

Abstract
Over a century since its development, the analytical technique of mass spectrometry is blooming more than ever, and applied in nearly all aspects of the natural and life sciences. In the last two decades mass spectrometry has also become amenable to the analysis of proteins and even intact protein complexes, and thus begun to make a significant impact in the field of structural biology. In this Review, we describe the emerging role of mass spectrometry, with its different technical facets, in structural biology, focusing especially on structural virology. We describe how mass spectrometry has evolved into a tool that can provide unique structural and functional information about viral‐protein and protein‐complex structure, conformation, assembly, and topology, extending to the direct analysis of intact virus capsids of several million Dalton in mass. Mass spectrometry is now used to address important questions in virology ranging from how viruses assemble to how they interact with their host.
Keywords: mass spectrometry, proteomics, structural biology, structure elucidation, viruses
Viruses on mass: Structural biology has profited greatly from modern biomolecular mass spectrometry (MS). In this Review a variety of mass spectrometry techniques, including proteomics, H/D exchange, chemical labeling, and native and ion mobility MS, are presented along with how they have contributed to structural‐biology investigations, in particular of virus structure, assembly, and dynamics (see picture).

1. Introduction
Mass spectrometry (MS) is a very powerful analytical technique known to and used by most researchers in the natural and life sciences. The technique behind MS dates back about a hundred years, when people like Thomson and Aston were amongst the first to separate particles of different mass to charge ratios (m/z), discovering isotopes of rare gases and other elements. For half a century, MS remained primarily in the hands of physicists, and often shrouded in secrecy, as it was also used to enrich uranium for the Manhattan project.1 Around the 1960s, the technique began to be adopted by chemists, for instance working in the petroleum industry, to investigate the chemical nature of the compounds formed in refinement processes. In the domain of chemistry, MS subsequently came to bloom particularly in the chemical analysis of unknown compounds (assisted by the coupling to separation technologies, such as gas and liquid chromatography) and as a research area by itself, termed organic MS, concerned with studying structures and fragmentation mechanisms of ions, and ion–molecule reactions.2
Up to the 1980s, the impossibility of transferring larger molecules as intact gaseous ions into the vacuum of the mass spectrometer represented a serious bottleneck in the analysis. This problem was gradually solved by new desorption techniques, cumulating in the introduction of matrix assisted laser desorption/ionization (MALDI)3 and electrospray ionization (ESI) MS.4 These new ionization methods, together with breakthrough innovations in instrumentation, really opened up applications in biology, nanotechnology, polymer science, and medicine, as well as many other fields in the natural and life sciences.
Focusing on the life sciences, MS became a key technology used for peptide sequencing,5 through which the identity of a protein can be revealed. The speed and sensitivity in MS allows the qualitative and quantitative analysis of the protein content of a whole cell or tissue, nowadays termed proteomics.6 Similarly, the chemical analysis of the small‐molecule content of a cell or body fluid can now be performed using MS, and is termed metabolomics.7 New desorption techniques and special dedicated mass spectrometers even allow MS‐based imaging by the position‐sensitive measurement of compound distributions (protein, neuropeptide, metabolite, drug molecule) in a tissue or organelle.8 Furthermore, ambient desorption technologies have been introduced to directly sample molecular compounds of surfaces and organisms.9
The importance of MALDI and ESI for these revolutionary developments was recognized by awarding the Nobel Prize in Chemistry to the late John B. Fenn and to Koichi Tanaka. Fenn entitled his Nobel Lecture “Electrospray Wings for Molecular Elephants”10 as ESI (and MALDI) expanded the mass regime attainable for MS at least 1000‐fold. Soon after the introduction of ESI, it became apparent that not only the mass of intact proteins but also the tertiary and quaternary structure of these proteins could be partially retained and therefore analyzed. This potential was evidenced by the early discovery that the noncovalent complex between myoglobin and its heme cofactor could be kept intact in the gas phase.11 Groundbreaking work of Standing et al.,12, 13 Smith, Loo, et al.,14, 15 Robinson et al.,16–19 and our own group20, 21 in the area of MS on intact macromolecular complexes led to a new very powerful tool in structural biology, now termed native mass spectrometry.22
Next to the well‐established peptide‐sequencing approach and native MS, modern techniques have many other facets relevant for the analysis of proteins in numerous ways. Especially, H/D (hydrogen/deuterium) exchange, and chemical labeling of solvent‐accessible amino acids in combination with MS or cross‐linking MS can add to our knowledge about protein structure–function relationships. In this Review, we will describe how such tools in MS can be used to study several aspects of protein structure and function, focusing in particular on the biochemical and biophysical properties of viruses and viral particles.
Viruses are ideal model systems to study the assembly of protein complexes, since the viral protein shells, capsids, often have the amazing ability to self‐organize their folding and assembly even in vitro without the help of chaperones.23 Moreover, their natural capacity of encapsulating material, that is, the viral genome, makes virus capsids an interesting target for nanotechnological applications that extend far beyond drug delivery.24 The detailed biophysical and biochemical characterization of the virus assembly and maturation processes is crucial, as such data may potentially be used to interfere with viral infection.25 Technically, studying virus assemblies is rather challenging as the structures formed can be very large, hampering analysis by conventional structural‐biology techniques such as protein X‐ray crystallography and NMR spectroscopy. Another problem is posed by the transient nature of the intermediates formed during assembly and/or maturation impeding their purification and analysis.26 Although virus structure, dynamics, and assembly has been studied for decades, in recent years MS has entered this area of research and tackled several important questions that were less accessible by other means.27 Proteomics approaches were early on used to study the “new” SARS virus,28 but MS is also an emerging method to reveal the constituents of a virus, the stoichiometry of the viral structural proteins, virus assembly and the corresponding intermediates.27 After introducing briefly a few general concepts in structural virology, we describe how modern MS can assist in virus characterization, especially focusing on structural aspects.
2. General Concepts in Structural Virology
Viruses are infectious agents that can replicate inside a host.29 With sizes from nm to μm, most viruses are invisible under the light microscope.30–32 Viruses are ubiquitous; they appear in archaea, bacteria, plants, and animals.33–38 Estimates suggest that after prokaryotes, viruses account for the second largest amount of biomass on earth.39 Amongst the viruses, those hosting in bacteria are most abundant and called phages. The viral genome encodes all viral proteins necessary for replication in the adequate host, which can range from several hundred to just a few different proteins. Owing to their inability to reproduce independently, viruses are often not regarded as a form of life.40 However, the discussion is still ongoing and was recently fueled by the discovery of giant mimiviruses.30, 41 Furthermore, the evolutionary ancestry of viruses is still unclear: did they arise from pieces of nucleic acids replicating in cells; or reduce from cellular organisms?42, 43
The ubiquity of viruses amongst all species already foreshadows a broad diversity complicating viral classification. Generally, viruses are distinguished based on either their host organism or morphogenetic characteristics and genome organization.44–47 The genome can be encoded by both RNA or DNA, in single‐ or double‐stranded (ss and ds) form as exemplified in Figure 1. The information can be located on (+) or (−) strands and in some cases requires reverse transcription for replication. Viruses can contain single or multiple pieces of nucleic acid in linear or circular form. The real evolutionary relationship between viruses is often difficult to obtain from one single feature, complicating virus taxonomy. The viral genome is typically enclosed in a protein shell termed the nucleocapsid. This capsid can be helical, icosahedral, or more complex in structure. The nucleocapsid alone can facilitate host‐cell attachment and entry, but especially eukaryotic viruses are often enveloped with a lipid bilayer containing the adaptor proteins.48
Figure 1.
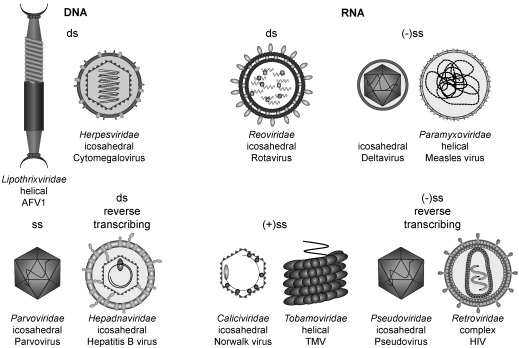
Virus classification is largely based on the type and organization of the nucleic acid. A selection of virus families and their corresponding morphologies are depicted.
2.1. Infection and Lifecycle
To replicate, viruses first have to recognize the host and introduce their genome into the cell. Next, the protein and nucleic acid machinery of the cell is taken over to produce the viral constituents, finally comes the assembly and release of the infectious virus. After successful attachment to the host cell, the nucleocapsid can enter the cytosol by various mechanisms (Figure 2), such as, membrane fusion or phagocytosis often used by eukaryotic viruses.37, 49, 50 Bacteriophages generally inject their genome directly into the cell,51, 52 whereas most other viruses release the genome from the capsid at the pore complexes of the cell nucleus.53 An exception are (+)ssRNA viruses, which also uncoat in the cytoplasm, but replicate their genome outside the nucleus in close proximity to the membrane.54 Some viruses are incorporated metastably into the host genome, for example retroviruses and lysogenic phages. Replication of such proviruses is then induced spontaneously or after a specific trigger.55–57 Generally following infection, viruses take control of the host‐cell machineries to facilitate their own replication.58 At late stages of infection, the structural proteins either assemble around new copies of nucleic acid or assemble independently and package the nucleic acid afterwards.59–61 Eventually, mature virions are released by budding like vesicles or the cell can rupture as a consequence of the viral load.62, 63 The virus can then spread and infect new cells, reinitiating the cycle. Also multiple ways of replication occur during the lifecycle of different viruses, an important step is usually maturation, signaling complete assembly of infectious particles that are ready for release. This process includes a variety of biochemical adaptations such as conformational changes triggered by the nucleic acid incorporated, attachment of auxiliary proteins, or protein posttranslational modifications, such as phosphorylation, glycosylation, proteolysis, and cross‐linking.64–68 Viruses represent a strong selective pressure on the host. Resistant cells have an evolutionary advantage by increased viability. As a consequence, viruses evolve by constantly altering their genome. RNA viruses, in particular, often show a high mutagenesis rate enforcing a constant struggle for survival.69 Another mechanism which results in an increased virulence and a switch in species specificity is the recombination of partial genomes from related viral strains, as is common in influenza viruses.70, 71
Figure 2.
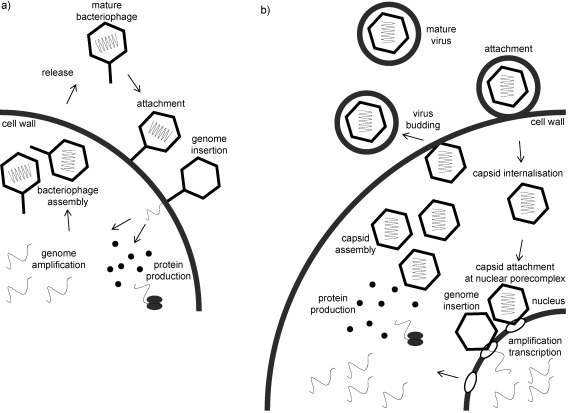
Viral lifecycle: a) Prokaryotic viruses (phages) attach to the host cell and directly inject their genome as shown in this case for a tailed phage. Then, the genome is amplified and proteins transcribed in the cytosol. In case of tailed phages, an empty capsid forms, then the genome is incorporated followed by maturation. The assembled phages accumulate in the cytoplasm until the cell ruptures. b) Eukaryotic viruses transfer their capsid into the cytoplasm. Internalization can occur by membrane fusion in the case of enveloped viruses. At the nucleus the capsid disintegrates and releases its genome, which is reproduced in the nucleus. Protein synthesis and assembly take place in the cytoplasm. For (+)ssRNA viruses the genome is also amplified in the cytoplasm. After assembly and possibly maturation, the virus is released from the cell through budding or destructing the cell.
2.2. Capsid Assembly and Structure
The viral nucleocapsid is of crucial importance in the viral lifecycle since it encapsulates and protects the viral genome. Especially their high efficiency to self‐assemble, their strength, and efficiency in nucleic acid packaging mark nucleocapsids as intriguing structures. Typically, the capsid shell is formed by multiple copies of one or a few different structural proteins,72, 73 although decorations with other proteins are common in non‐enveloped viruses.74 These attached proteins can increase the capsid stability, or serve a role during infection and genome packaging. The high copy number of a limited set of small proteins beneficially decreases the length of nucleic acid needed to encode for them, requiring less space for encapsulation.72 This illustrates some of the brilliant and efficient principles underlying virus structure and function.
Many capsid proteins (cp) can readily be produced recombinantly in high quantities and represent thus ideal model systems to study protein (self‐)assembly.23 Even though, the building blocks in capsid assembly are different for certain viruses, their formation is generally in agreement with nucleation theory as has also been proposed in amyloid assembly.73, 75, 76 First, an assembly nucleus has to be formed, which may be an oligomeric assembly or just a conformationally changed cp monomer. After formation of this nucleus, further building blocks attach to it until the capsid is completed (Figure 3). Under conditions of efficient assembly, the nucleation is generally the rate‐limiting step and only a small fraction of the proteins are in this intermediate state. The following elongation steps take place at a much higher rate leading to immediate propagation of the nucleus to a capsid. Therefore, the intermediate oligomeric species forming the assembly nucleus are typically only present in trace amounts under assembly conditions.26, 73, 76, 77 For some viruses, it is possible to change the solution conditions favoring over‐nucleation, whereby intermediates become kinetically trapped. Still, even under such conditions, processes such as protein mis‐folding and aggregation, can further hamper the detection of these intermediates.67, 78–80 A low nucleation rate ensures effective capsid assembly. Nucleation can be triggered by increasing concentration or posttranslational modifications to secure a sufficiently high titer of cp in the cell. Also, interactions with the nucleic acid can facilitate nucleus formation.78, 81, 82
Figure 3.
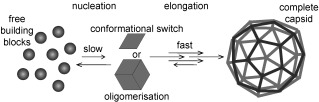
Model for nucleated assembly: Conformational change or oligomer formation can result in nucleation. The nucleus formation is a slow reaction, whereas the subsequent addition of building blocks proceeds fast towards capsid completion. In cases where the nucleation is a fast process, assembly intermediates accumulate because of overnucleation and few capsids are formed.73
The corresponding viral capsids are often highly stable towards changes in environment and can resist, for example, extreme pH values, high concentrations of denaturants and organic solvents, dilution to very low concentrations that don′t facilitate assembly, and even dehydration.83–87 This effect is reflected by a strong hysteresis observed in virus dissociation experiments. Theoretical and experimental results suggest that the interaction between individual building blocks is rather weak and cannot account for the high apparent stability.73, 88 However, in the capsid the binding energies of the subunits add up, explaining the pseudo‐stability of the capsids under conditions where assembly typically does not occur. Nevertheless, theory suggests that in such a pseudo‐equilibrium, there are always some free building blocks in solution that could dynamically exchange with proteins in the capsid. This process has been termed “capsid breathing” and some indirect experimental evidence has been described supporting this model.88–92 We like to note that the term “breathing” has also been used to describe the significant conformational changes that can occur in viral cp proteins, sometimes even resulting in transient externalization of domains that, according to structural models, are on the inside.91, 93
Most commonly, either helical or icosahedral capsid structures are observed, which both allow the formation of a regular shell with multiple copies of a single cp as a result of the high symmetry.72 The prototype of a helical virus is the tobacco mosaic virus (TMV). In this virus, cp monomers assemble around the RNA and the length of the genome defines the nucleocapsid size.94, 95 Icosahedral structures allow the complete closure of the shell using just one type of protein.72 Additionally, the almost spherical structure reduces the surface area relative to the enclosed volume. An icosahedron consists of 12 vertices, 20 faces, and 30 edges corresponding to the different symmetry axis (5‐, 3‐, and 2‐fold, respectively). At least 30, generally dimeric, building blocks are required to form the smallest possible icosahedron, where all proteins are located in pentamers. Larger capsids are formed by addition of hexamers. Only certain numbers of hexamers can be inserted to produce a perfect icosahedron reflected by the triangulation number (T): T=h 2 + h k + k 2 where h and k can be any positive integer and T=2 is therefore not allowed. The number of building blocks corresponds to 30 T. Even though viral capsids can be built up by a single cp, the surrounding contacts between subunits in hexamers and pentamers are different.96, 97 However, the conformational changes to compensate this are often marginal, yielding cp structures that are quasi‐equivalent. Additional hexamers may be introduced in a ring‐like fashion leading to prolate capsids as in bacteriophage Phi29.98, 99 Complex viruses often deviate from icosahedral symmetry, for example the HIV cp typically forms conical, but also rod‐shaped capsids.100
3. Mass Spectrometry in Structural Virology
Next, we describe how biochemical and biophysical properties of viruses can be studied by modern MS. Before focusing on some case studies we first summarize some of the key technologies used, in four boxes: “proteomics”, “native and ion mobility mass spectrometry”, “H/D‐exchange mass spectrometry”, and “chemical‐labeling approaches coupled to mass spectrometry”. These refer the readers to background Reviews on these individual methods. When applied to studying virus structure, dynamics and assembly, the information obtained by these four approaches is schematically summarized in Figure 4.
Figure 4.

Applications of mass spectrometry in structural virology: Illustrative examples on how modern MS has recently contributed to structural virology. Examples include the applications of MS for virus protein identification, including their posttranslational modifications and the structural analysis of viruses using H/D‐exchange MS and native ion mobility MS. Left: from top to bottom is illustrated how proteomics approaches can be used to probe the composition of a virus. Proteins of the SARS coronavirus were identified early on by MS, the detected peptides of the spike protein S1 are shown (mapped onto the structure model 1Q4Z).143 Beneath, schematic illustrations of how proteomics was used to map the precise disposition of heterogeneous glycosylation patterns in the major HIV surface protein,65 multiple phosphorylation sites in the HBV cp,157 proteolytic cleavage sites in bacteriophage P22 gp4,193 and the location of cross‐links like in HK97 maturation.67, 165 Right: How MS can be used to obtain more structural information, from top to bottom: how H/D exchange and chemical labels, for example, cross‐linking were used to dissect a crucial step in HIV capsid maturation.64 How native and ion‐mobility MS provide information on the stoichiometry of an assembly, in favorable cases even the binding affinity,160 the amount of material encapsulated,184 and the shape of a viral protein complex.172
Box 1: Proteomics MS‐based proteomics is currently the most powerful tool to obtain sequence information of proteins.6 Common practice in proteome analyses is the sequencing of proteolytic peptides obtained through fragmentation by collision‐induced dissociation (CID) and/or electron transfer dissociation (ETD).101, 102 These fragmentation spectra are then searched against large protein‐sequence databases containing the predicted spectra, derived from continuously actualized genome/protein databases.103 Besides identifying proteins, MS‐based proteomics can also be used to identify and localize naturally occurring posttranslational modifications (such as glycosylation,104 phosphorylation,105 lysine and N‐terminal acetylation)106 or protein chemical modifications (induced by cross‐linking reactions, surface mapping by oxidation,107 acetylation,108 or deuterium incorporation.107) For peptide/protein identification the very selective protease trypsin is the enzyme of choice, although other enzymes are becoming popular especially in conjunction with the use of ETD.109 Box 2: Native and Ion Mobility Mass Spectrometry In ESI‐MS, molecules are ionized by a combined process of desolvation and (de)protonation. ESI is most sensitive at low flow rates (nl min−1) and optimal conditions can be obtained by bringing the analyte into a 1:1 mixture of water and organic solvent. Such solutions are typically acidified to promote protonation. Although these solvent conditions allow ultra‐sensitive detection, they typically denature and unfold proteins. However, the goal of native MS21, 22, 110 is to preserve higher order protein structures to enable the investigation of protein conformation, protein complex topology, and dynamics. Therefore, the samples need to be held as close as possible to physiological conditions. An ESI‐MS compatible “volatile buffer” is provided by aqueous ammonium acetate, whereby salt concentrations can be varied from approximately 5 mm to 1 m, retaining a neutral pH value. From numerous biophysical validation studies it has become apparent that many quaternary protein structures can be preserved under these conditions. During the ESI process the volatile buffer easily evaporates, leaving “naked” protein ions, albeit substantially less charged than when sprayed from organic ESI solvents, as the surface is more compact in these folded species. Since larger protein assemblies may attain m/z values exceeding a few thousand, dedicated/modified time of flight (ToF) mass analyzers are required for detection.111, 112 Retaining quaternary structures in the gas phase opens up ways to measure the mass of intact protein complexes and sub‐complexes, from which information about stoichiometry and topology can be extracted.19, 113 To probe the structure of the protein complexes, sub‐complexes may also be formed intentionally using either a low concentration of denaturant, a shift in pH value and/or ionic strength114 or through CID.115–117 By in vitro reconstitution of membrane protein complexes in micelles, even membrane‐embedded protein complexes can be studied by native MS.118 A further strength of native MS is its high sensitivity, allowing even the analysis of endogenously expressed protein complexes.119, 120 The available toolbox has recently been extended by the coupling of ion mobility (IM) separation to MS (IMMS).121 In IMMS, ions are separated not only on the basis of their m/z but also, inside a gas‐filled ion‐mobility chamber, according to their drift time, which depends on their overall shape or collision cross section (Ω). Typically, molecules with larger Ω values, that is, larger apparent volumes, exhibit longer drift times. Using IMMS data, the Ω value or average projected area of a protein or protein complex can be determined. Early results have revealed that solution‐phase structures can be, in particular for larger protein complexes, mostly retained in the gas phase.121, 122 IMMS is nowadays used in conjunction with computational modeling to generate refined structural models for protein complexes.114, 123 For instance, by having high‐resolution structures (from X‐ray crystallography or NMR spectroscopy) of the protein complex constituents, the Ω value of the intact complex and/or sub‐complexes can be used to predict structural models. Box 3: H/D‐Exchange Mass Spectrometry In H/D‐exchange MS, the incorporation of deuterium atoms into proteins is monitored over time.124–128 The method is based on the exchange of solvent‐accessible backbone hydrogen atoms with deuterium atoms when a protein is placed in deuterated water (D2O). The subsequent increase in protein mass over time is measured with MS. Using intact or native MS global exchange in a protein or protein complex can be monitored providing information on major conformational changes, for example upon ligand binding.129 The more detailed location of the deuterium incorporation can be determined by monitoring the mass shift in peptic fragments that are produced after the H/D‐exchange reaction. Therefore, the samples are incubated in D2O over different times and then typically diluted to acidic solution conditions (pH≈2.5) at low temperatures (0 °C) to slow down back‐exchange processes. However, few proteases can efficiently digest proteins at such pH values. Pepsin has an optimum activity at low pH values and is the preferred enzyme for H/D‐exchange applications. Even though pepsin is regarded an unspecific protease, the detected peptides are reproducible.130 After digestion, the peptides obtained are subjected to MS analysis and identified by exact mass and fragmentation pattern. The mass shift owing to deuterium incorporation can then be monitored over time, elucidating which peptides are engaged in structural changes occurring upon protein complex formation or, for instance virus maturation.66, 131 H/D exchange coupled to MS has become a valuable analytical tool for the study of protein dynamics. By combining this information with classical functional data, a more thorough understanding of protein function can be obtained. The H/D‐exchange MS approach is comparable to, and has been used in conjunction with, NMR spectroscopic experiments in which the H/D exchange is monitored over time. Although H/D exchange analysis was, for a long time, somewhat limited to small proteins or protein domains, improved resolution and sensitivity in mass analyzers combined with better software for data interpretation now also allow large proteins, such as whole antibodies, to be investigated.132 Box 4: Chemical‐Labeling Approaches Coupled to Mass Spectrometry Next to H/D exchange, there are a couple of alternative chemical approaches to probe the surface accessibility and interconnectivity of proteins in protein complexes which are frequently used in combination with MS.133 In these approaches specific amino acids are rapidly and efficiently chemically labeled under pseudo‐physiological conditions. MS is then used to probe the chemically induced mass shifts in the peptides/amino acids affected by the label. The idea is that only the accessible amino acids will be modified, allowing conformational changes in proteins to be monitored, for instance upon ligand binding or protein‐complex formation. Most popular in the field of structural biology are oxidative labeling by hydroxyl radicals134–136 or labeling of free accessible amine groups, for instance by acetylation.108, 133, 137 Chemical labeling using molecules with at least two reactive groups can also be used for cross‐linking specific amino acids that are in close proximity to each other. Using such cross‐linking approaches, intra‐ and intermolecular interactions can be identified in a protein complex.138–140 The most commonly used bifunctional chemical cross‐linkers target lysine residues, whereby the linker rigidity and spacing in between the two reactive groups defines the range of interactions that can be probed, providing distance restraints for computational modeling. After modification of the proteins, residual reagent needs to be removed or inactivated. Following proteolysis, peptides originating from specific cross‐linked regions of the proteins need to be filtered out of the background of unmodified peptides, for which dedicated software is usually a prerequisite.139, 141 For instance, chemical cross‐linking and MS were applied to probe subunit–subunit interactions in the bacteriophage P22 procapsid.138
3.1. Identification of Viral Proteins and Their Posttranslational Modifications
New viral infectious pathogens are most efficiently characterized by genotyping.142 However, such experiments do not directly provide information about the expressed viral proteins. Using standard proteomics experiments (Box 1), viral proteins can easily be identified even from complex samples such as host‐cell lysates. Such data provides direct information about the composition of the virion and may reveal its structural and accompanied proteins.143 An illustrative example is provided by the human coronavirus causing SARS, which emerged around 2003 in Asia. After the outbreak, all the predicted structural proteins were soon identified by MS (Figure 4), and also several glycosylation and phosphorylation sites could be mapped.28, 143, 144 With a time delay of a few years, some of these proteins could be structurally analyzed using high‐resolution techniques, such as X‐ray crystallography.145
Quantitative proteomics experiments have been used to study dynamic temporal changes in the host proteome upon infection by the pathogen, with the aim to identify the host–pathogen interactome.146, 147 Therefore, often some form of isotope labeling is used enabling the identification of proteins whose expression level is most affected by the infection. For example, isotope‐labeling strategies in combination with proteomics provided information on the cellular changes upon SARS infection and allowed the identification of host‐cell factors putatively involved in virus replication.148, 149
Surface‐exposed viral proteins or protein domains usually mediate the attachment of the virus to the host. For enveloped viruses, such as HIV, those proteins are located in the lipid bilayer and are typically highly glycosylated. These glycans are likely involved in antigenicity, shielding the virus from the immune system. The sites and in particular types of glycosylation are heavily affected by mutations, hampering vaccine development against HIV.150 Proteomics methods have been used to reveal major changes in the glycosylation patterns between different viral strains.151–153 After peptide digestion, the mass discrepancy between glycosylated and enzymatically deglycosylated samples determines the size of the carbohydrate. Using tandem MS, the modified amino acid and the composition of the glycans can be determined.65, 151, 152 Viral protein decoration by glycans may be very complex, as attached carbohydrates typically vary in length and type, whereby all three structural classes, high mannose, complex, and hybrid, occur and have been detected on the HIV gp120 protein (Figure 4). Presumably because of the high structural flexibility and heterogeneity of the glycan layer, only a deglycosylated variant of gp120 could be crystallized to date. Binding of CD4 and chemokine receptors on human T‐cells to gp120 is apparently influenced by the glycosylation pattern, making this sort of analysis important.
Although virus capsids may self‐assemble, the assembly process in vivo is much more complex, and the host cellular machinery can regulate the assembly process. For example, phosphorylation of HBV cp enhances the formation of DNA from RNA in the assembling capsid.154 This is likely connected to the enhanced capsid assembly and encapsulation of RNA observed after phosphorylation of multiple HBV cp amino acid residues.155–157 Proteomics and mutagenesis studies have revealed that the host kinases PKA and PKC each phosphorylate one of the two central serine residues on HBV cp (Figure 4).156, 157
3.2. Monitoring Virus Maturation
Proteins involved in viral assembly and maturation processes are less prone to mutational changes than the surface proteins, and therefore offer potential targets for interference. MS has been used extensively to elucidate not only virus structure, but also to monitor dynamic changes therein throughout the viral lifecycle. Such modifications and rearrangements, which can take place during virus assembly and maturation, and also upon infection, have traditionally been mapped using fluorescent labels or globally monitored by spectroscopic techniques.158, 159 Moreover, mutagenesis studies, such as alanine scanning, have been very valuable to characterize and localize posttranslational modifications occurring during maturation.67, 156, 160, 161
In an elegant example, several MS‐based technologies have been used and combined with electron microscopy (EM) and X‐ray crystallography to investigate capsid formation and maturation of the icosahedral bacteriophage HK97. This lambdoid phage stores its dsDNA under high pressure and thus demands a stable capsid structure and irreversible assembly to ensure protection of the genome.68, 162 Initially, 420 cp subunits organize into pentamers and hexamers. These capsomers then build the spherical, thick‐walled prohead I (Figure 5). The N‐terminal Δ‐domain functions as a scaffold and is cleaved off during maturation leading to prohead II. Such examples of proteolytic cleavage have been observed in many viruses and the exact location of cleavage can be identified using proteomics based methods (Figure 4).163 A critical step in HK97 maturation is the conformational change leading to a thin‐walled icosahedral capsid or phagehead. This expansion is usually accompanied by DNA packaging, but can be triggered in vitro in the absence of DNA and of the packaging machinery. Major structural changes associated with this transition were recognized in EM and crystallography studies. However, the resolution achieved was limited for some intermediate structures. H/D‐exchange MS (Box 3) on the different intermediates in the head assembly, from capsomers to the nearly mature head I, could precisely demonstrate which amino acids were involved in the changes accompanying maturation (Figure 5).66, 131, 164 The next step in maturation is the autocatalytic cross‐linking of the cp subunits locking the capsid in the expanded conformation. The specific peptide carrying a rather unusual lysine–asparagine cross‐link could be identified by MS after proteolysis by trypsin (Figure 4).67
Figure 5.
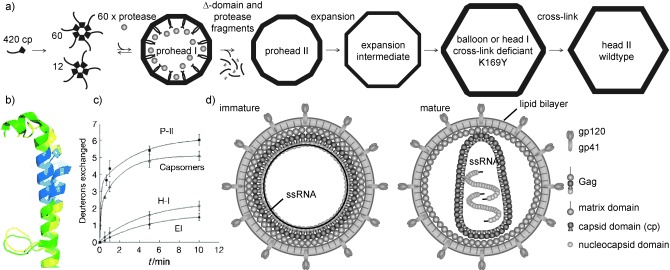
HK97 and HIV virus‐maturation studied by mass spectrometry. a) The cp of bacteriophage HK97 preassembles into hexamers and pentamers. These then form the prohead I together with the viral protease. Assembly is facilitated by the cp Δ‐domain, which functions as a scaffold. The protease cleaves the Δ‐domain and itself leading to prohead II marking the first irreversible step in capsid formation. Under certain conditions expansion occurs followed by the second irreversible step, auto‐catalytic cross‐linking of cp subunits. The head I of a cross‐link deficient mutant closely resembles the mature head II.66 b) The spine helix from the crystal structures of one subunit in prohead II (yellow) was aligned with the straightened helix in head II of the same subunit. A peptide analyzed by H/D exchange (c) is shown in blue. As revealed by the exchange data in (c), the straightening of the helix occurs upon transition from the prohead II (P‐II) to the expansion intermediate (EI). Also shown are the deuterons incorporated in the corresponding peptide fragments for the free capsomers and head I (H‐I). Reprinted by permission from Macmillan Publishers Ltd: Nature,66 2009. d) In HIV, the immature capsid of the Gag polyprotein encloses the (+)ssRNA and becomes enveloped. After release from the cell, the viral protease cleaves the Gag into three major proteins and some small peptides. The matrix domain stays bound to the lipid envelope through a myristoyl residue and the nucleocapsid domain is associated with the ssRNA. A conformational change in cp leads to a collapse of the spherical towards a conical capsid. At this stage the virus is infective. Other proteins, including the protease and peptide fragments, are omitted for clarity.64
More typical inter‐ and intramolecular cross‐links are provided by disulfide bonds, which are commonly observed in mature viruses. For instance, stabilization of the Cytomegalovirus glycoprotein B involves extensive disulfide bonds, which have largely been mapped using MS and such disulfide linkages are apparently common amongst Herpesviridae (Figure 4).165
Another approach commonly used to identify and map structural changes or binding surfaces between viral proteins is chemical cross‐linking coupled to MS (Box 4).141, 160, 166 H/D‐exchange and chemical cross‐linking experiments64, 166–168 have been combined to analyze the pleiomorphic HIV capsids. Their variable appearance obstructs classical high‐resolution structural analysis, since this generally relies on averaging multiple particles.100
In HIV, the Gag polyprotein assembles around the (+)ssRNA at the plasma membrane. After budding of the enveloped virus, the viral protease residing in the spherical immature capsid cleaves the Gag protein. The released domains reorganize, leading to mature virions. The nucleocapsid domain is associated with the RNA in the now collapsed and conical core formed by the capsid domain (from here on cp), whereas the matrix domain stays bound to the envelope by an N‐terminal myristoylation (Figure 5). This myristoyl residue was also detected by MS in the matrix domain in virus‐like particles demonstrating their similarity to the fully infectious virus.167 Alanine scanning had already identified certain sites in the N‐ and C‐terminal domain of the cp involved in intersubunit binding between these homotypic domains. However, only by using H/D exchange, a previously unknown site in the N‐terminal region of cp was found to be highly protected for in‐vitro assembled and mature particles, whereas immature capsids showed behavior similar to the free cp subunit.167, 168 In general, the cp arrangement is similar for different HIV capsid appearances. Moreover, chemical cross‐linking elucidated an unknown interaction between the N‐ and C‐terminal domains of adjacent monomers (Figure 4). This contact probably drives maturation and is probably protected or inhibited in the Gag polyprotein.
Another example focusing on virus assembly used H/D‐exchange MS to locate regions in the MS2 bacteriophage cp dimer that exhibit conformational changes upon binding to a stem‐loop from genomic ssRNA known to initiate assembly.169 The data revealed specific areas within the cp dimer that altered their exchange kinetics in the presence of the RNA, including the known RNA‐binding sites.
3.3. Mass Analysis of Intact Viral Assemblies
ESI‐MS has expanded the attainable mass range for the analysis of biomolecules tremendously, and viruses and their capsids have provided showcase benchmarks. The successful transfer of the TMV into the gas phase using ESI was demonstrated as early as 1996.83 In these pioneering studies, the exact mass determination was precluded by the limitations of the mass analyzer employed. However, collection of the electrosprayed TMV particles and subsequent EM analysis disclosed that the viral structures had been largely retained. The TMV harvested after MS was even infective. Since then, multiple instrumental setups have been applied to estimate the mass of virus particles. A combination of m/z and charge detection in a ToF analyzer resulted in an estimated mass of 40 MDa for TMV, with a substantial uncertainty of 15 %.170 ESI can thus be readily used to ionize viruses and viral particles, but accurate mass analysis is not straightforward when these particles become too big or heterogeneous. ESI has been combined with gas‐phase electrophoretic mobility molecular analyzers (GEMMA).171, 172 The high charge of particles produced by ESI is reduced to obtain singly charged ions, which are subsequently separated and sized by their electrophoretic mobility. GEMMA analysis was successfully applied on the 4.6 MDa cowpea chlorotic mottle virus (CCMV). Although the mass resolution of such an instrument is still too low to enable accurate mass measurement, GEMMA does provide in parallel information about the electrophoretic mobility diameter of the analyzed particle. Such analysis indicated that the gas‐phase CCMV particle had largely retained its quaternary structure (Figure 4).171, 172
A first more‐accurate mass assessment was performed on intact MS2 particles, whereby ESI‐ToF analysis allowed the identification of partly resolved charge states.173 Unprecedented high‐resolution data on intact viral capsids of HBV were obtained using a modified Q‐ToF instrument.111, 174 HBV capsids are rather unique in exhibiting two distinct icosahedral morphologies even in vivo, composed of 90 and 120 dimers with masses of approximately 3 and 4 MDa, respectively. The mass spectra displayed well‐separated charge‐state distributions (Figure 6) for both capsids enabling a mass assignment within 0.1 %, revealing that both lattices were complete. The HBV capsids were surprisingly stable during transfer into the gas phase and through the vacuum of the mass spectrometer. Measuring the Ω values of the capsids by IMMS allowed an estimation of the capsid radii in good agreement with the dimensions of the particles in EM, verifying a largely retained capsid morphology in the gas phase.175 Although the HBV capsid turned out to be very stable, models have suggested that dimers in the particles are exchanging with very low abundant “free” cp dimers in solution. Native MS combined with CID on the intact HBV capsids was used to monitor the incorporation of isotopically labeled cp dimers into preassembled unlabeled HBV capsids. Slow exchange could be observed over a timeframe of months, albeit only for the 3 MDa particles and exclusively at low temperatures176 providing experimental evidence for the theoretically predicted “capsid breathing”.88
Figure 6.
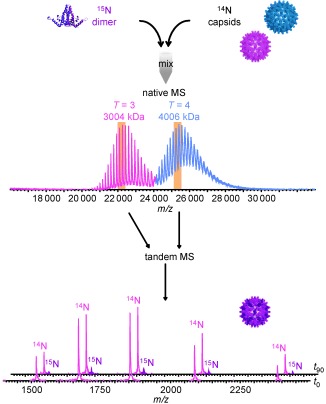
Capsid breathing in HBV monitored by tandem MS: 15N‐labeled cp dimers of HBV were incubated with preassembled unlabeled capsids. At certain times native MS spectra were recorded and either T=3 or T=4 capsids of HBV were selected for CID. Subunit exchange was detected in the T=3 capsids after prolonged times as judged by the growth of a signal assigned to labeled monomers, which were ejected from the capsids in tandem MS. Reproduced by permission of the PCCP Owner Societies from Ref. 176.
The even larger (ca. 10 MDa) norovirus capsid proved much less stable and prone to dissociation when altering pH value and ionic strength as monitored by native MS.177 At pH 6 in solution only the intact T=3 capsid was detected, although with insufficient resolution to allow an accurate mass assignment. More acidic pH values resulted in chiefly cp dimers. Intriguingly at basic pH values, higher order oligomers were formed preferentially at high ionic strength. Remarkably, the transition between T=3 particles and other oligomers was fully reversible. The larger oligomers most likely arose from initial dissociation of the T=3 capsids into dimers and subsequent reassembly. The main products of this pathway were identified as 60‐ and 80‐mers, although also smaller oligomers were present under certain conditions (Figure 7). Atomic force microscopy (AFM)177 and earlier EM studies178 confirmed that T=1 norovirus capsids consisting of 60 cp subunits can be formed at basic pH values. Ionic strength and pH value could thus drive the norovirus capsid into various morphologies rendering this a particularly ideal model system to study capsid (dis)assembly.
Figure 7.
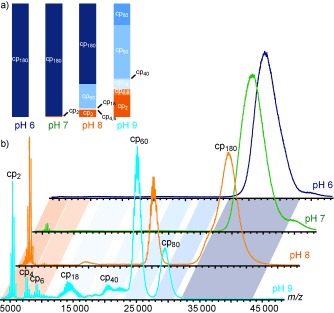
Capsid stability and morphology of norovirus depend on pH value: ESI‐MS analysis of norovirus capsids incubated at pH 6–9. The abundance of the different cp oligomers are depicted as bar graph (a). The graphs and mass spectra (b) indicate that the capsid (cp180) is instable at alkaline pH values. The dissociated dimers form larger oligomers (up to 80 subunits) at alkaline pH values and high ionic strength. Adapted from Ref. 177 the American Society for Biochemistry and Molecular Biology.
Owing to their low abundance and transient nature, it is extremely hard to probe potential oligomeric intermediates occurring during virus assembly in between the stage of the usually dimeric building block and the intact capsid. The nucleating intermediate is thought to form rather slowly, whereas the subsequent addition of building blocks proceeds fast towards capsid completion (Figure 3). The high sensitivity of native IMMS was used to detect and structurally characterize a wide variety of intermediate oligomers of the norovirus and HBV coexisting with the intact capsid forms.179 In combination with computational modeling, the shape of these intermediates could be assessed which revealed that they all exhibited extended sheet‐like structures as would be expected for on‐pathway products. Moreover, the anticipated assembly nuclei for norovirus (decamer) and HBV capsids (hexamer) could be confirmed from the MS data. An assembly pathway common for both viruses was proposed.179
In case of the MS2 bacteriophage, the assembly pathways are restricted by the bound RNA. Introducing longer RNA stretches favors assembly along the threefold axis (C 3 axis). Importantly not only the polyanionic character, but also the sequence of the RNA affects the assembly efficiency.180 The abundance of two major intermediates was monitored by MS over time in presence of various RNAs and interpreted by kinetic modeling.181 In conclusion, the common protein‐centric view of capsid assembly seems rather simplified. Furthermore, the structure of the two intermediates was deduced by IMMS of the intact and CID fragmented species.182 Both exhibit an extended, ring‐like topology as found in the norovirus and HBV oligomers.
For applications in nanotechnology the integrity of the capsids is important, but also their dynamic properties, such as reversible assembly. Knowledge about such abilities is important for particle modification and to support reactions of encapsulated materials. Limited proteolysis in combination with MS on intact capsids revealed the dynamic nature of the cp in flock‐house virus and CCMV in which cp regions located at the inner face become transiently exposed to the exterior.91, 183 The incorporation of material into the caspids and modification of the capsids and other protein cages utilizes such dynamic properties. Mass shifts induced by functionalizing virus particles or creation of mixed assemblies can be readily measured with MS providing a tool to monitor the reaction and control the product quality (Figure 4).184–186 Dendrimers covalently attached to the inner face of a protein cage can be nurtured to subsequent generations. Thereby, the particle may increase its stability enabling applications in more extreme environments. The possibility to specifically modify the enclosed dendrimers further increases the utility in imaging.184
4. Outlook
MS has evolved into a technology that contributes meaningful insights into structural and functional aspects of proteins and protein complexes, such as the viral capsids and viruses described herein. While focusing herein on structural virology, the application areas are not limited, and elegant work has also been described studying ribosomes,17 RNA polymerases,115 and proteasomes.187, 188 Although further improvement in mass‐spectrometric analyzers and ionization techniques will develop the field, most progress in the near future will come from the linking of MS with other technologies, such as computational modeling, microscopy, and spectroscopy.
Structural modeling of protein complexes is still largely underdeveloped partly because of the size of the particles to be modeled. However, advances made in recent years, for instance in modeling of the yeast nuclear pore complex,189 may be suitable for wider application and benefit from Ω value and topology data of protein (sub‐)complexes obtained by native IMMS.
Coupling of MS with EM or AFM can be envisaged to be bi‐directional. The mass analyzer can be used as sample purification step, using soft‐landing approaches to select particles of interest that may be further structurally characterized by EM or AFM.190 In this way, maybe low abundant viral‐assembly intermediates can be trapped and studied by microscopic techniques. Conversely, EM‐ and AFM‐based methods can be used to “grap” selective organelles or even protein complexes out of their natural cellular environment, and subsequently bring those to the mass spectrometer for identification and/or structural analysis.191
Another exciting development that may become available in the near future uses ultra‐short and intense X‐ray laser pulses to obtain high‐resolution structures of individual protein complexes in the gas phase.192 Such a method would be highly complementary to the herein described native MS and IMMS approaches and could provide higher resolution structural information, which may be directly compared with solution‐phase structural data obtained by more conventional structural‐biology techniques, such as NMR spectroscopy, X‐ray crystallography and EM. These are exciting times in biomolecular mass spectrometry.
Abbreviations
- CID
collision induced dissociation
- cp
capsid protein
- ds
double‐stranded
- EM
electron microscopy
- ESI
electrospray ionization
- HBV
Hepatitis B virus
- HIV
Human Immunodeficiency Virus
- TMV
Tobacco Mosaic Virus
- CCMV
Cowpea Chlorotic Mottle Virus
- IM
ion mobility
- IMMS
ion mobility mass spectrometry
- MALDI
matrix assisted laser desorption/ionization
- MS
mass spectrometry
- m/z
mass to charge ratio
- NVLP
norovirus‐like particle
- Q
quadrupole
- ss
single‐stranded
- ToF
time of flight
Biographical Information
Charlotte Uetrecht, born 1982, studied biochemistry at the University of Potsdam, Germany. From 2006 to 2010, she was a PhD student at Utrecht University under supervision of Albert Heck. Her research there focused on the mass spectrometric analyses of viral assemblies. Since 2011 she is an EMBO long‐term fellow at the Molecular Biophysics department, Uppsala University in Sweden, investigating viral and other large structures/particles with MS and X‐ray free electron lasers in vacuum.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
Albert J. R. Heck, born 1964, has held the chair of Biomolecular Mass Spectrometry & Proteomics at Utrecht University since 1998. Since 2003 he has been director of the Netherlands Proteomics Centre, and since 2006 director of the Bijvoet Centre for Biomolecular Research. His research interests comprise all aspects of protein mass spectrometry. He has received awards including the gold medal of the Dutch Royal Chemical Society, the Descartes Huygens Award from the Republique Francaise, and the Life Science Award of the German Mass Spectrometry Society.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Acknowledgements
We thank all colleagues in the Heck‐lab, especially the native MS group, who contributed to some of the research described herein. We would like to take this opportunity to thank all our collaborators through whom we entered the exciting field of structural virology. With the danger of accidently leaving people out, we mention in particular Norman R. Watts, Paul T. Wingfield, and Alasdair C. Steven from NIAMS, Mary K. Estes, B. V. Venkataram Prasad from Baylor College of Medicine, Peter E. Prevelige from the University of Alabama, Gino Cingolani from Thomas Jefferson University, Jack E. Johnson from the Scripps Research Institute, and Wouter H. Roos and Gijs J. L. Wuite from the Vrije Universiteit Amsterdam. This work was supported by the Netherlands Proteomics Centre, and by the Netherlands Organisation for Scientific Research ALW‐ECHO (819.02.10) to A.J.R.H.
References
- 1. Nier A. O., J. Chem. Educ. 1989, 66, 385. [Google Scholar]
- 2. Budzikiewicz H., Djerassi C., Williams D. H., Interpretation of mass spectra of organic compounds., Holden‐Day, San Francisco, 1964. [Google Scholar]
- 3. Karas M., Hillenkamp F., Anal. Chem. 1988, 60, 2299. [DOI] [PubMed] [Google Scholar]
- 4. Fenn J. B., Mann M., Meng C. K., Wong S. F., Whitehouse C. M., Science 1989, 246, 64. [DOI] [PubMed] [Google Scholar]
- 5. Chowdhury S. K., Katta V., Chait B. T., Biochem. Biophys. Res. Commun. 1990, 167, 686. [DOI] [PubMed] [Google Scholar]
- 6. Aebersold R., Mann M., Nature 2003, 422, 198. [DOI] [PubMed] [Google Scholar]
- 7. Dettmer K., Aronov P. A., Hammock B. D., Mass Spectrom. Rev. 2007, 26, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chughtai K., Heeren R. M., Chem. Rev. 2010, 110, 3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takats Z., Wiseman J. M., Gologan B., Cooks R. G., Science 2004, 306, 471. [DOI] [PubMed] [Google Scholar]
- 10. Fenn J. B., Angew. Chem. 2003, 115, 3999; [Google Scholar]; Angew. Chem. Int. Ed. 2003, 42, 3871. [Google Scholar]
- 11. Katta V., Chait B. T., J. Am. Chem. Soc. 1991, 113, 8534. [Google Scholar]
- 12. Fitzgerald M. C., Chernushevich I., Standing K. G., Whitman C. P., Kent S. B., Proc. Natl. Acad. Sci. USA 1996, 93, 6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang X. J., Brewer C. F., Saha S., Chernushevich I., Ens W., Standing K. G., Rapid Commun. Mass Spectrom. 1994, 8, 750. [DOI] [PubMed] [Google Scholar]
- 14. Light‐Wahl K. J., Loo J. A., Edmonds C. G., Smith R. D., Witkowska H. E., Shackleton C. H., Wu C. S., Biol. Mass Spectrom. 1993, 22, 112. [DOI] [PubMed] [Google Scholar]
- 15. Loo J. A., Mass Spectrom. Rev. 1997, 16, 1. [DOI] [PubMed] [Google Scholar]
- 16. Rostom A. A., Fucini P., Benjamin D. R., Juenemann R., Nierhaus K. H., Hartl F. U., Dobson C. M., Robinson C. V., Proc. Natl. Acad. Sci. USA 2000, 97, 5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benjamin D. R., Robinson C. V., Hendrick J. P., Hartl F. U., Dobson C. M., Proc. Natl. Acad. Sci. USA 1998, 95, 7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benesch J. L., Robinson C. V., Curr. Opin. Struct. Biol. 2006, 16, 245. [DOI] [PubMed] [Google Scholar]
- 19. Sharon M., Robinson C. V., Annu. Rev. Biochem. 2007, 76, 167. [DOI] [PubMed] [Google Scholar]
- 20. van Berkel W. J., van den Heuvel R. H., Versluis C., Heck A. J., Protein Sci. 2000, 9, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heck A. J., Van den Heuvel R. H., Mass Spectrom. Rev. 2004, 23, 368. [DOI] [PubMed] [Google Scholar]
- 22. van den Heuvel R. H., Heck A. J., Curr. Opin. Chem. Biol. 2004, 8, 519. [DOI] [PubMed] [Google Scholar]
- 23. Steinmetz N. F., Lin T., Lomonossoff G. P., Johnson J. E., Curr. Top. Microbiol. Immunol. 2009, 327, 23. [DOI] [PubMed] [Google Scholar]
- 24. Singh P., Gonzalez M. J., Manchester M., Drug Dev. Res. 2006, 67, 23. [Google Scholar]
- 25. Mitragotri S., Lahann J., Nat. Mater. 2009, 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dokland T., Structure 2000, 8, R157. [DOI] [PubMed] [Google Scholar]
- 27. Morton V. L., Stockley P. G., Stonehouse N. J., Ashcroft A. E., Mass Spectrom. Rev. 2008, 27, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krokhin O., Li Y., Andonov A., Feldmann H., Flick R., Jones S., Stroeher U., Bastien N., Dasuri K. V., Cheng K., Simonsen J. N., Perreault H., Wilkins J., Ens W., Plummer F., Standing K. G., Mol. Cell. Proteomics 2003, 2, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pe’ery T., Mathews M. B. in Fields Virology, Vol. 1, 5th ed. (Eds.: D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, S. E. Straus), Lippincott Williams & Wilkins, Philadelphia, 2007. [Google Scholar]
- 30. Claverie J. M., Abergel C., Annu. Rev. Genet. 2009, 43, 49. [DOI] [PubMed] [Google Scholar]
- 31. Steven A. C., Conway J. F., Cheng N., Watts N. R., Belnap D. M., Harris A., Stahl S. J., Wingfield P. T., Adv. Virus Res. 2005, 64, 125. [DOI] [PubMed] [Google Scholar]
- 32. Beijerinck M. W., Verh. Akad. Wet. Amsterdam Afd. Natuurkd. Sect. 1 1898, 6, 3. [Google Scholar]
- 33. Lawrence C. M., Menon S., Eilers B. J., Bothner B., Khayat R., Douglas T., Young M. J., J. Biol. Chem. 2009, 284, 12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pearson M. N., Beever R. E., Boine B., Arthur K., Mol. Plant Pathol. 2009, 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ackermann H. W., Kropinski A. M., Res. Microbiol. 2007, 158, 555. [DOI] [PubMed] [Google Scholar]
- 36. Nelson R. S., Citovsky V., Plant Physiol. 2005, 138, 1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mercer J., Schelhaas M., Helenius A., Annu. Rev. Biochem. 2010, 79, 803. [DOI] [PubMed] [Google Scholar]
- 38. Yamada T., Onimatsu H., Van Etten J. L., Adv. Virus Res. 2006, 66, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suttle C. A., Nature 2005, 437, 356. [DOI] [PubMed] [Google Scholar]
- 40. Moreira D., Lopez‐Garcia P., Nat. Rev. Microbiol. 2009, 7, 306. [DOI] [PubMed] [Google Scholar]
- 41. La Scola B., Desnues C., Pagnier I., Robert C., Barrassi L., Fournous G., Merchat M., Suzan‐Monti M., Forterre P., Koonin E., Raoult D., Nature 2008, 455, 100. [DOI] [PubMed] [Google Scholar]
- 42. Koonin E. V., Ann. N. Y. Acad. Sci. 2009, 1178, 47.19845627 [Google Scholar]
- 43. Forterre P., Prangishvili D., Res. Microbiol. 2009, 160, 466. [DOI] [PubMed] [Google Scholar]
- 44. Condit R. C. in Fields Virology, Vol. 1, 5th ed. (Eds.: D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, S. E. Straus), Lippincott Williams & Wilkins, Philadelphia, 2007. [Google Scholar]
- 45. Fauquet C. M., Fargette D., Virol. J. 2005, 2, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rohwer F., Edwards R., J. Bacteriol. 2002, 184, 4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Regenmortel M. H., Mahy B. W., Emerging Infect. Dis. 2004, 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harrison S. C. in Fields Virology, Vol. 1, 5th ed. (Eds.: D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, S. E. Straus), Lippincott Williams & Wilkins, Philadelphia, 2007. [Google Scholar]
- 49. White J. M., Delos S. E., Brecher M., Schornberg K., Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huiskonen J. T., Butcher S. J., Curr. Opin. Struct. Biol. 2007, 17, 229. [DOI] [PubMed] [Google Scholar]
- 51. Marvin D. A., Curr. Opin. Struct. Biol. 1998, 8, 150. [DOI] [PubMed] [Google Scholar]
- 52. Grayson P., Molineux I. J., Curr. Opin. Microbiol. 2007, 10, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whittaker G. R., Kann M., Helenius A., Annu. Rev. Cell Dev. Biol. 2000, 16, 627. [DOI] [PubMed] [Google Scholar]
- 54. Denison M. R., PLoS Biol. 2008, 6, e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brussow H., Canchaya C., Hardt W. D., Microbiol. Mol. Biol. Rev. 2004, 68, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oppenheim A. B., Kobiler O., Stavans J., Court D. L., Adhya S., Annu. Rev. Genet. 2005, 39, 409. [DOI] [PubMed] [Google Scholar]
- 57. Bushman F., Lewinski M., Ciuffi A., Barr S., Leipzig J., Hannenhalli S., Hoffmann C., Nat. Rev. Microbiol. 2005, 3, 848. [DOI] [PubMed] [Google Scholar]
- 58. Netherton C., Moffat K., Brooks E., Wileman T., Adv. Virus Res. 2007, 70, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun S., Rao V. B., Rossmann M. G., Curr. Opin. Struct. Biol. 2010, 20, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Patton J. T., Carpio R. Vasquez‐Del, Tortorici M. A., Taraporewala Z. F., Adv. Virus Res. 2007, 69, 167. [DOI] [PubMed] [Google Scholar]
- 61. Patient R., Hourioux C., Roingeard P., Cell. Microbiol. 2009, 11, 1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen B. J., Lamb R. A., Virology 2008, 372, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang I. N., Smith D. L., Young R., Annu. Rev. Microbiol. 2000, 54, 799. [DOI] [PubMed] [Google Scholar]
- 64. Lanman J., P. E. Prevelige, Jr. , Adv. Virus Res. 2005, 64, 285. [DOI] [PubMed] [Google Scholar]
- 65. Cutalo J. M., Deterding L. J., Tomer K. B., J. Am. Soc. Mass Spectrom. 2004, 15, 1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gertsman I., Gan L., Guttman M., Lee K., Speir J. A., Duda R. L., Hendrix R. W., Komives E. A., Johnson J. E., Nature 2009, 458, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Duda R. L., Hempel J., Michel H., Shabanowitz J., Hunt D., Hendrix R. W., J. Mol. Biol. 1995, 247, 618. [DOI] [PubMed] [Google Scholar]
- 68. Steven A. C., Heymann J. B., Cheng N., Trus B. L., Conway J. F., Curr. Opin. Struct. Biol. 2005, 15, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Estes M. K., Prasad B. V., Atmar R. L., Curr. Opin. Infect. Dis. 2006, 19, 467. [DOI] [PubMed] [Google Scholar]
- 70. Casjens S. R., Curr. Opin. Microbiol. 2005, 8, 451. [DOI] [PubMed] [Google Scholar]
- 71. Yen H. L., Webster R. G., Curr. Top. Microbiol. Immunol. 2009, 333, 3. [DOI] [PubMed] [Google Scholar]
- 72. Caspar D. L., Klug A., Cold Spring Harbor Symp. Quant. Biol. 1962, 27, 1. [DOI] [PubMed] [Google Scholar]
- 73. Zlotnick A., J. Mol. Recognit. 2005, 18, 479. [DOI] [PubMed] [Google Scholar]
- 74. Johnson J. E., Chiu W., Curr. Opin. Struct. Biol. 2007, 17, 237. [DOI] [PubMed] [Google Scholar]
- 75. Wetzel R., Acc. Chem. Res. 2006, 39, 671. [DOI] [PubMed] [Google Scholar]
- 76. Zandi R., van der Schoot P., Reguera D., Kegel W., Reiss H., Biophys. J. 2006, 90, 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hagan M. F., Elrad O. M., Biophys. J. 2010, 98, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stockley P. G., Rolfsson O., Thompson G. S., Basnak G., Francese S., Stonehouse N. J., Homans S. W., Ashcroft A. E., J. Mol. Biol. 2007, 369, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zlotnick A., J. Mol. Biol. 2007, 366, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zlotnick A., Ceres P., Singh S., Johnson J. M., J. Virol. 2002, 76, 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kivenson A., Hagan M. F., Biophys. J. 2010, 99, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hagan M. F., J. Chem. Phys. 2009, 130, 114902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Siuzdak G., Bothner B., Yeager M., Brugidou C., Fauquet C. M., Hoey K., Chang C. M., Chem. Biol. 1996, 3, 45. [DOI] [PubMed] [Google Scholar]
- 84. Flenniken M. L., Uchida M., Liepold L. O., Kang S., Young M. J., Douglas T., Curr. Top. Microbiol. Immunol. 2009, 327, 71. [DOI] [PubMed] [Google Scholar]
- 85. Newman M., Suk F. M., Cajimat M., Chua P. K., Shih C., J. Virol. 2003, 77, 12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Singh S., Zlotnick A., J. Biol. Chem. 2003, 278, 18249. [DOI] [PubMed] [Google Scholar]
- 87. Roos W. H., Wuite G. L., Adv. Mater. 2009, 21, 1187. [Google Scholar]
- 88. Ceres P., Zlotnick A., Biochemistry 2002, 41, 11525. [DOI] [PubMed] [Google Scholar]
- 89. Hilmer J. K., Zlotnick A., Bothner B., J. Mol. Biol. 2008, 375, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Reisdorph N., Thomas J. J., Katpally U., Chase E., Harris K., Siuzdak G., Smith T. J., Virology 2003, 314, 34. [DOI] [PubMed] [Google Scholar]
- 91. Bothner B., Dong X. F., Bibbs L., Johnson J. E., Siuzdak G., J. Biol. Chem. 1998, 273, 673. [DOI] [PubMed] [Google Scholar]
- 92. Parent K. N., Suhanovsky M. M., Teschke C. M., J. Mol. Biol. 2007, 365, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bothner B., Schneemann A., Marshall D., Reddy V., Johnson J. E., Siuzdak G., Nat. Struct. Biol. 1999, 6, 114. [DOI] [PubMed] [Google Scholar]
- 94. Klug A., Philos. Trans. R. Soc. London Ser. B 1999, 354, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kegel W. K., van der Schoot P., Biophys. J. 2006, 91, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang X., Jin L., Fang Q., Hui W. H., Zhou Z. H., Cell 2010, 141, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Vellinga J., Van der Heijdt S., Hoeben R. C., J. Gen. Virol. 2005, 86, 1581. [DOI] [PubMed] [Google Scholar]
- 98. Choi K. H., Morais M. C., Anderson D. L., Rossmann M. G., Structure 2006, 14, 1723. [DOI] [PubMed] [Google Scholar]
- 99. Moody M. F., J. Mol. Biol. 1999, 293, 401. [DOI] [PubMed] [Google Scholar]
- 100. Ganser‐Pornillos B. K., von Schwedler U. K., Stray K. M., Aiken C., Sundquist W. I., J. Virol. 2004, 78, 2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zubarev R. A., Horn D. M., Fridriksson E. K., Kelleher N. L., Kruger N. A., Lewis M. A., Carpenter B. K., McLafferty F. W., Anal. Chem. 2000, 72, 563. [DOI] [PubMed] [Google Scholar]
- 102. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F., Proc. Natl. Acad. Sci. USA 2004, 101, 9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sadygov R. G., Cociorva D., Yates J. R., 3rd, Nat. Methods 2004, 1, 195. [DOI] [PubMed] [Google Scholar]
- 104. Tissot B., North S. J., Ceroni A., Pang P. C., Panico M., Rosati F., Capone A., Haslam S. M., Dell A., Morris H. R., FEBS Lett. 2009, 583, 1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Grimsrud P. A., Swaney D. L., Wenger C. D., Beauchene N. A., Coon J. J., ACS Chem. Biol. 2010, 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mischerikow N., Heck A. J., Proteomics 2011, 11, 571. [DOI] [PubMed] [Google Scholar]
- 107. Konermann L., Simmons D. A., Mass Spectrom. Rev. 2003, 22, 1. [DOI] [PubMed] [Google Scholar]
- 108. Scholten A., Visser N. F., van den Heuvel R. H., Heck A. J., J. Am. Soc. Mass Spectrom. 2006, 17, 983. [DOI] [PubMed] [Google Scholar]
- 109. Taouatas N., Drugan M. M., Heck A. J., Mohammed S., Nat. Methods 2008, 5, 405. [DOI] [PubMed] [Google Scholar]
- 110. Heck A. J., Nat. Methods 2008, 5, 927. [DOI] [PubMed] [Google Scholar]
- 111. van den Heuvel R. H., van Duijn E., Mazon H., Synowsky S. A., Lorenzen K., Versluis C., Brouns S. J., Langridge D., van der Oost J., Hoyes J., Heck A. J., Anal. Chem. 2006, 78, 7473. [DOI] [PubMed] [Google Scholar]
- 112. Benesch J. L., Ruotolo B. T., Sobott F., Wildgoose J., Gilbert A., Bateman R., Robinson C. V., Anal. Chem. 2009, 81, 1270. [DOI] [PubMed] [Google Scholar]
- 113. Taverner T., Hernandez H., Sharon M., Ruotolo B. T., Matak‐Vinkovic D., Devos D., Russell R. B., Robinson C. V., Acc. Chem. Res. 2008, 41, 617. [DOI] [PubMed] [Google Scholar]
- 114. Geiger S. R., Lorenzen K., Schreieck A., Hanecker P., Kostrewa D., Heck A. J., Cramer P., Mol. Cell 2010, 39, 583. [DOI] [PubMed] [Google Scholar]
- 115. Lorenzen K., Vannini A., Cramer P., Heck A. J., Structure 2007, 15, 1237. [DOI] [PubMed] [Google Scholar]
- 116. van Duijn E., Simmons D. A., van den Heuvel R. H., Bakkes P. J., van Heerikhuizen H., Heeren R. M., Robinson C. V., van der Vies S. M., Heck A. J., J. Am. Chem. Soc. 2006, 128, 4694. [DOI] [PubMed] [Google Scholar]
- 117. Benesch J. L., Aquilina J. A., Ruotolo B. T., Sobott F., Robinson C. V., Chem. Biol. 2006, 13, 597. [DOI] [PubMed] [Google Scholar]
- 118. Barrera N. P., Di Bartolo N., Booth P. J., Robinson C. V., Science 2008, 321, 243. [DOI] [PubMed] [Google Scholar]
- 119. Synowsky S. A., van den Heuvel R. H., Mohammed S., Pijnappel P. W., Heck A. J., Mol. Cell. Proteomics 2006, 5, 1581. [DOI] [PubMed] [Google Scholar]
- 120. Synowsky S. A., van Wijk M., Raijmakers R., Heck A. J., J. Mol. Biol. 2009, 385, 1300. [DOI] [PubMed] [Google Scholar]
- 121. Uetrecht C., Rose R. J., van Duijn E., Lorenzen K., Heck A. J., Chem. Soc. Rev. 2010, 39, 1633. [DOI] [PubMed] [Google Scholar]
- 122. Ruotolo B. T., Giles K., Campuzano I., Sandercock A. M., Bateman R. H., Robinson C. V., Science 2005, 310, 1658. [DOI] [PubMed] [Google Scholar]
- 123. Pukala T. L., Ruotolo B. T., Zhou M., Politis A., Stefanescu R., Leary J. A., Robinson C. V., Structure 2009, 17, 1235. [DOI] [PubMed] [Google Scholar]
- 124. Wang L., Lane L. C., Smith D. L., Protein Sci. 2001, 10, 1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang L., Smith D. L., Protein Sci. 2005, 14, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Maier C. S., Deinzer M. L., Methods Enzymol. 2005, 402, 312. [DOI] [PubMed] [Google Scholar]
- 127. Engen J. R., Smith D. L., Anal. Chem. 2001, 73, 256 A. [DOI] [PubMed] [Google Scholar]
- 128. Wales T. E., Engen J. R., Mass Spectrom. Rev. 2006, 25, 158. [DOI] [PubMed] [Google Scholar]
- 129. Alverdi V., Mazon H., Versluis C., Hemrika W., Esposito G., van den Heuvel R., Scholten A., Heck A. J., J. Mol. Biol. 2008, 375, 1380. [DOI] [PubMed] [Google Scholar]
- 130. Sinz A., ChemMedChem 2007, 2, 425. [DOI] [PubMed] [Google Scholar]
- 131. Gertsman I., Fu C. Y., Huang R., Komives E., Johnson J. E., Mol. Cell. Proteomics 2010, 9, 1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Houde D., Peng Y., Berkowitz S. A., Engen J. R., Mol. Cell. Proteomics 2010, 9, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mendoza V. L., Vachet R. W., Mass Spectrom. Rev. 2009, 28, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Xu G., Chance M. R., Chem. Rev. 2007, 107, 3514. [DOI] [PubMed] [Google Scholar]
- 135. Konermann L., Stocks B. B., Pan Y., Tong X., Mass Spectrom. Rev. 2010, 29, 651. [DOI] [PubMed] [Google Scholar]
- 136. Gau B. C., Sharp J. S., Rempel D. L., Gross M. L., Anal. Chem. 2009, 81, 6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Suckau D., Mak M., Przybylski M., Proc. Natl. Acad. Sci. USA 1992, 89, 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kang S., Hawkridge A. M., Johnson K. L., Muddiman D. C., P. E. Prevelige, Jr. , J. Proteome Res. 2006, 5, 370. [DOI] [PubMed] [Google Scholar]
- 139. Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., Aebersold R., Mol. Cell. Proteomics 2010, 9, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sinz A., Mass Spectrom. Rev. 2006, 25, 663. [DOI] [PubMed] [Google Scholar]
- 141. Kang S., Mou L., Lanman J., Velu S., Brouillette W. J., P. E. Prevelige, Jr. , Rapid Commun. Mass Spectrom. 2009, 23, 1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Belák S., Thoren P., LeBlanc N., Viljoen G., Expert Rev. Mol. Diagn. 2009, 9, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zeng R., Ruan H. Q., Jiang X. S., Zhou H., Shi L., Zhang L., Sheng Q. H., Tu Q., Xia Q. C., Wu J. R., J. Proteome Res. 2004, 3, 549. [DOI] [PubMed] [Google Scholar]
- 144. Ying W., Hao Y., Zhang Y., Peng W., Qin E., Cai Y., Wei K., Wang J., Chang G., Sun W., Dai S., Li X., Zhu Y., Li J., Wu S., Guo L., Dai J., Wan P., Chen T., Du C., Li D., Wan J., Kuai X., Li W., Shi R., Wei H., Cao C., Yu M., Liu H., Dong F., Wang D., Zhang X., Qian X., Zhu Q., He F., Proteomics 2004, 4, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bartlam M., Xu Y., Rao Z., J. Struct. Funct. Genomics 2007, 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Radtke K., Kieneke D., Wolfstein A., Michael K., Steffen W., Scholz T., Karger A., Sodeik B., PLoS Pathog. 2010, 6, e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Munday D., Emmott E., Surtees R., Lardeau C. H., Wu W., Duprex W. P., Dove B. K., Barr J. N., Hiscox J. A., Mol. Cell. Proteomics 2010, 9, 2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Jiang X. S., Tang L. Y., Dai J., Zhou H., Li S. J., Xia Q. C., Wu J. R., Zeng R., Mol. Cell. Proteomics 2005, 4, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhang L., Zhang Z. P., Zhang X. E., Lin F. S., Ge F., J. Virol. 2010, 84, 6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Vigerust D. J., Shepherd V. L., Trends Microbiol. 2007, 15, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhu X., Borchers C., Bienstock R. J., Tomer K. B., Biochemistry 2000, 39, 11194. [DOI] [PubMed] [Google Scholar]
- 152. Liedtke S., Geyer R., Geyer H., Glycoconjugate J. 1997, 14, 785. [DOI] [PubMed] [Google Scholar]
- 153. Go E. P., Chang Q., Liao H. X., Sutherland L. L., Alam S. M., Haynes B. F., Desaire H., J. Proteome Res. 2009, 8, 4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Melegari M., Wolf S. K., Schneider R. J., J. Virol. 2005, 79, 9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gazina E. V., Fielding J. E., Lin B., Anderson D. A., J. Virol. 2000, 74, 4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kang H., Yu J., Jung G., Biochem. J. 2008, 416, 47. [DOI] [PubMed] [Google Scholar]
- 157. Kang H. Y., Lee S., Park S. G., Yu J., Kim Y., Jung G., Biochem. J. 2006, 398, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Masi A., Cicchi R., Carloni A., Pavone F. S., Arcangeli A., Adv. Exp. Med. Biol. 2010, 674, 33. [DOI] [PubMed] [Google Scholar]
- 159. Lakowicz J. R., J. Biochem. Biophys. Methods 1980, 2, 91. [DOI] [PubMed] [Google Scholar]
- 160. Fu C., Uetrecht C., Kang S., Morais M., Heck A. J., Walter M. R., Prevelige P. E., Mol. Cell. Proteomics 2010, 9, 1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Moreira I. S., Fernandes P. A., Ramos M. J., Proteins Struct. Funct. Genet. 2007, 68, 803. [DOI] [PubMed] [Google Scholar]
- 162. Duda R. L., Ross P. D., Cheng N., Firek B. A., Hendrix R. W., Conway J. F., Steven A. C., J. Mol. Biol. 2009, 391, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Pepinsky R. B., Papayannopoulos I. A., Campbell S., Vogt V. M., J. Virol. 1996, 70, 3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Gertsman I., Komives E. A., Johnson J. E., J. Mol. Biol. 2010, 397, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lopper M., Compton T., J. Virol. 2002, 76, 6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lanman J., Lam T. T., Barnes S., Sakalian M., Emmett M. R., Marshall A. G., P. E. Prevelige, Jr. , J. Mol. Biol. 2003, 325, 759. [DOI] [PubMed] [Google Scholar]
- 167. Lanman J., Lam T. T., Emmett M. R., Marshall A. G., Sakalian M., P. E. Prevelige, Jr. , Nat. Struct. Mol. Biol. 2004, 11, 676. [DOI] [PubMed] [Google Scholar]
- 168. Lam T. T., Lanman J. K., Emmett M. R., Hendrickson C. L., Marshall A. G., Prevelige P. E., J. Chromatogr. A 2002, 982, 85. [DOI] [PubMed] [Google Scholar]
- 169. Morton V. L., Burkitt W., O’Connor G., Stonehouse N. J., Stockley P. G., Ashcroft A. E., Phys. Chem. Chem. Phys. 2010, 12, 13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Fuerstenau S. D., Benner W. H., Thomas J. J., Brugidou C., Bothner B., Siuzdak G., Angew. Chem. 2001, 113, 559; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001, 40, 541. [Google Scholar]
- 171. Thomas J. J., Bothner B., Traina J., Benner W. H., Siuzdak G., Spectrosc. Int. J. 2004, 18, 31. [Google Scholar]
- 172. Kaddis C. S., Lomeli S. H., Yin S., Berhane B., Apostol M. I., Kickhoefer V. A., Rome L. H., Loo J. A., J. Am. Soc. Mass Spectrom. 2007, 18, 1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Tito M. A., Tars K., Valegard K., Hajdu J., Robinson C. V., J. Am. Chem. Soc. 2000, 122, 3550. [Google Scholar]
- 174. Uetrecht C., Versluis C., Watts N. R., Roos W. H., Wuite G. J., Wingfield P. T., Steven A. C., Heck A. J., Proc. Natl. Acad. Sci. USA 2008, 105, 9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Uetrecht C., Versluis C., Watts N. R., Wingfield P. T., Steven A. C., Heck A. J., Angew. Chem. 2008, 120, 6343; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008, 47, 6247. [Google Scholar]
- 176. Uetrecht C., Watts N. R., Stahl S. J., Wingfield P. T., Steven A. C., Heck A. J., Phys. Chem. Chem. Phys. 2010, 12, 13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Shoemaker G. K., van Duijn E., Crawford S. E., Uetrecht C., Baclayon M., Roos W. H., Wuite G. J., Estes M. K., Prasad B. V., Heck A. J., Mol. Cell. Proteomics 2010, 9, 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. White L. J., Hardy M. E., Estes M. K., J. Virol. 1997, 71, 8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Uetrecht C., Barbu I. M., Shoemaker G. K., van Duijn E., Heck A. J., Nat. Chem. 2011, 3, 126. [DOI] [PubMed] [Google Scholar]
- 180. Basnak G., Morton V. L., Rolfsson O., Stonehouse N. J., Ashcroft A. E., Stockley P. G., J. Mol. Biol. 2010, 395, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Morton V. L., Dykeman E. C., Stonehouse N. J., Ashcroft A. E., Twarock R., Stockley P. G., J. Mol. Biol. 2010, 401, 298. [DOI] [PubMed] [Google Scholar]
- 182. Knapman T. W., Morton V. L., Stonehouse N. J., Stockley P. G., Ashcroft A. E., Rapid Commun. Mass Spectrom. 2010, 24, 3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Liepold L. O., Revis J., Allen M., Oltrogge L., Young M., Douglas T., Phys. Biol. 2005, 2, S166. [DOI] [PubMed] [Google Scholar]
- 184. Abedin M. J., Liepold L., Suci P., Young M., Douglas T., J. Am. Chem. Soc. 2009, 131, 4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kang S., Oltrogge L. M., Broomell C. C., Liepold L. O., Prevelige P. E., Young M., Douglas T., J. Am. Chem. Soc. 2008, 130, 16527. [DOI] [PubMed] [Google Scholar]
- 186. Kang S., Jolley C. C., Liepold L. O., Young M., Douglas T., Angew. Chem. 2009, 121, 4866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 4772. [Google Scholar]
- 187. Sharon M., Witt S., Glasmacher E., Baumeister W., Robinson C. V., J. Biol. Chem. 2007, 282, 18448. [DOI] [PubMed] [Google Scholar]
- 188. Loo J. A., Berhane B., Kaddis C. S., Wooding K. M., Xie Y., Kaufman S. L., Chernushevich I. V., J. Am. Soc. Mass Spectrom. 2005, 16, 998. [DOI] [PubMed] [Google Scholar]
- 189. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni‐Schmidt O., Williams R., Chait B. T., Sali A., Rout M. P., Nature 2007, 450, 695. [DOI] [PubMed] [Google Scholar]
- 190. Benesch J. L., Ruotolo B. T., Simmons D. A., Barrera N. P., Morgner N., Wang L., Saibil H. R., Robinson C. V., J. Struct. Biol. 2010, 172, 161. [DOI] [PubMed] [Google Scholar]
- 191. Nickell S., Kofler C., Leis A. P., Baumeister W., Nat. Rev. Mol. Cell Biol. 2006, 7, 225. [DOI] [PubMed] [Google Scholar]
- 192. Neutze R., Wouts R., van der Spoel D., Weckert E., Hajdu J., Nature 2000, 406, 752. [DOI] [PubMed] [Google Scholar]
- 193. Lorenzen K., Olia A. S., Uetrecht C., Cingolani G., Heck A. J., J. Mol. Biol. 2008, 379, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]