

Abstract
Glycoproteins in animal cells contain a variety of glycan structures that are added co‐ and/or posttranslationally to proteins. Of over 20 different types of sugar–amino acid linkages known, the two major types are N‐glycans (Asn‐linked) and O‐glycans (Ser/Thr‐linked). An abnormal mucin‐type O‐glycan whose expression is associated with cancer and several human disorders is the Tn antigen. It has a relatively simple structure composed of N‐acetyl‐d‐galactosamine with a glycosidic α linkage to serine/threonine residues in glycoproteins (GalNAcα1‐O‐Ser/Thr), and was one of the first glycoconjugates to be chemically synthesized. The Tn antigen is normally modified by a specific galactosyltransferase (T‐synthase) in the Golgi apparatus of cells. Expression of active T‐synthase is uniquely dependent on the molecular chaperone Cosmc, which is encoded by a gene on the X chromosome. Expression of the Tn antigen can arise as a consequence of mutations in the genes for T‐synthase or Cosmc, or genes affecting other steps of O‐glycosylation pathways. Because of the association of the Tn antigen with disease, there is much interest in the development of Tn‐based vaccines and other therapeutic approaches based on Tn expression.
Keywords: antigens, carbohydrates, chaperone proteins, mucins, transferases
An abnormal sugar: The expression of the abnormal O‐glycan called Tn antigen (see structure) in animal glycoproteins typically represents a disease condition. This Review discusses a broad range of chemical and biological studies on the Tn antigen that could lead to new diagnostics and therapeutics.
1. Introduction
1.1. Definition and History of the Tn Antigen
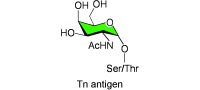
The discovery of the Tn antigen was a milestone in the history of the glycosciences, and its unusual expression was among the first to be associated with a human disorder. The Tn syndrome or Tn polyagglutinability syndrome was first described in 1957 by Moreau et al.1 They observed an unusual antigen on the erythrocytes of a patient with a rare, acquired hemolytic anemia that was associated with the polyagglutinability of erythrocytes in the cold and independent of the ABO(H) blood‐group status.1 It had been shown earlier in 1925 by Huebner2 and also in 1927–1928 by Thomsen3–5 that stored erythrocytes from normal donors can occasionally become agglutinated. However, that phenomenon was caused by bacterial glycosidases (neuraminidases) that uncovered the carbohydrate T antigen (Galβ1‐3GalNAcα1‐O‐Ser/Thr; Scheme 1), also termed the Thomsen–Friedenreich or TF antigen. This observation itself was seminal in the field, since it led to the discovery of neuraminidase and of the bacterium Vibrio cholera as a source for the enzyme. The exposed T antigen could be bound by preexisting antibodies, as defined in 1930 by Friedenreich.6 By contrast, the Tn syndrome was characterized by the immediate agglutination in the cold of freshly collected blood samples. This agglutination is now thought to result from the presence of circulating cold agglutinins of the immunoglobulin M type. The Tn antigen was subsequently shown to be a truncated form of a major type of glycosylation in animal glycoproteins with the simple structure GalNAcα1‐O‐Ser/Thr (Scheme 1). The historical naming of the Tn antigen derived from the observation by Moreau et al. that it was similar yet distinct from the T antigen; hence, the designation “T antigen nouvelle” or Tn antigen.1, 7 In the “cluster of differentiation” nomenclature, the Tn antigen is designated CD175, and its sialylated version (sialyl Tn) CD175s. The disaccharide T antigen is termed CD176.8–11
Scheme 1.

Structures of the Tn antigen and O‐glycan derivatives of the Tn antigen. The Tn antigen is a precursor for many types of O‐glycans, which are designated as core 1, core 2, and core 3 structures. These structures may be further modified, as shown in Scheme 3.
Dahr et al. reported in 1974 that the Tn antigen occurs in glycoproteins of human erythrocytes as a cryptic GalNAc‐containing antigen.12 The structure of the Tn antigen was then defined in 1975 by Dahr et al., who used gas–liquid chromatography to show the presence of Ser/Thr‐linked GalNAc in tryptic glycopeptides of erythrocytes from Tn‐syndrome patients.13 They also showed that the treatment of glycoproteins with low concentrations of NaOH in the presence of sodium borohydride (NaBH4) caused the so‐called β‐elimination reaction14, 15 and released N‐acetylgalactosaminitol from many different erythrocyte membrane glycoproteins. The chemical structure of the T antigen as the disaccharide Galβ1‐3GalNAcα1‐O‐Ser/Thr had been elucidated earlier in 1960 by Klenk and Uhlenbruck.16 Interestingly, the disaccharide Galβ1‐3GalNAc determinant was first demonstrated in studies on human brain gangliosides.17–19 For example, GM1, asialoGM1, and the nonsialylated GA1 all contain terminal Galβ1‐3GalNAcβ1‐R; however, GalNAc is β‐ rather than α‐linked in this case, and thus distinguishable from the T antigen.
1.2. Chemical Synthesis of the Tn Antigen and Related Structures
The chemical synthesis of both Tn and T antigens linked to l‐serine and l‐threonine was first described by Kaifu and Osawa in 1977,20 and subsequently by several other groups, including Lemieux and co‐workers21 and Paulsen and Hölck.22 Each group took a different synthetic approach. An elegant strategy for Tn‐antigen synthesis was devised by Paulsen and Hölck, who pioneered the use of a nonparticipating azido group at C2 of the GalNAc donor. Thus, 3,4,6‐tri‐O‐acetyl‐2‐azido‐2‐deoxy‐β‐d‐galactopyranosyl chloride underwent a Koenigs–Knorr type of reaction with N‐benzyloxycarbonyl derivatives of serine or threonine in the presence of silver carbonate and silver perchlorate as promoters to form the α anomer stereoselectively (Scheme 2 a).22 The C2 azido group was then converted through reductive acetylation into the naturally occurring acetamide group, and the protecting esters were cleaved to generate GalNAcα1‐O‐Ser and GalNAcα1‐O‐Thr, respectively. Similar reactions with the Fmoc derivatives of Ser and Thr were described later by Kunz and Birnbach.23 The use of the C2‐azido halo donor promotes a highly selective α‐glycosylation reaction. To generate the disaccharide T antigen, the azido group of the first product in Scheme 2 a was reduced, and the sugar was selectively O‐deacetylated and benzylidenated to give a derivative that reacted with tetra‐O‐acetyl‐α‐d‐galactosylpyranosyl bromide to yield the disaccharide. Subsequent removal of the protecting groups provided the N‐acetylated Gal‐GalNAc‐Ser/Thr building block.
Scheme 2.

Chemical synthesis of Tn antigen by α glycosylation of Ser or Thr building blocks. For details on the synthetic strategies shown, see references and the text. In (a), an azidosugar halogen donor is used,22, 32–40, 334, 335 whereas in (b), an azidosugar thiophenyl donor is used.24 Bn=benzyl, Fmoc=9‐fluorenylmethoxycarbonyl, Tf2O=trifluoromethanesulfonic anhydride.
Another elegant synthesis of the Tn antigen (Scheme 2 b) involved the use of the acetylated 2‐azido‐2‐deoxy thiophenyl donor and the Ph2SO/Tf2O promoter system, which promoted efficient stereoselective glycosylation of Fmoc‐Thr(OH)‐OBn (or Fmoc‐Ser(OH)‐OBn) to specifically generate the α‐glycoside.24 Modern syntheses of glycopeptides containing the Tn antigen have been conducted by various strategies.24–40 An interesting solid‐phase synthetic approach in which glycosyl trichloroacetimidates are used to stereoselectively modify the Ser or Thr hydroxy group of a resin‐bound peptide was developed for peptides with a single or multiple Tn antigens.41 Many techniques for glycopeptide synthesis now utilize N α‐Fmoc‐Thr‐(AcO3‐α‐d‐GalNAc) or the corresponding Ser derivative, which are commercially available. Furthermore, glycopeptides with the Tn antigen may also be chemoenzymatically generated by using synthetic peptide acceptors and glycosyltransferases that transfer GalNAc to Ser/Thr residues.42 These strategies have been important in the development of potential vaccines against the Tn antigen, as discussed in Section 6.1.
1.3. Detection of the Tn Antigen
The Tn antigen does not occur in abundance in normal cells and tissues of adult animals, and there is no evidence that it is an oncofetal antigen. Over the years, a variety of approaches have been used to detect the Tn antigen. These approaches, each of which has advantages and limitations, may be broadly subdivided into three categories: chemical, lectin‐based, and antibody‐based. The direct chemical approach typically involves the β‐elimination strategy,14, 15 in which N‐acetylgalactosaminitol is generated by alkaline degradation of glycopeptides in the presence of NaBH4 with the simultaneous modification of the linking amino acids (Ser to 2‐aminoacrylic acid or dehydroalanine and Thr to 2‐aminocrotonic acid). The identification of N‐acetylgalactosaminitol by gas chromatography following acetylation, often in association with the detection of modified Ser or Thr amino acids, is taken as chemical evidence for the presence of Tn antigen on glycopeptides or glycoproteins. Although such approaches are appropriate when sufficient amounts of material are available, it is difficult to perform this type of analysis on the small amounts present in cells or tissues. A variation on this strategy has been to use galactose oxidase without prior neuraminidase treatment to oxidize potential nonreducing terminal GalNAc residues in the Tn antigen at the C6 hydroxy group, and then to reduce with radiolabeled NaB[3H]4.43 The presence of radiolabeled GalNAc is then indicative of the Tn antigen. Of course, radiolabeled Gal can also be produced if the nonsialylated T antigen is present.
However, historically the dominant strategies for detecting Tn antigen are lectin‐ and antibody‐based. A huge number of different GalNAc‐binding lectins from plants and animals have been used to explore expression of the Tn antigen. The lectins used include the plant lectins Dolichos biflorus agglutinin (DBA),44 Marrubium candidissimum agglutinin,45 Maclura pomifera lectin (MPL),46 Salvia horminum agglutinin (SHA),47 Salvia sclarea agglutinin (SSA),48 Salvia bogotensis lectin,49 Artocarpus integrifolia lectin (jacalin),50 Bauhinia purpurea agglutinin (BPA),51, 52 the B4 isolectin of Vicia villosa agglutinin (VVA‐B4),53 Soja hispida lectin,54 Moluccella laevis lectin (MLL),55 and the snail‐derived Helix pomatia agglutinin (HPA).48, 56, 57 The absence of binding of some lectins has also been used to explore the potential expression of the Tn/STn antigens (STn antigen=sialyl‐Tn antigen, Neu5Acα2‐6GalNAcα1‐O‐Ser/Thr). For example, Springer et al. showed that erythrocytes from patients with Tn syndrome had diminished binding to the lectins Vicia graminea and Arachis hypogoea (peanut agglutinin, PNA), which bind to sialic acid and the T antigen,58 respectively. Furthermore, Tn‐expressing cell lines, such as Jurkat leukemic cells and human colon carcinoma LSC cells,59, 60 bind well to HPA, but do not bind well to PNA even after desialylation;61 this behavior is consistent with the expression of Tn antigen in those cells. Two of the most common plant lectins used to explore Tn expression are VVA‐B4 and HPA. For example, VVA‐B4 has been used to explore the expression of Tn antigen in many different cancer cells, including those of the pancreas, breast, lung, and prostate.62, 63 HPA has been widely used since the early 1980s to study Tn‐antigen expression. For example, the clonality of the expression of Tn antigen in blood cells was established by Vainchenker et al.,64 who used fluorescently labeled HPA to show expression of the Tn antigen by erythroid and granulocytic cell colonies derived from a patient with Tn syndrome. Tn expression in platelets of a patient with Tn syndrome was also studied by using binding by HPA along with galactose oxidase/NaB[3H]4 reduction to identify Tn expression on platelet glycoprotein GPIb.43 However, the use of HPA to identify Tn‐antigen expression is problematic, because HPA also binds to the Cad antigen NeuAcα2‐3(GalNAcβ1‐4)Galβ1‐R, blood group A antigen Fucα1‐2(GalNAcα1‐3)Galβ1‐R, and Forssman glycolipid.65, 66 In general, most of the plant and animal lectins that bind GalNAc cross‐react with nonreducing and terminal GalNAc residues that are either α‐ or β‐linked, as seen for VVA‐B4, which also binds Cad (Sda) antigen.67 Thus, studies in which lectins are used to define the expression of the Tn antigen should be carried out cautiously, and evidence should be supplemented by corroborating chemical, immunological, and/or genetic/molecular data.
One of the earliest immunological approaches to the detection of Tn antigen was based on anti‐Tn antisera,68, 69 which were generated naturally upon its recognition as an antigen. The Springer research group used human anti‐Tn antibodies occurring in people as a reagent for Tn identification,70 following their observations that small levels of anti‐Tn antibody occur in the sera of most people.6, 7, 71 The formation of circulating anti‐Tn antibodies was shown to be partly due to exposure to Enterobacteriaceae expressing the Tn antigen.72 Springer and Tegtmeyer showed that sera from White Leghorn chicks grown in germ‐free environments did not contain anti‐Tn antibodies, whereas chicks grown in normal hatcheries did express such antibodies. Moreover, the feeding of germ‐free chicks with bacteria expressing the Tn antigen elicited the formation of anti‐Tn antibodies.72 The inhalation or ingestion of bacteria expressing the Tn antigen by human infants or adults also caused the expression of circulating anti‐Tn antibodies.73
Springer et al.74, 75 defined the immunogenicity of the Tn antigen as part of their studies on human blood group MN antigens and discovered that T and Tn antigens are expressed as carcinoma‐associated antigens in human tumor cells. In 1985, they reported the development, for the first time, of murine monoclonal antibodies (mAbs) to the Tn antigen.70 Shortly thereafter, the first murine mAbs to sialyl‐Tn antigen were generated by Hakomori and co‐workers.76 Through the use of these mAbs it was shown that both Tn and sialyl‐Tn antigens are coexpressed on erythrocytes of patients with Tn syndrome but are not present to significant extents on erythrocytes from control donors.77 Hakomori and co‐workers also developed several mAbs to the Tn antigen (NCC‐LU‐35 and NCC‐LU‐81) by immunizing mice with lung‐carcinoma Lu65 cells and screening histologically for hybridomas that made mAbs to tumor‐tissue specimens.78 A problem with the use of different mAbs to the Tn antigen has been the specificity of the antibodies. Some mAbs to the Tn antigen clearly cross‐react with other GalNAc‐containing glycans, such as blood group A antigen.78 Examples of the use of anti‐Tn monoclonal antibodies are in references 61, 78–85. Some of these mAbs appear to be specific and do not cross‐react with blood group A antigen.81
One of the most useful mouse anti‐Tn mAbs is an IgM (CA3638, clone 12A8‐C7‐F5) originally developed by Springer and co‐workers and now used widely in immunohistochemistry and flow cytometry to detect the Tn antigen.79 Following the death of Dr. Springer, his assistant Herta Tegtmeyer shipped all stocks of purified mAbs, including the above‐mentioned IgM to the Tn antigen, to the Cummings laboratory. Unfortunately, all hybridomas from the Springer laboratory were lost earlier; however, samples of the mAb to the Tn antigen can be obtained upon request from the Cummings research group and have been used by others, for example, in studies on Tn‐antigen expression in Drosophila.86, 87 The most useful mouse anti‐STn mAb is an immunoglobulin G1 (IgG1; clone HB‐STn1) commercially obtainable from several sources, including DakoCytomation. Additionally, mAbs to the Tn antigen (CD175) and sialyl‐Tn antigen (CD175s) are commercially available from several sources.
2. Molecular Mechanisms for Expression of the Tn Antigen
2.1. Biosynthesis of the Tn Antigen and O‐Glycosylation Pathways for Mucin‐Type Glycoproteins
The Tn antigen on glycoproteins is synthesized by a family of enzymes, polypeptide α‐N‐acetylgalactosaminyltransferases (ppGalNAcTs), which transfer a GalNAc unit from the donor UDP‐GalNAc to a Ser or Thr residue of a protein (Scheme 3).88 This process the first step in the biosynthesis of all mucin‐type O‐glycans. It is estimated that 24 unique ppGalNAcT genes occur in the human genome and 18 in that of mice.89–91 The human genes are located on chromosomes 1–4, 7–9, 12, and 18. The ppGalNAcTs are transmembrane proteins consisting of 600–800 amino acids and are thus larger than most other glycosyltransferases. The transferase activity is located in the N‐terminal domain, and the C‐terminal domain contains an R‐type lectin domain.92 Although all ppGalNAcTs (EC 2.4.1.41) seem to catalyze the same general reaction, expression of ppGalNAcT isoforms or isozymes is tissue‐specific in adult mammals, and the ppGalNAcTs display distinct, yet overlapping substrate preferences.93 The activity of all ppGalNAcTs (except for T10) appears to be enhanced if there is a Pro residue at the +3 position relative to Ser/Thr. Pro residues at the −3, −1, and +1 positions enhance activity to various degrees depending on the ppGalNAcT isoforms.94–96 The C‐terminal R‐type lectin domain has some homology to the lectin domain of ricin, which recognizes and binds carbohydrates. It was suggested that the catalytic and the lectin domains of ppGalNAcTs function in concert to direct the selection of glycosylation sites. This proposal is in agreement with a hierarchical addition of core GalNAc residues to the protein.90 Mucin 1 (MUC1), for example, is a protein composed of 1000 amino acids whose molecular mass is increased to over 2 million Da by the addition of hundreds of O‐glycans. The extreme efficiency of its O‐glycosylation is not only due to the high activity of ppGalNAcTs and their wide distribution along the Golgi apparatus of epithelial cells, but also to their lectin domain. Binding to previously formed glycans by their R‐type lectin domain enables these enzymes to slide along the protein and thus more efficiently attach GalNAc to one Ser/Thr residue after another.97
Scheme 3.

Biosynthesis of the Tn antigen and different O‐glycan core structures derived from the Tn precursor.
The ppGalNAcTs are localized to the Golgi apparatus as shown in Hela cells98 and are hence presumed to act on folded proteins that have exited correctly from the endoplasmic reticulum (ER). Following its formation in the Golgi apparatus, the Tn antigen can serve as an acceptor for at least three other Golgi glycosyltransferases: 1) core 1 β1,3‐galactosyltransferase (core 1 β3GalT or T‐synthase), which synthesizes the core 1 structure or T antigen, 2) core 3 β1,3‐N‐acetylglucosaminyltransferase (core 3 β3GlcNAcT), which synthesizes the core 3 structure, and 3) the sialyltransferase ST6GalNAc‐I, which synthesizes the sialyl‐Tn antigen (Scheme 3). The most common modification of the Tn antigen is the formation of the core 1 disaccharide by the action of the T‐synthase.99, 100 This key enzyme in mucin‐type O‐glycan biosynthesis is expressed by all cells in humans and mice (Scheme 3). Core 1 glycans are usually either sialylated by ST3Gal‐I to yield sialyl‐T antigen,101 which can then be further sialylated by ST6GalNAc‐III or IV to yield disialyl‐T antigen (DiSia3,6Core 1),102 or alternatively, the core 1 O‐glycan disaccharide can be modified by ST6GalNAc‐II to the 2,6‐sialylated core 1 O‐glycan (Sia6Core 1),103 or elongated to form extended core 1 O‐glycans, or branched by the core 2 β6‐GlcNAcT104 to form core 2 O‐glycans. The core 3 structure GlcNAcβ1‐3GalNAcα1‐Ser/Thr is mostly found in the gastrointestinal tract and is usually elongated to form complex core 3 O‐glycans.105 ST6GalNAc‐I is expressed at high levels in the bovine submaxillary gland, but is hardly found in other tissues.106 Therefore, STn is not detectable in normal tissues other than the submaxillary gland of cows and sheep. If formed, STn is a terminal structure that is not an acceptor for other glycosyltransferases.
2.2. The T‐synthase Utilizes the Tn Antigen To Synthesize the T Antigen
The major enzyme that modifies the Tn antigen in normal tissues is the UDP‐Gal:GalNAcα1‐O‐Ser/Thr glycopeptide β3‐galactosyltransferase (core 1 β3GalT, T‐synthase, C1GALT1, EC2.4.1,122). The T‐synthase transfers Gal from the donor UDP‐Gal in the presence of a divalent cation to GalNAcα1‐O‐Ser/Thr in glycoproteins through the formation of a β‐glycosidic bond to form the core 1 structure, Galβ1‐3GalNAcα1‐O‐Ser/Thr or T antigen. Thus, the T‐synthase is an inverting glycosyltransferase, since it forms a β linkage from the α‐linked Gal residue in the donor UDP‐Gal.107 The enzyme was assayed originally in BHK cell homogenates,108 in crude preparations of pig and rat colonic mucosa,109 and in partly purified form from rat liver.110 The enzyme was purified to homogeneity from rat liver in 2002.100 The purified enzyme is an 84/86 kDa disulfide‐bound homodimer, but the 42/43 kDa monomer was observed and was active as well. The reason for the two homodimer forms, which differ in their molecular mass, is unclear. On the basis of the N‐terminal sequence of the rat T‐synthase, the cDNA for the human T‐synthase was cloned.99 It encodes a 363 amino acid (aa) type‐II transmembrane protein. Northern‐blot analysis revealed that there are two different transcripts of human T‐synthase with sizes of 2.0 and 7.0 kb. The transcript of 2.0 kb is probably the mature and active form, as only the 2.0 kb transcript is found in rat testes. Whether the transcript of larger size is just an incomplete splicing form or has its own function is unclear. The orthologues from various species, including chimpanzee, cow, dog, rat, mouse, bird, frog, and zebra fish, as well as the invertebrates Drosophila and C. elegans, were either cloned or identified.111 From their aligned protein sequences, a high homology, especially between the mammalian proteins, was observed. T‐synthase is unique in that it shares minimal homology with conserved motifs of other β3‐galactosyltransferases. Remarkably, there is only a single gene for T‐synthase in humans and other mammals. The human gene (C1GALT1) resides on chromosome 7p14‐p13 and consists of three exons, of which the second exon is the largest. Interestingly, mammalian T‐synthase is not N‐glycosylated, unlike most other glycosyltransferases, which contain N‐glycosylation sites in their primary amino acid sequences; by contrast, the T‐synthases from C. elegans 111 and Drosophila 99, 112 contain N‐glycosylation sites.
Even though there is only a single functional gene for T‐synthase in humans, a BLAST homology search revealed four highly homological DNA sequences related to C1GALT1 on chromosomes 5, 12, 8, and 15. We termed these sequences human C1GALT1 pseudogenes 1, 2, 3, and 4, respectively (pC1GALT‐1, ‐2, ‐3, and ‐4; see the Supporting Information for sequence alignments). In contrast to the functional gene, all pC1GALTs consist of a single “exon” with many missense mutations and DNA‐fragment deletions. The sequence of pC1GALT‐1 on chromosome 5 is the most conserved; it exhibits 93 % identity to the cDNA of C1GALT1. pC1GALT‐2 on chromosome 12 is 91 % identical to the cDNA of C1GALT1. pC1GALT‐3 on chromosome 8 is only 80 % identical to the cDNA of C1GALT1. The first 130 bases of pC1GALT‐3 do not match exon I (220 bp) of C1GALT1, which accounts for the major overall differences between pC1GALT‐3 and C1GALT1. pC1GALT‐4 on chromosome 15 is the least conserved with 71 % homology to the functional gene. It lacks the first approximately 100 bp at the 5′ end of exon I and 200 bp in the 3′ portion of exon II. The mutations and deletions in the four pseudogenes result in many stop codons and open‐reading‐frame (ORF) shifts, which lead to non‐ORF DNA sequences. Neither reverse nor complementary sequences of the pseudogenes contain any ORF, which indicates that they are not other genes or parts of other genes and further suggests that they are pseudogenes. Searches of human expressed sequence tags (ESTs) in the database gave three hits that perfectly matched pC1GALT‐1 and two matches to pC1GALT‐2. These matches indicate that pC1GALT‐1 and pC1GALT‐2 on chromosomes 5 and 12, respectively, are probably transcriptionally active in certain tissues. Given the similarities of the four pseudogenes to human C1GALT1, it is likely that pC1GALT‐3 and 4 on chromosome 8 and 15 are evolutionarily the oldest, whereas pC1GALT‐1 on chromosome 5 is the most recent. The “single‐exon” nature of these pseudogenes suggests that they resulted from reverse transcription of the mRNA of human C1GALT1 and subsequent integration into the human genome. The existence of four nonfunctional genes may not only testify to the evolution of human C1GALT1, but may also be important for the regulation of human C1GALT1 expression, particularly as some of these pseudogenes appear to be transcriptionally active.
2.3. Other Glycosyltransferases That Utilize the Tn Antigen as an Acceptor
Besides the T‐synthase, there are two other well‐known glycosyltransferases that also use the Tn antigen as an acceptor: ST6GalNAc‐I and the core 3 β3‐GlcNAcT (Scheme 3). CMP‐NeuAc:GalNAcα1‐O‐Ser/Thr glycopeptide α2,6‐sialyltransferase‐I (ST6GalNAc‐I, EC2.4.99.3)113 transfers a sialic acid (N‐acetyl‐5‐neuraminic acid, Neu5Ac) from CMP‐Neu5Ac (CMP=cytidine monophosphate) to the 6‐position of GalNAcα1‐O‐Ser/Thr to generate the STn antigen Neu5Acα2‐6GalNAcα1‐O‐Ser/Thr. Human ST6GalNAc‐I is encoded by a gene on 17q25.1. It belongs to the human ST6GalNAc family,114 which has six members that differ in their acceptor–substrate specificity. ST6GalNAc‐I can act on Tn antigen, T antigen, and α2,3 sialyl‐T antigen (Neu5Acα2‐3Galβ1‐3GalNAcα1‐O‐Ser/Thr).113 ST6GalNAc‐II mainly acts on the core 1 O‐glycan to form the α2,6‐sialylated T antigen (Sia6Core 1; Schemes 1 and 3).115 Although one study116 showed that both ST6GalNAc‐I and II could be sialyl‐Tn synthases, others115, 117 have demonstrated that ST6GalNAc‐I is the major enzyme responsible for sialyl‐Tn formation in human tumors. Interestingly, some studies showed that ST6GalNAc‐II transcript, but not ST6GalNAc‐I, was upregulated in immortalized B cells from patients with IgA nephropathy.118, 119 Clearly, much more needs to be done to clarify the role(s) of these enzymes in STn formation.
UDP‐GlcNAc:GalNAcα1‐O‐Ser/Thr glycopeptide β3‐N‐acetylglucosaminyltransferase (core 3 β3‐GlcNAcT, C3GnT, β3GnT6, EC2.4.1.146) catalyzes the transfer of GlcNAc from UDP‐GlcNAc to GalNAcα1‐O‐Ser/Thr (Tn antigen) to form the core 3 structure (GlcNAcβ1‐3GalNAcα1‐O‐Ser/Thr).105 Human C3GnT is located on chromosome 11q13.4. C3GnT expression is restricted to mucus‐secreting tissues. The level of C3GnT‐transcript expression in various human tissues, as measured by real‐time PCR, revealed that it was highest in the stomach, followed by the colon and small intestine. O‐Glycans, including both core 3 and core 1, are the primary components of the intestinal mucus layer that covers the gastrointestinal epithelium. The mucus layer is a dense, carbohydrate‐rich matrix that consists primarily of mucins containing multiple serine and threonine residues modified by O‐glycans, which account for 80–90 % of the mucin mass. The mucus layer and epithelial cells form an intestinal barrier that protects epithelial and intestinal mucosal immune cells from potentially harmful luminal microflora and food components.
3. Requirement of the Molecular Chaperone Cosmc for the Generation of Active T‐Synthase
3.1. Discovery of Cosmc
Core 1 β3 galactosyltranferase‐specific molecular chaperone, abbreviated Cosmc, is required for the formation of active T‐synthase in vivo.60 Human Cosmc is encoded by a single exon gene on Xq24.60 In contrast to T‐synthase, which is found in both vertebrates and invertebrates, Cosmc orthologues occur only in vertebrates. Cosmc was first partially copurified with T‐synthase from rat liver and displayed an apparent molecular mass in SDS‐PAGE of 36–38 kDa, which is very similar to the size of T‐synthase. On the basis of its protein sequence, the 1.5 kb cDNA was cloned, and it could be predicted that the ORF encoded a 318 amino acid type‐II transmembrane protein. It has now been demonstrated that Cosmc localizes to the endoplasmic reticulum (ER), where it functions as a chaperone in the folding of T‐synthase,83 as discussed in Section 3.3.
As mentioned in Section 1.3, the human T‐leukemic Jurkat cell line expresses the Tn antigen. Jurkat cells are deficient in T‐synthase activity.120 They therefore generate primarily the Tn antigen and lack larger mucin‐type O‐glycans. The T‐synthase gene and transcript are normal in Jurkat cells, but the Cosmc cDNA is mutated and contains a T insertion at position 473, which results in an ORF shift and a premature stop codon.60, 61 The introduction of functional, wild‐type Cosmc into Jurkat cells not only restores T‐synthase activity, but also corrects the O‐glycan structure on the cell‐surface glycoproteins.60, 61
We recently engineered mice with complete or partial deletion of Cosmc.121 Similar to the human gene, mouse Cosmc is a single‐exon gene on the X chromosome (Xc3). Animals with complete or major loss of Cosmc died as embryos by day E10.5–E12.5 of embryonic development with robust expression of the Tn antigen in virtually every identifiable cell. Mice with partial deletion of Cosmc exhibited variable phenotypes dependent on the extent of the deletion and the sex, whereby male mice survived much more rarely than female mice owing to the X‐chromosomal location of Cosmc. The observation of Tn and STn antigens in many tissues, such as those of the stomach, intestine, kidney, liver, spleen, lung, and pancreas, further confirmed that Cosmc is required for T‐synthase function.121
Cosmc orthologues with high homology to human Cosmc were found in many vertebrates, including chimpanzee, cow, mouse, rat, dog, bird, frog, and zebra fish. Unlike other mammalian Cosmc proteins, which comprise 318 amino acids, rodent Cosmc contains 316 amino acids. It has more than 95 % sequence identity to the human protein, but lacks the two amino acids at positions 33 and 34 (human sequence order). These two amino acids are located at the beginning of the luminal C‐terminal domain that accounts for the function of Cosmc, but appear not to influence its localization in the endoplasmic reticulum. Indeed, mouse Cosmc functions as well as human Cosmc in promoting both human and mouse T‐synthase activity.
In contrast to vertebrates, invertebrates, such as C. elegans or Drosophila, have no Cosmc orthologues. However, T‐synthase is expressed in both vertebrates and invertebrates. Whereas vertebrate T‐synthase lacks conserved N‐glycosylation sites, invertebrate T‐synthases have multiple N‐glycosylation sites. It may therefore be that in vertebrates, Cosmc compensates for the failure of non‐N‐glycosylated T‐synthase to be recognized and folded by the calnexin/calreticulin system in the endoplasmic reticulum (ER). In contrast, invertebrate T‐synthases with their multiple N‐glycosylation sites may not require Cosmc for folding control and formation of the active protein.111
Human Cosmc shares approximately 20 % sequence identity with T‐synthase,60 which contributed to its original misidentification as another T‐synthase.122 This mistake was subsequently corrected.123 Homologies mostly concern the luminal domain and the six cysteine residues, which suggests that Cosmc may have evolved from T‐synthase or that the two proteins had a common origin. More indirect evidence comes from the four pseudogenes of human T‐synthase on chromosomes 5, 8, 12, and 15, all of which, like Cosmc, are found as “single‐exon” genes. Therefore, it appears that Cosmc may have evolved from T‐synthase through the pseudogenes.
3.2. Other Factors That Could Lead to Expression of the Tn Antigen
It is now clear that two major mechanisms for expression of the Tn antigen result from genetic changes that lead to decreased expression of either functional Cosmc and/or T‐synthase. However, there are other possible factors that could influence the O‐glycosylation pathways described above and lead to expression of the Tn antigen. The decreased expression of the C3GnT, which synthesizes the core 3 structure in the gastrointestinal tract, can lead to Tn/STn expression, and loss of C3GnT is associated with colon cancer.124–126 Chinese hamster ovary (CHO) cells lacking the transporter for UDP‐Gal, the so‐called Lec8 cell line,127, 128 lack galactose residues on both N‐ and O‐glycans129 and consequently synthesize the Tn antigen. Krieger and co‐workers isolated a CHO cell line which lacks the UDP‐Glc/GlcNAc 4‐epimerase. These cells, termed ldlD cells, are thus unable to synthesize UDP‐Gal or UDP‐GalNAc unless supplied exogenously with Gal/GalNAc or glycoproteins containing Gal/GalNAc.130 In the absence of added galactose, ldlD cells synthesize glycoproteins that lack galactose and thus express truncated N‐glycans as well as Tn‐antigen O‐glycans. Therefore, both the nucleotide sugar UDP‐Gal and the UDP‐Gal transporter are essential for the synthesis of normal O‐glycans that lack the Tn antigen. Defects in the biosynthetic and transport pathways can lead to abnormal Tn expression.
Furthermore, the overexpression of ST6GalNAc‐I can lead to production of the sialyl‐Tn antigen, which is commonly found in colon and breast tumors,131 as a result of competition with the endogenous T‐synthase.117–119, 132, 133 Finally, it is possible that normal O‐glycans could be degraded to expose the Tn antigen. For example, the T antigen can be converted into the Tn antigen by treatment with bovine testicular β‐d‐galactosidase.75 Thus, in cells in which the Tn and/or STn antigens are expressed, it is important to define the pathways for their expression both biochemically and genetically.
3.3. Mechanism of Action of Cosmc
During the biosynthesis of the T‐synthase in the ER, Cosmc is predicted to bind to newly synthesized T‐synthase and prevent its aggregation and subsequent degradation in the ER‐associated degradation (ERAD) proteasome pathway (Figure 1).60, 83, 134, 135 In cells lacking Cosmc, T‐synthase is still synthesized, but it is truncated at its N terminus, is misfolded, and can be bound in the ER to Bip (binding protein: the major chaperone in the ER, also called 78 kDa glucose‐regulated protein, Grp78) and perhaps other chaperones that bind to misfolded proteins. The misfolded T‐synthase is then retrotranslocated to the cytosol, where it is polyubiquitinated and subsequently degraded by the proteasome. Without Cosmc, the T‐synthase appears within large disulfide‐linked oligomeric complexes that are inactive and trapped within the lumen of the ER. The process of the separation and retrotranslocation of the inactive T‐synthase from the ER to the cytoplasm, and then ubiquitination of the inactive T‐synthase for its targeted destruction, is poorly understood. The treatment of such cells with a proteasome inhibitor leads to the accumulation of inactive T‐synthase protein aggregates. Interestingly, this inactive T‐synthase can be co‐immunoprecipitated with Grp78, an ER chaperone that binds exposed hydrophobic regions in misfolded proteins. However, the binding of T‐synthase to ER chaperones such as Grp78 does not lead to correct folding, and the presence of the functional Cosmc chaperone remains essential.
Figure 1.

Molecular basis for the production of normal O‐glycans and for the expression of Tn and sialyl‐Tn antigen in cells lacking functional T‐synthase owing to loss of function of Cosmc.61 Left: Cosmc is expressed in the endoplasmic reticulum, where it binds to newly synthesized T‐synthase and assists in folding of the enzyme and its acquisition of activity. The dimeric T‐synthase can then move to the Golgi apparatus, where it functions to add galactose residues from the donor UDP‐Gal to the Tn‐antigen precursor of O‐glycans. Normal O‐glycans may be sialylated to generate the sialylated core 1 O‐glycans, or may be elongated to extended core 1 structures or core 2 O‐glycans (see Scheme 3). Right: In the absence of functional Cosmc, the newly synthesized T‐synthase in the ER is misfolded, retrotranslocated to the cytoplasm, ubiquitinated, and degraded in the 26S proteasome. The lack of functional T‐synthase in the Golgi apparatus leads to expression of the Tn and sialyl‐Tn antigens on the cell surface and secreted glycoproteins.61, 83, 134
In recent studies, it was shown that recombinant T‐synthase, when denatured by heat or treatment with guanidinium hydrochloride, can reacquire activity in vitro when incubated with recombinant Cosmc in the absence of other protein factors. The binding or hydrolysis of adenosine 5′‐triphosphate (ATP) does not affect this renaturation process.134 This finding is interesting, since Cosmc was shown to bind ATP,83 and suggests that ATP may have some role in vivo in Cosmc interactions or functions. Clearly, there is much to be learned about the biological role of Cosmc and the need for a specific chaperone for the T‐synthase.
4. The Functions of Normal Mucin‐Type O‐Glycans Derived from Tn‐Antigen Precursors
4.1. O‐Glycans in Leukocyte Trafficking
It is not surprising that expression of the Tn antigen is a pathological indicator, since the Tn antigen is a precursor to many important complex O‐glycans. These complex O‐glycans usually have extended structures based on core 1 or core 2 O‐glycans and are found in numerous cell‐surface glycoproteins. Glycoproteins containing core 1 or 2 O‐glycans include the low‐density‐lipoprotein (LDL) receptor,136–138 the transferrin receptor,129, 139–141 glycophorin A,142–144 podoplanin,145–147 PSGL‐1,30, 148–150 CD34,151 endoglycan,152 zona pellucida glycoprotein ZP3,153, 154 CD43,155–157 CD45,158–161 the TRAIL receptor family,162 the neurotrophin receptor,163, 164 the MUC family of mucins (both membrane‐bound and secreted),165–169 platelet glycoprotein GPIbα,170 and platelet integrin GPIIb/IIIa (αIIbβ3).171 Such core 1 and/or 2 O‐glycans are also found in many secreted and serum glycoproteins, including human IgA1172–177 and IgD,178–180 erythropoietin,181–184 von Willebrand factor (vWF),185 blood coagulation factor X,186 serum apoC‐III,187–189 human chorionic gonadotropin (HCG),190–193 interleukin‐6 (IL‐6),194, 195 Tamm–Horsfall glycoprotein/uromodulin,196–199 Gc macrophage activating factor (GcMAF, a naturally derived form of human vitamin D binding protein),200 human urinary thrombomodulin,201 and human milk bile‐salt‐stimulated lipase.202–205 Furthermore, O‐glycans are found in virtually all mucin secretions of epithelial cells and on the surface of all animal cells, including erythrocytes. Thus, it is reasonable to conclude that core 1 O‐glycans are expressed by all mammalian cells and can be found on many membrane and secreted glycoproteins. Alterations in the expression of the O‐glycans on many of the glycoproteins listed above are associated with molecular changes in the glycoprotein, and these molecular changes are often associated in turn with changes in cellular signaling, metabolism, or receptor function. Thus, expression of the Tn antigen on glycoproteins may lead to or reflect an altered function.
One of the best‐understood functions of the core 2 O‐glycans is selectin recognition and especially binding of the P‐selectin glycoprotein ligand 1 (PSGL‐1) to P‐, L‐, and E‐selectins.206–210 Human PSGL‐1 is a mucin with 16 repeating mucin motifs and contains sialyl Lewis x (SLex) antigen on the core 2 O‐glycans.150, 211 Such glycans are not common on PSGL‐1, and only the one at the extreme N terminus, along with adjacent sulfated tyrosine residues, serves as the key carbohydrate recognition ligand for P‐ and L‐selectins,30, 212 whereas E‐selectin binding is not dependent on sulfation. Thus, the loss of O‐glycan biosynthetic pathways as a result of the loss of T‐synthase or Cosmc would lead to the loss of functional PSGL‐1 ligands on leukocytes. In addition, extended core 1 O‐glycans are expressed by specialized endothelial cells within the high endothelial venules and these also contain sulfated GlcNAc residues within the SLex motif (6‐sulfo‐SLex).213, 214 A loss of fucosylation215 or 6‐O‐sulfate residues216, 217 leads to loss of functional recognition by L‐selectin on circulating leukocytes and compromises lymphocyte homing and recirculation. Thus, the pathways regulated by the T‐synthase in the synthesis of core 1, extended core 1, and core 2 O‐glycans are essential in leukocyte trafficking, inflammatory responses, and expression of the Tn antigen in blood cells and can be expected to have biological consequences in inflammation, thrombosis, and hemostasis.
4.2. O‐Glycans in Vascular Biology, Angiogenesis, and Lymphangiogenesis
The potential roles of O‐glycans in vascular formation, including angiogenesis and lymphangiogenesis, was not clear until it was shown that deletion of the T‐synthase gene in mice and expression of the Tn antigen caused the failure of both angiogenesis and lymphangiogenesis.124, 218, 219 More interestingly, a lack of T‐synthase in endothelial and hematopoietic cells causes a misconnection between blood and lymphatic vessels. This observation indicates the important role of O‐glycans in lymphatic‐vessel development (lymphangiogenesis).145 Although the precise mechanistic role of O‐glycans has yet to be defined, it was shown that deletion of the heavily O‐glycosylated glycoprotein podoplanin, which contains dozens of O‐glycans and lacks N‐glycans, phenocopies the phenotype of mice deficient in the gene for T‐synthase. Interestingly, partial loss of the T‐synthase in mice results in a phenotype of thrombocytopenia and kidney disease with distorted glomerular–tubular architecture and other renal lesions along with progressive proteinuria and premature death.220 The precise pathological consequences of Tn expression on angiogenesis and lymphangiogenesis are unknown; however, they may be dually caused by the loss of glycoprotein function through the truncation of O‐glycans as well as by recognition of the Tn antigen by host immune cells and other pathological cell–cell interactions, as discussed in Sections 5.2 and 6.2.
4.3. O‐Glycans in Mucins of Mucosal Tissue
A characteristic of mucosal epithelial cells is the production of mucins and other O‐glycosylated glycoproteins, whose expression is often altered in different cancers.131, 221–231 At least 21 different mucin genes have been identified (MUC1, 2, 3A, 3B, 4, 5AC, 5B, 6, 7, 8, 9, 11, 12, 13, 15, 16, 17, 18, 19, 20, and 21—note that MUC15 and MUC18 lack tandem repeats), and although they are not related in sequence, they share the features of having repeating motifs rich in Ser/Thr/Pro residues and containing large amounts of O‐glycans.228, 232 These mucins can contain 377 to more than 11 000 aa residues in a single polypeptide;228 the largest allele of porcine submaxillary mucin encodes a protein with 13 288 aa.233 Several mucins in body fluids, such as the epitope of CA125,234 have been studied as tumor markers. Cloning of the gene encoding the tumor marker CA125 showed that it is the MUC16 gene.235 Several mucins have been studied in regard to Tn‐ and/or STn‐antigen expression and cancer (Figure 2). MUC1 is highly expressed in many breast, ovarian, and other types of carcinomas236–238 and has been associated with expression of the Tn and STn antigens in breast, colorectal, and hepatocellular carcinomas and other neoplasms.239–248 Related studies have also shown expression of Tn/STn on MUC2, MUC4, and MUC6, as well as other mucins in different carcinomas.249–255 As discussed in Section 6.1, a number of research groups are attempting to use this information for the synthesis of mucin‐based glycopeptide vaccines to target tumors expressing those epitopes. Tumor colonic mucins, such as MUC‐1 and MUC‐2, carry other types of tumor‐associated carbohydrate antigens,221 including SLex[256] and sialyl Lewis a.257
Figure 2.
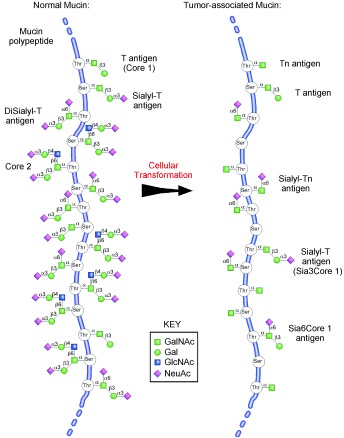
Mucins and the expression of Tn and sialyl‐Tn antigens. Typical mucins contain large numbers of Ser, Thr, and Pro residues in the backbone polypeptide and an abundance of GalNAcα1‐O‐Ser/Thr modifications. In normal mucins, the GalNAcα1‐O‐Ser/Thr residues are modified to contain different core glycans, as shown in Scheme 3. However, upon cellular transformation, the modification of GalNAcα1‐O‐Ser/Thr may be compromised, and cells may generate tumor‐associated mucins containing the tumor‐associated carbohydrate antigens (TACAs) Tn, sialyl‐Tn, and T. One mechanism for the expression of the Tn and sialyl‐Tn antigens on mucins is the loss of function of the chaperone Cosmc (Figure 1), but other factors could also lead to elevated expression of these TACAs, as discussed in the text.
In 1977, Newman and Uhlenbruck observed that bovine fat globule membrane glycoproteins were bound by both anti‐Tn serum and HPA48 and contained the T and Tn antigens. In contrast, human fat globule membrane glycoproteins were only bound by HPA. Bovine submaxillary mucin (BSM) also contains considerable amounts of both Tn and sialyl‐Tn antigens.258 Thus, bovine fat globule membrane glycoproteins and BSM are rare examples of glycoproteins in mammals in which the Tn antigen may occur naturally on a glycoprotein that is neither linked to cancer nor to another disorder, such as Tn syndrome.
The common pathway for mucin glycosylation in most cells is the generation of core 1 O‐glycans, followed by their elongation or modification to core 2 O‐glycans. Indeed, the transcript for the T‐synthase can be found in most cells and tissues.60, 121 A notable exception is the biosynthesis of core 3 O‐glycans on mucins of the gastrointestinal tract, where the C3GnT transcript is specifically expressed.105 Thus, any compromise in the expression of the T‐synthase or C3GnT can lead to elevated expression of the Tn/STn antigens on mucins from a wide variety of cells.
Whereas much is known about the functions of O‐glycans, and especially of core 1 and core 2 O‐glycans in membrane glycoproteins, little is really known about the specific functions of glycans on MUC gene products, such as MUC1, MUC2, or MUC4, in normal biology. Some have suggested that in cancer, O‐glycans on these large mucins may have an antiadhesive function259, 260 and thus indirectly support tumor‐cell metastasis by favoring the detachment of cells from the extracellular matrix (ECM). Overexpression of MUC1 can interfere with integrin‐mediated adhesion to the ECM.261 Alternatively, the glycosylation of MUCs, such as epiglycanin (MUC21),262 may lead to masking of other tumor cell‐surface antigens263 so that the immune system does not recognize the cells and tumor proliferation is therefore promoted. Loss of C3GnT in engineered mice leads to decreased expression of MUC2 and susceptibility to colitis and colorectal cancer.124 This observation indicates that mucin glycosylation may have a barrier and protective function. However, it is disturbing that we know so little about the specific requirements and biological roles of distinct glycan structures (core 1 versus core 3) in these large mucins. In airway mucins, such as MUC5AC and MUC5B, the glycosylation is altered under disease conditions and may contribute, as in the case of patients with cystic fibrosis, to enhanced adhesion and biofilm formation by pneumonia‐causing pathogens, such as Pseudomonas aeruginosa.264 But again, although O‐glycans may be recognized by pathogens in that case, the normal physiological functions of specific glycan species in mucins are not well understood.
5. Expression of the Tn Antigen in Human Diseases
5.1. Tn Syndrome
Tn syndrome is a rare hematological disorder in which subpopulations of blood cells in all lineages carry the Tn antigen. As described in Section 1.1., it was in a patient with Tn syndrome that the Tn antigen was first discovered.1 Clinically, such patients usually appear healthy and do not require treatment.265 Laboratory tests may uncover moderate hemolytic anemia and reduced numbers of thrombocytes and leukocytes.
In 1975, Dahr et al.13 reported that reduced amounts of sialic acid and galactose in erythrocyte glycoproteins of a patient with Tn syndrome led to the exposure of GalNAc α‐linked to hydroxy groups of serine or threonine. They postulated that the exposure of α‐GalNAc might be due to a defective galactosyltransferase. As blood cells in all lineages were affected, but many cells were normal, the defect probably arose from a somatic mutation at the pluripotent‐stem‐cell level. Interestingly, Bird suggested in 1974 that the Tn antigen is formed spontaneously in patients with Tn syndrome through a somatic mutation.266 His suggestion came in response to a question from Springer regarding Bird’s presentation on the specificity of plant agglutinins for erythrocyte membrane antigens at the Annual Meeting of the New York Academy of Sciences.
After the definition of the structure of the Tn antigen as a truncated form of the T antigen, it was proposed by Bird and co‐workers that its expression was due to a defect in a β‐galactosyltransferase in cells expressing Tn antigen of patients with Tn syndrome.267, 268 In 1982, it was noted that platelets from Tn‐syndrome patients in which more than 80 % of platelets expressed the Tn antigen exhibited defective glycosylation of platelet glycoprotein GPIb; this finding correlated with the thrombocytopenia observed in those individuals.43 In fact, a substantial number of T‐cell clones from a patient affected by Tn syndrome were later shown to be devoid of T‐synthase activity.269 The underlying genetic changes turned out to be somatic mutations in Cosmc, as summarized in Table 1. These mutations either cause an ORF shift and/or stop codon or cause the Cosmc gene to not be transcribed and result in total or severe impairment of the chaperone function. The fact that the immortalized leukocytes from patient Tn4 (Table 1) had no transcript of Cosmc was probably due to a change in its promoter region. Thurnher et al.270 hypothesized that this change may be hypermethylation of the promoter region and that this modification explains the suppression of the gene for T‐synthase in Tn‐syndrome patients. It would thus be interesting to examine the whole Cosmc gene from the immortalized leukocytes of patient Tn4 to see whether the loss of transcript is due to a mutation or to hypermethylation in the promoter.
Table 1.
Summary of the somatic mutations in Cosmc identified in patients with Tn syndrome.
|
Patient (gender) |
Tn/STn expression |
Mutation of Cosmc gene |
Change in Cosmc protein |
Activity |
Ref. |
|---|---|---|---|---|---|
|
C.C. (male) |
Tn and STn |
C202T |
R68* |
2–5 % |
|
|
C.L. (male) (Tn1) |
Tn and STn |
G454A |
E152K |
0 |
|
|
Tn2 (female) |
Tn and STn |
T577C |
S193P |
NT[a] |
|
|
Tn3 (male) |
Tn and STn |
G3C |
M1I |
NT[a] |
|
|
Tn4 (male) |
Tn and STn |
no transcript (in gene C428T) |
no protein |
0 |
[a] Not tested. * indicates a stop codon.
The mechanisms that occasionally lead to hemolytic anemia and reduced thrombocyte and leukocyte counts in patients with Tn syndrome appear to be multifactorial and are not fully understood. However, it is likely that auto‐antibodies directed against the Tn antigen are formed. These antibodies may be of the IgM cold agglutinin type, as shown for auto‐antibodies against the carbohydrate large “I” antigen present on adult erythrocytes.271
5.2. Cancer
Terminal α‐linked GalNAc on human tumor cells was first described in 1969 on the basis of the observation that these cells bound agglutinins from the snail‐lectin HPA that recognize Tn antigen.56 Springer et al. reported that the Tn antigen was present at high levels in around 90 % of breast carcinomas.75 Cumulative studies showed that 70–90 % of cancers of the colon, lung, bladder, cervix, ovary, stomach, and prostate express the Tn antigen.272–274 In contrast, little or no expression was observed in normal adult tissues. In many cancers, including cervical cancer,275, 276 lung adenocarcinomas,277 colorectal carcinomas,278 breast carcinomas,279 and gastric carcinomas,280 Tn‐antigen expression correlates with metastatic potential and poor prognosis. Expression of sialyl‐Tn antigen is also often observed in many human cancers and could also arise by multiple mechanisms. A likely mechanism for enhanced expression of the sialyl‐Tn antigen is an increase in the expression or activity of ST6GalNAc‐I.115, 281 The enzyme ST6GalNAc‐II, which can also form sialyl‐Tn antigen in vitro, may be more important in vivo in forming the alternatively sialylated structure Sia6Core 1 (Scheme 1).115 Truncated O‐glycans in cancer mucins may also arise from changes in the relative expression of core 2 β6‐GlcNAcT and ST3Gal‐I.282
The mechanisms linking Tn‐antigen expression to cancer progression are only starting to be understood. Tn antigen present on MUC1 of colon carcinoma cells was found to be bound by the macrophage galactose‐type lectin (MGL) expressed by macrophages and dendritic cells (see also Section 6.2.). As MGL is expressed on human immature dendritic cells and particularly on tolerogenic dendritic cells, Tn‐antigen binding may lead to immunosuppressive effects and enable the tumor to escape immunosurveillance.283 Additionally, it can be hypothesized that, in analogy with observations regarding the low‐density‐lipoprotein (LDL) receptor,137, 138 the transferrin receptor,284 P‐selectin glycoprotein ligand‐1 (PSGL‐1),285 or dystroglycan,286 the incomplete O‐glycosylation of receptors present on cancer cells may impair their expression and/or biological activity. As a consequence, proliferation control and the adhesive properties of cancer cells may be altered.
One molecular basis for Tn‐antigen expression by human tumor cells was recently shown to be the loss of functional Cosmc.61 The exposure of the Tn antigen in two specimens of human cervical carcinoma as well as in colon cancer and melanoma‐derived cell lines was due to either somatic mutations in Cosmc or an absence of Cosmc transcript (Table 2). Consistent with the findings in human tumors, mouse fibrosarcoma express Tn antigen as a result of deletion of the 26 aa between positions 170 and 195 within the luminal domain of Cosmc. The mouse neuroblastoma cell line287 Neuro‐2a (also known as C1300)287, 288 is Tn‐positive because its Cosmc contains a G301T mutation, which results in a premature stop codon. With the discovery of somatic Cosmc mutations in cancer cells, the first example was found of the regulation of the global expression of a tumor‐specific carbohydrate antigen by a single genetic locus in diverse types of human neoplastic disease.
Table 2.
Summary of the mutations in Cosmc identified in human tumor cell lines and mouse fibrosarcoma and neuroblastoma cells.
|
Human cancer and tumor cell lines |
Tn/STn expression |
Mutation of Cosmc gene |
Change in Cosmc protein |
Activity |
Ref. |
|---|---|---|---|---|---|
|
human cervical cancer |
Tn and STn |
deletion of functional allele (LOH) |
no protein made |
0 |
|
|
human Jurkat (E6.1, I2.1, I9.2) |
Tn |
deletion of T at position 473 |
ORF shift; N‐terminal 168 aa peptide |
2–5 % |
60 and unpublished data for I2.1 and I9.2 |
|
human LSC |
Tn and STn |
insertion of T at position 53 |
ORF shift; N‐terminal 28 aa peptide |
0 |
|
|
human LS174T clone I |
Tn and STn |
deletion of A at position 482 |
ORF shift; N‐terminal 170 aa peptide |
5 % |
|
|
human LS174T clone II |
Tn and STn |
G553T |
G185* |
5 % |
|
|
human LOX |
Tn and STn |
deletion of the promoter (no transcript) |
no protein made |
0 |
|
|
mouse fibrosarcoma |
Tn and STn |
deletion of C509–A587 |
deletion of T170–L195 |
NT[a] |
|
|
mouse neuroblastoma (Neuro2a, C1300) |
Tn and STn |
G301T |
E101* |
NT[a] |
[a] Not tested. * indicates a stop codon.
Remarkably, Dahr et al. wrote in 197412 that they were tempted to speculate that the same pathogenic mechanism may lead to the generation of the Tn antigen in tumors and in Tn syndrome. This speculation was shown to be true 34 years later with a common pathogenic mechanism being loss‐of‐function mutations in Cosmc (Tables 1 and 2). Of course, as indicated above, there are alternative pathways for the generation of Tn antigen, and thus, each type of tumor must be carefully examined to distinguish which of the potential pathways is affected.
5.3. IgA Nephropathy
IgA nephropathy (IgAN) is the most common primary glomerulonephritis289 and leads to terminal renal failure in 20–40 % of patients over 20–25 years.290 IgAN is most frequent in Chinese and Japanese populations and relatively rare in persons of African descent, which suggests a role of genetic factors in its pathogenesis.291 However, to date no causal gene has been identified,292–294 and additional physiological and environmental factors appear to be required for clinical manifestation of the disease. The only method to unambiguously diagnose IgA nephropathy is based on renal biopsy.295 IgAN is characterized by deposition of polymeric IgA, mostly of the IgA1 subclass, in the mesangium. These deposits elicit a glomerular inflammation, which leads to progressive renal injury.
IgA1 isolated from the kidneys of IgA‐nephropathy patients typically displays galactose‐deficient O‐glycans in its hinge region (Figure 3). The hinge region of IgA1 has a high content of proline, serine, and threonine and about five O‐linked glycan chains among the nine potential Ser/Thr residues available for O‐glycan modification.173 In normal IgA1, the hinge O‐glycans consist of the core 1 disaccharide (Galβ1‐3GalNAcα1‐O‐Ser/Thr or T antigen) carrying one or two sialic acids.295 In IgA nephropathy, IgA1 O‐glycosylation includes the expression of Tn and STn antigens, possibly as a result of a B‐cell‐restricted reduction in T‐synthase activity.296
Figure 3.

IgA1 and O‐glycosylation of the hinge region. Whereas normal IgA1 contains core 1 type O‐glycans at about five sites in the hinge region, IgA1 from patients with IgA nephropathy displays O‐glycans with reduced amounts of galactose, and many chains are truncated and contain the Tn and STn antigens.
Whether Cosmc plays a role in the pathogenesis of IgAN is uncertain. Several studies suggested that the transcript levels of Cosmc and T‐synthase were reduced in the B cells of patients with IgAN.119, 297–299 Other studies linked IgAN to polymorphisms of the genes that encode Cosmc and T‐synthase.300 Yet another report concluded that there are no compromising Cosmc mutations in patients with IgAN.301 A key problem in clarifying the role of Cosmc in IgAN is that it is likely that only a minor fraction of plasma cells that secrete the IgA1 are involved in this disease. Identification and isolation of this population of plasma cells is extremely difficult, but is crucial to the elucidation of a potential role of Cosmc and/or T‐synthase in IgAN.
The abnormal glycosylation of the IgA1 hinge region appears to contribute to the pathogenesis of IgA nephropathy. In the absence of galactose, the terminal GalNAc residue of hinge‐region glycopeptides can be recognized by naturally occurring IgA1 or IgG antibodies, which results in the formation of circulating immune complexes.295 Therefore, the propensity of an individual to develop autoimmune antiglycan antibodies may be a cofactor in disease manifestation.291 Alternatively, the aberrantly glycosylated hinge regions may render IgA1 molecules prone to self‐aggregation and the formation of macromolecular complexes by a nonimmunological mechanism.
The macromolecular IgA1 complexes may escape hepatic catabolism because they are too large to pass through endothelial fenestrae and reach the hepatocytes.302 Instead, they are shunted to the renal circulation, where endothelial fenestrae overlying the glomerular mesangia are larger. The mesangial cells bind high‐molecular‐weight IgA1 with high affinity by an as yet unknown mechanism.303 The activation of mesangial cells by IgA1 immune complexes is considered the initiating event in the pathogenesis of IgA nephropathy.
5.4. Other Disorders and Diseases in Which Tn Antigen Is Expressed
Tn antigen is expressed by various parasites. It was first described in Schistostoma mansoni schistosomula, which produce O‐glycans consisting of simple monosaccharides—mostly O‐GlcNAc, but also O‐GalNAc.304 The cestode Echinococcus granulosus expresses the Tn antigen in its larval and adult stages. The highest levels of Tn antigen are found in the secretions of the adult worm.305 Echinococcus granulosus causes disease in livestock as well as in people. Human cystic echinococcosis is a chronic condition and may prove fatal. Tn antigen can be detected in serum samples of affected patients. The role of Tn antigen in the parasitism of Echinococcus granulosus has not been determined. However, the antigens in the secretions of the worm represent the main immunogenic challenge to the host and are therefore likely to be crucial in determining whether successful parasitism is established.
Cryptosporidium parvum is a waterborn enteric coccidian that causes human and animal diarrheal disease. In immunocompetent hosts, the infection is asymptomatic and/or self‐limited. However, in immunocompromised patients, it may be severe, chronic, and fatal. Infection with Cryptosporidium occurs when oocysts are ingested with food or contaminated water. The oocysts release sporozoites that can specifically adhere to and infect the epithelial cells of the small intestine. Infection depends on mucin‐like glycoproteins (gp), namely, gp15/40 and gp900, expressed and/or secreted by the sporozoites. These glycoproteins were found to carry multiple O‐linked α‐GalNAc epitopes306 that appeared to be crucial for infection, as sporozoite infectivity was blocked by GalNAc‐specific lectins. Cryptosporidium furthermore expresses a multivalent Gal/GalNAc‐binding lectin termed p30,307 which associates with gp40 and gp900 through protein–carbohydrate interactions. As p30, gp900, and gp40 are all implicated in the attachment of sporozoites to intestinal epithelial cells, these proteins may form a functional adhesive complex. Being a multivalent Gal/GalNAc‐specific lectin, p30 may engage in multiple interactions with both parasite and host carbohydrates, thereby bridging parasite and host cells during the invasion process.
Interestingly, it was shown that anti‐Tn antibodies can block the infection of lymphocytes by human immunodeficiency virus type 1 (HIV‐1)308, 309 and that both gp120 and gp160 contain the Tn antigen. Several viral‐envelope glycoproteins contain not only N‐glycans but also O‐glycans. Some examples of viruses and viral glycoproteins that contain O‐glycans are herpes simplex virus type 1,310–312 vaccinia virus HA protein,313 respiratory syncytial virus GP1,314 mouse hepatitis virus,315 murine leukemia virus gp70,316 coronavirus E1,315, 317 and HIV gp160/120.308, 309, 318 In the case of HIV, it has been proposed that HIV infection of T cells may induce Tn‐antigen expression,309 but the potential mechanism and generality of the phenomenon is not yet clear. A complication in this regard is that the commonly used T cell line Jurkat expresses the Tn antigen in its glycoproteins60, 120 as a result of defective synthesis of the T‐synthase.60 Expression of the Tn antigen by Jurkat cells has been used as a target for improved gene transfer through Tn‐mediated endocytosis of antibody‐coupled DNA.85 Interestingly, it was recently shown that the infection of ferrets with H1N1 influenza virus causes upregulated expression of the sialyl‐Tn antigen.319
6. Targeting Tn Antigen To Treat Diseases
6.1. Development of Vaccines Based on the Tn Antigen
The finding of Tn antigen at the cell surfaces of neoplastic lesions has encouraged considerable effort toward the development of Tn‐antigen‐based antitumor vaccines. Early experiments were carried out by Springer et al., who used erythrocyte glycoproteins as antigens that they had isolated from outdated, banked blood and treated with enzymes to expose the Tn antigen.320 On the basis of skin hypersensitivity tests with this reagent, Springer and co‐workers claimed to be able to diagnose various carcinomas years before biopsies or X‐ray examination gave a positive result.320, 321 Furthermore, Springer and co‐workers used the preparation for long‐term anticarcinoma vaccination and reported the successful treatment of breast carcinoma. Of 16 patients, all survived more than 5 years and 10 of them more than 10 years. Comparing these results to the 1990 standard PDQ (physician data query) data of the National Cancer Institute, they claimed that the probability that their results for the five‐year survival were based on chance was surprisingly minimal (p<10−8).320 Even though, from our perspective today, the work of Springer and co‐workers may have been pioneering, it is impossible to judge the validity of their data owing to the enigmatic quality of their erythrocyte glycoprotein preparation, the nonvalidated diagnostic procedures, and the lack of multiple controls.
Nevertheless, attempts to generate Tn‐antigen‐based vaccines are ongoing, and the results have been encouraging. In contrast to the approach of Springer and co‐workers, recent attempts to generate vaccines aim at generating well‐defined and pure Tn‐antigen preparations. Pure Tn antigen cannot be isolated from natural sources in sufficient amounts and hence needs to be produced by enzymatic and/or chemical methods.322, 323
For the production of antibodies against a carbohydrate antigen, helper T lymphocytes need to be activated and cooperate with B lymphocytes. In general, carbohydrates alone, however, do not activate helper T lymphocytes and have limited immunogenicity.25 Therefore, for the production of vaccines, carbohydrates are usually coupled to a carrier protein, which enhances presentation to the immune system and provides epitopes that can activate helper T cells.324 When such a glycoprotein is processed and presented by antigen‐presenting cells, a strong T‐cell immune response is mounted, leading to the release of cytokines that increase antibody response. These antibodies are not only directed against the protein, but also against the less immunogenic carbohydrate antigen, and may lead to the production of IgG and other antibody classes. The increased immunogenicity of different presentations of Tn antigens linked to the protein keyhole limpet haemocyanin (KLH) suggested that the essential immunogenic structure for Tn antigen is a cluster of three to four consecutive monosaccharides.325
Another glycopeptide vaccine carrying clustered Tn antigens was generated by either chemically or enzymatically transferring GalNAc to Ser/Thr residues of MUC1 peptide fragments.323, 326 Because the MUC1 glycoprotein is overexpressed and aberrantly glycosylated in the majority of epithelial cancers (see also Sections 4.3 and 5.2), this approach is regarded as particularly promising. It was shown that MUC1 glycopeptides (consisting of three tandem repeats and carrying nine Tn antigens) were specifically internalized by dendritic cells through binding of their Tn antigens by the MGL receptor.323 Subsequently, the glycopeptides entered the HLA‐I and HLA‐II compartments, which are associated with the induction of Th1 immunity.
In addition to the presentation of several clustered carbohydrate antigens on a carrier peptide, the immune response can be enhanced through coadministration of a third component: an immunological adjuvant, such as the saponin QS21.327–329 Several three‐component anticancer vaccines have advanced to clinical trials. They appeared to be safe, showed no evidence of autoimmunity, and produced an immune response to the Tn antigen in all patients.84 Of 15 prostate cancer patients who had received the three‐component vaccine Tn‐KLH with QS21, 33 % achieved a favorable response as measured by a decrease in prostate‐specific antigen (PSA) slopes.325 Whether decreased PSA slopes translate into a slower progression of prostate cancer and improved survival of patients remains, however, to be validated.
The effectiveness of a Tn‐antigen vaccine clearly depends on the presence of Tn antigen on the carcinoma cell surface to enable targeting and to elicit cell killing, but results on Tn expression in prostate tumors are conflicting. By using a combination of immunohistochemistry with four different antibodies and carbohydrate microarray profiling of each antibody, Li et al. recently found that of 77 prostate tumors, only 4–26 % expressed the Tn antigen.84 Therefore, a preselection of patients with Tn‐positive tumors may significantly increase the success rate of Tn‐antigen vaccines.
6.2. Removal of Cells Expressing the Tn Antigen
The Tn antigen is a nonphysiological glycan structure in humans; thus, it is not surprising that it may be recognized as foreign by the immune system. The MGL expressed on myeloid antigen‐presenting cells is known to specifically bind terminal α‐ and β‐linked GalNAc.330 MGL‐positive antigen‐presenting cells were found in the small intestine and lymph nodes.331 Upon ligand binding, MGL rapidly internalizes, and the endocytosed ligand is transported along the endosomal–lysosomal pathway and presented in major histocompatibility complex class II (MHC II) molecules. MGL can regulate Toll‐like receptor signaling and thus influence the outcome of an immune response. Depending on whether additional dendritic‐cell activation occurs, Tn binding may lead either to an immune response and cell killing or to immune tolerance and inhibition of immune responses.332, 333 Unraveling how the balance between immune response and tolerance to Tn antigen is regulated is key to understanding the pathogenic mechanisms underlying Tn syndrome, IgA nephropathy, and the escape of Tn‐antigen‐positive cancer cells from immunosurveillance; it is also necessary for the design of effective Tn‐antigen‐based anticancer vaccines.
7. Summary and Outlook
The Tn antigen, though small in size and simple in structure, has created huge interest since its discovery over 50 years ago. Apparently, the biology or pathophysiology of Tn antigen is complex. The abnormal expression of Tn antigen in human tissues, associated with pathology, as well as expression of the Tn antigen by human pathogens, indicate that it is a key target of immunity and regulation. The discovery that expression of the Tn antigen in human and animal pathologies may result from altered expression or mutation of the key molecular chaperone Cosmc, which regulates T‐synthase folding, has created a new direction of research aimed at uncovering the genetic and potentially epigenetic regulation of protein O‐glycosylation. However, much remains to be learned about the factors contributing to the pathological expression of the Tn antigen and about the complex biosynthetic controls that prevent its expression in normal cells. New strategies aimed at creating vaccines containing peptide conjugates of the Tn/STn antigen are promising and may lead to effective therapies against tumors expressing those antigens. Molecular strategies aimed at restoring normal O‐glycan expression in cells compromised in O‐glycan biosynthesis might also prove fruitful, now that the specific molecular targets have been identified. There is a dearth of knowledge, however, about the normal functions of O‐glycans derived from elongation and extension of the Tn antigen, and that knowledge could help in defining the pathological consequences of Tn expression. It is hoped that a combination of approaches in synthetic chemistry, immunology, cell biology, biochemistry, and human and animal genetics will lead to breakthroughs in defining the structures and functions of O‐glycans and in understanding the roles of the Tn/STn antigens in disease.
Dedicated to Professor Hans Paulsen on the occasion of his 80th birthday and in memory of Toshi Osawa
Biographical Information
Tongzhong Ju received his MD in 1986 from Qingdao Medical College and his PhD in biochemistry in 1994 from the Medical School of Fudan University Shanghai (China). Following postdoctoral studies in biochemistry/glycobiology with Richard D. Cummings and William M. Canfield (1997–1999), he worked as a Research Associate (1999–2001) and Research Assistant Professor (2002–2006) at the University of Oklahoma Health Sciences Center (OUHSC). Since 2006, he has been an Assistant Professor in the Department of Biochemistry at Emory University School of Medicine in Atlanta, Georgia.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
Vivianne I. Otto received her MSc in pharmaceutical sciences in 1987 and her PhD in pharmaceutical chemistry in 1997 from ETH Zurich (Switzerland). After postdoctoral research in neuroinflammation at the University Hospital of Zurich with M. C. Morganti‐Kossmann (1997–2001), she joined the research group of Richard D. Cummings at the OUHSC as a research fellow (2001–2003). She completed her habilitation in 2006 in the Department of Chemistry and Applied Biosciences of ETH Zurich, where she is currently a lecturer in medicinal chemistry and glycobiology.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
Richard D. Cummings received his PhD in biochemistry from The Johns Hopkins University in 1980. After postdoctoral research with Stuart Kornfeld at the Washington University School of Medicine (1980–1983), he took up a position at the University of Georgia and the Complex Carbohydrate Research Center (1983–1992). He later moved to the OUHSC and formed the Oklahoma Center for Medical Glycobiology. Since 2006 he has been the William Patterson Timmie Professor and Chair of the Department of Biochemistry at Emory University School of Medicine, where he is founder and codirector of the Glycomics Center.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Supporting information
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer‐reviewed, but not copy‐edited or typeset. They are made available as submitted by the authors.
miscellaneous_information
Acknowledgements
We thank Dr. Jamie Heimburg‐Molinaro and Sandra Cummings for helpful comments during the writing of the manuscript. This work was supported by an NIH RO1 grant (RO1 GM068559) to R.D.C. and by an NIH RO1 grant (RO1K80876) to T.J.
References
- 1. Moreau R., Dausset J., Bernard J., Moullec J., Bull. Mem. Soc. Med. Hop. Paris 1957, 73, 569–587. [PubMed] [Google Scholar]
- 2. Huebner G., Z. Immunitaetsforsch. Exp. Ther. 1926, 45, 223–248. [Google Scholar]
- 3. Thomsen O., Z. Immunitaetsforsch. Exp. Ther. 1927, 52, 85–107. [Google Scholar]
- 4. Thomsen O., C. R. Seances Soc. Biol. Ses Fil. 1927, 96, 556–558. [Google Scholar]
- 5. Thomsen O., Z. Immunitaetsforsch. Exp. Ther. 1928, 57, 301–319. [Google Scholar]
- 6. Friedenreich V., The Thomsen Hemagglutination Phenomenon, Vol. 1, Levin and Munskgaard, Copenhagen, 1930. [Google Scholar]
- 7. Dausset J., Moullec J., Bernard J., Blood 1959, 14, 1079–1093. [PubMed] [Google Scholar]
- 8. Muroi K., Suda T., Nakamura M., Okada S., Nojiri H., Amemiya Y., Miura Y., Hakomori S., Blood 1994, 83, 84–91. [PubMed] [Google Scholar]
- 9. Kawano‐Yamamoto C., Muroi K., Nagatsuka Y., Higuchi M., Kikuchi S., Nagai T., Hakomori S. I., Ozawa K., Leuk. Res. 2006, 30, 829–839. [DOI] [PubMed] [Google Scholar]
- 10. Jass J. R., Allison L. M., Edgar S., Pathology 1994, 26, 418–422. [DOI] [PubMed] [Google Scholar]
- 11. Cao Y., Merling A., Karsten U., Goletz S., Punzel M., Kraft R., Butschak G., Schwartz‐Albiez R., Int. J. Cancer 2008, 123, 89–99. [DOI] [PubMed] [Google Scholar]
- 12. Dahr W., Uhlenbruck G., Bird G. W., Vox Sang. 1974, 27, 29–42. [DOI] [PubMed] [Google Scholar]
- 13. Dahr W., Uhlenbruck G., Gunson H. H., Hart M. Van Der, Vox Sang. 1975, 29, 36–50. [DOI] [PubMed] [Google Scholar]
- 14. Iyer R. N., Carlson D. M., Arch. Biochem. Biophys. 1971, 142, 101–105. [DOI] [PubMed] [Google Scholar]
- 15. Carlson D. M., J. Biol. Chem. 1968, 243, 616–626. [PubMed] [Google Scholar]
- 16. Klenk E., Uhlenbruck G., Hoppe Seylers Z. Physiol. Chem. 1960, 319, 151–160. [DOI] [PubMed] [Google Scholar]
- 17. Uhlenbruck G., Immunol. Commun. 1981, 10, 251–264. [DOI] [PubMed] [Google Scholar]
- 18. Klenk E., Hendricks U. W., Gielen W., Hoppe Seylers Z. Physiol. Chem. 1962, 330, 140–144. [DOI] [PubMed] [Google Scholar]
- 19. Kim Z., Uhlenbruck G., Z. Immunitaetsforsch. Allerg. Klin. Immunol. 1966, 130, 88–99. [PubMed] [Google Scholar]
- 20. Kaifu R., Osawa T., Carbohydr. Res. 1977, 58, 235–239. [DOI] [PubMed] [Google Scholar]
- 21. Ratcliffe R. M., Baker D. A., Lemieux R. U., Carbohydr. Res. 1981, 93, 35–41. [DOI] [PubMed] [Google Scholar]
- 22. Paulsen H., Hölck J.‐P., Carbohydr. Res. 1982, 109, 89–107. [DOI] [PubMed] [Google Scholar]
- 23. Kunz H., Birnbach S., Angew. Chem. 1986, 98, 354–355; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1986, 25, 360–362. [Google Scholar]
- 24. Cato D., Buskas T., Boons G. J., J. Carbohydr. Chem. 2005, 24, 503–516. [Google Scholar]
- 25. Buskas T., Ingale S., Boons G. J., Glycobiology 2006, 16, 113R–136R. [DOI] [PubMed] [Google Scholar]
- 26. Kunz H., Birnbach S., Wernig P., Carbohydr. Res. 1990, 202, 207–223. [DOI] [PubMed] [Google Scholar]
- 27. Grogan M. J., Pratt M. R., Marcaurelle L. A., Bertozzi C. R., Annu. Rev. Biochem. 2002, 71, 593–634. [DOI] [PubMed] [Google Scholar]
- 28. Huang K. T., Wu B. C., Lin C. C., Luo S. C., Chen C., Wong C. H., Lin C. C., Carbohydr. Res. 2006, 341, 2151–2155. [DOI] [PubMed] [Google Scholar]
- 29. Koeller K. M., Smith M. E., Wong C. H., Bioorg. Med. Chem. 2000, 8, 1017–1025. [DOI] [PubMed] [Google Scholar]
- 30. Leppanen A., Mehta P., Ouyang Y. B., Ju T., Helin J., Moore K. L., van Die I., Canfield W. M., McEver R. P., Cummings R. D., J. Biol. Chem. 1999, 274, 24838–24848. [DOI] [PubMed] [Google Scholar]
- 31. Pratt M. R., Bertozzi C. R., Chem. Soc. Rev. 2005, 34, 58–68. [DOI] [PubMed] [Google Scholar]
- 32. Liebe B., Kunz H., Angew. Chem. 1997, 109, 629–631; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1997, 36, 618–621. [Google Scholar]
- 33. Kuduk S. D., Schwarz J. B., Chen X.‐T., Glunz P. W., Sames D., Ragupathi G., Livingston P. O., Danishefsky S. J., J. Am. Chem. Soc. 1998, 120, 12474–12485. [Google Scholar]
- 34. Elofsson M., Salvador L. A., Kihlberg J., Tetrahedron 1997, 53, 369–390. [Google Scholar]
- 35. Winans K. A., King D. S., Rao V. R., Bertozzi C. R., Biochemistry 1999, 38, 11700–11710. [DOI] [PubMed] [Google Scholar]
- 36. Svarovsky S. A., J. J. Barchi, Jr. , Carbohydr. Res. 2003, 338, 1925–1935. [DOI] [PubMed] [Google Scholar]
- 37. Elofsson M., Kihlberg J., Tetrahedron Lett. 1995, 36, 7499–7502. [Google Scholar]
- 38. Paulsen H., Rauwald W., Weichert U., Liebigs Ann. Chem. 1988, 75–86. [Google Scholar]
- 39. Nakahara Y., Iijima H., Shohei S., Ogawa T., Tetrahedron Lett. 1990, 31, 6897–6900. [Google Scholar]
- 40. Chen X. T., Sames D., Danishefsky S. J., J. Am. Chem. Soc. 1998, 120, 7760–7769. [Google Scholar]
- 41. Schleyer A., Meldal M., Manat R., Paulsen H., Angew. Chem. 1997, 109, 2064–2067; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1997, 36, 1976–1978. [Google Scholar]
- 42. Wandall H. H., Blixt O., Tarp M. A., Pedersen J. W., Bennett E. P., Mandel U., Ragupathi G., Livingston P. O., Hollingsworth M. A., Taylor‐Papadimitriou J., Burchell J., Clausen H., Cancer Res. 2010, 70, 1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nurden A. T., Dupuis D., Pidard D., Kieffer N., Kunicki T. J., Cartron J. P., J. Clin. Invest. 1982, 70, 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Etzler M. E., Gupta S., Borrebaeck C., J. Biol. Chem. 1981, 256, 2367–2370. [PubMed] [Google Scholar]
- 45. Bird G. W., Wingham J., Rev. Fr. Transfus. Immuno‐Hematol. 1981, 24, 347–348. [DOI] [PubMed] [Google Scholar]
- 46. Wu A. M., J. Biomed. Sci. 2005, 12, 135–152. [DOI] [PubMed] [Google Scholar]
- 47. Moore B. P., Marsh S., Transfusion 1975, 15, 132–134. [DOI] [PubMed] [Google Scholar]
- 48. Newman R. A., Uhlenbruck G. G., Eur. J. Biochem. 1977, 76, 149–155. [DOI] [PubMed] [Google Scholar]
- 49. Vega N., Pérez G., Phytochemistry 2006, 67, 347–355. [DOI] [PubMed] [Google Scholar]
- 50. Tachibana K., Nakamura S., Wang H., Iwasaki H., Tachibana K., Maebara K., Cheng L., Hirabayashi J., Narimatsu H., Glycobiology 2006, 16, 46–53. [DOI] [PubMed] [Google Scholar]
- 51. Wu A. M., Wu J. H., Liu J. H., Singh T., Life Sci. 2004, 74, 1763–1779. [DOI] [PubMed] [Google Scholar]
- 52. Uhlenbruck G., Dahr W., Vox Sang. 1971, 21, 338–351. [DOI] [PubMed] [Google Scholar]
- 53. Tollefsen S. E., Kornfeld R., J. Biol. Chem. 1983, 258, 5172–5176. [PubMed] [Google Scholar]
- 54. Dahr W., Uhlenbruck G., Blut 1971, 22, 128–132. [DOI] [PubMed] [Google Scholar]
- 55. Teneberg S., Leonardsson I., Angström J., Ehrlich‐Rogozinski S., Sharon N., Glycoconjugate J. 1994, 11, 418–423. [DOI] [PubMed] [Google Scholar]
- 56. Prokop O., Uhlenbruck G., Med. Welt 1969, 46, 2515–2519. [PubMed] [Google Scholar]
- 57. Hammarstrom S., Murphy L. A., Goldstein I. J., Etzler M. E., Biochemistry 1977, 16, 2750–2755. [DOI] [PubMed] [Google Scholar]
- 58. Bird G. W., Vox Sang. 1964, 9, 748–749. [DOI] [PubMed] [Google Scholar]
- 59. Brockhausen I., Yang J., Dickinson N., Ogata S., Itzkowitz S. H., Glycoconjugate J. 1998, 15, 595–603. [DOI] [PubMed] [Google Scholar]
- 60. Ju T., Cummings R. D., Proc. Natl. Acad. Sci. USA 2002, 99, 16613–16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ju T., Lanneau G. S., Gautam T., Wang Y., Xia B., Stowell S. R., Willard M. T., Wang W., Xia J. Y., Zuna R. E., Laszik Z., Benbrook D. M., Hanigan M. H., Cummings R. D., Cancer Res. 2008, 68, 1636–1646. [DOI] [PubMed] [Google Scholar]
- 62. Konska G., Guerry M., Caldefie‐Chezet F., De Latour M., Guillot J., Oncol. Rep. 2006, 15, 305–310. [DOI] [PubMed] [Google Scholar]
- 63. Kulkarni K. A., Sinha S., Katiyar S., Surolia A., Vijayan M., Suguna K., FEBS Lett. 2005, 579, 6775–6780. [DOI] [PubMed] [Google Scholar]
- 64. Vainchenker W., Testa U., Deschamps J. F., Henri A., Titeux M., Breton‐Gorius J., Rochant H., Lee D., Cartron J. P., J. Clin. Invest. 1982, 69, 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Piller V., Piller F., Cartron J. P., Eur. J. Biochem. 1990, 191, 461–466. [DOI] [PubMed] [Google Scholar]
- 66. Lescar J., Sanchez J.‐F., Audfray A., Coll J.‐L., Breton C., Mitchell E. P., Imberty A., Glycobiology 2007, 17, 1077–1083. [DOI] [PubMed] [Google Scholar]
- 67. Conzelmann A., Lefrancois L., J. Exp. Med. 1988, 167, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gunson H. H., Stratton F., Mullard G. W., Br. J. Haematol. 1970, 18, 309–316. [DOI] [PubMed] [Google Scholar]
- 69. Issitt P. D., Issitt C. H., Moulds J., Berman H. J., Transfusion 1972, 12, 217–221. [DOI] [PubMed] [Google Scholar]
- 70. Springer G. F., Taylor C. R., Howard D. R., Tegtmeyer H., Desai P. R., Murthy S. M., Felder B., Scanlon E. F., Cancer 1985, 55, 561–569. [DOI] [PubMed] [Google Scholar]
- 71. Springer G. F., Desai P. R., Murthy M. S., Scanlon E. F., J. Surg. Oncol. 1979, 11, 95–106. [DOI] [PubMed] [Google Scholar]
- 72. Springer G. F., Tegtmeyer H., Br. J. Haematol. 1981, 47, 453–460. [DOI] [PubMed] [Google Scholar]
- 73. Springer G. F., Horton R. E., J. Clin. Invest. 1969, 48, 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Springer G. F., Desai P. R., Banatwala I., J. Natl. Cancer Inst. 1975, 54, 335–339. [PubMed] [Google Scholar]
- 75. Springer G. F., Desai P. R., Banatwala I., Naturwissenschaften 1974, 61, 457–458. [DOI] [PubMed] [Google Scholar]
- 76. Kjeldsen T., Clausen H., Hirohashi S., Ogawa T., Iijima H., Hakomori S., Cancer Res. 1988, 48, 2214–2220. [PubMed] [Google Scholar]
- 77. Kjeldsen T., Hakomori S., Springer G. F., Desai P., Harris T., Clausen H., Vox Sang. 1989, 57, 81–87. [DOI] [PubMed] [Google Scholar]
- 78. Hirohashi S., Clausen H., Yamada T., Shimosato Y., Hakomori S., Proc. Natl. Acad. Sci. USA 1985, 82, 7039–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Avichezer D., Springer G. F., Schechter B., Arnon R., Int. J. Cancer 1997, 72, 119–127. [DOI] [PubMed] [Google Scholar]
- 80. Numata Y., Nakada H., Fukui S., Kitagawa H., Ozaki K., Inoue M., Kawasaki T., Funakoshi I., Yamashina I., Biochem. Biophys. Res. Commun. 1990, 170, 981–985. [DOI] [PubMed] [Google Scholar]
- 81. Takahashi H. K., Metoki R., Hakomori S., Cancer Res. 1988, 48, 4361–4367. [PubMed] [Google Scholar]
- 82. Ju T., Cummings R. D., Nature 2005, 437, 1252. [DOI] [PubMed] [Google Scholar]
- 83. Ju T., Aryal R. P., Stowell C. J., Cummings R. D., J. Cell Biol. 2008, 182, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li Q., Anver M. R., Butcher D. O., Gildersleeve J. C., Mol. Cancer Ther. 2009, 8, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thurnher M., Wagner E., Clausen H., Mechtler K., Rusconi S., Dinter A., Birnstiel M. L., Berger E. G., Cotten M., Glycobiology 1994, 4, 429–435. [DOI] [PubMed] [Google Scholar]
- 86. Tian E., Ten Hagen K. G., Glycobiology 2007, 17, 820–827. [DOI] [PubMed] [Google Scholar]
- 87. Tian E., Ten Hagen K. G., J. Biol. Chem. 2007, 282, 606–614. [DOI] [PubMed] [Google Scholar]
- 88. Hang H. C., Bertozzi C. R., Bioorg. Med. Chem. 2005, 13, 5021–5034. [DOI] [PubMed] [Google Scholar]
- 89. Miwa H. E., Gerken T. A., Jamison O., Tabak L. A., J. Biol. Chem. 2010, 285, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ten Hagen K. G., Fritz T. A., Tabak L. A., Glycobiology 2003, 13, 1R–16R. [DOI] [PubMed] [Google Scholar]
- 91. Tarp M. A., Clausen H., Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 546–563. [DOI] [PubMed] [Google Scholar]
- 92. Fritz T. A., Raman J., Tabak L. A., J. Biol. Chem. 2006, 281, 8613–8619. [DOI] [PubMed] [Google Scholar]
- 93. Pratt M. R., Hang H. C., Ten Hagen K. G., Rarick J., Gerken T. A., Tabak L. A., Bertozzi C. R., Chem. Biol. 2004, 11, 1009–1016. [DOI] [PubMed] [Google Scholar]
- 94. Gerken T. A., Raman J., Fritz T. A., Jamison O., J. Biol. Chem. 2006, 281, 32403–32416. [DOI] [PubMed] [Google Scholar]
- 95. Gerken T. A., Ten Hagen K. G., Jamison O., Glycobiology 2008, 18, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Perrine C., Ju T., Cummings R. D., Gerken T. A., Glycobiology 2009, 19, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Raman J., Fritz T. A., Gerken T. A., Jamison O., Live D., Liu M., Tabak L. A., J. Biol. Chem. 2008, 283, 22942–22951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rottger S., White J., Wandall H. H., Olivo J. C., Stark A., Bennett E. P., Whitehouse C., Berger E. G., Clausen H., Nilsson T., J. Cell Sci. 1998, 111, 45–60. [DOI] [PubMed] [Google Scholar]
- 99. Ju T., Brewer K., D′Souza A., Cummings R. D., Canfield W. M., J. Biol. Chem. 2002, 277, 178–186. [DOI] [PubMed] [Google Scholar]
- 100. Ju T., Cummings R. D., Canfield W. M., J. Biol. Chem. 2002, 277, 169–177. [DOI] [PubMed] [Google Scholar]
- 101. Gillespie W., Kelm S., Paulson J. C., J. Biol. Chem. 1992, 267, 21004–21010. [PubMed] [Google Scholar]
- 102. Lee Y. C., Kaufmann M., Kitazume‐Kawaguchi S., Kono M., Takashima S., Kurosawa N., Liu H., Pircher H., Tsuji S., J. Biol. Chem. 1999, 274, 11958–11967. [DOI] [PubMed] [Google Scholar]
- 103. Kurosawa N., Kojima N., Inoue M., Hamamoto T., Tsuji S., J. Biol. Chem. 1994, 269, 19048–19053. [PubMed] [Google Scholar]
- 104. Bierhuizen M. F., Fukuda M., Proc. Natl. Acad. Sci. USA 1992, 89, 9326–9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Iwai T., Inaba N., Naundorf A., Zhang Y., Gotoh M., Iwasaki H., Kudo T., Togayachi A., Ishizuka Y., Nakanishi H., Narimatsu H., J. Biol. Chem. 2002, 277, 12802–12809. [DOI] [PubMed] [Google Scholar]
- 106. Kurosawa N., Takashima S., Kono M., Ikehara Y., Inoue M., Tachida Y., Narimatsu H., Tsuji S., J. Biochem. 2000, 127, 845–854. [DOI] [PubMed] [Google Scholar]
- 107. Lairson L. L., Henrissat B., Davies G. J., Withers S. G., Annu. Rev. Biochem. 2008, 77, 521–555. [DOI] [PubMed] [Google Scholar]
- 108. Stojanovic D., Vischer P., Hughes R. C., Eur. J. Biochem. 1984, 138, 551–562. [DOI] [PubMed] [Google Scholar]
- 109. Brockhausen I., Möller G., Merz G., Adermann K., Paulsen H., Biochemistry 1990, 29, 10206–10212. [DOI] [PubMed] [Google Scholar]
- 110. Brockhausen I., Möller G., Pollex‐Krüger A., Rutz V., Paulsen H., Matta K. L., Biochem. Cell Biol. 1992, 70, 99–108. [DOI] [PubMed] [Google Scholar]
- 111. Ju T., Zheng Q., Cummings R. D., Glycobiology 2006, 16, 947–958. [DOI] [PubMed] [Google Scholar]
- 112. Müller R., Hülsmeier A. J., Altmann F., Ten Hagen K., Tiemeyer M., Hennet T., FEBS J. 2005, 272, 4295–4305. [DOI] [PubMed] [Google Scholar]
- 113. Ikehara Y., Kojima N., Kurosawa N., Kudo T., Kono M., Nishihara S., Issiki S., Morozumi K., Itzkowitz S., Tsuda T., Nishimura S. I., Tsuji S., Narimatsu H., Glycobiology 1999, 9, 1213–1224. [DOI] [PubMed] [Google Scholar]
- 114. Harduin‐Lepers A., Vallejo‐Ruiz V., Krzewinski‐Recchi M. A., Samyn‐Petit B., Julien S., Delannoy P., Biochimie 2001, 83, 727–737. [DOI] [PubMed] [Google Scholar]
- 115. Marcos N. T., Pinho S., Grandela C., Cruz A., Samyn‐Petit B., Harduin‐Lepers A., Almeida R., Silva F., Morais V., Costa J., Kihlberg J., Clausen H., Reis C. A., Cancer Res. 2004, 64, 7050–7057. [DOI] [PubMed] [Google Scholar]
- 116. Kono M., Tsuda T., Ogata S., Takashima S., Liu H., Hamamoto T., Itzkowitz S. H., Nishimura S., Tsuji S., Biochem. Biophys. Res. Commun. 2000, 272, 94–97. [DOI] [PubMed] [Google Scholar]
- 117. Sewell R., Bäckström M., Dalziel M., Gschmeissner S., Karlsson H., Noll T., Gätgens J., Clausen H., Hansson G. C., Burchell J., Taylor‐Papadimitriou J., J. Biol. Chem. 2006, 281, 3586–3594. [DOI] [PubMed] [Google Scholar]
- 118. Raska M., Moldoveanu Z., Suzuki H., Brown R., Kulhavy R., Andrasi J., Hall S., Vu H. L., Carlsson F., Lindahl G., Tomana M., Julian B. A., Wyatt R. J., Mestecky J., Novak J., J. Mol. Biol. 2007, 369, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Suzuki H., Moldoveanu Z., Hall S., Brown R., Vu H. L., Novak L., Julian B. A., Tomana M., Wyatt R. J., Edberg J. C., Alarcon G. S., Kimberly R. P., Tomino Y., Mestecky J., Novak J., J. Clin. Invest. 2008, 118, 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Piller V., Piller F., Fukuda M., J. Biol. Chem. 1990, 265, 9264–9271. [PubMed] [Google Scholar]
- 121. Wang Y., Ju T., Ding X., Xia B., Wang W., Xia L., He M., Cummings R. D., Proc. Natl. Acad. Sci. USA 2010, 107, 9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kudo T., Iwai T., Kubota T., Iwasaki H., Takayma Y., Hiruma T., Inaba N., Zhang Y., Gotoh M., Togayachi A., Narimatsu H., J. Biol. Chem. 2002, 277, 47724–47731. [DOI] [PubMed] [Google Scholar]
- 123. Kudo T., Iwai T., Kubota T., Iwasaki H., Takayma Y., Hiruma T., Inaba N., Zhang Y., Gotoh M., Togayachi A., Narimatsu H., J. Biol. Chem. 2006, 281, 24999–25000. [DOI] [PubMed] [Google Scholar]
- 124. An G., Wei B., Xia B., McDaniel J. M., Ju T., Cummings R. D., Braun J., Xia L., J. Exp. Med. 2007, 204, 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yang J. M., Byrd J. C., Siddiki B. B., Chung Y. S., Okuno M., Sowa M., Kim Y. S., Matta K. L., Brockhausen I., Glycobiology 1994, 4, 873–884. [DOI] [PubMed] [Google Scholar]
- 126. Vavasseur F., Dole K., Yang J., Matta K. L., Myerscough N., Corfield A., Paraskeva C., Brockhausen I., Eur. J. Biochem. 1994, 222, 415–424. [DOI] [PubMed] [Google Scholar]
- 127. Stanley P., Sundaram S., Sallustio S., Glycobiology 1991, 1, 307–314. [DOI] [PubMed] [Google Scholar]
- 128. Deutscher S. L., Hirschberg C. B., J. Biol. Chem. 1986, 261, 96–100. [PubMed] [Google Scholar]
- 129. Do S. I., Cummings R. D., J. Biochem. Biophys. Methods 1992, 14, 153–165. [DOI] [PubMed] [Google Scholar]
- 130. Kingsley D. M., Kozarsky K. F., Hobbie L., Krieger M., Cell 1986, 44, 749–759. [DOI] [PubMed] [Google Scholar]
- 131. Burchell J. M., Mungul A., Taylor‐Papadimitriou J., J. Mammary Gland Biol. Neoplasia 2001, 6, 355–364. [DOI] [PubMed] [Google Scholar]
- 132. Julien S., Krzewinski‐Recchi M. A., Harduin‐Lepers A., Gouyer V., Huet G., Le Bourhis X., Delannoy P., Glycoconjugate J. 2001, 18, 883–893. [DOI] [PubMed] [Google Scholar]
- 133. Julien S., Lagadec C., Krzewinski‐Recchi M. A., Courtand G., Le Bourhis X., Delannoy P., Breast Cancer Res. Treat. 2005, 90, 77–84. [DOI] [PubMed] [Google Scholar]
- 134. Aryal R. P., Ju T., Cummings R. D., J. Biol. Chem. 2010, 285, 2456–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Narimatsu Y., Ikehara Y., Iwasaki H., Nonomura C., Sato T., Nakanishi H., Narimatsu H., Biochem. Biophys. Res. Commun. 2008, 366, 199–205. [DOI] [PubMed] [Google Scholar]
- 136. Cummings R. D., Kornfeld S., Schneider W. J., Hobgood K. K., Tolleshaug H., Brown M. S., Goldstein J. L., J. Biol. Chem. 1983, 258, 15261–15273. [PubMed] [Google Scholar]
- 137. Kozarsky K., Kingsley D., Krieger M., Proc. Natl. Acad. Sci. USA 1988, 85, 4335–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kuwano M., Seguchi T., Ono M., J. Cell Sci. 1991, 98, 131–134. [DOI] [PubMed] [Google Scholar]
- 139. Do S. I., Cummings R. D., Glycobiology 1992, 2, 345–353. [DOI] [PubMed] [Google Scholar]
- 140. Hayes G. R., Enns C. A., Lucas J. J., Glycobiology 1992, 2, 355–359. [DOI] [PubMed] [Google Scholar]
- 141. Rutledge E. A., Root B. J., Lucas J. J., Enns C. A., Blood 1994, 83, 580–586. [PubMed] [Google Scholar]
- 142. Gahmberg C. G., Autero M., Hermonen J., J. Cell. Biochem. 1988, 37, 91–105. [DOI] [PubMed] [Google Scholar]
- 143. Jokinen M., Andersson L. C., Gahmberg C. G., J. Biol. Chem. 1985, 260, 11314–11321. [PubMed] [Google Scholar]
- 144. Gahmberg C. G., Ekblom M., Andersson L. C., Proc. Natl. Acad. Sci. USA 1984, 81, 6752–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Fu J., Gerhardt H., McDaniel J. M., Xia B., Liu X., Ivanciu L., Ny A., Hermans K., Silasi‐Mansat R., McGee S., Nye E., Ju T., Ramirez M. I., Carmeliet P., Cummings R. D., Lupu F., Xia L., J. Clin. Invest. 2008, 118, 3725–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kaneko M., Kato Y., Kunita A., Fujita N., Tsuruo T., Osawa M., J. Biol. Chem. 2004, 279, 38838–38843. [DOI] [PubMed] [Google Scholar]
- 147. Kaneko M. K., Kato Y., Kameyama A., Ito H., Kuno A., Hirabayashi J., Kubota T., Amano K., Chiba Y., Hasegawa Y., Sasagawa I., Mishima K., Narimatsu H., FEBS Lett. 2007, 581, 331–336. [DOI] [PubMed] [Google Scholar]
- 148. Leppanen A., White S. P., Helin J., McEver R. P., Cummings R. D., J. Biol. Chem. 2000, 275, 39569–39578. [DOI] [PubMed] [Google Scholar]
- 149. Li F., Wilkins P. P., Crawley S., Weinstein J., Cummings R. D., McEver R. P., J. Biol. Chem. 1996, 271, 3255–3264. [PubMed] [Google Scholar]
- 150. Wilkins P. P., McEver R. P., Cummings R. D., J. Biol. Chem. 1996, 271, 18732–18742. [DOI] [PubMed] [Google Scholar]
- 151. Satomaa T., Renkonen O., Helin J., Kirveskari J., Makitie A., Renkonen R., Blood 2002, 99, 2609–2611. [DOI] [PubMed] [Google Scholar]
- 152. Fieger C. B., Sassetti C. M., Rosen S. D., J. Biol. Chem. 2003, 278, 27390–27398. [DOI] [PubMed] [Google Scholar]
- 153. Chen J., Litscher E. S., Wassarman P. M., Proc. Natl. Acad. Sci. USA 1998, 95, 6193–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Florman H. M., Wassarman P. M., Cell 1985, 41, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Fukuda M., Glycobiology 1991, 1, 347–356. [DOI] [PubMed] [Google Scholar]
- 156. Fukuda M., Carlsson S. R., Med. Biol. 1986, 64, 335–343. [PubMed] [Google Scholar]
- 157. van den Berg T. K., Nath D., Ziltener H. J., Vestweber D., Fukuda M., van Die I., Crocker P. R., J. Immunol. 2001, 166, 3637–3640. [DOI] [PubMed] [Google Scholar]
- 158. Earl L. A., Bi S., Baum L. G., J. Biol. Chem. 2010, 285, 2232–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Fernsten P., Shaw M., Hocker S., Fulghum R., Winfield J., Immunol. Invest. 1998, 27, 323–338. [DOI] [PubMed] [Google Scholar]
- 160. Lefebvre J. C., Giordanengo V., Doglio A., Cagnon L., Breittmayer J. P., Peyron J. F., Lesimple J., Virology 1994, 199, 265–274. [DOI] [PubMed] [Google Scholar]
- 161. Pulido R., Sánchez‐Madrid F., Eur. J. Immunol. 1990, 20, 2667–2671. [DOI] [PubMed] [Google Scholar]
- 162. Wagner K. W., Punnoose E. A., Januario T., Lawrence D. A., Pitti R. M., Lancaster K., Lee D., von Goetz M., Yee S. F., Totpal K., Huw L., Katta V., Cavet G., Hymowitz S. G., Amler L., Ashkenazi A., Nat. Med. 2007, 13, 1070–1077. [DOI] [PubMed] [Google Scholar]
- 163. Chapman B. S., Eckart M. R., Kaufman S. E., Lapointe G. R., J. Neurochem. 1996, 66, 1707–1716. [DOI] [PubMed] [Google Scholar]
- 164. Yeaman C., Le Gall A. H., Baldwin A. N., Monlauzeur L., Le Bivic A., Rodriguez‐Boulan E., J. Cell Biol. 1997, 139, 929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Baldus S. E., Engelmann K., Hanisch F. G., Crit. Rev. Clin. Lab. Sci. 2004, 41, 189–231. [DOI] [PubMed] [Google Scholar]
- 166. Brockhausen I., EMBO Rep. 2006, 7, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Guzman‐Aranguez A., Argueso P., Immunol. Lacrimal Gland Tear Film Ocul. Surf. 2010, 8, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Hanisch F. G., Biol. Chem. 2001, 382, 143–149. [DOI] [PubMed] [Google Scholar]
- 169. Tian E., Ten Hagen K. G., Glycoconjugate J. 2009, 26, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kieffer N., Debili N., Wicki A., Titeux M., Henri A., Mishal Z., Breton‐Gorius J., Vainchenker W., Clemetson K. J., J. Biol. Chem. 1986, 261, 15854–15862. [PubMed] [Google Scholar]
- 171. Calvete J. J., Muñiz‐Diaz E., FEBS Lett. 1993, 328, 30–34. [DOI] [PubMed] [Google Scholar]
- 172. Allen A., Feehally J., Adv. Exp. Med. Biol. 1998, 435, 175–183. [DOI] [PubMed] [Google Scholar]
- 173. Baenziger J., Kornfeld S., J. Biol. Chem. 1974, 249, 7270–7281. [PubMed] [Google Scholar]
- 174. Hiki Y., Odani H., Takahashi M., Yasuda Y., Nishimoto A., Iwase H., Shinzato T., Kobayashi Y., Maeda K., Kidney Int. 2001, 59, 1077–1085. [DOI] [PubMed] [Google Scholar]
- 175. Xu L. X., Yan Y., Zhang J. J., Zhang Y., Zhao M. H., Clin. Exp. Immunol. 2005, 142, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Xu L. X., Zhao M. H., Kidney Int. 2005, 68, 167–172. [DOI] [PubMed] [Google Scholar]
- 177. Barratt J., Smith A. C., Feehally J., Nephrology 2007, 12, 275–284. [DOI] [PubMed] [Google Scholar]
- 178. Mellis S. J., Baenziger J. U., J. Biol. Chem. 1983, 258, 11557–11563. [PubMed] [Google Scholar]
- 179. Rudd P. M., Fortune F., Patel T., Parekh R. B., Dwek R. A., Lehner T., Immunology 1994, 83, 99–106. [PMC free article] [PubMed] [Google Scholar]
- 180. Swenson C. D., Patel T., Parekh R. B., Tamma S. M., Coico R. F., Thorbecke G. J., Amin A. R., Eur. J. Immunol. 1998, 28, 2366–2372. [DOI] [PubMed] [Google Scholar]
- 181. Sasaki H., Bothner B., Dell A., Fukuda M., J. Biol. Chem. 1987, 262, 12059–12076. [PubMed] [Google Scholar]
- 182. Tsuda E., Goto M., Murakami A., Akai K., Ueda M., Kawanishi G., Takahashi N., Sasaki R., Chiba H., Ishihara H., Mori M., Tejima S., Endo S., Arata Y., Biochemistry 1988, 27, 5646–5654. [DOI] [PubMed] [Google Scholar]
- 183. Tsuda E., Kawanishi G., Ueda M., Masuda S., Sasaki R., Eur. J. Biochem. 1990, 188, 405–411. [DOI] [PubMed] [Google Scholar]
- 184. Wasley L. C., Timony G., Murtha P., Stoudemire J., Dorner A. J., Caro J., Krieger M., Kaufman R. J., Blood 1991, 77, 2624–2632. [PubMed] [Google Scholar]
- 185. Carew J. A., Quinn S. M., Stoddart J. H., Lynch D. C., J. Clin. Invest. 1992, 90, 2258–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Inoue K., Morita T., Eur. J. Biochem. 1993, 218, 153–163. [DOI] [PubMed] [Google Scholar]
- 187. Maeda H., Hashimoto R. K., Ogura T., Hiraga S., Uzawa H., J. Lipid Res. 1987, 28, 1405–1409. [PubMed] [Google Scholar]
- 188. Spaapen L. J., Bakker J. A., van der Meer S. B., Sijstermans H. J., Steet R. A., Wevers R. A., Jaeken J., J. Inherited Metab. Dis. 2005, 28, 707–714. [DOI] [PubMed] [Google Scholar]
- 189. Wopereis S., Abd Hamid U. M., Critchley A., Royle L., Dwek R. A., Morava E., Leroy J. G., Wilcken B., Lagerwerf A. J., Huijben K. M., Lefeber D. J., Rudd P. M., Wevers R. A., Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 598–607. [DOI] [PubMed] [Google Scholar]
- 190. Cole L. A., Perini F., Birken S., Ruddon R. W., Biochem. Biophys. Res. Commun. 1984, 122, 1260–1267. [DOI] [PubMed] [Google Scholar]
- 191. Hanover J. A., Elting J., Mintz G. R., Lennarz W. J., J. Biol. Chem. 1982, 257, 10172–10177. [PubMed] [Google Scholar]
- 192. Kessler M. J., Mise T., Ghai R. D., Bahl O. P., J. Biol. Chem. 1979, 254, 7909–7914. [PubMed] [Google Scholar]
- 193. Parsons T. F., Bloomfield G. A., Pierce J. G., J. Biol. Chem. 1983, 258, 240–244. [PubMed] [Google Scholar]
- 194. May L. T., Santhanam U., Sehgal P. B., J. Biol. Chem. 1991, 266, 9950–9955. [PubMed] [Google Scholar]
- 195. Parekh R. B., Dwek R. A., Rademacher T. W., Opdenakker G., Van Damme J., Eur. J. Biochem. 1992, 203, 135–141. [DOI] [PubMed] [Google Scholar]
- 196. Afonso A. M., Marshall R. D., Biochem. Soc. Trans. 1979, 7, 170–173. [DOI] [PubMed] [Google Scholar]
- 197. Easton R. L., Patankar M. S., Clark G. F., Morris H. R., Dell A., J. Biol. Chem. 2000, 275, 21928–21938. [DOI] [PubMed] [Google Scholar]
- 198. van Rooijen J. J., Voskamp A. F., Kamerling J. P., Vliegenthart J. F., Glycobiology 1999, 9, 21–30. [DOI] [PubMed] [Google Scholar]
- 199. Williams J., Marshall R. D., van Halbeek H., Vliegenthart J. F., Carbohydr. Res. 1984, 134, 141–155. [DOI] [PubMed] [Google Scholar]
- 200. Rehder D. S., Nelson R. W., Borges C. R., Protein Sci. 2009, 18, 2036–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Wakabayashi H., Natsuka S., Honda M., Naotsuka M., Ito Y., Kajihara J., Hase S., J. Biochem. 2001, 130, 543–552. [DOI] [PubMed] [Google Scholar]
- 202. Baba T., Downs D., Jackson K. W., Tang J., Wang C. S., Biochemistry 1991, 30, 500–510. [DOI] [PubMed] [Google Scholar]
- 203. Landberg E., Huang Y., Stromqvist M., Mechref Y., Hansson L., Lundblad A., Novotny M. V., Pahlsson P., Arch. Biochem. Biophys. 2000, 377, 246–254. [DOI] [PubMed] [Google Scholar]
- 204. Landberg E., Pahlsson P., Krotkiewski H., Stromqvist M., Hansson L., Lundblad A., Arch. Biochem. Biophys. 1997, 344, 94–102. [DOI] [PubMed] [Google Scholar]
- 205. Wang C. S., Dashti A., Jackson K. W., Yeh J. C., Cummings R. D., Tang J., Biochemistry 1995, 34, 10639–10644. [DOI] [PubMed] [Google Scholar]
- 206.R. P. McEver, Ernst Schering Res. Found. Workshop 2004, 137–147. [DOI] [PubMed]
- 207. McEver R. P., Cummings R. D., J. Clin. Invest. 1997, 100, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Karamatic Crew V., Singleton B. K., Green C., Parsons S. F., Daniels G., Anstee D. J., Br. J. Haematol. 2008, 142, 657–667. [DOI] [PubMed] [Google Scholar]
- 209. Zarbock A., Müller H., Kuwano Y., Ley K., J. Leukocyte Biol. 2009, 86, 1119–1124. [DOI] [PubMed] [Google Scholar]
- 210. Rosen S. D., Annu. Rev. Immunol. 2004, 22, 129–156. [DOI] [PubMed] [Google Scholar]
- 211. Sako D., Chang X.‐J., Barone K. M., Vachino G., White H. M., Shaw G., Veldman G. M., Bean K. M., Ahern T. J., Furie B., Cumming D. A., Larsen G. R., Cell 1993, 75, 1179–1186. [DOI] [PubMed] [Google Scholar]
- 212. Somers W. S., Tang J., Shaw G. D., Camphausen R. T., Cell 2000, 103, 467–479. [DOI] [PubMed] [Google Scholar]
- 213. Mitoma J., Petryniak B., Hiraoka N., Yeh J. C., Lowe J. B., Fukuda M., J. Biol. Chem. 2003, 278, 9953–9961. [DOI] [PubMed] [Google Scholar]
- 214. Yeh J. C., Hiraoka N., Petryniak B., Nakayama J., Ellies L. G., Rabuka D., Hindsgaul O., Marth J. D., Lowe J. B., Fukuda M., Cell 2001, 105, 957–969. [DOI] [PubMed] [Google Scholar]
- 215. Maly P., Thall A., Petryniak B., Rogers C. E., Smith P. L., Marks R. M., Kelly R. J., Gersten K. M., Cheng G., Saunders T. L., Camper S. A., Camphausen R. T., Sullivan F. X., Isogai Y., Hindsgaul O., von Andrian U. H., Lowe J. B., Cell 1996, 86, 643–653. [DOI] [PubMed] [Google Scholar]
- 216. Kawashima H., Petryniak B., Hiraoka N., Mitoma J., Huckaby V., Nakayama J., Uchimura K., Kadomatsu K., Muramatsu T., Lowe J. B., Fukuda M., Nat. Immunol. 2005, 6, 1096–1104. [DOI] [PubMed] [Google Scholar]
- 217. Uchimura K., Kadomatsu K., El‐Fasakhany F. M., Singer M. S., Izawa M., Kannagi R., Takeda N., Rosen S. D., Muramatsu T., J. Biol. Chem. 2004, 279, 35001–35008. [DOI] [PubMed] [Google Scholar]
- 218. Xia L., Ju T., Westmuckett A., An G., Ivanciu L., McDaniel J. M., Lupu F., Cummings R. D., McEver R. P., J. Cell Biol. 2004, 164, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Xia L., McEver R. P., Methods Enzymol. 2006, 416, 314–331. [DOI] [PubMed] [Google Scholar]
- 220. Alexander W. S., Viney E. M., Zhang J. G., Metcalf D., Kauppi M., Hyland C. D., Carpinelli M. R., Stevenson W., Croker B. A., Hilton A. A., Ellis S., Selan C., Nandurkar H. H., Goodnow C. C., Kile B. T., Nicola N. A., Roberts A. W., Hilton D. J., Proc. Natl. Acad. Sci. USA 2006, 103, 16442–16447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Szajda S. D., Jankowska A., Zwierz K., Dis. Markers 2008, 25, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Desseyn J. L., Tetaert D., Gouyer V., Gene 2008, 410, 215–222. [DOI] [PubMed] [Google Scholar]
- 223. Gendler S. J., Spicer A. P., Lalani E. N., Duhig T., Peat N., Burchell J., Pemberton L., Boshell M., Taylor‐Papadimitriou J., Am. Rev. Respir. Dis. 1991, 144, S42–47. [DOI] [PubMed] [Google Scholar]
- 224. Taylor‐Papadimitriou J., Burchell J., Miles D. W., Dalziel M., Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1455, 301–313. [DOI] [PubMed] [Google Scholar]
- 225. Cozzi P. J., Wang J., Delprado W., Perkins A. C., Allen B. J., Russell P. J., Li Y., Clin. Exp. Metastasis 2005, 22, 565–573. [DOI] [PubMed] [Google Scholar]
- 226. Li Y., Cozzi P. J., Curr. Cancer Drug Targets 2007, 7, 259–271. [DOI] [PubMed] [Google Scholar]
- 227. Singh P. K., Hollingsworth M. A., Trends Cell Biol. 2006, 16, 467–476. [DOI] [PubMed] [Google Scholar]
- 228. Rose M. C., Voynow J. A., Physiol. Rev. 2006, 86, 245–278. [DOI] [PubMed] [Google Scholar]
- 229. Byrd J. C., Bresalier R. S., Cancer Metastasis Rev. 2004, 23, 77–99. [DOI] [PubMed] [Google Scholar]
- 230. Ringel J., Lohr M., Mol. Cancer 2003, 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Higuchi T., Orita T., Nakanishi S., Katsuya K., Watanabe H., Yamasaki Y., Waga I., Nanayama T., Yamamoto Y., Munger W., Sun H. W., Falk R. J., Jennette J. C., Alcorta D. A., Li H., Yamamoto T., Saito Y., Nakamura M., J. Biol. Chem. 2004, 279, 1968–1979. [DOI] [PubMed] [Google Scholar]
- 232. Gendler S. J., Spicer A. P., Annu. Rev. Physiol. 1995, 57, 607–634. [DOI] [PubMed] [Google Scholar]
- 233. Eckhardt A. E., Timpte C. S., DeLuca A. W., Hill R. L., J. Biol. Chem. 1997, 272, 33204–33210. [DOI] [PubMed] [Google Scholar]
- 234. Perez B. H., Gipson I. K., Exp. Eye Res. 2008, 87, 400–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Yin B. W., Dnistrian A., Lloyd K. O., Int. J. Cancer 2002, 98, 737–740. [DOI] [PubMed] [Google Scholar]
- 236. Burchell J., Gendler S., Taylor‐Papadimitriou J., Girling A., Lewis A., Millis R., Lamport D., Cancer Res. 1987, 47, 5476–5482. [PubMed] [Google Scholar]
- 237. Lloyd K. O., Burchell J., Kudryashov V., Yin B. W., Taylor‐Papadimitriou J., J. Biol. Chem. 1996, 271, 33325–33334. [DOI] [PubMed] [Google Scholar]
- 238. Taylor‐Papadimitriou J., Peterson J. A., Arklie J., Burchell J., Ceriani R. L., Bodmer W. F., Int. J. Cancer 1981, 28, 17–21. [DOI] [PubMed] [Google Scholar]
- 239. Ho J. J., Bi N., Siddiki B., Chung Y. S., Yuan M., Kim Y. S., Cancer Res. 1993, 53, 884–890. [PubMed] [Google Scholar]
- 240. Jass J. R., Allison L. J., Edgar S. G., J. Pathol. 1995, 176, 143–149. [DOI] [PubMed] [Google Scholar]
- 241. Cao Y., Schlag P. M., Karsten U., Virchows Arch. 1997, 431, 159–166. [DOI] [PubMed] [Google Scholar]
- 242. Hanisch F. G., Tumor Biol. 1998, 19, 111–117. [Google Scholar]
- 243. Sasaki M., Yamato T., Nakanuma Y., Pathol. Int. 1999, 49, 325–331. [DOI] [PubMed] [Google Scholar]
- 244. Cao Y., Karsten U., Otto G., Bannasch P., Virchows Arch. 1999, 434, 503–509. [DOI] [PubMed] [Google Scholar]
- 245. Amaya S., Sasaki M., Watanabe Y., Tsui W. M., Tsuneyama K., Harada K., Nakanuma Y., Histol. Histopathol. 2001, 38, 550–560. [DOI] [PubMed] [Google Scholar]
- 246. Flucke U., Zirbes T. K., Schroder W., Monig S. P., Koch V., Schmitz K., Thiele J., Dienes H. P., Holscher A. H., Baldus S. E., Anticancer Res. 2001, 21, 2189–2193. [PubMed] [Google Scholar]
- 247. Kim G. E., Bae H. I., Park H. U., Kuan S. F., Crawley S. C., Ho J. J., Kim Y. S., Gastroenterology 2002, 123, 1052–1060. [DOI] [PubMed] [Google Scholar]
- 248. Freire T., Medeiros A., Reis C. A., Real F. X., Osinaga E., Oncol. Rep. 2003, 10, 1577–1585. [PubMed] [Google Scholar]
- 249. Conze T., Carvalho A. S., Landegren U., Almeida R., Reis C. A., David L., Soderberg O., Glycobiology 2010, 20, 199–206. [DOI] [PubMed] [Google Scholar]
- 250. Tashiro Y., Yonezawa S., Kim Y. S., Sato E., Hum. Pathol. 1994, 25, 364–372. [DOI] [PubMed] [Google Scholar]
- 251. Robbe‐Masselot C., Herrmann A., Carlstedt I., Michalski J. C., Capon C., Glycoconjugate J. 2008, 25, 213–224. [DOI] [PubMed] [Google Scholar]
- 252. Robbe‐Masselot C., Herrmann A., Maes E., Carlstedt I., Michalski J. C., Capon C., J. Proteome Res. 2009, 8, 702–711. [DOI] [PubMed] [Google Scholar]
- 253. De Bolos C., Garrido M., Real F. X., Gastroenterology 1995, 109, 723–734. [DOI] [PubMed] [Google Scholar]
- 254. Pereira M. B., Dias A. J., Reis C. A., Schmitt F. C., J. Clin. Pathol. 2001, 54, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Freire T., Bay S., von Mensdorff‐Pouilly S., Osinaga E., Cancer Res. 2005, 65, 7880–7887. [DOI] [PubMed] [Google Scholar]
- 256. Hanski C., Drechsler K., Hanisch F.‐G., Sheehan J., Manske M., Ogorek D., Klussmann E., Hanski M.‐L., Blank M., Xing P.‐X., McKenzie I. F. C., Devine P. L., Riecken E.‐O., Cancer Res. 1993, 53, 4082–4088. [PubMed] [Google Scholar]
- 257. Lloyd K. O., Glycoconjugate J. 2000, 17, 531–541. [DOI] [PubMed] [Google Scholar]
- 258. Tsuji T., Osawa T., Carbohydr. Res. 1986, 151, 391–402. [DOI] [PubMed] [Google Scholar]
- 259. Carraway K. L., Theodoropoulos G., Kozloski G. A., Carothers Carraway C. A., Future Oncol. 2009, 5, 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Carraway K. L., Price‐Schiavi S. A., Komatsu M., Jepson S., Perez A., Carraway C. A., J. Mammary Gland Biol. Neoplasia 2001, 6, 323–337. [DOI] [PubMed] [Google Scholar]
- 261. Wesseling J., van der Valk S. W., Vos H. L., Sonnenberg A., Hilkens J., J. Cell Biol. 1995, 129, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Itoh Y., Kamata‐Sakurai M., Denda‐Nagai K., Nagai S., Tsuiji M., Ishii‐Schrade K., Okada K., Goto A., Fukayama M., Irimura T., Glycobiology 2008, 18, 74–83. [DOI] [PubMed] [Google Scholar]
- 263. Codington J. F., Cooper A. G., Miller D. K., Slayter H. S., Brown M. C., Silber C., Jeanloz R. W., J. Natl. Cancer Inst. 1979, 63, 153–161. [PubMed] [Google Scholar]
- 264. Lamblin G., Degroote S., Perini J. M., Delmotte P., Scharfman A., Davril M., Lo‐Guidice J. M., Houdret N., Dumur V., Klein A., Rousse P., Glycoconjugate J. 2001, 18, 661–684. [DOI] [PubMed] [Google Scholar]
- 265. Berger E. G., Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1455, 255–268. [DOI] [PubMed] [Google Scholar]
- 266. Bird G. W., Ann. N. Y. Acad. Sci. 1974, 234, 129–144. [DOI] [PubMed] [Google Scholar]
- 267. Cartron J. P., Andreu G., Cartron J., Bird G. W., Salmon C., Gerbal A., Eur. J. Biochem. 1978, 92, 111–119. [DOI] [PubMed] [Google Scholar]
- 268. Cartron J. P., Cartron J., Andreu G., Salmon C., Bird G. W., Lancet 1978, 311, 856–857. [DOI] [PubMed] [Google Scholar]
- 269. Thurnher M., Clausen H., Fierz W., Lanzavecchia A., Berger E. G., Eur. J. Immunol. 1992, 22, 1835–1842. [DOI] [PubMed] [Google Scholar]
- 270. Thurnher M., Rusconi S., Berger E. G., J. Clin. Invest. 1993, 91, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Vigorito E., Robles A., Balter H., Nappa A., Goni F., Appl. Radiat. Isot. 1995, 46, 975–979. [DOI] [PubMed] [Google Scholar]
- 272. Springer G. F., J. Mol. Med. 1997, 75, 594–602. [DOI] [PubMed] [Google Scholar]
- 273. Springer G. F., Science 1984, 224, 1198–1206. [DOI] [PubMed] [Google Scholar]
- 274. Desai P. R., Transfus. Med. Rev. 2000, 14, 312–325. [DOI] [PubMed] [Google Scholar]
- 275. Numa F., Tsunaga N., Michioka T., Nawata S., Ogata H., Kato H., J. Obstet. Gynaecol. 1995, 21, 385–389. [DOI] [PubMed] [Google Scholar]
- 276. Inoue M., Ogawa H., Nakanishi K., Tanizawa O., Karino K., Endo J., Obstet. Gynecol. 1990, 75, 1032–1036. [PubMed] [Google Scholar]
- 277. Laack E., Nikbakht H., Peters A., Kugler C., Jasiewicz Y., Edler L., Hossfeld D. K., Schumacher U., Am. J. Pathol. 2002, 160, 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Konno A., Hoshino Y., Terashima S., Motoki R., Kawaguchi T., Clin. Exp. Metastasis 2002, 19, 61–70. [DOI] [PubMed] [Google Scholar]
- 279. Fernández Madrid F., Tang N., Alansari H., Karvonen R. L., Tomkiel J. E., Autoimmun. Rev. 2005, 4, 230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Kakeji Y., Tsujitani S., Mori M., Maehara Y., Sugimachi K., Cancer 1991, 68, 2438–2442. [DOI] [PubMed] [Google Scholar]
- 281. Julien S., Adriaenssens E., Ottenberg K., Furlan A., Courtand G., Vercoutter‐Edouart A. S., Hanisch F. G., Delannoy P., Le Bourhis X., Glycobiology 2006, 16, 54–64. [DOI] [PubMed] [Google Scholar]
- 282. Dalziel M., Whitehouse C., McFarlane I., Brockhausen I., Gschmeissner S., Schwientek T., Clausen H., Burchell J. M., Taylor‐Papadimitriou J., J. Biol. Chem. 2001, 276, 11007–11015. [DOI] [PubMed] [Google Scholar]
- 283. Saeland E., van Vliet S. J., Bäckström M., van den Berg V. C. M., Geijtenbeek T. B. H., Meijer G. A., van Kooyk Y., Cancer Immunol. Immunother. 2007, 56, 1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Byrne S. L., Leverence R., Klein J. S., Giannetti A. M., Smith V. C., MacGillivray R. T., Kaltashov I. A., Mason A. B., Biochemistry 2006, 45, 6663–6673. [DOI] [PubMed] [Google Scholar]
- 285. Liu W., Ramachandran V., Kang J., Kishimoto T. K., Cummings R. D., McEver R. P., J. Biol. Chem. 1998, 273, 7078–7087. [DOI] [PubMed] [Google Scholar]
- 286. Endo T., Acta Myol. 2007, 26, 165–170. [PMC free article] [PubMed] [Google Scholar]
- 287. Schietinger A., Philip M., Yoshida B. A., Azadi P., Liu H., Meredith S. C., Schreiber H., Science 2006, 314, 304–308. [DOI] [PubMed] [Google Scholar]
- 288. Lundström M., Jeansson S., Olofsson S., Virology 1987, 161, 395–402. [DOI] [PubMed] [Google Scholar]
- 289. Levy M., Berger J., Am. J. Kidney Dis. 1988, 12, 340–347. [DOI] [PubMed] [Google Scholar]
- 290. D’Amico G., Am. J. Kidney Dis. 2000, 36, 227–237. [DOI] [PubMed] [Google Scholar]
- 291. Kiryluk K., Gharavi A. G., Izzi C., Scolari F., Nephrol. Dial. Transplant 2010, 25, 336–338. [DOI] [PubMed] [Google Scholar]
- 292. Paterson A. D., Liu X.‐Q., Wang K., Magistroni R., Song X., Kappel J., Klassen J., Cattran D., George‐Hyslop P. St., Pei Y., J. Am. Soc. Nephrol. 2007, 18, 2408–2415. [DOI] [PubMed] [Google Scholar]
- 293. Gharavi A. G., Yan Y., Scolari F., Schena F. P., Frasca G. M., Ghiggeri G. M., Cooper K., Amoroso A., Viola B. F., Battini G., Caridi G., Canova C., Farhi A., Subramanian V., Nelson‐Williams C., Woodford S., Julian B. A., Wyatt R. J., Lifton R. P., Nat. Genet. 2000, 26, 354–357. [DOI] [PubMed] [Google Scholar]
- 294. Bisceglia L., Cerullo G., Forabosco P., Torres D. D., Scolari F., Di Perna M., Foramitti M., Amoroso A., Bertok S., Floege J., Mertens P. R., Zerres K., Alexopoulos E., Kirmizis D., Ermelinda M., Zelante L., Schena F. P., Am. J. Hum. Genet. 2006, 79, 1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Julian B. A., Novak J., Curr. Opin. Nephrol. Hypertens. 2004, 13, 171–179. [DOI] [PubMed] [Google Scholar]
- 296. Allen A. C., Topham P. S., Harper S. J., Feehally J., Nephrol. Dial. Transplant. 1997, 12, 701–706. [DOI] [PubMed] [Google Scholar]
- 297. Qin W., Zhou Q., Yang L. C., Li Z., Su B. H., Luo H., Fan J. M., J. Intern. Med. 2005, 258, 467–477. [DOI] [PubMed] [Google Scholar]
- 298. Inoue T., Sugiyama H., Hiki Y., Takiue K., Morinaga H., Kitagawa M., Maeshima Y., Fukushima K., Nishizaki K., Akagi H., Narimatsu Y., Narimatsu H., Makino H., Clin. Immunol. 2010, 136, 447–455. [DOI] [PubMed] [Google Scholar]
- 299. Yamada K., Kobayashi N., Ikeda T., Suzuki Y., Tsuge T., Horikoshi S., Emancipator S. N., Tomino Y., Nephrol. Dial. Transplant. 2010, 25, 3890–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Li G. S., Zhang H., Lv J. C., Shen Y., Wang H. Y., Kidney Int. 2007, 71, 448–453. [DOI] [PubMed] [Google Scholar]
- 301. Malycha F., Eggermann T., Hristov M., Schena F. P., Mertens P. R., Zerres K., Floege J., Eitner F., Nephrol. Dial. Transplant. 2009, 24, 321–324. [DOI] [PubMed] [Google Scholar]
- 302. Novak J., Julian B. A., Tomana M., Mesteck J., J. Clin. Immunol. 2001, 21, 310–327. [DOI] [PubMed] [Google Scholar]
- 303. Leung J. C., Tsang A. W., Chan L. Y., Tang S. C., Lam M. F., Lai K. N., J. Lab. Clin. Med. 2002, 140, 398–406. [DOI] [PubMed] [Google Scholar]
- 304. Nyame K., Cummings R. D., Damian R. T., J. Parasitol. 1988, 74, 562–572. [PubMed] [Google Scholar]
- 305. Alvarez Errico D., Medeiros A., Míguez M., Casaravilla C., Malgor R., Carmona C., Nieto A., Osinaga E., Exp. Parasitol. 2001, 98, 100–109. [DOI] [PubMed] [Google Scholar]
- 306. O’Connor R. M., Kim K., Khan F., Ward H. D., Infect. Immun. 2003, 71, 6027–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Bhat N., Joe A., PereiraPerrin M., Ward H. D., J. Biol. Chem. 2007, 282, 34877–34887. [DOI] [PubMed] [Google Scholar]
- 308. Hansen J. E., Clausen H., Nielsen C., Teglbjaerg L. S., Hansen L. L., Nielsen C. M., Dabelsteen E., Mathiesen L., Hakomori S. I., Nielsen J. O., J. Virol. 1990, 64, 2833–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Hansen J. E., Nielsen C., Arendrup M., Olofsson S., Mathiesen L., Nielsen J. O., Clausen H., J. Virol. 1991, 65, 6461–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Johnson D. C., Spear P. G., Cell 1983, 32, 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Olofsson S., Blomberg J., Lycke E., Arch. Virol. 1981, 70, 321–329. [DOI] [PubMed] [Google Scholar]
- 312. Wenske E. A., Courtney R. J., J. Virol. 1983, 46, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Shida H., Dales S., Virology 1981, 111, 56–72. [DOI] [PubMed] [Google Scholar]
- 314. Gruber C., Levine S., J. Gen. Virol. 1985, 66 ( Pt 3), 417–432. [DOI] [PubMed] [Google Scholar]
- 315. Niemann H., Klenk H. D., J. Mol. Biol. 1981, 153, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Pinter A., Honnen W. J., J. Virol. 1988, 62, 1016–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Holmes K. V., Doller E. W., Sturman L. S., Virology 1981, 115, 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Bernstein H. B., Tucker S. P., Hunter E., Schutzbach J. S., Compans R. W., J. Virol. 1994, 68, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Kirkeby S., Martel C. J. M., Aasted B., Virus Res. 2009, 144, 225–232. [DOI] [PubMed] [Google Scholar]
- 320. Springer G. F., Desai P. R., Tegtmeyer H., Spencer B. D., Scanlon E. F., Ann. N. Y. Acad. Sci. 1993, 690, 355–357. [DOI] [PubMed] [Google Scholar]
- 321. Springer G. F., Desai P. R., Wise W., Carlstedt S. C., Tegtmeyer H., Stein H., Scanlon E. F. in Immunodiagnosis of Cancer, 2nd ed. (Eds.: R. B. Herberman, D. W. Mercer), Marcel Dekker, New York, 1990, pp. 587–612. [Google Scholar]
- 322. Galonić D. P., Gin D. Y., Nature 2007, 446, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Napoletano C., Rughetti A., Agervig Tarp M. P., Coleman J., Bennett E. P., Picco G., Sale P., Denda‐Nagai K., Irimura T., Mandel U., Clausen H., Frati L., Taylor‐Papadimitriou J., Burchell J., Nuti M., Cancer Res. 2007, 67, 8358–8367. [DOI] [PubMed] [Google Scholar]
- 324. Helling F., Shang A., Calves M., Zhang S., Ren S., Yu R. K., Oettgen H. F., Livingston P. O., Cancer Res. 1994, 54, 197–203. [PubMed] [Google Scholar]
- 325. Slovin S. F., Ragupathi G., Musselli C., Olkiewicz K., Verbel D., Kuduk S. D., Schwarz J. B., Sames D., Danishefsky S., Livingston P. O., Scher H. I., J. Clin. Oncol. 2003, 21, 4292–4298. [DOI] [PubMed] [Google Scholar]
- 326. Kaiser A., Gaidzik N., Westerlind U., Kowalczyk D., Hobel A., Schmitt E., Kunz H., Angew. Chem. 2009, 121, 7688–7692; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 7551–7555. [DOI] [PubMed] [Google Scholar]
- 327. Kensil C. R., Barrett C., Kushner N., Beltz G., Storey J., Patel U., Recchia J., Aubert A., Marciani D., J. Am. Vet. Med. Assoc. 1991, 199, 1423–1427. [PubMed] [Google Scholar]
- 328. Helling F., Zhang S., Shang A., Adluri S., Calves M., Koganty R., Longenecker B. M., Yao T. J., Oettgen H. F., Livingston P. O., Cancer Res. 1995, 55, 2783–2788. [PubMed] [Google Scholar]
- 329. Ragupathi G., Cancer Immunol. Immunother. 1996, 43, 152–157. [DOI] [PubMed] [Google Scholar]
- 330. van Vliet S. J., Saeland E., van Kooyk Y., Trends Immunol. 2008, 29, 83–90. [DOI] [PubMed] [Google Scholar]
- 331. van Vliet S. J., Gringhuis S. I., Geijtenbeek T. B., van Kooyk Y., Nat. Immunol. 2006, 7, 1200–1208. [DOI] [PubMed] [Google Scholar]
- 332. Steinman R. M., Hawiger D., Nussenzweig M. C., Annu. Rev. Immunol. 2003, 21, 685–711. [DOI] [PubMed] [Google Scholar]
- 333. Geijtenbeek T. B. H., van Vliet S. J., Engering A., ’t Hart B. A., van Kooyk Y., Annu. Rev. Immunol. 2004, 22, 33–54. [DOI] [PubMed] [Google Scholar]
- 334. Kunz H., Wernig P., Schilling M., Marz J., Unverzagt C., Birnbach S., Lang U., Waldmann H., Environ. Health Perspect. 1990, 88, 247–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Paulsen H., Bielfeldt T., Peters S., Meldal M., Bock K., Liebigs Ann. Chem. 1994, 369–379. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer‐reviewed, but not copy‐edited or typeset. They are made available as submitted by the authors.
miscellaneous_information