

Abstract
Photogenic: The synthesis of linderaspirone A has been accomplished in only three steps by a Darzens cyclopentenedione synthesis and dioxygen‐assisted photochemical dimerization. Moreover, the thermal isomerization of linderaspirone A into bi‐linderone has been discovered, which may give clues to the biosynthetic pathway for bi‐linderone.
Keywords: bi‐linderone, Darzens reaction, linderaspirone A, photochemistry, total synthesis
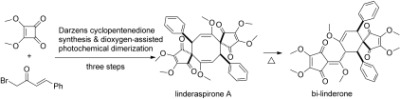
As one of the well‐known traditional medicine sources in China and Japan, the Lindera plant species has a long history of being used as an analgesic and antispasmodic.1 To date, a number of interesting bioactivities have been discovered from pharmacological studies on this plant, which include antioxidation,2 protection against postischemic myocardial dysfunction,3 antiviral (severe acute respiratory syndrome (SARS)‐associated coronavirus) activity,4 cytotoxicity,1 and slowing down the progression of diabetic nephropathy in db/db mice.5 Various natural products, ranging from sesquiterpenoids to alkaloids, flavonoids, lignans, butanolides, and cyclopentenedione derivatives, have been isolated from Lindera species.6 In 2010, Liu and co‐workers reported the isolation of linderaspirone A and bi‐linderone (1 and 2, Scheme 1) from the root of Lindera aggregata (Sims) kosterm.7 These compounds are two spirocyclopentenedione derivatives that contain highly congested eight‐ or six‐membered ring skeletons, respectively. More importantly, both of them are significantly active against glucosamine‐induced insulin resistance in bioactivity tests with HepG2 cells. Because of the unique architecture, remarkable bioactivity, and very limited natural availability (15 mg 1 and 4 mg 2 from 800 g of air‐dried powdered roots of the plant), 1 and 2 are attractive targets for organic chemists.
Scheme 1.
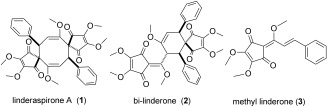
Linderaspirone A, bi‐linderone, and methyl linderone.
Through a 2,2′‐azobisisobutyronitrile (AIBN)‐promoted dimerization of the homologous natural product methyl linderone (3),6a, 8 Liu and co‐workers developed a one‐step biomimetic approach to 1 and 2 last year.9 At almost the same time, Wang and co‐workers reported a biomimetic total syntheses of 1 and 2 through photochemical [2+2] cycloaddition/Cope rearrangement and photochemical [2+2] cycloaddition/radical rearrangement, respectively. These syntheses also offered an important contribution to the understanding of the biosynthetic pathways of the dimerization of 3 and the formation of these compounds.10
However, the development of an efficient route to these valuable targets is still an important topic. Herein, we present another efficient approach to 1 through a Darzens cyclopentenedione synthesis and dioxygen‐assisted photochemical dimerization. Furthermore, the thermal isomerization of 1 is an efficient and biomimetic route for the synthesis of 2.
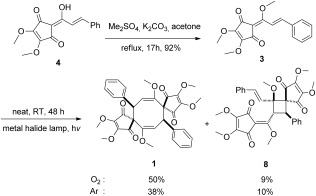
Our work started initially with the pursuit of concise access to 3. So far, several different strategies have been used to synthesize 3 and its natural analogues, which include linderone, methyl lucidone, and coruscanone A.10, 11 The most efficient synthesis of 3 was developed by Wang and co‐workers.10 Through the strategy of rearrangement of 4‐ylidenebutenolides,11f the synthesis of 3 has been accomplished in five steps in 45 % overall yield. Inspired by the Darzens reaction and its versatile following transformations,12 we envisioned that the cyclopentenedione skeleton of 3 could be constructed by the Darzens reaction and a subsequent ring expansion of cyclobutenedione (Scheme 2). To our surprise, linderone (4), the precursor of 3, is directly formed after the treatment of α‐bromoketone 6 13 and dimethyl squarate (5, 1.5 equivalents with respect to 6) with strong base. This is understandable, as the in situ expansion of the four‐membered ring is preferred through the release of the intense ring strain that exists in epoxide 7. The best result was obtained by using lithium hexamethyldislazide (LiHMDS) as the base. Compound 4 was formed in 62 % yield at 0.5 gram scale (based on the recovery of 23 % of the starting material). The subsequent methylation with dimethyl sulfate furnished 3 in good yield (Scheme 3). As a result, a Darzens cyclopentenedione synthesis of 3 has been developed. Starting from dimethyl squarate, the synthesis of 3 was accomplished in only two steps and 57 % overall yield.14
Scheme 2.
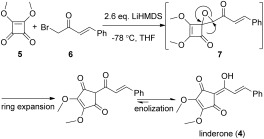
The Darzens cyclopentenedione synthesis.
Scheme 3.
Synthesis of linderaspirone A.
With the efficient strategy to cyclopentenediones established, 3 can be easily prepared on a multigram scale. Then, we turned our attention to the syntheses of 1 and 2. Solid 3 can be slowly transformed into 1 and 2 when exposed to ambient light, which matches the report of Wang and co‐workers that sunlight is sufficient to initiate the dimerization. Obviously, light irradiation provides favorable dimerization conditions. To our surprise, the presence of dioxygen can increase the yield of 1 when a metal halide lamp15 is used. As shown in Scheme 3, 1 was obtained in 50 % yield in the presence of dioxygen. The spectroscopic data of our synthetic linderaspirone A are in complete agreement with those of the natural product.7b The known [2+2] cycloaddition product 8 10 was also formed in 9 % yield. The reaction irradiated under argon afforded 1 in 38 % yield and 8 in 10 % yield. No clear signal for 2 was detected in the NMR spectra of the crude products from both reactions. We noticed that all photochemical dimerizations of 3 in Wang and co‐workers′ study of 1 and 2 had been carried out under inert atmosphere. There are also other studies on the photochemical dimerization of butadiene derivatives and the subsequent rearrangement.16 However, no dioxygen effect on the photochemical process has been reported to date.
The synthesis of 1 was accomplished in only three steps. However, 2 was always a minor product formed in the reported dimerization of 3, in which the best yield for 2 was only 20 %.10 No formation of 2 was detected in our dioxygen‐assisted photochemical dimerization approach. Because of our curiosity about a possible thermal isomerization of 1, a solution of 1 in p‐xylene was heated to reflux. Surprisingly, 2 was obtained in 51 % yield (Scheme 4). The NMR spectra and single crystal X‐ray crystallographic data of the synthesized bi‐linderone are identical to the published data for the naturally occurring compound.7a The thermal isomerization of 1 has thus opened up an efficient biomimetic approach for the synthesis of 2. In light of Wang and co‐workers′ contribution to the biomimetic synthesis of these important targets,10 a plausible mechanism for the formation of 2 is proposed in Scheme 4. Compound 1 is first transformed into 8 through a Cope rearrangement process. Then, 2 is generated from a subsequent radical rearrangement of 8. Notably, epi‐bi‐linderone (9)10 reported from the radical rearrangement of 8 is not obtained under these conditions.
Scheme 4.
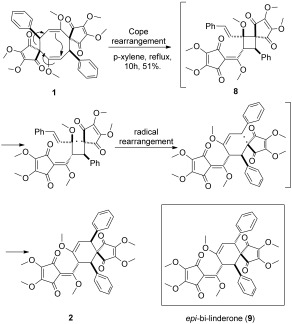
Concise synthesis of 2 and the plausible mechanism.
In conclusion, a concise synthesis of linderaspirone A has been accomplished in only three steps. The newly developed Darzens cyclopentenedione synthesis provides, so far, the most efficient approach to methyl linderone. The dioxygen‐assisted photochemical dimerization, which could be considered biomimetic conditions, has been disclosed as a better approach to linderaspirone A. Moreover, our study on the thermal isomerization of linderaspirone A revealed an interesting thermal Cope/radical rearrangement cascade of linderaspirone A. This is an important discovery not only of relevance for the synthesis, but also for the biosynthetic pathway to bi‐linderone.
Experimental Section
General
All reagents were obtained commercially and used without further purification unless otherwise noted. Anhydrous tetrahydrofuran (THF) was distilled from sodium/benzophenone until a deep blue color persisted. Anhydrous CH2Cl2 was distilled from CaH2. p‐Xylene was distilled from sodium sand. Acetone was distilled from phosphorus pentoxide. Diethyl ether was dried and distilled from CaH2. N,N‐dimethylformamide (DMF) was distilled from CaH2 under reduced pressure. Yields refer to chromatographically purified products unless otherwise stated. 1H NMR (400 MHz) spectra were referenced to CDCl3 (7.26 ppm) and 13C NMR (100 MHz) spectra were referenced to CDCl3 (77.0 ppm). All 13C NMR spectra were measured with complete proton decoupling. Peak multiplicities are designated by the following abbreviations: s=singlet; d=doublet; t=triplet; m=multiplet; br=broad; d=doublet and J=coupling constant in Hz. Mass spectrometry data were obtained in electrospray ionization mode (ESI MS).
Compound 6 was prepared according to the reported procedure:17 1H NMR (CDCl3, 400 MHz): δ=4.08 (s, 2 H), 6.94 (d, J=16 Hz, 1 H), 7.40–7.41 (m, 3 H), 7.56–7.58 (m, 2 H), 7.69 ppm (d, J=16 Hz, 1 H); 13C NMR (CDCl3, 100 MHz): δ=33.1, 122.1, 128.5, 128.9, 131.0, 133.8, 145.2, 190.9 ppm; HRMS (ESI+): calcd for C10H9BrNaO+: 246.9729 [M+Na+]; found: 246.9730.
Preparation of Linderone (4)
Compound 6 (500 mg, 2.22 mmol) and dimethyl squarate (473.5 mg, 3.33 mmol) were dissolved in anhydrous THF (25 mL). The resulting solution was cooled to −78 °C. Meanwhile, a solution of LiHMDS (1 m, 5.8 mL, 5.77 mmol) in THF (6 mL) was also cooled to −78 °C. The two resulting solutions were transferred to another empty flask via cannula at the same time. The reaction mixture was maintained at −78 °C for about 20 min and then quenched with HCl (36 % solution). The reaction system was quickly filtered through a short silica column and concentrated. The resulting crude product was purified by chromatography on a silica gel column (diethyl ether/petroleum ether=1:10–1:3) to afford 4 (304.9 mg, 48 %) as a yellow crystal with the recovery of 23 % of compound 6 (115 mg). 4: IR (Neat):  =1710, 1634, 1593, 982, 759, 693 cm−1. 1H NMR (CDCl3, 400 MHz): δ=4.16 (s, 3 H), 4.20 (s, 3 H), 7.38–7.39 (m, 3 H), 7.59–7.61 (m, 2 H), 7.65 (s, 2 H), 11.54 ppm (s, 1 H); 13C NMR (CDCl3, 100 MHz): δ=59.8, 59.9, 101.7, 117.7, 128.4, 128.8, 130.3, 134.9, 141.4, 145.3, 148.1, 164.5, 184.6, 193.2 ppm; HRMS (ESI+): m/z: calcd for C16H14NaO5
+: 309.0733 [M+Na+]; found: 309.0728.
=1710, 1634, 1593, 982, 759, 693 cm−1. 1H NMR (CDCl3, 400 MHz): δ=4.16 (s, 3 H), 4.20 (s, 3 H), 7.38–7.39 (m, 3 H), 7.59–7.61 (m, 2 H), 7.65 (s, 2 H), 11.54 ppm (s, 1 H); 13C NMR (CDCl3, 100 MHz): δ=59.8, 59.9, 101.7, 117.7, 128.4, 128.8, 130.3, 134.9, 141.4, 145.3, 148.1, 164.5, 184.6, 193.2 ppm; HRMS (ESI+): m/z: calcd for C16H14NaO5
+: 309.0733 [M+Na+]; found: 309.0728.
Synthesis of Methyl Linderone (3)8b
Dimethyl sulfate (2 mL, 2.66 g) and anhydrous potassium carbonate (2.52 g) were added to a solution of linderone (1.043 g, 3.6 mmol) in acetone (40 mL) in small portions over 10 min. The reaction mixture was heated to reflux for 17 h and the solvent was then removed under vacuum. Water (20 mL) was added to the resulting crude product and the system was extracted with ethyl acetate three times. The combined organic layer was washed with brine and dried with anhydrous Na2SO4. The filtrate was concentrated and the residue was purified by chromatography on a silica gel column (diethyl ether/petroleum ether=1:3) to afford 3 (993.9 mg, 92 %) as a luminous yellow solid. IR (Neat):  =1745, 1668, 1627, 1576, 970, 757, 692 cm−1. 1H NMR (CDCl3, 400 MHz): δ=4.10 (s, 3 H), 4.19 (s, 6 H), 7.37–7.41 (m, 3 H), 7.52 (d, J=15.6, 1 H), 7.59–7.61 (m, 2 H), 7.94 ppm (d, J=15.6, 1 H); 13C NMR (CDCl3, 100 MHz): δ=59.8, 59.9, 64.3, 109.3, 121.1, 128.3, 128.8, 130.0, 135.5, 141.2, 147.7, 148.9, 165.4, 184.7, 187.2 ppm; HRMS (ESI+): m/z: calcd for C17H16NaO5
+: 323.0890 [M+Na+]; found: 323.0896.
=1745, 1668, 1627, 1576, 970, 757, 692 cm−1. 1H NMR (CDCl3, 400 MHz): δ=4.10 (s, 3 H), 4.19 (s, 6 H), 7.37–7.41 (m, 3 H), 7.52 (d, J=15.6, 1 H), 7.59–7.61 (m, 2 H), 7.94 ppm (d, J=15.6, 1 H); 13C NMR (CDCl3, 100 MHz): δ=59.8, 59.9, 64.3, 109.3, 121.1, 128.3, 128.8, 130.0, 135.5, 141.2, 147.7, 148.9, 165.4, 184.7, 187.2 ppm; HRMS (ESI+): m/z: calcd for C17H16NaO5
+: 323.0890 [M+Na+]; found: 323.0896.
Synthesis of Linderaspirone A (1)
Methyl linderone (172.1 mg) was dissolved in CH2Cl2 (2 mL) in a silica tubular reactor and then the solvent was blow‐dried with dioxygen. The reaction was then irradiated for 48 h at room temperature by using a 400 W metal halide lamp. The resulting crude product was purified by chromatography on a silica gel column (acetone/petroleum ether=1:6) to afford 1 (86.2 mg, 50 %), 8 (15.5 mg, 9 %), and recovered 3 (17.3 mg, 10 %). 1: IR (Neat):  =3008, 2954, 1688, 1647, 1624, 1496, 1463, 1433, 1330, 1195, 1145 cm−1. 1H NMR (CDCl3, 400 MHz): δ=3.50 (s, 6 H), 3.89 (s, 6 H), 3.95 (s, 6 H), 5.43 (d, J=10.4 Hz, 2 H), 5.55 (d, J=10.4 Hz, 2 H), 7.29–7.30 ppm (m, 10 H); 13C NMR (CDCl3, 100 MHz): δ=43.2, 55.4, 59.5, 66.5, 102.0, 127.7, 128.4, 129.6, 137.9, 151.8, 153.2, 153.9, 192.2, 194.7 ppm. HRMS (ESI+): m/z: calcd for C34H32NaO10
+: 623.1888 [M+Na+]; found: 623.1892. 8: 1H NMR (CDCl3, 400 MHz): δ=3.33 (s, 3 H), 4.03 (s, 3 H), 4.06 (s, 3 H), 4.09 (s, 3 H), 4.13 (s, 3 H), 4.23 (s, 3 H), 4.41 (d, J=12 Hz, 1 H), 6.12 (d, J=12 Hz, 1 H), 6.55 (d, J=16.4 Hz, 1 H), 6.96 (d, J=16 Hz, 1 H), 7.04–7.47 ppm (m, 10 H). HRMS (ESI+): m/z: calcd for C34H32NaO10
+: 623.1888 [M+Na+]; found: 623.1891.
=3008, 2954, 1688, 1647, 1624, 1496, 1463, 1433, 1330, 1195, 1145 cm−1. 1H NMR (CDCl3, 400 MHz): δ=3.50 (s, 6 H), 3.89 (s, 6 H), 3.95 (s, 6 H), 5.43 (d, J=10.4 Hz, 2 H), 5.55 (d, J=10.4 Hz, 2 H), 7.29–7.30 ppm (m, 10 H); 13C NMR (CDCl3, 100 MHz): δ=43.2, 55.4, 59.5, 66.5, 102.0, 127.7, 128.4, 129.6, 137.9, 151.8, 153.2, 153.9, 192.2, 194.7 ppm. HRMS (ESI+): m/z: calcd for C34H32NaO10
+: 623.1888 [M+Na+]; found: 623.1892. 8: 1H NMR (CDCl3, 400 MHz): δ=3.33 (s, 3 H), 4.03 (s, 3 H), 4.06 (s, 3 H), 4.09 (s, 3 H), 4.13 (s, 3 H), 4.23 (s, 3 H), 4.41 (d, J=12 Hz, 1 H), 6.12 (d, J=12 Hz, 1 H), 6.55 (d, J=16.4 Hz, 1 H), 6.96 (d, J=16 Hz, 1 H), 7.04–7.47 ppm (m, 10 H). HRMS (ESI+): m/z: calcd for C34H32NaO10
+: 623.1888 [M+Na+]; found: 623.1891.
Thermal Isomerisation of Linderaspirone A for the Synthesis of Bi‐linderone (2)
A solution of 1 (50 mg) in freshly distilled p‐xylene (5 mL) was heated to reflux. After about 10 h, the solvent was removed under reduced pressure. The residue was purified by chromatography on a silica gel column (acetone/petroleum ether=1:5) to afford 2 (25.5 mg, 51 %) and recovered 1 (2.4 mg, 4.8 %). 2: IR (Neat):  =3006, 2953, 1687, 1672, 1639, 1620, 1601, 1492, 1462, 1432, 1326, 1210, 1200, 1122 cm−1. 1H NMR (CDCl3, 400 MHz): δ=3.59 (s, 3 H), 3.62 (s, 3 H), 3.76 (s, 3 H), 3.89 (d, J=11.6 Hz, 1 H), 4.00 (s, 3 H), 4.05 (s, 3 H), 4.12 (s, 3 H), 4.32 (br s, 1 H), 4.84 (s, 1 H), 6.14 (d, J=11.6 Hz, 1 H), 7.04–7.27 ppm (m, 10 H); 13C NMR (CDCl3, 100 MHz): δ=43.1, 45.8, 48.2, 54.9, 58.8, 59.3, 59.4, 59.5, 59.9, 65.0, 96.3, 110.9, 127.4, 127.6, 128.0, 128.2, 136.1, 140.1, 129.0, 146.7, 147.6, 152.8, 153.4, 154.6, 172.6, 183.9, 186.6, 194.7, 196.1 ppm. HRMS (ESI+): m/z: calcd for C34H32NaO10
+: 623.1888 [M+Na+]; found: 623.1860.
=3006, 2953, 1687, 1672, 1639, 1620, 1601, 1492, 1462, 1432, 1326, 1210, 1200, 1122 cm−1. 1H NMR (CDCl3, 400 MHz): δ=3.59 (s, 3 H), 3.62 (s, 3 H), 3.76 (s, 3 H), 3.89 (d, J=11.6 Hz, 1 H), 4.00 (s, 3 H), 4.05 (s, 3 H), 4.12 (s, 3 H), 4.32 (br s, 1 H), 4.84 (s, 1 H), 6.14 (d, J=11.6 Hz, 1 H), 7.04–7.27 ppm (m, 10 H); 13C NMR (CDCl3, 100 MHz): δ=43.1, 45.8, 48.2, 54.9, 58.8, 59.3, 59.4, 59.5, 59.9, 65.0, 96.3, 110.9, 127.4, 127.6, 128.0, 128.2, 136.1, 140.1, 129.0, 146.7, 147.6, 152.8, 153.4, 154.6, 172.6, 183.9, 186.6, 194.7, 196.1 ppm. HRMS (ESI+): m/z: calcd for C34H32NaO10
+: 623.1888 [M+Na+]; found: 623.1860.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (NSF‐21002078), the Ph.D. Programs Foundation for young teachers of Ministry of Education of China (20106101120004), the Education Department of Shaanxi Province (2010JK873), Northwest University (PR11025), and the Open Foundation from the Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education.
References
- 1. Ohno T., Nagatsu A., Nakagawa M., Inoue M., Li Y. M., Minatoguchi S., Mizukami H., Fujiwara H., Tetrahedron Lett. 2005, 46, 8657–8660. [Google Scholar]
- 2. Gu L. H., Wu T., Zhang Z. J., Chou G. X., Wang Z. T., Acta Pharm. Sin. 2006, 41, 956–962. [PubMed] [Google Scholar]
- 3. Wang N. Y., Minatoguchi S., Arai M., Uno Y., Hashimoto K., Chen X. H., Fukuda K., Akao S., Takemura G., Fujiwara H., Am. J. Chin. Med. 2004, 32, 587–598. [DOI] [PubMed] [Google Scholar]
- 4. Li S. Y., Chen C., Zhang H. Q., Guo H. Y., Wang H., Wang L., Zhang X., Hua S. N., Yu J., Xiao P. G., Li R. S., Tan X. H., Antiviral Res. 2005, 67, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ohno T., Takemura G., Murata I., Kagawa T., Akao S., Minatoguchi S., Fujiwara T., Fujiwara H., Life Sci. 2005, 77, 1391–1403. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Gan L. S., Zheng Y. L., Mo J. M., Liu X., Li X. H., Zhou C. X., J. Nat. Prod. 2009, 72, 1497–1501; [DOI] [PubMed] [Google Scholar]
- 6b. Chang S. Y., Cheng M. J., Peng C. F., Chang H. S., Chen I. S., Chem. Biodiversity 2008, 5, 2690–2698; [DOI] [PubMed] [Google Scholar]
- 6c. Wang S. Y., Lan X. Y., Xiao J. H., Yang J. C., Kao Y. T., Chang S. T., Phytother. Res. 2008, 22, 213–216; [DOI] [PubMed] [Google Scholar]
- 6d. Zhang C. F., Sun Q. S., Zhao Y. Y., Wang Z. T., Chin. Chem. Lett. 2003, 14, 1033–1036; [Google Scholar]
- 6e. Zhang C. F., Sun Q. S., Zhao Y. Y., Wang Z. T., Chin. J. Med. Chem. 2001, 11, 274–276; [Google Scholar]
- 6f. Kwon H. C., Baek N. I., Choi S. U., Lee K. R., Chem. Pharm. Bull. 2000, 48, 614–616; [DOI] [PubMed] [Google Scholar]
- 6g. Kozuka M., Yoshikawa M., Sawada T., J. Nat. Prod. 1984, 47, 1063–1063. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Wang F., Gao Y., Zhang L., Liu J. K., Org. Lett. 2010, 12, 2354–2357; [DOI] [PubMed] [Google Scholar]
- 7b. Wang F., Gao Y., Zhang L., Bai B., Hu Y. N., Dong Z. J., Zhai Q. W., Zhu H. J., Liu J. K., Org. Lett. 2010, 12, 3196–3199. [DOI] [PubMed] [Google Scholar]
- 8. Kiang A. K., Lee H. H., Sim K. Y., J. Chem. Soc. 1962, 4338–4345. [Google Scholar]
- 9. Wang G. Q., Wei K., Zhang L., Feng T., Wang F., Wang Q. A., Liu J. K., Tetrahedron Lett. 2011, 52, 2719–2721. [Google Scholar]
- 10. Tan H., Zheng C., Liu Z., Wang D. Z., Org. Lett. 2011, 13, 2192–2195. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Bose G., Langer P., Synlett 2005, 6, 1021–1023; [Google Scholar]
- 11b. Li X. C., Ferreira D., Jacob M. R., Zhang Q., Khan S. I., EI Sohly H. N., Nagle D. G., Smillie T. J., Khan I. A., Walker L. A., Clark A. M., J. Am. Chem. Soc. 2004, 126, 6872–6873; [DOI] [PubMed] [Google Scholar]
- 11c. Aoyama Y., Konoike T., Kanda A., Naya N., Nakajima M., Bioorg. Med. Chem. Lett. 2001, 11, 1695–1697; [DOI] [PubMed] [Google Scholar]
- 11d. Bennett G. J., Lee H. H., J. Chem. Soc. Perkin Trans. 1 1986, 633–638; [Google Scholar]
- 11e. Lee H. H., Que Y. T., J. Chem. Soc. Perkin Trans. 1 1985, 453–455; [Google Scholar]
- 11f. Clemo N. G., Gedge D. R., Pattenden G., J. Chem. Soc. Perkin Trans. 1 1981, 1448–1453. [Google Scholar]
- 12.
- 12a. Sweeney J. B., Cantrill A. A., Drew M. G. B., McLAaren A. B., Thobhani S., Tetrahedron 2006, 62, 3694–3703; [Google Scholar]
- 12b. Davis F. A., Ramachandar T., Wu Y., J. Org. Chem. 2003, 68, 6894–6898; [DOI] [PubMed] [Google Scholar]
- 12c. Aldous D. J., Dalencon A. J., Steel P. G., Org. Lett. 2002, 4, 1159–1162; [DOI] [PubMed] [Google Scholar]
- 12d. Schwartz A., Madan P. B., Mohacsi E., O’Brien J. P., Todaro L. J., Coffen D. L., J. Org. Chem. 1992, 57, 851–856; [Google Scholar]
- 12e. Deyrup J. A., J. Org. Chem. 1969, 34, 2724–2727. [Google Scholar]
- 13.The corresponding alpha‐chloro/iodo ketones gave worse results. Please see the Supporting Information for details.
- 14.The new approach to methyl linderone is the subject of an application for a Patent Chinese (no. 201110267743.1).
- 15.The metal halide lamp (400 W) was purchased from Xian Bilon Biological Technology Co., Ltd. in China. Please see the Supporting Information for the relative spectral energy distribution curve of the lamp.
- 16.
- 16a. Row T. N. G., Swamy H. R., Acharya K. R., Ramamurthy V., Venkatesan K., Rao C. N. R., Tetrahedron Lett. 1983, 24, 3263–3266; [Google Scholar]
- 16b. Inhülsen I., Chin K., Göwert M., Margaretha P., Helv. Chim. Acta 2011, 94, 1030–1034. [Google Scholar]
- 17. Onizawa Y., Kusama H., Iwasawa N., J. Am. Chem. Soc. 2008, 130, 802–803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information