

Abstract
Dissipative self‐assembly processes in nature rely on chemical fuels that activate proteins for assembly through the formation of a noncovalent complex. The catalytic activity of the assemblies causes fuel degradation, resulting in the formation of an assembly in a high‐energy, out‐of‐equilibrium state. Herein, we apply this concept to a synthetic system and demonstrate that a substrate can induce the formation of vesicular assemblies, which act as cooperative catalysts for cleavage of the same substrate.
Keywords: amphiphiles, biomimetic catalysis, cooperative catalysis, dissipative self-assembly, systems chemistry
Self‐destruction for good: A substrate is shown to induce the self‐assembly of a cooperative catalyst, leading to its own destruction. Nature exploits the same strategy to obtain high‐energy structures such as microtubules and to drive non‐equilibrium phenomena.

Supramolecular chemistry is transitioning from the study of systems under thermodynamic or kinetic control towards the study of systems that operate out‐of‐equilibrium.1, 2 These systems require the continuous consumption of energy to keep the functional high‐energy state populated.3, 4 Compared to systems at equilibrium, this offers exciting new possibilities for the development of molecular machines, smart materials and complex reaction networks.3, 4, 5, 6 Energy dissipating processes play a key role in living organisms, for example, for controlling the structure and dynamics of the cytoskeleton,7, 8 and have recently also been linked to evolutionary processes.9 This has sparked strong interest in the design of chemical‐fuel driven out‐of‐equilibrium systems, in particular related to self‐assembly.3, 10, 11, 12 The majority of reported examples rely on the covalent modification of building blocks, which changes their propensity to form assemblies.13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 However, driven self‐assembly processes in Nature, that is, fuel‐driven processes that lead to a population of a high‐energy state,12 rely exquisitely on the use of noncovalent interactions for building block activation.7 This provides advantages typically associated with molecular recognition processes, such as high selectivity and fast activation rates. It is exemplified by microtubule formation (Figure 1 a), which initiates with the activation of tubulin dimers for self‐assembly upon complexation with guanosine triphosphate (GTP).8 Critically, tubulin dimers act also as a catalyst for the hydrolysis of GTP to GDP, and importantly, catalysis is significantly accelerated in the assembled state.25, 26 This leads to formation of a GDP‐rich high‐energy structure which collapses when the stabilizing caps are lost.27
Figure 1.

Reaction schemes of non‐equilibrium systems; in all figures the red arrows indicate the preferred reaction pathway. a) Reaction scheme for the dissipative formation of microtubules. Guanosine triphosphate (GTP) activates tubulin towards self‐assembly and the enhanced catalytic activity in the assembled states affords a high‐energy tubular structure. b) Example of the commonly used strategy for the formation of noncovalent assemblies under dissipative conditions: a high‐energy small molecule (here adenosine triphosphate, ATP) templates the assembly of vesicular structures, but is subsequently consumed in an independent process (here as a consequence of the enzymatic hydrolysis of ATP by potato apyrase) reverting the system to its initial state. c) Reaction scheme investigated in the present work: hydroxypropyl p‐nitrophenyl phosphate (HPNPP) templates the formation of assemblies that act as cooperative catalysts for its transphosphorylation.
Synthetic chemical‐fuel driven self‐assembly processes have been reported that also rely on noncovalent interactions between the building blocks and a chemical fuel.28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38 However, while most cases allude to similarities with microtubule formation or related biological dissipative processes, it turns out that in all the cases reported so far, a fundamentally different mechanism is operative (Figure 1 b).28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38 Contrary to what happens in Nature, energy dissipation, intended as the release of energy stored in the chemical fuel, is not catalysed by the building blocks, but rather by external elements such as an enzyme. This difference is of crucial importance, as to chemically drive an assembly process away from equilibrium using a chemical fuel, two fundamental prerequisites are that (1) the fuel‐to‐waste conversion is catalysed by the building blocks and that (2) fuel conversion is more efficient in the assembled state. These insights have emerged from a recent theoretical analysis of chemical‐fuel driven self‐assembly processes.12
Numerous research groups are currently pursuing synthetic dissipative self‐assembly processes that mimic by design the mechanism of microtubules.16, 21, 23, 30, 33 The system that comes closest to meeting the above‐cited criteria is reported by Otto et al. whom described the substrate‐induced structural reconfiguration of a dynamic covalent library in favour of the library component best adapted to catalyse the conversion of the substrate.15 Herein we show, to the best of our knowledge, the first example of substrate‐induced templation of a noncovalent assembly, which simultaneously acts as a catalyst for its cleavage by exhibiting cooperativity. This represents the first steps in the operating scheme of the driven self‐assembly of microtubules (Figure 1 c).
We recently demonstrated that the assembly behaviour of amphiphiles can be regulated by the addition of small oligoanions.30, 31 Amphiphilic C16TACN⋅Zn2+ (Figure 1 b), containing Zn2+‐complexed 1,4,7‐triazacyclononane (TACN) head groups were observed to form stabilised vesicular assemblies in the presence of ATP. These studies relied on the ability of charged counterions to effectively stabilize the assembly of surfactants containing charged head groups.39, 40, 41 Such counterions have been shown to significantly decrease the critical assembly concentration (cac) of amphiphiles in solution.30, 31, 39 Importantly, the TACN⋅Zn2+ moiety featured in the above studies has also been utilised as catalysts for the cleavage of phosphodiester bonds.42, 43, 44 This transphosphorylation reaction is known to require at least two metal ions acting cooperatively to achieve productive levels of activity. Manea et al. demonstrated that the complexation of Zn2+ by TACN units immobilised on the surface of gold nanoparticles allows cooperativity to occur between proximal catalytic moieties, leading to remarkable efficiencies in the cleavage of phosphate esters.42 These nanoparticles covered with an organic monolayer of TACN⋅Zn2+ groups have been termed “nanozymes”, as they possess many key features of natural enzymes, including Michaelis–Menten kinetics and cooperativity.42, 45 Our goal was to investigate whether these two effects—templation and catalysis—could be combined in a single system in which the phosphodiester substrate would induce assembly of TACN⋅Zn2+‐containing amphiphiles, and in this way, also generate the catalyst for its destruction (Figure 1 c). The model substrate typically used in the study of RNA phosphodiester hydrolysis is 2‐hydroxypropyl p‐nitrophenyl phosphate (HPNPP).42, 43 HPNPP is negatively charged and contains a phosphate group which we have shown previously to exhibit high affinities for the TACN⋅Zn2+ moiety.46 We thus had reason to believe that HPNPP would be able to act as an efficient counterion and to have a significant effect on the assembly behaviour of C16TACN⋅Zn2+. The cac of C16TACN⋅Zn2+ in the absence of substrate was first measured by titrating increasing amounts of the surfactant to an aqueous solution buffered at pH 7.0 containing the fluorescent apolar probe Nile red (5 μm, λ ex=570 nm, λ em=643 nm). This probe is solubilised by the apolar compartment of the assemblies, leading to an increase in fluorescence intensity after the cac has been reached (Figure 2 a). The cac was determined to be approximately 93 μm under these conditions, which is in close agreement with previous studies.30 The cac was next determined in the presence of different concentrations of HPNPP substrate. Figure 2 b shows the significant decrease in the cac of C16TACN⋅Zn2+ with increasing concentrations of HPNPP. The initial drop in cac is steep, with substrate binding shifting the cac down to 34 μm in the presence of 125 μm of HPNPP. Further increases in the concentration of HPNPP resulted in additional decreases in the cac, eventually levelling off at around 13 μm in the presence of 1 mm of HPNPP. The decrease in cac demonstrates that the presence of HPNPP increases the thermodynamic stability of the formed assemblies. The induced formation of assemblies below the native cac was further supported by dynamic light scattering (DLS) experiments, in which assemblies of 24±15 nm were detected (Figure 2 c). Objects of comparable size were also observed by (cryo) transmission electron microscopy (TEM/cryoTEM) and scanning laser confocal microscopy images (Figure 2 d), supporting the fact that HPNPP promotes the formation of vesicular assemblies.
Figure 2.
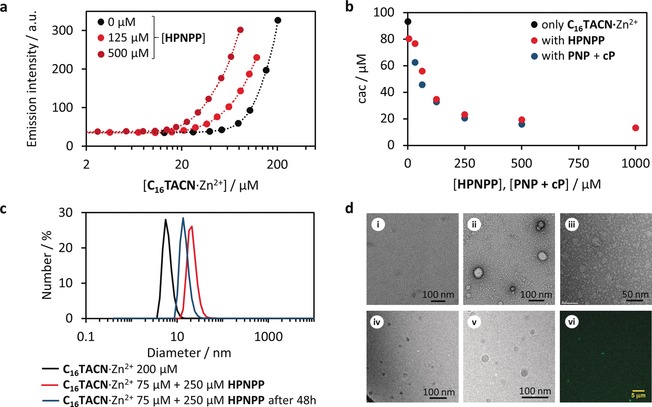
HPNPP templating ability. a) Selected emission intensity profiles for Nile red (5 μm, λ ex=570 nm, λ em=643 nm) at increasing C16TACN⋅Zn2+ concentrations, in the absence (black dots) and in the presence of HPNPP (125 μm, light red dots and 500 μm, dark red dots); the dotted lines serve as guide for the eye; b) Critical assembly concentration of C16TACN⋅Zn2+ measured in the presence of different concentrations of HPNPP or waste products (PNP + cP) with Nile red as a fluorescent probe. c) Hydrodynamic diameter of assemblies measured with dynamic light scattering (DLS) in the absence of HPNPP (black line) and in the presence of HPNPP (red line) and in the presence of waste (PNP + cP). d) Representative transmission electron microscopy (TEM) images of (i) [C16TACN⋅Zn2+]=50 μm in the absence of substrate HPNPP; (ii) vesicles obtained with [C16TACN⋅Zn2+]=50 μm in the presence of HPNPP (250 μm) and (iii) structures formed with [C16TACN⋅Zn2+]=50 μm in the presence of waste; (iv) and (v) show representative cryoTEM images with [C16TACN⋅Zn2+]=50 μm and [HPNPP]=250 μm; (vi) shows a representative image of vesicles with confocal microscopy for samples prepared with [C16TACN⋅Zn2+]=75 μm, [HPNPP] 250 μm and [coumarin 153]=1 μm. All experiments were performed in aqueous buffer solution (HEPES, 10 mm, pH 7.0) at 25 °C and standard TEM images were stained with 1 % uranyl acetate solution.
Satisfied that the substrate was effective in promoting assembly, we proceeded to examine the ability of the formed assemblies to promote catalysis. Using HPNPP as the substrate, the transphosphorylation reaction results in the formation of a cyclic phosphate (cP) and the release of p‐nitrophenolate (PNP), which allows the reaction rate to be conveniently measured spectrophotometrically. Figure 3 b shows a plot of the initial rates of reaction with varying concentrations of C16TACN⋅Zn2+ in the presence of HPNPP (62 μm) in aqueous buffer ([HEPES]=10 mm, pH 7). At low concentrations of C16TACN⋅Zn2+ (0–50 μm), very low reaction rates are observed. At a concentration of around 50 μm, however, the measured initial rates started to increase significantly, with the reaction rate being directly proportional to the surfactant concentration. Several important conclusions can be drawn from this experiment. The observation that the change of slope is observed at a concentration (54 μm), which closely matches the cac under these conditions (≈55 μm, see SI, section 4b), indicates that assembly formation facilitates catalysis. In addition, comparison of the slopes below and above the cac shows that the assemblies have a significantly higher activity compared to the monomeric surfactant. The significant rate enhancement upon assembly‐formation is confirmed by additional studies that are described below. Independent evidence for catalysis was obtained from 31P‐NMR spectroscopy. A fixed amount of C16TACN⋅Zn2+ (75 μm) was added to a constant concentration of HPNPP (1.0 mm) and changes in the 31P NMR spectra were monitored as a function of time (see SI, section 4c). The intensity of the signal due to HPNPP (−5.69 ppm) decreases, while a new signal at 17.14 ppm originating from the cyclic phosphate waste product appears.
Figure 3.

a) Cooperative catalysis induced by neighbouring TACN⋅Zn2+ complexes upon assembly. b) Initial speed of HPNPP hydrolysis at increasing C16TACN⋅Zn2+ concentrations ([HPNPP]=62 μm), the dotted lines are the linear fit to the first three and last three data points. c) Initial speed of HPNPP hydrolysis at a fixed concentration of C16TACN and varying Zn2+ concentrations ([HEPES buffer]=5 mm, [C16TACN]=50 μm, [HPNPP]=500 μm, 40 °C); the solid line represents the sigmoidal fit of the experimental data. d) Initial rates of HPNPP hydrolysis after successive additions of HPNPP (125 μm each addition) in the presence [HEPES buffer]=5 mm and [C16TACN⋅Zn2+]=50 μm at 40 °C. Black data points represent the rate directly after each addition, Grey data points represent the rate after 48 hours just before addition of the new batch of fuel.
The observed rate acceleration upon assembly strongly suggests that a cooperative mechanism is operative (Figure 3 a), similar to that observed previously in catalytic nanoparticles containing the same TACN⋅Zn2+ complex. Strong support that this is indeed the case came from a study in which the transphosphorylation activity of the assemblies was measured in the presence of varying concentrations of Zn2+ ions (Figure 3 c). Experiments were performed at 50 μm of surfactant and 500 μm of HPNPP, at conditions where the system is in the assembled state both in the presence and absence of Zn2+ ions (see SI, section 4d). The sigmoidal curve observed for the initial rate as a function of Zn2+ concentration is characteristic of cooperative catalysis by metal centres.42, 43 At low concentrations of Zn2+ metal ions (up to around 30 mol % saturation of the head groups) low reaction rates are observed, because of the low number of catalytic pockets formed by neighbouring Zn2+ complexes. At higher Zn2+‐loadings, the amount of catalytic pockets rapidly increases and, consequently, the rate increases significantly until a maximum is reached when the system is fully saturated with Zn2+ ions.
The observation of cooperative catalysis by C16TACN⋅Zn2+ assemblies was not an obvious result. Indeed, it had been previously reported that an analogous surfactant with a shorter chain showed very low catalytic activity for the same reaction, which was attributed to the highly dynamic nature of the assemblies.42 To investigate this aspect in more detail, we decided to prepare and study a series of surfactant molecules with hydrophobic chains of varying lengths, from ethyl (C2TACN) through to stearyl (C18TACN) as the kinetic stability of surfactant‐based assemblies is known to increase with the lengthening of the alkyl chain.47, 48 Figure 4 a shows the transphosphorylation activity at different concentrations of surfactants and equimolar Zn2+ in a buffered solution containing excess HPNPP (500 μm). Analysis of the data shows, as expected, a general increase in reaction rate with increasing concentrations of CnTACN⋅Zn2+. However, differences in reactivity of multiple orders of magnitude are observed between catalysts with hydrophobic chains of different lengths measured at the same head group concentration. For example, the rate of reaction with C18TACN⋅Zn2+ is roughly double the rate of reaction with C16TACN⋅Zn2+ at 100 μm, which is over a magnitude higher than that of C14TACN⋅Zn2+ (note the logarithmic scale on both axes). At the same concentration, the activities of C12TACN⋅Zn2+ or C2TACN⋅Zn2+ were negligible. Importantly, comparison of the onset of catalytic activity with the cac value (42 μm) of C14TACN⋅Zn2+ in the presence of 500 μm HPNPP confirms that assembly is a prerequisite for observing catalysis (see SI, section 4e for details). Indeed, the poor catalytic activity of C12TACN⋅Zn2+ and C2TACN⋅Zn2+ is in agreement with the fact that these surfactants do not assemble at the concentration regime studied.
Figure 4.

Effect of chain length on catalytic activity. a) Initial speed of HPNPP hydrolysis at increasing C n TACN⋅Zn2+ concentration (n=2 to 18, see legend, [HPNPP]=500 μm, [HEPES buffer]=5 mm), the dotted lines serve as a guide for the eye. b) Initial speed of HPNPP hydrolysis at increasing HPNPP concentration, and fixed C n TACN⋅Zn2+ concentration ([C n TACN⋅Zn2+]=50 μm, [HEPES buffer]=50 mm), the solid line are the data fit according to a Michaelis–Menten mechanism. Experiments were performed in aqueous buffer at pH 7, at 40 °C.
Interestingly, one might expect the reaction rates to be in a similar range for the different catalysts once the cac is reached, as above these concentrations similar assemblies are expected to be formed. Yet, the observed large difference between for example C14TACN⋅Zn2+ and C18TACN⋅Zn2+ indicates clearly that this is not the case. To gain further insight into this difference, the catalytic activity of C18TACN⋅Zn2+ and C16TACN⋅Zn2+ was measured at varying concentrations of HPNPP (Figure 4 b). Fitting of the saturation profiles to the Michaelis–Menten equation yielded similar V max values for C18TACN⋅Zn2+ (7.8±0.3×10−8 mol s−1) and C16TACN⋅Zn2+ (7.2±0.6×10−8 mol s−1), but a significantly lower K M value for C18TACN⋅Zn2+ (0.53±0.06 mm) compared to C16TACN⋅Zn2+ (1.0±0.1 mm). The nearly identical V max values indicate that no intrinsic difference exists between the catalytic pockets in the formed assemblies, whereas the different K M values indicate that the difference originates from the binding interaction between the surfactants and HPNPP. It is important to emphasize that, in contrast to a regular covalent catalyst such as an enzyme or nanozyme, in this system the substrate also affects the amount of catalyst present by acting on the equilibrium between monomeric and assembled states. The observation that the cac values in the presence of the same amount of HPNPP are inversely correlated to the hydrophobicity of the surfactant, implies that a higher amount of catalyst is present at the same concentration of HPNPP. This explains the higher observed rate at lower substrate concentrations and, consequently, the difference in apparent K M. Taken together, the three‐order of magnitude increase in activity upon self‐assembly (comparing C18TACN⋅Zn2+ and C2TACN⋅Zn2+) suggests that the enhanced activity with increasing the chain length could arise from the thermodynamic interplay between the surfactant and the substrate.
The fate of the self‐assembled structures following HPNPP hydrolysis was investigated by observing samples after 48 h, when nearly all of the HPNPP had been converted to cP and PNP. Data from both DLS and TEM experiments revealed the presence of assemblies that were smaller in size than in the presence of HPNPP (see Figure 2 c, 2d(iii)). The observation that the waste products of HPNPP cleavage are able to stabilise the formation of assemblies was supported by measuring the cac of the system in the presence of cP and PNP (see Figure 2 b). These experiments suggest that cP and PNP have similar affinity for the assemblies as the substrate HPNPP. Regrettably, it implies that in the current system we are not able to observe the spontaneous dissociation of assembled structures after fuel consumption. Yet, this state is still a responsive one as shown by refuelling experiments in which successive batches of chemical fuel are added at 48 h intervals (Figure 3 d). The catalytic activity can be reactivated, but with successively lower rates due to the build‐up of waste products which compete with the HPNPP substrate for binding to the surface of the assembled structures. This shows that binding is reversible and that the catalyst does not become irreversibly inhibited by the accumulated waste.
This study highlights the importance of cooperative catalysis for the design of energy driven processes. We have demonstrated the substrate‐induced self‐assembly of a supramolecular cooperative catalyst, a common mechanism of action in natural systems, that has so far not been exploited in synthetic systems.7, 12 In analogy with microtubule formation, the substrate promotes the formation of a noncovalent assembly and activates a cooperative catalytic pathway leading to its degradation. Cooperativity is connected to the assembled state and is able to induce rate accelerations of multiple orders of magnitude. The cooperative catalytic mechanism demonstrated is of utmost importance for the development of dissipative self‐assembling systems as it provides a tool to install kinetic asymmetry in energy consumption pathways.12 It paves the way for the preparation of high‐energy assemblies through energy‐dissipating processes and eliminates the necessity for external elements to dissipate energy. It is important to note that the substrate‐induced self‐assembly of cooperative catalysts is also exploited in natural systems for regulatory purposes, including the activation of protease caspase‐149 and of the main protease of SARS coronavirus,50 which points to a common underlying mechanism widely exploited by nature. Current efforts are aimed at developing alternative substrates with substantially higher affinity for the catalyst compared to the waste products which would cause spontaneous disassembly after fuel‐to‐waste conversion.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from a Catalyst: Seeding Grant (CSG‐AUT1701) from the Royal Society of New Zealand and New Zealand's Ministry of Business, Innovation and Employment and the European Research Council (ERC StG 239898) is acknowledged. We thank Dr Adrian Turner for assistance with TEM imaging, Dr Ilaria Fortunati for the confocal microscopy images, and Sushmitha Chandrabhas for carrying out preliminary experiments.
P. Solís Muñana, G. Ragazzon, J. Dupont, C. Z.-J. Ren, L. J. Prins, J. L.-Y. Chen, Angew. Chem. Int. Ed. 2018, 57, 16469.
Contributor Information
Prof. Leonard J. Prins, Email: leonard.prins@unipd.it.
Dr. Jack L.‐Y. Chen, Email: jack.chen@aut.ac.nz.
References
- 1. Grzybowski B. A., Huck W. T. S., Nat. Nanotechnol. 2016, 11, 585–592. [DOI] [PubMed] [Google Scholar]
- 2. Mattia E., Otto S., Nat. Nanotechnol. 2015, 10, 111–119. [DOI] [PubMed] [Google Scholar]
- 3. van Rossum S. A. P., Tena-Solsona M., van Esch J. H., Eelkema R., Boekhoven J., Chem. Soc. Rev. 2017, 46, 5519–5535. [DOI] [PubMed] [Google Scholar]
- 4. Ashkenasy G., Hermans T. M., Otto S., Taylor A. F., Chem. Soc. Rev. 2017, 46, 2543–2554. [DOI] [PubMed] [Google Scholar]
- 5. Kassem S., van Leeuwen T., Lubbe A. S., Wilson M. R., Feringa B. L., Leigh D. A., Chem. Soc. Rev. 2017, 46, 2592–2621. [DOI] [PubMed] [Google Scholar]
- 6. Merindol R., Walther A., Chem. Soc. Rev. 2017, 46, 5588–5619. [DOI] [PubMed] [Google Scholar]
- 7. Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P., Molecular Biology of the Cell, 4th ed., Garland, New York, 2002. [Google Scholar]
- 8. Hess H., Ross J. L., Chem. Soc. Rev. 2017, 46, 5570–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. England J. L., Nat. Nanotechnol. 2015, 10, 919–923. [DOI] [PubMed] [Google Scholar]
- 10. Della Sala F., Neri S., Maiti S., Chen J. L.-Y., Prins L. J., Curr. Opin. Biotechnol. 2017, 46, 27–33. [DOI] [PubMed] [Google Scholar]
- 11. De S., Klajn R., Adv. Mater. 2018, 30, 1706750. [DOI] [PubMed] [Google Scholar]
- 12. Ragazzon G., Prins L. J., Nat. Nanotechnol. 2018, 13, 882–889. [DOI] [PubMed] [Google Scholar]
- 13. Boekhoven J., Brizard A., Kowlgi K., Koper G., Eelkema R., van Esch J., Angew. Chem. Int. Ed. 2010, 49, 4825–4828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4935–4938. [Google Scholar]
- 14. Dambenieks A. K., Vu P. H. Q., Fyles T. M., Chem. Sci. 2014, 5, 3396–3403. [Google Scholar]
- 15. Fanlo-Virgós H., Alba A. R., Hamieh S., Colomb-Delsuc M., Otto S., Angew. Chem. Int. Ed. 2014, 53, 11346–11350; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11528–11532. [Google Scholar]
- 16. Boekhoven J., Hendriksen W., Koper G., Eelkema R., van Esch J., Science 2015, 349, 1075–1079. [DOI] [PubMed] [Google Scholar]
- 17. Tena-Solsona M., Rieß B., Grötsch R., Löhrer F., Wanzke C., Käsdorf B., Bausch A., Mueller-Buschbaum P., Lieleg O., Boekhoven J., Nat. Commun. 2017, 8, 15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kariyawasam L. S., Hartley C. S., J. Am. Chem. Soc. 2017, 139, 11949–11955. [DOI] [PubMed] [Google Scholar]
- 19. Sawczyk M., Klajn R., J. Am. Chem. Soc. 2017, 139, 17973–17978. [DOI] [PubMed] [Google Scholar]
- 20. Sorrenti A., Leira-Iglesias J., Sato A., Hermans T. M., Nat. Commun. 2017, 8, 15899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Ravensteijn B. G. P., Hendriksen W. E., Eelkema R., van Esch J. H., Kegel W. K., J. Am. Chem. Soc. 2017, 139, 9763–9766. [DOI] [PubMed] [Google Scholar]
- 22. Che H., Cao S., van Hest J. C. M., J. Am. Chem. Soc. 2018, 140, 5356–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colomer I., Morrow S. M., Fletcher S. P., Nat. Commun. 2018, 9, 2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tena-Solsona M., Wanzke C., Riess B., Bausch A. R., Boekhoven J., Nat. Commun. 2018, 9, 2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. David-Pfeuty T., Erickson H. P., Pantaloni D., Proc. Natl. Acad. Sci. USA 1977, 74, 5372–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caplow M., Shanks J., J. Biol. Chem. 1990, 265, 8935–8941. [PubMed] [Google Scholar]
- 27. Bowne-Anderson H., Zanic M., Kauer M., Howard J., Bioessays 2013, 35, 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pezzato C., Prins L. J., Nat. Commun. 2015, 6, 7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wood C. S., Browne C., Wood D. M., Nitschke J. R., ACS Cent. Sci. 2015, 1, 504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maiti S., Fortunati I., Ferrante C., Scrimin P., Prins L. J., Nat. Chem. 2016, 8, 725–731. [DOI] [PubMed] [Google Scholar]
- 31. Chen J. L. Y., Maiti S., Fortunati I., Ferrante C., Prins L. J., Chem. Eur. J. 2017, 23, 11549–11559. [DOI] [PubMed] [Google Scholar]
- 32. della Sala F., Maiti S., Bonanni A., Scrimin P., Prins L. J., Angew. Chem. Int. Ed. 2018, 57, 1611–1615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1627–1631. [Google Scholar]
- 33. Dhiman S., Jain A., George S. J., Angew. Chem. Int. Ed. 2017, 56, 1329–1333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1349–1353. [Google Scholar]
- 34. Dhiman S., Jain A., Kumar M., George S. J., J. Am. Chem. Soc. 2017, 139, 16568–16575. [DOI] [PubMed] [Google Scholar]
- 35. Hao X., Sang W., Hu J., Yan Q., ACS Macro Lett. 2017, 6, 1151–1155. [DOI] [PubMed] [Google Scholar]
- 36. Mishra A., Korlepara D. B., Kumar M., Jain A., Jonnalagadda N., Bejagam K. K., Balasubramanian S., George S. J., Nat. Commun. 2018, 9, 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang G., Sun J., An L., Liu S., Biomacromolecules 2018, 9, 2542–2548. [DOI] [PubMed] [Google Scholar]
- 38. Del Grosso E., Amodio A., Ragazzon G., Prins L. J., Ricci F., Angew. Chem. Int. Ed. 2018, 57, 10489–10493; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10649–10653. [Google Scholar]
- 39. Sasaki R., Murata S., Langmuir 2008, 24, 2387–2394. [DOI] [PubMed] [Google Scholar]
- 40. Köstereli Z., Severin K., Chem. Commun. 2012, 48, 5841–5843. [DOI] [PubMed] [Google Scholar]
- 41. Li G. C., Zhang S. Y., Wu N. J., Cheng Y. Y., You J. S., Adv. Funct. Mater. 2014, 24, 6204–6209. [Google Scholar]
- 42. Manea F., Houillon F. B., Pasquato L., Scrimin P., Angew. Chem. Int. Ed. 2004, 43, 6165–6169; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 6291–6295. [Google Scholar]
- 43. Zaupa G., Mora C., Bonomi R., Prins L. J., Scrimin P., Chem. Eur. J. 2011, 17, 4879–4889. [DOI] [PubMed] [Google Scholar]
- 44. Gruber B., Kataev E., Aschenbrenner J., Stadlbauer S., König B., J. Am. Chem. Soc. 2011, 133, 20704–20707. [DOI] [PubMed] [Google Scholar]
- 45. Wei H., Wang E., Chem. Soc. Rev. 2013, 42, 6060–6093. [DOI] [PubMed] [Google Scholar]
- 46. Pezzato C., Scrimin P., Prins L. J., Angew. Chem. Int. Ed. 2014, 53, 2104–2109; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2136–2141. [Google Scholar]
- 47. Hoffmann H., Ber. Bunsen-Ges. 1978, 82, 988–1001. [Google Scholar]
- 48. Zana R. in Encyclopedia of Surface and Colloid Science, 2nd ed, Vol. 5 (Ed.: A. T. Hubbard), Taylor & Francis, New York, 2002. [Google Scholar]
- 49. Datta D., McClendon C. L., Jacobson M. P., Wells J. A., J. Biol. Chem. 2013, 288, 9971–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheng S., Chang G., Chou C., Biophys. J. 2010, 98, 1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary