

 This work is licensed under a
This work is licensed under a Abstract
Summary
Sodium/glucose co-transporter 2 (SGLT2) inhibitors are novel oral hypoglycaemic agents that are increasingly used in the management of type 2 diabetes mellitus (T2DM). They are now recommended as second-line pharmacotherapy (in conjunction with metformin) in patients with type 2 diabetes and established atherosclerotic heart disease, heart failure or chronic kidney disease due to their favourable effects on cardiovascular and renal outcomes. We report a case of a 69-year-old man who developed muscle pain, weakness and wasting after commencing the SGLT2 inhibitor empagliflozin. This persisted for 1 year before he underwent resistance testing, which confirmed muscle weakness. His symptoms resolved within weeks of ceasing empagliflozin, with improvement in muscle strength on clinical assessment and resistance testing and reversal of MRI changes. No other cause of myopathy was identified clinically, on biochemical assessment or imaging, suggesting that empagliflozin was the cause of his myopathy.
Learning points:
Empagliflozin, a commonly used SGLT2 inhibitor, was associated with myopathy.
A high degree of suspicion is required to diagnose drug-induced myopathy, with a temporal relationship between starting the medication and symptom onset being the main indicator.
Recognition of drug-induced myopathy is essential, as discontinuation of the offending drug typically improves symptoms.
Patient Demographics: Adult, Male, White, Australia
Clinical Overview: Pancreas, Diabetes, Insulin, Diabetes mellitus type 2, Myopathy*, Iatrogenic disorder, Myositis
Diagnosis and Treatment: Diabetes mellitus type 2, Myopathy, Muscle atrophy, Fatigue, Oedema, Myalgia, Myasthaenia, Weight loss, Polyuria, MRI, Resistance testing*, Exercise tolerance, Empagliflozin, SGLT2 inhibitors, Insulin, Insulin Aspart, Atorvastatin
Publication Details: Unusual effects of medical treatment, April, 2020
Background
Sodium-glucose co-transporter 2 (SGLT2) inhibitors such as empagliflozin, dapagliflozin, canagliflozin and ertugliflozin are increasingly used in the management of type 2 diabetes mellitus (T2DM), largely owing to their beneficial effects on atherosclerotic cardiovascular disease, heart failure and diabetic kidney disease. SGLT2 is located in the proximal tubule of the kidney and is responsible for the majority of renal glucose reabsorption; SGLT2 inhibitors decrease glucose reabsorption, thereby lowering blood glucose levels by promoting glycosuria.
More recently, SGLT2 inhibitors have been shown to improve cardiovascular outcomes in high risk patients with T2DM by reduced cardiovascular death and admissions for heart failure (1) and have also been shown to reduce progression rates of kidney disease (2). These findings have been confirmed in a recent meta-analysis (3) and have resulted in the most recent American Diabetes Association guidelines recommending the addition of SGLT2 inhibitors in patients with established atherosclerotic cardiovascular disease, heart failure or chronic kidney disease who are not meeting glycaemic targets or to consider switching to SGLT2 inhibitors in those already on multiple glucose lowering agents (4). The decrease in HbA1c with SGLT2 inhibitors is fairly modest, suggesting that the cardiovascular benefits may be mediated, in part, via other actions, such as decreased blood pressure, plasma volume and sympathetic nervous system activity, together with weight loss (5).
Although generally well tolerated, a number of adverse effects may occur with SGLT2 inhibitors, most commonly genital candidiasis due to glycosuria. Other side effects include transient renal dysfunction and hypovolaemia. Rare but serious adverse effects include euglycaemic ketoacidosis and necrotising fasciitis of the perineum. Additionally, an increased risk of bone fractures and amputations has been described with canagliflozin but not with other SGLT2 inhibitors (3).
The beneficial cardiovascular and renal effects combined with their safety profile (including low risk of hypoglycaemia) make SGLT2 inhibitors an attractive option in the armamentarium of medications to treat T2DM, typically as an adjunct to metformin in patients not meeting glycaemic targets. Here, we describe a case of myopathy secondary to empagliflozin.
Case presentation
A 69-year-old man with a 6-year history of well-controlled T2DM (HbA1c 6.7%) on small doses of twice daily pre-mixed insulin aspart and insulin aspart protamine was commenced on empagliflozin 10 mg daily after reading about its beneficial cardiovascular and renal effects. He was intolerant of metformin and was not taking any other oral hypoglycaemic agents at the time, having previously been trialed on sitagliptin. He had also been taking atorvastatin 40 mg for approximately 10 years. He initially ceased insulin after commencing empagliflozin, but restarted a small dose (4–5 units) pre-dinner due to high blood post-prandial glucose levels (7–9 mmol/L). He did not experience any hypoglycaemic episodes. He was a very active man who had completed many multi-day hiking trips over many years. Soon after starting empagliflozin, he developed decreased energy, muscle aches and decreased exercise tolerance. This was associated with weight loss of 5.1 kg to 66.1 kg (BMI 20.4) and polyuria, but not nocturia. He initially managed these symptoms by stopping empagliflozin prior to planned vigorous exercise. At outpatient review after 2 months, he elected to continue empagliflozin despite these symptoms.
Approximately 1 year after commencing empagliflozin, he commenced Kieser strength training and underwent baseline resistance testing at a gym which showed leg extension strength in the 13th percentile and elbow flexion in the 27th percentile compared to a reference group composed of people who had been undertaking this training for at least a year. At this stage, he was also reviewed by a rheumatologist. Examination revealed obvious wasting of supraspinatus and infraspinatus, with profound weakness in hip flexion and shoulder abduction, as well as neck flexion. No other cause of myopathy was identified on biochemistry or imaging.
Investigation
Creatine kinase (CK) was minimally elevated at 210 U/L (reference range 40–200). Antinuclear antibodies (ANA), extractable nuclear antigen (SM, RNP, SS-A (Ro), SS-B (La), Scl-70 and Jo-1) antibodies and muscle specific kinase antibodies were negative. Thyroid function tests were normal (TSH 1.28 mU/L (0.5–5.5), fT4 12.2 pM (9.0–19.0)), as was parathyroid hormone, Vitamin D, renal and liver function and full blood count. HbA1c remained stable at 7.4% after starting empagliflozin. CT of his chest, abdomen and pelvis was normal, apart from minimal fibrotic change at the lung bases posteriorly. MRI of his upper and lower limbs showed patchy, asymmetric oedema in muscles of both calves, with the remaining muscles being normal (Fig. 1).
Figure 1.
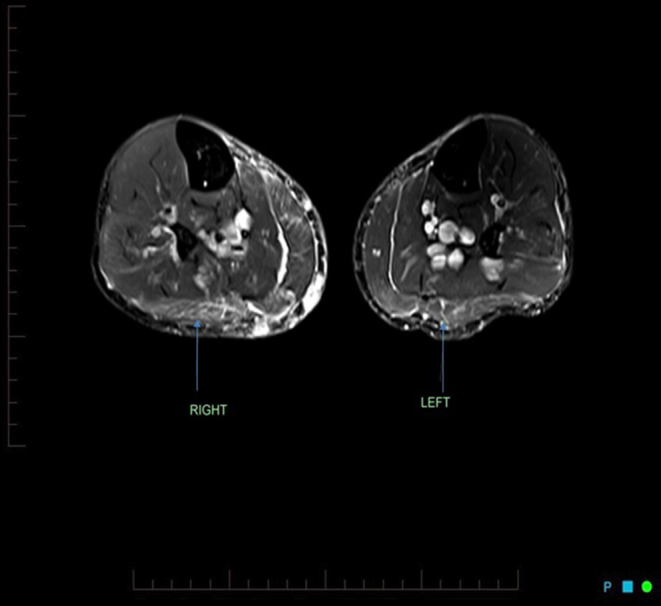
Initial MRI demonstrating patchy, asymmetric oedema in the muscles of both calves, consistent with myositis (arrows).
Treatment
Empagliflozin was discontinued and he remained on insulin monotherapy, with the dose increased to 7–8 units in the morning and 7 units at night. He continued to maintain good glycaemic control after stopping empagliflozin.
Outcome and follow-up
Muscle pain and weakness improved within 6 weeks of stopping empagliflozin. Clinical examination revealed normal strength in hip flexion and shoulder abduction following empagliflozin cessation, although he remained somewhat weak in neck flexion. Repeat resistance testing 1 month after stopping empagliflozin demonstrated an improvement in leg extension from the 13th to the 37th percentile and elbow flexion from the 27th to the 53rd percentile. At this stage, he had only been undertaking resistance training for 1 month, which was considered inadequate to explain the degree of improvement in his strength. He has not developed any further issues with muscle weakness and remains physically active 8 months post cessation of empagliflozin. His muscle wasting fully reversed and strength continues to improve. He continues to maintain good glycaemic control on insulin alone. Repeat MRI, 8 months after stopping empagliflozin, demonstrated complete resolution of the changes previously seen (Fig. 2).
Figure 2.
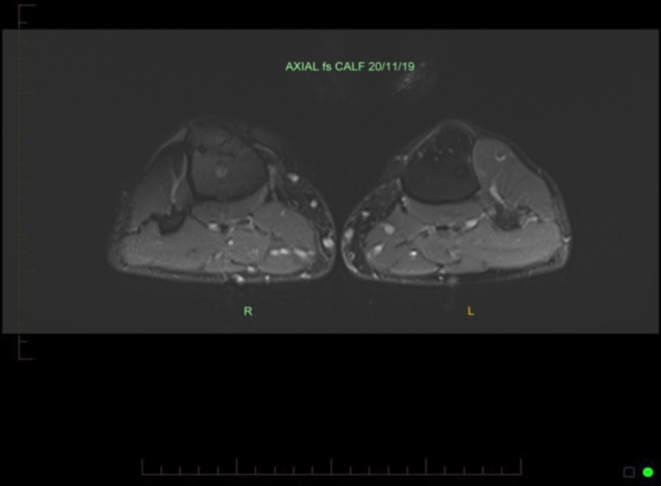
Resolution of previously demonstrated myositis after stopping empagliflozin.
Discussion
Drug-induced myopathies may present with varying severity, ranging from asymptomatic CK elevation to myalgia, exercise intolerance, weakness and myoglobuinuria and, at its most severe, rhabdomyolysis with acute kidney injury requiring dialysis and permanent disability. Commonly implicated agents include statins and glucocorticoids, although a very wide range of drugs have been shown to cause muscle damage (6). The symptoms of drug-induced myopathy typically subside following discontinuation of the offending medication, but they may persist in a minority of cases (7). Recognition of drug-induced myopathies is therefore essential.
A number of mechanisms for drug-induced myopathy have been delineated, including direct toxicity to muscle organelles, immunologic or inflammatory myopathy, necrosis of muscle fibres and indirect muscle damage that may occur through a number of effects such as electrolyte imbalance (6). Often a diagnosis of exclusion, it is important to rule out other causes of myopathy, including endocrine disease (such as thyroid or parathyroid disease), muscular dystrophies, metabolic disorders and inflammatory or immune disease. A drug-induced myopathy should be considered when no other cause of myopathy is apparent and a temporal relationship with a particular drug can be established. In cases where the diagnosis is unclear, electrodiagnostic studies, imaging (such as MRI) or muscle biopsy can be considered. No muscle biopsy was performed in this case.
Although our patient was taking atorvastatin, a well-described cause of drug-induced myopathy (8), this was not thought to be causing his symptoms, as he had been taking this drug for 10 years and the temporal relationship between starting empagliflozin and the onset of symptoms, as well as the brisk resolution after ceasing empagliflozin, are more in keeping with this as the cause of the patient’s weakness.
There has been one previous case report of muscle wasting and weakness associated with fatigue within 2 weeks of commencing empagliflozin (9). In that case, proximal wasting and weakness of the upper and lower limbs were evident on history and examination, CK was elevated and the patient experienced an improvement in fatigue within 2 weeks of stopping the drug. Medication was changed to insulin with good glycaemic control, but muscle mass and strength had still not improved 12 months after ceasing empagliflozin despite complete recovery from fatigue. In contrast, our patient had a quick recovery in muscle strength after ceasing empagliflozin, with improvement within 6 weeks.
We are not aware of any case reports of myopathy secondary to any of the other SGLT2 inhibitors. Indeed, initiation of SGLT2 inhibitor treatment increased grip strength in patients with type 2 diabetes in one study (10). Although that study did not specifically examine the mechanism behind the improvement, proposed causes include improved mitochondrial function, decreased inflammation and protein turnover and increased energy intake secondary to chronic glycosuria.
In conclusion, we present the case of a patient with myopathy secondary to empagliflozin. It illustrates the importance of recognising drug-induced myopathy, as his symptoms dramatically improved post cessation of the offending medication. With the widespread use of SGLT2 inhibitors, it should be considered in patients developing fatigue and weakness.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this case report.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient for the publication of this article and accompanying images.
Author contribution statement
All authors contributed to the writing of this manuscript. S H and L B were involved in the clinical care of the patient.
References
- 1.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al Empagliflozin, cardiovascular outcomes, and mortality in Type 2 diabetes. New England Journal of Medicine 2015. 373 2117–2128. ( 10.1056/NEJMoa1504720) [DOI] [PubMed] [Google Scholar]
- 2.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B, et al Empagliflozin and progression of kidney disease in Type 2 diabetes. New England Journal of Medicine 2016. 375 323–334. ( 10.1056/NEJMoa1515920) [DOI] [PubMed] [Google Scholar]
- 3.Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RHM, et al SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019. 393 31–39. ( 10.1016/S0140-6736(18)32590-X) [DOI] [PubMed] [Google Scholar]
- 4.American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in Diabetes-2019. Diabetes Care 2019. 42 (Supplement 1) S90–S102. ( 10.2337/dc19-S009) [DOI] [PubMed] [Google Scholar]
- 5.van Baar MJB, van Ruiten CC, Muskiet MHA, van Bloemendaal L, IJzerman RG, van Raalte DH. SGLT2 inhibitors in combination therapy: from mechanisms to clinical considerations in Type 2 diabetes management. Diabetes Care 2018. 41 1543–1556. ( 10.2337/dc18-0588) [DOI] [PubMed] [Google Scholar]
- 6.Valiyil R, Christopher-Stine L. Drug-related myopathies of which the clinician should be aware. Current Rheumatology Reports 2010. 12 213–220. ( 10.1007/s11926-010-0104-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalakas MC. Toxic and drug-induced myopathies. Journal of Neurology, Neurosurgery, and Psychiatry 2009. 80 832–838. ( 10.1136/jnnp.2008.168294) [DOI] [PubMed] [Google Scholar]
- 8.Sathasivam S, Lecky B. Statin induced myopathy. BMJ 2008. 337 2286 ( 10.1136/bmj.a2286) [DOI] [PubMed] [Google Scholar]
- 9.Kabadi UM. Marked weight loss, muscle wasting and fatigue on administration of empagliflozin in a subject with Type 2 diabetes. British Journal of Medicine and Medical Research 2017. 21 1–7. ( 10.9734/BJMMR/2017/33253) [DOI] [Google Scholar]
- 10.Sano M, Meguro S, Kawai T, Suzuki Y. Increased grip strength with sodium-glucose cotransporter 2. Journal of Diabetes 2016. 8 736–737. ( 10.1111/1753-0407.12402) [DOI] [PubMed] [Google Scholar]