

Abstract
Multiple sclerosis is an immune‐mediated disease with an environmental component. According to a long‐standing but unproven hypothesis dating to initial descriptions of multiple sclerosis (MS) at the end of the 19th century, viruses are either directly or indirectly implicated in MS pathogenesis. Whether viruses in MS are principally causal or simply contributory remains to be proven, but many viruses or viral elements—predominantly Epstein‐Barr virus, human endogenous retroviruses (HERVs) and human herpesvirus 6 (HHV‐6) but also less common viruses such as Saffold and measles viruses—are associated with MS. Here, we present an up‐to‐date and comprehensive review of the main candidate viruses implicated in MS pathogenesis and summarize how these viruses might cause or lead to the hallmark demyelinating and inflammatory lesions of MS. We review data from epidemiological, animal and in vitro studies and in doing so offer a transdisciplinary approach to the topic. We argue that it is crucially important not to interpret “absence of evidence” as “evidence of absence” and that future studies need to focus on distinguishing correlative from causative associations. Progress in the MS‐virus field is expected to arise from an increasing body of knowledge on the interplay between viruses and HERVs in MS. Such interactions suggest common HERV‐mediated pathways downstream of viral infection that cause both neuroinflammation and neurodegeneration. We also comment on the limitations of existing studies and provide future research directions for the field.
Keywords: Epstein‐Barr virus, human endogenous retroviruses, human herpesvirus 6, multiple sclerosis, viruses
1. INTRODUCTION
The prevalence of multiple sclerosis (MS) has steadily increased over the last five decades. This high socio‐economic burden, together with its challenging management especially when chronic and progressive, underscores the need for further research to determine its exact aetiology. MS is a multifactorial disease that arises from a complex interaction between genetic (notably immunogenetic), autoimmune and environmental factors.1, 2, 3, 4, 5 Environmental‐immune system interactions are increasingly recognized as important in MS pathophysiology6, 7 and are likely to explain the discordant MS incidence in monozygotic twins that cannot be attributed to genomic, transcriptomic or epigenomic factors alone.8 Furthermore, the environment represents a modifiable factor in contrast to the genomic landscape, so it is of particular interest from the perspective of prevention.
MS was initially proposed to be of infectious origin at the end of the 19th century, but the development of the experimental allergic encephalitis (EAE) model in 1934 shifted attention away from microorganisms and towards an allergy‐related and then autoimmune basis for the disease. However, a myelin‐targeting autoimmune model does not fully explain the segmental distribution of lesions as myelin is ubiquitous in the central nervous system (CNS),9 and auto‐antigens are neither pathognomonic nor universal in MS.10 In addition, some authors have suggested that the EAE model might more closely represent immunologically induced encephalomyelitis rather than demyelination.11, 12
However, the microbial aetiological theory—in which viruses take centre stage—has not been abandoned but has flourished in the light of mainly indirect discoveries of different viruses in MS.13 Although direct evidence for causative viruses in MS has generally been lacking, accumulated evidence from human and animal studies supports a role for viruses as at least a trigger for MS.2 Epidemiological evidence in support of this theory includes observations of MS epidemics in the Faroe Islands in the 1980s, and more recently, MS clusters in Ottawa, Canada.14
Although the evidence for a causative viral aetiology for MS in humans remains inconclusive, viruses appear to play a role in modulating the neuro‐immunological system of genetically susceptible individuals to cause MS. For instance, IgG antibodies against several viruses including varicella zoster virus (VZV), cytomegalovirus (CMV), measles, rubella, mumps and herpes simplex virus (HSV‐1) have been identified in the cerebrospinal fluid (CSF) of patients with MS.15, 16 More recently, other viruses have attracted attention including Saffold virus (a novel human cardiovirus).17, 18 With this in mind, this review provides an in‐depth discussion of the viruses implicated in MS pathogenesis. We first consider viruses with the greatest evidence base, namely Epstein‐Barr virus (EBV), human herpesvirus (HHV‐6), VZV, human endogenous retroviruses (HERV) and then go on to describe the potential roles for “minor” viruses in MS. We focus on the connection between viruses and MS pathophysiology rather than its clinical progression, and we highlight the limitations of existing studies and possible future research directions.
2. SEARCH METHODS AND SELECTION CRITERIA
The PubMed and Google Scholar databases were searched for articles published (or appeared “Epub ahead of print”) between 1 January 2006 to 31 December 2016 and the bibliographies examined. For initial screening, “multiple sclerosis,” “infectious cause,” “virus” or “viral model” were applied through the Boolean operators “AND” and “OR”. More specific terms were applied to different sections of the review based on their relevance. If and when an infectious agent had more than one name, all relevant search terms were applied. Priority was given to original research articles and systematic reviews/meta‐analyses over case reports or hypothesis/viewpoint articles and the most recent papers as applicable. Some references prior to the above time period were included given their historical importance. Studies referring to paediatric MS, infectious agents other than viruses and those not published in English were excluded.
3. EPSTEIN‐BARR VIRUS (EBV)
There is a lively ongoing debate on the role of EBV, the prevailing MS infectious risk factor and MS pathogenesis.19, 20, 21 One hypothesis suggests that MS is caused by a genetically predisposed deficiency in eliminating previous EBV infection; EBV then persistently accumulates or even establishes itself in the brains of such patients.7, 22 Consistent with this theory, EBV might exercise a strong influence on the number of naïve and/or memory B cells and their differentiation status.23 A competing hypothesis is that abnormal responses to EBV infection are secondary to and not a cause of MS.24
At the epidemiological level, several systematic reviews clearly support an association between MS and EBV seropositivity.3, 25, 26 Practically, all MS patients are EBV seropositive, raising the question of whether EBV‐seronegative MS patients even exist.27 EBV seropositivity confers double the risk of MS than infectious mononucleosis (IM) (OR=4.56 vs OR=2.17, respectively),3 and IM appears to have a stronger genetic component than EBV infection.28 However, the reasons for this difference in risk between EBV seropositivity and IM might be due to: (i) reporting bias for IM; (ii) the molecular stochasticity of EBV‐induced downstream events; (iii) the role of EBV latency; or, importantly, (iv) subclinical infection. High Epstein‐Barr virus nuclear antigen (EBNA) IgG titres are associated with other MS risk factors such as non‐HLA gene loci and the HLADRB1*15 allele (the most important genetic factor in MS).7, 29 T cells restricted to the HLADRB1*15 allele and linked to MS‐related antigens seem to cross‐react with the immunological response induced by the EBNA‐1 sequence.30 However, the latest meta‐analysis revealed an additive but not synergistic effect between the two risk factors, corroborating that HLADRB1*15 carriage is not a confounding factor for EBV and MS.31, 32
A highly synergistic increase (14‐fold) in MS risk was reported for EBV detection or IM combined with obesity, notably during adolescence.7 However, there are conflicting results on the interaction between EBV and other well‐established MS risk factors (reviewed also in33). For instance, a prospective study found a positive association between smoking and MS development only in older patients and a negative one in patients less than around 30 years old,34 whereas a later case‐control study reported a negative, multiplicative interaction between IM history and a prior history of smoking on MS risk.35 With regard to vitamin D, some studies have failed to detect a statistically significant interaction,32, 36 while others have reported an interaction with either EBV antibodies or DNA load.37 Mechanistically, observations that there is overlap between EBNA‐2a and vitamin D receptor (VDR) binding sites within MS‐associated genomic regions and that EBNA‐3 binds to the VDR may provide further insights.24, 38
In neuroimaging studies, MRI (magnetic resonance imaging) markers of MS activity and grey matter atrophy were found to be associated with anti‐EBV antibody levels.39, 40 At the cellular level, CD8+ T cells specific for EBV lytic and latent antigens were more frequent in patients with active and inactive MS, respectively.41 Deep sequencing of T‐cell receptor‐β genes (“immunosequencing”) showed intrathecally enriched EBV‐reactive CD8+ T cells that were specific to patients with MS.42 Furthermore, in animal models using lymphocryptovirus (LCV), which is a close relative of EBV, LCV‐infected B cells lost their ability to process the extrinsic pathogenic CD8+ T‐cell epitope in myelin oligodendrocyte glycoprotein (MOG). In doing so, they cross‐presented this epitope to auto‐aggressive cytotoxic T lymphocytes, a reaction that can initiate an autoimmune reaction and demyelination.43
With regard to humoral immunity, the high antibody titres against EBNA proteins in patients with MS might be due to intrathecal synthesis but it has yet to be clarified whether they result from high‐frequency latent EBV‐infected cells or, alternatively, have a concrete pathogenic role.44 Conversely, patients with IM showed activation of MOG‐specific memory B cells.45
Furthermore, EBV genetic material has been identified in the CSF and perivenular infiltrates of brain and spinal cord white matter, and, more recently, in the cortical grey matter and cervical lymph nodes of patients with MS.44, 46 EBV brain infection is likely to be limited to only a small number of B cells (approximately 5‐3000 per 107 memory B cells).23, 47 This could explain why histological studies for their detection are difficult, and it underscores the need for technologies such as massively parallel single‐cell sequencing to detect these rare events in the future.48 Dual infection with EBV types 1 and 2 is more common in patients with MS compared to single infection.49
Mechanistically, EBV might act as an environmental trigger or by attacking the CNS.50 With respect to the former, an EAE model with the murine EBV homologue gamma‐herpesvirus 68 showed more pronounced MS‐like clinicopathological features that were dependent on the latent life cycle of the virus.51 There are a number of theories with regard to the latter mechanism of direct CNS destruction by the virus: (i) cross‐reactivity of EBV‐infected T cells with self‐antigens (“molecular mimicry”) causes destruction of CNS tissue but does not explain the presence of EBV‐infected B cells in the brain; (ii) the bystander damage hypothesis proposes that immune responses in the CNS are directed towards EBV antigens but does not explain the autoimmune component of the disease and the failure to eliminate these cells; (iii) MS results from EBV infection of autoreactive B cells, which in turn produces pathogenic autoantibodies44; (iv) the “mistaken self” hypothesis based on proteomic analyses shows a higher frequency of a peptide corresponding to an EBNA‐1 region sharing homology with the N‐terminus of αB‐crystallin in patients with MS.52 Overall, understanding these mechanisms paves the way for novel anti‐MS strategies, notably EBV‐specific adoptive immunotherapy.22
It is also mechanistically intriguing how EBV plays a role in both cancer—a disorder of cellular proliferation—and MS—a disease characterized by neuronal cell death; however, recent reports of a genetic overlap between the EBV‐related Hodgkin lymphoma and MS could shed some light on this.53, 54 In parallel, dogma that EBV cannot possibly be found in glial cells or neurons, the host immune response must remain the focus of studies,53 or that EBV latency status underpins virus‐mediated pathogenesis24 should be re‐examined in the light of recent observations that EBV can cause lytic infection in human primary neurons.55
To summarize, in the context of discordance between the high rates of EBV infection vs low rates of MS worldwide, EBV is likely to be necessary but not sufficient to cause MS.19 Future studies on shared polygenic risk from genomewide association studies on MS cases with those with markers of increased EBV levels (eg EBNA‐156) are likely to shed further light on such host‐pathogen interactions.
4. HUMAN HERPESVIRUS 6 (HHV‐6)
A recent, inconclusive, non‐systematic summary of evidence on the role of HHV‐6 in MS57 highlighted the need for a formal meta‐analysis on this topic. Furthermore, although HHV‐6 has been detected mostly in acute demyelinating brain lesions in MS, detection rates are highly variable (HHV‐6 DNA in the CSF ranging from 3% to 46% of patients).58 Additionally, other markers such as B‐ or T‐cell reactivity, higher antibody responses or higher viral loads have not been consistently observed in MS patients’ serum in different ethnic groups or prospective studies.57, 59
Some specific single nucleotide polymorphisms (SNPs; eg in CD46 and MHC2TA) are strongly associated with active replication of HHV‐6 and, together, with worse clinical prognosis in MS.60 At the edge of such gene‐environment interactions lie the HERVs (see below). One of their subtypes (HERV‐K18) was shown to be activated by HHV‐6A, mainly in cell lines productively infected with the virus and followed by those with latently infected virus. These observations reinforce the notion that there is a common HERV‐mediated pathway downstream of viral infection in MS,61 which might be therapeutically exploitable.62
The marmoset (Callithrix jacchus) HHV‐6 model has been used to study viral neurotropism.63 Interestingly, in contrast to how the virus seems to gain entry to the human CNS via olfactory pathways,64 findings in marmosets revealed that only those with intravenous (and not intranasal) inoculation of HHV‐6A (and not HHV‐6B) developed neurological disease.63 Furthermore, in contrast to the global seroprevalence of >95% for HHV‐6B, HHV‐6A is more frequent in patients with MS than HHV‐6B, which is certainly worthy of further investigation.57 HHV‐6A infection leads to apoptosis in the brain, induces autoimmunity in several ways65 and activates antiviral genes in human astrocytes including some genes upregulated in MS.66
5. VARICELLA ZOSTER VIRUS (VZV)
Varicella zoster virus (VZV)‐induced encephalomyelitis is characterized by demyelination similar to that seen in MS, so VZV is suggested as an MS‐triggering factor.67 However, while some epidemiological studies reported no association between a history of varicella infection in childhood and MS risk,68 others have observed an association, most notably for relapse‐remitting (RR) and secondary progressive types.69 A fourfold increase in MS risk in the year following herpes zoster infection has been observed in a region with a low MS prevalence.70
Regrettably, serological and molecular studies have not helped much in this area. VZV seropositivity was not significantly higher in patients with MS vs controls in two studies.71, 72 Moreover, while VZV DNA was identified in the CSF of patients with MS (particularly of RR type) in some studies,73, 74 others failed to confirm these findings in the CSF, blood or in acute MS lesions.58, 75, 76, 77 More consistently, however, are the observations that the high levels of VZV DNA in CSF and PBMCs during relapse ultimately disappear during clinical remission.78, 79 Interestingly, the progressive MS type has been associated with VZV DNA at levels between those found during the relapse and remission periods of the RR‐MS type.79, 80
The median fraction of intrathecal VZV‐specific IgG of total IgG can differentiate patients with MS from those with VZV reactivation (35‐fold higher in the latter).81 This observation implies that low‐level infection is present in at least some MS cases. It also helps address whether or not VZV detection in MS is due to reactivated, previously latent VZV infection; that is, “centripetal infection” from the neural ganglia towards the CNS.82 Another theory suggests that VZV in MS is purely epiphenomenal due to leakage from destroyed sensory neurons; however, experimental evidence is lacking.83 Also, VZV has not been identified in “traditional” autoimmune diseases, implying a more specific connection with MS.82 Finally, VZV antigens induced and maintained activity of HERVs in peripheral lymphocytes from patients with MS compared to controls; retroviruses, as explained below, are implicated as causal in MS.84
6. HUMAN ENDOGENOUS RETROVIRUSES (HERV)
Although initially both were implicated in MS pathology since the 1980s, subsequent studies have continued to support a role for endogenous rather than exogenous retroviruses in MS.85 HERVs were integrated into the human genome relatively recently in evolutionary terms, that is some 30‐40 million years ago, as a result of ancestral retroviral infections. In humans, they form up to 8% of the genome and constitute a notable category of long terminal repeat (LTR) retrotransposons. These transposable elements, also known as “jumping genes,” change position within a genome and have repetitive sequences, explaining why it is more difficult to investigate their inheritance with classical genetics approaches.86 Despite these difficulties, the estimated 320 000 transcription factor binding sites (TFBSs) regulated by HERVs underscore their genomewide role. Deciphering their pathophysiological roles will offer further insights into the molecular basis of disease beyond that offered by focusing exclusively on the exome.87, 88
Putative mechanisms of HERV‐related pathophysiology in MS are illustrated in Figure 1. For example, several SNPs are associated with MS corresponding to genes implicated in immunological or vitamin D regulation. These SNPs occur more often in the vicinity of HERV‐related open reading frames (ORFs) than non‐MS‐related SNPs.89 Conversely, SNPs in regions around HERVs (such as the X‐linked HERV‐Fc1) are associated with MS, primarily the RR and secondary progressive types.90, 91 Furthermore, HERVs might “bridge” the environmental‐genetic interaction in MS given that any trigger (including viruses) may reactivate HERVs and enhance their expression.92 Of recent note—given that interferon signalling is implicated in MS—is the observation that HERVs contain binding sites for interferon‐γ‐induced transcription factors and therefore affect the expression of other genes, notably ones with immune function.93
Figure 1.
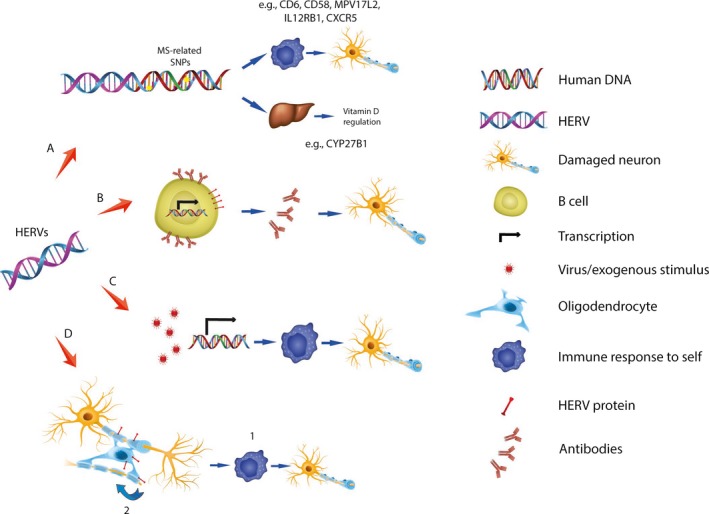
Putative mechanisms of human endogenous retrovirus (HERV)‐related autoimmunity in multiple sclerosis. (A) HERV‐encoded RNAs with intact open reading frames (ORFs) can be translated into proteins. Some of these (eg HERV‐K, HCML‐ARV) are in close proximity to SNPs shown to be associated with MS in genomewide association studies and representing genes involved in immune responses and vitamin D metabolism. (B) Some HERV proteins, notably MASP‐3, HERV‐H and HERV‐W, are expressed on the surface of normal cells including B cells. This serological response may be associated with autoimmunity, although causality has yet to be established. (C) HERVs are integral to the human genome but are epigenetically inactivated under normal conditions. HERV expression may be induced by environmental triggers including HSV‐1, HHV‐6, VZV and EBV viruses to stimulate an immune response and autoimmunity. (D) The MRSV‐Env protein has been identified in MS plaques and is brain selective and immunopathogenic so may directly stimulate an autoimmune response.1 Furthermore, this protein inhibits differentiation of oligodendrocyte precursors so may have a negative feedback effect in the brains of patients with MS 2
There is also an established body of evidence that the envelope protein of the “MS‐associated retrovirus” (MSRV‐Env) in the HERV‐W family is causal for MS.94 Initially observed in leptomeningeal cells, the MRSV‐Env protein has been detected in MS plaques containing macrophages, microglia and perivascular cells in actively demyelinating lesions and in the astrocytes of inactive areas but not in control brains.95 High MRSV‐Env DNA copy number, transcript and antigen levels have recently been detected in the blood of over 70% of patients with MS96; the increased DNA copy number is indicative of HERV‐related reverse transcriptase activity. Earlier studies suggested potential MRSV‐Env selectivity for the MS brain after observations of viral genetic material present at higher levels in the brain than in the blood of the same patients.97
HERV‐W Env expression is also increased on the surface of B cells and monocytes during the active phase of MS and parallels MS exacerbations.98 This protein, a Toll‐like receptor 4 (TLR4) agonist, stimulates immune cells and enhances expression of markers of leucocyte adhesion to endothelial cells. The above raises interesting questions about the effect of MRSV‐Env on blood‐brain barrier integrity.99 In parallel, HERV‐W Env impairs remyelination by inhibiting the differentiation of oligodendrocyte precursors to myelin‐producing oligodendrocytes, potentially due to nitrosative stress.100 The HERV‐W glycoprotein syncytin‐1 also seems to be implicated in MS via a similar mechanism; it causes an endoplasmic reticulum stress sensor to induce inducible nitric oxygen synthase and, concomitantly, the release of oligodendrocyte cytotoxins by astrocytes (for further details, see101). Also, HERVs can induce EAE in mice, implying a role upstream of other mediators.102 Therefore, HERVs seem to be implicated in both the neuroinflammatory and neurodegenerative components of the disease, rendering them promising therapeutic targets.
The autoimmune mechanism may also lie in the fact that common viruses (including but not limited to HSV‐1, HHV‐6, EBV or influenza) can activate HERV proteins84, 103 (Table 1). As a “dual infection,” EBV may be an exogenous and delayed cause for MS, with HERV‐W acting as a precipitant.104 However, the mechanisms of transcriptional activation of HERVs are generally obscure, as are the downstream events in human cells. A general framework might be that HERVs and, more broadly, endogenous transposons act as a genomic defence response to external stimuli.105 Only a few studies have failed to find differences in the presence of HERV nucleic acids or antibodies between MS cases and controls.106, 107 In,108 no difference was detected in HERV‐K113 levels between patients with MS and healthy controls, but this study did not investigate the retroviral families most related to MS. However, other studies favour a relationship between HERVs and MS pathobiology. For instance, MOG shares similarities with five regions in the envelope protein (ERVWE2), with one region consisting of B‐ and T‐cell epitopes capable of mediating antibody production and T‐cell function in vivo, respectively.109
Table 1.
The association between environmental viruses and HERV elements and the downstream effects
| Virus | HERV element | Downstream effect | References |
|---|---|---|---|
| HSV‐1 |
Matrix protein Gag protein |
Oligodendrotoxic and immunopathogenic | Ruprecht et al.,144 |
| HHV‐6 | HERV‐W Env and pol proteins | Synergy; interaction with HHV‐6 U94/rep and DNA‐pol | Nexo et al.,145 Perron et al.,146 |
| EBV | HERV‐W genes | Increased HERV‐W Env transcripts in PBMCs of IM patients; correlation of EBNA IgG levels with HERV gene expression levels in healthy, latently infected individuals (ie with anti‐EBNA‐1 titres >600) | Perron et al.,146 |
| EBV | HERV‐K18 env protein | Endothelial permeability; Proinflammatory reactions | Tai et al.,147 |
7. HUMAN IMMUNODEFICIENCY VIRUS (HIV)
Human immunodeficiency virus (HIV) is an exogenous retrovirus and HIV infection contributes to HERV activation, possibly via TLR‐4 stimulation.110 This association is exemplified by post‐mortem studies of brains from HIV patients and their epitope cross‐reactivity in T‐cell responses to HIV.111 There have been, to our knowledge, less than twenty HIV cases reported that describe demyelinating CNS diseases including MS, with a disturbance in the CD8+ cytotoxic T‐cell and CD4+ T regulatory cell ratio implicated as causal.112, 113
This rarity of documented cases of HIV and MS is consistent with the largest relevant record linkage study, in which HIV patients—all presumed to have undergone highly active antiretroviral therapy (HAART) therapy—were at a statistically significant reduced risk (relative risk=0.38)) for developing MS, with this relative risk including all recorded time intervals from first HIV record to the first MS record.114 One explanation for this finding could be that HIV‐induced immunodeficiency is protective against MS. Alternatively, HAART usually employs competitive or non‐competitive reverse transcriptase (RT) inhibitors, and due to suspected similarity between the HIV RTs and those of other viruses like HERVs, these inhibitors might suppress expression of the latter.115
8. CYTOMEGALOVIRUS (CMV)
The majority of epidemiological studies on CMV in MS are underpowered and inconclusive.7, 116 Two synchronous but different meta‐analyses suggested a protective role for CMV seropositivity in MS.117, 118
At the molecular level, it seems that CMV is present in the CNS including in some MS cases, but both exacerbating and protective roles are proposed.116 For example, CMV‐ and brain‐specific B cells are correlated in patients with MS,119 while concurrently CMV infection might indirectly exacerbate MS by inducing specific T cells with proinflammatory properties.116 Conversely, some studies have shown that higher anti‐CMV antibody titres in patients with MS are positively associated with improved MS‐related neuroimaging and disability status markers.120 In addition, human CMV‐induced natural killer cell expansion reduces the risk of disability progression in patients with MS.121
In animal models, cross‐reactivity between human CMV peptide and MOG has been detected, while secondary CMV infection following vaccinia virus infection can worsen T‐cell autoreactivity and white matter lesions. In contrast, murine CMV infection prior to Theiler's murine encephalomyelitis virus (TMEV) infection in the TMEV murine model of MS appears to improve symptoms both clinically (ie motor performance) and histologically (ie the severity of the inflammatory cell infiltrate).122
Finally, CMV (betaherpesvirinae subfamily) and EBV (gammaherpesvirinae subfamily) might oppose each other with regard to the downstream immune cascade (the so‐called “immune response competition”), which might explain their inverse epidemiological patterns in MS.118 It has been also suggested that these herpesviridae viruses could both be required to elicit a “primate‐specific autoimmune pathway”.116
9. MEASLES AND OTHER MORBILLIVIRUSES
The association between the measles virus and MS has been investigated for over 50 years, with MS postulated to be a host response to later measles infection. However, measles vaccination is not associated with MS, indicating that the measles virus is probably not connected with MS and supporting the evidence that measles vaccines are safe despite unjustified and well‐publicized claims to the contrary.123 However, it is worth mentioning that two CNS complications of measles virus infection manifest with features of demyelination: acute disseminated encephalomyelitis, a differential diagnosis of paediatric MS and the very rare subacute sclerosing panencephalitis.124
To our knowledge, recent research in this area has focused on the association between virus‐specific CSF‐to‐serum antibody indices (AIs) and MS, not on virus detection using molecular techniques. The AIs for measles, rubella and VZV, which form the “MRZ reaction”—high‐specificity markers for “ruling‐in” MS (reviewed in125)—are twofold higher than that for EBV.126 In particular, the measles AI is higher in patients with ≥6 lesions on MRI than those with fewer lesions in early MS.127 Another study showed that antimeasles virus antibody titres in the serum and CSF of patients with MS increase according to the age and duration of the disease.128
The phylogenetically close rinderpest virus has not been shown to be demyelinating or even neurotropic in its ruminant hosts.124 In contrast, infection with the more distant canine distemper virus (CDV) causes CDV demyelinating leukoencephalitis and serves as an established animal model of MS. In that model, demyelinating lesions and initial and later phases are characterized by direct infection of astrocytes and excess inflammation with myelin loss, respectively.124, 129 Interestingly, axonal damage precedes demyelination, prompting questions on the role of inflammation and astrocytes as intermediate players.130
10. LYMPHOCYTIC CHORIOMENINGITIS VIRUS (LCMV)
Lymphocytic choriomeningitis virus (LCMV) can affect the human CNS to cause paralysis and reduced consciousness. However, investigating its role in MS is more difficult due to low titres and short presence of LCMV in the CSF.131 In our opinion, this might indicate a “hit‐and‐run” mechanism. On the other hand, recent in silico predictions show high sequence and structural similarity between LCMV's nucleoprotein and specific myelin basic protein (MBP) residues.132
Murine models of chronic LCMV infection have given rise to two Nobel Prizes.131 The virus is thought to activate microglia and astrocytes in the CNS via a TLR2‐mediated cascade.133 Moreover, LCMV blocks induction of type 1 interferon and consequential upregulation of HLA class II. This observation supports a potential virus‐induced disturbance in the interferon‐tumour necrosis factor balance, which is already known to trigger autoimmunity.132
Interestingly, LCMV infection limited to the periphery with concurrent CNS measles virus infection can induce CNS pathology via LCMV‐specific CD8+ T‐cell recruitment to the brain without the need for LCMV replication. The underlying reason why the brain, broadly considered “immuno‐privileged”, attracts these mis‐recruited cells needs further exploration.134
11. CORONAVIRUS
In rodents, certain coronavirus‐family mouse hepatitis virus strains are neurotropic, disrupt the blood‐brain barrier and cause immune‐mediated demyelinating‐like lesions.135 Human coronaviruses (HCoV) predominantly cause upper respiratory tract infections and are also neurotropic. Recent epidemiological studies are lacking, while molecular analyses have shown the HCoV‐specific surface glycoprotein acts as a trigger for programmed cell death in a murine model of neurodegeneration. In addition, HCoV‐229E/MBP cross‐reactive T cells have been isolated from patients with MS in single‐cell analyses, implying a molecular mimicry mechanism (for a review, see136). In a mouse model of encephalomyelitis/demyelination induced by gliatropic murine coronavirus, the initial activation and accumulation of self‐reactive CD4+ T cells were followed by a mechanism of host‐mediated suppression that consequentially led to their decline, thus diminishing autoimmune phenotypes.137
12. SAFFOLD VIRUS
Saffold virus (SAFV), a picornaviridae family member identified in 2007, was the first human virus in the Cardiovirus genus to be described.138 SAFV has a seroprevalence of over 90% in the adult population and is known to cause infection early in life.139 SAFV is associated with both enteric and extra‐intestinal diseases and, due to homology with TMEV, is implicated in MS.138
However, its ubiquity has created difficulties in deciphering any association between SAFV and MS.17 SAFV was not detected in CSF samples from patients with MS.138 One hypothesis is that SAFV might cause low‐grade persistent infection followed by inflammation rather than act as a “hit‐and‐run” trigger for autoimmunity. However, a recent study failed to find any SAFV in MS brains and only rare SAFV‐specific oligoclonal bands in patients with MS and not different from controls.17
13. LIMITATIONS OF EXISTING DATA
Several methodological issues could explain the described inconsistencies between studies in the MS‐virus arena: (i) not choosing appropriate healthy matched controls following a specific study design but instead samples simply available at the time of study (ie an “opportunistic” approach); (ii) even though quantification of viral load by real‐time PCR is helpful, there seems to be a failure to use positive PCR or serology to distinguish active from latent infection (ie earlier infection during childhood in the case of serology). The enigmatic nature of MS poses challenges in the interpretation of the results as, according to some authors, detecting some antibodies under certain circumstances, that is in worsening MS, could be due to a hyperactivated immune system and not real infection140; (iii) conversely, interpreting absence of evidence of virus infection as evidence of absence, especially in the genomic era, may be a mistake.141
14. CONCLUSIONS AND SUGGESTIONS FOR FUTURE RESEARCH
There is, therefore, accumulated evidence that viruses may trigger or cause MS, with these organisms and the immune system interacting in several, potentially overlapping, ways. Deciphering the epidemiological contribution of viruses to MS along with their pathogenic mechanisms may help in the development of effective targeted therapies to develop vaccines, treat the disease, prevent relapses and maintain remission.
Possible future research avenues include prospectively studying and monitoring carefully defined groups of patients, such as comparing patients with clinically isolated syndrome (CIS) who went on to convert to MS with those that did not. Although EBV has been studied in such cases, a broader causative role for viruses would be strengthened if any marker of viral presence (ie increased viral load and/or higher antiviral response) was observed in the first category. Furthermore, the B‐ and T‐cell receptor repertoires in MS samples need to be fully characterized, preferably in relation to viral detection and burden and perhaps using newer high‐throughput technologies such as deep sequencing. This would be facilitated by the enrichment of immunosequencing databases with extensive experimental data on the repertoires induced by different human viruses. It would also be sensible to examine latent‐to‐lytic switching of potentially existing viruses in MS biopsies. To complement previous efforts focusing on EBV‐specific markers,23 it would be interesting to analyse more recently proposed markers of cellular antiviral response with respect to the above switch.142 Finally, given that many viruses, not least EBV, express several proteins during different viral life cycle stages, the full spectrum of antibody responses to viruses over their infective course needs further exploration, perhaps using protein arrays methods for novel antigen discovery to overcome the limitations of current techniques.143
CONFLICT OF INTEREST
No financial conflict of interest exists.
ACKNOWLEDGEMENTS
A.‐F.A.M. has been supported through an educational scholarship from the Onassis Public Benefit Foundation. The latter played no role in the design of the study, collection and/or interpretation of data, or writing of the review article.
Mentis A‐FA, Dardiotis E, Grigoriadis N, Petinaki E, Hadjigeorgiou GM. Viruses and endogenous retroviruses in multiple sclerosis: From correlation to causation. Acta Neurol Scand. 2017;136:606–616. 10.1111/ane.12775
REFERENCES
- 1. Procaccini C, de Rosa V, Pucino V, Formisano L, Matarese G. Animal models of multiple sclerosis. Eur J Pharmacol. 2015;759:182‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tselis A. Evidence for viral etiology of multiple sclerosis. Semin Neurol. 2011;31:307‐316. [DOI] [PubMed] [Google Scholar]
- 3. Belbasis L, Bellou V, Evangelou E, Ioannidis JPA, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta‐analyses. Lancet Neurol. 2015;14:263‐273. [DOI] [PubMed] [Google Scholar]
- 4. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545‐558. [DOI] [PubMed] [Google Scholar]
- 5. Grigoriadis N, van Pesch V, Paradig MSG. A basic overview of multiple sclerosis immunopathology. Eur J Neurol. 2015;22(Suppl 2):3‐13. [DOI] [PubMed] [Google Scholar]
- 6. Brodin P, Jojic V, Gao T, et al. Variation in the human immune system is largely driven by non‐heritable influences. Cell. 2015;160:37‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. 2017;13:25‐36. [DOI] [PubMed] [Google Scholar]
- 8. Baranzini SE, Mudge J, van Velkinburgh JC, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sotelo J, Corona T. Varicella zoster virus and relapsing remitting multiple sclerosis. Mult Scler Int. 2011;2011:214763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse‐translational research. Lancet Neurol. 2016;15:317‐331. [DOI] [PubMed] [Google Scholar]
- 11. Behan PO, Chaudhuri A. EAE is not a useful model for demyelinating disease. Mult Scler Relat Disord. 2014;3:565‐574. [DOI] [PubMed] [Google Scholar]
- 12. Brandle SM, Obermeier B, Senel M, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self‐proteins. Proc Natl Acad Sci U S A. 2016;113:7864‐7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Venkatesan A, Johnson RT. Infections and multiple sclerosis. Handb Clin Neurol. 2014;122:151‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown C. Aetiology: neighbourhood watch. Nature. 2016;540:S4‐S6. [DOI] [PubMed] [Google Scholar]
- 15. Brettschneider J, Tumani H, Kiechle U, et al. IgG antibodies against measles, rubella, and varicella zoster virus predict conversion to multiple sclerosis in clinically isolated syndrome. PLoS One. 2009;4:e7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grigoriadis N, Hadjigeorgiou GM. Virus‐mediated autoimmunity in multiple sclerosis. J Autoimmune Dis. 2006;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galama JM, Zoll JG, Lanke KH, et al. Saffold cardiovirus and multiple sclerosis: no evidence for an association. Ann Clin Transl Neurol. 2014;1:618‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lamberto I, Gunst K, Muller H, Zur Hausen H, De Villiers EM. Mycovirus‐like DNA virus sequences from cattle serum and human brain and serum samples from multiple sclerosis patients. Genome Announc. 2014;2: pii: e00848‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pakpoor J, Ramagopalan SV. Epstein‐Barr virus is a necessary causative agent in the pathogenesis of multiple sclerosis: yes. Mult Scler. 2013;19:1690‐1691. [DOI] [PubMed] [Google Scholar]
- 20. Salzer J, Myhr KM. Epstein‐Barr virus is a necessary causative agent in the pathogenesis of multiple sclerosis: no. Mult Scler. 2013;19:1692‐1693. [DOI] [PubMed] [Google Scholar]
- 21. ‘t Hart BA, Gran B, Weissert R. EAE: imperfect but useful models of multiple sclerosis. Trends Mol Med. 2011;17:119‐125. [DOI] [PubMed] [Google Scholar]
- 22. Fernandez‐Menendez S, Fernandez‐Moran M, Fernandez‐Vega I, Perez‐Alvarez A, Villafani‐Echazu J. Epstein‐Barr virus and multiple sclerosis. From evidence to therapeutic strategies. J Neurol Sci. 2016;361:213‐219. [DOI] [PubMed] [Google Scholar]
- 23. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM, Neuropromise EBVWG. Epstein‐Barr virus in the multiple sclerosis brain: a controversial issue‐report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011;134:2772‐2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pakpoor J, Giovannoni G, Ramagopalan SV. Epstein‐Barr virus and multiple sclerosis: association or causation? Expert Rev Neurother. 2013;13:287‐297. [DOI] [PubMed] [Google Scholar]
- 25. Mckay KA, Jahanfar S, Duggan T, Tkachuk S, Tremlett H. Factors associated with onset, relapses or progression in multiple sclerosis: a systematic review. Neurotoxicology. 2016. DOI: 10.1016/j.neuro.2016.03.020 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 26. McKay KA, Kwan V, Duggan T, Tremlett H. Risk factors associated with the onset of relapsing‐remitting and primary progressive multiple sclerosis: a systematic review. Biomed Res Int. 2015;2015:817238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Almohmeed YH, Avenell A, Aucott L, Vickers MA. Systematic review and meta‐analysis of the sero‐epidemiological association between Epstein Barr virus and multiple sclerosis. PLoS One. 2013;8:e61110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hwang AE, Hamilton AS, Cockburn MG, et al. Evidence of genetic susceptibility to infectious mononucleosis: a twin study. Epidemiol Infect. 2012;140:2089‐2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wergeland S, Myhr KM, Loken‐Amsrud KI, et al. Vitamin D, HLA‐DRB1 and Epstein‐Barr virus antibody levels in a prospective cohort of multiple sclerosis patients. Eur J Neurol. 2016;23:1064‐1070. [DOI] [PubMed] [Google Scholar]
- 30. Tschochner M, Leary S, Cooper D, et al. Identifying patient‐specific Epstein‐Barr nuclear antigen‐1 genetic variation and potential autoreactive targets relevant to multiple sclerosis pathogenesis. PLoS One. 2016;11:e0147567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao D, Ye X, Zhang N, et al. A meta‐analysis of interaction between Epstein‐Barr virus and HLA‐DRB1*1501 on risk of multiple sclerosis. Sci Rep. 2015;5:18083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salzer J, Nystrom M, Hallmans G, Stenlund H, Wadell G, Sundstrom P. Epstein‐Barr virus antibodies and vitamin D in prospective multiple sclerosis biobank samples. Mult Scler. 2013;19:1587‐1591. [DOI] [PubMed] [Google Scholar]
- 33. Tao C, Simpson S Jr, Taylor BV, van der Mei I. Association between human herpesvirus & human endogenous retrovirus and MS onset & progression. J Neurol Sci. 2017;372:239‐249. [DOI] [PubMed] [Google Scholar]
- 34. Salzer J, Stenlund H, Sundstrom P. The interaction between smoking and Epstein‐Barr virus as multiple sclerosis risk factors may depend on age. Mult Scler. 2014;20:747‐750. [DOI] [PubMed] [Google Scholar]
- 35. Bjornevik K, Riise T, Bostrom I, et al. Negative interaction between smoking and EBV in the risk of multiple sclerosis: the EnvIMS study. Mult Scler. 2016. DOI: 10.1177/1352458516671028 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 36. Munger KL, Levin LI, O'Reilly EJ, Falk KI, Ascherio A. Anti‐Epstein‐Barr virus antibodies as serological markers of multiple sclerosis: a prospective study among United States military personnel. Mult Scler. 2011;17:1185‐1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nejati A, Shoja Z, Shahmahmoodi S, et al. EBV and vitamin D status in relapsing‐remitting multiple sclerosis patients with a unique cytokine signature. Med Microbiol Immunol. 2016;205:143‐154. [DOI] [PubMed] [Google Scholar]
- 38. Ricigliano VA, Handel AE, Sandve GK, et al. EBNA2 binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin D receptor occupancy. PLoS One. 2015;10:e0119605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Latham LB, Lee MJ, Lincoln JA, Ji N, Forsthuber TG, Lindsey JW. Antivirus immune activity in multiple sclerosis correlates with MRI activity. Acta Neurol Scand. 2016;133:17‐24. [DOI] [PubMed] [Google Scholar]
- 40. Kvistad S, Myhr KM, Holmoy T, et al. Antibodies to Epstein‐Barr virus and MRI disease activity in multiple sclerosis. Mult Scler. 2014;20:1833‐1840. [DOI] [PubMed] [Google Scholar]
- 41. Angelini DF, Serafini B, Piras E, et al. Increased CD8+ T cell response to Epstein‐Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog. 2013;9:e1003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lossius A, Johansen JN, Vartdal F, et al. High‐throughput sequencing of TCR repertoires in multiple sclerosis reveals intrathecal enrichment of EBV‐reactive CD8+ T cells. Eur J Immunol. 2014;44:3439‐3452. [DOI] [PubMed] [Google Scholar]
- 43. Jagessar SA, Holtman IR, Hofman S, et al. Lymphocryptovirus infection of nonhuman primate B cells converts destructive into productive processing of the pathogenic CD8 T cell epitope in myelin oligodendrocyte glycoprotein. J Immunol. 2016;197:1074‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pender MP, Burrows SR. Epstein‐Barr virus and multiple sclerosis: potential opportunities for immunotherapy. Clin Transl Immunology. 2014;3:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kakalacheva K, Regenass S, Wiesmayr S, et al. Infectious mononucleosis triggers generation of IgG auto‐antibodies against native myelin oligodendrocyte glycoprotein. Viruses. 2016;8:pii: E51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Magliozzi R, Serafini B, Rosicarelli B, et al. B‐cell enrichment and Epstein‐Barr virus infection in inflammatory cortical lesions in secondary progressive multiple sclerosis. J Neuropathol Exp Neurol. 2013;72:29‐41. [DOI] [PubMed] [Google Scholar]
- 47. Thart BA, Kap YS, Morandi E, Laman JD, Gran B. EBV infection and multiple sclerosis: lessons from a marmoset model. Trends Mol Med. 2016;22:1012‐1024. [DOI] [PubMed] [Google Scholar]
- 48. Lodato MA, Woodworth MB, Lee S, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Santon A, Cristóbal E, Aparicio M, Royuela A, Villar LM, Alvarez‐Cermeño JC. High frequency of co‐infection by Epstein‐Barr virus types 1 and 2 in patients with multiple sclerosis. Mult Scler. 2011;17:1295‐1300. [DOI] [PubMed] [Google Scholar]
- 50. Owens GP, Bennett JL. Trigger, pathogen, or bystander: the complex nexus linking Epstein‐Barr virus and multiple sclerosis. Mult Scler. 2012;18:1204‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Casiraghi C, Marquez AC, Shanina I, Horwitz MS. Latent virus infection upregulates CD40 expression facilitating enhanced autoimmunity in a model of multiple sclerosis. Sci Rep. 2015;5:13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hecker M, Fitzner B, Wendt M, et al. High‐density peptide microarray analysis of IgG autoantibody reactivities in serum and cerebrospinal fluid of multiple sclerosis patients. Mol Cell Proteomics. 2016;15:1360‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sugden B. Epstein‐Barr virus: the path from association to causality for a ubiquitous human pathogen. PLoS Biol. 2014;12:e1001939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khankhanian P, Cozen W, Himmelstein DS, et al. Meta‐analysis of genome‐wide association studies reveals genetic overlap between Hodgkin lymphoma and multiple sclerosis. Int J Epidemiol. 2016;45:728‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jha HC, Mehta D, Lu J, et al. Gammaherpesvirus infection of human neuronal cells. MBio. 2015;6:e01844‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou Y, Zhu G, Charlesworth JC, et al. Genetic loci for Epstein‐Barr virus nuclear antigen‐1 are associated with risk of multiple sclerosis. Mult Scler. 2016;22:1655‐1664. [DOI] [PubMed] [Google Scholar]
- 57. Leibovitch EC, Jacobson S. Evidence linking HHV‐6 with multiple sclerosis: an update. Curr Opin Virol. 2014;9:127‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Álvarez‐Lafuente R, García‐Montojo M, Heras VDL, et al. Herpesviruses and human endogenous retroviral sequences in the cerebrospinal fluid of multiple sclerosis patients. Mult Scler. 2008;14:595‐601. [DOI] [PubMed] [Google Scholar]
- 59. Simpson S Jr, Taylor B, Burrows J, et al. EBV & HHV6 reactivation is infrequent and not associated with MS clinical course. Acta Neurol Scand. 2014;130:328‐337. [DOI] [PubMed] [Google Scholar]
- 60. Garcia‐Montojo M, Martinez A, De Las Heras V, et al. Herpesvirus active replication in multiple sclerosis: a genetic control? J Neurol Sci. 2011;311:98‐102. [DOI] [PubMed] [Google Scholar]
- 61. Tai AK, Luka J, Ablashi D, Huber BT. HHV‐6A infection induces expression of HERV‐K18‐encoded superantigen. J Clin Virol. 2009;46:47‐48. [DOI] [PubMed] [Google Scholar]
- 62. Curtin F, Perron H, Faucard R, Porchet H, Lang AB. Treatment against human endogenous retrovirus: a possible personalized medicine approach for multiple sclerosis. Mol Diagn Ther. 2015;19:255‐265. [DOI] [PubMed] [Google Scholar]
- 63. Leibovitch E, Wohler JE, Cummings Macri SM, et al. Novel marmoset (Callithrix jacchus) model of human Herpesvirus 6A and 6B infections: immunologic, virologic and radiologic characterization. PLoS Pathog. 2013;9:e1003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Harberts E, Yao K, Wohler JE, et al. Human herpesvirus‐6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci U S A. 2011;108:13734‐13739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Broccolo F, Fusetti L, Ceccherini‐Nelli L. Possible role of human herpesvirus 6 as a trigger of autoimmune disease. ScientificWorldJournal. 2013;2013:867389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shao Q, Lin Z, Wu X, et al. Transcriptome sequencing of neurologic diseases associated genes in HHV‐6A infected human astrocyte. Oncotarget. 2016;7:48070‐48080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Berth S, Carbunar O, Yang NS, Fredericks B, Lipton HL, Valyi‐Nagy T. Varicella‐zoster virus encephalomyelitis with a prominent demyelinating component. Neuropathology. 2015;35:587‐591. [DOI] [PubMed] [Google Scholar]
- 68. Ahlgren C, Toren K, Oden A, Andersen O. A population‐based case‐control study on viral infections and vaccinations and subsequent multiple sclerosis risk. Eur J Epidemiol. 2009;24:541‐552. [DOI] [PubMed] [Google Scholar]
- 69. Rodriguez‐Violante M, Ordonez G, Bermudez JR, Sotelo J, Corona T. Association of a history of varicella virus infection with multiple sclerosis. Clin Neurol Neurosurg. 2009;111:54‐56. [DOI] [PubMed] [Google Scholar]
- 70. Kang JH, Sheu JJ, Kao S, Lin HC. Increased risk of multiple sclerosis following herpes zoster: a nationwide, population‐based study. J Infect Dis. 2011;204:188‐192. [DOI] [PubMed] [Google Scholar]
- 71. Najafi S, Ghane M, Yousefzadeh‐Chabok S, Amiri M. The high prevalence of the varicella zoster virus in patients with relapsing‐remitting multiple sclerosis: a case‐control study in the north of Iran. Jundishapur J Microbiol. 2016;9:e34158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yoshimura S, Isobe N, Matsushita T, et al. Genetic and infectious profiles influence cerebrospinal fluid IgG abnormality in Japanese multiple sclerosis patients. PLoS One. 2014;9:e95367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sotelo J, Ordonez G, Pineda B. Varicella‐zoster virus at relapses of multiple sclerosis. J Neurol. 2007;254:493‐500. [DOI] [PubMed] [Google Scholar]
- 74. Mancuso R, Delbue S, Borghi E, et al. Increased prevalence of varicella zoster virus DNA in cerebrospinal fluid from patients with multiple sclerosis. J Med Virol. 2007;79:192‐199. [DOI] [PubMed] [Google Scholar]
- 75. Burgoon MP, Cohrs RJ, Bennett JL, et al. Varicella zoster virus is not a disease‐relevant antigen in multiple sclerosis. Ann Neurol. 2009;65:474‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hon GM, Erasmus RT, Matsha T. Low prevalence of human herpesvirus‐6 and varicella zoster virus in blood of multiple sclerosis patients, irrespective of inflammatory status or disease progression. J Clin Neurosci. 2014;21:1437‐1440. [DOI] [PubMed] [Google Scholar]
- 77. Mancuso R, Hernis A, Cavarretta R, et al. Detection of viral DNA sequences in the cerebrospinal fluid of patients with multiple sclerosis. J Med Virol. 2010;82:1051‐1057. [DOI] [PubMed] [Google Scholar]
- 78. Sotelo J, Martínez‐Palomo A, Ordoñez G, Pineda B. Varicella‐zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann Neurol. 2008;63:303‐311. [DOI] [PubMed] [Google Scholar]
- 79. Sotelo J, Ordonez G, Pineda B, Flores J. The participation of varicella zoster virus in relapses of multiple sclerosis. Clin Neurol Neurosurg. 2014;119:44‐48. [DOI] [PubMed] [Google Scholar]
- 80. Ordoñez G, Martinez‐Palomo A, Corona T, et al. Varicella zoster virus in progressive forms of multiple sclerosis. Clin Neurol Neurosurg. 2010;112:653‐657. [DOI] [PubMed] [Google Scholar]
- 81. Otto C, Hofmann J, Finke C, Zimmermann M, Ruprecht K. The fraction of varicella zoster virus‐specific antibodies among all intrathecally‐produced antibodies discriminates between patients with varicella zoster virus reactivation and multiple sclerosis. Fluids Barriers CNS. 2014;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sotelo J. On the viral hypothesis of multiple sclerosis: participation of varicella‐zoster virus. J Neurol Sci. 2007;262:113‐116. [DOI] [PubMed] [Google Scholar]
- 83. Torkildsen O, Power O, Storstein A. Detection of varicella‐zoster virus DNA during medullary and brainstem relapses in multiple sclerosis. BMJ Case Rep. 2016;2016: pii: bcr2016214555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Brudek T, Luhdorf P, Christensen T, Hansen HJ, Moller‐Larsen A. Activation of endogenous retrovirus reverse transcriptase in multiple sclerosis patient lymphocytes by inactivated HSV‐1, HHV‐6 and VZV. J Neuroimmunol. 2007;187:147‐155. [DOI] [PubMed] [Google Scholar]
- 85. Nissen KK, Laska MJ, Hansen B, et al. Endogenous retroviruses and multiple sclerosis‐new pieces to the puzzle. BMC Neurol. 2013;13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Varade J, Garcia‐Montojo M, de la Hera B, et al. Multiple sclerosis retrovirus‐like envelope gene: role of the chromosome 20 insertion. BBA Clin. 2015;3:162‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dolei A, Perron H. The multiple sclerosis‐associated retrovirus and its HERV‐W endogenous family: a biological interface between virology, genetics, and immunology in human physiology and disease. J Neurovirol. 2009;15:4‐13. [DOI] [PubMed] [Google Scholar]
- 88. Suntsova M, Garazha A, Ivanova A, Kaminsky D, Zhavoronkov A, Buzdin A. Molecular functions of human endogenous retroviruses in health and disease. Cell Mol Life Sci. 2015;72:3653‐3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Brutting C, Emmer A, Kornhuber M, Staege MS. A survey of endogenous retrovirus (ERV) sequences in the vicinity of multiple sclerosis (MS)‐associated single nucleotide polymorphisms (SNPs). Mol Biol Rep. 2016;43:827‐836. [DOI] [PubMed] [Google Scholar]
- 90. Nexo BA, Christensen T, Frederiksen J, et al. The etiology of multiple sclerosis: genetic evidence for the involvement of the human endogenous retrovirus HERV‐Fc1. PLoS One. 2011;6:e16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. de la Hera B, Varade J, Garcia‐Montojo M, et al. Human endogenous retrovirus HERV‐Fc1 association with multiple sclerosis susceptibility: a meta‐analysis. PLoS One. 2014;9:e90182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Brodziak A, Ziółko E, Muc‐Wierzgoń M, Nowakowska‐Zajdel E, Kokot T, Klakla K. Endogenous retroviruses_Auto‐immune disease. Med Sci Monit. 2012;18:RA80‐RA88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co‐option of endogenous retroviruses. Science. 2016;351:1083‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Morandi E, Tarlinton RE, Gran B. Multiple sclerosis between genetics and infections: human endogenous retroviruses in monocytes and macrophages. Front Immunol. 2015;6:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. van Horssen J, van der Pol S, Nijland P, Amor S, Perron H. Human endogenous retrovirus W in brain lesions: rationale for targeted therapy in multiple sclerosis. Mult Scler Relat Disord. 2016;8:11‐18. [DOI] [PubMed] [Google Scholar]
- 96. Perron H, Germi R, Bernard C, et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult Scler. 2012;18:1721‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Antony JM, Izad M, Bar‐Or A, et al. Quantitative analysis of human endogenous retrovirus‐W env in neuroinflammatory diseases. AIDS Res Hum Retroviruses. 2006;22:1253‐1259. [DOI] [PubMed] [Google Scholar]
- 98. Brudek T, Christensen T, Aagaard L, Petersen T, Hansen HJ, Moller‐Larsen A. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV‐H Env and HERV‐W Env, accompanied by increased seroreactivity. Retrovirology. 2009;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Duperray A, Barbe D, Raguenez G, et al. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int Immunol. 2015;27:545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Madeira A, Burgelin I, Perron H, Curtin F, Lang AB, Faucard R. MSRV envelope protein is a potent, endogenous and pathogenic agonist of human toll‐like receptor 4: relevance of GNbAC1 in multiple sclerosis treatment. J Neuroimmunol. 2016;291:29‐38. [DOI] [PubMed] [Google Scholar]
- 101. Antony J, Ellestad KK, Hammond R, et al. The human endogenous retrovirus envelope glycoprotein, syncytin‐1, regulates neuroinflammation and its receptor expression in multiple sclerosis. J Immunol. 2007;179:1210‐1224. [DOI] [PubMed] [Google Scholar]
- 102. Perron H, Dougier‐Reynaud HL, Lomparski C, et al. Human endogenous retrovirus protein activates innate immunity and promotes experimental allergic encephalomyelitis in mice. PLoS One. 2013;8:e80128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Li F, Nellaker C, Sabunciyan S, et al. Transcriptional derepression of the ERVWE1 locus following influenza A virus infection. J Virol. 2014;88:4328‐4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mameli G, Poddighe L, Mei A, et al. Expression and activation by Epstein Barr virus of human endogenous retroviruses‐W in blood cells and astrocytes: inference for multiple sclerosis. PLoS One. 2012;7:e44991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mavragani CP, Sagalovskiy I, Guo Q, et al. Expression of long interspersed nuclear element 1 retroelements and induction of type I interferon in patients with systemic autoimmune disease. Arthritis Rheumatol. 2016;68:2686‐2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Schmitt K, Richter C, Backes C, Meese E, Ruprecht K, Mayer J. Comprehensive analysis of human endogenous retrovirus group HERV‐W locus transcription in multiple sclerosis brain lesions by high‐throughput amplicon sequencing. J Virol. 2013;87:13837‐13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Laufer G, Mayer J, Mueller BF, Mueller‐Lantzsch N, Ruprecht K. Analysis of transcribed human endogenous retrovirus W env loci clarifies the origin of multiple sclerosis‐associated retrovirus env sequences. Retrovirology. 2009;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Moyes DL, Goris A, Ban M, et al. HERV‐K113 is not associated with multiple sclerosis in a large family‐based study. AIDS Res Hum Retroviruses. 2008;24:363‐365. [DOI] [PubMed] [Google Scholar]
- 109. Do Olival GS, Faria TS, Nali LH, et al. Genomic analysis of ERVWE2 locus in patients with multiple sclerosis: absence of genetic association but potential role of human endogenous retrovirus type W elements in molecular mimicry with myelin antigen. Front Microbiol. 2013;4:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Uleri E, Mei A, Mameli G, Poddighe L, Serra C, Dolei A. HIV Tat acts on endogenous retroviruses of the W family and this occurs via Toll‐like receptor 4: inference for neuroAIDS. AIDS. 2014;28:2659‐2670. [DOI] [PubMed] [Google Scholar]
- 111. Christensen T. Human endogenous retroviruses in neurologic disease. APMIS. 2016;124:116‐126. [DOI] [PubMed] [Google Scholar]
- 112. Chin JH. Multiple sclerosis and HIV‐1 infection: case report of a HIV controller. J Neurovirol. 2015;21:464‐467. [DOI] [PubMed] [Google Scholar]
- 113. Delgado SR, Maldonado J, Rammohan KW. CNS demyelinating disorder with mixed features of neuromyelitis optica and multiple sclerosis in HIV‐1 infection. Case report and literature review. J Neurovirol. 2014;20:531‐537. [DOI] [PubMed] [Google Scholar]
- 114. Gold J, Goldacre R, Maruszak H, Giovannoni G, Yeates D, Goldacre M. HIV and lower risk of multiple sclerosis: beginning to unravel a mystery using a record‐linked database study. J Neurol Neurosurg Psychiatry. 2015;86:9‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kop MLVD. Does antiretroviral therapy for HIV reduce the risk of developing multiple sclerosis? Neurol Neurosurg Psychiatry. 2015;86:3. [DOI] [PubMed] [Google Scholar]
- 116. Vanheusden M, Stinissen P, Stinissen P, ‘t Hart BA, Hellings N. Cytomegalovirus: a culprit or protector in multiple sclerosis? Trends Mol Med. 2015;21:16‐23. [DOI] [PubMed] [Google Scholar]
- 117. Pakpoor J, Pakpoor J, Disanto G, Giovannoni G, Ramagopalan SV. Cytomegalovirus and multiple sclerosis risk. J Neurol. 2013;260:1658‐1660. [DOI] [PubMed] [Google Scholar]
- 118. Sundqvist E, Bergstrom T, Daialhosein H, et al. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult Scler. 2014;20:165‐173. [DOI] [PubMed] [Google Scholar]
- 119. Wunsch M, Hohmann C, Milles B, et al. The correlation between the virus‐ and brain antigen‐specific B cell response in the blood of patients with multiple sclerosis. Viruses. 2016;8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zivadinov R, Nasuelli D, Tommasi MA, et al. Positivity of cytomegalovirus antibodies predicts a better clinical and radiological outcome in multiple sclerosis patients. Neurol Res. 2006;28:262‐269. [DOI] [PubMed] [Google Scholar]
- 121. Martinez‐Rodriguez JE, Cobo‐Calvo A, Villar LM, et al. Adaptive natural killer cell response to cytomegalovirus and disability progression in multiple sclerosis. Mult Scler. 2016;22:741‐752. [DOI] [PubMed] [Google Scholar]
- 122. Pirko I, Cardin R, Chen Y, et al. CMV infection attenuates the disease course in a murine model of multiple sclerosis. PLoS One. 2012;7:e32767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ahlgren C, Oden A, Toren K, Andersen O. Multiple sclerosis incidence in the era of measles‐mumps‐rubella mass vaccinations. Acta Neurol Scand. 2009;119:313‐320. [DOI] [PubMed] [Google Scholar]
- 124. Sips GJ, Chesik D, Glazenburg L, Wilschut J, de Keyser J, Wilczak N. Involvement of morbilliviruses in the pathogenesis of demyelinating disease. Rev Med Virol. 2007;17:223‐244. [DOI] [PubMed] [Google Scholar]
- 125. Jarius S, Eichhorn P, Franciotta D, et al. The MRZ reaction as a highly specific marker of multiple sclerosis: re‐evaluation and structured review of the literature. J Neurol. 2016;264:453‐466. [DOI] [PubMed] [Google Scholar]
- 126. Pohl D, Rostasy K, Jacobi C, et al. Intrathecal antibody production against Epstein‐Barr and other neurotropic viruses in pediatric and adult onset multiple sclerosis. J Neurol. 2010;257:212‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Rosche B, Laurent S, Conradi S, Hofmann J, Ruprecht K, Harms L. Measles IgG antibody index correlates with T2 lesion load on MRI in patients with early multiple sclerosis. PLoS One. 2012;7:e28094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ahlgren C, Oden A, Bergstrom T, Lycke J. Serum and CSF measles antibody levels increase over time in patients with multiple sclerosis or clinically isolated syndrome. J Neuroimmunol. 2012;247:70‐74. [DOI] [PubMed] [Google Scholar]
- 129. Beineke A, Puff C, Seehusen F, Baumgartner W. Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet Immunol Immunopathol. 2009;127:1‐18. [DOI] [PubMed] [Google Scholar]
- 130. Lempp C, Spitzbarth I, Puff C, et al. New aspects of the pathogenesis of canine distemper leukoencephalitis. Viruses. 2014;6:2571‐2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wilson MR, Peters CJ. Diseases of the central nervous system caused by lymphocytic choriomeningitis virus and other arenaviruses. Handb Clin Neurol. 2014;123:671‐681. [DOI] [PubMed] [Google Scholar]
- 132. Hogeboom C. Peptide motif analysis predicts lymphocytic choriomeningitis virus as trigger for multiple sclerosis. Mol Immunol. 2015;67:625‐635. [DOI] [PubMed] [Google Scholar]
- 133. Shenghua Z, Halle A, Kurt‐Jones EA, et al. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2‐MyD88/Mal‐dependent inflammatory responses. J Neuroimmunol. 2008;194:70‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Matullo CM, O'Regan KJ, Curtis M, Rall GF. CNS recruitment of CD8+ T lymphocytes specific for a peripheral virus infection triggers neuropathogenesis during polymicrobial challenge. PLoS Pathog. 2011;7:e1002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bleau C, Filliol A, Samson M, Lamontagne L. Brain invasion by mouse hepatitis virus depends on impairment of tight junctions and beta interferon production in brain microvascular endothelial cells. J Virol. 2015;89:9896‐9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Desforges M, le Coupanec A, Brison E, Meessen‐Pinard M, Talbot PJ. Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans. Adv Exp Med Biol. 2014;807:75‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Savarin C, Bergmann CC, Gaignage M, Stohlman SA. Self‐reactive CD4(+) T cells activated during viral‐induced demyelination do not prevent clinical recovery. J Neuroinflammation. 2015;12:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Chiu CY, Greninger AL, Kanada K, et al. Identification of cardioviruses related to Theiler's murine encephalomyelitis virus in human infections. Proc Natl Acad Sci U S A. 2008;105:14124‐14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zoll J, ErkensHulshof S, Lanke K, et al. Saffold virus, a human Theiler's‐like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 2009;5:e1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Voumvourakis KI, Kitsos DK, Tsiodras S, Petrikkos G, Stamboulis E. Human herpesvirus 6 infection as a trigger of multiple sclerosis. Mayo Clin Proc. 2010;85:1023‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Simmonds P, Adams MJ, Benko M, et al. Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Micro. 2017;15:161‐168. [DOI] [PubMed] [Google Scholar]
- 142. Lieberman PM. Epigenetics and genetics of viral latency. Cell Host Microbe. 2016;19:619‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dooley MM, de Gannes SL, Fu KA, Lindsey JW. The increased antibody response to Epstein‐Barr virus in multiple sclerosis is restricted to selected virus proteins. J Neuroimmunol. 2016;299:147‐151. [DOI] [PubMed] [Google Scholar]
- 144. Ruprecht K, Obojes K, Wengel V, et al. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: implications for multiple sclerosis. J Neurovirol. 2006;12:65‐71. [DOI] [PubMed] [Google Scholar]
- 145. Nexo BA, Jensen SB, Nissen KK, Hansen B, Laska MJ. Two endogenous retroviral loci appear to contribute to multiple sclerosis. BMC Neurol. 2016;16:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Perron H, Lang A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin Rev Allergy Immunol. 2010;39:51‐61. [DOI] [PubMed] [Google Scholar]
- 147. Tai AK, O'Reilly EJ, Alroy KA, et al. Human endogenous retrovirus‐K18 Env as a risk factor in multiple sclerosis. Mult Scler. 2008;14:1175‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]