

Summary
Coronaviruses are a diverse group of viruses that infect mammals and birds. Bats are reservoirs for several different coronaviruses in the Alphacoronavirus and Betacoronavirus genera. They also appear to be the natural reservoir for the ancestral viruses that generated the severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus outbreaks. Here, we detected coronavirus sequences in next‐generation sequence data created from Eonycteris spelaea faeces and urine. We also screened by PCR urine samples, faecal samples and rectal swabs collected from six species of bats in Singapore between 2011 and 2014, all of which were negative. The phylogenetic analysis indicates this novel strain is most closely related to lineage D Betacoronaviruses detected in a diverse range of bat species. This is the second time that coronaviruses have been detected in cave nectar bats, but the first coronavirus sequence data generated from this species. Bat species from which this group of coronaviruses has been detected are widely distributed across SE Asia, South Asia and Southern China. They overlap geographically, often share roosting sites and have been witnessed to forage on the same plant. The addition of sequence data from this group of viruses will allow us to better understand coronavirus evolution and host specificity.
Keywords: disease ecology, virus diversity, evolution, distribution
Introduction
Over 70% of emerging or re‐emerging infectious diseases originate in animals, with zoonotic RNA viruses responsible for the majority of these cross‐species spillover events (Woolhouse et al., 2005). Zoonotic outbreaks are amplified or dampened by a number of ecological, environmental and anthropogenic factors (Karesh et al., 2012). Change in land use is a principal driver for emerging infectious diseases by modifying the wildlife–human interface (Jones et al., 2008). Increase in human populations has caused wild animals to lose their natural habitat and diminished resource availability, while bushmeat hunting and consumption have presented further opportunities for cross‐species exposure (Wolfe et al., 2005).
In South‐East Asia, severe habitat loss has impacted the natural ecology of bats and occasionally resulted in zoonotic virus spillover, most notably in the Nipah outbreaks in Malaysia and Bangladesh (Chua et al., 2000; Hsu et al., 2004). Bats are reservoirs for many notable pathogenic viruses, including Hendra virus in Australia (Halpin et al., 2000), the recent outbreaks of Ebola virus in sub‐Saharan Africa (Pourrut et al., 2009; Olival and Hayman, 2014; Ogawa et al., 2015), rabies and several other lyssaviruses (Banyard et al., 2014). Furthermore, zoonotic coronaviruses (e.g. SARS‐CoV and MERS‐CoV) and the human coronavirus HCoV‐229E, a causal agent of the common cold, appear to have resulted from zoonotic transfer from bats, either directly or through an intermediate amplifying host (Drexler et al., 2014). Indeed, bat coronaviruses are exceptionally diverse and genetic evidence provides further support that group 1 and group 2 mammalian coronaviruses originated in bats (Vijaykrishna et al., 2007; Hu et al., 2015), highlighting the importance of bats as a potential source of new human diseases.
Coronaviruses cause a wide variety of symptoms, with human coronaviruses causing respiratory infections in humans, while other animals suffer respiratory, gastroenteric and more severe manifestations of disease (Peiris et al., 2003; Saif, 2004). Coronaviruses can be highly pathogenic, especially when they spill over into incidental hosts, as evidenced by the high fatality rates seen in the severe acute respiratory syndrome (SARS) in 2003 and the ongoing Middle East respiratory syndrome (MERS) outbreak, 9.6% and 40%, respectively (Zumla et al., 2015).
Coronaviruses are enveloped single‐stranded, positive sense RNA viruses that belong to the Order Nidovirales, the Family Coronaviridae and the subfamily Coronavirinae (de Groot et al., 2012). There are four genera: Alphacoronavirus, Betacoronavirus, Deltacoronavirus and Gammacoronavirus (Adams and Carstens, 2012). Viruses of the Deltacoronavirus and Gammacoronavirus genera primarily infect birds, with avian infectious bronchitis virus a prominent member of the latter that causes systemic infections of chickens, resulting in major economic losses (Jackwood, 2012). Alphacoronavirues infect humans (HCoV‐229E and HCoV‐NL63), pigs, felines, canines and a number of different bat species (Drexler et al., 2014). An alphacoronavirus, that forms a sister group to HCoV‐229E, has also been recently found to commonly infect camels (Sabir et al., 2016). Betacoronavirus, which is further classified into lineages A–D, is found in a wide variety of hosts including multiple bat species, humans and numerous domestic and peridomestic animals (Woo et al., 2009). Of note, this genus contains both SARS‐CoVs (lineage B) and MERS‐CoVs (lineage C).
The large genomes and high rates of recombination witnessed in coronaviruses are suggested reasons for the ability of coronaviruses to jump into other species (Woo et al., 2009). Interestingly, coronaviruses have some proof‐reading mechanisms and are not subject to the infidelity of the RNA polymerase reading mechanism like other RNA viruses, such as influenza (Drake, 1993; Minskaia et al., 2006). There is a deep phylogenetic divergence between the avian coronavirus‐specific genera and the mammalian coronaviruses and a lack of evidence of avian–mammalian cross‐species transmission (Woo et al., 2012a); however, there is prolific evidence of these events occurring between different mammal species (Lau et al., 2012) and between different bird species (Chu et al., 2011).
Detecting viruses that possess the capacity to effectively transmit across species greatly enhances our knowledge on the evolution and ecology of coronaviruses. Little is known about bat–virus interactions in Singapore, and although this country lacks endemic bat species, it shares the geographical range of several South‐East Asian bat species. Here, we use both RT‐PCR and next‐generation sequencing methods to detect coronaviruses in the bats of Singapore and examine their phylogenetic relatedness.
Materials and Methods
Sample collection
Fresh faecal and urine samples were collected from a colony of Eonycteris spelaea on plastic sheets every 2 weeks from April 2011 until June 2015. Samples were placed into virus transport media at 4°C until transport to Duke‐NUS where they were placed into a −80°C freezer until screening. Additionally, a total of 431 individual bats from six species: E. spelaea (n = 169), Cynopterus brachyotis (n = 144), Penthetor lucasi (n = 79), Macroglossus minimus (n = 2), Rhinolophus lepidus (n = 36) and Myotis sp. (n = 1) were captured from nine locations throughout Singapore from April 2011 to March 2014. Collections were undertaken with approval from the National Parks Board (NP/RP11‐011‐3a) and the National University of Singapore Institutional Animal Care and Use Committee (IACUC Permit # B01/12). Bats were captured in‐flight using a ground level or elevated mist net or a harp trap. Oral and rectal swabs were collected and treated in the same manner as the faecal and urine samples.
Next‐generation sequencing
We performed next‐generation sequencing on pooled faecal material and pooled urine collected from a colony of E. spelaea for virus discovery. Ten grams of faecal matter pooled from collections taken from plastic sheets laid under the colony on 14 March, 28 March and 11 April 2013 was vortexed in TBS buffer (25 mm Tris, 150 mm NaCl and Roche ULTRA protease inhibitor cocktail tablet) to resuspend the material into solution. The samples were then homogenized using silica beads (MP FastPrep – 24 bead beater; MP Biomedicals, Santa Ana, CA, USA). After homogenization, the samples underwent a series of centrifugations where the supernatant was transferred to new tubes (full protocol available upon request). After the final centrifugation step, the supernatant was discarded and the pellet was resuspended in 500 μl of TBS buffer with protease inhibitors and kept at −80°C. A library was prepared from the faecal processing and the serological capture extracted RNA/DNA. The SMARTer Universal Low Input RNA Kit (Clontech Laboratories, Mountain View, CA, USA) used with low concentration nucleic acids. The double‐stranded cDNA library was purified with AMPure beads (Agencourt, Beckman Coulter, Brea, CA, USA), adaptors were removed with a digestion mix, and the library was amplified with an Advantage 2 PCR kit (Clontech Laboratories). The libraries were analysed on a bioanalyser (Agilent Technologies, Santa Clara, CA, USA), and the reaction was run on both the Illumina HiSeq and MiSeq machines.
Urine samples from an E. spelaea colony in Singapore on 24 April, 8 May and 20 May 2014 were pooled and centrifuged at 10 000 g for 3 min. TRI‐Reagent was added to urine viral supernatant and RNA extracted using Direct‐zol™ RNA MiniPrep (#R2050; Zymo Research Corporation, Irvine, CA, USA) as per manufacturer's instruction. RNA was subjected to in‐column DNase I (#M0303S; New England BioLabs Inc., Ipswich, MA, USA) digestion as instructed in Direct‐zol™ RNA MiniPrep manual. Extracted and DNase I digested RNA was treated with Ribo‐Zero™ Gold rRNA Removal Kit (Epidemiology) (#MRZE706; Epicentre, Madison, WI, USA) as described in the instruction manual.
Two 500‐bp next‐generation sequencing cDNA libraries (urine‐MiSeq‐25 and urine‐MiSeq‐27) were constructed from the same sample using NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (#E7420S; New England BioLabs Inc.) as per manufacturer's instruction. cDNA libraries were visualized on 1.5% agarose gel, excised and purified using Zymoclean Gel DNA Recovery Kit (#D4007; Zymo Research Corporation) as instructed.
Fastq read files generated from the Illumina MiSeq and HiSeq runs were analysed using the DIAMOND alignment tool (Buchfink et al., 2015), against reference sequences in a custom protein database generated from RefSeq virus and neighbour nucleotide records (http://www.ncbi.nlm.nih.gov/genome/viruses/) (Brister et al., 2015) using a custom Perl script. DIAMOND alignment results were imported into MEGAN 6 (Huson and Mitra, 2012), using a naive LCA algorithm to perform taxonomic assignment and visualization of aligned reads. All reads taxonomically assigned to family Coronaviridae were extracted and pooled, and SPAdes 3.6.2 (Bankevich et al., 2012) was used to perform de novo assembly of pooled reads.
PCR screening
Oral and rectal swabs or fresh faeces from each individual were pooled and screened for coronaviruses. A total of 974 urine samples (947 samples from individuals and 27 samples pooled by collection period) collected biweekly from the E. spelaea colony over 4 years from May 2011 to May 2015 in addition to 150 fresh faecal pellets from the above‐mentioned bat colony collected during 3 time points over 6 weeks (April–May 2014) were included. RNA was extracted from these samples using the QIAxtractor automated purification system (QIAGEN, Hilden, Germany) following the manufacturer's instructions. cDNA was synthesized using Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and screened using coronavirus family‐specific primer (PanCor IN‐6 5′– GGTTGGGACTATCCTAAGTGTGA –3′; PanCor IN‐7 5′– CCATCATCAGATAGAATCATCATA –3′) (Drosten et al., 2003) that targeted the RNA‐dependent RNA polymerase gene. The PCR protocol used an initial denaturation step of 95°C for 10 min, 40 cycles of denaturation at 95°C for 1 min, annealing at 56°C for 1 min, extension at 72°C for 1 min, a final extension at 72°C for 5 min. PCR products were screened on a 1.5% agarose gel and visualized on a gel doc machine. RNA from cultured Coronaviruses 229E, OC43 and NL63 was extracted manually, and cDNA was synthesized to serve as positive controls.
Phylogenetic analysis
Coronavirus sequences representing diverse groups were downloaded from GenBank for three genes used in this study: RdRp (RNA‐dependent RNA polymerase genes), Hel (helicase: nsp13), E (envelope) and N (nucleocapsid), including α‐coronavirus, β‐coronavirus (comprised of lineages A–D), and MERS‐coronavirus, obtained from a wide range of hosts (bats, felines, human, avian and camels). Two Indonesian bat coronavirus that were recently detected from Moluccan naked‐backed fruit bats (Dobsonia moluccensis) were also included (Stamatakis, 2014; Anindita et al., 2015). Partial sequences of the novel bat coronavirus generated were aligned with this data set and alignments manually edited in Geneious v9.0.3 (Biomatters, Auckland, New Zealand). Final aligned data sets comprise 82 RdRp sequences (2814 bp in length), 69 Hel sequences (1827 bp), 67 E sequences (240 bp) and 49 N sequences (578 bp) that were analysed separately (see accession numbers in Table S1). Phylogenetic trees were reconstructed using maximum likelihood implemented in RAxML v8.0.14 (Scientific Computing Group, Heidelberg Institute for Theoretical Studies, Heidelberg, Germany) (Stamatakis, 2014), and branch support was assessed with 1000 nonparametric bootstrap replicates, with only values greater than 50% indicated at the major nodes. Trees were rooted with one representative of the gammacoronaviruses (γ‐CoV, accession number: AY338732).
Host distribution mapping
Bat species distribution and country shapefiles were downloaded from the International Union for Conservation of Nature (IUCN, 2016) and imported into qGIS to create a map displaying the distribution of known Betacoronavirus lineage D reservoirs from South Asia and South‐East Asia (QGIS Development Team, 2016). A map was also made of the overlapping distribution of the three cave roosting bat species.
Results
The Illumina NGS data sets from libraries generated from E. spelaea faeces and urine produced a total of 772 coronavirus reads (Table 1). The libraries constructed from the urine samples produced 712 coronavirus reads, while the HiSeq and MiSeq runs from the faecal samples generated 60 reads. The MiSeq run on the pooled faeces produced the fewest reads. The read lengths were from a minimum of 76 bp to a maximum of 251 bp, with a mean of 240 bp (stdv 34.7 bp). A total of nine contigs were generated from the de novo assembly (min. 392 bp and max. 1092 bp). All 431 pooled oral–rectal samples from the six bat species were PCR‐negative. The urine and faecal samples (n = 1124) collected from the E. spelaea colony were also PCR‐negative.
Table 1.
Total number of coronavirus reads from four NGS data sets on pooled faeces and urine from a colony of Eonycteris spelaea
| Total reads | Total corona reads | |
|---|---|---|
| Urine MiSeq | 18 547 895 | 712 |
| Faeces HiSeq | 68 584 413 | 26 |
| Faeces MiSeq | 4 952 973 | 34 |
We analysed four individual data sets (RdRp, Hel, E and N) in the phylogenetic analysis, with each data set including alpha‐ and beta‐coronaviruses. Maximum‐likelihood phylogenies show Betacoronavirus is composed of four distinct and well‐supported lineages (A–D), although the intrageneric relationships of beta‐coronaviruses vary between the four gene trees (Figs 1 and 2). The RdRp phylogeny places lineage C basal to lineages A, B and D, which form a monophyletic group with only moderate support (bootstrap, BS = 58%). This interlineage relationship is consistent with a recent bat CoV study from Thailand (Wacharapluesadee et al., 2015). In contrast, in the Hel phylogeny, lineage A is basal to the monophyletic lineages B, C and D (BS = 88%), with lineages B and D forming a strongly supported sister group (BS = 90%). This intrageneric relationship based on the Hel gene is also concordant with previous studies (Lau et al., 2012; Woo et al., 2012b). The E gene phylogeny resembles that of the RdRp in grouping lineages A and D as sister taxa but with low support (BS = 51%). We also compared the genetic divergence of the RdRp gene within the different lineages: lineage D exhibits a higher level of sequence variation with only 58.4% nucleotide identity, in contrast to lineages A (83–100% nucleotide identity), B (92–100%) and C (82–100%) that show a higher percentage similarity. The N gene phylogeny indicated lineage D is basal to other lineages A, B and C; however, this interlineage relationship was statistically insignificant.
Figure 1.
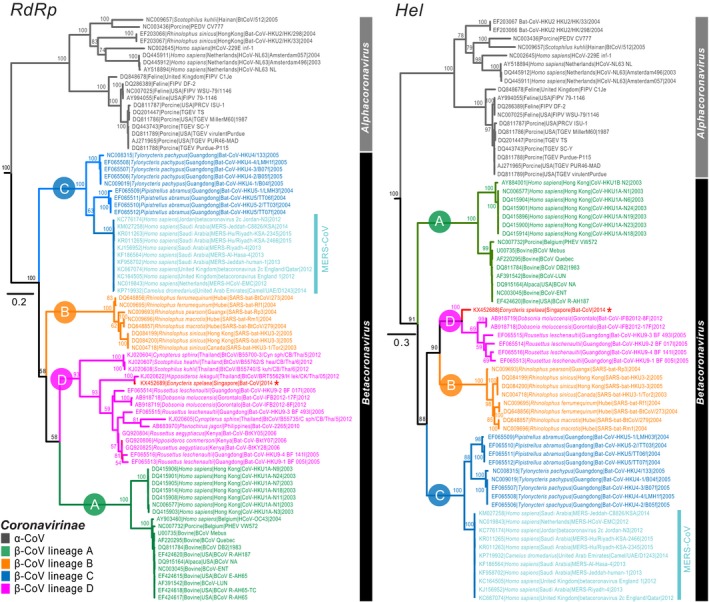
Phylogenetic trees of the RdRp and Hel nucleotide sequences of alpha‐ and beta‐coronaviruses reconstructed using maximum‐likelihood method in RAxML, with 1000 bootstrap replicates. Bootstrap support values greater than 50% are indicated at major nodes. The trees were rooted with one representative of the gammacoronavirus (γ‐CoV, accession number: AY338732). Coloured branches denote different betacoronavirus lineages (i.e. A–D). New CoV sequence collected from a bat species (Eonycteris spelaea) in this study is marked by an asterisk. The scale bar indicates the number of nucleotide substitutions per site. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Figure 2.
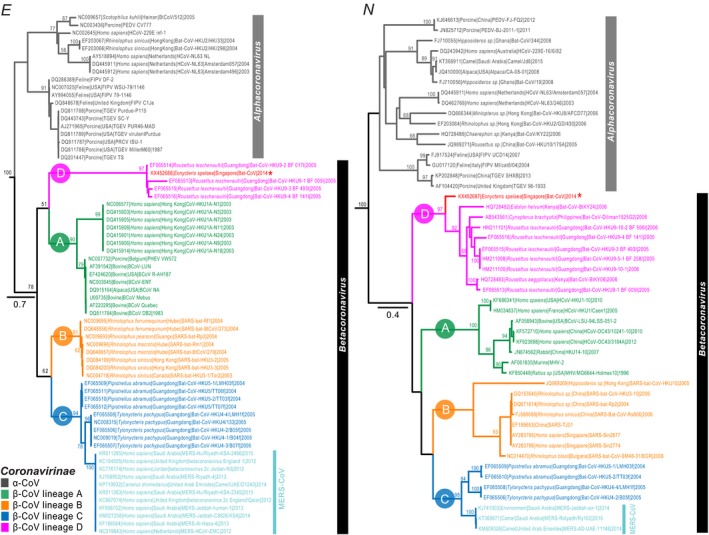
Phylogenetic tree of the E and N nucleotide sequences of alpha‐ and beta‐coronaviruses reconstructed using maximum‐likelihood method in RAxML, with 1000 bootstrap replicates. Bootstrap support values greater than 50% are indicated at major nodes. The tree was rooted with one representative of the gammacoronavirus (γ‐CoV, accession number: AY338732). Coloured branches denote different betacoronavirus lineages (i.e. A–D). New CoV sequence collected from a bat species (Eonycteris spelaea) in this study is marked by an asterisk. The scale bar indicates the number of nucleotide substitutions per site. [Colour figure can be viewed at http://wileyonlinelibrary.com]
The RdRp, Hel, E and N gene phylogenies all place the novel CoV identified from E. spelaea unequivocally in the well‐supported monophyletic Betacoronavirus lineage D (RdRp and Hel: 100% BS; E: 91% BS; N: 97%). The HKU9 prototype virus for lineage D Betacoronavirus was reported from Rousettus leschenaultii in Hong Kong in 2005 (Woo et al., 2007). Within lineage D, partial RdRp sequence of the novel CoV possesses 77.2–97.4% nucleotide sequence identity with the remaining lineage D bat‐CoVs. In contrast, the partial Hel and E genes of the E. spelaea CoV sequences exhibit greater similarity (Hel: 80–82.5%; E: 70.6–97.9%) with other lineage D viruses, although this may due to the limited number of lineage D Hel and E gene sequences that are available. The N gene indicated the CoV of E. spelaea showed more distant relationship with the remaining lineage D bat‐CoVs (52–60% similarity).
The RdRp phylogeny indicates that the E. spelaea CoV is most closely related to a virus from a large Asian roundleaf bat (Hipposideros lekaguli), strongly supported with a bootstrap value of 100%. These two sequences form a sister group (bootstrap 93%) with coronaviruses from Cynopterus sphinx, D. moluccensis, Hipposideros commersoni, Ptenochirus jagori, R. leschenaultii and Rousettus aegypticus that comprise the majority of lineage D betacoronavirus sequences available.
Discussion
This is the first coronavirus detected from a bat in Singapore, and our study provides the first evidence of cave nectar bat E. spelaea harbouring a lineage D betacoronavirus. Coronaviruses were previously detected in E. spelaea in the Philippines, but no sequence data were generated (Watanabe et al., 2010). Eonycteris spelaea has also previously been reported to harbour a paramyxovirus in China (Yuan et al., 2014), Phnom‐Penh bat virus (Queen et al., 2015), Issyk‐kul virus (Calisher et al., 2006) and were also seropositive for Nipah virus in Malaysia (Yob et al., 2001).
In South‐East Asia, lineage D betacoronaviruses (also referred to as Ro‐BatCoV HKU9: Woo et al., 2012a,b) have been detected in eight different bat species from three bat families belonging to two suborders, Yinpterochiroptera and Yangochiroptera: C. sphinx, D. moluccensis, E. spelaea, H. lekaguli, P. jagori, R. leschenaultii, Scotophilus heathi and Scotophilus kuhli (Woo et al., 2007; Tsuda et al., 2012; Anindita et al., 2015). A next‐generation sequencing approach in Yunnan province China detected HKU9 coronavirus sequences in a community dominated by Hipposideros armiger, but the host identity of the faeces tested was not confirmed (Ge et al., 2012). The bat species known to harbour lineage D Betacoronavirus are widely distributed across South Asia and South‐East Asia, and there are large areas where these distributions overlap (Fig 3 , Table S2). Lineage D betacoronaviruses were also detected in two bat species in Kenya, Rousettus aegyptiacus and H. commersoni, and one in Madagascar, Pteropus rufus (Tong et al., 2009; Razanajatovo et al., 2015).
Figure 3.
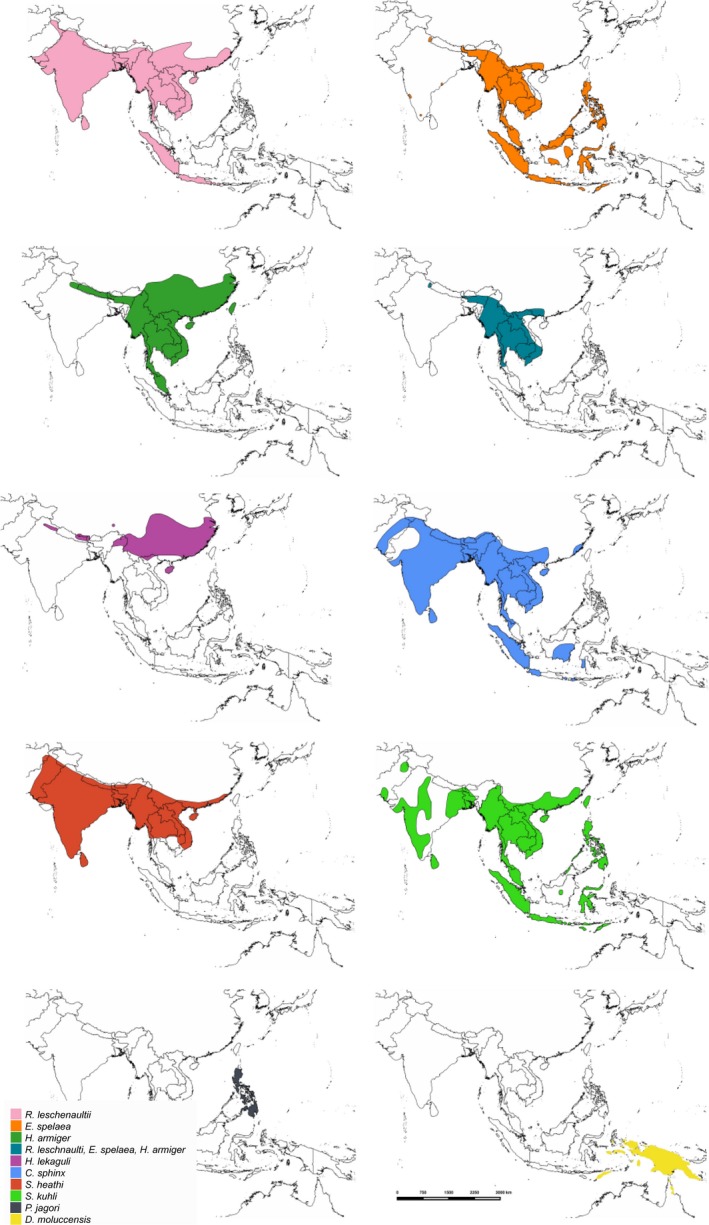
Bat distribution map for each species in South‐East Asia from which lineage D betacoronaviruses have been detected. These maps were generated from IUCN maps in QGIS. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Interestingly, the pooled urine samples where coronavirus was detected in the Illumina NGS were negative with traditional RT‐PCR, indicating a low amount of virus. These screening results possibly indicate that there is a low prevalence in the bats in Singapore, low virus titres in the samples and/or we missed shedding foci during our sampling. The only bat species we detected coronaviruses in were the cave nectar bats (E. spelaea), and this was using deep sequencing where the majority of the reads were detected in the urine sample. We did not detect this in the other insectivorous or frugivorous bat species with this well‐validated primer set; however, we did not perform NGS on samples from other species in Singapore. The majority of coronavirus detections have been from insectivorous bats, but this may have been spurred by the discovery of SARS‐like CoV in Rhinolophus bat species in China (Drexler et al., 2014). Different coronaviruses can infect the same species of bats, even very divergent viruses (Wacharapluesadee et al., 2015). The close evolutionary relationship of lineage D betacoronaviruses detected from geographically separated bat species indicates that the spread of lineage D infections in bats is not host‐restricted.
In Germany, bat parturition peaks appear correlated with bat coronavirus shedding (Drexler et al., 2011); it would be beneficial to determine the pregnancy cycles of bats in the tropics, which is largely unknown or based on studies with limited geographical scope. In the tropics, the constant availability of resources means there is less dependence on temporal windows of food. In Malaysia, E. spelaea has been found with dependent young in 8 of 12 months, indicating that individuals are reproducing throughout the year (Kingston et al., 2006), with two peak periods of pregnancy in June and September. In the Philippines, it was also found that E. spelaea has two annual peaks in reproduction although with some variation (Krutzsch, 2005). The first peak centred on March or April while the second centred on or near August (Heideman and Utzurrum, 2003). These bats typically roost in large numbers in caves, although the studied colony in Singapore roosts under an overpass and numbers over 3000 individuals (Lee, unpublished data). This opens up the possibility that there is a persistent source of virus in the colonies and immunologically naïve individuals have the opportunity for exposure from infected bats as they are tightly packed and may fight for specific sites in a roosting area. This is especially true for larger and older males because it is hypothesized that E. spelaea exhibits a resource defence polygynous mating system, where males spend significantly more time and energy for roost defence and surveillance (Bumungsri et al., 2013).
In addition, E. spelaea, R. leschenaultii and H. armiger co‐roost in caves in India and in SE Asia and multispecies roosts may facilitate the transmission and recombination of coronaviruses (Mendenhall, unpublished data; Struebig et al., 2005; Furey et al., 2011). Hipposideros lekaguli is thought to be associated with Rousettus species and even E. spelaea in SE Asia (Alviola et al., 2015). These species also have a large, overlapping distribution across SE Asia, which may facilitate the emergence of novel coronaviruses (Fig. 2). The foraging ecology of these bats E. spelaea is known to feed on a wide range of plant species and their high visitation frequency to the inflorescence of a few keystone plant species such as Durio zibethinus and Parkia speciosa (Heideman and Utzurrum, 2003; Krutzsch, 2005). Both C. sphinx and R. leschnaulti have also been found to feed on the same tree (Singaravelan and Marimuthu, 2004, 2008).
The discovery of this novel coronavirus demonstrates the unsampled diversity of this virus family in bats. Betacoronaviruses are responsible for two of the major coronavirus cross‐species spillover events (SARS and MERS virus) from bats to incidental hosts, ultimately resulting in sustained transmission chains. The bat species where lineage D betacoronaviruses have been detected share roosting sites, do so in large numbers and high density and also forage on the same plants. These may be factors that ultimately lead to the emergence of novel virus strains (Table 2). Even though the lineage D betacoronaviruses are not implicated as viruses capable of infecting humans, understanding their evolution can help us understand the evolution and ecological aspects of this medically important virus family. Furthermore, as little is known about the evolutionary factors affecting the transmission of CoVs between hosts, additional studies, such as host cell receptor usage, are required to assess the potential risks of zoonotic transmission to humans.
Table 2.
Roosting and foraging behaviour of lineage D Betacoronavirus‐positive bat species from South Asia and South‐East Asia
| Bat species | Roost site (cave/tree) | Food (no. of plant species) | Numbers in single roost | References |
|---|---|---|---|---|
| Eonycteris spelaea | Caves in forested areas and man‐made structure in urban areas | Flowers/nectar | Several thousands. Co‐habits with other bat species | (Francis et al., 2008) |
| Cynopterus sphinx | Foliage (tree and palms) only | Fruits (wild and cultivated) | 3–7 bats per roost | (Bates et al., 2008b) |
| Dobsonia moluccensis | Caves, sinkholes, boulder piles, old mines, disused buildings, dense vegetation | Fruits | Several thousands | (Hutson et al., 2008) |
| Hipposideros lekaguli | Mainly limestone caves | Insects | 300 individuals (per colony) (Bumrungsri per comm.) | (Csorba et al., 2008) |
| Rousettus leschenaultii | Caves, old buildings, forts and disused tunnels | Fruits and flowers/nectar | A few to several thousand individuals | (Bates and Helgen, 2008) |
| Hipposideros armiger | Caves and man‐made structures | Insects | Hundreds of individuals. Co‐habits with Rhinolophus and other bat species | (Bates et al., 2008a) |
| Ptenochirus jagori | Mainly tree cavities, also caves and sheltered rock crevices | Fruits (145), flowers/nectar (2) and leaves (7) | Singly or in small groups in caves | (Mickleburgh, 1992; Reiter and Curio, 2001; Ong et al., 2008) |
| Scotophilus kuhli | Tree cavities, foliage and man‐made structures (in forest and human‐modified environments) | Insects (hymenopterans and dipterans) | Several hundred individuals | (Bates et al., 2008d) |
| Scotophilus heathii | Tree cavities, foliage and man‐made structures (in forest and human‐modified environments) | Insects | Singly or in colonies of up to 50 bats | (Bates et al., 2008c) |
Supporting information
Table S1: Accession numbers and details for coronavirus sequences used in the phylogenetic analysis.
Acknowledgements
This study was supported by the Duke‐NUS Signature Research Program funded by the Agency of Science, Technology and Research, Singapore, and the Ministry of Health, Singapore, and by the NUS‐Global Asia Institute grant NIHA‐2011‐1‐005. Bat sampling and collection was supported by the grant NMRC/BNIG/2005/2013 from the National Medical Research Council (IHM). L‐FW is partially funded by the NRF‐CRP grant (NRF2012NRF‐CRP001‐056). BPYH Lee was supported by a National Parks Board Postgraduate Scholarship and the Wildlife Reserves Singapore Conservation Fund. We thank U. Joseph, J. Gan and YF Chung for their assistance in the field.
Accession numbers for new sequences: KX452686–KX452686.
Contributor Information
I. H. Mendenhall, Email: ian.mendenhall@duke-nus.edu.sg.
G. J. D. Smith, Email: gavin.smith@duke-nus.edu.sg.
References
- Adams, M. J. , and Carstens E. B., 2012: Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 157, 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alviola, P. A. , Macasaet J. P. A., Afuang L. E., Cosico E. A., and Eres E. G., 2015: Cave‐Dwelling Bats of Marinduque Island. Museum Publications in Natural History, Philippines: 4. [Google Scholar]
- Anindita, P. D. , Sasaki M., Setiyono A., Handharyani E., Orba Y., Kobayashi S., Rahmadani I., Taha S., Adiani S., Subangkit M., Nakamura I., Sawa H., and Kimura T., 2015: Detection of coronavirus genomes in Moluccan naked‐backed fruit bats in Indonesia. Arch. Virol. 160, 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., Lesin V. M., Nikolenko S. I., Pham S., Prjibelski A. D., Pyshkin A. V., Sirotkin A. V., Vyahhi N., Tesler G., Alekseyev M. A., and Pevzner P. A., 2012: SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyard, A. C. , Evans J. S., Luo T. R., and Fooks A. R., 2014: Lyssaviruses and bats: emergence and zoonotic threat. Viruses 6, 2974–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, P. , and Helgen K., 2008: Rousettus leschenaultii. The IUCN Red List of Threatened Species 2008: e.T19756A9011055. Available at: 10.2305/IUCN.UK.2008.RLTS.T19756A9011055.en (accessed 10 February 2016). [DOI]
- Bates, P. , Bumrungsri S., Francis C., and Csorba G., 2008a: Hipposideros armiger. The IUCN Red List of Threatened Species 2008: e.T10110A3162617. Available at: 10.2305/IUCN.UK.2008.RLTS.T10110A3162617.en (accessed February 10, 2016). [DOI]
- Bates, P. , Bumrungsri S., Molur S., and Srinivasulu C., 2008b: Cynopterus sphinx. The IUCN Red List of Threatened Species 2008: e.T6106A12427966. Available at: 10.2305/IUCN.UK.2008.RLTS.T6106A12427966.en (accessed February 10, 2016). [DOI]
- Bates, P. , Csorba G., Molur S., and Srinivasulu C., 2008c: Scotophilus heathii. The IUCN Red List of Threatened Species 2008: e.T20067A9142155. Available at: 10.2305/IUCN.UK.2008.RLTS.T20067A9142155.en (accessed February 10, 2016). [DOI]
- Bates, P. , Kingston T., Francis C., Rosell‐Ambal G., Heaney L., Gonzales J.‐C., Molur S., and Srinivasulu C., 2008d: Scotophilus kuhlii. The IUCN Red List of Threatened Species 2008: e.T20068A9142479. Available at: 10.2305/IUCN.UK.2008.RLTS.T20068A9142479.en (accessed February 10, 2016). [DOI]
- Brister, J. R. , Ako‐Adjei D., Bao Y., and Blinkova O., 2015: NCBI viral genomes resource. Nucleic Acids Res. 43, D571–D577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink, B. , Xie C., and Huson D. H., 2015: Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Bumungsri, S. , Lang D., Harrower C., Sripaoraya E., Kitpipit K., and Racey P. A., 2013: The dawn bat, Eonycteris spelaea Dobson (Chiroptera: Pteropodidae) feeds mainly on pollen of economically important food plants in Thailand. Acta Chiropt. 15, 95–104. [Google Scholar]
- Calisher, C. H. , Childs J. E., Field H. E., Holmes K. V., and Schountz T., 2006: Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19, 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, D. K. , Leung C. Y., Gilbert M., Joyner P. H., Ng E. M., Tse T. M., Guan Y., Peiris J. S., and Poon L. L., 2011: Avian coronavirus in wild aquatic birds. J. Virol. 85, 12815–12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, K. B. , Bellini W. J., Rota P. A., Harcourt B. H., Tamin A., Lam S. K., Ksiazek T. G., Rollin P. E., Zaki S. R., Shieh W., Goldsmith C. S., Gubler D. J., Roehrig J. T., Eaton B., Gould A. R., Olson J., Field H., Daniels P., Ling A. E., Peters C. J., Anderson L. J., and Mahy B. W., 2000: Nipah virus: a recently emergent deadly paramyxovirus. Science 288, 1432–1435. [DOI] [PubMed] [Google Scholar]
- Csorba, G. , Bumrungsri S., Francis C., Bates P., Gumal M., and Kingston T., 2008: Hipposideros lekaguli. The IUCN Red List of Threatened Species 2008: e.T10144A3174182. Available at: 10.2305/IUCN.UK.2008.RLTS.T10144A3174182.en (accessed February 10, 2016). [DOI]
- Drake, J. W. , 1993: Rates of spontaneous mutation among RNA viruses. Proc. Natl Acad. Sci. USA 90, 4171–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler, J. F. , Corman V. M., Wegner T., Tateno A. F., Zerbinati R. M., Gloza‐Rausch F., Seebens A., Muller M. A., and Drosten C., 2011: Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 17, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler, J. F. , Corman V. M., and Drosten C., 2014: Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 101, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten, C. , Gunther S., Preiser W., van der Werf S., Brodt H. R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R. A., Berger A., Burguiere A. M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J. C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H. D., Osterhaus A. D., Schmitz H., and Doerr H. W., 2003: Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976. [DOI] [PubMed] [Google Scholar]
- Francis, C. , Rosell‐Ambal G., Tabaranza B., Carino P., Helgen K., Molur S., and Srinivasulu C., 2008: Eonycteris spelaea. The IUCN Red List of Threatened Species 2008: e.T7787A12850087. Available at: 10.2305/IUCN.UK.2008.RLTS.T7787A12850087.en (accessed February 10, 2016). [DOI]
- Furey, N. M. , Mackie I. J., and Racey P. A., 2011: Reproductive phenology of bat assemblages in Vietnamese karst and its conservation implications. Acta Chiropt. 13, 341–354. [Google Scholar]
- Ge, X. , Li Y., Yang X., Zhang H., Zhou P., Zhang Y., and Shi Z., 2012: Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 86, 4620–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, R. J. , Baker S. C., Baric R., Enjuanes L., Gorbalenya A. E., Holmes K. V., Perlman S., Poon L., Rottier P. J. M., Talbot P. J., Woo P. C. Y., and Ziebuhr J., 2012: Family – Coronaviridae A2. In: King A. M. Q., M. J Adams, E. B Carstens, and E. J Lefkowitz. (eds), Virus Taxonomy, pp. 806–828. Elsevier, San Diego. [Google Scholar]
- Halpin, K. , Young P. L., Field H. E., and Mackenzie J. S., 2000: Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J. Gen Virol. 81, 1927–1932. [DOI] [PubMed] [Google Scholar]
- Heideman, P. D. , and Utzurrum R. C., 2003: Seasonality and synchrony of reproduction in three species of nectarivorous Philippines bats. BMC Ecol. 3, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, V. P. , Hossain M. J., Parashar U. D., Ali M. M., Ksiazek T. G., Kuzmin I., Niezgoda M., Rupprecht C., Bresee J., and Breiman R. F., 2004: Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis. 10, 2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B. , Ge X., Wang L. F., and Shi Z., 2015: Bat origin of human coronaviruses. Virol. J. 12, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , and Mitra S., 2012: Introduction to the analysis of environmental sequences: metagenomics with MEGAN. Methods Mol. Biol. 856, 415–429. [DOI] [PubMed] [Google Scholar]
- Hutson, A. M. , Suyanto A., Helgen K., Maryanto I., and Sinaga U., 2008: Dobsonia moluccensis. The IUCN Red List of Threatened Species 2008: e.T6779A12805284. Available at: 10.2305/IUCN.UK.2008.RLTS.T6779A12805284.en (accessed February 10, 2016). [DOI]
- IUCN , 2016: The IUCN Red List of Threatened Species. Version 2016‐1. Available at http://www.iucnredlist.org (accessed February 10, 2016). [Google Scholar]
- Jackwood, M. W. , 2012: Review of infectious bronchitis virus around the world. Avian Dis. 56, 634–641. [DOI] [PubMed] [Google Scholar]
- Jones, K. E. , Patel N. G., Levy M. A., Storeygard A., Balk D., Gittleman J. L., and Daszak P., 2008: Global trends in emerging infectious diseases. Nature 451, 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karesh, W. B. , Dobson A., Lloyd‐Smith J. O., Lubroth J., Dixon M. A., Bennett M., Aldrich S., Harrington T., Formenty P., Loh E. H., Machalaba C. C., Thomas M. J., and Heymann D. L., 2012: Ecology of zoonoses: natural and unnatural histories. Lancet 380, 1936–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston, T. , Lim B. L., and Akbar Z., 2006: Bats of Krau Wildlife Reserve. Penerbit Universiti Kebangsaan Malaysia, Bangi, Malaysia. [Google Scholar]
- Krutzsch, P. H. , 2005: Reproductive anatomy and cyclicity of the bat Eonycteris spelaea Dobson (Chiroptera: Pteropodidae) in West Malaysia. Acta Chiropt. 7, 51–64. [Google Scholar]
- Lau, S. K. , Li K. S., Tsang A. K., Shek C. T., Wang M., Choi G. K., Guo R., Wong B. H., Poon R. W., Lam C. S., Wang S. Y., Fan R. Y., Chan K. H., Zheng B. J., Woo P. C., and Yuen K. Y., 2012: Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault's rousettes to pomona leaf‐nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 86, 11906–11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickleburgh, S. P. , 1992: Old World Fruit Bats: an Action Plan for Their Conservation. IUCN, Gland, Switzerland. [1992] ©1992. [Google Scholar]
- Minskaia, E. , Hertzig T., Gorbalenya A. E., Campanacci V., Cambillau C., Canard B., and Ziebuhr J., 2006: Discovery of an RNA virus 3′‐>5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl Acad. Sci. USA 103, 5108–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, H. , Miyamoto H., Nakayama E., Yoshida R., Nakamura I., Sawa H., Ishii A., Thomas Y., Nakagawa E., Matsuno K., Kajihara M., Maruyama J., Nao N., Muramatsu M., Kuroda M., Simulundu E., Changula K., Hang'ombe B., Namangala B., Nambota A., Katampi J., Igarashi M., Ito K., Feldmann H., Sugimoto C., Moonga L., Mweene A., andTakada A., 2015: Seroepidemiological prevalence of multiple species of filoviruses in fruit bats (Eidolon helvum) Migrating in Africa. J. Infect. Dis. 212(Suppl 2), S101–S108. [DOI] [PubMed] [Google Scholar]
- Olival, K. J. , and Hayman D. T., 2014: Filoviruses in bats: current knowledge and future directions. Viruses 6, 1759–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong, P. , Rosell‐Ambal G., Tabaranza B., Heaney L., Pedregosa M., Paguntalan L. M., Cariño A. B., Ramayla S., Duya P., Warguez D., Alcala E., Garcia H., Pamaong R., Gonzalez J. C., and Lorica R. P., 2008: Ptenochirus jagori. The IUCN Red List of Threatened Species 2008: e.T18653A8504028. Available at: 10.2305/IUCN.UK.2008.RLTS.T18653A8504028.en (accessed Feburary 10, 2016). [DOI]
- Peiris, J. S. , Chu C. M., Cheng V. C., Chan K. S., Hung I. F., Poon L. L., Law K. I., Tang B. S., Hon T. Y., Chan C. S., Chan K. H., Ng J. S., Zheng B. J., Ng W. L., Lai R. W., Guan Y., Yuen K. Y., andH. U. S. S. Group , 2003: Clinical progression and viral load in a community outbreak of coronavirus‐associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourrut, X. , Souris M., Towner J. S., Rollin P. E., Nichol S. T., Gonzalez J. P., and Leroy E., 2009: Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect. Dis. 9, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QGIS Development Team , 2016: QGIS Geographic Information System. Open Source Geospatial Foundation Project. Available at http://www.qgis.org/ (accessed February 10, 2016). [Google Scholar]
- Queen, K. , Shi M., Anderson L. J., and Tong S., 2015: Other bat‐borne viruses In: Wang, L. and Cowled C. (eds), Bats and Viruses: A New Frontier of Emerging Infectious Diseases, pp. 217–247. Wiley‐Blackwell, Hoboken, NJ, USA. [Google Scholar]
- Razanajatovo, N. H. , Nomenjanahary L. A., Wilkinson D. A., Razafimanahaka J. H., Goodman S. M., Jenkins R. K., Jones J. P., and Heraud J. M., 2015: Detection of new genetic variants of Betacoronaviruses in Endemic Frugivorous Bats of Madagascar. Virol. J. 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter, J. , and Curio E., 2001: Home range, roost switching, and foraging area in a Philippine fruit bat (Ptenochirus jagori). Ecotropica 7, 109–113. [Google Scholar]
- Sabir, J. S. , Lam T. T., Ahmed M. M., Li L., Shen Y., Abo‐Aba S. E., Qureshi M. I., Abu‐Zeid M., Zhang Y., Khiyami M. A., Alharbi N. S., Hajrah N. H., Sabir M. J., Mutwakil M. H., Kabli S. A., Alsulaimany F. A., Obaid A. Y., Zhou B., Smith D. K., Holmes E. C., Zhu H., and Guan Y., 2016: Co‐circulation of three camel coronavirus species and recombination of MERS‐CoVs in Saudi Arabia. Science 351, 81–84. [DOI] [PubMed] [Google Scholar]
- Saif, L. J. , 2004: Animal coronavirus vaccines: lessons for SARS. Dev. Biol. (Basel) 119, 129–140. [PubMed] [Google Scholar]
- Singaravelan, N. , and Marimuthu G., 2004: Nectar feeding and pollen carrying from Ceiba pentandra by pteropodid bats. J. Mammal. 85, 1–7. [Google Scholar]
- Singaravelan, N. , and Marimuthu G., 2008: In situ feeding tactics of short‐nosed fruit bat (Cynopterus sphinx) on mango fruits: evidence of extractive foraging in a flying mammal. J. Ethol. 26, 1–7. [Google Scholar]
- Stamatakis, A. , 2014: RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struebig, M. J. , Rossiter S. J., Bates P. J., Kingston T., Lin Oo S. S., Nwe A. A., Aung M. M., Win S. S., and Mya K. M., 2005: Results of a recent bat survey in Upper Myanmar including new records from the Kachin forests. Acta Chiropt. 7, 147–163. [Google Scholar]
- Tong, S. , Conrardy C., Ruone S., Kuzmin I. V., Guo X., Tao Y., Niezgoda M., Haynes L., Agwanda B., Breiman R. F., Anderson L. J., and Rupprecht C. E., 2009: Detection of novel SARS‐like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda, S. , Watanabe S., Masangkay J. S., Mizutani T., Alviola P., Ueda N., Iha K., Taniguchi S., Fujii H., Kato K., Horimoto T., Kyuwa S., Yoshikawa Y., and Akashi H., 2012: Genomic and serological detection of bat coronavirus from bats in the Philippines. Arch. Virol. 157, 2349–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykrishna, D. , Smith G. J., Zhang J. X., Peiris J. S., Chen H., and Guan Y., 2007: Evolutionary insights into the ecology of coronaviruses. J. Virol. 81, 4012–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee, S. , Duengkae P., Rodpan A., Kaewpom T., Maneeorn P., Kanchanasaka B., Yingsakmongkon S., Sittidetboripat N., Chareesaen C., Khlangsap N., Pidthong A., Leadprathom K., Ghai S., Epstein J. H., Daszak P., Olival K. J., Blair P. J., Callahan M. V., and Hemachudha T., 2015: Diversity of coronavirus in bats from Eastern Thailand. Virol. J. 12, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, S. , Masangkay J. S., Nagata N., Morikawa S., Mizutani T., Fukushi S., Alviola P., Omatsu T., Ueda N., Iha K., Taniguchi S., Fujii H., Tsuda S., Endoh M., Kato K., Tohya Y., Kyuwa S., Yoshikawa Y., and Akashi H., 2010: Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 16, 1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, N. D. , Daszak P., Kilpatrick A. M., and Burke D. S., 2005: Bushmeat hunting, deforestation, and prediction of zoonoses emergence. Emerg. Infect. Dis. 11, 1822–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Wang M., Lau S. K., Xu H., Poon R. W., Guo R., Wong B. H., Gao K., Tsoi H. W., Huang Y., Li K. S., Lam C. S., Chan K. H., Zheng B. J., and Yuen K. Y., 2007: Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 81, 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau S. K., Huang Y., and Yuen K. Y., 2009: Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 234, 1117–1127. [DOI] [PubMed] [Google Scholar]
- Woo, P. C. , Lau S. K., Lam C. S., Lau C. C., Tsang A. K., Lau J. H., Bai R., Teng J. L., Tsang C. C., Wang M., Zheng B. J., Chan K. H., and Yuen K. Y., 2012a: Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86, 3995–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau S. K., Li K. S., Tsang A. K., and Yuen K. Y., 2012b: Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 1, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse, M. E. , Haydon D. T., and Antia R., 2005: Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol. Evol. 20, 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yob, J. M. , Field H., Rashdi A. M., Morrissy C., van der Heide B., Rota P., bin Adzhar A., White J., Daniels P., Jamaluddin A., and Ksiazek T., 2001: Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg. Infect. Dis. 7, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, L. , Li M., Li L., Monagin C., Chmura A. A., Schneider B. S., Epstein J. H., Mei X., Shi Z., Daszak P., and Chen J., 2014: Evidence for retrovirus and paramyxovirus infection of multiple bat species in china. Viruses 6, 2138–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla, A. , Hui D. S., and Perlman S., 2015: Middle East respiratory syndrome. Lancet 386, 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Accession numbers and details for coronavirus sequences used in the phylogenetic analysis.