

Summary
A diet deficient in the amino acid methionine has previously been shown to extend lifespan in several stocks of inbred rats. We report here that a methionine‐deficient (Meth‐R) diet also increases maximal lifespan in (BALB/cJ × C57BL/6 J)F1 mice. Compared with controls, Meth‐R mice have significantly lower levels of serum IGF‐I, insulin, glucose and thyroid hormone. Meth‐R mice also have higher levels of liver mRNA for MIF (macrophage migration inhibition factor), known to be higher in several other mouse models of extended longevity. Meth‐R mice are significantly slower to develop lens turbidity and to show age‐related changes in T‐cell subsets. They are also dramatically more resistant to oxidative liver cell injury induced by injection of toxic doses of acetaminophen. The spectrum of terminal illnesses in the Meth‐R group is similar to that seen in control mice. Studies of the cellular and molecular biology of methionine‐deprived mice may, in parallel to studies of calorie‐restricted mice, provide insights into the way in which nutritional factors modulate longevity and late‐life illnesses.
Keywords: biomarkers, diet, longevity, mice, migration inhibition factor, oxidative stress
Introduction
The demonstration that caloric restriction (CR) extends lifespan and delays many aspects of aging in rodents (Weindruch & Walford, 1988; Weindruch & Sohal, 1997) has been a major source of new ideas and discoveries in biogerontology, and it is commonly asserted that the CR diet is the only well‐authenticated method for delaying aging in mammals. In the last 10 years, however, studies of F344 and other rat strains (Orentreich et al., 1993; 2004, 1994; Zimmerman et al., 2003) have shown that maximal lifespan can be extended by diets in which cystine and cysteine are absent and methionine levels are as low as possible. Rats fed this methionine‐restricted (Meth‐R) diet are light in weight, but consume more total calories per gram body weight than control rats (Meth‐C) fed a diet with higher levels of methionine (Orentreich et al., 1993). Maximum lifespan of F344 inbred rats is extended by about 45% in at least two independent studies (Orentreich et al., 1993; Richie et al., 1994), and there is suggestive preliminary evidence (Zimmerman et al., 2003) that the Meth‐R diet extends lifespan in at least three other rat stocks that are not subject to the nephropathy and testicular tumors that characterize the F344 stock. Pair‐feeding studies, in which rats are fed the Meth‐C diet at the levels consumed by the Meth‐R rat group, have shown that the lifespan extension seen in the Meth‐R rats is not attributable to their slightly lower per‐rat energy intake. It is thus an open question as to what degree the mechanisms that lead to lifespan extension in Meth‐R rats overlap the pathways that extend life and delay age‐associated pathology in CR rodents. Nor is it clear whether the ability of low methionine diets to extend lifespan is limited to rats, or in contrast can be shown in other laboratory species of interest.
We have therefore conducted a methionine‐restriction experiment in a non‐inbred but genetically homogeneous mouse strain, CB6F1, to evaluate the effects of this dietary regimen on lifespan, hormone levels and several age‐sensitive traits.
Results
Improved survival on Meth‐R diet
To test the idea that low methionine diets would extend lifespan in mice, as has been reported in rats (Orentreich et al., 1993; Richie et al., 1994; Zimmerman et al., 2003), we placed a group of CB6F1 female mice on a semi‐purified diet containing 0.1% methionine at 6 weeks of age. Control mice were fed a semi‐purified diet similar in all respects except that it contained 0.43% methionine by weight. Methionine levels for the experimental group were increased to 0.12% when the mice reached 4 months of age, and again to 0.15% when the mice were 6 months of age, in an effort to reduce the proportion of experimental mice dying as a consequence of rectal prolapse. The experimental diet remained at 0.15% for the duration of the experiment. To test the hypothesis that reduction of methionine content from 0.43% to 0.15% reduced the palatability of the food, we conducted a short‐term experiment in which food consumption was measured daily for five consecutive days in two cages of young adult female mice on the control diet, and in two cages of mice receiving the experimental diet; five cage‐days were omitted when food shredding made it impossible to measure consumption accurately in a specific cage. Mice given the control diet consumed an average of 3.8 ± 0.7 g day−1 (mean ± SD), as compared with 4.3 ± 1.1 g day−1 for mice receiving the restricted diet. These data provide no evidence for the idea that the low‐methionine food is less palatable than the control diet. Because both groups of mice were equivalent in body weight at the start (29.3 ± 2.3 g) and end (28.8 ± 1.9 g) of this 5‐day period, the data are consistent with previous reports, from rat studies, that animals on the low‐methionine diet consume at least as much food per gram body weight as controls.
Figure 1 shows survival curves for the experimental and control groups. The left panel includes all of the mice entered into the study, and the right panel shows survival patterns for mice that were alive at the age of 365 days. It is clear that the experimental group had a higher risk of death in the first year of the protocol, but that this effect was diminished when methionine levels were raised to 0.15%. Survival at ages beyond 365 days was much higher in mice receiving the Meth‐R diet. The log‐rank test found the difference between the two groups significant at P = 0.02 when all mice were considered, and at P < 0.0002 when only those surviving more than 365 days were considered. At the time of writing (February 2005), 2/40 mice (5%) from the Meth‐R group are still alive, and none of the 40 mice in the control group. The longest‐lived mouse in the control group died at the age of 1150 days, an age at which 12/40 (30%) of the mice in the Meth‐R group were still alive. Maximum lifespan of the control mice, estimated as the mean age of the oldest 10% to die (1144 ± 26 days, mean ± SD, n = 4), is significantly less (P < 0.002 by Student's t‐test) than the corresponding value for the restricted mice (1261 ± 32 days, a minimum estimate with 5% of the mice still living).
Figure 1.
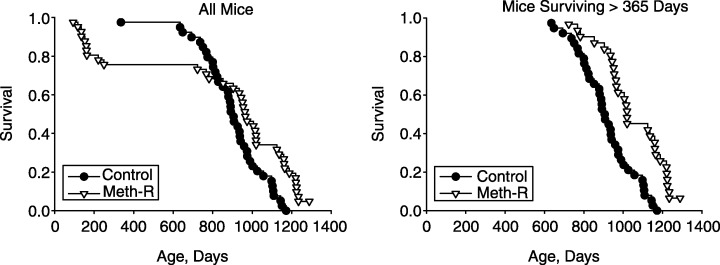
Survival curves for control and methionine‐restricted (Meth‐R) groups. Each symbol represents one mouse dying at the age indicated. The left panel shows all mice and the right panel shows only those mice that survived at least 365 days. At the time of writing, there are 2/40 survivors in the Meth‐R group, and 0/40 survivors in the control group.
Body weight
Figure 2 shows mean body weight levels in mice of the control and experimental groups. Mean weight in the control mice rose steadily to a plateau level of 44 g at ∼18 months, and then declined steadily from 22 months onward, consistent with previous studies. Mice in the Meth‐R group reached their highest average weight at 24 months of age, and were lighter than controls at all ages, averaging ∼65% of control weight between 6 and 18 months of age.
Figure 2.
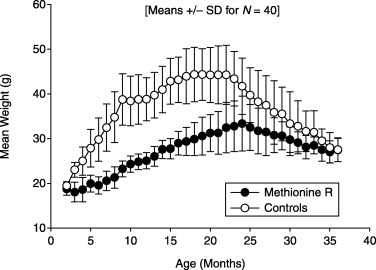
Body weight in control and methionine‐restricted mice, shown as mean and standard deviation at monthly intervals; n = 40 mice per group at the outset.
Hormone levels
Table 1 shows the results of serum hormone and fasting glucose levels measured at 16 months of age. Mice in the Meth‐R group are significantly lower in serum IGF‐I, and thyroxine (T4) levels. Serum insulin is approximately 25% of controls, and fasting glucose is reduced by about 50%. Differences between groups are significant at P < 0.01 for all four measures.
Table 1.
Hormone and glucose levels
| Measure | Control | Methionine‐restricted |
|---|---|---|
| IGF‐I (ng mL−1) | 397 ± 125 (12) | 257 ± 66 (12) |
| T4 (µg dL−1) | 3.5 ± 1.1 (37) | 2.7 ± 0.7 (30) |
| Insulin (ng mL−1) | 1.6 ± 1.1 (12) | 0.4 ± 0.2 (11) |
| Glucose (mg dL−1) | 64.7 ± 22.6 (11) | 32.9 ± 14.9 (12) |
Values shown are means ± SD; number of mice tested is given in parentheses.
Liver concentrations of mRNA for MIF
Microarray data have previously shown significantly increased levels of macrophage migration inhibition factor (MIF) mRNA in liver cell lysates from young adult mice of the long‐lived Snell dwarf stock and also in young adult mice on a CR diet (Miller et al., 2002a). MIF mRNA was also found to be elevated by microarray testing in liver of young adult mice of a long‐lived growth hormone receptor knockout stock (Miller et al., 2002a), and by RT‐PCR in liver of mice of the long‐lived wild‐derived Idaho stock (Y. Chang and R. A. Miller, unpublished data.) Because MIF mRNA was elevated in each of these four varieties of long‐lived mice, we used RT‐PCR to estimate MIF mRNA levels in female CB6F1 mice after 5 months of exposure to the methionine‐deficient diet. CR mice, and controls for both diets, were evaluated in parallel. The results are shown in the upper panel of Fig. 3. Liver of Meth‐R mice has higher concentrations of MIF mRNA (P < 0.004) than liver of age‐matched mice on the control diet. The CR diet also leads to an increase in liver MIF mRNA concentration, confirming the original array‐based analyses. (These data do not permit valid comparisons between CR and Meth‐R mice, because the CR and Meth‐R samples were extracted using different methods and were not evaluated at the same time.) In addition, serum samples from the Meth‐R mice were tested for MIF protein by an ELISA method, and were found to be elevated approximately two‐fold above the levels seen in controls (Fig. 3, lower panel).
Figure 3.
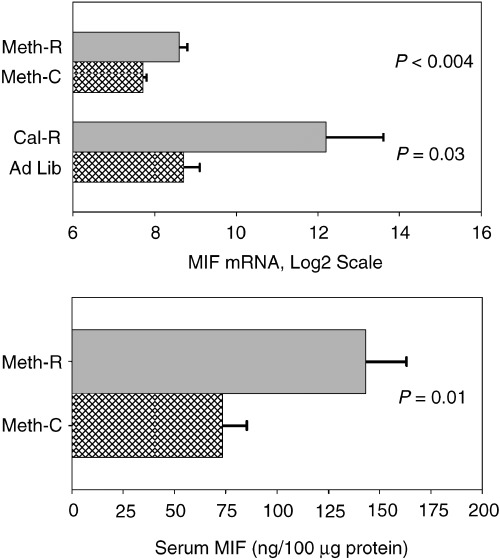
Levels of MIF mRNA in liver cell lysates (upper panel) of Meth‐R, CR and their respective controls, and (lower panel) of serum MIF protein concentrations in Meth‐R and control mice. Bars show mean levels ± SEM; mRNA is in arbitrary units. There were seven or eight mice per group for the Meth‐R samples and four mice per group for the CR samples. Significance tests for the serum protein used the Student's t‐test, and the Mann–Whitney test was used (with log2‐transformed values) for the mRNA comparisons.
Cataract testing
Each mouse was evaluated for the presence of lens opacity by slit lamp examination at the ages of 18 and 24 months. Using a scale where 0 represented absence of turbidity, 4 represented severe occlusive cataract, and 1 indicated a just‐detectable level of opacity, mean cataract score for the Meth‐R group at 18 months of age was 0.45 ± 0.06 (mean ± SEM, n = 30), as compared with a score of 0.68 ± 0.06 (n = 39) for mice on the control diet. The difference between the two groups is significant at P = 0.014 by Student's t‐test. When evaluated at 24 months of age, there was no difference between the two groups (mean score = 1.06 for Meth‐R and 1.07 for controls, n = 29 and 34, respectively). Thus the Meth‐R diet seems to retard, slightly but significantly, the development of lens opacity in these mice.
T‐cell subsets
Blood levels of six T‐cell subsets were evaluated when the mice were 18 months of age. The results are shown in Table 2. Five of these T‐cell subsets (all except CD4 cells) have been shown in previous studies (Miller, 1995, 1997, 1999) to increase with age in several stocks of mice. For each of these five subsets, mice in the Meth‐R group had lower subset levels; the difference was statistically significant for the CD8, CD8M, CD4P and CD8P cells, but not for the CD4M subset. Blood CD4 cells have been shown in previous studies to decline with age in mice, and values in the Meth‐R group were slightly, but not significantly (P = 0.11), higher than those of the control mice. Thus data for all six subsets were consistent with the hypothesis that the Meth‐R diet slightly retarded the rate of age‐dependent change in the T‐cell subset pattern.
Table 2.
T‐cell subsets
| Subset | Meth‐R | Control | P‐value |
|---|---|---|---|
| CD4 | 49 ± 0.81 (30) | 47.1 ± 0.8 (38) | 0.11 |
| CD8 | 49.7 ± 0.7 (30) | 51.8 ± 0.8 (37) | 0.05 |
| CD4M | 51.7 ± 1.3 (30) | 53.4 ± 1.2 (38) | 0.33 |
| CD8M | 52.0 ± 1.5 (30) | 56.5 ± 1.4 (38) | 0.03 |
| CD4P | 14.8 ± 0.8 (30) | 17.7 ± 0.97 (35) | 0.03 |
| CD8P | 51.4 ± 1.7 (30) | 62.3 ± 1.5 (37) | 0.001 |
T‐cell subset levels measured in peripheral blood at 18 months of age. Values shown are means ± SD; number of mice tested is given in parentheses. The P‐value column represents the result of unpaired, two‐tailed Student's t‐test comparison. CD4 and CD8 cells are presented as a percentage of CD3 cells, CD4M and CD4P cells as a percentage of CD4 cells, and CD8M and CD8P cells as a percentage of CD8 cells.
Resistance of liver cells to oxidative stress
To evaluate the effect of the Meth‐R diet on resistance to oxidative damage, we injected acetaminophen (APAP) intraperitoneally into a group of 10‐month‐old mice and evaluated the levels of lactate dehydrogenase (LDH) and alanine aminotransferase (ALT) enzymes in serum as an index of liver cell death. These experiments were performed on a group of female CB6F1 mice that were not part of the longevity study, and that had been exposed to the Meth‐R or control diet from the age of 6 weeks. The results are shown in Fig. 4. APAP injection led to a rapid increase in serum levels of both LDH and ALT, consistent with previous demonstrations that this agent induces liver cell injury and release of hepatic enzymes. The damage is easily detectable at 8 h, maximal at 24 h, and resolved by 48 h after APAP exposure. Figure 4 shows that Meth‐R mice were dramatically less sensitive to APAP‐induced liver damage. A group of mice on a low‐calorie standard chow diet (60% restriction in caloric intake with respect to controls) were found to be resistant to APAP‐induced liver damage when tested in parallel with the Meth‐R animals (data not shown).
Figure 4.
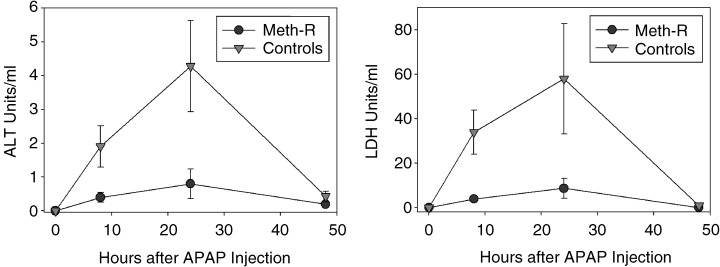
Meth‐R diet diminishes liver cell vulnerability to acetaminophen (APAP) toxicity. Symbols show mean and standard error for n = 6 mice in each group at the indicated time points. Student's t‐test indicates P < 0.05 for the 8‐h and 24‐h time points for each enzyme, except for LDH at 24 h, for which P = 0.08.
Necropsy analysis
A detailed histological analysis was performed at necropsy for each animal, to determine the most likely cause of death. In some cases autolysis was too far advanced to permit a diagnosis, and in one case two disease processes were deemed contributory to death. Of the remaining 20 cases in the control group, four were deemed to have died of histiocytic sarcoma, six of lymphoma, four of other neoplasias (hemangiosarcoma, mammary adenocarcinoma, pulmonary adenocarcinoma and Schwann cell tumor), and six of non‐neoplastic diseases (inanition, peritonitis, pituitary adenoma, pyometra, heart failure and amyloidosis). Among the 11 diagnosable cases in the methionine‐restricted group, there were three cases of histiocytic sarcoma, five of lymphoma, one gastric adenocarcinoma and two deaths from non‐neoplastic causes (thrombus and hemorrhage). The necropsy series excluded mice dying prior to 1 year of age; thus mean age of death in the diagnosable cases was 876 days for the control mice and 921 days for the methionine‐restricted mice. Although this small case series does not permit a comprehensive picture of late life illness in methionine‐restricted mice, it supports the inference that mice on the restricted diet succumb to the same spectrum of illnesses seen in the control mice, although at later ages. The extent of lesions at the glomerular basement membrane could be evaluated in 27 control and 15 Meth‐R mice, and were scored on a scale of 0–3, with high scores representing more severe lesions. The mean score in the Meth‐R group was 0.47 ± 0.17 (mean ± SEM), slightly lower than that seen in controls (0.89 ± 0.19), but the difference is not statistically significant (P < 0.1).
Discussion
Caloric restriction and methionine restriction are the only two interventions that have now been shown to extend maximum lifespan in rats and mice. Our data suggest strongly that the lifespan extension induced by Meth‐R diets, like that seen in the CR regimen, represents a retardation or perhaps a delay in the effects of aging on multiple tissues and organ systems, including for example T‐cell subsets and lens cataract development as well as in the lethal illnesses reflected in the mortality curves.
Work reported by others has established the important point that rats on the Meth‐R diet are in fact not calorie restricted to a significant degree, if at all. Food intake is higher per gram of body mass in Meth‐R rats than in control rats (Orentreich et al., 1993); in contrast, food intake per gram of body mass is not altered by the CR diet (Masoro et al., 1992). Rats on the Meth‐R diet weigh 40% less than controls, but consume only 10% less food, and rats provided with a control diet in the same amount (per rat) as consumed by the Meth‐R rats show only modest weight loss and minimal lifespan extension (Orentreich et al., 1993). Our own informal observations of Meth‐R mice suggest that they eat more food than mice on the control diet, and urine output of the Meth‐R mice is much higher than for controls, presumably due to the osmotic demands of excreting nutrients that cannot be incorporated into protein due to limiting levels of methionine and/or cyst(e)ine. A short‐term food consumption trial showed that mice placed on the low‐methionine diet eat as much food as mice placed on the (control) diet with higher levels of methionine, refuting the idea that the low‐methionine food source was so unpalatable that the mice lost weight because of voluntary food restriction.
Clearly a great deal more work will need to be done to see what aspects of aging are retarded by the Meth‐R diet, and to learn about the ways in which Meth‐R and CR rodents do and do not resemble one another. It is clear that the physiological responses to both diets have several elements in common, including declines in IGF‐I (Sonntag et al., 1999) and T4 levels (Weindruch & Walford, 1988), glucose and insulin (Masoro et al., 1992), as well as increases in mRNA for MIF in liver. Hepatocytes of both CR and Meth‐R mice are also both resistant to oxidative damage induced by APAP, consistent with the idea that resistance of multiple cell types to trauma might contribute to the disease resistance and longevity seen in both kinds of mice.
There is a need for more empirical work on the way in which alterations of dietary methionine might modulate aging. Many basic questions remain unanswered: (1) Are male mice as susceptible to Meth‐R diets as females? (2) Are mice of other strains, and genetically heterogeneous mice, also susceptible to lifespan extension by Meth‐R? (3) Is the longevity effect seen only in mice whose genes have been selected for laboratory confinement, or would it be demonstrable as well in wild‐derived mouse stocks, which are longer‐lived than laboratory‐adapted strains (2000, 2002b)? (4) What is the optimal regimen for lifespan extension by Meth‐R diets? For example, can longevity be improved by methionine‐restriction that is initiated not at 6 weeks of age but at later ages, and would such a system mitigate the early life mortality risks noted in Fig. 1? Would there be advantages to a protocol in which methionine was limited in the first half of the lifespan, but then provided at ‘normal’ levels as the mice enter old age? (5) Are the effects of the Meth‐R diet additive to those produced by CR? Would a diet that combined methionine restriction (at some level) with caloric restriction produce a longevity effect that exceeded what could be produced with either intervention alone?
The mechanisms by which methionine restriction leads to slower aging are not known, of course, but there are several possibilities worth considering. One set of models would emphasize the similarities in endocrine profiles, and perhaps cellular properties, shared by Meth‐R and CR rodents. Thus, for example, declines in IGF‐I and T4, lower glucose, lower insulin and (by hypothesis) improved insulin sensitivity might be induced by either diet and lead, through unknown intermediate steps, to slower aging and resistance to multiple diseases. More generally, each diet might be imagined to induce a mild stress response, perhaps accompanied by mild increases in chronic levels of glucocorticoids (Sabatino et al., 1991) and compensatory increases in MIF that along with other unknown factors modulate aging and disease risks.
A second class of explanations might involve the role of sulfur‐containing amino acids and their metabolic derivatives in the synthesis of S‐adenosylmethionine (SAM) and its derivatives. Regulation of polyamine biosynthesis and modulation of homocysteine levels by methionine and its derivatives would also be expected to be disordered by diminution of methionine, SAM and their metabolic products (James et al., 2002; Niculescu & Zeisel, 2002; Poirier, 2002), many of which are thought to play a role in the pathogenesis of neoplastic, cardiovascular and other late‐life abnormalities. Changes in SAM levels might also interfere with patterns of DNA methylation in ways that influence risk and timing of neoplastic disease (Richardson, 2002).
A third possibility emphasizes diet‐induced alterations in levels and distribution of glutathione, a key defense against oxidation damage. Studies in methionine‐restricted rats have shown a dramatic increase (84%) in blood glutathione levels in methionine‐deficient F344 rats, with a corresponding 66% decline in liver glutathione levels (2004, 1994). The authors note that these changes are not seen in CR animals, and postulate that the changes may reflect compensatory adjustments to keep glutathione levels within a relatively narrow intracellular range in non‐hepatic tissues.
It is also worth considering models that focus on specific alterations in the control of protein synthesis and degradation. There is some evidence that dietary restriction of tryptophan can also extend lifespan and delay some signs of aging in rats (Segall & Timiras, 1976; Ooka et al., 1988), an effect unlikely to reflect the alterations in sulfur metabolism produced by diets low in methionine. Rates of protein synthesis and degradation both decline with age, a decline that is itself diminished by a CR diet (Van Remmen et al., 1995). Diets low in methionine or in tryptophan might lead to a decline in protein synthesis rate, which in turn might induce compensatory mechanisms that protect against some forms of age‐related cellular dysfunctions. Studies of the effects of amino‐acid‐deficient diets, as well as long‐lived mutant mouse stocks (Miller, 2001), on rates of protein turnover and post‐translational status could be very rewarding.
It will require a good deal of additional effort to elucidate the ways in which chronic nutritional stress, DNA methylation, alterations in glutathione and other sulfur‐containing metabolites, alterations in protein synthesis and degradation, and potentially other consequences of prolonged methionine shortage lead to postponement of aging and late‐life diseases. It seems likely that some of the connections between methionine restriction and aging overlap with the still‐obscure mechanisms of caloric restriction, and others, perhaps, with the pathways that lengthen life in IGF‐I‐deficient mouse mutants. We are currently undertaking studies of gene expression patterns and DNA methylation patterns to improve our picture of how Meth‐R and CR mice may differ from one another, and to provide insights into the way in which these interventions might alter aging rate in mammals.
Experimental procedures
Mice and diets
The main longevity experiment involved a group of 80 female (BALB/cJ × C57BL/6 J)F1 mice, born on 8 January 2001, purchased from the Jackson Laboratories (Bar Harbor, ME, USA). Room temperature was maintained at 22 ± 2 °F, with 10–15 fresh air changes per hour and a 12 : 12‐h light–dark cycle. To evaluate the health status of the mice, groups of sentinel mice were exposed to spent bedding from the study population on a quarterly basis, and were later evaluated serologically for the presence of specific viral and bacterial pathogens. The mice were negative in all tests at the end of December 2002, but in March 2003 half of the cages, including equal numbers of control and Meth‐R mice, showed serologic evidence of exposure to mouse hepatitis virus (MHV, coronavirus). Tests repeated in June 2003 were uniformly negative, but the mice cannot be considered to have been specific pathogen‐free throughout their lives.
When the mice were 6 weeks old, half of them were placed on a diet containing 0.43% methionine (‘control’ mice) at age 6 weeks, and the other half were placed on a diet containing lower levels of methionine, initially 0.1%, but then increased at 4 months of age to 0.12% and again at 6 months of age to 0.15% to diminish the incidence of rectal prolapse and early death. The control diet, prepared by Purina Test Diets, Inc. (Richmond, IN, USA), was based upon the AIN‐76 formulation, and contained 0.43% methionine by weight. It was compounded using 43% corn starch, 20% sucrose, 8% corn oil, 5% dextrin, 5% cellulose, 3.6% glutamic acid, vitamin and mineral mix, choline, and a set of defined amino acids in lieu of a protein source. Amino acid concentrations were as follows: 0.93% arginine, 0% cystine, 2.31% glycine, 0.27% histidine, 0.82% isoleucine, 1.11% leucine, 1.15% lysine, 0.43% methionine, 1.16% phenylalanine, 0% tyrosine, 0.82% threonine, 0.18% tryptophan and 0.82% valine. Related diets containing 0.1–0.15% methionine were also prepared by the same company for our use, in which cornstarch was substituted for the diminished methionine; the diet containing 0.15% methionine is available as Catalogue no. 52501 (58MK).
Venous blood samples from this group were taken at age 16 months for hormone evaluation, and again at approximately 18 months for T‐cell subset testing. Cages were inspected daily and mice found dead were transferred to the pathology suite for necropsy study. Mice found to be moribund, i.e. judged by an experienced caretaker to be unlikely to survive more than another 3 days based on a checklist including unresponsiveness, inability to eat or drink, or rapid sustained weight loss, were humanely killed and submitted for necropsy.
Two other groups of CB6F1 mice were exposed to the Meth‐R diet for briefer time intervals and used as described for analysis of MIF mRNA or of hepatocyte resistance to acetaminophen toxicity. CR CB6F1 mice were also produced as controls for these determinations, using a standard lab chow diet administered either ad libitum or in diminished amounts (80% of ad libitum food intake for 2 weeks, then 70% for 2 weeks, then 60% for the remaining period) starting at 6 weeks of age.
Measurement of T‐cell subsets
Two‐color flow cytometry analyses were used in the evaluation of individual T‐cell subset levels in blood samples collected from each stock using previously described methods (Miller et al., 1997; Miller, 1997).
Assessment of lens opacity
Mice were tested at the ages of 18 and 24 months for lens turbidity using a hand‐held slit lamp after pupillary dilation with a solution containing equal parts of 1% tropicamide and 2.5% phenylephrine. Turbidity was scored on a scale from 0 (no evidence of cataract) to 4 (severe) for each eye separately, and the mean score from both eyes was used as an index of cataract severity.
Hormone and glucose determinations
Blood samples for measurements of hormones and glucose were taken by tail venipuncture between 07:00 and 11:00 h after an overnight (18‐h) fast. Serum glucose levels were quantified in 12.5 µL of serum using a glucose oxidase‐based assay system (Procedure no. 510; Sigma Diagnostics, St. Louis, MO, USA). All samples were processed in duplicate according to the manufacturer's instructions at one‐half volume except that a linear standard curve (range 25–400 mg dL−1) was used to calculate the glucose concentration of the samples using simple linear regression. Serum insulin levels were quantified via a double antibody rat insulin radioimmunoassay (RIA) kit (Linco, St. Charles, MO, USA). Each serum sample (50 µL) was assayed in duplicate according to the manufacturer's instructions using a 1 : 2 dilution with assay buffer if necessary to achieve adequate sample volume. Serum T4 levels were determined using a monoclonal solid‐phase RIA kit (ICN Pharmaceuticals, Costa Mesa, CA, USA) run at one‐quarter volume according to the manufacturer's instructions.
Serum IGF‐I levels were quantified via a double‐antibody RIA kit (Diagnostic Systems Laboratories, Webster, TX, USA) run at one‐quarter volume according to the manufacturer's instructions. Prior to assay, 10 µL of serum from each individual was subjected to an acid‐ethanol extraction procedure using the materials provided in the kit. In each case, sera from every individual were assayed at the same time to avoid interassay variability. The inclusion of pooled serum controls run within each assay indicated that the intra‐assay variability was ≤ 12% dependent upon the control and assay.
Exposure to acetaminophen (APAP)
A group of female CB6F1 mice not included in the longevity experiment was evaluated for liver damage in response to APAP injection. The mice were 10 months old when tested, and had been on the methionine‐deficient diet (0.15% methionine), or the control diet with 0.43% methionine, since the age of 6 weeks. Mice were deprived of food for 16 h prior to APAP administration to reduce hepatic stores of glutathione. APAP was injected intraperitoneally in sterile phosphate‐buffered saline, at a dose of 150 mg kg−1 body weight. CR mice and their ad libitum‐fed controls were also tested, using a dose of 250 mg kg−1 APAP. Food was made available to the mice immediately after APAP injection, and venous blood samples were withdrawn at 8, 24 and 48 h for comparison with blood withdrawn just prior to APAP administration.
Measurement of ALT and LDH in serum samples of APAP‐exposed mice
ALT levels were estimated by the conversion of alanine and 2‐oxoglutarate to pyruvate and glutamate, with subsequent NADH‐dependent reduction of pyruvate, using a 20‐µL serum sample at 37 °C in a 3‐min reaction. LDH levels were estimated by NAD‐dependent conversion of lactate to pyruvate, using a 13‐µL serum sample in a 3‐min reaction at 37 °C.
Measurement of MIF mRNA by RT‐PCR
For Meth‐R mice and their controls, total RNA from 6‐month‐old animals was prepared from livers using Trizol reagent (GibcoBRL) following the manufacturer's protocol. For CR and their ad libitum controls, total RNA was extracted from livers using the Atlas Pure Total RNA Isolation Kit (Clontech, CA, USA) following the vendor's protocol. cDNA synthesis, target amplification and DNA double strandspecific fluorescent dye incorporation was accomplished in a single step, starting with 300–500 ng of total RNA by using the LightCycler‐RNA Amplification Kit SYBR Green I (Roche, Indianapolis, IN, USA) following the manufacturer's protocols. Primers for MIF (forward: CAGAACCGCAACTACAGTAAGC, reverse: GGTGGATAAACACAGAACACTACG) were at 0.5 µm each in total, and target amplification reactions were cycled 50 times with 56 °C annealing for 8 s, and extension at 72 °C for 12 s. Levels of mRNA expression were determined by degree of fluorescence of each sample detected by the LightCycler System (Roche) and analyzed with software provided by Roche.
Necropsy methods
Mice were examined for clinical signs at least daily. Mice suspected to be ill (because of weight loss, poor grooming or visible tumor) were observed twice daily except on weekends where they were inspected once daily. Mice judged by an experienced technician to be moribund were humanely killed and necropsied. Mice found dead were also submitted for necropsy. The necropsy protocol has been described in detail elsewhere (Chrisp et al., 1996), and involved both gross inspection and histological examination of sections from 37 organs and any additional tissues that had gross lesions.
Acknowledgments
This work was supported by NIH grants AG08808 and AG13283. We thank Dr Richard Bucala for advice on the MIF assay, and Dr Cory Hogaboam for advice on the assessment of acetaminophen toxicity. We thank Maggie Vergara and Jessica Sewald for technical assistance.
References
- Chrisp CE, Turke P, Luciano A, Swalwell S, Peterson J, Miller RA (1996) Lifespan and pathology in genetically heterogeneous (four‐way cross) mice: a new model for aging research. Vet. Pathol. 33, 735–743. [DOI] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA (2002) Elevation in S‐adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine‐related pathology. J. Nutrition 132, 2361S–2366S. [DOI] [PubMed] [Google Scholar]
- Masoro EJ, McCarter RJ, Katz MS, McMahan CA (1992) Dietary restriction alters characteristics of glucose fuel use. J. Gerontol. 47, B202–B208. [DOI] [PubMed] [Google Scholar]
- Miller RA (1995) Immune system In Handbook of Physiology. Section 11: Physiology of Aging (Masoro E, ed.). New York: Oxford University Press, pp. 555–590. [Google Scholar]
- Miller RA (1997) Age‐related changes in T cell surface markers: a longitudinal analysis in genetically heterogeneous mice. Mech. Ageing Dev. 96, 181–196. [DOI] [PubMed] [Google Scholar]
- Miller RA (1999) Aging and immune function In Fundamental Immunology (Paul WE, ed.). Philadelphia: Lippincott‐Raven, pp. 947–966. [Google Scholar]
- Miller RA (2001) Genetics of increased longevity and retarded aging in mice In Handbook of the Biology of Aging (Masoro EJ, Austad SN, eds). San Diego: Academic Press, pp. 369–395. [Google Scholar]
- Miller RA, Chang Y, Galecki AT, Al‐Regaiey K, Kopchick JJ, Bartke A (2002a) Gene expression patterns in calorically restricted mice: partial overlap with long‐lived mutant mice. Mol. Endocrinol. 16, 2657–2666. [DOI] [PubMed] [Google Scholar]
- Miller RA, Chrisp C, Galecki A (1997) CD4 memory T cell levels predict lifespan in genetically heterogeneous mice. FASEB J. 11, 775–783. [DOI] [PubMed] [Google Scholar]
- Miller RA, Dysko R, Chrisp C, Seguin R, Linsalata L, Buehner G, Harper JM, Austad S (2000) Mouse (Mus musculus) stocks derived from tropical islands: new models for genetic analysis of life history traits. J. Zool. 250, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Harper JM, Dysko RC, Durkee SJ, Austad SN (2002b) Longer life spans and delayed maturation in wild‐derived mice. Exp. Biol. Med. 227, 500–508. [DOI] [PubMed] [Google Scholar]
- Niculescu MD, Zeisel SH (2002) Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J. Nutrition 132, 2333S–2335S. [DOI] [PubMed] [Google Scholar]
- Ooka H, Segall PE, Timiras PS (1988) Histology and survival in age‐delayed low‐tryptophan‐fed rats. Mech. Ageing Dev. 43, 79–98. [DOI] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA (1993) Low methionine ingestion by rats extends life span. J. Nutrition 123, 269–274. [DOI] [PubMed] [Google Scholar]
- Poirier LA (2002) The effects of diet, genetics and chemicals on toxicity and aberrant DNA methylation: an introduction. J. Nutrition 132, 2336S–2339S. [DOI] [PubMed] [Google Scholar]
- Richardson BC (2002) Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J. Nutrition 132, 2401S–2405S. [DOI] [PubMed] [Google Scholar]
- Richie JP Jr, Komninou D, Leutzinger Y, Kleinman W, Orentreich N, Malloy V, Zimmerman JA (2004) Tissue glutathione and cysteine levels in methionine‐restricted rats. Nutrition 20, 800–805. [DOI] [PubMed] [Google Scholar]
- Richie JP Jr, Leutzinger Y, Parthasarathy S, Malloy V, Orentreich N, Zimmerman JA (1994) Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J. 8, 1302–1307. [DOI] [PubMed] [Google Scholar]
- Sabatino F, Masoro EJ, McMahan CA, Kuhn RW (1991) Assessment of the role of the glucocorticoid system in aging processes and in the action of food restriction. J. Gerontol. Biol. Sci. 46, B171–B179. [DOI] [PubMed] [Google Scholar]
- Segall PE, Timiras PS (1976) Patho‐physiologic findings after chronic tryptophan deficiency in rats: a model for delayed growth and aging. Mech. Ageing Dev. 5, 109–124. [DOI] [PubMed] [Google Scholar]
- Sonntag WE, Lynch CD, Cefalu WT, Ingram RL, Bennett SA, Thornton PL, Khan AS (1999) Pleiotropic effects of growth hormone and insulin‐like growth factor (IGF‐1) on biological aging: inferences from moderate caloric‐restricted animals. J. Ger. Biol. Sci. 54A, B521–B538. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Ward WF, Sabia RV, Richardson A (1995) Gene expression and protein degradation In Handbook of Physiology. Section 11: Aging (Masoro EJ, ed.). New York: Oxford University Press, pp. 171–234. [Google Scholar]
- Weindruch R, Sohal RS (1997) Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. New Engl. J. Med. 337, 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL (1988) The Retardation of Aging and Disease by Dietary Restriction. Springfield, IL: Charles C. Thomas. [Google Scholar]
- Zimmerman JA, Malloy V, Krajcik R, Orentreich N (2003) Nutritional control of aging. Exp. Gerontol. 38, 47–52. [DOI] [PubMed] [Google Scholar]