

Abstract
A potential anti‐SARS drug has been developed by dynamic ligation screening (DLS), by which nucleophilic fragments are directed to the protein's active site by reversible reaction with an aldehyde inhibitor. Their inhibitory effect is detected by competition with a fluorogenic enzyme substrate. With this concept, low‐affinity fragments binding specifically to the active site are quickly identified in a functional enzyme assay.
Keywords: combinatorial chemistry, dynamic chemistry, enzyme catalysis, high‐throughput screening, medicinal chemistry
The conventional approach to identify biologically active, druglike small molecules is based on high‐throughput screening (HTS) of chemical libraries. However, the composition of large chemical libraries and their screening are time‐consuming and expensive endeavors; the success relies heavily on the quality of the available libraries, and even the largest library can span only a minute section of the virtual chemical space. Therefore, over the past decade several strategies have been proposed to facilitate the development process by using the protein target as a template for ligand assembly.1–3 The binding of low‐molecular‐weight fragments has been detected “directly” by NMR spectroscopy2a, b or X‐ray crystallography.2c, d These biophysical methods have been demonstrated to provide low‐affinity ligands as rational starting points for the iterative development of potent protein binders. Alternatively, protein‐binding molecules have been identified from mixtures of compounds formed in dynamic equilibria. In the presence of a protein the equilibrium was shifted, and the best binding products were concentrated in the mixture and could be detected by chromatography, mass spectrometry, or NMR spectroscopy.3a, b The reported fragment‐based methods have in common that they detect binding, not biological activity. Moreover, all these methods require large amounts of protein and test compounds and suffer from the difficult, time‐consuming, and expensive detection of active compounds.
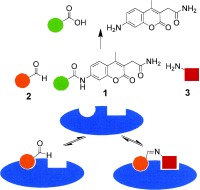
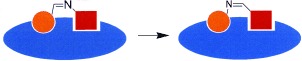
We envisioned that the detection of bioactive ligands should be sensitized considerably if reversibly formed ligation products compete in dynamic equilibrium with a fluorogenic reporter substrate for an enzyme (Figure 1). This approach would combine dynamic, target‐assisted formation of inhibitory species and detection by a fluorescence‐based screening methodology; thus, we designated it dynamic ligation screening (DLS). In DLS, the application of chemically reactive inhibitors as directing probes should enable the testing of inhibitory fragments for a defined binding site on the protein surface. Using an enzymatic reaction for fragment detection amplifies the signals and thus reduces the required amount of protein drastically. Finally, enzymatic detection with a fluorescent reporter molecule should enable high‐throughput screening (HTS) in microtiter plates (MTPs); thus, for the first time conventional HTS methodology could be employed in fragment‐based dynamic ligand development.
Figure 1.
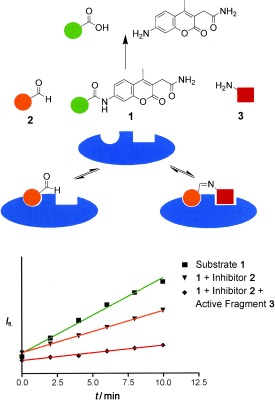
The concept of dynamic ligation screening (DLS). Substrate 1 competes with peptide aldehyde inhibitor 2 for the SARS‐CoV main protease (blue). Active fragment 3 leads to an increased inhibition through the binding of the imine ligation product to the active site.
The SARS coronavirus main protease (SARS‐CoV Mpro; SARS=severe acute respiratory syndrome) was selected as the protein target to demonstrate the DLS approach. SARS‐CoV Mpro is a cysteine protease that is essential for replication of the virus inside the infected host cell. Thus, it has been proposed as a drug target for SARS and—owing to the reported high homology among coronaviral main proteases—also for other coronaviral infections.5 Several irreversible (covalent) peptide‐based inhibitors of SARS‐CoV have been prepared and cocrystallized with the enzyme; however, only a few reversible,6 non‐peptidic7 inhibitors have been reported to date.
To establish DLS for site‐directed identification of inhibitory fragments, at first a fluorescence‐based assay4 for SARS‐CoV Mpro activity was developed by employing the substrate Ac‐TSAVLQ‐AMCA (1). Enzymatic cleavage of 1 released 2‐(7‐amino‐4‐methyl‐3‐coumarinyl)acetamide, which was excited at 380 nm for fluorescence detection at a wavelength of 460 nm. Second, a peptide aldehyde inhibitor 2 was selected for the DLS and synthesized on the protected oxazolidine resin.6 This peptide aldehyde contains a C‐terminal glutamine residue and thus forms an equilibrium between the aldehyde and its cyclic condensation product in aqueous solution.6 Treatment of aryl aldehydes with an excess of various primary amines has been reported to form imines as major components of the equilibrium in aqueous solution, whereas aliphatic aldehydes such as 2 are not converted into the imines as the major product.8 Thus, it remained to be tested whether the hypothetical ligation products of peptide aldehyde 2 and nucleophiles are stabilized on a protein surface and consequently can be detected by substrate competition.
For this purpose a collection of 234 nucleophiles was assembled comprising aromatic and aliphatic amines, thiols, and hydrazines. Aldehyde 2 as the directing probe was incubated with an eightfold excess of one nucleophilic fragment per well and in the presence of enzyme on a 384‐well microtiter plate. After the addition of reporter substrate 1, rate differences in the turnover of the substrate were quantified to identify active inhibitory fragments (Figure 1, Table 1). None of the selected fragments alone showed activity as SARS‐CoV Mpro inhibitor in a control experiment at a concentration of 400 μm; thus, their affinity is in the millimolar range or lower. For seven nucleophiles, however, a stronger inhibition than with the inhibitor 2 alone was observed (Table 1).
Table 1.
Observed initial velocities v 0 of the substrate conversion in the presence of the SARS main protease, substrate, peptide aldehyde 2, and active nucleophilic fragments.[a]
|
Electrophile |
Nucleophile |
v 0 [μm min−1] |
|---|---|---|
|
– |
– |
5.5±0.2 |
|
2 |
– |
2.8±0.1 |
|
2 |
|
1.0±0.1 |
|
2 |
|
1.0±0.1 |
|
2 |
|
1.6±0.1 |
|
2 |
|
1.9±0.1 |
|
2 |
|
2.1±0.1 |
|
2 |
|
2.2±0.1 |
|
2 |
|
2.2±0.1 |
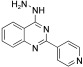
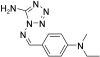
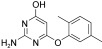
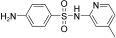
[a] For reaction conditions, see the Experimental Section.
In the next step, the specific binding of identified hit compounds to the active site of the SARS‐CoV main protease, and not, for example, to an allosteric site, had to be confirmed. 3‐Amino‐(N‐3‐aminophenyl)benzamide (3) was the most active and was selected for exemplary verification of the binding site by combining chemical synthesis and modeling. The imine formed from 2 and 3 was expected to be the active species. To test this hypothesis, at first the reduced amination product (4, Scheme 1) was synthesized. Tested in the HPLC assay described by Tan et al.,11 4 displayed a K I value of 50.3 μm. Comparison of the inhibitory activity of 4 with that of reduced amide 5 and those of the peptides Ac‐DSFDQ‐OH, DSFDQ‐OH, and Ac‐DSFDQ‐NH2, which all were completely inactive at 500 μm, supported the directing effect of peptide aldehyde 2 and the binding of fragment 3 to the S1′ site. The lower inhibition by 4 compared to peptide aldehyde 2 can be attributed to the absence of the electrophilic carbonyl group interacting favorably with the active‐site cysteine residue of SARS‐CoV Mpro. Furthermore, the complexes of peptide aldehyde 2 and of the imine formed with fragment 3 with SARS‐CoV main protease were modeled, which suggested a possible binding mode of fragment 3 (Figure 2).
Scheme 1.
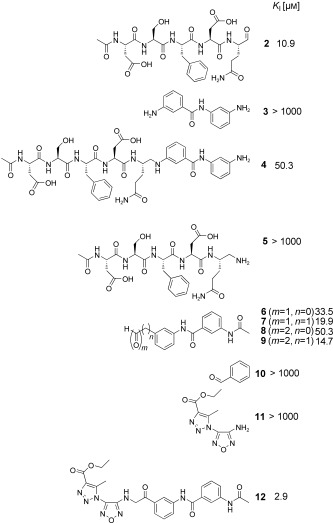
Development of a non‐peptidic SARS‐CoV Mpro inhibitor through dynamic ligation screening. Active fragment 3, which binds to the S1′site of the protein, has been transformed into electrophilic derivatives 6–9, which were employed iteratively in reverted DLS, yielding the non‐peptidic inhibitor 12.
Figure 2.
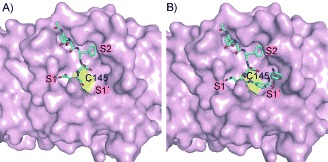
Molecular model of the aldehyde Ac‐DSFDQ‐H (A; residues P5–P1) and the imine ligation product of Ac‐DSFDQ‐H with 3 (B) docked into the substrate‐binding site of the SARS‐CoV Mpro (C cyan, N blue, O red). The active site Cys145 is shown as a yellow surface.10
Additional evidence for the binding of fragment 3 in the S1′ pocket was provided by the synthesis and testing of aldehydes and 2‐ketoaldehydes 6–9, which are all electrophilic derivatives of 3 (Scheme 1). Compounds 6–9 were designed with an electrophilic group to interact with the active site cysteine of the protease. While 6 and 7 were obtained by oxidation of the respective alcohols, the 2‐ketoaldehydes 8 and 9 were prepared by polymer‐supported C‐acylation, decarboxylation, and oxidative cleavage from a phosphane resin.12 Indeed all designed mono‐ and bis‐electrophiles were active inhibitors of SARS‐CoV Mpro (Scheme 1). Inhibitors 7 and 9, which are expected to position the active fragment in the same place relative to the cysteine residue as in the initial ligation product, were more potent inhibitors than compounds 6 and 8. Benzaldehyde (10), used as a control, was completely inactive, again indicating that the fragments detected by DLS bind specifically to the S1′ pocket of SARS‐CoV Mpro.
To obtain an entirely non‐peptidic inhibitor of SARS‐CoV Mpro targeting both the S1′ and S1 pockets, the dynamic ligation screening was conducted iteratively in a “reverted” mode (Table 2). Instead of peptide aldehyde 2, which binds to the S side of the binding cleft, 2‐ketoaldehyde 9, which presumably binds to the S′ side, was employed as a directing probe. For this experiment, 110 amines selected by diversity analysis were screened. Compound 9 was incubated with one amine per well, the protease, and the fluorogenic substrate Ac‐TSAVLQ‐AMCA (1). In this second screen, three fragments were identified which were active in the presence of the directing probe 9 (Table 2). The most active was 11, which was selected for verification of the inhibitor binding by chemical synthesis. Using the 2‐ketoaldehydes 8 and 9 for the covalent linking appeared to be advantageous, as the aldehyde could undergo reductive amination while the 2‐keto functionality remained intact for interaction with cysteine 145. Amine 11 was prepared as reported,13 employed for reductive amination of 2‐ketoaldehyde 9 with trichlorosilane as reducing agent,14 and yielded successfully 2‐aminoketone 12 as the covalent ligation product. Compound 12 inhibited SARS‐CoV Mpro in the HPLC assay11 with a K I value of 2.9 μm.
Table 2.
From nucleophilic fragment 3, the electrophilic 2‐ketoaldehyde inhibitor 9 was designed and used for “reverted” dynamic ligation screening (observed initial rates of substrate conversion (v
0) in the presence of active nucleophiles).[a]
|
Nucleophile |
Electrophile |
v 0 [μm min−1] |
|---|---|---|
|
– |
9 |
4.3±0.1 |
|
11 |
9 |
2.0±0.05 |
|
|
9 |
2.5±0.05 |
|
|
9 |
3.7±0.1 |
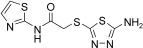
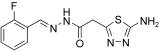
[a] For reaction conditions, see the Experimental Section.
Thus, we can conclude that dynamic ligation screening (DLS) enables the sensitized and site‐directed detection of low‐affinity fragments with inhibition constants in the millimolar range that are difficult or impossible to detect with previous dynamic strategies and conventional fragment‐based methods. The method was operated in high‐throughput format, and only very small amounts of protein were used by exploiting the amplification effect of the enzyme‐catalyzed detection. No additional equipment was required besides a standard microtiter plate reader. Most importantly, DLS was operated iteratively in an evolutionary process and succeeded in the transformation of a moderately active peptidic inhibitor to an entirely non‐peptidic inhibitor with an inhibition constant in the low micromolar range. Dynamic ligation screening has been demonstrated for protease inhibitor development in this work; it is currently being extended to other proteases, other enzyme classes, and to protein–protein interactions.
Experimental Section
The activity of SARS‐CoV Mpro was determined by measuring the release of AMCA from the fluorogenic substrate Ac‐TSAVLQ‐AMCA (1). The excitation wavelength was set to 380 nm, and the emission was recorded at 460 nm. Relative fluorescence units (RFUs; λ em 460 nm) were determined as 63.861 RFU μm(AMCA)−1. Reaction mixtures for cleavage were incubated at 298 K and contained 1 μm SARS‐CoV Mpro, 100 mm β‐morpholinoethanesulfonic acid (MES) buffer pH 7.0, and different concentrations of the fluorogenic substrate (0.25 mm–2.5 mm) in a total volume of 20 μL. All measurements were carried out on a TECAN SAFIRE fluorescence plate reader (Crailsheim, Germany).
Dynamic ligation screening for the S1′ site was performed for a library of 234 nucleophilic fragments using 1 μm of SARS‐CoV Mpro, 200 μm 1, 400 μm of one nucleophilic fragment per well, and 50 μm of the peptide aldehyde inhibitor Ac‐DSFDQ‐H (2) on a 384‐well microtiter plate. The initial rates were observed and compared with the initial rate without any nucleophilic fragment. Dynamic ligation screening for the S1 site was performed for a library of 110 nucleophilic fragments using 1 μm of SARS‐CoV Mpro, 200 μm 1, 200 μm of a nucleophilic fragment, and 20 μm of the non‐peptidic inhibitor 9 in a total volume of 20 μL MES buffer (100 mm, pH 7.0) on a 384‐well microtiter plate. The initial rate of product formation was observed and compared with the initial rate of the controls.
Dedicated to Professor Günther Jung on the occasion of his 70th birthday
Supporting information
Supporting information for this article is available on the WWW under http://www.wiley-vch.de/contents/jc_2002/2008/z704594_s.pdf or from the author.
We wish to thank Samuel Beligny, Angelika Ehrlich, Franziska Hinterleitner, Dagmar Krause, Jörn Saupe, Bernhard Schmikale, and Walter Verheyen for technical support. We also acknowledge Jeroen R. Mesters and Koen H. Verschueren for discussions. Work at the FMP was supported by the DFG (Ra 895/2‐5), by the government of Catalunya (stipend to A.I.L.), and by Boehringer Ingelheim Pharma. Work at Lübeck University was supported by the DFG (Hi 611/4‐1), the Schleswig‐Holstein Innovation Fund, the Sino–German Center for the Promotion of Research, Bejing (GZ 233‐202/6), and the European Commission through its SEPSDA project (SP22‐CT‐2004‐003831). J.R. and R.H. thank the Fonds der Chemischen Industrie for continuous support.
References
- 1.Reviews:
- 1a. Hajduk P. J., Greer J., Nat. Rev. Drug Discovery 2007, 6, 211–219; [DOI] [PubMed] [Google Scholar]
- 1b. Rademann J., Angew. Chem. 2004, 116, 4654–4656; [Google Scholar]; Angew. Chem. Int. Ed. 2004, 43, 4554–4556; [DOI] [PubMed] [Google Scholar]
- 1c. Corbett P. T., Leclaire J., Vial L., West K. R., Wietor J. L., Sanders J. K. M., Otto S., Chem. Rev. 2006, 106, 3652–3711; [DOI] [PubMed] [Google Scholar]
- 1d. Erlanson D. A., Wells J. A., Braisted A. C., Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [DOI] [PubMed] [Google Scholar]
- 2a. Shuker S. B., Hajduk P. J., Meadows R. P., Fesik S. W., Science 1996, 274, 1531–1534; [DOI] [PubMed] [Google Scholar]
- 2b. Nienhaber V. L., Richardson P. L., Klinghofer V. J., Bousha J. J., Greer J., Nat. Biotechnol. 2000, 18, 1105–1108; [DOI] [PubMed] [Google Scholar]
- 2c. Blundell T. L., Jhoti H., Abell C., Nat. Rev. Drug Discovery 2002, 1, 45–54; [DOI] [PubMed] [Google Scholar]
- 2d. Sharff A., Jhoti H., Curr. Opin. Chem. Biol. 2003, 7, 340–345. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Huc I., Lehn J. M., Proc. Natl. Acad. Sci. USA 1997, 94, 2106‐ 2110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Lehn J. M., Eliseev A., Science 2001, 291, 2331–2332; [DOI] [PubMed] [Google Scholar]
- 3c. Sanders W. J., Nienhaber V. L., Lerner C. G., McCall J. O., Merrick S. M., Swanson S. J., Harlan J. E., Stoll V. S., Stamper G. F., Betz S. F., Condroski K. R., Meadows R. P., Severin J. M., Walter K. A., Magdalinos P., Jakob C. G., Wagner R., Beutel B. A., J. Med. Chem. 2004, 47, 1709–1718; [DOI] [PubMed] [Google Scholar]
- 3d. Vongvilai P., Angelin M., Larrson R., Ramström O., Angew. Chem. 2007, 119, 966–968; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007, 46, 948–950. [DOI] [PubMed] [Google Scholar]
- 4. Meldal M., Svendsen I., J. Chem. Soc. Perkin Trans. 1 1995, 1591–1596. [Google Scholar]
- 5. Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., Hilgenfeld R., Science 2003, 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 6. Al‐Gharabli S. I., Shah S. T. Ali, Weik S., Schmidt M. F., Mesters J. R., Kuhn D., Klebe G., Hilgenfeld R., Rademann J., ChemBioChem 2006, 7, 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G. F., Anand K., Bartlam M., Hilgenfeld R., Rao Z., Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K. Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z., PLoS Biol. 2005, 3, e324; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Lu I. L., Mahindroo N., Liang P. H., Peng Y. H., Kuo C. J., Tsai K. C., Hsieh H. P., Chao Y. S., Wu S. Y., J. Med. Chem. 2006, 49, 5154–5161; [DOI] [PubMed] [Google Scholar]
- 7d. Yang S., Chen S. J., Hsu M. F., Wu J. D., Tseng C. T., Liu Y. F., Chen H. C., Kuo C. W., Wu C. S., Chang L. W., Chen W. C., Liao S. Y., Hung H. H., Shr H. L., Liu C. Y., Huang Y. A., Chang L. Y., Hsu J. C., Peters C. J., Wang A. H., Hsu M. C., J. Med. Chem. 2006, 49, 4971–4980. [DOI] [PubMed] [Google Scholar]
- 8. Godoy‐Alcántar C., Yatsimirsky A. K., Lehn J.‐M., J. Phys. Org. Chem. 2005, 18, 979–985. [Google Scholar]
- 9.The deviation was determined by plotting the observed fluorescence rate of three independent measurements in the program “Prism Graph Pad”.
- 10.For the modeling, the crystal structure of the reaction product of SARS‐CoV Mpro with a chloromethylketone was used as a template (molecule A of PDB coordinate set 1K4; reference [7a]). Subsites S2, S1, and S1′ are labeled. Note that the aspartate side chain in position P2 of the inhibitor is oriented towards the solvent, while the phenylalanine residue in P3 occupies the hydrophobic S2 pocket of the enzyme. A similar binding mode has been seen in the 1K4 structure by X‐ray crystallography (reference [7a]). The models were energy refined with the program Sybyl. The figure was prepared using Pymol (DeLano Scientific LLC, San Carlos).
- 11. Tan J., Verschueren K. H., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J. R., Chen K., Shen X., Jiang H., Hilgenfeld R., J. Mol. Biol. 2005, 354, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. El‐Dahshan A., Weik S., Rademann J., Org. Lett. 2007, 9, 949–952; [DOI] [PubMed] [Google Scholar]
- 12b. Weik S., Luksch T., Evers A., Böttcher J., Hasilik A., Löffler H.‐G., Klebe G., Rademann J., ChemMedChem 2006, 1, 445–457; [DOI] [PubMed] [Google Scholar]
- 12c. Weik S., Rademann J., Angew. Chem. 2003, 115, 2595–2598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003, 42, 2491–2494. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Tselinski V., Mel'nikova S. F., Vergizov S. N., Transl. Org. Khim. 1981, 17, 1123–1124; [Google Scholar]
- 13b. Batog L. V., Rozhkov V. Y., Struchkuva M. I., Mendeleev Commun. 2002, 12, 159–162. [Google Scholar]
- 14. Groarke M., Hartzoulakis B., McKervey M. A., Walker B., Williams C. H., Bioorg. Med. Chem. Lett. 2000, 10, 153–155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under http://www.wiley-vch.de/contents/jc_2002/2008/z704594_s.pdf or from the author.