

Abstract
The evolution of drug resistance is one of the most fundamental problems in medicine. In HIV/AIDS, the rapid emergence of drug‐resistant HIV‐1 variants is a major obstacle to current treatments. HIV‐1 protease inhibitors are essential components of present antiretroviral therapies. However, with these protease inhibitors, resistance occurs through viral mutations that alter inhibitor binding, resulting in a loss of efficacy. This loss of potency has raised serious questions with regard to effective long‐term antiretroviral therapy for HIV/AIDS. In this context, our research has focused on designing inhibitors that form extensive hydrogen‐bonding interactions with the enzyme’s backbone in the active site. In doing so, we limit the protease’s ability to acquire drug resistance as the geometry of the catalytic site must be conserved to maintain functionality. In this Review, we examine the underlying principles of enzyme structure that support our backbone‐binding concept as an effective means to combat drug resistance and highlight their application in our recent work on antiviral HIV‐1 protease inhibitors.
Keywords: antiviral agents, backbone binding, drug resistance, enzymes, proteins
Impeding the evolution of drug resistance: HIV protease inhibitors are critical to antiretroviral treatment regimens. However, the rapid onset of drug resistance limits the effectiveness of most approved inhibitors. The structure‐based design of inhibitors targeting atoms in the protein backbone is an attractive strategy for maintaining drug efficacy. This approach limits the enzyme's ability to evolve resistance without sacrificing its catalytic activity.
1. Introduction
It has taken half a century for the selection of antibiotic‐resistant bacteria to represent a widespread threat to humans, and yet it takes only weeks to months to select inhibitor‐resistant immunodeficiency viruses in treated patients.
Esteban Domingo, Christof Biebricher, Manfred Eigen, and John Holland[1]
1.1. A Brief History of Viruses
Viruses have been causing disease in humans since ancient times.2 As early as 1150 B.C., viral diseases were recorded in the hieroglyphics of ancient Egypt and evidence of smallpox infection was found in the pockmark‐scarred remains of Pharaoh Ramses V. In the 15th century, early writings describing preventive inoculations against the smallpox virus began to appear in China.3 However, it wasn’t until the pioneering work of Adolf Mayer, Dimitri Ivanovsky, and Martinus Beijerinck, with the tobacco mosaic virus in the early 1900s, that viruses were recognized as distinct pathogenic microorganisms.4 Throughout history, viruses have primarily played a detrimental role in human health. The smallpox and influenza viruses caused worldwide epidemics resulting in millions of deaths.5 To prevent the spread of the aphthovirus pathogen responsible for foot‐and‐mouth disease, millions of animals were slaughtered resulting in significant economical losses.6 Recently, the SARS coronavirus made headlines when an outbreak spread rapidly across the world in near‐pandemic fashion killing 11 % of infected individuals.7 Also, the highly pathogenic influenza A virus, subtype H5N1, often known as avian influenza virus, is a potential pandemic threat and has become a major global concern.8 The world witnessed the emergence of human immunodeficiency virus (HIV) in the 1980s. Since then, HIV infection leading to acquired immunodeficiency syndrome (AIDS) has become a global crisis of catastrophic proportion infecting nearly 2.6 million new individuals per year.9 There are effective treatments for HIV and AIDS that can slow the course of the disease, but there is no cure or vaccine to date.
1.2. HIV Emergence
The HIV time line began early in 1981 and the virus was first isolated in 1983.10, 11 Since then, millions of individuals throughout the world have been infected with HIV. The resulting onset of AIDS has swept across the continents causing an estimated 25 million deaths and leaving millions of children orphaned.9 Recent estimates from the joint United Nations program on HIV/AIDS (UNAIDS) indicate that worldwide, over 33 million adults and children are currently living with the disease and 1.8 million AIDS‐related deaths occur each year.9 While these statistics are alarming, significant advancements in both HIV treatment and prevention have appeared to be turning the tide in the fight against AIDS, as evidenced by the steadily decreasing number of annual deaths.9 Intensive research into the development of novel antiviral agents and the use of multidrug combination therapies have resulted in a significant increase in life expectancy for those with access to therapy.12, 13 Unfortunately, the rapid emergence of drug resistance has rendered many treatments ineffective and continues to be a formidable challenge for molecular design and drug discovery.14, 15 Moreover, the consequences of drug resistance may unravel the progress made toward HIV/AIDS management. Today, there is still an urgent need for the development of novel anti‐HIV therapeutics and drug‐design tools for combating drug resistance.
1.3. The Advent of Protease Inhibitors
Biochemical events critical to HIV replication have suggested a number of drug‐design targets for therapeutic intervention. Among them, the HIV protease enzyme was quickly recognized as an important therapeutic target.16 It has been demonstrated that an effective HIV protease is essential to the production of mature, infectious HIV virions.17 Logically, inhibition of the HIV protease became the subject of much pharmaceutical research. Subsequent drug‐development efforts led to the advent of the first generation of protease inhibitors (PIs) marking a new era in AIDS chemotherapy.18 The use of PIs in combination with reverse transcriptase inhibitors proved to be an extremely effective treatment regimen which suppresses viral reproduction and reduces the possibility of viral mutations.19 Despite their early success, PIs were plagued with several drawbacks including low metabolic stability, poor bioavailability, debilitating side effects, and drug toxicity.20 Perhaps the most concerning obstacle has been the rapid development of drug‐resistant viral strains which render the PIs ineffective.21 Currently, 40–50 % of the patients who achieve initial viral suppression will eventually experience treatment failure.22 Additionally, these drug‐resistant viral strains can be transmitted to new individuals.23 Success in the future management of HIV/AIDS depends on the development of new antiviral agents that maintain efficacy against drug‐resistant viral strains.
1.4. Mechanism of Drug Resistance
HIV‐1 has an astonishing capacity for genetic evolution which is a major driving force for drug resistance. Its relentless ability to mutate arises from a high mismatch error rate (10−3 to 10−5 nucleotide bases per cycle) of the virus’s reverse transcriptase enzyme and the absence of exonuclease‐based proofreading activity.24, 25 These factors, in conjunction with the virus’s rapid replication cycle (1010 virions per day) and genetic recombination ability, result in seemingly endless genetic diversification.26, 27 However, the number of variants within the quasi‐species at a given time is limited by natural selection, deleterious mutations, and limited host cell availability, and by inactivation from the host’s immune system response.28 As a result, the virus population primarily consists of single‐mutation strains and relatively few double‐mutation strains. Antiretroviral treatment creates a new selection pressure resulting in the amplification of drug‐resistant strains. Viral evolution continues over time adding new mutations that restore viral fitness while maintaining drug resistance. This leads to a resurgence of the viral load and eventually treatment failure. Combating drug resistance remains a formidable challenge that must be considered during the design of new antiretroviral agents.
It may be more effective to develop therapies that limit the emergence or growth of HIV‐1 variants than to combat these variants once they have already evolved. The development of new classes of antiretroviral drugs with new mechanisms of action, showing durable effects, and causing minimal side effects, are important therapeutic objectives. There is reason to be optimistic as tremendous progress has been made in terms of new drugs which are very potent and with a high genetic barrier to resistance. Recently a number of new approved drugs with novel mechanisms of action, including an integrase inhibitor,29 and a virus‐entry inhibitor,30 have shown efficacy against resistant strains. However, these drugs quickly succumb to the development of resistance rendering them ineffective.31, 32 Also, maturation inhibition and small‐molecule inhibition of HIV pre‐mRNA splicing holds considerable promise.33, 34
1.5. Protease Evolution in Response to Inhibitor Pressure
The evolution of HIV protease in response to therapeutic pressures has been reviewed in great detail and will be presented here only briefly.35–39 HIV protease mutations that arise in response to treatment conditions must by definition provide a replication advantage. Primary mutations typically include D30N, G48V, I50L/V, V82A/F/T, I84V, and L90M and are commonly found near the active site.40 They affect hydrophobic, van der Waals, and electrostatic interactions between the enzyme and inhibitor, resulting in a loss of binding affinity that decreases an inhibitor drug’s effectiveness.41, 42 However, the mutations also interfere with substrate processing, conferring a fitness cost and negatively affecting the replication of HIV.43, 44 As a result, additional mutations accumulate in a stepwise fashion that restore catalytic efficiency while maintaining drug resistance.45 Often, these secondary mutations occur further away from the active site causing long‐range structural perturbations that compensate for the primary mutation’s functional deficit. Additionally, Gag and Gag‐Pol may co‐evolve with the protease incurring mutations that enhance their ability to be processed by the mutant protease.46, 47 Ultimately, ten or more mutations can accumulate resulting in viable viruses with resistance to multiple drugs.
2. Targeting the Protein Backbone to Combat Drug Resistance
2.1. The Underlying Principle behind the Backbone‐Binding Strategy
We have developed a novel structure‐based concept for drug design to address the problem of drug resistance. Our structural analysis and comparison of the X‐ray structures of various mutant HIV‐1 proteases with the X‐ray structure of wild‐type HIV‐1 protease revealed that the backbone conformation in the active site of mutant proteases is only minimally distorted.48, 49 Conceivably, if we design an inhibitor that maximizes interactions in the HIV protease active site, particularly extensive hydrogen‐bonding interactions with the protein backbone of the wild‐type HIV‐1 protease, such an inhibitor will likely maintain these contacts with mutant proteases. In essence, by targeting the protein backbone, the development of drug‐resistant HIV should be hindered, as mutations that alter the backbone conformation would most likely reduce catalytic capacity.50 We view this designed inhibitor as a “molecular crab” capable of tightly gripping the protein backbone and holding on in the enzyme active site.
This backbone‐binding design strategy led to the development of new PIs with enhanced active‐site interactions, in particular inhibitor–backbone hydrogen‐bonding interactions. Fundamentally, for drug resistance to occur, viral mutations must arise that diminish drug binding but retain viral fitness. Mutations that occur within the active site or result in structural distortions are limited because they produce impaired proteolysis of the natural polyprotein substrates.43, 44 This was reinforced during our reviews of X‐ray structures of mutant HIV‐1 proteases which revealed the structural changes associated with drug‐resistant mutants.51, 52 Based upon this backbone‐binding strategy, we have focused our molecular design efforts on promoting extensive hydrogen‐bonding interactions with the protein backbone atoms contributing to the S2–S2′ subsites. The S1 and S1′ subsites are largely formed by hydrophobic residues, while both hydrophobic and hydrophilic residues contribute to the S2 and S2′ subsites.53 In addition, we planned to fill the hydrophobic pockets throughout the protease active site and thus further limit the ability of the virus to develop drug resistance. Furthermore, we have sought to improve bioavailability by decreasing the peptidic character of our inhibitors through the design of heterocyclic or cyclic‐polyether‐derived templates and ligands. In this Review, we highlight the molecular basis for this targeted protein‐backbone‐binding concept and feature our designed inhibitors that have emerged from this novel design concept. Our extensive X‐ray crystallographic studies and a detailed analysis of antiviral data strongly corroborate our protein‐backbone‐binding strategy to combat drug resistance.
2.2. Protein Structure Defines Catalytic Activity
Investigations into the factors controlling enzyme activity have revealed that protein structure and enzyme function are closely related.54–56 Most enzymes are proteins consisting of strands of amino acids combined to form distinct polypeptide chains. These proteins fold spontaneously, generating local secondary structures (α helix, β strands, etc.) that lead to the formation of a defined three‐dimensional (3D) tertiary structure. With many enzymes, multiple protein chains combine through noncovalent interactions forming a quaternary structure possessing multiple subunits. Protein structures are stabilized by a collection of weak intramolecular forces that can be disrupted and reformed allowing a limited range of dynamic movement. The folding process creates cavities near the surface of the enzyme that may serve as substrate binding sites. Interestingly, a relatively small volume of the overall enzyme constitutes the active catalytic site with the remainder of the protein serving as a structural scaffold.57 For effective catalysis to occur, the amino acid residues within the active site require a specific 3D configuration in which the transition state of the chemical reaction can be attained more readily than in the absence of the enzyme.58–60 Perturbations of this configuration can have a deleterious effect resulting in a loss of catalytic function. Hence, viral mutations are limited by natural selection requirements to maintain the key structural elements of an enzyme’s active site.
2.3. HIV‐1 Protease’s Substrate Binding Site and Conserved Interactions
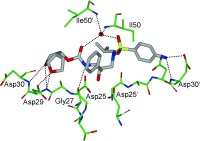
HIV‐1 protease is an aspartic protease containing two catalytic aspartic acid residues in the active site that share an acidic proton and interact with a water molecule in the absence of a substrate or inhibitor. The catalytically active enzyme is a homo dimer and each monomer comprises 99 amino acids. The active site is formed by two catalytic aspartic acids and each residue is located in each domain (monomer). The scissile bond of the peptide substrate is in close proximity to the active site. A pair of flaps, one from each monomer, is located at the entrance to the active site.61 The flaps fold down over substrates upon binding and act as a solvent shield that excludes water and creates a local environment conducive to catalysis. The flaps are flexible, showing an open conformation in the apoenzyme.53, 62 The peptide substrate contains at least seven residues extending from P4 to P3′, where the scissile bond lies between P1 and P1′. The side chains of the substrate lie in subsites S4 to S3′ formed by protease residues. Hydrophobic residues occupy the S1 and S1′ subsites, and hydrophobic or hydrophilic residues can be accommodated by the S2 and S2′ subsites. A series of conserved hydrogen‐bonding interactions connect the backbone of the protease and the substrate and serve as a major contribution to binding affinity.63 These interactions are shown in Figure 1. The X‐ray structure of HIV protease bound to the peptide analogue of the p2/NC cleavage site (PDB code 2AOD) demonstrates these interactions.64 A number of amino acid residues in the active site, such as Asp25, Gly27, Ala28, Asp29, and Gly48, are highly conserved; therefore, inhibitor design strategies targeting these residues have led to potent PIs.50, 65–68
Figure 1.
a) Comparison of unliganded protease with open conformation flaps (PDB code 1HHP64 in cyan) and protease in complex with a substrate analogue (PDB code 2AOD64 in magenta) showing closed conformation flaps. The catalytic residues Asp25 and Asp25′ and the peptide analogue are shown as stick models. b) Interactions of the protease with the peptide analogue of the p2/NC cleavage site (ace‐Thr‐Ile‐Nle‐r‐Nle‐Gln‐Arg, where Nle is norleucine substituting for Met in the natural peptide sequence and r indicates the reduced peptide bond) (PDB code 2AOD).64 The conserved hydrogen‐bonding interactions are shown as green dotted lines, and nonconserved hydrogen bonds are indicated by black dashed lines.
2.4. Structural Evidence of Minimal Backbone Distortion in Mutant HIV‐1 Proteases
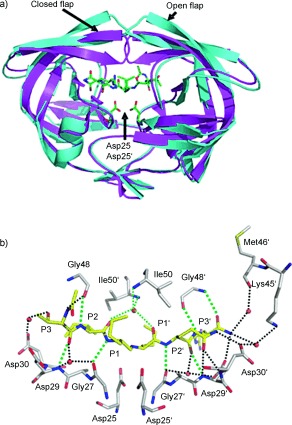
During our efforts in structure‐based drug design to develop novel antiviral HIV‐1 protease inhibitors, we have compared the X‐ray crystal structures of inhibitor‐bound HIV‐1 protease (wild type) with the crystal structures of drug‐resistant mutants of the protease.48, 49 As stated earlier, superimposition of these structures indicates only a minimal deviation in the backbone atoms around the active site. These structural comparisons provided insight into the molecular design strategy for combating drug resistance as reviewed recently.50 Mutations can be divided into two main categories. First, mutations of active‐site residues can directly alter protease interactions with an inhibitor, as shown by examples of mutants containing I84V or I50V that reduce interactions with a number of PIs.69–71 Alternatively, distal mutations can act indirectly to diminish protease stability and interactions with an inhibitor, as shown for mutants with L24I, F53L, and L90M.70, 72, 73 The majority of these mutants show minimal changes in the backbone structure around the active site. Even the flexible flaps generally show changes of less than 1 Å for the backbone atoms. One exception is mutation V82A, which produces shifts in the loop comprising residues 79–82 with compensating hydrophobic contacts.71, 73, 74 The loop can adjust by 1–2 Å to accommodate the different‐sized hydrophobic groups at P1 and P1′ of the inhibitors.75 As depicted in Figure 2, even structures of drug‐resistant HIV‐1 proteases with 10–14 mutations, and HIV‐2 protease which differs in about 40 different residues, superimpose with only a minimal deviation in the backbone atoms around the active site.51, 76–79 Mutations producing drug resistance cannot significantly alter the overall structure of the active site that is essential for protease function. Viable mutant strains will show minimal distortions in the structure of the protease active site as expected in order to maintain catalytic activity and viral replication fitness.50, 52 Based on these observations, we hypothesized that inhibitors that maximize hydrogen‐bonding interactions with backbone NH or C=O atoms in the active site would retain these interactions in viral mutants. Therefore, these compounds would maintain potency despite viral mutation, providing a viable solution to the problem of drug resistance in HIV‐1 treatments. Using this original design concept, we have actively designed and synthesized a variety of inhibitors that form extensive binding interactions with the protease backbone and are capable of maintaining efficacy against panels of clinically relevant drug‐resistant HIV‐1 viral strains.50, 67, 68, 80
Figure 2.

Overlay of HIV‐1 protease with multiple mutants (green: PDB code 2FDD;76, 77 yellow: PDB code 1SGU78), HIV‐2 protease (red: PDB code 1HSH79) with HIV‐1 protease (blue: PDB code 2IEN69) showing minimal backbone deviation.
3. Structure‐Based Design Targeting the Protein Backbone
3.1. PIs with High‐Affinity Ligands Derived from Cyclic Ethers
In an effort to combat drug resistance, we have been involved in the design and synthesis of conceptually novel protease inhibitors based upon the X‐ray structure of HIV‐1 protease bound to saquinavir (1, Figure 3). Our major strategy in structure‐based design is to maximize inhibitor interactions in the protease active site. Particularly, we planned to promote hydrogen bonding with the protease backbone atoms in the S2 to S2′ subsites.50 These efforts culminated in the discovery of a wide range of exceedingly potent PIs with impressive resistance profiles. One of these PIs was darunavir (2, TMC‐114, UIC‐94017), which has been approved by the FDA for the treatment of HIV/AIDS patients harboring drug‐resistant HIV.81, 82 We have described the development of darunavir in a number of recent reviews.67, 83, 84 Here, we will briefly highlight those early structure‐based efforts and focus mainly on darunavir’s unique binding properties using X‐ray crystallographic studies and we will analyze drug‐resistance properties in light of the structural information. We will then provide highlights of our subsequent efforts into the design of a variety of PIs using the backbone‐binding strategy to develop a new generation of PIs to withstand emerging multidrug‐resistant HIV‐1 variants.
Figure 3.
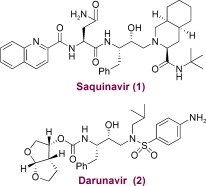
Structures of saquinavir and darunavir.
Initially, our design of PIs focused on reducing the peptide‐like features and improving the druglike properties of saquinavir‐based protease inhibitors.67 Saquinavir (1) is a potent FDA‐approved inhibitor; established structure–activity studies and known X‐ray structural information have provided important molecular insight into the ligand‐binding interactions.85, 86 On the basis of this structural information, we were particularly interested in reducing the molecular weight and eliminating peptide‐like bonds. We drew inspiration from nature and began incorporating cyclic ether features inherent to bioactive natural products.68 We developed a series of molecular scaffolds featuring conformationally constrained cyclic ether and heterocyclic structures that mimic the binding modes of peptide/amide bonds in the S2 subsite of the HIV‐1 protease active site.
3.2. Development of 3S‐THF and Bis‐THF P2 Ligands
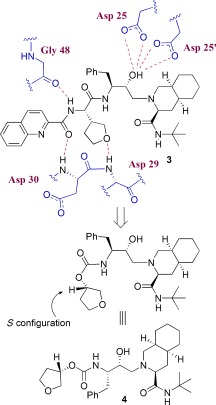
Although saquinavir is a potent PI, its oral bioavailability is poor, possibly because of the presence of multiple amide/peptide bonds. Based upon the X‐ray structure of saquinavir‐bound HIV‐1 protease, we attempted to replace two amide carbonyls (P2/P3) with cyclic ether and sulfone templates. Particularly, we planned to position the ether or sulfone such that the oxygen atom could form interactions with the protease similar to those seen for the P2 and P3 amide/peptide carbonyl functions of saquinavir. We were interested in cyclic ether features because numerous bioactive natural products contain such structural subunits, and natural products such as monensin and ginkolide do not suffer from the absorption problems inherent to peptidelike drugs.87, 88 As shown in Figure 4, replacement of the P2‐asparagine of saquinavir with 3R‐tetrahydrofuranyl glycine resulted in the very potent PI 3 (enzyme IC50=0.05 nm; antiviral CIC95=8 nm). The R configuration appeared to be critical to its potency.89 We then removed the P3‐quinaldic ligand and designed the corresponding stereochemically defined urethane derivative 4 (IC50=160 nm; concentration for 95 % inhibition in cell culture (CIC95)=800 nm).90 This lead structure was quite important as the molecular weight of 4 (515 Da) was much less than that of saquinavir (670 Da).
Figure 4.
Inhibitors containing a cyclic ether as a P2 ligand.
The 3S‐THF urethane 4 was significantly more potent (more than 18‐fold) than the corresponding N‐Boc derivative. Incorporation of this functionality in the hydroxyethylene‐derived inhibitor 5 resulted in a marked enhancement of enzyme inhibitory and antiviral activity over that of the corresponding N‐Boc derivative.90 The cyclopentyl derivative 6 (Figure 5) was significantly less active even though this PI presumably fills the substrate binding site in the same fashion as inhibitor 5. This result indicated that the cyclic ether oxygen is very important. A preliminary X‐ray structure of 4‐bound HIV‐1 protease showed that the THF‐ring oxygen is involved in a weak hydrogen‐bonding interaction with the NH groups of Asp29 and Asp30.90 Introduction of this 3S‐THF urethane in the hydroxyethylamine sulfonamide isostere developed by Vasquez et al.91 and Tung et al.92 resulted in PI 7 (Figure 5, VX‐478).93 This was subsequently developed into the FDA‐approved inhibitor amprenavir/fosamprenavir. The X‐ray structure of 7‐bound HIV‐1 protease indicated that the ring fills the S2 subsite and the ring oxygen is involved in a weak interaction with the Asp29 and Asp30 backbone amides (distances of 3.4 and 3.5 Å, respectively).93
Figure 5.
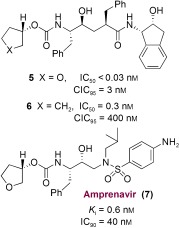
Potent PIs based on 3S‐THF‐urethane.
Based upon our preliminary development of 3S‐THF urethane as a possible substitute for both the P2 and the P3 ligands of saquinavir, we became interested in further enhancing the binding‐site interactions in the S2 subsite. This objective ultimately led us to design the stereochemically defined bicyclic (3R,3aS,6aR) tetrahydrofuran (bis‐THF) ligand shown in Figure 6.94 Inhibitor 8 with the (3R,3aS,6aR) bis‐THF ligand was significantly more potent than inhibitor 9 containing the (3S,3aR,6aS) bis‐THF ligand. Inhibitor 8 was also considerably more potent than inhibitor 4 with 3S‐THF as the P2 ligand. Our X‐ray crystallographic studies revealed that the bis‐THF oxygens form effective hydrogen bonds with the backbone NH groups of Asp29 and Asp30.94 Furthermore, the X‐ray structure showed that the bicyclic ring in 8 fills the hydrophobic pocket in the S2 site more effectively than the monocycle in inhibitor 4. Interestingly, however, inhibitor 8 does not form any hydrogen bonds with the protein backbone in the S2′ site.86, 94 As stated earlier, to combat drug resistance, the main emphasis of our backbone‐binding strategy is to maximize ligand binding site interactions, especially to promote hydrogen‐bond formation with the backbone atoms from the S2 to S2′ subsites of the protease.50
Figure 6.
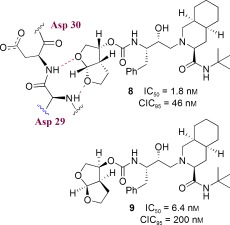
Design of PIs containing a bis‐THF ligand.
3.3. Design of TMC‐126 and Its Relevance to the Backbone‐Binding Concept
Following the design of the high‐affinity and nonpeptidic bis‐THF ligand, our next objective was to design an inhibitor that could form robust hydrogen bonds throughout the S2 to S2′ subsites.50 We investigated the effect of a P2 bis‐THF ligand with a number of different isosteres, including (R)‐(hydroxyethyl)sulfonamide isosteres91, 92 with a p‐methoxysulfonamide as the P2′ ligand.50, 81 Our initial choice of p‐methoxysulfonamide was based upon the presumption that the methoxy oxygen would form effective hydrogen bonds with the Asp29′ and Asp30′ backbone NH groups in the S2′ subsite. As shown in Figure 7, inhibitor 10 (UIC‐PI or UIC94003 and later TMC‐126) exhibited marked enzyme inhibitory potency (K i=14 pm) and antiviral activity (ID50=1.4 nm) in CEM cell lines.82 To obtain molecular insight into the ligand binding site interactions, a high‐resolution X‐ray structure of 10‐bound HIV‐1 protease was determined.95 As shown in Figure 8, both oxygen atoms of the P2 bis‐THF ligand form strong hydrogen bonds with the backbone NH groups of Asp29 and Asp30 in the S2 subsite. In the S2′ subsite, the p‐methoxy oxygen also forms strong hydrogen bonds with the backbone NH group of Asp30′ as well as with carboxylate of the the Asp30′ side chain.95 The inhibitory potency of 10 against numerous mutant HIV proteases was determined. As shown in Table 1, this inhibitor maintained very impressive potency (K i<100 pm) and the K i mut/K i wt ratios were no greater than 5. This indicated that proteases with multiple mutations, which were shown to be highly resistant to approved first‐generation PIs, displayed a low level of resistance against 10.96
Figure 7.
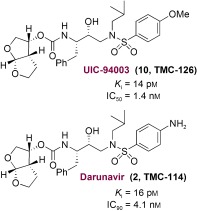
Potent bis‐THF PIs, TMC‐126 and darunavir.
Figure 8.
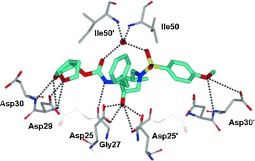
X‐ray crystal structure of 10‐bound HIV‐1 protease.
Table 1.
Enzyme inhibitory potency of 10 against wild‐type and mutant proteases.
|
Enzyme |
K i [pm] |
K i mut/K i wt |
Vitality |
|---|---|---|---|
|
wild type |
14 |
1 |
1 |
|
D30N |
<5 |
0.33 |
0.3 |
|
V32I |
8 |
0.57 |
0.5 |
|
I84V |
40 |
2.85 |
1 |
|
V32I/I84V |
70 |
5 |
0.7 |
|
M46F/V82A |
<5 |
0.33 |
0.1 |
|
G48V/L90M |
<5 |
0.33 |
0.1 |
|
V82F/I84V |
7 |
0.5 |
0.1 |
|
V82T/I84V |
22 |
1.57 |
0.1 |
|
V32I/K45I/F53L/A71V/I84V/L89M |
31 |
2.2 |
0.1 |
|
V32I/L33F/K45I/F53L/A71V/I84V |
46 |
3.3 |
0.1 |
|
20R/36I/54V/71V/82T |
31 |
2.2 |
0.1 |
Inhibitor 10 also maintained excellent potency against a wide spectrum of drug‐resistant HIV‐1 variants with IC50 values ranging from 0.3 to 0.5 nm.82 As shown in Table 2, a detailed drug‐sensitivity evaluation with 10 demonstrated significant advantages compared to structurally related amprenavir and other approved PIs in terms of the emergence of drug resistance. Interestingly, viral acquisition of resistance to 10 was substantially delayed. Furthermore, 10‐resistant HIV remained sensitive to all approved PIs except amprenavir. Notably, inhibitor 10 retained impressive potency (IC50=0.5 to 5.5 nm) against multi‐PI‐resistant HIV‐1 strains isolated from patients who were harboring drug‐resistant HIV‐1.82 We speculated that the impressive activity of 10 against a wide spectrum of drug‐resistant HIV variants is because of its robust binding properties in the active site, particularly its extensive interactions with the backbone NH groups of aspartates in the S2 to S2′ subsites.50 Thus, the backbone‐binding strategy promoting extensive hydrogen bonds throughout the active site (S2 to S2′ subsites) may be an intriguing conceptual framework for the design of a new generation of PIs to combat drug resistance.
Table 2.
Sensitivities of 10 (TMC‐126) against HIV‐1 isolated from individuals having previous extensive PI treatment.
|
Virus[a] |
IC50 [μm] (fold change) |
|||||
|---|---|---|---|---|---|---|
|
RTV |
IDV |
SQV |
NFV |
APV |
10 (TMC‐126) |
|
|
wild type |
0.044 (1) |
0.013 (1) |
0.010 (1) |
0.023 (1) |
0.025 (1) |
0.0007 (1) |
|
1 |
>1 (>23) |
>1 (>77) |
0.27 (27) |
>1 (>43) |
0.27 (11) |
0.004 (6) |
|
2 |
>1 (>23) |
0.49 (38) |
0.037 (4) |
0.33 (14) |
0.28 (11) |
0.0013 (2) |
|
3 |
>1 (>23) |
0.49 (38) |
0.036 (4) |
>1 (>43) |
0.26 (10) |
0.001 (1) |
|
4 |
>1 (>23) |
0.21 (16) |
0.033 (3) |
0.09 (4) |
0.31 (12) |
0.0016 (2) |
|
5 |
>1 (>23) |
>1 (>77) |
0.31 (31) |
0.41 (18) |
0.67 (27) |
0.0024 (3) |
|
6 |
>1 (>23) |
0.30 (23) |
0.19 (19) |
>1 (>43) |
0.16 (6) |
0.0005 (1) |
|
7 |
>1 (>23) |
>1 (>77) |
0.12 (12) |
>1 (>43) |
0.49 (20) |
0.0055 (8) |
|
8 |
>1 (>23) |
0.55 (42) |
0.042 (4) |
>1 (>43) |
0.15 (6) |
0.001 (1) |
[a] Amino acid substitutions identified in the protease‐encoding regions of viruses compared to the consensus sequence cited from the Los Alamos database. See reference 82 for details.
4. Backbone‐Binding Strategy Leading to the Clinical Development of Darunavir to Combat Drug Resistance
4.1. Structural Optimization Leading to Darunavir
Based upon the results of inhibitor 10 (TMC‐126), we then explored the combination of the bis‐THF ligand and (R)‐(hydroxyethyl)sulfonamide isosteres with a variety of P2′ sulfonamide functionalities. These ligands were chosen to interact with the backbone atoms in the S2′ site. These efforts led to the design and synthesis of a number of exceptionally potent PIs. However, only inhibitor 2 (Figure 7, later named TMC‐114 and then darunavir) exhibited improved pharmacological properties and drug‐resistance profiles.81, 97–99 We attributed the unique binding profile of 10 (UIC‐94003 or TMC‐126) and 2 (UIC‐94017 or TMC‐114) as the main contributing factor for the antiviral profile which led us to establish the design concept of protein‐backbone binding as a promising strategy to overcome drug resistance.50, 100 The following section takes a closer look at the binding of darunavir and its unique antiviral profile.
4.2. Darunavir’s Extensive Interactions with the Protease Backbone
Darunavir’s enhanced binding affinity (K i=16 pm) is likely related to its ability to form an extensive network of hydrogen‐bonding interactions within the HIV‐1 protease active site. A high‐resolution X‐ray crystal structure of darunavir‐bound HIV‐1 protease revealed a number of key interactions between darunavir and the protease’s backbone atoms.69 As shown in Figure 9, the bis‐THF P2 ligand forms strong hydrogen bonds with the backbone amide NH groups of Asp29 and Asp30, which anchor darunavir to the S2 subsite. On the opposite end, darunavir’s p‐aminosulfonamide interacts with the amide of Asp30′ and the carboxylic acid side chain of Asp30′ thereby stabilizing darunavir within the active site. The hydroxy group of the (hydroxyethyl)sulfonamide isostere serves as a transition‐state mimic forming hydrogen bonds to the catalytic residues Asp25 and Asp25′. In addition, the urethane NH group interacts with the Gly27 carbonyl, while a tetracoordinated water molecule forms hydrogen bonds between flap residues Ile50 and Ile50′ and the urethane carbonyl and sulfonamide oxygen of darunavir. The P1′ isobutyl and P1 benzyl groups of darunavir further enhance binding through hydrophobic interactions.69 These multiple binding interactions allow darunavir to act as a “molecular crab” tightly clutching the protein backbone.
Figure 9.
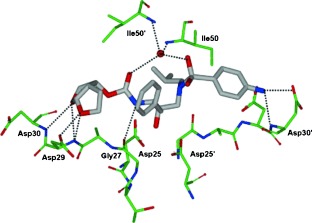
Darunavir binding to HIV‐1 protease like a “molecular crab” (PDB code 2IEN).69
Unlike many first‐generation PIs, the binding of darunavir to HIV‐1 protease is unique. For example, while the binding of many PIs is driven by entropic gain, the binding of darunavir is highly enthalpically favored, possibly because of its numerous hydrogen‐bonding interactions.101 Another noticeable difference lies in the kinetics of darunavir′s binding to the protease which shows a high association rate and very slow dissociation rate, much lower than that of other PIs.102 Together, these attributes provide darunavir with a specific, high‐affinity binding profile and an exceptional ability to accommodate protease mutations.103
A final distinguishing characteristic of darunavir is that it is capable of binding to the protease at a second location, as indicated by a recent X‐ray analysis.69 The second binding site lies on the surface of the protease on one of its flexible flaps. Allosteric binding at this location may contribute to darunavir’s exceptional antiviral activity by further inhibiting the function of the HIV‐1 protease. Further studies examined the kinetics of darunavir binding and suggested a mixed‐type competitive–uncompetitive inhibition model in contrast to first‐generation PIs which exhibit strictly competitive inhibition.104 These results were consistent with a second binding site for darunavir and likely contribute to darunavir’s heightened antiviral activity profile.
4.3. Darunavir’s Robust Potency against Multidrug‐Resistant HIV‐1 Variants
Darunavir has demonstrated remarkable antiviral potency across a broad range of HIV‐1 viral strains. As depicted in Table 3, against a panel of HIV‐1 isolates, darunavir outperformed many other approved PIs at inhibiting viral replication and infectivity (IC50=3–6 nm).105 Darunavir’s potent antiviral activity combined with its relatively low cytotoxicity provides it with an elevated selectivity index (>20 000 CC50/EC50).105 More importantly, darunavir has consistently retained its impressive antiviral activity against a host of viral strains with resistance‐related mutations. Notably, darunavir exerted very impressive activity against highly multi‐PI‐resistant clinical HIV‐1 variants isolated from patients with AIDS who did not respond to existing antiviral regimens (results are shown in Table 4). Darunavir (2) exhibited excellent antiviral activity with IC50 values ranging from 3 to 30 nm while APV, IDV, NFV, and RTV, were virtually ineffective in blocking the replication of all multi‐PI‐resistant strains.105
Table 3.
Sensitivities of 2 and selected anti‐HIV agents against HIV‐1Ba‐L, HIV‐2ROD, and HIV‐2EHO.
|
Virus |
Mean IC50 [nm][a] |
|||||||
|---|---|---|---|---|---|---|---|---|
|
Cell type |
SQV |
RTV |
IDV |
NFV |
APV |
AZT |
DRV (2) |
|
|
HIV‐1Ba‐L |
PBMC |
18 |
39 |
25 |
17 |
26 |
9 |
3 |
|
HIV‐2ROD |
MT‐2 |
3 |
130 |
14 |
19 |
230 |
18 |
3 |
|
HIV‐2EHO |
MT‐2 |
6 |
240 |
11 |
29 |
170 |
11 |
6 |
[a] All assays were conducted in duplicate or triplicate; the data represent mean IC50 values from three independent experiments. IC50 were evaluated with PHA‐PBMC and the inhibition of p24 Gag protein production by the drug as an end point. MT‐2 cells were exposed to the virus and cultured, and IC50 values were determined by MTT assay. See references 82 and 105 for details.
Table 4.
Activity of inhibitor 2 against HIV‐1 clinical isolates in PHA‐PBMCs.
|
Virus[a] |
IC50 values [μm] |
|||||
|---|---|---|---|---|---|---|
|
SQV |
APV |
IDV |
NFV |
RTV |
DRV (2) |
|
|
HIV‐1ERS104pre (wt X4) |
0.010 |
0.023 |
0.018 |
0.019 |
0.027 |
0.003 |
|
HIV‐1MOKW (wt R5) |
0.004 |
0.011 |
0.018 |
0.033 |
0.032 |
0.003 |
|
HIV‐1TM (MDR X4) |
0.23 (23) |
0.39 |
>1 (>56) |
0.54 (28) |
>1 (>37) |
0.004 (1) |
|
HIV‐1MM (MDR R5) |
0.30 (30) |
0.34 |
>1 (>56) |
>1 (>53) |
>1 (>37) |
0.02 (7) |
|
HIV‐1JSL (MDR R5) |
0.35 (35) |
0.75 (33) |
>1 (>56) |
>1 (>53) |
>1 (>37) |
0.029 (10) |
|
HIV‐1A (MDR X4) |
0.14 (14) |
0.16 (7) |
>1 (>56) |
0.36 (19) |
>1 (>37) |
0.004 (1) |
|
HIV‐1B (MDR X4) |
0.31 (31) |
0.34 (15) |
>1 (>56) |
>1 (>53) |
>1 (>37) |
0.013 (4) |
|
HIV‐1C (MDR X4) |
0.037 (4) |
0.28 (12) |
>1 (>56) |
0.44 (23) |
>1 (>37) |
0.003 (1) |
|
HIV‐1G (MDR X4) |
0.029 (3) |
0.25 (11) |
0.39 (22) |
0.32 (17) |
0.44 (16) |
0.004 (1) |
[a] Amino acid substitutions identified in the protease‐encoding regions of viruses compared to the consensus sequence cited from the Los Alamos database. See reference 105 for details.
Also, when surveyed against a panel of laboratory HIV‐1 strains with selected resistance against other PIs, darunavir maintained excellent activity (Table 5). Only APV‐resistant viral strains displayed cross‐resistance to darunavir; this can be explained by the fact that APV contains a sulfonamide isostere similar to that in darunavir.105 More elaborate studies utilizing a broad range of clinical isolates (1500+) further confirmed darunavir’s remarkable properties. Darunavir maintained an EC50 of less than 10 nm against 75 % of the variants and showed less than a tenfold change in EC50 compared to the wild‐type against 90 % of the strains.106 In contrast, APV, SQV, IDV, RTV, NFV, and LPV displayed ED50 values below 10 nm against less than 30 % of the viral strains and showed significantly higher levels of variability in the EC50 values as compared to the wild‐type.106
Table 5.
Activity of DRV against PI‐resistant HIV‐1 laboratory strains.
|
Virus |
Amino acid substitution |
EC50 [μm][a] |
|||||
|---|---|---|---|---|---|---|---|
|
SQV |
RTV |
IDV |
NFV |
APV |
DRV |
||
|
HIV‐1NL4‐3 |
wild type |
0.009 |
0.018 |
0.011 |
0.020 |
0.027 |
0.003 |
|
HIV‐1SQV 5μm |
L10I, G48V, I54V, L90M |
>1 (>111) |
>1 (>56) |
>1 (>91) |
0.30 (15) |
0.17 (6) |
0.005 (2) |
|
HIV‐1RTV 5μm |
M46I, V82F, I84V |
0.013 (1) |
>1 (>56) |
0.31 (28) |
0.24 (12) |
0.61 (23) |
0.025 (8) |
|
HIV‐1IDV5 μm |
L10F, L24I, M46I, L63P, A71V, G73S, V82T |
0.015 (2) |
>1 (>56) |
>1 (>91) |
0.74 (37) |
0.33 (12) |
0.029 (10) |
|
HIV‐1NFV 5μm |
L10F, D30N, K45I, A71V, T74S |
0.031 (3) |
0.09 (5) |
0.28 (25) |
>1 (>50) |
0.093 (3) |
0.003 (1) |
|
HIV‐1APV 5μm |
L10F, V32I, M46I, I54M, A71V, I84V |
0.020 (2) |
>1 (>56) |
0.31 (28) |
0.21 (11) |
>1 (>37) |
0.22 (73) |
[a] MT‐4 cells were exposed to each HIV‐1 strain (100×TCID50), and the inhibition of p24 Gag protein production by the drug was used as an end point. Numbers in parentheses represent the fold changes of the IC50 values for each isolate relative to that of HIV‐1NL4‐3. See reference 105.
A major challenge in the treatment of HIV remains the rapid emergence of drug resistance which reduces the effectiveness of antiviral treatments. The most prominent attribute of darunavir that sets it apart from other PIs is its high genetic barrier to the development of viral resistance. Early attempts to select for darunavir‐resistant HIV viruses in vitro proved difficult; resistance developed very slowly after multiple passages and only at concentrations of less than 200 nm of darunavir (Figure 10).106 Later studies showed that although the wild‐type HIV virus did not propagate darunavir resistance easily, HIV‐1 isolates from antiretroviral‐experienced patients were capable of acquiring resistance‐related mutations.107 During the POWER clinical trials, 11 amino acid substitutions were correlated to darunavir resistance including V11I, V32I, L33F, I47V, I50V, I54L/M, G73S, L76V, I84V, and L89V, of which I50V, I54M/L, L76V, and I84V are considered the major mutations.108, 109 A28S was later identified as an amino acid substitution distinctly associated with darunavir and not caused by other PIs.110 By itself, A28S results in a significant reduction in enzyme fitness which can be restored in part by the secondary mutation I50V. Darunavir resistance to A28S is believed to occur from a shift in position of the P2 sulfonamide that alters its ability to hydrogen bond with the protease causing a decrease in binding affinity.
Figure 10.
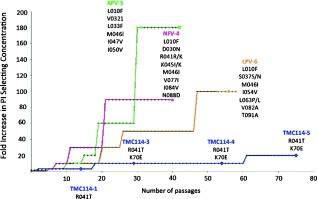
In vitro selection of resistant HIV strains in the presence of NFV, APV, LPV, and TMC‐114(DRV). The figure is modified from Figure 4 in reference 106.
4.4. Darunavir Inhibits Dimerization of HIV‐1 Protease
Darunavir’s impressive antiviral profile can be attributed in part to its small flexible conformation and its ability to form extensive hydrogen‐bonding interactions with the protease backbone, which imparts a high binding affinity. Another contributing factor is darunavir’s unique ability to act as a dual inhibitor, blocking not only the cleavage of the natural peptide substrate but also inhibiting dimerization of the HIV‐1 protease. An active HIV‐1 protease consists of two chains of 99 amino acids each that combine or dimerize into a single catalytically active quaternary structure. Dimerization of the monomer subunits is essential for activity and thus its inhibition represents a distinct mechanism for inhibiting viral replication.111 Darunavir blocks this dimerization process at concentrations as low as 0.01 μm.112 Further investigation is currently ongoing. TPV is the only other PI besides darunavir that has been shown to possess this property.112 While capable of blocking the dimerization of individual monomers, neither darunavir nor TPV is able to cause disassociation of an assembled protease unit.
5. Retaining Backbone Binding and Designing Exceptionally Potent Bis‐THF‐Derived PIs
5.1. The Effect of Benzodioxolane Sulfonamide at the S2′Site
We continued to explore structural modifications that would form additional hydrogen bonds with the protease backbone residues leading to inhibitors with higher affinity. We have incorporated a benzodioxolane sulfonamide as the P2′ ligand and this has provided inhibitor 11 (GRL‐98065) shown in Figure 11. This turned out to be an exceedingly potent inhibitor with significant antiviral activity (K i=11 pm and IC50=1.1 nm).113 As can be seen in Table 6, inhibitor 11 was evaluated against a wide spectrum of multidrug‐resistant clinical isolates and inhibitor 11 outperformed other approved PIs including darunavir. It maintained significant antiviral activity (6–12‐fold change) similar to darunavir. Furthermore, 11 was evaluated against PI‐resistant HIV‐1 variants and was found to have a unique antiviral activity profile (Table 7). Against the PI‐resistant variants, cross‐resistance to APV was observed. Interestingly, SQV and ATV remained active against viral strains selected against 11 which contain the A28S mutation. This was linked to TMC‐126 resistance and resulted in a significant loss in fitness of the protease.113
Figure 11.
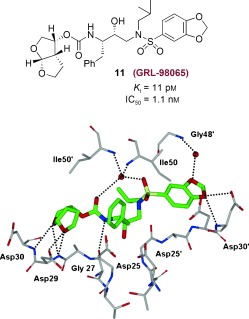
Structure of 11 and the X‐ray crystal structure of 11‐bound HIV‐1 protease.
Table 6.
Antiviral activities of GRL‐98065 (11) against multidrug‐resistant clinical isolates.
|
Virus[a] |
EC50 [nm][b] |
|||||
|---|---|---|---|---|---|---|
|
SQV |
RTV |
NFV |
APV |
DRV |
11 (GRL‐98065) |
|
|
HIV‐1ERS104pre (wild‐type X4) |
8 |
25 |
15 |
29 |
3.8 |
0.5 |
|
HIV‐1MDR/TM (X4) |
180 (23) |
>1000 (>40) |
>1000 (>67) |
300 (10) |
4.3 (1) |
3.2 (6) |
|
HIV‐1MDR/MM (R5) |
140 (18) |
>1000 (>40) |
>1000 (>67) |
480 (17) |
16 (4) |
3.8 (8) |
|
HIV‐1MDR/JSL (R5) |
290 (36) |
>1000 (>40) |
>1000 (>67) |
430 (15) |
27 (7) |
6 (12) |
|
HIV‐1MDR/B (X4) |
270 (34) |
>1000 (>40) |
>1000 (>67) |
360 (12) |
40 (11) |
3.9 (8) |
|
HIV‐1MDR/C (X4) |
35 (4) |
>1000 (>40) |
420 (28) |
250 (9) |
9 (2) |
2.7 (5) |
|
HIV‐1MDR/G (X4) |
33 (4) |
>1000 (>40) |
370 (25) |
320 (11) |
7 (2) |
3.4 (7) |
[a] The amino acid substitutions identified in the protease‐encoding region compared to the consensus type B sequence cited from the Los Alamos database. See reference 113 for details. [b] Effective concentration by 50 %.
Table 7.
Antiviral activities of 11 against laboratory PI‐resistant HIV‐1 variants.
|
Virus |
EC50 [μm] of drug[a] |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
SQV |
RTV |
IDV |
NFV |
APV |
LPV |
ATV |
DRV |
11 (GRL‐98065) |
|
|
HIV‐1NL4‐3 |
0.007 |
0.033 |
0.034 |
0.033 |
0.026 |
0.031 |
0.0042 |
0.0030 |
0.0003 |
|
HIV‐1SQV 5μm |
>1 (>143) |
>1 (>30) |
>1 (>29) |
0.48 (15) |
0.33 (13) |
0.27 (9) |
0.326 (78) |
0.0058 (2) |
0.006 (20) |
|
HIV‐1RTV 5μm |
0.010 (1) |
>1 (>30) |
0.25 (7) |
0.21 (6) |
0.28 (11) |
0.16 (5) |
0.018 (4) |
0.018 (6) |
0.0025 (8) |
|
HIV‐1IDV 5μm |
0.059 (8) |
>1 (>30) |
>1 (>29) |
0.47 (14) |
0.17 (7) |
0.26 (8) |
0.06 (14) |
0.015 (5) |
0.0037 (12) |
|
HIV‐1NFV 5μm |
0.024 (3) |
0.051 (2) |
0.27 (8) |
>1 (>30) |
0.060 (2) |
0.024 (1) |
0.021 (5) |
0.0033 (1) |
0.0024 (8) |
|
HIV‐1APV 5μm |
0.031 (4) |
0.29 (9) |
0.200 (6) |
0.27 (8) |
>1 (>38) |
0.23 (7) |
0.003 (1) |
0.33 (110) |
0.032 (107) |
|
HIV‐1LPV 1μm |
0.032 (5) |
>1 (>30) |
>1 (>29) |
0.49 (15) |
0.31 (12) |
0.31 (10) |
0.040 (10) |
n.d. |
0.0075 (25) |
|
HIV‐1ATV 1μm |
0.037 (5) |
0.12 (4) |
0.388 (11) |
0.22 (7) |
0.20 (8) |
0.033 (1) |
0.33 (79) |
0.0034 (1) |
0.0015 (5) |
|
HIV‐1GRL98065p40 |
0.032 (5) |
0.38 (12) |
0.28 (8) |
0.34 (10) |
>1 (>38) |
0.19 (6) |
0.011 (3) |
0.21 (70) |
0.18 (600) |
[a] MT‐4 cells were exposed to 100 TCID50 (dose for 50 % infection in cell culture) of each HIV‐1, and inhibition of p24 Gag protein production by each drug was used as an end point. Numbers in parentheses represent n‐fold changes in the EC50 values for each isolate compared to the EC50 values for wild‐type HIV‐1NL4‐3. All assays were conducted in duplicate or triplicate, and data shown are derived from the results of three independent experiments. n.d.=not determined. See reference 113 for details.
We determined the crystal structure of 11‐bound HIV‐1 protease at 1.6 Å resolution. This structure has provided important molecular insight into the inhibitor potency and drug‐resistance profile.113 Structural analyses revealed that 11 is involved in extensive interactions with the backbone atoms of the amino acids in the protease active site (Asp29 and Asp30, Figure 11) in the S2 to S2′ subsites. These interactions are important for its potency and wide‐spectrum activity against multi‐PI‐resistant HIV‐1 variants. Moreover, these interactions are maintained in crystal structures of 11‐bound drug‐resistant mutants.114 Comparison of the crystal structure of 11 with the crystal structure of darunavir (Figure 9) showed that the interactions with the S2 site are similar, but the nature of the hydrogen bonds with residues differs in the S2′ region. A water‐mediated interaction of one of the benzodioxolane oxygens with flap residue Gly48′ is not observed for darunavir (2). These differences in interactions may account for the improvement of IC50 values of 11 compared to those of darunavir.113
5.2. Design and Clinical Development of Bis‐THF PIs with Novel P1 Functionalities
Numerous potent PIs have been designed based upon the privileged bis‐THF ligand.67, 84 As shown in Figure 12, brecanavir (12; BCV/GW0385), which was developed by Glaxo Smith Kline, contains a bis‐THF P2 ligand, a benzodioxolane P2′ ligand, and a substituted P1 ligand.115, 116 It showed femtomolar enzyme inhibitory potency (K i=15 fm) and subnanomolar antiviral activity with an IC50 value of 0.7 nm (wild‐type virus). Also, BCV exhibited IC50 values of 1.1 nm and 4.8 nm against two MDR viral strains, EP13 HIV‐1 and D545701 HIV‐1, respectively.115 BCV exhibited sub‐ to low nanomolar IC50 values with low cross‐resistance against a panel of 10 highly resistant and specifically PI‐resistant HIV‐1 isolates.115 Furthermore, BCV was tested against a panel of 55 clinical isolates from PI‐experienced patients and it maintained low nanomolar IC50 values (0.1–14.9 nm) for all isolates. The majority of isolates (80 %) displayed IC50 values at or below 0.8 nm.117, 118 This inhibitor had undergone clinical development at the phase III level. However, brecanavir’s clinical trials were terminated because of formulation issues.119
Figure 12.
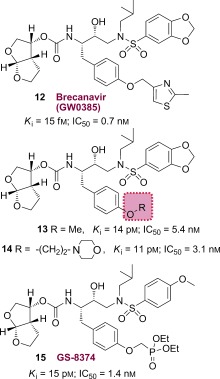
Structures of PIs 12–15.
We have investigated structure‐based modifications of the P1 side chain of inhibitor 11. Of particular interest, we attempted to incorporate a basic amine or a cyclic ether functionality to improve aqueous solubility and other pharmacological properties. Both PIs 13 and 14 have shown very potent antiviral activity.120
Inhibitor 15 (GS‐8374) containing a P2 bis‐THF unit, a P2′ p‐methoxybenzesulfonamide ligand, and a diethylphosphonylmethoxy group attached to the P1 phenyl ligand was developed by researchers at Gilead Sciences.121 The phosphonate functionality was designed to promote better intracellular retention without interfering with the protease binding site of 10 (TMC‐126). This PI (15) displayed an excellent resistance profile.121, 122 It exhibited a mean 6.2‐fold change in EC50 values (range 0.6–26 nm) from the wild‐type HIV. Inhibitor 15 displayed a mean 29.8‐fold change (1.0–157 nm) and 23.6‐fold change of EC50 values (1.2–121 nm) compared to darunavir and BCV, respectively. Towards a possible explanation of this marked resistance suppression, it was proposed that the phosphonate moiety acted as an anchor point in the solvent medium and enhanced the degeneracy of the binding state of the inhibitor by providing favorable entropic compensation following protease mutations. Interestingly, selection of HIV‐1 strains exposed to 15 did not exhibit any signs of RAM (resistance‐associated mutation) even after six months.123 GS‐8374 has been reported to show more favorable pharmacological and metabolic profiles than other PIs. In the X‐ray crystal structure of 15‐bound HIV‐1 protease the binding profiles for the P2 bis‐THF and the P2′ methoxysulfonamide ligands are similar to those observed in the X‐ray structure of the 10‐bound HIV‐1 protease. A comparison of the two structures showed that one of the phosphonate ethyl moieties of 15 is bound in a hydrophobic cleft on the surface of protease.121 GS‐8374 with a p‐diethylphosphonate at the P1 phenyl residue exhibited a better resistance profile than to BCV, which contains a substituted methylthiazole at P1.123
5.3. The Effect of a C4‐Methoxy Bis‐THF Ligand at the S2 Site
The HIV‐1 protease flaps are flexible in the apoenzyme form, but they are closed when inhibitors bind and show minimal change in their backbone conformation.53, 124 As can be seen in Figure 11, a novel water‐mediated interaction with Gly48’ through the benzodioxolane oxygen may be responsible for its superb antiviral and drug‐resistance profile. Based upon the X‐ray structure of 10‐bound HIV‐1 protease, we envisioned that heteroatom‐containing substituents at the C4 position of the bis‐THF ligand would be ideally positioned to interact with the backbone NH group of Gly48.95 Therefore, we synthesized a series of new PIs incorporating C4‐alkoxy‐substituted bis‐THF ligands.125 As shown in Figure 13, inhibitor 16 with a 4R‐methoxy group has better enzyme inhibitory potency (K i=2.9 pm) than inhibitor 10 (K i=14 pm). The 4R isomer 16 was 12‐fold more potent than the corresponding 4S isomer 17. Larger alkyl groups at C4, such as benzyloxy substituents, led to significant reduction in potency. An X‐ray structure of 16‐bound HIV‐1 protease (Figure 13) showed extensive interactions of the inhibitor with the protease active site similar to those of inhibitor 10.125 However, it appears that the oxygen of the 4R‐methoxy group forms a unique water‐mediated hydrogen bond with the NH group of Gly48. The improvement in binding affinity of 16 may be due to this water‐mediated hydrogen bond with the backbone NH group of Gly48.125
Figure 13.
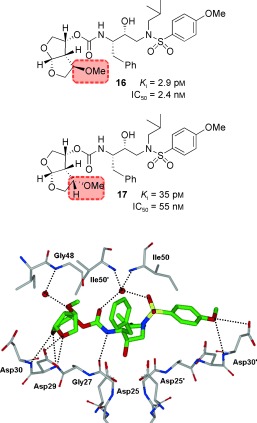
Structures of PIs 16 and 17 and the X‐ray structure of 16‐bound HIV‐1 protease.
5.4. Design of Macrocyclic Inhibitors with a Bis‐THF Unit at the S2 Site
In an effort to fill the hydrophobic pocket in the S1′–S2’ subsites with flexible macrocycles, we investigated bis‐THF‐derived macrocyclic inhibitors involving P1′–P2′ ligands that can retain all major hydrogen‐bonding interactions with the protein backbone similar to those of inhibitor 10 and darunavir but effectively fill the hydrophobic pocket in the S1′ and S2′ subsites.95 The design perception for these macrocycles evolved from the observation that certain mutations lead to decreased van der Waals interactions and an increase in size of the hydrophobic pocket in the S1′ subsite. The structural studies of A‐77003126 indicated that the V82A mutant results in decreased van der Waals interactions with the phenyl rings in both the S1 and S1′ subsites.127 Also, there was evidence of the repacking of the inhibitor side chain and protease atoms in the S1 subsite. Based upon this insight, we envisioned that 11‐ to 15‐membered saturated and unsaturated macrocycles would effectively fill the S1′–S2′ subsites. As shown in Figure 14, macrocyclic inhibitors 20 and 21 displayed excellent enzyme inhibitory and antiviral activity; however, their acyclic homologues were significantly less potent. Also, saturated inhibitors were less active than their unsaturated analogues.95
Figure 14.
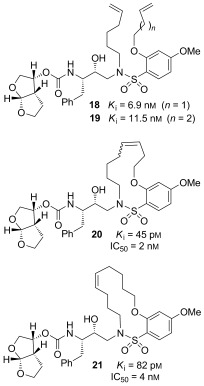
Structures of acyclic and macrocyclic PIs 18–21.
To ascertain if the structural effects led to improved drug‐resistance properties, inhibitors 20 and 21 were evaluated against a panel of clinical wild‐type X4‐HIV‐1 isolates (HIV‐1ERS104pre) along with various multidrug‐resistant clinical X4‐ and R5‐HIV‐1 isolates using PBMCs as target cells.95, 105 As shown in Table 8, the potency of both inhibitors against HIV‐1ERS104pre (IC50=7 and 5 nm, respectively) was superior to that of the approved inhibitors IDV, APV, and LPV but nearly twofold less potent than darunavir (IC50=3 nm).95 Inhibitor 20 showed better potency than amprenavir against HIV‐1MDR/C, HIV‐1MDR/G, HIV‐1MDR/TM, and HIV‐1MDR/JSL and was six times more potent against HIV‐1MDR/MM. Inhibitor 21 also displayed superior potency against HIV‐1MDR/C and HIV‐1MDR/G (greater than 12‐ and 15‐fold, respectively) compared to amprenavir.95, 128 Furthermore, both macrocyclic PIs prevented the replication of HIV‐1NL4‐3 variants selected against up to 5 μm of saquinavir, lopinavir, and indinavir with IC50 values of 20 nm to 46 nm. We have determined an X‐ray crystal structure of 20‐bound HIV‐1 protease at 1.17 Å resolution. As can be seen in Figure 15, both P2 and P2′ ligands are involved in extensive hydrogen‐bonding interactions with the protein backbone atoms in both the S2 and S2′ subsites, similar to inhibitor 10. The crown‐shaped P1′–P2′macrocycle nicely fills the S1′ pocket. Interestingly, the macrocycle acts more or less like a spring and pushes against the P1 phenyl ring. This causes a rotation about 30° towards Asp29′ along the backbone which is absent in the X‐ray structure of 10‐bound HIV‐1 protease. Both macrocyclic PIs were able to maintain excellent potency against multidrug‐resistant clinical isolates possibly because of their ability to make extensive hydrogen bonds with the protease backbone as well as their hydrophobic interactions in the S1′–S2′ subsites.95
Table 8.
Antiviral activity of macrocyclic inhibitors against multidrug‐resistant clinical isolates in PHA‐PBMCs.
|
Virus[a] |
IC50 value [μm] |
||||||
|---|---|---|---|---|---|---|---|
|
SQV |
IDV |
APV |
LPV |
DRV |
21 |
20 |
|
|
HIV‐1ERS104pre (WT X4) |
0.008 |
0.043 |
0.030 |
0.034 |
0.003 |
0.007 |
0.005 |
|
HIV‐1MDR/B (X4) |
0.27 (34) |
>1 (>23) |
>1 (>33) |
>1 (>29) |
0.019 (6) |
0.089 (13) |
0.037 (7) |
|
HIV‐1MDR/C (X4) |
0.032 (11) |
>1 (>23) |
0.37 (12) |
>1 (>29) |
0.008 (3) |
0.029 (4) |
0.044 (9) |
|
HIV‐1MDR/G (X4) |
0.030 (4) |
0.34 (5) |
0.43 (14) |
0.26 (8) |
0.023 (5) |
0.028 (4) |
0.057 (11) |
|
HIV‐1MDR/TM (X4) |
0.26 (33) |
>1 (>23) |
0.32 (11) |
>1 (>29) |
0.004 (1) |
0.072 (10) |
0.027 (6) |
|
HIV‐1MDR/MM (R5) |
0.19 (24) |
>1 (>23) |
0.21 (7) |
>1 (>29) |
0.011 (4) |
0.055 (8) |
0.033 (7) |
|
HIV‐1MDR/JSL (R5) |
0.30 (37) |
>1 (>23) |
0.62 (21) |
>1 (>29) |
0.027 (9) |
0.21 (30) |
0.073 (15) |
[a] The amino acid substitutions identified in the protease‐encoding region compared to the consensus type B sequence cited from the Los Alamos database. See reference 95 for details.
Figure 15.
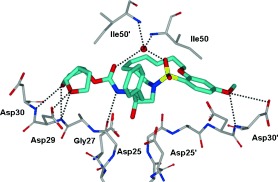
X‐ray structure of HIV‐1 protease bound to the macrocyclic inhibitor 20. All strong hydrogen‐bonding interactions are shown as dotted lines.
6. Probing the Backbone‐Binding Concept as a Design Strategy to Combat Drug Resistance
6.1. Development of Cyclopentanyltetrahydrofuran (Cp‐THF) as a Novel P2 Ligand
To further investigate the merit of targeting the protein backbone as a design strategy, based upon various protein–ligand X‐ray structures, we decided to design structurally different cyclic‐ether‐derived ligands that were not related to bis‐THF ligand. This effort led to the design of a stereochemically defined bicyclic hexahydrocyclopentanofuran (Cp‐THF) as the P2 ligand. Incorporation of this ligand in the hydroxyethylaminosulfonamide isostere provided a series of exceptionally potent PIs.51 We positioned the cyclic ether oxygen in the Cp‐THF ring to form hydrogen bonds with the backbone NH groups of Asp29 and Asp30. As can be seen in Figure 16, replacing the bis‐THF in darunavir with a new Cp‐THF ligand provided inhibitor 22, which exhibited sub‐nanomolar enzyme inhibitory potency nearly ten times less than that of darunavir (K i=16 pm). We believed that binding of the Cp‐THF ligand in the S2 subsite was distinct from the bis‐THF ligand and may have caused a slight shift in position of the remainder of the inhibitor structure within the active site.51 We then speculated that modifications of the P2′ aniline could allow improved interactions with the NH groups of Asp29′ and Asp30′ in the S2′ subsite. As shown, we have incorporated a hydroxymethylsulfonamide as the P2′ ligand and the resulting inhibitor 23 showed a 30‐fold improvement of enzyme inhibition (K i=4.5 pm) compared to 22. In addition, it has shown very impressive antiviral potency (IC50=1.8 nm) similar to that of inhibitor 10. In order to probe the importance of the Cp‐THF ring oxygen, we synthesized inhibitor 24 in which the oxygen is replaced with a methylene group. Interestingly, 24 displayed a more than 1100‐fold loss of enzyme inhibitory potency compared to 23. Furthermore, 24 exhibited a drastic loss in antiviral activity (IC50>1000 nm). This result indicated that the Cp‐THF ring oxygen is involved in critical interactions in the active site.
Figure 16.
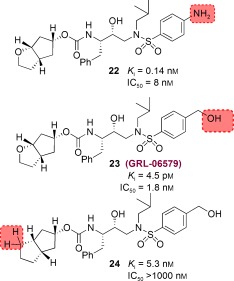
Structures of Cp‐THF‐related PIs.
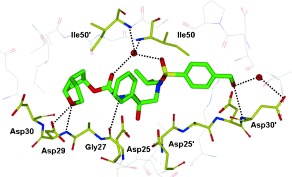
We determined the X‐ray crystal structure of 23‐bound HIV‐1 protease at a 1.35 Å resolution and this high‐resolution structure provided critical molecular insight into the interactions at the ligand binding site.51 As shown in Figure 17, inhibitor 23 makes extensive interactions in the active site similar to darunavir. The P2′ hydroxy group forms a strong hydrogen bond to the backbone NH group of Asp30′and a water‐mediated contact with the side chain oxygen of Asp30′. The ring oxygen of the P2 Cp‐THF ligand forms a strong hydrogen bond with the backbone NH group of Asp29 and a weak hydrogen bond with Asp30. These interactions cannot occur for inhibitor 24 which lacks the ring oxygen and this may explain why 24 is significantly less potent than 23. This result illustrates the importance of forming hydrogen bonds with the protease backbone in the S2 subsite and suggests that simply filling the binding space of this site is not sufficient to induce tight binding and elicit a biological response.
Figure 17.
X‐ray structure of 23‐bound HIV‐1 protease.
We superimposed the X‐ray structure of 23‐bound HIV‐1 protease (wild‐type) with the three most highly mutated drug‐resistant proteases.51 These structures showed minimal root‐mean‐square deviation of the α‐carbon backbone atoms (0.5 to 1.1 Å) suggesting inhibitor 23 should retain good to excellent contacts with the backbone of the mutant proteases. As it turned out, inhibitor 23 exerted very potent activity against HIV‐1 isolates (HIV‐1LAI and HIV‐1Ba‐L) in both MT‐2 cells and PHA‐PBMC (Table 10). Furthermore, as evident in Table 9, 9, inhibitor 23 retained significant antiviral activity against a panel of HIV‐1 drug‐resistant viral strains. Inhibitor 23 displayed the most potent activity (IC50=3 nm) against HIV‐1 clinical strain HIV‐1ET, which had been isolated from a drug‐naive patient. Furthermore, six drug‐resistant clinical strains containing 10–12 amino acid substitutions associated with protease inhibitor resistance (HIV‐1B, HIV‐1C, HIV‐1G, HIV‐1TM, HIV‐1EV, and HIV‐1ES) were isolated from patients with HIV‐1 infection having received 7–11 different antiviral agents for 24 to 81 months.82, 105 All tested approved PIs were highly resistant. However, inhibitor 23 exerted highly potent activity against all of these six variants with IC50 values ranging from 4 nm to 52 nm. Inhibitor 23 was also highly potent against HIV‐1K with an IC50 value as low as 3 nm. This data indicate that inhibitor 23 is highly active against a wide spectrum of drug‐resistant variants.51
Table 10.
Antiviral activity (IC50) of 23 in PBMC and MT‐2 cells.
|
Virus |
IC50 [nm][a] |
|||||
|---|---|---|---|---|---|---|
|
SQV |
RTV |
INV |
NFV |
APV |
23 |
|
|
HIV‐1LAI |
14 |
43 |
32 |
14 |
34 |
1.8 |
|
HIV‐1Ba‐L |
18 |
36 |
24 |
7 |
29 |
2.0 |
|
HIV‐1LAI |
24 |
34 |
26 |
10 |
24 |
1.8 |
|
HIV‐2EHO |
1.9 |
290 |
13 |
20 |
440 |
21 |
[a] Data represent the mean value of three determinations. See reference 51 for details.
Table 9.
Antiviral activity of 23 against a panel of HIV‐1 viral strains.
|
Virus |
IC50 [nm] values |
||||||
|---|---|---|---|---|---|---|---|
|
SQV |
RTV |
IDV |
NFV |
APV |
DRV |
23 |
|
|
HIV‐1ET |
17 |
15 |
30 |
32 |
23 |
n.d. |
3 |
|
HIV‐1B |
230 |
>1000 |
>1000 |
>1000 |
290 |
10.2 |
15 |
|
HIV‐1C |
100 |
>1000 |
500 |
310 |
300 |
3.5 |
5 |
|
HIV‐1G |
59 |
>1000 |
500 |
170 |
310 |
3.7 |
20 |
|
HIV‐1TM |
250 |
>1000 |
>1000 |
>1000 |
220 |
3.5 |
4 |
|
HIV‐1EV |
>1000 |
>1000 |
>1000 |
>1000 |
>1000 |
n.d. |
52 |
|
HIV‐1ES |
>1000 |
>1000 |
>1000 |
>1000 |
>1000 |
n.d. |
31 |
|
HIV‐1K |
20 |
58 |
260 |
>1000 |
68 |
3 |
3 |
[a] Amino acid substitutions identified in the protease‐encoding region of HIV‐1B (B), HIV‐1C (C), HIV‐1G (G), HIV‐1TM (TM), HIV‐1EV (EV), HIV‐1ES (ES), HIV‐1ET (ET), HIV‐1K (NFVR) as compared to the consensus B sequence cited from the Los Alamos data base. All values were determined in triplicate. The IC50 values were determined by employing PHA‐PBMC as target cells and the inhibition of p24 Gag protein production as the endpoint. See reference reference 51 for details.
6.2. Design of meso‐Hexahydrocyclopenta‐1,3‐dioxolane as a P2 Ligand
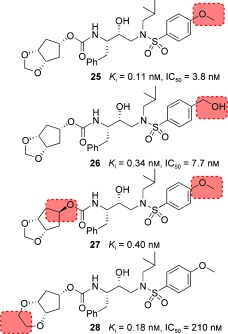
As we have seen, the oxygen atom in the Cp‐THF ring of 23 is critical to its superb antiviral and anti‐drug‐resistance properties. Based upon the X‐ray structure of 23‐bound HIV‐1 protease, we then speculated that a corresponding meso‐hexahydrocyclopenta‐1,3‐dioxolane ligand would be able to maintain interactions similar to those of the Cp‐THF ligand. Essentially, we would insert an oxygen atom into the Cp‐THF ring and form a meso‐hexahydrocyclopenta‐1,3‐dioxolane ligand which would greatly reduce the stereochemical complexity and allow for a simplified synthetic pathway. In addition to the synthetic advantage, we postulated that the additional ether oxygen may engage in hydrogen‐bonding interactions with the protease thereby enhancing the potency of the PIs. Figure 18 depicts the structure and potency of a number of PIs incorporating a meso ligand.129
Figure 18.
Enzyme K i values and antiviral potency of PIs 25–28.
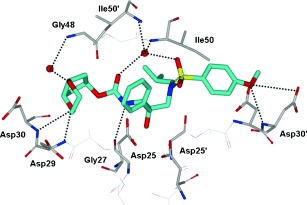
The syn isomer 25 demonstrated enzyme inhibitory potency and antiviral activity comparable to that of the Cp‐THF‐derived PI 23, whereas the anti isomer 27 showed a threefold decrease in potency. Unlike the results with the Cp‐THF ligand, incorporation of a hydroxymethyl group in the P2′ ligand resulted in a slight reduction in potency. We next explored a 1,4‐dioxane P2 ligand; the resulting inhibitor 28 exhibited a significant reduction in antiviral potency. A larger trioxepane system also provided a less active PI. We evaluated 25 against a panel of multidrug‐resistant HIV‐1 variants, and the results are shown in Table 11. Inhibitor 25 exhibited antiviral activity comparable to that of the approved PIs SQV and APV, while it outperformed IDV. However, 25 was not as active as darunavir against the wild‐type or drug‐resistant HIV‐1 clinical variants. We determined an X‐ray structure of 28‐bound HIV‐1 protease at 1.07 Å resolution (Figure 19). The inhibitor binds with extensive interactions in the protease active site. Interestingly, one of the dioxane oxygens forms a hydrogen bond with the backbone NH group of Asp29. The other oxygen is involved in a water‐mediated hydrogen bond with the amide NH group of Gly48. These interactions with Gly48 were similar to those reported for several peptide substrate analogues.15, 64 However, this interaction with Gly48 has not been previously utilized in ligand design. Based upon this X‐ray structure, we created an active model of 25. It appears that smaller the 1,3‐dioxolane forms an additional hydrogen bond with the backbone NH group of Asp30.129 This additional hydrogen bond may explain the increased antiviral activity of 25 relative to 28.
Table 11.
Antiviral activity of inhibitor 25 against clinical HIV‐1 isolates in PBMC cells.
|
Virus[a] |
IC50 values [nm] |
||||
|---|---|---|---|---|---|
|
SQV |
IDV |
APV |
DRV |
25 |
|
|
HIV‐1ERS104pre (wild‐type: X4) |
12 |
26 |
33 |
3.5 |
29 |
|
HIV‐1MDR/MM (R5) |
190 (16) |
>1000 (>38) |
300 (9) |
17 (5) |
150 (5) |
|
HIV‐1MDR/JSL (R5) |
330 (28) |
>1000 (>38) |
430 (13) |
26 (7) |
550 (19) |
|
HIV‐1MDR/C (X4) |
36 (3) |
>1000 (>38) |
230 (7) |
7 (2) |
300 (10) |
|
HIV‐1MDR/G (X4) |
29 (2) |
290 (11) |
340 (10) |
7 (2) |
340 (12) |
|
HIV‐1MDR/A (X4) |
81 (7) |
>1000 (>38) |
100 (3) |
3 (1) |
21 (1) |
[a] Amino acid substitutions identified in the protease‐encoding region compared to the consensus type B sequence cited from the Los Alamos database, see reference 129 for details.
Figure 19.
X‐ray structure of inhibitor 28 bound to the active site of wild‐type HIV‐1 protease.
6.3. Alkoxy/Hydroxy‐Cp‐THF Ligands and Their Effect on Drug‐Resistance Properties
As described above, the meso‐dioxolane‐derived inhibitor exhibited very potent enzyme inhibitory and antiviral activity.129 As shown in Figure 19, we speculated that both oxygens of the dioxolane ring in 25 form hydrogen bonds with backbone Asp29 and Asp30 NH groups and also form a water‐mediated hydrogen bond with the Gly48 backbone NH group. Based upon these possible interactions in the ligand binding site, we subsequently designed a 3‐hydroxy‐Cp‐THF derivative to interact with the Gly48 NH group in the flap.130 We synthesized a stereochemically defined alkoxy‐Cp‐THF derivative and converted it into the PIs shown in Figure 20. As can be seen, inhibitor 30 with a 3R‐hydroxy group showed the most potent antiviral activity comparable to that of darunavir. The related inhibitor 29 with a 3S‐hydroxy group was also quite potent. We prepared the corresponding 3‐methoxy‐Cp‐THF ligands and the resulting inhibitors 31 and 32 showed stereochemical preferences and potencies similar to those of the corresponding hydroxy derivatives.130
Figure 20.
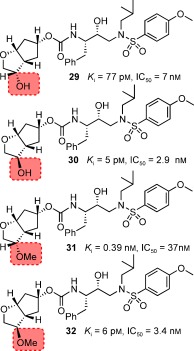
Structures of PIs 29–32 with alkoxy/hydroxy‐Cp‐THF ligands.
We then determined the X‐ray crystal structure of 30‐bound HIV‐1 protease at 1.23 Å resolution. As shown in Figure 21, the Cp‐THF ring oxygen forms a strong hydrogen bond with the Asp29 NH group and a rather weak hydrogen bond with the Asp29 carboxylate. The 3‐hydroxy group appears to form a nice water‐mediated hydrogen bond with the Gly48 backbone NH group.
Figure 21.
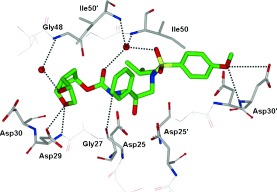
X‐ray structure of 30‐bound HIV‐1 protease.
PIs 29 and 30 were evaluated against a panel of multidrug‐resistant HIV‐1 variants and compared with the approved PIs darunavir and APV (Table 12). The activity of inhibitor 30 against various multidrug‐resistant HIV‐1 variants is similar to that of darunavir.130 The changes in the IC50 values with 30 were similar to those with darunavir. In contrast, PI 29 with a 3S‐hydroxy ligand lost potency significantly. Also, APV showed high IC50 values and lower resilience against the drug‐resistant HIV‐1 strains examined. The X‐ray structure of 30‐bound HIV‐1 protease and its resistance profile further supported the backbone‐binding strategy for combating drug resistance.
Table 12.
Comparison of the antiviral activity of 29, 30 , and of other PIs against multidrug‐resistant HIV‐1 variants.
|
Virus[a] |
IC50 [μm] (fold change) |
|||
|---|---|---|---|---|
|
APV |
DRV |
29 |
30 |
|
|
HIV‐1ERS104pre (wild type) |
0.030 |
0.0037 |
0.020 |
0.0029 |
|
HIV‐1MDR/B |
0.93 (31) |
0.036 (10) |
>1 (>50) |
0.029 (10) |
|
HIV‐1MDR/C |
0.26 (9) |
0.013 (4) |
>1 (>50) |
0.022 (7) |
|
HIV‐1MDR/G |
0.38 (12) |
0.0023 (1) |
0.27 (13) |
0.0045 (2) |
|
HIV‐1MDR/TM |
0.19 (6) |
0.0019 (1) |
0.041 (2) |
0.0031 (1) |
[a] Amino acid substitutions identified in the protease‐encoding region compared to the consensus type B sequence cited from the Los Alamos database, see reference 130 for details.
6.4. Further Enhancing the Backbone Interactions of Cp‐THF‐Derived PIs at the S1′ Site and Probing the Effect on Drug‐Resistance Properties
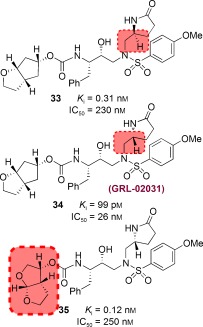
In addition to ligand design to enhance interactions with the protein backbone in the S2 subsite, we have also expanded our design concept in other regions of the protease active site. We particularly planned to design PIs with new P1′ ligands in place of the isobutyl group of 23 that could interact with backbone atoms as well as fill the hydrophobic pocket in S1′ subsite. We explored the incorporation of stereochemically defined 2‐pyrrolidinone and oxazolidinone functionalities so that the pyrrolidinone NH group could form a hydrogen bond with Gly27′ and its carbonyl group could interact with Arg8′ in the S1′ site.131 Our initial plan was to examine the potential of the new P1′ ligand in combination with Cp‐THF and bis‐THF ligands. We also wanted to address the question of whether enhancement of backbone‐binding interactions would lead to PIs with improved drug‐resistance profiles. The results of this investigation are summarized in Figure 22. Inhibitor 33 with (S)‐methyl‐2‐pyrrolidinone as the P1′ ligand showed good enzyme inhibitory potency but its antiviral IC50 value was 230 nm. The (R)‐methyl‐2‐pyrrolidinone derivative 34 showed improvement in both the K i value and antiviral activity (IC50=26 nm) relative to S‐pyrrolidine derivative 33. For the combination of bis‐THF as P2 and (R)‐methyl‐2‐oxazolidinone as P1′ ligands, however, the antiviral activity was significantly less than that of 10. The antiviral potency of 34 was nearly ten times less than that of 23. This is possibly a result of the poor cellular permeability of the polar 2‐oxazolidinone functionality. Nevertheless, inhibitor 34 is a very potent inhibitor with antiviral activity comparable to that of FDA‐approved PIs such as IDV, APV and LPV.131
Figure 22.
Structures and potency of PIs 33–35.
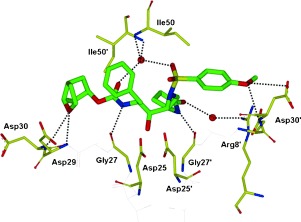
To obtain molecular insight into various ligand binding site interactions, we determined a high‐resolution X‐ray crystal structure of 34‐bound HIV‐1 protease at 1.29 Å resolution.131 As shown in Figure 23 the interactions between the inhibitor and the active site are quite extensive. Most strikingly, the P1′‐pyrrolidinone exists in two conformations. In one conformation, the pyrrolidinone NH group is engaged in a hydrogen bond with the Gly27′ carbonyl and the pyrrolidinone carbonyl forms a water‐mediated hydrogen bond with the Arg8′ side chain. In another conformation, the pyrrolidinone ligand fills the hydrophobic pocket and the carbonyl group makes a weak hydrogen bond with the Val82′ backbone NH group. Other binding interactions of the Cp‐THF ligand in the S2 site are similar to those of 23 and the methoxy oxygen in S2′ forms a strong hydrogen bond with the backbone NH group of Asp30′ and the side‐chain carboxylate group.131
Figure 23.
X‐ray structure of 34‐bound HIV‐1 protease.
Inhibitor 34 was evaluated against a wide spectrum of laboratory and clinical wild‐type and multidrug‐resistant HIV‐1 strains. Table 13 shows its anti‐HIV activity against selected clinical isolates highly resistant to multiple PIs.131, 132 As can be seen, inhibitor 34 was highly potent against various clinical isolates tested. Except darunavir, all other approved PIs failed to exert comparable activity. However, inhibitor 34, like darunavir, potently inhibited all seven primary strains. Particularly, 34 maintained nearly full potency except with the R5 phenotype where it lost potency slightly (by less than twofold). Overall, the interactions of inhibitor 34 (GRL‐02031) with backbone atoms particularly in the S1′ subsite were enhanced compared to those with inhibitor 23. These polar interactions and the conformational flexibility of the P1′ oxazolidinone most likely contributed to its robust activity against multidrug‐resistant HIV‐1 variants.132
Table 13.
Anti‐HIV activity of 34 against selected clinical isolates highly resistant to multiple protease inhibitors.
|
Virus[a] |
Phenotype |
EC50 [μm] |
||||
|---|---|---|---|---|---|---|
|
IDV |
APV |
LPV |
DRV |
GRL‐02031 (34) |
||
|
HIV‐1ERS104pre (wild‐type) |
X4 |
0.028 |
0.025 |
0.03 |
0.0036 |
0.028 |
|
HIV‐1TM (MDR) |
X4 |
>1 (>36) |
0.25 (10) |
0.73 (24) |
0.0036 (1) |
0.029 (1) |
|
HIV‐1MM (MDR) |
R5 |
>1 (>36) |
0.32 (13) |
0.72 (24) |
0.019 (5) |
0.042 (2) |
|
HIV‐1C (MDR) |
X4 |
>1 (>36) |
0.35 (14) |
0.32 (11) |
0.015 (4) |
0.023 (1) |
|
HIV‐1G (MDR) |
X4 |
0.29 (10) |
0.33 (13) |
0.14 (5) |
0.014 (4) |
0.027 (1) |
7. Conformationally Flexible P2 Ligands Capable of Forming Extensive Interactions with the Backbone
7.1. Design of Flexible Cyclic Polyethers as P2 Ligands and Their Effect on Drug‐Resistance Properties
Following exploration of our PIs based on meso P2 ligands, we continued to examine ways in which we could reduce the stereochemical complexity of the bis‐THF ligand while maintaining key backbone interactions and accommodating variations in the amino acid side chains within the active site occurring after viral mutations. To probe this, we turned to cyclic‐polyether‐derived P2 ligand systems possessing flexible rings capable of repacking within the binding pocket in response to mutational changes.133 Based on this proposition, we removed the shared C—C bond from bis‐THF producing the flexible eight‐membered‐ring inhibitor 36 shown in Figure 24. Unfortunately, 36 displayed significantly lower enzyme inhibitory potency than darunavir. Reducing the ring size to a seven‐membered ring restored enzyme inhibitory activity, and a preference for the R stereoisomer was revealed as the corresponding PI with epimeric P2 ligand displayed significant loss in potency (K i=0.16 nm, IC50=30 nm). Further reductions in ring size led to the six‐membered‐ring inhibitor 38 which was also highly potent. In general, expanding the ring to larger polycyclic ethers (ten‐membered rings and larger) resulted in a drastic loss in potency. The ether oxygens within these ring systems are critical for maintaining high levels of enzyme inhibition activity. The removal of either oxygen from 37 resulted in a significant loss in activity.
Figure 24.
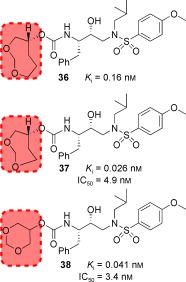
Structures and potency of PIs containing cyclic polyethers.
An X‐ray crystal structure of 37‐bound HIV‐1 protease was determined at 1.00 Å resolution. The majority of binding interactions within the active site are similar in nature to those with inhibitor 10 (TMC‐126) except for interactions in the S2 site. As shown in Figure 25, one of the oxygens of the 1,3‐dioxepane ligand is involved in hydrogen bonding with Asp29 and Asp30 NH groups. The other oxygen is involved in a unique interaction with the Gly48 NH group through a water molecule.133
Figure 25.
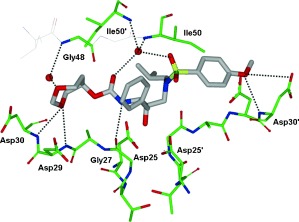
X‐ray structure of 37‐bound HIV‐1 protease.
Both PIs 37 and 38 were further evaluated for their antiviral activity against a panel of clinically relevant HIV‐1 isolates (Table 14). While they were less potent than darunavir, both compounds outperformed the approved PIs RTV and IDV and were comparable in antiviral activity to APV. These results suggested that the ability of the P2 ligand in 37 to maintain hydrogen‐bonding interactions with the protein backbone may be responsible for the improved drug‐resistance profiles of 37 over other PIs examined. The design of PIs using the concept of maximized backbone binding has led to PIs characterized by high potency against both wild‐type and multidrug‐resistant HIV‐1 strains.50
Table 14.
Anti‐HIV activity of 37 and 38 against selected clinical isolates highly resistant to multiple protease inhibitors.
|
Virus[a] |
IC50 [nm] values |
|||||
|---|---|---|---|---|---|---|
|
IDV |
RTV |
APV |
DRV |
37 |
38 |
|
|
ERS104pre (wild‐type) |
26 |
34 |
33 |
3.5 |
20 |
6 |
|
MDR/TM |
>1000 (>38) |
>1000 (>29) |
290 (9) |
4 (1) |
220 (11) |
64 (10) |
|
MDR/MM |
>1000 (>38) |
>1000 (>29) |
300 (9) |
17 (5) |
250 (13) |
110 (5) |
|
MDR/JSL |
>1000 (>38) |
>1000 (>29) |
430 (13) |
26 (7) |
500 (25) |
330 (55) |
|
MDR/B |
>1000 (>38) |
>1000 (>29) |
320 (10) |
26 (7) |
340 (17) |
230 (38) |
|
MDR/C |
>1000 (>38) |
>1000 (>29) |
230 (7) |
7 (2) |
210 (11) |
160 (27) |
|
MDR/G |
290 (11) |
>1000 (>29) |
340 (10) |
7 (2) |
360 (18) |
300 (50) |
|
MDR/A |
>1000 (>38) |
>1000 (>29) |
100 (3) |
3 (1) |
20 (1) |
13 (2) |
[a] Amino acid substitutions identified in the protease‐encoding region compared to the consensus type B sequence cited from the Los Alamos database, see reference 133.
7.2. Further Optimization of Bis‐THF Ligands and the Design of a P2 TP‐THF Ligand
We subsequently evaluated options to improve upon the bis‐THF ligand of darunavir. An analysis of the X‐ray structure of darunavir‐bound HIV‐1 protease (Figure 9) revealed that the bis‐THF ether oxygens are involved in hydrogen‐bonding interactions with the amide N‐H groups of Asp29 and Asp30 at a distance of 2.9 and 3.1 Å, respectively. We conceptualized that incorporation of a larger ring system might promote closer more effective hydrogen bonding to these backbone residues and result in a more favorable alignment between the cyclic ether oxygen and the Asp30 amide N‐H bond. These factors might result in stronger hydrogen bonds and higher affinity inhibitors. In addition, a larger ring size may promote favorable hydrophobic interactions within the S2 subsite and allow additional flexibility to better accommodate steric changes caused by protease mutations. Therefore, we synthesized and evaluated a series of PIs containing a tetrahydropyranyl‐THF (Tp‐THF) P2 ligand.134 As shown in Figure 26, consistent with bis‐THF, the bicyclic ligand in 39 (GRL‐0476) with its 4S configuration is more effective than the epimeric ligand. Like the bis‐THF ligand, both cyclic ether oxygens are critical for binding as their respective replacement with methylene groups resulted in a significant loss in potency. We have also prepared PIs 40 and 41 incorporating a p‐methoxybenzyl side chain as the P1 ligand and p‐methoxysulfonamide and p‐aminophenylsulfonamide as the P2′ ligands, respectively. Our detailed drug‐resistance studies of 40 and 41 showed that both PIs were very potent against multi‐PI‐resistant HIV‐1 variants.135
Figure 26.
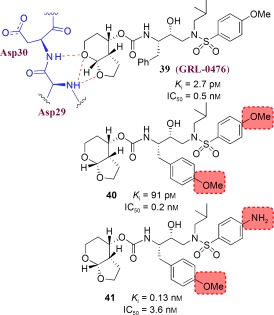
Structure and potency of Tp‐THF‐derived PIs.
To obtain molecular insight, we have created an active model of 39 starting from the X‐ray crystal structure of 10 (TMC‐126). The conformation of 39 was optimized using the MMFF94 force field.136 It appeared that the cyclic ether oxygens of Tp‐THF are within hydrogen‐bonding distance to Asp29 and Asp30 backbone NH groups in the S2 subsite. Other active‐site interactions are similar to those in the X‐ray structure of 10‐bound HIV‐1 protease.95 As depicted in Table 15, against a panel of multidrug‐resistant clinical isolates, inhibitor 39 outperformed two other approved PIs (APV and LPV) displaying a high level of antiviral activity against all the strains with EC50 values ranging from 2.6 to 27.5 nm. These results are comparable to those of 10 (TMC‐126), and 39 is more potent than darunavir in absolute terms; however, the fold changes in efficacy factors between viral strains are similar.
Table 15.
Comparison of the antiviral activity of 39 against multidrug‐resistant clinical isolates.
|
Virus[a] |
Phenotype |
EC50 [μm] |
|||
|---|---|---|---|---|---|
|
ATV |
LPV |
DRV |
39 (GRL‐0476) |
||
|
HIV‐1ERS104pre (wild type) |
X4 |
0.0027 |
0.031 |
0.004 |
0.0019 |
|
HIV‐1MDR/B |
X4 |
0.470 (174) |
>1 (>32) |
0.034 (9) |
0.0145 (8) |
|
HIV‐1MDR/C |
X4 |
0.039 (14) |
0.437 (14) |
0.009 (2) |
0.0037 (2) |
|
HIV‐1MDR/G |
X4 |
0.019 (7) |
0.181 (6) |
0.026 (7) |
0.0026 (1) |
|
HIV‐1MDR/TM |
X4 |
0.075 (28) |
0.423 (14) |
0.022 (6) |
0.0275 (14) |
|
HIV‐1MDR/MM |
R5 |
0.205 (76) |
0.762 (25) |
0.017 (4) |
0.0050 (3) |
|
HIV‐1MDR/JSL |
R5 |
0.293 (109) |
>1 (>32) |
0.023 (6) |
0.0275 (14) |
8. Further Improvement of Drug Resistance by Targeting Protein Backbone and Protein–Ligand Interactions
In our efforts to target the protein backbone as a design strategy to combat drug resistance, we have developed a variety of intriguing ligands and scaffolds and generated diverse inhibitors with exceptional potency and drug‐resistance profiles. Our next objective was to further optimize a ligand structure that could maintain critical backbone interactions and at the same time effectively fill the hydrophobic pocket in the active site and maximize protein–ligand interactions. Towards this objective, we elected to append functionalities to the bis‐THF ligand to further improve the drug‐resistance properties of the PIs. As shown in Figure 27, based upon the overlay of the X‐ray structures of darunavir‐bound69 and SQV‐bound86 HIV‐1 protease, we planned to fill the hydrophobic pocket occupied by the quinaldic moiety of SQV. Particularly, we have speculated that the fusion of another tetrahydrofuran ring on the bis‐THF ligand would provide additional ligand binding site interactions. While such an oxatricyclic ligand could have a number of possible stereochemical motifs, including syn‐syn‐syn (SSS‐type) and syn‐anti‐syn (SAS‐type) isomers, our model based upon overlay structures in Figure 27 suggested that the SAS‐type ligand‐based inhibitor would make enhanced interactions in the S2 subsite. We subsequently synthesized both SAS‐ and SSS‐oxatricyclic ligands in a stereoselective manner and prepared the respective PIs 42 and 43 shown in Figure 28.137
Figure 27.
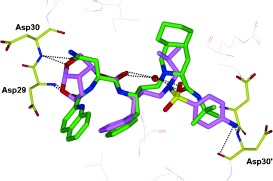
Overlay of the X‐ray structures of darunavir‐bound and SQV‐bound HIV‐1 protease.
Figure 28.
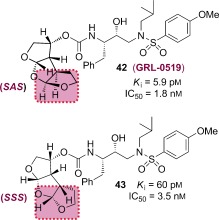
Structures and potency of PIs 42 and 43.
Inhibitor 42 (GRL‐0519A) with the syn‐anti‐syn configuration of the tris‐THF rings exhibited a tenfold better enzyme inhibitory potency over the syn‐syn‐syn derivative 43. Inhibitor 42 also displayed better antiviral activity than 43. An X‐ray structure of 42‐bound HIV‐1 protease was determined at 1.27 Å (Figure 29).137 Analysis of this structure revealed a number of additional interactions within the S2 subsite not seen with the bis‐THF unit of darunavir or TMC‐126. The two top THF ring oxygens are involved in hydrogen‐bonding interactions with the backbone NH groups of Asp29 and Asp30. The second THF oxygen appears to form a hydrogen bond with the carboxylate side chain of Asp29. As expected, the third THF ring fills the S2 subsite very nicely. The third THF ring also participates in a semicircular hydrogen‐bonding network with three conserved water molecules that surround the guanidine side chain of Arg8.
Figure 29.
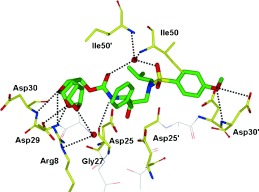
X‐ray structure of 42‐bound HIV protease (PDB code 30K9).137
Inhibitor 42 proved to be extremely potent against various multidrug‐resistant HIV‐1 variants, with IC50 values ranging from 0.6–4.3 nm, nearly a 10‐fold improvement over the potency of darunavir (Table 16). The emergence of GRL‐0519A‐resistant HIV‐1 in vitro was substantially delayed compared to selected approved PIs.137 Also, very strikingly, GRL‐0519A more potently blocked protease dimerization by at least a factor of 10 compared to darunavir as examined in the fluorescence resonance energy transfer based HIV‐1 expression assay employing cyan and yellow fluorescent protein tagged protease monomers.112 The present data suggested that the GRL‐0519 class of PIs may be further developed as potential therapeutic agents for the treatment of primary and multidrug‐resistant HIV‐1 infections.
Table 16.
Antiviral activity of 42, amprenavir (APV), and darunavir (DRV) against multidrug‐resistant clinical isolates in PHA‐PBMCs.
|
Virus[a] |
EC50 [μm] |
||
|---|---|---|---|
|
APV |
DRV |
42 (GRL‐0519) |
|
|
HIV‐1ERS104pre (wild‐type) |
0.032 |
0.005 |
0.0006 |
|
HIV‐1MDR/B |
0.521 (16) |
0.028 (6) |
0.0043 (7) |
|
HIV‐1MDR/C |
0.357 (11) |
0.011 (2) |
0.0009 (2) |
|
HIV‐1MDR/G |
0.485 (15) |
0.031 (6) |
0.0027 (5) |
|
HIV‐1MDR/TM |
0.488 (15) |
0.031 (6) |
0.0022 (4) |
|
HIV‐1MDR/MM |
0.291 (9) |
0.016 (3) |
0.0027 (5) |
|
HIV‐1MDR/JSL |
0.419 (13) |
0.024 (5) |
0.0028 (5) |
[a] See reference 137 for details.
9. Summary and Outlook
Our specific interest in the chemistry and biology of natural products brought a unique perspective to our design and synthesis of HIV‐1 protease inhibitors for the treatment of HIV infection and AIDS. Our initial academic pursuit was focused on addressing the question of whether we could design natural product derived ligands or templates that could mimic the biological mode of action of peptide bonds and alleviate problems inherent to peptide‐based drugs. Nature has been optimizing various cyclic ether/polycyclic ether templates for millions of years in various biological microenvironments involving biosynthetic enzymes. Inspired by nature and based upon X‐ray structures of protein–ligand complexes, we invoked the idea of designing stereochemically defined cyclic ether or polyether‐like molecular features to replace peptide bonds and effectively fill the hydrophobic pockets in the active site of HIV‐1 protease. We envisioned positioning cyclic ether oxygen to mimic the biological action of a peptide carbonyl group and the cyclic functionality would make necessary van der Waals interactions in the hydrophobic pocket. These research efforts led to the creation of a variety of conceptually novel molecular templates that are entirely nonpeptidic but interact with HIV‐1 protease with remarkable affinity. Our many X‐ray structural studies of inhibitor‐bound HIV‐1 proteases provided strong evidence that such a cyclic ether/polyether oxygen indeed serves as an effective mimic of the carbonyl of a peptide/amide functionality. Also, such cyclic units nicely fill the hydrophobic pockets in the enzyme active site.
Following the development of various nonpeptide high‐affinity ligands, we turned toward addressing the issue of drug resistance. We were interested in optimizing inhibitor structures against wild‐type HIV‐1 protease as well as against known mutant proteases. This objective led us to examine X‐ray structures of inhibitor‐bound wild‐type HIV‐1 proteases as well as the X‐ray structures of a number of mutant proteases. Superimposition of these X‐ray structures evidenced only minimal distortion of the backbone conformation. This led to our proposition of targeting the protein backbone as a strategy to evade drug resistance. By maximizing hydrogen‐bonding interactions with the protein backbone, we have essentially created a “molecular crab” capable of latching on and holding tightly in the enzyme active site. Using the combination of our in‐depth antiviral studies, along with drug‐resistance, and X‐ray crystallographic studies, we have documented the practicality and usefulness of the backbone‐binding design strategy to combat drug resistance. The combination of our ligand‐design efforts inspired by polyether natural products, and subsequent inhibitor design efforts targeting the protease backbone to combat drug resistance, culminated in the discovery and ultimate development of darunavir, the first FDA‐approved treatment for patients with multidrug‐resistant HIV‐1 variants. Its indications were later generalized for all patients with HIV infection and AIDS. Furthermore, we discovered that darunavir possesses a dual mechanism of action and is a potent inhibitor of HIV‐1 protease dimerization.
The discovery of darunavir marked an important turning point in the paradigm of designing HIV PIs. Our work has led us to develop the backbone‐binding concept as an effective means to mitigate viral adaptability. We have continued to apply our backbone‐binding design strategy resulting in the design and synthesis of a variety of exceedingly potent HIV‐1 protease inhibitors with intriguing structural features. Interestingly, GRL‐02031 retained near full potency against a panel of multidrug‐resistant HIV‐1 variants. Also, the design of GRL‐0519 marked a tenfold improvement in antiviral activity over that of darunavir with retention of potency against a wide range of clinically relevant multidrug‐resistant strains. The backbone‐binding concept may prove useful as a guide for the design of antiretroviral agents in other areas as well. We will continue to utilize and develop this concept in our future designs as we strive to meet the challenges of today’s medicine.
Biographical Information
Arun K. Ghosh received his BS and MS in Chemistry from the University of Calcutta and Indian Institute of Technology, Kanpur, respectively. He obtained his PhD in 1985 at the University of Pittsburgh. He pursued postdoctoral research with Professor E. J. Corey at Harvard University (1985–1988). He was a research fellow at Merck Research Laboratories prior to joining the University of Illinois‐Chicago as an Assistant Professor in 1994. In 2005 he moved to Purdue University, where he is currently the Ian P. Rothwell Distinguished Professor in Chemistry and Medicinal Chemistry. His research interests are in the areas of organic, bioorganic, and medicinal chemistry.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
David Anderson received his BS in chemistry from the University of Wisconsin‐Madison. He joined Eli Lilly and Co. in 2001 as an analytical chemist supporting the commercial process development of clinical drug candidates. In 2005 he received his MS from Indiana University‐Purdue University at Indianapolis and later moved to Purdue University to study medicinal chemistry with Professor Ghosh. His research focuses on the design and synthesis of HIV‐1 protease inhibitors and the total synthesis of pladienolide B.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
Irene T. Weber received her BS and MS from Cambridge University (UK) and obtained her PhD in 1978 from Oxford University (UK). She pursued postdoctoral research with Professor Thomas Steitz at Yale University. In 1991 she accepted a position as Professor of Microbiology and Immunology at Thomas Jefferson University in Philadelphia. In 2001 she moved to Georgia State University, Atlanta, where she is Professor of Biology and Chemistry and Georgia Cancer Coalition Distinguished Cancer Scientist. Her research focuses on the structure and activity of enzymes.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Biographical Information
Hiroaki Mitsuya received his MD and PhD from National Kumamoto University School of Medicine, Japan. In 1982 following training in oncology/hematology/immunology, he joined the National Cancer Institute in Bethesda, Maryland (USA), where he has been Principal Investigator & Chief of the Experimental Retrovirology Section since 1991. Since 1997, he has also served as Professor of Medicine and Chairman of the Departments of Hematology, Clinical Immunology/Rheumatology, and Infectious Diseases at the Kumamoto University School of Medicine.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Acknowledgements
This work was supported by the National Institute of Health (GM53386). We thank Dr. K. V. Rao (Purdue University) for helpful discussions.
The frontispiece depicts a “molecular crab” tightly gripping the protein backbone of HIV‐1 protease. We thank Dr. Xiaoming Xu for his help in creating this artwork.
References
- 1. Domingo E., Biebricher C. K., Eigen M., Holland J. J. in Quasispecies and RNA Virus Evolution: Principles and Consequences, Eurekah, Georgetown, 2001, preface. [Google Scholar]
- 2. Burnet M., White D. O. in Natural History of Infectious Disease, Cambridge University Press, London, 1972. [Google Scholar]
- 3. Needham J., Gwei‐djen L. in Science and Civilization in China, Vol. 6 (Ed.: N. Sivin), Cambridge University Press, Cambridge, 1999, p. 134. [Google Scholar]
- 4. Waterson A., Wilkinson L. in An Introduction to the History of Virology, Cambridge University Press, Cambridge, 1978, pp. 23–34. [Google Scholar]
- 5. Fenner F., Burnett F. M. in Portraits of Viruses: A History of Virology (Eds.: F. Fenner, A. Gibbs), S. Karger AG, Basel, 1988, pp. 1–37. [Google Scholar]
- 6. Brooksby J. B. in Portraits of Viruses: A History of Virology (Eds.: F. Fenner, A. Gibbs), S. Karger AG, Basel, 1988, pp. 124–146. [Google Scholar]
- 7.WHO, Summary of SARS Cases, http://www.who.int/csr/sars/country/country2003_08_15.pdf, 2003.
- 8.WHO, Cumulative Number of Confirmed Human Cases of Avian Influenza A/(H5N1) Reported to WHO, http://www.who.int/csr/disease/avian_influenza/country/cases_table_2011_02_02/en/index.htm, 2011.
- 9.UNAIDS Report on the global HIV/AIDS epidemic, http://www.unaids.org/en/media/unaids/contentassets/documents/unaidspublication/2010/20101123_globalreport_en.pdf, 2010.
- 10. Barre‐Sinoussi F., Chermann J., Rey F., Nugeyre M., Chamaret S., Gruest J., Dauguet C., Axler‐Blin C., et al., Science 1983, 220, 868–871. [DOI] [PubMed] [Google Scholar]
- 11. Gallo R. C., Sarin P. S., Gelmann E. P., Robert‐Guroff M., Richardson E., Kalyanaraman V. S., Mann D., Sidhu G. D., Stahl R. E., Zolla‐Pazner S., Leibowitch J., Popovic M., Science 1983, 220, 865–867. [DOI] [PubMed] [Google Scholar]
- 12. Lyle T. in Comprehensive Medicinal Chemistry II, Vol. 7 (Eds.: J. Taylor, D. Triggle), Elsevier Science Maryland Heights, 2007, pp. 329–371. [Google Scholar]
- 13. Mehellou Y., De Clercq E., J. Med. Chem. 2010, 53, 521–538. [DOI] [PubMed] [Google Scholar]
- 14. Menéndez‐Arias L., Antiviral Res. 2010, 85, 210–231. [DOI] [PubMed] [Google Scholar]
- 15. Flexner C., Nat. Rev. Drug Discovery 2007, 6, 959–966. [DOI] [PubMed] [Google Scholar]
- 16. Mitsuya H., Broder S., Nature 1987, 325, 773–778. [DOI] [PubMed] [Google Scholar]
- 17. Kohl N. E., Emini E. A., Schleif W. A., Davis L. J., Heimbach J. C., Dixon R. A. F., Scolnick E. M., Sigal I. S., Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Virgil S. in Methods and Principles in Medicinal Chemistry, Vol. 45 (Ed.: A. K. Ghosh), Wiley‐VCH, Weinheim, 2010, pp. 139–168. [Google Scholar]
- 19. Mitsuya H., Erickson J. in Textbook of AIDS Medicine (Eds.: T. Merigan, J. Bartlett, D. Bolgnesi), Williams & Wilkis, Baltimore, 1999, pp. 751–780. [Google Scholar]
- 20. Glesby M. in Protease Inhibitors in AIDS Therapy, (Eds.: R. Ogden, C. Flexner), Marcel Dekker, New York, 2001, pp. 237–256. [Google Scholar]
- 21. Wensing A., van Maarseveen N. M., Nijhuis M., Antiviral Res. 2010, 85, 59–74. [DOI] [PubMed] [Google Scholar]
- 22. Grabar S., Pradier C., Le Corfec E., Lancar R., Allavena C., Bentata M., Berlureau P., Dupont C., Fabbro‐Peray P., Poizot‐Martin I., Costagliola D., AIDS 2000, 14, 141–149. [DOI] [PubMed] [Google Scholar]
- 23. Wainberg M., Friedland G., JAMA J. Am. Med. Assoc. 1998, 279, 1977–1983. [DOI] [PubMed] [Google Scholar]
- 24. Mansky L., Temin H., J. Virol. 1995, 69, 5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Drake J., Holland J., Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perelson A., Neumann A., Markowitz M., Leonard J., Ho D., Science 1996, 271, 1582–1586. [DOI] [PubMed] [Google Scholar]
- 27. Robertson D., Hahn B., Sharp P., J. Mol. Evol. 1995, 40, 249–259. [DOI] [PubMed] [Google Scholar]
- 28. Leigh Brown A., Proc. Natl. Acad. Sci. USA 1997, 94, 1862–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steigbigel R., Cooper D., Kumar P., et al., N. Engl. J. Med. 2008, 359, 339–354. [DOI] [PubMed] [Google Scholar]
- 30. Stephenson J., J. Am. Med. Assoc. 2007, 297, 1535–1536. [Google Scholar]
- 31. Cane P., Antimicro J., Chemoth 2009, Suppl. 1, i37–i40. [Google Scholar]
- 32. Dau B., Holodniy M., Drugs 2009, 69, 31–50. [DOI] [PubMed] [Google Scholar]
- 33. Stoddart C., Joshi P., Sloan B., Bare J., Smith P., Allaway G., Wild C., Martin D., PLoS ONE 2007, 2, e1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tazi J., Bakkour N., Marchand V., Ayadi L., Aboufirassi A., Branlant C., FEBS J. 2010, 277, 867–876. [DOI] [PubMed] [Google Scholar]
- 35. Mitsuya H., Ghosh A. in Aspartic Acid Proteases as Therapeutic Targets (Ed.: A. Ghosh), Wiley‐VCH, Weinheim, 2010, pp. 245–262. [Google Scholar]
- 36. Domingo E., Webster R., Holland J. in Origins and Evolution of Viruses, Academic Press, London, 1999, pp. 197–224. [Google Scholar]
- 37. Ali A., Bandaranayake R., Cai Y., King N., Kolli M., Mittal S., Murzycki J., Nalam M., Nalivaika E., Ozen A., Prabu‐Jeyabalan M., Thayer K., Schiffer C., Viruses 2010, 2, 2509–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Menéndez‐Arias L., Antiviral Res. 2010, 85, 210–231. [DOI] [PubMed] [Google Scholar]
- 39. Wensing A., Maarseveen N., Nijhuis M., Antiviral Res. 2010, 59–74. [DOI] [PubMed] [Google Scholar]
- 40. Gulnik S. V., Suvorov L. I., Liu B., Yu B., Anderson B., Mitsuya H., Erickson J. W., Biochemistry 1995, 34, 9282–9287. [DOI] [PubMed] [Google Scholar]
- 41. Kantor R., Fessel W. J., Zolopa A. R., Israelski D., Shulman N., Montoya J. G., Harbour M., Schapiro J. M., Shafer R. W., Antimicrob. Agents Chemother. 2002, 46, 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yusa K., Song W., Bartelmann M., Harada S., J. Virol. 2002, 76, 3031–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Croteau G., Doyon L., Thibeault D., McKercher G., Pilote L., Lamarre D., J. Virol. 1997, 71, 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martinez‐Picado J., Savara A. V., Sutton L., D’Aquila R. T., J. Virol. 1999, 73, 3744–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Condra J., Schleif W., Blahy O., Gabryelski L., Graham D. J., Quintero J. C., Rhodes A., Robbins H. L., Roth E., Shivaprakash M., Titus D., Yang T., Tepplert H., Squires K. E., Deutsch P. J., Emini E. A., Nature 1995, 374, 569–571. [DOI] [PubMed] [Google Scholar]
- 46. Tamiya S., Mardy S., Kavlick M., Yoshimura K., Mitsuya H., J. Virol. 2004, 78, 12030–12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mammano F., Petit C., Clavel F., J. Virol. 1998, 72, 7632–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hong L., Zhang X., Hartsuck J. A., Tang J., Protein Sci. 2000, 9, 1898–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laco G. S., Schalk‐Hihi C., Lubkowski J., Morris G., Zdanov A., Olson A., Elder J. H., Wlodawer A., Gustchina A., Biochemistry 1997, 36, 10696–10708. [DOI] [PubMed] [Google Scholar]
- 50. Ghosh A. K., Chapsal B., Weber I., Mitsuya H., Acc. Chem. Res. 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- 51. Ghosh A. K., Sridhar P. R., Leshchenko S., Hussain A. K., Li J., Kovalevsky A. Y., Walters D. E., Wedekind J., Grum‐Tokars V., Das D., Koh Y., Maeda K., Gatanaga H., Weber I. T., Mitsuya H., J. Med. Chem. 2006, 49, 5252–5261. [DOI] [PubMed] [Google Scholar]
- 52. Agniswamy J., Weber I. T., Viruses 2009, 1, 1110–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wlodawer A., Vondrasek J., Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [DOI] [PubMed] [Google Scholar]
- 54. Chothia C., Lesk A., Cold Spring Harbor Symp. Quant. Biol. 1987, 52, 399–405. [DOI] [PubMed] [Google Scholar]
- 55. Worth C., Gong S., Blundell T., Nat. Rev. Mol. Cell Biol. 2009, 10, 709–720. [DOI] [PubMed] [Google Scholar]
- 56. Todd A., Orengo C., Thornton J., Curr. Opin. Chem. Biol. 1999, 3, 548–556. [DOI] [PubMed] [Google Scholar]
- 57. Liang J., Edelsbrunner H., Woodward C., Protein Sci. 1998, 7, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wolfenden R., Snider M. J., Acc. Chem. Res. 2001, 34, 938–945. [DOI] [PubMed] [Google Scholar]
- 59. Benkovic S. J., Hammes‐Schiffer S., Science 2003, 301, 1196–1202. [DOI] [PubMed] [Google Scholar]
- 60. Garcia‐Viloca M., Gao J., Karplus M., Truhlar D. G., Science 2004, 303, 186–195. [DOI] [PubMed] [Google Scholar]
- 61. Wlodawer A., Miller M., Jaskolski M., Sathyanarayana B. K., Baldwin E., Weber I. T., Selk L. M., Clawson L., Schneider J., Kent S. B., Science 1989, 245, 616–621. [DOI] [PubMed] [Google Scholar]
- 62. Gustchina A., Weber I. T., FEBS Lett. 1990, 269, 269–272. [DOI] [PubMed] [Google Scholar]
- 63. Gustchina A., Sansom C., Prevost M., Richelle J., Wodak S. Y., Wlodawer A., Weber I. T., Protein Eng. 1994, 7, 309–317. [DOI] [PubMed] [Google Scholar]
- 64. Tie Y., Boross P. I., Wang Y. F., Gaddis L., Liu F., Chen X., Tozser J., Harrison R. W., Weber I. T., FEBS J. 2005, 272, 5265–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wlodawer A., Gustchina A., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1477, 16–34. [DOI] [PubMed] [Google Scholar]
- 66. Wang W., Kollman P. A., Proc. Natl. Acad. Sci. USA 2001, 98, 14937–14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ghosh A. K., Sridhar P. R., Kumaragurubaran N., Koh Y., Weber I. T., Mitsuya H., ChemMedChem 2006, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- 68. Ghosh A. K., J. Med. Chem. 2009, 52, 2163–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kovalevsky A. Y., Liu F., Leshchenko S., Ghosh A. K., Louis J. M., Harrison R. W., Weber I. T., J. Mol. Biol. 2006, 363, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu F., Boross P. I., Wang Y. F., Tozser J., Louis J. M., Harrison R. W., Weber I. T., J. Mol. Biol. 2005, 354, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kovalevsky A. Y., Tie Y., Liu F., Boross P. I., Wang Y. F., Leshchenko S., Ghosh A. K., Harrison R. W., Weber I. T., J. Med. Chem. 2006, 49, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu F., Kovalevsky A. Y., Louis J. M., Boross P. I., Wang Y. F., Harrison R. W., Weber I. T., J. Mol. Biol. 2006, 358, 1191–1199. [DOI] [PubMed] [Google Scholar]
- 73. Mahalingam B., Wang Y. F., Boross P. I., Tozser J., Louis J. M., Harrison R. W., Weber I. T., Eur. J. Biochem. 2004, 271, 1516–1524. [DOI] [PubMed] [Google Scholar]
- 74. Tie Y., Kovalevsky A. Y., Boross P., Wang Y. F., Ghosh A. K., Tozser J., Harrison R. W., Weber I. T., Proteins Struct. Funct. Bioinf. 2007, 67, 232–242. [DOI] [PubMed] [Google Scholar]
- 75. Martin P., Vickrey J. F., Proteasa G., Jimenez Y. L., Wawrzak Z., Winters M. A., Merigan T. C., Kovari L. C., Structure 2005, 13, 1887–1895. [DOI] [PubMed] [Google Scholar]
- 76. Shen C. H., Wang Y. F., Kovalevsky A. Y., Harrison R. W., Weber I. T., FEBS J. 2010, 277, 3699–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miller J. F., Andrews C. W., Brieger M., Furfine E. S., Hale M. R., Hanlon M. H., Hazen R. J., Kaldor I., et al., Bioorg. Med. Chem. Lett. 2006, 16, 1788–1794. [DOI] [PubMed] [Google Scholar]
- 78. Clemente J. C., Moose R. E., Hemrajani R., Whitford L. R., Govindasamy L., Reutzel R., McKenna R., Agbandje‐McKenna M., Goodenow M. M., Dunn B. M., Biochemistry 2004, 43, 12141–12151. [DOI] [PubMed] [Google Scholar]
- 79. Chen Z., Li Y., Chen E., Hall D. L., Darke P. L., Culberson C., Shafer J. A., Kuo L. C., J. Biol. Chem. 1994, 269, 26344–26348. [PubMed] [Google Scholar]
- 80. Ghosh A. K., Gemma S., Simoni E., Baldridge A., Waters D. E., Ide K., Tojo Y., Koh Y., Mitsuya H., Bioorg. Med. Chem. Lett. 2010, 20, 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ghosh A. K., Kincaid J. F., Cho W., Walters D. E., Krishnan K., Hussain K. A., Koo Y., Cho H., Rudall C., Holland L., Buthod J., Bioorg. Med. Chem. Lett. 1998, 8, 687–690. [DOI] [PubMed] [Google Scholar]
- 82. Yoshimura K., Kato R., Kavlck M. F., Nguyen A., Maroun V., Maeda K., Hussain K. A., Ghosh A. K., Gulnik S. V., Erickson J. W., Mistuya H., J. Virol. 2002, 76, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ghosh A. K., Dawson Z. L., Mitsuya H., Bioorg. Med. Chem. 2007, 15, 7576–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ghosh A. K., Chapsal B. D., Mitsuaya H. in Aspartic Acid Proteases as Therapeutic Targets (Ed.: A. K. Ghosh), Wiley‐VCH, Weinheim, 2010, pp. 205–235. [Google Scholar]
- 85. Roberts N. A., Martin J. A., Kinchington D., Broadhurst A. V., Craig J. C., Duncan I. B., Galpin S. A., Handa B. K., Kay J., Krohn A., Lambert R. W., Merrett J. H., Mills J. S., Parkes K. E. B., Redshaw S., Ritchie A. J., Taylor D. L., Thomas G. J., Machin P. J., Science 1990, 248, 358–361. [DOI] [PubMed] [Google Scholar]
- 86. Krohn A., Redshaw S., Ritchie J. C., Graves B. J., Hatada M. H., J. Med. Chem. 1991, 34, 3340–3342. [DOI] [PubMed] [Google Scholar]
- 87. Nakanishi K., Bioorg. Med. Chem. 2005, 13, 4987–5000. [DOI] [PubMed] [Google Scholar]
- 88. Donoho A. L., J. Anim. Sci. 1984, 58, 1528–1539. [DOI] [PubMed] [Google Scholar]
- 89. Ghosh A. K., Thompson W. J., Holloway M. K., McKee S. P., Duong T. T., Lee H. Y., Munson P. M., Smith A. M., Wai J. M., Darke P. L., et al., J. Med. Chem. 1993, 36, 2300–2310. [DOI] [PubMed] [Google Scholar]
- 90. Ghosh A. K., Thompson W. J., McKee S. P., Duong T. T., Lyle T. A., Chen J. C., Darke P. L., Zugay J. A., Emini E. A., Schleif W. A., et al., J. Med. Chem. 1993, 36, 292–294. [DOI] [PubMed] [Google Scholar]
- 91. Vazquez M. L., Bryant M. L., Clare M., DeCrescenzo G. A., Doherty E. M., Freskos J. N., Getman D. P., Houseman K. A., Julien J. A., Kocan G. P., J. Med. Chem. 1995, 38, 581–584. [DOI] [PubMed] [Google Scholar]
- 92. Tung R. D., Livingston D. J., Rao B. G., Kim E. E., Baker C. T., Boger J. S., Chambers S. P., Deininger D. D., Dwyer M., Elsayed L., Fulghum J., Li B., Murcko M. A., Navia M. A., Novak P., Pazhanisamy S., Stuver C., Thomson J. A. in Protease Inhibitors in AIDS Therapy (Eds.: R. C. Ogden, C. W. Flexner), Marcel Dekker, New York, 2001, pp. 101–137. [Google Scholar]
- 93. Kim E. E., Baker C. T., Dwyer M. D., Murcko M. A., Rao B. G., Tung R. D., Navia M. A., J. Am. Chem. Soc. 1995, 117, 1181–1182. [Google Scholar]
- 94. Ghosh A. K., Kincaid J. F., Walters D. E., Chen Y., Chaudhuri N. C., Thompson W. J., Culberson C., Fitzgerald P. M., Lee H. Y., McKee S. P., Munson P. M., Duong T. T., Darke P. L., Zugay J. A., Schleif W. A., Axel M. G., Lin J., Huff J. R., J. Med. Chem. 1996, 39, 3278–3290. [DOI] [PubMed] [Google Scholar]
- 95. Ghosh A. K., Kulkarni S., Anderson D. D., Hong L., Baldridge A., Wang Y.‐F., Chumanevich A. A., Kovalevsky A. Y., Tojo Y., Amano M., Koh Y., Tang J., Weber I. T., Mitsuya H., J. Med. Chem. 2009, 52, 7689–7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Erickson J. W., Gulnik S. V., Mitsuya H., Ghosh A. K. (Fitness Assay and Associated Methods), US Patent 7470506B1, 2008.
- 97.S. De Meyer, M. Peters, Abstracts 533 and 620, 11th Conference on Retroviruses and Opportunistic Infections (CROI), February 8–11 2004, San Francisco, CA (USA).
- 98.R. Hoetelmans, I. van der Sandt, M. De Pauw, K. Struble, M. Peeters, R. van der Geest, Abstract 549, 10th Conference on Retroviruses and Opportunistic Infections (CROI), February 2003, Boston, MA (USA).
- 99. Surleraux D. L., Tahri A., Verschueren W. G., Pille G. M., De Kock H. A., Jonckers T. H., Peeters A., De Meyer S., Azjin H., Pauwels R., de Bethune M. P., King N. M., Prabu‐Jeyabalan M., Schiffer C. A., Wigerinck P. B., J. Med. Chem. 2005, 48, 1813–1822. [DOI] [PubMed] [Google Scholar]
- 100. Tie Y., Boross P., Wang Y., Gaddis L., Hussain A., Leshchenko S., Ghosh A., Louis J., Harrison R., Weber I., J. Mol. Biol. 2004, 338, 341–352. [DOI] [PubMed] [Google Scholar]
- 101. King N., Prabu‐Jeyabalan M., Nalivaika E., Wigerinck P., de Bethune M., Schiffer C., J. Virol. 2004, 78, 12012–12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Dierynck I., Keuleers I., De Wit M., Tahri A., Surleraux D., Peeters D. A., Hertogs K., Antiviral Res. 2005, 10, S71. [Google Scholar]
- 103. Lefebvre E., Schiffer C., AIDS Rev. 2008, 10, 131–142. [PMC free article] [PubMed] [Google Scholar]
- 104. Kovalevsky A., Ghosh A. K., Weber I. T., J. Med. Chem. 2008, 51, 6599–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Koh Y., Nakata H., Maeda K., Ogata H., Bilcer H. G., Devasamudram T., Kincaid J. F., Boross P., Wang Y. F., Tie Y., Volarath P., Gaddis L., Harrison R. W., Weber I. T., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother. 2003, 47, 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. De Meyer S., Azijn H., Surleraux D., Jochmans D., Tahri A., Pauwels R., Wigerinck P., de Bethune M., Antimicrob. Agents Chemother. 2005, 49, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Koh Y., Amano M., Towata T., Danish M., Leshchenko‐Yashchuk S., Das D., Nakayama M., Tojo Y., Ghosh A. K., Mitsuya H., J. Virol. 2010, 84, 11961–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. De Meyer S., Hill A., De Baere I., Rimsky I., Azijin H., Van Baelen B., De Paepe E., Vangeneugden T., et al., Antiviral Ther. 2006, 11, S73. [Google Scholar]
- 109. Wolfe C., Hicks C., HIV/AIDS 2009, 1, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Saskova K., Kozisek M., Rezacova P., Brynda J., Yashina T., Kagan R., Konvalinka J., J. Virol. 2009, 83, 8810–8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wlodwaer A., Miller M., Jaskolski M., Sathyanarayana B., Baldwin E., Weber I., Selk L., Clawson L., Science 1989, 245, 616–621. [DOI] [PubMed] [Google Scholar]
- 112. Koh Y., Matsumi S., Das D., Amano M., Davis D., Li J., Leschenko S., Baldridge A., et al., J. Biol. Chem. 2007, 282, 28709–28720. [DOI] [PubMed] [Google Scholar]
- 113. Amano M., Koh Y., Das D., Li J., Leschenko S., Wang Y. F., Boross P. I., Weber I. T., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother. 2007, 51, 2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang Y. F., Tie Y., Boross P. I., Tozser J., Ghosh A. K., Harrison R. W., Weber I. T., J. Med. Chem. 2007, 50, 4509–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Miller J. F., Furfine E. S., Hanlon M. H., Hazen R. J., Ray J. A., Robinson L., Samano V., Spaltenstein A., Bioorg. Med. Chem. Lett. 2004, 14, 959–963. [DOI] [PubMed] [Google Scholar]
- 116. Hazen R., Harvey R., Ferris R., Craig C., Yates P., Griffin P., Miller J., Kaldor I., Ray J., Samano V., Furfine E., Spaltenstein A., Hale M., Tung R., Clair M. St., Hanlon M., Boone L., Antimicrob. Agents Chemother. 2007, 51, 3147–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ford S. L., Reddy Y. S., Anderson M. T., Murray S. C., Fernandez P., Stein D. S., Johnson M. A., Antimicrob. Agents Chemother. 2006, 50, 2201–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lalezari J. R., Ward D. J., Tomkin S. A., Garges H. R., J. Antimicrob. Chemother. 2007, 60, 170–174. [DOI] [PubMed] [Google Scholar]
- 119.Corresponding press release online: “GlaxoSmithKline Discontinues Clinical Development of Investigational Protease Inhibitor Brecanavir (640 385)”. http://www.gsk.com/media/pressreleases/2006/2006_12_18_GSK945.htm.
- 120.A. K. Ghosh, J. Li, H. Mitsuya, unpublished work, Purdue University and National Cancer Institute.
- 121. Cihlar T., He G. X., Liu X., Chen J. M., Hatada M., Swaminathan S., McDermott M. J., Yang Z. Y., et al., J. Mol. Biol. 2006, 363, 635–647. [DOI] [PubMed] [Google Scholar]
- 122.C. Callebaut, K. Stray, L. Tsai, L. H. Xu, G. X. He, A. Mulato, T. Priskich, N. Parkin, et al., 20th International Conference on Antiviral Research; Palm Spring, CA, April 29 to May 3, 2007, p. 2.
- 123. Callebaut C., Stray K., Tsai L., Williams M., Yang Z., Cannizzaro C., Leavitt S. A., Liu X., Wang K., Murray B. P., Mulato A., Hatada M., Priskich T., Parkin N., Swaminathan S., Lee W., He G., Xu L., Cihlar T., Antimicrob. Agents Chemother. 2011, 55, 1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gustchina A., Weber I. T., FEBS Lett. 1990, 269, 269–272. [DOI] [PubMed] [Google Scholar]
- 125. Ghosh A. K., Martyr C. D., Steffey M., Wang Y.‐F., Agniswamy J., Amano M., Weber I. T., Mitsuya H., ACS Med. Chem. Lett. 2011, 2, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kempf D. J., Marsh K. C., Paul D. A., Knige M. F., Norbeck D. W., Kohlbrenner W. E., Codacovi L., Vasavanonda S., Bryant P., Wang X. C., Wideburg N. E., Clement J. J., Plattner J. J., Erickson J., Antimicrob. Agents Chemother. 1991, 35, 2209–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Baldwin E. T., Bhat T. N., Liu B., Pattabriaman N., Erickson J. W., Struct. Biol. 1995, 2, 244–249. [DOI] [PubMed] [Google Scholar]
- 128. Tojo Y., Koh Y., Amano M., Aoki M., Das D., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother. 2010, 54, 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ghosh A. K., Gemma S., Takayama J., Baldridge A., Leshchenko‐Yashchuk S., Miller H. B., Wang Y.‐F., Kovalevsky A. Y., Koh Y., Weber I. T., Mitsuya H., Org. Biomol. Chem. 2008, 6, 3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ghosh A. K., Chapsal B., Parham G. L., Steffey M. P., Agniswamy J., Wang Y.‐F., Amano M., Weber I. T., Mitsuya H., J. Med. Chem. 2011, 54, 5890–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ghosh A. K., Leshchenko‐Yashchuk S., Anderson D. D., Baldridge A., Noetzel M., Miller H. B., Tie Y., Wang Y.‐F., Koh Y., Weber I. T., Mitsuya H., J. Med. Chem. 2009, 52, 3902–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Koh Y., Das D., Leshchenko S., Nakata H., Ogata‐Aoki H., Amano M., Nakayama M., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother. 2009, 53, 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ghosh A. K., Gemma S., Baldridge A., Wang Y. F., Kovalevsky A. Y., Koh Y., Weber I. T., Mitsuya H., J. Med. Chem. 2008, 51, 6021–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ghosh A. K., Chapsal B., Baldridge A., Steffey M. P., Walters D. E., Koh Y., Amano M., Mitsuya H., J. Med. Chem. 2011, 54, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ide K., Aoki M., Amano M., Koh Y., Yedidi R. S., Das D., Leschenko S., Chapsal B., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother. 2011, 55, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Halgren T. A., J. Comput. Chem. 1999, 20, 730–748. [DOI] [PubMed] [Google Scholar]
- 137. Ghosh A. K., Xu C. X., Rao K. V., Baldridge A., Agniswamy J., Wang Y. F., Weber I. T., Aoki M., Miguel S. G. P., Amano M., Mitsuya H., ChemMedChem 2010, 5, 1850–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]