicate and assemble. The origin, the protein composition and the function of these structures are not well established. To shed further light on these structures, we have performed a time‐course experiment in which the mouse hepatitis virus (MHV)‐induced membrane rearrangements were examined qualitatively and quantitatively by (immuno)‐electron microscopy. With our approach we were able to confirm the appearance of 6, previously reported, membranous structures during the course of a complete infection cycle. These structures include the well‐characterized double‐membrane vesicles (DMVs), convoluted membranes (CMs) and virions but also the more enigmatic large virion‐containing vacuoles (LVCVs), tubular bodies (TBs) and cubic membrane structures (CMSs). We have characterized the LVCVs, TBs and CMSs, and found that the CoV‐induced structures appear in a strict order. By combining these data with quantitative analyses on viral RNA, protein synthesis and virion release, this study generates an integrated molecular and ultrastructural overview of CoV infection. In particular, it provides insights in the role of each CoV‐induced structure and reveals that LVCVs are ERGIC/Golgi compartments that expand to accommodate an increasing production of viral particles.
Introduction
Viruses require cellular membranes in one or more steps of their infection cycle for replication, assembly and/or release, and therefore they have developed sophisticated mechanisms to opportunistically rearrange host membranes for their needs. For example, a common feature among positive (+) strand RNA viruses is the assembly of their replication–transcription complexes (RTCs) in association with cytoplasmic membranes (Salonen et al., 2005; Miller and Krijnse‐Locker, 2008). The potential benefit of anchoring the RTCs to lipid bilayers is still unclear, but it may facilitate and co‐ordinate different steps of the viral life cycle, and/or delay induction of the host immune response (Ahlquist, 2006; Haller et al., 2006). Enveloped viruses are another example; they generate their new virions by budding through cellular membranes (Garoff et al., 1998).
CoV are enveloped (+) strand RNA viruses (Weiss and Navas‐Martin, 2005; Gorbalenya et al., 2006). They are pathogens of veterinary importance, but the relevance of this family of viruses has increased considerably due to the recent emergence of new human viruses such as the CoV NL63 and the severe acute respiratory syndrome‐CoV (SARS‐CoV). These viruses cause severe respiratory tract diseases and patients often have evidence of other organ dysfunctions (Godfraind and Coutelier, 1998; Peiris et al., 2003; Saif, 2004).
After fusion of the viral and cellular membranes, CoV disassemble thereby releasing their genomic RNA, which allows the production of 16 non‐structural proteins (nsp's) (Brian and Baric, 2005; Sawicki et al., 2005). These nsp's collectively form the RTCs and induce the formation of cytoplasmic DMVs into which the RTCs are anchored (Ziebuhr et al., 2000). The mechanism underlying the biogenesis of CoV‐induced DMVs is completely unknown even if experimental evidences indicate an endoplasmic reticulum (ER) origin (Harcourt et al., 2004; Kanjanahaluethai et al., 2007; 2007, 2008; Knoops et al., 2008).
The CoV genomes encode for a common set of four structural proteins: the envelope (E), the membrane (M),the nucleocapsid (N) and the spike (S) proteins (de Haan and Rottier, 2006). E, M and S proteins are integral membrane components and after insertion in the ER limiting membrane, they are transported to the ER‐to‐Golgi intermediate compartment (ERGIC), where together with the N protein and the genomic RNA, they assemble into virions by inducing the invagination and luminal pinching off of the limiting membrane of this organelle (Tooze et al., 1984; Ng et al., 2003; Goldsmith et al., 2004; Stertz et al., 2007). The resulting luminal virions subsequently reach the extracellular environment following the conventional secretory pathway (Tooze et al., 1987).
The first ultrastructural analyses of CoV‐infected cells by electron microscopy (EM) were already preformed in the 1960s (Svoboda et al., 1962; David‐Ferreira and Manaker, 1965; Ruebner et al., 1967). The presence of DMVs and virions was the obvious morphological changes described at that time. These initial observations have successively been corroborated by numerous studies. More recently, other membranous rearrangements have been described. An electron tomography analysis of SARS‐CoV‐infected cells has confirmed the presence of reticular inclusions in between DMVs (Knoops et al., 2008), which were already reported in one of the original works (David‐Ferreira and Manaker, 1965). The recent study has proposed that these reticular inclusions, re‐named CMs, are the precursors of DMVs (Knoops et al., 2008). In addition to CMs, it has been shown that the CoV triggers the formation of highly organized crystalloid conformations, tubular rearrangements and vacuoles enclosing viral particles that have been named CMSs, TBs and LVCVs respectively (David‐Ferreira and Manaker, 1965; Ruebner et al., 1967; Tooze et al., 1984; Ng et al., 2003; Goldsmith et al., 2004; Almsherqi et al., 2005; Knoops et al., 2008).
The function of CMs, CMSs, TBs and LVCVs in CoV infection is largely unknown. A major difficulty in understanding the role of these different structures has been the absence of a quantitative and qualitative EM analyses over time that could help ordering them during the infection cycle. We have now filled this gap by performing a time‐course EM and immunoelectron microscopy (IEM) examination of MHV‐infected cells. By combining these qualitative and quantitative data with the measurement of viral RNA synthesis, viral protein production and progeny virus release, we have, for the first time, integrated ultrastructural analyses with molecular information. This approach has allowed us to establish that MHV induces the formation of six membranous rearrangements in the following order: DMVs, CMs, virions, LVCVs, TBs and CMSs. Importantly, we were able to show that most membrane rearrangements (LVCVs, TBs, CMSs and possibly CMs) observed in addition to the key structures in the infection (DMVs and virions) actually appear to be the consequence of a massive synthesis of viral proteins. In particular, LVCVs are ERGIC/Golgi compartments that expand to accommodate an increasing production of virions. All together, our study provides an overall comprehensive picture of the ultrastructural events taking place inside a cell in the course of a CoV infection.
Results
Time‐course MHV infection and measurement of CoV life cycle parameters
To understand the relationship between the different membranous structures induced by MHV and their role during the infection, we infected the cells at high multiplicity of infection (moi) and we analysed, in a time‐course manner, their ultrastructure by EM as well as various other infection parameters during a period of 10 h as described under Experimental procedures. In order to be able to correlate our EM and IEM analyses with the progression of a CoV infection inside the host cells, we first measured important known parameters that reflect the CoV life cycle: viral RNA replication/transcription, viral protein synthesis and secretion of progeny virus.
To study the RNA replication/transcription rate during MHV infection, the amount of genomic RNA (gRNA) and that of subgenomic RNA encoding for the N protein (sgRNA N) was determined at each time point by RT‐PCR as described in Experimental procedures. Both gRNA and sgRNA N were already detected at 2 h post infection (p.i.; Fig. 1A). Their amount gradually increased until the 9 h time point, after which the levels remained constant, resulting from a decline in RNA synthesis (Sawicki et al., 2007). At each time, the amount of sgRNA was about 100‐fold higher than that of gRNA. This analysis allowed us to ascertain that the observed changes in the levels of gRNA and sgRNA synthesis are identical to those measured using similar or different assays in various cell lines infected with diverse CoV (Sethna and Brian, 1997; Versteeg et al., 2006; Sawicki et al., 2007).
Figure 1.
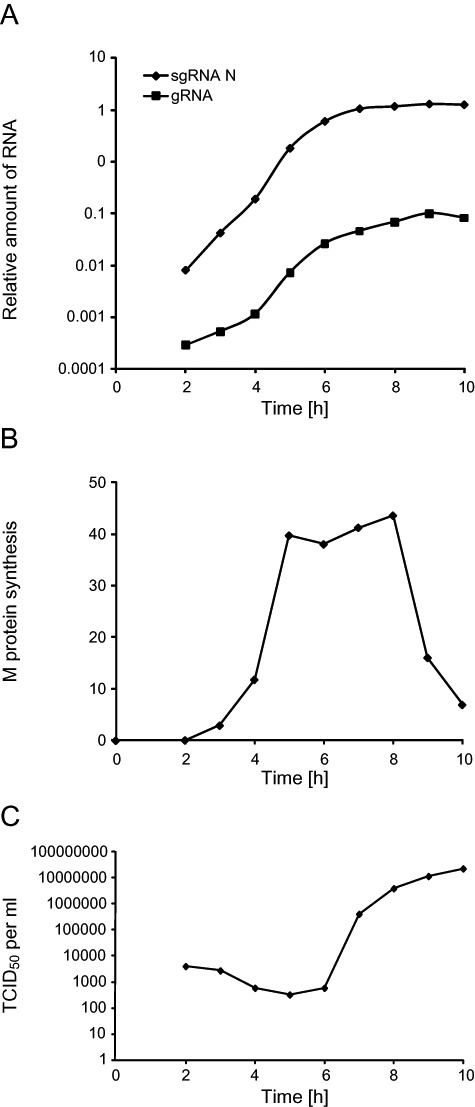
Analysis of the MHV infection progression by monitoring the viral RNA and protein synthesis, and the extracellular release of viral particles. HeLa‐CEACAM1a cells were infected with MHV‐A59 and the culture supernatants and cell lysates were collected at different p.i. time points as described in Experimental procedures. A. The total RNA was isolated from the infected cells and the amount of gRNA and sgRNA was quantified by RT‐PCR. Results are expressed using arbitrary units. B. Infected cells were metabolically labelled 30 min prior of being collected and lysed. Cell lysates were then immunoprecipitated with an antiserum again the complete MHV in combination with anti‐M protein antibodies. Finally, immuno‐complexes were resolved by SDS‐PAGE and the amount of radioactive M protein was quantified. Results are expressed using arbitrary units. C. The production of the progeny virus was assessed by determining the virus titre of the culture supernatants by end‐point dilutions on LR7 cells and then calculating the TCID50 units per ml of supernatant.
Next, we analysed the synthesis of the structural proteins during the infection. To this end, we measured the production rate of the M protein at the different time points by short pulse radio‐labelling experiments followed by SDS‐PAGE analysis of the crude cell lysates. The synthesis of the M protein was already detectable at 3 h p.i. and continued to increase until 8 h p.i. with the main increase occurring between 4 and 6 h p.i. (Fig. 1B). Essentially, identical results were obtained when we analysed the production of the structural proteins S and N in the same way (data not shown). This correlates perfectly with the synthesis kinetics of the sgRNA (Fig. 1A) and consequently indicates that these mRNAs are immediately available for translation. After 8 h of infection, we observed a drop in the production of the M protein (Fig. 1B). These results again correspond with those of others (Rottier et al., 1981; Hilton et al., 1986; Tahara et al., 1994).
To monitor the assembly and release of MHV over time, the infectivity levels in the culture supernatants collected at the different p.i. times were determined. Secretion of MHV virions was first detected at 6 h p.i. and increased until 9 h p.i. before slowing down (Fig. 1C). This observation matches with the analysis of the viral RNA and structural protein production (Fig. 1A and B) because as expected, it shows that MHV assembly and release are processes that follow intracellular MHV replication.
All together, these measurements demonstrate that MHV infection in HeLa‐CEACAM1a cells progresses following the typical, established dynamics, thereby validating the use of this cell line. In addition, they also show that the 10 h time window used in our time‐course analysis comprises all the phases of a CoV infection and therefore our examinations allow obtaining a complete overview of a CoV life cycle.
MHV induces the formation of multiple membranous structures
As a first ultrastructural analysis, we compared the morphology of cells at 0 h p.i. with that of those at 8 h p.i. by EM in order to make a repertoire of all the membranous rearrangements that MHV induces. We identified six different structures. The most abundant of them were large vesicles with an average diameter of 200–350 nm, which are limited by a double‐membrane and often were clustered together (Fig. 2A and B; and Fig. S1A, arrows). These are the characteristic DMVs induced by CoV (Svoboda et al., 1962; David‐Ferreira and Manaker, 1965; Ruebner et al., 1967; Pedersen et al., 1999; Gosert et al., 2002; Snijder et al., 2006; Knoops et al., 2008). Surprisingly, most of the observed DMVs appeared to have an invagination, which, from time to time, was associated to what looked as a small vesicle with a diameter of 50–100 nm (Fig. S1B, arrow). This structural peculiarity is cell type‐specific because it was also observed in MHV‐A59‐infected mouse embryonic fibroblasts (data not shown) but not in mouse LR7 cells (Knoops et al., 2008). In the centre of the DMV clusters, we frequently observed a small network of membranes with a diameter varying from 200 to 600 nm, which have recently been described in SARS‐CoV‐infected cells and called CMs [Fig. 2A and B, arrowheads; (Knoops et al., 2008). The CMs were often in close proximity of the ER (< 10–20 nm distance), and sometimes appeared to be connected with this organelle (Fig. S1C, arrow). Virions were the third structure that we identified. These dark circular structures with a diameter of 70–110 nm were found in the lumen of either a stack of adjacent cisternae, very likely the Golgi, or what appeared to be secretory vesicles (Fig. 2C) as well as extracellularly (Fig. S1D) in complete agreement with their known assembly and secretion mechanisms. The virions were also observed inside large circular organelles with a diameter of approximately 450–750 nm (Fig. 2D, arrow). Interestingly, we observed virion particles assembling by invagination at the limiting membrane of these compartments through a process identical to the one occurring at the ERGIC (Fig. 2D, arrow). Because similar organelles have previously been described (Ng et al., 2003; Goldsmith et al., 2004; Knoops et al., 2008), we called them in the same way: the LVCVs. The fifth conformation that we detected was a condensed rearrangement of membranes with a diameter of approximately 300–650 nm but without an apparent ultrastructural organization, which seemed to be connected to the ER (Fig. 2E). These structures have already been described a long time ago and called TBs (David‐Ferreira and Manaker, 1965). The sixth classified structure was a rectangular, extended (up to 850 nm in length) and highly organized membranous conformation always continuous to what appeared to be a swollen ER cisterna (Fig. 2F, arrow; the arrowhead indicates the swollen ER). A morphologically similar subcellular arrangement has been observed in SARS‐CoV infected cell sections and consequently, we also called these structures CMS (Almsherqi et al., 2005; Almsherqi et al., 2009).
Figure 2.
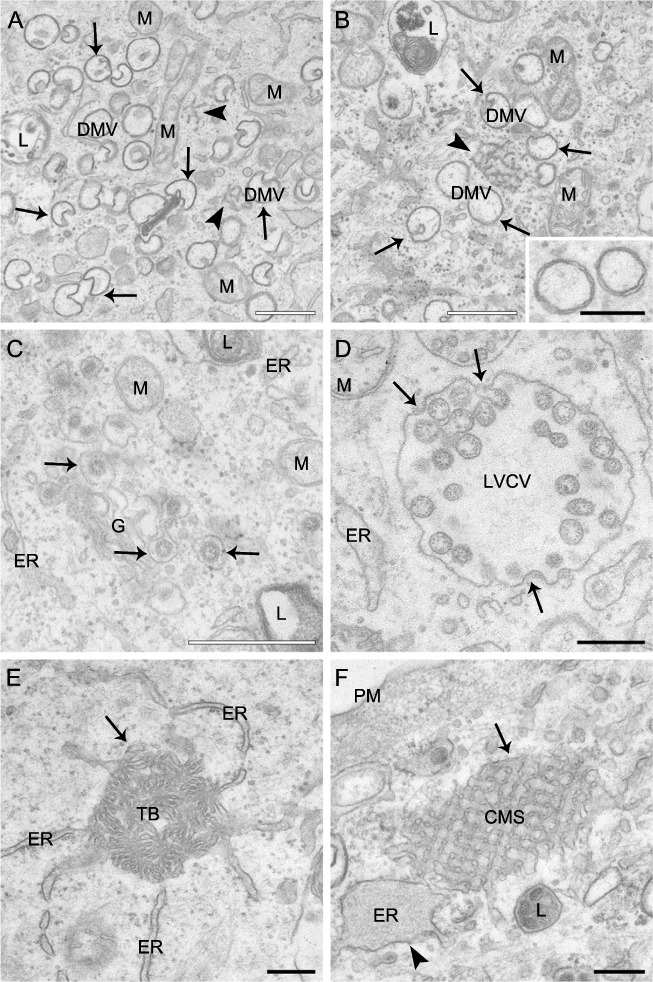
Ultrastructure of membranous structures induced by MHV in host cells. HeLa‐CEACAM1a cells inoculated with MHV‐A59 were fixed at 8 h p.i. and processed for conventional EM as described in Experimental procedures. A and B. DMVs are cytoplasmic double‐membrane vesicles (arrows) that frequently possess an invagination. DMVs are often found clustered together in close proximity of a small network of membranes, the CMs (arrowheads). The inset in (B) shows a magnification of two DMVs to highlight the two lipid bilayers characterizing these vesicles. C. Newly made virions (arrows) present in the lumen of the Golgi complex. D. LVCVs are large circular organelles with a diameter of approximately 450–750 nm that contain numerous virions in their interior. In addition, viral particles can be observed that are assembling at the limiting membrane of this structure by invagination and successive pinching off (arrows). E. The TBs (arrow) are ball of wool‐like membranous rearrangements with a diameter of approximately 300–650 nm that appear to be continuous with the ER. F. CMSs are extended (up to 850 nm in length), geometrical and highly organized conformations, which are often seen connected to a swollen ER (arrowhead). ER, endoplasmic reticulum; G, Golgi complex; M, mitochondria; PM, plasma membrane; L, lysosome. White bar, 500 nm; black bar, 200 nm.
Quantitative analysis of the appearance of the various MHV‐induced structures
To see whether the changes observed at the ultrastructural level correlate with the other measured infection parameters (Fig. 1), we first morphologically determined the number of cell sections at each time point that demonstrate visible signs of infection. To this end, the number of cell sections demonstrating at least one of the six structures induced by the MHV was determined at each time point. At 2 h p.i., 10% of the cell sections showed visible signs of infection and this percentage gradually increased during time until reaching 84% at 10 h p.i. (Fig. 3A). Importantly, the percentages of cell sections with visible signs of infection as determined with the EM analysis were very similar to those as evaluated by IF (data not shown) and correlated well with the rest of the measured parameters, demonstrating that this is a reliable alternative approach to follow the MHV infection.
Figure 3.
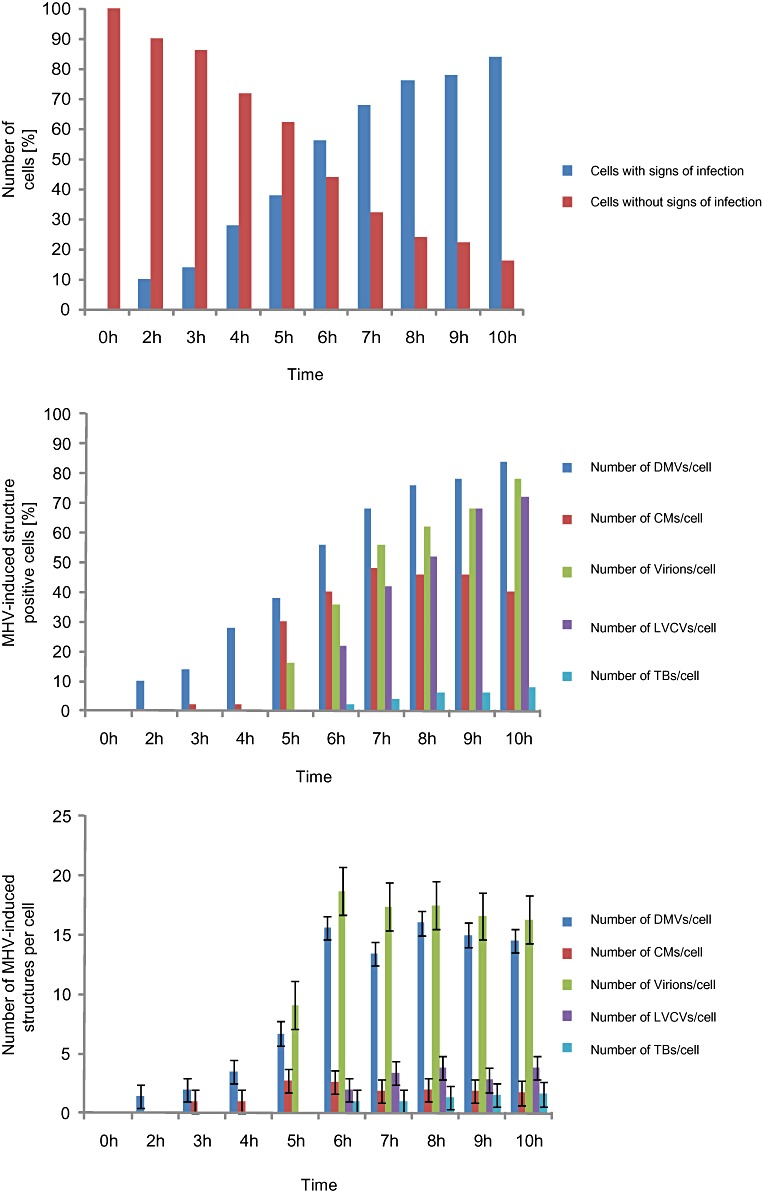
Quantitative analysis of the appearance of the various MHV‐induced structures during the course of an infection. HeLa‐CEACAM1a cells infected with MHV‐A59 were collected at different time points p.i. as described in Experimental procedures before being processed for conventional EM. For the statistical analyses, 50 cell sections were randomly selected for each time point p.i. and used to determine the percentage of cells containing a specific structure (B) and the average number of the same structure per cell section (C). The counted structures were DMVs, CMs, virions, LVCVs and TBs. In addition, the number of cells containing at least one of the six MHV‐induced structures was counted to assess morphologically the proportion of infected cells (A). Error bars in (C) represent the standard deviation of the number of the same structure per cell section.
To understand the role of the six MHV‐induced structures during an infection and to unravel their relationship, we the quantitatively analysed the EM sections obtained at the different p.i. time points. Two values were calculated: (i) the percentage of cell profiles containing a specific structure and (ii) the average number of a given structure per cell section. Our analysis revealed that the DMVs are the first membrane rearrangement to be detected in the infected cells. DMVs were already observed at 2 h p.i. in about 10 % of the cell sections and the number of cell sections positive for these vesicles gradually increased over time (Fig. 3B). The average number of DMVs per cell section reached a maximum, e.g. 16 DMVs/cell section, at 8 h p.i. (Fig. 3C). Interestingly, the localization and morphology of the DMVs changed during the infection. At early time points, from 2 to 4 h p.i., DMVs were small (190–250 nm diameters) with a regular circular shape and distributed throughout the cytoplasm. From 5 h onwards, DMVs organized in clusters mostly found in the perinuclear region of the cell. The DMV invaginations became more pronounced at 6 h p.i. after which the small vesicles located in their interior also became more prominent (Fig. S1B, arrow). Two hours later, the shape of the DMVs started to change acquiring a less circular form and with protuberances emerging from their surface that entered invaginations of adjacent DMVs (Fig. S1E, arrows).
The next structures to be detected during the MHV infection were the CMs, which became apparent at 3 h p.i. The CMs were always found in close proximity to at least one DMV (< 15–60 nm distance). At the early infection time points, e.g. 2–4 h p.i., the CMs had small sizes (80–100 nm diameters) and were present in only 2% of the cells (Fig. 3D). Their number per cell and their size, however, increased during the progression of the infection reaching a plateau at 7 h p.i. (Fig. 3E; 4 CM/cell section, 200–600 nm in diameter). A dramatic change in the percentage of CMs was observed at 5 h p.i. when the number of CM positive cells had increased from 2% to 30%. Overall, these data suggested that the CMs are structures that are functionally connected with DMVs as suggested (Knoops et al., 2008).
Virions appeared at 5 h p.i. and their intracellular number became constant (8 virions/cell section) already at 6 h p.i., probably upon reaching an equilibrium between synthesis and secretion (Fig. 3F and G). The virions were mostly observed in the Golgi cisternae at 5 h p.i. in agreement with the fact that MHV particles assemble at the ERGIC and are released into the extracellular space by passing through the secretory pathway (Ng et al., 2003; Stertz et al., 2007). LVCVs became detectable at 6 h p.i. (Fig. 3H) in coincidence with a more than twofold increase in the number of virions per cell profile (Fig. 3I). This observation suggested that the formation of LVCVs is probably induced by a higher production of virions in the cells. The TBs also became visible at 6 h p.i. (Fig. 3J). The number of TB‐positive cell sections increased during time as well as the size of these structures (Fig. 3K), with an average diameter of 200–300 nm at 6 h p.i. to one of about 650 nm at 10 h p.i. Initially, each infected cell profile contained only one TB but, after 8 h p.i., we occasionally observed more than one TB per cell section. The CMSs, in contrast, were even more rare (observed in only 1% of the cells) and only detectable after 9 h p.i.; hence a significant statistical analysis could not be performed. We concluded that the TBs and the CMSs are not required for the early steps of the MHV infection cycle but rather the result of an advanced infection.
The MHV‐induced membranous rearrangements have different viral protein compositions
To further understand the role of the MHV‐induced structures, we explored by IEM the presence or absence of viral non‐structural and structural proteins in the six identified structures. The antibodies used were recognizing either nsp2/nsp3, nsp4, nsp8, the N, the M or the E proteins. We immunolabelled cryo‐sections obtained from cells fixed at 6 and 10 h p.i. to be able to detect compositional changes. However, the labelling profiles at these two infection time points were identical; the only major difference was the higher immunoreactivity of the 10 h p.i. samples due to the higher amounts of viral proteins.
As expected (Gosert et al., 2002; Snijder et al., 2006; Stertz et al., 2007), the nsp2/nsp3, nsp4 and nsp8 proteins were decorating the surface of the DMVs (Fig. 4A–C). Importantly, we discovered that these nsp's were also present in the CMs but not in the other MHV‐induced structures (Fig. 4A–C and Fig. S3A–H). The N protein was also distributed on both DMVs and CMs (Fig. 4D) suggesting a possible direct relationship between these two structures. Like the two other structural proteins, i.e. M and E, the N protein was found in virions present in the Golgi complex and LVCVs (Fig. 4E–H). The M and E proteins were additionally observed in the limiting membrane of the Golgi cisternae but not on other MHV induced membranous rearrangements (Fig. S3). Interestingly, we discovered that the TBs contain the E protein but are negative for the other tested viral proteins (Fig. 4I, Fig. S3A and C). This result, plus the fact that the TBs appear at the late stage of the MHV infection (Fig. 3J and K), indicates that they could be generated by self‐assembly, possibly in the ER, of high levels of E protein. This hypothesis is sustained by the observation that the individual expression of the E protein induces the formation of a complex of tubular and smooth membranes with morphology reminiscent to that of the TBs (Raamsman et al., 2000).
Figure 4.
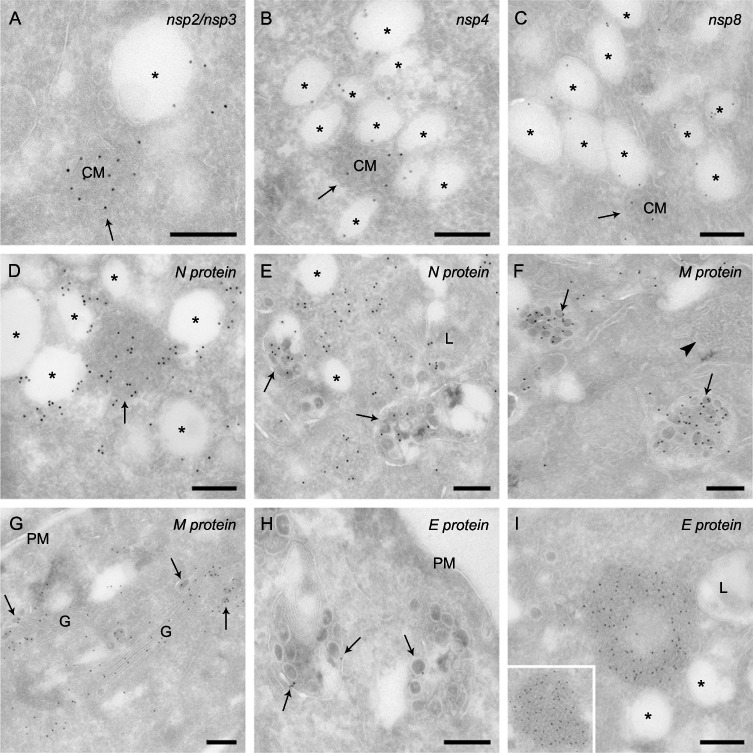
Non‐structural and structural protein localization in MHV‐infected cells. HeLa‐CEACAM1a cells infected with MHV‐A59 fixed at 10 h p.i. were processed for IEM and immunolabelled as described in Experimental procedures. (A) Nsp2/nsp3, (B) nsp4 and (C) nsp8 are distributed on the limiting membrane of DMVs and on the CMs. D. The N protein is distributed onto the DMVs and the CMs (arrow). E. The N nucleocapsid is also present in the viral particles, in this particular image contained in the LVCVs (arrows). F. The M protein localizes to virions, in this specific case present in the LVCV lumens (arrows), but not to the TBs (arrowhead). G. The M protein is present in the Golgi complex, both individually or incorporated into luminal virions (arrows). H. Virions (arrows) are positive for the E protein. I. The E protein is concentrated into the TBs. The inset shows an additional example. Arrows point to CMs, asterisks mark the DMVs. M, mitochondria, G, Golgi complex; L, lysosomes; PM, plasma membrane. Bar, 200 nm.
None of the tested antibodies labelled the CMSs. Because these structures appear at the late stage of the infection (data not shown), we speculate that they are induced by aggregation of the S protein for which we do not have an antibody compatible with our IEM procedure.
The MHV‐induced membranous rearrangements have different origins
To acquire information about the host organelle origins of the MHV‐induced structures, cryosections were obtained from cells harvested at 0 and 10 h p.i., and labelled with antibodies recognizing the protein disulfide isomerize PDI (ER), ERGIC53 (ERGIC), GM130 (cis‐Golgi), TGN‐46 (trans‐Golgi network, TGN) and LAMP1 (late endosomes and lysosomes). These antibodies localized to the expected compartments in non‐infected cells (Fig. S4). None of the employed organelle protein markers labelled the DMVs or the CMs (data not shown), even though the latter ones were often observed in proximity of the ER (Fig. S1C). As previously reported, forming and completed virions were observed in the ERGIC and Golgi cisternae (Fig. 5A and data not shown respectively). Importantly, ERGIC53, GM130 and TGN46 were found on the LVCVs as well, albeit at very low levels (Fig. 5B–D). Notably, despite its close proximity and almost clear continuity with the ER, TBs were not positive for PDI (Fig. 5E). In contrast, the CMSs contained this ER protein marker (Fig. 5F) in agreement with morphological connection with this organelle (Fig. 2F).
Figure 5.
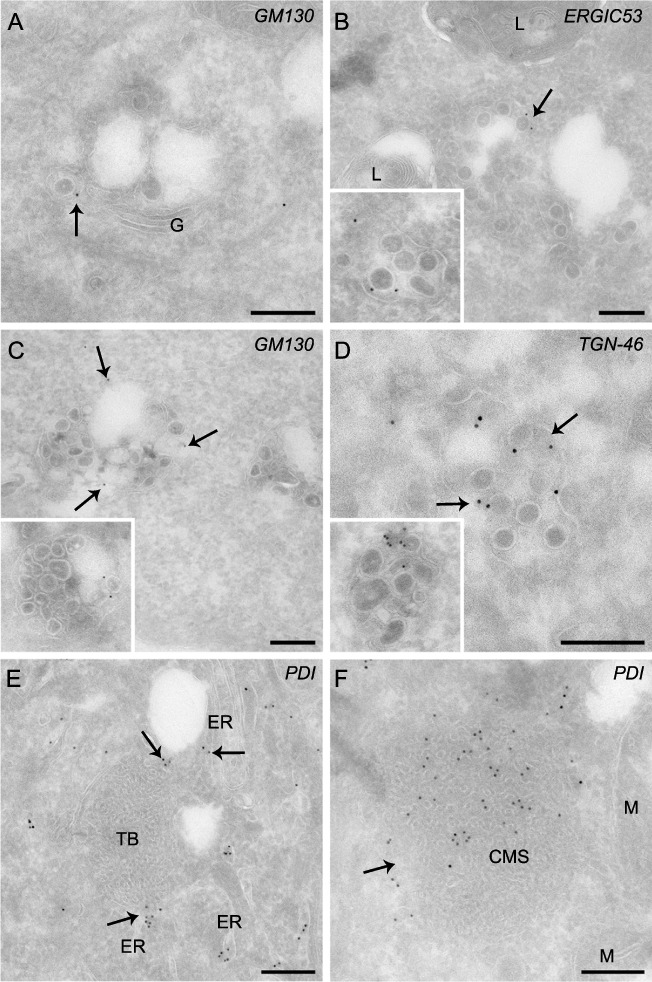
The distribution of various organelle protein markers on MHV‐induced structures. HeLa‐CEACAM1a cells infected with MHV‐A59 were fixed at 10 h p.i., processed for IEM and immunolabelled as described in Experimental procedures. A. Virions (arrows) present in the Golgi labelled with anti‐GM130 antibodies. B–D. The LVCVs are positive for ERGIC and Golgi protein markers, e.g. (B) ERGIC53, (C) GM130 and (D) TGN‐46. The insets show additional examples. The arrows highlight the labelling on LVCVs. E. The ER protein marker PDI localizes to the ER adjacent to the TB but not on this membranous rearrangement. F. The CMSs (arrow) are derived from the ER because they are positive for PDI. ER, endoplasmic reticulum; G, Golgi complex; L, lysosomes; M, mitochondria. Bar, 200 nm.
LVCVs are expanded ERGIC/Golgi cisternae
The low labelling of LVCVs with antibodies against ERGIC53, GM130 and TGN‐46 indicated that these organelles are derived from the ERGIC and/or Golgi complex. It has been shown that Golgi cisternae can increase in size in order to accommodate large luminal cargo proteins such as collagen (Bonfanti et al., 1998). Therefore, we hypothesized that LVCVs are Golgi cisternae that have expanded to increase their capacity to contain a higher number of viral proteins and/or forming virions. To sustain this notion, we first examined if the Golgi changes its organization and subcellular distribution during the course of an MHV infection. We took advantage of the HeLa‐GalNAcT2‐GFP stable cell line (Storrie et al., 1998), which expresses the fluorescent Golgi protein marker GalNActT2‐GFP, and inoculated these cells with MHV‐Srec before analysing them by IF at 0, 8 and 16 h p.i. The presence of viral proteins and virions in the Golgi was assessed using anti‐M protein antibodies. In non‐infected cells, the Golgi appeared as a juxtanuclear concentration of ribbon‐like structures as expected (Fig. 6A, upper panel; Storrie et al., 1998). The MHV infection caused three major changes, which were already detectable at 8 h p.i. but became more prominent at 16 h p.i. (Fig. 6A, middle and lower panels). First, the Golgi lost its compact organization; the organelle was scattered throughout the cytoplasm. Second, the intensity of the GalNAcT2‐GFP signal in the Golgi decreased. Third, GalNAcT2‐GFP appeared to partially localize to the ER. To unravel the effects of MHV infection on the Golgi at the ultrastructural level, the same samples were also processed in parallel for IEM and immunolabelled with anti‐GFP antibodies. At 0 h p.i the GalNAcT2‐GFP was exclusively concentrated in the Golgi complex, where it distributes into several cisternae (Fig. 6B). In contrast, at 16 h p.i., the labelling was found onto two different types of structures. The first were fragmented Golgi complexes (Fig. 6C). This morphological change during an MHV infection phenomenon has previously been reported (Lavi et al., 1996). The second types of labelled structures were LVCVs, in keeping with our notion that these compartments have an ERGIC/Golgi origin (Fig. 6D).
Figure 6.
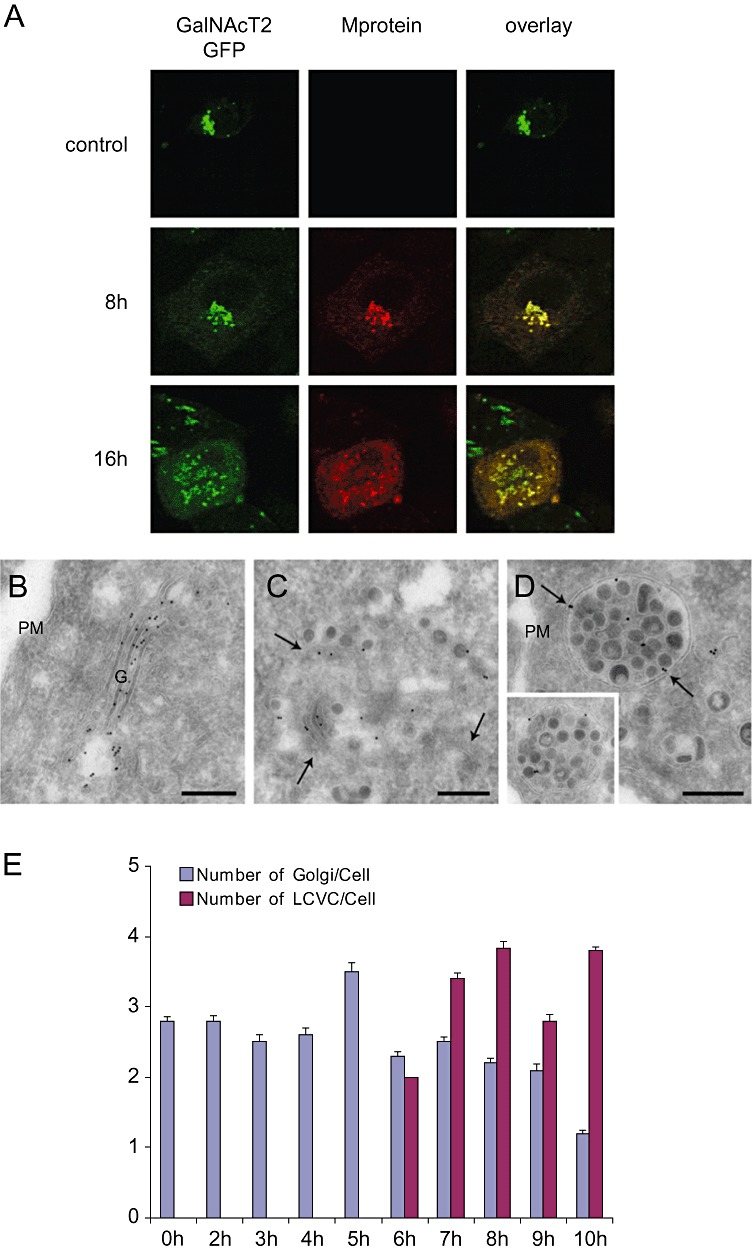
MHV infection leads to morphological alterations of the Golgi complex. HeLa‐GalNAcT2‐GFP cells were inoculated with MHV‐Srec and successively fixed at 0, 8 and 16 h p.i. A. Golgi viral proteins and virions were detected by IF using antibodies against the M protein for IF. The 16 h p.i. time point was selected because the number of infected cells and the morphological changed induced by MHV are similar to those of HeLa‐CEACAM1a cells infected with MHV‐A59 at 8 h p.i. (not shown). B–D. The same cells examined in (A) at 0 and 16 h p.i., were also analysed by IEM using anti‐GFP antibodies. A typical Golgi in non‐infected cells is shown in (B). MHV induces the fragmentation of the Golgi cisternae (C, arrows highlight the fragmented cisternae) and the formation of LVCVs (D, arrows), which are positive for the Golgi protein marker GalNAcT2‐GFP. The inset shows an additional example. E. The EM preparations described in Fig. 2 were used to statistically determine the average number of Golgi, defined as a stack of flattened sacs fragmented or not, and LVCVs per cell profile at the different p.i. time points. Error bars represent standard deviations. G; Golgi, PM, plasma membrane. Bar, 200 nm.
To substantiate that LVCVs derive from the Golgi, we statistically evaluated whether there is a numeric relationship between these two compartments and whether the appearance of LVCVs correlates with a decrease of Golgi complexes. To this end, we determined the number of LVCVs and Golgi complexes per cell profile at the different p.i. time points by counting these two organelles in the EM preparations of the MHV infection time‐course experiment. As shown in Fig. 6E, the LVCVs were first detected at 6 h p.i. and their number subsequently increased concomitant with a reduction of Golgi complexes.
All together, our data strongly suggest that LVCVs are ERGIC/Golgi cisternae that expand as a consequence of a large local production of virions.
Discussion
While DMVs and complete virions have been observed in cells infected with all CoV studied so far, such as MHV, SARS‐CoV, NL63, infectious bronchitis virus (IBV) and transmissible gastroenteritis virus (TGEV) (Alonso‐Caplen et al., 1984; David‐Ferreira and Manaker, 1965; Ruebner et al., 1967; Salanueva et al., 1999; Escorcia et al., 2002; Gosert et al., 2002; Snijder et al., 2006; Stertz et al., 2007; Knoops et al., 2008; Banacha et al. 2009), CMs, LVCVs and CMSs have exclusively been described in SARS‐CoV‐infected cells (Goldsmith et al., 2004; Almsherqi et al., 2005; Knoops et al., 2008) and TBs only in cells inoculated with MHV (David‐Ferreira and Manaker, 1965). Importantly, not all the ultrastructural analyses on SARS‐CoV‐and MHV‐infected cells have detected CMs, LVCVs and CMSs, and TB's, respectively (Table 1). This apparent discrepancy is probably due to various reasons, including the rarity of some of these structures, the examination of a single infection time point and the use of EM procedures lacking high resolution. In our study, we have detected all these types of membrane rearrangements during the course of an MHV infection because we performed a time‐course ultrastructural analysis using state‐of‐the‐art EM and IEM procedures. Combining our data with the previous ones, we can assert that SARS‐CoV is inducing the same type of structures and therefore we hypothesize that these membrane rearrangements are also formed during infections with other CoV. As a result, the data presented in this study generate a model that could be applicable to all CoV, even if some minor differences could exist.
Table 1.
Appearance time and categorization of the MHV‐induced structures.
| MHV‐induced structure | Appearance time (h) | nsp's | N protein | E protein | M protein | Organelle protein marker |
|---|---|---|---|---|---|---|
| DMVs | 2 | + | + | − | − | − |
| CMs | 3 | + | + | − | − | − |
| virions | 5 | − | + | + | + | − |
| LVCVs | 6 | − | + | + | + | ERGIC/Golgi |
| TBs | 6 | − | − | + | − | (ER) |
| CMSs | 9 | − | − | − | − | ER |
Overview of the MHV‐induced structure composition established by IEM. The presence (+) or absence (−) of nsp's, the various structural proteins and the analysed organelle protein markers in the DMVs, CMs, virions, LVCVs, TBs and CMSs is illustrated. The p.i. appearance time of these MHV‐induced structures is also indicated.
A major difficulty in understanding and studying the role of DMVs, CMs, virions, LVCVs, TBs and CMSs in the CoV life cycle has been the absence of information concerning their appearance and fate in the course of an infection. Another obstacle has also been the very limited characterization of LVCVs, TBs and CMSs but also in part of CMs. Our time‐course qualitative and quantitative examination of the MHV infection fills these two gaps (Table 1), and thus generates a more comprehensive chronological picture of the ultrastructural transformations occurring in the host cells that are induced by this virus.
MHV replication (Fig. 7A )
Figure 7.
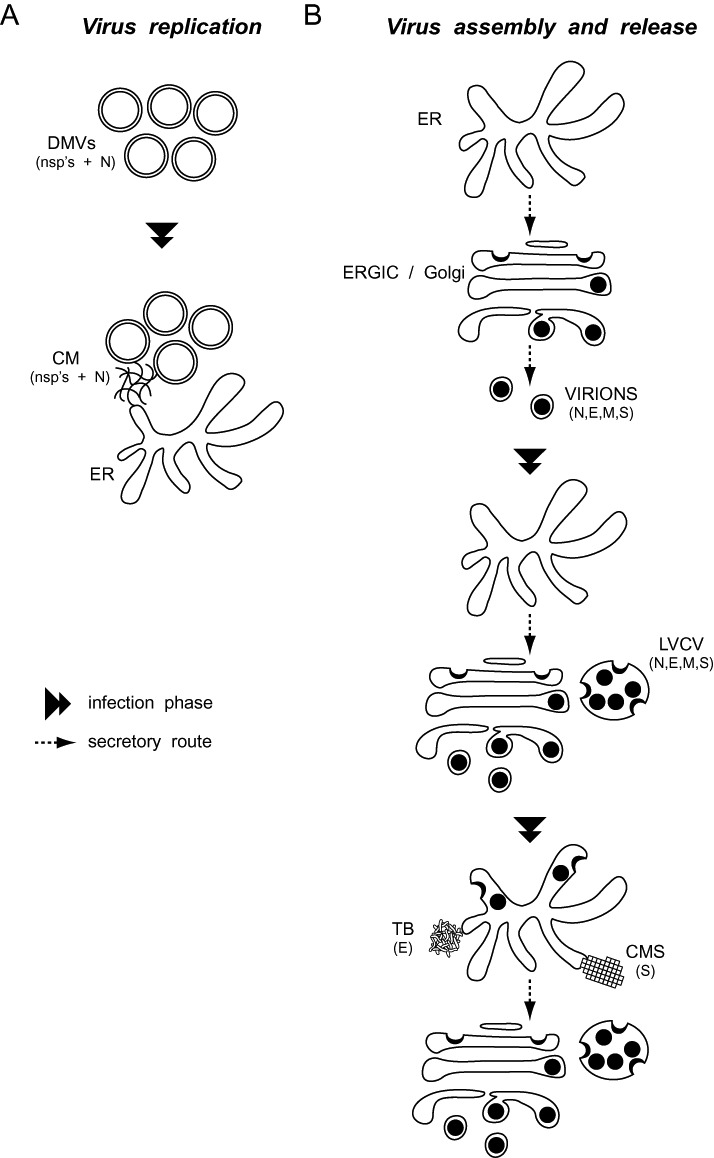
Schematic representation of the morphological changes induced by CoV in host cells. A. CoV replication. DMVs are the first membranous rearrangements that CoV induce. Subsequently, CMs become visible in their proximity. CMs appear to be connected with the ER and their size increases with the aggravation of the infection. B. CoV virion assembly and release. At the early stages of a CoV infection, viral particles principally assemble in the ERGIC and Golgi compartments. An increased synthesis of viral components triggers the expansion of these organelles into LVCVs, which are an additional site where complete virions are produced. A sustained infection and the consequent overflow of the secretory system with viral components results in the assembly of virions in the ER as well, but also in the formation of structures such as the TBs and the CMSs, which are composed by excess of self‐aggregating viral proteins. The composition in viral proteins of each CoV‐induced structure is indicated in between brackets.
Double‐membrane vesicles were the first structure (Knoops et al., 2008) that we observed after inoculating the cells with MHV (2 h p.i.; Fig. 3B and C). These vesicles are known to play a crucial role in viral RNA synthesis (Gosert et al., 2002; Prentice et al., 2004; Ahlquist, 2006; Haller et al., 2006; Sawicki et al., 2007). In agreement with this notion but also with previous studies (Gosert et al., 2002; Snijder et al., 2006; Stertz et al., 2007; Knoops et al., 2008), we found that the tested components of the RTCs, i.e. nsp2, nsp3, nsp4 and nsp8, localize to DMVs (Fig. 4) and that viral RNA production becomes detectable simultaneous with the appearance of DMVs (2 h p.i.; Fig. 1A). Moreover, the RNA levels correlate with the number of DMVs throughout the course of the MHV infection (Fig. S5). A particularly interesting observation was that the production of RNA reaches a steady‐state coincident with a diminution in the generation of DMVs (Fig. 1A, Fig. 3C and D, Fig. S5). This observation could support a new concept in which viral RNA synthesis is dictated by the number of DMVs (and thus RTCs) rather than by, or in addition to, the regulation of the rate of RNA replication and transcription. Alternatively, depletion of one or more host factors required for DMV biogenesis could lead to an identical outcome. Obviously, future studies are necessary to address this issue.
A remarkable new feature that we observed while studying DMVs generated in HeLa cells was the presence of an invagination, which often contained a small vesicle (Fig. 2A and B, Fig. S1B). This characteristic has not previously been documented perhaps because it is cell type specific. Yet, it may provide valuable information about the DMV biogenesis. The small vesicles could carry newly synthesized viral proteins and/or host components to DMVs. Alternatively; these profiles may also represent the fusion events between DMVs described for the SARS‐CoV (Knoops et al., 2008). Unfortunately, the Tokuyasu cryosectioning technique that we have used for our IEM analyses does not allow the preservation of inner content of the DMVs and the small vesicles intimately associated with them (Fig. 4D, E and I, Fig. S2B and H, Fig. S3B, C and E; Gosert et al., 2002; Snijder et al., 2006; Stertz et al., 2007). Hence, we could not determine the presence of viral proteins and double‐stranded RNA (dsRNA) in these small vesicles.
It has recently been proposed that CMs are the site of generation of DMVs (Knoops et al., 2008). Our data showing that the CMs have the same viral protein composition as the DMVs may support this hypothesis (Fig. 4A–E, Table 1). In addition, they reinforce the model (Knoops et al., 2008) that CMs are also involved in the replication and transcription of the viral RNA. As already observed (Knoops et al., 2008), however, the CMs appear after DMV formation (Fig. 3B and D). Because of this observation, we favour the idea that CMs are unlikely to be the precursor structure of DMVs. Potentially, they could originate from the DMVs but we never observed continuity between these two structures. Another possibility is that CMs are structures generated either for nsp's storage or by accumulation of excess nsp's that cannot be incorporated into DMVs. The possibility that the DMV biogenesis could require host components makes it possible that depletion of these components causes an accumulation of the newly synthesized transmembrane nsp's in the ER (Harcourt et al., 2004; Kanjanahaluethai et al., 2007; Oostra et al., 2007; Oostra et al., 2008), which leads to a clustering of RTCs at this site that results in the formation of CMs. In accordance with this hypothesis, CMs are frequently connected with the ER (Fig. S1C; Knoops et al., 2008). In addition, their size increases over time especially after 7 h p.i. when DMV formation slows down. It remains to be investigated whether CMs are able to synthesize viral RNA and to eventually pack dsRNA into their interior, which seems not to be the case so far (Knoops et al., 2008).
Virion assembly and release (Fig. 7B )
As expected, the virions were positive for the structural proteins, M, E and N, but not for the nsp's (Fig. 4 and Fig. S2, Table 1). Their immunolabelling with the anti‐E protein antiserum, however, was weak, consistent with the low number of E proteins per virion (Vennema et al., 1996; de Haan and Rottier, 2006). In agreement with the previous literature about their formation and release, virions were seen assembling in the ERGIC/Golgi (Fig. 2C) and complete viral particles were present in these organelles but also in secretory vesicles and the extracellular space (Fig. 4G, Fig. 5A, Fig. 6C, Fig. S1D). These observations were first recorded at 5 h p.i. (Fig. 5F and G). From 6 h p.i., virions were also forming and contained in the LVCVs (Fig. 2D, Fig. 3H and I, Fig. 4E, F and H, Fig. 5B–D, Fig. 6D, Fig. S2A and C, Table 1). LVCVs have already been described in SARS‐infected cells (Ng et al., 2003; Goldsmith et al., 2004; Knoops et al., 2008) and the observation that viral particles assemble at the limiting membrane of these compartments has led us to hypothesize and to show that they have a Golgi origin (5, 6). The enlargement of the ERGIC/Golgi compartments that leads to the formation of the LVCVs is probably a consequence of the accumulation of massive amounts of viral proteins and/or luminal virions that require an expansion of this organelle to accommodate them. This phenomenon has already been reported in the case of large cargo molecules passing through the Golgi (Bonfanti et al., 1998). It cannot be excluded a priori, however, that an overloading in viral proteins causes a fusion of the Golgi cisternae, which results in the LVCV formation. The fact that the secretory pathway is flooded by viral components is also emphasized by the formation of virions in the ER from 8 h p.i. (Fig. S6). The labelling efficiency of LVCVs for ERGIC and Golgi protein markers, however, was reduced as compared with that of the same organelles in non‐infected cells (Fig. 6B and Fig. S4B–D). These observations indicate that some of the standard Golgi functions of this compartment are probably altered by the high content of viral components. This is exemplified by the observation that the Golgi cisternae fragment and the Golgi protein marker GalNAcT2‐GFP partially localizes to the ER during the course of an MHV infection (Fig. 6A and C). In addition, the secretion of Gaussia luciferase is reduced during MHV infection (Verheije et al., 2008). Nevertheless, the LVCVs are functional structures because the extracellular release of MHV is still effective at the advanced stages of the MHV infection when numerous LVCVs are present in each cell (Fig. 1C, Fig. 3H and I). LVCVs are probably not an organelle exclusively induced by CoVs, as LVCV‐like structures have also been observed in cells infected with the (+) RNA rubellavirus (Risco et al., 2003; Novoa et al., 2005), which also assembles its progeny virions in association with the Golgi compartment.
Our data show that TBs and CMSs are infrequent structures (Fig. 3J and data not shown), that they appear at the late stages of the CoV infection (Fig. 3J and K, and data not shown) and that they probably contain a single viral protein (Fig. 4I, Table 1). These observations indicate that they are the result of the massive production of viral proteins that cannot be incorporated into the virions (Fig. 7B). The TBs are only positive for the E protein but not for the nsp's, the N and the M protein (Fig. S2D–E, Fig. S3A and C; Table 1). We cannot formally exclude that they also contain the S protein but it is unlikely because the individual expression of the E protein is sufficient to induce the formation of a complex of smooth tubular membranes with morphology reminiscent to that of the TBs (Raamsman et al., 2000). The E protein is a transmembrane component that is inserted in the ER before being transported to the ERGIC/Golgi (Lim and Liu, 2001; Corse and Machamer, 2003) that is also able to self‐interact (Raamsman et al., 2000; Von brunn et al., 2007). Importantly, even if not positive for the ER resident chaperone PDI, the TBs have clear membrane continuity with the ER (Fig. 2E, Fig. 5E, Fig. S2D and F). Consequently, a very likely scenario is that the TBs are generated by excess E protein that self‐aggregates in the ER. The CMSs were negative for any of the tested viral proteins (Fig. S2G and H, Fig. S3D and F, Table 1), and therefore we speculate that they are induced by the S protein, for which we do not have an antibody compatible with our IEM procedure. The CMSs are clearly connected with ER and positive for PDI (2, 5). Interestingly, it has previously been shown that overexpression of transmembrane proteins that have the capacity to self‐interact can lead to the formation of geometrical and crystalloid conformations in the ER that have been called either ‘cubic membranes’ or ‘organised smooth ER’, which resemble the CMSs (Snapp et al., 2003; 2006, 2009). Therefore, the simplest model that we propose is that CMSs are formed by the self‐assembly of excess S protein in the ER.
In conclusion, our study has ordered and characterized the different membranous rearrangements induced during a CoV infection. This information paves the way for future investigations about the biogenesis and the function of these structures, which is crucial to understanding the CoV life cycle and eventually for the development of more effective therapies against these pathogens.
Experimental procedures
Cells, virus and time‐course analysis of MHV infection
HeLa‐CEACAM1a cells (Verheije et al., 2008), the HeLa‐GalNAcT2‐GFP stable cell line (Storrie et al., 1998) and LR7 cells, used to propagate and titrate MHV‐A59 and MHV‐Srec, were maintained in Dulbecco's Modified Eagle Medium (DMEM; Cambrex Bioscience, Walkersville, MD, USA) containing 10% fetal calf serum (Bodinco, Alkmaar, the Netherlands), 100 IU of penicillin·per millilitre and 100 µg ml−1 of streptomycin (both from Life Technologies, Rochester, NY, USA).
HeLa cells expressing the mouse CEACAM1a receptor (HeLa‐CEACAM1a cells), which allows infecting these human cells with MHV‐A59 (Verheije et al., 2006), were used for the time‐course MHV infection. We decided to employ HeLa cells because many antibodies against human proteins are available and those cells have previously been successfully used for the study of CoV Replication (Gosert et al., 2002). The HeLa‐CEACAM1a cells were inoculated with MHV‐A59 at an moi of 30 Tissue Culture Infection Dose (TCID50) as determined on LR7 cells, in phosphate‐buffered saline (PBS) containing 50 µg ml−1 diethylaminoethyl‐dextran (PBS‐DEAE). After 30 min, HeLa‐CEACAM1a cells were washed and maintained in complete DMEM containing 10 µM of the fusion inhibitor mHR2 peptide (GenScript, Piscataway, NJ; Bosch et al., 2003). The mHR2 peptide prevents fusion of adjacent cells upon interaction of expressed S protein and mCEACAM1a receptor as well as additional infection by the residual inoculum in the medium or, at later times, secreted by the infected cells (Bosch et al., 2003). As a result, the MHV infection is synchronized. Subsequently, aliquots of infected cells and culture supernatants were collected for analysis at 0, 2, 3, 4, 5, 6, 7, 8, 9 and 10 h p.i.
The HeLa‐GalNAcT2‐GFP stable cell line was infected with MHV‐Srec, a recombinant MHV strain with an extended host range (de Haan et al., 2005). These cells were then fixed at 0, 8 and 12 h p.i. before being processed for IF and IEM.
Isolation of the total RNA and TaqMan analysis
The total RNA was isolated from the infected cells using the TRIzol reagent (Invitrogen, San Diego, CA, USA). RNA was further purified using the RNeasy mini‐kit (Qiagen, Hilden, Germany) according to the manufacturer instructions with subsequent DNaseI treatment on the column. RNA integrity was determined by spectrometry using a UV‐mini1240 device (Shimadzu, Kyoto, Japan). TaqMan single‐tube reverse transcription‐PCR (RT‐PCR) assay (PE Biosystems, Foster City, CA) was performed as described (de Haan et al., 2004) and used to analyse both the Orf1b region (de Haan et al., 2004) and the N protein‐encoding region (Raaben et al., 2007) of the viral genome. The reactions were performed in triplicate according to the manufacturer's instructions using an ABI Prism 7700 sequence detector (Foster City, CA, USA).
Metabolic labelling
Thirty minutes before the indicated time points, cells were starved for 30 min in cysteine‐and methionine‐free modified Eagle's medium containing 5% fetal calf serum and 10 mM Hepes, pH 7.2. Cells were subsequently radiolabelled in the same medium containing 100 µCi of 35S in vitro cell‐labelling mixture (Amersham Pharmacia Biotech, Freiburg, Germany) for 15 min and lysed in the TESV lysis buffer (20 mM Tris‐HCl, pH 7.3, 1 mM EDTA, 100 mM NaCl, 1 mM PMSF, 1% Triton X‐100). The lysates were analysed by SDS‐PAGE. Due to the preferential and sustained synthesis of viral proteins during the course of an infection, the radioactive bands corresponding to the M protein (22–27 kDa) and the structural proteins S and N were the most prominent on the autoradiographs. The M protein was quantified using a PhosphorImager system (Molecular Dynamics, Uppsala, Sweden).
Monitoring the extracellular release of the virus
The amount of virions present in the culture supernatants was determined by end‐point dilutions on LR7 cells and then calculating the TCID50 values.
Conventional electron microscopy
Cells were fixed with Karnovsky (2% para‐formaldehyde, 2.5% glutaraldehyde, 5 mM CaCl2, 10 mM MgCl2 in 0.1 M sodium cacodylate pH 7.4), first at room temperature for 30 min and then overnight at 4°C. Cell pellets were then post fixed with 1% OsO4/1. Then 5% KCNFe in 0.1 M cacodylate buffer for 1 h on ice and dehydrated stepwise with increasing concentrations of ethanol. The dehydrated pellets were rinsed with propylene oxide at room temperature and then embedded in Epon (Degtyarev et al., 2008). After resin polymerization, 65–80 nm sections were cut using an ultra‐microtome (Leica) and contrasted with uranyl acetate and lead citrate before being viewed in a JEOL 1010 or a JEOL 1200 electron microscope (JEOL, Tokyo, Japan).
For the quantitative analyses, 50 cell profiles were randomly selected at each p.i. time point and three different quantifications were performed. First, the number of cells containing at least one of the six MHV‐induced structures was counted to determine morphologically the proportion of infected cells. Second, we calculated the percentage of cell sections positive for each MHV‐induced structure. Third, we determined the average number of each structure per cell section and calculated the standard deviation. The standard deviation values were subsequently used to perform the t‐test, which revealed the significance of the data about DMVs (P = 0.0001), CMs (P = 0.0003), virions (P = 0.0003), LVCVs (P = 0.0005) and Golgi (P = 10−11) but not for the TB (P = 0.05). The calculation of the standard deviation and the t‐test could not be performed for the CMSs because of the rarity of these structures.
Immunoelectron microscopy
Cells were directly fixed by adding double‐strength fixative (4% para‐formaldehyde/0.4% glutaraldehyde or 4% para‐formaldehyde, both in 0.1 M phosphate buffer, pH 7.4) to the culture for 5 min at room temperature. This fixative was replaced by fresh standard strength fixative (2% paraformaldehyde/0.2% glutaraldehyde or 2% para‐formaldehyde, both in 0.1 M phosphate buffer, pH 7.4) and incubated overnight at 4°C. Fixation was subsequently continued for 1 h at room temperature before washing the cells 5 times with 0.1 M phosphate buffer, pH 7.4 and scraping them from the plate in the same buffer containing 1% gelatin. Cells were successively centrifuged and embedded in 12% gelatin (Slot and Geuze, 2007). The obtained pellets were cut into small cubes, which were cryo‐protected in 2.3 M sucrose and subsequently frozen in liquid nitrogen. Finally, after trimming to a suitable block shape, 45–60 nm ultrathin sections were cut at −120°C on dry diamond knives (Diatome AG, Biel, Switzerland) using either an UC6 or an UCT ultra‐microtome (Leica).
Cryo‐sections were labelled with rabbit antisera recognizing nsp2 and nsp3 [a kind gift of S. Baker (Schiller et al., 1998)], nsp4 [a kind gift from S. Baker (Schiller et al., 1998)], nsp8 [a kind gift of M. Denison (Lu et al., 1995)], M protein (Krijnse‐Locker et al., 1994), E protein (de Haan et al., 2002), GM130 [a kind gift of E.S. Sztul (Styers et al., 2008)], GFP (Abcam, Cambridge, UK) or mouse monoclonal antibodies against the nucleocapsid N (a kind gift from Stuart Siddell), PDI (Stressgen, Ann Arbor, MI, USA), ERGIC53 (Alexis Biochemicals, Lausen, Switzerland) or sheep anti‐TGN‐46 (Serotec, Oxford, UK) antiserum. The specificity of the antibodies against viral proteins was tested on mockinfected cells (data not shown). Antibodies were visualized with 10 or 15 nm gold particles conjugated to protein A (Slot and Geuze, 2007). Rabbit antibodies were directly detected with protein A‐gold particles whereas mouse and sheep antibodies where bridged with polyclonal anti‐mouse (DAKO, Carpinteria, CA, USA) and anti‐sheep (NORDIC, Tady, Sweden) IgG antibodies. Labelled cryo‐sections were finally contrasted with an uranyl acetate‐uranyl acetate methyl cellulose mixture. All specimens were imaged as described for the conventional EM.
Supporting information
Fig. S1. Additional morphological details of the preparations presented in Fig. 2. A. DMVs showing the presence of two lipid bilayers (arrows). B. DMVs with invaginations that appear to contain a small vesicle. C. A CM (arrow) that appears to be connected with the ER. D. Virions (arrows) released in the extracellular space. E. DMVs with protuberances emerging from their surface entering the invagination of an adjacent DMV (arrows). ER, endoplasmic reticulum; M, mitochondria; N, nucleus; PM, plasma membrane. White bar, 500 nm; black bar, 200 nm.
Fig. S2. MHV‐induced structures negative for non‐structural proteins. HeLa‐CEACAM1a cells infected with MHV‐A59 were fixed at 10 h p.i. before being processed for IEM and immunolabelled as described in Experimental procedures. A–C. Nsp2/nsp3, nsp4 and nsp8 do not localize to the LVCVs (arrows). D–F. Nsp2/nsp3, nsp4 and nsp8 are not present in the TBs (arrow). G and H. The CMCs (arrows) are not positive for nsp4 and nsp8. Asterisks mark the DMVs. M, mitochondria; N, nucleus; PM, plasma membrane. Bar, 200 nm.
Fig. S3. MHV‐induced structures negative for structural proteins. HeLa‐CEACAM1a cells inoculated with MHV‐A59 were fixed at 10 h p.i. before being processed for IEM and immunolabelled as described in Fig. 4. A. The N protein does not localize to the TBs. B. The M protein is not present on both DMVs and CMs. C and D. The TBs and the CMSs are not positive for the M protein. E and F. The E protein is not distributed on the DMVs, CMs and CMSs. Arrows and arrowheads indicate TBs and CMSs, respectively, whereas asterisks mark the DMVs. CM, convoluted membranes; ER, endoplasmic reticulum; G, Golgi; L, lysosome; LVCV, large virion‐containing vacuole; M, mitochondria. White bar, 500 nm; black bar, 200 nm.
Fig. S4. Specific immunolabelling of intracellular organelles. Mock‐infected cells were fixed and processed with antibodies against (A) PDI (ER), (B) ERGIC53 (ERGIC), (C) GM130 (cis‐Golgi cisternae), (D) TGN46 (trans‐Golgi) and (E) Lamp1 (late endosomes and lysosomes). ER, endoplasmic reticulum; L, lysosome; M, mitochondria; N, nucleus; PM, plasma membrane. White bar, 500 nm; black bar, 200 nm.
Fig. S5. Correlation between the viral RNA synthesis and DMV biogenesis. The number of DMVs/100 cells at each time point of the MHV infection was calculated by multiplying the percentage of DMV‐containing cells with the average number of DMVs per cell. Results are plotted on a log graph together with the gRNA and sgRNA N amounts expressed using arbitrary units.
Fig. S6. Virion assembly can occur in the ER. Immunolabelling of Hela‐CEACAM1a infected cells with anti‐PDI antibodies at 10 h p.i. Assembly of virions (arrows) can be observed in the ER. ER, endoplasmic reticulum; M, mitochondria. Bar, 200 nm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
The authors thank S. Baker, M. Denison, T. Nilsson, S. Siddell and E.S. Sztul for reagents, Matthijs Raaben and Eddie te Lintelo for technical assistance, Peter Rottier and Judith Klumperman for the critical reading of the manuscript, and Marc van Peski and René Scriwanek for assistance with the preparation of the figures. F.R. and C.A.M.H. are supported by the Utrecht University (High Potential grant).
References
- Ahlquist, P. (2006) Parallels among positive‐strand RNA viruses, reverse‐transcribing viruses and double‐stranded RNA viruses. Nat Rev Microbiol 4: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almsherqi, Z.A. , McLachlan, C.S. , Mossop, P. , Knoops, K. , and Deng, Y. (2005) Direct template matching reveals a host subcellular membrane gyroid cubic structure that is associated with SARS virus. Redox Rep 10: 167–171. [DOI] [PubMed] [Google Scholar]
- Almsherqi, Z.A. , Kohlwein, S.D. , and Deng, Y. (2006) Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J Cell Biol 173: 839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almsherqi, Z.A. , Landh, T. , Kohlwein, S.D. , and Deng, Y. (2009) Chapter 6: cubic membranes the missing dimension of cell membrane organization. Int Rev Cell Mol Biol 274: 275–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso‐Caplen, F.V. , Matsuoka, Y. , Wilcox, G.E. , and Compans, R.W. (1984) Replication and morphogenesis of avian coronavirus in Vero cells and their inhibition by monensin. Virus Res 1: 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banacha, S.B. , Orenstein, J.M. , Fox, L.M. , Randell, S.H. , Rowley, A.H. , and Baker, S.C. (2009) Human airway epithelial cell culture to identify new respiratory viruses: coronavirus NL63 as a model. J Virol Methods 156: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfanti, L. , Mironov, A.A., Jr , Martinez‐Menarguez, J.A. , Martella, O. , Fusella, A. , Baldassarre, M. , et al. (1998) Procollagen traverses the Golgi stack without leaving the lumen of cisternae: evidence for cisternal maturation. Cell 95: 993–1003. [DOI] [PubMed] [Google Scholar]
- Bosch, B.J. , van der Zee, R. , de Haan, C.A. , and Rottier, P.J. (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77: 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian, D.A. , and Baric, R.S. (2005) Coronavirus genome structure and replication. Curr Top Microbiol Immunol 287: 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Brunn, A. , Teepe, C. , Simpson, J.C. , Pepperkok, R. , Friedel, C.C. , Zimmer, R. , et al. (2007) Analysis of intraviral protein‐protein interactions of the SARS coronavirus ORFeome. PLoS ONE 2: e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse, E. , and Machamer, C.E. (2003) The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 312: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David‐Ferreira, J.F. , and Manaker, R.A. (1965) An electron microscope study of the development of a mouse hepatitis virus in tissue culture cells. J Cell Biol 24: 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyarev, M. , De Maziere, A. , Orr, C. , Lin, J. , Lee, B.B. , Tien, J.Y. , et al. (2008) Akt inhibition promotes autophagy and sensitizes PTEN‐null tumors to lysosomotropic agents. J Cell Biol 183: 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escorcia, M. , Fortoul, T.I. , Petrone, V.M. , Galindo, F. , Lopez, C. , and Tellez, G. (2002) Gastric gross and microscopic lesions caused by the UNAM‐97 variant strain of infectious bronchitis virus after the eighth passage in specific pathogen‐free chicken embryos. Poult Sci 81: 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff, H. , Hewson, R. , and Opstelten, D.J. (1998) Virus maturation by budding. Microbiol Mol Biol Rev 62: 1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfraind, C. , and Coutelier, J.P. (1998) Morphological analysis of mouse hepatitis virus A59‐induced pathology with regard to viral receptor expression. Histol Histopathol 13: 181–199. [DOI] [PubMed] [Google Scholar]
- Goldsmith, C.S. , Tatti, K.M. , Ksiazek, T.G. , Rollin, P.E. , Comer, J.A. , Lee, W.W. , et al. (2004) Ultrastructural characterization of SARS coronavirus. Emerg Infect Dis 10: 320–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya, A.E. , Enjuanes, L. , Ziebuhr, J. , and Snijder, E.J. (2006) Nidovirales: evolving the largest RNA virus genome. Virus Res 117: 17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert, R. , Kanjanahaluethai, A. , Egger, D. , Bienz, K. , and Baker, S.C. (2002) RNA replication of mouse hepatitis virus takes place at double‐membrane vesicles. J Virol 76: 3697–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan, C.A. , and Rottier, P.J. (2006) Hosting the severe acute respiratory syndrome coronavirus: specific cell factors required for infection. Cell Microbiol 8: 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan, C.A. , Masters, P.S. , Shen, X. , Weiss, S. , and Rottier, P.J. (2002) The group‐specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 296: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan, C.A. , Stadler, K. , Godeke, G.J. , Bosch, B.J. , and Rottier, P.J. (2004) Cleavage inhibition of the murine coronavirus spike protein by a furin‐like enzyme affects cell‐cell but not virus‐cell fusion. J Virol 78: 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan, C.A. , Li, Z. , te Lintelo, E. , Bosch, B.J. , Haijema, B.J. , and Rottier, P.J. (2005) Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J Virol 79: 14451–14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller, O. , Kochs, G. , and Weber, F. (2006) The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt, B.H. , Jukneliene, D. , Kanjanahaluethai, A. , Bechill, J. , Severson, K.M. , Smith, C.M. , et al. (2004) Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain‐like protease activity. J Virol 78: 13600–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton, A. , Mizzen, L. , MacIntyre, G. , Cheley, S. , and Anderson, R. (1986) Translational control in murine hepatitis virus infection. J Gen Virol 67 (Pt 5): 923–932. [DOI] [PubMed] [Google Scholar]
- Kanjanahaluethai, A. , Chen, Z. , Jukneliene, D. , and Baker, S.C. (2007) Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology 361: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops, K. , Kikkert, M. , Worm, S.H. , Zevenhoven‐Dobbe, J.C. , van der Meer, Y. , Koster, A.J. , et al. (2008) SARS‐coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6: e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse‐Locker, J. , Ericsson, M. , Rottier, P.J. , and Griffiths, G. (1994) Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J Cell Biol 124: 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi, E. , Wang, Q. , Weiss, S.R. , and Gonatas, N.K. (1996) Syncytia formation induced by coronavirus infection is associated with fragmentation and rearrangement of the Golgi apparatus. Virology 221: 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K.P. , and Liu, D.X. (2001) The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre‐Golgi compartments and physical interaction between the envelope and membrane proteins. J Biol Chem 276: 17515–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y. , Lu, X. , and Denison, M.R. (1995) Identification and characterization of a serine‐like proteinase of the murine coronavirus MHV‐A59. J Virol 69: 3554–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S. , and Krijnse‐Locker, J. (2008) Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, M.L. , Tan, S.H. , See, E.E. , Ooi, E.E. , and Ling, A.E. (2003) Proliferative growth of SARS coronavirus in Vero E6 cells. J Gen Virol 84: 3291–3303. [DOI] [PubMed] [Google Scholar]
- Novoa, R.R. , Calderita, G. , Arranz, R. , Fontana, J. , Granzow, H. , and Risco, C. (2005) Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 97: 147–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra, M. , te Lintelo, E.G. , Deijs, M. , Verheije, M.H. , Rottier, P.J. , and de Haan, C.A. (2007) Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J Virol 81: 12323–12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra, M. , Hagemeijer, M.C. , van Gent, M. , Bekker, C.P. , te Lintelo, E.G. , Rottier, P.J. , and de Haan, C.A. (2008) Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol 82: 12392–12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, K.W. , van der Meer, Y. , Roos, N. , and Snijder, E.J. (1999) Open reading frame 1aencoded subunits of the arterivirus replicase induce endoplasmic reticulum‐derived double‐ membrane vesicles which carry the viral replication complex. J Virol 73: 2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris, J.S. , Lai, S.T. , Poon, L.L. , Guan, Y. , Yam, L.Y. , Lim, W. , et al. (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361: 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice, E. , Jerome, W.G. , Yoshimori, T. , Mizushima, N. , and Denison, M.R. (2004) Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 279: 10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaben, M. , Einerhand, A.W. , Taminiau, L.J. , van Houdt, M. , Bouma, J. , Raatgeep, R.H. , et al. (2007) Cyclooxygenase activity is important for efficient replication of mouse hepatitis virus at an early stage of infection. Virol J 4: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raamsman, M.J. , Locker, J.K. , de Hooge, A. , de Vries, A.A. , Griffiths, G. , Vennema, H. , and Rottier, P.J. (2000) Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E. J Virol 74: 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risco, C. , Carrascosa, J.L. , and Frey, T.K. (2003) Structural maturation of rubella virus in the Golgi complex. Virology 312: 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier, P.J. , Horzinek, M.C. , and van der Zeijst, B.A. (1981) Viral protein synthesis in mouse hepatitis virus strain A59‐infected cells: effect of tunicamycin. J Virol 40: 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruebner, B.H. , Hirano, T. , and Slusser, R.J. (1967) Electron microscopy of the hepatocellular and Kupffer‐cell lesions of mouse hepatitis, with particular reference to the effect of cortisone. Am J Pathol 51: 163–189. [PMC free article] [PubMed] [Google Scholar]
- Saif, L.J. (2004) Animal coronavirus vaccines: lessons for SARS. Dev Biol (Basel) 119: 129–140. [PubMed] [Google Scholar]
- Salanueva, I.J. , Carrascosa, J.L. , and Risco, C. (1999) Structural maturation of the transmissible gastroenteritis coronavirus. J Virol 73: 7952–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen, A. , Ahola, T. , and Kaariainen, L. (2005) Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 285: 139–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki, S.G. , Sawicki, D.L. , Younker, D. , Meyer, Y. , Thiel, V. , Stokes, H. , and Siddell, S.G. (2005) Functional and genetic analysis of coronavirus replicase‐transcriptase proteins. PLoS Pathog 1: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki, S.G. , Sawicki, D.L. , and Siddell, S.G. (2007) A contemporary view of coronavirus transcription. J Virol 81: 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller, J.J. , Kanjanahaluethai, A. , and Baker, S.C. (1998) Processing of the coronavirus MHV‐JHM polymerase polyprotein: identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a. Virology 242: 288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna, P.B. , and Brian, D.A. (1997) Coronavirus genomic and subgenomic minus‐strand RNAs copartition in membrane‐protected replication complexes. J Virol 71: 7744–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot, J.W. , and Geuze, H.J. (2007) Cryosectioning and immunolabeling. Nat Protoc 2: 2480–2491. [DOI] [PubMed] [Google Scholar]
- Snapp, E.L. , Hegde, R.S. , Francolini, M. , Lombardo, F. , Colombo, S. , Pedrazzini, E. , et al. (2003) Formation of stacked ER cisternae by low affinity protein interactions. J Cell Biol 163: 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E.J. , van der Meer, Y. , Zevenhoven‐Dobbe, J. , Onderwater, J.J. , van der Meulen, J. , Koerten, H.K. , and Mommaas, A.M. (2006) Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol 80: 5927–5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz, S. , Reichelt, M. , Spiegel, M. , Kuri, T. , Martinez‐Sobrido, L. , Garcia‐Sastre, A. , et al. (2007) The intracellular sites of early replication and budding of SARS‐coronavirus. Virology 361: 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie, B. , White, J. , Rottger, S. , Stelzer, E.H. , Suganuma, T. , and Nilsson, T. (1998) Recycling of golgi‐resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole‐induced Golgi scattering. J Cell Biol 143: 1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styers, M.L. , O'Connor, A.K. , Grabski, R. , Cormet‐Boyaka, E. , and Sztul, E. (2008) Depletion of beta‐COP reveals a role for COP‐I in compartmentalization of secretory compartments and in biosynthetic transport of caveolin‐1. Am J Physiol Cell Physiol 294: C1485–C1498. [DOI] [PubMed] [Google Scholar]
- Svoboda, D. , Nielson, A. , Werber, A. , and Higginson, J. (1962) An electron microscopic study of viral hepatitis in mice. Am J Pathol 41: 205–224. [PMC free article] [PubMed] [Google Scholar]
- Tahara, S.M. , Dietlin, T.A. , Bergmann, C.C. , Nelson, G.W. , Kyuwa, S. , Anthony, R.P. , and Stohlman, S.A. (1994) Coronavirus translational regulation: leader affects mRNA efficiency. Virology 202: 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze, J. , Tooze, S. , and Warren, G. (1984) Replication of coronavirus MHV‐A59 in sac‐cells: determination of the first site of budding of progeny virions. Eur J Cell Biol 33: 281–293. [PubMed] [Google Scholar]
- Tooze, J. , Tooze, S.A. , and Fuller, S.D. (1987) Sorting of progeny coronavirus from condensed secretory proteins at the exit from the trans‐Golgi network of AtT20 cells. J Cell Biol 105: 1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema, H. , Godeke, G.J. , Rossen, J.W. , Voorhout, W.F. , Horzinek, M.C. , Opstelten, D.J. , and Rottier, P.J. (1996) Nucleocapsid‐independent assembly of coronavirus‐like particles by co‐expression of viral envelope protein genes. EMBO J 15: 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije, M.H. , Raaben, M. , Mari, M. , Te Lintelo, E.G. , Reggiori, F. , van Kuppeveld, F.J. , et al. (2008) Mouse hepatitis coronavirus RNA replication depends on GBF1‐mediated ARF1 activation. PLoS Pathog 4: e1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije, M.H. , Wurdinger, T. , van Beusechem, V.W. , de Haan, C.A. , Gerritsen, W.R. , and Rottier, P.J. (2006) Redirecting coronavirus to a nonnative receptor through a virus‐encoded targeting adapter. J Virol 80: 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg, G.A. , Slobodskaya, O. , and Spaan, W.J. (2006) Transcriptional profiling of acute cytopathic murine hepatitis virus infection in fibroblast‐like cells. J Gen Virol 87: 1961–1975. [DOI] [PubMed] [Google Scholar]
- Weiss, S.R. , and Navas‐Martin, S. (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69: 635–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr, J. , Snijder, E.J. , and Gorbalenya, A.E. (2000) Virus‐encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81: 853–879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Additional morphological details of the preparations presented in Fig. 2. A. DMVs showing the presence of two lipid bilayers (arrows). B. DMVs with invaginations that appear to contain a small vesicle. C. A CM (arrow) that appears to be connected with the ER. D. Virions (arrows) released in the extracellular space. E. DMVs with protuberances emerging from their surface entering the invagination of an adjacent DMV (arrows). ER, endoplasmic reticulum; M, mitochondria; N, nucleus; PM, plasma membrane. White bar, 500 nm; black bar, 200 nm.
Fig. S2. MHV‐induced structures negative for non‐structural proteins. HeLa‐CEACAM1a cells infected with MHV‐A59 were fixed at 10 h p.i. before being processed for IEM and immunolabelled as described in Experimental procedures. A–C. Nsp2/nsp3, nsp4 and nsp8 do not localize to the LVCVs (arrows). D–F. Nsp2/nsp3, nsp4 and nsp8 are not present in the TBs (arrow). G and H. The CMCs (arrows) are not positive for nsp4 and nsp8. Asterisks mark the DMVs. M, mitochondria; N, nucleus; PM, plasma membrane. Bar, 200 nm.
Fig. S3. MHV‐induced structures negative for structural proteins. HeLa‐CEACAM1a cells inoculated with MHV‐A59 were fixed at 10 h p.i. before being processed for IEM and immunolabelled as described in Fig. 4. A. The N protein does not localize to the TBs. B. The M protein is not present on both DMVs and CMs. C and D. The TBs and the CMSs are not positive for the M protein. E and F. The E protein is not distributed on the DMVs, CMs and CMSs. Arrows and arrowheads indicate TBs and CMSs, respectively, whereas asterisks mark the DMVs. CM, convoluted membranes; ER, endoplasmic reticulum; G, Golgi; L, lysosome; LVCV, large virion‐containing vacuole; M, mitochondria. White bar, 500 nm; black bar, 200 nm.
Fig. S4. Specific immunolabelling of intracellular organelles. Mock‐infected cells were fixed and processed with antibodies against (A) PDI (ER), (B) ERGIC53 (ERGIC), (C) GM130 (cis‐Golgi cisternae), (D) TGN46 (trans‐Golgi) and (E) Lamp1 (late endosomes and lysosomes). ER, endoplasmic reticulum; L, lysosome; M, mitochondria; N, nucleus; PM, plasma membrane. White bar, 500 nm; black bar, 200 nm.
Fig. S5. Correlation between the viral RNA synthesis and DMV biogenesis. The number of DMVs/100 cells at each time point of the MHV infection was calculated by multiplying the percentage of DMV‐containing cells with the average number of DMVs per cell. Results are plotted on a log graph together with the gRNA and sgRNA N amounts expressed using arbitrary units.
Fig. S6. Virion assembly can occur in the ER. Immunolabelling of Hela‐CEACAM1a infected cells with anti‐PDI antibodies at 10 h p.i. Assembly of virions (arrows) can be observed in the ER. ER, endoplasmic reticulum; M, mitochondria. Bar, 200 nm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item